Submitted:
27 January 2026
Posted:
03 February 2026
You are already at the latest version
Abstract
The goal of this review is to provide a comprehensive overview of the alterations that occur in the transition from acute to chronic pain states in peripheral and central nervous structures. Chronic or persistent pain is a devastating condition for affected individuals and to the society loaded with immense costs, as it is often difficult or impossible to treat. Unlike acute pain, chronic pain is maladaptive and is usually associated with severe changes in brain structures and functions, potentially including psychiatric diseases like anxiety and depressive disorders. Unfortunately, and for still ill-known reasons, certain acute pain states tend to become chronic. As will turn out throughout this review, chronic pain and the underlying neuronal systems are highly complex. There are many reasons for this complexity, exhibiting several layers, the first sub-cellular layer not even being touched upon here. The next higher level consists of a multiplicity of involved macroscopic structures: nociceptors, various nuclei, cortices etc., which have themselves complicated internal structures with diverse cell types of different morphologies, functions and internal non-linear interactions. Third, individual neuronal nodes usually receive multiple intputs from, and send multiple outputs to, other nodes, thus creating an extended macroscopic network. Fourth, the operation of this network is modulated and changed by many diverse neuromodulators, many of which in addition work on more than one receptors. Fifth, multiple parallel effects may be exerted on anyone structure, for example, the spinal dorsal horn (DH). In brief, the pain-processing networks are multi-functional, fluid and dependent on many external influences, reflecting a general principle or “Bauplan” of the nervous system. This review adds yet another layer: the impact of multifarious diseases that disturb the workings of the networks under `normal´ conditions and promote maladaptive plasticity. It is difficult to envision the interactions between the multitudes of time- and condition-dependent factors and influences. After a characterization of chronic pain, the discussion will follow a path from the periphery to the uppermost central structures that is from the nociceptors to the cerebral cortex, integrating peripheral sensitization, central sensitization, neuroinflammatory processes, and large-scale brain reorganization.
Keywords:
chronic pain
; pain-processing structures
; peripheral and central sensitization
; maladaptive neuroplasticity
; pain-related brain networks
"Lasciate ogne speranza, voi ch'intrate“
“All hope abandon, ye who enter here“
(Dante Alighieri: La Divina Commedia, 1307-1320)
1. Introduction
Even though acute pain may appear unbearable, it is tansitory, largely treatable and offers the hope for fading away after the initial injury has healed. By contrast, chronic, persistent or frequently recurring severe pain is a devastating condition to the partient suffering from it and to the society burdened with immense costs because it is hard or impossible to treat. Chronic pain is maladaptive and is usually associated with severe changes in brain structures and functions, and often with psychiatric diseases like anxiety and depressive disorders, with the risk of suicide. Unfortunately, and for still ill-known reasons, certain acute pain states tend to become chronic. What remains to be done for researchers is to try and find out what the reasons for chronicity are and how one or the other of them might be a target of therapeutical attack.
Pain is a multi-dimensional process that may include sensory-discriminative aspects and emotional-aversive components, all of which involve activation of different brain areas and neuronal ensembles. In addition to pain, noxious stimuli may have other effects, such as vocalizations, withdrawal reflexes, vegetative (cardio-vascular, respiratory) and hormonal responses (Sandkühler 2009; Windhorst and Dibaj 2025a, 2025b).
1.1. Definition
Chronic pain is defined as pain lasting longer than the expected healing period, which is often practically defined as pain lasting longer than three to six months (Tsay et al. 2015). Chronic pain is common in developed countries. Epidemiological surveys have reported prevalence rates of 16–22 % (Zouikr et al. 2016). Chronic pain typically results from long-term inflammatory tissue damage (inflammatory pain) or from nervous tissue damage (neuropathic pain), metabolic dysfunction, pathogenic infections, cancer growth, auto-immune disorders, anti-viral treatment and chemotherapy, modulated by interactions between environmental factors, inherited genetic risk factors, sex, developmental and medical history (Kuner and Kuner 2021; Maletic and Raison 2009; Price and Ray 2019). Chronic pain syndromes cover a broad spectrum, including headache, temporo-mandibular joint pain, osteoarthritis, chronic low back pain (cLBP), fibromyalgia (FM), musculo-skeletal pain, chronic fatigue syndrome (CFS), irritable bowel syndrome (IBS), endometriosis and others (Sluka and Clauw 2016).
1.2. Manifestations, Origins, Causes, and Consequences
Risk Profiles for Chronic Pain. Despite similar pain characteristics, some people with chronic pain recover, whereas others do not. Possibly, contributions and interactions of biological, social, and psychological perturbations underlie the evolution of treatment-resistant chronic pain. Potential mechanisms that produce or exacerbate persistent pain remain relatively unclear. Different risk profiles for disease development, pain severity and chronicity may be produced in different people, contributors being factors such as genetics, age stress, environment, and immune responsivity. The neurobiology of reward and aversion may play roles, as well as alterations in synaptic complexity, neural networks and systems (e.g., opioidergic and dopaminergic (DA)) (Borsook et al. 2018).
Chronic pain manifests itself in various ways, e.g., spontaneous pain, enhanced sensitivity to painful stimuli (hyperalgesia), pain in response to normally innocuous stimuli, e.g. gentle brushing of skin (mechanical allodynia) or mild cool temperatures (cold allodynia), and aberrant referral of pain to unaffected body parts, and is associated with sensory abnormalities such as dysesthesias and paresthesias (Kuner and Kuner 2021; Tsantoulas and McMahon 2014). Chronic pain states have different pathophysiological, neurochemical and clinical characteristics, as expressed, for example, in the different susceptibility to analgesics (Apkarian et al. 2009; Costigan et al. 2009; Hunt and Mantyh 2001; Kuner and Flor 2016). For the suffering from chronic pain, motivational and emotional influences appear to be of particular importance, with an inability to extinguish the associated pain memory trace (Mansour et al. 2014). Chronic pain can drive the individual to immobility, psychological distress, depression, disruption of family relationships, potentially loss of employment, and suicide. Widespread hyperalgesia and dysfunctional endogenous pain inhibition have been identified as characteristics of many musculo-skeletal and neuropathic pain conditions. These similarities suggest common central nervous system (CNS) abnormalities in pain processing among many chronic pain conditions.
1.2.1. Hyperalgesia and Allodynia
Chronic pain patients show several deviations from normal nociception and pain sensitivity, ranging from hyperalgesia, allodynia, dysesthesias (abnormal sensations, often abnormal unpleasant perceptions of touch), paresthesias (tingling, tickling, pricking, numbness, burning, ‘pins and needles’, ‘falling-asleep’-sensations in cutaneous dermatomes) and spontaneously occurring pain (Naser and Kuner 2018).
- Allodynia
Allodynia means a pain or unpleasant sensation in response to a non-noxious stimulus, e.g., a tactile stimulus (mechanical allodynia or tactile allodynia) or mild change in temperature (usually cold) stimulus (cold allodynia) (Mills et al. 2021; Sandkühler 2009).
- Hyperalgesia
Hyperalgesia is the increased pain perception in the area of injury or exposure to a noxious stimulus, e.g., a local skin injury. In the injured zone, hyperalgesia develops to mechanical of heat stimuli (primary hyperalgesia), whereas the adjacent non-injured healthy zone shows only mechanical hyperalgesia (secondary hyperalgesia) with little heat hyperalgesia. Secondary hyperalgesia can also develop as `mirror pain´ in the respective skin area of the contralateral limb, which – together with modality dfifferenc suggest a central mechanism (Sandkühler 2009; You et al. 2022).
Hyperalgesia and to some extent allodynia may be adaptations to protect vulnerable tissues. Enhanced sensitivity may outlast the intial cause of injury. In some animal models, cellular elements required for the expression of hyperalgesia and/or allodynia include capsaicin-sensitive group IV (C) and isolectin B4 (IB4)-sensitive and vagal afferents; spinal dorsal horn (DH) neurons expressing the neurokinin-1 receptor (NK1R), microglia and astrocytes; spinal fiber tracts including the dorsal columns, anterior lateral quadrant and lateral funiculus; nuclei including the rostral ventro-medial medulla (RVM), nucleus reticularis gigantocellularis (NGc), ventro-basal (thalamus (THAL) complex, anterior cingulate cortex (ACC), ventro-lateral orbito-frontal cortex (vlOFC); and sympathetic post-ganglionic efferents. Hyperalgesia and allodynia are influenced by genotype, sex, age, and diet (Sandkühler 2009).
1.2.2. Origins
- Chronic Cutaneous Pain
Pain is a common condition in dermatology. Cutaneous nociception is altered following diseases that affect peripheral nerves innervating the skin. Some skin diseases cause pain; e.g. ulcers, pyoderma gangrenosum, herpes zoster (shingles), psoriasis or atopic dermatitis. Some cause neuropathic pain and/or pruritus, without visible primary lesions: e.g. the neuro-cutaneous diseases, including small-fiber neuropathies. Several acquired and hereditary diseases and disorders cause painful or insensate (lack of sensation) cutaneous peripheral neuropathies. Non-neuronal skin cells, particularly keratinocytes, are implicated in cutaneous nociception and peripheral neuropathies (Hayoun-Vigouroux and Misery 2022; Stucky and Mikesell 2021).
- Chronic Pruritus
Pruritus has been defined as an autonomous, pain-independent sensation, depending on itch-specific peripheral neurons, mediators, spinal neurons and cortical areas. Itch-specific receptors and afferent fibers respond to various pruritogenic mediators including histamine (HIST), substance P (SP), vasoactive intestinal peptide (VIP), calcitonin gene-related peptide (CGRP), and opioids (Metz et al. 2011). Chronic pruritus may result from primary dermatoses, systemic diseases, psychogenic pruritus, idiopathic pruritus, prurigo nodularis and/or lichen simplex chronicus, end-stage renal disease and hepato-biliary disease. In primary dermatoses, sleep disorders are a common comorbidity interrelated with pruritus, anxiety and depressive symptoms. Psychogenic pruritus, lichen simplex chronicus and some primary dermatoses are linked with personality characteristics (Ferreira and Misery 2023). Molecular mechanisms underlying pruritus, the cross-talk between the immune and nervous systems that regulate itch, and CNS pathways and projections affected by itch are being revealed (Patel and Dao 2018).
- Chronic Muscle Pain
While chronic muscle pain, like cutaneous pain, arises in nociceptor activation, the central processing of these categories differ. At the spinal level, the excitatory effects of un-myelinated group IV (C) afferents from skeletal muscle underlie strong segmental inhibition by myelinated afferent fibers, which is largely absent in the effects of cutaneous group IV (C) fibers. At the cortical level, experimental muscle pain excites other regions than does cutaneous pain. At the level of descending pain-modulating pathways, interruption of the activity in these pathways leads to higher activity of nociceptive neurons caudal to the site of interruption. The activity was higher in neurons with input from deep nociceptors than in cells mediating cutaneous nociception. All this demonstrates that at all CNS levels, the connections and processing of nociceptive information from muscle and skin differ (Mense 2003, 2004, 2008).
- Chronic Visceral Pain
Visceral pain is diffusely localized, referred to other tissues, frequently not correlated with visceral traumata, preferentially accompanied by autonomic and somatomotor reflexes, and associated with strong negative affective feelings. Together with the somatic pain sensations and non-painful body sensations, it belongs to the interoception of the body. Visceral pain is correlated with the excitation of spinal (thoraco-lumbar, sacral) visceral afferents and (with a few exceptions) not with the excitation of vagal afferents. Together with other visceral sensations and nociceptive as well as non-nociceptive somatic body sensations, visceral pain is presumably primarily represented in the posterior dorsal insular cortex (IC; primary interoceptive cortex). In primates, this cortex receives its spinal synaptic inputs mainly from lamina I tract neurons via the ventro-medial posterior nucleus of the THAL The transmission of activity from visceral afferents to second-order neurons in spinal cord is modulated in an excitatory and inhibitory way by endogenous anti- and pro-nociceptive control systems in the lower and upper brainstem, which in turn are under cortical control. Visceral pain is referred to deep somatic tissues, to the skin and to other visceral organs. This referred pain consists of spontaneous pain and mechanical hyperalgesia (Jänig 2014).
1.2.3. Causes
Several classes of hypotheses have been put forward that chronic pain results from: (i) persistent noxious signaling in the periphery; (ii) enduring maladaptive neuroplastic changes at the spinal DH and/or higher CNS structures reflecting a multiplicity of factors, including peripherally released neurotrophic factors (NFs) and interactions between neurons and microglia; (iii) compromized inhibitory modulation of noxious signaling in medullary-spinal pathways; (iv) descending facilitatory modulation; and (v) maladaptive brain re-modeling in function, structure, and connectivity (Chapman and Vierck 2017).
Genetic and Epigenetic Influences. Chronic pain is unevenly distributed among sexes, with women experiencing more pain and suffering. Genetic and epigenetic influences trigger chronic neuro-inflammatory changes, which are involved in transitioning from acute to chronic pain. Pain (and suffering) can be regarded as the consequence of an imbalance between the two ascending nociceptive pathways and the descending pain modulatory pathways (De Ridder et al. 2021). Persistent pain is associated with de novo gene expression (Khoutorsky and Price 2018). In the context of chronic post-surgical pain (CPSP), three genes involved in DA neurotransmission have been associated with variability in pain sensitivity, development of CPSP, and analgesic requirement (Van Reij et al. 2019).
Changes in Activity Patterns. Transition from acute to chronic pain entails considerable changes in brain function. Brain activations in acute pain were in areas more related to the sensory aspect of noxious stimulation, including primary somatosensory cortex (S1), IC, cingulate cortex (CC), THAL, retro-splenial cortex, and midbrain peri-aqueductal gray (PAG). On the other hand, in chronic pain models, brain activity was observed in regions commonly associated with emotion and motivation, including prefrontal cortex (PFC), anterior cingulate cortex (ACC), hippocampus (HIPP), amygdala (AMY), basal ganglia (BG) and nucleus accumbens (NAc) (Da Silva and Seminowicz 2019; Kuner and Kuner 2021). In a mouse model of neuropathic pain, the activity of pyramidal neurons in the S1 was persistently increased. This increase in activity was caused in part by increases in synaptic activity and NMDA-receptor-dependent Ca2+ discharges in apical tuft dendrites and by shifts of local inhibitory activity in favor of pyramidal neuron (Cichon et al. 2017).
Changes in Descending Pain Control. Most probably, dysfunction in descending pain modulatory systems may contribute to pain chronification. Disruption of the balance of descending modulatory circuits to favor facilitation may promote and maintain chronic pain (Ossipov et al. 2014). Cortical-spinal top-down facilitation, including those relayed through brainstem neurons, powerfully controls nociceptive transmission in the spinal cord. The brainstem-spinal descending facilitation may promote chronic pain (Zhuo 2017).
1.2.4. Chronic Inflammatory Pain (with Case Report)
This pain type is a response to transient and chronic inflammation evoked by tissue damage of various origins. The inflammatory tissue reaction involves increased vascular permeability, leukocyte infiltration, glia-cell activation and the production of inflammatory mediators such as protons (H+), SP, prostaglandins, bradykinin, serotonin (5-HT), HIST, tumor necrosis factor (TNF), interleukin-1 (IL-1), interleukin-6 (IL-6), interleukin-1 (IL-1), NFs, nitric oxide (NO) that cause vasodilation as well as oxygen free radicals and lysosomal enzymes which are related to tissue injury, and other endogenous chemicals (Binshtok et al. 2008; Costigan et al. 2009; Dibaj et al. 2024; Gebhart 2009; Hucho and Levine 2007; Julius and Basbaum 2001; Nicol and Vasko 2007; Pezet and McMahon 2006; Ren and Dubner 2007; Scholz and Woolf 2002, 2007; Stein et al. 2009; Wang et al. 2006). Many inflammatory chemical agents excite nociceptors and/or modulate sensory receptor channels and voltage-gated ion channels. Conversely, nociceptive afferents contribute to inflammation. Inflammatory pain supports healing and tissue repair by promoting immobility and rest (Costigan et al. 2009; Gao et al. 2022).
Neuroinflammation. Activation of glial and immune cells leads to increased production of pro-inflammatory mediators. Neuro-inflammation is a fundamental mechanism in the genesis of acute pain and its transition to neuropathic and chronic pain. A noxious event that stimulates peripheral afferent nerve fibers may also activate pro-nociceptive receptors situated at the dorsal-root ganglion (DRG) and DH, as well as peripheral glial cells, setting off the so-called peripheral sensitization and spreading neuro-inflammation to the brain. Once activated, microglia produce cytokines, chemokines, and neuropeptides that can increase the sensitivity and firing properties of second-order neurons, increasing nociceptive signals to the cerebral cortex. Immune-neuronal interactions are also implicated in the complex regulatory relationship between pain and opioids. Activated immune and glial cells may alter neuronal function, induce and maintain pathological pain, and disrupt the analgesic effects of opioids by contributing to the development of tolerance and dependence, even causing paradoxical hyperalgesia. Such alterations may occur when the neuronal environment is impacted by trauma, inflammation, and immune-derived molecules, or when opioids induce pro-inflammatory glial activation (Echeverria-Villalobos et al. 2023).
Microglia. Depending on the challenges, the microglia reaction varies between activated-toxic-neuro-inflammatory to non-activated-protective-tissue re-modeling. Increased inflammatory reactions result from brain damage, such as stroke, encephalitis, as well as chronic dysfunctions, including pain and stress (Hoffmann and Beyer 2020). Microglia may directly contribute to altering pain circuits and synaptic re-modeling, or indirectly contribute to neuroplasticity through property changes, including the secretion of growth factors. Activated microglia release and respond to various chemokines and cytokines, which regulate neuro-inflammation and mediate chronic neuropathic pain (CNP). The mechanisms underlying neuroplasticity can occur in the somatosensory circuit of the spinal DH, THAL and cortex (Hiraga et al. 2022). Central sensitization can be associated with changes in membrane excitability, synaptic plasticity and inhibition of neurons. A main underlying mechanism is the activation of the N-methyl-D-aspartate receptors (NMDARs) for glutamate. This activation increases synaptic efficiency and causes Ca2+ influx. This process in turn also relies on NA-β ligand, calcitonin gene-related peptide (CGRP), brain-derived neurotrophic factor (BDNF), and SP (Cui et al. 2023).
- Case Report: Transition from Acute Inflammatory Pain to Chronic Pain
Case Presentation: A 46-year-old female office worker presented with persistent pain of the right knee lasting 18 months. The pain initially developed after a minor twisting injury during recreational jogging. Magnetic resonance imaging (MRI) at the time of injury showed a small medial meniscal tear without ligamentous damage. Arthroscopic partial meniscectomy was performed four weeks later. Postoperative wound healing was uncomplicated, and no signs of infection or structural instability were observed. Despite adequate postoperative rehabilitation, the patient reported increasing pain intensity beginning approximately six weeks after surgery. Pain was described as deep, burning, and aching, accompanied by intermittent sharp exacerbations. Pain intensity averaged 7/10 on the numeric rating scale (NRS), was present at rest, and was aggravated by minimal mechanical stimuli such as light touch or clothing contact around the knee.
Clinical Manifestations:Hyperalgesia and Allodynia: On examination, the patient exhibited pronounced primary mechanical and thermal hyperalgesia localized to the peri-articular region, as well as secondary mechanical hyperalgesia extending to the ipsilateral thigh and calf. Gentle brushing of the skin evoked pain (mechanical allodynia), and exposure to mildly cool surfaces triggered unpleasant pain sensations (cold allodynia). Mirror pain phenomena were observed, with increased sensitivity to pinprick stimuli over the contralateral knee. The patient also reported spontaneous pain episodes without identifiable triggers, as well as dysesthetic sensations described as “electric buzzing” and “burning pressure,” consistent with abnormal nociceptive processing.
Functional and Psychosocial Impact: The persistent pain led to progressive reduction in physical activity and avoidance of weight-bearing tasks. The patient developed fear-avoidance behavior, discontinued exercise, and reported sleep disturbances. Psychological assessment revealed moderate depressive symptoms, pain catastrophizing, and heightened stress related to occupational demands and family responsibilities. She reported feelings of helplessness and a perceived inability to “turn off” the pain, suggesting impaired extinction of pain-related memory traces. There was no prior psychiatric history.
Diagnostic Evaluation: Repeated imaging showed no progression of structural pathology. Laboratory parameters, including inflammatory markers, were within normal limits. Quantitative sensory testing demonstrated reduced mechanical and thermal pain thresholds locally and at remote sites, indicating widespread pain sensitization. Conditioned pain modulation testing revealed deficient endogenous pain inhibition.
Pathophysiological Interpretation: The clinical course suggests a transition from acute inflammatory pain to chronic pain, driven by persistent neuroinflammatory and neuroplastic mechanisms rather than ongoing peripheral tissue damage. The initial tissue injury and surgery likely triggered a local inflammatory response involving immune cell infiltration and release of pro-inflammatory mediators (e.g., prostaglandins, cytokines, bradykinin), leading to peripheral sensitization of nociceptors. Continued nociceptive input, combined with individual risk factors — female sex, psychosocial stress, and heightened affective vulnerability — may have facilitated the development of central sensitization. At the spinal level, sustained afferent input plausibly induced NMDA receptor–dependent synaptic plasticity, glial activation, and loss of inhibitory control in the DH. Supraspinally, maladaptive re-organization within pain-related networks — including the ACC, IC, AMY, and PFC — likely contributed to enhanced pain perception, emotional distress, and impaired descending inhibition. The presence of mirror pain and widespread hyperalgesia further supports a dominant central mechanism involving altered connectivity and excitability across distributed CNS structures.
Clinical Course and Treatment Response: Treatment with non-steroidal anti-inflammatory drugs and opioids provided minimal relief. Gabapentinoids partially reduced spontaneous pain but had limited effect on mechanical allodynia. Multimodal therapy combining graded physical rehabilitation, cognitive-behavioral therapy, and antidepressant medication resulted in modest functional improvement but persistent pain.
Discussion: This case illustrates how chronic inflammatory pain can outlast its initial peripheral cause and become a self-sustaining pathological state. It exemplifies the interaction between peripheral inflammation, neuro-immune activation, maladaptive plasticity, and psychosocial factors in shaping chronic pain phenotypes. Despite resolution of tissue injury, alterations in nociceptive processing at spinal and supraspinal levels maintained pain and disability. Importantly, the case highlights individual risk profiles that predispose to pain chronicity, including biological vulnerability, stress exposure, impaired endogenous pain inhibition, and affective dysregulation. These features align with growing evidence that chronic pain reflects a disorder of distributed neural networks rather than a persistent peripheral lesion.
Conclusion: This case underscores the complexity of chronic pain as a maladaptive neurobiological condition emerging from inflammatory injury. It demonstrates how hyperalgesia, allodynia, spontaneous pain, and emotional distress can arise from persistent alterations in nociceptive processing and CNS function, reinforcing the need for mechanism-based, individualized therapeutic strategies.
1.2.5. Chronic Neuropathic Pain (CNP) (with Case Report)
CNP affects some 7-10% of the general population globally, predominantly in patients above 50 years of age (Cui et al. 2023; Saadé and Jabbur 2008; Szok et al. 2019).
- Causes
The IASP classification of CNP contains: CNP after peripheral nerve injury, painful polyneuropathy, post-herpetic neuralgia, painful radiculopathy, trigeminal neuralgia (TN; tic douloureux), spinal cord injury (SCI), brain injury, post-stroke pain, multiple sclerosis (MS) (Szok et al. 2019). CNP may also arise from tumor invasion, or toxic, neuro-degenerative or metabolic diseases (e.g., from chemotherapy, diabetic neuropathy, alcoholism) and can be accompanied by neuropathic itch, e.g., in post-herpetic states or small-fiber neuropathy (Borsook 2012; Cao and DeLeo 2009; Cevikbas and Lerner 2020; Cui et al. 2023; Dibaj and Schomburg 2017; Nadrigny et al. 2017; Saab et al. 2008; Saadé and Jabbur 2008; Windhorst and Dibaj 2023). Specifically, peripheral neuropathic pain can be caused by many insults that directly affect peripheral sensory neurons, including mechanical trauma, metabolic imbalance (e.g., diabetes), auto-immune diseases, chemotherapeutic agents, viral infections (e.g., shingles). These insults cause acquired neuropathies such as small-fiber neuropathies, diabetic neuropathy, chemotherapy-induced peripheral neuropathy, and post-herpetic neuralgia. Peripheral neuropathic pain can also be caused by genetic factors and result in hereditary neuropathies that include Charcot-Marie-Tooth disease, rare channelopathies and Fabry disease (Stucky and Mikesell 2021).
- Symptoms
Negative symptoms of CNP include numbness, weakness, and loss of deep tendon reflexes in the affected neural area. Positive symptoms of CNP include spontaneous pain or burning, bursts of `pins and needles´, stimulus-dependent pain (e.g., excruciating pain when clothes touch the skin), and other symptoms such as paresthesia, dysesthesia, allodynia, hyperalgesia or hyperpathia (Cerveco 2009; Cui et al. 2023). Post-traumatic neural damage may also lead to more extensive complex regional pain syndromes (CRPSs) characterized by sensory disturbances, skeletal motor and automomic nervous system (ANS) dysfunctions. Neuropathic pain is often associated with affective mood disorders, anxiety, depression and insomnia (Baron 2006; Borsook 2012; Ji et al. 2019; Yalcin et al. 2014; Wasner et al. 2003).
- Mechanisms
No consensus has been reached on how CNP occurs or develops. Certain is, however, that there is no single, but several, mechanisms that contribute to CNP, even in the same patient or affected animal. CNP arises consequent to nerve injury either of the peripheral nervous system (PNS) or CNS. Following peripheral nerve injury, a cascade of events in the primary afferents leads to peripheral sensitization resulting in spontaneous nociceptor activity, decreased threshold and increased response to supra-threshold stimuli (Ren and Dubner 2010; Scholz and Woolf 2007). Initially, nervous-tissue damage induces neuro-inflammation that activates microglia such as Schwann cells (Saab et al. 2008). The accumulation of inflammatory cells in injured nerves could contribute to the early occurrence of peripheral neuropathic pain. Further mechanisms may be peripheral sensitization, central sensitization, dysfunction in descending nociceptive modulatory systems, oxidative stress response and the activation of glia cells. Moreover, neuropathic pain may reflect central sensitization, as a result of mitochondrial dysfunction induced by oxidative and nitrosative stress, as well as inflammatory signals and the overload in intracellular Ca2+, in which transient receptor potential (TRP) channels may play a role. Thus, it has been suggested that neuropathic pain could be a consequence of the imbalance between reactive oxygen species (ROS) and endogenous anti-oxidants (Carrasco et al. 2018). CNP has also been linked to psychosocial factors (Cui et al. 2023).
- Neuroplasticity
In neuropathic pain, alterations of neuronal function can be a direct consequence of nerve damage or a result of neuroplasticity secondary to the damage to tissues or to neurons. A series of molecular changes in the spinal cord and in brain centers are associated with central sensitization, which is responsible for the pain to non-injured extra-territory regions (extra-territorial pain) and contralateral parts (mirror-image pain). The peripheral nerve injury induces neuroplastic changes in different brain regions including the ACC, IC, vlOFC, AMY, striatum, THAL, hypothalamus (HYP), locus coeruleus (LC), red nucleus, PAG and RVM (Jaggi and Singh 2011).
- Case Report: Chronic Peripheral Neuropathic Pain with Central Sensitization
Case Presentation: A 62-year-old woman was referred to a tertiary pain clinic with a 4-year history of persistent neuropathic pain affecting both feet and distal lower legs. The pain began gradually following a 15-year history of type 2 diabetes mellitus complicated by poor glycemic control. There was no history of traumatic nerve injury, spinal cord disease, or malignancy. The patient described continuous burning and electric shock–like pain, rated 7–9/10 on a numerical rating scale, accompanied by paroxysmal stabbing sensations, especially at night. She also reported severe mechanical allodynia, stating that even light contact with bed sheets provoked excruciating pain. Intermittent paresthesias (“pins and needles”) and dysesthesias were present in a stocking distribution.
Clinical Examination: Neurological examination revealed:
- Reduced pinprick and temperature sensation in a bilateral stocking distribution
- Preserved vibration sense proximally
- Absent ankle reflexes
- Marked dynamic mechanical allodynia to light brushing
- Secondary hyperalgesia extending beyond the initially affected dermatomes
Motor strength was intact. Autonomic symptoms included episodic foot discoloration, hyperhidrosis, and cold intolerance, suggesting involvement of sympathetic post-ganglionic efferents.
Diagnostic Workup: Nerve conduction studies were affected. Quantitative sensory testing demonstrated lowered heat-pain thresholds and exaggerated pain responses to suprathreshold mechanical stimuli. MRI of the spine excluded compressive radiculopathy. Laboratory testing (inclusive cerebrospinal fluid (CSF) testing) ruled out vitamin deficiencies, autoimmune disease, and paraproteinemia.
Pain Phenotype and Classification: The patient was diagnosed with chronic peripheral neuropathic pain due to painful diabetic polyneuropathy. The clinical picture was dominated by:
- Positive neuropathic symptoms (burning pain, electric shocks, allodynia)
- Negative symptoms (thermal hypoesthesia)
- Evidence of central sensitization, indicated by mirror-image pain and extraterritorial hyperalgesia
Pathophysiological Considerations: The patient’s pain was interpreted as resulting from multiple interacting mechanisms, consistent with contemporary models of CNP:
- Peripheral sensitization, driven by metabolic injury to nociceptive fibers, resulting in spontaneous activity and reduced activation thresholds.
- Neuroinflammation, initiated by nerve injury and sustained by immune–neuronal interactions involving Schwann cells and macrophages, leading to persistent peripheral input.
- Central sensitization, reflected by exaggerated pain responses, expanded receptive fields, and allodynia, likely mediated by NMDA-receptor–dependent synaptic plasticity in spinal DH neurons.
- Glial activation, with microglial and astrocytic release of cytokines and neurotrophic factors contributing to altered excitability and maintenance of pain states.
- Maladaptive neuroplasticity, extending to supraspinal structures including the THAL, ACC, AMY, and PFC, reinforcing the emotional–motivational dimension of pain
Psychosocial and Affective Comorbidities: The patient reported sleep fragmentation, depressive symptoms, anxiety, and social withdrawal. She expressed fear of movement due to anticipated pain and demonstrated pain catastrophizing during clinical interviews. These affective and cognitive factors were considered to amplify pain perception via dysfunctional descending pain modulation and altered meso-limbic processing.
Treatment Course and Outcome: The patient had failed multiple first-line treatments, including gabapentinoids, 5-HT–NA re-uptake inhibitors, and topical lidocaine. Opioid therapy was avoided due to limited efficacy. A multimodal treatment approach was initiated, combining optimized metabolic control, low-dose tricyclic antidepressants, cognitive-behavioral therapy, and graded physical activity. Although pain intensity decreased modestly, allodynia and spontaneous pain persisted, consistent with treatment-resistant CNP driven by central mechanisms.
Discussion: This case illustrates how CNP emerges from the interaction of peripheral nerve injury, immune activation, and central neuroplastic changes. Despite the resolution of acute injury mechanisms, pain persisted due to enduring alterations in neuronal excitability, synaptic transmission, and brain network connectivity. The coexistence of sensory, affective, and autonomic symptoms highlights the multi-dimensional nature of chronic neuropathic pain, as well as the limitations of purely peripheral or pharmacological treatment strategies.
Conclusion: The present case exemplifies a chronic neuropathic pain syndrome characterized by persistent positive sensory symptoms, central sensitization, neuro-inflammatory mechanisms, and maladaptive neuroplasticity. It underscores the need for integrative treatment approaches addressing not only peripheral nerve pathology but also central pain processing and psychosocial dimensions of suffering.
1.2.6. Peripheral and Central Sensitization
Increased pain sensitivity is a characteristic of chronic pain and may develop through peripheral mechanisms (peripheral sensitization) or consequent to neuroplastic changes in the CNS (central sensitization), or both (Staud 2012).
Peripheral Sensitization refers to changes in nociceptor sensitivity and responses to noxious stimuli manifesting as reduced thresholds, increased supra-threshold responses to stimuli in their receptive fields, and spontaneous activity (Saab 2012; Sandkühler 2009; Treede 2016). A plethora of molecular mechanisms may be involved. Peripheral sensitization requires persistent nociceptive inputs that can trigger a prolonged but reversible increase in the excitability and synaptic efficacy of neurons in central nociceptive pathways (Finnerup et al. 2021).
Central Sensitization is defined as an increased responsiveness of CNS nociceptive neurons to their normal or sub-threshold afferent input (Treede 2016). It manifests as pain hypersensitivity, particularly dynamic tactile allodynia, secondary punctate or pressure hyperalgesia, after-sensations, and enhanced temporal summation. It can be readily and rapidly elicited in human volunteers by diverse experimental noxious conditioning stimuli to skin, muscles or viscera, and, in addition to producing pain hypersensitivity, entails secondary changes in brain activity. Changes in pain sensitivity have been interpreted as contributions of central sensitization in patients with neuropathic pain, visceral pain hypersensitivity disorders, osteoarthritis, musculo-skeletal disorders with generalized pain hypersensitivity, temporo-mandibular joint disorders, dental pain, headache, FM, and post-surgical pain (Woolf 2011).
Loci. Pain-related sensitization has been recorded in many CNS regions involved in pain processing. A selection of such regions receiving nociceptive inputs and modulating nociceptive processing is depicted in Figure 1 and Figure 2, respectively. Central sensitization is an over-excited state of the CNS and occurs when intense nociceptive stimulation induces CNS neurons to respond to normal or subliminal afferent signals. During central sensitization, nociceptive sensory neurons can produce or increase spontaneous activity, decrease the threshold to peripheral stimulation that can activate neurons, increase the response to supra-threshold stimulation and enlarge the receptive field. In this process, the nociceptive-specific (NS) neurons are converted into wide-dynamic-range (WDR) neurons that can respond to both nociceptive and non-nociceptive stimuli, their response to repeated non-nociceptive stimuli gradually increasing and their receptive field expanding (Cui et al. 2023). The mechanisms underlying central hypersensitivity are complex and manifold, and range from channelopathies to dysfunctional neuronal networks to involvement of the immune system (Alles and Smith 2018; Saab 2012).
Excitability. Central sensitization can be associated with changes in membrane excitability, synaptic plasticity and inhibition of neurons. It can also increase neuronal function while creating loops in pain pathways to cause pain. A main underlying mechanism is the activation of the NMDARs for glutamate. This activation increases synaptic efficiency and causes Ca2+ influx. This process also relies on NA-β ligand, CGRP, BDNF, and SP (Cui et al. 2023).
1.2.7. Neuroplasticity
Neuroplasticity is an important, but fairly general notion because it encompasses a variety of processes, from changes in neuronal networks and their internal and external connections, in cell properties, in synaptic processes to neurotransmitters and neuromodulators, influenced by genetic predispositions and epigenetic factors. Various forms of nociceptive system dysfunction and circuitry plasticity result in heightened circuit activity, such as central sensitization, synaptic plasticity, homeostatic plasticity, and excitation/inhibition balance (Chen and Tang 2024).
The brain network dealing with nociception and pain might contribute to the transition from acute pain to chronic pain. Chronic pain is associated with an extensive re-organization in brain activity such as alterations in cortical thickness, gray matter density and activity in several brain regions, including somatosensory, motor, IC, and PFC, as well as in the THAL, AMY, BG, and the HIPP (Labrakakis 2023; Ong et al. 2019). In neuropathic pain, alterations of neuronal function can be a direct consequence of nerve damage or a result of neuroplasticity secondary to the damage to tissues or to neurons.
- Human Brain Imaging
Neuroimaging methods, e.g., magneto-encephalography (MEG), positron emission tomography (PET) and functional magnetic resonance imaging (fMRI), may provide non-invasive means to reveal pain-related neural structures, and specific pain modulation mechanisms within the somatosensory (diffuse noxious inhibitory controls, acupuncture, movement), affective (depression, anxiety, catastrophizing, stress) and cognitive (anticipation/placebo, attention/distraction, hypnosis) domains. Results of imaging studies are complex reflecting activation or de-activation in numerous brain areas. A number of pain-control mechanisms include the PAG, which is one area that is consistently activated across the majority of pain mechanisms. Activity in RVM relays descending modulation from the PAG, and also occurs both during acupuncture analgesia and anxiety-induced hyperalgesia. Other brain areas involved in a number of mechanisms are the PFC, orbito-frontal cortex (OFC), ACC, and NAc in the BG (Knudsen et al. 2018).
- Central Nervous System (CNS) Alterations
Persistent inflammation as well as peripheral nerve and spinal cord injuries, along with other painful syndromes such as FM, diabetic neuropathy, chemotherapeutic neuropathy, TN, CRPS, and/or IBS, cause several neuroplasticity changes along its entire neuraxis affecting the different neuronal nuclei, including the supraspinal structures that are involved in the processing and modulation of pain, including the primary and secondary somatosensory cortex (S1, and S2, respectively), motor cortex, PFC, HIPP, septum, AMY, CC, and BG, THAL, habenula (Hb), HYP, red nucleus, cerebellum, LC, PAG, and RVM. Alteration of gray matter, changes in dendritic spines, neural circuit re-modeling, and up-regulation of pro-inflammatory mediators (e.g., cytokines) by re-activation of astrocytes and microglial cells are the main functional, structural, and molecular neuroplasticity changes observed in the above supraspinal structures, associated with pathological pain. Hyperexcitability of neuronal structures is caused by modification of postsynaptic receptor expression, and potentiation of presynaptic delivery of neurotransmitters, as well as the reduction of inhibitory inputs (Boadas-Vaello et al. 2017).
- Structural Alterations
Functional and structural alterations of various kinds in association with inflammatory and neuropathic pain occur in numerous supraspinal structures, such as the PAG, RVM, nucleus ruber (NR), midbrain DA neurons, LC, cerebellum, HYP, THAL, HIPP, BG, AMY, somatosensory cortices, motor cortex, PFC, ACC, IC, and Hb (Boadas-Vaello et al. 2017; Costigan et al. 2009; Kuner and Flor 2016; Mitsi and Zachariou 2016; Saab 2012; Saadé and Jabbur 2008; Xiao and Zhang 2018). Chronic pain is associated with structural alterations including reduction (less often increase) in gray matter in patients suffering from persistent pain (Brodal 2017). Brain volume decreased following pain induction in a number of brain regions that form a part of the nociceptive network as well as meso-limbic pathways (Da Silva and Seminowicz 2019; Kuner and Kuner 2021). The affected regions depend on the pain syndromes investigated and the methods used (Bushnell et al. 2013; Doan et al. 2015; Ong et al. 2019; Yang and Chang 2019). Furthermore, there are changes in dendritic and synaptic structure, changes in neurochemistry, inflammatory responses, long-term plastic changes and functional re-organization in the brain (Bushnell et al. 2013; Kuner and Flor 2016; Saab 2012).
- Changes in Connectivity
There are also changes in connectivity betwee brain regions. In humans, an acute, transient and moderately noxious stimulus evokes blood-oxygen-concentration-dependent responses in regions including the THAL, S1, PFC, ACC, IC, and cerebellum. Pathological pain conditions are associated with sensitization along this circuit and dysfunctional connectivity (Saab 2012). In chronic pain patients, the connectivity between the PFC and HIPP decreased, consistent with the de-activation of the medial PFC (mPFC) as well as deficits in working memory. On the other hand, in an animal neuropathic pain model, an electro-encephalographic (EEG) study showed increased coherence between S1 and PFC at a late, but not early stage of pain, suggesting that chronic pain increases the connectivity between regions related to sensory-discriminative (S1) and negative-aversive (PFC) dimensions of pain. In rats with neuropathic pain, allodynia-related brain activity did not depend on area S1 and instead involved the NAc and PFC. The NAc acquired a more prominent role in connectivity, consistent with the hypotheses on emerging meso-limbic dominance over pain chronicity. But a cold stimulus eliciting cold allodynia (a debilitating problem in neuropathic pain), evoked sustained changes in multiple brain areas, including the THAL, the somatosensory and cingulate cortices, and the PAG over the course of chronic cold allodynia. Hence, there are individual points of divergence (Da Silva and Seminowicz 2019; Kuner and Kuner 2021). Changes in connectivity between nociceptive afferents and brain regions are illustrated by c-fos markers in rats to study the effects of chronic inflammatory pain in the spinal DH (laminae I-V) and supraspinal pain-control centers intrinsically connected with the DH [caudal ventro-lateral medulla (CVLM), nucleus raphé dorsalis (NRD), ventral reticular nucleus (VRt), nucleus tractus solitarii (NTS), RVM] (Pinto et al. 2007). In a mouse model of neuropathic pain, the activity of pyramidal neurons in the S1 was persistently increased. This increase in activity was caused in part by increases in synaptic activity and NMDA-receptor-dependent Ca2+ discharges in apical tuft dendrites and by shifts of local inhibitory activity in favor of pyramidal neuron hyper-activity (Cichon et al. 2017).
- Immune System Involvement
Neuroplasticity relevant to chronic pain is modulated by microglia. Microglia are immune cells resident in the CNS, which survey the brain parenchyma for pathogens, initiate inflammatory responses, secrete inflammatory mediators, and phagocyte debris. In addition, they contribute to the regulation of brain ion homeostasis and to pruning synaptic contacts. Central sensitization involves: up-regulation of sensory neuron-specific Na+ channels; change in NMDA and transient receptor potential vanilloid (TRPV) receptors; phenotype switching of large myelinated axons; axon sprouting within the DH; and loss of inhibitory interneurons (INTs) (Staud 2012).
- Cognitive and Affective Changes
Many of the altered brain regions and networks in chronic pain patients are not only involved in pain processing, but also in other sensory and particularly cognitive tasks. MRI studies have provided information on the association of brain alterations with pain catastrophizing, fear-avoidance, anxiety and depressive symptoms. Pain catastrophizing is related to brain areas involved in pain processing, attention to pain, emotion and motor activity, and to reduced top-down pain inhibition. In contrast to pain catastrophizing, there are no clear associations with brain characteristics of anxiety and depressive symptoms. All cognitive or emotional factors show significant associations with data from resting-state fMRI, indicating that even at rest the brain reserves a certain activity for these pain-related factors (Malfliet et al. 2017).
2. The Functional Structures of Chronic Pain
- Peripheral Sensitization
Chronic pain is associated with alterations in the sensory functions of nociceptive afferents, their central connections and subsequent processing stages. In response to injury, resident immune cells are activated and blood-borne immune cells are recruited to the injury site. Immune cells not only contribute to immune protection but also initiate the sensitization of peripheral nociceptors (Ren and Dubner 2010). Changes in nociceptor functions have been well studied in inflammatory pain and involve interactions with the immune cells including monocytes, lymphocytes, leukocytes, macrophages, T cells and mast cells which not only signal other components of the immune system, but also the brain to control the inflammatory response and to maintain its homeostasis. Involved are a plethora of pro-inflammatory cytokines including interleukins IL-1β, IL-6, IL-8, IL-12, interferon-α (IFN-α) and IFN-δ as well as tumor necrosis factor α (TNF-α). Pro-inflammatory cytokines released by immune cells induce hyperalgesia when administered at the peripheral or central level (Zouikr et al. 2016) (Figure 1).
2.1. Changes in Nociceptors
Afferent nociceptive fibers can be divided into several group IV (C) sub-types: non-peptidergic NP1-3, peptidergic PEP1, TH [corresponding to low-threshold group IV (C) mechano-receptors]. Many CGRP-positive are thinly myelinated group III (Aδ) fibers (PEP2). Peptidergic afferents innervate superficial DH layers I and outer II (Iio). Non-peptidergic afferents innervate the dorsal portion of the inner lamina II (IIi). Nociceptive groups III (Aδ) and IV (C) contact projection neurons in laminae I-II, most of them being nociceptive-specific (Artola et al. 2020).
- Cutaneous Nociceptors
Cutaneous nociceptors can be excited or modulated by „A plethora of painful molecules“ (Lewin et al. 2004), as detailed below).
- Muscle Nociceptors
Muscle nociceptors can be sensitized to chemical and mechanical stimuli. The sensitization is caused by endogenous algesic substances binding to highly specific receptor molecules in the membrane of the nociceptive ending. For example, animal studies showed that 5-HT sensitizes muscle nociceptors to chemical and mechanical stimuli. In humans, 5-HT combined with bradykinin to induce muscle hyperalgesia to pressure. The sensitization process by endogenous substances that are likely to be released during trauma or inflammatory injury is probably the best established peripheral mechanism for muscle tenderness and hyperalgesia (Graven-Nielsen and Mense 2001).
Nociceptors have afferent fibers in groups III (Aδ) and IV (C). Sub-populations in these groups react to noxious stimuli or metabolic changes or mechanical events, with these properties being differentially distributed between the groups. These different sub-populations may therefore subserve not only nociception and pain perception. The first role played by group III and group IV afferent fibers from skeletal muscle is to transmit nociceptive information from muscle to the CNS. The second role is to induce cardio-vascular and respiratory adjustments during muscular exercise (Decherchi et al. 2004). Moreover, in both healthy and pathological populations, group III and IV muscle afferents exert actions on central motor drive during physical exercise, possibly contributing to improve muscle performance by regulating the peripheral fatigue development and by avoiding excessive muscle impairments (Laurin et al. 2015).
A sub-population of muscle free nerve endings in cats is sensitive to mechanical events, such as muscle contraction and stretch. Afferents were excited only by large stretches that produced significant passive force. Isometric contraction produced by electrical stimulation of the muscle nerve consistently excited free nerve endings. Stimulation of free nerve endings by squeezing the Achilles tendon exhibiting the clasp-knife reflex evoked powerful, homonymous inhibition and a flexion-withdrawal pattern of reflex action -- that is, inhibition of extensor and excitation of flexor muscles throughout the hindlimb. Intra-thecal application of capsaicin, which preferentially blocks the reflex actions of small afferent fibers, blocked clasp-knife inhibition in decerebrated, dorsal hemisectioned cats (Cleland et al. 1990). -- In cats, spinal INTs that were excited by squeezing the Achilles tendon or manipulation of the muscle surfaces were extracellularly recorded in lamina V-VII of the L5-S1 spinal cord. INTs were uniformly excited by increases in muscular length and force. Responses to isometric contraction induced by electrical stimulation of motor axons was prolonged after contraction. For similar increases in force, stretch evoked greater excitation than contraction, indicating that both stretch and contraction contributed to interneuronal activity. INTs were also excited by cutaneous receptors and only occasionally by primary muscle spindle or Golgi tendon organ (GTO) afferents, which suggests that activation of muscular free nerve endings mediated the interneuronal responses to stretch and contraction. A small number of INTs were only weakly excited by muscular free nerve endings but strongly excited by group I afferents (Cleland and Rymer 1993). Hence, INTs in deep DH laminae V-VII receive inputs from muscle free nerve endings.
A sub-population of cutaneous group IV (C tactile, CT) afferents senses gentle or pleasant or affective touch (brushing, stroking) and sends the signals to the contralateral IC with a somatotopical projection similar to noxious and cooling stimuli (Björnsdotter et al. 2009).
- Visceral Nociceptors
Visceral pain is correlated with the excitation of spinal (thoraco-lumbar, sacral) visceral afferents and (with a few exceptions) not with the excitation of vagal afferents. Spinal visceral afferents are polymodal and activated by adequate mechanical and chemical stimuli. All groups of spinal visceral afferents can be sensitized (e.g., by inflammation). Silent mechano-insensitive spinal visceral afferents are recruited by inflammation. Spinal visceral afferent neurons project into the laminae I, II (outer part IIo) and V of the spinal DH over several segments, medio-lateral over the whole width of the DH and contralateral. Their activity is synaptically transmitted in laminae I, IIo and deeper laminae to viscero-somatic convergent neurons that receive additionally afferent synaptic (mostly nociceptive) input from the skin and from deep somatic tissues of the corresponding dermatomes, myotomes and sclerotomes (Jänig 2014).
- Peripheral Plasticity
During chronic pain, the sensory functions of nociceptive afferents, their central connections and subsequent processing stages are altered (Finnerup et al. 2021). These alterations produce plastic changes in nociceptive signal transmission and processing on short- to long-term time scales, and create a pain memory (Price and Inyang 2015). The sensitivity of nociceptors in skin and deep tissues can increase under a variety of pathophysiological conditions, such as tissue injury and inflammation. Increased sensitivity manifests itself as lowering of receptor threshold, increased and more sustained responsiveness to supra-threshold stimuli and expansion of receptive fields. Tissue injury or nociceptive stimulation can induce the release of a large number of chemicals from non-nerve cells and primary afferent terminals in local tissues, which participate in the activation and sensitization of nociceptors. Such agents include arachidonic acid metabolites, 5-HT, bradykinin, nerve growth factor (NGF), nucleotides, and others. It appears that sensitization of nociceptive group III (A) and group IV (C) fiber endings seldom outlasts the primary cause of pain and is confined to the area of injury. The persistence of pain thus requires enduring central changes in the processing of nociceptive signals (Cui et al. 2023; Sandkühler 2009).
- Neuromodulation
The activity of nociceptive afferents can be modulated during pain.
In behavioral and electrophysiological experiments, oxytocin (OXT) has been shown to be a mediator of endogenous analgesia. OXT receptors (OXTRs) in the spinal DH participate in a selective inhibition of the neuronal activity mediated by group III (Aδ) and group IV (C) fibers but not group II (Aβ) fibers. OXTRs are expressed in the terminal nerve endings and are able to inhibit nociceptive neuronal firing. Local peripheral OXT blocked the first sensorial activity of group III (Aδ) and group IV (C) fibers recorded in the spinal neurons. In formalin behavioral nociceptive tests, only ipsilateral OXTR activation inhibited pain behavior. This was reinforced by the fact that the OXTR protein is expressed in the sciatic nerve. Immuno-fluorescence of primary afferent fibers suggests that OXTRs could be located in nociceptive-specific terminals of the skin. Hence, OXTRs could be found in nociceptive terminals and on activation, they were able to inhibit nociceptive input (González-Hernández et al. 2017).
- Ion-channel Changes
Neuronal ion-channels can change their properties in the PNS and CNS, but for convenience are assembled here. Voltage-gated ion channels altered by inflammatory or neuropathic pain include Na+ (Nav) channels, K+ (Kv) channels, Ca2+ (Cav) channels, Ca2+-dependent K+ channels and hyperpolarization-activated cyclic nucleotide-gated (HCN) channels (Baron 2006; Bennett et al. 2019; Carbone 2009; Costigan et al. 2009; Dib-Hajj and Waxman 2019; Finnerup et al. 2021; Levinson 2009; Mathie and Veale 2009; Rogers et al. 2006; Tsantoulas and McMahon 2014). The precise patterns of ion-channel changes can vary widely in inflammatory pain or in various etiologies of neuropathic pain, and are influenced by several other factors including genetic mutations (Finnerup et al. 2021). Following nerve injury, Nav, Kv, and Cav channels play important roles in modulating neuronal excitability and pain-signal transmission (Felix et al. 2025).
Sodium (Na+) Channels. There are many molecular and cellular mechanisms by which dysregulation in the expression, localization, and function of specific Nav channel sub-types (mainly Nav1.7 and Nav1.8) and their auxiliary sub-units contributes to aberrant neuronal activation, the generation of ectopic discharges, and sensitization in neuropathic pain (Felix et al. 2025). The array of Na+ channels changes properties, leading to spontaneous discharge and higher than normal firing rates in response to stimuli. For example, one responsible ion channel is the Nav1.3 channel that is over-expressed in DH neurons, as well in ventro-posterior lateral (VPL) THAL neurons following spinal injury (Waxman and Hains 2006).
Potassium (K+) Channels. Kv channels (particularly Kv7 channels) function as brakes on neuronal excitability, and their dysregulation facilitates the development and maintenance of neuropathic pain (Felix et al. 2025). Changes in intrinsic plasticity in the superficial DH involves phosphorylation of Kv4.2. This would reduce IA currents, leading to an increase in excitability. Firing patterns associated with IA currents are largely restricted to excitatory INTs in lamina II. Increasing transmission in excitatory INTs could enhance activation of lamina I projection cells through polysynaptic pathways, contributing to the hyperalgesia arising in inflammatory pain states (Todd 2010).
Calcium (Ca2+) Channels. A crucial role is also played by Cav channels, particularly Cav2.2 and the auxiliary sub-unit Cavα2δ, whose over-expression increases Ca2+ influx into the cell, neurotransmitter release, and neuronal hyperexcitability, thus maintaining persistent pain states (Felix et al. 2025).
Transient Receptor Potential Ankyrin (TRPA) Channel. TRPA1 is involved in acute and chronic pain as well as inflammation, plays key roles in the pathophysiology of nearly all organ system. TRPA channels are Ca2+-permeable non-selective cation channels. Mammals have only one member, TRPA1, which is widely expressed in sensory neurons and in non-neuronal cells. TRPA1 is involved in the detection of a wide variety of exogenous stimuli that may produce cellular damage. TRPA1 is activated by cold, heat, and mechanical stimuli, and its function is modulated by multiple factors, including Ca2+, pH, and reactive oxygen (Talavera et al. 2020).
Adenosine Triphosphate (ATP)-sensitive Potassium [K(ATP)] Currents. In neuropathic and control rats, whole-cell voltage-clamp recordings were performed in DRG neurons. Normal primary afferent neurons expressed K(ATP) channels that conducted current which was eliminated by peripheral nerve injury (Sarantopoulos et al. 2003).
Spontaneous Pain. Most of the patients with peripheral neuropathies complain mostly about spontaneous forms of pains. Upon nerve section, axotomized but also intact fibers develop ectopic spontaneous activity. A proportion of axotomized fibers might present receptive fields in the skin far beyond the site of damage, indicative of a functional cross talk between neuromatose and intact fibers (Roza and Bernal 2022).
2.1.1. Peripheral Sensitization to Inflammation
A tissue injury entails inflammation, which induces a complex, self-reinforcing sequence of events. As to nociceptor sensitization, there is a close reciprocal cross-talk between the immune system, in particular with mast cells (Figure 1). In response to injury, nociceptors release various mediators from their terminals (e.g., SP, CGRP) that potently activate and recruit immune cells (e.g., mast cells), whereas infiltrated immune cells in turn release plenty of mediators that further promote sensitization of nociceptors and the transition from acute to chronic pain by producing cytokines, chemokines, lipid mediators and growth factors (Julius and Basbaum 2001; Liu et al. 2021b; Nelissen et al. 2013; Wang and Ma 2016). This ensemle of agents has been dubbed `inflammatory soup´.
More specifically, activation of resident mast cells leads to the release of pro-inflammatory chemokines, cytokines, growth factors (GFs), lipids, and ROS and reactive nitrogen species (RNS). Inflammatory agents so far identified also include protons (H+), prostaglandins, SP, bradykinin, 5-HT, IL-1, and other endogenous chemicals (Binshtok et al. 2008; Costigan et al. 2009; Hucho and Levine 2007; Julius and Basbaum 2001; Nicol and Vasko 2007; Pezet and McMahon 2006; Ren and Torres 2009; Scholz and Woolf 2002, 2007; Stein et al. 2009; Wang et al. 2006). Some agents induce local degenerative processes, sensitize nociceptors, recruit silent nociceptors, and lead to expression of new receptors and ion channels (Finnerup et al. 2021; Grace et al. 2016; McMahon et al. 2015; Pinho-Ribeiro et al. 2017). Many inflammatory chemicals that excite nociceptors activate intracellular signal transduction pathways and modulate sensory receptor channels and voltage-gated ion channels. Moreover, heat can render cutaneous group III (Aδ) mechanical nociceptors sensitive to heat (Willis 1996). Pro-inflammatory influences also spread from the peripheral injury site to the dorsal roots and spinal cord (Cao and DeLeo 2009; Moalem and Tracey 2006; Watkins et al. 2007; White et al. 2005).
Contribution of the Immune System. In response to injury, resident immune cells are activated and blood-borne immune cells are recruited to the injury site. Immune cells not only contribute to immune protection but also initiate the sensitization of peripheral nociceptors (Ren and Dubner 2010). Changes in nociceptor functions have been well studied in inflammatory pain and involve interactions with the immune cells including monocytes, lymphocytes, leukocytes, macrophages, T cells and mast cells which not only signal other components of the immune system, but also the brain to control the inflammatory response and to maintain its homeostasis. Involved are a plethora of pro-inflammatory cytokines including interleukins IL-1β, IL-6, IL-8, IL-12, interferon-α (IFN-α) and IFN-δ as well as TNF-α. Pro-inflammatory cytokines released by immune cells induce hyperalgesia when administered at the peripheral or central level (Zouikr et al. 2016) (Figure 1).
Neurogenic Inflammation by Axon Reflexes. Local excitation of nociceptors elicits axon reflexes mediated by antidromic discharges, i.e., backfiring into peripheral axon branches (Figure 1, curved arrow). Excitation of the branches releases neural mediators, such as SP, CGRP, VIP, and gastrin-releasing peptide (GRP), which contribute to neurogenic inflammation and primary hyperalgesia (Kanashiro et al. 2020).
- Substance P (SP)
SP is synthesized by small-diameter sensory `pain´ fibers, and release of the peptide into the DH following intense peripheral stimulation promotes central hyperexcitability and increased sensitivity to pain. Mice with a disrupted gene encoding the NK1Rs were healthy and fertile, but the characteristic amplification (`wind up´) and intensity coding of nociceptive reflexes was absent. Although SP did not mediate the signalling of acute pain or hyperalgesia, it was essential for the full development of SIA and for an aggressive response to territorial challenge, demonstrating that the peptide plays an unexpected role in the adaptive response to stress (De Felice et al. 1998).
- Protons (H+)
Modest decreases in extracellular pH (increases in H+ concentration) that occur within the physiological range.can be sensed by acid-sensing ion channels (ASICs). ASICs detect tissue acidosis occurring at tissue injury, inflammation, ischemia, stroke, and tumors as well as fatiguing muscle to activate pain-sensing nerves in the periphery and transmit pain signals to the brain. Various types of cutaneous nociceptors and mechano-receptors (some of which also function as proprioceptors) express ASIC2 and ASIC3. Injection of acidic saline into a muscle produces enhanced nociceptive behaviors in animals and pain in human subjects. Tissue acidity is a major risk factor in the development of chronic musculo-skeletal pain. This can occur as a direct result of injury and is associated with injury and inflammation. Some inflammatory conditions persist, such as rheumatoid arthritis, and lead to long-lasting pain and disability. In most cases, the acute injury resolves, but in some cases, pain persists despite the lack of peripheral tissue injury or inflammation. Associated with tissue acidosis may be activation of ASICs, TRP channels, and two-pore K+ (K2P) channels). In pain-behavioral studies, Asic1a-/- knock-out mice responded to muscle inflammation and developed secondary (or referred) mechanical hyperalgesia in distal tissues but failed to develop primary mechanical hyperalgesia in muscle. Of the different types of ASICs, ASIC3 and ASIC1 have been implicated in transmission of nociceptive information from the musculo-skeletal system (Cheng et al. 2018; Sluka and Gregory 2015).
- Glutamate
In rodent models of pain, group II mGluRs were efficacious when activated. Positive immuno-reactivity for mGlu2 was present in DRG, peripheral fibers in skin, and centrally in the DH. mGlu2-positive immuno-reactivity also occurred in human neonatal and adult DRG. In rodent sensory neurons under basal conditions, activation of group II mGluRs with a selective group II agonist produced no changes to membrane excitability. In human sensory neurons from donors without a history of chronic pain, prostaglandin E2 (PGE2) produced hyperexcitability that was similarly blocked by group II mGluR activation (Davidson et al. 2016).
2.1.2. Peripheral Sensitization after Nerve Injury
Nerve injury can be followed by several events: (i) immediate injury firing followed by abnormal discharge in damaged or intact nerve fibers, which may persist for a long time; (ii) secretion of neuromodulators and pro-inflammatory mediators; (iii) sensitization of nociceptors and activation of silent nociceptors or expression of new receptors; (iv) changes in gene expression; (v) abnormal sprouting of peripheral and central fibers; (vi) changes in receptive fields and sensory modilities of injured or intact peripheral fibers (Saadé and Jabbur 2008); (vii) changes in receptors and ion channels.
