Submitted:
30 January 2026
Posted:
02 February 2026
You are already at the latest version
Abstract
Mitochondrial dysfunction is increasingly recognized as a central, integrative driver of biological aging and a convergent mechanism underlying multiple age-associated pathologies. This review synthesizes current evidence identifying a coordinated network of mitochondrial “drivers of aging” that collectively erode cellular homeostasis and organismal resilience. Core processes include decline in ATP production, impaired electron transport chain efficiency and supercomplex assembly, excessive reactive oxygen species generation, accumulation of mitochondrial DNA damage and mutations, rising heteroplasmy, reduced DNA repair capacity, and progressive loss of mitochondrial DNA copy number. These genomic and bioenergetic failures are compounded by dysregulated mitochondrial dynamics, diminished biogenesis, and defective mitophagy, leading to the persistence of dysfunctional organelles and amplification of inflammatory and senescence-associated signaling. We propose a conceptual mitochondrial lifespan clock model in which the cumulative imbalance among these interdependent mechanisms accelerates functional decline across tissues, particularly in post-mitotic systems such as muscle, heart, and brain. Importantly, multiple drivers remain plastic and responsive to metabolic, genetic, and pharmacological interventions, highlighting mitochondria not only as biomarkers but as actionable targets for extending healthspan. Understanding the hierarchy, interaction, and reversibility of these mitochondrial determinants provides a unifying framework for translational strategies aimed at delaying aging and mitigating age-related disease.
Keywords:
mitochondrial dysfunction
; aging biology
; bioenergetics
; mitophagy
; mitochondrial DNA mutations
; heteroplasmy
; mitochondrial biogenesis
; reactive oxygen species
; electron transport chain
; ATP decline
; mitochondrial dynamics
; healthspan
; cellular senescence
; oxidative phosphorylation
; mitochondrial quality control
I. Introduction
All organisms age. Aging is a natural physiological process governed by various biological and genetic pathways that are closely linked to lifespan. For many centuries, humankind has had a unique interest in this natural process, looking for ways to stop, slow down, or reverse aging [1]. Aging is characterized by a gradual deterioration of physiological functions, which ultimately results in various age-associated conditions, including cardiovascular diseases, musculoskeletal disorders like arthritis, neurodegenerative diseases (Alzheimer’s, Parkinson’s, Huntington’s, ELA), and cancer [2]. These conditions impose a high economic cost and psychological strain on patients, their families, the healthcare systems and society at large long-term care costs, this contributes to disability and diminishes quality of life [3,4].
Specifically in the US demographic research showed that the health of elderly people has worsened the past 40 years [5]. In the European Union it is projected that there will be close to half a million centenarians by 2050. The population of older adults (65 and above) is expected to grow substantially, increasing from 90.5 million in early 2019 to 129.8 million by 2050. Within this timeframe, the group aged 75–84 years is projected to experience a 56.1% rise [6]. Such facts are likely to have profound implications, not only for individuals, but also for governments, business and civil society, impacting, among others: health and social care systems, labor markets, public finances and pension entitlements.
In the present, humanity worldwide has a significantly higher life expectancy compared to previous centuries due to the significant increase of medical knowledge and improvements in quality-of-life services [7], accompanied by lowered fertility, which has generated higher proportions of older populations throughout the world [8], referred as “Population Aging”, which describes the increase in the proportion of the older population aged above 59 years, plus the decrease of the proportion of the young population aged below 16 years old [8]. Even more, the proportion of the global population aged 65 and older is projected to rise significantly, increasing from 10% in 2000 to 21.8% by 2050, eventually reaching 32.2% by 2100 [7]. By 2030, one sixth of the population will be aged 60 or older, and projections suggest that the population of individuals over 80 years old will triple between 2020 and 2050 [9]. By 2025 China’s elderly population will increase by 1 million per year [10]. By 2058 the elderly population in China should reach a peak of 31.54% increase [11]. In America population aging is occurring more rapidly [12]. In Europe the population is also aging rapidly [13]. Europeans are living longer than ever before and population aging is rapidly increasing [6].
Given that lifespan has greatly increased while, conversely, healthspan has not, and considering that aging is a process regulated by numerous biological and genetic pathways, this results in a strong force driving the development of a wide range of age-related diseases [14]. It is important to note that common characteristics of aging include symptoms like senile dementia, delirium, blood disorders, anemia, bone fragility, muscle weakness, alopecia, and diseases such as Parkinson’s, Alzheimer’s, cancer, cardiovascular diseases, and diabetes [15].
There are several aging theories classifications: due to Programmation, internal or external causes, due to damage, of systemic nature, due to mutations, and finally a combination of these theories [16]. Internal aging theories are cell-centric: due to somatic mutations, free radicals, DNA or microRNAs damage [17], due to hyper functions of the cell [18], mutations accumulation [19,20] and caused by mitochondria, referred to as chemiosmotic mitochondrial aging [21]. Combined aging theories comprise evolutionary mechanisms as antagonistic pleiotropy [18,19,20], stochastic molecular damage, the developmental and disposable soma theories [17,18,19]. Besides several hallmarks of aging have been defined over the decades comprising 12 main causes of aging: epigenetic alterations, stem cell exhaustion, cellular senescence, genomic instability, telomere attrition, loss of proteostasis, impaired autophagy, deregulated nutrient sensing, altered intracellular communication, dysbiosis, chronic inflammation and mitochondrial dysfunction [22,23]. Since the 1950s after the “ROS theory of aging”, mitochondria have been found to be the main source and first afflicted target of free radicals’ attacks, which contribute to the aging process [23]. The decline in mitochondrial function accelerates aging and is associated with several aging diseases like cancer, Parkinson's and Alzheimer's [22,23,24,25,26,27].
II. Mitochondrial Function and Aging
Nowadays mitochondria are recognized as a metabolic hub, integrating and modulating intracellular signaling to control cell fate, maintenance, proliferation, differentiation, and death [28]. Cell energy production, metabolism, and biosynthesis involve mitochondrial processes. ATP/ADP production and oxidative phosphorylation (OXPHOS) are linked to mitochondrial: biogenesis, fission, fusion, translation and protein synthesis. Fatty acid β-oxidation and amino acid metabolism depend on mitochondrial lipid metabolism and protein import/export. The urea cycle, NADH/NAD and FADH2/FAD production are supported by mitochondrial biogenesis, fission and fusion. The steroid hormone and heme synthesis are connected to mitochondrial translation, protein synthesis, import and export. Phospholipid and pyrimidine synthesis are connected with mitochondrial lipid metabolism and protein import/export. Metabolite transport is integral to mitochondrial biogenesis and lipid metabolism. The previous processes produce: Acetyl-CoA, Pyruvate, Citrate, Succinate, Ketone bodies, Carnitine derivatives, Alpha ketoglutarate, Ubiquinone (Coenzyme Q), Cardiolipin, and Metabolites (e.g., citrate, glutamate, succinate) necessary for cell functions [28]. Cell regulation, signaling, and maintenance involve mitochondrial processes. Calcium buffering is connected to the maintenance of mitochondrial membrane potential. Apoptosis regulation is linked with mitophagy and mitochondrial permeability transition pore (mPTP) regulation. Thermogenesis is related to inter-organellar communication and mitochondrial unfolded protein response (mtUPR), which depends on mtDNA maintenance, replication and repair. Redox signaling depends on ROS production and is linked to innate immune responses. Immune signaling and responses are linked to mPTP regulation. Stem cell maintenance, function and differentiation are associated with mitochondrial biogenesis (mtBio). The products of the mentioned processes include Ions, Gases, and Pro-inflammatory cytokines (e.g., IL-9, IL-6) [28].
Remarkably, disruptions in mitochondrial functions and processes are associated with the onset of age-related diseases and accelerated aging [28,29,30,31,32,33,34,35,36,37,38,39,40,41]. Several cases proof the latter. For instance, a low amount of NAD+ causes mitochondrial dysfunction which promotes the development of cellular senescence [42]. Defects such as mitochondrial: impaired metabolism, swelling, shrinkage, DNA mutations accumulation are associated with age-related phenotypes and diseases such as cancer, neurodegenerative, cardiovascular, and metabolic disorders [43].
Mitochondrial defective generation of ATP via OXPHOS in the ETC is associated with aging [44,45,46,47,48,49]. Mitochondrial regulation of intracellular calcium levels is essential for various cellular processes, including muscle contraction and neurotransmitters release [27,50,51]. Synthesis of lipids like cardiolipin is essential for maintaining mitochondrial membrane integrity [52]. Mitochondrial release cytochrome c initiates the intrinsic apoptosis pathway, crucial in both development and disease [27,49,53,54,55]. Mitochondrial import of nuclear-encoded proteins is critical for their functions [56]. Mitochondrial iron and sulfur synthesis for Fe/S clusters assembly is essential for various enzymatic reactions [57]. Mitochondria are the primary source of ROS, which play a role in cell signaling but also contribute to oxidative stress and aging [27,54,58,59,60]. Mitochondria are involved in the synthesis of steroids, including hormones like cortisol and testosterone [27,56,61]. The selective degradation of mitochondria through autophagy (mitophagy) is crucial for maintaining mitochondrial quality and is associated with duration of lifespan [49,62,63,64,65]. mtUPR stress response maintains mitochondrial function and is linked to longevity and resistance to age-related diseases [64,66,67,68]. Mitochondria influence epigenetic regulation through the tricarboxylic acid (TCA) cycle, providing key metabolites like alpha-ketoglutarate and acetyl-CoA, which modulate histone modifications and DNA methylation, affecting aging processes [69]. Mitochondria associated membranes (MAMs) are involved in endoplasmic reticulum-mitochondria communication, crucial for calcium homeostasis and implicated in aging [70]. Mitochondrial derived peptides such as humanin and MOTS-c have systemic roles and implications in aging [71,72]. The formation of new mitochondria decreases with age reducing cellular energy capacity [49,73,74]. Mitochondria can trigger inflammatory responses, which are linked to aging and age-related conditions like type 2 diabetes [27,60,75]. Heat production by mitochondria regulates energy consumption and cell homeostasis [76]. Mitochondria Cytochrome c protein released during apoptosis activates caspases, leading to cell death [27,49,53,55] Mitochondria are key players in maintaining cellular NAD+ level, which is critical for metabolic processes and declines with aging [77,78].
Mitochondria are known to generate approximately 90% of cellular ROS [79], as they are the cellular centers for ATP production via OXPHOS and are essential for metabolite production, fatty acid β-oxidation, ROS manufacture, apoptosis, and innate immunity, being vital that mitochondrial homeostasis and quality must be maintained [80]. The organization of protein complexes within the ETC is explained by various models. The fluid model suggests that each complex is freely distributed throughout the inner mitochondrial membrane and can diffuse within it according to the mitochondrial requirements [81]. Similarly, the organization of individual enzymatic complexes has also been proposed through different models. Recently, the assembly of respiratory chain supercomplexes (SCs) has been observed. These are functional quaternary structures in which the enzymatic complexes are assembled together [81]. Not only are these SCs responsible for mitochondrial respiration, but they also enhance the efficiency of electron transfer reactions and simultaneously limit the production of ROS. These SCs are embedded in the inner mitochondrial membrane, forming a continuous reaction system where the movement and transfer of protons and electrons generate the energy necessary for the phosphorylation of ADP [81].
The OXPHOS system in mammals consists of five multi-enzymatic protein complexes, two electron carriers, and ATP synthase, all located in the inner mitochondrial membrane. The mitochondrial respiratory chain (MRC) includes the first four complexes (Complexes I-IV), which transfer electrons to create a proton gradient. This gradient is then utilized by the Complex V to drive ATP synthesis. During the TCA cycle, nutrients are catabolized to produce NADH and succinate, which are further converted to NADH2 and FADH2. These molecules donate electrons to Complex I and Complex II, respectively. Coenzyme Q receives these electrons and transfers them to Complex III, then to Cytochrome C, and finally to Complex IV, where they are passed to molecular oxygen [82,83]. Since the proposal of the fluid model for the electron transport chain, it has been observed that the assembly of SCs allows for more efficient electron transfer by enabling the diffusion of coenzyme Q and cytochrome C over shorter distances. Indeed, there is evidence suggesting the presence of channels within these SCs that facilitate the movement of electron carriers between different complexes [84]. This mechanism not only reduces electron leakage but also decreases the formation of ROS, particularly between complexes I and III, where electron leakage is most prevalent [85]. The electron transfer facilitates the pumping of protons from the mitochondrial matrix to the intermembrane space via Complexes I, III, and IV, generating the electrochemical gradient used by Complex V during oxidative phosphorylation, which involves the reduction of molecular oxygen and electron transfer [83].
The F1F0 subunits of ATP synthase are energized by the proton motive force (PMF), which is the sum of the proton gradient and the membrane potential. This PMF drives protons through the F0 part of the enzyme, coupled with the rotation of the F1 part, producing ATP from ADP and inorganic phosphate (Pi) [86]. The transmembrane potential, formed by the proton gradient, is crucial for the discriminatory removal of dysfunctional mitochondria, thereby maintaining mitochondrial homeostasis. ATP levels and mitochondrial membrane polarization are kept within stable ranges to ensure normal cellular function, such as the transport of essential charged compounds into the mitochondria [87].
The primary function of mitochondrial metabolism is ATP production through aerobic substrate oxidation [83]. ATP is synthesized from ADP and inorganic phosphate by ATP synthase, driven by the proton gradient established by glycolysis, the TCA cycle, and OXPHOS. ATP concentrations must remain within a narrow range to maintain cell viability; deviations can lead to degenerative processes. Concurrently, the MRC reduces O2 to H2O in the mitochondrial matrix, where ROS are produced in small quantities, accounting for less than 0.1% of the electrons transferred under normal conditions. A drop in ATP, caused by inadequate generation or excessive consumption, can lead to intracellular acidosis and depletion of pH buffers. This acidosis can activate proteases, nucleases, and lipases, resulting in excessive degradation of cellular components [87].
ROS are generated under specific conditions: (i) when electron transfer is blocked up in the sequence of Fe-S clusters, especially when Complexes I, III, or IV are inhibited by quinone-binding site inhibitors; (ii) during reverse electron transfer from Complex II to Complex I via cytochrome C; and (iii) at the Flavin and Q-binding sites of enzymes involved in electron transfer flavoprotein and cytochrome C oxidoreductase [82]. Additionally, ROS serve as signaling molecules in mitochondria, regulating biological responses such as adaptation to stress, differentiation, and proliferation, thereby influencing the pathological and physiological outcomes of aging, cancer, and immunity [88].
Mitochondrial metabolism exhibits suppressed flexibility in the aged heart, with less capacity for fatty acid oxidation and a dependence on glucose, leading to oxidative damage and promoting aging. The decrease in mitochondrial respiration during aging is caused by reduced activity of respiratory complexes III and IV of the ETC. Additionally, myofibrils reduce their mitochondrial content with approximately 50% reduction in function with aging. This results in increased oxidant production, oxidative damage, and cell death activation via oxidant signaling due to the persistently deficient mitochondria in the aged heart [89]. Cell death results from the final step in the oxidant activation signaling pathway, preceded by injury to mitochondrial components and damage to contiguous organelles, all originating from higher amounts of ROS due to mitochondrial alterations with age. The higher ROS levels could be due to the dysregulation of transcription elements of mitochondrial metabolism during aging [89]. Accordingly, OXPHOS defects lead to hypermetabolism and shorten lifespan in both cells and individuals with mitochondrial disorders [90]. Remarkably, genetic alterations affecting the assembly and organization of MRC complexes can lead to severe mitochondrial disorders, including neurodegenerative diseases. Such genetic changes are linked to aging, as deterioration of these complexes is often observed with age [83]. For example, chronic exposure to rotenone in the brain leads to dopaminergic neuron degeneration, and Leigh syndrome, which is caused by hereditary mutations affecting the OXPHOS system [91].
Mitochondria possess their own DNA, a circular and double-stranded molecule distinct from nuclear DNA, with each mitochondrion containing between 3 and 10 copies. [92]. The mtDNA contains 16.6 kilobases that code for 37 genes involved in mitochondrial translation, oxidative phosphorylation, and has a role in aging and age-associated diseases [93]. mtDNA genes are housed in the mitochondrial matrix and are crucial for several mitochondrial metabolic pathways [94]. Each cell contains thousands of copies of mtDNA, with the number of copies (CN) being subject to changes due to physiological and environmental stimuli [95]. This number is regulated by mitochondrial dynamics processes, such as fission and fusion [96]. As mitochondria proliferate, they increase their biomass and DNA in response to energy demands and coordination with the cell cycle [97]. mtDNA replication and repair ensure its integrity protecting it against mutations [98]. The activity of proteins: TWINKLE helicase, mitochondrial transcription factor A (TFAM) and DNA polymerase gamma, are crucial for the transcription, replication, and protection of the mtDNA [99,100,101]. These proteins have been independently associated with aging. For instance, mutations in TWINKLE are linked to mitochondrial dysfunction, neurodegeneration, and premature aging [102].
mtDNA can accumulate mutations, which impairs mitochondrial function, contributing to cellular aging and degenerative diseases [78]. An accrual of mtDNA mutations in old experimental animals and humans has been proposed to contribute to the development of aging phenotypes [103]. Cell-free mtDNA extracellular release increase has been associated with age-related chronic inflammation which supports the central role of mitochondria in the aging process and age conditions [104].
Mitochondria undergo regular fusion and fission, fundamental processes for energy adaptation and homeostasis [105]. Aging affects mitochondrial quality control due to changes in fusion and fission [78]. The loss of mitochondrial fission in satellite cells due to aging disrupts the ETC, resulting in inefficient OXPHOS, impaired mitophagy, and increased oxidative stress [106]. Optimizing the regulation of mitochondrial quality control processes plays a crucial role in addressing skin aging issues [107].
Mitophagy is a conserved cellular process that removes injured and dysfunctional mitochondria, thereby maintaining mitochondrial quality and homeostasis [105,108,109,110,111,112,113,114,115,116,117]. Mitochondrial homeostasis is strongly linked to age-related disorders and longevity, involving the removal of dysfunctional mitochondria through mitophagy [118]. A key cause of mitochondrial homeostasis loss in aging is the buildup of dysfunctional mitochondria, resulting from cells' reduced ability to clear faulty organelles through mitophagy [119]. The disruption in balance among mitophagy, ROS generation, and mitochondrial damage can initiate, propel, or hasten the aging process, whether in normal aging or in pathological age-associated conditions [33]. Mitophagy is notably compromised in various human conditions, including aging and age-related illnesses such as neurodegenerative diseases, cardiovascular disorders, and cancer [120,121,122,123]. During aging, mitophagy levels decline and negatively impact skeletal muscle performance [124]. Mitophagy is linked to the development of several age-related diseases, including AD, PD, HD, and ALS, with amyloid beta and phosphorylated tau, supporting that mitophagy is a pro-longevity mechanism [122,125,126]. Diminished mitophagy in the vasculature contributes to oxidative stress and the vascular aging phenotype associated with hypertension [127,128,129]. Mitochondrial dysfunction is involved in multiple joint degenerative diseases [108,109,110,111,112,114,115,116,120].
Mitophagy addresses all hallmarks of aging: It neutralizes mitochondrial dysfunction, providing an adaptive quality control strategy that sustains metabolic homeostasis as an inducible stress response in model organisms [58,130]. It reduces mtDNA mutations and DNA damage, supporting genomic stability [131]. Mitophagy lowers oxidative stress, thus reducing harmful epigenetic changes [132]. By reducing ROS production and preventing the accumulation of dysfunctional mitochondria, mitophagy slows telomere shortening and cellular senescence [132,133,134]. Mitophagy maintains protein quality by clearing dysfunctional mitochondria [135]. Mitophagy helps preserve mitochondrial health in stem cells, supporting their regenerative potential [135,136]. Mitophagy supports proper cell signaling and reduces inflammation [137,138].
Enhancement of mitophagy preserves cardiac homeostasis and extends lifespan [139]. Caloric restriction activates mitophagy, which optimizes mitochondrial function and promotes healthy aging [140,141]. In old mice Urolithin A induction of mitophagy attenuates cGAS/STING activation, senescence inflammatory pathway, and diminishes deterioration of neurological function [36]. The mitochondrial drivers of aging are represented in Figure 1.
III. Mitochondrial Drivers of Aging
Mitochondrial Driver of Aging Nº1: ATP Crisis
ATP levels decline with age, as seen in the elderly and patients with Alzheimer’s disease, largely due to mitochondrial dysfunction [142]. In line with this, F1F0 ATP synthase activity decreases in an age-dependent manner in the brain mitochondria of mice, characterized by reduced catalytic activity and increased complex uncoupling. Consequently, there is a lower ATP ratio in the brains of aged mice, attributable to modifications in the MRC. Mitochondria from older mice (16-24 months) show a reduced capacity to convert ADP into ATP compared to younger mice [143]. Furthermore, neurons from a 30-year-old can generate sufficient ATP to survive a short infarct, but by age 70, their diminished ATP capacity can lead to failure of Na+ and Ca2+ plasma membrane pumps, resulting in rapid necrotic cell death. Although the exact mechanisms are not fully understood, mitochondrial bioenergetic capacity declines with age [144]. As a result, the potential for an ATP crisis in neurons increases with age due to the decreased capacity for ATP production [145].
The primary cause of ATP reduction is the decline in OXPHOS efficiency with aging. Aging decreases the expression of both nuclear and mitochondrial genes coding for Complex IV subunits, correlating with a reduced rate of ATP production. This is accompanied by significantly higher concentrations of ADP and Pi in the elderly compared to younger individuals, indicating an age-associated decline in ATP synthase activity [146]. Notably, ATP levels decline significantly with age in the cardiac muscles of humans, mice, and rats, with human calf muscle ATP levels decreasing by approximately 8% per decade. Similarly, older Drosophila melanogaster exhibit about a 50% reduction in ATP levels compared to younger flies. In conditions like progeria and Werner syndrome, which exhibit accelerated aging, fibroblasts show a 50% and 40% reduction in ATP levels, respectively, compared to healthy individuals [147]. This decrease in mitochondrial ATP production impairs homeostasis and can eventually trigger apoptosis, contributing to aging and the progression of age-related disorders [148].
