Submitted:
29 January 2026
Posted:
30 January 2026
You are already at the latest version
Abstract
NLRP6 is a member of the NOD-like receptor family that was initially characterized as an inflammasome-forming sensor in the intestine. However, accumulating evidence over the past decade has revealed that the functions of NLRP6 extend far beyond this canonical role. NLRP6 operates in a wide range of tissues, including the intestine, liver, lung, and immune system, where it exerts context-dependent effects that can be either protective or detrimental. In the intestine, NLRP6 is most consistently associated with host protection, contributing to antiviral defense, epithelial barrier integrity, and the maintenance of microbial and metabolic homeostasis through both inflammasome-dependent and -independent mechanisms. In contrast, in systemic infection models and in certain inflammatory settings, NLRP6 can also promote pathology by suppressing NF-κB signaling, inducing IL-18–mediated lymphocyte death, or enhancing inflammatory cell death pathways. Moreover, studies using both conventional and tissue-specific knockout models have highlighted the importance of the gut–organ axis, particularly the gut–liver axis, in shaping NLRP6-dependent disease outcomes. Here, we summarize recent advances in understanding the upstream regulation, downstream signaling, and tissue-specific functions of NLRP6.
Keywords:
NLRP6
; innate immunity
; adaptive immunity
; infectious diseases
; cancer
Introduction
NLRP6, a member of the NLR (nucleotide-binding oligomerization domain (NOD)–like receptor) family, monitors the cytosol to identify the presence of microbe-associated molecular patterns [1,2]. NLRs exhibit a conserved domain architecture consisting of a central NBD (nucleotide-binding domain), also known as the NACHT domain, a C-terminal LRR (leucine-rich repeat) domain, and an N-terminal effector domain, either a CARD (caspase activation and recruitment domain) or a PYD (pyrin domain) [3,4] (Figure 1). Upon ligand recognition by the LRR and NACHT-mediated oligomerization, NLRP6 utilizes its N-terminal PYD to assemble and activate the inflammasome complex for downstream IL-18 release and secretion of anti-microbial peptides [5].
NLRP6 is preferentially expressed in the intestine, particularly in intestinal epithelial cells and goblet cells [5,6,7,8,9,10]; however, its expression has also been reported in other tissues, including the lung, liver, kidney, brain, and in immune cells such as neutrophils, T cells, and eosinophils [11,12,13,14,15,16]. In these organs, NLRP6 has been implicated in the regulation of intestinal homeostasis and in the sensing of microbes and cancer cells through the inflammasome pathway or through inflammasome-independent regulation of IFN (interferon) signaling, NF-κB (nuclear factor-κB), and MAPK (mitogen-activated protein kinase) signaling pathways [4,5,6,10,15,17,18,19] (Figure 2). In this review, we summarize recent advances in NLRP6 research reported over the past several years, together with earlier findings obtained in various tissues and immune cell types, and organize current knowledge regarding the upstream pathways that regulate NLRP6 and the downstream pathways influenced by NLRP6 (Table 1).
1. Intestinal Disease Models
The NLRP6 inflammasome is most highly expressed in the intestine and has been identified as a critical regulator of colonic homeostasis [6,10,20]. NLRP6 is predominantly expressed in intestinal epithelial cells, including goblet cells [6,10,21], and has been shown to be essential for mucosal self-renewal, epithelial proliferation, and mucus secretion [8,9,10].
1.1. Dextran Sulfate Sodium (DSS)-Induced Models (Colitis)
Dextran sulfate sodium (DSS) is a water-soluble sulfated polysaccharide that induces colonic inflammation through interactions with commensal microbiota and is widely used to generate experimental models of colitis. In early studies using Nlrp6 knockout (KO), these mice were characterized by spontaneous intestinal hyperplasia, inflammatory cell infiltration, and exacerbated DSS-induced colitis [6]. Nlrp6 KO mice exhibited an altered fecal microbiota composition characterized by an expansion of Bacteroidetes and TM7 (also known as Saccharibacteria). Notably, these bacterial taxa have been reported to be significantly enriched in human colorectal adenoma tissues [22]. Nlrp6 deficiency was also associated with reduced IL-18 production and induction of CCL5, and a similar increase in CCL5 was observed in IL-18-deficient mice. Bone marrow chimera experiments suggested that NLRP6 expressed in the non-hematopoietic compartment of the colon, most likely in epithelial cells, is a major source of active IL-18. In contrast, no apparent changes were observed in KC (CXCL1) or MCP-1 (CCL2). Furthermore, cohousing experiments demonstrated that the colitogenic microbiota of Nlrp6 KO mice could be transmitted to neonatal or adult wild-type (WT) mice, resulting in exacerbation of DSS-induced colitis (Box1).
Another group reported that Nlrp6-deficient mice developed more severe colitis and colitis-associated tumorigenesis following DSS administration [8]. In the intestinal epithelium of Nlrp6-deficient mice, the production of inflammatory cytokines, including TNFα, IL-6, IL-1β, MIP-2 (CXCL2), CXCL1, and IFNγ, was increased at days 10–13 after DSS treatment. These mice failed to efficiently repair damaged epithelium, and epithelial proliferative activity remained elevated for a prolonged period. IL-18 levels were reduced in both serum and colonic tissues, which was proposed to contribute to the increased tumor susceptibility observed in these mice. In contrast, a reduction in IL-1β was not observed, suggesting that the production of mature IL-1β and IL-18 may be differentially regulated in Nlrp6-deficient mice. In contrast to earlier findings [6], analysis of intestinal epithelium in bone marrow chimera mice generated using WT and Nlrp6 KO donors suggested that NLRP6 activity in hematopoietic cells is important for protection against inflammation-associated colorectal tumorigenesis.
Subsequent work[23] further investigated these chimera experiments and demonstrated that NLRP6-dependent regulation of IL-18 in intestinal Ly6Chi inflammatory monocytes is critical for promoting barrier function and suppressing bacterial inflammation through the regulation of TNFα production. During DSS-induced inflammation, NLRP6 expression was upregulated in Ly6Chi inflammatory monocytes infiltrating the colon. Adoptive transfer of wild-type Ly6Chi inflammatory monocytes into Nlrp6−/− mice significantly reduced tumor size and protected mice from lethality. In contrast, Nlrp6-deficient inflammatory monocytes were defective in TNFα production, whereas IL-1β production was unchanged, suggesting that TNFα production by Ly6Chi monocytes is at least partially dependent on IL-18. IL-18 is classically associated with T helper 1 (Th1) responses, particularly the upregulation of IFNγ [24], but it has also been shown to activate monocytes and promote TNFα production [25]. Similar to IL-18, early TNFα production is critical for tissue repair and the maintenance of intestinal homeostasis [26,27]. Indeed, administration of recombinant TNFα (rTNFα) to Nlrp6−/− mice during the early phase of DSS treatment was sufficient to ameliorate disease severity and mortality, demonstrating that early TNFα production by inflammatory monocytes plays a protective role. In Nlrp6-deficient mice, TNFα levels were reduced, whereas IL-16, IL-22, CCL2, and CXCL10 were increased.