- De-afferentation Pain
Subjects with trauma to spinal roots or the trigeminal system can experience burning pain projected to thede-afferented skin region, a phenomenon called anesthesia dolorosa, or de-afferentation pain (Baron 2006). The pain is described as constant, burning, aching or severe.
Animal models of neuropathic pain focussed on spinal-cord mechanisms and described a sequence that begins with the formation of an amputation neuroma at the site of nerve damage, followed by erratic mechano-sensitivity in DRG and the DH; long-term potentiation (LTP) of synaptic transmission; and attenuation of central pain inhibitory mechanisms including loss of opioid-mediated anti-nociception. Hyperalgesia is maintained by a high rate of discharge in small afferent fibers that release supra-normal levels of glutamate and aspartate that act on NMDARs, and degeneration of presumed inhibitory INTs in DH laminae I-III (Zimmermann 2001).
- Centralization of Primary Afferent Activity
Sciatic nerve lesions can invoke CNP that is accompanied by persistent, spontaneous activity in primary afferents. This activity reflects changes in the properties and functional expression of Na+, K+, and Ca2+ channels, persists for many weeks, and can hypothetically be propagated to the spinal DH. This centralization probably involves the inappropriate release of peptidergic neuromodulators from primary afferents. Peptides such as SP, neuropeptide Y (NPY), CGRP, and BDNF, which may promote enduring changes in excitability as a consequence of neurotrophic actions on ion channel expression in the DH (Abdulla et al. 2003).
- Mechanisms
The sensitivity of peripheral nociceptors is differentially altered in neuropathic pain (Finnerup et al. 2021). Nerve fibers develop abnormal ectopic excitability at or near the site of nerve injury, caused by unusual distributions of Na+ channels and abnormal responses to endogenous pain-producing substances and cytokines such as TNF-α. Any local nerve injury tends to spread to distant parts of the PNS and CNS. This includes erratic mechano-sensitivity along the injured nerve including DRG cell as well as ongoing activity in the DH. The spread of pathophysiology includes an up-regulation of nitric oxide synthase (NOS) in axotomized neurons, de-afferentation hypersensitivity of spinal neurons following afferent apoptosis, LTP of spinal synaptic transmission and suppression of central pain-inhibitory mechanisms. Repeated or prolonged noxious stimulation and the persistent abnormal input following nerve injury activate a number of intracellular second messenger systems. Apoptosis seems to induce neuronal sensitization and loss of inhibitory systems (Zimmermann 2001). Other mechanisms underlying neuropathic pain are mitochondrial dysfunction, inflammatory processes, intracellular Ca2+ overload, peripheral and central sensitization, and nerve terminal sprouting (Alles and Smith 2018; Baron 2006; Carrasco et al. 2018; Costigan et al. 2009; Devor 2018; Finnerup et al. 2021; Kuner and Flor 2016; Saadé and Jabbur 2008; Sun et al. 2020b; Tsantoulas et al. 2016; Watkins et al. 2007).
- Oligodendrocytes
Oligodendrocytes play important roles in neuropathic pain following peripheral nerve injury, SCI, and chemotherapy. A decrease in the number of oligodendrocytes and increased cytokine production by oligodendroglia in response to injury can induce or exacerbate pain. An increase in endogenous oligodendrocyte precursor cells may be a compensatory response to repair damaged oligodendrocytes. Moreover, oligodendrocyte apoptosis in brain regions such as the mPFC is connected to opioid-induced hyperalgesia (OIH). Chemotherapeutic agents disrupt oligodendrocyte differentiation, leading to persistent pain, while HIV-associated neuropathy involves up-regulation of oligodendrocyte lineage cell markers (Kim and Angulo 2025).
2.1.3. Changes in Sympathetic Modulation of Nociceptors
Sympathetically mediated pain can result from several complex mechanisms. The ANS controls the heart rate, blood pressure, respiration, digestion, pupillary reactivity, urination, pupillary reactivity, sexual arousal, and regulates the functions of internal organs. The ANS has three main divisions: The sympathetic nervous system (SNS), the parasympathetic nervous system (PaNS), and the enteric nervous system (ENS). Each region belonging to the `pain matrix´ interacts with ANS. Pain patients offen also suffer from dysfunction of the ANS (Arslan and Çevik 2022).
The SNS induces, facilitates, or potentiates chronic pain, characterized by increased responsiveness of injured sensory nerves to catecholamines, increased expression of α1 adreno-receptors on the primary afferent nociceptors, sensitization of group II (Aβ) mechanoreceptors, enhanced discharge and sympathetic sprouting in DRG, central sensitization, and dysfunction of the pain modulation (Arslan and Çevik 2022).
- Catecholamines
The SNS and its peripheral neurotransmitters normally modulate the discharge of a number of sensory receptors involved in tactile, stretch, and nociceptive sensation, and can change the membrane properties of nerve fibers (Passatore and Roatta 2009). Following peripheral nerve lesions and previous sensitization of the sensory receptors, catecholamines released from postganglionic sympathetic nerve terminals or into the blood circulation from the adrenal glands can evoke or aggravate pain by exciting skin nociceptors and enhancing their responsiveness to noxious stimuli (Arslan and Çevik 2022).
- Sprouting of Postganglionic Sympathetic Fibers within Dorsal-Root Ganglion (DRG)
Nerve injury can cause non-injured postganglionic sympathetic fibers to sprout around large axotomized DRG cells, where they form basket-like structures (Arslan and Çevik 2022; Ramer et al. 1999). The aberrant sympathetic input to DRG cells amplifies spontaneous and evoked activity and recruits silent neurons (Millan 1999), leading to sympathetically mediated pain through interactions between peripheral group II (Aβ) afferents and sympathetic efferents (Chung et al. 1997; Michaelis et al. 1996; Zimmermann 2001). It has also been suggested that releases of ATP, in addition to NA, from sympathetic nerve endings may contribute to sympathetically mediated pain.
In humans, trauma to a peripheral nerve may be followed by chronic pain syndromes, which are only relieved by blockade of the effects of sympathetic impulse traffic. It is presumed that, after the lesion, noradrenaline (NA) released by activity of sympathetic postganglionic axons excites primary afferent neurons by activating α-adrenoceptors. In some patients, local anesthesia of the relevant peripheral nerve does not alleviate pain, implying that ectopic impulses arise either within the CNS or in proximal parts of the primary afferent neurons. In lesioned rats, activity can originate within the DRG. In rats subjected to sciatic nerve ligation, NA peri-vascular axons sprout into DRG and form basket-like structures around large-diameter axotomized sensory neurons. Sympathetic stimulation can activate such neurons repetitively. These unusual connections could be an origin for abnormal discharge following peripheral nerve damage. In contrast to the sprouting of intact nerve terminals into nearby denervated effector tissues in skin, muscle, sympathetic ganglia and sweat glands, the axons sprout into a target which has not been partially denervated (McLachlan et al. 1993).
Animal’s experiments have shown that NGF and neurotrophin-3 (NT-3) synthesis is up-regulated in satellite cells surrounding neurons in lesioned DRG as early as 48 hours after nerve injury. This response lasts for at least two months. Noradrenergic sprouting around the axotomized neurons was associated with p75-immunoreactive satellite cells. Further, antibodies specific to NGF or NT3, delivered by an osmotic mini-pump to the DRG via the lesioned L5 spinal nerve, significantly reduced NA sprouting (Zhou et al. 1999).
Sympathetic sprouting into the DRG may be stimulated by NGF (Ramer and Bisby 1999). In addition to NGF, other neurotrophic factors might cause similar effects. In rats, chronic constriction injury (CCI) of the sciatic nerve induced significant increases in the percentage of small, medium and large BDNF-immuno-reactive neurons in the ipsilateral L4 and L5 DRG. After spinal nerve ligation, the percentage of large BDNF-immuno-reactive neurons increased significantly, and that of small BDNF-immuno-reactive neurons decreased markedly in the ipsilateral L5 DRG, while that of BDNF-immuno-reactive L4 DRG neurons of all sizes showed marked increase. Both CCI and spinal nerve ligation induced significant increases in the number of BDNF-immunoreactive axonal fibers in the superficial and deeper laminae of the L4/5 DH and the gracile nuclei on the ipsilateral side. Considering that BDNF may modulate nociceptive sensory inputs and that injection of antiserum to BDNF significantly reduces the sympathetic sprouting in the DRG and allodynic response following sciatic nerve injury (SNI), endogenous BDNF plays an important role in the induction of neuropathic pain after CCI and spinal nerve ligation (Ha et al. 2001).
In the rat spinal nerve ligation (SNL) model, an isolated whole DRG preparation was used to investigate sympathetic-sensory connections. Three days after ligation of the ventral ramus of the spinal nerve, sympathetic fibers sprouting into the DRG originated largely in the intact dorsal ramus of the spinal nerve. In whole DRG isolated three days after SNL, micro-electrode recordings of sensory neurons showed that repeated stimulation of the dorsal ramus enhanced spontaneous activity in large- and medium-diameter neurons and reduced rheobase in large neurons. These effects were slow and long-lasting and were attributed to stimulation of the sympathetic sprouts because stimulation had no effect in un-injured DRG and effects could be reduced or eliminated by a cocktail of antagonists of NA and ATP receptors, by pre-treatment with the sympathetic release blocker bretylium, or by pre-cutting the gray ramus through which sympathetic fibers coursed to the ligated DRG. The latter treatment, a relatively minimal form of sympathectomy, was also highly effective in reducing mechanical pain ipsilateral to the SNL (Xie et al. 2010).
2.2. Changes in the Spinal Cord
Rather than being conveyed unchanged, the signals from nociceptive afferents undergo extensive processing in the spinal DH and spinal trigeminal nucleus (SpV). This is done primarily by INTs, of which there is mind-boggling variety (Braz et al. 2014; Figure 2). Within the spinal cord, maladaptive plasticity is an important substrate for the development of neuropathic pain. These processes can be viewed at multiple levels including morphological and electrophysiological changes which occur both to primary afferent inputs as well as spinal neurons, synaptic re-modeling including enhanced excitatory and reduced inhibitory drive, and ultimately circuit-level re-wiring, which leads to altered connectivity and aberrant processing of sensory inputs in the DH. The DH is subject to important descending modulation from the brainstem, which is also dysregulated by nerve injury (Costigan et al. 2009; Dibaj and Schomburg 2017; Moore et al. 2000; Nadrigny et al. 2017; Sandkühler 2009; Todd 2015, 2017; West et al. 2015; Woolf and Salter 2000). In addition, the immune system is activated, which has consequences for nociceptive processing.
Nerve Injury entails several changes in spinal-cord neurons that contribute to hyperalgesia arising from the injured tissue. In this process, synaptic inhibition plays an important role. Neuropathy and inflammation are associated with reduced spinal inhibition, including presynaptic inhibition (PSI). Spinal neurons become hypersensitive in association with inflammatory and neuropathic pain, the hypersensitivity can be reproduced by blocking inhibition. The neurons exhibit sponaneous injury discharges and after discharges. Spinal neurons can also change their receptive fields (Prescott 2015; Saadé and Jabbur 2008).
Inflammation. In models of unilateral hindpaw inflammation, changes in the neurochemistry and electrophysiology of DH neurons ipsilateral, and to a much lesser extent contralateral, to the inflamed paw occurred. The excitability of DH neurons increased, receptive field size increased, and the content of various proteins and neuropeptides in the DH [e.g. fos, dynorphin (Dyn), enkephalin (ENK)] are affected following peripheral inflammatory insult (Solodkin et al. 1992).
Myositis. At spinal level, myositis changed the connectivity of DH neurons, reflected in an expansion of the input (target) region of the muscle nerve. Such central sensitization can explain the hyperalgesia and spread of pain in patients. The transition from acute to chronic pain is accompanied by structural changes. In rats with muscle inflammation or nerve lesions, such morphological changes become apparent within a few hours after the lesion (Mense 2004). In anesthetized rats with an experimentally induced acute or chronic myositis, chronic myositis was associated with a higher innervation density of the tissue with putative nociceptive free nerve endings that contained the SP. The nociceptive signals from muscle to the spinal cord were largely conveyed by un-myelinated fibers with tetrodotoxin (TTX)-resistant Na+-channels (Mense 2004). In mice lacking SP signaling by deletion of the tachykinin precursor 1 (Tac1) gene or co-administration of neurokinin-1 (NK1) receptor antagonists, a single intramuscular acid injection produced long-lasting hyperalgesia rather than the transient hyperalgesia seen in control animals. The inhibitory effect of SP occurred exclusively in neurons expressing acid-sensing ion channel 3 (ACID3), where SP enhanced M-channel-like K+ currents through the NK1 receptor in a G-protein-independent but tyrosine kinase-dependent manner. The SP signaling could alter action-potential thresholds and modulate the expression of TTX-resistant Na+ currents in medium-sized muscle nociceptors. Thus, intramuscular.SP mediated an unconventional NK1 receptor signal pathway to inhibit acid activation in muscle nociceptors, resulting in an unexpected anti-nociceptive effect against chronic mechanical hyperalgesia, here induced by repeated intramuscular acid injection (Lin et al. 2012).
Neuroplasticity. At the spinal and medullar levels, painful muscle lesions induce marked neuroplastic changes that result in hyperexcitability and hyper-activity of nociceptive neurons to their normal or sub-threshold afferent input (Sandkühler 2009). This central sensitization is the basis for the spontaneous pain and hyperalgesia of patients. The transition from acute to chronic muscle pain is complete when the initially functional changes are transformed into structural ones. Patients with morphologic alterations in their nociceptive system are difficult to treat because the changes need time to normalize (Mense 2003).
Central Sensitization of Viscero-somatic Dorsal-Horn (DH) Neurons. This sensitization depends on the sensitization of spinal visceral afferent neurons, local spinal excitatory and inhibitory INTs and supraspinal endogenous control systems (Jänig 2014).
Potential spinal mechanisms causing enhanced neuronal responsiveness include direct facilitation along the chain of excitation or alterations in physiological modulation of spinal nociception, i.e., less than normal inhibition, conversion from inhibition to excitation, or stronger than normal excitation (Sandkühler 2009).
2.2.1. Loss of Inhibition
Inhibitory neurotransmission plays a crucial role in the maintenance of a physiologically meaningful state of pain sensitivity and helps separating innocuous and noxious signal relay. Following injury and inflammation, inhibition is reduced (Figure 2), which contributes to hyperalgesia and allodynia. Under unfortunate conditions, both phenomena outlast the healing process. The spinal DH is a major site for endogenous pain control and maladaptive plasticity, which underlies many chronic pain conditions. At this site and in corresponding brainstem areas, various processes triggered by peripheral inflammation or nerve damage compromise synaptic inhibition. Among these processes are alterations in the excitatory drive to inhibitory DH neurons, a compromised electro-chemical gradient of Cl- ions, and altered responsiveness of inhibitory neurotransmitter receptors. While the first two mechanisms are triggered by peripheral nerve damage and affect both GABAergic and glycinergic inhibition, peripheral inflammation has a specific impact on the function of DH Gly receptors (GlyRs). General aspects of glycinergic neurotransmission are illustrated in diminished inhibitory control of spinal nociception is one of the major mechanisms underlying chronic pain states (Zeilhofer et al. 2021).
Loss of Inhibitory Neurons. It has been suggested that neuropathic pain is supported by loss of function of DH inhibitory INTs and that several mechanisms are involved, including apoptosis or diminished activity of inhibitory INTs, decreased transmitter release and reduced effectiveness of γ-amino-butyric acid (GABA) and glycine (Gly) as inhibitory transmitters (Gradwell et al. 2020; Hughes and Todd 2020; Todd 2015).
In both inflammatory and neuropathic pain, inhibition mediated by GABA gets lost. In several animal models of chronic pain, both pre- and postsynaptic inhibitions become less efficient, leading the DH to a state of hyperexcitability (Comitato and Bardoni 2021). In neuropathic pain with allodynia resulting from ischemic injury of the spinal cord, GABA content of the DH is decreased, and GABAergic presynaptic inhibition is impeded (Wiesenfeld-Hallin et al. 1997). Presynaptic inhibition is also reduced in inflammatory pain (Guo and Hu 2014). Inhibitory modulation of spinal nociceptive signal transmission exerted by descending pathways may also be disrupted via a number of mechanisms (Costigan et al. 2009; Sandkühler 2009). Still, the precise situation appears not quite clear (Todd 2015).
2.2.2. Changes in Presynaptic Inhibition (PSI)
Right at the entrance to the DH and SpV, sensory afferents are met by a powerful filter called PSI. PSI acts by decreasing the efficacy of synaptic transmission from presynaptic terminals of sensory afferents to spinal neurons, the effects being mediated via inhibitory GABAergic or Gly INTs contacting the terminals of sensory afferents. PSI is modulated by many sensory and spinally desending inputs (Figure 2). The input-output patterns are comlex (Quevedo 2009; Rudomin 2009; Rudomin and Schmidt 1999; Zimmerman et al. 2019). Importantly, group IV (C) input to lamina I neurons is also under control of the afferent-driven GABAergic PSI, by recruitment of groups II (Aβ), III (Aδ) and IV (C) afferents, as shown in an isolated spinal-cord preparation. The occurrence of PSI of group IV (C) fibers and its extent are quite heterogeneous (Fernandes et al. 2020). It could be expected that PSI (including self-inhibition) of nociceptive afferents would mitigate the effects of increased nociceptive inputs, but this could be counteracted by the loss of GABAergic inhibition.
Stimulation of primary sensory afferents generates depolarization of sensory nerve terminals (primary afferent depolarization: PAD), which is electrotonically conducted into the dorsal roots and can here be measured as dorsal-root potential (Hochman et al 2010; Quevedo 2009).
In inflammatory pain, a switch from presynaptic inhibition to excitation occurred. After tissue injury, the amplitude of PAD was increased in both nociceptive and non-nociceptive fibers, which could be sufficient to generate action-potential firing at primary afferent terminals and dorsal root reflexes (DRRs). Orthodromically propagated action potentials may enhance glutamate and peptide release from central nociceptive terminals, causing excitation of spinal second-order neurons. In experimental arthritis or after intra-dermal injection of capsaicin, action potentials propagated antidromically to the periphery and may be involved in neurogenic inflammation. Following nerve injury, PAD amplitude was generally reduced and PSI was diminished. Several factors appear to contribute to PAD modifications in neuropathic pain: (i) decrease of GABA synthesis in DH inhibitory INTs; (ii) depolarizing shift of ECl; and (iii) reduction of GABAA conductance and down-regulation of γ2, α2, and α1 sub-units on DRG neurons, observed after nerve ligation, chronic constriction, and crush-nerve injury. The selective down-regulation of the α2 sub-unit in DRG neurons worsened thermal and mechanical hypersensitivity in crush-injured animals and induced pain hypersensitivity in sham animals, while up-regulation of endogenous GABA alleviated neuropathic pain (Comitato and Bardoni 2021).
- Inhibitory Parvalbumin (PV) INTs (INTs)
Parvalbumin (PV) INTs in laminae IIi and III of the spinal DH are a source of axo-axonic inputs onto the central terminals of myelinated low-threshold mechano-receptive (LTMR) afferents from both hairy and glabrous skin. These INTs gated the passage of LTMR afferent input both by GABA-mediated presynaptic inhibition and by inhibiting the postsynaptic targets of these afferents through the release of GABA and Gly. The postsynaptic targets of LTMRs under inhibitory control from PV-expressing INTs included vertical cells. Under normal circumstances, PV-cell-mediated inhibition of vertical cells, and of their LTMR input, is likely to play a central role in segregating LTMR afferent input from pain circuits. Under pathological conditions, however, the features of vertical cells enable them to act as a potential route for LTMR input into lamina I, where the loss of PV-cell-mediated inhibition could unmask this relay circuit, leading to the polysynaptic activation of pain circuits. In a neuropathic model, PV-cell excitability was down-regulated, which would compromise the pre- and postsynaptic inhibitory gating mediated by these cells. Under such circumstances, the aberrant recruitment of vertical cells following LTMR input would help explain the cellular basis of tactile allodynia associated with neuropathic pain (Boyle et al. 2019).
Activation of low-threshold cutaneous afferents evoke a GABAA-receptor-dependent PSI form that inhibits similar afferent sub-types, whereas small-diameter afferents predominantly evoke an NMDA-receptor-dependent form of PSI that inhibits large-diameter fibers. Behaviorally, loss of either GABAA receptors (GABAARs) or NMDARs in primary afferents leads to tactile hypersensitivity across skin types, and loss of GABAARs, but not NMDARs, leads to impaired texture discrimination (Zimmerman et al. 2019).
2.2.3. Increase in Dorsal-Root Reflexes (DRRs)
Antidromic discharges in sensory afferents can also be generated by PSI. A model is based on the notion that group II (Aß) mechano-receptor afferents can gain access to spinal nociceptive neurons by means of a presynaptic link between LTMR afferents and nociceptive afferents. The excitation of nociceptor afferents provoked by a peripheral injury activates the spinal INTs that mediate PAD between LTMRs and nociceptor afferents. As a consequence of the increased and persistent barrage driving these neurons, their excitability is increased such that, when activated by LTMR afferents from areas surrounding the injury site, they produce a very intense PAD in the nociceptive afferents which is capable of generating spike activity. This activation would be conducted antidromically in the form of DRRs but would also be conducted forward activating the second-order neurons normally driven by nociceptor afferents. The sensory consequence of this mechanism is pain evoked by the activation of LTMR afferents from an area surrounding an injury site (allodynia) (Carlton 2014; Willis 1999; Zeilhofer et al. 2012). This model has been formulated for cutaneous group II (Aß) afferents. However, there are other afferents, for example muscle spindles from skeletal muscles and others. It woud be interesting to know whether the model also applies to muscle pain.
Neurogenic Inflammation by Dorsal-Rroot Reflexes (DRRs). DRR-generated antidromic action potentials can peripherally release neuropeptides such as SP and CGRP from peripheral sensory nerve endings (Figure 1). The consequences of this increase in DRRs may include exacerbation of hyperalgesia as well as of peripheral inflammation. The conversion of an inhibitory process, PSI, to an excitatory one by DRRs can thus lead to pathological consequences (Willis 1999).
2.2.4. Shift in Anion Gradients
Since synaptic inhibition depends not only on neurotransmitter (e.g., Gly and GABA) release and receptor density, but also on transmembrane chloride (Cl-) concentration gradients, changes in these gradients will alter the potency of inhibition. Cl- gradients are maintained by transmembrane co-transporters, and the expression and/or function of such co-transporters are indeed altered in chronic pain states (Price et al. 2009).
Peripheral nerve injury can shift transmembrane anion gradients in lamina-I cells by down-regulating the K+-Cl- exporter KCC2 with a subsequent increase in the intracellular Cl- concentration. Consequently, GABA and Gly release from INTs will depolarize the cells rather than hyperpolarize them, causing an overall increase in excitability (Baron 2006; Costigan et al. 2009; Lewin et al. 2004; Price et al. 2009).
2.2.5. Changes in Intrinsic Neuron Properties
Peripheral nerve injury and SCI generate particularly debilitating and hard-to-manage chronic pain states, which are due at least in part to changes in intrinsic neuron properties, prominently in ion channels.
- Injury Discharge
Immediately after nerve injury, spinal neurons (DH and SpV) start firing spontaneously and become hypersensitive to nociceptive inputs (hyperalgesia) and other sensory inputs (allodynia), the hyperexcitability resulting from massive repeated nociceptive input from group IV (C) afferents. In rats with mono-neuropathy, the spinal neurons show abnormal firing and after-discharges. The sustained firing has been likened to LTP. Metabolic activity, fos-expression and glutamate release are increased (Saadé and Jabbur 2008). Moreover, intrinsic neuronal changes such as those in ion channels play a role.Changes in Neuron Sensitivity
The DH hosts a plethora of neuron types, which are being characterized by modern genetic methods in rodents. At least five populations of excitatory neurons are involved in neuropathic pain. For example, one group propagates signals underlying mechanical pain and are sufficient and necessary to mediate mechanical hyper-ensitivity in neuropathic pain. Another group of excitatory neurons in the deeper laminae is involved in mechanical pain and mechanical allodynia; it receives group II (Aβ) fiber input and projects to lamina I and II. The genetic ablation of yet another group of excitatory neurons selectively attenuated neuropathic mechanical and cold allodynia without affecting basal mechanical or thermal processing. In the DH, inhibitory Gly neurons are more abundant in the more superficial laminae and GABAergic INTs are more prevalent in the deeper laminae, both groups receiving inputs from peripheral LMTRs. Ablation of Gly inhibitory neurons broadly induces mechanical, heat and cold hypersensitivity as well as spontaneous pain-related behavior in naive mice, and their chemogenetic activation alleviated neuropathic allodynia (Finnerup et al. 2021). Moreover, intrinsic neuronal changes such as those in ion channels play a role.
- Wide-dynamic Range (WDR) Neurons
WDR neurons are one of the two spinal neuron types that project rostrally (Figure 2). The activity of WDR neurons is considered as necessary and sufficient for pain sensation. They respond with progressively greater discharge rates to brushing hair, light touch, pressure, pinch and squeeze, and also respond to graded noxious heat, cold and inputs from visceral and deep tissues such as muscles and joints. WDR neurons have large receptive fields, and are not somatotopically organized (Craig 2003). Nociceptive inputs, in association with neuropathy-induced pain hypersensitivity, can also change the intrinsic response properties of WDR projection neurons in the deep DH (Berger et al. 2011; Sandkühler 2009). The response patterns of WDR neurons are of three types: tonic slowly adapting discharge, rhythmic bursting, and plateau potentials (Sandkühler 2009). The expression of any one of the patterns depends on a dynamic balance between excitatory mGluR and inhibitory G-protein-coupled GABAB metabotropic receptors (Benke 2022; Goudet et al. 2009). Normally, inhibition predominates and most cells show a tonic slowly adapting discharge pattern. A switch to the plateau state might be an important factor in central sensitization (Derjean et al. 2003).
In preclinical models of both peripheral and central neuropathic pain, DH neurons increased their discharge rate following innocuous and noxious skin stimulation. The number of DH WDR neurons increased, which in part could be due to a functional conversion of nociceptive into WDR neurons (Berger et al. 2011).
Windup is a progressive, frequency-dependent increase in the excitability of nociceptive trigeminal and spinal DH WDR neurons to repetitive stimulation of primary afferent nociceptive group IV (C) fibers. Superficial DH NK1R-expressing neurons regulate the sensitization of nociceptive WDR neurons through activation of a defined spino-bulbo-spinal loop. However, the windup of such WDR neurons was not regulated through this loop. The alternative circuit activated by DH NK1Rs that mediates WDR neuron windup was identified in the rat SpV, in which the sub-nucleus oralis (SpVo) contains a pool of WDR neurons that receive their nociceptive group IV (C) indirectly via INTs located in the medullary DH. It was concluded that, in contrast to central sensitization that is controlled by a spino-bulbo-spinal loop, Sp5o WDR neuron windup is regulated through a local circuit activated by medullary DH lamina III NK1Rs (Coste et al. 2008).
Mice with a deletion of the pre-protachykinin-A (pptA) gene, from which SP and neurokinin A (NKA) are derived, exhibited reduced behavioral responses to intense stimuli, but behavioral hypersensitivity after injury was unaltered. In urethane-anesthetized wild-type and ppt-A null mutant (-/-) mice, single-unit activity from WDR neurons in lamina V of the lumbar DH were recorded. Intensity coding to thermal stimuli was largely preserved in the ppt-A-/- mice. Neither the peak stimulus-evoked firing nor the neuronal activity during the initial phase (0-4 s) of the 41-49o C thermal stimuli differed between the genotypes. However, responses during the late phase of the stimulus (5-10 s) and post-stimulus (11-25 s) were significantly reduced in ppt-A -/- mice. To activate group IV (C) fibers and to sensitize the DH neurons, mustard oil was applied topically to the hindpaw. Neither total mustard oil-evoked activity nor sensitization to subsequent stimuli differed between the wild-type and ppt-A -/- mice. However, the time course of the sensitization and the magnitude of the post-stimulus discharges were reduced in ppt-A -/- mice. Hence, SP and/or NKA are not required for intensity coding or sensitization of spinal nociceptive neurons, but these peptides prolong thermal stimulus-evoked responses. Whereas behavioral hypersensitivity after injury is preserved in ppt-A -/- mice, the magnitude and duration of these behavioral responses would be reduced in the absence of SP and/or NKA (Martin et al. 2004).
2.2.6. Changes in Receptive-Fields
Central hypersensitivity induced by group IV (C) fiber activation makes previously sub-threshold inputs generate action potentials within the receptive fields of spinal neurons. The receptive fields can also change more drastically (Wall 1988; Koerber and Brown 1995). In the rat DH, dynamic receptive-field expansion up to nearly 400% could be caused by relatively brief (20 s) barrages of group IV fiber input (Cook et al. 1987). DH neurons with receptive fields adjacent to a cutaneous region exposed to heat injury can expand their receptive field into the traumatized area (McMahon and Wall 1984). Receptive-field expansion linked to mechanical, chemical and inflammatory injuries and during polyarthritis has also been documented in neurons of the ventro-basal THAL (Coderre and Katz 1997). The appearance of new receptive fields sometimes results in faulty interpretation of the source of pain (Hoheisel et al. 1993).
Central sensitization also manifests itself in the enlargement of receptive fields for nociceptive withdrawal reflexes in humans. Injections of capsaicin dramatically expanded the reflex receptive fields in both intact and spinal cord transected individuals (Biurrun Manresa et al. 2014). Moreover, patients with chronic pelvic pain exhibited a generalized expansion of reflex receptive fields in comparison with pain-free volunteers (Neziri et al. 2010).
The underlying mechanisms include collateral sprouting, unmasking of normally inactive silent synapses, reduction in pre- and postsynaptic inhibition and increased postsynaptic discharge activity.
2.2.7. Sensory Axonal Sprouting
Among the mechanisms associated with neuropathic pain is sprouting which could occur at the distal and central terminals of nociceptive afferents.
- Peripheral Sprouting
Nerve injury leads to chronic pain and exaggerated sensitivity to gentle touch (allodynia) as well as a loss of sensation in the areas in which injured and non-injured nerves come together. In mice after nerve injury, genetically labeled populations of afferent fibers that sense noxious stimuli (nociceptive afferents) and gentle touch (low-threshold afferents) peripherally in the skin were longitudinally and non-invasively imaged for longer than 10 months, while in parallel tracking pain-related behavior. Fully denervated areas of skin initially lost sensation, gradually recovered normal sensitivity and developed marked allodynia and aversion to gentle touch several months after injury. This re-innervation-induced neuropathic pain involved nociceptive afferents that sprouted into denervated territories, precisely reproducing the initial pattern of innervation, and showed irregular terminal connectivity in the skin and lowered activation thresholds mimicking low-threshold afferents. Genetic ablation of nociceptive afferents fully eliminated re-innervation allodynia. This demonstrared the emergence of a form of CNP that is driven by structural plasticity, abnormal terminal connectivity and malfunction of nociceptors during re-innervation (Gangadharan et al. 2022).
- Central Sprouting
DH lamina II contains NS neurons that generally are not stimulated by myelinated afferent input from mechanical receptors. After partial DRG crush injury, the mechanical force causing footpad withdrawal was measured with von Frey hairs, and myelinated primary afferents were labeled with horseradish peroxidase. After partial DRG injury, mechanical allodynia developed in the corresponding footpad within three days and persisted throughout the experimental period. At two and four weeks after the injury, horseradish peroxidase-positive fibers, presumably myelinated afferents, sprouted into DH lamina II on the injured side, but not on the contralateral control side (Nakamura and Myers 2000).
Following peripheral nerve section, injured sensory group A afferents project into DH lamina II and form aberrant functional synapses. Such changes may underlie some of the sensory abnormalities in nerve-injured patients, including neuropathic pain. In two rat models of neuropathic pain, the ability to sprout of intact and injured group A afferents was compared. Rats received either a unilateral CCI of the sciatic nerve or the L5 SNL. The central distribution of the injured and un-injured afferents was examined at different post-operative survival times. In both models, the contralateral un-injured side, used for control nerve or DRG injections, showed labeling of the L3-6 spinal segments in laminae I, III-V, leaving lamina II un-labelled. In CCI rats, injured sciatic afferents sprouted in lamina II of the L4-5 DH by 10 days post-injury. In SNL rats, injured L5 afferents sprouted into lamina II of the L4-5 DH by 24 hour post-injury and were robust from three to ten days. In both models, the labelling in lamina II was absent by 4 months. Labeling of the adjacent un-injured saphenous or intact L4 spinal nerve afferents did not reveal group A afferent sprouting. The time-course of sprouting of injured group A afferents paralleled the behavior (Shortland et al. 1997).
In the thoracic spinal cord of naïve adult mouse, all myelinated nociceptive afferents gave rise to terminal projections throughout the superficial DH laminae I-II. Most (70%) of these fibers had large-diameter axons with recurving flame-shaped central arbors that projected throughout the DH laminae I-V. After peripheral nerve axotomy, LTMR afferents with narrow, un-inflected somal action potentials did not sprout into superficial DH laminae. Only myelinated nociceptive afferents with broad, inflected somal action potentials gave rise to recurving collaterals and projected into superficial `pain-specific´ laminae after axotomy. Hence, the central morphology of large, myelinated cutaneous nociceptors may account for the morphological findings previously thought to require sprouting of LTMRs (Woodbury et al. 2008).
Tactile allodynia is a sign of chronic pain in which innocuous touch becomes painful. Data suggested that, after injury, aberrant axon sprouting from touch sensory afferents into pain-processing spinal laminae might contribute to mechanical allodynia. This hypothesis was later challenged by experiments using intra-axonal labeling of group A afferents, as well as single-neuron labeling of electrophysiologically identified sensory neurons. In genetically modified mice, group II (Aβ) trigeminal touch afferents were specifically labeled by timed taxomifen injection prior to inflammation or infra-orbital nerve (IoN) transection). The peripheral and central projections of labeled axons into the brainstem caudalis nucleus were examined after injuries vs controls. There was no evidence for ectopic sprouting labeled group II (Aβ) axons into the superficial trigeminal noci-receptive laminae. There was also no evidence for peripheral sprouting. Hence, touch allodynia in chronic orofacial pain is unlikely caused by ectopic sprouting of group II (Aβ) trigeminal afferents (Zhang et al. 2015).
2.2.8. Changes in Network Structures
Under healthy conditions, pathways processing noxious stimuli and innocuous tactile stimuli are crudely separated. During noxious activation, the superficial DH layers receive excitation from nociceptive group IV (C) afferents, which activate neurokinin 1 (NK1)-projection neurons located in lamina I. Tactile input is conveyed by myelinated group II (Aβ) afferents, which activate projection neurons in the deep DH (layers III-V), in which most cells are WDR. These neurons respond to many different types of stimuli, possess very large receptive fields and code for stimulus intensity among other features (Peirs et al 2015). It has been suggested that these different pathways are differentially recruited in inflammatory of neuropathic pain states. Impaired segregation of signal relay in the superficial versus deep DH is thought to underlie allodynia, which may result from the abnormal activation of lamina I neurons by group II (A(β) afferents. Lamina I projection neurons normally become activated only in response to noxious input. After blockade of GABAA receptors and GlyRs, these neurons become excitable also by input from group II (A(β) afferents through a polysynaptic pathway (Zeilhofer et al. 2021).
Polysynaptic Connections between both pathways have various configurations, of which only one will be described here. These connections are normally silenced by several types of inhibitory INTs, among which are PV-positive neurons, dynorphin (Dyn)-positive INTs and islet cells (Figure 2). Dyn and PV neurons evoke postsynaptic responses with a strong Gly component. About half of the more superficially located islet cells release Gly in addition to GABA. Many of these inhibitory INTs are activated by input from nociceptors or non-nociceptive myelinated group II (Aβ) afferents and thereby provide feed-forward inhibition (Zeilhofer et al. 2021). However, further INTs may contribute, namely excitatory INTs in the inner lamina II, which express the isoform γ of protein kinase C (PKC-γ). They receive inputs from low-threshold afferents such as group II (Aß) and from group IV (C) afferents, and innocuous stimuli could thus activate PKC-γ INTs. However, PKC-γ INTs are normally inhibited by Gly and GABA neurons. Loss of inhibition would gate low-threshold afferent inputs to NS projection neurons (Berger et al. 2011).
Inflammation. In inflammation, the pathway starts with cholecystokinin (CCK)-positive excitatory INTs that are activated by group II (A(β) afferents and project to calretinin (CR)-positive neurons, which then excite NK1 projection neurons in lamina I (Zeilhofer et al. 2021). During inflammation, the `gate´ that controls CR neurons is opened resulting in the activation of the pathway involving vesicular glutamate transporter 3 (VGLUT3), CR-vertical cell (VC), and NK1R cell (Peirs et al. 2015).
Neuropathy. In neuropathy, the pathway also begins with CCK-positive INTs, which activate PKCγ positive INTs. The PKCγ INTs then project to vertical cells, which connect to lamina I NK1 projection neurons. PKCγ-positive neurons can also be directly activated by group II (Aβ) afferents. Several types of inhibitory INTs silence these polysynaptic pathways, among them are PV-positive neurons, dynorphin positive INTs (Dyn), and so-called islet cells. Dyn and PV neurons evoke postsynaptic responses with a strong glycinergic component. About half of the more superficially located islet cells release Gly in addition to GABA. Many of these inhibitory INTs are activated by input from nociceptors or non-nociceptive Aβ fibers and thereby provide feed-forward inhibition. Schemes are based on (Zeilhofer et al. 2021). In rats with inflamed paws, the glycinergic input to lamina I neurons is reduced. Altered heat hyperalgesia in the α3 GlyR-deficient mice may reflect changes in the glycinergic control of superficial DH neurons, as heat stimuli are primarily processed in this area (Zeilhofer et al. 2021).
Isolated circuits have been suggested to play a role in neuropathy. Some examples follow.
(i) After nerve injury, group II (Aß) inputs gain access to lamina I projection neurons by two mechanisms. Inhibitory vertical cells (VCs) show reduced inhibitory drive to excitatory VCs removing the inhibitory gate. Inhibitory Dyn cells show reduced inhibitory tone, resulting in increased access of group Aß inputs to excitatory lamina II, resulting in increased activation of projection neurons (West et al. 2015).
(ii) Under normal conditions, inhibitory PV INTs mediate presynaptic inhibtion of group A LTMR inputs and postsynaptic inhibition of vertical cells and PKCγ-expressing cells. Peripheral nerve injury reduces PV excitability leading to spinal disinhibition (Hughes and Todd 2020).
(iii) The DH hosts a subset of inhibitory INTs operated by adeno-associated viral vectors incorporating an NPY promoter (AAV-NpyP+), whose specific ablation or silencing converted touch-sensing group II (Aβ) fiber-derived signals to morphine-resistant pain-like behavioral responses. AAV-NpyP+ neurons received excitatory inputs from group II (Aβ) and transmitted inhibitory GABA signals to lamina I neurons projecting to the brain. In a model of neuropathic pain developed by peripheral nerve injury, AAV-NpyP+ neurons exhibited deeper resting membrane potentials, and their excitation by group II (Aβ) fibers was impaired. Conversely, chemogenetic activation of AAV-Npy P+ neurons in nerve-injured rats reversed group II (Aβ) fiber-derived neuropathic pain-like behavior that was morphine-resistant and reduced pathological neuronal activation of superficial DH including lamina I (Tashima et al. 2021).
(iv) During chronic pain, allodynia involves the excitation of lamina I, II and IIIo projection neurons by tactile inputs. The mechanisms cross-connecting touch and nociceptive pathways are diverse. One involves excitatory PKCγ INTs that express the PKCγ. One type of PKCγ INTs located in laminae I and IIo could receive nociceptive and thermoreceptive inputs from groups III (Aδ) and IV (C) fibers. PKCγ INTs in laminae IIi to III could recieive inputs from groups II (Aß), III (Aδ) and LTMR IV (C) fibers. In physiological conditions, LTMR inputs activate inhibitory DH INTs, which may control activation of second-order nociceptive neurons. During chronic pain, LTMR inputs now activate PKCγ neurons that forward the message to second-order nociceptive neurons, turning thus tactile inputs into pain. Several mechanisms may contribute to opening this gate, including disinhibition, activation of local astrocytes, release of diffusible factors, such as ROS, and alteration of the descending 5-HT control on PKCγ neurons through 5-HT2A receptors (Artola et al 2020).
Vesicular Glutamate Transporters 3 (VGLUTs) package glutamate into synaptic vesicles for regulated release. VGLUT3 is required specifically for acute mechanical pain and the persistent mechanical pain that develops in various models including inflammatory and neuropathic pain. The transient expression of VGLUT3 by a discrete population of neurons in the deep DH is required for mechanical pain and activation of the cells in the adult conveys mechanical hypersensitivity. Analysis of c-fos revealed that the circuit extends dorsally to nociceptive lamina I projection neurons, and includes lamina II CR neurons, which also convey mechanical allodynia. In inflammatory and neuropathic pain models, multiple micro-circuits in the DH encode this form of pain. The scheme for mechanical allodynia is as follows. Transient VGLUT3 cells located in lamina III transmit the input from myelinated fibers to the more dorsal neural network underlying mechanical hypersensitivity. Neighboring cells in lamina III receive input from transient VGLUT3 neurons and relay the signal to lamina II cells including excitatory PKCγ-positive and CR-positive INTs that act to refine the excitability of the circuit. Vertical cells in lamina IIo, which receive input from transient VGLUT3 cells and lamina II cells, integrate the signal and send an output to the nociceptive NK1R-positive lamina I projection neurons. In persistent pain induced by nerve injury, the `gate´ that controls PKCγ is opened resulting in activation of the VGLUT3-PKCγ-CR-vertical cell-NK1R pathway (Peirs et al. 2015).
In mice, dynamic mechanical hypersensitivity induced by nerve injury or inflammation was reduced by ablating a group of adult spinal neurons defined by developmental co-expression of vesicular glutamate transporter 3 (VGLUT3) and Lbx1 (VT3Lbx1 neurons). The mice lost brush-evoked nocifensive responses and conditional place aversion. Electrophysiological recordings showed that VT3Lbx1 neurons form morphine-resistant polysynaptic pathways relaying inputs from low-threshold group II (Aβ) mechano-afferents to lamina I output neurons. The subset of somatostatin (STT)-lineage neurons preserved in VT3Lbx1-neuron-ablated mice was largely sufficient to mediate morphine-sensitive and morphine-resistant forms of von Frey filament-evoked punctate mechanical hypersensitivity. Moreover, acute silencing of VT3Lbx1 neurons attenuated pre-established dynamic mechanical hypersensitivity induced by nerve injury (Cheng et al. 2017).
Longitudinal Ca2+ imaging of DH projection neurons was applied to determine whether and how the representation of somatosensory stimuli in the antero-lateral tract changes between distinct pain states. In healthy mice, stable outputs selective for cooling or warming and a neuronal ensemble activated by noxious thermal and mechanical stimuli were identified. Induction of acute peripheral sensitization by topical capsaicin transiently re-tuned nociceptive output neurons to encode low-intensity stimuli. In contrast, peripheral nerve injury resulted in a persistent suppression of innocuous spinal outputs coupled with persistent activation of a normally silent population of high-threshold neurons. This demonstrate differential modulation of spinal outputs to the brain during nociceptive and neuropathic pain states (Yarmolinsky et al. 2025).
Acupuncture diminishes pain by exciting somatic afferent nerve fibers. In rats, the mechanism was explored by which electro-puncture (EA) relieves inflammatory muscle pain, which was associated with activation of the spontaneous firing of LTMR neurons and inhibition of WDR neuronal activities in the DH. Inflammatory muscle pain was induced by injecting complete Freund´s adjuvant (CFA) into the right biceps femoris muscle. EA with intensity at threshold of group A fibers (Ta) in Liangqiu (ST34) muscle considerably inhibited the abnormal spontaneous activities of electromyography (EMG) due to muscle inflammation. While EA with intensity of group IV(C)-fiber threshold (Tc) increased the abnormal activities of EMG, EA with Ta also ameliorated the imbalance of weight-bearing behavior. A microelectrode array with 750-μm depth covering 32 channels was used to record the neuronal activities of WDR and LTMR in different DH layers. The spontaneous firing of LTMR neurons was enhanced by EA-Ta, while the spontaneous firing of WDR neurons was inhibited. Moreover, EA-Ta led to a significant inverse correlation between changes in the firing rate of WDR and LTMR neurons. Hence, EA could alleviate inflammatory muscle pain, which was associated with facilitation of the spontaneous firing of LTMR neurons and inhibition of WDR neuronal activities (Duan-Mu et al. 2021).
2.2.9. Synaptic Changes
Synaptic processes in the DH might help initiate the transition from acute to chronic pain. Low-level stimulation of nociceptor afferents triggers release of glutamate from the central terminals terminating in DH laminae I, II and V. During intense or sustained nociceptive activity, SP and glutamate are co-released from afferent terminals, enabling temporal summation of second pain through activation of NMDARs. Hence, intracellular Ca2+ concentrations increase, which activate signaling cascades that lower the firing threshold of DH neurons (Staud 2012). Hyperalgesia and allodynia following inflammation or nerve injury are in part due to activity-dependent pre- and postsynaptic changes, such as LTP and increases in the density of synaptic spines (Baumbauer et al. 2009; Costigan et al. 2009; Grau 2014; Kuner and Flor 2016; Li et al. 2019; Merighi 2024; Sandkühler 2009).
- Spinal Short-term Plasticity (STP)
The efficient processing of somatosensory messages within the DH involves frequency-tuned synapses, a phenomenon linked to their ability to display activity-dependent forms of short-term plasticity (STP). These STP properties allow for a powerful gain control in DH neuronal networks that may be critical for the integration of nociceptive messages. Moreover, these STPs can be finely modulated by endogenous signaling molecules, such as neurosteroids, adenosine, or GABA. The STP properties of DH inhibitory synapses might also, at least in part, participate in the pain-relieving effect of non-pharmacological analgesic procedures. The properties of target-specific STP at inhibitory DH synapses might possibly contribute to electrical stimulation-induced reduction of hyperalgesic and allodynic states in chronic pain (Cathenaut et al. 2023).
- Long-term Potentiation (LTP)
High-rate electrical stimulation of group IV (C) afferents induces LTP in spinal neurons (Treede 2016). In rat DH neurons of laminae I-III, LTP of excitatory postsynaptic potentials (EPSPs) can be evoked by brief, high-rate tetanization of dorsal-root fibers in the group III (Aδ) to group IV (C) range. LTP is also seen in WDR cells in the rat DH after high-frequency stimulation of the sciatic nerve (Svendsen et al. 1997) and probably contributes to sensitization of spino-thalamic tract (STTr) neurons (Willis 2002).
Spinal LTP requires the activation of a sub-set of superficial DH neurons expressing the NK1-R that mediate certain forms of hyperalgesia. These neurons participate in local sensory processing, but are also the origin of a spino-bulbo-spinal loop driving a 5-HT3 receptor (5HT3R)-mediated descending facilitation of spinal pain processing. NK1R-expressing cells in the superficial DH are crucial for the generation of LTP-like changes in neuronal excitability in deep DH neurons, and this was modulated by descending 5HT3R-mediated facilitatory controls. Hence, following peripheral injury, the generation of LTP in DH neurons may be one mechanism whereby acute pain can be transformed into a long-term pain state (Rygh et al. 2006).
In NK1R-expressing projection neurons in lamina I, two different forms of LTP occur following dorsal-root stimulation at group IV (C) fiber strength and are considered to operate through Ca2+-dependent postsynaptic mechanisms. One form, induced by high-frequency (100 Hz) stimulation, was restricted to neurons retrogradely labelled from lateral parabrachial nucleus (lPBN), while the other, induced only by low-frequency (2 Hz) stimulation, occurred in cells labelled from the PAG. As NK1R-expressing projection neurons have an important role in the development of chronic pain states, LTP of their nociceptive inputs may play a significant part in this phenomenon (Todd 2010).
- Windup
When group IV (C) fibers are stimulated at frequencies between 0.5 and 5 Hz, some postsynaptic neurons respond with an increasing discharge rate to the first 10–30 stimuli (i.e., in the first few seconds of an ongoing noxious stimulation). Thereafter, the response reaches a plateau or may decline. Windup occurs under normal experimental conditions, i.e., in the absence of any intentional inflammation, trauma, or nerve injury and thus constitutes a normal coding property of some nociceptive spinal DH neurons. Windup per se is thus not a mechanism of hyperalgesia. However, some form of signal amplification can be induced, for example, LTP of synaptic strength between primary group IV (C) afferents and spinal DH neurons (Sandkühler 2009). WDR neurons with inputs from nociceptive and non-nociceptive sensory afferents (Figure 2) also show windup, but group A fiber inputs do not show this phenomenon. Prolonged low-rate stimulation turns windup into long-term depression (LTD) (Treede 2016).
- Nitric Oxide (NO)
NO is a diffusible gas and appears to act as an anterograde neurotransmitter between primary afferent neurons and non-neuronal cells in DRG and between different neuronal cell types in the spinal cord. Although not important for normal nociceptive processing, NO plays an essential role in central sensitization during inflammatory and neuropathic pain in conjunction with cyclic guanosine monophosphate (cGMP), and several signaling pathways (Dibaj et al. 2010; Schmidtko et al. 2009). NO can activate both pro-nociceptive and anti-nociceptive mechanisms (Schmidtko 2015).
- Changes in Synaptic Connections
In the SNL mouse model of neuropathic pain, whole-cell electrophysiological recordings with laser-scanning photo-stimulation was used to test whether peripheral nerve injury leads to alterations in the functional connectivity of spinal cord circuits including lamina II excitatory INTs. Indeed, SNL enhanced excitation and decreased inhibition to lamina II excitatory INTs along with their increased glutamate-evoked excitability. The enhanced excitatory postsynaptic input and connectivity evoked by SNL eventually returned to normal levels concurrently with the resolution of the neuropathic pain states. This supports the notion that SNL induces functional changes in synaptic input and connectivity to lamina II excitatory INTs that code for pain perception (Gong et al. 2019).
2.2.10. Activation of the Immune System
Inflammatory agents and mediators have a role not only in nociceptor sensitization, but also in the spinal cord and higher centers. For example, in a rat model of mono-neuropathy, increased concentrations of IL-6 contribute to hyperalgesia and allodynia (Saadé and Jabbur 2008).
Within the DH, peripheral nerve injury activates microglia, which up-regulates the expression of P2X and P2Y purinergic receptors, e.g., P2X4, which in turn leads to the release of BDNF and activation of its receptor, TrkB (Inoue 2021; Inoue and Tsuda 2018; Jarvis 2010; Tam and Salter 2021). Among other released substances are prostaglandins, TNF, IL-1, IL-6 and NO, which in turn change neuronal and synaptic functions via a multitude of mechanisms, and reduce the pain-suppressive effects of opioid drugs by contributing to opioid tolerance (Watkins et al. 2007). Paradoxically, opioids can even induce hyperalgesia (OIH) via a spectrum of mechanisms (Roeckel et al. 2016). Glia cells also influence synaptic transmission including LTD, and immune mediators modulate glutamatergic and GABAergic signaling (Austin and Fiore 2019). Finally, immune-system activation affects neuronal excitability at multiple sites, from peripheral nerve to DRG to spinal cord and beyond (Basbaum et al. 2009; Costigan et al. 2009; Dibaj et al. 2010, 2024; Pinho-Ribeiro et al. 2017; Saab et al. 2008; Saab and Hains 2009).
- Myositis
At spinal level, myositis changed the connectivity of DH neurons, reflected in an expansion of the input (target) region of the muscle nerve. Such central sensitization can explain the hyperalgesia and spread of pain in patients. The transition from acute to chronic pain is accompanied by structural changes. In rats with muscle inflammation or nerve lesions, such morphological changes become apparent within a few hours after the lesion (Mense 2004).
- Chronic Stress
Chronic stress contributes to the maintenance of chronic pain states, and vice versa. Stress elevates the concentrations of glucocorticoids. Since astrocytes and microglia express high concentrations of glucocorticoid receptors (GRs), prolonged glucocorticoid exposure entails the activation and hypertrophy of glia cells, which in turn leads to maladaptive neuronal and glial plasticity with structural and functional changes and maintenance of neuropathic pain (Madalena and Lerch 2017).
2.3. The PAG-Triad Connection
Besides abnormal sensitization in the periphery and spinal cord, neural plasticity in the brain is critical for the development and maintenance of neuropathic pain. Pathological neural plasticity occurs in many pain-related brain areas (Figure 3). Neuropathic pain is associated with adaptations in the descending modulatory pain systems involved in NA and 5-HT neurochemical control, some of which are mediated by local opioidergic effects. Major changes occur at the LC, RVM, and dorsal reticular nucleus (DReN) and affect top-down modulation of nociceptive transmission at the superficial DH (Tavares et al. 2021).
2.3.1. Peri-Aqueductal Gray (PAG)
The midbrain PAG is a cell-dense region surrounding the midbrain aqueduct. Its anatomical and functional organization is characterized by longitudinal columns of afferent inputs, output neurons and intrinsic INTs (Bandler and Shipley 1994; Koutsikou et al. 2017). The PAG receives nociceptive inputs from the STTr (Kuner and Kuner 2021). Direct and indirect inputs come from the mPFC, ACC, AMY and dorso-medial nucleus of HYP (DMH) (Heinricher et al. 2008). The ventro-lateral peri-aqueductal gray (vlPAG) receives inputs from regions that are targets of the ascending nociceptive fibers, including the parabrachial nucleus (PBN) and spinal cord. Conversely, the lateral peri-aqueductal gray (lPAG) receives direct inputs from the DH and SpV organized in a roughly somatotopic map, with orofacial afferents terminating rostrally and afferents from the legs terminating caudally (Mills et al. 2021). The PAG sends pain-modulatory signals to the RVM via its reciprocal connections (Bak et al. 2021).
Together with other descending pain-modulating pathways, the PAG and the triad of RVM, CVLM and DReT forms the final stage of an influential axis. Dysregulation of this axis could result from two broad classes of changes: (i) changes in inputs, and (ii) changes in internal properties. The first icludes inputs from mPFC and ACC (Figure 4).
Imbalance between Descending Inhibition and Facilitation. During chronic pain states, changes may occur in the balance between descending inhibition and descending facilitation of nociception via the axis from the PAG and RVM to the spinal cord (Heinricher et al. 2008; Kuner and Kuner 2021; Lima and Almeida 2002; Martins and Tavares 2017; Porreca et al. 2002; Ren and Dubner 2007; Zhou 2017).
Changes in Connectivity. After nerve injury, connections of the PAG to other brain regions undergo plastic changes, these changes being strongly associated with the development of neuropathic pain. In brain slices of mice with neuropathic pain induced by CCI, connections between the PAG and the mPFC changed. Cortico-PAG neurons showed a significant reduction in intrinsic excitability. Descending visceral nociception control that may contribute to gut injury-associated visceral hyperalgesia (Lyubashina et al. 2022). PAG. This highlights the cellular and molecular changes in PAG after neuropathic pain interfering with their descending modulatory role (Boadas-Vaello et al. 2017). In patients with CNP, stronger functional connectivity between the PAG and the THAL occurred, possibly reflecting changes in ascending nociceptive pathway activity. In animals, neuropathic pain was associated with morphological and functional changes in the mPFC, including reduced inhibitory drive from the mPFC to the PAG. This might suggest a shift in the excitatory/inhibitory balance in this pathway, which may contribute to a pro-nociceptive shift in projections descending from the PAG and thus the persistence of ongoing pain following nerve injury (Mills et al. 2021).
Stress-induced Hyperalgesia (SIH). SIH is induced by repeated or chronic exposure to stressful or uncomfortable environments. Modulatory effects of the PAG and its associated circuits might play a role in SIH development during chronic restraint stress (CRS). CRS-induced hyperalgesia was manipulated by altering neuronal activity via a pharmaco-genetic approach. Activation of PAG neurons alleviated CRS-induced hyperalgesia. Conversely, inhibition of PAG neurons facilitated CRS-induced hyperalgesia. This modulatory effect was achieved by the neurons, which project to the RVM. Hence, the PAG-RVM circuit has a functional role in SIH (Chen et al. 2024b).
Optogenetic approaches combined with pharmacology revealed that the pathway from the baso-lateral amygdala (BLA) via PFC to PAG regulated neuropathic pain behaviors, such as mechanical and thermal hypersensitivity. SNI enhanced synaptic inputs from the BLA to inhibitory neurons in the mPFC, resulting in decreased analgesic modulation by inhibiting at protein projections from the mPFC to the PAG. Projection neurons in the central medial (CeM) AMY to the PAG underwent plastic changes due to inflammatory pain (Bak et al. 2021).
Intrinsic Peri-aqueductal Gray (PAG) Changes. In gut inflammation-triggered chronic abdominal pain, an association of structural and neuronal alterations within the PAG exists. In urethane-anesthetized male Wistar rats, c-fos immuno-histochemistry and extracellular micro-electrode recording were used to evaluate the colitis-induced changes in visceral pain-related neuronal properties of the PAG and its descending outflow to visceral nociceptive neurons of the CVLM. Analysis of c-fos protein expression in inflamed animals showed diminished activation of the lPAG and vlPAG columns by noxious colo-rectal distension (CRD), although the non-stimulated c-fos labeling in these PAG sub-divisions was enhanced compared with that in controls (Lyubashina et a. 2022).
Cellular and Molecular Changes in Peri-aqueductal Gray (PAG). After CCI in rats, glial reactivity increased in the PAG, and immuno-histo-chemistry showed the concentrations of TNF-α, IL−1ß, and IL-6 were significantly increased in the PAG of CCI. In addition, the PAG of CCI rats significantly increased the expression of GR mRNA and protein. Consequently, PAG neurons are sensitive to the increase in corticosterone occurring after CCI, whereas endocannabinoid (eCB) receptor type 1 (CB1) decreased in both CCI rats and SCI rats, despite the fact that the concentrations of eCBs increased in PAG after CCI, suggesting a down-regulation of the eCB system that modulates pain at PAG. Peripheral nerve ligation-induced chronic pain was associated with an increased NMDA/non-NMDA ratio in 5-HT neurons of PAG. These changes in the expression of NMDARs in PAG entail an alteration of the descending modulation of nociception, which might be an underlying mechanism for peripheral nerve injury-evoked persistent pain. Likewise, diabetic neuropathy is accompanied by a progressive increase in spontaneous neuronal activity in the spinal cord and spinal signal transmission. 5-HT from the RVM and NA from the LC to the spinal DH are major outputs for these pain modulation system projections from the mPFC to the PAG (Bak et al. 2021).
2.3.2. RVM-CVLM-DReN Triad
The medulla oblongata contains a number of structures that are intimately related to pain, but also to associated regulatory functions like cardio-vascular and ventilatory ones. As to pain, a so-called `triad´ has been proposed to be of great importance in pain processing and modulation. Based on the importance of spino-bulbo-spinal loops in central sensitization, the reciprocal connections of the RVM, CVLM, and DReN with the spinal cord (Figure 4) provide an important anatomical background for the participation of those areas in central sensitization during chronic pain. Stimulation in the CVLM strongly inhibits behavioral and DH nociceptive responses, whereas lesions result in apparent disinhibition. This suggests that the CVLM exerts a tonic inhibitory control of DH nociception. Nevertheless, the CVLM may, like the RVM, exert a facilitatory influence, as neurons with features of ON- and OFF-cells have been identified in this region (Heinricher et al. 2008).
The endogenous pain modulatory system is a complex network of brain areas that control nociceptive transmission at the spinal cord by inhibitory and facilitatory actions. The balance between these actions ensures effective modulation of acute pain, while during chronic pain the pro-nociceptive effects appear to dominate (Tavares and Lima 2007). Descending facilitation is activated after nerve injury and its neurotransmitter 5-HT acts through the 5-HT3 receptor (Treede 2016).