The interplay between ATP and ROS levels significantly impacts the aging process. First, an increase in superoxide levels can occur as a harmful ROS-mediated retrograde response due to ATP synthase inhibition, as seen with ATP5A knockdown in mitochondria [142]. Second, aged mice exhibit decreased F1F0 synthase activity in brain mitochondria due to defective coupling within the protein complex, resulting in lower ATP production and higher superoxide generation, which is linked to aging brain pathology [143]. Third, mitochondrial dysfunction, combined with hormone deficiencies and reduced physical activity in aging, further reduces ATP production, exacerbating these conditions and accelerating aging [149,150]. Fourth, aging is accompanied by toxin accumulation, partly due to ATP synthase inhibition through altered AMPK/mTOR pathway modulation, damaging the brain [142]. Fifth, the inhibition of adenine nucleotide translocase during aging decreases the exchange of ADP for ATP, which typically promotes mitophagy. As mitophagy declines with age, dysfunctional mitochondria accumulate, leading to a further decline in ATP levels [151]. Sixth, the reduced release of ATP in bone cells as an early response to mechanical stimulation impairs the activation of repair processes, resulting in critical damage and membrane injury, thus hindering cellular repair mechanisms essential for maintaining tissue integrity during aging [152].
Higher ATP levels support longer lifespan and age-related muscle function by enhancing repair and proteostasis. However, ATP production declines with age due to accumulating mtDNA mutations, which accelerate aging. Reduced ATP is linked to over 250 pathogenic mtDNA mutations [147].
Mitochondrial Driver of Aging N°2: ETC-SCs Assembly Defects
Aging leads to mitochondrial dysfunction, with reduced complex I and IV levels and loss of supercomplexes, impairing energy production [153]. Incorrect assembly of ETC-SCs fosters the onset of neurodegenerative diseases, genetic abnormalities, heart failure, and cancer, highlighting its essential role in maintaining mitochondrial homeostasis and cellular functions [85]. Also, the coordination of SCs, protein expression, formation and activity, is affected in the aging of skeletal muscle [154]. Aging-related loss of the MICOS complex disrupts mitochondrial structure in muscle, reducing oxidative capacity and contributing to sarcopenia. These structural changes impair cellular metabolism and membrane integrity, impacting muscle function [155].
ETC-SCs structure and function is affected by aging and oxidative stress, which is linked to neurodegenerative diseases [156]. SC assembly decreases in the cortex with age, reducing mitochondrial efficiency in aged brains [157]. In PD models, SCs, particularly complexes I and IV, show lower abundance and activity during neurodegeneration [158]. Mutations in PINK1 and DJ-1 reduce SC assembly and activity, leading to cellular energy deficits [159]. AD studies reveal lower levels of specific SC subunits: CII, CIII and CV [160].
Mitochondrial Driver of Aging N°3: ROS
Elevated ROS levels can cause oxidative stress and severe damage to the cell, organelle membranes, DNA, lipids, and proteins. This damage contributes to the aging phenotype. Oxidative damage has been linked to premature aging and neurodegenerative diseases [161]. ROS triggers aging events, including imbalanced ECM homeostasis, accumulation of senescent fibroblasts, loss of cell identity, and chronic inflammation mediated [162]. The depletion of oxidative stress-responsive serine-rich protein 1 leads to a shorter lifespan [163].
According to mitochondrial aging and ROS theories, ROS contribute to human aging by accumulating mtDNA mutations [23]. Extrinsic factors also increase ROS production, including UV radiation, air pollutants, alcohol consumption, cigarette smoking, and xenobiotics. All these factors promote ROS generation [23].
Mitochondrial Driver of Aging N°4: mtDNA Damage
The primary intrinsic causes of mtDNA damage are ROS and replication errors. Accumulated mtDNA mutations result in deficiencies in the respiratory chain, which increases the production of DNA-damaging free radicals, contributing to the aging process in humans [23]. mtROS promotes mitochondrial dysfunction by suppressing TFAM-mediated mtDNA maintenance, leading to reduced mitochondrial energy metabolism and increased cytokine release [164]. Specific mtDNA damage leads to exacerbated mtDNA turnover, independent of canonical macroautophagy, but dependent on lysosomal function and ATG5 [165]. Increased mtDNA mutational load, including the common 4977 bp deletion, disrupts mitochondrial function and is linked to aging [89,166,167,168]. Oxidative stress and damaged DNA binding are strongly associated with Parkinson's disease [169]. Oxidative stress from ROS near mtDNA drives deletions, causing genomic instability in aging tissues such as brain, heart, muscle, and liver [80,105,170,171,172,173,174,175,176].
Over time, these factors introduce new mtDNA variants, elevating the mutation rate. This interaction forms a tightly coupled cycle where ROS produce mtDNA mutations that, in turn, generate more ROS. Similarly, replication errors by POLGA mutate mtDNA, affecting the ETC and producing more ROS. These two intrinsic factors interact to enhance ROS in a vicious cycle of mtDNA mutations [177].
Mitochondria from mtDNA mutator mice show elevated ROS production under physiological substrates and during active oxidative phosphorylation, correlating with increased oxidative damage markers in vivo and directly linking mtDNA mutations to premature aging [178]. Thus, a vicious cycle of mtDNA mutations and ROS generation ensues [177]. Furthermore, ROS not only induce mutations in mtDNA but also in nuclear DNA, exacerbating cellular damage [179]. While ROS can be deleterious, they also play a role in cell signaling, gene expression, and apoptosis [180].
When cells divide with unrepaired or miss repaired damage, this can lead to mutations. These mutations often manifest as Guanine to Thymine transversions, oxidized pyrimidines and purines, single-strand breaks, and abasic sites [181].
ROS can induce large-scale deletions up to 8 kb, and may affect multiple genes. Such mutations can occur early in development, leading to the propagation of the same deletion across all cells in affected tissues [182]. ROS can cause non-conventional single-strand breaks, abasic sites, intra- and interstrand DNA crosslinks, protein-DNA adducts, and both bulky and non-bulky base changes [181]. ROS can also affect nuclear DNA, proteins, RNA, and lipids, contributing to the physiology of aging [183]. At high concentrations, ROS are extremely damaging and can cause cell damage or death. Oxidative stress can lead to lipid peroxidation, DNA damage, protein oxidation, and enzyme inhibition [184]. Hydroperoxyl radicals, which are more reactive than superoxide radicals, can initiate the oxidation of polyunsaturated phospholipids, impairing membrane function [185].
Evidence robustly links the age-associated increase of oxidative damage to lipids, proteins, and DNA observed across various organisms, including humans. With age, imbalances in ROS generation and elimination cause oxidative stress, leading to the progressive accumulation of oxidative damage, which declines the efficiency of cellular processes [22]. This association between ROS and aging extends to age-related diseases such as Familial Alzheimer's where elevated ROS generation is a key factor [186], as well as frailty, sarcopenia, cardiovascular diseases, chronic obstructive pulmonary disease, neurodegenerative diseases, and chronic kidney disease. Reactive oxygen-nitrogen species (RONS) induce cellular senescence and the expression of matrix metalloproteinases, which are linked to age-related and chronic diseases like lung emphysema, cancer, osteoarthritis, Alzheimer's, and atherosclerosis [22].
Mitochondrial Driver of Aging N°5: mtDNA Mutations
mtDNA is transcriptionally and physically distinct from nuclear DNA. It is circular, spans 16.5 kb, and exists in thousands of copies per cell, encoding 13 proteins essential for oxidative phosphorylation, along with 2 rRNAs and 22 tRNAs [187]. Unlike nuclear DNA, mtDNA lacks chromatin protection and is not enclosed by a nuclear envelope. Instead, it associates with TFAM and contains minimal noncoding sequences (~3%) [188]. Due to its structure and exposure to ROS, mtDNA is highly susceptible to damage from replication errors, oxidative stress, and faulty repair mechanisms. This results in the accumulation of mutations with age, disease, and environmental insults [189]. Over 400 mtDNA mutations have been linked to human diseases, including metabolic, neuromuscular, and degenerative disorders [92].
mtDNA mutations promote a proliferative advantage in some cells, allowing mutant variants to outcompete wild-type mtDNA through replicative segregation [190]. For instance, mtDNA loss in T47D and MCF-7 breast cancer cells impairs proliferation, anchorage-independent growth, and tumorigenicity [191]. Dysfunction is exacerbated by large-scale deletions (~5 kb), frequently observed in aging tissues and mitochondrial diseases. These deletions contribute to pathologies such as sarcopenia and cytochrome c oxidase deficiency, as seen in ragged red fibers in aging muscle. In tissues like colonic epithelium, clonal expansion of pre-existing mutations—rather than de novo mutation—drives dysfunction [92]. The accumulation of somatic mtDNA mutations with age has been documented across multiple tissues, especially in postmitotic cells like neurons. Single-nucleotide variants rise linearly with age in brain regions such as the prefrontal cortex and hippocampus, mirroring changes seen in neurodegeneration [192]. Even with active repair, lifelong mutation accrual generates cellular mosaicism and promotes age-related decline [193,194,195] Similar expansion patterns have been noted in blood and skin fibroblasts, increasing both heteroplasmic and homoplasmic mutation loads [92].
Replication errors and ROS establish a feedback loop that drives mitochondrial decline during aging. While some mutations arise early, most accumulate gradually, increasing heteroplasmy and impairing function. Even low-frequency de novo mutations can contribute to aging and disease without reaching the clinical threshold [177]. Cytochrome c oxidase mutations, for instance, accelerate liver aging in mice [196]. Dysfunctional nuclear-encoded mtDNA replication factors—including PolG2, RNase H1, TFAM, MGME1, Twinkle, and polymerase γ—further intensify this decline [177].
The causative role of mtDNA mutations in aging was definitively demonstrated in the mutator mouse model, which carries a proofreading-deficient polymerase γ (POLGA). These mice accumulate random point mutations in mtDNA, leading to early-onset aging phenotypes by six months, including weight loss, infertility, alopecia, blindness, deafness, anemia, muscular atrophy, and kyphosis [43,197]. Notably, increased mtDNA mutation burden in these mice exacerbates small intestinal aging via mtUPR activation—a phenotype reversible by NAD+ precursor NMN supplementation [198].
Co-replication of mutant and wild-type mtDNA generates heteroplasmy, leading to mitochondrial dysfunction, shortened lifespan, and premature aging due to respiratory chain failure [43]. POLGA mutations further accelerate mtDNA damage by introducing replication and proofreading defects. Twinkle helicase mutations, single-stranded breaks, and ROS compound this effect, linking stochastic mtDNA damage to cardiac aging [199].
Ultimately, mtDNA mutations drive age-related pathology by disrupting metabolism, fragmenting mitochondria, amplifying mutation loads, activating inflammatory responses, and promoting cellular senescence. These processes underlie diverse diseases, including cancer, neurodegeneration, cardiovascular disease, and metabolic syndrome [43]. Impaired mitochondrial bioenergetics—a hallmark of aging—is marked by reduced electron transport chain (ETC) function, elevated ROS, increased mtDNA mutagenesis, enhanced proapoptotic signaling, and defective cytosolic calcium buffering [166].
Large-scale mtDNA deletions (~5 kb) accumulate with age, especially in postmitotic tissues such as brain, skeletal muscle, and heart, and are linked to mitochondrial diseases, COX deficiency in ragged red fibers, and sarcopenia. In aging colon, mtDNA dysfunction results primarily from clonal expansion of pre-existing mutations rather than increased de novo mutations. Similar clonal expansions occur in blood and skin fibroblasts of elderly individuals, increasing both homoplasmic and heteroplasmic mtDNA variant loads [92]. The functional consequences of mtDNA deletions depend on their expansion dynamics. A low-frequency deletion with reduced mtDNA copy number is less impactful than a pathogenic variant that has undergone clonal amplification [200]. Such expansion produces heteroplasmic populations that progressively disrupt ETC function, elevate ROS, and shorten lifespan [43]. Mitochondrial deletions that impair function but evade mitophagy may confer a replicative advantage, expanding clonally and outcompeting healthy mitochondria [201]. This clonal dominance results in cellular energy deficits, particularly in high-demand tissues [43].
Clonal expansion of mtDNA deletions is especially evident in neurodegeneration. In PD, neurons from the substantia nigra show increased mtDNA deletions per cell, and heteroplasmic divergence within neurons indicates somatic clonal expansion of these deletions [202]. In AD patients under age 75, mtDNA deletions across multiple brain regions are up to 15-fold higher compared to age-matched controls without dementia [203]. In the context of tumorigenesis, a mouse model of intestinal cancer demonstrates that elevated mtDNA mutagenesis, along with metabolic network remodeling, promotes growth advantages in transformed cells [204].
mtDNA deletions also originate from defective replication fidelity. Mutations in POLGA impair proofreading and promote accumulation of deletions, contributing to aging phenotypes, cardiac hypertrophy, and ETC dysfunction [199]. ROS exacerbate this by oxidizing POLGA and decreasing its fidelity, leading to replication-induced mtDNA damage [199]. Additional disruptions in mtDNA maintenance—such as those involving Twinkle helicase and single-strand breaks—amplify mutagenesis under oxidative conditions [199].
These findings collectively support that mtDNA deletions expand through replication errors and selective pressures. This process—not just the initial mutational event—constitutes a key mechanism of mitochondrial dysfunction during aging [43,92,199,200,202,203,204].
mtDNA accumulates mutations with age across tissues, contributing to mitochondrial dysfunction and age-related decline. In mice, mutation burden rises steadily with age, with quantifiable increases across tissues [205]. Pedigree-based and phylogenetic analyses corroborate age-dependent increases in mutation frequency [206]. The mtGenome mutation rate is estimated at 0.058 mutations/site/Myr, with detectable heteroplasmy in 5.8% of individuals—both markers of mitochondrial aging [207]. In the mtDNA control region, mutation rates reach up to 0.32 mutations/site/Myr, highlighting this region’s vulnerability [208]. Mutations in the D-loop and other control elements impair mtDNA transcription and replication, destabilizing mitochondrial homeostasis [147,209].
Polymerase γ (PolG) fidelity provides a baseline for estimating mtDNA mutation rates. Error rates during replication, when modeled, show exponential mutation accumulation with age—from 162 mutations by age 10 to over 1,000 by age 70 [177]. Spontaneous replication errors also generate deletions and point mutations [143,210]. ROS further exacerbates these errors by oxidizing PolG and compromising proofreading [211].
mtDNA mutation accumulation alters mitochondrial protein expression, particularly affecting respiratory complexes I and IV, leading to progressive ETC dysfunction [212]. A COX3 mutation, for example, increases ROS and accelerates liver aging via fibrosis and metabolic impairment [196]. Differences in mtDNA mutation burden across tissues may explain cellular aging heterogeneity [189]. Cross-species comparisons indicate that lower mtDNA mutation rates are associated with larger body mass and longer lifespans, as seen in birds [213]. In humans, mutation rates correlate strongly with neurodegeneration; age-related mtDNA mutations are implicated in AD and other disorders [43,177,214].
Ultimately, the accumulation of mtDNA mutations, driven by replication errors, oxidative damage, and impaired repair capacity, is central to mitochondrial aging. These mutations disrupt ETC integrity, reduce ATP production, and propagate dysfunction at the cellular and tissue levels [98,215]. The mutations rate and accumulation are represented in Figure 2.
Mitochondrial Driver of Aging N°6: mtDNA Heteroplasmy
Heteroplasmy is the coexistence of multiple mtDNA variants within the same cell or mitochondrion, often associated with unfavorable conditions, diseases, and aging [216,217,218,219,220]. Wild-type and mutated mtDNA coexist within the same mitochondria, creating heteroplasmy. Once heteroplasmy surpasses around 60%, mitochondrial dysfunction is triggered, causing cellular atrophy or death and contributing to aging and related diseases [221,222,223]. Heteroplasmy indicates that a mutation is present in a subset of all mtDNA copies, and when mutant mtDNA surpasses a threshold of 60–80%, pathological phenotypes emerge [43].
The clinical severity of mitochondrial disorders depends heavily on heteroplasmy extent and mutation type, with thresholds typically ranging from 60% to 90% mutant mtDNA before symptoms appear [224]. Increasing heteroplasmy disrupts mitochondrial metabolism by reducing acetyl-CoA and altering α-ketoglutarate levels, thereby affecting histone acetylation and methylation. These epigenetic changes induce gene expression shifts linked to aging and related diseases, especially at heteroplasmy levels above 70% [225]. Heteroplasmic somatic SNVs sharply increase after age 70, correlating with oxidative stress and accumulating through somatic mutagenesis, while indels remain stable, maternally inherited, and regulated by nuclear genes controlling mtDNA maintenance, suggesting rising heteroplasmy drives aging via cumulative cellular damage [99]. Moreover, polymorphic mtDNA variants are linked to higher risks of cancer and neurodegenerative diseases like Parkinson’s and Alzheimer’s, accumulating in tissues during aging in both animals and humans [103].
The regulation and control of mtDNA heteroplasmy are critical, with cell-specific heteroplasmy patterns sustaining metabolic capacity throughout normal aging [226,227,228,229,230,231]. Disruption of this control worsens during aging and cancer progression, exacerbating mtDNA mutation effects [170,232,233,234,235,236]. Differences in disease progression and aging rates among cells within an organism may be partly explained by heterogeneous mtDNA mutation levels and distributions [189]. Heteroplasmy plays a key role in aging by impairing mitochondrial function; as mutant mtDNA rises, mitochondrial dysfunction accelerates, promoting aging [237]. Aging-related mtDNA damage, including deletions, contributes to sarcopenia in humans, with clonal expansion of deletions mainly observed in long-lived species but not in short-lived ones like C. elegans [221]. These deletions accumulate especially in post-mitotic tissues, causing bioenergetic defects once pathogenic mtDNA surpasses a threshold [180,185,211,224,238,239].
Aging and mtDNA maintenance disorders cause deletions that clonally expand within muscle fibers, leading to respiratory chain deficiency and impaired mitochondrial function, promoting muscle dysfunction and age-related decline [222]. Heteroplasmy, with increasing mtDNA mutations over time, contributes to aging by causing mitochondrial dysfunction, especially in muscle, when mutant mtDNA exceeds 70–90%. This accumulation accelerates conditions like sarcopenia [223]. mtDNA haplogroups represent heteroplasmic variants that correlate with lifespan and risk for age-related diseases, including Parkinson’s, sarcopenia, heart failure, diabetes, and cancer [92]. For example, haplogroup J links to reduced mtDNA damage and longevity beyond 85 years, while D4b2b, D4a, and D5 are associated with exceptional longevity. Additionally, MOTS-c polymorphisms regulate insulin sensitivity and metabolic balance, potentially promoting longevity by preventing insulin resistance and obesity [71,240].
Mitochondrial Driver of Aging N°7: mtDNA Repair Mechanisms
Mitochondria replicate throughout life, but replication errors and repair inefficiencies increase with age, leading to a progressive accumulation of mtDNA mutations. The diminished activity of mitochondrial DNA repair systems further accelerates this mutational burden, making mtDNA more vulnerable than nuclear DNA during aging [241]. These repair mechanisms are essential for correcting specific mitochondrial lesions. When these pathways are compromised, mitochondrial function deteriorates and the risk of cell death rises. Defective repair has been directly linked to age-associated pathologies, particularly cancer [242]. Faulty repair also facilitates the formation of large-scale mtDNA deletions, which eliminate critical genes required for oxidative phosphorylation. These deletions severely impair mitochondrial bioenergetics and are associated with aging, cancer, and mitochondrial diseases [176]. The scenario worsens when mitophagy—the mechanism for clearing defective mitochondria—also declines with age. In such conditions, damaged mitochondria harboring unrepaired or deleted mtDNA persist within cells, amplifying dysfunction and contributing to the progression of neurodegeneration, cancer, inflammation, and metabolic disorders [118].
Mitochondrial DNA sustains more point mutations and deletions than nuclear DNA, likely due to its limited repair capacity. Unlike the nucleus, mitochondria lack nucleotide excision repair (NER), a key pathway for removing bulky lesions such as pyrimidine dimers, and they appear to have a less developed mismatch repair (MMR) system [243,244,245]. This makes base excision repair (BER) the principal repair mechanism for mtDNA, responsible for correcting oxidative base lesions and single-strand breaks via DNA glycosylases and end-processing enzymes [232].
BER efficiency has been directly linked to lifespan. In a comparative study across eight mammalian species differing 13-fold in longevity, only mitochondrial—not nuclear—BER activity correlated with species lifespan, highlighting its role in promoting mitochondrial stability and organismal longevity [246]. However, BER activity must be tightly regulated. Incomplete BER leads to the buildup of toxic intermediates and DNA breaks, which drive neurodegeneration with age. For example, in Parkinson’s disease models, NTH-1 deficiency reduced neuronal loss by lowering BER intermediates and improving mitochondrial function [247]. Excessive BER activity can also cause mitochondrial stress and dysfunction, while moderate reductions may preserve transcriptional activity and delay aging-related decline [248].
Experimental models reinforce these links. In D-galactose-treated rats, aging suppressed mtDNA repair enzymes pol γ and OGG1, leading to a rise in the 4834 bp mtDNA deletion associated with presbycusis and impaired auditory processing [249]. Further, TFAM overexpression in these models increased mtDNA replication but also amplified mutational burden, exacerbating age-related hearing loss [250]. When BER is insufficient, mitochondria rely on backup mechanisms to manage damage. One such pathway is microhomology-mediated end joining (MMEJ), which repairs double-strand breaks using short homologous sequences. This process depends on mitochondrial Mre11, a protein involved in recombination and telomere stability [233,251]. Together, these repair pathways—while essential—are inherently error-prone, and their dysfunction with age accelerates mitochondrial decline, contributing to the broader aging process [252].
mtDNA is particularly vulnerable to oxidative stress, which intensifies with age and drives the accumulation of molecular damage in post-mitotic tissues. Oxidative lesions to cellular macromolecules, including lipids, proteins, and DNA, compromise mitochondrial integrity and promote functional decline [253]. This ongoing oxidative burden contributes to a progressive accumulation of somatic mtDNA mutations over the lifespan. Evidence suggests that the rate of mutation accumulation may correlate with organismal lifespan, implying that quantitative features of the genome are linked to aging trajectories and longevity potential [254]. In specific tissues, such as the auditory system, aging-related oxidative stress has been shown to upregulate p66Shc and its phosphorylated form Ser36-P-p66Shc. These alterations are associated with increased levels of a specific 3873-bp mtDNA deletion in the inner ear, implicating redox-sensitive pathways in tissue-specific mitochondrial genome instability [255].