Box1. Impact of Nlrp6 deficiency on gut microbiota and its transmissibility
In the above-mentioned analyses of the intestines of Nlrp6-deficient mice, it was reported that a disease-promoting dysbiotic microbiota outcompetes the previously stable WT microbiota upon fecal transfer, resulting in the transmissibility of disease susceptibility [6]. Consistent with these observations, another study that mechanistically linked the gut microbiota to inflammation-driven colorectal cancer (CRC) demonstrated that colitis-associated CRC observed in Nlrp6 KO, Asc (Pycard) KO, and Il18 KO mice could be transmitted to co-housed WT mice via fecal microbiota exchange between mice of different genotypes [20]. Thus, deficiency of inflammasome-related genes in the intestine can give rise to a transmissible dysbiotic microbiota, which in turn affects the function of multiple organs within the same host [6]. These studies highlights an important caveat: when analyzing the function of NLRP6 or immune responses in tissues outside the intestine using conventional Nlrp6-deficient mice, it is critical to distinguish whether the observed phenotypes are due to (i) indirect effects mediated by alterations in the gut microbiota or (ii) direct, cell-intrinsic consequences of NLRP6 deficiency in the tissue or cell type under investigation. Methodologically, experimental designs using co-housing of wild-type and inflammasome-deficient mice or the use of littermate controls are now widely adopted and are considered standard approaches in mouse studies that take the gut microbiota into account. In addition, approaches based on fecal microbiota transplantation (FMT) or transient depletion of the microbiota by antibiotic treatment followed by recolonization have also been employed to dissect microbiota-dependent and -independent effects [28].
1.2. Intestinal Viral Infections
NLRP6 has also been implicated in the regulation of inflammation during enteric viral infections, including norovirus, rotavirus, and encephalomyocarditis virus (EMCV). In the gastrointestinal tract of Nlrp6−/− mice, increased viral loads and higher mortality were observed following infection with these enteric viruses[17]. Mechanistically, NLRP6 binds viral RNA via the RNA helicase DHX15 and, in an inflammasome-independent manner, promotes type I and type III interferon (IFN) responses through the mitochondrial antiviral signaling protein (MAVS), an adaptor protein localized on the outer mitochondrial membrane. This pathway induces the expression of interferon-stimulated genes (ISGs), including Nlrp6 itself. In addition, TRIM29 (tripartite motif-containing protein 29) has been identified as a negative regulator of NLRP6 during rotavirus and EMCV infections. TRIM29-mediated suppression of NLRP6 attenuates downstream MAVS-dependent IFN-λ signaling as well as IL-18 production and pyroptosis, thereby compromising antiviral defense [29]. Using intestinal organoid systems, it has been shown that TRIM29 promotes ubiquitination of NLRP6, leading to its degradation during rotavirus and EMCV infection.
1.3. Regulation by Commensal Microbiota–Derived Metabolites Under Homeostatic Conditions
NLRP6 is also regulated by microbial metabolites present under homeostatic conditions. A regulatory mechanism has been described in which NLRP6 controls the expression of IL-18 and downstream antimicrobial peptides (AMPs), including ITLN1, RELMβ, and angiogenin-4 (Ang4), forming a “metabolite–inflammasome–AMP axis.” In a study performing metabolomic screening of mouse cecal contents, taurine was identified as an activator of NLRP6, whereas histamine and spermine were identified as inhibitors of NLRP6 [5]. Taurine induced activation of the intestinal inflammasome and promoted caspase-1 cleavage, thereby upregulating the IL-18–AMP pathway. In contrast, colitogenic microbiota transmitted by fecal transfer suppressed inflammasome activation through these metabolites, resulting in an imbalance of AMP expression. In wild-type mice co-housed with Asc-deficient mice, the levels of procaspase-1 were comparable to those in control wild-type mice, whereas caspase-1 cleavage was markedly impaired, indicating that a dysbiotic microbiota derived from Asc-deficient mice can suppress intestinal inflammasome activation even in a genetically intact host. Bone marrow chimera experiments using WT and Il18−/− mice demonstrated that, under homeostatic conditions, intestinal IL-18 is predominantly produced by the non-hematopoietic compartment.
Another study demonstrated that sphingolipids derived from Bacteroides in the gut induce NLRP6-dependent goblet cell responses [30]. Colonization with the protist Tritrichomonas triggered a marked alteration in the gut microbiota characterized by rapid expansion of Bacteroides species, leading to increased release of sphingolipids. These sphingolipids activated the NLRP6 inflammasome and promoted protective mucus secretion by sentinel goblet cells. Using a combination of gnotobiotic mouse models and ex vivo mucus analysis, the authors further showed that wild-type Bacteroides thetaiotaomicron, but not a sphingolipid-deficient mutant, was sufficient to induce NLRP6-dependent sentinel goblet cell function.
1.4. Bacterial and Fungal Pathogens
A deubiquitinating enzyme, CYLD, has been identified as a negative regulator of the NLRP6–IL-18 axis in intestinal inflammation [31]. Although the upstream mechanisms controlling CYLD itself remain unclear, CYLD negatively regulates NLRP6 inflammasome activity by removing K63-linked polyubiquitin chains from NLRP6, which are required for inflammasome stabilization. Studies using Cyld-deficient mice demonstrated that loss of CYLD results in excessive IL-18 production in the colonic mucosa, indicating that CYLD serves as an important checkpoint to prevent aberrant activation of the NLRP6–IL-18 pathway during intestinal inflammation.
Although limited to in vitro studies, there is also a report analyzing the response to infection with Candida albicans (C. albicans), an opportunistic fungal pathogen that colonizes the human gastrointestinal mucosa [32]. In this study, a monolayer model of intestinal epithelial cells (Caco-2 cells) was infected with C. albicans, and molecular changes were examined. Infection with C. albicans resulted in decreased levels of NLRP3, NLRP6, ASC, tight junction proteins (occludin and ZO-1), antimicrobial peptides (AMPs; BD-2 and BD-3), and IL-1β. In contrast, IL-18 expression was not altered. Notably, similar results were observed with heat-inactivated C. albicans, suggesting that this pathogen disrupts intestinal homeostasis through mechanisms independent of active fungal invasion.
2. Systemic Infection Models
Studies using experimental systems in which bacteria are administered intravenously or intraperitoneally have also investigated the role of NLRP6 in systemic infections.
2.1. Listeria Monocytogenes, Salmonella Typhimurium, and Escherichia coli (Systemic Infection Models)
An early study[15] demonstrated that, in an inflammasome-independent manner, NLRP6 negatively regulates NF-κB signaling and thereby suppresses inflammatory responses. In this study, intravenous or intraperitoneal administration of both Gram-negative and Gram-positive bacteria (S. typhimurium, E. coli, and L. monocytogenes) into Nlrp6-deficient mice resulted in exacerbated intracellular and extracellular bacterial infection. These findings indicate that, in the context of systemic infection, NLRP6 plays a detrimental role for the host, in striking contrast to its protective function in the intestine. Following infection, both the proportion and absolute number of circulating monocytes and neutrophils were markedly increased in the peripheral blood of Nlrp6-deficient mice. In contrast, the numbers of circulating lymphocytes and infiltrating T cells in the peritoneal cavity, as well as the induction of T cell responses, were not significantly altered. In Nlrp6-deficient mice, TLR-induced NF-κB and ERK signaling pathways were hyperactivated, accompanied by increased production of TNFα, IL-6, CXCL1, and interferon-β. These effects were considered to be independent of inflammasome activation. Bone marrow chimera experiments further suggested that both hematopoietic and non-hematopoietic compartments contribute to NLRP6-mediated suppression of bacterial clearance. Moreover, wild-type mice co-housed with Nlrp6-deficient mice also exhibited increased bacterial burdens in the spleen and liver, indicating that NLRP6 regulates systemic bacterial infection independently of its effects on the composition of the gut microbiota.