2.3.3. Rostral Ventro-medial Medulla (RVM)
Functionally, the RVM is defined as the area in which electrical stimulation or opioid micro-injection produces analgesia. It consists of several nuclei, including the midline nucleus raphé magnus (NRM) and lateral NGc, as well as GABAergic and glycinergic cell populations, all of which project diffusely to the SpV and to superficial and deep DH layers (Heinricher et al. 2008; Mills et al. 2021; Ossipov et al. 2014). The RVM receives nociceptive inputs via the PBN, a dense input from the PAG, and further inputs from the THAL, HYP and a variety of other cortical and sub-cortical areas as well as from the NA LC (Bouchet and Ingram 2020; Ossipov et al. 2014).
- Input/Output
The RVM receives nociceptive inputs via the PBN, a dense input from the PAG, and further inputs from the THAL, DMH and a variety of other cortical and sub-cortical areas as well as from the NA LC (Bouchet and Ingram 2020; Ossipov et al. 2014). In humans, fMRI studies showed that somatic and visceral noxious stimulation led to activation of brainstem regions including the PAG and RVM, and at the primary nociceptive synapse, either at the SpV or DH, during noxious muscle and cutaneous inputs (Mills et al. 2021). The central nucleus of the amygdala (CeA) output targets the RVM and PAG, which are crucial for mediating behavioral coping responses in the face of threat (Bak et al. 2021; Kuner and Kuner 2021). The RVM bi-directionally modulates nociceptive processing through pro-nociceptive `ON-cells´ and anti-nociceptive `OFF-cells´ that project to the spinal DH as well as the trigeminal nucleus caudalis (SpVc), including superficial and deep DH layers that receive nociceptor primary afferents (Bak et al. 2021; Heinricher et al. 2008; Mills et al. 2021).
- ON- and OFF-cells
The DH status is co-determined by neural circuits involving inputs and outputs, which affect ON- and OFF-cell excitability. Many neuromodulators/neurotransmitters/ion channels are involved, including amino acids, SP, CCK, 5-HT, cannabinoids, opioids, and their dynamic impact on both ON and OFF cell activities in modulating pain transmission (Peng et al. 2023).
- Imbalance between Inhibition and Facilitation
It has been suggested that an imbalance between the inhibitory and facilitatory descending pain modulatory systems may underlie pain chronification into a pathological pain state. Both ON- and OFF-cells are simultaneously activated in the early stages of pain. However, during pain chronification, ON-cells showed a stronger signal strength, overcoming the OFF-cells to promote chronic pain. Some data point to chronic stress as a trigger for pain chronification and related psychiatric comorbidities, e.g., anxiety and depression, which are associated with maladaptive responses to stress (Pagliusi and Gomes 2023).
- μ-Opioid RVM Cells
It has been shown that, in male mice subjected to partial sciatic nerve ligation, activity of spinally projecting μ-opioid RVM cells is required for expression and manifestation of both sensory and affective dimensions of established neuropathic pain and to promote descending facilitation that overcomes apparently intact descending inhibition to maintain chronic pain. Enhanced descending facilitation likely regulates the output signal from the spinal cord to the brain to shape the pain experience and may provide a mechanism for nonopioid management of pain. (Dogrul et al. 2025).
- Wide-dynamic Range (WDR)Neurons
Models of neuropathic and inflammatory pain have shown that the hyperexcitation of specific nociceptive and WDR neurons in the DH caused hyperexcitability and sensitization of the neurons in the RVM that facilitate the descending pain modulation of the DH. In the RVM, which contains the NRM, ON-neuron responses were strengthened and OFF-neuron response weakened. ON-cells exerted a pro-nociceptive effect, whereas OFF-cells produced an anti-nociceptive effect. The preferential activation of RVM ON-cells caused hyperalgesia, whereas hypoalgesia entailed from the activation of OFF-cells. ON-neurons did not anymore respond to inhibitory signals from PAG, whereas they were strongly stimulated by ascending inputs (Boadas-Vaello et al. 2017).
2.3.4. Caudal Ventro-Lateral Medulla (CVLM)
In several species including mouse, rat, cat, monkey and man, the CVLM is located in the ventro-lateral quadrant of the caudal-most aspect of the medulla oblongata (Figure 3).
- Nociceptive Inputs
The ascending projections from the spinal cord to the CVLM are anatomically segregated, with the lateral CVLM (CVLMlat) receiving mainly afferents from the superficial DH, namely from nociceptively responsive neurons located in lamina I. An important proportion of VLMlat-projecting neurons is located at lamina II, which appears to be a special feature of this spino-fugal pathway. Circuits capable of conveying CVLM-elicited anti-nociception include a direct reciprocal CVLM-spinal loop (Figure 4). Additionally, the CVLM is also bi-directionally connected with both the RVM and DReN, building a cooperative `triad´ (Martins and Tavares 2017).
In pentobarbitone-anesthetized control and mono-arthritic rats, electrophysiological recordings were used to characterize neuronal responses to noxious pinch, heat, cold and colo-rectal distension. CVLM neurons gave excitatory, inhibitory or no response to noxious test stimulation. Response patterns for part of the neurons varied with sub-modality of test stimulation; e.g., a cell with an excitatory response to heat could give no or an inhibitory response to cold (Pinto-Ribeiro et al. 2011). Somatic and visceral of pain exhibit crucial differences not only in the experience, but also in their peripheral and central processing. -- In urethane-anesthetized adult male Wistar rats, responses of CVLM neurons were investigated to visceral (CRD) and somatic (squeezing of the tail) noxious stimulations. The CVLM of healthy control rats, along with hosting cells excited by both stimulations (23.7%), contained neurons that were activated by either visceral (31.9%) or somatic noxious stimuli (44.4%). In inflamed animals, the percentages of the visceral and somatic nociceptive cells were decreased (to 18.3% and 34.3%, correspondingly) and the number of bimodal neurons was increased (up to 47.4%). These changes were associated with substantially enhanced responses of both the modality-specific and convergent CVLM neurons not only to CRD, but also to squeezing of the tail (Lyubashina et al. 2019). -- In anesthetised rats, following both direct muscle stimulation and L5 ventral-root stimulation, fatigue-related c-fos gene expression was most prominent in the DH of the ipsilateral L2-L5 segments and within the ipsilateral NTS, the CVLM and RVL, and the intermediate reticular nucleus, and contralaterally (Maisky et al. 2002). Note that muscle fatigue activates group III/IV afferents.
- Other Inputs
The CVLMlat receives inputs from the somatosensory and motor cortices, the IL PFC, insular (IC), and limbic cortices, the CeA, lateral (lHyP), posterior HYP (pHYP), HYP paraventricular nucleus (PVN), PAG, red nucleus, PBN, NRM, NTS, lateral reticular nucleus (LRN), dorsal and ventral medullary reticular formation (RF), and the lateral cerebellar nucleus (Cobos et al. 2003). The CVLMlat is also activated in response to increases in blood pressure. Increases in blood pressure are a feature of the `fight or flight”´ response. Altogether, the VLM is an integrative center which is involved in producing the adequate pain, motor and cardio-vascular responses (Martins and Tavares 2017).
- Outputs
The CVLMlat projects to the PAG, red nucleus or lateral cerebellar nucleus (Cobos et al. 2003). The projections to spinal DH laminae involved in nociceptive transmission originate exclusively in the CVLMlat. The CVLMlat is integrated in a disynaptic pathway involving spinally projecting pontine NA A5 cells (Sect 2.8.2), which appears to convey α2-adreno-receptor-mediated analgesia produced from the VLM. The descending CVLMlat-spinal pathway targets lamina I, IV–V and X. Electrophysiological mapping of the VLM has shown that it contains inhibitory neurons (OFF-like neurons) along with excitatory cells (ON-like cells) which indicates that the descending modulation from the VLM may include facilitatory modulation, along with the inhibitory effects. Neurons in the CVLMlat and in DH lamina I are reciprocally connected by a closed loop that is likely to mediate feedback control of supraspinal nociceptive transmission (Martins and Tavares 2017).
Acute and chronic Inflammation. Inflammation induced by intra-articular injection of a solution of PGE2 and bradykinin induced a strong activation both at the VLM and the spinal cord. This suggests that at the initial phases of inflammation, descending inhibition from the VLM fails to inhibit the strong nociceptive transmission arising from the spinal cord. With chronic inflammation induced by CFA into the knee joint, innocuous stimulation of the affected paw gives rise to an inverse correlation between c-fos expression in the VLM and DH, suggesting that descending inhibition is sufficient to suppress spinal activation. However, when intense pinch is applied to the same limb, strong c-fos expression is seen at both levels, implying either that the c-fos expression represents activated pain-facilitating neurons or, if expression is in pain-inhibiting neurons, that descending inhibition is insufficient to suppress activation of the DH neurons (Heinricher et al. 2008).
The Spino-CVLM Loop in Chronic Pain. Normally, stimulation in the CVLM strongly inhibits behavioral and DH nociceptive responses, whereas lesions result in apparent disinhibition. This suggests that the CVLM exerts a tonic inhibitory control of DH nociception. Nevertheless, the CVLM may, like the RVM, exert a facilitatory influence, as neurons with features of ON- and OFF-cells have been identified in this region (Heinricher et al. 2008). It has been hypothesized that, in chronic pain, the CVLM changes its spinal actions from inhibitory to excitatory. While in acute pain, the ascending branch in the spino-CVLM loop is under potent excitation of SP released from primary afferents, which is likely to trigger the intense descending inhibition, during chronic pain, the activity in the lateral reticular formation of the CVLM changes, so that the action of the CVLM upon SP-responsive spinal neurons shifts from inhibitory to excitatory. The mechanisms of this modulatory shift are unknown but probably relate to the decreased expression of μ-opioid, δ-opioid and GABAB receptors (Tavares and Lima 2007).
As noted above, colonic inflammation caused a shift in the effects of vlPAG electro-stimulation on CRD-excited CVLM neurons from being mostly inhibitory under normal conditions to excitatory in colitis (Lyubashina et al. 2022).
2.3.5. Dorsal Reticular Nucleus (DReN)
The DReN is located in the caudal-most aspect of the medulla oblongata in several species including rat, cat, monkey and man. It is located in the most caudal, dorso-lateral portion of the medulla (Figure 3; Martins and Tavares 2017). DReN neurons are reciprocally connected with DH lamina I neurons, thus forming a reverberative nociceptive circuit.
- Inputs
The DReN is targeted from fibers originating from deeper layers in the spinal cord, namely from laminae IV–V, which specifically terminate in the lateral part of the DReN, and from lamina VII which terminate at the medial part of the nucleus. DReN neurons are activated only or mainly by noxious stimulation, whose intensity is reflected in the firing rate. They have large receptive fields that often cover the whole body surface. The DReN also receives projections from the A1 and C1 cell groups (Sect 2.8.2) (Lima and Almeida 2002; Martins and Tavares 2017).
- Outputs
The DReN is reciprocally connected with sensory medullary nuclei (e.g., nucleus cuneatus and SpV, pars caudalis), and with many brainstem and diencephalic nuclei involved in anti-nociception and/or autonomic control. Anterograde tracing showed that fibers and terminal boutons labeled from the DReN were located predominately in the brainstem, although extending also to the forebrain. In the rat medulla oblongata, anterograde labeling appeared in the RVM, CVLM, NTS, orofacial motor nuclei, and inferior olive (IO). Labeling was also present in the LC, NA A5 and A7 cell groups (Sect 2.8.2), PBN and deep cerebellar nuclei. In the midbrain, it was located in the PAG, substantia nigra (SN), deep mesencephalic, oculomotor and anterior pretectal nuclei. In the diencephalon, fibers and terminal boutons occurred mainly in THAL nuclei, and in the HYP ARC, PVN, lateral, posterior, peri- and paraventricular areas. Telencephalic labeling was less intense and concentrated in the septal nuclei, globus pallidus (BG) and AMY. This suggests that the DReN is possibly implicated in the modulation of: (i) the ascending nociceptive transmission involved in the motivational-affective dimension of pain; (ii) the endogenous supraspinal pain control system centered in the PAG-RVM-spinal cord circuitry; (iii) the motor reactions associated with pain (Leite-Almeida et al. 2006). The medullary DReN is also reciprocally connected with the spinal DH (Leite-Almeida et al. 2006; Martins and Tavares 2017).
- Dorsal Horn (DH)-Dorsal Reticular Nucleus (DReN)-Cerebellum Connections
The cerebellum receives input from nociceptors which may serve to adjust motor programs in response to pain and injury. A significant proportion of spino-reticular DH cells projecting to the DReN respond to noxious mechanical stimuli. One of the functions of this pathway may be to provide the cerebellum with nociceptive information (Huma et al. 2015).
The DReN is a distinctive pain-facilitatory area and an important player in the diffuse noxious inhibitory control (DNIC) paradigm. Maladaptive processes within the signaling of the µ-opioid receptor, which entail de-sensitization and a switch to excitatory signaling, contribute to tolerance and OIH. In chronic pain, the alterations are complex and depend on the area and model of chronic pain. For example, during neuropathic pain, the down-regulation of µ-opioid receptors and δ-opioid receptors in some areas, including the DReN, likely contributes to the inefficacy of opioids. However, in inflammatory pain models, µ-opioid receptors and δ-opioid receptors are up-regulated in the RVM. The diversity and complexity of alterations in the brainstem is likely provided by the alternative splicing of opioid receptors and the heteromerization of µ-opioid receptors (Costa et al. 2024).
- Dorsal Reticular Nucleus (DReN)and Wide-dynamic Range (WDR) Neurons
In neuropathic-pain rats with CCI of one sciatic nerve and in sham-operated rats, recordings were made from ipsilateral lumbar WDR neurons and simultaneously the DReN neuron activity was monitored. In particular, the spinal-neuron spontaneous activities and the activities evoked by noxious stimulations of ipsi- and contralateral sciatic supplied areas before and during DReN activity blockade were recorded. In sham-operated rats, WDR activity was modulated by iontophoretic NMDA to mimic CCI WDR hyper-activity without peripheral damage. During DReN activity blockade in CCI rat neurons and in sham-operated rats NMDA-treated neurons, the spontaneous activity was significantly reduced, the responses to contralateral sciatic area stimulation were reduced or suppressed, the responses to ipsilateral sciatic area were poorly affected (slightly reduced or unaffected), except for the post-stimulus afterdischarges that were mostly suppressed. In sham-operated rats, the neuronal activity was not affected by DReN blockade. The finding that during the DReN nucleus blockade some expressions of spinal neurons sensitization, seemingly associated with sensory disorders in neuropathic pain, faded or extinguished designates a likely facilitatory role of DReN in the maintenance of neuronal sensitization and thus a contribution to neuropathic pain state (Sotgiu et al. 2008).
- Dorsal Reticular Nucleus (DReN) and Wide-dynamic Range (WDR) Neurons in Diffuse Noxious Inhibitory Control (DNIC)
DNICs are a mechanism of endogenous descending pain modulation and are deficient in a large proportion of chronic pain patients. The roles of the DreN and the IL region of the mPFC were examined in DNIC. In anesthetized sham-operated and L5/L6 SNL rats, in vivo electrophysiology was performed to record from DH laminae V/VI WDR neurons with left hindpaw receptive fields. Evoked neuronal responses were quantified in the presence and absence of a conditioning stimulus (left-ear clamp). In sham rats, DNIC were reproducibly recruited by a heterotopically applied conditioning stimulus, an effect that was absent in neuropathic rats. Intra-DReN naloxone had no effect on spinal neuronal responses to dynamic brush, punctate mechanical, evaporative cooling and heat stimuli in sham and SNL rats. Intra-DReN naloxone blocked DNIC in sham rats, but had no effect in SNL rats. Intra-IL lidocaine had no effect on spinal neuronal responses to dynamic brush, punctate mechanical, evaporative cooling and heat stimuli in sham and SNL rats. Differential effects occurred in relation to the expression of DNIC; intra-IL lidocaine blocked activation of DNIC in sham rats but restored DNIC in SNL rats. This suggests that the IL is not directly involved in mediating DNIC but can modulate its activation, and that DReN involvement in DNIC requires opioidergic signaling (Patel and Dickenson 2020).
2.3.6. The RVM-VLM-DReN Triad in Chronic Pain
- Activity Changes
During the installation of neuropathy, the RVM suffered from neurodegeneration. In initial phases of neuropathy, the RVM exhibited plastic changes in its involvement in descending modulation, namely by increases in the activity of pain facilitatory ON-cells and opposite for OFF-cells, along with an enhancement of pain facilitatory actions at the spinal cord mediated by local 5-HT3 receptors. These changes may lead to increases in descending pain facilitation, which may account for chronic pain installation. During the progression of pain, and due to the continuous barrage of nociceptive input from the spinal cord, a disruption of local RVM circuits occurs with the appearance of massive oxidative-stress damage and hyperactivation of glial cells. The subsequent neuro-inflammation may lead to neuro-degeneration, associated with neuronal loss, which represents a non-plastic effect of chronic pain installation in the RVM. Hence, the alterations occurring in the RVM-VLM-DReN triad during chronic pain are fundamental to its maintenance (Martins and Tavares 2017).
- Changes in Peri-aqueductal Gray (PAG) and Rostral Ventro-medial Medulla (RVM)
Prolonged pain goes along with a switch in the RVM output from inhibition to facilitation of pain, indicating that the circuit is bi-directional in terms of pain modulation (Bouchet and Ingram 2020; Mills et al. 2021; Ossipov et al. 2014). Neuropathic pain was associated with persistent glial activation, in particular astrogliosis in the PAG and RVM. Following nerve injury, there were also changes in the firing properties of RVM ON- and OFF-cells. Both ON- and OFF-cells exhibited lowered thresholds and altered responsivity to mechanical and thermal stimuli in a stimulus intensity graded manner. The spontaneous discharge rates of inhibitory RVM OFF-cells were reduced and ON-cell firing rates were increased in models of neuropathic pain, and de-activation of either the PAG or RVM or disrupting the RVM-DH pathway could reduce allodynia and hyperalgesia (Mills et al. 2021). In rats demonstrating allodynia due to nerve injury, blockade of RVM activity with lidocaine reversed both evoked hypersensitivity and produced conditioned place preference (CPP), revealing the presence of ongoing pain. In SNL rats that were not demonstrating evoked hypersensitivity (i.e. presumably `pain-free´), RVM lidocaine precipitated allodynia and produced conditioned place aversion (CPA). Moreover, selective inhibition of pain-inhibitory RVM neurons with the κ-opioid agonist U69593 or spinal administration of the α2-adrenergic antagonist yohimbine also unmasked signs of enhanced pain in asymptomatic nerve-injured rats. Electrophysiological studies suggested that these `pain-free´ injured rats had a reduced functioning of RVM ON-cells and enhanced function of RVM OFF-cells (Ossipov et al. 2014).
- Chronic Inflammatory Pain
Chronic inflammatory pain is associated with activity changes of the triad. In chronic monoarthitic rats, increased activity in the CVLM and DReN occurred in a functional study based in the consumption of 2-D-glucose. In the specific case of the DReN, this could be related both to increased spinal excitatory inputs to the DReN in addition to a loss of inhibitory opioidergic tone into the DReN. The former was suggested to occur due to an imbalance between excitatory and inhibitory actions exerted upon spino-DReN neurons. Spinal DH neurons projecting to the DReN were under direct inhibitory GABAergic modulation, as they expressed mainly GABAB receptors, but in the spinal DH of mono-arthritic animals there were lower percentages of GABAB-expressing neurons and higher percentages of NK1 neurons, which were subjected to excitatory actions exerted by SP. A loss of inhibitory opioidergic tone likely occurred at the DReN since mono-arthritic rats showed decreased expression levels of of μ- and δ-opioid receptors at the DReN. These hypotheses are consistent with data showing the involvement of the DReN in formalin-induced secondary allodynia and hyperalgesia through a tonic glutamatergic excitatory tone. The administration of glutamate antagonists into the DReN reduced both secondary allodynia and hyperalgesia and their development was prevented by pretreatment with glutamate antagonists injected into the DReN before formalin injection (Martins and Tavares 2017).
- Locus Coeruleus→Dorsal Reticular Nucleus (DReN) Pathway
The DReN is subject to NA modulation. In rats subjected to the spared-nerve injury model of neuropathic pain, chronic pain induced brainstem NA activation that enhanced descending facilitation from the DReN (Martins et al. 2015).
In male Sprague-Dawley rats with CCI of the sciatic nerve and neuropathic pain, time-dependent plasticity of LC neurons related to the site of injury were identified, ipsilateral (LCipsi) or contralateral (LCcontra) to the lesion, suggesting that the LC to DReN pathway was involved in chronic pain development. LCipsi inactivation with lidocaine increased cold allodynia two days after nerve injury but not later. However, similar blockade of LCcontra reduced cold allodynia seven and 30 days after inducing neuropathy but not earlier. Lidocaine blockade of the LCipsi or LCcontra reversed pain-induced depression 30 days after neuropathy. Moreover, inactivation of the LCcontra to DReNcontra pathway produced consistent analgesia in evoked and spontaneous pain 30 days post-injury. This analgesia was similar to that produced by spinal activation of α2-adrenoreceptors. Chemogenetic inactivation of the Lccontra to DRtcontra pathway induced depressive-like behavior in naïve animals, but it did not modify long-term pain-induced depression. Overall, nerve damage activated the LCipsi, which temporally dampened the neuropathic phenotype. However, the ensuing activation of an Lccontra to DReNcontra facilitatory pain projection contributed to chronic pain, whereas global bilateral LC activation contributed to associated depressive-like phenotype (Camarena-Delgado et al. 2022).
- Spinal Hypersensitivity
Neuropathic pain is associated with an activity-dependent hypersensitivity of spinal neurons. Electrophysiological studies suggest a contribution of the DReN to the maintenance of spinal sensitization. During neuropathic pain, an NA modulation of the DReN has been implicated in the enhancement of DReN pain facilitation. Nociceptive stimulation increased NA release in the DReN, which enhanced pain facilitation from the DReN through activation of α1-adrenoreceptors. The feedback inhibitory function of α2-adrenoreceptors in the DReN during neuropathic pain was impaired, which likely further contributed to enhance the NA input to the DreN (Martins and Tavares 2017).
- Impact of Chronic Stress
Under mild and persistent stress, hyperalgesia induced by DMH stimulation recruited RVM ON-cells: stress-induced hyperalgesia (SIH). However, micro-injection of lidocaine into the RVM potentiated HYP-mediated analgesia, as observed by the increase in the pain threshold and inhibition of the tail-flick reflex. The direct HYP-RVM projection thus influences the activities of ON- and OFF-cells, exerting bi-directional pain modulation (Peng et al. 2023).
2.4. Nucleus Tractus Solitarii (NTS)
The NTS (or nucleus of the solitary tract) is a complex of sub-nuclei aligned in a vertical slice located in the dorso-medial medulla oblongata. The NTS has been divided cytoarchitectonically into various sub-nuclei, which are partly correlated with the areas of projection of peripheral afferent endings. Gustatory and somatic afferents from the oro-pharyngeal region project to the rostral part of the NTS (rNTS) and visceral afferents from cardio-vascular, digestive, respiratory and renal systems terminate viscero-topically within the caudal NTS (cNTS) (Jean 1991).
- Inputs
The NTS receives fibers from the superficial laminae (I-III) of the spinal DH terminating bilaterally in the cNTS, and fibers from the deeper DH laminae (IV-V) terminating ipsilaterally, mostly in the lateral areas of the cNTS (Gamboa-Esteves et al. 2001). In rats, NTS cells receive hindlimb somatosensory inputs from low- and high-threshold cutaneous mechano-receptors, respond to capsaicin delivered into the hindlimb arterial supply, lack thermal sensitivity, and respond to activation of mechano-sensitive as well as metabo-sensitive endings in skeletal muscle. Visceral sensory information is conveyed via the afferent glossopharyngeal (IX) and vagus (X) nerves (Toney and Mifflin 2000). The NTS receives cardio-pulmonary vagal inputs from small-diameter afferents responding to mechanical distension (lung stretch, group III (Aδ) fibers) and noxious stimuli/immune processes (lung irritants/cytokines, via group IV (C)-fibers/nociceptors) leading to efferent vagal activity that evokes airway defensive reflexes (Zyuzin and Jendzjowsy 2022). The NTS also receives telencephalic inputs from a large array of structures (Holt 2022; Toney and Mifflin 2000).
- Outputs
The cNTS processes and conveys peripheral viscero-sensory information to CNS regions concerned with autonomic-affective and other interoceptive reflex functions (Holt 2022). The NTS projects to multiple brain regions via short connections to bulbo-ponto-mesencephalic structures, e.g., PBN, ventro-lateral reticular formation, raphé nuclei (RN), motor nuclei of several cranial nerves, and others, and long connections to the spinal cord and diencephalic and telencephalic structures, in particular the HYP and some limbic structures. Most of the structures that receive a direct projection from the NTS project back to it. The NTS has extensive connections with the vestibular nuclei, both directly and via the PBN; whereby the vestibular nuclei could also receive nociceptive inputs (Saman et al. 2020). The NTS contains a great diversity of neuroactive substances. Indeed, most of the substances identified within the CNS have also been detected in the NTS and may act, at this level, as classical transmitters and/or neuromodulators (Jean 1991).
- Functions
The extensive connections indicate that the NTS is a key structure for autonomic and neuro-endocrine functions as well as for integration of somatic and autonomic responses in certain behaviors. Painful stimuli can evoke dramatic responses in the cardio-vascular and respiratory systems. The NTS has a major role as a site for integrating nociceptive and cardio-respiratory afferents for mediating tachycardia evoked by somatic noxious stimulation. Similar noxious stimulation attenuates the cardiac component of the peripheral chemo-receptor reflex and inhibits the peripheral chemoreceptor-evoked excitatory synaptic response of some NTS neurons. Hence, by depressing homeostatic reflexes in the NTS, noxious stimulation-evoked cardio-respiratory changes can be expressed and maintained, which may be essential for the survival of the animal (Boscan et al. 2002).
- Nucleus Tractus Solitarii (NTS) and Chronic Pain
In rats with chronic pancreatitis (CPan), excitatory transmission within the cNTS was enhanced and thus contributed to pancreatic pain, emphasizing the central sensitization of painful chronic pain (Bai et al. 2019). Also, after trigeminal nerve injury-elicited neuropathic pain, rats exhibited heat and mechanical hypersensitivity in the ipsilateral upper lip. The rats then received capsaicin or noxious mechanical stimulation to the upper lip. In the NTS contralateral to nerve injury, the number of PBN projection neurons increased. Hence, enhanced noxious inputs from the NTS to the PBN after trigeminal nerve injury modulates PBN neuron activity, which accompanies the affective components of oro-facial neuropathic pain (Okada et al. 2019).
2.5. Parabrachial Nucleus (PBN)
The PBN surrounds the superior cerebellar peduncles in the dorso-lateral pons. It is a collection of cell groups that, in rodents, can be divided into more than a dozen sub-nuclei on the basis of cytoarchitecture (Chiang et al. 2019). The PBN receives nociceptive inputs from the STTr (Kuner and Kuner 2021). Particularly the lPBN, is the primary supraspinal target of nociceptive, pruritic and thermal signals transmitted via the spino-parabrachial tract (SPT) from the trigeminal and spinal DHs (Chiang et al. 2019). The PBN is reciprocally connected with CeA, bed nucleus of the stria terminalis (BNST), and multiple HYP nuclei, including the preoptic area (POA).
The PBN responds to bodily threats including noxious stimuli and transmits alarm signals to the forebrain. PBN neuron activity is enhanced during chronic pain, and in mice the inactivation of PBN neurons prevents the establishment of CNP symptoms. Chemogenetic or optogenetic activation of all glutamatergic PBN neurons is sufficient to establish pain phenotypes, including long-lasting tactile allodynia (Palmiter 2024).
- Parabrachial Nucleus (PBN)-Peri-aqueductal Gray (PAG) Connection
In patients with chronic pain, resting-state functional connectivity was used to assess functional brain alterations. Right PBN-PAG functional connectivity during pain-free rest positively correlated with subsequently experienced pain. During pain, this connection´s functional connectivity was diminished (Meeker et al. 2022).
- Parabrachial Nucleus (PBN)-Rostral Ventro-medial Medulla (RVM)Connection
PBN projects directly to the RVM. Under physiological conditions and when exposed to acute pain stimuli, the contralateral PBN transmits signals to the RVM ON- and OFF-cells and then triggers acute hyperalgesia, while the ipsilateral PBN is involved in the RVM ON-and OFF-cells-induced modulation of persistent inflammation and chronic pain (Peng et al. 2023).
- Parabrachial Nucleus (PBN)-Central Nucleus of Amygdala (CeA) Connection
The lPBN relays nociceptive signals to the AMY, which is important for tagging pain with the emotional-affective dimension and for pain modulation (Neugebauer et al. 2023; Sun et al. 2020a). In rodent models of persistent pain, excitatory synaptic transmission in the PBN to CeA pathway is potentiated. In male and female mice, activation of CeA-projecting PBN neurons contributed to injury-induced behavioral hypersensitivity but not baseline nociception. Peripheral noxious stimulation increased the expression of the neuronal activity marker c-fos in CeA-projecting PBN neurons and chemogenetic inactivation of these cells reduced behavioral hypersensitivity in models of neuropathic and inflammatory pain without affecting baseline nociception. Chemogenetic activation of CeA-projecting PBN neurons is sufficient to induce bilateral hypersensitivity without injury. Hence, activation of this pathway contributes to injury-induced hypersensitivity, directly demonstrating a critical function of the PBN-CeA circuit in pain modulation (Torres-Rodriguez et al. 2023). lPBN neurons exhibited a reduction in inhibitory GABAergic inputs showing that the CeA-LPB pathway is critically involved in pain regulation, and in the pathogenesis of chronic pain (Chiang et al. 2019; Raver et al. 2020).
The lPBN makes a limited contribution to acute pain responses, but a larger one to hyperalgesia and persistent pain. The lPBN receives its primary nociceptive input from NK1-expressing neurons in the superficial DH, a population associated with hyperalgesia in persistent pain due to inflammation or nerve injury. Thus, in persistent inflammatory pain models, the connection between lPBN and descending control systems appear to be re-organized, and blocking lPBN reversed behavioral hypersensitivity under these conditions. In rodent models of arthritic and neuropathic pain, lPBN neurons increased their excitability. In a mouse model of neuropathic pain, the inhibition of lPBN neurons by the CeA was suppressed. Animals with pain after CCI of the IoN displayed higher spontaneous and evoked activity in PBN neurons, and a dramatic increase in after-discharges, responses that far outlast the stimulus, compared with controls (Chiang et al. 2019; Raver et al. 2020).
In lightly anesthetized rats, recordings were made of behavioral withdrawal, evoked by mechanical stimulation of the hindpaw, and of the activity of identified pain-modulating neurons, ON-cells and OFF-cells in the RVM. This was done before and after the inactivation of PBN, contralateral or ipsilateral to an inflamed paw 5-6 days after intra-plantar injection of CFA. The inactivation of contralateral, but not ipsilateral, PBN interfered with nociceptive input to RVM under basal conditions, as well as in acute inflammation. By contrast, blocking ipsilateral, but not contralateral, PBN in established inflammation interfered with behavioral hyperalgesia and ON-cell and OFF-cell responses. The lesioning of contralateral PBN before CFA injection prevented this recruitment of ipsilateral PBN in persistent inflammation. This shows that contralateral PBN is required to initiate hyperalgesia, which is then maintained by ipsilateral PBN, most likely in both cases via the engagement of pain-modulating neurons of the RVM (Chen and Heinricher 2019).
Neuropathic pain-like hypersensitivity induced by common peroneal nerve (CPN) ligation increased nociceptive stimulation-induced responses in glutamatergic lPBN neurons. Optogenetic activation of GABAergic lPBN neurons did not affect basal nociception, but alleviated neuropathic pain-like behavior. In naïve mice, optogenetic activation of glutamatergic or inhibition of GABAergic lPBN neurons induced neuropathic pain-like behavior. Inhibition of glutamatergic lPBN neurons alleviated both basal nociception and neuropathic pain-like hypersensitivity. Repetitive pharmacogenetic activation of glutamatergic or GABAergic LPBN neurons respectively mimicked or prevented the development of CPN ligation-induced neuropathic pain-like hypersensitivity. This indicates that the development and maintenance of neuropathic pain depends on a delicate balance between excitatory and inhibitory lPBN neuronal activity (Sun et al. 2020a).
In naive or neuropathic pain rats, CGRP induced anti-nociception in PBN, in which CGRP receptors mediated this effect. Neuropathic pain decreased the expression of CGRP receptors as well as in CGRP-induced anti-nociception in PBN (Wang et al. 2021). While, under physiological conditions and when exposed to acute pain stimuli, the contralateral PBN triggers acute hyperalgesia, the ipsilateral PBN is involved in the RVM ON- and OFF-cells-induced modulation of persistent inflammation and chronic pain (Peng et al. 2023).
2.6. Cerebellum
The cerebellum has a role in pain processing and/or modulation, possibly due to its extensive connections with the PFC and brainstem regions involved in descending pain control (Adamaszek et al. 2017; Ong et al. 2019). The cerebellum is one of the most responsive brain structures to painful stimuli, as shown by neuroimaging. Animal studies suggested spinally projecting multi-sensory inputs from the skin, including tactile group III (Aδ) and nociceptive group III (Aδ) and IV (C) fiber (Welman et al. 2018). The cerebellum connects to many pain-related CNS regions: S1, premotor cortex (PM), mPFC, ACC, IC, HIPP, AMY, THAL, HYP, PAG, RF, NR, SpV, and spinal cord.
Patients with TN showed a reduction in the volume of cerebellar gray matter. In rats with spinal nerve ligation, PET revealed a decrease in connections in the cerebellum and certain prefrontal regions, suggesting a potential association between neuropathic pain and connectional plasticity of the resting-state brain. Neuropathic rats after tibial and sural nerve transection showed increased mechanical sensitivity of the injured hindpaw. Longitudinal micro-PET scan of brains from neuropathic rat models showed sequential cerebellar activity in accordance with results from behavioral test responses, thus supporting a role for the cerebellum in the development of neuropathic pain (Boadas-Vaello et al. 2017).
Compared to healthy controls, patients with cerebellar infarction limited to the cerebellum displayed hyperalgesia to thermal and repeated mechanical stimuli, which were applied to the forearm. In patients with cerebellar infarction, offset analgesia (defined as a form of endogenous pain inhibition characterized by disproportionately large reductions in pain intensity ratings evoked by small decreases in stimulus intensity) was reduced, suggesting deficient descending pain inhibition. In addition, children with cerebellar resection extending into Crus I/II showed decreased cold pain tolerance compared to healthy controls. This suggests that cerebellar impairment could lead to hyperalgesia (Wang et al. 2022).
2.7. Midbrain Dopamine (DA) Cell Groups
The midbrain DA complex comprises the substantia nigra pars compacta (SNc), ventral tegmental area (VTA) and retro-rubral field (RRF), which contain the A9, A10 and A8 groups of nigro-striatal, meso-limbic and meso-cortical DA neurons, respectively. Additionally, there are dorsal-caudal A10dc and rostro-ventral A10 extensions into the vlPAG and supra-mammillary nucleus, respectively (Yetnikoff et al. 2014). The meso-limbic DA system indirectly receives somatosensory inputs, including nociceptive inputs, from the lHYP mediated by the LHb. The lHYP-LHb pathway is necessary for nociceptive modulation of the DA system (Dai et al. 2022; Ogawa and Watabe-Uchida 2018). In anesthetized rats, many DA neurons exhibited a short-latency response to noxious stimuli, which appears to be mediated by the nociceptive-recipient PBN (Coizet et al. 2010). The DA brain reward system includes neurons of the VTA and their projections to the NAc and several other brain regions, including the PFC (Kuner and Kuner 2021).
DA has been associated with several chronic pain disorders (Schomburg et al. 2011a, 2011b, 2012, 2013, 2015) including chronic post-surgical pain (CPSP). DA has emerged as a good modulator of CPSP. Pharmacotherapy focused on DA neurotransmission has potential in both prevention (via D1-like receptors) and treatment (via D2-like receptors and DA re-uptake inhibitors) of CPSP (Van Reij et al. 2019). The mPFC could induce pain chronification via its cortico-striatal projection, possibly depending on the level of DA receptor activation (or lack of) in the reward pathway from the VTA to NAc. There is a role for the PFC during placebo analgesia, and in establishing links between pain and anxiety, depression, and loss of cognition. The mPFC is also involved in modulation of pain catastrophizing, reduction of pain-induced sympathetic activity, and decrease in facial expressions of pain (Ong et al. 2019).
In mice, when DH neurokinin 1 (NK1) receptor-positive neurons or descending 5-HT neurons were ablated before hyperalgesic priming, IL-6- and carrageenan-induced mechanical hypersensitivity was impaired, and subsequent PGE2 response was blunted. However, when these neurons were lesioned after the induction of priming, they had no effect on the PGE2 response, reflecting differential mechanisms driving plasticity in a primed state. By contrast, animals with a spinally applied DA lesion showed intact IL-6- and carrageenan-induced mechanical hypersensitivity, but the subsequent PGE2 injection failed to cause mechanical hypersensitivity. Ablating spinally projecting DA neurons after the resolution of the IL-6- or carrageenan-induced response also reversed the maintenance of priming as assessed through mechanical hypersensitivity and the mouse grimace scale. Pharmacological antagonism of spinal DA D1/D5 receptors reversed priming, whereas D1/D5 agonists induced mechanical hypersensitivity exclusively in primed mice. This demonstrated a novel role for descending DA neurons in the maintenance of pathological pain plasticity (Kim et al. 2015).
- Neuronal Plasticity and Functional Connectivity
Chronic pain states go along with changes in neuronal plasticity and functional connectivity in several parts of the brain reward system, including the VTA, NAc and the PFC. In chronic pain patients, brain imaging has shown neuronal activation of the meso-limbic DA system. The circuitry between the VTA and SN and NAc is modulated by several limbic and cortical inputs involved in remembering past experiences, values, expectations, and salience (Mitsi and Zachariou 2016).
- Dorsal Raphé Nucleus (DRN)-Ventral Tegmental Area (VTA) Pathway
In mouse CNP, a pathway was described from VGLUT3 neurons in the Dorsal Raphé Nucleus (DRN) to DA neurons in the VTA (VgluT3DRN→DAVTA), wherein population-level activity in response to innocuous mechanical stimuli and sucrose consumption was inhibited by CNP. Mechanistically, neuropathic pain dampens the glutamatergic transmission from VgluT3DRN to DAVTA and neural excitability in the VTA DA neurons. VgluT3DRN → DAVTA activation alleviated neuropathic pain and comorbid anhedonia-like behavior by releasing glutamate, which subsequently promoted DA release in the NAc medial shell and produced analgesic and anti-anhedonia effects via D2 and D1 receptors, respectively. In intact mice, VgluT3DRN → DAVTA inhibition produced pain-like reflexive hypersensitivity and anhedonia-like behavior. This has been suggested to reveal an important role for VgluT3DRN → DAVTA → D2/D1NAcMed pathway in establishing and modulating chronic pain (Wang et al. 2023).
- Ventral Tegmental Area (VTA)-Nucleus Accumbens (NAc) Connection
In rodents, noxious stimuli affect the release of DA in the brain reward center. Chronic pain states in rodents lead to a DA reduction in NAc, and pain relief is associated with increased DA concentrations in the NAc shell. Hypo-DA states in rodent models of neuropathic pain are associated with depression-like behaviors and reduced motivation. These affective deficits are linked to LTD of excitatory synaptic transmission in the medium-spiny neurons (MSNs) of the indirect BG pathway. When mice were conditioned to environmental cues, compounds with pain-alleviating properties promoted place preference. Pain relief promoted place preference in the spared-nerve injury model of neuropathic pain, the CFA model of inflammatory pain, as well as models of post-surgical, cancer, and osteo-arthritic pain (Serafini et al. 2020).
The meso-limbic DA projections possibly contribute to promoting allodynia related to neuropathic and cancer pain. Mice with ligation of the sciatic nerve or treated with intra-femoral osteo-sarcoma cells showed allodynia to a thermal stimulus applied to the paw on the injured side. Patch-clamp electrophysiology revealed that the intrinsic neuronal excitability of VTA DA neurons projecting to the NAc was significantly reduced in those mice. Optogenetic activation of these cells produced a significant but transient anti-allodynic effect in nerve injured or tumor-bearing mice without increasing response thresholds to thermal stimulation in sham-operated animals. Suppressed activity of meso-limbic DA neurons is likely to contribute to decreased inhibition of NAc. Output neurons and to neuropathic or cancer pain-induced allodynia suggesting strategies for modulation of pathological pain states (Watanabe et al. 2018).
- Ventral Tegmental Area (VTA)-Prefrontal Cortex (PFC) Connection
Dysfunction in the meso-cortical pathway from the VTA to the PFC has been implicated in chronic pain. In a mouse model of neuropathic pain, the use of in vivo Ca2+ imaging revealed diminished VTA glutamatergic activity targeting the pre-limbic cortex (PL). Optogenetic activation of VTA glutamatergic terminals in the PL alleviated neuropathic pain, while inhibiting these terminals in naïve mice induced pain-like responses. This pain-modulating effect was independent of DA co-release. VTA neurons primarily projected to excitatory neurons in the PL, and their activation restored PL outputs to the ACC. All this revealed a distinct meso-cortical glutamatergic pathway that modulated neuropathic pain independent of DA signaling (Li and Yang 2024).
2.8. Locus Coeruleus (LC) and Other Noradrenergic (NA) Cell Groups
It has been suggested that the activity of the NA system descending to the spinal cord is augmented in conditions of nerve injury in an effort to compensate for enhanced nociceptive inputs. Injury is associated with increased synthesis and release of NA along with an enhanced efficacy of spinal α2-adrenergic receptors. Enhanced spinal NA efficiency in injury or inflammation also provides a mechanistic basis for the clinical success of the 5-HT/NA re-uptake inhibitors in diabetic neuropathy, FM and osteoarthritis (Ossipov et al. 2014). Selective activation of NA fibers descending from the LC alleviates neuropathic pain in mice by increasing the release of NA and reducing neuro-inflammation of astrocytes and microglia in the DH (Li et al. 2022).
2.8.1. Locus Coeruleus (LC) and Subnucleus Reticularis Dorsalis (SRD)
The LC consists of a dense, cell-rich `core´, where NA cell bodies and processes reside, and a peri-coeruleus (peri-LC) `shell´ into which LC-NA dendrites extend and ramify. The nucleus paragigantocellularis (PG) is a major excitatory afferent input to the LC. In anesthetized rats, LC neurons were potently activated by foot-shock, this effect being indirect because attenuated or blocked by pharmacologic blockade of te PG (Chiang and Aston-Jones 1993). NA was originally thought to be released uniformly and act simultaneously on cells and circuits throughout the forebrain, brainstem, cerebellum and spinal cord (Pertovaara 2006, 2013; Poe et al. 2020). However, matters are more complex (Figure 4).
- Human Brain Imaging
In chronic oro-facial neuropathic pain patients, brain imaging showed enhanced resting-state fMRI signal coupling between the LC and both the RVM and SRD. There was no change in lateral LC (lLC) signal coupling with the SpV, suggesting that a direct descending NA projection from the LC to the DH/SpV may not be involved in the maintenance of ongoing neuropathic pain, in contrast to a recent preclinical finding. Furthermore, there was also a reduced (near-zero) connectivity between the LC and PAG in patients, suggesting that altered resting interactions between the LC and the PAG, SRD, and RVM may collectively contribute to the pro-nociceptive effects often observed in patients with CNP (Mills et al. 2021).
- Affective Dimensions
The ascending NA projections have also been implicated in affective dimensions of pain. The BNST has been suggested to play important roles in pain-induced CPA. NA afferents to the ventral domain of the BNST activate neuronal circuits with the BNST, ultimately resulting in reduced BNST output to the VTA. Reduced activity of VTA DA neurons may underlie place aversion downstream of NA signaling (Kuner and Kuner 2021).
- Pain Chronification
Injury is associated with increased synthesis and release of NA along with an enhanced efficacy of spinal α2-adrenergic receptors. Enhanced spinal NA efficiency in injury or inflammation also provides a basis for the clinical success of the 5-HT/NA re-uptake inhibitors (SNRIs) in diabetic neuropathy, FM and osteoarthritis (Ossipov et al. 2014). Selective activation of NA fibers descending from the LC alleviates neuropathic pain in mice by increasing the release of NA and reducing neuro-inflammation of astrocytes and microglia in the DH (Li et al. 2022).
While acute pain promotes aversion, vigilance, and threat detection through LC´s ascending efferents and produces spinal cord-mediated endogenous analgesia, the protective biological system fails in chronic pain, and LC activity produces pain facilitation, anxiety, increased aversive memory, and behavioral despair, acting at the levels of the medulla oblongata, AMY and PFC. The activation/deactivation of specific LC projections contributes to different behavioral outcomes in the shift from acute to chronic pain (Suárez-Pereira al. 2022). Thus, it has been hypothesized that, with time after injury, the balance of LC function shifts from pain inhibition to pain facilitation effected by simultaneous activation of supraspinal facilitatory systems dependent on α1-adrenoreceptor in the mPFC, DReN, as well as α2-adrenoreceptors in the LC (Taylor and Westlund 2017). At early stages of nerve injury, LC neurons show higher electrophysiological responses evoked by noxious stimulation, but spontaneous activity does not change. By contrast, in long-term traumatic neuropathy, higher spontaneous activity and enhanced responses of LC neurons to noxious stimulation occur. The other brainstem NA cell groups also contribute to the maintenance of neuropathic pain. Moreover, nerve injury increases the density of NA fibers in the spinal cord, which is associated with increased BDNF (Tavares et al. 2021).
- Locus Coeruleus (LC) and Descending Pain Modulation
Besides the direct input to the spinal cord, the alterations of the NA system also affect the brainstem pain-modulatory system. Interactions among brainstem pathways and their receptors may modulate both pain inhibition and facilitation, however. After nerve injury, LC NA neurons and their terminals in the mPFC, DReN, SpV and DH may participate in the development and maintenance of allodynia and hyperalgesia (Taylor and Westlund 2017). The increased activation of NA LC and A5 neurons in traumatic neuropathic models led to increased release of NA into DReN, which was proposed to enhance descending facilitation of nociceptive transmission from that medullary area (Tavares et al. 2021).
While in baseline conditions the NA system may have little effect, sustained pain induces NA feedback inhibition of pain. NA systems may also contribute to top-down control of pain, such as induced by a change in the behavioral state. Following injury or inflammation, the central as well as peripheral NA system are subject to various plastic changes that influence its anti-nociceptive efficacy (De Felice and Ossipov 2016; Pertovaara 2006, 2013). Altered functioning of the LC and SRD are involved in the development and/or maintenance of CNP. The LC effects on pain following nerve injury have been difficult to investigate due to the vastly different effects of NA on α1- and α2-adrenoreceptors. On the one hand, following nerve injury, the LC continued to inhibit nociception at the DH level since the disruption of NA locally in the spinal cord led to enhanced mechanical sensitivity. By contrast, other evidence indicates that the LC played an overall pro-nociceptive role and contributed to the maintenance of sensory and affective behavioral responses (Mills et al. 2021). Thus, while in a rat neuropathic pain model, activation of the spinally descending LC projection normally reduced spontaneous pain behavior, increased withdrawal thresholds and produced a positive affective bias, activation of the projection to the PFC produces aversion and increases spontaneous pain behavior. These differential effects have been suggested to argue for a modular functional LC architecture (Chandler et al. 2019).
In rats with tibial nerve transection, descending NA inhibition delayed the expression and extent of enhanced pain. Blockade of spinal α2-adrenergic receptors accelerated the onset of behavioral sensitization as well as the onset of contralateral allodynia and enhanced c-fos expression in the DH. Hence, an imbalance between pain inhibition and facilitation could lead to enhanced abnormal pain (Ossipov et al. 2014). There is also preclinical evidence of pro-nociceptive changes in SRD control over DH firing following nerve injury. In rats with neuropathic pain, blocking SRD activity significantly reduced noxious-evoked and spontaneous activity of spinal WDR neurons, which may result from inputs to the SRD from the LC. Following nerve injury, noxious stimulation increased NA release in the SRD, and blocking this release with a selective viral vector targeting SRD, NA afferents could significantly attenuate allodynia and hyperalgesia. These effects were likely not due to changes in tonic drive since basal LC activity levels were unchanged following nerve injury (Mills et al. 2021).
In a rat model of chronic orofacial neuropathic pain, trigeminal neuropathy (TN) was induced by chronic constrictive injury (CCI) of the IoN (CCI-IoN). Orofacial neuropathic pain was indicated by development of whisker-pad mechanical hypersensitivity. Using a neurotoxin (micro-injected either intra-cerebro-ventricularly (ICV) or into SpVc, hypersensitivity was alleviated by selective elimination of NA neurons, including the LC A6 cell group. The GABAA receptor antagonist bicuculline, administered directly into LC (week 8) inhibited hypersensitivity. This indicated a valence shift in which increased GABAA signaling in LC after trigeminal nerve injury produced excitatory facilitation of the chronic pain state. Micro-injection of a NA α1-receptor antagonist into mPFC attenuated whisker-pad hypersensitivity, while a NA α2-receptor antagonist was ineffective. Thus, GABAA-mediated activation of NA neurons during CCI-ION can facilitate hypersensitivity through NA α1-receptors in the mPFC (Kaushal et al. 2016).
- Diffuse Noxious Inhibitory Control (DNIC)
Some neurons in the DH are strongly inhibited when a nociceptive stimulus is applied to any part of the body, distinct from their excitatory receptive fields. DNIC is mediated by the SRD in the dorsal medulla, which receives NA input from the LC, and is able to modulate nociceptive transmission via direct projections to the DH/SpV. In human acute-pain studies, the SRD was activated by noxious thermal stimuli, and also received signals from the PAG during and after noxious thermal stimulation (Bouhassira et al. 1990; Mills et al. 2021; Villanueva and Le Bars 1995). In rats, the loss of DNIC application of a noxious stimulus reduced or abolished an initial nociceptive response (Ossipov et al. 2014).
2.8.2. A1-A7 (NA) and Adrenergic Cell Groups
The brainstem contains scattered NA cell groups: A1, A5, A6 (LC) and A7. These NA neurons receive dissimilar afferents via various circuits to coordinate organismal responses to internal and environmental challenges, including pain. A1, A5, A6, and A7 receive nociceptive inputs from the lateral STTr (Dostrovsky 2000; Kuner and Kuner 2021). Brainstem NA neurons project to local segmental (brainstem), cephalic (telencephalon and diencephalon), and caudal (spinal cord anterior, lateral, and DH) regions of the CNS, to the LC (A6), the primary source of NA in the brain, and to spinal pre-ganglionic neurons (Delbono et al.2022).
- A1, A2, C1, C2 Cell Groups
In the rat sciatic-nerve CCI model of chronic pain, the same nerve injury produced a range of behavioral outcomes, each associated with distinctive adaptations to the hypothalamic-pituitary-adrenal (HPA) axis to achieve stable plasma corticosterone levels. Corticotropin-releasing hormone (CRH) and glucocorticoid receptor (GR) expression in the HYP PVN was increased in rats that showed persistent changes to their social behaviors during resident-intruder testing (`Persistent Effect´ rats) when compared to rats that showed no behavioral changes (`No Effect´ rats). These changes could be driven in part by altered sensitivity of the brainstem catecholaminergic pathways (known to regulate the PVN) to glucocorticoids. GR expression in NA A1, A2 cells and adrenergic C1,C2 cells was determined using immuno-histochemistry in behaviorally tested CCI rats and in un-injured controls. There were no differences between `Persistent Effect´ and `No Effect´ rats in (i) the glucocorticoid sensitivity of these cells, or (ii) the numbers of NA or adrenergic cells in each region. However, GR expression was reduced in the non-catecholaminergic cells of these regions in both experimental groups when compared to un-injured controls, most likely attributable to the repeated Resident-Intruder testing. These data suggest that brainstem mechanisms are unlikely to play a key role in the re-balancing of the HPA axis triggered by CCI, increasing the probability that these changes are driven by supra-HYP regions (Sosa et al. 2023). An NA A1 cell group receives direct inputs from STTr collaterals and projects further to the HYP (Figure 4).
- A5 Cell Group
These cells receive a wide range of inputs, which include connections from neurons of spinal lamina I (Dostrovsky 2000; Kuner and Kuner 2021), and from the ventral region of the medulla, more specifically from the CVLM (Rocha et al. 2024). The A5 group projects segmentally to A6 (LC), The A5 group is supposed to contribute to the regulation of nociceptive messages at the spinal cord level (Kwiat et al. 1992).
- A7 Cell Group
Electrical lHYP stimulation produced anti-nociception, which was partially blocked by intra-thecal α-adrenergic antagonists. SP-immuno-reactive neurons in the lHYP project near the NA A7 cell group, which effects anti-nociception in the DH. However, while some A7 cells inhibit nociception through the action of α2-adrenoceptors in the spinal DH, other A7 cells increase nociception through the action of α1-adrenoceptors in the spinal DH (Holden and Naleway 2001).
2.9. Raphé Nuclei (RN)
The RN are a collection of functionally and anatomically diverse cell groups that span the brainstem and contain the majority of the 5-TH-producing neurons in the CNS (Brodal 1981). From caudal to rostral, the raphé nuclei are, in the medulla oblongata: nucleus raphé obscurus (NRO), NRM, nucleus raphé pallidus (NRP); in the pons: nucleus raphé pontis (NRPo), nucleus centralis inferior (NRI); in the midbrain: nucleus centralis superior (NCS), raphé nucleus medianus (RNM), DRN, caudal linear nucleus (CLN) (Nieuwenhuys et al. 1978).
As in the NA system, the descending 5-HT pain modulation changes during chronic pain. In normal adult rats, descending 5-HT modulation of nociceptive signaling in the DH is mainly inhibitory and mediated by 5-HT1a, 5-HT1b, 5-HT2c, 5-HT3 and 5-HT4 receptors. Upon injury and in neuropathic rats, this modulation becomes facilitatory via activation of the 5-HT2a, 5-HT2b and 5-HT3 receptors. Neuromodulatory intervention like spinal-cord stimulation restores the inhibitory function and involves 5-HT2, 5-HT3 and 5-HT4 receptors (Heijmans et al. 2021).
2.9.1. Dorsal Raphé Nucleus (DRN)
The DRN, located in the ventro-medial PAG (vmPAG), has a fan-shaped structure that is symmetrically distributed along the midline. It is highly heterogeneous in anatomical distribution, molecular markers, synaptic connectivity and function, and consists of various cell types, mainly including 5-HT neurons, GABA neurons, DA neurons, glutamatergic neurons, and peptidergic neurons. Among these, the 5-HT neurons are the most abundant, constituting approximately two-thirds of the neurons in the DRN. The DRN is an important hub for transmitting nociceptive signals from the spinal cord, ascending through the specific pathways to the cerebral cortex via the THAL, and it plays a crucial role in inhibiting pain transmission (Zhang et al. 2024). Its vast fiber connections to other parts of the CNS provide a morphological basis for its pain modulating function. Its descending projections, directly or via the NRM, modulate the responses caused by noxious stimulation of the spinal DH neurons. In ascending projections, it directly modulates the responses of pain-sensitive neurons in the THAL. It can also be involved in analgesia effects induced by the HYP ARC. Neurophysiological and neuro-pharmacological data suggest that 5-HT neurons and ENK neurons in the DRN inhibit pain, and GABA neurons do the opposite (Wang and Nakai 1994).
In the rat, the DRN, exclusive of the lateral wing regions, has a predominantly ipsilateral projection with decreasing numbers of cells projecting to frontal, parietal, and occipital cortex. Overlapping cell groups within the DRN project differentially to these three cortical areas. The percentage of cortically projecting 5-HT cells is 80% for the DRN, 60% in the RNM and 33% in the B9 cell group. The differential organization of the RN suggests that groups of cells within these three RN are likely to innervate different combinations of cortical targets and thus to have different functional effects (O´Hearn and Molliver 1984).
- Dorsal Raphé Nucleus (DRN) in Pain Regulation
There is an ascending facilitatory pathway, from the DRN to the meso-accumbal DA circuit, for regulating pain. In male mice, chronic pain increased the activity of DRN glutamatergic, but not 5-HT, neurons projecting to the VTA (DRNGlu-VTA). Optogenetic activation of the DRNGlu -VTA pathway induced a pain-like response in naive male mice, and its inhibition produced an analgesic effect in male mice with neuropathic pain. The DRN ascending pathway regulated pain through strengthened excitatory transmission onto the VTA DA neurons projecting to the ventral part of NAc medial shell, thereby activating the meso-accumbal DA neurons. Hence, optogenetic manipulation of this three-node pathway bilaterally regulated pain behaviors (Liu et al. 2024).
- Anterior Cingulate Cortex (ACC)-Dorsal Raphé Nucleus (DRN)Connection
Patients with neuropathic pain show a strong association between neuropathic pain and psychiatric conditions such as anxiety. Electro-acupuncture (EA) effectively alleviated anxiety-like behaviors induced by CNP. In animal models of spared-nerve injury, EA stimulation impacted mechanical allodynia and anxiety-like behaviors. In mice with spared-nerve injury, EA plus chemogenetic manipulation of glutamatergic (Glu) neurons projecting from the rostral anterior cingulate cortex (rACC) to the DRN were used to explore the changes of mechanical allodynia and anxiety-like behaviors. EA significantly alleviated both mechanical allodynia and anxiety-like behaviors with increased activities of glutamatergic neurons in the rACC and 5-HT neurons in the DRN. Chemogenetic activation of the rACCGlu-DRN projections attenuated both mechanical allodynia and anxiety-like behaviors at day 14 after spared-nerve injury. The role of the rACCGlu -DRN circuit may differ during the progression of CNP, and these changes may be related to the DRN 5-HT neurons (Xu et al. 2023).
2.9.2. Nucleus Raphé Obscurus (NRO) and Nucleus Raphé Pallidus (NRP)
Pain in response to physical activity is common in people with chronic musculo-skeletal pain. Because the NRO and NRP not only modulate motor output but also respond to noxious stimuli, it was hypothesized that the NRO and NRP were important nuclei in the interaction between pain and exercise. In a model of exercise-induced pain that is associated with increased activation of neurons in the medullary RN, i.e., the NRO and NRP, it was tested whether exercise enhances hyperalgesia through activation of NMDARs in the NRO/NRP. Muscle insult was induced by two injections of pH 5.0 saline five days apart into one gastrocnemius muscle (GM). It was initially tested whether hyperalgesia developed in mice injected with acidic saline (pH 5.0) into the GM immediately after a 30-minutes or 2-hours exercise task or two hours after a two-hour exercise task. It was then tested whether blockade of NMDARs in the NRO/NRP during the exercise task prevented the development of exercise-induced hyperalgesia. Changes were then evaluated in phosphorylation of the NR1 sub-unit of the NMDAR (pNR1) after the exercise task at times in which muscle insult was given in behavioral experiments, i.e., immediately after a 30-minute or two-hour exercise task or two hours after the two-hour exercise task. All exercise conditions enhanced nociception (hyperalgesia) after combining with two injections of pH 5.0 saline. Micro-injection of 2-amino-5-phophonopenanoate (AP5) dose-dependently prevented the development of exercise-induced hyperalgesia. All exercise conditions increased pNR1 in the NRO and NRP. Thus, exercise-induced pain in sedentary mice is associated with increased phosphorylation and activation of NMDARs in the NRO/NRP, suggesting that changes in central excitability mediate an interaction between un-accustomed exercise and pain (Sluka et al. 2012).