Mitochondrial Driver of Aging N°8: mtDNA Copy Number Decrease
mtDNA-CN is a critical regulator of cellular metabolism and aging. High-energy-demand tissues such as the brain, heart, and skeletal muscle maintain elevated mtDNA-CN relative to low-demand tissues like blood [256,257]. Declines in mtDNA-CN reduce oxidative phosphorylation (OXPHOS) capacity [258], impairing ATP synthesis and altering reactive oxygen species (ROS) levels [259]. These shifts influence energy homeostasis and link mtDNA-CN directly to age-associated metabolic dysfunction. Regulatory factors such as TFAM and PGC-1α modulate mtDNA-CN, but their dysregulation, particularly TFAM overactivity, contributes to age-related sarcopenia [260]. Experimental models offer strong evidence connecting mtDNA-CN and mutation load with aging phenotypes. In mtDNA “depleter mice,” inducible expression of a dominant-negative polymerase reduces mtDNA-CN, producing phenotypes of premature aging—hair loss, dermal wrinkling, and altered expression of aging biomarkers including IGF1R, VEGF, KLOTHO, and MRPS5 [258,261,262]. Complementarily, mtDNA “mutator mice” harbor defective polymerase gamma, leading to random point mutations and deletions. These mice exhibit accelerated aging traits—muscle atrophy, weight loss, infertility—and a median lifespan of just 13 months [43,92,197]. Mutation loads in mutator mice reach 20-fold above normal with 25–30% deletions [173], exceeding mitochondrial tolerance thresholds and disrupting function even before the canonical 60–90% dysfunction level is reached.
Threshold effects are especially evident in postmitotic tissues. In muscle and brain, mtDNA deletions below 30% are often tolerated, but deletions above 70% induce respiratory chain failure. Aged muscle fibers can accumulate mutation burdens between 49–94%, reinforcing the concept of a mutation threshold for mitochondrial decline [263]. In parallel, reductions in mtDNA-CN contribute to aging-related cellular dysfunction, independent of heteroplasmy [199]. Clinically, altered mtDNA-CN is implicated in numerous age-related diseases. In mtDNA depletion syndromes, sharply reduced copy number correlates with disease severity and progression [264,265,266], while higher mtDNA-CN is often associated with milder phenotypes and compensatory OXPHOS activity [267]. mtDNA-CN alterations are also found in cancer, although trends vary across tumor types, with both increases and decreases reported [101]. Broadly, aging individuals with lower mtDNA-CN exhibit worse clinical outcomes. Population studies show that older adults with higher mtDNA-CN display improved health and survival [89]. In middle age, reduced leukocyte mtDNA-CN strongly predicts increased all-cause mortality, particularly from cancer and cardiovascular disease—two major age-related causes of death [268].
Reversal of mitochondrial DNA depletion restores function and mitigates aging phenotypes. In the mtDNA depleter mouse model, deactivating the dominant-negative polymerase reestablishes mtDNA copy number, reversing hair loss, skin aging, and inflammatory markers—effectively rejuvenating the organism [258,261,262]. These results indicate that mtDNA damage alone may not be the primary driver of aging. Heterozygous mutator mice, despite increased mtDNA mutations, display normal lifespan and phenotype, while homozygous mutants exhibit early organ failure due to bioenergetic collapse, not accelerated aging [92]. Notably, aged humans accumulate fewer mtDNA mutations than wild-type aged mice, further supporting energy deficit as the dominant factor in functional decline.
Restoration of mtDNA copy number occurs via endogenous replication or horizontal mitochondrial transfer, accompanied by proportional increases in mtDNA variants and broad nuclear epigenomic and transcriptomic shifts—reflecting mito-nuclear signaling [269]. Functionally, mtDNA-CN restoration improves mitochondrial performance and slows vascular aging in mice [270].
A progressive decline in mtDNA-CN is a defining feature of mitochondrial aging across multiple tissues. Blood-based studies show modest yet significant reductions in mtDNA-CN with age, particularly beginning in the fifth decade of life, continuing at a rate of several percent per decade [89,271,272,273]. Individuals over 58 with low mtDNA-CN exhibit increased mortality and deteriorated cognitive and physical function [89], while in middle-aged cohorts, lower mtDNA-CN significantly predicts higher mortality and reduced cumulative survival [268]. mtDNA-CN is also linked to age-associated physiological traits, accounting for 22.6% of deletion frequency and 10.4% of lean mass variability [274]. In participants under 65, mtDNA-CN slightly increases with age, but declines after 65, where lower levels are associated with greater incidence of obesity, hypertension, diabetes, and hyperlipidemia [275].
In neurodegenerative conditions, mtDNA-CN reductions are particularly severe. Alzheimer’s disease patients show a 30–50% decrease in the frontal cortex [276,277], while Parkinson’s disease is marked by mtDNA depletion in dopaminergic neurons, correlating with mitochondrial bioenergetic deficits [278,279]. Similar mtDNA loss occurs in zebrafish retinas during age-related neurodegeneration, impairing mitochondrial mass, function, and energy output [199].
Cardiovascular aging is also tied to mtDNA-CN. Lower mtDNA-CN associates with increased risk of coronary artery disease and sudden cardiac death, particularly among smokers, where elevated ROS production correlates with diminished mtDNA-CN [280,281,282]. In murine pressure-overload heart failure models, overexpression of TFAM or Twinkle restores mtDNA-CN, attenuates ventricular fibrosis, reduces oxidative stress, and enhances cardiac performance [283,284]. Moreover, mtDNA-CN positively correlates with telomere length and endothelial function; regression analyses confirm an inverse relationship between age, mtDNA-CN, and vascular health [285]. Mitochondrial dysfunction in aging hearts includes mtDNA damage (mutations, deletions, reduced mtDNA-CN), strongly linked to cardiovascular disease and sudden cardiac death [273,282,286,287]. Increasing mtDNA-CN via TFAM/Twinkle enhances cardiac function and reduces fibrosis [283,284].
Mechanistically, disrupting adenosine N6-methylation in mtDNA reduces mtDNA-CN and transcript levels, impairing oxidative phosphorylation, increasing oxidative stress, and shortening lifespan [288]. Aging-related ROS accumulation further drives a decline in mtDNA-CN by inducing deletions and point mutations, contributing to mitochondrial dysfunction, energetic failure, and cell death [199]. Experimental reduction of mitochondrial single-stranded DNA-binding protein (mtSSB), crucial for replication, accelerates reproductive aging and reduces mtDNA-CN, emphasizing the importance of genome maintenance under oxidative stress [289].
Mitochondrial Driver of Aging N°9 & 10: Mito Dynamics Deficiency
Mitochondrial dynamics—fusion and fission—are critical for maintaining mitochondrial integrity, bioenergetics, and tissue homeostasis during aging. Disruption of these remodeling processes contributes to mitochondrial dysfunction, oxidative stress, and cellular senescence across multiple organs [121,189,290]. Impaired dynamics are observed in hyperglycemic models, where they trigger vascular complications and represent therapeutic targets for diabetic pathology [291].
In aging skeletal muscle, a marked dysregulation of fusion-fission balance is evident. Mfn2 content drops by 20–30% in aged rodents (35 months) compared to young (5 months) [292,293,294], and OPA1 levels are significantly lower in older humans (~67 years) [295,296]. Drp1 expression rises in aged muscle (22–24 months), indicating excessive fission [297,298], while Drp1 deletion leads to 40–50% muscle mass loss [299,300,301]. Parkin levels decline by up to 50% in aged rodent muscle, and the Parkin/VDAC ratio is significantly reduced in older human muscle [302,303,304,305,306,307,308,309,310]. Skin aging similarly reflects dynamic imbalance. Aged keratinocytes display reduced mitochondrial connectivity and a fragmented network [311]. UVB exposure upregulates Drp1 and downregulates Mfn1/2, promoting mitochondrial fragmentation [312], while TFAM-depleted keratinocytes show decreased ATP, increased ROS, and dysfunctional morphology [313]. Dysfunctional mitochondrial dynamics increases ROS production in aged skin cells, leading to oxidative damage and accelerating cellular senescence [314]. Aged melanocytes from sun-exposed areas exhibit 1.5–2x higher ROS levels [189,290], and UV exposure reduces ΔΨM and mitochondrial mass [315]. Aged fibroblasts show increased proton leakage, baseline respiration, and impaired complex II activity, contributing to energetic inefficiency [316,317]. In cardiac tissue, aging leads to decreased Mfn1/2, increased OPA1 and Drp1, impaired mitophagy (via Parkin loss), and increased ROS, resulting in cardiac dysfunction [318,319,320,321]. Fis1 knockdown or MARCH5 loss promotes senescence, which is rescued by Drp1 overexpression, reinforcing the need for balanced [322,323,324]. Mitochondrial dysfunction in aging hearts includes mtDNA damage (mutations, deletions, reduced mtDNA-CN), strongly linked to cardiovascular disease and sudden cardiac death [273,282,286,287]. Increasing mtDNA-CN via TFAM/Twinkle enhances cardiac function and reduces fibrosis [283,284].
In Drosophila, age-related increases in Fis1 (especially LL isoform) induce mitochondrial enlargement, 20–25% decreased respiration and ATP production, reduced complex IV activity, elevated ROS, and protein aggregates—demonstrating conserved dynamic deficiencies in aging [325]. Similar disruptions occur in ischemic models of stroke and peripheral artery disease, with excessive fission and respiratory defects [326,327].
Brain and immune aging are also impacted. Unregulated dynamics compromise neuronal self-renewal and contribute to neurodegeneration via mtDNA damage [328]. In T-cells, aging promotes mitochondrial STAT3 (mitoSTAT3) localization, altering dynamics, function, and cytokine production [329].
Therapeutically, mitochondrial dynamics remain plastic. One week of exercise restores OPA1 levels in aged mouse muscle [295]. AMPK activation mimics exercise, preserving dynamic function and muscle responsiveness [330]. In satellite cells, fusion inhibition or fission activation reverses aging-related regenerative decline [106]. PGAM5 deficiency, though initially increasing mitochondrial length, mass, mtDNA, and ATP, ultimately triggers ROS, mTOR, and senescence markers (p16^Ink4a ↑5x, γH2AX ↑2x), correlating with reduced survival in 50% of 18-month-old mice [331].
Together, these data confirm that loss of mitochondrial dynamic homeostasis is a core mechanism of mitochondrial aging, driving multi-system dysfunction and frailty.
Mitochondrial Driver of Aging N°11: Mito-Biogenesis Deficiency
Aging is strongly associated with a progressive decline in mitochondrial biogenesis (MB), contributing to functional deterioration across multiple organ systems. This is evidenced by a 20–30% reduction in canonical MB markers—such as citrate synthase activity, cytochrome c protein levels, and mtDNA content—along with a 15–40% decrease in respiratory capacity, reflecting impaired mitochondrial function relative to biogenic capacity [332]. More severe impairments include a 40–60% reduction in regulatory proteins such as PGC-1α, NRF1, NRF2, and TFAM; a ~50% decline in mtDNA copy number; a ~30% reduction in ATP production; and a 2-fold increase in oxidative stress [333]. Additional studies show reduced expression of PGC-1α and Sirt3, increased mitochondrial fragmentation, and elevated ROS in aged tissues, effects partially mitigated by calorie restriction [334,335].
The aging-associated decline in SIRT1, both transcriptionally and post-transcriptionally, is a key factor impairing mitochondrial biogenesis, thereby accelerating the onset of age-related diseases [148]. This loss of mitochondrial integrity is compounded by increased oxidative damage and mtDNA instability [336,337]. Biogenesis itself entails tightly regulated mtDNA replication and coordinated expression of both nuclear- and mitochondrial-encoded proteins that form the oxidative phosphorylation (OXPHOS) complexes [174]. This process is orchestrated by PGC-1α, which activates NRF1 and NRF2, leading to the expression of TFAM and other transcriptional coactivators essential for mtDNA replication and mitochondrial protein synthesis [338]. Fusion and fission events are integral to this process, allowing the incorporation of lipids and proteins into newly formed mitochondrial sub-compartments [339].
Importantly, a disruption in the balance between MB and mitophagy results in the accumulation of dysfunctional mitochondria, contributing to cellular degeneration. For example, in aging C57BL/6 mouse cochlea, MB-mitophagy imbalance induces oxidative stress and damages cochlear hair cells [172]. Inhibition of MB signaling pathways impairs mitophagy, and reduced PGC-1α—often secondary to SIRT1 downregulation—hinders PINK1/Parkin-mediated mitophagy, thus accelerating the aging process [23,105]. Tissue-specific consequences of MB decline are increasingly recognized. In cardiac tissue, reduced mitochondrial biogenesis underlies myocardial injury and heart failure, with PGC-1α and NRF1/2 playing central roles in regulating mitochondrial turnover and mtDNA repair [340,341]. Similarly, in the aging kidney, GLIS1 has been identified as a modulator of mitochondrial quality control via PGC-1α regulation, offering a potential therapeutic target for preventing senescence and renal fibrosis [342].
Interventions aimed at restoring MB hold therapeutic promise. For instance, pentoxifylline improves antioxidant defenses and stimulates mitochondrial biogenesis in D-galactose-induced aging models via activation of the cAMP-CREB pathway, upregulating Nrf2 and PGC-1α [343]. Likewise, overexpression of TOM70 increases mtDNA content by 33% and protein biogenesis by 40%, resulting in enhanced lifespan, whereas TOM70 deletion leads to decreased mitochondrial membrane potential (–50%) and protein levels (–60%), accelerating cellular aging [344]. Exercise training also exerts protective effects by improving myocardial energy metabolism and promoting mitochondrial biogenesis [341,345,346].
Finally, understanding the intricate molecular crosstalk between mitochondrial biogenesis and quality control processes is essential for designing strategies to preserve cellular homeostasis during aging and to combat age-related pathologies [347].
Mitochondrial Driver of Aging N°12: Mitophagy Deficiency
Mitophagy, the selective removal of damaged mitochondria, is essential for mitochondrial quality control and cellular homeostasis, and its efficiency varies with age and cell type [347]. It operates through two primary pathways:
1. Ubiquitin-Dependent Mitophagy: Upon mitochondrial depolarization, PINK1 accumulates on the outer mitochondrial membrane, recruits cytosolic PARKIN, and triggers ubiquitination of mitochondrial proteins. These are recognized by ubiquitin-binding adaptors such as p62/SQSTM1 and OPTN, which link to LC3 and the autophagy machinery for degradation [214].
2. Ubiquitin-Independent Mitophagy: Mitochondrial receptors such as NIX and BNIP3 interact directly with LC3, especially under hypoxia or during erythrocyte maturation [348].
Defective mitophagy leads to ROS accumulation, mtDNA mutations, and mitochondrial dysfunction—hallmarks observed in aging and age-related diseases including Parkinson’s, Alzheimer’s, sarcopenia, and chronic kidney disease [80,122,123,125]. Genetic defects in PINK1, PARKIN, or nuclear-encoded regulators disrupt mitophagy, increasing pathological mtDNA heteroplasmy and accelerating mitochondrial decline [118,139,171].
With aging, mitophagy declines across multiple tissues. In skeletal muscle, mitophagy-related markers increase paradoxically, with PINK1 and PARKIN rising 3.6-fold and 1.4-fold, respectively, and mitochondrial ubiquitination increasing 1.5-fold, while mitochondrial function deteriorates—evidenced by a 64% reduction in citrate synthase activity and 85% loss of COXIV content. Fis1 increases 14-fold, suggesting altered fission dynamics. PGC-1α overexpression mitigates these effects, reducing PINK1 and PARKIN levels by ~50–60%, lowering ubiquitination by 20%, and restoring mitochondrial function and antioxidant capacity [166]. Experimental models show that impaired mitophagy accelerates aging: in yeast and C. elegans, mitophagy deficiency leads to ROS accumulation and shortened lifespan, while its enhancement in flies prolongs life, and in mice mitigates sarcopenia and oxidative stress—effects also seen with caloric restriction and spermidine supplementation [33].
Mitophagy is regulated by AMPK, SIRT1, and mTOR signaling, all of which decline with age. Loss-of-function in PINK1, PARKIN, or SIRT1 worsens neurodegeneration and muscle atrophy. Age-related reduction in lysosomal function and autophagic flux in muscle and liver further compromise mitophagy, highlighting its essential role in tissue maintenance and systemic aging [179].
Mitophagy efficiency changes with age and varies across tissues and species. Loss-of-function of HKDC1 accelerates DNA damage-induced senescence via accumulation of hyperfused mitochondria and damaged lysosomes, highlighting its dual role in maintaining mitochondrial and lysosomal homeostasis [349]. In the dentate gyrus, mitophagy increases with age but is impaired by huntingtin overexpression, whereas it declines with age in C. elegans intestinal cells and human muscle satellite cells [221]. In murine tibialis anterior muscle, aging elevates mitophagic markers: PINK1 by 3.6-fold, Parkin by 40%, and mitochondrial ubiquitination by 60% [166]. Similarly, Drosophila flight muscle shows an age-dependent rise in mitophagy, though increased fission may compensate for declining biogenesis, resulting in dysfunctional mitochondria and neuromuscular degeneration [350]. In the liver, mitophagy is not significantly affected by oxidative capacity or lipid peroxidation with age, but elevated autophagy and mitophagy markers are observed; muscle-derived PGC-1α influences hepatic mitophagy by regulating Parkin translocation [351]. Satellite cells exhibit disrupted fission due to aging or genetic loss, impairing OXPHOS, reducing mitophagy, and increasing oxidative stress, which impairs muscle regeneration; restoring fission or enhancing mitophagy can reverse this degeneration [106].
System-wide profiling reveals that reduced mitophagy correlates with impaired muscle bioenergetics and atrophy. In older adults, transcriptional profiling shows an inverse relationship between mitophagy gene expression and both muscle volume and physical performance, consistent with the role of mitochondrial quality control (MQC) in sarcopenia and sterile inflammation [352]. Aging brains retain some mitophagic activity; however, cell-type-specific profiling across neuronal subtypes, astrocytes, and glial cells reveals distinct declines in flux with age, particularly in dopaminergic neurons and Purkinje cells [353]. BNIP3-mediated mitophagy in Drosophila neurons improves systemic aging markers in muscle and gut, underscoring cell non-autonomous benefits and therapeutic potential [354]. Broad declines in mitophagy have been linked to progressive dysfunction in heart and brain [355]. Evidence suggests that reduced autophagy and mitophagy accelerate aging, whereas their enhancement supports cardiac homeostasis and promotes longevity [139].
Impaired mitophagy plays a major role in aging-related diseases. In skeletal muscle, it contributes to sarcopenia through accumulation of dysfunctional mitochondria, disrupted dynamics, and reduced turnover [356]. In neurodegeneration, mitophagy is dysregulated in Parkinson’s and Alzheimer’s, exacerbated by mutations in PINK1, Parkin, and autophagy regulators [353]. In joint tissues, mitophagy dysfunction promotes intervertebral disc degeneration and osteoarthritis [117]. Similarly, disrupted mitophagy may facilitate autoimmunity via defective mtDNA clearance and type I interferon activation, with genes like IRGM, ATG5, and ATG7 implicated in autoimmune pathogenesis [357]. In vascular aging, the accumulation of ROS-producing mitochondria is a key driver in hypertension-associated endothelial decline [129]. Cardiac aging is marked by reduced mitophagy and declining function. Increased Shank3 expression suppresses mitophagy and worsens heart function with age; its ablation restores Parkin-mediated mitophagy, reduces ROS, and protects cardiac tissue [266]. In osteoarthritis, stabilization of HIF-1α enhances mitophagy, alleviating joint degeneration and positioning mitophagy as a therapeutic target [358]. In p17/PERMIT⁻/⁻ mice, ceramide-mediated mitophagy suppression causes mitochondrial dysfunction and sensorimotor decline, reversible by treatment with a ceramide–selenium analog (LCL768), which restores mitophagy and reduces metabolic dysregulation [359]. Similarly, the natural compound Kanglexin enhances Parkin-mediated mitophagy more effectively than emodin in aged cardiomyocytes, improving mitochondrial function and offering anti-aging potential [10].
Recent evidence highlights the therapeutic relevance of mitophagy enhancement. Disruption of mitophagy in muscle leads to mosaic respiratory chain deficiencies and pathogenic mtDNA accumulation. Enhancing mitophagy ameliorates muscle mitochondrial dysfunction in both normal aging and adult-onset mitochondrial disease, reinforcing mitophagy’s role in preserving tissue integrity and systemic homeostasis [360]. Mitophagy, the selective degradation of dysfunctional mitochondria, is critical for maintaining mitochondrial quality and cellular homeostasis. Aging is associated with a decline in mitophagy across species including yeast, flies, nematodes, and mammals [65,168,361], contributing to mitochondrial dysfunction, increased oxidative stress, and tissue [33,129]. In mammals, reduced mitophagy flux is observed in aged tissues such as the dentate gyrus, heart, and skeletal muscle satellite cells, with decreased expression of mitophagy regulators including BNIP3 and Parkin in elderly individuals with low physical activity [125,130,362]. In aged mice and humans, mitophagy can paradoxically increase in certain tissues, such as the retina and brain, yet still fail to counterbalance mitochondrial damage [36,170]. This dysregulated activity results in elevated cytosolic mtDNA, activating the cGAS/STING pathway and driving inflammation [36]. Reduced mitochondrial content and efficiency, increased ROS production, and mtDNA mutations are hallmarks of mitochondrial aging [363], compounded by impaired mitophagy.
Oxidative stress further inhibits mitophagy by damaging autophagy regulators such as PINK1, Parkin, and LC3 through oxidation and S-nitrosylation [147,148,364]. S-nitrosoglutathione reductase (GSNOR) activity decreases with age, promoting protein nitrosylation and impairing mitophagy [365]. NAD+ depletion also contributes to mitophagy failure and accelerated aging, as seen in Werner syndrome models, with supplementation restoring mitophagy via ULK-1 and DCT-1 activation [167,366].
Inflammatory pathways such as NLRP3 activation suppress autophagy and exacerbate aging phenotypes. NLRP3-deficient mice exhibit improved healthspan, reduced cardiac fibrosis, and extended lifespan [152,238]. Parkin-mediated mitophagy inhibits NLRP3 inflammasome activation by preventing the release of mtDNA from damaged mitochondria [367], while the PINK1/Parkin pathway reduces STING-induced inflammation [368]. Rubicon, a negative regulator of Beclin1, increases with age and its inhibition extends lifespan in model organisms [23].
Lysosomal dysfunction—central to autophagic flux—also contributes to mitophagy impairment with age, correlating with neurodegeneration and reduced lifespan [180,185,211,239]. Neural stem cells from aged individuals show reduced lysosome levels, limiting mitophagic capacity [139].
In neurodegenerative diseases, mitophagy disruption is a core pathogenic driver. In Alzheimer's disease (AD), hippocampal mitophagy is reduced by 30–50% compared to controls [369], with lower Parkin levels in serum and fibroblasts from AD patients [370,371]. Tau impairs mitophagy by interacting with and sequestering Parkin, leading to reduced mitochondrial clearance and increased oxidative damage [126,372,373]. Phospho-tau inhibits complex I activity, elevating ROS and lipid peroxidation, impairing membrane potential and detoxification enzymes such as SOD [374,375]. Drp1 activity, elevated in AD, promotes mitochondrial fragmentation, and its interaction with Aβ and tau accelerates mitochondrial dysfunction [376,377]. Aβ-treated cells show reduced mitophagy, reversible with Parkin overexpression, which increases mitochondrial LC3-II and reduces p62 accumulation [333]. Resveratrol and NAD+ precursors restore mitophagy in AD models [118]. Exercise enhances mitophagy and reduces insoluble Aβ species, reversing cognitive decline in APP/PS1 mice [167,378]. In Parkinson’s disease (PD), mutations in PINK1 and Parkin impair mitophagy, leading to ROS accumulation and neuronal death [379,380]. PINK1 knockout causes dopaminergic neuron loss in the substantia nigra [381], and mitophagic activity is reduced in PD-affected brain regions [382]. Mitochondrial damage accumulates, disrupting network integrity [122,383,384,385]. Similar disruptions are seen in Huntington’s and ALS models, where mutations interfere with Parkin-mediated mitophagy [118].