2.2. Salmonella Typhimurium (Systemic Dissemination Following Intestinal Infection)
An additional mechanism underlying the disease-promoting role of NLRP6 during systemic infection has been reported to involve necroptosis [33]. In this study, using both in vitro and in vivo models of Salmonella Typhimurium infection, NLRP6 was shown to exacerbate disease by promoting bacterial systemic dissemination through the induction of RIP1 kinase–dependent necroptosis, a form of lytic programmed cell death. Mechanistically, NLRP6 downregulated the phosphorylation of TAK1(TGF-β activated kinase 1), thereby inhibiting NF-κB signaling, and simultaneously suppressed the phosphorylation of the downstream p38 MAPK/MK2 pathway. Reduced MK2 phosphorylation led to decreased phosphorylation of RIP1 at Ser321, which in turn resulted in enhanced phosphorylation of MLKL, the executioner molecule of necroptosis. Through this signaling cascade, NLRP6 was proposed to promote necroptosis and facilitate bacterial dissemination, ultimately worsening disease severity.
Another study demonstrated that the circadian clock component REV-ERBα negatively regulates Nlrp6 transcription, leading to rhythmic expression of NLRP6 and oscillatory secretion of IL-18 in intestinal epithelial cells [34]. This mechanism contributes to the time-of-day–dependent differences in host responses to Salmonella Typhimurium infection mediated by NLRP6. REV-ERBα, a nuclear receptor, is also recognized as an important regulator in various diseases, including inflammatory disorders and cancer, and has been reported to directly control the expression of multiple immune-related genes such as Il1b, Tlr4, Il6, Ccl2, and Cx3cr1 [35,36]. Pharmacological activation of REV-ERBα using the agonist SR9009 in WT mice reduced NLRP6 levels and alleviated S. Typhimurium infection. In contrast, siRNA-mediated knockdown of REV-ERBα efficiently upregulated Nlrp6 transcription. These findings suggest that the circadian clock enables the host to anticipate the timing of pathogen exposure and to accordingly tune immune activity.
2.3. Listeria Monocytogenes and Staphylococcus aureus (In Vitro Macrophage Infection and Systemic Infection Models)
Subsequent studies using in vitro macrophage infection models demonstrated that, in addition to caspase-1, caspase-11 also functions as an important downstream effector of NLRP6 [18]. Both Nlrp6⁻/⁻ and Casp11⁻/⁻ mice exhibited enhanced resistance to Listeria monocytogenes infection, accompanied by reduced pathogen burdens and decreased IL-18 production. Administration of recombinant IL-18 to Nlrp6⁻/⁻ or Casp11⁻/⁻ mice restored susceptibility to Listeria infection to levels comparable to those observed in WT mice. Furthermore, this study demonstrated that LTA (lipoteichoic acid) derived from Gram-positive bacteria directly binds to and activates NLRP6. In contrast to LPS (lipopolysaccharide) stimulation, LTA did not induce cell death through GSDMD (gasdermin D) cleavage. Although the precise reason for this difference remains unclear, one possible explanation is that GSDMD is accessible to the LPS-induced active form of caspase-11, but not to the cleaved form of caspase-11 activated by LTA.
2.4. Sepsis Model
In a study using a cecal ligation and puncture (CLP)–induced polymicrobial sepsis mouse model, Nlrp6⁻/⁻ mice exhibited reduced cytokine and chemokine production, decreased bacterial burdens in organs, and significantly improved survival compared with wild-type mice [37]. In the spleens of Nlrp6⁻/⁻ mice after sepsis induction, the numbers of CD3⁺, CD4⁺, and CD8⁺ T cell subsets were increased, accompanied by reduced T cell death. Consistently, the levels of IL-10, IL-1β, TNF-α, CCL2, and CXCL1 were lower than those in WT mice, and fewer neutrophils were recruited into the peritoneal cavity. Furthermore, antibody-mediated depletion experiments demonstrated that bacterial clearance after sepsis was mediated by CD8⁺ T cells rather than CD4⁺ T cells. Administration of recombinant IL-18 induced excessive inflammation and reversed the survival advantage observed in Nlrp6⁻/⁻ mice, thereby exacerbating disease severity. These findings indicate that NLRP6 promotes hyperinflammation through IL-18 and reveal that NLRP6-driven IL-18, rather than IL-1β, contributes to sepsis-induced lymphocyte death. In addition, cohousing of wild-type and Nlrp6⁻/⁻ mice after sepsis did not improve bacterial burdens in WT mice compared with singly housed controls, suggesting that this phenotype is independent of the intestinal microbiota and gut metabolism. Several studies have reported the expression and function of NLRP6 in T cells despite the fact that endogenous activators of NLRP6 remain unidentified (Box 2).
Box 2. NLRP6 expression and function in T cells
In a study using in vitro–differentiated T cells from murine lymphoid tissues under Th2-polarizing conditions (anti-CD3ε/CD28 stimulation in the presence of IL-4), NLRP6 was shown to be expressed in naïve T cells and to suppress the differentiation of CD4⁺ Th2 cells as well as the production of Th2 cytokines [16]. In the absence of NLRP6, the expression of the transcription factor GATA3 was increased, resulting in enhanced activation of both Th2 and Th17 cells. Because these in vitro differentiation experiments were performed in the absence of infection, it remains unclear what the endogenous ligands of NLRP6 are in this context, or whether the observed phenotypes are mediated by inflammasome-dependent responses to microbiota-derived metabolites.
The function of NLRP6 in CD4⁺ T cells has also been examined in another study [38]. When naïve CD4⁺ T cells isolated from WT spleens were cultured under Th1-polarizing conditions for five days, Nlrp6 expression gradually increased and appeared to be higher in Th1 cells than in Th2 or Th17 cells. Upon stimulation with PMA and ionomycin, Nlrp6-deficient Th1 cells produced less IFN-γ than WT Th1 cells. In contrast, Asc-deficient Th1 cells exhibited IFN-γ production comparable to that of WT cells, suggesting that the impaired IFN-γ production observed in Nlrp6-deficient Th1 cells is independent of ASC and inflammasome activation. Consistently, no difference in IL-18 concentrations was detected in the culture supernatants of WT and Nlrp6-deficient Th1 cells. Notably, these experiments were also conducted in the absence of defined infectious stimuli. Although it has been reported that fetal calf serum (FCS) used in culture media may contain microbiota-derived metabolites such as spermidine and taurine [5], which could potentially activate NLRP6, whether the observed T cell phenotypes are mediated through inflammasome-dependent mechanisms remains to be determined.