2.10. Hypothalamus (HYP)
The HYP is a diencephalic structure in the basal forebrain (BFB), consisting of many nuclei that are critical for integrated autonomic and neuro-endocrine responses for homeostasis and adaptation to internal or external stimuli (Takayanagi and Onaka 2021). The HYP receives major sensory inputs from the vagus nerve via the NTS as well as converging nociceptive and visceral inputs from the spinal and trigeminal DHs (Benarroch 2006; Jänig 2014; Saadé and Jabbur 2008), and direct and indirect (via PBN and NA A1 cells, Figure 3) nociceptive inputs from the STTr (Kuner and Kuner 2021).
2.10.1. Hypothalamic-Pituitary-Adrenal (HPA) Axis
The majority of clinical studies have found reduced activity and impaired feedback sensitivity of the HPA axis in chronic pain conditions, mostly characterized by low basal concentrations of cortisol as well as a blunted cortisol responses to a variety of stressors and dynamic tests. Yet, chronic pain conditions such as rheumatoid arthritis and FM are associated with profound dysfunction in the HPA axis, which may exacerbate the symptoms of chronic pain. Possibly, some pain syndromes are associated with changes in the HPA axis, whereas other pain syndromes do not cause any functional changes on the HPA axis, but nonetheless affect the limbic structures involved in the processing of pain and the HYP function (Boadas-Vaello et al. 2017).
HPA axis dysfunction occurred in animal models of chronic inflammatory pain. By contrast, in models of neuropathic pain after CCI, the basal HPA axis function remained unchanged, and the increased nociceptive sensitivity during CCI-associated pain was linked to alterations in the limbic system, but it was nonetheless dissociated from HPA axis activation. Data indicated that, in CCI rats, the medial nucleus of AMY (MeA), CeA and the HIPP were affected, suggesting that increased nociceptive sensitivity during chronic pain was associated with alterations in the limbic system, without HPA axis activation. In the animal model of adjuvant-induced arthritis (AA), associated with hyperalgesia and allodynia in response to hindpaw sensory stimulation, the HPA axis displayed dysfunction characterized by increased basal plasma concentrations of adreno-corticotropic hormone (ACTH) and corticosterone. After intra-plantar injection of carrageenan, the HPA axis was also altered with an over-secretion of corticosterone (Boadas-Vaello et al. 2017).
2.10.2. Hypothalamic-Pituitary-Thyroid (HPT) Axis
Hypophysiotropic thyrotropin-releasing hormone (TRH) is synthesized by neurons that reside in the HYP PVN and are the central regulators of the HPT axis. TRH synthesis and release from these neurons are primarily under negative feedback regulation by thyroid hormones (THs). Feedback regulation of TRH neurons may also be modified by local tissue concentrations of TH. During infection, endotoxin or endotoxin-induced cytokines increase D2 activity in the medio-basal HYP, which by inducing local hyperthyroidism, may play an important role in infection-induced inhibition of hypophysiotropic TRH neurons (Fekete and Lechan 2007).
Of a population of male Sprague-Dawley rats with neuropathic pain following ligation of the sciatic nerve, 20% showed persistently decreased social dominance. The mean plasma thyroxine (T4), free thyroxine (fT4) and triiodothyronine (T3) concentrations decreased significantly post-injury in rats with persistently changed behavior compared to rats with unchanged behavior. There was a correlation between decreased dominance behavior and decrease in both T4 and fT4, but no correlation with thyroid-stimulating hormone (TSH) (Kilburn-Watt et al. 2010).
2.10.3. Hypothalamic-Pituitary-Gonadal (HPG) Axis
Estrogen receptors α and ß (ERα and ERß) mediate different physiological functions. In wild-type and ERß knockout (ERß KO) mice of both sexes, ERß is possibly involved in acute and pain as well as on endogenous pain inhibitory mechanisms. In female groups, ovariectomies followed by estrogen and progesterone replacement were performed to insure comparable sex hormone levels. Nociceptive responses were lower in ERß KO female than in wild-type female mice during the interphase and early tonic phase II of the formalin test but not during acute and late tonic phases. Behavioral and spinal c-fos differences occurred only in females. ERß KO females showed lower c-fos expression in DH laminae I-II and IV-V than wild-type females. This suggests that estrogen, through its actions on ERß, dampens the efficacy of endogenous pain modulation mechanisms during the interphase and/or inflammation process in the early phase II, triggering an increase in spinal nociceptive neuronal activity (Spooner et al. 2007).
2.11. Basal Ganglia (BG)
In higher mammals, the BG consist of bilateral sub-cortical nuclei in the basement of the brain. The dorso-lateral BG contains the striatum (consisting of nucleus caudatus and putamen), the subthalamic nucleus (STN), globus pallidus externus (GPe), globus pallidus internus (GPi) and substantia nigra pars reticularis (SNr). The STN is integrated into the network of BG nuclei (Grillner 2025; Grillner et al. 2020; Groenewegen 2003; Humphries and Prescott 2010; Tewari et al. 2016).
The BG receive nociceptive information from ascending spinal projections, albeit not directly. It has been suggested that the BG may be involved in the (i) sensory-discriminative dimension of pain, (ii) affective dimension of pain, (iii) cognitive dimension of pain, (iv) modulation of nociceptive information and (v) sensory gating of nociceptive information to higher motor areas (Chudler and Dong 1995). The BG are a major site for adaptive plasticity, affecting a broad range of normal behaviors and neurological and psychiatric conditions. The BG integrate incoming nociceptive information to contribute to coordinated, graded motor responses in complex and spatially guided pain avoidance/nocifensive behaviors (Bak et al. 2021).
2.11.1. Striatum
The striatum is a major BG input station and is associated with the development of chronic back pain (CBP). PET studies showed presynaptic dysfunction of the nigro-striatal DA pathway (Bak et al. 2021). The effects of striatal DA on pain are thought to be mediated by the D2 receptor (D2R), since the striatal administration of selective D2R agonists reduced pain responses in animal models of persistent pain, whereas D2R antagonists enhanced this effect. In animal models of chronic pain, both the D2R expression and the excitatory drive in D2R-expressing neurons in the NAc were reduced (Boadas-Vaello et al. 2017). In rats with neuropathic pain, the BG participated in chronic pain as shown by reduced opioid receptor availability in the caudate/putamen. A PET study showed a positive correlation between the deficit of opioid receptor expression and anhedonia/depression-like behavior, suggesting the contribution of the opioid system to the comorbid depression in chronic pain (Da Silva and Seminowicz 2019).
The dorsal striatum not only controls voluntary movement, but also inhibits pain. It is connected to the descending pain modulatory system and in particular to the RVM through the DReN. BG diseases, such as Parkinson´s disease (PD), are associated with pain and hyperactivation of excitatory transmission. Glutamatergic hyperactivation might be counteracted by the activation of group III mGluRs, which are located on presynaptic terminals inhibiting neurotransmitter release. In the dorsal striatum, mGluR7 and mGluR8 are located at glutamatergic cortico-striatal terminals and their stimulation inhibits pain in pathological conditions such as neuropathic pain. In normal conditions, though, the two receptors in the dorsal striatum have a different role in pain control (Boccella et al. 2020).
2.11.2. Nucleus Accumbens (NAc)
The NAc is the ventro-medial input station of the BG and is widely considered to be made up of a shell sub-region and a lateral core section. These regions are distinguished by their connectivity to cortical and sub-cortical regions.
- Inputs/Outputs
The NAc receives inputs from sensorimotor areas, ACC, limbic as well as entorhinal/perirhinal cortices, HIPP, THAL, lHYP, SNc, VTA, and other brainstem areas. Output projections target the CC, BNST, lateral septum, BG, AMY, THAL, Hb, lHYP, Snc/SNR, VTA and brainstem areas (Salgado and Kaplitt 2015).
- Functions
The NAc is a central component of the brain´s reward circuits and has been implicated in a wide range of behaviors and emotional states. It integrates cortical and affective information in order to assign motivation and value for selection of appropriate behavioral responses to outside stimuli. In addition, the NAc shell is thought to evaluate impending pain and utilize spatial information from the HIPP for appetitive learning, and the core activates with expectation of relief of an aversive stimulus and signals the reward value of pain cessation, as well as utilizes information from the BLA for appetitive learning. Dysfunctions of brain networks involved in assigning salience and value are considered to be involved in the transition to chronic pain. The NAc has emerged as an important mediator of this pain-related dysfunction (Thompson and Neugebauer 2019; Xu et al. 2024).
- Alterations ofNucleus Accumbens (NAc)
Changes of NAc circuitry and connectivity occurred as independent risk factors for the development of chronic pain. The connectivity between the NAc and PFC was predictive of the chronic pain progression in patients presenting with low back pain. This was accompanied by decreased gray-matter density in the NAc in patients that progressed to have persistent back pain. In freely moving rats, decreased NAc activity occurred with thermal and electrical noxious stimuli (Thompson and Neugebauer 2019). In rats subjected to SNI, DA levels were reduced in the NAc, and SNI led to up-regulation of GABAergic indirect spiny projection neurons (iSPN), which is related to mechanical hypersensitivity. In rats with SNI, fMRI scans have shown a decrease in functional connectivity in the NAc core. These rats had behavioral signs of persistent neuropathic pain, including tactile allodynia (Boadas-Vaello et al. 2017). Chemogenetic excitation of MSNs of the indirect pathway worsened SNI-induced mechanical allodynia, whereas inhibition alleviated allodynia. In models of neuropathic pain, pharmacological blockade of Ca2+-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors in the NAc core increased depression-like behaviors (Serafini et al.2020).
A select population of NAc neurons exhibited increased activity either upon nociceptive stimulation or during wakefulness. Experimental activation of the population neurons exacerbated pain-like (nociceptive) responses and reduces NREM sleep, while inactivation of these neurons produced the opposite effects. The NAc population primarily consisted of D1 neurons and projected divergently to the VTA and POA. This suggests a common NAc population that encodes chronic pain and controls sleep, and achieves the modality specificity through its divergent down-stream circuit targets (Sun et al. 2023).
- Cortico-Nucleus Accumbens (NAc)Connections
The cerebral cortex is connected to the BG nuclei and THAL by several cortico-basal nuclei-thalamo-cortical loops. A longitudinal brain imaging study of patients with CBP showed that increased functional connectivity of the PFC to the NAc is predictive of pain persistence. In addition, the strength of synchrony or functional connectivity between the mPFC and NAc is predictive of individuals who subsequently transit to chronicity one year later. Moreover, greater functional connections between the dorsal mPFC-AMY-NAc circuit contributed to risk of chronic pain in sub-acute back pain patients. In awake rats, a longitudinal fMRI study indicated that the PFC and NAc displayed abnormal activity to normally innocuous stimuli at 28 days after peripheral nerve injury, coincident with the development of tactile allodynia (Ong et al. 2019).
In a rat model of peripheral neuropathy, activation of PFC neurons projecting to the NAc core increased EPSPs and alleviated sensory hypersensitivity symptoms. Light activation of neurons, infected with adeno-associated viruses encoding channelrhodopsin-2, induced action potential (AP) spikes within the PL PFC of rats. This optogenetic activation alleviated both mechanical and cold allodynia. Optical activation of these neurons relieved pain and promoted a preference for the associated compartment. Photo-activation of channelrhodopsin-2-expressing NAc MSNs confirmed that the activation of projections from the PL PFC to the NAc was responsible for these effects. By contrast, inhibiting rat pyramidal mPFC neurons projecting to the NAc core by halorhodopsin exacerbated acute pain symptoms, suggesting a role of this circuitry in the endogenous antinociceptive pathway (Serafini et al. 2020).
In SNI rats, DA concentrations were reduced in the NAc, and SNI induced an up-regulation of GABAergic iSPN, which was related to mechanical hypersensitivity (Thompson and Neugebauer 2019). In rats, long neuropathic pain states were associated with a reduction in dendritic number and size, as well as with a decrease in extracellular DA levels. Prolonged SNI also led to elevated intrinsic excitability of NAc-shell MSNs of the indirect pathway. Furthermore, peripheral nerve injury reduced the spontaneous spiking of VTA DA cells projecting to NAc MSNs (Serafini et al. 2020).
2.11.3. Subthalamic Nucleus (STN)
The STN is an integral part of the BG network but also a BG input and output station by receiving external inputs and sending glutamatergic projections to external targets, respectively. Thus, the STN receives major inputs from the frontal cortex, THAL, GPe, LC, NRD, SNc and pedunculo-pontine nucleus (PPN) (Brodal 1981), and sends major projections to the GPe and GPi, SNr (Brodal 1981; Wichmann and DeLong 2016) and pontine nuclei which relay inputs to the cerebellum (Bostan and Strick 2018).
Nociceptive STN responses are abnormal in a rat model of PD, suggesting that they depend on the integrity of the nigro-striatal DA system (Pautrat et al. 2018).
STN is closely associated with the pain that is a prevalent non-motor symptom in PD. In rat PD models induced by injection of 6-hydroxydopamine (6-OHDA) into the SNc, STN cells exhibited longer responses with greater amplitude in response to painful foot-shock stimuli, indicating that abnormal neural plasticity in the STN could mediate the pain symptoms in PD. In 6-OHDA-induced PD model mice, the hyper-activity of STN neurons and pain hypersensitivity were blocked by optogenetic inhibition of STN neurons. Optogenetic inhibition of STN-SNr projections attenuated both mechanical and thermal hypersensitivity, while optogenetic inhibition of STN-GPi or STN-ventral pallidum (VP) projections attenuated only mechanical hypersensitivity in PD mice (Bak et al. 2021).
- Anterior Cingulate Cortex (ACC)-Subthalamic Nucleus (STN)Pathway
In mice, neurons in the STN become hyperactive following nerve injury and promote pain-related responses. In chronic pain, neurons in the ACC, which project to the STN, may contribute to STN neuron hyper-activity. Indeed, in mice in chronic pain states following spared nerve injury of the SNI, ACC neurons showed enhanced activity in response to noxious stimuli and alterations in emotional states. When ACC neurons were inhibited, pain responses of STN neurons were attenuated in both naïve and SNI mice. Optogenetic activation of the ACC-STN pathway induced bilateral hyperalgesia and depression-like behaviors in naive mice. Conversely, inhibition of this pathway was sufficient to attenuate hyperalgesia and depression-like behaviors in SNI mice and naïve mice subjected to stimulation of STN neurons. Hence, hyper-activity in the ACC-STN pathway may play an important role in comorbid chronic pain and depression (Wang et al. 2024b).
- Substantia Nigra Reticularis (SNr)-Subthalamic Nucleus (STN)-Parabrachial Nucleus (PBN) Pathway
Finally, in mice, a pathway, consisting of GABAergic neurons in the SNr and glutamatergic neurons in the STN and the lPBN modulated acute and persistent pain states. In these states, the activity of STN neurons was enhanced. This enhancement was accompanied by hypoactivity in SNr neurons and strengthening of the glutamatergic STN-lPBN projection. Reversing the dysfunction in the SNr-STN-PBN pathway attenuated activity of lPBN neurons and mitigated pain-like behaviors (Jia et al. 2022).
2.11.4. Substantia Nigra Pars Reticularis (SNr)
As a BG output nucleus, the SNr may be a candidate for mitigating hyperalgesia in pain states induced by peripheral nociceptive stimuli. In inflammatory and neuropathic pain states in freely behaving male mice, stimulation of either SNr GABAergic neurons or their projections to the STN significantly alleviated nociceptive responses in all pain states on the contralateral side and comorbid depression in chronic pain. However, SNr modulation did not affect baseline pain thresholds. The GABAergic SNr-STN projection was attenuated in pain states, resulting in disinhibition of STN neurons. Thus, impairment of the SNr-STN pathway may contribute to the maintenance of hyperalgesia in both inflammatory and neuropathic pain states and the comorbid depression in chronic pain (Yin et al. 2022).
2.12. Amygdala (AMY)
The AMY is an almond-shaped, heterogeneous nuclear complex embedded deep within the rostral pole of the cerebral temporal lobe, and is a limbic region (Neugebauer et al. 2020). It is comprised of different nuclei; the lateral amygdala (LA), BLA and CeA nuclei and in between, the intercalated cells (ITC) (Figure 5). Altogether, it has nine sub-nuclei (Vogt 2019). The latero-capsular division of the CeA is now defined as the `nociceptive amygdala´ and integrates nociceptive information with poly-modal information about the internal and external bodily environment (Neugebauer et al. 2004). The CeA receives direct and indirect nociceptive inputs from the STTr and from the PBN through the spino-parabrachio-amygdaloid pathway (Allen et al. 2021; Kuner and Kuner 2021).
- Amygdalar Role in Pain Processing
The AMY has a role in the processing of pain, emotions and attaching emotional valence to memories and other experiences, in fear conditioning and affect (Corder et al. 2019; Serafini et al. 2020). It forms the association between an aversive response and the environment in which it occurs (Kissiwaa and Bagley 2018). Its operation changes in chronic pain. Persistent pain and negative affective states such as fear, anxiety and depression are reciprocally related. In persistent pain, electrophysiological, pharmacological, and biochemical neuroplastic changes were shown in the nociceptive AMY. Depending on environmental conditions and affective states, the AMY appears to play a dual facilitatory and inhibitory role in the modulation of pain behavior and nociceptive processing at different levels of the pain neuraxis (Neugebauer et al. 2004).
- Unpleasant Pain Representation in Baso-lateral Amygdala (BLA)
In freely behaving mice encountering noxious stimuli, time-lapse in vivo Ca2+ imaging and neural activity manipulation enabled to identify a distinct neural ensemble in the BLA that encodes the negative affective valence of pain. Silencing this nociceptive ensemble alleviated pain affective-motivational behaviors without altering the detection of noxious stimuli, withdrawal reflexes, anxiety, or reward. After peripheral nerve injury, innocuous stimuli activated this nociceptive ensemble to drive dysfunctional perceptual changes associated with neuropathic pain, including pain aversion to light touch (allodynia) (Corder et al. 2019).
- Amygdalar Hypertrophy and Changed Activity in Rats
Rats with SNI displayed an increase in AMY volume and signs of depressive-like behavior. The AMY hypertrophy was associated with an increased cell proliferation in the CeA and BLA, suggesting that neuropathic pain promotes the generation of new neurons. In the rat, following carrageenan-induced inflammatory pain, BLA slow-firing neurons (supposedly GABAergic) decreased activity, and BLA fast-firing neurons (supposedly glutamatergic) remained hyper-active after four hours following intra-plantar micro-injection of carrageenan (Boadas-Vaello et al. 2017). The CeA appears able to modulate nociception bi-directionally. These opposing functions are associated with two distinct classes of GABAergic neurons that receive inputs from the PBN: protein kinase C-δ (PKC-δ)-expressing neurons enhancing nociception, and Somatostatin (STT)-expressing neurons having anti-nociceptive effects. Following nerve injury, the activity of the first is enhanced, while that of STT neurons is suppressed, leading to a net increase in nociceptive sensitivity (Kuner and Kuner 2021)
- Amygdalar Activation or Hyper-activity in Humans
In experimental and clinical pain states, neuroimaging data showed AMY activation or hyper-activity. Aberrant AMY function also increased the risk of developing chronic pain as increased white-matter connectivity within the mPFC-AMY-HIPP circuit and reduced AMY size are independent risk factors for persistence of back pain (Serafini et al. 2020; Thompson and Neugebauer 2019). Pediatric patients with CRPS showed changes in the functional connectivity of the AMY with cortical and sub-cortical regions (Boadas-Vaello et al. 2017). The AMY appears to modulate affective abnormalities associated with chronic pain, such as catastrophizing. This is important because catastrophizing can further exacerbate chronic pain conditions and increase the likelihood of poor clinical outcomes (Serafini et al. 2020).
- Neuropathic Pain
In rats subjected to CCI of the sciatic nerve, peripheral neuropathy induced a significant decrease in the nociceptive threshold, inducing mechanical hyperalgesia and mechanical allodynia in the right hind-paw in comparison with the baseline measurements. The CCI animals showed more labeling for c-fos-immuno-reactivity in the contralateral BLA and posterior BLA, or the entire BLA than in the naive and sham animals, and higher c-fos-immuno-reactivity in the contralateral central CeA and whole CeA. Hence, the anterior and posterior portions of the BLA and the central portion of the CeA were involved in controlling neuropathic pain (Seno et al. 2018).
- Disturbed Interactions between Amygdala (AMY) and Cortical Regions
Dysfunction in the interactions between AMY and cortical regions such as mPFC (Figure 5) is critical for acute and chronic pain-related plasticity in the brain. Interactions between the BLA, ITC and IL mPFC are important mediators of pain-related dysfunction of cognitive control of AMY output in the pain state. Decreased activity of the infra-limbic PFC (IL-mPFC) projection neurons accounts for decreased ITC-mediated feedforward inhibitory control of CeL activity, and thus for decreased cognitive control of AMY output (Thompson and Neugebauer 2019). An imbalance between excitatory and inhibitory mechanisms enhances excitatory inputs from the brainstem (parabrachial area, PBA) and from the BLA hyper-activity to generate abnormally enhanced feedforward inhibition of principal cells in the mPFC. Impaired cortical control allows the un-controlled persistence of AMY pain mechanisms. In neuropathic animal models, the PBN-CeA synapses and the BLA-CeA synapses are potentiated, whereby the excitability of the CeA neurons is increased (Neugebauer 2015; Yalcin et al. 2014).
- Baso-lateral Amygdala (BLA)-Prefrontal Cortex (PFC)-Peri-aqueductal Gray (PAG)-Spinal Cord Pathway
A BLA-PFC-PAG-spinal cord pathway may be important for the development of mechanical and thermal hypersensitivity after peripheral nerve injury. Nerve injury strengthened synaptic input from the BLA onto inhibitory INTs located in the PL mPFC, by virtue of reduced eCB modulation. These enhanced synaptic connections mediated a feedforward inhibition of projections from the PFC to the vlPAG and its downstream targets. Optogenetic approaches combined with in vivo pharmacology revealed that these BLA-PFC-PAG connections altered pain behaviors by reducing descending NA and 5-HT modulation of spinal pain signals (Huang et al. 2019).
- Amygdala (AMY)-Nucleus Accumbens (NAc) Circuit
While in pain-naïve mice, the AMY-NAc circuit did not have a prominent role in modulating nociceptive thresholds, it played a major role in modulating affective pain symptoms in mice suffering from prolonged peripheral nerve injury. Inhibiting this pathway with opsins alleviated the aversive components of pain without impacting mechanical allodynia (Serafini et al.2020).
- Neuroplasticity in Central Nucleus of Amygdala (CeA)
Neuroplasticity in the CeA may be a mechanism for sensory and emotional-affective aspects of injury-induced pain. In a rat SNL model with neuropathic pain, paw-withdrawal thresholds were measured one week (acute phase) or four weeks (chronic phase) after ligation. Neuropathic animals showed significant mechanical hypersensitivity at both acute and chronic phases of SNL compared to sham controls. In neuropathic pain models, corticotropin-releasing hormone (CRH) neurons in the CeA exhibited differences in excitability and PBN-driven synaptic transmission between acute and chronic phases of neuropathic pain models using brain slice patch-clamp recordings. In genetically distinct and projection-specific CeA neurons, time-dependent changes in neuropathic pain. Hyperexcitability of CRH projection neurons and synaptic plasticity of PBN input at the acute stage shifted to hyperexcitability without synaptic plasticity in non-CRH neurons at the chronic phase. Accordingly, chemogenetic inhibition of the PBN-to-CeA pathway mitigated pain-related behaviors in acute, but not chronic, neuropathic pain (Kiritoshi et al. 2024).
CRH in the CeA is an important modulator of pain and affective disorders. In adult rats tested one week and four weeks after spinal nerve ligation and in sham surgery (control), optical silencing of CeA-CRH neurons decreased the emotional-affective responses in theacute and chronic phases of the neuropathic pain model, but had anxiolytic effects only at the chronic stage and no effect on mechano-sensitivity. Optogenetic activation of CeA-CRH neurons increased the emotional-affective responses and induced anxiety-like behaviors, but had no effect on mechano-sensitivity in control rats. Hence, CeA-CRH neurons contribute to pain-related behaviors under normal conditions (Mazzitelli et al. 2022).
Synaptic Plasticity. In the acute arthritis pain state, the excitatory transmission at the synapses from PBN to lateral central nucleus of the AMY (lCeA), from BLA to lCeA, and from LA to BLA was enhanced. The degree of synaptic potentiation at the PBN-lCeA synapse was determined by a mechanism involving group IV (C)-fiber afferents and correlated with the degree of mechanical hypersensitivity in neuropathic pain. The PBN input is uniquely characterized by its content of CGRP, and CGRP receptor activation in the lCeA was critically involved in pain-related plasticity (Thompson and Neugebauer 2019).
In the rat, a brief nociceptive stimulus with no ongoing injury was able to produce long-lasting synaptic plasticity at the PBN-AMY synapse. This plasticity was caused by an increase in postsynaptic AMPA receptors with a transient change in the AMPA receptor sub-unit, similar to LTP. Moreover, this synaptic potentiation primed the synapse so that a subsequent noxious stimulus caused prolonged potentiation of the nociceptive signal flow into the AMY. Hence, a second injury could have an increased negative emotional value and promote associative learning that results in pain-related avoidance (Kissiwaa and Bagley 2018).
2.13. Habenula (Hb)
The Hb is a phylogenetically old small bilateral epi-thalamic structure in the posterior-medial aspect of the dorsal THAL (Shelton et al. 2012). The Hb can be divided into two major areas, the lateral habenula (LHb) and medial habenula (MHb), which can be further subdivided, resulting in six main subregions (Antunes et al. 2022).
The Hb has been implicated in a large variety of acute and chronic processes, such as pain and analgesia, olfaction, ingestion, mating, endocrine functions, circadian rhythms, reward processing, social behavior, behavioral flexibility, decision-making, cognitive flexibility, contextual memory, as well as in the neurobiology of several psychiatric disorders and neuropsychiatric symptoms (Baker et al. 2022; Dai et al. 2022; Gouveia and Ibrahim 2022; Hu et al. 2020; Metzger et al. 2021; Shelton et al. 2012).
Inputs/Outputs. The Hb receives diverse inputs that conyey information about the sensory world and the animal´s internal state (Fore et al. 2018). The primary input and output region of the LHb is the VTA, through a bi-directional connection. The LHb receives inhibitory inputs from the mPFC, lateral septum, entopeduncular nucleus, diagonal band of Broca, BG (globus pallidus, ventral pallidum), HYP [lateral pre-optic area (lPOA), BNST], supra-chiasmatic nucleus (SCN), and pineal gland (reciprocal). The MHb receives mostly excitatory inputs from the BFB (septum, diagonal band of Broca), pineal gland, and NAc. The LHb projects to the ventro-lateral septum, THAL, HYP, PBN, dorsal raphé, median raphé, SNc, VTA, rostro-medial tegmental nucleus (rmTN), PAG, and LC. In addition, the MHb sends projections to the commissural septum and LHb (Baker and Mizumori 2017; Gouveia and Ibrahim 2022; Hu et al. 2020; Metzger et al. 2021; Shelton et al. 2012). Eletrical stimulation of the LHb inhibited the activity of DA neurons (Shelton et al. 2012).
Chronic Pain is associated with several alterations including a deficient reward state due to changes in reward–aversion processing; stress; in co-morbid conditions such as anxiety, depression or addiction; and in more complex processes such as defensive behaviors and prediction errors. Stimulation of the Hb may reverse some of these behaviors including depression or potentially addiction. Given the Hb connections to DA regions in the VTA and SNc, Hb likely plays a role in reward-related signals. Analgesia may be a process driving these reward related activities. Consistent with a putative role of the Hb in negative re-inforcement or responses to aversive stimuli (such as pain), stimulation of the LHb results in a reward decrease as measured by sucrose self administration (Shelton et al. 2012).
Pain is a stressor, and the Hb acts as a behavioral mediator to stressors. Implications of pain as a stressor are particularly prominent if chronic or repeated (e.g., in migraine). The MHb has been shown to be involved in neuro-endocrine and immunological responses to various kinds of stress. The Hb seems to have a role in promoting decision-making and survival driven reaction to aversive stressors including pain (Shelton et al. 2012).
Rats with peripheral neuropathy (CCI) showed decreased mechanical nociceptive threshold and increased depressive-like behavior compared to control groups. The CCI group presented decreased cFos immunoreactivity in the total Hb, total LHb and lateral Hb sub-regions, compared to controls. This suggests an involvement of the Hb in neuropathic pain and accompanying depressive-like behavior (Antunes et al. 2022).
Aberrant activity of the LHb has been suggested to be associated with depressive symptoms characterized by excessive negative focus, leading to high-level cognitive dysfunctions. There are complex interactions between the LHb and VTA, which may have a crucial role in behavioral regulation and be potentially involved in the pathological impact of chronic pain on cognitive functions (Pereira et al. 2023).
2.14. Thalamus (THAL)
The THAL forms the larger dorsal sub-division of the diencephalon located medially to the capsula interna and nucleus caudatus. It is a large mass of gray matter made up of a multitude of nuclei, through which sensory information is relayed and processed on its way to the cerebral cortex. The THAL also transmits and processes signals from the BG and cerebellum. The main spinally ascending pathway conveying nociceptive signals is the STTr. The STTr directly targets the posterior, lateral and medial THAL, the latter also indirectly via the RF, but also several other regions, including brainstem areas such as group A1 NA cells, PBN, inter-collicular area, and the PAG (Kuner and Kuner 2021).
The THAL has a pro-nociceptive role. Chronic pain was associated with a reduced THAL volume and transmitter imbalances consistent with the loss of neurons, and loss of THAL somatosensory neurons affected the activity of THAL reticular nucleus neurons, thereby affecting pain perception. In neuropathic patients, medial thalamotomy produces pain relief. The VPL showed increases in spontaneous discharge and burst activity (Bak et al. 2021). Furthemore, CNP was associated with an ongoing decrease of THAL activity accompanied by an altered thalamo-cortical rhythm. THAL micro-stimulation revealed pain sensations in both non-pain and chronic pain patients (Kuner and Kuner 2021).
Moreover, some midline THAL nuclei appear to function as `nociceptive discriminators´ and form two distinct endogenous systems that control noxious responses: the THAL medio-dorsal nucleus (MD) that participates in the descending facilitation underlying the development of mechanical allodynia and the ventro-medial nuclei of the THAL that specifically mediates the descending inhibition of thermal hyperalgesia in rats (Liang et al. 2020).
Rats subjected to sciatic CCI showed thermal hyperalgesia and microglial reactivity in THAL VPL nucleus. In rats subjected to spinal cord contusion, chemokine expression was increased. SCI suppressed the activity in the GABAergic nucleus of the zona incerta (ZI), and concomitantly increased the activity in one of its main targets: the posterior nucleus of the THAL (PO). SCI also caused a similar pathological increase in other THAL nuclei, specifically the MD, involved in emotional-affective aspects of pain. The activity of neurons in the two THAL ventro-posterior medial (VPM) nuclei also increased in rats subjected to CCI. Other experimental studies have revealed that neuropathic pain caused central sensitization of neurons in the VPL nucleus, which are responsible for this hyperexcitability to neural injuries. Hyperexcitability of THAL neurons has also been linked to a reduction of inhibitory inputs (Boadas-Vaello et al. 2017).
- Thalamic (THAL) Re-organization
In response to peripheral injuries and inflammation, the THAL undergoes acute and chronic plastic changes. THAL lesions can entail pain referred to the periphery, such as allodynia, hyperalgesia and hyperpathia. There also alterations of somatotopic organization (Saadé and Jabbur 2008).
- Thalamic (THAL) Discharge
Neuropathic patients showed slow rhythmic bursting activity but decreased metabolic hypo-activation in somatosensory THAL. However, in neuropathic animal models, the THAL ventro-basal complex exhibited bi-lateral hyper-activity to moderate mechanical stimuli, changed responses to natural stimuli, and abnormal activation during the processing of somatic inputs (Saadé and Jabbur 2008). In a rat model of trigeminal neuropathic pain (TNP), the activity of 120 neurons was recorded in the two VPM nuclei two weeks after a CCI of the IoN. The rats exhibited strong pain-related reactions to extremely weak mechanical stimuli applied to the lesioned and, to a lesser extent, un-lesioned IoN territories. The activities of neurons recorded in the contralateral VPM (VPMc) and ipsilateral VPM (VPMi) were compared with those of 62 neurons recorded in the VPM of ten normal rats (VPMn). The neuronal background activity was higher in the VPM of CCI-IoN rats than in normal rats (about 4 vs 1 spikes/s). The effective stimulus modality differed significantly between the normal and the lesioned rats. In particular, the number of neurons driven by vibrissa- or guard-hair movements dramatically decreased in the VPM of CCI-IoN rats, mainly in the VPMc. Inversely, a consistent number of neurons in both the VPMc and VPMi were driven by other stimulus modalities applied to the IoN territory (moderate pressure for VPMc neurons, pinch or pinprick for both VPMc and VPMi neurons). The responses so induced were especially intense and included after-discharges (Vos et al. 2000).
- Posterior Paraventricular Nucleus (pPVT) Neurons
In male rats, neurons in the posterior portion of the paraventricular nucleus of THAL (nPVT) and their downstream pathway were involved in descending nociceptive facilitation regarding the development of neuropathic pain conditions. Lesions or inhibition of pPVT neurons alleviated mechanical allodynia induced by SNI. The excitability of pPVT neurons projecting to the CeA was significantly increased in rats with SNI (Liang et al. 2020).
- Paraventricular Nucleus of THAL (PVT) to Baso-lateral Amygdala (BLA) Connection
In a mouse model of neuropathic pain, the THAL PVT played a critical role in chronic pain-induced anxiety-like behavioral abnormalities. Fiber photometry and electrophysiology demonstrated that chronic pain increased the activities in PVT glutamatergic neurons. Chemogenetic manipulation revealed that suppression of PVT glutamatergic neurons relieved pain-like behavior and anxiety-like behaviors. Conversely, selective activation of PVT glutamatergic neurons showed algesic and anxiogenic effects. The elevated excitability of PVT glutamatergic neurons resulted in increased excitatory inputs to the BLA neurons. Optogenetic manipulation of the PVT-BLA pathway bilaterally modulated both the pain-like behavior and anxiety-like phenotypes (Tang et al. 2024).
- Thalamo-cortical Connections
Under pathological conditions, increased thalamo-cortical input not only to the somatosensory cortex or IC but also the PFC might contribute to chronic pain. In patients with CNP, THAL GABA content was significantly reduced. Increased thalamo-cortical activity could increase the activity of the IC and the constant perception of pain (Mills et al. 2021).
- Nucleus Accumbens (NAc) to THAL Connection
In female Sprague-Dawley rats, L4-5 DRG root compression was employed, and the animals received unilateral injections of optogenetic virus in the NAc core. Reflexive pain tests were performed to assess the alterations between the groups at the light on/off states. To determine THAL firing, extracellular single-unit in vivo recording were performed. Compared to sham-operated rats, chronic compressed DRG rats showed elevated behavioral sensitivity and sustained neuronal hyperexcitability in the THAL. NAc optic stimulation improved pain behaviors and lowered THAL discharge from ventral postero-lateral THAL nuclei. Hence, the NAc core impacts the reward and motivational aspects of CNP influenced by limbic behaviors to THAL discharge. Increased THAL firing activity may result in chronic compressed DRG-induced neuropathic pain (Kc et al. 2020).
High-frequency stimulation of the sciatic nerve at intensities recruiting group IV (C) afferents induced LTP in the DH for 4–9 hours, which could be at the origin of chronic pain arising after an initial painful event. High-frequency stimulation of the sciatic nerve also caused supraspinal modifications as evident by the development of enhanced and long-lasting neuron excitability in the VPL nucleus and cortical synchronization, and it also enhanced the excitability of neurons in the THAL posterior triangular nucleus (PoT), which is one of the main targets of spinal and trigeminal lamina I neurons. PoT neurons project to the posterior insular cortex (pIC) involved in pain perception in humans and nociceptive processing in rodents (Boadas-Vaello et al. 2017).
2.15. Cerebral Cortex
Since the cerebral cortex is anatomically and functionally divided into a large number of areas that are connected into a complex network, many chronic pain syndromes have repercussions on several brain areas.
Besides abnormal sensitization in the periphery and spinal cord, neural plasticity in the brain contributes the development and maintenance of neuropathic pain. Peripheral nerve injury triggers maladaptive plastic changes along the somatosensory system, altering nociceptive signal processing. Maladaptive plastic changes occur in the S1, ACC, PAG and the BG (Bak et al. 2021).
At cortical levels, several areas including somatosensory cortices (S1 and S2), motor cortex, PFC, vlOFC, ACC and IC, are involved in modulation of nociception. This occurs via various mechanisms: (i) reorganization; (ii) cortico-cortical or cortico-sub-cortical interactionsa; (iii) descending modulatory effect at spinal level by direct cortico-spinal projections or mostly by activation of brainstem structures (i.e. PAG, LC, RN, and RVM) (Gamal-Eltrabily et al. 2021).
- Re-organization
The adult brain is capable of substantial plastic change in such areas as the S1. For example, neuropathic and musculo-skeletal pain are associated with substantial re-organization of S1, motor cortex, ACC and IC. In patients with cLBP and FM, the amount of re-organizational change increases with chronicity. In phantom-limb pain and other neuropathic pain syndromes, cortical re-organization is correlated with the amount of pain. Cortical plasticity related to chronic pain can be modified by behavioral interventions that provide feedback to the brain areas that were altered by somatosensory pain memories or by pharmacological agents that prevent or reverse maladaptive memory formation (Flor 2003).
- Gray Matter Loss
Central post-stroke pain might be associated with cortical structural plasticity. MRI and voxel-based morphometry were used in 45 patients suffering from chronic sub-cortical sensory stroke with and without post-stroke pain, and healthy matched controls. In comparison to healthy controls, post-stroke pain patients showed decreases in gray-matter volume, involving S2, anterior insular cortex (aIC) and pIC, ventro-lateral prefrontal cortex (vlPFC) and OFC, temporal cortex, and NAc. Comparing post-stroke pain patients to non-pain patients revealed a similar but more restricted pattern of atrophy comprising S2, vlPFC and temporal cortex. Gray-matter volume in the ventro-medial prefrontal cortex (vmPFC) negatively correlated to pain intensity ratings. Thus, post-stroke pain is accompanied by a unique pattern of widespread structural plasticity, which involves the sensory-discriminative areas of IC and somatosensory cortex, but also expands into PFC and ventral striatum, where emotional aspects of pain are processed (Krause et al. 2016).
2.15.1. Somatosensory Cortices
S1 and S2 receive nociceptive inputs from the STTr mediated via the THAL (Kuner and Kuner 2021). In functional brain imaging studies, nerve-injured patients and animals showed enhanced excitation, somatotopic re-organization, and changes in cortical thickness in S1, the amount of which highly correlated with the degree of allodynia. Structural changes have also been seen in patients with TN who showed a reduction in the volume of gray matter in S1, S2, as well as in the dorso-lateral prefrontal cortex (dlPFC), OFC, IC, ACC, THAL and cerebellum (Boadas-Vaello et al. 2017).
Functional neuroimaging has demonstrated a relationship between the intensity of de-afferentation pain and the degree of de-afferentation-related re-organization of the S1. De-afferentation pain is also associated with hyperactivity of the somatosensory THAL and cortex. The cortical hyper-activity and re-organization can be mitigated by using transcranial magnetic stimulation (TMS). The first studies demonstrated a statistically significant pain suppression in all patients and a clinically significant pain suppression in 80% of them (De Ridder et al. 2007).
- Activity of S1
In a mouse model of neuropathic pain, the activity of pyramidal neurons in the S1 was persistently increased (Cichon et al. 2017).
S1 activity is closely associated with the development and maintenance of chronic pain. Human fMRI showed that neuropathic patients displayed increased S1 neural activity in response to painful or allodynic stimuli. This could result from several functional and structural changes elucidated in animal studies. Nerve injury led to an increase in gain rate of axonal boutons. Bouton turnover in S1 appeared only in the development phase of neuropathic pain. Up-regulation of metabotropic glutamate receptor 5 (mGluR5) signaling in S1 astrocytes appeared after partial sciatic nerve ligation injury. Chronic pain could thus be induced by functional and structural changes in S1 without changes in the spinal cord or peripheral nerves, and activation of S1 astrocytes plays a role in neuropathic pain. The transient spinal cord ischemia (tSCI) model mice showed bilateral mechanical allodynia and increased spontaneous Ca2+ activity of S1 pyramidal neurons. The number of APs increased in response to current injections; the number of spontaneous AP firing rate increased, and EPSP frequency increased. STT-expressing inhibitory neurons and PV-expressing inhibitory neurons showed decreased Ca2+ activity after spared nerve injury, whereas VIP-expressing INTs increased their activity. These changes synergistically move pyramidal neurons into hyper-activity (Bak et al. 2021).
2.15.2. Primary Motor Cortex (M1)
- Primary Motor Cortex (M1) Stimulation
Epidural stimulation of the motor cortex involves activation of multiple cortico-subcortical areas initiated in the THAL, with involvement of endogenous opioids and descending inhibition toward the spinal cord. In neuropathic pain, repetitive high-frequency dlPFC has not proven efficacious for pain, so far.. Transcranial direct current stimulation (tDCS) is applied upon similar targets as rTMS and does not elicit APs but modulates the neuronal resting membrane state. Patients responsive to tDCS may differ from those improved by rTMS. Both invasive and non-invasive procedures exert their effects through multipledistributed brain networks influencing the sensory, affective and cognitive aspects of chronic pain. Their effects are mainly exerted upon abnormally sensitized pathways, rather than on acute physiological pain (Garcia-Larrea and Quesada 2022).
Transcranial direct current stimulation (tDCS) targeting both M1 and dlPFC may modulate the descending pain inhibitory system. The effects of anodic stimulation of M1 and dlPFC were explored on conditioned pain modulation (CPM) and pain thresholds. Twenty-six healthy adults were randomly assigned to M1-tDCS, dlPFC-tDCS, or sham-tDCS groups. During three sessions, each participant received an active or sham stimulation of 2 mA for 20 minutes, with at least three-day intervals between sessions. Quantitative sensory tests were performed to obtain pressure-pain threshold, cold-pain threshold, and CPM before and after the tDCS intervention. Only M1-tDCS significantly increased CPM in healthy individuals compared with sham control. No statistically significant difference was found in pressure pain threshold and cold-pain threshold between tDCS vs. sham control. This supports a role of M1 as a target in pain regulation (Jiang et al. 2022).
2.15.3. Prefrontal Cortex (PFC) and Associates
The PFC is an important neural substrate for executive functions and decision-making. Reduced PFC function leads to increased impulsivity, reduced control over social behaviors and impaired decision making. It is composed of multiple structures and exhibits species-specific differences between rodents, primates, and humans in connectivity, cytoarchitecture, electrophysiological properties, protein expression, and responses following damage (Thompson and Neugebauer 2019).
The PFC is on top of a complex network of interconnecte areas involved in the representation and modulation of nociception and pain, which include the ACC, vlOFC, IC, HIPP, motor cortex, somatosensory cortices (S1, S2), AMY, and NAc. .Any changes of its structure, connectivity and functions should thus have significant consequences for pain processing and could contribute to the genesis and maintenance of chronic pain. This also depends on PFC connections to other CNS regions, including HIPP, PAG, AMY, BG, and THAL. During acute and chronic pain, alterations occur in gene expression, glial cells, neurotransmitters, and neuro-inflammation, which affect its structure, activity, and connectivity (Ong et al. 2019; Thompson and Neugebauer 2019).
- Epigenetic Re-programming
In rodents, widespread epigenetic re-programming occurs in the PFC many months after nerve injury. Epigenetic modifications, including DNA methylation, can drive changes in gene expression without modifying DNA sequences. It has been hypothesized that acute pain after injury results in rapid and persistent epigenetic re-modelling in the PFC that evolves as pain becomes chronic. In the mouse PFC, epigenome-wide analysis was performed one day, two weeks, six months, and one year after peripheral injury using the spared nerve injury.
Spared nerve injury resulted in rapid and persistent changes in DNA methylation, with robust differential methylation observed between spared nerve injury and sham-operated control mice at all time points. Hundreds of differentially methylated genes were identified, including many with known function in pain. Pathway analysis revealed enrichment in genes related to stimulus response at early time points, immune function at later time points, and actin and cytoskeletal regulation throughout the time course (Topham et al. 2020).
- Prefrontal Cortex (PFC)-Nucleus Accumbens (NAc) Connection
Activation of the projections from the PFC to the NAc can produce inhibition of both sensory and affective components of pain. An optogenetic strategy selectively inhibited the axonal projection from PFC pyramidal neurons to neurons in the NAc core. In naïve rats, the inhibition of the PFC or its projection to the NAc enhanced both sensory and affective symptoms of acute pain. Inhibition of this pathway also increased nociceptive sensitivity and the aversive response in a CNP model. PFC-NAc inhibition also entailed a similar aversive phenotype as chronic pain. Hence, the PFC-NAc projection plays an important role in endogenous pain regulation, and its impairment contributes to the pathology of chronic pain (Zhou et al. 2018).
- Prefrontal Cortex (PFC)-Amygdala (AMY) Connections
The AMY is reciprocally connected to the PFC and is involved in fear and fear conditioning. Pain patients exhibited increased functional connectivity between the left AMY and multiple cortical, subcortical and cerebellar regions, whereas decreased hyper-connectivity between the left AMY to the motor cortex, parietal lobe, and CC may occur after pain rehabilitation treatment. Animal studies showed that the BLA evoked excitatory and inhibitory responses in cortico-PAG neurons in layer V of both the pre-limbic (PL) and infra-limbic (IL) PFC cortices. High-frequency stimulation induced long-lasting suppression of specific high-threshold responses of nociceptive neurons in the PFC, and micro-injection of NMDAR antagonists or mGluR antagonists prevented the induction of long-lasting suppression. In a rat model of arthritic pain, direct excitatory transmission from the AMY to the PFC was not changed, but inhibitory transmission increased in the mPFC. This could not only result in abnormally enhanced inhibition of principal cells in the mPFC, resulting in reduced output from the mPFC to the PAG and reduced anti-nociception, but also reduced inhibition of the AMY itself, possibly contributing to un-controlled AMY pain mechanisms. The IL mPFC evoked strong synaptic inhibition of neurons in the latero-capsular division of the CeA, and this inhibition was impaired in an arthritis pain model (Ong et al. 2019).
- Medial Prefrontal Cortex (mPFC)
In rodents, the mPFC is composed of the PL PFC, IL, and ACC (Kummer et al. 2020; Thompson and Neugebauer 2019). It has been suggested to process the emotional and cognitive components of pain and to be a `central hub for mental comorbidities associated with chronic pain´, such as depressive mood and impaired cognition (Kummer et al. 2020). The vmPFC has rich reciprocal connections with limbic areas and the HYP, and modulates AMY and HIPP activity (Maletic and Raison 2009; Thompson and Neugebauer 2019). The mPFC mediates anti-nociceptive effects via its connections with other cortical areas and via its inputs to the PAG for modulation of pain (Ong et al. 2019).
- Cortical Re-organization
MRI has revealed gray-matter loss in chronic pain conditions, including a decrease in mPFC volume in patients with chronic pain (Serafini et al. 2020; Yang and Chang 2019). Anatomically, the anterior cerebral cortex, HIPP and AMY are implicated in experiential aspects of pain. In humans, longitudinal imaging studies showed that the brain activity is spatio-temporally re-organized during the transition to chronic pain, during which process the representation of pain gradually shifts from sensory to emotional and limbic structures. The state of the limbic-cortical network determines whether nociceptive signals are transient or chronic by extinguishing pathways or amplifying signals that intensify the emotional component of nociceptive inputs. Thus, chronic pain has been interpreted as the persistence of the memory of pain and/or the inability to extinguish painful memories (McCarberg and Peppin 2019). Considering its importance in extinction of fear behaviors, impaired mPFC activation could lead to a failure in the elimination of sub-cortically driven fear behaviors, thereby resulting in pain chronification. The expanse and degree of cortical re-organization and changes in cortical activation are notable in humans suffering from persistent pain, as well as phantom pain, a difficult to treat type of neuropathic pain (Aternali and Katz 2019; Giummarra et al. 2008). A resting-state fMRI study with rheumatoid arthritis patients revealed that prolonged pain states are associated with increased connectivity between the PFC and IC. CBP resultsed in increased PFC activity and this activity is strongly related to pain intensity (Serafini et al. 2020).
- Re-arrangement of Excitatory Inputs
The excitability of the mPFC depends on the integrated effects of intrinsic excitability and excitatory and inhibitory synaptic inputs. The main extra-cortical sources of excitatory input to the mPFC originate in the HIPP, THAL, and AMY. In the rodent mPFC uncder chronic pain conditions, the synaptic properties of these main glutamatergic inputs are altered, and these alterations contribute to pain-associated mPFC de-activation (Jefferson et al. 2021).
- Changes inMedial Prefrontal Cortex (mPFC)Activity
The excitability of the mPFC depends on the integrated effects of intrinsic excitability and excitatory and inhibitory inputs. The main extra-cortical excitatory inputs to the mPFC originate in the HIPP, THAL and AMY. The mPFC is severely impaired in chronic pain and shows pain-associated de-activation. The PL PFC and IL PFC exhibit reduced activity, while the ACC appears hyperactive (Jefferson et al. 2021; Kummer et al. 2020; (Figure 4). The degree of change in the the BOLD (blood-oxygen-level-dependent) signal in the mPFC and the ACC were highly correlated with the severity of spontaneous pain in chronic back patients (Serafini et al. 2020). While, in chronic pain, the ACC appeared hyper-active, PL and IL regions showed reduced activity. Accordingly, factors such as distraction, positive mood, and anticipation of pain relief such as placebo can ameliorate pain by affecting mPFC function (Kummer et al. 2020). In chronic pain states, hyper-activity of neurons in the BLA led to deactivation of the neurons in the mPFC (Kuner and Kuner 2021).
It has been hypothesized that pain-related diminution of mPFC activation (Figure 4) could contribute to pain chronification. In the anesthetized rat subjected to acute arthritis pain, electrophysiological studies demonstrated decreased evoked and background activity in the PL and IL mPFC. The spared-nerve injury model of neuropathic pain reduced glutamatergic transmission in PL mPFC layer V neurons, and reduced pyramidal cell firing in brain slice experiments. The reduction in neuronal mPFC activity can in part be explained by increased feedforward inhibition exerted by by PV-expressing GABAergic INTs in the AMY. In this model, projection neurons in the BLA synapse on PV-GABAergic INTs as well as on mPFC projection neurons in the IL and and PL cortices. PV-GABAergic INTs in turn project to mPFC projection neurons, resulting in BLA-driven feedforward inhibition of mPFC projection-neuron activity. BLA activity is increased in arthritis pain, resulting in pain-related cortical dysfunction. Projection neurons from the IL cortex synapse on BLA projection neurons, as well as on GABAergic ITC neurons. GABAergic ITC neurons and BLA projection neurons synapse on CeL projection neurons. Reduced cortical drive onto BLA and ITC neurons in the arthritis pain state results in reduced feedforward inhibition onto CeL projection neurons. This, coupled with increased drive from the BLA, results in pain-related AMY hyper-activity (Thompson and Neugebauer 2019).
Data obtained in animal models support reduced activity of layer V mPFC neurons in acute and chronic pain states. Reduced mPFC activity has been linked to pain behaviors in a series of experiments using manipulations of mPFC activity. In rats with neuropathic pain due to SNI, optogenetic activation of layer V mPFC neurons significantly inhibited tonic pain responses in the CPP test and mechanical and thermal sensitivity on the von Frey and Hargreaves tests, respectively. Such studies show the importance of GABAergic INT activity for the regulation of mPFC output (Neugebauer et al. 2009).
In rodents, PL cortex neurons showed increased firing rates after noxious stimulation, while maintained nociceptive input suppressed both basal spontaneous and pain-evoked activity. Neuropathic pain caused disruption of working memory and PL neuronal firing activity, which was reversed by selective inhibition of PL pyramidal cells using optogenetic interventions. In the IL cortex of monoarthritic rats, both metabotropic glutamate receptor 1 (mGluR1) and mGluR5 activation induced pro-nociceptive behavior, which is based on increased activation of DReN neurons. Peripheral inflammation decreased BDNF levels in the IL but not PL, and infusion of BDNF into the IL alleviated inflammatory pain and accelerated long-term recovery from inflammatory pain (Kummer et al. 2020).
- Medial Prefrontal Cortex (mPFC)-Thalamus (THAL) Connections
Under pathological pain conditions, increased thalamo-cortical input to not only the somatosensory cortex or IC, but also the PFC might be a contributing factor in chronic pain. In patients with CNP, magnetic resonance spectroscopy showed that the THAL GABA content was significantly reduced. Chronic pain also induced a higher coherence between the medio-dorsal THAL nucleus and mPFC field potential activities. In animals, pharmaco-genetic inhibition of the PVT attenuated visceral pain induced by pancreatitis. This suggested a pro-nociceptive effect of PVT or MD THAL inputs that might occur via activation of GABAergic neurons and feedforward inhibition of principal neurons in the mPFC that project to the PAG for the suppression of pain. Gabapentin, a GABA analogue, exerts an analgesic effect by reversing SNI-induced increases in connectivity between the THAL and the cortex (Ong et al. 2019).
- Medial Prefrontal Cortex (mPFC)-Peri-aqueductal Gray (PAG) Connections
A major sub-population of mPFC neurons projects to the PAG. In mice having undergone CCI of the sciatic nerve, whole-cell recordings revealed a significant reduction in excitability for layer 5 (L5) PFC-PAG neurons contralateral to CCI in the PL, but not IL, region of mPFC. Excitatory inputs to L5 PFC-PAG neurons in both PL and IL mPFC arose primarily from layer 2/3 (L2/3) and were significantly reduced in CCI mice, as was the excitability and L2/3 inhibitory input to PFC-PAG neurons ipsilateral to CCI. This showed region- and laminar-specific changes to mPFC-PAG neurons (Cheriyan and Sheets 2018). In mice with SNI, the use of chemogenetic methods activated glutamatergic projections from the rACC to the vlPAG and induced both hyperalgesia and anxiety-like behaviors in sham mice, while its inhibition of the rACC-vlPAG pathway reduced anxiety-like behaviors and hyperalgesia. These symptoms were alleviated by EA. While chemogenetic activation of the rACC-vlPAG pathway blocked the analgesic effect of EA in the SNI mice, it did not affect the chronic pain-induced negative emotions (Zhu et al. 2022).
- Dorso-medial Prefrontal Cortex (dmPFC)
The dmPFC has been recognized as an important cortical area for nociceptive modulation. Lesions in the dmPFC induced an algesic and anxious state. There is an excitatory descending neural pathway from the dmPFC to the vlPAG. In a mouse model of chronic pain, activation of the dmPFC/vlPAG neural pathway by optogenetic manipulation produced analgesic and anti-anxiety effects. Inhibitory neurons in the dmPFC were specifically activated using a chemogenetic approach, which logically produced an algesic and anxious state under both normal and chronic pain conditions. Application of antagonists of the GABAAR or mGluR1 to the dmPFC produced analgesic and anti-anxiety effects. This suggests that the dmPFC/vlPAG neural pathway might participate in the maintenance of pain thresholds and anti-anxiety behaviors under normal conditions, while silencing or suppressing the dmPFC/vlPAG pathway might be involved in the initial stages and maintenance of chronic pain and the emergence of anxiety-like behaviors (Yin et al. 2020).
- Ventro-medial Prefrontal Cortex (vmPFC)
The ANS and pain interact, which might involve descending pain-modulatory mechanisms. The PAG is involved both in descending pain modulation and ANS, but its role in mediating this relationship has not yet been explored. Thirty participants underwent CPM assessments, in which they rated painful pressure stimuli applied to their thumbnail, either alone or with a painful cold contralateral stimulation. Differences in pain ratings between `pressure-only´ and `pressure+cold´ stimuli provided a measure of descending pain control. In 18 of the 30 participants, structural scans and two functional MRI assessments, one pain-free and one during cold-pain were acquired. Heart-rate variability (HRV) was simultaneously recorded. Normalized low-frequency HRV (LF-HRVnu) and the CPM score were negatively correlated. Individuals with higher LF-HRVnu during pain reported reductions in pain during CPM. PAG-vmPFC and PAG-RVM functional connectivity correlated negatively with the CPM. PAG-vmPFC functional connectivity mediated the strength of the LF-HRVnu-CPM association. CPM response magnitude was also negatively correlated with vmPFC gray-mass volume. ANS dysregulation and dysfunctional descending pain modulation are characteristics of chronic pain (Makovac et al. 2021).
Brain resting-state PAG functional connectivity (FC) differences were studied between patients with cLBP in low pain or high pain condition and matched healthy controls. PAG seed-based FC analysis of the fMRI data was performed to investigate the difference among the connectivity maps in the cLBP in the low or high pain condition and healthy groups as well as within the cLBP at differing endogenous back pain intensities. FC between the PAG and the vmPFC/rACC increased in cLBP patients compared to matched controls. There were also significant negative correlations between pain ratings and PAG-vmPFC/rACC FC in cLBP patients after pain-inducing maneuver. This agrees with the impairments of the descending pain modulation reported in patients with cLBP (Yu et al. 2014).
- Dorso-Lateral Prefrontal Cortex (dlPFC)
The dlPFC is a region in the frontal lobe of the brain, specifically located on the lateral (side) surface, above the inferior frontal sulcus and anterior to the PM (Figure 4). The dlPFC is a functionally and structurally heterogeneous region and implicated in sensory, cognitive and affective processing. The dlPFC is commonly activated in experimental pain, and shows abnormally increased function in chronic pain populations. Some chronic pains are associated with diminished left dlPFC gray matter. The dlPFC is involved in encoding and modulating acute pain and functional and structural abnormalities were reversed after successful interventions for chronic pain. Non-invasive stimulation of the left dlPFC effectively treated some chronic pains (Seminowicz and Moayedi 2017).
Connections. The dlPFC has extensive top-down projections, e.g., to the posterior association cortices to regulate attention and to the sub-genual CC via the rostral and medial PFC to regulate emotional responses (Joyce et al. 2025).
Connections between Right dlPFC and Left NAc. Animal and human studies suggest that the NAc and its connections play a critical role in the transition from acute to cLBP. The identified connections were correlated with cLBP intensity. Compared with healthy controls, individuals with cLBP exhibited hyper-connectivity between the NAc shell and core and the PFC. Although several NAc-PFC connections were linked to higher cLBP intensity, only the connections between the left NAc shell and core to the right dlPFC were reproduced in validation cohorts. Connections between NAc and right dlPFC achieved 84% classification accuracy. Overall, NAc-PFC connectivity consistently distinguishes people with cLBP from healthy controls and suggest an abnormal interaction between the NAc and brain regions involved in motivation, decision-making, and pain regulation (Sunavsky et al. 2025).
Functions. The dlPFC is activated in response to nociceptive stimuli in healthy subjects, and exhibits differential activation between chronic pain patients and controls. Its role in pain remains ambiguous. Left dlPFC activity was negatively related to pain unpleasantness. The dlPFC may also be involved in placebo modulation of pain. A role of dlPFC in pain detection is supported by the observation that, in a sample of healthy subjects, the dlPFC exhibited binary (all-or-none) activity in response to pain regardless of the stimulus or reported pain intensities. Moreover, neuroimaging studies of experimental persistent pain, and experimental models of hyperalgesia and allodynia have revealed a parametric relationship between pain sensitization and dlPFC activity, suggesting a role in pathological pain (Seminowicz and Moayedi 2017).
Gray-Matter Reduction occurs often in chronic pain states, but may vary between different brain areas and pain syndromes. However, there is one region that shows gray-matter loss across various syndromes: As compared to healthy subjects, patients with CBP, migraine, TNP, hypnic headache, chronic post-traumatic headache, hip osteo-arthritis, CRPS have reduced dlPFC gray matter, suggesting a particular importance (Seminowicz and Moayedi 2017).