Genetic studies affirm that mitophagy supports longevity. Enhancing mitophagy through PINK1/Parkin or BNIP3 overexpression improves mitochondrial turnover and extends lifespan [354,386]. Parkin-deficient mice display accelerated cardiac aging, whereas cardiac-specific Parkin overexpression delays aging, despite potential fibrosis with excessive overexpression [86,91,387]. NIX/BNIP3 double knockout mice accumulate mitochondrial defects, confirming their essential role in basal mitophagy [142]. In Drosophila, PINK1 or Parkin deficiency causes flight muscle degeneration and reduced lifespan [82,83,87], and Drp1-driven fission supports mitophagy and healthspan [143]. Pharmacological inducers like Urolithin A and spermidine boost mitophagy and extend lifespan [108,388]. In aging mice, Urolithin A enhances mitophagy, reduces cytosolic mtDNA, and improves cognition and visual function [36]. Caloric restriction (CR) activates mitophagy via AMPK and inhibits mTORC1 and IGF-1 signaling [389], while dietary restriction increases BNIP3/Parkin expression and reduces oxidative damage [33]. Short-term starvation enhances protein hydrolysis, reducing the need for protein turnover during aging. Exercise-induced mitophagy likewise reverses cognitive deficits in neurodegenerative models [167,378].
Mitophagy impairment underlies aging-related diseases beyond neurodegeneration. In senile osteoporosis, BMSCs senescence and mitochondrial dysfunction are linked to defective mitophagy, while Sirt3 overexpression reverses these effects [390]. In intervertebral disc degeneration, mitophagy disruption in NP cells exacerbates oxidative stress, and agents like salidroside, melatonin, and Urolithin A restore function [117]. In diabetic cardiomyopathy, mitophagy is suppressed by reduced Sirt3-Foxo3A-Parkin signaling [118], while in aged hearts, Parkin deficiency worsens mitochondrial damage [199].
The inability of mitophagy to clear damaged mitochondria leads to cellular energy deficits, increased apoptosis, and inflammation, which collectively contribute to sarcopenia, cardiovascular dysfunction, and frailty [124,391]. Cells tolerate mtDNA mutations up to a threshold (~80%), beyond which mitochondrial respiration fails, contributing to cell death and tissue degeneration [43,118]. The accumulation of dysfunctional mitochondria, increased ROS, and defective clearance is a shared mechanism in aging and chronic diseases such as Alzheimer’s, Parkinson’s, Huntington’s, and chronic kidney disease [171,392].
IV. Discussion
The Vicious Cycle of Mitophagy, Biogenesis, ROS, and Mutations
The mitochondrial "vicious cycle" hypothesis of aging refines the free radical theory [58], positing that mtDNA mutations, accumulating as byproducts of respiration, impair oxidative phosphorylation and elevate ROS production, which in turn exacerbates mtDNA damage — establishing a self-perpetuating loop leading to cellular dysfunction and death [201,393]. Elevated oxidative stress, driven by superoxide, hydrogen peroxide, and ROS, contributes to mitochondrial fragmentation, inhibits fusion-fission dynamics, disrupts the ETC, reduces ATP output, and suppresses mitochondrial biogenesis — all while intensifying ROS accumulation [394,395]. Dysfunctional mitochondria containing large deletions can evade mitophagy and continue replication [201], furthering this vicious cycle. ROS elevation and impaired antioxidant responses compound mitochondrial dysfunction, as seen in aging, neurodegeneration, and metabolic syndromes [23,245,396]. Lipid overload induces a similar feedback loop where lipid surplus elevates ROS, driving mitochondrial damage, which in turn impairs lipid oxidation and worsens lipid accumulation [397]. In NAFLD, mitochondrial dysfunction escalates oxidative stress, fueling inflammation and hepatocyte death [398], while in MS, myelinic Ca²⁺ influx activates cathepsin-calpain axes and MOMP, initiating mitochondrial destabilization [399]. In neurons, glutamate excitotoxicity impairs mitochondrial dynamics, further increasing oxidative stress and NMDA receptor activation — establishing yet another degenerative cycle [400].
Mitophagy disruption with age, often due to excessive S-nitrosylation of Parkin and Drp1 from decreased S-nitrosoglutathione reductase activity, impairs mitochondrial quality control and promotes accumulation of damaged organelles [80]. This ROS-driven mitophagy inhibition, combined with mtDNA/nDNA mutations, accelerates aging and disease pathogenesis, including neurodegeneration, cardiovascular disease, and sarcopenia [23,171,245,392]. Biogenesis and mitophagy must be tightly coordinated to maintain mitochondrial homeostasis, yet their balance is perturbed with age. An imbalance—whether from excessive mitophagy or inhibited biogenesis—disrupts mitochondrial content and function [23,172]. In aging cochleae, both pathways are upregulated, but their mismatch contributes to oxidative damage [172]. Inhibited biogenesis impairs mitophagy, as seen when PGC-1α levels decline due to SIRT1 inhibition [23], while its overexpression restores autophagy and mitochondrial integrity [105]. Intriguingly, mitophagy also limits excessive biogenesis, implying a reciprocal regulatory loop that safeguards longevity by preventing mitochondrial overload and dysfunction [23]. Disruption of this mitophagy-biogenesis axis contributes to age-related phenotypes across tissues, including the skin [107]. Thus, the interplay among mitophagy, biogenesis, ROS, and mtDNA mutations forms an interconnected network — a vicious cycle at the core of mitochondrial aging. The associations and cross-interactions among the mitochondrial drivers of aging are illustrated in Figure 3.
V. Acknowledgments and Conflicts of Interest
Both authors are partners in “Elyxia Vita”, a biotechnology startup developing personalized aging-assessment and lifestyle optimization services. The authors received no financial compensation for the preparation of this review, which was completed entirely on personal time. No external funding influenced the content, analysis, or conclusions presented.
Soffia JP contributed with conceptualization, investigation, resources, writing—original draft preparation, writing—review and editing, visualization, supervision, project administration. Dawson AP contributed with editing. An artificial intelligence large language model (LLM) was used to assist with language editing and improvement of clarity. All scientific content, data interpretation, and conclusions were developed and verified solely by the authors.
The authors thank their startup partner, Matias Fernández, for his investigation support in the early stage of this review. They also thank Professor Dr. Álvaro Elorza for having reviewed a draft manuscript and providing feedback.
References
- D. N. J. De Grey and Michael. Rae, “Ending aging: the rejuvenation breakthroughs that could reverse human aging in our lifetime,” p. 433, 2013.
- J. P. De Magalhães, M. Stevens, and D. Thornton, “The Business of Anti-Aging Science,” Trends Biotechnol., vol. 35, no. 11, pp. 1062–1073, Nov. 2017. [CrossRef]
- M. A. Lee R, “Cost of Aging -- Finance & Development.” Accessed: Nov. 14, 2025. [Online]. Available: https://www.imf.org/external/pubs/ft/fandd/2017/03/lee.htm.
- S. Ogura and M. M. Jakovljevic, “Editorial: Global population aging - health care, social and economic consequences,” Front. Public Health, vol. 6, no. NOV, p. 430033, Nov. 2018. [CrossRef]
- W. James, J. Cossman, and J. K. Wolf, “Persistence of death in the United States: The remarkably different mortality patterns between America’s Heartland and Dixieland,” Demogr. Res., vol. 39, no. 1, pp. 897–910, Oct. 2018. [CrossRef]
- European Union Stats, “Ageing Europe-statistics on population developments Statistics Explained,” 2020, Accessed: Nov. 14, 2025. [Online]. Available: https://ec.europa.eu/eurostat/statisticsexplained/.
- W. Lutz, W. Sanderson, and S. Scherbov, “The coming acceleration of global population ageing,” Nature 2007 451:7179, vol. 451, no. 7179, pp. 716–719, Jan. 2008. [CrossRef]
- Z. Ismail, W. I. W. Ahmad, S. H. Hamjah, and I. K. Astina, “The Impact of Population Ageing: A Review,” Iran. J. Public Health, vol. 50, no. 12, pp. 2451–2460, 2021. [CrossRef]
- United Nations, “World Population Prospects 2022 World Population Prospects 2022 Summary of Results,” 2022.
- S. jin Li et al., “Population aging and trends of pulmonary tuberculosis incidence in the elderly,” BMC Infect. Dis., vol. 21, no. 1, Dec. 2021. [CrossRef]
- L. Wu, Z. Huang, and Z. Pan, “The spatiality and driving forces of population ageing in China,” PLoS One, vol. 16, no. 1, p. e0243559, Jan. 2021. [CrossRef]
- N. Glasgow and E. H. Berry, “Rural aging in 21st century America,” Rural Aging in 21st Century America, pp. 1–384, Jan. 2013. [CrossRef]
- G. Marois, A. Bélanger, and W. Lutz, “Population aging, migration, and productivity in Europe,” Proc. Natl. Acad. Sci. U. S. A., vol. 117, no. 14, pp. 7690–7695, Apr. 2020. [CrossRef]
- Z. Li et al., “Aging and age-related diseases: from mechanisms to therapeutic strategies,” Biogerontology 2021 22:2, vol. 22, no. 2, pp. 165–187, Jan. 2021. [CrossRef]
- Y. Cai et al., “The landscape of aging,” Sci. China Life Sci., vol. 65, no. 12, pp. 2354–2454, Dec. 2022. [CrossRef]
- J. P. Da Costa, R. Vitorino, G. M. Silva, C. Vogel, A. C. Duarte, and T. Rocha-Santos, “A synopsis on aging-Theories, mechanisms and future prospects,” Ageing Res. Rev., vol. 29, pp. 90–112, Aug. 2016. [CrossRef]
- S. Tabibzadeh, “Cell-centric hypotheses of aging,” Front. Biosci. (Landmark Ed)., vol. 26, no. 1, pp. 1–49, Jan. 2021. [CrossRef]
- D. Gems, “The hyperfunction theory: an emerging paradigm for the biology of aging,” Ageing Res. Rev., vol. 74, p. 101557, Feb. 2022. [CrossRef]
- A. A. Johnson, M. N. Shokhirev, and B. Shoshitaishvili, “Revamping the evolutionary theories of aging,” Ageing Res. Rev., vol. 55, Nov. 2019. [CrossRef]
- M. I. Brengdahl, C. M. Kimber, V. N. Shenoi, M. Dumea, A. Mital, and U. Friberg, “Age-specific effects of deletions: implications for aging theories,” Evolution, vol. 77, no. 1, pp. 254–263, Jan. 2023. [CrossRef]
- P. R. Rich, “A perspective on Peter Mitchell and the chemiosmotic theory,” J. Bioenerg. Biomembr., vol. 40, no. 5, pp. 407–410, Oct. 2008. [CrossRef]
- López-Otín, M. A. Blasco, L. Partridge, M. Serrano, and G. Kroemer, “Hallmarks of aging: An expanding universe,” Cell, vol. 186, no. 2, pp. 243–278, Jan. 2023. [CrossRef]
- S. K. Bhutia, P. P. Naik, D. P. Panigrahi, C. S. Bhol, and K. K. Mahapatra, “Mitophagy, Diseases, and Aging,” Models, Molecules and Mechanisms in Biogerontology, pp. 177–191, 2019. [CrossRef]
- A. Dillin et al., “Rates of Behavior and Aging Specified by Mitochondrial Function During Development,” Science (1979)., vol. 298, no. 5602, pp. 2398–2401, Dec. 2002. [CrossRef]
- C. Wallace, “Mitochondria and Cancer: Warburg Addressed,” Cold Spring Harb. Symp. Quant. Biol., vol. 70, pp. 363–374, Jan. 2005. [CrossRef]
- C. Wallace, “A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine,” Annu. Rev. Genet., vol. 39, no. Volume 39, 2005, pp. 359–407, Dec. 2005. [CrossRef]
- S. Bartman, G. Coppotelli, and J. M. Ross, “Mitochondrial Dysfunction: A Key Player in Brain Aging and Diseases,” Curr. Issues Mol. Biol., vol. 46, no. 3, pp. 1987–2026, Mar. 2024. [CrossRef]
- M. Picard and O. S. Shirihai, “Mitochondrial signal transduction,” Cell Metab., vol. 34, no. 11, pp. 1620–1653, Nov. 2022. [CrossRef]
- S. van der Rijt, M. Molenaars, R. L. McIntyre, G. E. Janssens, and R. H. Houtkooper, “Integrating the Hallmarks of Aging Throughout the Tree of Life: A Focus on Mitochondrial Dysfunction,” Front. Cell Dev. Biol., vol. 8, Nov. 2020. [CrossRef]
- B. Kothe, S. Klein, and S. N. Petrosky, “Urolithin A as a Potential Agent for Prevention of Age-Related Disease: A Scoping Review,” Cureus, vol. 15, no. 7, Jul. 2023. [CrossRef]
- S. B. Poudel et al., “Effects of GH/IGF on the Aging Mitochondria,” Cells, vol. 9, no. 6, Jun. 2020. [CrossRef]
- Maldonado, S. Morales-Pison, F. Urbina, and A. Solari, “Aging Hallmarks and the Role of Oxidative Stress,” Antioxidants 2023, Vol. 12, Page 651, vol. 12, no. 3, p. 651, Mar. 2023. [CrossRef]
- A. De Gaetano, L. Gibellini, G. Zanini, M. Nasi, A. Cossarizza, and M. Pinti, “Mitophagy and Oxidative Stress: The Role of Aging,” Antioxidants, vol. 10, no. 5, p. 794, May 2021. [CrossRef]
- M. C. Sanchez, S. Lancel, E. Boulanger, and R. Neviere, “Targeting Oxidative Stress and Mitochondrial Dysfunction in the Treatment of Impaired Wound Healing: A Systematic Review,” Antioxidants (Basel)., vol. 7, no. 8, Aug. 2018. [CrossRef]
- Y. Li, L. Berliocchi, Z. Li, and L. J. Rasmussen, “Interactions between mitochondrial dysfunction and other hallmarks of aging: Paving a path toward interventions that promote healthy old age,” Aging Cell, vol. 23, no. 1, Jan. 2024. [CrossRef]
- J. I. Jiménez-Loygorri et al., “Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging,” Nat. Commun., vol. 15, no. 1, Dec. 2024. [CrossRef]
- J. Tian et al., “Hippocampal transcriptome-wide association study and pathway analysis of mitochondrial solute carriers in Alzheimer’s disease,” Translational Psychiatry 2024 14:1, vol. 14, no. 1, pp. 250-, Jun. 2024. [CrossRef]
- S. Xie, S. C. Xu, W. Deng, and Q. Tang, “Metabolic landscape in cardiac aging: insights into molecular biology and therapeutic implications,” Signal Transduct. Target. Ther., vol. 8, no. 1, Dec. 2023. [CrossRef]
- L. Holper, D. Ben-Shachar, and J. Mann, “Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease,” Neuropsychopharmacology, vol. 44, no. 5, pp. 837–849, Apr. 2019. [CrossRef]
- S. Chen et al., “TOMM40 genetic variants associated with healthy aging and longevity: a systematic review,” BMC Geriatrics 2022 22:1, vol. 22, no. 1, pp. 667-, Aug. 2022. [CrossRef]
- J. I. Escrig-Larena, S. Delgado-Pulido, and M. Mittelbrunn, “Mitochondria during T cell aging,” Semin. Immunol., vol. 69, p. 101808, Sep. 2023. [CrossRef]
- C. C. S. Chini, H. S. Cordeiro, N. L. K. Tran, and E. N. Chini, “NAD metabolism: Role in senescence regulation and aging,” Aging Cell, vol. 23, no. 1, Jan. 2024. [CrossRef]
- X. Yang, R. Zhang, K. Nakahira, and Z. Gu, “Mitochondrial DNA Mutation, Diseases, and Nutrient-Regulated Mitophagy,” Annu. Rev. Nutr., vol. 39, no. Volume 39, 2019, pp. 201–226, Aug. 2019. [CrossRef]
- A. Suomalainen and J. Nunnari, “Mitochondria at the crossroads of health and disease,” Cell, vol. 187, no. 11, pp. 2601–2627, May 2024. [CrossRef]
- D. A. Harris and A. M. Das, “Control of mitochondrial ATP synthesis in the heart,” Biochem. J., vol. 280 (Pt 3), no. Pt 3, pp. 561–573, 1991. [CrossRef]
- D. J. Pagliarini and J. Rutter, “Hallmarks of a new era in mitochondrial biochemistry,” Genes Dev., vol. 27, no. 24, pp. 2615–2627, Dec. 2013. [CrossRef]
- R. Kerr, S. Jabbari, and I. G. Johnston, “Intracellular Energy Variability Modulates Cellular Decision-Making Capacity,” Scientific Reports 2019 9:1, vol. 9, no. 1, pp. 20196-, Dec. 2019. [CrossRef]
- Fernandez-Vizarra and M. Zeviani, “Mitochondrial disorders of the OXPHOS system,” FEBS Lett., vol. 595, no. 8, pp. 1062–1106, Apr. 2021. [CrossRef]
- J. A. Amorim, G. Coppotelli, A. P. Rolo, C. M. Palmeira, J. M. Ross, and D. A. Sinclair, “Mitochondrial and metabolic dysfunction in ageing and age-related diseases,” Nat. Rev. Endocrinol., vol. 18, no. 4, pp. 243–258, Apr. 2022. [CrossRef]
- M. Patron, H. G. Sprenger, and T. Langer, “m-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration,” Cell Res., vol. 28, no. 3, pp. 296–306, Mar. 2018. [CrossRef]
- R. Rizzuto et al., “Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses,” Science, vol. 280, no. 5370, pp. 1763–1766, Jun. 1998. [CrossRef]
- R. H. Houtkooper et al., “Mitonuclear protein imbalance as a conserved longevity mechanism,” Nature, vol. 497, no. 7450, pp. 451–457, 2013. [CrossRef]
- J. C. Goldstein, N. J. Waterhouse, P. Juin, G. I. Evan, and D. R. Green, “The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant,” Nat. Cell Biol., vol. 2, no. 3, pp. 156–162, 2000. [CrossRef]
- X. Liu, C. N. Kim, J. Yang, R. Jemmerson, and X. Wang, “Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c,” Cell, vol. 86, no. 1, pp. 147–157, Jul. 1996. [CrossRef]
- M. Feric, T. G. Demarest, J. Tian, D. L. Croteau, V. A. Bohr, and T. Misteli, “Self-assembly of multi-component mitochondrial nucleoids via phase separation,” EMBO J., vol. 40, no. 6, Mar. 2021. [CrossRef]
- A. S. Monzel, M. Levin, and M. Picard, “The energetics of cellular life transitions,” Life metabolism, vol. 3, no. 3, Jun. 2024. [CrossRef]
- R. Lill, R. Lill, and S. A. Freibert, “Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis,” Annu. Rev. Biochem., vol. 89, pp. 471–499, Jun. 2020. [CrossRef]
- D. HARMAN, “Aging: a theory based on free radical and radiation chemistry,” J. Gerontol., vol. 11, no. 3, pp. 298–300, 1956. [CrossRef]
- W. Yang and S. Hekimi, “A Mitochondrial Superoxide Signal Triggers Increased Longevity in Caenorhabditis elegans,” PLoS Biol., vol. 8, no. 12, p. e1000556, Dec. 2010. [CrossRef]
- C. Fang, X. Wei, and Y. Wei, “Mitochondrial DNA in the regulation of innate immune responses,” Protein Cell, vol. 7, no. 1, pp. 11–16, Jan. 2016. [CrossRef]
- A. Fairbrother-Browne et al., “Mitochondrial-nuclear cross-talk in the human brain is modulated by cell type and perturbed in neurodegenerative disease,” Commun. Biol., vol. 4, no. 1, Dec. 2021. [CrossRef]
- R. J. Youle and D. P. Narendra, “Mechanisms of mitophagy,” Nat. Rev. Mol. Cell Biol., vol. 12, no. 1, pp. 9–14, Jan. 2011. [CrossRef]
- B. M. Baker, A. M. Nargund, T. Sun, and C. M. Haynes, “Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2,” PLoS Genet., vol. 8, no. 6, Jun. 2012. [CrossRef]
- J. Yun and T. Finkel, “Mitohormesis,” Cell Metab., vol. 19, no. 5, pp. 757–766, May 2014. [CrossRef]
- K. Palikaras, E. Lionaki, and N. Tavernarakis, “Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans,” Nature, vol. 521, no. 7553, pp. 525–528, May 2015. [CrossRef]
- E. Owusu-Ansah, W. Song, and N. Perrimon, “XMuscle mitohormesis promotes longevity via systemic repression of insulin signaling,” Cell, vol. 155, no. 3, p. 699, Oct. 2013. [CrossRef]
- Q. Zhao, J. Wang, I. V. Levichkin, S. Stasinopoulos, M. T. Ryan, and N. J. Hoogenraad, “A mitochondrial specific stress response in mammalian cells,” EMBO J., vol. 21, no. 17, pp. 4411–4419, Sep. 2002. [CrossRef]
- R. D. Martinus et al., “Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome,” Eur. J. Biochem., vol. 240, no. 1, pp. 98–103, Jan. 1996. [CrossRef]
- K. M. Berendzen et al., “Neuroendocrine Coordination of Mitochondrial Stress Signaling and Proteostasis,” Cell, vol. 166, no. 6, pp. 1553-1563.e10, Sep. 2016. [CrossRef]
- N. S. Chandel, “Mitochondria as signaling organelles,” BMC Biology 2014 12:1, vol. 12, no. 1, pp. 34-, May 2014. [CrossRef]
- C. Lee et al., “The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance,” Cell Metab., vol. 21, no. 3, pp. 443–454, Mar. 2015. [CrossRef]
- Y. Hashimoto, S. Ookuma, and E. Nishida, “Lifespan extension by suppression of autophagy genes in Caenorhabditis elegans,” Genes Cells, vol. 14, no. 6, pp. 717–726, 2009. [CrossRef]
- R. C. Scarpulla, R. B. Vega, and D. P. Kelly, “Transcriptional integration of mitochondrial biogenesis,” Trends in Endocrinology and Metabolism, vol. 23, no. 9, pp. 459–466, Sep. 2012. [CrossRef]