2.5. Sepsis Model (Klebsiella Pneumoniae–Induced)
In contrast to the systemic infection models described above, a more recent report proposed a protective role for NLRP6 in host defense [39]. Using a Klebsiella pneumoniae (Kp)–induced pneumonia–derived sepsis model, the authors observed increased expression of NLRP6 in the lungs of infected mice, particularly in Ly6G⁺ neutrophils, F4/80⁺ macrophages, and cytokeratin⁺ epithelial cells. NLRP6 expression was upregulated at 6, 24, and 48 hours after infection. In Nlrp6⁻/⁻ mice, levels of CXCL1, CXCL2, and CXCL5 in bronchoalveolar lavage fluid (BALF), as well as neutrophil numbers and function, were markedly reduced, accompanied by increased bacterial burdens in the lung and spleen and decreased survival. Consistently, in vitro analyses showed that Nlrp6⁻/⁻ neutrophils exhibited impaired formation of neutrophil extracellular traps (NETs) and reduced NET-mediated bacterial killing. Administration of recombinant CXCL1 restored host defense, granulopoietic responses, and defective NETosis in Kp-infected Nlrp6⁻/⁻ mice, suggesting that the profound defect in neutrophil homeostasis observed in these mice is largely attributable to reduced CXCL1 production. In addition, Kp-infected Nlrp6⁻/⁻ mice exhibited reduced levels of inflammatory cytokines, including TNF-α, IL-6, G-CSF, and IL-1β, supporting the notion that an NLRP6–IL-1β axis functions upstream of CXCL1 production. Notably, although neutrophil numbers were also decreased in the previously described sepsis model [37], the disease outcomes were opposite. The authors speculated that when the pathogen does not elicit a sufficiently strong inflammatory or pathological response, as in this experimental setting, NLRP6-mediated inflammatory responses may instead be beneficial for bacterial clearance.
2.6. Sepsis Model (Platelet-Specific Nlrp6 Deletion)
Another study has demonstrated a protective role of platelet-expressed NLRP6 in sepsis-associated microvascular thrombosis [40]. Using platelet-specific Nlrp6 KO mice in a CLP–induced sepsis model, the authors showed that loss of NLRP6 in platelets resulted in increased mortality, exacerbated microvascular thrombosis in the lung and liver, and enhanced platelet activation, platelet–neutrophil interactions, and NET formation after sepsis induction. In vitro platelet functional assays further revealed that NLRP6 deficiency augmented platelet aggregation, activation, and granule release. Mechanistically, NLRP6 was shown to promote the interaction between TRIM21 and TAB1 in activated platelets in an inflammasome-independent manner, thereby inducing K48-linked polyubiquitination and subsequent degradation of TAB1. In the absence of NLRP6, sustained TAB1 expression led to enhanced NF-κB signaling in platelets. Importantly, pharmacological inhibition of NF-κB signaling abolished the exacerbation of intravascular microthrombosis and NET formation caused by platelet-specific NLRP6 deficiency and significantly improved overall survival in septic mice.
2.7. Sepsis Model (Effect of Gut Microbiota on Sepsis)
Another study investigated the impact of sepsis on the gut microbiota, short-chain fatty acid (SCFA) levels, intestinal barrier integrity, and NLRP6 inflammasome activity using a CLP-induced sepsis model [41]. Septic mice exhibited marked gut dysbiosis, characterized by an increased Firmicutes/Proteobacteria ratio and reduced α-diversity, along with decreased SCFA levels and impaired intestinal barrier function, as evidenced by reduced expression of the tight junction proteins ZO-1 and occludin. Notably, supplementation with SCFAs restored microbial homeostasis, enhanced intestinal tight junction protein expression (particularly ZO-1), and ameliorated barrier dysfunction. These findings suggest that SCFAs promote NLRP6 activation during sepsis and highlight a potentially protective role of the SCFA–NLRP6 axis in maintaining gut homeostasis and targeting the gut microbiota during systemic inflammation.
3. Lung Infection and Lung Cancer Models
NLRP6 is also expressed in epithelial and immune cells in the lung, where it has been implicated in the regulation of pulmonary immune responses.
3.1. Staphylococcus aureus Pneumonia
In a model of intratracheal Staphylococcus aureus (S. aureus) infection [11], Nlrp6 KO mice were shown to be protected from pulmonary S. aureus infection, as evidenced by improved survival and reduced bacterial burdens in the lung and extrapulmonary organs. In the absence of NLRP6, neutrophil recruitment to the lung was markedly increased after infection, and depletion of neutrophils abolished this protective phenotype. Neutrophils from Nlrp6 KO mice exhibited enhanced intracellular bacterial killing, accompanied by increased myeloperoxidase (MPO) activity and reactive oxygen species (ROS) production. In addition, increased NK cell–mediated IFN-γ production was observed in the lungs of Nlrp6 KO mice following infection. Consistent with these findings, both pyroptosis and necroptosis were reduced in the lungs of Nlrp6 KO mice after infection. The lungs of Nlrp6 KO mice displayed elevated MAPK activity and reduced TNF-α levels, suggesting that NLRP6 may promote necroptosis through a TNF-α–dependent pathway. In bone marrow–derived macrophages (BMDMs) lacking NLRP6, the production of IL-1β and IL-18 was also decreased, and in wild-type mice, NLRP6 was found to colocalize with ASC and caspase-1 under infectious conditions. Bone marrow chimera experiments further indicated that NLRP6 derived from both hematopoietic and non-hematopoietic compartments contributes to impaired bacterial clearance during S. aureus pneumonia. Moreover, wild-type mice cohoused with Nlrp6 knockout mice still exhibited significantly higher bacterial burdens in the lung and bronchoalveolar lavage fluid (BALF), supporting the notion that NLRP6 regulates pulmonary S. aureus infection independently of the gut microbiota.
3.2. Streptococcus pneumoniae Infection
In a study examining NLRP6 activation during Streptococcus pneumoniae infection, Nlrp6−/− mice exhibited significantly attenuated pulmonary inflammation, reduced bacterial colonization, and improved survival compared with wild-type mice, indicating a detrimental role of NLRP6 in host defense against pneumococcal pneumonia [42]. Mechanistically, NLRP6 was shown to mediate the expression of inflammatory cytokines in S. pneumoniae–infected macrophages, including IL-1β, IL-1α, IL-6, and IL-12, whereas TNF-α levels were not affected. At the same time, NLRP6 negatively regulated the recruitment of neutrophils and macrophages to the lung. Using infected WT and Nlrp6−/− macrophages, the authors further demonstrated that NLRP6 promotes the activation of both caspase-1 and caspase-11, thereby contributing to IL-1β maturation and GSDMD cleavage. In addition, NLRP6 was found to suppress NF-κB and ERK signaling pathways in macrophages during S. pneumoniae infection.