Pain following normally non-painful heat stimuli on chemically irritated skin (heat allodynia) uniquely engages extensive areas of the bilateral dlPFC, ventral orbito-frontal cortex (vOFC) and peri-genual ACC. On data obtained during heat allodynia in 14 male healthy subjects, principal component analysis (PCA) and multiple regression analysis were applied to evaluate the covariance structure of the volumes of interest (VOI) activated specifically and the relationship of these VOI to ratings of pain intensity and affect. There was a primary principal component (PC) that correlated positively with intensity and unpleasantness and accounted for activity in the peri-genual ACC, bilateral aIC, bilateral vOFC, ventral striatum, and medial THAL. Right dlPFC activity was associated with a weakened relationship of the aIC with both pain intensity and affect. Thus, dlPFC may exert active control on pain perception by modulating cortico-cortical and cortico-subcortical pathways (Lorenz et al, 2003).
Controllable and Un-controllable Pain. Compared with controllable, un-controllable painful stimulation can lead to increased pain perception and activation in pain-processing brain regions. When pain is controllable, the lateral PFC seems to inhibit pain processing. During controllable pain trials, healthy voluntees significantly down-regulated the temperature to keep their sensation constant. During the un-controllable pain trials, intensity ratings increased despite receiving the identical nociceptive input. This additional sensitization was mirrored in increased fMRI activation of pain-processing regions such as ACC, IC, and THAL. Moreover, the connectivity increased between the aIC and mPFC in the uncontrollable task, and negative connectivity increased between dlPFC and IC in the controllable task. This suggests a pain-facilitating role of the mPFC during un-controllable pain and a pain-inhibiting role of the dlPFC during controllable pain, both exerting their respective effects via the aIC (Bräscher et al. 2016).
Descending Pain Modulation. The dlPFC is functionally linked to the descending pain modulation system and has been implicated in top-down pain inhibition, including placebo analgesia. Functions of the dlPFC may thus be impaired in patients with chronic pain. Post-herpetic neuralgia is one of several syndromes with CNP. In a conditioning phase, healthy controls and patients with post-herpetic neuralgia were exposed to low-frequency (LF) and high- frequency (HF) tones associated with noxious stimuli: weak (WS) and strong (SS) electrical stimulation, respectively. After the conditioning, cerebral hemodynamic activity was recorded from the bilateral dlPFC while the subjects were subjected to the cue tone-noxious electrical stimulation, in which incorrectly cued noxious stimuli were sometimes delivered to induce placebo and nocebo effects. Compared to the healthy controls, hemodynamic responses to the LF tone in the right dlPFC was significantly lower in patients with post-herpetic neuralgia. The same hemodynamic responses in the right dlPFC were correlated with placebo effects. Clinical symptoms of post-herpetic neuralgia were negatively correlated with cerebral hemodynamic responses in the right dlPFC and magnitudes of the placebo effects. This suggests that the right dlPFC, which is closely associated with the descending pain-modulation system, is disturbed in post-herpetic neuralgia (Hibi et al. 2020).
In 16 patients with cLBP and 16 healthy controls, fMRI during mechanical pain stimulation was performed on the lower back followed by anatomical imaging. Voxel-based morphometry and functional connectivity analysis performed from the seeds with altered gray-matter volume. Compared with controls, cLBP patients showed decreases in gray-matter volume at the right dlPFC, middle occipital gyrus, and cerebellum, and increases at the bilateral S1, left fusiform gyrus, and right cerebellum. dlPFC and fusiform volumes showed negative associations with affective comorbidity, whereas motor-cortex volume with impaired daily activity. Connectivity was decreased between the cerebellar and limbic, and increased between the bilateral sensorimotor regions. Hence, cLBP patients showed a decreased volume of cortical centers for descending pain modulation and an increased volume of sensorimotor network, in association with suppressed descending pain modulatory and cerebellum-limbic networks and enhanced sensorimotor network during pain (Li et al. 2018).
In many patients, the intensity of this ongoing pain fluctuates intensely. In individuals with painful TN, fMRI revealed co-variations between moment-to-moment fluctuations in pain intensity and the PAG, RVM, and SpV. The dlPFC-PAG connection anti-correlated with perceived pain intensity over a 12 minute period, revealing cortical systems underlying moment-to-moment changes in perceived pain in PTN, which likely cause dysregulation in the brainstem circuits, and consequently alter the appraisal of pain across time (Meylakh et al. 2024).
Mental Disorders. Most mental disorders involve dysfunction of the dlPFC. For example, schizophrenia, depression, long COVID, and Alzheimer´s disease are all associated with dlPFC dysfunction. The dlPFC is particularly dependent on arousal state and is very vulnerable to stress and inflammation. For example, the layer III dlPFC circuits that generate working memory-related neuronal firing have unusual neurotransmission, depending on NMDA receptor and nicotinic α7 receptor actions that are blocked under inflammatory conditions by kynurenic acid. Stress rapidly weakens layer III connectivity by driving feedforward Ca2+-cAMP (cyclic adenosine monophosphate) opening of K+ channels on spines (Joyce et al. 2025).
- Ventro-Lateral Prefrontal Cortex (vlPFC)
Analgesic effects co-occur with reduced anxiety and increased activity in the vlPFC. In a pilot fMRI study, the neural bases were compared of the analgesic and anxiolytic effect of two types of threat modulation: a `behavioral control´ paradigm, which involved the ability to terminate a noxious stimulus, and a `safety signaling´ paradigm, which involved visual cues signaling the threat that a subsequent noxious stimulus might be of unusually high intensity. Analgesia was paralleled by vlPFC activity during behavioral control. Safety signaling recruited elements of the descending pain control system, including the rACC that showed increased functional connectivity with the PAG and vlPFC. By contrast, anxiety reduction scaled with dlPFC activation during behavioral control but not during safety signaling. This suggests that analgesic and anxiolytic effects are produced in distinguishable neural mechanisms and differ between distinct stress- and pain-modulatory approaches, supporting the notion of multiple pathways subserving top-down modulation of the pain experience (Wiech et al. 2014).
fMRI was used to examine the neural activation associated with individual differences in the impact of perceived controllability on self-reported pain perception. Subjects with greater activation in response to un-controllable rather than controllable (C) pain in the pIC/S2, pre-genual anterior cingulate cortex (pACC) and PAG reported higher levels of pain during the un-controllable versus controllable conditions. Conversely, subjects with greater activation in the vlPFC in anticipation of pain in the un-controllable versus controllable conditions reported less pain in response to un-controllable versus controllable pain. Activation in the vlPFC was significantly correlated with the acceptance and denial subscales of the COPE inventory, supporting the interpretation that this anticipatory activation was associated with an attempt to cope with the emotional impact of un-controllable pain. A regression model containing the vlPFC and pACC predicted 64% of the variance in pain rating difference, with activation in the IC/S2 and PAG predicting almost no additional variance. This suggests that these effects are primarily top-down, driven by processes in the PFC previously associated with cognitive modulation of pain and emotion regulation (Salomons et al. 2007).
- Social Pain
Social pain is an emotional response to inter-personal rejection or criticism. The right vlPFC has been implicated in mitigating social pain. Right vlPFC facilitation induced by TMS significantly reduced self-reported social pain. Functional connectivity analyses demonstrated enhanced interactions between the right vlPFC and the dlPFC. Negative social feedback led to negative social attitudes, whereas rvlPFC activation countered this detrimental effect (Li et al. 2024).
- Ventro-Lateral Orbito-Frontal Cortex (vlOFC)
Normally, the vlOFC has a nociceptive role. Ablation of DA terminals or DA receptor blockade attenuate neuropathic pain (Bak et al. 2021). In awake rats, the vlOFC is possibly involved in the modulation of neuropathic manifestations. Several groups of rats were subjected to mono-neuropathy following the spared nerve injury model, known to produce evident tactile and cold allodynia and heat hyperalgesia. The activity of the vlOFC was selectively blocked by using either chronic or acute injection of lidocaine, electrolytic lesion, or chemical lesion with kainic acid or 6-OHDA. The effects of these manipulations were compared with those following lesion of the somatic sensorimotor cortical areas. Local injection of lidocaine resulted in a reversible depression of all neuropathic manifestations while electrolytic or chemical lesions elicited transient attenuation affecting mainly the heat hyperalgesia and to a lesser extent the cold allodynia. The effects were transient despite the permanence of the lesions while lesion of the somatosensorimotor cortices produced sustained reduction of the neuropathic manifestations. This correlates well with the vlOFC connections with the THAL nucleus subnucleus involved in the procession of thermal nociception (Baliki et al. 2003).
2.15.4. Anterior: Cingulate Cortex (ACC)
The ACC covers the anterior part of the cingulate cortex (CC), which is a long gyrus (gyrus cinguli) on the medial hemispheric side. In primates, the CC has three major regions: an agranular ACC; a dysgranular mid-cingular cortex (MCC); and a granular posterior cingulate cortex (PCC)//retro-splenial cortex (RSC). All regions are related to emotion and motivation, and memory insofar as they are connected to the HIPP (Oane et al. 2023; Vogt 2016, 2019).
Neuropathic pain is often accompanied by negative aspects involving a broad network of brain regions. In particular, the ACC is a part of the limbic system that has highly interconnected structures involved in processing components of pain. The ACC is an important region for acute pain perception as well as the development of neuropathic pain, characterized by LTP induced in pain pathways. The exact mechanism for neuropathic pain in the ACC is unclear (Moon and Park 2022).
ACC pyramidal neurons receive nociceptive inputs from the THAL and the S1. The ACC output projections target the PAG. Functional connectivity analysis showed a significant correlation between ACC and PAG in pain states. Additionally, neurons in the ACC project to the IC or spinal DH (Figure 2), and these projections play a role in the top–down system for rapid modulation of spinal sensory transmission (Bak et al. 2021).
- Anterior Cingulate Cortex (ACC)Functions
The ACC plays an important role in cognitive functions such as error detection, decision-making, affect and emotion, particularly in pain processing (Jaggi and Singh 2011) The ACC and MCC are involved in the processing and modulation of nociception and pain and additionally concerned with its affective and motivational aspects. The ACC is also supposed to sustain other functions such as pain-related attention, arousal, pain modulation, engagement of endogenous pain control system, and the motor withdrawal reflex. The ACC contains a rich density of opioid receptors. Thus, the peri-genual sub-division has been suggested to participate in top-down controls of pain, (including the placebo effects known to be opioid mediated), mainly (but not exclusively) through the connection between the orbito-frontal/sub-genual ACC and the PAG (Peyron et al. 2019). Indeed, the ACC and the PAG form an axis that is primarily involved in spontaneous pain, and the connectivity between these regions is increased in chronic pain patients (Serafini et al. 2020).
- Nociceptive Inputs
The CC receives nociceptive inputs from the STTr mediated via the medial THAL (Kuner and Kuner 2021). Nociceptive inputs to the ACC are mediated via a pathway through the midline and intra-laminar THAL nuclei (including the PVT), which also project to other limbic cortices, robustly to the AMY, and to the PAG. In humans, both pACC and MCC were activated when noxious heat was applied to the back of the hand when controlled for innocuous heating to the same skin, while there were fewer activation sites in pACC. Imaging studies also showed coding for the intensity of noxious stimulation in pACC and MCC. Nociceptive visceral responses showed a preference for pACC and to a lesser extent anterior MCC (aMCC). In rabbits, neurons in ACC did not signal the location of the noxious stimulus on the body surface because stimulation anywhere on their body can evoke a discharge. These neurons respond mainly to noxious stimuli including pressure and temperatures over 46°C. By and large, these responses reflect those of their input THAL neurons that have large and bilateral receptive fields (Vogt 2016, 2019).
- Anterior Cingulate Cortex (ACC)Connections
In chronic pain, the ACC modulates the different pain aspects by increased connectivity to the striatum, medio-dorsal nucleus of the THAL (MD), and HYP. The ACC also interacts with pain neuro-circuitry in the PAG, which accounts for the activation of the ACC and PAG in the presence of noxious stimuli. The activation of ACC-PFC-PAG networks and increased activity in the ACC is associated with negative emotions (Yang and Chang 2019). Glutamate signaling through α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) in the ACC and AMY has been suggested to increase synaptic plasticity and confer hyperalgesia. Thus, in chronic pain, AMPAR signaling plays both pro-nociceptive and anti-nociceptive roles, depending on the target CNS regions (Doan et al. 2015). A number of neuromodulators control the activity of the ACC, ncluding the monoamines DA, NA, 5-HT, and others, whose actions are altered by chronic pain in ACC circuits to promote pathological hyperexcitability (Lançon and Séguéla 2023).
- Anterior Cingulate Cortex (ACC) during Neuropathic Pain
ACC lesions in rats who underwent nerve ligation did not change mechanical hypersensitivity but reduced escape/avoidance behavior, which reflects damage to the affective component of pain. Neuropathic pain is accompanied by several changes. Morphological changes in spine morphology, which are also reflected in the increased miniature excitatory postsynaptic current (mEPSC) amplitudes of ACC pyramidal neurons. PKMζ, a key molecule for maintaining LTP, plays a role in neuropathic pain-associated neural plasticity in the ACC. In mice subjected to CCI, multiple whole-cell recordings in ACC layer 5 neurons showed a loss of functional connections between excitatory and inhibitory neurons as well as a decrease in mEPSC and miniature inhibitory postsynaptic current (mIPSC) frequencies. CCI potentiates the intrinsic excitability of pyramidal neurons, but not that of inhibitory neurons. In mouse ACC layer 5 pyramidal neurons, Ca2+ activity was enhanced in the absence or presence of pain stimuli in SNI-induced neuropathic pain. In mouse brain slices, spontaneous EPSC frequency was increased and intrinsic excitability of ACC layer 2/3 pyramidal neurons was elevated after SNI (Bak et al. 2021).
In neuropathic pain models, ACC lesions alleviate the affective aspects of pain, reduce escape and avoidance behaviors, and decrease the floating time in the forced-swim test (FST). Projections from the MD to layer L5 pyramidal neurons elicit both excitation and inhibition, the ratio of which would favor inhibition in a sub-population of pyramidal neurons that project to sub-cortical areas. The inhibition of the neurons projecting to the contralateral side to a nerve injury promoted analgesia without influencing aversive behavior. Inhibiting the excitatory ACC neuronal sub-population resulted in aversive behavior during neuropathic pain. Direct excitatory projections to neurons in laminae I-III of the spinal DH were activated in models of neuropathic and cancer pain. Electrical or chemical activation of the ACC facilitates the tail-flick reflex, supporting the existence of descending facilitation (Kummer et al. 2020).
The ACC may also facilitate spinal sensory transmission, which is believed to contribute to chronic pain. Little is known about whether there might be direct cortico-spinal modulation circumventing the RVM. Stimulation in the ACC potentiated spinal excitatory synaptic transmission, and this modulation was independent of the RVM. Peripheral nerve injury enhanced the spinal synaptic transmission and occluded the ACC-spinal cord facilitation. Inhibition of ACC reduced the enhanced spinal synaptic transmission caused by nerve injury. By the use of optogenetics, selective activation of ACC-spinal cord projecting neurons caused behavioral pain sensitization, while inhibiting the projection induced analgesic effects. This may indicate that ACC stimulation facilitates spinal sensory excitatory transmission in an RVM-independent manner, and that such top-down facilitation may contribute to the process of CNP (Chen et al. 2018).
- Anterior Cingulate Cortex (ACC) Hyperexcitability
Neuronal hyperexcitability in the ACC is considered as one of the most important pathological changes responsible for the chronification of neuropathic pain. Electrophysiological studies have indicated that increased excitability of ACC neurons mainly results from the increased excitatory afferent activity altered by long-term peripheral sensitization in the nociceptive system. Following CCI, the firing activity of ACC layer V neurons, measured by whole-cell current–clamp recordings in brain slices, has provided evidence that mGluR1 is up-regulated and activated. CCI nerve injury also strengthened the intrinsic excitability of pyramidal neurons in ACC. Inflammatory pain enhanced presynaptic glutamate release in ACC neurons, mediated by an increase in neuronal cAMP (Boadas-Vaello et al. 2017). Nerve injury-induced pain hypersensitivity involves aberrant excitability in spinal DH neurons as a consequence of dysfunction of inhibitory INTs and of hyper-activity of glial cells, especially microglia. The pathologically altered somatosensory signals in the DH are then transmitted to various brain regions, including the ACC. The descending projection pathways from the ACC directly and indirectly to the DH (the top-down cortico-spinal network) regulate nociceptive sensory transmission in the DH (Tsuda et al. 2017).
- Synaptic Transmission
Synaptic transmission in ACC is also modified after neuropathy. In the rodent ACC, different forms of LTP contribute to chronic pain, in particular affective aspects and anxiety (Bliss et al. 2016; Zhuo 2024). Nerve injury produces pre- and postsynaptic long-term plasticity, which contributes to negative emotions and anxiety associated with chronic pain conditions. The descending projection pathways from the ACC directly and indirectly to the DH regulate nociceptive sensory transmission in the DH (Tsuda et al. 2017). For example, in rodents, the common peroneal ligation model of neuropathic pain increased presynaptic transmitter release and postsynaptic responses. Loss of inhibitory synapses on excitatory pyramidal neurons led to a local disinhibition of the cortex network and may explain the increased activity of ACC in patients with nerve injury (Yalcin et al. 2014).
- Anterior Cingulate Cortex (ACC) and Dorsal Horn (DH)
The ACC has direct descending connections to the DH. Nerve injury-induced pain hypersensitivity involves aberrant excitability in spinal DH neurons as a consequence of dysfunction of inhibitory INTs and of hyper-activity of glial cells, especially microglia, the immune cells of the CNS. The pathologically altered somatosensory signals in the DH are then transmitted to the brain regions, including the ACC (Tsuda et al. 2017).
fMRI in humans showed an increased ACC activity during high-intensity nociceptive inputs in chronic pain, Activation of ACC neurons has been associated with pain-like aversive responses (fear memory). However, in an L5 spinal-nerve ligation model, electrical ACC stimulation attenuated the aversive quality of noxious cutaneous hindpaw stimuli without an anti-allodynic effect, suggesting that ACC activity differentially affected the affective and sensory pain components. However, in a rat neuropathic pain model, electrical ACC stimulation attenuated mechanical allodynia (Jaggi and Singh 2011).
- Stress-induced Pain Facilitation
Chronic stress exacerbates neuropathic pain. The ACC and its descending projections to the DH may be involved in mediating stress-induced pain facilitation. In male Sprague-Dawley rats, a chronic unpredictable mild stress (CUMS) model combined with a CCI model was used to assess behavioral changes, neuronal activity, and molecular alterations. CUMS significantly exacerbated mechanical allodynia in CCI rats, correlating with increased c-fos expression in both the ACC and spinal cord, indicative of heightened neuronal activity. Chemogenetic inhibition of ACC-DH projection neurons alleviated mechanical allodynia without affecting depressive-like behaviors, suggesting a specific role for this pathway in pain modulation. Furthermore, BDNF signaling in the ACC-DH pathway is crucial for the facilitation of neuropathic pain under chronic stress conditions. BDNF levels were elevated in the ACC of CUMS+CCI rats, and knockdown of BDNF in ACC-DH projecting neurons attenuated stress-induced pain sensitivity (Yang et al. 2025).
2.15.5. Insular Cortex (IC)
- Structure and Connections
In humans, the IC (or island of Reil) is a region of the cerebral cortex located in the center of the cerebral hemisphere, within the lateral fissure (Sylvian fissure) and covered by parts of the frontal, temporal and parietal cortices (Nagai et al. 2007). (In rodents, the arrangement is somewhat different; Lu et al. 2016). The IC is parcellated into various areas, which, in different species from rodents to primates, show roughly graded differences in macro- and micro-anatomy, cyto-architectonics, connectivity and function,
The IC is an extended cortical area where multi-modal inputs from different brain areas converge. In primates, it is located within a fold of the lateral sulcus (Figure 4), while in rodents, it lies at the lateral surface of the brain. It can be divided into a posterior (pIC) and an anterior part (aIC), with cytoarchitecturally diverse areas with differential connectivity to other brain areas.
The IC is thought to be a hub for the integration of multi-modal information. Thus, several functions and processes have been ascribed to the IC. These functions encompass information of the internal physiological and homeostatic condition of the body, so that the IC serves as the primary interoceptive cortex. The IC may also form the neurobiological substrate for the recognition and awareness of one´s `self´. The strong interconnection between the IC and the limbic system indicates an essential role of the IC in emotion (Labrakais 2023). The IC maintains reciprocal connections with the PFC, ACC, AMY and somatosensory cortex (Jaggi and Singh 2011).
- Inputs
The pIC receives nociceptive inputs from the STTr mediated via the posterior THAL (Kuner and Kuner 2021). In the macaque, the posterior dorsal fundus of the IC responds to innocuous and noxious thermal stimuli as well as to noxious mechanic pinch stimuli, with a somatotopic representation of the foot, hand, and face from posterior to anterior (Evrard 2019). The pIC participates in the somatosensory features of pain, while the aIC is more involved in mediating its affective aspects (Lu et al. 2016). The pIC receives inputs from THAL nuclei, some of which are indirect, nociceptive and thermo-ceptive sensory inputs from spinal cord lamina I neurons, and processed sensory information from the S1 and S2, which fits well with the role of the pIC in sensory-discriminative pain coding. The aIC receives THAL inputs mainly from the medial THAL nuclei. Integration of these with sensory inputs from ventro-posterior THAL, S1 and S2 inputs, together with inputs from the PFC and AMY, suggests a main role of the aIC in the affective and cognitive functions of pain. Both the pIC and aIC receive inputs from the AMY, mainly from the BLA but not from the CeA. Other limbic regions that send afferents to the pIC are the ACC and MCC. Many of the inputs to the pIC and aIC are reciprocal connections with the target brain areas (Labrakakis 2023).
- Outputs
IC outputs project to the THAL, S1 and S2, and the AMY. The pIC outputs to the AMY are stronger than those of the aIC. The aIC sends a major output to the BG (caudate/putame) and NAc, indicating a role in the motivational response to pain by engaging the reward/aversion system. In addition, these outputs, in coordination with outputs to the PBN and feedback loops from the motor cortex, might be involved in nocifensive behaviors (Labrakakis 2023). The aIC is connected to the vlPFC and OFC, while the pIC strongly connects to the S1 and S2. These areas show abnormal bilateral recruitment in response to innocuous stimuli during allodynia and neuropathic pain, possibly as a result of re-organization of thalamo-cortical inputs (Ong et al. 2019).
- Insula´s Role in Pain
The IC has an important role in pain processing. Human imaging and electrophysiological studies showed insula (IC) activation after a noxious stimulus. It is the brain area activated most frequently in fMRI pain studies, being one of the brain regions that activates with the shortest latencies to nociceptive stimulation. IC activation correlated with the intensity of the noxious stimulus, indicating involvement in intensity coding. Its activation also correlated with the perceived magnitude of pain. Stimulation and imaging studies showed that the pIC is involved mainly in coding pain intensity. While individuals with lesions in the pIC showed higher pain-intensity ratings, individuals with lesions in the aIC showed its involvement mostly correlated with the emotional processing of pain, cognitive evaluation of pain, and empathy for pain (Labrakakis 2023). In patients with nerve injury, PET revealed IC activation in brush-evoked allodynia. Patients with IC lesions showed increased heat-pain thresholds. The rostral agranular IC plays an important part in the modulation of inflammatory and neuropathic pain (Jaggi and Singh 2011). During allodynia and neuropathic pain, IC areas showed abnormal bilateral recruitment in response to innocuous stimuli, possibly as a result of re-organization of thalamo-cortical inputs, from lateral-posterior to anterior-medial THAL nuclei (Ong et al. 2019).
IC lesions alter pain perception. Higher pain ratings occurred in lesioned individuals, as well as a lack of motor withdrawal to painful stimuli, absent emotional response, and indifference to pain, termed pain asymbolia. In strokes involving the pIC, dissociated sensory loss with neuropathic pain occurred. Clinical studies also emphasized the IC role in chronic pain, showing a relationship between alterations in IC activity, structure, and pain chronification. In cLBP patients, changes of the IC connectivity with the somatosensory cortex and the mPFC occurred. Increased IC excitability as a cause of pain hypersensitivity syndromes is also supported by the finding that FM patients have higher glutamate levels in the pIC (Labrakakis 2023).
- Structural Changes
Chronic pain can lead to anatomical and functional alterations in the IC, which are correlated with cognitive and affective disorders. Structural and connectional properties of the IC may change differentially during chronic pain. For example, gray-matter volume in patients with TNP was reduced in the aIC, but increased in the pIC. In this particular case, altered gray matter was nearly reversed to normal after effective therapy for TNP. Just as distinct neuro-degenerative diseases involve the atrophy and degradation of their corresponding resting-state networks, the specific condition of chronic pain results in unique patterns of network re-organization (Lu et al. 2016).
- Activity Changes
In patients with neuropathic pain, brain imaging showed that brush-evoked allodynia mainly activated the aIC (Lu et al. 2016). In humans and animals, increased IC activity is associated with increased pain perception and pain hypersensitivity in pathological conditions. It has been suggested that the increased IC activity contributes to pain hypersensitivity through activation of pro-nociceptive descending pathways, which would place the IC in a central position on the descending pain modulatory network. Placebo analgesia reduced IC activity during a noxious stimulus (Labrakakis 2023). In naive and neuropathic-pain male mice subjected to spared nerve injury, pIC neurons were easily activated by mechanical, thermal, aversive, stressful and appetitive stimulation (Chen et al. 2024a).
- Insular Cortex (IC) Stimulation
In neuropathic patients, IC TMS was analgesic. Similar results have recently been described in rodent models. In a mouse model of neuropathic pain, repeated transcranial current stimulation of the pIC diminished mechanical pain hypersensitivity for two–three weeks. In the rat, repeated electrical stimulation of the pIC reduced neuropathic hypersensitivity. The seemingly contradictory anti-nociceptive effects of repeated IC stimulation to the increased pain sensation with increased IC activation could be explained by the mixed effects of deep-brain stimulation. Thus, it could involve sub-areas of the IC or cellular sub-populations (e.g.,, GABAergic neurons); incoming fibers, including neuromodulatory 5-HT, DA, and acetylcholine (ACh) fibers; or descending outputs (Labrakakis 2023).
- Chronic Constriction Injury (CCI) of Infra-orbital Nerve
Rats showed a strong bilateral trigeminal mechanical allodynia in the IC after CCI of the IoN (IoN-CCI). Light, moving stroking of the infra-orbital skin resulted in strong, bilateral up-regulation of extracellular-signal regulated kinase phosphorylation (pERK-1/2) in the IC of IoN-CCI animals but not sham rats, in whom the concentrations levels were similar to those of un-stimulated IoN-CCI rats. pERK-1/2 was located in neuronal cells only. Stimulus-evoked pERK-1/2 immuno-positive cell bodies displayed a rostro-caudal gradient and layer-selective distribution, being predominant in the rostral IC and in layers II-III of the dysgranular and to a lesser extent, of the agranular IC In layers II-III of the rostral dysgranular IC cortex, intense pERK also extended into distal dendrites, up to layer I. Hence, trigeminal nerve injury induced a significant alteration in the IC processing of tactile stimuli and ERK phosphorylation contributed to the mechanisms underlying abnormal pain perception under this condition (Alvarez et al. 2009).
- Insular Cortex (IC)Connections to Thalamus (THAL) and Amygdala (AMY)
It has been hypothesized that the pIC and its projections may contribute to the pathophysiology of comorbid chronic pain and depression. In naive mice, optogenetic activation of pIC neurons induced hyperalgesia and CPA, whereas inhibition of these neurons led to analgesia, CPP, and anti-depressant effect in both naive and SNI mice. The monosynaptic glutamatergic projections from the pIC to the BLA and the ventro-medial nucleus (VM) of the THAL mimicked pIC neurons in pain modulation in naive mice. In SNI mice, both projections were enhanced accompanied by hyper-activity of pIC, BLA, and VM neurons and inhibition of these projections led to analgesia, CPP, and anti-depressant-like effect. This suggests that potentiation of the pIC→BLA and pIC→VM projections may be important pathophysiological bases for hyperalgesia and depression-like behavior in neuropathic pain (Chen et al. 2024a).
- Desending Pain Control
Stimulation of the pIC evokes spinal DH potentials via several paths. One descending pathway passes through the S1. This pathway is thought to support the maintenance of allodynia during neuropathic pain since pIC lesions alleviate long-term allodynia in rats. An additional descending pathway connects the pIC to the spinal cord via the NRM and exerts its pro-algesic action by 5-HT modulation of the spinal circuitry. Indeed, the aIC sends outputs to the NRM, and inhibition of the aIC by increasing its local GABA concentration increases nociceptive heat thresholds in a spinal adrenergic-dependent manner. Since the NRM also connects to the LC, the aIC modulates nociception via a NRM–LC–spinal cord descending pathway. In neuropathic rats, the functional connectivity between the aIC and the LC was enhanced. At the aIC level, this pathway is regulated by opioidergic, DA, and OXT systems. There is also a direct connection of the IC to the SpV that facilitates nociceptive and WDR neurons and has pro-nociceptive actions. Moreover, there are direct pIC outputs to the PAG. The aIC could also indirectly influence descending pathways by outputs to the frontal cortex, which, through top-down descending pathways from the mPFC exerts analgesic control (Labrakakis 2023).
2.15.6. Hippocampus (HIPP)
- Structure
The HIPP is a structure with the shape of a seahorse located in the medial temporal lobe of the brain. The HIPP formation includes the dentate gyrus (DG), C1-C4 sectors, pro-subiculum, subiculum), and pre-subiculum. It has been proposed that the HIPP is composed of an anterior, middle and posterior division (Vogt 2019). The HIPP connects extensively with other brain regions. Its major input and output structure is the entorhinal cortex (EC), which receives additonal inputs from the peri-rhinal and para-HIPP cortices from cortical and sensory sources (Garcia and Buffalo 2020; Lavenex and Amaral 2000; Witter et al. 2017).
- Pain Processing
The HIPP takes part in both the processing and modulation of nociceptive signals. The HIPP is activated during pain processing and involved in the modification of nociceptive stimuli (Vasic and Schmidt 2017). By regulating memory processes, the HIPP may play an active role in modulating the perception of pain itself (Kuner and Kuner 2021). Nociceptive signals reach the HIPP indirectly via the STTr and PBN. Septo-HIPP neurons receive direct input from the spinal cord and respond to intense thermal stimuli. Neurons in the CA1 region and the DG react ro painful stimuli. Injection of lidocaine directly into the DG produces analgesia. A HIPP lesion can alter the perception of noxious stimuli and partially alleviate pain (Fasick et al. 2015).
- Changes in Hippocampus (HIPP)Volume and Connectivity
Functional and structural changes in the HIPP and in HIPP connectivity to other limbic or cortical structures could contribute to learning and memory deficits, as well as to aversive cognitive and affective states associated with chronic pain. In both patients and rodent models of chronic pain, HIPP volume was reduced (Kuner and Kuner 2021). Reduced HIPP volumes occurred in patients suffering from CBP and CRPS, while this effect was absent in knee osteoarthritis patients, possibly due to the type of pain. Patients with CBP had smaller HIPP and stronger phasic pain responses in the bilateral anterior para-HIPP gyrus. In these patients, the level of basal cortisol and the clinical pain intensity were associated with increased pain-related responses in the anterior HIPP formation (Vasic and Schmidt 2017). In rats, SNI causing CNP affected fronto-HIPP functional connectivity, impairing spatial memory performance (Boadas-Vaello et al. 2017). In the freely moving rat with SNI, pain reduced the information flow in the mPFC to dorsal CA1 circuit (Thompson and Neugebauer 2019).
- Changes in Firing Patterns
In rat neuropathic pain models, chronic multi-channel electrophysiological recordings of HIPP place cells became instable. This was complemented by reduced performance in spatial memory tasks and impairment of connectivity and coherence of activity between the HIPP and the PFC (Cardoso-Cruz et al. 2011; Kuner and Kuner 2021).
- Inflammation
Neuropathic pain models caused a robust expression of IL-1β in the HIPP contralateral to the lesion site that correlated with neuropathic pain behavior. The formalin model of inflammatory pain decreased the number of c-fos-positive cells in whole CA1, CA3 and DG. In inflammatory models of pain, the expression of both the NK-1 receptor and BDNF genes was down-regulated in the HIPP (Boadas-Vaello et al. 2017).
- Neurogenesis
HIPP neurogenesis contributes to learning and memory, but may also trigger the development of chronic pain. Functional and structural changes in the HIPP, such as decreased HIPP neurogenesis, are closely associated with memory deficits and aversive affective states in patients with chronic pain (McEwen et al. 2016). Adult neurogenesis was reduced in animals with neuropathic pain (Kuner and Kuner 2021). Chronic pain interfered with HIPP mossy fiber to CA3 synaptic plasticity and DG neurogenesis. In neuropathic animals, reduction in HIPP neurogenesis was exacerbated by chronic stress (Fasick et al. 2015; Mutso et al. 2012; Romero-Grimaldi et al. 2015; Vasic and Schmidt 2017).
- Plasticity
Rats with SNI-induced neuropathic pain had short-term memory deficits, which corresponded to increased IL-1β in the sciatic nerve, serum, PFC, NAc, AMY, and HIPP. HIPP plasticity could contribute to both memory and averse affective deficits in the chronic pain states. In a neuropathic pain mouse model (SNI), short-term plasticity and LTP at the CA3-CA1 synapses were impaired, the density of presynaptic boutons in CA1 synapses was reduced, and TNF-α levels were increased in CSF, plasma, and the HIPP. Intra-HIPP injection of TNF-α in naïve rats mimicked the behavioral effects associated with SNI, whereas deletion of the TNF receptor 1 in SNI rats prevented their development, suggesting a role for neuro-inflammation in these effects (Thompson and Neugebauer 2019).
2.15.7. Bed Nucleus of the Stria Terminalis (BNST)
Chronic pain is often associated with mental dysfunctions such as anxiety and depression. In a mouse model of neuropathic pain, neuronal plasticity in the BNST occurred. Electrophysiology demonstrated that chronic pain increased inhibitory inputs to lHYP-projecting BNST neurons. Chemogenetic manipulation also revealed that sustained suppression of lHYP-projecting BNST neurons played a crucial role in chronic pain-induced anxiety. Molecular genetic approaches demonstrated that chronic pain elevated the excitability of a specific sub-population of BNST neurons, which express cocaine- and amphetamine-regulated transcript (CART). The elevated excitability of CART-positive neurons caused the increased inhibitory inputs to lHYP-projecting BNST neurons, thereby inducing anxiety-like behavior (Yamauchi et al. 2022).
3. Changes in the Motor Changes in the Motor and Associated Systems
Pain may affect the motor and associated systems like the cardio-vascular and respiratory systems.
In healthy humans, the effects of experimental (sub)cutaneous pain, joint pain, muscle pain and tendon pain on the motor system affected many components of motor processing at various levels of the nervous system, the effects being largely irrespective of its source. Pain was associated with inhibition of muscle activity in the (painful) agonist and its non-painful synergists and antagonists, especially at higher intensities of muscle contraction. Despite the influence of pain on muscle activation, only subtle alterations occurred in movement kinetics and kinematics. The performance of various motor tasks mostly remained unimpaired, presumably as a result of a re-distribution of muscle activity, both within the (painful) agonist and among muscles involved in the task. Cutaneous pain caused amplification of the nociceptive withdrawal reaction, whereas other spinal reflexes were hardly modulated. At higher levels of motor control, pain was associated with decreased cortico-spinal excitability. Hence, short-lasting experimentally induced limb pain may induce immediate changes at all levels of motor control, irrespective of the source of pain. These changes support protective and compensatory motor behavior (Bank et al. 2013).
The Hoffmann reflex (H-reflex) was largely not influenced by the presence of either clinical or experimental pain. While inhibitory effects on cortico-spinal excitability and motor-unit (MU) behavior were evident under experimental pain conditions, more variable responses were observed for patients with painful musculo-skeletal disorders. Experimental pain consistently reduced MU discharge rate, which was not consistent with data obtained from patients. In tonic pain, induced via experimental pain models, inhibitory effects on motoneuron (MN) behavior were evident. However, in chronic-pain patients, more varied responses were evident likely reflecting individual adaptations to chronic symptoms (Sanderson et al. 2021).
3.1. Changes in Proprioceptive Functions
“You can only control what you sense” (McCloskey and Prochazka 1994).
Motor control requires sensory signals for guidance and corrections. One important type of sensory signals derives from so-called `proprioceptors´. These include muscle spindles and GTOs (and other receptors) in skeletal muscles, joint receptors and some cutaneous mechano-receptors. Very important are the former two. Muscle spindle signals are essential for motor purposes (Windhorst 2007; 2021) and kinesthesia (Proske and Gandevia 2018). If, in chronic pain conditions, muscle spindle operation is disturbed, so should be motor control and kinesthesia. This should also apply to GTOs, whose actions at spinal level have been investigated intensely (Jankowska 1992; Windhorst 2021). Under chronic pain conditions, however, disruptions of the functions of these two classes could occur at the peripheral level, by the changes in ion channels (below), and centrally. Proprioceptive deficits occur in chronic pain syndromes, such as cLBP and FM (Lee and Chen 2023).
At the spinal level, group III (Aδ) and IV (C) afferents exert effects on MNs and INTs (Figure 2), thus evoking and modulating complex reflex effects (Windhorst 2021; Windhorst and Dibaj 2025a). In cats and rats, group III/IV muscle afferents, activated chemically or metabolically or by muscle fatigue, have polysynaptic effects on α-MNs, γ-MNs and other spinal neurons (reviewed in Windhorst 2021). But these effects may differ between human and animal preparations. While in the cat, data have shown that muscle pain caused marked changes in the firing of muscle spindles, attributed to a nociceptor-driven fusimotor reflexes, in humans, experimental muscle pain elicited by intra-muscular injection of hypertonic saline failed to increase the firing of muscle-spindle afferents during weak voluntary contractions, when fusimotor drive sufficient to increase their firing was present (Birznieks et al. 2008; Smith et al. 2019).
During mastication, jaw muscle spindles play an important role in monitoring and regulating the chewing cycle and the bite forces generated during mastication. Both acute and chronic orofacial pain disorders are associated with changes in proprioceptive feedback and motor function. It has been proposed that altered motor function and proprioceptive input result from group III muscle afferent modulation by the fusimotor system, which alters spindle afferent sensitivity in limb muscles. The response to nociceptive stimuli may enhance or reduce the response of spindle afferents to proprioceptive stimuli. Nociceptive stimulation of the masseter muscle primarily influences the amplitude sensitivity of spindle afferents with relatively little effect on the dynamic sensitivity. Reversible inactivation of the caudal trigeminal nuclei attenuates the nociceptive modulation of spindle afferents. Functionally identified γ-MNs in the trigeminal motor nucleus are modulated by intra-muscular injection with algesic substances. This suggests that pain-induced modulation of spindle afferent responses are mediated by small-diameter muscle afferents and that this modulation depends, in part, on the relay of muscle nociceptive information from trigeminal sub-nucleus caudalis onto trigeminal γ-MNs (Capra et al. 2007). During chronic pain, these influences might change, and if fusimotor function is disturbed, so would be spindle function. Beside the fusimotor system, sympathetic effects and ion channels influence muscle spindle discharge.
3.1.1. Changes in Muscle Spindle Afferents
- Sympathetic Innervation of Muscle Spindles
The sympathetic innervation of muscle spindles has been controversial. Anatomical evidence (Bombardi et al. 2006; Radovanovic et al. 2015) and more indirect physiological evidence for it were presented (Hellström et al. 2005; Kamibayashi et al. 2009; Roatta et al. 2002, 2005). This was interpreted as a role for the activated SNS in chronic pain in modulating muscle spindle discharges so as to change motor control, maintain pain, and stress. This concept was cast in doubt by the lack of enhanced sympathetic drive on muscle spindle resting discharge in humans (Macefield et al. 2003).
- Ion Channels
Another way by which chronic pain might change muscle spindle properties would be at the molecular level of primary sensory afferents. Ion-channels in neuron membranes can change widely in inflammatory pain or in various etiologies of neuropathic pain, and are influenced by several other factors including genetic mutations. However, there are some special channels involved in sensory receptor functions. The exact molecular mechanism by which muscle-spindle afferents transduce internal muscle movement into APs is incompletely understood. Recent evidence suggests the participation of several ion channels. Piezo2 is an essential mechanically sensitive ion channel in muscle-spindle afferents and vesicle-released glutamate contributes to maintaining afferent excitability during the static phase of stretch. Other mechanically gated ion channels, voltage-gated Na+ channels, other ion channels, regulatory proteins, and interactions with the intrafusal fibers are also important for muscle-spindle afferent mechano-sensation (Wilkinson 2022).
Mechano-sensitive Piezo Channels are ion channels activated by mechanical stimuli and play a crucial role in mechano-transduction processes and mechanical hypersensitivity. When these channels are subjected to mechanical loading, membrane currents rise instantaneously, depolarizing and activating voltage-gated Ca2+ channels. This leads to an increase in intracellular Ca2+, which contributes to heightened sensitivity to mechanical stimuli. While Piezo1 is predominantly found in non-neuronal cells, Piezo2 is mainly located in DRG neurons. It is critical for sensory functions, such as proprioceptive feedback, gentle touch perception, and tactile allodynia. Piezo channels are important for the occurrence and development of chronic pain, including inflammatory pain, neuropathic pain, visceral pain, musculo-skeletal pain, headache or orofacial pain (Wan et al. 2024).
Piezo2 has been claimed to be the main mechano-transducer in mouse proprioceptors, and its deletion in knock-out mice causes impairment of stretch-activated discharge in muscle afferents and of coordination in all limbs. Piezo2 is expressed in sensory endings of muscle spindles and GTOs (Anderson et al. 2017; Woo et al. 2015).
It may be presumed that, during chronic pain, Piezo2 up-regulation may also alter muscle spindle and GTO actions and thereby motor control and kinesthesia.
Acid-sensing Ion Channels (ASICs) in Muscle-Spindle Sensory Endings. Surprisingly, muscle-spindle afferents express ASIC1b, ASIC2, ASIC2b, and ASIC3. The generation of Asic3-knockout/eGFPf-knockin mice was used to characterize heterogeneous expression of ASIC3 in the DRG. ASIC3 is expressed in PV+ proprioceptor axons innervating muscle spindles. Targeted knockout of Asic3 disrupted spindle afferent sensitivity to dynamic stimuli and impaired mechano-transduction in PV+ DRG neurons. In behavioral tasks, global knockout (Asic3(-/-)) and PV-Cre::Asic3(f/f) mice produced similar deficits in grid and balance beam walking tasks. Hence, at least in mice, ASIC3 is a molecular determinant contributing to dynamic mechano-sensitivity in proprioceptors (Lin et al. 2016). Why usually non-nociceptive proprioceptive afferents express the acid-sensing property and whether muscle afferents are involved in pain hypersensitivity of deep tissues is unclear. A potential role has been hypothesized for proprioceptive afferents in producing `non-nociceptive pain´ associated with peripheral and central neuropathy, FM, trauma-induced pain, idiopathic low back pain (LBP) and CRPS. ASIC3 is a dual-function protein for H+-sensing and mechano-sensing in proprioceptors that can be activated by eccentric muscle contraction or lactic acidosis. In chronic musculo-skeletal pain, proprioceptors may play a role in non-nociceptive unpleasantness, associated with their acid-sensing properties (Lee and Chen 2023; Lee et al. 2025).
Masticatory muscle afferent fibers express ASICs. Experiments showed that both ASIC1 and ASIC3 were expressed by predominantly larger masticatory muscle ganglion neurons, but the frequency of ASIC3 expression was significantly greater than ASIC1. No sex-related differences in expression were identified. Injection of pH 5.8, but not pH 6.8, phosphate-buffered saline evoked afferent discharges that were significantly greater than those evoked by pH 7.4 buffer (control). Since ASIC3 channels were not activated until the pH is around 6, this indicates that activation of both channels contributes to excitation of masticatory muscle afferent fibers. Moreover, many masticatory muscle afferent fibers, which respond to low pH, were LTMRs (Gazerani and Cairns 2018).
Inflammation of a rodent paw increased ASIC currents and the expression of ASIC1, 2 and 3 mRNA in DRG neurons. Similarly, inflaming a mouse knee with carrageenan increased ASIC3 protein expression in nociceptive DRG neurons innervating the joint, and at the peripheral terminals of the neurons. In a rat model of spinal disk herniation, compression of the lumbar nerve root increased ASIC3 protein expression in DRG neurons (Sluka et al. 2009).
Other ASIC Effects on Muscle-spindle Afferents. In several models of neuropathic pain, the phenotype of large-diameter afferent neurons changes. In rats, acidic saline (AS, pH 4.0) injections into the masseter muscle were used to induce persistent myalgia. Nocifensive responses of the experimental rats to applications of von Frey filaments to the masseters were above control levels 1-38 days post-injection. This effect was bilateral. Expression of c-fos in the trigeminal mesencephalic nucleus (NVmes), which contains the somata of masseter muscle spindle afferents, was above baseline levels one and four days after acidic-saline injections. The resting membrane potentials of neurons exposed to acidic-saline injections were hyperpolarized when compared to their control counterparts, as were their thresholds for firing, high-frequency membrane-potential oscillations, bursting, inward and outward rectification, and ectopic firing. These changes appeared within the same time period as the observed nocifensive behavior. Annulo-spiral endings of masseter muscle-spindle afferents expressed the vesicular glutamate transporter 1 (VGLUT1), indicating that they can release glutamate. Many spindle capsules also contained fine fibers that were labelled by markers associated with nociceptors (SP, CGRP, P2X3 receptors and TRPV1 receptors) and that expressed the mGluR), mGluR5. Hence, low pH leads to changes in several electrical properties of muscle-spindle afferents, including initiation of ectopic APs which could propagate centrally but could also invade the peripheral endings causing glutamate release and activation of nearby nociceptors within the spindle capsule. This peripheral drive could contribute both to the transition to, and maintenance of, persistent muscle pain as seen in some functional pain syndromes (Lund et al. 2010). Strangely, equivalent data as for muscle spindle afferents do not appear to exist for GTOs afferents.
Ectopic Firing. In conditions leading to neuropathic pain, large-diameter primary sensory afferents, including muscle-spindle afferents, become hyperexcitable and develop spontaneous ectopic firing. Ectopic APs can travel centrally, but also antidromically to the peripheral terminals of muscle-spindle afferents where they could cause neurotransmitter release and activation of adjacent fiber terminals. In a model of chronic orofacial myalgia, muscle-spindle afferents of jaw-closing muscles became hyperexcitable. In these afferents, discharges emerged from fast membrane-potential oscillations that were supported by a persistent Na+ current (INaP) mediated by Na+ channels containing the α-subunit Nav1.6. The current flowing through Nav1.6 channels increased when the extracellular Ca2+ concentration decreased, andt his may happen when INaP -driven firing is increased by an astrocytic protein. Indeed, astrocytes, which are activated in pain conditions, regulate extracellular Ca2+ and might contribute to the generation of ectopic firing in muscle spindle afferents. Hypothetically, ectopic firing in muscle spindle afferents might cause pain by cross-talk between peripheral proprioceptive and nociceptive pathways (Sas et al. 2024). Tachykinin 1 (Tac1), encoding neurokinin A and SP, are expressed in group II muscle spindle afferents, which may hint at a direct role for muscle afferents in inflammatory muscle pain (Oliver et al. 2021).
3.1.2. Changes in Golgi Tendon Organ (GTO) Afferents
In comparison to muscle spindles, little is known about changes in GTO function associated with pain. It appears as if GTOs had been forgotten by pain researchers.
- Ion Channels
In mice, the mechanically activated non-selective cation channel Piezo2 was expressed in sensory endings of proprioceptors innervating muscle spindles and GTOs. Two independent mouse lines that lack Piezo2 in proprioceptive neurons showed severely un-coordinated body movements and abnormal limb positions. The mechano-sensitivity of parvalbumin (PV)-expressing neurons that predominantly mark proprioceptors was dependent on Piezo2 expression in vitro, and the stretch-induced firing of proprioceptors in muscle-nerve recordings was markedly reduced in Piezo2-deficient mice. This would indicate that Piezo2 is the major mechano-transducer of mammalian proprioceptors (Woo et al. 2015).
3.1.3. Intermezzo
Even under healthy conditions, the central actions of muscle spindle and GTO afferents are anything but simple (Jankowska 1992; Windhorst 2021). Under chronic pain conditions, this situation is aggravated. As noted above, inflammation and nerve injury increase the expression of Piezo2 and ASIC (Sluka et al. 2009). If this also occurs at `nociceptive´ muscle spindle endings, as appears probable, the operation of the latter would change in chronic pain, which in turn would impact motor control and kinesthesia. Moreover, functional interactions between nociceptive and proprioceptive afferent inputs occur in the spinal cord and change in chronic pain (Figure 2). Like all vertebrate primary mechano-sensory nerve terminals, primary muscle-spindle endings contain small clear synaptic-like vesicles that release glutamate during stretch in a Ca2+-dependent manner, the glutamate in turn activating an unusual glutamate receptor. This glutamate system maintains and increases muscle-spindle responsiveness in a positive feedback manner (Bewick 2015; Bewick and Banks 2015, 2021). All this should have consequences for motor control and kinesthesia at the spinal and higher levels.
3.2. Changes in Spinal Somatic Reflexes
- Presynaptic (PSI) and Recurrent Inhibition
Under acute conditions, activation of group III (Aδ) and group IV (C) afferents elicited by fatiguing muscle contractions changed PSI and recurrent inhibition in the cat. PSI increased and recurrent inhibition decreased, which could contribute in part to decrease the homonymous monosynaptic H-reflex (Kalezic et al. 2004). This was supported by suppressive effects on Renshaw-cell (RC) responses to antidromic motor axon stimulation when small-diameter calf muscle afferents were excited by intra-arterially injected algesic substances (Windhorst et al. 1997).
The pattern is different in chronic pain elicited by diabetic polyneuropathy. In spinalized streptozotocin (STZ)-induced diabetic rats with a reduced withdrawal threshold to mechanical stimuli, the mono- and disynaptic reflexes (MSR and DSR, respectively) were recorded from L5 ventral roots in response to stimulation of the ipsilateral L5 dorsal root. The diabetic rats generally showed larger spinal reflex amplitudes, the DSR being influenced in particular. PSI and recurrent inhibition of the spinal reflexes were altered in STZ-treated animals. The recurrent inhibition of the MSR and DSR elicited by preceding antidromic conditioning stimulation delivered to the recorded L5 ventral root was markedly suppressed in diabetic rats. By contrast, the PSI of the MSR and DSR elicited by preceding conditioning stimulation to the ipsilateral L4 dorsal root was not impaired (Tanabe et al. 2005).
In humans, soleus spinal reflex excitability, PSI and recurrent inhibition were compared between chronic ankle instability (CAI), acute Lateral Ankle Sprain coper (LAS-coper) and healthy cohorts. Twelve individuals with CAI, twelve `copers´ and twelve healthy age, limb and gender-matched controls participated. Soleus H-reflex recruitment curves, presynaptic excitability and recurrent inhibition of the spinal reflex pathway were examined during static double- and single-leg stance. Reporting of pain and perceived instability were used to perform a regression analysis on measures of soleus spinal excitability in people with CAI, LAS-coper and healthy controls. Soleus spinal reflex excitability was greater during single-leg stance in CAI compared to healthy and coper individuals. PSI was three times less in CAI participants compared to both healthy controls and copers. There were no differences between healthy and coper participants in spinal measures of sensory-motor control. Reports of pain explained 15-16% of the variance in soleus spinal reflex excitability and PSI during single and double-leg stance, while perceived instability explained 20% of the variance in spinal reflex during single leg stance only. CAI participants presented with an inability to suppress soleus spinal reflexes during tasks with increased postural threat, likely due to disinhibition of presynaptic mechanisms. Pain and perceived instability may contribute to changes in spinal sensory-motor control in CAI (Thompson et al. 2019).
In the spinal cord, neuronal activity is controlled by the balance between excitatory and inhibitory neurotransmission, mediated mainly by glutamate and GABA/Gly, respectively. Alterations of this equilibrium have been associated with spinal MN hyperexcitability and degeneration, which can be induced by excitotoxicity or by decreasing inhibitory neurotransmission. Whereas glutamate-mediated excitotoxicity appears to be an important factor, recent experimental and histopathological evidence argue in favor of a decreased activity of the inhibitory circuits controlling MN excitability, mainly the recurrent inhibition exerted by RCs. A decreased RC activity may be caused by cell loss or by a reduction of its inhibitory action secondary to a decreased excitation from ACh INTs. Inhibitory failure by either mechanism might lead to MN degeneration (Ramírez-Jarquín et al. 2014).
Under chronic pain conditions, the central effects of GOTs should change because GTO group Ib afferents in a very complex pattern project to diverse excitatory and inhibitory INTs which most probably are affected during pain (Windhorst 2021). These INTs may also include `hidden´ WDR neurons because many receive nociceptive inputs (Jankowska 1992). – In passing, it is mentioned here that some cutaneous mechano-receptors also are proprioceptors.
- Withdrawal Reaction
The nociceptive withdrawal reflex (NWR) has been widely used as a measure of spinal hyperexcitability, which is believed to contribute to chronic musculo-skeletal pain and headache. In patients compared to controls, moderate-quality evidence was found indicating lower NWR threshold, larger NWR area, and shorter NWR latency. Low-quality evidence demonstrated facilitated temporal summation of NWR threshold in patients compared to controls. Spinal hyperexcitability as evidenced by lowered NWR threshold values and temporal summation of the NWR was present in patients with chronic MSKP and headache. No evidence existed for alterations in NWR duration and NWR magnitude (Van Oosterwijck et al. 2025).
3.3. Pain and Posture
Postural control deficits are a potential cause of persistent and recurrent pain in patients with cLBP. The dlPFC contributes to pain regulation in cLBP, but its precise role in the postural control of cLBP patients remains unclear. Twenty patients with cLBP and 20 healthy controls performed upright stance tasks under three conditions: Task-1 was static balance with eyes open. Task-2 was static balance with eyes closed. Task-3 involved dynamic balance on an unstable surface with eyes open. A wireless functional near-infrared spectroscopy system measured cortical activity, including the bilateral dlPFC, pre-motor cortex (PM) with supplementary motor area (SMA), the M1, the S1, and a force platform measured balance parameters during upright stance. There were significant interactions in bilateral PM/SMA activation. Moreover, cLBP patients had significantly increased right dlPFC activation and higher postural sway and velocity than healthy controls during upright stance. This implies that PM/SMA and dlPFC maintain standing balance. The cLBP patients had higher cortical activity and upright-stance control deficits, which may indicate that the cLBP patients have low neural efficiency and need more motor resources to maintain balance (Li et al. 2023).
Multi-sensory afferent inputs to the cervical spinal cord assist in neck-muscle control. Chronic neck pain and muscle fatigue are factors that disturb somatosensory function. The effects of chronic neck pain and neck-flexor muscle fatigue on muscle-control strategy and postural control were investigated in young patients performing voluntary shoulder flexion movements. Twenty-five patients with chronic neck pain and 25 age-matched asymptomatic controls participated. The postural sway, muscle onset time, and activation level of the erector spinae, rectus abdominal, semispinalis capitis (SSC), and sternocleidomastoid (SCM) muscles were recorded. The chronic neck pain group exhibited significantly larger body sway, greater neck-muscle activation, and longer onset time of neck-flexor muscle compared with the control group. The chronic neck pain group showed a trend of greater postural sway and shorter muscle onset under the eyes-closed condition than those under the eyes-open condition. After muscle fatigue, the chronic neck pain group further exhibited (i) greater body sway during the eyes-open condition but decreased body sway during the eyes-closed condition, (ii) higher activation of the neck flexor (SCM) and lower activation of the trunk extensor (erector spinae), and (iii) early onset of the neck muscles. Hence, chronic neck pain causes poor postural control and altered neck muscle control patterns. The addition of neck-flexor muscle fatigue further decreases balance stability and provokes a protective neck muscle control strategy during the shoulder flexion movement (Hsu et al. 2020).
In female patients with chronic idiopathic neck pain, the relationship was determined between joint-position sense and static and dynamic. As compared to healthy controls, the patients with chronic neck pain showed a deviation in cervical joint-position sense that was greater in extension, right rotation, and left lateral rotation. The performance of the patients was worse in the Single-Leg Balance Test with both eyes open and eyes closed. The patients showed worse dynamic balance only in the anterior direction reach of the left leg. Hence, cervical joint-position sense and static balance were worse in female patients with chronic idiopathic neck pain than in asymptomatic individuals. Dynamic balance in all other directions except for the anterior direction was not negatively affected in individuals with chronic idiopathic neck pain (Ashyüce et al. 2022).
3.4. Pain and Locomotion
Locomotor Burst Activity in the mature intact spinal cord alternates between flexor and extensor MNs through reciprocal inhibition and between left and right sides through commisural inhibitory INTs. The alternating pattern disappears after neonatal spinal cord transection which suppresses supraspinal influences upon the locomotor networks, emphasizing the plasticity of spinal-cord locomotor networks 5-HT, in particular 5-HT2 receptors, helps shape the alternating pattern. The action of 5-HT2 receptors is mediated, at least in part, through a modulation of Cl- homeostasis. The postsynaptic action of GABA and Gly depends on the intracellular concentration of Cl- ions which is regulated by a protein in the plasma membrane, the K+-Cl- cotransporter (KCC2) extruding both K+ and Cl-. Absence or reduction of KCC2 expression leads to a depolarizing action of GABA and Gly and a reduction in the strength of postsynaptic inhibition (Gackière and Vinay 2014).
Walking and Prehension. LBP can affect performance in the combined task (CT) of gait and prehension, since it increases muscle-activity amplitude during voluntary movements, impairs the anticipatory postural adjustments and reduces gait speed. The effects were compared of adding the prehension movement toward a dowel located at three different heights (80, 100 and 120% of the lower limb length) on gait of individuals with and without LBP. The CT caused anticipatory adjustments, showing that gait changes started during the approach phase and continued on the step corresponding to grasping, especially for the lowest dowel height. Individuals with LBP reduced walking speed, increased the width of the base of support, increased EMG activity of low back trunk muscles, and increased the margin of dynamic stability compared with control group. This suggests that individuals with LBP used a strategy to reduce threat to body stability due to addition of the manual task (Abbári Rossi Manciopi et al. 2017).
Voluntary Wheel Running. C57BL/6J and DBA/2J mice received unilateral intra-plantar injections of 100% CFA, paclitaxel, or CCI surgery to compare their distance traveled in a voluntary wheel running assay, paw edema diameter, and mechanical sensitivity. Mechanical withdrawal thresholds were lower in both strains of mice that received CFA when compared to their vehicle. However, a decrease in distance traveled was observed in CFA-treated C57BL/6J but not DBA/2J mice. In a separate group, chemotherapy agent paclitaxel was administered intra-peritoneally to both strains of mice to induce CIPN, which was confirmed by lower mechanical thresholds in paclitaxel-treated mice compared to vehicle-treated mice. Only female C57BL/6J mice showed attenuation of distance traveled following treatment, whereas male C57BL/6J and DBA/2J mice did not. To compare the impact of another CNP model, C57BL/6J mice underwent CCI or sham surgery in the wheel-running assay. CCI mice showed a gradual decrease in mechanical withdrawal threshold and a decrease in distance traveled compared to sham five days following the procedure (Contreras et al. 2021).
3.5. Musculo-Skeletal Pain Disorders
Muscle hyperalgesia, referred pain, referred hyperalgesia, and widespread hyperalgesia play an important role in chronic musculo-skeletal pain. Peripheral sensitization is involved in increased sensitivity of deep tissue, but central sensitization may be equally important and may play a role in the persistence, amplification, and spread of pain (Graven-Nielsen and Arendt-Nielsen 2002). In musculo-skeletal pain conditions, a re-organized motor control system is an important factor. The interaction between muscle pain and motor control depends on the specific motor task. Muscle pain causes no increase in EMG activity at rest and reduces maximal voluntary contraction and endurance time during sub-maximal contractions. Muscle pain is also associated with an adaptive change in the coordination during dynamic exercises. Increased muscle activity reflecting re-organized muscle coordination and strategy is a component of the functional adaption to muscle pain (Graven-Nielsen and Arendt-Nielsen 2008).