- E. Nisoli et al., “Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS,” Science, vol. 310, no. 5746, pp. 314–317, Oct. 2005. [CrossRef]
- A. M. Pickrell and R. J. Youle, “The roles of PINK1, Parkin, and mitochondrial fidelity in parkinson’s disease,” Neuron, vol. 85, no. 2, pp. 257–273, Jan. 2015. [CrossRef]
- B. Cannon and J. Nedergaard, “Brown adipose tissue: function and physiological significance,” Physiol. Rev., vol. 84, no. 1, pp. 277–359, Jan. 2004. [CrossRef]
- S. H. Du et al., “Nicotinamide mononucleotide ameliorates acute lung injury by inducing mitonuclear protein imbalance and activating the UPRmt,” Exp. Biol. Med., vol. 247, no. 14, pp. 1264–1276, Jul. 2022. [CrossRef]
- R. Liang, L. Zhu, Y. Huang, J. Chen, and Q. Tang, “Mitochondria: fundamental characteristics, challenges, and impact on aging,” Biogerontology, vol. 25, no. 6, pp. 923–941, Nov. 2024. [CrossRef]
- Tirichen, H. Yaigoub, W. Xu, C. Wu, R. Li, and Y. Li, “Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress,” Front. Physiol., vol. 12, Apr. 2021. [CrossRef]
- L. Liu, X. Liao, H. Wu, Y. Li, Y. Zhu, and Q. Chen, “Mitophagy and Its Contribution to Metabolic and Aging-Associated Disorders,” Antioxid. Redox Signal., vol. 32, no. 12, pp. 906–927, Apr. 2020. [CrossRef]
- S. Javadov, S. Jang, X. R. Chapa-Dubocq, Z. Khuchua, and A. K. Camara, “Mitochondrial respiratory supercomplexes in mammalian cells: structural versus functional role,” J. Mol. Med. (Berl)., vol. 99, no. 1, pp. 57–73, Jan. 2021. [CrossRef]
- V. Larosa and C. Remacle, “Insights into the respiratory chain and oxidative stress,” Biosci. Rep., vol. 38, no. 5, Oct. 2018. [CrossRef]
- T. Lobo-Jarne and C. Ugalde, “Respiratory chain supercomplexes: Structures, function and biogenesis,” Semin. Cell Dev. Biol., vol. 76, pp. 179–190, Apr. 2018. [CrossRef]
- J. A. Letts, K. Fiedorczuk, and L. A. Sazanov, “The architecture of respiratory supercomplexes,” Nature 2016 537:7622, vol. 537, no. 7622, pp. 644–648, Sep. 2016. [CrossRef]
- S. Guan, L. Zhao, and R. Peng, “Mitochondrial Respiratory Chain Supercomplexes: From Structure to Function,” Int. J. Mol. Sci., vol. 23, no. 22, Nov. 2022. [CrossRef]
- A. Toth et al., “Kinetic coupling of the respiratory chain with ATP synthase, but not proton gradients, drives ATP production in cristae membranes,” Proc. Natl. Acad. Sci. U. S. A., vol. 117, no. 5, pp. 2412–2421, Feb. 2020. [CrossRef]
- L. D. Zorova et al., “Mitochondrial membrane potential,” Anal. Biochem., vol. 552, pp. 50–59, Jul. 2018. [CrossRef]
- Martínez-Reyes et al., “TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions,” Mol. Cell, vol. 61, no. 2, p. 199, Jan. 2015. [CrossRef]
- Mengel-From, M. Thinggaard, C. Dalgård, K. O. Kyvik, K. Christensen, and L. Christiansen, “Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly,” Hum. Genet., vol. 133, no. 9, pp. 1149–1159, Sep. 2014. [CrossRef]
- G. Sturm et al., “OxPhos defects cause hypermetabolism and reduce lifespan in cells and in patients with mitochondrial diseases,” Commun. Biol., vol. 6, no. 1, Dec. 2023. [CrossRef]
- X. Zheng et al., “Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation,” Elife, vol. 5, no. JUN2016, p. e13374, Jun. 2016. [CrossRef]
- E. S. Chocron, E. Munkácsy, and A. M. Pickering, “Cause or casualty: The role of mitochondrial DNA in aging and age-associated disease,” Biochim. Biophys. Acta Mol. Basis Dis., vol. 1865, no. 2, pp. 285–297, Feb. 2019. [CrossRef]
- U. Basu, A. M. Bostwick, K. Das, K. E. Dittenhafer-Reed, and S. S. Patel, “Structure, mechanism, and regulation of mitochondrial DNA transcription initiation,” Journal of Biological Chemistry, vol. 295, no. 52, pp. 18406–18425, Dec. 2020. [CrossRef]
- W. Wei et al., “Nuclear-embedded mitochondrial DNA sequences in 66,083 human genomes,” Nature, vol. 611, no. 7934, pp. 105–114, Nov. 2022. [CrossRef]
- G. De Benedittis et al., “Alteration of Mitochondrial DNA Copy Number and Increased Expression Levels of Mitochondrial Dynamics-Related Genes in Sjögren’s Syndrome,” Biomedicines, vol. 10, no. 11, Nov. 2022. [CrossRef]
- E. S. Ramos et al., “Mitochondrial fusion is required for regulation of mitochondrial DNA replication,” PLoS Genet., vol. 15, no. 6, Jun. 2019. [CrossRef]
- P. Katajisto et al., “Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness,” Science, vol. 348, no. 6232, pp. 340–343, Apr. 2015. [CrossRef]
- D. C. Wallace, “A mitochondrial bioenergetic etiology of disease,” J. Clin. Invest., vol. 123, no. 4, pp. 1405–1412, Apr. 2013. [CrossRef]
- R. Gupta et al., “Nuclear genetic control of mtDNA copy number and heteroplasmy in humans,” Nature, vol. 620, no. 7975, pp. 839–848, Aug. 2023. [CrossRef]
- N. A. Bonekamp et al., “High levels of TFAM repress mammalian mitochondrial DNA transcription in vivo,” Life Sci. Alliance, vol. 4, no. 11, 2021. [CrossRef]
- S. M. A. Radzak, S. Z. N. M. Khair, F. Ahmad, A. Patar, Z. Idris, and A. A. M. Yusoff, “Insights regarding mitochondrial DNA copy number alterations in human cancer (Review),” Int. J. Mol. Med., vol. 50, no. 2, Aug. 2022. [CrossRef]
- B. Peter and M. Falkenberg, “TWINKLE and Other Human Mitochondrial DNA Helicases: Structure, Function and Disease,” Genes (Basel)., vol. 11, no. 4, Apr. 2020. [CrossRef]
- A. Picca et al., “The contribution of mitochondrial DNA alterations to aging, cancer, and neurodegeneration,” Exp. Gerontol., vol. 178, Jul. 2023. [CrossRef]
- Picca et al., “Circulating Mitochondrial DNA and Inter-Organelle Contact Sites in Aging and Associated Conditions,” Cells, vol. 11, no. 4, Feb. 2022. [CrossRef]
- N. N. Wu, Y. Zhang, and J. Ren, “Mitophagy, Mitochondrial Dynamics, and Homeostasis in Cardiovascular Aging,” Oxid. Med. Cell. Longev., vol. 2019, 2019. [CrossRef]
- X. Hong et al., “Mitochondrial dynamics maintain muscle stem cell regenerative competence throughout adult life by regulating metabolism and mitophagy,” Cell Stem Cell, vol. 29, no. 9, pp. 1298-1314.e10, Sep. 2022. [CrossRef]
- Zhang et al., “The role of mitochondrial quality surveillance in skin aging: Focus on mitochondrial dynamics, biogenesis and mitophagy,” Ageing Res. Rev., vol. 87, Jun. 2023. [CrossRef]
- G. Chen, G. Kroemer, and O. Kepp, “Mitophagy: An Emerging Role in Aging and Age-Associated Diseases,” Front. Cell Dev. Biol., vol. 8, Mar. 2020. [CrossRef]
- W. N. Xu et al., “Mitochondrial NDUFA4L2 attenuates the apoptosis of nucleus pulposus cells induced by oxidative stress via the inhibition of mitophagy,” Exp. Mol. Med., vol. 51, no. 11, Nov. 2019. [CrossRef]
- L. Kang, S. Liu, J. Li, Y. Tian, Y. Xue, and X. Liu, “Parkin and Nrf2 prevent oxidative stress-induced apoptosis in intervertebral endplate chondrocytes via inducing mitophagy and anti-oxidant defenses,” Life Sci., vol. 243, Feb. 2020. [CrossRef]
- L. Xie et al., “CircERCC2 ameliorated intervertebral disc degeneration by regulating mitophagy and apoptosis through miR-182-5p/SIRT1 axis,” Cell Death Dis., vol. 10, no. 10, Oct. 2019. [CrossRef]
- Huang et al., “Compression-induced senescence of nucleus pulposus cells by promoting mitophagy activation via the PINK1/PARKIN pathway,” J. Cell. Mol. Med., vol. 24, no. 10, pp. 5850–5864, May 2020. [CrossRef]
- Liu, J. Wang, and Y. Zhou, “Upregulation of BNIP3 and translocation to mitochondria in nutrition deprivation induced apoptosis in nucleus pulposus cells,” Joint Bone Spine, vol. 79, no. 2, pp. 186–191, Mar. 2012. [CrossRef]
- Liu, C. Yuan, L. Pu, and J. Wang, “Nutrient deprivation induces apoptosis of nucleus pulposus cells via activation of the BNIP3/AIF signalling pathway,” Mol. Med. Rep., vol. 16, no. 5, pp. 7253–7260, Nov. 2017. [CrossRef]
- Z. He, L. Pu, C. Yuan, M. Jia, and J. Wang, “Nutrition deficiency promotes apoptosis of cartilage endplate stem cells in a caspase-independent manner partially through upregulating BNIP3,” Acta Biochim. Biophys. Sin. (Shanghai)., vol. 49, no. 1, pp. 25–32, Jan. 2017. [CrossRef]
- H. J. Shin et al., “Pink1-Mediated Chondrocytic Mitophagy Contributes to Cartilage Degeneration in Osteoarthritis,” J. Clin. Med., vol. 8, no. 11, Nov. 2019. [CrossRef]
- Sun, X. Jing, J. Guo, X. Yao, and F. Guo, “Mitophagy in degenerative joint diseases,” Autophagy, vol. 17, no. 9, pp. 2082–2092, 2021. [CrossRef]
- Babbar, S. Basu, B. Yang, D. L. Croteau, and V. A. Bohr, “Mitophagy and DNA damage signaling in human aging,” Mech. Ageing Dev., vol. 186, Mar. 2020. [CrossRef]
- A. Diot, K. Morten, and J. Poulton, “Mitophagy plays a central role in mitochondrial ageing,” Mamm. Genome, vol. 27, no. 7–8, pp. 381–395, Aug. 2016. [CrossRef]
- Y. Chen et al., “Mfn2 is involved in intervertebral disc degeneration through autophagy modulation,” Osteoarthritis Cartilage, vol. 28, no. 3, pp. 363–374, Mar. 2020. [CrossRef]
- K. Palikaras, I. Daskalaki, M. Markaki, and N. Tavernarakis, “Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover,” Pharmacol. Ther., vol. 178, pp. 157–174, Oct. 2017. [CrossRef]
- Bakula and M. Scheibye-Knudsen, “MitophAging: Mitophagy in Aging and Disease,” Front. Cell Dev. Biol., vol. 8, Apr. 2020. [CrossRef]
- T. A. Banarase et al., “Mitophagy regulation in aging and neurodegenerative disease,” Biophys. Rev., vol. 15, no. 2, pp. 239–255, Apr. 2023. [CrossRef]
- J. Faitg, D. D’Amico, C. Rinsch, and A. Singh, “Mitophagy Activation by Urolithin A to Target Muscle Aging,” Calcif. Tissue Int., vol. 114, no. 1, pp. 53–59, Jan. 2024. [CrossRef]
- J. Guo and W. C. Chiang, “Mitophagy in aging and longevity,” IUBMB Life, vol. 74, no. 4, pp. 296–316, Apr. 2022. [CrossRef]
- Tran and P. H. Reddy, “Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease,” Front. Neurosci., vol. 14, Jan. 2021. [CrossRef]
- T. J. LaRocca, R. A. Gioscia-Ryan, C. M. Hearon, and D. R. Seals, “The autophagy enhancer spermidine reverses arterial aging,” Mech. Ageing Dev., vol. 134, no. 7–8, pp. 314–320, Jul. 2013. [CrossRef]
- C. G. McCarthy et al., “Reconstitution of autophagy ameliorates vascular function and arterial stiffening in spontaneously hypertensive rats,” Am. J. Physiol. Heart Circ. Physiol., vol. 317, no. 5, pp. H1013–H1027, 2019. [CrossRef]
- Z. J. Schreckenberger, C. F. Wenceslau, B. Joe, and C. G. McCarthy, “Mitophagy in Hypertension-Associated Premature Vascular Aging,” Am. J. Hypertens., vol. 33, no. 9, pp. 804–812, Sep. 2020. [CrossRef]
- A. Rappe and T. G. McWilliams, “Mitophagy in the aging nervous system,” Front. Cell Dev. Biol., vol. 10, Oct. 2022. [CrossRef]
- B. Lombard, K. F. Chua, R. Mostoslavsky, S. Franco, M. Gostissa, and F. W. Alt, “DNA repair, genome stability, and aging,” Cell, vol. 120, no. 4, pp. 497–512, Feb. 2005. [CrossRef]
- C. McMahon et al., “TRIBE: Hijacking an RNA-Editing Enzyme to Identify Cell-Specific Targets of RNA-Binding Proteins,” Cell, vol. 165, no. 3, pp. 742–753, Apr. 2016. [CrossRef]
- L. Hayflick and P. S. Moorhead, “The serial cultivation of human diploid cell strains,” Exp. Cell Res., vol. 25, no. 3, pp. 585–621, 1961. [CrossRef]
- M. Zhou et al., “Acupoint catgut embedding improves senescence in a rat model of ageing by regulating mitophagy via the PINK1 pathway,” J. Cell. Mol. Med., vol. 25, no. 8, pp. 3816–3828, Apr. 2021. [CrossRef]
- M. Zatyka, S. Sarkar, and T. Barrett, “Autophagy in Rare (NonLysosomal) Neurodegenerative Diseases,” J. Mol. Biol., vol. 432, no. 8, pp. 2735–2753, Apr. 2020. [CrossRef]
- S. Salvioli et al., “Genes, ageing and longevity in humans: problems, advantages and perspectives,” Free Radic. Res., vol. 40, no. 12, pp. 1303–1323, Dec. 2006. [CrossRef]
- J. Zhang and P. A. Ney, “Reticulocyte mitophagy: monitoring mitochondrial clearance in a mammalian model,” Autophagy, vol. 6, no. 3, pp. 405–408, Apr. 2010. [CrossRef]
- T. Ito et al., “Regulation of myeloid leukaemia by the cell-fate determinant Musashi,” Nature, vol. 466, no. 7307, pp. 765–768, Aug. 2010. [CrossRef]
- W. J. Liang and Å. B. Gustafsson, “The Aging Heart: Mitophagy at the Center of Rejuvenation,” Front. Cardiovasc. Med., vol. 7, Feb. 2020. [CrossRef]
- S. A. Detmer and D. C. Chan, “Functions and dysfunctions of mitochondrial dynamics,” Nat. Rev. Mol. Cell Biol., vol. 8, no. 11, pp. 870–879, Nov. 2007. [CrossRef]
- R. Bernardi and P. P. Pandolfi, “Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies,” Nat. Rev. Mol. Cell Biol., vol. 8, no. 12, pp. 1006–1016, Dec. 2007. [CrossRef]
- Yetkin-Arik et al., “The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis,” Sci. Rep., vol. 9, no. 1, Dec. 2019. [CrossRef]
- Gauba, L. Guo, and H. Du, “Cyclophilin D Promotes Brain Mitochondrial F1FO ATP Synthase Dysfunction in Aging Mice,” J. Alzheimers Dis., vol. 55, no. 4, pp. 1351–1362, 2017. [CrossRef]
- G. Nicholls, “Oxidative stress and energy crises in neuronal dysfunction,” Ann. N. Y. Acad. Sci., vol. 1147, pp. 53–60, 2008. [CrossRef]
- G. Nicholls, “Spare respiratory capacity, oxidative stress and excitotoxicity,” Biochem. Soc. Trans., vol. 37, no. Pt 6, pp. 1385–1388, 2009. [CrossRef]
- C. Desler, T. L. Hansen, J. B. Frederiksen, M. L. Marcker, K. K. Singh, and L. Juel Rasmussen, “Is There a Link between Mitochondrial Reserve Respiratory Capacity and Aging?,” J. Aging Res., vol. 2012, 2012. [CrossRef]
- S. N. Chaudhari and E. T. Kipreos, “The Energy Maintenance Theory of Aging: Maintaining Energy Metabolism to Allow Longevity,” Bioessays, vol. 40, no. 8, Aug. 2018. [CrossRef]
- Y. Yuan, V. F. Cruzat, P. Newshome, J. Cheng, Y. Chen, and Y. Lu, “Regulation of SIRT1 in aging: Roles in mitochondrial function and biogenesis,” Mech. Ageing Dev., vol. 155, pp. 10–21, Apr. 2016. [CrossRef]
- C. Abboudi, “The Effects of Aging on Skeletal Muscle ATP Production,” The Science Journal of the Lander College of Arts and Sciences, vol. 12, no. 2, p. 10, Jan. 2019, Accessed: Nov. 15, 2025. [Online]. Available: https://touroscholar.touro.edu/sjlcas/vol12/iss2/10.
- Distefano and B. H. Goodpaster, “Effects of Exercise and Aging on Skeletal Muscle,” Cold Spring Harb. Perspect. Med., vol. 8, no. 3, Mar. 2018. [CrossRef]
- A. Hoshino et al., “The ADP/ATP translocase drives mitophagy independent of nucleotide exchange,” Nature, vol. 575, no. 7782, pp. 375–379, Nov. 2019. [CrossRef]
- Mikolajewicz, E. A. Zimmermann, B. M. Willie, and S. V. Komarova, “Mechanically stimulated ATP release from murine bone cells is regulated by a balance of injury and repair,” Elife, vol. 7, Oct. 2018. [CrossRef]
- V. Warnsmann, J. Meisterknecht, I. Wittig, and H. D. Osiewacz, “Aging of Podospora anserina Leads to Alterations of OXPHOS and the Induction of Non-Mitochondrial Salvage Pathways,” Cells, vol. 10, no. 12, Dec. 2021. [CrossRef]
- L. M. de Smalen et al., “Impaired age-associated mitochondrial translation is mitigated by exercise and PGC-1α,” Proc. Natl. Acad. Sci. U. S. A., vol. 120, no. 36, 2023. [CrossRef]
- Z. Vue et al., “3D reconstruction of murine mitochondria reveals changes in structure during aging linked to the MICOS complex,” Aging Cell, vol. 22, no. 12, Dec. 2023. [CrossRef]
- M. Frenzel, H. Rommelspacher, M. D. Sugawa, and N. A. Dencher, “Ageing alters the supramolecular architecture of OxPhos complexes in rat brain cortex,” Exp. Gerontol., vol. 45, no. 7–8, pp. 563–572, Aug. 2010. [CrossRef]
- Lopez-Fabuel, M. Resch-Beusher, M. Carabias-Carrasco, A. Almeida, and J. P. Bolaños, “Mitochondrial Complex I Activity is Conditioned by Supercomplex I-III2-IV Assembly in Brain Cells: Relevance for Parkinson’s Disease,” Neurochem. Res., vol. 42, no. 6, pp. 1676–1682, Jun. 2017. [CrossRef]
- A. Grünewald, K. R. Kumar, and C. M. Sue, “New insights into the complex role of mitochondria in Parkinson’s disease,” Prog. Neurobiol., vol. 177, pp. 73–93, Jun. 2019. [CrossRef]
- M. R. Cookson, “Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways,” Cold Spring Harb. Perspect. Med., vol. 2, no. 9, 2012. [CrossRef]
- M. Kenney and J. P. Bennett, “Alzheimer’s Disease Frontal Cortex Mitochondria Show a Loss of Individual Respiratory Proteins but Preservation of Respiratory Supercomplexes,” Int. J. Alzheimers Dis., vol. 2019, 2019. [CrossRef]
- S. K. Sagwal and S. Bekeschus, “ROS Pleiotropy in Melanoma and Local Therapy with Physical Modalities,” Oxid. Med. Cell. Longev., vol. 2021, 2021. [CrossRef]
- Zhang, H. Yu, M. Q. Man, and L. Hu, “Aging in the dermis: Fibroblast senescence and its significance,” Aging Cell, vol. 23, no. 2, Feb. 2024. [CrossRef]
- Song et al., “FOXO-regulated OSER1 reduces oxidative stress and extends lifespan in multiple species,” Nat. Commun., vol. 15, no. 1, Dec. 2024. [CrossRef]
- M. Zhao et al., “Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance,” Theranostics, vol. 11, no. 4, pp. 1845–1863, Jan. 2021. [CrossRef]
- A. Sen et al., “Mitochondrial membrane proteins and VPS35 orchestrate selective removal of mtDNA,” Nat. Commun., vol. 13, no. 1, Dec. 2022. [CrossRef]
- Yeo, C. Kang, M. C. Gomez-Cabrera, J. Vina, and L. L. Ji, “Intensified mitophagy in skeletal muscle with aging is downregulated by PGC-1alpha overexpression in vivo,” Free Radic. Biol. Med., vol. 130, pp. 361–368, Jan. 2019. [CrossRef]
- F. Fang et al., “Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease,” Nat. Neurosci., vol. 22, no. 3, pp. 401–412, Mar. 2019. [CrossRef]
- N. Sun et al., “Measuring In Vivo Mitophagy,” Mol. Cell, vol. 60, no. 4, pp. 685–696, Nov. 2015. [CrossRef]
- E. Tresse et al., “Mitochondrial DNA damage triggers spread of Parkinson’s disease-like pathology,” Mol. Psychiatry, vol. 28, no. 11, pp. 4902–4914, Nov. 2023. [CrossRef]
- A. T. Erlich and D. A. Hood, “Mitophagy Regulation in Skeletal Muscle: Effect of Endurance Exercise and Age,” Journal of Science in Sport and Exercise 2019 1:3, vol. 1, no. 3, pp. 228–236, Nov. 2019. [CrossRef]
- M. Villanueva-Paz et al., “Parkin-mediated mitophagy and autophagy flux disruption in cellular models of MERRF syndrome,” Biochim. Biophys. Acta Mol. Basis Dis., vol. 1866, no. 6, Jun. 2020. [CrossRef]
- Xiong et al., “Modulation of miR-34a/SIRT1 signaling protects cochlear hair cells against oxidative stress and delays age-related hearing loss through coordinated regulation of mitophagy and mitochondrial biogenesis,” Neurobiol. Aging, vol. 79, pp. 30–42, Jul. 2019. [CrossRef]
- M. Vermulst et al., “Mitochondrial point mutations do not limit the natural lifespan of mice,” Nat. Genet., vol. 39, no. 4, pp. 540–543, Apr. 2007. [CrossRef]
- A. R. Lee et al., “Involvement of mitochondrial biogenesis during the differentiation of human periosteum-derived mesenchymal stem cells into adipocytes, chondrocytes and osteocytes,” Arch. Pharm. Res., vol. 42, no. 12, pp. 1052–1062, Dec. 2019. [CrossRef]
- Sarangarajan, S. Meera, R. Rukkumani, P. Sankar, and G. Anuradha, “Antioxidants: Friend or foe?,” Asian Pac. J. Trop. Med., vol. 10, no. 12, pp. 1111–1116, Dec. 2017. [CrossRef]
- A. Fontana and H. L. Gahlon, “Mechanisms of replication and repair in mitochondrial DNA deletion formation,” Nucleic Acids Res., vol. 48, no. 20, pp. 11244–11258, Nov. 2020. [CrossRef]
- Li, J. Slone, L. Fei, and T. Huang, “Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations,” Cells, vol. 8, no. 6, 2019. [CrossRef]
- G. Shabalina et al., “Enhanced ROS Production in Mitochondria from Prematurely Aging mtDNA Mutator Mice,” Biochemistry (Mosc)., vol. 89, no. 2, pp. 279–298, Feb. 2024. [CrossRef]
- E. Vernucci et al., “Mitophagy and Oxidative Stress in Cancer and Aging: Focus on Sirtuins and Nanomaterials,” Oxid. Med. Cell. Longev., vol. 2019, 2019. [CrossRef]
- Adam Held, “An Introduction to Reactive Oxygen Species - Measurement of ROS in Cells,” 2012.