3.3. Birch Pollen Exposure and Helminth Infection Models
In a model of allergic airway inflammation induced by two rounds of intraperitoneal sensitization with birch pollen extract (BPE) followed by intranasal challenge, NLRP6 was shown to function as a negative regulator of type 2 immune responses and, consequently, as a suppressor of parasite expulsion [16]. In Nlrp6−/− mice, birch pollen exposure resulted in a markedly enhanced type 2 immune profile, characterized by increased numbers of eosinophils and elevated eosinophil peroxidase (EPO) activity in bronchoalveolar lavage (BAL) fluid, increased neutrophil infiltration, enhanced induction of group 2 innate lymphoid cells (ILC2s), and increased production of type 2 cytokines and chemokines, including IL-5 and IL-13. In parallel, macrophage-derived IL-18 production was reduced, whereas the Th2-attracting chemokine CCL17 and the neutrophil-attracting chemokine CXCL1 were significantly increased. Consistent with these changes, both total serum IgE levels and BPE-specific IgE levels were markedly elevated in Nlrp6−/− mice. Notably, a similar enhancement of type 2 immune responses was observed in Asc−/− and Caspase-1/11−/− mice, including increased eosinophil and lymphocyte counts, elevated IL-4 and IL-5 levels in BAL fluid, enhanced peribronchial leukocyte infiltration, and increased numbers of mucus-producing goblet cells in the airway epithelium, indicating that this phenotype is largely inflammasome-dependent. Furthermore, administration of recombinant mouse IL-18 (rIL-18) suppressed IL-4 and IL-5 levels, whereas treatment with a neutralizing anti–IL-18 antibody increased neutrophil numbers in the BAL fluid of Nlrp6−/− mice, supporting the notion that the regulatory effect of NLRP6 in this model is mediated, at least in part, through IL-18.
4. Hepatitis and Hepatocellular Carcinoma Models
Hepatocellular carcinoma (HCC) most commonly develops in the context of chronic liver disease (CLD), with chronic hepatitis B and C virus infection, alcoholic steatohepatitis, and non-alcoholic steatohepatitis (NASH) being the major etiological factors [43]. These conditions are characterized by persistent hepatic inflammation and ongoing liver injury, which lead to hepatocyte death, compensatory proliferation, and ultimately hepatocarcinogenesis [44]. Therefore, a deeper understanding of the inflammatory processes underlying chronic liver disease is essential for the development of novel therapeutic strategies. The liver receives approximately two-thirds of its blood supply from the portal vein. Consequently, it is constantly exposed to large amounts of pathogens and microbe-associated molecular patterns (MAMPs), as well as pathogen-associated molecular patterns (PAMPs). These microbial-derived molecules engage pattern-recognition receptors (PRRs) and trigger NF-κB activation in both hepatocytes and non-parenchymal cells, thereby promoting the progression of liver disease [45]. This continuous microbial input critically shapes the inflammatory microenvironment of the liver. Using Nlrp6−/− mice, which harbor a dysbiotic gut microbiota, several studies have investigated how intestinal dysbiosis modulates the hepatic tumor microenvironment and influences anti-tumor immune responses during the progression of steatohepatitis and hepatocarcinogenesis.
4.1. Non-Alcoholic Steatohepatitis (NASH)
Accumulating evidence indicates that NLRP6 exerts a protective role against liver inflammation through both gut inflammasome–dependent [7] and inflammasome-independent, liver-intrinsic mechanisms [46]. Feeding mice a methionine- and choline-deficient diet (MCDD) for four weeks starting at 8 weeks of age induces key pathological features of human NASH, including hepatic steatosis, inflammatory cell infiltration, and fibrosis. An early study using this model demonstrated that Nlrp6 deficiency–induced alterations in the gut microbiota increase the portal influx of TLR4 and TLR9 ligands, thereby exacerbating hepatic steatosis and liver inflammation [7]. This phenotype was attenuated in Tlr4−/−, Tlr9−/−, and Tnf−/− mice, indicating a critical role of downstream TNF-α signaling in the progression of NASH. Similar phenotypes were also observed in Asc−/− and Casp1−/− mice, supporting the notion that this effect is inflammasome-dependent. Notably, co-housing inflammasome-deficient mice with wild-type mice resulted in the transfer of steatosis and obesity phenotypes to WT animals, providing strong evidence that gut microbiota alterations are sufficient to drive liver pathology.
In contrast, a study using hepatocyte-specific Nlrp6 knockout mice demonstrated a gut-independent, cell-intrinsic role of NLRP6 in hepatocytes under high-fat diet (HFD) or MCDD conditions [46]. In this model, NLRP6 was shown to promote lysosome-dependent degradation of TAB2/3, thereby suppressing NF-κB activation. Accordingly, livers from Nlrp6-deficient mice exhibited accumulation of TAB2/3, increased phosphorylation of p65, elevated expression of Tnf, Ccl2, IL-1β, and IL-6, increased infiltration of F4/80-positive macrophages, and exacerbated hepatic inflammation. In addition, enhanced fatty acid uptake via CD36 was suggested in Nlrp6-deficient hepatocytes. No evidence for the involvement of inflammasome-related pathways was provided in this model.
4.2. Alcoholic Hepatitis (AH) / Alcoholic Liver Disease (ALD)
A protective role of NLRP6 in the progression of alcoholic hepatitis (AH) has been reported [47]. In ethanol (EtOH)-fed mice, hepatic NLRP6 expression was markedly reduced, which was accompanied by enhanced activation of the NF-κB pathway, attenuation of hepatoprotective mechanisms, and subsequent development of AH. This was associated with increased macrophage infiltration into the liver, and among a broad panel of cytokines and chemokines, CCL20 emerged as one of the most strongly upregulated chemokines. CCL20 has been implicated in the recruitment and activation of hepatic stellate cells, thereby promoting liver fibrosis during AH. Conversely, adeno-associated virus (AAV)-mediated overexpression of NLRP6 in EtOH-fed mice significantly attenuated hepatic lipid accumulation and neutrophil infiltration. In these mice, serum ALT and AST levels were significantly reduced, accompanied by decreased expression of phosphorylated p65 and phosphorylated IκBα, indicating suppression of NF-κB signaling. Moreover, hepatic CCL20 expression was reduced, together with diminished macrophage accumulation as assessed by F4/80 staining. Collectively, these findings suggest that NLRP6 exerts a hepatoprotective role in AH by suppressing NF-κB signaling, macrophage infiltration, and CCL20 production.
In contrast, another study investigated the impact of Nlrp6 deficiency in a model of chronic alcohol consumption [48]. Chronic ethanol feeding in wild-type mice induced hepatic steatosis, liver injury, and neutrophil infiltration, thereby establishing a model of alcoholic liver disease (ALD). This model recapitulated key features of alcohol-associated gut dysbiosis observed in humans, including an increased relative abundance of Bacteroidetes and Campylobacterota and a reduction in Firmicutes. Notably, however, this ALD regimen exerted only minor effects on the gut microbiota composition of Nlrp6-deficient mice. The hepatic phenotype observed in Nlrp6-deficient mice in this model differed from that reported in other hepatitis models. Nlrp6 deficiency significantly reduced immune cell infiltration into the liver, including monocyte-derived macrophages, B lymphocytes, and NK cells. At the level of soluble mediators, hepatic Il1b expression was significantly decreased in Nlrp6-deficient mice, whereas the expression of other inflammatory markers such as Tnf, Il6, and Ccl2 was not significantly affected by the loss of NLRP6 signaling.