3.6. Cardio-Vascular Reactions
Whether the association between chronic pain and hypertension is either causally linked or influenced by other shared risk factors is not completely known. Mendelian randomization (MR) was employed to examine the potential causal relationship between risk of chronic pain and hypertension. Data were derived from the pooled dataset of the genome-wide association study, enabling the evaluation of the causal effects of hypertension on various types of chronic pain including chronic headache, chest, abdominal, joint, back, limb, and multisite chronic pain. Genetically predicted essential hypertension was associated with an increased risk of chronic headache and limb pain. No potential causal associations were identified between chronic pain and essential hypertension in the reverse direction MR. There was also no potential causal association between secondary hypertension and chronic pain (Wang et al. 2024a).
In healthy individuals, elevated blood pressure is associated with diminished acute pain sensitivity. These interactions between the cardio-vascular and pain regulatory systems appear altered in patients with chronic pain, i.e., elevated blood pressure is associated with increased acute and chronic pain respnsiveness. A retrospective review was conducted on randomly selected records of 300 patients with chronic pain (Pain) evaluated at a tertiary care pain center and 300 non-pain internal medicine (Medicine) patients seen at the same institution. 39% of the Pain group was diagnosed with clinical hypertension, compared with 21% of the Medicine group. Analyses by sex revealed similar group differences in males and females, although the difference in females was double in magnitude compared with males. In contrast to more frequent male hypertension in the general population and the Medicine sample, females were more often diagnosed with hypertension than males in the Pain group. Similar group differences were obtained for anti-hypertensive use. In the Pain group, chronic-pain intensity was a significant predictor of hypertension independent of the effects of age, race/ethnicity, and parental hypertension. This suggests that chronic pain may be associated with increased risk of hypertension (Bruehl et al. 2005).
HRV and baroreflex sensitivity (BRS) are indexes reflecting the ability to maintain cardio-vascular homeostasis in changing conditions. Data have suggested that both HRV and BRS may be reduced in individuals with chronic pain (CP), with potential implications for cardio-vascular risk. HRV and BRS were compared between individuals with CP (broadly defined) and pain-free controls in a large unselected population sample. Participants were 1143 individuals reporting clinically meaningful CP and 5640 pain-free controls who completed a 106-second cold-pressor test (CPT). Participants self-reported hypertension status. Resting HRV and BRS were derived from continuous beat-to-beat blood-pressure recordings obtained before and after the CPT. Hierarchical regressions for the pre-CPT period indicated that beyond effects of age, sex, and body-mass index, the CP group displayed significantly lower HRV in both the time domain and frequency domain, as well as lower BRS. Results were somewhat weaker for the post-CPT period. For six of seven HRV and BRS measures tested, there were significant indirect (mediated) effects of CP status on the presence of comorbid hypertension via reduced HRV or BRS. In the largest and broadest sample tested to date, results confirm that the presence of CP is linked to impaired cardio-vascular regulation and for the first time provide support for the hypothesis that links between CP and comorbid hypertension may be due in part to CP-related decrements in cardio-vascular regulation (Bruehl et al. 2018).
3.7. Respiratory Reactions
Chronic Neck Pain. Cervical musculo-skeletal and respiratory parameters in patients with chronic neck pain were reviewed. Compared to asymptomatic subjects, patients with chronic neck pain showed significant differences in maximum inspiratory and expiratory pressures. Some of the respiratory volumes were lower in patients with chronic neck pain. Muscle strength and endurance, cervical range of motion, and psychological states were significantly correlated with respiratory parameters. Patients also showed lower PCo2 and significant relationship between chest expansion and neck pain. Respiratory retraining was effective in improving some cervical musculo-skeletal and respiratory impairment. Hence, functional pulmonary impairments accompany chronic neck pain (Kahlaee et al. 2017).
Efficacy of Opioids in Chronic Pain. Clinical investigations have revealed that opioid-induced respiratory depression is less severe in patients with chronic pain. Respiration was quantified by double-chamber, flow-through whole-body plethysmography. Respiratory frequency was dose-dependently and significantly decreased after morphine administration. This effect peaked at 30 minutes after administration and lasted three hours. In contrast, tidal volume was increased. Minute volume was significantly decreased by morphine at a higher dose, but not a lower dose. In nerve-ligated mice, a morphine-induced decrease in respiratory frequency was observed, whereas the increase of tidal volume was more prominent. Minute volume was not decreased in nerve-ligated mice. This attenuation of the morphine-induced decrease in minute volume in nerve-ligated mice was reversed by treatment with the 5-HT4a receptor antagonist GR125487. Moreover, treatment with the 5-HT4 receptor agonist mosapride antagonized the morphine-induced decrease in minute volume, due to the enhancement of tidal volume. The expression of 5-HT4a receptors in the brainstem was enhanced in nerve-ligated mice compared to that in sham-operated mice. This suggests that the decrease in morphine-induced respiratory depression under chronic pain is mediated by the enhancement of 5-HT4a receptor systems in the brainstem (Kamei et al. 2011).
4. Clinical Syndromes
Chronic pain syndromes represent the clinical manifestation of persistent maladaptive changes in peripheral and CNS structures. Although the initiating events vary widely — ranging from tissue injury and inflammation to nerve damage or central lesions — the resulting pain states share common pathophysiological features, including peripheral and central sensitization, altered descending modulation, neuro-immune activation, and maladaptive plasticity across distributed brain networks. This section highlights selected clinical syndromes that exemplify how chronic pain emerges from alterations of functional structures discussed in the preceding chapters.
Nociceptive, ergoceptive and mechano-receptive group III and IV afferents have far-reaching effects on pain sensation and perception, motor control, cardio-vascular and respiratory functions. For example, stimulation of the vanilloid receptors in dorsal-neck muscles with capsaicin evoked c-fos expression and NADPH-diaphorase reactivity with distinctive patterns in the cervical (C1-C8) and lumbar (L1-L7) segments (Pilyavskii et al. 2005). In anesthetized rats, following both direct muscle stimulation and L5 ventral-root stimulation, fatigue-related c-fos expression was most prominent in the DH of the ipsilateral L2-L5 segments and within the ipsilateral NTS, the CVLM and rostral ventro-lateral medulla (RVL), the intermediate reticular nucleus (Maisky et al. 2002). In rats, unilateral injections of algesic solutions (6% hypertonic saline or 0.05% capsaicin) into the gastrocnemius muscle elicited mostly c-fos-labeled neurons in the spinal cord in laminae IV-V, VI, VII and X, with fewer labeled neurons in laminae I and II, as well as in the brainstem, predominantly in the lateral reticular formation (LRF), bi-laterally in CVLM, where also neurons responsive to noxious stimulation of cutaneous and visceral structures lie. Labeled neurons, many of them catecholaminergic, also occurred bilaterally in the gracile nucleus, NTS, A1 area, A5 area, CVLM and RVL, LC, NRM in the pons, as well as the PBN. The rostral ventro-medial medulla (RVM) was labeled consistently (Panneton et al. 2015). The conspicuous difference here is the predominantly unilateral c-fos cell labelling during muscle fatigue (Maisky et al. 2002), while the injection of algesic substances into the gastrocnemius muscle labelled many cells bilaterally (Panneton et al. 2015).
In acute pain, concerted actions should occur to rapidly adapt motor, cardio-vascular and respiratory functions to the new situation. It may be presumed, therefore, that these concerted actions may be disrupted in chronic pain conditions. In the following, we will briefly deal with these effects.
4.1. Phantom Pain
Phantom pain is a paradigmatic example of centrally generated chronic pain, occurring after limb amputation or de-afferentation. Patients experience pain sensations seemingly originating from the missing body part, often described as burning, cramping, stabbing, or electric.
At the peripheral level, neuroma formation and ectopic discharges in transected nerves may contribute to ongoing nociceptive input. However, phantom pain persists even in the absence of peripheral input, underscoring the dominant role of central mechanisms (Granata et al. 2024). Profound re-organization occurs in the spinal DH, THAL, and somatosensory cortex, including expansion of adjacent receptive fields, loss of inhibitory control, and enhanced excitatory transmission. Maladaptive cortical plasticity, particularly within primary somatosensory cortex (S1), motor cortex (M1), and associated parietal areas, correlates with pain intensity (Chen et al. 2002).
Alterations in thalamo-cortical rhythms, changes in the PAG–RVM descending modulatory system, and increased engagement of limbic structures such as the AMY and ACC contribute to the affective and distressing components of phantom pain. Thus, phantom pain exemplifies how loss of sensory input can paradoxically lead to persistent pain through network-level re-organization (Bao et al. 2022).
4.2. Post-Herpetic Neuropathy
Post-herpetic neuropathy (PHN) is a CNP condition that develops following re-activation of varicella-zoster virus in sensory ganglia. Pain persists long after the resolution of the acute herpetic rash and is often severe, spontaneous, and stimulus-evoked.
PHN is characterized by damage to primary afferent neurons in DRG, leading to spontaneous ectopic activity, altered ion-channel expression, and increased excitability. In the spinal DH, loss of inhibitory INTs, microglial and astrocytic activation, and enhanced NMDA receptor-dependent transmission promote central sensitization. These changes result in allodynia and hyperalgesia that are disproportionate to peripheral stimuli (Finnerup et al. 2021).
Supraspinally, altered processing within the THAL, IC, and ACC amplifies pain perception and emotional suffering. The persistence of pain despite viral clearance illustrates how immune-triggered nerve injury can initiate long-lasting maladaptive plasticity throughout nociceptive pathways (Tang et al. 2021).
4.3. Trigeminal Neuropathy (TN)
TN encompasses a group of chronic facial pain syndromes arising from injury or dysfunction of the trigeminal nerve or its central projections. Pain may be continuous or paroxysmal and is frequently accompanied by sensory deficits or dysesthesias. TN is triggered by mild mechanical stimulation in the orofacial area. Pathophysiological changes occur at multiple levels, including ectopic activity in trigeminal-ganglion neurons, altered processing within the trigeminal spinal nucleus, and abnormal thalamo-cortical signaling. Compared to spinal neuropathic pain, trigeminal pain shows particularly strong engagement of limbic and autonomic centers, reflecting the high affective salience of facial pain. Altered descending modulation from the PAG, RVM, and monoaminergic brainstem nuclei further contributes to pain persistence. TN highlights the regional specificity of chronic pain syndromes and the importance of brainstem and cortical integration in shaping pain experience (Finnerup et al. 2021).
Piezo2 mediates tactile allodynia in neuropathic pain. In a rat model of TN after trigeminal nerve compression injury, it was investigated whether Piezo2-mediated mechano-transduction contributes to peripheral sensitization. In rats subjected to chronic compression of the trigeminal nerve root (CCT) for 21 days, the expression of Piezo2 in the trigeminal ganglion increased. Purinergic P2 receptors P2X4, P2X7, P2Y1, and P2Y2 were also significantly up-regulated after compression injury. Hence, in the rat compression model, Piezo2 may play a crucial role in peripheral mechanical allodynia (Luo et al. 2022).
In rats subjected to CCI of IoN, a significant up-regulation of Piezo2 and IL-6 occurred during trigeminal neuropthic pain (TNP). The Piezo2-positive neurons accounted for 72.3±9.5% in those IL-6 positive neurons. The Piezo2 co-localized with CGRP, and IB4. After administration of GsMTx4 or anti-rat IL-6 antibody in the TNP model, the dynamic allodynia and pinprick hyperalgesia scores as well as the mechanical threshold changed significantly. In the sham-operation rates, with local administration of IL-6, an up-regulation of Piezo2 also occurred. Hence, the up-regulation of Piezo2 in the pain afferent neurons following trigeminal nerve injury may play a role in the development of the neuralgia. Also, the expression of Piezo2 may be modulated by inflammatory cytokines, such as IL-6 (Liu et al 2021a).
4.4. Pain After SCI
Chronic pain following SCI is a common and debilitating condition that may occur below, at, or above the level of injury. SCI pain can include neuropathic pain, musculo-skeletal pain, and visceral pain components.
SCI disrupts ascending and descending pathways, leading to profound re-organization of spinal and supraspinal circuits (Hulsebosch et al. 2009). Below the lesion, de-afferentation induces hyperexcitability in DH neurons, enhanced spontaneous activity, and altered inhibitory control. Above the lesion, cortical and THAL re-organization occurs, often accompanied by increased activity in pain-related networks (Endo et al. 2008).
Neuro-immune activation within the spinal cord, including microglial and astrocytic responses, plays a central role in maintaining chronic pain after SCI. Impaired descending inhibition from the PAG–RVM system further exacerbates pain, while maladaptive plasticity in motor and sensory systems contributes to abnormal sensorimotor integration (Shoraka et al. 2024).
4.5. Chronic Widespread Non-Inflammatory Muscle Pain (CWP): Fibromyalgia, Myofascial Syndromes, Chronic Fatigue Syndrome (CFS)
CWP as exemplified by FM, myofascial syndromes and CFS (related to myalgic encephalomyelitis (ME)) is characterized by diffuse musculo-skeletal pain, fatigue (CFS), sleep disturbances, and cognitive symptoms. Several hypotheses have been proposed regarding the underlying pathophysiology: muscular dysfunction/ischemia, central sensitization, and a deficit in endogenous pain-modulating systems. Peripheral tissue pathology is minimal or absent, indicating a predominantly central origin. In human subjects and animal models, CWP is associated with changes in the CNS, which likely reflect alterations in supraspinal modulation of nociception, and include increases in excitatory and decreases in inhibitory modulation pathways. These alterations in excitation and inhibition likely drive changes also in the spinal cord to result in central sensitization, and the consequent pain and hyperalgesia (DeSantana and Sluka 2008; Dibaj et al. 2020,2021).
Patients exhibit enhanced pain sensitivity, widespread hyperalgesia, and reduced pain thresholds, consistent with generalized central sensitization (Lesnak and Sluka 2019). Functional imaging studies reveal altered activity and connectivity within the IC, ACC, PFC, and somatosensory cortices. Dysregulation of descending inhibitory systems, including 5-HT and NA pathways, contributes to deficient endogenous analgesia (Liang et al. 2025).
Neuro-endocrine disturbances involving the HPA axis and altered autonomic regulation further modulate pain perception. CWP illustrates how chronic pain can arise from distributed network dysfunction without clear peripheral pathology. CWP, fibromyalgia, and CFS/ME can be conceptualized as overlapping syndromes of maladaptive central network regulation, in which pain and fatigue emerge as parallel expressions of a system persistently biased toward heightened sensory gain, reduced inhibitory capacity, and diminished physiological resilience (Dibaj and Windhorst 2024a, 2024b).
4.6. Chronic or Recurrent Low Back Pain (cLBP)
Chronic low back pain is one of the most prevalent and costly pain conditions worldwide (Safavi-Abbasi et al. 2025). Although often initiated by peripheral tissue injury or degeneration, pain persistence frequently becomes dissociated from structural abnormalities. 85% percent of LBP disorders are not diagnosed. LBP disorders are multi-factorial. At the spinal level, sustained nociceptive input leads to DH sensitization, altered reflexes, and changes in proprioceptive processing. Supraspinal changes include re-organization of sensorimotor cortices, reduced representation of trunk muscles in M1, and altered connectivity between motor, sensory, and limbic regions.
It has been proposed that three broad sub-groups of LBP disorders exist. The first group presents where underlying pathological processes drive the pain, and the patients´ motor responses in the disorder are adaptive. A second group represents psychological and/or social factors as the primary mechanism underlying the disorder, and where the patient´s coping and motor control strategies are maladaptive in nature. A third group comprises a large group of LBP disorders in which patients present with either movement impairments (characterized by pain avoidance behavior) or control impairments (characterized by pain provocation behavior). These pain disorders are predominantly mechanically induced and patients typically present with maladaptive primary physical and secondary cognitive compensations for their disorders that become a mechanism for ongoing pain. These subjects present either with an excess or deficit in spinal stability (O´Sullivan 2005).
Caudal LBP (cLBP) is associated with impaired motor control, maladaptive movement patterns, and altered body perception. Emotional and cognitive factors, mediated by prefrontal and limbic circuits, strongly influence pain persistence and disability. Thus, cLBP exemplifies the tight coupling between pain, motor systems, and higher-order brain functions (Greenwals and Shafritz 2018).
4.7. Complex Regional Pain Syndrome (CRPS)
CRPS is a severe chronic pain condition typically developing after limb trauma or surgery and characterized by disproportionate pain, autonomic disturbances, trophic changes, and motor dysfunction.
CRPS involves a combination of peripheral inflammation, neurogenic inflammation, autonomic dysregulation, and central sensitization (Borchers and Gershwin 2017). Increased sympathetic–sensory coupling, altered immune responses, and changes in spinal and supraspinal processing contribute to pain persistence.
Functional and structural changes are observed in the somatosensory cortex, motor cortex, insula, and limbic regions, often accompanied by distorted body representation and neglect-like symptoms. CRPS represents an extreme example of how injury-induced alterations across multiple functional systems can culminate in a highly complex chronic pain syndrome (Doan and Chang 2025).
4.8. Summary
The clinical syndromes briefly presented here demonstrate that chronic pain is not a unitary condition but rather a spectrum of disorders arising from diverse yet overlapping alterations in functional structures. Despite differences in etiology, these syndromes converge on common mechanisms, including maladaptive plasticity, disrupted inhibition, neuro–immune interactions, and altered network dynamics extending from the periphery to the cerebral cortex. Understanding these syndromes through the lens of functional structure alterations provides a framework for developing more targeted and mechanism-based therapies.
5. Concluding Remarks
Not only is pain a double-edged sword, but it also shows a double face in fighting with itself. While acute pain is indispensable for survival and tissue protection, chronic pain represents a maladaptive state in which the very systems designed to limit injury become sources of persistent suffering.
Widespread hyperalgesia and dysfunctional endogenous pain inhibition have been identified as characteristics of many musculo-skeletal and neuropathic pain conditions. These similarities suggest common CNS abnormalities in pain processing among many chronic pain conditions. Increased pain sensitivity is a characteristic of chronic pain and may develop through peripheral mechanisms (peripheral sensitization) or consequent to neuroplastic changes in the CNS (central sensitization), or both. Central sensitization involves: up-regulation of sensory neuron-specific Na+ channels; change in NMDA and TRPV receptors; phenotype switching of large myelinated axons; axon sprouting within the DH; and loss of inhibitory INTs (Staud 2012). Together, these processes shift nociceptive circuits toward a state of persistent hyperexcitability and reduced inhibitory control.
Many of these altered brain regions and networks in chronic pain patients are not only involved in pain processing, but also in other, especially motor, cardio-vascular and respiratory functions, as well as in sensory and particularly cognitive tasks. MRI studies have provided information on the association of brain alterations with pain catastrophizing, fear-avoidance, anxiety and depressive symptoms. Pain catastrophizing is related to brain areas involved in pain processing, attention to pain, emotion and motor activity, and to reduced top-down pain inhibition. In contrast to pain catastrophizing, there are no clear associations with brain characteristics of anxiety and depressive symptoms. All cognitive or emotional factors show significant associations with data from resting-state fMRI, indicating that even at rest the brain reserves a certain activity for these pain-related factors (Malfliet et al. 2017). Pain (and suffering) can be regarded as the consequence of an imbalance between the two ascending nociceptive pathways and the descending pain modulatory pathways (De Ridder et al. 2021).
This review has outlined the extensive functional and structural changes in nervous structures and functions in chronic pain. It has emerged that chronic pain and the underlying neuronal mechanism are complex. Sub-cellular signaling cascades, molecular plasticity, cellular phenotype changes, local circuit re-organization, long-range network remodeling, and neuromodulatory influences converge to produce a dynamic and unstable system state. Chronic pain thus emerges from interacting mechanisms operating across multiple organizational levels. Macroscopic structures — nociceptors, spinal and brainstem nuclei, THAL relays, and cortical areas — are internally complex, interconnected in large-scale networks and subject to modulation by diverse neurotransmitters and neuromodulators acting on multiple receptor systems. Multiple parallel processes may affect a single structure, such as the DH, underscoring the multi-functional and fluid nature of pain networks. This organization reflects a general principle or “Bauplan” of the nervous system (Windhorst and Dibaj 2025a, 2025b). Now another layer has been added on top, consisting of the multifarious effects and influences of a multitude of diseases.
The question remains open as to what influential factors play a role in the transition from acute to chronic pain. Almost certainly, there is no single one. One theory explaining the origin of chronic pain is that of priming and the accumulation of events that can be predictors along a continuum before chronic pain becomes apparent. `Chronification´ cannot explain all cases of chronic/persistent pain. The plastic changes in the pain-processing system have been envisaged as a continuum where at some point an acute pain event is only one of several possible tipping points that changes potential pain into perceived pain (Butler 2022).
In summary, chronic pain is associated with a mosaic of underlying structural and functional changes distributed across the neuraxis – and remains a mystery. Enigmata tend to persist. But they stimulate dedicated problem solvers to continue. Stay calm and carry on.
Funding
This research received no external funding.
Acknowledgments
UW is grateful to his wife Siggi for occasional help and much patience.
Conflicts of Interest
The authors declare no conflict of interest.
Ethics Approval and Consent to Participate
Not applicable.
Abbreviations:
5-HT: 5-hydroxy-tryptophan, serotonin; 5-HT2: serotonin receptor 2; AA: adjuvant-induced arthritis; ACC: anterior cingulate cortex; ACh: acetylcholine; ACID3: acid-sensing ion channel 3; ACTH: adreno-corticotropic hormone; aIC: anterior insular cortex; α-MN: alpha motoneuron; aMCC: anterior mid-cingulate cortex; AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; AMY: amygdala; ANS: autonomic nervous system; AP: action potential; AP5: 2-amino-5-phophonopenanoate; ASIC: acid-sensing ion channel; ATP: adenosine triphosphate; BDNF: brain-derived neurotrophic factor; BFB: basal forebrain; BG: basal ganglia; BLA: baso-lateral nucleus of amygdala; BNST: bed nucleus of the stria terminalis; BOLD: blood-oxygen-level-dependent; CBP: chronic back pain; CC: cingulate cortex; CCI: chronic constriction injury; CCK: cholecystokinin; CeA: central nucleus of the amygdala; CeM: medial amygdala nucleus; CFA: complete Freund´s adjuvant; CFS: chronic fatigue syndrome; cGMP: cyclic guanosine monophosphate; CGRP: calcitonin gene-related peptide; cLBP: chronic low back pain; CLN: caudal linear nucleus; CNP: chronic neuropathic pain; CNS: central nervous system; cNTS: caudal nucleus tractus solitarii; CP: chronic pain; CPan: chronic pancreatitis; CPA: conditioned place aversion; CPM: conditioned pain modulation; CPN: common peroneal nerve (ligation); CPP: conditioned place preference; CPSP: chronic post-surgical pain; CR: calretinin; CRD: colo-rectal distension; CRH: corticotropin-releasing hormone; CRPS: complex regional pain syndrome; CRS: chronic restraint stress; CSF: cortico-spinal fluid; CUMS: chronic unpredictable mild stress; CVLM: caudal ventro-lateral medulla; CWP: chronic widespread pain; D1: dopamine receptor1; D2: dopamine receptor 2; D5: dopamine receptor 5; DA: dopamine; DG: dentate gyrus; DH: dorsal horn; DSR; disynaptic reflex; DNIC: diffuse noxious inhibitory control; dlPFC: dorso-lateral prefrontal cortex; DMH: dorso-medial nucleus of hypothalamus; dmPFC: dorsal medial prefrontal cortex; DRG: dorsal-root ganglion; DReN: dorsal reticular nucleus; DRN: dorsal raphé nucleus; DRR: dorsal root reflex; Dyn: dynorphin; EA: electro-acupuncture; EC: entorhinal cortex; eCB: endocannabinoid; EEG: electro-encephalogram, electro-encephalography; EMG: electromyography; ENK: enkephalin; ENS: enteric nervous system; EPSC: excitatory postsynaptic current; EPSP: excitatory postsynaptic potential; ERK: extracellular-signal regulated kinase; ERα: estrogen receptor α; ERß: estrogen receptor ß; FM: fibromyalgia; fMRI: functional magnetic resonance imaging; FST: forced-swim test; GABA: γ-amino-butyric acid; GABAAR: γ-amino-butyric acid receptor; γ-MN: gamma motoneuron; Gly: glycine; GlyR: glycine receptor; GM: gastrocnemius muscle; GPe: globus pallidus externus; GPi: globus pallidus internus; GR: glucocorticoid receptor; GRP : gastrin-releasing peptide; GTO: Golgi tendon organ; Hb: Habenula; HCN: hyperpolarization-activated cyclic nucleotide-gated channels; HIPP: hippocampus; HIST: histamine; HPA: hypothalamic-pituitary-adrenal (axis); HPT: hpothalamic-pituitary-thyroid (axis); H-reflex: Hoffmann reflex; HRV: heart-rate variability; HYP: hypothalamus; HYP ARC: hypothalamic arcuate nucleus; HYP PVN: hypothalamic paraventricular nucleus; IB4: isolectin B4; IBS: irritable bowel syndrome; IC: insular cortex (insula); ICV: intra-cerebro-ventricular; IFN-α: interferon-α; IFN-δ: interferon-δ; IL: infra-limbic; IL-1: interleukin-1; IL-1β: interleukin-1β; IL-6: interleukin-6; IL-8: interleukin-8; IL-12: interleukin-12; INaP : persistent sodium current; INT: interneuron; IO: inferior olive; IoN: infra-orbital nerve; iSPN: indirect spiny projection neuron; ITC: intercalated cell (in amygdala); KCC2: K+-Cl- exporter; LA: lateral amygdala; lCeA: lateral central nucleus of the amygdala; LHb: lateral Habenula; lHYP: lateral hypothalamus; lPAG: lateral peri-aqueductal gray; lPBN: lateral parabrachial nucleus; LC: locus coeruleus; lLC: lateral locus coeruleus; lPOA: lateral pre-optic area; LRN: lateral reticular nucleus; LTD: long-term depression; LTMR: low-threshold mechano-receptor; LTP: long-term potentiation; M1: primary motor cortex; MCC: mid-cingulate cortex; MD: medio-dorsal nucleus of the thalamus; ME: Myalgic encephalomyelitis; MeA: medial Amygdala; MEG: magneto-encephalography; mEPSC: miniature excitatory synaptic current; mGluR: metabotropic glutamate receptor; mGluR1: metabotropic glutamate receptor 1; mGluR5: metabotropic glutamate receptor 5; MHb: medial Habenula; mIPSC: miniature inhibitory postsynaptic current; MN: motoneuron; mPFC: medial prefrontal cortex; MRI: magnetic resonance imaging; MS: multiple sclerosis; MSN: medium-spiny neuron; MSR: monosynaptic reflex; MU: motor-unit; NA: noradrenaline, noradrenergic; NAc: nucleus Accumbens; NCS: nucleus centralis superior; NF: neurotrophic factor; NGc: nucleus reticularis gigantocellularis; NGF: nerve growth factor; NKA: neurokinin A; NK1: neurokinin-1; NK1R: neurokinin-1 receptor; NMDA: N-methyl-D-aspartate; NMDAR: N-methyl-D-aspartate receptor; NO: nitric oxide; NOS: nitric oxide synthase; NPY: neuropeptide Y; NR: nucleus ruber; NRD: nucleus raphé dorsalis; NRI: nucleus centralis inferior; NRM: nucleus raphé magnus; NRO: nucleus raphé obscurus; NRP: nucleus raphé pallidus; NRPo: nucleus raphé pontis; NS: nociceptive-specific (neuron); NT-3: neurotrophin 3; NTS: nucleus tractus solitarii; NVmes: trigeminal mesencephalic nucleus; NWR: nociceptive withdrawal reflex; 6-OHDA: 6-hydroxydopamine; OFC: orbito-frontal cortex; OIH: opioid-induced hyperalgesia; OXT: oxytocin; OXTR: oxytocin receptor; pACC: pre-genual anterior cingulate cortex; PAD: primary afferent depolarization; PAG: peri-aqueductal gray; PaNS: parasympathetic nervous system; PBN: parabrachial nucleus; pCC: posterior cingulate cortex; PD: Parkinson´s disease; pERK-1/2: extracellular-signal regulated kinase phosphorylation; PET: positron emission tomography; PFC: prefrontal cortex; PG: nucleus paragigantocellularis; PGE2: prostaglandin E2; PHN: post-herpetic neuropathy; pHYP: posterior HYP; pIC: posterior insular cortex; PKC: protein kinase C; PKC-γ: protein kinase C γ; PKC-δ: protein kinase C δ; PL: pre-limbic cortex; PM: premotor cortex; PNS: peripheral nervous system; PO: posterior nucleus (of the thalamus); POA: preoptic area; PoT: posterior triangular nucleus (of thalamus); PPN: pedunculo-pontine nucleus; pptA: pre-protachykinin-A gene; pPVT: posterior portion of the paraventricular nucleus of thalamus; PSI: presynaptic inhibition; PV: parvalbumin; PVN: paraventricular nucleus (of hypothalamus); PVT: paraventricular nucleus of thalamus; rACC: rostral anterior cingulate cortex; RC: Renshaw cell; RF: reticular formation; rmTN: rostro-medial tegmental nucleus; RN: raphé nuclei; RNM: raphé nucleus medianus; RNS: reactive nitrogen species; rNTS: rostral part of the nucleus tractus solitarii; RRF: retro-rubral field; RSC: retro-splenial cortex; rTMS: repetitive transcranial magnetic stimulation; RVM: rostral ventro-medial medulla; ROS: reactive oxygen species; S1: primary somatosensory cortex; S2: secondary somatosensory cortex; SCI: spinal cord injury; SCN: supra-chiasmatic nucleus; SMA: supplementary motor area; SN: substantia nigra; SNc: substantia nigra pars compacta; SNr: substantia nigra pars reticularis; SNI: sciatic nerve injury; SNL: spinal nerve ligation; SNS: sympathetic nervous system; SP: substance P; SpV: spinal trigeminal nucleus; SpVc: trigeminal nucleus caudalis; SpVo: spinal trigeminal sub-nucleus oralis; SRD: subnucleus reticularis dorsalis; SPT: spino-parabrachial tract; STN: subthalamic nucleus; STP: short-term plasticity; STT: somatostatin; STTr: spino-thalamic tract; T3: triiodothyronine; T4: thyroxine; tDCS: transcranial direct current stimulation; TH: thyroid hormone; THAL: thalamus; TMS: transcranial magnetic stimulation; TN: trigeminal neuralgia; TNP: trigeminal neuropathic pain; TNF: tumor necrosis factor; TNF-α: tumor necrosis factor α; TRH: thyrotropin-releasing hormone; TrkB: tropomyosine receptor kinase B; TRPA: transient receptor potential ankyrin; TRP: transient receptor potential ion channel; TRPV: transient receptor potential vanilloid; TRPV1: transient receptor potential vanilloid 1; tSCI: transient spinal cord ischemia; TSH: thyroid-stimulating hormone; TTX: tetrodotoxin; VC: vertical cell; VGLUT: vesicular glutamate transporter; VGLUT1: vesicular glutamate transporter 1; VGLUT3: vesicular glutamate transporter 3; VIP: vasoactive intestinal peptide; VLM: ventro-lateral medulla; VLMlat: lateral ventro-lateral medulla; vlOFC: ventro-lateral orbito-frontal cortex; vlPAG: ventro-lateral peri-aqueductal gray; vlPFC: ventro-lateral prefrontal cortex; VM: ventro-medial nucleus of the thalamus; vmPAG: ventro-medial peri-aqueductal gray; vmPFC: ventro-medial prefrontal cortex; VPL: ventro-posterior lateral (nucleus of thalamus); VPM: ventro-posterior medial (nucleus of thalamus); VRt: ventral reticular nucleus; VTA: ventral tegmental area; WDR: wide-dynamic-range (neuron); ZI: zona incerta.
References
- Abbári Rossi Manciopi, P; Rinaldi, NM; Moraes, R. Prehension combined with gait in individuals with chronic low back pain. Motor Control 2017, 21, 90–111. [Google Scholar] [CrossRef]
- Abdulla, FA; Moran, TD; Balasubramanyan, S; Smith, PA. Effects and consequences of nerve injury on the electrical properties of sensory neurons. Can J Physiol Pharmacol 2003, 81, 663–682. [Google Scholar] [CrossRef]
- Adamaszek, M; D´Agata, F; Ferrucci, R; Habas, C; Keulen, S; Kirkby, KC; Leggio, M; Mariёn, P; Molinari, M; Moulton, E; Orsi, L; Van Overwalle, F; Papadelis, C; Priori, A; Sacchetti, L; Schutter, DJ; Styliadis, C; Verhoeven, J. Consensus Paper: Cerebellum and emotion. Cerebellum 2017, 16, 552–576. [Google Scholar] [CrossRef] [PubMed]
- Allen, HN; Bobnar, HJ; Kolber, BJ. Left and right hemispheric lateralization of the amygdala in pain. Prog Neurobiol 2021, 196, 101891. [Google Scholar] [CrossRef]
- Alles, SRA; Smith, PA. Etiology and pharmacology of neuropathic pain. Pharmacol Rev 2018, 70, 315–347. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, P; Dieb, W; Hafidi, A; Voisin, DL; Dallel, R. Insular cortex representation of dynamic mechanical allodynia in trigeminal neuropathic rats. Neurobiol Dis 2009, 33, 89–95. [Google Scholar] [CrossRef]
- Anderson, EO; Schneider, ER; Bagriantsev, SN. Piezo2 in cutaneous and proprioceptive mechanotransduction in vertebrates. Curr Top Membr 2017, 79, 197–217. [Google Scholar] [PubMed]
- Antunes, GF; Pinheiro Campos, AC; Varin de Assis, D; Gouveia, FV; Dias de Jesus Seno, M; Pagano, RL; Ruiz Martinez, RC. Habenula activation patterns in a preclinical model of neuropathic pain accompanied by depressive-like behaviour. PloS One 2022, 17, e0271295. [Google Scholar] [CrossRef]
- Apkarian, AV; Baliki, MN; Geha, PY. Towards a theory of chronic pain. Prog Neurobiol 2009, 87, 81–97. [Google Scholar] [CrossRef]
- Arslan, D; Çevik, IÜ. Interactions between the painful disorders and the autonomic nervous system. Agri 2022, 34, 155–165. [Google Scholar] [CrossRef]
- Artola, A; Voisin, D; Dallel, R. PKCγ interneurons, a gateway to pathological pain in the dorsal horn. J Neural Transm (Vienna) 2020, 127, 527–540. [Google Scholar] [CrossRef]
- Ashyüce, YÖ; Demirel, A; Ülger, Ö. Investigation of joint position sense and balance in individuals with chronic idiopathic neck pain: A cross-sectional study. J Manipulative Physiol Ther 2022, 45, 188–195. [Google Scholar]
- Aternali, A; Katz, J. Recent advances in understanding and managing phantom limb pain. F1000Research 2019, 8, 1167. [Google Scholar] [CrossRef]
- Austin, J; Fiore, NT. Supraspinal neuroimmune crosstalk in chronic pain states. Curr Opin Physiol 2019, 11, 7–15. [Google Scholar] [CrossRef]
- Bai, Y; Chen, Y-B; Qiu, X-T; Chen, Y-B; Ma, L-T; Li, Y-Q; Sun, H-K; Zhank, M-M; Zhang, T; Chen, T; Fan, B-Y; Li, H; Li, Y-Q. Nucleus tractus solitarius mediates hyperalgesia induced by chronic pancreatitis in rats. World J Gastroenterol 2019, 25, 6077–6093. [Google Scholar] [CrossRef]
- Bak, MS; Park, H; Kim, SK. Neural plasticity in the brain during neuropathic pain. Biomedicines 2021, 9, 624. [Google Scholar] [CrossRef] [PubMed]
- Baker, PM; Mathis, V; Lecourtier, L; Simmons, SC; Nugent, FS; Hill, S; Mizumori, SJY. Lateral habenula beyond avoidance: Roles in stress, memory, and decision-making with implications for psychiatric disorders. Front Syst Neurosci 2022, 16, 826475. [Google Scholar] [CrossRef]
- Baliki, M; Al-Amin, HA; Atweh, SF; Jabei, M; Hawwa, N; Jabbur, SJ; Apkarian, AV; Saadé, NE. Attenuation of neuropathic manifestations by local block of the activities of the ventrolateral orbito-frontal area in the rat. Neuroscience 2003, 120, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Bandler, R; Shipley, MT. Columnar organization in the midbrain periaqueductal gray: modules for emotional expression? Trends Neurosci 1994, 7, 379–389. [Google Scholar] [CrossRef]
- Bank, PJM; Peper, CE; Marinus, J; Beek, PJ; van Hilten, JJ. Motor consequences of experimentally induced limb pain: a systematic review. Eur J Pain 2013, 17, 145–157. [Google Scholar] [CrossRef]
- Bao, BB; Zhu, HY; Wei, HF; Li, J; Wang, ZB; Li, YH; Hua, XY; Zheng, MX; Zheng, XY. Altered intra- and inter-network brain functional connectivity in upper-limb amputees revealed through independent component analysis. Neural Regen Res 2022, 17, 2725–2729. [Google Scholar] [PubMed]
- Baron, R. Mechanisms of disease: neuropathic pain – a clinical perspective. Nature Clinical Practice Neurol 2006, 2, 95–106. [Google Scholar] [CrossRef]
- Basbaum, AI; Bautista, DM; Scherrer, G; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Baumbauer, KM; Young, EE; Joynes, RL. Pain and learning in a spinal system: contradictory outcomes from common origins. Brain Res Rev 2009, 61, 124–143. [Google Scholar] [CrossRef]
- Benarroch, EE. Pain-autonomic interactions. Neurol Sci 2006, 27, S130–123. [Google Scholar] [CrossRef]
- Bennett, DL; Clark, AJ; Huang, J; Waxman, SG; Dib-Hajj, SD. The role of voltage-gated sodium channels in pain signaling. Physiol Rev 2019, 99, 1079–1151. [Google Scholar] [CrossRef]
- Benke, D. GABAB receptors and pain. Curr Top Behav Neurosci 2022, 52, 213–239. [Google Scholar]
- Berger, JV; Knaepen, L; Janssen, SPM; Jaken, RJP; Marcus, MAE; Joosten, EAJ; Deumens, R. Cellular and molecular insights into neuropathy-induced pain hypersensitivity for mechanism-based treatment approaches. Brain Res Rev 2011, 67, 282–310. [Google Scholar] [CrossRef]
- Bewick, GS. Synaptic-like vesicles and candidate transduction channels in mechanosensory terminals. J Anat 2015, 227, 194–213. [Google Scholar] [CrossRef]
- Bewick, GS; Banks, RW. Mechanotransduction in the muscle spindle. Pflügers Arch - Eur J Physiol 2015, 467, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Bewick, GS; Banks, RW. Mechanotransduction channels in proprioceptive sensory nerve terminals: still an open question? Curr Opin Physiol 2021, 21, 90–104. [Google Scholar] [CrossRef]
- Binshtok, AM; Wang, H; Zimmermann, K; Amaya, F; Vardeh, D; Shi, L; Brenner, GJ; Ji, RR; Bean, BP; Woolf, CJ; Samad, TA. Nociceptors are interleukin-1beta sensors. J Neurosci 2008, 28, 14062–14073. [Google Scholar] [CrossRef]
- Birznieks, I; Burton, AR; Macefield, VG. The effects of experimental muscle and skin pain on the static stretch sensitivity of human muscle spindles in relaxed leg muscles. J Physiol (Lond) 2008, 586, 2713–2723. [Google Scholar] [CrossRef]
- Biurrun Manresa, JA; Brix Finnerup, NS; Johannesen, IL; Biering-Sørensen, F; Staehelin-Jensen, T; Arendt-Nielsen, L; Kæseler-Andersen, O. Central sensitization in spinal cord injured humans assessed by reflex receptive fields. Clin Neurophysiol 2014, 125, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Björnsdotter, M; Löken, L; Olausson, H; Vallbo, A; Wessberg, J. Somatotopic organization of gentle touch processing in the posterior insular cortex. J Neurosci 2009, 29, 9314–9320. [Google Scholar] [CrossRef] [PubMed]
- Bliss, TVP; Collingridge, GL; Kaang, B-K; Zhuo, M. Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat Rev Neurosci 2016, 17, 485–946. [Google Scholar] [CrossRef]
- Boadas-Vaello, P; Homs, J; Reina, F; Carrera, A; Verdú, E. Neuroplasticity of supraspinal structures associated with pathological pain. Anat Rec 2017, 300, 1481–1501. [Google Scholar] [CrossRef]
- Boccella, S; Marabese, I; Guida, F; Luongo, L; Maione, S; Palazzo, E. The modulation of pain by metabotropic glutamate receptors 7 and 8 in the dorsal striatum. Curr Neuropharmacol 2020, 18, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Bombardi, C; Grandis, A; Chiocchetti, R; Bertolami, R; Johansson, H; Lucchl. Immunohistochemical localization of alpha1a-adrenoreceptors in muscle spindles of rabbit masseter muscle. Tissue and Cell 2006, 38, 121–125. [Google Scholar] [CrossRef]
- Borchers, AT; Gershein, ME. The clinical relevance of complex regional pain syndrome type I: The emperorˈs new clothes. Autoimmun Rev 2017, 16, 22–33. [Google Scholar] [CrossRef]
- Borsook, D. Neurological diseases and pain. Brain 2012, 135, 320–344. [Google Scholar] [PubMed]
- Borsook, D; Youssef, AM; Simons, L; Elman, I; Ecclestone, C. When pain gets stuck: the evolution of pain chronification and treatment resistance. Pain 2018, 159, 2421–2436. [Google Scholar] [CrossRef]
- Boscan, P; Pickering, AE; Paton, JFR. The nucleus of the solitary tract: an integrating station for nociceptive and cardiorespiratory afferents. Exp Physiol 2002, 87, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Bostan, AC; Strick, PL. The basal ganglia and the cerebellum nodes in an integrated network. Nat Rev Neurosci 2018, 19, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Bouchet, CA; Ingram, SL. Cannabinoids in the descending pain modulatory circuit: Role in inflammation. Pharmacol Ther 2020, 209, 107495. [Google Scholar] [CrossRef]
- Bouhassira, D; Bing, Z; Le Bars, D. Studies of the brain structures involved in diffuse noxious inhibitory controls: the mesencephalon. J Neurophysiol 1990, 64, 1712–1723. [Google Scholar] [CrossRef]
- Boyle, KA; Gradwell, MA.; Yasaka, T; Dickie, AC; Polgár, E; Ganley, RP; Orr, DPH; Watanabe, M; Abraira, VE; Kuehn, ED; Zimmerman, AL; Ginty, DD; Callister, RJ; Graham, BA, DI. Defining a spinal microcircuit that gates myelinated afferent Input: Implications for tactile allodynia. Cell Rep 2019, 28, 526–540.e6. [Google Scholar] [CrossRef] [PubMed]
- Bräscher, A-K; Becker, S; Hoeppli, M-E; Schweinhardt, P. Different brain circuitries mediating controllable and uncontrollable pain. J Neurosci 2016, 36, 5013–5025. [Google Scholar] [CrossRef]
- Braz, J; Solorzano, C; Wang, X; Basbaum, AI. Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron 2014, 82, 522–536. [Google Scholar] [CrossRef]
- Brodal, A. Neurological anatomy. In relation to clinical medicine, 3rd edn; Oxford University Press: New York, 1981. [Google Scholar]
- Brodal, P. A neurobiologist´s attempt to understand persistent pain. Scand J Pain 2017, 15, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Bruehl, S; Bjørkholt Olsen, R; Tronstad, C; Sevre, K; Burns, JW; Schirmer, H; Nielsen, CS; Stubhaug, A; Rosseland, LA. Chronic pain-related changes in cardiovascular regulation and impact on comorbid hypertension in a general population: the Tromsø study. Pain 2018, 159, 119–127. [Google Scholar] [CrossRef]
- Bruehl, S; Chung, OY; Jirjis, JN; Biridepalli, S. Prevalence of clinical hypertension in patients with chronic pain compared to nonpain general medical patients. Clin J Pain 2005, 21, 147–153. [Google Scholar] [CrossRef]
- Bushnell, MC; Čeko, M; Low, LA. Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci 2013, 14, 502–511. [Google Scholar] [CrossRef]
- Butler, SH. Pain “chronification“: what is the problem with this model? Scand J Pain 2022, 23, 419–423. [Google Scholar] [CrossRef]
- Camarena-Delgado, C; Llorca-Torralba, M; Suárez-Pereira, I; Bravo, L; López-Martín, C; Garcia-Partida, J; Mico, JA; Berrosco, E. Nerve injury induces transient locus coeruleus activation over time: role of the locus coeruleus-dorsal reticular nucleus pathway. Pain 2022, 163, 943–954. [Google Scholar] [CrossRef]
- Cao, L; DeLeo, JA. Immune system and pain. In Encyclopedia of neuroscience; Binder, MD, Hirokawa, N, Windhorst, U, Eds.; Springer-Verlag: Berlin Heidelberg, 2009; pp. pp 1919–1922. [Google Scholar]
- Capra, NF; Hisley, CK; Masri, RM. The influence of pain on masseter spindle afferent discharge. Arch Oral Biol 2007, 52, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Carbone, E. Calcium channels – an overview. In Encyclopedia of neuroscience; Binder, MD, Hirokawa, N, Windhorst, U, Eds.; Springer-Verlag: Berlin Heidelberg, 2009; pp. pp 545–550. [Google Scholar]
- Cardoso-Cruz, H; Lima, D; Galhardo, V. Instability of spatial encoding by CA1 hippocampal place cells after peripheral nerve injury. Eur J Neurosci 2011, 33, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Carlton, SM. Nociceptive primary afferents: they have a mind of their own. J Physiol (Lond) 2014, 592, 3403–3411. [Google Scholar] [CrossRef]
- Carrasco, C; Naziroğlu, M; Rodríguez, A; Patiente, JA. Neuropathic pain: Delving into the oxidative origin and the possible implication of transient receptor potential channels. Front Physiol 2018, 9, 95. [Google Scholar] [CrossRef]
- Cathenaut, L; Schlichter, R; Hugel, S. Short-term plasticity in the spinal nociceptive system. Pain 2023, 164, 2411–2424. [Google Scholar] [CrossRef] [PubMed]
- Cerveco, F. Hyperalgesia and allodynia. In Encyclopedia of neuroscience; Binder, MD, Hirokawa, N, Windhorst, U, Eds.; Springer-Verlag: Berlin Heidelberg, 2009; pp. pp 1876–1880. [Google Scholar]
- Cevikbas, F; Lerner, EA. Physiology and pathophysiology of itch. Physiol Rev 2020, 100, 945–982. [Google Scholar] [CrossRef]
- Chandler, DJ; Jensen, P; McCall, JG; Pickering, AE; Schwarz, LA; Totah, NK. Redefining NA neuromodulation of behavior: impacts of a modular locus coeruleus architecture. J Neurosci 2019, 39, 8239–8249. [Google Scholar] [CrossRef]
- Chapman, CR; Vierck, CJ. The transition of acute postoperative pain to chronic pain: An integrative overview of research on mechanisms. J Pain 2017, 18, 359.e1–359.e38. [Google Scholar] [CrossRef]
- Chen, J; Gao, Y; Bao, S-T; Wang, Y-D; Jia, T; Yin, C; Xiao, C; Zhou, C. Insula→amygdala and insula→thalamus pathways are involved in comorbid chronic pain and depression-like behavior in mice. J Neurosci 2024, 44, e2062232024. [Google Scholar] [CrossRef]
- Chen, Q; Zhao, M; Dong, J; Yang, K. Chronic restraint stress-induced hyperalgesia is modulated by the periaqueductal gray neurons projecting to the rostral ventromedial medulla in mice. Biochem Biophys Res Commun 2024, 710, 149875. [Google Scholar] [CrossRef]
- Chen, QL; Heinricher, MM. Plasticity in the link between pain-transmitting and pain-modulating systems in acute and persistent inflammation. J Neurosci 2019, 39, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Chen, R; Cohen, LG; Halett, M. Nervous system reorganization following injury. Neurosci 2002, 111, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Chen, T; Taniguchi, W; Chen, Q-Y; Tozaki-Saitoh, H; Song, Q; Liu, R-H; Koga, K; Matsuda, T; Kaito-Sugimura, Y; Wang, J; Li, Z-H; Lu, Y-C; Inoue, K; Tsuda, M; Li, Y-Q; Nakatsuka, T; Zhuo, M. Top-down descending facilitation of spinal sensory excitatory transmission from the anterior cingulate cortex. Nature Commun 2018, 9, 1886. [Google Scholar] [CrossRef]
- Chen, X; Tang, S-J. Neural circuitry polarization in the spinal dorsal horn (SDH): A novel form of dysregulated circuitry plasticity during pain pathogenesis. Cells 2024, 13, 398. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L; Duan, B; Huang, T; Zhang, Y; Chen, Y; Britz, O; Garcia-Campmany, L; Ren, X; Vong, L; Lowell, BB; Goulding, M; Wang, Y; Ma, Q. Identification of spinal circuits involved in touch-evoked dynamic mechanical pain. Nat Neurosci 2017, 20, 804–814. [Google Scholar] [CrossRef]
- Cheng, Y-R; Jiang, B-Y; Chen, C-C. Acid-sensing ion channels: dual function proteins for chemo-sensing and mechano-sensing. J Biomed Sci 2018, 25, 46. [Google Scholar] [CrossRef]
- Cheriyan, J; Sheets, PL. Altered excitability and local connectivity of mPFC-PAG neurons in a mouse model of neuropathic pain. J Neurosci 2018, 38, 4829–4839. [Google Scholar] [CrossRef]
- Chiang, C; Aston-Jones, G. Response of locus coeruleus neurons to foot shock stimulation is mediated by neurons in the rostral ventral medulla. Neuroscience 1993, 53, 705–715. [Google Scholar] [CrossRef]
- Chiang, MC; Bowen, A; Schier, LA; Tupone, D; Uddin, O; Heinricher, MM. Parabrachial complex: A hub for pain and aversion. J Neurosci 2019, 39, 8225–8230. [Google Scholar] [CrossRef] [PubMed]
- Chudler, EH; Dong, WK. The role of the basal ganglia in nociception and pain. Pain 1995, 60, 3–38. [Google Scholar] [CrossRef]
- Chung, K; Yoon, YW; Chung, JM. Sprouting sympathetic fibers form synaptic varicosities in the dorsal root ganglion of the rat with neuropathic injury. Brain Res 1997, 751, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Cichon, J; Blanck, TJJ; Gan, WB; Yang, G. Activation of cortical somatostatin interneurons prevents the development of neuropathic pain. Nat Neurosci 2017, 20, 1122–1132. [Google Scholar] [CrossRef]
- Cleland, CL; Hayward, L; Rymer, WZ. Neural mechanisms underlying the clasp-knife reflex in the cat. II. Stretch-sensitive muscular-free nerve endings. J Neurophysiol 1990, 64, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Cleland, CL; Rymer, WZ. Functional properties of spinal interneurons activated by muscular free nerve endings and their potential contributions to the clasp-knife reflex. J Neurophysiol 1993, 69, 1181–1191. [Google Scholar] [CrossRef]
- Cobos, A; Lima, D; Almeida, A; Tavares, I. Brain afferents to the lateral caudal ventrolateral medulla: a retrograde and anterograde tracing study in the rat. Neuroscience 2003, 120, 485–498. [Google Scholar] [CrossRef]
- Coderre, TJ; Katz, J. Peripheral and central hyperexcitability: Differential signs and symptoms in persistent pain. Beh Brain Sci 1997, 20, 404–419. [Google Scholar] [CrossRef]
- Coizet, V; Dommett, EJ; Klop, EM; Redgrave, P; Overton, PG. The parabrachial nucleus is a critical link in the transmission of short latency nociceptive information to midbrain dopaminergic neurons. Neuroscience 2010, 168, 263–272. [Google Scholar] [CrossRef]
- Comitato, A; Bardoni, R. Presynaptic inhibition of pain and touch in the spinal cord: From receptors to circuits. Int J Mol Sci 2021, 22, 414. [Google Scholar] [CrossRef]
- Contreras, KM; Caillaud, M; Neddenriep, B; Bagdas, D; Roberts, JL; Ulker, E; White, AB; Aboulhosn, R; Toma, W; Khalefa, T; Adel, A; Mann, JA; Damaj, MI. Deficit in voluntary wheel running in chronic inflammatory and neuropathic pain models in mice: Impact of sex and genotype. Behav Brain Res 2021, 399, 113009. [Google Scholar] [CrossRef]
- Cook, AJ; Woolf, CJ; Wall, PD; MacMahon, SB. Dynamic receptive field plasticity in rat spinal cord dorsal horn following C-primary afferent input. Nature 1987, 325, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Corder, G; Ahanonu, B; Grewe, BF; Wang, D; Schnitzer, MJ; Scherrer, G. An amygdalar neural ensemble that encodes the unpleasantness of pain. Science 2019, 363, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Costa, AR; Tavares, I; Martins, I. How do opioids control pain circuits in the brainstem during opioid-induced disorders and in chronic pain? Implications for the treatment of chronic pain. Pain 2024, 165, 324–336. [Google Scholar] [CrossRef]
- Coste, J; Voisin, DL; Miraucourt, L; Dallel, R; Luccarini, P. Dorsal horn NK1-expressing neurons control windup of downstream trigeminal nociceptive neurons. Pain 2008, 137, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Costigan, M; Scholz, J; Woolf, CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci 2009, 32, 1–32. [Google Scholar] [CrossRef]
- Craig, AD. Pain mechanisms: labeled lines versus convergence in central processing. Annu Rev Neurosci 2003, 26, 1–30. [Google Scholar] [CrossRef]
- Cui, C-X; Liu, H-Y; Yue, N; Du, Y-R; Che, L-M; Yu, J-S. Research progress on the mechanism of chronic neuropathic pain. IBRO Neurosci Rep 2023, 14, 80–85. [Google Scholar] [CrossRef]
- Dai, D; Li, W; Chen, A; Gao, X-F; Xiong, L. Lateral habenula and its potential roles in pain and related behaviors. ACS Chem Neurosci 2022, 13, 1108–1118. [Google Scholar] [CrossRef]
- Da Silva, JT; Seminowicz, DA. Neuroimaging of pain in animal models: a review of recent literature. Pain Rep 2019, 4, e732. [Google Scholar] [CrossRef]
- Davidson, S; Golden, JP; Copits, BA; Ray, PR; Vogt, SK; Valtcheva, MV; Schmidt, RE; Ghetti, A; Price, TJ; Gereau, 4th RW. Group II mGluRs suppress hyperexcitability in mouse and human nociceptors. Pain 2016, 157, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Decherchi, P; Dousset, E; Grélot, L. Metabolic stability and physiological adaptation of muscle under conditions of exercise. Rev Neurol (Paris) 2004, 160, 297–305. [Google Scholar] [CrossRef]
- Article in French.
- De Felice, C; Herrero, JF; O´Brien, JA; Palmer, JA; Doyle, CA; Smith, AJ; Laird, JM; Belmonte, C; Cervero, F; Hunt, SP. Altered nociception, analgesia and aggression in mice lacking the receptor for substance P. Nature 1998, 392, 394–397. [Google Scholar] [CrossRef] [PubMed]
- De Felice, M; Ossipov, MH. Cortical and subcortical modulation of pain. Pain Manag 2016, 6, 111–120. [Google Scholar] [CrossRef]
- Delbono, O; Wang, Z-M; Messi, ML. Brainstem noradrenergic neurons: Identifying a hub at the intersection of cognition, motility, and skeletal muscle regulation. Acta Physiol (Oxf) 2022, 236, e13887. [Google Scholar] [CrossRef]
- De Ridder, D; Adhia, D; Vanneste, S. The anatomy of pain and suffering in the brain and its clinical implications. Neurosci Biobehav Rev 2021, 130, 125–1463. [Google Scholar] [CrossRef]
- De Ridder, D; De Mulder, G; Verstraeten, E; Sunaert, S; Moller, A. Somatosensory cortex stimulation for deafferentation pain. Acta Neurochir Suppl 2007, 97, 67–74. [Google Scholar]
- Derjean, D; Bertrand, S; Le Masson, G; Landry, M; Morisset, V; Nagy, F. Dynamic balance of metabotropic inputs causes dorsal horn neurons to switch functional states. Nat Neurosci 2003, 6, 274–281. [Google Scholar] [CrossRef] [PubMed]
- DeSantana, JM; Sluka, KA. Central mechanisms in the maintenance of chronic widespread noninflammatory muscle pain. Curr Pain Headache Rep 2008, 12, 338–343. [Google Scholar] [CrossRef]
- Devor, M. Rethinking the causes of pain in herpes zoster and postherpetic neuralgia: the ectopic pacemaker hyposthesis. Pain Rep 2018, 3, e702. [Google Scholar] [CrossRef] [PubMed]
- Dibaj, P; Brockmann, K; Gärtner, J. Dopamine-mediated yawning-fatigue syndrome with specific recurrent initiation and responsiveness to opioids. JAMA Neurol 2020, 77, 254. [Google Scholar] [CrossRef] [PubMed]
- Dibaj, P; Nadrigny, F; Steffens, H; Scheller, A; Hirrlinger, J; Schomburg, ED; Neusch, C; Kirchhoff, F. NO mediates microglial response to acute spinal cord injury under ATP control in vivo. Glia 2010, 58, 1133–44. [Google Scholar] [CrossRef]
- Dibaj, P; Safavi-Abbasi, S; Asadollahi, E. In vivo spectrally unmixed multi-photon imaging of longitudinal axon-glia changes in injured spinal white matter. Neurosci Lett 2024, 841, 137959. [Google Scholar] [CrossRef]
- Dibaj, P; Schomburg, ED. In vivo recording of nerve conduction velocity of spinal CNS fibers in the mouse. Physiol Res 2017, 66, 545–548. [Google Scholar] [CrossRef]
- Dibaj, P; Seeger, D; Gärtner, J; Petzke, F. Follow-up of a case of dopamine-mediated yawning-fatigue-syndrome responsive to opioids, successful desensitization via graded activity treatment. Neurol Int 2021, 13, 79–84. [Google Scholar] [CrossRef]
- Dibaj, P; Windhorst, U. Preprints 2024091515; Muscle fatigue in health and disease. 2024b.