- G. Waris and H. Ahsan, “Reactive oxygen species: role in the development of cancer and various chronic conditions,” J. Carcinog., vol. 5, p. 14, May 2006. [CrossRef]
- H. A. L. Tuppen, E. L. Blakely, D. M. Turnbull, and R. W. Taylor, “Mitochondrial DNA mutations and human disease,” Biochim. Biophys. Acta Bioenerg., vol. 1797, no. 2, pp. 113–128, Feb. 2010. [CrossRef]
- “Understanding the Process of Aging: The Roles of Mitochondria: Free Radicals, and Antioxidants,” Understanding the Process of Aging, Jan. 1999. [CrossRef]
- N. Ghosh, A. Das, S. Chaffee, S. Roy, and C. K. Sen, “Reactive oxygen species, oxidative damage and cell death,” Immunity and Inflammation in Health and Disease: Emerging Roles of Nutraceuticals and Functional Foods in Immune Support, pp. 45–55, Jan. 2017. [CrossRef]
- A. Ayala, M. F. Muñoz, and S. Argüelles, “Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal,” Oxid. Med. Cell. Longev., vol. 2014, 2014. [CrossRef]
- B. K. Kennedy et al., “Geroscience: Linking aging to chronic disease,” Cell, vol. 159, no. 4, pp. 709–713, Nov. 2014. [CrossRef]
- C. Heidi Chial, “mtDNA and Mitochondrial Diseases | Learn Science at Scitable,” Nature Education, 2008, Accessed: Nov. 22, 2025. [Online]. Available: https://www.nature.com/scitable/topicpage/mtdna-and-mitochondrial-diseases-903/.
- W. Gray, “Mitochondrial evolution,” Cold Spring Harb. Perspect. Biol., vol. 4, no. 9, Sep. 2012. [CrossRef]
- Hahn and S. Zuryn, “The Cellular Mitochondrial Genome Landscape in Disease,” Trends Cell Biol., vol. 29, no. 3, pp. 227–240, Mar. 2019. [CrossRef]
- P. Burgstaller, I. G. Johnston, and J. Poulton, “Mitochondrial DNA disease and developmental implications for reproductive strategies,” Mol. Hum. Reprod., vol. 21, no. 1, p. 11, Jun. 2014. [CrossRef]
- Yu, “Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers,” Life Sci., vol. 89, no. 3–4, pp. 65–71, Jul. 2011. [CrossRef]
- A. Lodato et al., “Aging and neurodegeneration are associated with increased mutations in single human neurons,” Science, vol. 359, no. 6375, pp. 555–559, Feb. 2018. [CrossRef]
- A. Risques and S. R. Kennedy, “Aging and the rise of somatic cancer-associated mutations in normal tissues,” PLoS Genet., vol. 14, no. 1, Jan. 2018. [CrossRef]
- H. Ma et al., “Germline and somatic mtDNA mutations in mouse aging,” PLoS One, vol. 13, no. 7, p. e0201304, Jul. 2018. [CrossRef]
- A. M. Orogo et al., “Accumulation of Mitochondrial DNA Mutations Disrupts Cardiac Progenitor Cell Function and Reduces Survival,” J. Biol. Chem., vol. 290, no. 36, p. 22061, Sep. 2015. [CrossRef]
- J. Niemann et al., “An mtDNA mutation accelerates liver aging by interfering with the ROS response and mitochondrial life cycle,” Free Radic. Biol. Med., vol. 102, pp. 174–187, Jan. 2017. [CrossRef]
- A. Trifunovic et al., “Premature ageing in mice expressing defective mitochondrial DNA polymerase,” Nature, vol. 429, no. 6990, pp. 417–423, May 2004. [CrossRef]
- Yang et al., “NAD+ dependent UPRmt activation underlies intestinal aging caused by mitochondrial DNA mutations,” Nature Communications 2024 15:1, vol. 15, no. 1, pp. 546-, Jan. 2024. [CrossRef]
- Y. Quan, Y. Xin, G. Tian, J. Zhou, and X. Liu, “Mitochondrial ROS-Modulated mtDNA: A Potential Target for Cardiac Aging,” Oxid. Med. Cell. Longev., vol. 2020, no. 1, p. 9423593, Jan. 2020. [CrossRef]
- L. Samstag et al., “Deleterious mitochondrial DNA point mutations are overrepresented in Drosophila expressing a proofreading-defective DNA polymerase γ,” PLoS Genet., vol. 14, no. 11, Nov. 2018. [CrossRef]
- A. D. N. J. De Grey, “A proposed refinement of the mitochondrial free radical theory of aging,” Bioessays, vol. 19, no. 2, pp. 161–166, 1997. [CrossRef]
- G. S. Nido et al., “Ultradeep mapping of neuronal mitochondrial deletions in Parkinson’s disease,” Neurobiol. Aging, vol. 63, pp. 120–127, Mar. 2018. [CrossRef]
- A. Dabravolski, N. K. Sadykhov, A. G. Kartuesov, E. E. Borisov, V. N. Sukhorukov, and A. N. Orekhov, “The Role of Mitochondrial Abnormalities in Diabetic Cardiomyopathy,” Int. J. Mol. Sci., vol. 23, no. 14, Jul. 2022. [CrossRef]
- A. L. M. Smith et al., “Age-associated mitochondrial DNA mutations cause metabolic remodelling that contributes to accelerated intestinal tumorigenesis,” Nat. Cancer, vol. 1, no. 10, pp. 976–989, Oct. 2020. [CrossRef]
- J. Sturgis et al., “Modeling aging and retinal degeneration with mitochondrial DNA mutation burden,” bioRxiv, Dec. 2023. [CrossRef]
- E. R. Árnadóttir et al., “The rate and nature of mitochondrial DNA mutations in human pedigrees,” Cell, vol. 187, no. 15, pp. 3904-3918.e8, Jul. 2024. [CrossRef]
- J. R. Connell et al., “Pedigree derived mutation rate across the entire mitochondrial genome of the Norfolk Island population,” Sci. Rep., vol. 12, no. 1, Dec. 2022. [CrossRef]
- Sigurdardóttir, A. Helgason, J. R. Gulcher, K. Stefansson, and P. Donnelly, “The mutation rate in the human mtDNA control region,” Am. J. Hum. Genet., vol. 66, no. 5, pp. 1599–1609, 2000. [CrossRef]
- Santos et al., “Understanding Differences Between Phylogenetic and Pedigree-Derived mtDNA Mutation Rate: A Model Using Families from the Azores Islands (Portugal),” Mol. Biol. Evol., vol. 22, no. 6, pp. 1490–1505, Jun. 2005. [CrossRef]
- J. Wen et al., “Role of mismatch repair in aging,” Int. J. Biol. Sci., vol. 17, no. 14, p. 3923, 2021. [CrossRef]
- R. Filipovic and W. H. Koppenol, “The Haber-Weiss reaction – The latest revival,” Free Radic. Biol. Med., vol. 145, pp. 221–222, Dec. 2019. [CrossRef]
- L. Mao et al., “Estimation of the mtDNA mutation rate in aging mice by proteome analysis and mathematical modeling,” Exp. Gerontol., vol. 41, no. 1, pp. 11–24, Jan. 2006. [CrossRef]
- B. Nabholz, S. Glémin, and N. Galtier, “The erratic mitochondrial clock: variations of mutation rate, not population size, affect mtDNA diversity across birds and mammals,” BMC Evol. Biol., vol. 9, no. 1, 2009. [CrossRef]
- G. Lou, K. Palikaras, S. Lautrup, M. Scheibye-Knudsen, N. Tavernarakis, and E. F. Fang, “Mitophagy and Neuroprotection,” Trends Mol. Med., vol. 26, no. 1, pp. 8–20, Jan. 2020. [CrossRef]
- S. Schaack, E. K. H. Ho, and F. MacRae, “Disentangling the intertwined roles of mutation, selection and drift in the mitochondrial genome,” Philos. Trans. R. Soc. Lond. B Biol. Sci., vol. 375, no. 1790, 2020. [CrossRef]
- Lane, “Mitonuclear match: optimizing fitness and fertility over generations drives ageing within generations,” Bioessays, vol. 33, no. 11, pp. 860–869, Nov. 2011. [CrossRef]
- Lane, “The Problem with Mixing Mitochondria,” Cell, vol. 151, no. 2, pp. 246–248, Oct. 2012. [CrossRef]
- M. S. Sharpley et al., “Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition,” Cell, vol. 151, no. 2, pp. 333–343, Oct. 2012. [CrossRef]
- J. B. Stewart and P. F. Chinnery, “The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease,” Nat. Rev. Genet., vol. 16, no. 9, pp. 530–542, Aug. 2015. [CrossRef]
- F. Ghiselli et al., “Natural Heteroplasmy and Mitochondrial Inheritance in Bivalve Molluscs,” Integr. Comp. Biol., vol. 59, no. 4, pp. 1016–1032, Oct. 2019. [CrossRef]
- L. N. Lakshmanan, Z. Yee, L. F. Ng, R. Gunawan, B. Halliwell, and J. Gruber, “Clonal expansion of mitochondrial DNA deletions is a private mechanism of aging in long-lived animals,” Aging Cell, vol. 17, no. 5, Oct. 2018. [CrossRef]
- A. E. Vincent et al., “Subcellular origin of mitochondrial DNA deletions in human skeletal muscle,” Ann. Neurol., vol. 84, no. 2, pp. 289–301, Aug. 2018. [CrossRef]
- M. Li, A. Schönberg, M. Schaefer, R. Schroeder, I. Nasidze, and M. Stoneking, “Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes,” Am. J. Hum. Genet., vol. 87, no. 2, pp. 237–249, Aug. 2010. [CrossRef]
- Nissanka and C. T. Moraes, “Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches,” EMBO Rep., vol. 21, no. 3, Mar. 2020. [CrossRef]
- K. Kopinski et al., “Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy,” Proc. Natl. Acad. Sci. U. S. A., vol. 116, no. 32, pp. 16028–16035, Aug. 2019. [CrossRef]
- J. A. Irwin et al., “Investigation of heteroplasmy in the human mitochondrial DNA control region: a synthesis of observations from more than 5000 global population samples,” J. Mol. Evol., vol. 68, no. 5, pp. 516–527, May 2009. [CrossRef]
- A. D. Jayaprakash et al., “Stable heteroplasmy at the single-cell level is facilitated by intercellular exchange of mtDNA,” Nucleic Acids Res., vol. 43, no. 4, p. 2177, Feb. 2015. [CrossRef]
- J. Naue et al., “Evidence for frequent and tissue-specific sequence heteroplasmy in human mitochondrial DNA,” Mitochondrion, vol. 20, no. 1, pp. 82–94, 2015. [CrossRef]
- Y. He et al., “Heteroplasmic mitochondrial DNA mutations in normal and tumour cells,” Nature, vol. 464, no. 7288, pp. 610–614, Mar. 2010. [CrossRef]
- C. Samuels et al., “Recurrent tissue-specific mtDNA mutations are common in humans,” PLoS Genet., vol. 9, no. 11, Nov. 2013. [CrossRef]
- H. Goto et al., “Dynamics of mitochondrial heteroplasmy in three families investigated via a repeatable re-sequencing study,” Genome Biol., vol. 12, no. 6, Jun. 2011. [CrossRef]
- L. A. Zinovkina, “Mechanisms of Mitochondrial DNA Repair in Mammals,” Biochemistry (Mosc)., vol. 83, no. 3, pp. 233–249, Mar. 2018. [CrossRef]
- Sharma and H. Sampath, “Mitochondrial DNA Integrity: Role in Health and Disease,” Cells, vol. 8, no. 2, Feb. 2019. [CrossRef]
- M. Ding et al., “Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway,” J. Pineal Res., vol. 65, no. 2, Sep. 2018. [CrossRef]
- C. Driver, K. D. S. Bamitale, A. Kazi, M. Olla, N. A. Nyane, and P. M. O. Owira, “Cardioprotective Effects of Metformin,” J. Cardiovasc. Pharmacol., vol. 72, no. 2, pp. 121–127, Aug. 2018. [CrossRef]
- G. B. Stefano and R. M. Kream, “Mitochondrial DNA heteroplasmy in human health and disease,” Biomed. Rep., vol. 4, no. 3, p. 259, Mar. 2016. [CrossRef]
- A. A. Elorza and J. P. Soffia, “mtDNA Heteroplasmy at the Core of Aging-Associated Heart Failure. An Integrative View of OXPHOS and Mitochondrial Life Cycle in Cardiac Mitochondrial Physiology,” Front. Cell Dev. Biol., vol. 9, Feb. 2021. [CrossRef]
- J. S. L. Berg Jeremy, Biochemistry, Fifth Edition. W.H. FREEMAN AND COMPANY, 2013.
- Kivrak, K. Yurt, A. Kaplan, I. Alkan, and G. Altun, “Effects of electromagnetic fields exposure on the antioxidant defense system,” J. Microsc. Ultrastruct., vol. 5, no. 4, p. 167, 2017. [CrossRef]
- N. Fuku et al., “The mitochondrial-derived peptide MOTS-c: a player in exceptional longevity?,” Aging Cell, vol. 14, no. 6, pp. 921–923, Dec. 2015. [CrossRef]
- H. Kobayashi and S. Imanaka, “Mitochondrial DNA Damage and Its Repair Mechanisms in Aging Oocytes,” Int. J. Mol. Sci., vol. 25, no. 23, Dec. 2024. [CrossRef]
- C. Tang, J. Cai, X. M. Yin, J. M. Weinberg, M. A. Venkatachalam, and Z. Dong, “Mitochondrial quality control in kidney injury and repair,” Nat. Rev. Nephrol., vol. 17, no. 5, pp. 299–318, May 2021. [CrossRef]
- A. Bohr and G. L. Dianov, “Oxidative DNA damage processing in nuclear and mitochondrial DNA,” Biochimie, vol. 81, no. 1–2, pp. 155–160, 1999. [CrossRef]
- N. B. Larsen, M. Rasmussen, and L. J. Rasmussen, “Nuclear and mitochondrial DNA repair: Similar pathways?,” Mitochondrion, vol. 5, no. 2, pp. 89–108, Apr. 2005. [CrossRef]
- A. Hiona and C. Leeuwenburgh, “The role of mitochondrial DNA mutations in aging and sarcopenia: Implications for the mitochondrial vicious cycle theory of aging,” Exp. Gerontol., vol. 43, no. 1, pp. 24–33, Jan. 2008. [CrossRef]
- Gredilla, I. Sánchez-Román, A. Gómez, M. López-Torres, and G. Barja, “Mitochondrial base excision repair positively correlates with longevity in the liver and heart of mammals,” Geroscience, vol. 42, no. 2, pp. 653–665, Apr. 2020. [CrossRef]
- Gredilla, L. Weissman, J. L. Yang, V. A. Bohr, and T. Stevnsner, “Mitochondrial base excision repair in mouse synaptosomes during normal aging and in a model of Alzheimer’s disease,” Neurobiol. Aging, vol. 33, no. 4, p. 694, Apr. 2010. [CrossRef]
- SenGupta et al., “Base excision repair causes age-dependent accumulation of single-stranded DNA breaks that contribute to Parkinson disease pathology,” Cell Rep., vol. 36, no. 10, Sep. 2021. [CrossRef]
- A. Chen et al., “Increased mitochondrial DNA damage and decreased base excision repair in the auditory cortex of D-galactose-induced aging rats,” Mol. Biol. Rep., vol. 38, no. 6, pp. 3635–3642, Aug. 2011. [CrossRef]
- Y. Zhong et al., “Mitochondrial transcription factor A overexpression and base excision repair deficiency in the inner ear of rats with D-galactose-induced aging,” FEBS J., vol. 278, no. 14, pp. 2500–2510, Jul. 2011. [CrossRef]
- S. K. Tadi, R. Sebastian, S. Dahal, R. K. Babu, B. Choudhary, and S. C. Raghavan, “Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions,” Mol. Biol. Cell, vol. 27, no. 2, pp. 223–235, Jan. 2016. [CrossRef]
- K. Allkanjari and R. A. Baldock, “Beyond base excision repair: an evolving picture of mitochondrial DNA repair,” Biosci. Rep., vol. 41, no. 10, p. BSR20211320, Oct. 2021. [CrossRef]
- Di Carlo and C. Sorrentino, “Oxidative Stress and Age-Related Tumors,” Antioxidants (Basel)., vol. 13, no. 9, Sep. 2024. [CrossRef]
- P. Volobaev, S. S. Kunizheva, L. I. Uralsky, D. A. Kupriyanova, and E. I. Rogaev, “Quantifying human genome parameters in aging,” Vavilovskii Zhurnal Genet. Selektsii, vol. 27, no. 5, pp. 495–501, 2023. [CrossRef]
- L. Wu et al., “Increased p66Shc in the Inner Ear of D-Galactose-Induced Aging Mice with Accumulation of Mitochondrial DNA 3873-bp Deletion: p66Shc and mtDNA Damage in the Inner Ear during Aging,” PLoS One, vol. 7, no. 11, p. e50483, Nov. 2012. [CrossRef]
- C. A. Castellani, R. J. Longchamps, J. Sun, E. Guallar, and D. E. Arking, “Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease,” Mitochondrion, vol. 53, pp. 214–223, Jul. 2020. [CrossRef]
- Zhang et al., “The distinct spatiotemporal distribution and effect of feed restriction on mtDNA copy number in broilers,” Scientific Reports 2020 10:1, vol. 10, no. 1, pp. 3240-, Feb. 2020. [CrossRef]
- B. Singh, T. R. Schoeb, P. Bajpai, A. Slominski, and K. K. Singh, “Reversing wrinkled skin and hair loss in mice by restoring mitochondrial function,” Cell Death & Disease 2018 9:7, vol. 9, no. 7, pp. 735-, Jul. 2018. [CrossRef]
- Z. Zhang et al., “Decrease of MtDNA copy number affects mitochondrial function and involves in the pathological consequences of ischaemic stroke,” J. Cell. Mol. Med., vol. 26, no. 15, pp. 4157–4168, Aug. 2022. [CrossRef]
- N. Sun, R. J. Youle, and T. Finkel, “The Mitochondrial Basis of Aging,” Mol. Cell, vol. 61, no. 5, pp. 654–666, Mar. 2016. [CrossRef]
- R. R. Thomas et al., “RhTFAM treatment stimulates mitochondrial oxidative metabolism and improves memory in aged mice,” Aging, vol. 4, no. 9, pp. 620–635, 2012. [CrossRef]
- D. T. Soltys et al., “Lower mitochondrial DNA content but not increased mutagenesis associates with decreased base excision repair activity in brains of AD subjects,” Neurobiol. Aging, vol. 73, pp. 161–170, Jan. 2019. [CrossRef]
- D. Edgar and A. Trifunovic, “The mtDNA mutator mouse: Dissecting mitochondrial involvement in aging,” Aging, vol. 1, no. 12, pp. 1028–1032, 2009. [CrossRef]