4.3. Liver Injury Model After Allogeneic Hematopoietic Stem Cell Transplantation
A protective role of NLRP6 in the liver has also been reported in a model of hepatic injury following allogeneic hematopoietic stem cell transplantation (allo-HSCT) [49], a clinical setting in which liver injury represents a frequent and serious complication. In this model, allo-HSCT induced marked hepatic injury, as evidenced by elevated serum AST, ALT, and total bilirubin levels, together with hepatocellular swelling, inflammatory cell infiltration, thrombosis, and fibrin deposition in the liver tissue. Notably, hepatic NLRP6 protein expression was upregulated after transplantation, peaked at day 14, and then gradually declined, suggesting a potential role for NLRP6 in the hepatic response to transplant-associated injury. In Nlrp6-deficient mice, both myeloperoxidase (MPO) and TGF-β1 levels in the liver were significantly higher than in WT controls at day 14 after transplantation. Consistently, Nlrp6−/− mice developed more severe liver injury and fibrosis following allo-HSCT. This aggravated phenotype was accompanied by increased levels of phosphorylated IκBα and phosphorylated p38 MAPK, elevated expression of procaspase-1 and its cleaved p20 fragment, and enhanced secretion of proinflammatory cytokines, including IL-1β, IL-18, IL-6, and TNF-α. Mechanistically, these findings indicate that NLRP6 protects against allo-HSCT–associated liver injury by restraining the activation of the NF-κB and p38 MAPK signaling pathways, thereby limiting inflammasome activation, inflammatory cytokine production, and subsequent fibrogenic responses.
4.4. Liver Cancer Models
Accumulating evidence indicates that disturbance of intestinal homeostasis caused by NLRP6 deficiency profoundly affects liver tumorigenesis. In this section, we summarize multiple, apparently conflicting roles of NLRP6 in hepatocellular carcinoma (HCC) progression, highlighting its strong context dependency.
4.4.1. Impact of Beneficial Gut Microbiota on Liver Cancer Progression
NF-κB essential modulator (NEMO) is a key regulatory component of the NF-κB signaling pathway controlling inflammatory and immune responses. Hepatocyte-specific NEMO KO mice (NEMOΔhepa) spontaneously develop steatohepatitis and hepatocellular carcinoma. A recent study analyzed double-KO mice lacking both hepatocyte NEMO and systemic NLRP6 (NEMOΔhepa /Nlrp6−/−) [28]. In this model, Nlrp6 deficiency–associated gut dysbiosis promoted the expansion of monocytic myeloid-derived suppressor cells (mMDSCs) in the liver and suppressed T cell accumulation, thereby exacerbating hepatocarcinogenesis. Intestinal analyses revealed profound alterations in microbial composition compared with NEMOΔhepa single-KO mice, accompanied by impaired gut barrier integrity, as evidenced by reduced ZO-1 and occludin expression, and increased expression of Ccl2, Ccl5, and Il1b. In contrast, Il18 was only mildly increased and Tnf expression was largely unchanged. In the liver, tumor progression was markedly accelerated and associated with increased expression of Tlr4, Il1b, and Tnf. While T cell infiltration was reduced, mMDSC accumulation was strongly enhanced. Importantly, this phenotype was transmissible via fecal microbiota transfer and reversible by antibiotic treatment, demonstrating a causal role of the gut microbiota. Among beneficial commensals, Akkermansia muciniphila (A. muciniphila) was markedly reduced in the double-knockout mice, which correlated with increased mMDSC accumulation. Reconstitution of A. muciniphila restored gut barrier function and substantially alleviated hepatic inflammation and fibrosis. Consistently, analyses of human cirrhotic livers revealed increased bacterial load within the liver tissue and pronounced transcriptional reprogramming associated with fibrotic inflammation and cancer-associated immunosuppressive pathways.
4.4.2. Chemically Induced and Transplantation HCC Models Using Conventional and Conditional Nlrp6 KO Mice
In contrast to the above findings, a recent study reported a tumor-promoting role of NLRP6 in HCC [12]. Using macrophage-specific (Cx3cr1-Cre+Nlrp6f/f), neutrophil-specific (S100a8-Cre+ Nlrp6f/f)), and hepatocyte-specific (Alb-Cre+Nlrp6f/f)) knockout mice, the authors demonstrated that NLRP6 expression in macrophages is specifically responsible for promoting HCC progression. Analysis of human transcriptomic datasets further revealed that NLRP6 expression co-localizes with the macrophage marker CD68 in human HCC tissues, and that patients with lower macrophage NLRP6 expression exhibit longer survival. Mechanistically, the authors identified a direct interaction between NLRP6 and extended synaptotagmin-1 (E-Syt1), a protein involved in macrophage phagocytosis. NLRP6 binding suppressed E-Syt1 function, indicating that tight quantitative control of NLRP6 is critical for macrophage anti-tumor activity.
4.4.3. Subcutaneous HCC Transplantation Model with Intestinal Fungal Overgrowth
Another study demonstrated that excessive intestinal fungal colonization converts NLRP6 into a tumor-promoting factor [50]. Oral administration of Candida albicans significantly enhanced the growth of subcutaneously transplanted Hepa1-6 HCC cells, and this effect was strictly dependent on the presence of NLRP6. Plasma metabolomic analyses revealed elevated levels of fungal-derived metabolites, including L-carnitine and L-acetylcarnitine. However, this study did not report detailed analyses of the host intracellular signaling pathways, inflammatory cytokines, or immune cell infiltration patterns responsible for this phenotype.
4.4.4. Colorectal Cancer Liver Metastasis Model
A metastasis-promoting role of NLRP6 has also been reported in colorectal cancer liver metastasis [51]. In this model, MC38 colon cancer cells are injected into the spleen, from where they are transported via the splenic vein and portal vein directly to the liver, thereby mimicking the main metastatic route in human colorectal cancer, although the early steps of invasion and intravasation are bypassed [52]. In WT mice, metastatic colonization of the liver induced a strongly immunosuppressive microenvironment characterized by accumulation of monocytic MDSC and a reduction in CD8⁺ T cells. This immunosuppressive state was markedly attenuated in Nlrp6−/− mice. One proposed mechanism was the reduced production of CCL2, which is mainly produced by hepatocytes and Kupffer cells. CCL2 is known to recruit and activate M2 macrophages, chemotactic Tregs, and MDSC, thereby establishing an immunosuppressive tumor microenvironment that promotes tumor growth, invasion, and metastasis. Notably, this study did not provide data on inflammasome activation or upstream NLRP6-activating signals. However, histological analyses of human metastatic lesions revealed high NLRP6 expression.
Exosome-mediated activation of NLRP6 (lung cancer metastasis model)
A similar metastasis-promoting function of NLRP6 has been reported in a lung cancer model [53]. In this study, exosomes released from small-cell lung cancer (SCLC) cells activated NLRP6 in macrophages and induced M2 polarization (Arg1⁺CD206⁺) via the NLRP6/NF-κB pathway (downregulation of NF-κB pathway by NLRP6), primarily demonstrated in vitro using BMDMs. In vivo, exosomes isolated from SCLC cells derived from a genetically engineered mouse model (Rb and p53 mutant) were intravenously injected together with SCLC cells. Mice receiving exosomes developed significantly more lung tumors and exhibited increased levels of NLRP6 and phosphorylated p65. Notably, SCLC-derived exosomes were enriched in PPAR-γ, which is known to induce NLRP6 transcription [54]. High NLRP6 expression was also detected in metastatic lesions from SCLC patients. The ability of host NLRP6 to promote the generation of immunosuppressive myeloid cells closely mirrors the phenotype observed in the colorectal cancer liver metastasis model [51].
5. Gastric Cancer
Several independent studies using gastric cancer cell lines and human clinical samples consistently support a tumor-suppressive role of NLRP6 in gastric cancer.