- Dibaj, P; WindhorstU. Motor-control notions in health and disease. Preprints 2024b, 2024031799. [Google Scholar]
- Dib-Hajj, SD; Waxman, SG. Sodium channels in human pain disorders: Genetics and pharmacogenomics. Annu Rev Neurosci 2019, 42, 87–106. [Google Scholar] [CrossRef]
- Doan, HN; Chang, MC. Comprehensive mechanisms and non-invasive treatment for complex regional pain syndrome: a narrative review. J Pain Res 2025, 18, 4699–4726. [Google Scholar] [CrossRef]
- Doan, L; Manders, T; Wang, J. Neuroplasticity underlying the comorbidity of pain and depression. Neural Plast 2015, 2015, 504691. [Google Scholar] [CrossRef]
- Dogrul, BN; Machado Kopruszinski, C; Dolatyari Eslami, M; Watanabe, M; Luo, S; Moreira de Souza, LH; Vizin, RL; Yue, X; Palmiter, RD; Navratilova, E; Porreca, F. Descending facilitation from rostral ventromedial medulla mu opioid receptor-expressing neurons is necessary for maintenance of sensory and affective dimensions of chronic neuropathic pain. Pain 2025, 166, 153–159. [Google Scholar] [CrossRef]
- Dostrovsky, JO. Role of thalamus in pain. Prog Brain Res 2000, 129, 245–257. [Google Scholar]
- Duan-Mu, C-L; Zhang, X-N; Shi, H; Su, Y-S; Wan, H-Y; Wang, Y; Qu, Z-Y; He, W; Wang, X-Y; Jing, X-H. Electroacupuncture-induced muscular inflammatory pain relief was associated with activation of low-threshold mechanoreceptor neurons and inhibition of wide dynamic range neurons in spinal dorsal horn. Front Neurosci 2021, 8, 687173. [Google Scholar] [CrossRef] [PubMed]
- Echeverria-Villalobos, M; Tortorici, V; Brito, BE; Ryskamp, D; Uribe, A; Weaver, T. The role of neuroinflammation in the transition of acute to chronic pain and the opioid-induced hyperalgesia and tolerance. Front Pharmacol 2023, 14, 1297931. [Google Scholar] [CrossRef]
- Endo, T; Spenger, C; Westman, E; Tominaga, T; Olson, L. Reorganization of sensory processing below the level of spinal cord injury as revealed by fMRI. Exp Neurol 2008, 209, 155–160. [Google Scholar] [CrossRef]
- Evrard, HC. The organization of the primate insular cortex. Front Neuroanat 2019, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Fasick, V; Spengler, RN; Samankan, S; Nader, ND; Ignatowski, TA. The hippocampus and TNF: Common links between chronic pain and depression. Neurosci Biobehav Rev 2015, 53, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Fekete, C; Lechan, RM. Negative feedback regulation of hypophysiotropic thyrotropin-releasing hormone (TRH) synthesizing neurons: role of neuronal afferents and type 2 deiodinase. Front Neuroendocrinol 2007, 28, 97–114. [Google Scholar] [CrossRef]
- Felix, R; Corzo-Lopez, A; Sandoval, A. Voltage-gated ion channels in neuropathic pain signaling. Life (Basel) 2025, 15, 888. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, EC; Pechincha, C; Luz, LL; Kokai, E; Szucs, P; Safronov, BV. Primary afferent-driven presynaptic inhibition of C-fiber inputs to spinal lamina I neurons. Prog Neurobiol 2020, 188, 101786. [Google Scholar] [CrossRef]
- Ferreira, BR; Misery, L. Psychopathology associated with chronic pruritus: A systematic review. Acta Derm Venereol 2023, 103, adv8488. [Google Scholar] [CrossRef]
- Finnerup, NB; Kuner, R; Jensen, TS. Neuropathic pain: from mechanisms to treatment. Physiol Rev 2021, 101, 259–301. [Google Scholar] [CrossRef]
- Flor, H. Cortical reorganisation and chronic pain: implications for rehabilitation. J Rehabil Med 2003, 41 Suppl, 66–72. [Google Scholar] [CrossRef]
- Fore, S; Palumbo, F; Pelgrims, R; Yaksi, E. Information processing in the vertebrate habenula. Semin Cell Dev Biol 2018, 8, 130–139. [Google Scholar] [CrossRef]
- Gackière, F; Vinay, L. Serotonergic modulation of post-synaptic inhibition and locomotor alternating pattern in the spinal cord. Front Neural Circuits 2014, 8, 102. [Google Scholar]
- Gamal-Eltrabily, M; Márquez-Morales, C; Martínez-Lorenzana, G; González-Hernández, A; Condés-Lara, M. Cortical modulation of nociception. Neuroscience 2021, 15(458), 256–270. [Google Scholar] [CrossRef] [PubMed]
- Gamboa-Esteves, FO; Tavares, I; Almeida, A; Batten, TF; McWilliam, PN; Lima, D. Projection sites of superficial and deep spinal dorsal horn cells in the nucleus tractus solitarii of the rat. Brain Res 2001, 921, 195–205. [Google Scholar] [CrossRef]
- Gangadharan, V; Zheng, H; Taberner, FJ; Landry, J; Nees, TA; Pistolec, J; Agarwal, N; Männich, D; Benes, V; Helmstaedter, M; Ommer, B; Lechner, SG; Kuner, T; Kuner, R. Neuropathic pain caused by miswiring and abnormal end organ targeting. Nature 2022, 606, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y; Mei, C; Chen, P; Chen, X. The contribution of neuro-immune crosstalk to pain in the peripheral nervous system and the spinal cord. Int Immunopharmacol 2022, 107, 108700. [Google Scholar] [CrossRef]
- Garcia, AD; Buffalo, EA. Anatomy and function of the primate entorhinal cortex. Annu Rev Vis Sci 2020, 6, 411–432. [Google Scholar] [CrossRef]
- Garcia-Larrea, L; Quesada, C. Cortical stimulation for chronic pain: from anecdote to evidence. Eur J Phys Rehabil Med 2022, 58, 290–305. [Google Scholar] [CrossRef] [PubMed]
- Gazerani, P; Cairns, BE. Activation of rat masticatory muscle afferent fibres by acidic pH. Somatosens Mot Res 2018, 35, 86–94. [Google Scholar] [CrossRef]
- Gebhart, GF. Pain. In Encyclopedia of neuroscience; Binder, MD, Hirokawa, N, Windhorst, U, Eds.; Springer-Verlag: Berlin Heidelberg, 2009; pp. pp 3063–3065. [Google Scholar]
- Giummarra, MJ; Gibson, SJ; Georgiou-Karistianis, N; Bradshaw, JL. Mechanisms underlying embodiment, disembodiment and loss of embodiment. Neurosci Biobehav Rev 2008, 32, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Gong, N; Hagopian, G; Holmes, TC; Luo, D; Xu, X. Functional reorganization of local circuit connectivity in superficial spinal dorsal horn with neuropathic pain states. eNeuro 2019, 6, ENEURO.0272–19. [Google Scholar] [CrossRef] [PubMed]
- González-Hernández, A; Manzano-García, A; Martínez-Lorenzana, G; Tello-García, IA; Carranza, M; Arámburo, C; Condés-Lara, M. Peripheral oxytocin receptors inhibit the nociceptive input signal to spinal dorsal horn wide-dynamic-range neurons. Pain 2017, 158, 2117–2128. [Google Scholar] [CrossRef]
- Goudet, C; Magnaghi, V; Landry, M; Nagy, F; Gereau, RW, 4th; Pin, JP. Metabotropic receptors for glutamate and GABA in pain. Brain Res Rev 2009, 60, 43–56. [Google Scholar] [CrossRef]
- Gouveia, FV; Ibrahim, GM. Habenula as a neural substrate for aggressive Behavior. Front Psychiatry 2022, 13, 817302. [Google Scholar] [CrossRef]
- Grace, PM; Gaudet, AD; Staikopoulos, V; Maier, SF; Hutchinson, MR; Salvemini, D; Watkins, LR. Nitroxidative signaling mechanisms in pathological pain. Trends Neurosci 2016, 39, 862–879. [Google Scholar] [CrossRef]
- Gradwell, MA; Callister, RJ; Graham, BA. Reviewing the case for compromised spinal inhibition in neuropathic pain. J Neural Transm (Vienna) 2020, 127, 481–503. [Google Scholar] [CrossRef] [PubMed]
- Granata, G; Di lorio, R; Ilari, S; Angeloni, BM; Tomasello, F; Cimmino, AT; Carrarini, C; Marrone, A; lodice, F. Phantom limb syndrome: from pathogenesis to treatment. A narrative review. Neurol Sci 2024, 45, 4741–4755. [Google Scholar] [CrossRef]
- Grau, JW. Learning from the spinal cord: How the study of spinal cord plasticity informs our view of learning. Neurobiol Learning Memory 2014, 108, 155–171. [Google Scholar] [CrossRef]
- Graven-Nielsen, T; Arendt-Nielsen, L. Peripheral and central sensitization in musculoskeletal pain disorders: an experimental approach. Curr Rheumatol Rep 2002, 4, 313–321. [Google Scholar] [CrossRef]
- Graven-Nielsen, T; Arendt-Nielsen, L. Impact of clinical and experimental pain on muscle strength and activity. Curr Rheumatol Rep 2008, 10, 475–481. [Google Scholar] [CrossRef]
- Graven-Nielsen, T; Mense, S. The peripheral apparatus of muscle pain: evidence from animal and human studies. Clin J Pain 2001, 17, 2–10. [Google Scholar] [CrossRef]
- Greenwald, JD; Shafritz, KM. An integrative neuroscience framework for the treatment of chronic pain: from cellular alterations to behavior. Front Integr Neurosci 2018, 12, 18. [Google Scholar] [CrossRef]
- Grillner, S. How circuits for habits are formed within the basal ganglia. Proc Natl Acad Sci U S A 2025, 122, e2423068122. [Google Scholar] [CrossRef]
- Grillner, S; Robertson, S; Hellgren Kotaleski, J. Basal ganglia – A motion perspective. Compr Physiol 2020, 10, 1241–1275. [Google Scholar] [CrossRef]
- Groenewegen, HJ. The basal ganglia and motor control. Neural Plast 2003, 10, 107–120. [Google Scholar] [CrossRef]
- Guo, D; Hu, J. Spinal presynaptic inhibition in pain control. Neurosci 2014, 283, 95–106. [Google Scholar] [CrossRef]
- Ha, SO; Kim, JK; Hong, HS; Kim, DS; Cho, HJ. Expression of brain-derived neurotrophic factor in rat dorsal root ganglia, spinal cord and gracile nuclei in experimental models of neuropathic pain. Neuroscience 2001, 107, 301–309. [Google Scholar] [CrossRef]
- Hayoun-Vigouroux, M; Misery, L. Dermatological conditions inducing acute and chronic pain. Acta Derm Venereol 2022, 102, 284. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, L; Mons, MR; Joosten, EA. A systematic review on descending serotonergic projections and modulation of spinal nociception in chronic neuropathic pain and after spinal cord stimulation. Mol Pain 2021, 17, 17448069211043965. [Google Scholar] [CrossRef] [PubMed]
- Heinricher, MM; Tavares, I; Leith, JL; Lumb, BM. Descending control of nociception: Specificity, recruitment and plasticity. Brain Res Rev 2008, 60, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Hellström, F; Roatta, S; Thunberg, J; Passatore, M; Djupsjöbacka, M. Responses of muscle spindles in feline dorsal neck muscles to electrical stimulation of the cervical sympathetic nerve. Exp Brain Res 2005, 165, 328–342. [Google Scholar] [CrossRef]
- Hibi, D; Takamoto, K; Iwama, Y; Ebina, S; Nishimaru, H; Matsumoto, J; Takamura, Y; Yamazaki, M; Nishijo, H. Impaired hemodynamic activity in the right dorsolateral prefrontal cortex is associated with impairment of placebo analgesia and clinical symptoms in postherpetic neuralgia. IBRO Rep 2020, 8, 56–64. [Google Scholar] [CrossRef]
- Hiraga, S-I; Itokazu, T; Nishibe, M; Yamashita, T. Neuroplasticity related to chronic pain and its modulation by microglia. Inflamm Regen 2022, 42, 15. [Google Scholar] [CrossRef]
- Hochman, S; Shreckengost, J; Kimura, H; Quevedo, J. Presynaptic inhibition of primary afferents by depolarization: observations supporting nontraditional mechanisms. Ann NY Acad Sci 2010, 1198, 140–152. [Google Scholar] [CrossRef]
- Hoffmann, S; Beyer, C. A fatal alliance between microglia, inflammasomes, and central pain. Int J Mol Sci 2020, 21, 3764. [Google Scholar] [CrossRef]
- Hoheisel, U; Mense, S; Simons, DG; Yu, XM. Appearance of new receptive fields in rat (DH) neurons following noxious stimulation of skeletal muscle: a model for referral of muscle pain? Neurosci Lett 1993, 153, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Holden, JE; Naleway, E. Microinjection of carbachol in the lateral hypothalamus produces opposing actions on nociception mediated by alpha(1)- and alpha(2)-adrenoceptors. Brain Res 2001, 911, 27–36. [Google Scholar] [CrossRef]
- Holt, MK. The ins and outs of the caudal nucleus of the solitary tract: An overview of cellular populations and anatomical connections. J Neuroendocrinol 2022, 34, e13132. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W-L; Chen, CPc; Nikkhoo, M; Lin, C-F; Ching, CT-S; Niu, C-C; Cheng, C-H. Fatigue changes neck muscle control and deteriorates postural stability during arm movement perturbations in patients with chronic neck pain. Spine J 2020, 20, 530–537. [Google Scholar] [CrossRef]
- Hu, H; Cui, Y; Yang, Y. Circuits and functions of the lateral habenula in health and in disease. Nat Rev Neurosci 2020, 21, 277–295. [Google Scholar] [CrossRef]
- Huang, J; Gadotti, VM; Chen, L; Souza, IA; Huang, S; Wang, D; Ramakrishnan, C; Deisseroth, K; Zhang, Z; Zamponi, GW. A neuronal circuit for activating descending modulation of neuropathic pain. Nat Neurosci 2019, 22, 1659–1668. [Google Scholar] [CrossRef]
- Hucho, T; Levine, JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron 2007, 55, 365–376. [Google Scholar] [CrossRef]
- Hughes, DI; Todd, AJ. Central nervous system targets: Inhibitory interneurons in the spinal cord. Neurotherapeutics 2020, 17, 874–885. [Google Scholar] [CrossRef]
- Hulsebosch, CE; Hains, BC; Crown, ED; Carlton, SM. Mechanisms of chronic central neuropathic pain aftre spinal cord injury. Brain Res Rev 2009, 60, 202–213. [Google Scholar] [CrossRef]
- Huma, Z; Ireland, K; Maxwell, DL. The spino-bulbar-cerebellar pathway: Activation of neurons projecting to the lateral reticular nucleus in the rat in response to noxious mechanical stimuli. Neurosci Lett 2015, 591, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Humphries, MD; Prescott, TJ. The ventral basal ganglia, a selection mechanism at the crossroads of space, strategy, and reward. Prog Neurobiol 2010, 90, 385–417. [Google Scholar] [CrossRef] [PubMed]
- Hunt, SP; Mantyh, PW. The molecular dynamics of pain control. Nat Rev Neurosci 2001, 2, 83–91. [Google Scholar] [CrossRef]
- Inoue, K. Nociceptive signaling of P2X receptors in chronic pain states. Purinergic Signalling 2021, 17, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K; Tsuda, M. Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat Rev Neurosci 2018, 19, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Jänig, W. Neurobiology of visceral pain. Schmerz in German. 2014, 28, 233–251. [Google Scholar] [CrossRef]
- Jaggi, AS; Singh, N. Role of different brain areas in peripheral nerve injury-induced neuropathic pain. Brain Res 2011, 381, 187–201. [Google Scholar] [CrossRef]
- Jankowska, E. Interneuronal relay in spinal pathways from proprioceptors. Prog Neurobiol 1992, 38, 335–378. [Google Scholar] [CrossRef]
- Jarvis, MF. The neural-glial purinergic receptor ensemble in chronic pain states. Trends Neurosci 2010, 33, 48–57. [Google Scholar] [CrossRef]
- Jean, A. The nucleus tractus solitarius: neuroanatomic, neurochemical and functional aspects. Arch Int Physiol Biochim Biophys in French. 1991, 99, A3–52. [Google Scholar]
- Jefferson, T; Kelly, CJ; Martina, M. Differential rearrangement of excitatory inputs to the medial prefrontal cortex in chronic pain models. Front Neural Circuits 2021, 15, 791043. [Google Scholar] [CrossRef]
- Ji, R-R; Donnelly, CR; Nedergaard, M. Astrocytes in chronic pain and itch. 2019, 20, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Jia, T; Wang, Y-D; Chen, J; Zhang, X; Cao, J-L; Xiao, C; Zhou, C. A nigro-subthalamo-parabrachial pathway modulates pain-like behaviors. Nat Commun 2022, 13, 7756. [Google Scholar] [CrossRef]
- Jiang, X; Wang, Y; Wan, R; Feng, B; Zhang, Z; Lin, Y; Wang, Y. The effect of high-definition transcranial direct current stimulation on pain processing in a healthy population: A single-blinded crossover controlled study. Neurosci Lett 2022, 767, 136304. [Google Scholar] [CrossRef]
- Joyce, MKP; Uchendu, S; Arnsten, AFT. Stress and inflammation target dorsolateral prefrontal cortex function: Neural mechanisms underlying weakened cognitive control. Biol Psychiatry 2025, 97, 359–371. [Google Scholar] [CrossRef]
- Julius, D; Basbaum, AI. Molecular mechanisms of nociception. Nature 2001, 413, 203–210. [Google Scholar] [CrossRef]
- Kahlaee, AH; Ghamkhar, L; Arab, AM. The association between neck pain and pulmonary function: A systematic review. Am J Phys Med Rehabil 2017, 96, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Kalezic, I; Bugaychenko, LA; Kostyukov, AI; PilyavskiiAI; Ljubisavljevid, M; Windhorst, U; Johansson, H. Fatigue-related depression of the feline monosynaptic gastrocnemius-soleus reflex. J Physiol 2004, 556, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Kamei, J; Ohsawa, M; Hayashi, S-S; Nakanishi, Y. Effect of chronic pain on morphine-induced respiratory depression in mice. Neuroscience 2011, 174, 224–233. [Google Scholar] [CrossRef]
- Kamibayashi, K; Nakazawa, K; Ogata, H; Obata, H; Akai, M; Shinohara, M. Invariable H-reflex and sustained facilitation of stretch reflex with heightened sympathetic outflow. J Electromyogr Kinesiol 2009, 19, 1053–1060. [Google Scholar] [CrossRef]
- Kanashiro, A; Hiroji Hiroki, C; Morais da Fonseca, D; Birbrair, A; Gomes Ferreira, R; Shimizu Bassi, G; Fonseca, MD; Kusuda, R; Martelossi Cebinelli, GC; Pinho da Silva, K; Wagner Wanderley, C; Menezes, GC; Alves-Fiho, JC; Oliveira, AG; Cunha, TM; Sampaio Pupo, A; Ulloa, L; Queiroz Cunha, F. The role of neutrophils in neuro-immune modulation. Pharmacol Res 2020, 151, 104580. [Google Scholar] [CrossRef]
- Kaushal, R; Taylor, BK; Jamal, AB; Zhang, L; Ma, F; Donahue, R; Westlund, KN. GABA-A receptor activity in the noradrenergic locus coeruleus drives trigeminal neuropathic pain in the rat; contribution of NAα1 receptors in the medial prefrontal cortex. Neuroscience 2016, 334, 148–159. [Google Scholar] [CrossRef]
- Kc, E; Moon, HC; Kim, S; Kim, HK; Won, SY; Hyun, S-H; Park, YS. Optical modulation on the nucleus accumbens core in the alleviation of neuropathica pain in chronic dorsal root ganglion compression rat model. Neuromodulation 2020, 23, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Kilburn-Watt, E; Banati, RB; Keay, KA. Altered thyroid hormones and behavioural change in a sub-population of rats following chronic constriction injury. J Neuroendocrinol 2010, 22, 960–970. [Google Scholar] [CrossRef]
- Kim, W; Angulo, MC. Unraveling the role of oligodendrocytes and myelin in pain. J Neurochem 2025, 169, e16206. [Google Scholar] [CrossRef]
- Kim, J-YV; Tillu, DV; Quinn, TL; Mejia, GL; Shy, A; Asiedu, MNK; Murad, E; Schumann, AP; Totsch, SK; Sorge, RE; Mantyn, PW; Dussor, G; Price, TJ. Spinal dopaminergic projections control the transition to pathological pain plasticity via a D1/D5-mediated mechanism. J Neurosci 2015, 35, 6307–6317. [Google Scholar] [CrossRef]
- Kiritoshi, T; Yakhnitsa, V; Singh, S; Wilson, TD; Chaudhry, S; Neugebauer, B; Torres-Rodriguez, JM; Lin, JL; Carrasquillo, Y; Neugebauer, V. Cells and circuits for amygdala neuroplasticity in the transition to chronic pain. Cell Rep 2024, 43, 114669. [Google Scholar] [CrossRef] [PubMed]
- Kissiwaa, SA; Bagley, EE. Central sensitization of the spino-parabrachial-amygdala pathway that outlasts a brief nociceptive stimulus. J Physiol 2018, 596, 4457–4473. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, L; Petersen, GL; Næsted NØrskov, K; Vase, L; Finnerup, N; Staehelin Jensen, T; Svensson, P. Review of neuroimaging studies related to pain modulation. Scand J Pain 2018, 2, 108–120. [Google Scholar] [CrossRef]
- Koerber, HR; Brown, PB. Quantitative analysis of (DH) cell receptive fields following limited deafferentation. J Neurophysiol 1995, 74, 2065–2076. [Google Scholar] [CrossRef]
- Khoutorsky, A; Price, TJ. Translational control mechanisms in persistemt pain. Trends Neurosci 2018, 41, 100–114. [Google Scholar] [CrossRef]
- Koutsikou, S; Apps, R; Lumb, BM. Top-down control of spinal sensorimotor circuits essential for survival. J Physiol (Lond) 2017, 595, 4151–4158. [Google Scholar] [CrossRef] [PubMed]
- Krause, T; Asseyer, S; Taskin, B; Flöel, A; Witte, AV; Mueller, K; Fiebach, JB; Villringer, K; Villringer, A; Jungehulsing, GJ. The cortical signature of central poststroke pain: Gray matter decreases in somatosensory, insular, and prefrontal Cortices. Cereb Cortex 2016, 26, 80–88. [Google Scholar] [CrossRef]
- Kummer, KK; Mitrić, M; Kalpachidou, T; Kress, M. The medial prefrontal cortex as a central hub for mental comorbidities associated with chronic pain. Int J Mol Sci 2020, 21, 3440. [Google Scholar] [CrossRef]
- Kuner, R; Flor, H. Structural plasticity and reorganisation in chronic pain. Nat Rev Neurosci 2016, 18, 20–30. [Google Scholar] [CrossRef]
- Kuner, R; Kuner, T. Cellular circuits in the brain and their modulation in acute and chronic pain. Physiol Rev 2021, 101, 213–258. [Google Scholar] [CrossRef]
- Kwiat, GC; Basbaum, AI. The origin of brainstem noradrenergic and serotonergic projections to the spinal cord dorsal horn in the rat. Somatosensory & motor research 1992, 9, 157–173. [Google Scholar]
- Labrakakis, C. The role of the insular cortex in pain. Int J Mol Sci 2023, 24, 5736. [Google Scholar] [CrossRef] [PubMed]
- Lançon, K; Séguéla, P. Dysregulated neuromodulation in the anterior cingulate cortex in chronic pain. Front Pharmacol 2023, 14, 128921. [Google Scholar] [CrossRef] [PubMed]
- Laurin, J; Pertici, V; Dousset, E; Marqueste, T; Decherchi, P. Group III and IV muscle afferents: role on central motor drive and clinical implications. Neuroscience 2015, 290, 543–551. [Google Scholar] [CrossRef]
- Lavenex, P; Amaral, DG. Hippocampal-neocortical interaction: a hierarchy of associativity. Hippocampus 2000, 10, 420–430. [Google Scholar] [CrossRef]
- Lee, C-H; Chen, C-C. Role of proprioceptors in chronic musculoskeletal pain. Exp Physiol 2023, 109, 45–54. [Google Scholar] [CrossRef]
- Lee, C-H; Lin, J-H; Lin, S-H; Chang, C-T; Wu, Y-W; Bewick, G; Banks, RW; Gründer, S; Hochgeschwender, U; Chen, C-C. A role for proprioceptors in sngception. Sci Adv 2025, 11, eabc5219. [Google Scholar] [CrossRef]
- Leite-Almeida, H; Valle-Fernandes, A; Almeida, A. Brain projections from the medullary dorsal reticular nucleus: an anterograde and retrograde tracing study in the rat. Neuroscience 2006, 140, 577–595. [Google Scholar] [CrossRef]
- Lesnak, J; Sluka, KA. Chronic non-inflammatory muscle pain: central and peripheral mediators. Curr Opin Physiol 2019, 11, 67–74. [Google Scholar] [CrossRef]
- Levinson, SR. Sodium channels. In Encyclopedia of neuroscience; Binder, MD, Hirokawa, N, Windhorst, U, Eds.; Springer-Verlag: Berlin Heidelberg, 2009; pp. pp 3759–3766. [Google Scholar]
- Lewin, GR; Lu, Y; Park, TJ. A plethora of painful molecules. Curr Opin Neurobiol 2004, 14, 443–449. [Google Scholar] [CrossRef]
- Li, J; Wei, Y; Zhou, J; Zou, H; Ma, L; Liu, C; Xiao, Z; Liu, X; Tan, X; Yu, T; Cao, S. Activation of locus coeruleus-spinal cord NA neurons alleviates neuropathic pain in mice via reducing neuroinflammation from astrocytes and microglia in spinal dorsal horn. J Neuroinflammation 2022, 19, 123. [Google Scholar] [CrossRef] [PubMed]
- Li, M; Yang, G. A mesocortical glutamatergic pathway modulates neuropathic pain independent of dopamine co-release. Nat Commun 2024, 15, 643. [Google Scholar] [CrossRef] [PubMed]
- Li, S; Cao, X; Li, Y; Tang, Y; Cheng, S; Zhang, D. Enhancing ventrolateral prefrontal cortex activation mitigates social pain and modifies subsequent social attitudes: Insights from TMS and fMRI. Neuroimage 2024, 292, 120620. [Google Scholar] [CrossRef]
- Li, T; Zhang, S; Kurata, J. Suppressed descending pain modulatory and enhanced sensorimotor networks in patients with chronic low back pain. J Anesth 2018, 32, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Li, X-H; Miao, H-H; Zhuo, M. NMDA receptor dependent long-term potentiation in chronic pain. Neurochem Res 2019, 44, 531–538. [Google Scholar] [CrossRef]
- Li, Y; Xu, Z; Xie, H; Fu, R; Lo, WLA; Cheng, X; Yang, J; Ge, L; Yu, Q; Wang, C. Changes in cortical activation during upright stance in individuals with chronic low back pain: An fNIRS study. Front Hum Neurosci 2023, 17, 1085831. [Google Scholar] [CrossRef]
- Liang, S-H; Zhao, W-J; Yin, J-B; Chen, Y-B; Li, J-N; Feng, B; Lu, Y-C; Wang, J; Dong, Y-L; Li, Y-Q. A neural circuit from thalamic paraventricular nucleus to central amygdala for the facilitation of neuropathic pain. J Neurosci 2020, 40, 7837–7854. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y; Luo, M; Xu, Q; Zhang, S; Niu, S; Li, X; Sun, W; Song, M; Wang, L; Xing, X; Wang, J; Feng, M; Zhao, L; Chen, H; Sheng, R; Wang, Y. Microglia-neuronal communication mediated by P2X4R-BDNF-TrkB promotes synaptic plasticity and anterior cingulate cortex hyperactivity in muscle pain chronicity. Br J Anaesth 2025, 135, 710–722. [Google Scholar] [CrossRef]
- Lima, D; Almeida, A. The medullary dorsal reticular nucleus as a pronociceptive centre of the pain control system. Prog Neurobiol 2002, 66, 81–108. [Google Scholar] [CrossRef] [PubMed]
- Lin, C-CJ; Chen, W-N; Chen, C-J; Lin, Y-W; Zimmer, A; Chen, C-C. An antinociceptive role for substance P in acid-induced chronic muscle pain. Proc Natl Acad Sci U S A 2012, 109, E76-83. [Google Scholar] [CrossRef]
- Lin, S-H; Cheng, Y-R; Banks, RW; Min, M-Y; Bewick, GS; Chen, C-C. Evidence for the involvement of ASIC3 in sensory mechanotransduction in proprioceptors. Nat Commun 2016, 7, 11460. [Google Scholar] [CrossRef]
- Liu, D; Hu, S-W; Wang, D; Zhang, Q; Zhang, X; Ding, H-L; Cao, J-L. An ascending excitatory circuit from the dorsal raphe for sensory modulation of pain. J Neurosci 2024, 44, e0869232023. [Google Scholar] [CrossRef]
- Liu, M; Li, Y; Zhong, J; Xia, L; Dou, N. The effect of IL-6/Piezo2 on the trigeminal neuropathic pain. Aging (Albany NY) 2021, 13, 13615–13625. [Google Scholar] [CrossRef]
- Liu, JA; Yu, J; Cheung, CW. Immune actions on the peripheral nervous system in pain. Int J Mol Sci 2021, 22, 1448. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, J; Minoshima, S; Casey, KL. Keeping pain out of mind: the role of the dorsolateral prefrontal cortex in pain modulation. Brain 2003, 126, 1079–1091. [Google Scholar] [CrossRef]
- Lu, C; Yang, T; Zhao, H; Zhang, M; Meng, F; Fu, H; Xie, Y; Xu, H. Insular cortex is critical for the perception, modulation, and chronification of pain. Neurosci Bull 2016, 32, 191–201. [Google Scholar] [CrossRef]
- Lund, JP; Sadeghi, S; Athanassiadis, T; Salas, NC; Auclair, F; Thivierge, B; Arsenault, I; Rompré, P; Westberg, K-G; Kolta, A. Assessment of the potential role of muscle spindle mechanoreceptor afferents in chronic muscle pain in the rat masseter muscle. PLoS One 2010, 5, e11131. [Google Scholar] [CrossRef]
- Luo, Z; Liao, X; Luo, L; Fan, Q; Zhang, X; Guo, Y; Wang, F; Ye, Z; Luo, D. Extracellular ATP and cAMP signaling promote Piezo2-dependent mechanical allodynia after trigeminal nerve compression injury. J Neurochem 2022, 160, 376–391. [Google Scholar] [CrossRef] [PubMed]
- Lyubashina, OA; Sivachenko, IB; Mikhalkin, AA. Impaired visceral pain-related functions of the midbrain periaqueductal gray in rats with colitis. Brain Res Bull 2022, 182, 12–25. [Google Scholar] [CrossRef]
- Lyubashina, OA; Sivachenko, IB; Sokolov, AY. Differential responses of neurons in the rat caudal ventrolateral medulla to visceral and somatic noxious stimuli and their alterations in colitis. Brain Res Bull 2019, 152, 299–310. [Google Scholar] [CrossRef]
- Macefield, VG; Sverrisdottir, YB; Wallin, BG. Resting discharge of human muscle spindles is not modulated by increases in sympathetic drive. J Physiol 2003, 551, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Madalena, KM; Lerch, JK. The effect of glucocorticoid and glucocorticoid receptor interactions on brain, spinal cord, and glial cell plasticity. Neural Plast 2017, 2017(2017), 8640970. [Google Scholar] [CrossRef]
- Maisky, VA; Pilyavskii, AI; Kalezic, I; Ljubisavljevic, M; Kostyukov, AI; Windhorst, U; Johansson, H. NADPH-diaphorase activity and c-fos expression in medullary neurons after fatiguing stimulation of hindlimb muscles in the rat. Auton Neurosci 2002, 101, 1–12. [Google Scholar] [CrossRef]
- Makovac, E; Venezia, A; Hohenschurz-Schmidt, D; Dipasquale, O; Jackson, JB; Medina, S; O´Daly, O; Williams, SCR; McMahon, SB; Howard, MA. The association between pain-induced autonomic reactivity and descending pain control is mediated by the periaqueductal grey. J Physiol 2021, 599, 5243–5260. [Google Scholar] [CrossRef]
- Maletic, V; Raison, CL. Neurobiology of depression, fibromyalgia and neuropathic pain. Front Biosci (Landmark Ed) 2009, 14, 5291–338. [Google Scholar] [CrossRef] [PubMed]
- Malfliet, A; Coppieters, I; Van Wilgen, P; Kregel, J; De Pauw, R; Dolphens, M; Ickmans, K. Brain changes associated with cognitive and emotional factors in chronic pain: A systematic review. Eur J Pain 2017, 21, 769–786. [Google Scholar] [CrossRef]
- Mansour, AR; Farmer, MA; Baliki, MN; Apkarian, AV. Chronic pain: The role of learning and brain plasticity. Restor Neurol Neurosci 2014, 32, 129–139. [Google Scholar] [CrossRef]
- Martin, WJ; Cao, Y; Basbaum, AI. Characterization of wide dynamic range neurons in the deep dorsal horn of the spinal cord in preprotachykinin-a null mice in vivo. J Neurophysiol 2004, 91, 1945–1954. [Google Scholar] [CrossRef]
- Martins, I; Carvalho, P; de Vries, MG; Teixeira-Pinto, A; Wilson, SP; Westerink, BHC; Tavares, I. Increased noradrenergic neurotransmission to a pain facilitatory area of the brain is implicated in facilitation of chronic pain. Anesthesiology 2015, 123, 642–653. [Google Scholar] [CrossRef]
- Martins, I; Tavares, I. Reticular formation and pain: The past and the future. Front Neuroanat 2017, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Mathie, A; Veale, EL. Neuronal potassium channels. In Encyclopedia of neuroscience; Binder, MD, Hirokawa, N, Windhorst, U, Eds.; Springer-Verlag: Berlin Heidelberg, 2009; pp. pp 2792–2797. [Google Scholar]
- Mazzitelli, M; Yakhnitsa, V; Neugebauer, B; Neugebauer, V. Optogenetic manipulations of CeA-CRF neurons modulate pain- and anxiety-like behaviors in neuropathic pain and control rats. Neuropharmacology 2022, 210, 109031. [Google Scholar] [CrossRef] [PubMed]
- McCarberg, B; Peppin, J. Pain pathways and nervous system plasticity: Learning and memory in pain. Pain Medv 2019, 20, 2421–2437. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, DI; Prochazka, A. The role of sensory information in the guidance of voluntary movement. Somatosens Mot Res 1994, 11, 21–37. [Google Scholar] [CrossRef]
- McEwen, BS; Nasca, C; Gray, JD. Stress effects on neuronal structure: hippocampus, amygdala, and prefrontal cortex. Neuropsychopharmacology 2016, 41, 3–23. [Google Scholar] [CrossRef]
- McLachlan, EM; Jänig, W.; Devor, M; Michaelis, M. Peripheral nerve injury triggers noradrenergic sprouting within dorsal root ganglia. Nature 1993, 363, 543–546. [Google Scholar] [CrossRef]
- McMahon, SB; La Russa, F; Bennett, DLH. Crosstalk between the nociceptive and immune systems in host defence and disease. Nat Rev Neurosci 2015, 16, 389–402. [Google Scholar] [CrossRef]
- McMahon, SB; Wall, PD. Receptive fields of rat lamina 1 projection cells move to incorporate a nearby region of injury. Pain 1984, 19, 235–247. [Google Scholar] [CrossRef]
- Meeker, TJ; Schmid, A-C; Keaser, ML; Khan, SA; Gullapalli, RP; Dorsey, SG; Greenspan, JD; Seminowicz, DA. Tonic pain alters functional connectivity of the descending pain modulatory network involving amygdala, periaqueductal gray, parabrachial nucleus and anterior cingulate cortex. Neuroimage 2022, 256, 1192783. [Google Scholar] [CrossRef] [PubMed]
- Mense, S. What is different about muscle pain? Schmerz Article in German. 2003, 17, 459–463. [Google Scholar] [CrossRef]
- Mense, S. Mechanisms of transition from acute to chronic muscle pain. Orthopade Article in German. 2004, 33, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Mense, S. Muscle pain: mechanisms and clinical significance. Dtsch Ärztebl Int. 2008, 105, 214–219. [Google Scholar] [CrossRef]
- Merighi, A. Brain-derived neurotrophic factor, nociception, and pain. Biomolecules 2024, 14, 539. [Google Scholar] [CrossRef] [PubMed]
- Metz, M; Grundmann, S; Ständer, S. Pruritus: an overview of current concepts. Vet Dermatol 2011, 22, 121–131. [Google Scholar] [CrossRef]
- Metzger, M; Souza, R; Lima, LB; Bueno, D; Gonçalves, L; Sego, C; Donato, Jr J; Shammah-Lagnado, SL. Habenular connections with the dopaminergic and serotonergic system and their role in stress-related psychiatric disorders. Eur J Neurosci 2021, 53, 65–88. [Google Scholar] [CrossRef]
- Meylakh, N; Crawford, LS; Mills, EP; Macefield, VG; Vickers, ER; Macey, PM; Keay, KA; Henderson, LA. Altered corticobrainstem connectivity during spontaneous fluctuations in pain intensity in painful trigeminal neuropathy. eNeuro 2024, 11, ENEURO.0522–23. [Google Scholar] [CrossRef]
- Michaelis, M; Devor, M; Jänig, W. Sympathetic modulation of activity in rat dorsal root ganglion neurons changes over time following peripheral nerve injury. J Neurophysiol 1996, 76, 753–763. [Google Scholar] [CrossRef]
- Millan, MJ. The induction of pain: an integrative review. Prog Neurobiol 1999, 57, 1–164. [Google Scholar] [CrossRef] [PubMed]
- Mills, EP; Keay, KA; Henderson, LA. Brainstem pain-modulation circuitry and its plasticity in neuropathic pain: Insights from human brain imaging investigations. Front Pain Res (Lausanne) 2021, 2, 705345. [Google Scholar] [CrossRef] [PubMed]
- Mitsi, V; Zachariou, V. Modulation of pain, nociception, and analgesia by the brain reward center. Neuroscience 2016, 338, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Moalem, G; Tracey, DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev 2006, 51, 240–264. [Google Scholar] [CrossRef]
- Moon, HC; Park, YS. Optogenetic stimulation of the anterior cingulate cortex modulates the pain processing in neuropathic pain: A review. J Mol Neurosci 2022, 72, 1–8. [Google Scholar] [CrossRef]
- Moore, KA; Baba, H; Woolf, CJ. Synaptic transmission and plasticity in the superficial dorsal horn. Prog Brain Res 2000, 129, 63–80. [Google Scholar]
- Mutso, AA; Radzicki, D; Baliki, MN; Huang, L; Banisadr, G; Centeno, MV; Radulovic, J; Martina, M; Miller, RJ; Apkarian, AV. Abnormalities in hippocampal functioning with persistent pain. J Neurosci 2012, 32, 5747–5756. [Google Scholar] [CrossRef]
- Nadrigny, F; Le Meur, K; Schomburg, ED; Safavi-Abbasi, S; Dibaj, P. Two-photon laser-scanning microscopy for single and repetitive imaging of dorsal and lateral spinal white matter in vivo. Physiol Res 2017, 66, 531–537. [Google Scholar] [CrossRef]
- Nagai, M; Kishi, K; Kato, S. Insular cortex and neuropsychiatric disorders: a review of recent literature. Eur Psychiatry 2007, 22, 387–394. [Google Scholar] [CrossRef]
- Nakamura, SI; Myers, RR. Injury to dorsal root ganglia alters innervation of spinal cord dorsal horn lamina involved in nociception. Spine (Phila Pa 1976) 2000, 25, 537–542. [Google Scholar] [CrossRef]
- Naser, PV; Kuner, R. Molecular, cellular and circuit basis of cholinergic modulation of pain. Neuroscience 2018, 387, 135–148. [Google Scholar] [CrossRef]
- Nelissen, S; Lemmens, E; Geurts, N; Kramer, P; Maurer, M; Hendriks, J; Hendrix, S. The role of mast cells in neuroinflammation. Acta Neuropathol 2013, 125, 637–650. [Google Scholar] [CrossRef]
- Neugebauer, V. Amygdala pain mechanisms. Handb Exp Pharmacol 2015, 227, 261–284. [Google Scholar] [PubMed]
- Neugebauer, V; Galhardo, V; Maione, S; Mackey, SC. Forebrain pain mechanisms. Brain Res Rev 2009, 60, 226–242. [Google Scholar] [CrossRef]
- Neugebauer, V; Li, W; Bird, GC; Han, JS. The amygdala and persistent pain. Neuroscientist 2004, 10, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, V; Mazzitelli, M; Cragg, B; Ji, G; Navratilova, E; Porreca, F. Amygdala, neuropeptides, and chronic pain-related affective behaviors. Neuropharmacology 2020, 170, 108052. [Google Scholar] [CrossRef]
- Neugebauer, V; Presto, P; Yakhnitsa, V; Antenucci, N; Mendoza, B; Ji, G. Pain-related cortico-limbic plasticity and opioid signaling. Neuropharmacology 2023, 231, 109510. [Google Scholar] [CrossRef] [PubMed]
- Neziri, AY; Haesler, S; Petersen-Felix, S; Müller, M; Arendt-Nielsen, L; Biurrun Manresa, J; Andersen, OK; Curatolo, M. Generalized expansion of nociceptive reflex receptive fields in chronic pain patients. Pain 2010, 151, 798–805. [Google Scholar] [CrossRef]
- Nicol, GD; Vasko, MR. Unraveling the story of NGF-mediated sensitization of nociceptive sensory neurons: ON or OFF the Trks? Mol Interv 2007, 7, 26–41. [Google Scholar] [CrossRef]
- Nieuwenhuys, R; Voogd, J; van Huijzen, C. The human central nervous system. A synopsis and atlas; Springer: Berlin Heidelberg New York, 1978. [Google Scholar]
- Oane, I; Barborica, A; Mindruta, IR. Cingulate cortex: anatomy, structural and functional connectivity. J Clin Neurophysiol 2023, 40, 482–490. [Google Scholar] [CrossRef]
- Ogawa, SK; Watabe-Uchida, M. Organization of dopamine and serotonin system: Anatomical and functional mapping of monosynaptic inputs using rabies virus. Pharmacol Biochem Behav 2018, 174, 9–22. [Google Scholar] [CrossRef]
- O´Hearn, E; Molliver, ME. Organization of raphe-cortical projections in rat: a quantitative retrograde study. Brain Res Bull 1984, 3, 709–726. [Google Scholar] [CrossRef] [PubMed]
- Okada, S; Katagiri, A; Saito, H; Lee, J; Ohara, K; Iinuma, T; Iwata, K. Functional involvement of nucleus tractus solitarii neurons projecting to the parabrachial nucleus in trigeminal neuropathic pain. J Oral Sci 2019, 61, 370–378. [Google Scholar] [CrossRef]
- Oliver, KM; Florez-Paz, DM; Badea, TC; Mentis, GZ; Menon, V; de Nooij, JC. Molecular correlates of muscle spindle and Golgi tendon organ afferents. Nat Commun 2021, 12, 1451. [Google Scholar] [CrossRef]
- Ong, WY; Stohler, CS; Herr, DR. Role of the prefrontal cortex in pain processing. Mol Neurobiol 2019, 56, 1137–1166. [Google Scholar] [CrossRef]
- Ossipov, MH; Morimura, K; Porreca, F. Descending pain modulation and chronification of pain. Curr Opin Support Palliat Care 2014, 8, 143–151. [Google Scholar] [CrossRef] [PubMed]
- O´Sullivan, P. Diagnosis and classification of chronic low back pain disorders: maladaptive movement and motor control impairments as underlying mechanism. Man Ther 2005, 10, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Pagliusi, M; Gomes, FV. The role of the rostral ventromedial medulla in stress responses. Brain Sci 2023, 13, 776. [Google Scholar] [CrossRef]
- Palmiter, RD. Parabrachial neurons promote nociplastic pain. Trends Neurosci S0166-2236 2024, 24)00127–9. [Google Scholar] [CrossRef]
- Panneton, WM; Gan, Q; Ariel, M. Injections of algesic solutions into muscle activate the lateral reticular formation: A nociceptive relay of the spinoreticulothalamic tract. PLoS One 2015, 10, e0130939. [Google Scholar] [CrossRef] [PubMed]
- Passatore, M; Roatta, S. Autonomic control of sensory receptors. In Encyclopedia of neuroscience; Binder, MD, Hirokawa, N, Windhorst, U, Eds.; Springer-Verlag: Berlin Heidelberg, 2009; pp. pp 245–250. [Google Scholar]
- Patel, JM; Dao, H. Chronic pruritus: A review of meurophysiology and associated immune neuromodulatory treatments. Skin Therapy Lett 2018, 23, 5–9. [Google Scholar]
- Patel, R; Dickenson, AH. A study of cortical and brainstem mechanisms of diffuse noxious inhibitory controls in anaesthetised normal and neuropathic rats. Eur J Neurosci 2020, 51, 952–962. [Google Scholar] [CrossRef]
- Pautrat, A; Rolland, M; Barthelemy, M; Baunez, C; Sinniger, V; Piallat, B; Savasta, M; Overton, PG; David, O; Coizet, V. Revealing a novel nociceptive network that links the subthalamic nucleus to pain processing. Elife 2018, 7, e36607. [Google Scholar] [CrossRef]
- Peirs, C; Williams, S-PG; Zhao, X; Walsh, CE; Gedeon, JY; Cagle, NE; Goldring, AC; Hioki, H; Liu, Z; Marell, PS; Seal, RP. Dorsal horn circuits for persistent mechanical pain. Neuron 2015, 87, 797–812. [Google Scholar] [CrossRef]
- Peng, B; Jiao, Y; Zhang, Y; Li, S; Chen, S; Xu, S; Gao, P; Fan, Y; Yu, W. Bulbospinal nociceptive ON and OFF cells related neural circuits and transmitters. Front Pharmacol 2023, 14, 1159753. [Google Scholar] [CrossRef]
- Pereira, AR; Alemi, M; Cerqueira-Nunes, M; Monteiro, C; Galhardo, V; Cardoso-Cruz, H. Dynamics of lateral habenula-ventral tegmental area microcircuit on pain-related cognitive dysfunctions. Neurol Int 2023, 15, 1303–1319. [Google Scholar] [CrossRef] [PubMed]
- Pertovaara, A. NA pain modulation. Prog Neurobiol 2006, 80, 53–83. [Google Scholar] [CrossRef]
- Pertovaara, A. The NA pain regulation system: a potential target for pain therapy. Eur J Pharmacol 2013, 716, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Peyron, R; Quesada, C; Fauchon, C. Cingulate-mediated approaches to treating chronic pain. Handb Clin Neurol 2019, 166, 317–326. [Google Scholar]
- Pezet, S; McMahon, SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci 2006, 29, 507–538. [Google Scholar] [CrossRef] [PubMed]
- Pilyavskii, AI; Maznychenko, AV; Maisky, VA; Kostyukov, AI; Hellström, F; Windhorst, U. Capsaicin-induced effects on c-fos expression and NADPH-diaphorase activity in the feline spinal cord. Eur J Pharmacol 2005, 521, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Pinho-Ribeiro, FA; Verri, WA, Jr.; Chiu, IM; Chiu, M. Nociceptor sensory neuron-immune interactions in pain and inflammation. Trends Neuroimmunol 2017, 38, 5–19. [Google Scholar] [CrossRef]
- Pinto, M; Lima, D; Tavares, I. Neuronal activation at the spinal cord and medullary pain control centers after joint stimulation: a c-fos study in acute and chronic articular inflammation. Neuroscience 2007, 147, 1076–1089. [Google Scholar] [CrossRef]
- Pinto-Ribeiro, F; Ansah, OB; Almeida, A; Pertovaara, A. Response properties of nociceptive neurons in the caudal ventrolateral medulla (CVLM) in monoarthritic and healthy control rats: modulation of responses by the paraventricular nucleus of the hypothalamus (PVN). Brain Res Bull 2011, 86, 82–90. [Google Scholar] [CrossRef]
- Poe, GR; Foote, S; Eschenko, O; Johansen, JP; Bouret, S; Aston-Jones, G; Harley, CW; Manahan-Vaughan, D; Weinshenker, D; Valentino, R; Berridge, C; Chandler, DJ; Waterhouse, B; Sara, SJ. Locus coeruleus: a new look at the blue spot. Nat Rev Neurosci 2020, 21, 644–659. [Google Scholar] [CrossRef]
- Porreca, F; Ossipov, MH; Gebhart, GF. Chronic pain and medullary descending facilitation. Trends Neurosci 2002, 25, 319–325. [Google Scholar] [CrossRef]
- Prescott, SA. Synaptic inhibition and disinhibition in the spinal dorsal horn. Prog Mol Biol Transl Sci 2015, 131, 359–383. [Google Scholar] [PubMed]
- Price, TJ; Cervero, F; Gold, MS; Hammond, DL; Prescott, SA. Chloride regulation in the pain pathway. Brain Res Rev 2009, 60, 149–170. [Google Scholar] [CrossRef]
- Price, TJ; Inyang, KE. Commonalities between pain and memory mechanisms and their meaning for understanding chronic pain. Prog Mol Biol Transl Sci 2015, 131, 409–434. [Google Scholar] [PubMed]
- Price, TJ; Ray, PR. Recent advances toward understanding the mysteries of the acute to chronic pain transition. Curr Opin Physiol 2019, 11, 42–50. [Google Scholar] [CrossRef]
- Proske, U; Gandevia, SC. Kinesthetic Senses. Compr Physiol 2018, 8, 1157–1183. [Google Scholar] [CrossRef]
- Quevedo, JN. Presynaptic inhibition. In Encyclopedia of neuroscience; Binder, MD, Hirokawa, N, Windhorst, U, Eds.; Springer-Verlag: Berlin Heidelberg, 2009; pp. pp 3266–3270. [Google Scholar]
- Radovanovic, D; Peikert, K; Lindström, M; Domelöff, FP. Sympathetic innervation of human muscle spindles. J Anat 2015, 226, 542–548. [Google Scholar] [CrossRef]
- Ramer, MS; Bisby, MA. Adrenergic innervation of rat sensory ganglia following proximal or distal painful sciatic neuropathy: distinct mechanisms revealed by anti-NGF treatment. Eur J Neurosci 1999, 11, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Ramer, MS; Thompson, SWN; McMahon, SB. Causes and consequences of sympathetic basket formation in dorsal root ganglia. Pain Suppl 1999, 82, S111–S120. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Jarquín, UN; Lazo-Gómez, R; Tovar-Y-Romo, LB; Tapia, R. Spinal inhibitory circuits and their role in motor neuron degeneration. Neuropharmacology 2014, 82, 101–107. [Google Scholar] [CrossRef]
- Raver, C; Uddin, O; Ji, Y; Li, Y; Cramer, N; Jenne, C; Morales, M; Masri, R; Keller, A. An amygdalo-parabrachial pathway regulates pain perception and chronic pain. J Neurosci 2020, 40, 3424–3442. [Google Scholar] [CrossRef]
- Ren, K; Dubner, R. Pain facilitation and activity-dependent plasticity in pain modulatory circuitry: role of BNDF-TrkB signaling and NMDA receptors. Mol Neurobiol 2007, 35, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Ren, K; Dubner, R. Interactions between the immune and nervous systems in pain. Nat Med 2010, 16, 1267–1276. [Google Scholar] [CrossRef]
- Ren, K; Torres, R. Role of interleukin-1 during pain and inflammation. Brain Res Rev 2009, 60, 57–64. [Google Scholar] [CrossRef]
- Roatta, S; Windhorst, U; Ljubisavljevic, M; Passatore, M. Sympathetic modulation of muscle spindle afferent sensitivity to stretch in rabbit jaw closing muscles. J Physiol 2002, 540, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Roatta, S; Windhorst, U; Djupsjöbacka, M; Lytvynenko, S; Passatore, M. Effects of sympathetic stimulation on the rhythmical jaw movements produced by electrical stimulation of the cortical masticatory areas of rabbits. Exp Brain Res 2005, 162, 14–22. [Google Scholar] [CrossRef]
- Rocha, I; González-García, M; Carillo-Franco, L; Dawid-Milner, MS; López-González, MV. Influence of brainstem’s area A5 on sympathetic outflow and cardiorespiratory dynamics. Biology (Basel) 2024, 13, 161. [Google Scholar] [CrossRef]
- Roeckel, L-A; Le Coz, G-M; Gavériaux-Ruff, C; Simonin, Fréderic. Opioid-induced hyperalgesia: cellular and molecular mechanisms. Neuroscience 2016, 338, 160–182. [Google Scholar] [CrossRef]
- Rogers, M; Tang, L; Madge, DJ; Stevens, EB. The role of sodium channels in neuropathic pain. Sem Cell&Devel Biol 2006, 17, 571–581. [Google Scholar]