- N. Keshavan et al., “The natural history of infantile mitochondrial DNA depletion syndrome due to RRM2B deficiency,” Genetics in Medicine, vol. 22, no. 1, pp. 199–209, Jan. 2020. [CrossRef]
- C. Viscomi and M. Zeviani, “MtDNA-maintenance defects: syndromes and genes,” J. Inherit. Metab. Dis., vol. 40, no. 4, pp. 587–599, Jul. 2017. [CrossRef]
- Wang et al., “Ablation of Shank3 alleviates cardiac dysfunction in aging mice by promoting CaMKII activation and Parkin-mediated mitophagy,” Redox Biol., vol. 58, p. 102537, Dec. 2022. [CrossRef]
- R. Filograna, M. Mennuni, D. Alsina, and N. G. Larsson, “Mitochondrial DNA copy number in human disease: the more the better?,” FEBS Lett., vol. 595, no. 8, pp. 976–1002, Apr. 2021. [CrossRef]
- Mizuno et al., “Low mitochondrial DNA copy number in peripheral blood mononuclear cells is associated with future mortality risk: a long-term follow-up study from Japan,” J. Nutr. Health Aging, vol. 28, no. 1, p. 100013, Jan. 2024. [CrossRef]
- Sun and J. C. St John, “Modulation of mitochondrial DNA copy number in a model of glioblastoma induces changes to DNA methylation and gene expression of the nuclear genome in tumours,” Epigenetics Chromatin, vol. 11, no. 1, Sep. 2018. [CrossRef]
- K. Foote et al., “Restoring mitochondrial DNA copy number preserves mitochondrial function and delays vascular aging in mice,” Aging Cell, vol. 17, no. 4, Aug. 2018. [CrossRef]
- J. Ding et al., “Assessing Mitochondrial DNA Variation and Copy Number in Lymphocytes of ~2,000 Sardinians Using Tailored Sequencing Analysis Tools,” PLoS Genet., vol. 11, no. 7, Jul. 2015. [CrossRef]
- J. Knez et al., “Correlates of Peripheral Blood Mitochondrial DNA Content in a General Population,” Am. J. Epidemiol., vol. 183, no. 2, pp. 138–146, Jan. 2016. [CrossRef]
- R. Zhang, Y. Wang, K. Ye, M. Picard, and Z. Gu, “Independent impacts of aging on mitochondrial DNA quantity and quality in humans,” BMC Genomics 2017 18:1, vol. 18, no. 1, pp. 890-, Nov. 2017. [CrossRef]
- A. Herbst et al., “Skeletal muscle mitochondrial DNA copy number and mitochondrial DNA deletion mutation frequency as predictors of physical performance in older men and women,” Geroscience, vol. 43, no. 3, pp. 1253–1264, Jun. 2021. [CrossRef]
- X. Liu et al., “Association of mitochondrial DNA copy number with cardiometabolic diseases,” Cell Genomics, vol. 1, no. 1, Oct. 2021. [CrossRef]
- E. Coskun, M. F. Beal, and D. C. Wallace, “Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication,” Proc. Natl. Acad. Sci. U. S. A., vol. 101, no. 29, pp. 10726–10731, Jul. 2004. [CrossRef]
- B. Rodríguez-Santiago, J. Casademont, and V. Nunes, “Is mitochondrial DNA depletion involved in Alzheimer’s disease?,” Eur. J. Hum. Genet., vol. 9, no. 4, pp. 279–285, 2001. [CrossRef]
- C. Dölle et al., “Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease,” Nat. Commun., vol. 7, Nov. 2016. [CrossRef]
- A. Pyle, H. Anugrha, M. Kurzawa-Akanbi, A. Yarnall, D. Burn, and G. Hudson, “Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease,” Neurobiol. Aging, vol. 38, pp. 216.e7-216.e10, Jul. 2015. [CrossRef]
- X. bin Wang, N. hua Cui, S. Zhang, Z. jin Liu, J. fen Ma, and L. Ming, “Leukocyte telomere length, mitochondrial DNA copy number, and coronary artery disease risk and severity: A two-stage case-control study of 3064 Chinese subjects,” Atherosclerosis, vol. 284, pp. 165–172, May 2019. [CrossRef]
- Zhang et al., “Association between mitochondrial DNA copy number and sudden cardiac death: findings from the Atherosclerosis Risk in Communities study (ARIC),” Eur. Heart J., vol. 38, no. 46, pp. 3443–3448, Dec. 2017. [CrossRef]
- F. N. Ashar et al., “Association of Mitochondrial DNA Copy Number With Cardiovascular Disease,” JAMA Cardiol., vol. 2, no. 11, pp. 1247–1255, Nov. 2017. [CrossRef]
- M. Ikeda et al., “Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress,” PLoS One, vol. 10, no. 3, Mar. 2015. [CrossRef]
- Filograna et al., “Modulation of mtDNA copy number ameliorates the pathological consequences of a heteroplasmic mtDNA mutation in the mouse,” Sci. Adv., vol. 5, no. 4, 2019. [CrossRef]
- K. Li et al., “Leukocyte telomere length and mitochondrial DNA copy number associate with endothelial function in aging-related cardiovascular disease,” Front. Cardiovasc. Med., vol. 10, 2023. [CrossRef]
- B. Pillai et al., “Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice,” Am. J. Physiol. Heart Circ. Physiol., vol. 310, no. 8, pp. H962–H972, Apr. 2016. [CrossRef]
- J. Marín-García, “Mitochondrial DNA repair: a novel therapeutic target for heart failure,” Heart Fail. Rev., vol. 21, no. 5, pp. 475–487, Sep. 2016. [CrossRef]
- A. Hahn et al., “Misregulation of mitochondrial 6mA promotes the propagation of mutant mtDNA and causes aging in C. elegans,” Cell Metab., vol. 36, no. 12, pp. 2528-2541.e11, Dec. 2024. [CrossRef]
- Long et al., “Maintaining mitochondrial DNA copy number mitigates ROS-induced oocyte decline and female reproductive aging,” Commun. Biol., vol. 7, no. 1, Dec. 2024. [CrossRef]
- Sreedhar, L. Aguilera-Aguirre, and K. K. Singh, “Mitochondria in skin health, aging, and disease,” Cell Death Dis., vol. 11, no. 6, Jun. 2020. [CrossRef]
- Sherkhane, G. Chayanika, A. Sood, D. K. Khatri, and S. B. Singh, “Mitochondrial remodelling-a vicious cycle in diabetic complications,” Mol. Biol. Rep., vol. 48, no. 5, pp. 4721–4731, May 2021. [CrossRef]
- Sebastián et al., “Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway,” EMBO J., vol. 35, no. 15, pp. 1677–1693, Aug. 2016. [CrossRef]
- Iqbal, O. Ostojic, K. Singh, A. M. Joseph, and D. A. Hood, “Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse,” Muscle Nerve, vol. 48, no. 6, pp. 963–970, 2013. [CrossRef]
- L. Zhao et al., “Evidence for association of mitochondrial metabolism alteration with lipid accumulation in aging rats,” Exp. Gerontol., vol. 56, pp. 3–12, 2014. [CrossRef]
- Tezze et al., “Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence,” Cell Metab., vol. 25, no. 6, pp. 1374-1389.e6, Jun. 2017. [CrossRef]
- A. M. Joseph et al., “The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals,” Aging Cell, vol. 11, no. 5, pp. 801–809, Oct. 2012. [CrossRef]
- J. Faitg, J. P. Leduc-Gaudet, O. Reynaud, G. Ferland, P. Gaudreau, and G. Gouspillou, “Effects of Aging and Caloric Restriction on Fiber Type Composition, Mitochondrial Morphology and Dynamics in Rat Oxidative and Glycolytic Muscles,” Front. Physiol., vol. 10, no. APR, 2019. [CrossRef]
- J. P. Leduc-Gaudet et al., “Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice,” Oncotarget, vol. 6, no. 20, pp. 17923–17937, 2015. [CrossRef]
- Favaro et al., “DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass,” Nature Communications 2019 10:1, vol. 10, no. 1, pp. 2576-, Jun. 2019. [CrossRef]
- M. Dulac et al., “Drp1 knockdown induces severe muscle atrophy and remodelling, mitochondrial dysfunction, autophagy impairment and denervation,” J. Physiol., vol. 598, no. 17, pp. 3691–3710, Sep. 2020. [CrossRef]
- M. Dulac et al., “Regulation of muscle and mitochondrial health by the mitochondrial fission protein Drp1 in aged mice,” J. Physiol., vol. 599, no. 17, pp. 4045–4063, Sep. 2021. [CrossRef]
- A. M. Joseph et al., “Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging,” PLoS One, vol. 8, no. 7, Jul. 2013. [CrossRef]
- L. García-Prat et al., “Autophagy maintains stemness by preventing senescence,” Nature, vol. 529, no. 7584, pp. 37–42, Jan. 2016. [CrossRef]
- C. A. McMullen, A. L. Ferry, J. L. Gamboa, F. H. Andrade, and E. E. Dupont-Versteegden, “Age-related changes of cell death pathways in rat extraocular muscle,” Exp. Gerontol., vol. 44, no. 6–7, pp. 420–425, Jun. 2009. [CrossRef]
- M. Gaugler, A. Brown, E. Merrell, M. DiSanto-Rose, J. A. Rathmacher, and T. H. Reynolds IV, “PKB signaling and atrogene expression in skeletal muscle of aged mice,” J. Appl. Physiol. (1985)., vol. 111, no. 1, pp. 192–199, Jul. 2011. [CrossRef]
- M. J. Drummond et al., “Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: a cross-sectional comparison,” J. Gerontol. A Biol. Sci. Med. Sci., vol. 69, no. 8, pp. 1040–1048, 2014. [CrossRef]
- W. Russ, A. M. Wills, I. M. Boyd, and J. Krause, “Weakness, SR function and stress in gastrocnemius muscles of aged male rats,” Exp. Gerontol., vol. 50, no. 1, pp. 40–44, Feb. 2014. [CrossRef]
- P. Leduc-Gaudet, O. Reynaud, S. N. Hussain, and G. Gouspillou, “Parkin overexpression protects from ageing-related loss of muscle mass and strength,” J. Physiol., vol. 597, no. 7, pp. 1975–1991, Apr. 2019. [CrossRef]
- Gouspillou et al., “Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans,” FASEB J., vol. 28, no. 4, pp. 1621–1633, 2014. [CrossRef]
- A. R. Konopka, M. K. Suer, C. A. Wolff, and M. P. Harber, “Markers of human skeletal muscle mitochondrial biogenesis and quality control: effects of age and aerobic exercise training,” J. Gerontol. A Biol. Sci. Med. Sci., vol. 69, no. 4, pp. 371–378, Apr. 2014. [CrossRef]
- D. Mellem et al., “Fragmentation of the mitochondrial network in skin in vivo,” PLoS One, vol. 12, no. 6, Jun. 2017. [CrossRef]
- D. Gupta, S. Archoo, S. H. Naikoo, and S. T. Abdullah, “Rosmarinic Acid: A Naturally Occurring Plant Based Agent Prevents Impaired Mitochondrial Dynamics and Apoptosis in Ultraviolet-B-Irradiated Human Skin Cells,” Photochem. Photobiol., vol. 98, no. 4, pp. 925–934, Jul. 2021. [CrossRef]
- R. B. Hamanaka et al., “Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development,” Sci. Signal., vol. 6, no. 261, Feb. 2013. [CrossRef]
- Miwa, S. Kashyap, E. Chini, and T. von Zglinicki, “Mitochondrial dysfunction in cell senescence and aging,” J. Clin. Invest., vol. 132, no. 13, Jul. 2022. [CrossRef]
- E. Oblong et al., “Metabolic dysfunction in human skin: Restoration of mitochondrial integrity and metabolic output by nicotinamide (niacinamide) in primary dermal fibroblasts from older aged donors,” Aging Cell, vol. 19, no. 10, p. e13248, Oct. 2020. [CrossRef]
- Rorteau et al., “Maintenance of Chronological Aging Features in Culture of Normal Human Dermal Fibroblasts from Old Donors,” Cells, vol. 11, no. 5, Mar. 2022. [CrossRef]
- A. Bowman and M. A. Birch-Machin, “Age-Dependent Decrease of Mitochondrial Complex II Activity in Human Skin Fibroblasts,” Journal of Investigative Dermatology, vol. 136, no. 5, pp. 912–919, 2016. [CrossRef]
- D. F. Dai, T. Chen, S. C. Johnson, H. Szeto, and P. S. Rabinovitch, “Cardiac aging: from molecular mechanisms to significance in human health and disease,” Antioxid. Redox Signal., vol. 16, no. 12, pp. 1492–1536, Jun. 2012. [CrossRef]
- Ljubicic, K. J. Menzies, and D. A. Hood, “Mitochondrial dysfunction is associated with a pro-apoptotic cellular environment in senescent cardiac muscle,” Mech. Ageing Dev., vol. 131, no. 2, pp. 79–88, Feb. 2010. [CrossRef]
- W. Dorn, “Parkin-dependent mitophagy in the heart,” J. Mol. Cell. Cardiol., vol. 95, pp. 42–49, Jun. 2016. [CrossRef]
- J. Gollmer, A. Zirlik, and H. Bugger, “Mitochondrial Mechanisms in Diabetic Cardiomyopathy,” Diabetes Metab. J., vol. 44, no. 1, pp. 33–53, Feb. 2020. [CrossRef]
- T. Breitzig, M. D. Alleyn, R. F. Lockey, and N. Kolliputi, “A mitochondrial delicacy: dynamin-related protein 1 and mitochondrial dynamics,” Am. J. Physiol. Cell Physiol., vol. 315, no. 1, pp. C80–C90, Jul. 2018. [CrossRef]
- Y. Y. Park, S. Lee, M. Karbowski, A. Neutzner, R. J. Youle, and H. Cho, “Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1,” J. Cell Sci., vol. 123, no. Pt 4, pp. 619–626, Feb. 2010. [CrossRef]
- T. J. Cahill et al., “Resistance of Dynamin-related Protein 1 Oligomers to Disassembly Impairs Mitophagy, Resulting in Myocardial Inflammation and Heart Failure,” Journal of Biological Chemistry, vol. 290, no. 43, pp. 25907–25919, Oct. 2015. [CrossRef]
- T. T. Lee et al., “Loss of Fis1 impairs proteostasis during skeletal muscle aging in Drosophila,” Aging Cell, vol. 20, no. 6, p. e13379, Jun. 2021. [CrossRef]
- G. Rontoyanni, O. N. Lopez, G. T. Fankhauser, Z. F. Cheema, B. B. Rasmussen, and C. Porter, “Mitochondrial Bioenergetics in the Metabolic Myopathy Accompanying Peripheral Artery Disease,” Front. Physiol., vol. 8, no. MAR, Mar. 2017. [CrossRef]
- He, Y. Ma, R. Wang, J. Zhang, L. Jing, and P. A. Li, “Deletion of Mitochondrial Uncoupling Protein 2 Exacerbates Mitochondrial Damage in Mice Subjected to Cerebral Ischemia and Reperfusion Injury under both Normo- and Hyperglycemic Conditions,” Int. J. Biol. Sci., vol. 16, no. 15, pp. 2788–2802, 2020. [CrossRef]
- P. Semadhi, D. Mulyaty, E. Halimah, and J. Levita, “Healthy mitochondrial DNA in balanced mitochondrial dynamics: A potential marker for neuro-aging prediction (Review),” Biomed. Rep., vol. 19, no. 3, Sep. 2023. [CrossRef]
- E. Zukowski et al., “STAT3 modulates CD4+ T mitochondrial dynamics and function in aging,” Aging Cell, vol. 22, no. 11, Nov. 2023. [CrossRef]
- J. C. Campos et al., “Exercise preserves physical fitness during aging through AMPK and mitochondrial dynamics,” Proc. Natl. Acad. Sci. U. S. A., vol. 120, no. 2, Jan. 2023. [CrossRef]
- T. Yu et al., “Premature aging is associated with higher levels of 8-oxoguanine and increased DNA damage in the Polg mutator mouse,” Aging Cell, vol. 21, no. 9, Sep. 2022. [CrossRef]
- J. F. Halling et al., “PGC-1α regulates mitochondrial properties beyond biogenesis with aging and exercise training,” Am. J. Physiol. Endocrinol. Metab., vol. 317, no. 3, pp. E513–E525, Sep. 2019. [CrossRef]
- J. Wang et al., “Spermidine alleviates cardiac aging by improving mitochondrial biogenesis and function,” Aging, vol. 12, no. 1, pp. 650–671, Jan. 2020. [CrossRef]
- L. D. Popov, “Mitochondrial biogenesis: An update,” J. Cell. Mol. Med., vol. 24, no. 9, pp. 4892–4899, May 2020. [CrossRef]
- Li and Z. Cai, “SIRT3 regulates mitochondrial biogenesis in aging-related diseases,” J. Biomed. Res., vol. 37, no. 2, pp. 77–88, 2022. [CrossRef]
- M. H. Muhammad and M. M. Allam, “Resveratrol and/or exercise training counteract aging-associated decline of physical endurance in aged mice; targeting mitochondrial biogenesis and function,” Journal of Physiological Sciences, vol. 68, no. 5, pp. 681–688, Sep. 2018. [CrossRef]
- Y. Kim, M. Triolo, and D. A. Hood, “Impact of Aging and Exercise on Mitochondrial Quality Control in Skeletal Muscle,” Oxid. Med. Cell. Longev., vol. 2017, 2017. [CrossRef]
- A. Li, X. Hou, and S. Hao, “Mitochondrial biogenesis in neurodegeneration,” J. Neurosci. Res., vol. 95, no. 10, pp. 2025–2029, Oct. 2017. [CrossRef]
- C. Ploumi, I. Daskalaki, and N. Tavernarakis, “Mitochondrial biogenesis and clearance: a balancing act,” FEBS J., vol. 284, no. 2, pp. 183–195, Jan. 2017. [CrossRef]
- Tian, W. He, R. Yang, and Y. Liu, “Dl-3-n-butylphthalide protects the heart against ischemic injury and H9c2 cardiomyoblasts against oxidative stress: involvement of mitochondrial function and biogenesis,” J. Biomed. Sci., vol. 24, no. 1, 2017. [CrossRef]
- L. Tao et al., “Exercise Training Protects Against Acute Myocardial Infarction via Improving Myocardial Energy Metabolism and Mitochondrial Biogenesis,” Cell. Physiol. Biochem., vol. 37, no. 1, pp. 162–175, Aug. 2015. [CrossRef]