5.1. Clinical Relevance and Functional Validation in Gastric Cancer Cell Lines
An initial study demonstrated that NLRP6 expression is reduced in 24 of 32 (75%) primary gastric tumor specimens and in the AGS gastric cancer cell line [55]. Immunohistochemical analysis of 80 paraffin-embedded gastric cancer tissues stratified into NLRP6-high (n = 20) and NLRP6-low (n = 60) groups revealed that low NLRP6 expression was significantly associated with poor prognosis, and patients with high NLRP6 expression exhibited significantly longer 5-year overall survival. Functional experiments further validated these clinical observations. Overexpression of NLRP6 significantly inhibited proliferation of MKN45 and AGS cells, whereas siRNA-mediated knockdown of NLRP6 in AGS cells promoted cell proliferation and colony formation. In a subcutaneous xenograft model using nude mice, tumors derived from NLRP6-overexpressing MKN45 cells exhibited significantly reduced tumor volume and weight. Mechanistically, loss of NLRP6 was associated with enhanced NF-κB activation, suggesting that NLRP6 suppresses gastric tumor growth at least in part by restraining NF-κB signaling.
5.2. Regulation of NLRP6 Expression by Helicobacter Pylori
Decreased NLRP6 expression has also been shown to correlate with Helicobacter pylori infection [56]. Forkhead box O3 (FOXO3), a transcription factor, is known to be negatively regulated by H. pylori [57]. Short-term (e.g., 1 h) H. pylori infection induced phosphorylation of AKT and FOXO3, leading to functional inactivation of FOXO3. FOXO3 knockdown significantly reduced NLRP6 protein levels, mRNA expression, and promoter activity, phenocopying the effects of H. pylori infection. Conversely, the PI3K/AKT inhibitor LY294002 suppressed FOXO3 phosphorylation and restored NLRP6 expression. Together, these findings indicate that H. pylori downregulates NLRP6 transcription via the AKT–FOXO3 signaling pathway, thereby potentially contributing to gastric carcinogenesis.
5.3. GRP78 as a Downstream Effector of NLRP6 Tumor-Suppressive Function
The tumor-suppressive function of NLRP6 has also been linked to its interaction with GRP78 (glucose-regulated protein 78), an endoplasmic reticulum chaperone known to exert pro-tumorigenic functions in multiple cancer types [58]. NLRP6 was identified as a binding partner of GRP78 and was shown to promote ubiquitin–proteasome–dependent degradation of GRP78. Overexpression of GRP78 reversed the inhibitory effects of NLRP6 on gastric cancer cell proliferation, cell cycle progression, apoptosis, and migration. Conversely, GRP78 knockdown abrogated the pro-proliferative and pro–cell cycle effects induced by NLRP6 silencing, indicating that GRP78 functions as a critical downstream effector of NLRP6. Consistent with these functional data, NLRP6 expression was found to be inversely correlated with GRP78 expression in human gastric cancer tissues. Given the well-established oncogenic roles of GRP78 in multiple cancer types [59,60], this NLRP6–GRP78 axis may represent a broadly relevant tumor-suppressive mechanism.
5.4. Epigenetic Suppression of NLRP6 by the OIP5-AS1/EZH2 Axis
Another study identified NLRP6 as a tumor suppressor gene epigenetically silenced by the OIP5-AS1/EZH2 axis in gastric cancer [61]. Although this study relied exclusively on in vitro overexpression and knockdown systems, it proposed a novel regulatory pathway controlling NLRP6 expression. OIP5-AS1 is a long non-coding RNA (lncRNA) increasingly recognized as an oncogenic driver in multiple cancer types [62]. In gastric cancer cells, OIP5-AS1 was shown to bind EZH2, which in turn mediated H3K27 trimethylation at the NLRP6 promoter, leading to transcriptional silencing of NLRP6. Importantly, the effects of OIP5-AS1 overexpression or knockdown on cell proliferation, migration, and apoptosis were reversed by genetic manipulation of either EZH2 or NLRP6, demonstrating that this pathway acts upstream of NLRP6. Further in vivo validation and cross-tissue analyses will be required to establish the general relevance of this epigenetic regulatory axis.
6. Acute Injury Models
In addition to the intestine and cancer, NLRP6 is also expressed in other organs, including the kidney and the brain, where it has been reported to play either protective or detrimental roles in acute injury settings.
6.1. Acute Kidney Injury (AKI) Model
Folic acid–induced acute kidney injury (AKI) in mice, which is triggered by a single injection of folic acid, is characterized by elevated serum creatinine levels, tubular cell death, and interstitial inflammation, and recapitulates key features of human AKI [63]. A study reporting that Nlrp6 expression is downregulated during AKI [13] demonstrated that Nlrp6−/− mice subjected to AKI exhibited significantly higher serum creatinine and blood urea nitrogen levels, accompanied by more severe nephrotoxicity and renal inflammation. In the kidneys of Nlrp6-deficient mice, dysregulated activation of ERK1/2 and p38 MAPK signaling pathways was observed, along with increased infiltration of interstitial macrophages and neutrophils compared with WT mice. In contrast, plasma IL-18 levels were lower in Nlrp6−/− mice with AKI. Although the factors responsible for the downregulation of Nlrp6 in this model were not identified, and the potential contribution of gut immunity was not examined, these findings suggest that NLRP6 also plays a homeostatic and protective role in the context of renal injury.
6.2. Acute Neural Injury (Sciatic Nerve Injury Model)
An early study investigating the role of NLRP6 in peripheral nerve injury [64] analyzed the expression profiles of major inflammasome components in the peripheral nervous system, including Schwann cells, motor neurons, and sensory neurons, by RT–qPCR. Within 24 hours after sciatic nerve axotomy, mRNA expression of Nlrp6 and other NLRs, as well as Asc, Il1a, Il1b, Mip-1, Mcp-1, Il6, and Tnf, was robustly induced, with Nlrp6 showing particularly rapid upregulation as early as 4 hours after injury. In contrast, Il18 expression was only weakly induced. Genetic ablation of Nlrp6 resulted in significantly impaired functional recovery after sciatic nerve crush injury. In contrast, deficiency of Asc, caspase-1, or caspase-11 did not affect sciatic nerve function, suggesting that NLRP6 contributes to peripheral nerve regeneration in an inflammasome-independent manner. Mechanistically, enhanced phosphorylation of ERK1/2 (MAPK signaling) was observed in Nlrp6-deficient mice, indicating that excessive inflammatory signaling in the absence of NLRP6 exacerbates peripheral nerve injury.
6.3. Intracerebral Hemorrhage (ICH) Model
In a mouse model of intracerebral hemorrhage (ICH), the expression profile and biological role of NLRP6 in perihematomal brain tissue during acute brain injury were examined, revealing a protective function of NLRP6 [14]. NLRP6 expression was increased in perihematomal regions from 6 hours to 3 days after ICH, peaking at 1-day post-injury. Immunofluorescence analysis showed that NLRP6 was predominantly colocalized with GFAP-positive astrocytes, whereas only limited colocalization was observed in NeuN-positive neurons. NLRP6 expression was not detected in CD11b-positive microglia or CD31-positive endothelial cells. Nlrp6−/− mice subjected to ICH exhibited significantly elevated levels of proinflammatory cytokines, including IL-6, IL-1β, and TNF (IL-18 was not reported), along with increased NF-κB activity, brain water content, and neurological deficit scores, indicating exacerbated neuroinflammation and brain injury in the absence of NLRP6.