- Romero-Grimaldi, C; Berrocoso, E; Alba-Delgado, C; Madrigal, JL; Perez-Nievas, BG; Leza, JC; Mico, JA. Stress increases the negative effects of chronic pain on hippocampal neurogenesis. Anesth Analg 2015, 121, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Roza, C; Bernal, L. Electrophysiological characterization of ectopic spontaneous discharge in axotomized and intact fibers upon nerve transection: a role in spontaneous pain? Pflugers Arch 2022, 474, 387–396. [Google Scholar] [CrossRef]
- Rudomin, P. In search of lost presynaptic inhibition. Exp Brain Res 2009, 196, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Rudomin, P; Schmidt, RF. Presynaptic inhibition in the vertebrate spinal cord revisited. Exp Brain Res 1999, 129, 1–37. [Google Scholar] [CrossRef]
- Rygh, LJ; Suzuki, R; Rahman, W; Wong, Y; Vonsy, JL; Sandhu, H; Webber, M; Hunt, S; Dickenson, AH. Local and descending circuits regulate long-term potentiation and zif268 expression in spinal neurons. Eur J Neurosci 2006, 24, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Saab, CY. Pain-related changes in the brain: diagnostic and therapeutic potentials. Trends Neurosci 2012, 35, 629–637. [Google Scholar] [CrossRef]
- Saab, CY; Hains, BC. Remote neuroimmune signaling: a long-range mechanism of nociceptive network plasticity. Trends Neurosci 2009, 32, 110–117. [Google Scholar] [CrossRef]
- Saab, CY; Waxman, SG; Hains, BC. Alarm or curse? The pain of neuroinflammation. Brain Res Rev 2008, 58, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Saadé, NE; Jabbur, SJ. Nociceptive behavior in animal models for peripheral neuropathy: Spinal and supraspinal mechanisms. Prog Neurobiol 2008, 86, 22–47. [Google Scholar] [CrossRef] [PubMed]
- Safavi-Abbasi, S; Venezia, E; Sughrue, M; Dibaj, P. Effectiveness of mindfulness mediationon the quality of life, pain intensity, mobility and physical function in adults with chronic low back pain: a systemic review and meta-analysis. Discov Psychol 2025, 5, 183. [Google Scholar] [CrossRef]
- Salgado, S; Kaplitt, MG. The nucleus accumbens: a comprehensive review. Stereotact Funct Neurosurg 2015, 93, 75–93. [Google Scholar] [CrossRef]
- Salomons, TV; Johnstone, T; Backonja, M-M; Shackman, AJ; Davidson, RJ. Individual differences in the effects of perceived controllability on pain perception: critical role of the prefrontal cortex. J Cogn Neurosci 2007, 19, 993–1003. [Google Scholar] [CrossRef]
- Saman, Y; Arshad, Q; Dutia, M; Rea, P. Stress and the vestibular system. Int Rev Neurobiol 2020, 152, 221–236. [Google Scholar]
- Sanderson, A; Wang, SF; Elgueta-Cancino, E; Martinez-Valdes, E; Sanchis-Sanchez, E; Liew, B; Falla, D. The effect of experimental and clinical musculoskeletal pain on spinal and supraspinal projections to motoneurons and motor unit properties in humans: A systematic review. Eur J Pain 2021, 25, 1668–1701. [Google Scholar] [CrossRef] [PubMed]
- Sandkühler, J. Models and mechanisms of hyperalgesia and allodynia. Physiol Rev 2009, 89, 707–758. [Google Scholar] [CrossRef]
- Sarantopoulos, C; McCallum, B; Sapunar, D; Kwok, W-M; Hogan, Q. ATP-sensitive potassium channels in rat primary afferent neurons: the effect of neuropathic injury and gabapentin. Neurosci Lett 2003, 343, 185–189. [Google Scholar] [CrossRef]
- Sas, D; Gaudel, F; Verdier, D; Kolta, A. Hyperexcitability of muscle spindle afferents in jaw-closing muscles in experimental myalgia: Evidence for large primary afferents involvement in chronic pain. Exp Physiol 2024, 109, 100–111. [Google Scholar] [CrossRef]
- Schmidtko, A. Nitric oxide-mediated pain processing in the spinal cord. Handb Exp Pharmacol 2015, 227, 103–117. [Google Scholar]
- Schmidtko, A; Tegeder, I; Geisslinger, G. No NO, no pain? The role of nitric oxide and cGMP in spinal pain processing. Trends Neurosci 2009, 32, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J; Woolf, CJ. Can we conquer pain? Nature Neurosci Suppl 2002, 5, 1062–1067. [Google Scholar] [CrossRef]
- Scholz, J; Woolf, CJ. The neuropathic pain triad: neurons, immune cells and glia. Nature Neurosci 2007, 10, 1361–1368. [Google Scholar] [CrossRef]
- Schomburg, ED; Dibaj, P; Steffens, H. Differentiation between Aδ and C fibre evoked nociceptive reflexes by TTX resistance and opioid sensitivity in the cat. Neurosci Res 2011, 69, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, ED; Dibaj, P; Steffens, H. Role of L-DOPA in spinal nociceptive reflex activity: higher sensitivity of Aδ versus C fibre-evoked nociceptive reflexes to L-DOPA. Physiol Res 2011, 60, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, ED; Kalezic, I; Dibaj, P; Steffens, H. Reflex transmission to lumbar α-motoneurones in the mouse similar and different to those in the cat. Neurosci Res 2013, 76, 133–140. [Google Scholar] [CrossRef]
- Schomburg, ED; Steffens, H; Dibaj, P; Sears, TA. Major contribution of Aδ-fibres to increased reflex transmission in the feline spinal cord during acute muscle inflammation. Neurosci Res 2012, 72, 155–162. [Google Scholar] [CrossRef]
- Schomburg, ED; Steffens, H; Pilyavskii, AI; Maisky, VA; Brück, W; Dibaj, P; Sears, TA. Long lasting activity of nociceptive muscular afferents facilitates bilateral flexion pattern in the feline spinal cord. Neurosci Res 2015, 95, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Seminowicz, DA; Moayedi, M. The dorsolateral prefrontal cortex in acute and chronic pain. Pain 2017, 18, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Seno, MDJ; Assis, DV; Gouveia, F; Antunes, GF; Kuroki, M; Oliveira, CC; Santos, LCT; Pagano, RL; Martinez, RCR. The critical role of amygdala subnuclei in nociceptive and depressive-like behaviors in peripheral neuropathy. Sci Rep 2018, 8, 13608. [Google Scholar] [CrossRef]
- Serafini, RA; Pryce, KD; Zachariou, V. The mesolimbic dopamine system in chronic pain and associated affective comorbidities. Biol Psychiatry 2020, 87, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Shelton, L; Becerra, L; Borsook, D. Unmasking the mysteries of the habenula in pain and analgesia. Prog Neurobiol 2012, 96, 208–219. [Google Scholar] [CrossRef]
- Shoraka, O; Syed, M; Mandloi, S; Thalheimer, S; Naghizadeh Kashani, S; Heller, JE; Mohamed, FB; Sharan, AD; Talekan, KS; Matias, CM; HArrop, JS; Krisa, L; Alizadeh, M. Periaquaeductal gray connectivity in spinal cord injury-induced neuropathic pain. J Neuroimaging 2024, 34, 704–719. [Google Scholar] [CrossRef]
- Shortland, P; Kinman, E.; Molander, C. Sprouting of A-fibre primary afferents into lamina II in two rat models of neuropathic pain. Eur J Pain 1997, 1, 215–227. [Google Scholar] [CrossRef]
- Sluka, KA; Clauw, DJ. Neurobiology of fibromyalgia and chronic widespread pain. Neuroscience 2016, 338, 114–129. [Google Scholar] [CrossRef]
- Sluka, KA; Danielson, J; Rasmussen, L; DaSilva, LF. Exercise-induced pain requires NMDA receptor activation in the medullary raphe nuclei. Med Sci Sports Exerc 2012, 44, 420–427. [Google Scholar] [CrossRef]
- Sluka, KA; Gregory, NS. The dichotomized role for acid sensing ion channels in musculoskeletal pain and inflammation. Neuropharmacology 2015, 94, 58–63. [Google Scholar] [CrossRef]
- Sluka, KA; Winter, OC; Wemmie, JA. Acid-sensing ion channels: A new target for pain and CNS diseases. Curr Opin Drug Discov Devel 2009, 12, 693–704. [Google Scholar]
- Smith, LJ; Macefield, VG; Birznieks, I; Burton, AR. Effects of tonic muscle pain on fusimotor control of human muscle spindles during isometric ankle dorsiflexion. J Neurophysiol 2019, 121, 1143–1149. [Google Scholar] [CrossRef]
- Solodkin, A; Traub, RJ; Gebhart, GF. Unilateral hindpaw inflammation produces a bilateral increase in NADPH-diaphorase histochemical staining in the rat lumbar spinal cord. Neuroscience 1992, 51, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Sosa, MKS; Boorman, DC; Keay, KA. The impact of sciatic nerve injury and social interactions testing on glucocorticoid receptor expression in catecholaminergic medullary cell populations. Brain Res 2023, 1819, 148542. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, ML; Valente, M; Storchi, R; Caramenti, G; Biella, GEM. Contribution by DRt descending facilitatory pathways to maintenance of spinal neuron sensitization in rats. Brain Res 2008, 1188, 69–75. [Google Scholar] [CrossRef]
- Spooner, M-F; Robichaud, P; Carrier, JC; Marchand, S. Endogenous pain modulation during the formalin test in estrogen receptor beta knockout mice. Neuroscience 2007, 150, 675–680. [Google Scholar] [CrossRef]
- Staud, R. Abnormal endogenous pain modulation is a shared characteristic of many chronic pain conditions. Exp Rev Neurother 2012, 12, 577–585. [Google Scholar] [CrossRef]
- Stein, C; Clark, JD; Oh, U; Vasko, MR; Wilcox, GL; Overland, AC; Vanderah, TW; Spencer, RH. Peripheral mechanisms of pain and analgesia. Brain Res Rev 2009, 60, 90–113. [Google Scholar] [CrossRef] [PubMed]
- Stucky, CL; Mikesell, AR. Cutaneous pain in disorders affecting peripheral nerves. Neurosci Lett 2021, 765, 136233. [Google Scholar] [CrossRef]
- Suárez-Pereira, I; Llorca-Torralba, M; Bravo, L; Camarena-Delgado, C; Soriano-Mas, C; Berroscoso, E. The role of the locus coeruleus in pain and associated stress-related disorders. Biol Psychiatry 2022, 91, 786–797. [Google Scholar] [CrossRef]
- Sun, H; Li, Z; Qiu, Z; Shen, Y; Guo, Q; Hu, S-W; Ding, H-W; An, S; Cao, J-L. A common neuronal ensemble in nucleus accumbens regulates pain-like behaviour and sleep. Nat Commun 2023, 14, 4700. [Google Scholar] [CrossRef]
- Sun, L; Liu, R; Guo, F; Wen, M-Q; Ma, X-L; Li, K-Y; Sun, H; Xu, C-L; Li, Y-Y; Wu, M-Y; Zhu, Z-G; Li, X-J; Yu, Y-Q; Chen, Z; Li, X-Y; Duan, S. Parabrachial nucleus circuit governs neuropathic pain-like behavior. Nat Commun 2020, 11, 5974. [Google Scholar] [CrossRef]
- Sun, Z-C; Ma, S-B; Chu, W-G; Jia, D; Luo, C. Canonical transient receptor potential (TRPC) channels in nociception and pathological pain. Neural Plast 2020, 3764193. [Google Scholar] [CrossRef] [PubMed]
- Sunavsky, A; Hashmi, MA; Robertson, JW; Veinot, J; Hashmi, JA. The nucleus accumbens-prefrontal connectivity as a predictor of chronic low back pain. Pain 2025. [Google Scholar] [CrossRef]
- Svendsen, F; Tjølsen, A; Hole, K. LTP of spinal A and C-fibre evoked responses after electrical sciatic nerve stimulation. NeuroReport 1997, 8, 3427–3430. [Google Scholar] [CrossRef]
- Szok, D; Tajti, J; Nyári, A; Véscei, L. Therapeutic approaches for peripheral and central neuropathic pain. Behav Neurol 2019, 2019, 8685954. [Google Scholar] [CrossRef]
- Takayanagi, Y; Onaka, T. Roles of oxytocin in stress responses, allostasis and resilience. Int J Mol Sci 2021, 23, 150. [Google Scholar] [CrossRef]
- Talavera, K; Startek, JB; Alvarez-Collazo, J; Boonen, B; Alpizar, YA; Sanchez, A; Naert, R; Nilius, B. Mammalian transient receptor potential TRPA1 channels: From structure to disease. Physiol Rev 2020, 100, 725–803. [Google Scholar] [CrossRef] [PubMed]
- Tam, TH; Salter, MW. Purinergic signalling in spinal pain processing. Purinergic Signal 2021, 17, 49-54, 49-543. [Google Scholar] [CrossRef]
- Tanabe, M; Shimizu, S; Takabayashi, K; Honda, M; Ono, H. Functional alteration of inhibitory influences on spinal motor output in painful diabetic neuropathy in rats. Neurosci Lett 2005, 389, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q-Q; Wu, Y; Tao, Q; Shen, Y; An, X; Liu, D; Xu, Z. Direct paraventricular thalamus-basolateral amygdala circuit modulates neuropathic pain and emotional anxiety. Neuropsychopharmacology 2024, 49, 455–466. [Google Scholar] [CrossRef]
- Tang, Y; Wang, M; Zheng, T; Xiao, Y; Wang, S; Han, F; Chen, G. Structural and functional brain abnormalities in postherpetic neuralgia: A systematic review of neuroimaging studies. Brain Res 2021, 1752, 147219. [Google Scholar] [CrossRef]
- Tashima, R; Koga, K; Yoshikawa, Y; Sekine, M; Watanabe, M; Tozaki-Saitoh, H; Furue, H; Yasaka, T; Tsuda, M. A subset of spinal dorsal horn interneurons crucial for gating touch-evoked pain-like behavior. Proc Natl Acad Sci U S A 2021, 118, e2021220118. [Google Scholar] [CrossRef]
- Tavares, I; Costa-Pereira, JT; Martins, I. Monoaminergic and opioidergic modulation of brainstem circuits: New insights into the clinical challenges of pain treatment? Front Pain Res (Lausanne) 2021, 2, 696515. [Google Scholar] [CrossRef] [PubMed]
- Tavares, I; Lima, D. From neuroanatomy to gene therapy: searching for new ways to manipulate the supraspinal endogenous pain modulatory system. J Anat 2007, 211, 261–268. [Google Scholar] [CrossRef]
- Taylor, BK; Westlund, KN. The NA locus coeruleus as a chronic pain generator. J Neurosci Res 2017, 95, 1336–1346. [Google Scholar] [CrossRef] [PubMed]
- Tewari, A; Jog, R; Jog, MS. The striatum and subthalamic nucleus as independent and collaborative structures in motor control. Front Syst Neurosci 2016, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Thompson, CS; Hiller, CE; Schabrun, SM. Altered spinal-level sensorimotor control related to pain and perceived instability in people with chronic ankle instability. J Sci Med Sport 2019, 22, 425–429. [Google Scholar] [CrossRef]
- Thompson, JM; Neugebauer, V. Cortico-limbic pain mechanisms. Neurosci Lett 2019, 702, 15–23. [Google Scholar] [CrossRef]
- Todd, AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci 2010, 11, 823–836. [Google Scholar] [CrossRef]
- Todd, AJ. Plasticity of inhibition in the spinal cord. Handb Exp Pharmacol 2015, 227, 171–190. [Google Scholar]
- Todd, AJ. Identifying functional populations among the interneurons in laminae I-III of the spinal dorsal horn. Mol Pain 2017, 1744806917693003. [Google Scholar] [CrossRef]
- Toney, GM; Mifflin, SW. Sensory modalities conveyed in the hindlimb somatic afferent input to nucleus tractus solitarius. J Appl Physiol (1985) 2000, 88, 2062–2073. [Google Scholar] [CrossRef]
- Topham, L; Gregoire, S; Kang, H; Salmon-Divon, M; Lax, E; Millecamps, M; Szyf, M; Stone, LS. The transition from acute to chronic pain: dynamic epigenetic reprogramming of the mouse prefrontal cortex up to 1 year after nerve injury. Pain 2020, 161, 2394–2409. [Google Scholar] [CrossRef]
- Torres-Rodriguez, JM; Wilson, TD; Singh, S; Chaudhry, S; Adke, AP; Becker, JJ; Lin, JL; Martinez Gonzalez, S; Soler-Cedeño, O; Carrasquillo, Y. The parabrachial to central amygdala circuit is a key mediator of injury-induced pain sensitization. bioRxiv [Preprint] 2023. [Google Scholar]
- Treede, R-D. Gain control mechanisms in the nociceptive system. Pain 2016, 157, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulas, C; McMahon, SB. Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci 2014, 37, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulas, C; Mooney, ER; McNaughton, PA. HCN2 ion channels: basic science opens up possibilities for intervention in neuropathic pain. Biochem J 2016, 473, 2717–1236. [Google Scholar] [CrossRef]
- Tsay, A; Allen, TJ; Proske, U; Giummarra, MJ. Sensing the body in chronic pain: A review of psychophysical studies implicating altered body representation. Neurosci Biobehav Rev 2015, 52, 221–2323. [Google Scholar] [CrossRef]
- Tsuda, M; Koga, K; Chen, T; Zhuo, M. Neuronal and microglial mechanisms for neuropathic pain in the spinal dorsal horn and anterior cingulate cortex. J Neurochem 2017, 141, 486–498. [Google Scholar] [CrossRef]
- Van Oosterwijck, S; Billens, A; Cnockaert, E; Danneels, L; Mertens, T; Dhondt, E; Van Oosterwijck, J. Spinal hyperexcitability in patients with chronic musculoskeletal pain or headache as evidenced by alterations in the nociceptive withdrawal reflex: a systematic review and meta-analysis. Pain 2025, 166, 1002–1029. [Google Scholar] [CrossRef] [PubMed]
- Van Reij, RRI; Joosten, EAJ; van der Hoogen, NJ. Dopaminergic neurotransmission and genetic variation in chronification of post-surgical pain. Br J Anaesth 2019, 123, 853–864. [Google Scholar] [CrossRef]
- Vasic, V; Schmidt, MHH. Resilience and vulnerability to pain and inflammation in the hippocampus. Int J Mol Sci 2017, 18, 739. [Google Scholar] [CrossRef]
- Villanueva, L; Le Bars, D. The activation of bulbo-spinal controls by peripheral nociceptive inputs: diffuse noxious inhibitory controls. Biol Res 1995, 28, 113–125. [Google Scholar]
- Vogt, BA. Midcingulate cortex: Structure, connections, homologies, functions and diseases. J Chem Neuroanat 2016, 74, 28–46. [Google Scholar] [CrossRef]
- Vogt, BA. Cingulate cortex in the three limbic subsystems. Handb Clin Neurol 2019, 166, 39–51. [Google Scholar]
- Vos, BP; Benoist, JM; Gautron, M; Guilbaud, G. Changes in neuronal activities in the two ventral posterior medial thalamic nuclei in an experimental model of trigeminal pain in the rat by constriction of one infraorbital nerve. Somatosens Mot Res 2000, 17, 109–122. [Google Scholar] [PubMed]
- Wall, PD. Recruitment of ineffective synapses after injury. In Functional Recovery in Neurological Disease. Advances in Neurology; Waxman, S.G., Ed.; Raven Press: New York, 1988; Vol. 47, pp. 387–400. [Google Scholar]
- Wan, Y; Zhou, J; Li, H. The role of mechanosensitive piezo channels in chronic pain. J Pain Res 2024, 17, 4199–4212. [Google Scholar] [CrossRef] [PubMed]
- Wang, H; Ehnert, C; Brenner, GJ; Woolf, CJ. Bradykinin and peripheral sensitization. Biol Chem 2006, 387, 11–14. [Google Scholar] [CrossRef]
- Wang, L-L; Wang, H-B; Fu, F-H; Yu, L-C. Role of calcitonin gene-related peptide in pain regulation in the parabrachial nucleus of naive rats and rats with neuropathic pain. Toxicol Appl Pharmacol 2021, 414, 115428. [Google Scholar] [CrossRef]
- Wang, M; Tutt, JO; Dorricott, NO; Parker, KL; Russo, AF; Sowers, LP. Involvement of the cerebellum in migraine. Front Syst Neurosci 2022, 16, 984406. [Google Scholar] [CrossRef]
- Wang, QP; Nakai, Y. The dorsal raphe: an important nucleus in pain modulation. Brain Res Bull 1994, 34, 575–785. [Google Scholar]
- Wang, S-L; Chen, W-Y; Liu, Z-J; Huang, Y-G. Genetic evidence for causal association between hypertension and chronic pain: A Bidirectional Two-Sample Mendelian Randomization Study. Chin Med Sci J 2024, 39, 155–161. [Google Scholar]
- Wang, T; Ma, C. Peripheral nociceptors as immune sensors in the development of pain and itch. Adv Exp Med Biol 2016, 904, 77–85. [Google Scholar] [PubMed]
- Wang, X-Y; Jia, W-B; Xu, X; Chen, R; Wang, L-B; Su, X-J; Xu, P-F; Liu, X-Q; Wen, J; Song, X-Y; Liu, Y-Y; Zhang, Z; Liu, X-F; Zhang, Y. A glutamatergic DRN-VTA pathway modulates neuropathic pain and comorbid anhedonia-like behavior in mice. Nat Commun 2023, 14, 5124. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y-D; Bao, S-T; Gao, Y; Chen, J; Jia, T; Yin, C; Cao, J-L; Xiao, C; Zhou, C. The anterior cingulate cortex controls the hyper-activity in subthalamic neurons in male mice with comorbid chronic pain and depression. PLoS Biol 2024, 22, e3002518. [Google Scholar] [CrossRef] [PubMed]
- Wasner, G; Schattschneider, J; Binder, A; Baron, R. Complex regional pain syndrome – diagnostic, mechanisms, CNS involvement and therapy. Spinal Cord 2003, 41, 61–75. [Google Scholar] [CrossRef]
- Watanabe, M; Narita, M; Hamada, Y; Yamashita, A; Tamura, H; Ikegami, D; Kondo, T; Shinzato, T; Shimizu, T; Fukuchi, Y; Muto, A; Okano, H; Yamanaka, A; Tawfik, VL; Kuzumaki, N; Navratilova, E; Porreca, F; Narita, M. Activation of ventral tegmental area dopaminergic neurons reverses pathological allodynia resulting from nerve injury or bone cancer. Mol Pain 2018, 14, 1744806918756406. [Google Scholar] [CrossRef]
- Watkins, LR; Hutchinson, MR; Milligan, ED; Maier, SF. “Listening” and “talking” to neurons: implications of immune activation for pain control and increasing the efficacy of opioids. Brain Res Rev 2007, 56, 148–169. [Google Scholar] [CrossRef]
- Waxman, SG; Hains, BC. Fire and phantoms after spinal cord injury: Na+ channels and central pain. Trends Neurosci 2006, 29, 207–215. [Google Scholar] [CrossRef]
- Welman, FHSM; Smit, AE; Jongen, JLM; Tibboel, D; van der Geest, JN; Holstege, JC. Pain experience is somatotopically organized and overlaps with pain anticipation in the human cerebellum. Cerebellum 2018, 17, 447–460. [Google Scholar] [CrossRef]
- West, SJ; Bannister, K; Dickenson, AH; Bennett, DL. Circuitry and plasticity of the dorsal horn--toward a better understanding of neuropathic pain. Neuroscience 2015, 300, 254–275. [Google Scholar] [CrossRef]
- White, FA; Bhangoo, SK; Miller, RJ. Chemokines: integrators of pain and inflammation. Nat Rev Drug Discov 2005, 4, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, T; DeLong, MR. Deep brain stimulation for movement disorders of basal ganglia origin: Restoring function or functionality? Neurotherapeutics 2016, 13, 264–283. [Google Scholar] [CrossRef]
- Wiech, K; Edwards, R; Moseley, LG; Berna, C; Ploner, M; Tracey, I. Dissociable neural mechanisms underlying the modulation of pain and anxiety? An FMRI pilot study. PLoS One 2014, 9, e110654. [Google Scholar] [CrossRef]
- Wiesenfeld-Hallin, Z; Aldskogius, H; Grant, G; Hao, J-X; Hoekfelt, T; Xu, X-J. Central inhibitory dysfunctions: mechanisms and clinical implications. Beh Brain Sci 1997, 20, 420–425. [Google Scholar] [CrossRef]
- Wilkinson, KA. Molecular determinants of mechanosensation in the muscle spindle. Curr Opin Neurobiol 2022, 74, 102542. [Google Scholar] [CrossRef] [PubMed]
- Willis, WD. Temperature perception and pain. In Comprehensive human physiology. From cellular mechanisms to integration; Greger, R, Windhorst, U, Eds.; Springer-Verlag: Berlin Heidelberg New York, 1996; vol 1, pp. pp 677–696. [Google Scholar]
- Willis, WD. Dorsal root potentials and dorsal root reflexes: a double-edged sword. Exp Brain Res 1999, 124, 395–421. [Google Scholar] [CrossRef] [PubMed]
- Willis, WD. Long-term potentiation in spinothalamic neurons. Brain Res Rev 2002, 40, 202–21. [Google Scholar] [CrossRef]
- Windhorst, U. Muscle proprioceptive feedback and spinal networks. Brain Res Bull 2007, 73, 155–202. [Google Scholar] [CrossRef]
- Windhorst, U. Spinal cord circuits: models and reality. Neurophysiology 2021, 53, 142–222. [Google Scholar] [CrossRef]
- Windhorst, U; Dibaj, P. Plastic spinal motor circuits in health and disease. J Integr Neurosci 2023, 22, 167. [Google Scholar] [CrossRef]
- Windhorst, U; Dibaj, P. Nociception and acute pain: ascending functional structure and descending modulation. Preprints 2025a, 2025050743. [Google Scholar]
- Windhorst, U; Dibaj, P. Nociception and acute pain: Neurotransmitters and neuromodulators. Preprints 2025b, 2025080487. [Google Scholar]
- Windhorst, U; Meyer-Lohmann, J; Kirmayer, D; Zochodne, D. Renshaw cell responses to intra-arterial injection of muscle metabolites into cat calf muscles. Neurosci Res 1997, 27, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Witter, MP; Doan, TP; Jacobsen, B; Nilssen, ES; Ohara, S. Architecture of the entorhinal cortex. A review of entorhinal anatomy in rodents with some comparative notes. Front Syst Neurosci 2017, 11, 46. [Google Scholar] [CrossRef]
- Woo, S-H; Lukacs, V; de Nooij, JC; Zaytseva, D; Criddle, CR; Francisco, A; Jessell, TM; Wilkinson, KA; Patapoutian, A. Piezo2 is the principal mechanotransduction channel for proprioception. Nat Neurosci 2015, 18, 1756–1762. [Google Scholar] [CrossRef]
- Woodbury, CJ; Kullmann, FA; McIlwrath, SL; Koerber, HR. Identity of myelinated cutaneous sensory neurons projecting to nocireceptive laminae following nerve injury in adult mice. J Comp Neurol 2008, 508, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Woolf, CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain 2011, 152, S2–S15. [Google Scholar] [CrossRef]
- Woolf, CJ; Salter, MW. Neuronal plasticity: increasing the gain in pain. Science 2000, 288, 1765–1768. [Google Scholar] [CrossRef]
- Xiao, X; Zhang, Y-Q. A new perspective on the anterior cingulate cortex and affective pain. Neurosci Biobehav Rev 2018, 90, 200–211. [Google Scholar] [CrossRef]
- Xie, W; Strong, JA; Zhang, J-M. Increased excitability and spontaneous activity of rat sensory neurons following in vitro stimulation of sympathetic fiber sprouts in the isolated dorsal root ganglion. Pain 2010, 151, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y; Lin, Y; Yu, M; Zhou, K. The nucleus accumbens in reward and aversion processing: insights and implications. Front Behav Neurosci 2024, 18, 1420028. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y; Zhu, X; Chen, Y; Chen, Y; Zhu, Y; Xiao, S; Wu, M; Wang, Y; Zhang, C; Wu, Z; He, X; Liu, B; Shen, Z; drn, X; Fang, J. Electroacupuncture alleviates mechanical allodynia and anxiety-like behaviors induced by chronic neuropathic pain via regulating rostral anterior cingulate cortex-dorsal raphe nucleus neural circuit. CNS Neurosci Ther 2023, 29, 4043–4058. [Google Scholar] [CrossRef]
- Yalcin, I; Barthas, F; Barrot, M. Emotional consequences of neuropathic pain: insight from preclinical studies. Neurosci Biobehav Rev 2014, 47, 154–164. [Google Scholar] [CrossRef]
- Yamauchi, N; Sato, K; Sato, K; Murakawa, S; Hamasaki, Y; Nomura, H; Amano, T; Minami, M. Chronic pain-induced neuronal plasticity in the bed nucleus of the stria terminalis causes maladaptive anxiety. Sci Adv 2022, 8, eabj5586. [Google Scholar] [CrossRef]
- Yang, S; Chang, MC. Chronic pain: Structural and functional changes in brain structures and associated negative affective states. Int J Mol Sci 2019, 20, 3130. [Google Scholar] [CrossRef]
- Yang, X; Xu, W; Hou, X; Ding, Z; Zhang, C; Guo, Q; Zou, W; Wang, J. Chronic stress enhances neuropathic pain through activation of the anterior cingulate cortex-spinal dorsal horn pathway in rats. Prog Neuropsychopharmacol Biol Psychiatry 2025, 138, 111352. [Google Scholar] [CrossRef]
- Yarmolinsky, DA; Zeng, X; MacKinnon-Booth, N; Greene, CA; Kim, C; Cheng, Y-T; Lenders Turnes, B; Woolf, CJ. Differential modification of ascending spinal outputs in acute and chronic pain states. Neuron 2025, 113, 1223–1239.e5. [Google Scholar] [CrossRef] [PubMed]
- Yetnikoff, L; Laezzi, HN; Reichard, RA; Zahm, DS. An update on the connections of the ventral mesencephalic dopaminergic complex. Neuroscience 2014, 282, 23–48. [Google Scholar] [CrossRef] [PubMed]
- Yin, C; Jia, T; Luan, Y; Zhang, X; Xiao, C; Zhou, C. A nigra-subthalamic circuit is involved in acute and chronic pain states. Pain 2022, 163, 1952–1966. [Google Scholar] [CrossRef] [PubMed]
- Yin, J-B; Liang, S-H; Li, F; Zhao, W-J; Bai, Y; Sun, Y; Wu, Z-Y; Ding, T; Sun, Y; Liu, H-X; Lu, Y-C; Zhang, T; Huang, J; Chen, T; Li, H; Chen, Z-F; Cao, J; Ren, R; Peng, Y-N; Yang, J; Zang, W-D; Li, X; Dong, Y-L; Li, Y-Q. dmPFC-vlPAG projection neurons contribute to pain threshold maintenance and antianxiety behaviors. J Clin Invest 2020, 130, 6555–6570. [Google Scholar] [CrossRef]
- You, H-J; Lei, J; Pertovaara, A. Thalamus: The 'promoter' of endogenous modulation of pain and potential therapeutic target in pathological pain. Neurosci Biobehav Rev 2022, 139, 104745. [Google Scholar] [CrossRef]
- Yu, R; Gollub, RL; Spaeth, R; Napadow, V; Wasan, A; Kong, J. Disrupted functional connectivity of the periaqueductal gray in chronic low back pain. Neuroimage Clin 2014, 23(6), 100–108. [Google Scholar] [CrossRef]
- Zeilhofer, HU; Werynska, K; Gingras, J; Yévenes, GE. Glycine receptors in spinal nociceptive control – An update. Biomolecules 2021, 11, 846. [Google Scholar] [CrossRef] [PubMed]
- Zeilhofer, HU; Wildner, H; Yévenes, GE. Fast synaptic inhibition in spinal sensory processing and pain control. Physiol Rev 2012, 92, 193–235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H; Li, L; Zhang, X; Ru, G; Zang, W. Role of the dorsal raphe nucleus in pain processing. Brain Sci 2024, 14, 982. [Google Scholar] [CrossRef]
- Zhang, Y; Chen, Y; Liedtke, W; Wang, F. Lack of evidence for ectopic sprouting of genetically labeled Aβ touch afferents in inflammatory and neuropathic trigeminal pain. Mol Pain 2015, 11, 18. [Google Scholar] [CrossRef]
- Zhou, H; Martinez, E; Lin, HH; Yang, R; Dale, JA; Liu, K; Huang, D; Wang, J. Inhibition of the prefrontal projection to the nucleus accumbens enhances pain sensitivity and affect. Front Cell Neurosci 2018, 12, 240. [Google Scholar] [CrossRef]
- Zhuo, M. Descending facilitation: From basic science to the treatment of chronic pain. Mol Pain 2017, 13, 1–12. [Google Scholar] [CrossRef]
- Zhou, XF; Deng, YS; Chie, E; Xue, Q; Zhong, JH; McLachlan, EM; Rush, RA; Xian, CJ. Satellite-cell-derived nerve growth factor and neurotrophin-3 are involved in noradrenergic sprouting in the dorsal root ganglia following peripheral nerve injury in the rat. Eur J Neurosci 1999, 11, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, M. Long-term plasticity of NMDA GluN2B (NR2B) receptor in anterior cingulate cortical synapses. Mol Pain 2024, 20, 17448069241230258. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X; Xu, Y; Shen, Z; Zhang, H; Xiao, S; Zhu, Y; Wu, M; Chen, Y; Wu, Z; Xu, Y; He, X; Liu, B; Liu, J; Liu, Y; Du, J; Sun, J; Fang, J; Shao, X. Rostral anterior cingulate cortex-ventrolateral periaqueductal gray circuit underlies electroacupuncture to alleviate hyperalgesia but not anxiety-like behaviors in mice with spared nerve injury. Front Neurosci 2022, 15, 757628. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, AL; Kovatsis, EM; Pozsgai, RY; Tasnim, A; Zhang, Q; Ginty, DD. Distinct modes of presynaptic inhibition of cutaneous afferents and their functions in behavior. Neuron 2019, 102, 420–434.e8. [Google Scholar] [CrossRef]
- Zimmermann, M. Pathobiology of neuropathic pain. Eur J Pharmacol 2001, 429, 23–37. [Google Scholar] [CrossRef]
- Zouikr, I; Bartholomeusz, MD; Hodgson, DM. Early life programming of pain: focus on neuroimmune to endocrine communication. J Transl Med 2016, 14, 123. [Google Scholar] [CrossRef]
- Zyuzin, J; Jendzjowsky, N. Neuroanatomical and neurophysiological evidence of pulmonary nociceptor and carotid chemoreceptor convergence in the nucleus tractus solitarius and nucleus ambiguus. J Neurophysiol 2022, 127, 1511–1518. [Google Scholar] [CrossRef]
Figure 1.
Simplified scheme of nociceptor activation and sensitization by agents in the ìnflammatory soup´. Upon activation, groups of nociceptive afferents, expressing substance P (SP) or calcitonin gene-releasing peptide (CGRP), release these substances, thereby contributing to the inflammatory soup (neurogenetic inflammation) by a positive feedback. SP activates mast cells, which release various agents depending on their location and environment. Each of the inflammatory mediators excites, or lowers the acticvation threshold, of nociceptor terminals. Green lines symbolize group III and IV nociceptive afferents, green circles below the sites where these afferents release agents like CGRP or SP. The upward green arrow beside the SC symbolizes enhanced and prolonged activation. Abbreviations: ATP: adenosine trisphosphate; BDNF: brain-derived neurotrophic factor; CGRP: calcitonin gene-releasing peptide; DRG: dorsal root ganglion; H+: proton; 5-HT: serotonin; HIST: histamine; IL-1ß: interleukin 1ß; NGF: nerve growth factor; PGE2: prostaglandin E2; SP: substance P (Data from papers cited in the text).
Figure 1.
Simplified scheme of nociceptor activation and sensitization by agents in the ìnflammatory soup´. Upon activation, groups of nociceptive afferents, expressing substance P (SP) or calcitonin gene-releasing peptide (CGRP), release these substances, thereby contributing to the inflammatory soup (neurogenetic inflammation) by a positive feedback. SP activates mast cells, which release various agents depending on their location and environment. Each of the inflammatory mediators excites, or lowers the acticvation threshold, of nociceptor terminals. Green lines symbolize group III and IV nociceptive afferents, green circles below the sites where these afferents release agents like CGRP or SP. The upward green arrow beside the SC symbolizes enhanced and prolonged activation. Abbreviations: ATP: adenosine trisphosphate; BDNF: brain-derived neurotrophic factor; CGRP: calcitonin gene-releasing peptide; DRG: dorsal root ganglion; H+: proton; 5-HT: serotonin; HIST: histamine; IL-1ß: interleukin 1ß; NGF: nerve growth factor; PGE2: prostaglandin E2; SP: substance P (Data from papers cited in the text).
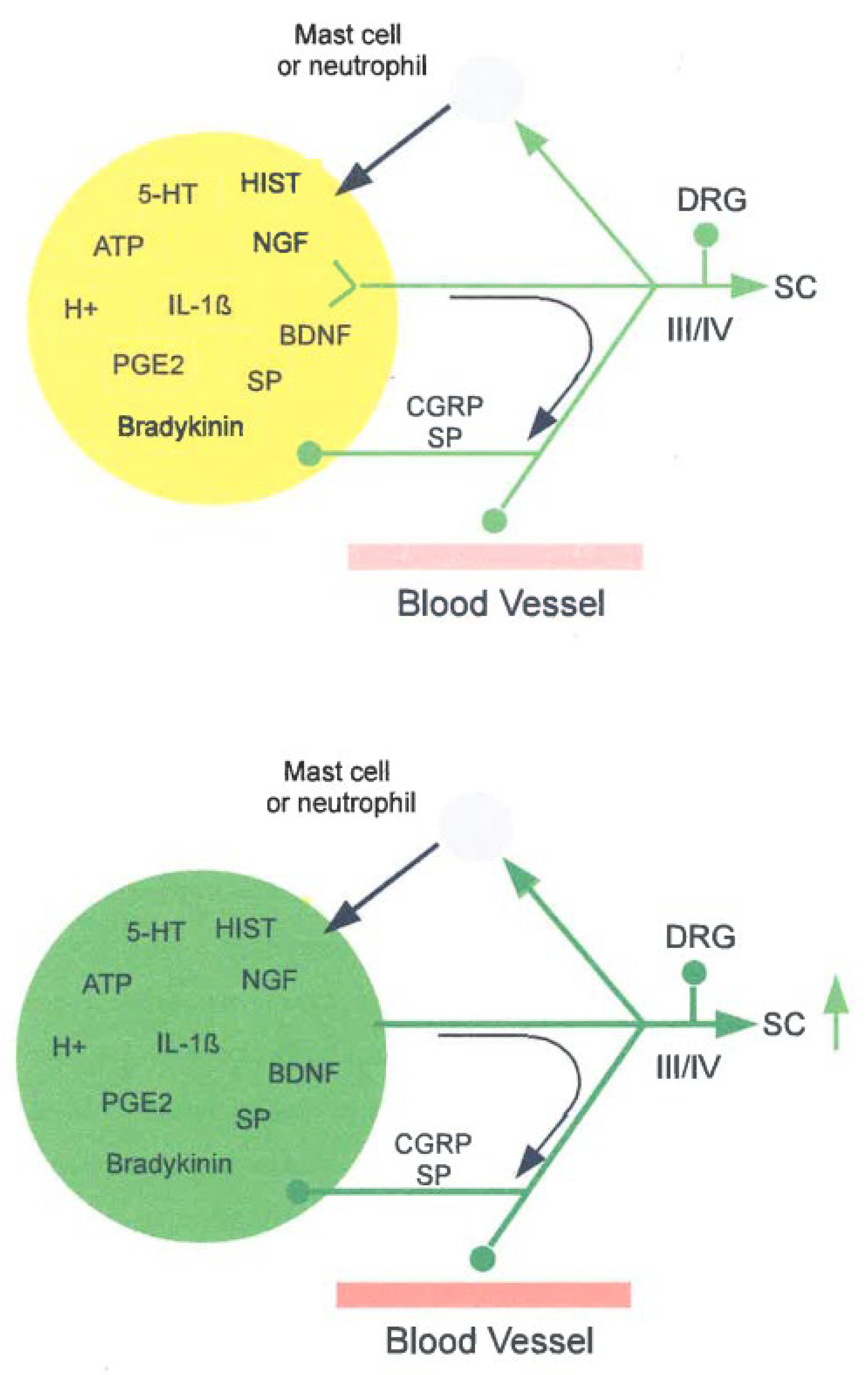
Figure 2.
Simplified scheme of presynaptic inhibition (PSI; left) and subsequent nociceptive networks (right) in the spinal dorsal horn (DH). Green ellipses and lines symbolize excitatory glutamatergic (Glu) neurons, with varying co-transmitters. Red ellipses and lines symbolize inhibitory neurons, expressing varying co-transmitters. Connections symbolized by lines are not necessarity monosynaptic. The PSI connection from the inhibitory neuron fed by group Ia afferents (lower left) to group III afferents is hypothetical as marked by a question mark (based on data from Björklund et al. 2004). Central cells (CCs) can be either excitatory (glutamatergic, fast-spiking) or inhibitory (GABAergic, tonically firing; not shown; Zeilhofer et al. 2012). Weak red ellipses and connections symbolize inhibitory neurons with decreased inhibitory strength. The upward green arrows beside the group III and IV inputs symbolize enhanced and prolonged activation. Nociceptive inputs enter the DH via myelinated group III (Aδ) and un-myelinated group IV (C) afferents. Mechano-sensitive inputs are conveyed in part by group III (Aδ) and by group II (Aß) afferents from cutaneous and muscle sources. Polysynaptic interactions between these modalities exist, but are normally supposed to be silenced by glycinergic and GABAergic inhibitory INTs and only unmasked during pathological, e.g., inflammatory and neuropathic pain, conditions (Zeilhofer et al. 2021). Abbreviations: CR: calretinin; BDNF: brain-derived neurotrophic factor; CC: central cell [CCs in Lam IIi can be GABAergic and tonic (not shown) or glutamatergic and transient, each with different connections (Zeilhofer et al. 2012)]; GTO: Golgi tendon organ; IC: islet cell; IIim VC: IIm vertical cell; INT: interneuron; LVC: large vertical cell (stalked cell); MNs: motoneurons; MS: muscle spindle; PN: projection neuron; PSI: presynaptic inhibition; SVC: small vertical cell; WDR: wide dynamic range neuron. (Data from papers cited in the text).
Figure 2.
Simplified scheme of presynaptic inhibition (PSI; left) and subsequent nociceptive networks (right) in the spinal dorsal horn (DH). Green ellipses and lines symbolize excitatory glutamatergic (Glu) neurons, with varying co-transmitters. Red ellipses and lines symbolize inhibitory neurons, expressing varying co-transmitters. Connections symbolized by lines are not necessarity monosynaptic. The PSI connection from the inhibitory neuron fed by group Ia afferents (lower left) to group III afferents is hypothetical as marked by a question mark (based on data from Björklund et al. 2004). Central cells (CCs) can be either excitatory (glutamatergic, fast-spiking) or inhibitory (GABAergic, tonically firing; not shown; Zeilhofer et al. 2012). Weak red ellipses and connections symbolize inhibitory neurons with decreased inhibitory strength. The upward green arrows beside the group III and IV inputs symbolize enhanced and prolonged activation. Nociceptive inputs enter the DH via myelinated group III (Aδ) and un-myelinated group IV (C) afferents. Mechano-sensitive inputs are conveyed in part by group III (Aδ) and by group II (Aß) afferents from cutaneous and muscle sources. Polysynaptic interactions between these modalities exist, but are normally supposed to be silenced by glycinergic and GABAergic inhibitory INTs and only unmasked during pathological, e.g., inflammatory and neuropathic pain, conditions (Zeilhofer et al. 2021). Abbreviations: CR: calretinin; BDNF: brain-derived neurotrophic factor; CC: central cell [CCs in Lam IIi can be GABAergic and tonic (not shown) or glutamatergic and transient, each with different connections (Zeilhofer et al. 2012)]; GTO: Golgi tendon organ; IC: islet cell; IIim VC: IIm vertical cell; INT: interneuron; LVC: large vertical cell (stalked cell); MNs: motoneurons; MS: muscle spindle; PN: projection neuron; PSI: presynaptic inhibition; SVC: small vertical cell; WDR: wide dynamic range neuron. (Data from papers cited in the text).
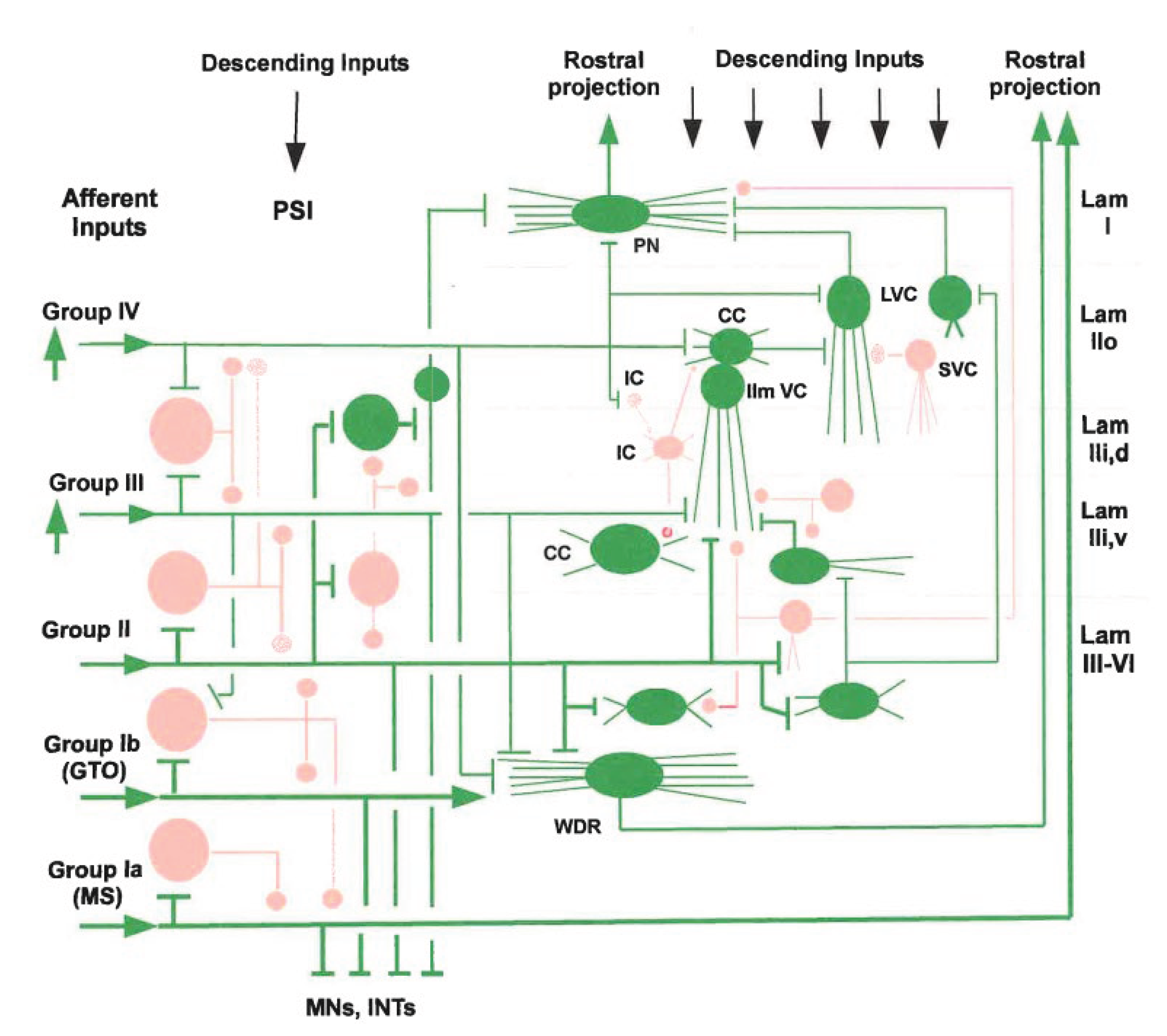
Figure 3.
Simplified scheme of locations of some nuclei and brain structures involved in nociceptives transmission from the periphery to the cerebral cortex. The sections are not scaled. The upper panel shows a coronal cross-section through the right prosencephalon containing the cerebrum (top) and diencephalon (bottom). The panels below show transverse cross-sections from the mesencephalon (midbrain) to the caudal medulla oblongata. Note that the marked structures may extend beyond the secitional planes so that they are projected into the respective planes. Green lines symbolize lumped ascending nociceptive connections except for the blue lines symbolizing NA connections from the LC. Connections need not be monosynaptic. The upwards green arrow beside SC sympolizes increased activity. Abbreviations: A1: noradrenergic (NA) A1 cell group; AMY: amygdala; BG: basal ganglia; CING: cingulate cortex; CMED: caudal medulla; CVLM: caudal ventro-lateral medulla, DH: dorsal horn; DReN: dorsal reticular nucleus; DRN: dorsal raphé nucleus (gray shade); HYP: hypothalamus; IC: insular cortex; LC: locus coeruleus; MES: mesencephalon; NRM: nucleus raphé magnus; NTS: nucleus tractus solitarii; PAG: peri-aqueductal gray; PBN: parabrachial nucleus; PROS: prosencephalon; RMED: rostral medulla; RVM: rostral ventro-medial medulla; S1: primary somatosensory cortex; SC: spinal cord; SNc: substantia nigra pars compacta; THAL: thalamus; VTA: ventral tegmental area (Data from papers cited in the text).
Figure 3.
Simplified scheme of locations of some nuclei and brain structures involved in nociceptives transmission from the periphery to the cerebral cortex. The sections are not scaled. The upper panel shows a coronal cross-section through the right prosencephalon containing the cerebrum (top) and diencephalon (bottom). The panels below show transverse cross-sections from the mesencephalon (midbrain) to the caudal medulla oblongata. Note that the marked structures may extend beyond the secitional planes so that they are projected into the respective planes. Green lines symbolize lumped ascending nociceptive connections except for the blue lines symbolizing NA connections from the LC. Connections need not be monosynaptic. The upwards green arrow beside SC sympolizes increased activity. Abbreviations: A1: noradrenergic (NA) A1 cell group; AMY: amygdala; BG: basal ganglia; CING: cingulate cortex; CMED: caudal medulla; CVLM: caudal ventro-lateral medulla, DH: dorsal horn; DReN: dorsal reticular nucleus; DRN: dorsal raphé nucleus (gray shade); HYP: hypothalamus; IC: insular cortex; LC: locus coeruleus; MES: mesencephalon; NRM: nucleus raphé magnus; NTS: nucleus tractus solitarii; PAG: peri-aqueductal gray; PBN: parabrachial nucleus; PROS: prosencephalon; RMED: rostral medulla; RVM: rostral ventro-medial medulla; S1: primary somatosensory cortex; SC: spinal cord; SNc: substantia nigra pars compacta; THAL: thalamus; VTA: ventral tegmental area (Data from papers cited in the text).
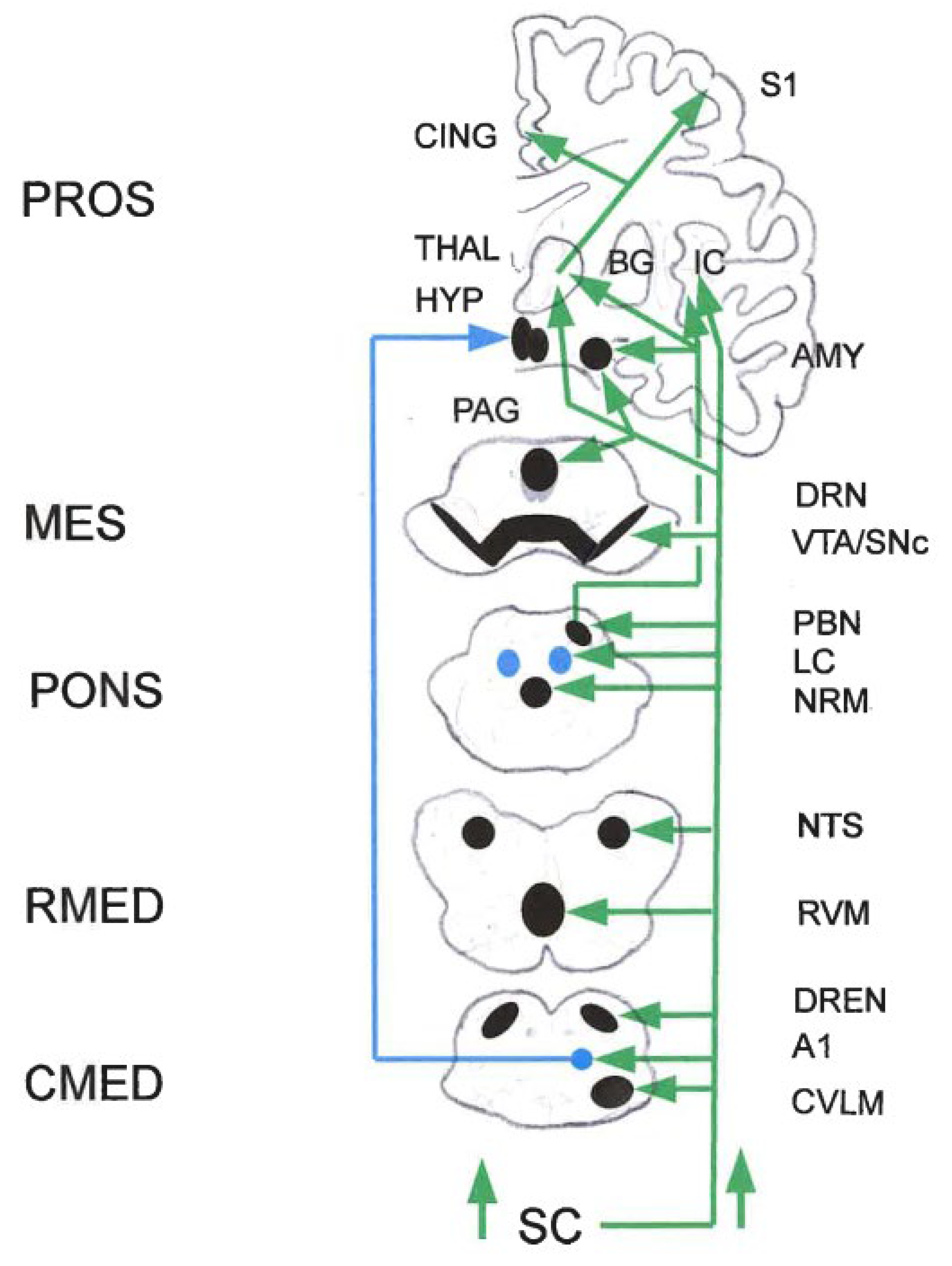
Figure 4.
Simplified scheme of the net changes in neuronal activity and connection strengths, indicated by upward green arrows (increase) or downward red arrows (decrease). As exemplified by the RVM-DH connections, inhibitory effects are diminished and facilitatory effects increased, leading to net change towards facilitation. The sections are not scaled. Some structures (e.g., raphé nuclei, RN) distribute quite far rostro-caudally and may occur in two cross-sections, which is not shown for graphical reasons. Connections symbolized by arrows may be excitatory (green lines) or inhibitory (red lines), and are not necessarily monosynaptic. For example and importantly, the connections from RVM to DH are both facilitory and inhibitory. Other arrowed lines, e.g., from HYP to DH, comprise DA and OXT influences. Therefore, and also for graphical reasons, most lines are black. Abbreviations: A1: noradrenergic (NA) A1 cell group; AMY: amygdala; BG: basal ganglia; CING: cingulate cortex; CMED: caudal medulla; CVLM: caudal ventro-lateral medulla; dlPFC: dorso-lateral prefrontal cortex; DH: dorsal horn; DReN: dorsal reticular nucleus; DRN: dorsal raphé nucleus; HYP: hypothalamus; IC: insular cortex; LC: locus coeruleus; MES: mesencephalon; mPFC: medial prefrontal cortex; NRM: nucleus raphé magnus; NTS: nucleus tractus solitarii; PAG: peri-aqueductal gray; PBN: parabrachial nucleus; PROS: prosencephalon; RMED: rostral medulla; RVM: rostral ventro-medial medulla; SC: spinal cord; SNc: substantia nigra pars compacta; THAL: thalamus; vlPFC: ventro-lateral prefrontal cortex; VTA: ventral tegmental area (Data from papers cited in the text).
Figure 4.
Simplified scheme of the net changes in neuronal activity and connection strengths, indicated by upward green arrows (increase) or downward red arrows (decrease). As exemplified by the RVM-DH connections, inhibitory effects are diminished and facilitatory effects increased, leading to net change towards facilitation. The sections are not scaled. Some structures (e.g., raphé nuclei, RN) distribute quite far rostro-caudally and may occur in two cross-sections, which is not shown for graphical reasons. Connections symbolized by arrows may be excitatory (green lines) or inhibitory (red lines), and are not necessarily monosynaptic. For example and importantly, the connections from RVM to DH are both facilitory and inhibitory. Other arrowed lines, e.g., from HYP to DH, comprise DA and OXT influences. Therefore, and also for graphical reasons, most lines are black. Abbreviations: A1: noradrenergic (NA) A1 cell group; AMY: amygdala; BG: basal ganglia; CING: cingulate cortex; CMED: caudal medulla; CVLM: caudal ventro-lateral medulla; dlPFC: dorso-lateral prefrontal cortex; DH: dorsal horn; DReN: dorsal reticular nucleus; DRN: dorsal raphé nucleus; HYP: hypothalamus; IC: insular cortex; LC: locus coeruleus; MES: mesencephalon; mPFC: medial prefrontal cortex; NRM: nucleus raphé magnus; NTS: nucleus tractus solitarii; PAG: peri-aqueductal gray; PBN: parabrachial nucleus; PROS: prosencephalon; RMED: rostral medulla; RVM: rostral ventro-medial medulla; SC: spinal cord; SNc: substantia nigra pars compacta; THAL: thalamus; vlPFC: ventro-lateral prefrontal cortex; VTA: ventral tegmental area (Data from papers cited in the text).
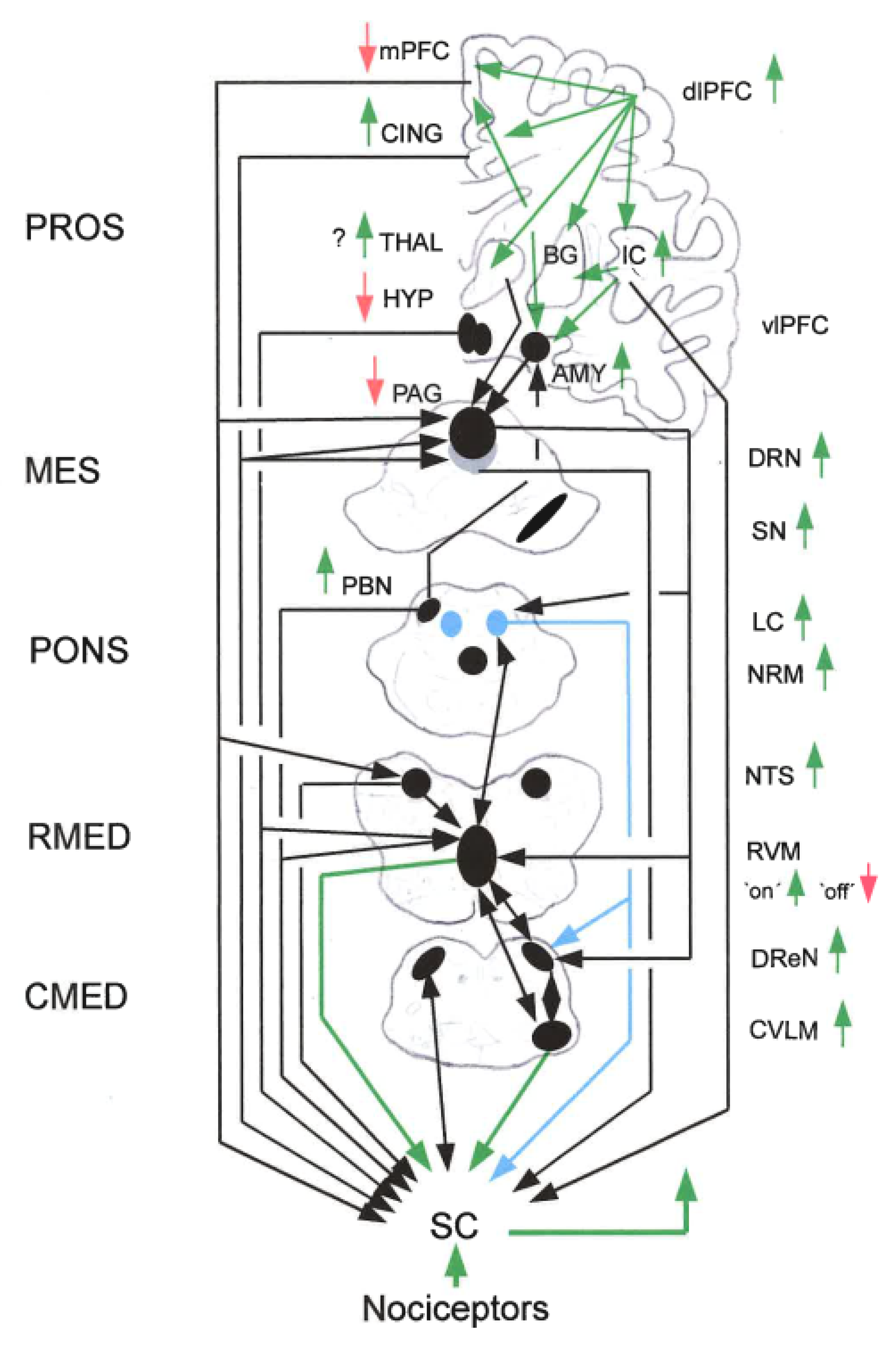
Figure 5.
Simplified scheme of the amygdala (AMY) showing pain-related changes in cortico-AMY interactions. Green circles and lines symbolize excitatory connections. Red circles and lines symbolize inhibitory connections. A: Normal conditions. B: Chronic high nociceptive inputs from the SC, for example in arthritic pain. Projection neurons in the baso-lateral amygdala (BLA) contact medial prefrontal cortex (mPFC) projection cells directly and indirectly via GABAergic INTs, entailing BLA-driven feedforward inhibition of mPFC neurons. BLA activity is increased in arthritic pain resulting in pain-related cortical dysfunction (B). Projection neurons in the infra-limbic PFC synapse on BLA cells and GABAergic intercalated cells (ITCs). ITCs and BLA projection neurons synapse on neurons in the central nucleus of the amygdala (CeA). In the arthritic condition (B), diminished cortical drive to BLA and ITC neurons results in reduced feedforward inhibition of CeA projection neurons. This, together with increased drive from the BLA, results in pain-telated AMY hyper-activity. Abbreviations: AMY: amygdala; BLA: baso-lateral nucleus; CeA: central nucleus of AMY; ITC: intercalated cell group; PFC: prefrontal cortex: SC: spinal cord (Data from Thompson and Neugebauer 2019).
Figure 5.
Simplified scheme of the amygdala (AMY) showing pain-related changes in cortico-AMY interactions. Green circles and lines symbolize excitatory connections. Red circles and lines symbolize inhibitory connections. A: Normal conditions. B: Chronic high nociceptive inputs from the SC, for example in arthritic pain. Projection neurons in the baso-lateral amygdala (BLA) contact medial prefrontal cortex (mPFC) projection cells directly and indirectly via GABAergic INTs, entailing BLA-driven feedforward inhibition of mPFC neurons. BLA activity is increased in arthritic pain resulting in pain-related cortical dysfunction (B). Projection neurons in the infra-limbic PFC synapse on BLA cells and GABAergic intercalated cells (ITCs). ITCs and BLA projection neurons synapse on neurons in the central nucleus of the amygdala (CeA). In the arthritic condition (B), diminished cortical drive to BLA and ITC neurons results in reduced feedforward inhibition of CeA projection neurons. This, together with increased drive from the BLA, results in pain-telated AMY hyper-activity. Abbreviations: AMY: amygdala; BLA: baso-lateral nucleus; CeA: central nucleus of AMY; ITC: intercalated cell group; PFC: prefrontal cortex: SC: spinal cord (Data from Thompson and Neugebauer 2019).
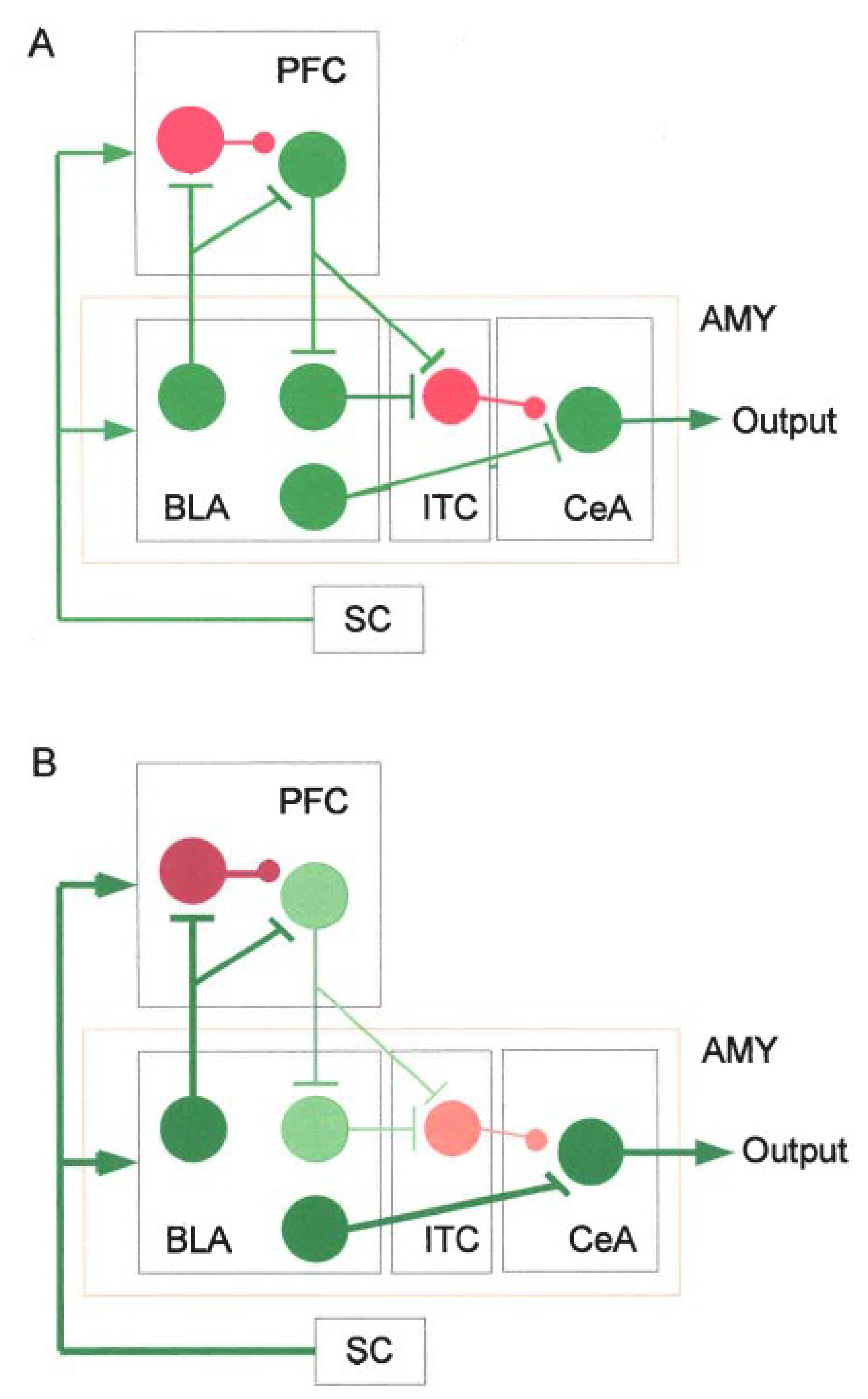
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