- L. Xu et al., “GLIS1 alleviates cell senescence and renal fibrosis through PGC1-α mediated mitochondrial quality control in kidney aging,” Free Radic. Biol. Med., vol. 209, no. Pt 1, pp. 171–184, Nov. 2023. [CrossRef]
- Wang et al., “Pentoxifylline Enhances Antioxidative Capability and Promotes Mitochondrial Biogenesis in D-Galactose-Induced Aging Mice by Increasing Nrf2 and PGC-1 α through the cAMP-CREB Pathway,” Oxid. Med. Cell. Longev., vol. 2021, 2021. [CrossRef]
- Liu, C. E. Chang, A. C. Wooldredge, B. Fong, B. K. Kennedy, and C. Zhou, “Tom70-based transcriptional regulation of mitochondrial biogenesis and aging,” Elife, vol. 11, Mar. 2022. [CrossRef]
- D’Aquila, D. Bellizzi, and G. Passarino, “Mitochondria in health, aging and diseases: the epigenetic perspective,” Biogerontology, vol. 16, no. 5, pp. 569–585, Oct. 2015. [CrossRef]
- M. Parodi-Rullán, X. R. Chapa-Dubocq, and S. Javadov, “Acetylation of Mitochondrial Proteins in the Heart: The Role of SIRT3,” Front. Physiol., vol. 9, no. AUG, Aug. 2018. [CrossRef]
- L. Liu, Y. Li, G. Chen, and Q. Chen, “Crosstalk between mitochondrial biogenesis and mitophagy to maintain mitochondrial homeostasis,” J. Biomed. Sci., vol. 30, no. 1, Dec. 2023. [CrossRef]
- K. Palikaras, E. Lionaki, and N. Tavernarakis, “Mechanisms of mitophagy in cellular homeostasis, physiology and pathology,” Nat. Cell Biol., vol. 20, no. 9, pp. 1013–1022, Sep. 2018. [CrossRef]
- M. Cui et al., “HKDC1, a target of TFEB, is essential to maintain both mitochondrial and lysosomal homeostasis, preventing cellular senescence,” Proc. Natl. Acad. Sci. U. S. A., vol. 121, no. 2, 2024. [CrossRef]
- M. Triolo, D. Bhattacharya, and D. A. Hood, “Denervation induces mitochondrial decline and exacerbates lysosome dysfunction in middle-aged mice,” Aging, vol. 14, no. 22, pp. 8900–8913, 2022. [CrossRef]
- N. M. Christensen, S. Ringholm, B. T. Buch, A. Gudiksen, J. F. Halling, and H. Pilegaard, “Muscle PGC-1α modulates hepatic mitophagy regulation during aging,” Exp. Gerontol., vol. 172, Feb. 2023. [CrossRef]
- E. Marzetti, R. Calvani, H. J. Coelho-Junior, and A. Picca, “Mitochondrial pathways and sarcopenia in the geroscience era,” Journal of Nutrition, Health and Aging, vol. 28, no. 12, Dec. 2024. [CrossRef]
- A. Rappe et al., “Longitudinal autophagy profiling of the mammalian brain reveals sustained mitophagy throughout healthy aging,” EMBO J., vol. 43, no. 23, pp. 6199–6231, Dec. 2024. [CrossRef]
- E. T. Schmid, J. H. Pyo, and D. W. Walker, “Neuronal induction of BNIP3-mediated mitophagy slows systemic aging in Drosophila,” Nat. Aging, vol. 2, no. 6, pp. 494–507, Jun. 2022. [CrossRef]
- Luo, R. Zhang, J. Krigman, A. McAdams, S. Ozgen, and N. Sun, “A Healthy Heart and a Healthy Brain: Looking at Mitophagy,” Front. Cell Dev. Biol., vol. 8, May 2020. [CrossRef]
- P. Leduc-Gaudet, S. N. A. Hussain, E. Barreiro, and G. Gouspillou, “Mitochondrial Dynamics and Mitophagy in Skeletal Muscle Health and Aging,” Int. J. Mol. Sci., vol. 22, no. 15, Aug. 2021. [CrossRef]
- P. Rai et al., “IRGM1 links mitochondrial quality control to autoimmunity,” Nature Immunology 2021 22:3, vol. 22, no. 3, pp. 312–321, Jan. 2021. [CrossRef]
- Hu et al., “Stabilization of HIF-1α alleviates osteoarthritis via enhancing mitophagy,” Cell Death & Disease 2020 11:6, vol. 11, no. 6, pp. 481-, Jun. 2020. [CrossRef]
- N. Oleinik et al., “Alterations of lipid-mediated mitophagy result in aging-dependent sensorimotor defects,” Aging Cell, vol. 22, no. 10, Oct. 2023. [CrossRef]
- Mito et al., “Mosaic dysfunction of mitophagy in mitochondrial muscle disease,” Cell Metab., vol. 34, no. 2, pp. 197-208.e5, Feb. 2022. [CrossRef]
- A. L. Hughes and D. E. Gottschling, “An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast,” Nature 2012 492:7428, vol. 492, no. 7428, pp. 261–265, Nov. 2012. [CrossRef]
- Shirakabe et al., “Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure,” Circulation, vol. 133, no. 13, pp. 1249–1263, Mar. 2016. [CrossRef]
- M. Schneider et al., “Age-Related Deterioration of Mitochondrial Function in the Intestine,” Oxid. Med. Cell. Longev., vol. 2020, p. 4898217, 2020. [CrossRef]
- M. Golpich, E. Amini, Z. Mohamed, R. Azman Ali, N. Mohamed Ibrahim, and A. Ahmadiani, “Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment,” CNS Neurosci. Ther., vol. 23, no. 1, pp. 5–22, Jan. 2017. [CrossRef]
- S. Rizza et al., “S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy,” Proc. Natl. Acad. Sci. U. S. A., vol. 115, no. 15, pp. E3388–E3397, 2018. [CrossRef]
- S. Lautrup, D. A. Sinclair, M. P. Mattson, and E. F. Fang, “NAD+ in Brain Aging and Neurodegenerative Disorders,” Cell Metab., vol. 30, no. 4, pp. 630–655, Oct. 2019. [CrossRef]
- E. F. Fang et al., “Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway,” Scientific Reports 2017 7:1, vol. 7, no. 1, pp. 46208-, Apr. 2017. [CrossRef]
- D. Ryu et al., “Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents,” Nat. Med., vol. 22, no. 8, pp. 879–888, Aug. 2016. [CrossRef]
- E. M. Fivenson et al., “Mitophagy in neurodegeneration and aging,” Neurochem. Int., vol. 109, pp. 202–209, Oct. 2017. [CrossRef]
- M. Castellazzi et al., “Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer’s disease and mild cognitive impairment,” Scientific Reports 2019 9:1, vol. 9, no. 1, pp. 20009-, Dec. 2019. [CrossRef]
- P. Martín-Maestro, R. Gargini, G. Perry, J. Avila, and V. García-Escudero, “PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease,” Hum. Mol. Genet., vol. 25, no. 4, pp. 792–806, Feb. 2016. [CrossRef]
- Götz, F. Chen, J. Van Dorpe, and R. M. Nitsch, “Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils,” Science, vol. 293, no. 5534, pp. 1491–1495, Aug. 2001. [CrossRef]
- L. Schulz et al., “A new link to mitochondrial impairment in tauopathies,” Mol. Neurobiol., vol. 46, no. 1, pp. 205–216, 2012. [CrossRef]
- R. K. Lane, T. Hilsabeck, and S. L. Rea, “The role of mitochondrial dysfunction in age-related diseases,” Biochim. Biophys. Acta Bioenerg., vol. 1847, no. 11, pp. 1387–1400, Nov. 2015. [CrossRef]
- Wang and R. L. Davis, “Early Mitochondrial Fragmentation and Dysfunction in a Drosophila Model for Alzheimer’s Disease,” Mol. Neurobiol., vol. 58, no. 1, pp. 143–155, Jan. 2021. [CrossRef]
- Manczak, M. J. Calkins, and P. H. Reddy, “Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage,” Hum. Mol. Genet., vol. 20, no. 13, pp. 2495–2509, Jul. 2011. [CrossRef]
- Manczak and P. H. Reddy, “Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage,” Hum. Mol. Genet., vol. 21, no. 11, pp. 2538–2547, Jun. 2012. [CrossRef]
- Zhao et al., “Treadmill Exercise Attenuates Aβ-Induced Mitochondrial Dysfunction and Enhances Mitophagy Activity in APP/PS1 Transgenic Mice,” Neurochem. Res., vol. 45, no. 5, pp. 1202–1214, May 2020. [CrossRef]
- R. J. Youle and D. P. Narendra, “Mechanisms of mitophagy,” Nat. Rev. Mol. Cell Biol., vol. 12, no. 1, pp. 9–14, Jan. 2011. [CrossRef]
- S. M. S. Yakhine-Diop et al., “Impaired Mitophagy and Protein Acetylation Levels in Fibroblasts from Parkinson’s Disease Patients,” Mol. Neurobiol., vol. 56, no. 4, pp. 2466–2481, Apr. 2019. [CrossRef]
- Cai et al., “Overexpression of PGC-1α influences the mitochondrial unfolded protein response (mtUPR) induced by MPP+ in human SH-SY5Y neuroblastoma cells,” Sci. Rep., vol. 10, no. 1, Dec. 2020. [CrossRef]
- M. W. Lin, C. C. Lin, Y. H. Chen, H. Bin Yang, and S. Y. Hung, “Celastrol Inhibits Dopaminergic Neuronal Death of Parkinson’s Disease through Activating Mitophagy,” Antioxidants (Basel)., vol. 9, no. 1, Jan. 2019. [CrossRef]
- M. D. Umare et al., “Interweaving of reactive oxygen species and major neurological and psychiatric disorders,” Ann. Pharm. Fr., vol. 80, no. 4, pp. 409–425, Jul. 2022. [CrossRef]
- A. H. V. Schapira and M. Gegg, “Mitochondrial contribution to Parkinson’s disease pathogenesis,” Parkinsons Dis., vol. 2011, 2011. [CrossRef]
- M. J. Quinn, P. I. Moreira, A. F. Ambrósio, and C. H. Alves, “PINK1/PARKIN signalling in neurodegeneration and neuroinflammation,” Acta Neuropathol. Commun., vol. 8, no. 1, p. 189, Nov. 2020. [CrossRef]
- L. Ma, J. Zhu, Q. Gao, M. J. Rebecchi, Q. Wang, and L. Liu, “Restoring Pharmacologic Preconditioning in the Aging Heart: Role of Mitophagy/Autophagy,” J. Gerontol. A Biol. Sci. Med. Sci., vol. 72, no. 4, pp. 489–498, 2017. [CrossRef]
- J. Ren et al., “Akt2 ablation prolongs life span and improves myocardial contractile function with adaptive cardiac remodeling: role of Sirt1-mediated autophagy regulation,” Aging Cell, vol. 16, no. 5, pp. 976–987, Oct. 2017. [CrossRef]
- A. Aliper et al., “Towards natural mimetics of metformin and rapamycin,” Aging, vol. 9, no. 11, pp. 2245–2268, 2017. [CrossRef]
- J. Wen et al., “Role of mitophagy in the hallmarks of aging,” J. Biomed. Res., vol. 37, no. 1, p. 1, 2022. [CrossRef]
- Y. Guo et al., “Sirt3-mediated mitophagy regulates AGEs-induced BMSCs senescence and senile osteoporosis,” Redox Biol., vol. 41, May 2021. [CrossRef]
- J. S. Harrington, S. W. Ryter, M. Plataki, D. R. Price, and A. M. K. Choi, “Mitochondria in health, disease, and aging,” Physiol. Rev., vol. 103, no. 4, pp. 2349–2422, 2023. [CrossRef]
- N. S. Swerdlow and H. M. Wilkins, “Mitophagy and the Brain,” Int. J. Mol. Sci., vol. 21, no. 24, Dec. 2020. [CrossRef]
- M. F. Alexeyev, S. P. LeDoux, and G. L. Wilson, “Mitochondrial DNA and aging,” Clin. Sci. (Lond)., vol. 107, no. 4, pp. 355–364, Oct. 2004. [CrossRef]
- O. S. Ademowo, O. Oyebode, R. Edward, M. E. Conway, H. R. Griffiths, and I. H. K. Dias, “Effects of carotenoids on mitochondrial dysfunction,” Biochem. Soc. Trans., vol. 52, no. 1, p. 65, Feb. 2024. [CrossRef]
- H. Alizadeh Pahlavani, I. Laher, B. Knechtle, and H. Zouhal, “Exercise and mitochondrial mechanisms in patients with sarcopenia,” Front. Physiol., vol. 13, Dec. 2022. [CrossRef]
- Fularski et al., “Statins in Chronic Kidney Disease-Effects on Atherosclerosis and Cellular Senescence,” Cells, vol. 12, no. 13, Jul. 2023. [CrossRef]
- M. Ge, F. Fontanesi, S. Merscher, and A. Fornoni, “The Vicious Cycle of Renal Lipotoxicity and Mitochondrial Dysfunction,” Front. Physiol., vol. 11, Jul. 2020. [CrossRef]
- A. Paoli and G. Cerullo, “Investigating the Link between Ketogenic Diet, NAFLD, Mitochondria, and Oxidative Stress: A Narrative Review,” Antioxidants (Basel)., vol. 12, no. 5, May 2023. [CrossRef]
- Poerwoatmodjo, G. J. Schenk, J. J. G. Geurts, and A. Luchicchi, “Cysteine Proteases and Mitochondrial Instability: A Possible Vicious Cycle in MS Myelin?,” Front. Cell. Neurosci., vol. 14, Dec. 2020. [CrossRef]
- D. Nguyen et al., “A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics,” Cell Death Dis., vol. 2, no. 12, Dec. 2011. [CrossRef]
Figure 1.
Mitochondrial Drivers of Aging: A Healthspan–Lifespan Clock Model. Schematic clock in which each hour position (1–12) represents a mitochondrial driver of aging, ordered by increasing contribution to functional decline based on frequency of appearance and mechanistic impact. The central gear represents mitochondria as the core regulator of cellular aging. The progressive clockwise arrangement illustrates how accumulating dysfunction across these processes accelerates biological aging and reduces healthspan.
Figure 1.
Mitochondrial Drivers of Aging: A Healthspan–Lifespan Clock Model. Schematic clock in which each hour position (1–12) represents a mitochondrial driver of aging, ordered by increasing contribution to functional decline based on frequency of appearance and mechanistic impact. The central gear represents mitochondria as the core regulator of cellular aging. The progressive clockwise arrangement illustrates how accumulating dysfunction across these processes accelerates biological aging and reduces healthspan.
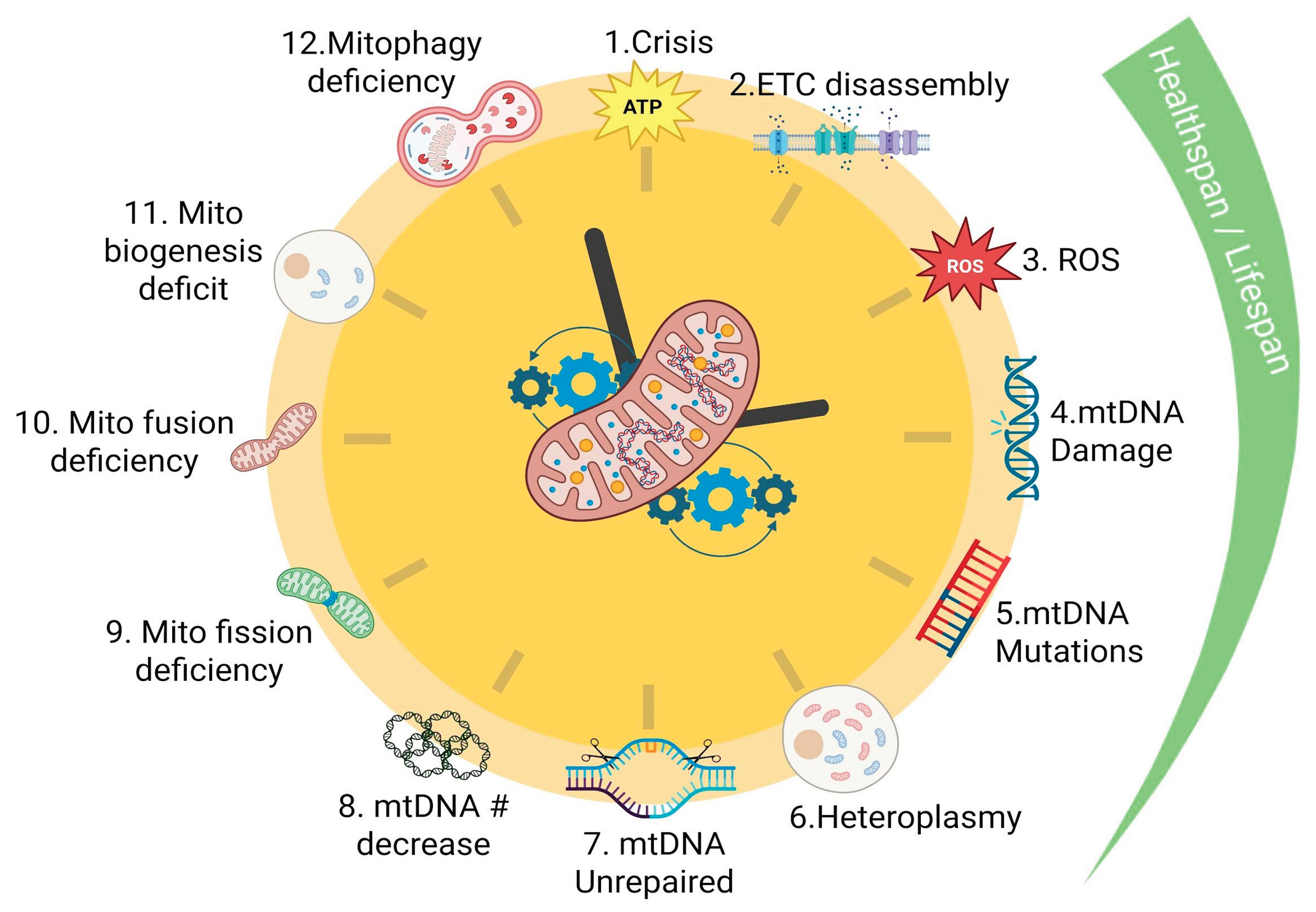
Figure 2.
Lifetime Accumulation Dynamics of Mitochondrial DNA Mutations. A) Table showing chronological age (10–70 years) and the estimated number of mtDNA mutations accumulated per decade, illustrating the progressive increase in mutational burden with aging. B) Linear plot of total mtDNA mutation count versus age, displaying a steady decade-to-decade rise with a marked inflection between 40 and 50 years, after which the upward trend resumes at a similar slope through age 70. C) Percentage-based model of clonal expansion dynamics comparing three scenarios of mutation frequency and proliferative advantage. The red curve (1% initial mutation frequency, 5% proliferative advantage) reaches the highest final burden due to strong selection. The green curve (10% frequency, 2% advantage) rises moderately despite higher starting frequency. The blue curve (1% frequency, 2% advantage) shows the lowest expansion and remains the most stable over the lifespan.
Figure 2.
Lifetime Accumulation Dynamics of Mitochondrial DNA Mutations. A) Table showing chronological age (10–70 years) and the estimated number of mtDNA mutations accumulated per decade, illustrating the progressive increase in mutational burden with aging. B) Linear plot of total mtDNA mutation count versus age, displaying a steady decade-to-decade rise with a marked inflection between 40 and 50 years, after which the upward trend resumes at a similar slope through age 70. C) Percentage-based model of clonal expansion dynamics comparing three scenarios of mutation frequency and proliferative advantage. The red curve (1% initial mutation frequency, 5% proliferative advantage) reaches the highest final burden due to strong selection. The green curve (10% frequency, 2% advantage) rises moderately despite higher starting frequency. The blue curve (1% frequency, 2% advantage) shows the lowest expansion and remains the most stable over the lifespan.
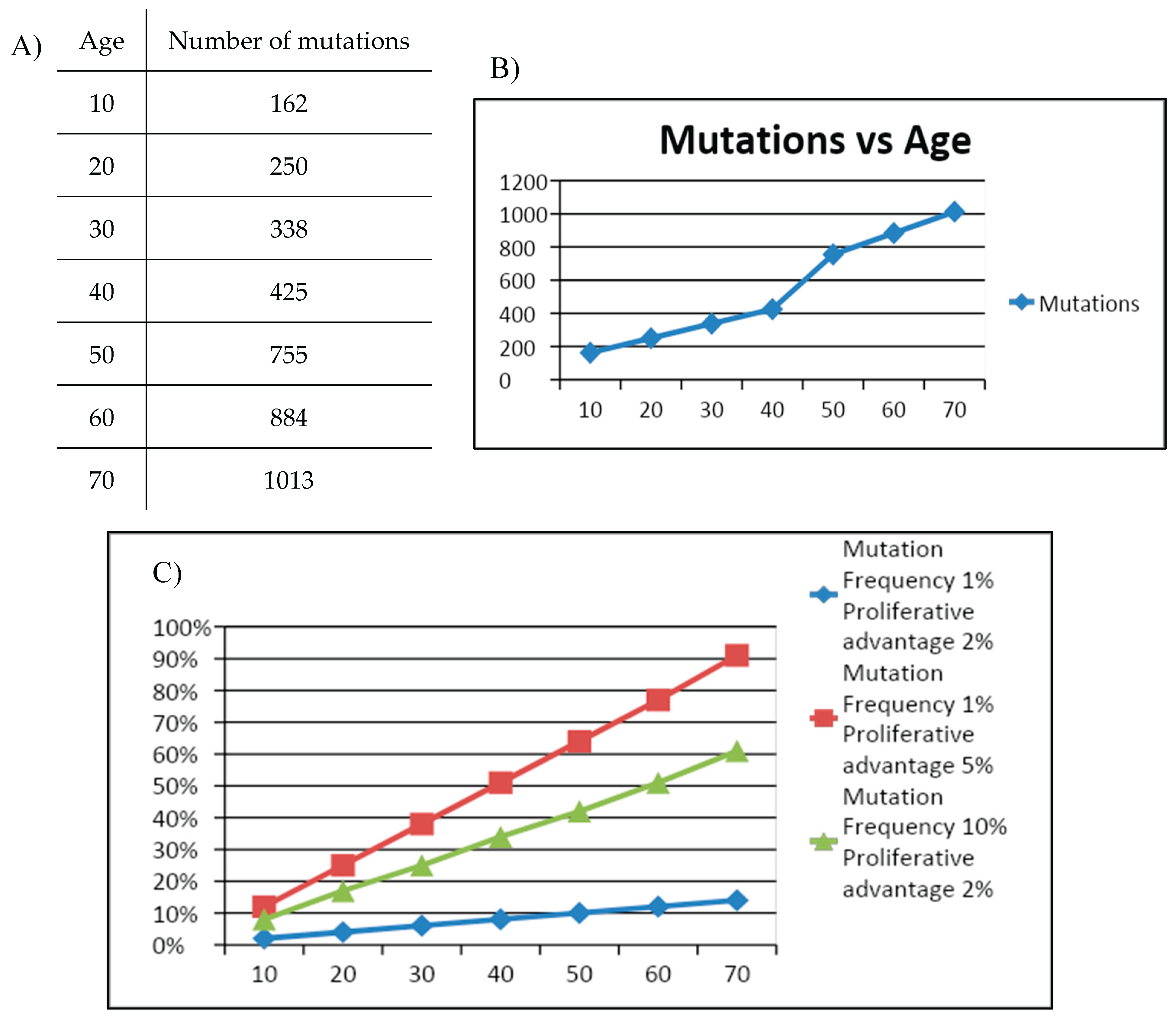
Figure 3.
Interdependent Network and Vicious-Cycle Architecture of Mitochondrial Drivers of Aging. Heatmap showing the strength of association (strong, moderate, weak, none) between the twelve mitochondrial drivers of aging. mtDNA-related processes (ROS, mtDNA damage, mutations, heteroplasmy, repair defects, copy-number loss) cluster together with strong interactions. Functional decline processes (ATP loss, ETC supercomplex disassembly, impaired dynamics, defective mitophagy, reduced biogenesis) also show strong cross-links. The map highlights that most drivers reinforce each other, forming an interconnected network rather than independent mechanisms.
Figure 3.
Interdependent Network and Vicious-Cycle Architecture of Mitochondrial Drivers of Aging. Heatmap showing the strength of association (strong, moderate, weak, none) between the twelve mitochondrial drivers of aging. mtDNA-related processes (ROS, mtDNA damage, mutations, heteroplasmy, repair defects, copy-number loss) cluster together with strong interactions. Functional decline processes (ATP loss, ETC supercomplex disassembly, impaired dynamics, defective mitophagy, reduced biogenesis) also show strong cross-links. The map highlights that most drivers reinforce each other, forming an interconnected network rather than independent mechanisms.
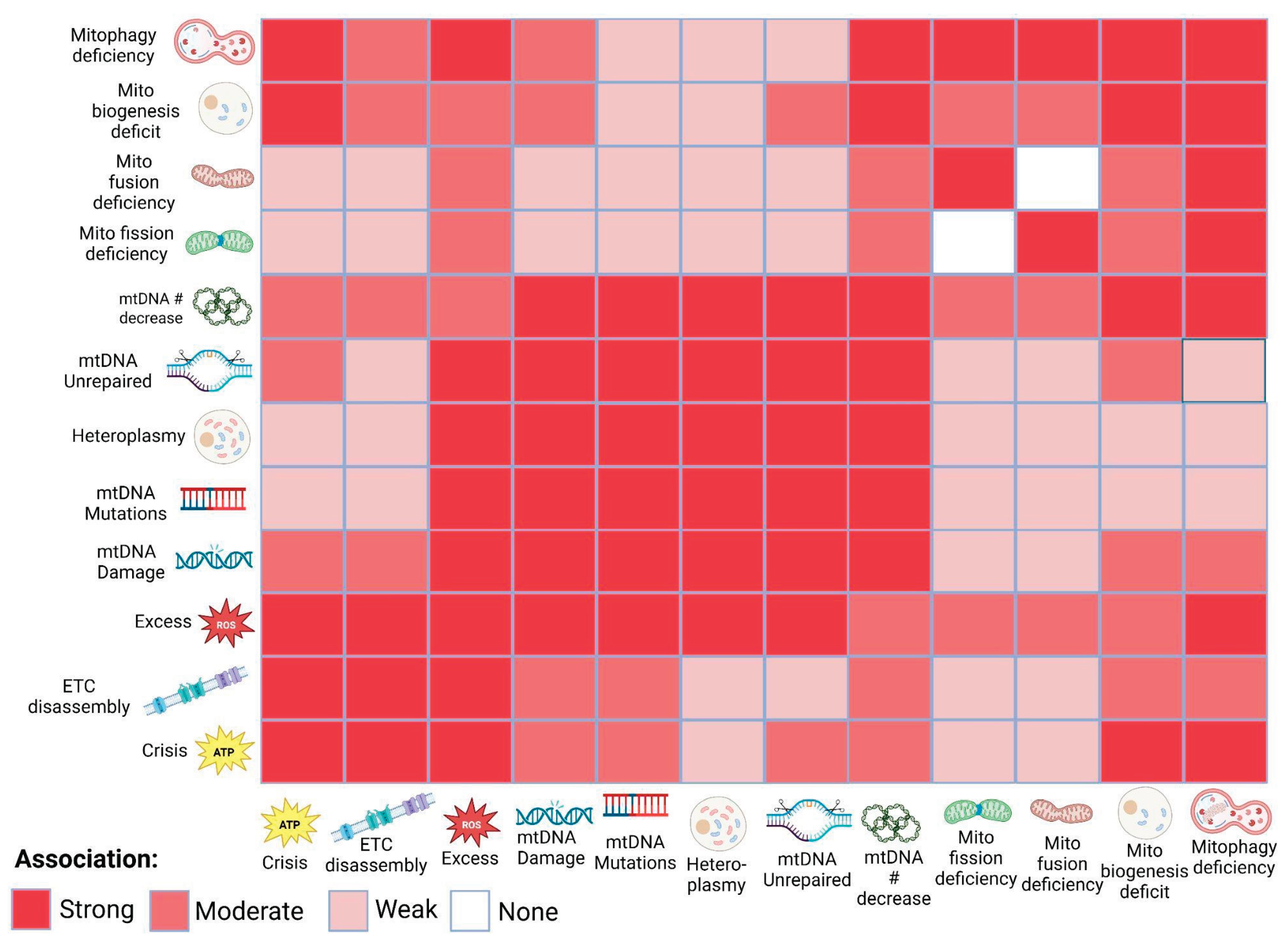
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