In a rat model of cerebral ischemia–reperfusion injury induced by middle cerebral artery occlusion (MCAO), NLRP6 knockdown using siRNA unexpectedly ameliorated histopathological brain injury, in contrast to the protective role reported in other acute brain injury models [65]. NLRP6 expression was significantly upregulated in brain tissue from 6 to 72 hours after I/R injury, peaking at 48 hours. Histopathological and biochemical analyses demonstrated that NLRP6 exerted a pro-inflammatory role in cerebral I/R injury. Following NLRP6 downregulation, the expression levels of IL-1β, IL-18, cleaved caspase-1, and myeloperoxidase (MPO) were markedly reduced. Suppression of NLRP6 attenuated inflammasome activation and downstream inflammatory signaling, leading to reduced infarct volume, decreased brain water content, and improved neurological outcomes.
In a subsequent study from the same group, NLRP6 expression was also found to be upregulated in a rat ICH model [66]. NLRP6 levels were increased from 6 to 72 hours after ICH, with a peak at 48 hours. Knockdown of NLRP6 significantly alleviated brain injury after ICH and was accompanied by reduced activation of NF-κB, decreased levels of cleaved caspase-1, IL-18, IL-1β, TNF, and MPO, as well as suppression of autophagy, as indicated by decreased Beclin 1 expression and a reduced LC3-II/I ratio. Notably, inflammation and autophagy induced by NLRP6 overexpression were suppressed by a caspase-1 inhibitor, suggesting that NLRP6 regulates both inflammation and autophagy in an inflammasome-dependent manner in this model.
In a more recent study, the same group employed both an in vivo mouse MCAO model and an in vitro oxygen–glucose deprivation/reoxygenation (OGD/R) model using HT22 cells to recapitulate cerebral ischemia–reperfusion injury [67]. The authors identified that the protein level of the deubiquitinase BRCC3 peaked at 24 hours after MCAO or OGD/R. BRCC3 is known to specifically cleave lysine 63 (K63)-linked ubiquitin chains and has been implicated in multiple pathological processes, including DNA damage repair, apoptosis, angiogenesis, tumorigenesis, myeloproliferation, cardiac injury, and inflammation. Mechanistically, BRCC3 was shown to interact with NLRP6 and to stabilize NLRP6 protein by reducing its ubiquitination, thereby exacerbating inflammatory responses. Knockdown of BRCC3 attenuated cerebral I/R-induced inflammation, reduced IL-1β expression and pyroptosis, and improved neurological deficits. Data on IL-18 levels were not reported in this study.
6.4. Neural Stem Cells and Stress Resilience
In addition to its roles in inflammation and tissue injury, NLRP6 has also been implicated in the regulation of neural stem cell (NSC) homeostasis and stress resilience. A recent study reported that both conventional Nlrp6 KO mice and NSC-specific conditional Nlrp6 KO (Nlrp6 CKO) mice exhibited increased stress susceptibility and depression-like behaviors [68]. In this study, NLRP6 was found to be highly enriched in NSCs within the dentate gyrus, with much higher expression levels than in neurons, astrocytes, or microglia. NLRP6 was required for the maintenance of hippocampal neurogenesis during development, enhancement of stress resilience, and proliferation of NSCs. Notably, NLRP6 expression was significantly reduced in the hippocampus of stress-exposed mice. RNA-seq analysis and qRT-PCR validation revealed a marked upregulation of Ecrg4 mRNA in the hippocampus and in primary NSCs from Nlrp6 KO mice. ECRG4 is a secreted factor associated with cellular senescence and growth inhibition and is known to be highly expressed in aging neural progenitor cells. Using the NE-4c NSC line exposed to the stress hormone corticosterone, co-overexpression experiments suggested that NLRP6 protects NSCs from mitochondrial stress in the hippocampus at least in part by suppressing ECRG4 expression. However, the molecular mechanism by which NLRP6 represses ECRG4 expression remains to be elucidated.
In a follow-up study by the same group, siRNA-mediated knockdown of Nlrp6 under corticosterone exposure was shown to suppress MAVS-mediated autophagy and to promote neural stem/progenitor cell (NSPC) death via ferroptosis [68]. Although the precise molecular mechanism remains unclear, the authors further demonstrated that short-chain fatty acids (SCFAs) increased Nlrp6 expression, enhanced autophagy, and suppressed corticosterone-induced cell death. SCFAs are known to regulate gene expression, metabolism, and immune function mainly through two mechanisms: activation of G protein–coupled receptors (GPR41/43) and inhibition of histone deacetylases (HDACs) [69].
6.5. miRNA-Mediated Regulation of NLRP6 in Astrocytes
One study identified miR-152 as an important upstream regulator of NLRP6[70]. In this work, the central nervous system stimulant methamphetamine was shown to suppress miR-152 expression in astrocytes and induce pyroptosis in an NLRP6 inflammasome–dependent manner. miR-152 inhibited NLRP6 expression and thereby attenuated NLRP6-dependent methamphetamine-induced pyroptotic signaling and neuroinflammation.
In another study by the same group, using an in vitro system with primary human astrocytes, miR-339 was shown to target the 3′-UTR of NLRP6 mRNA and suppress its expression under stress conditions [71]. Suppression of miR-339 expression in astrocytes by ethanol exposure or siRNA increased the expression of NLRP6 inflammasome signaling mediators, including NLRP6, caspase-1, and the inflammatory cytokines IL-1β and IL-18. These findings suggest one possible mechanism by which alcohol-induced neuroinflammation is associated with activation of central nervous system cells and the release of pro-inflammatory cytokines.
6.6. Glioma and Glioblastoma
In glioma cell models, NLRP6 has been reported to promote tumor growth through a non-inflammasome mechanism involving autophagy-mediated degradation of the PI3K regulatory subunit p85α [72]. In this study, NLRP6 was shown to directly interact with p85α and thereby enhance PI3K/AKT signaling, leading to increased proliferation of glioma cells. Mechanistically, NLRP6 recruits the E3 ligase RBX1 and promotes ubiquitination of p85α at lysine 256 (K256). This ubiquitinated p85α is subsequently recognized by the cargo receptor optineurin (OPTN) and targeted for selective autophagic degradation. Notably, the NLRP6–RBX1 complex was proposed to function as a non-canonical E3 ubiquitin ligase complex distinct from the classical Cullin–RBX1 E3 ligase complexes. Consistent with these molecular findings, pathological and biochemical analyses of human glioblastoma specimens revealed that NLRP6 expression was inversely correlated with p85α and PTEN expression levels. Furthermore, xenograft experiments using LN229 human glioma cells with NLRP6 overexpression or knockout demonstrated that NLRP6 overexpression significantly enhanced, whereas NLRP6 deficiency suppressed, tumor growth in nude mice.
In a separate transcriptome-based study aimed at identifying differentially expressed genes in glioma, gene ontology enrichment and gene interaction network analyses suggested that the transcription factor SP1 and NLRP6 are highly expressed in glioma cells and positively correlate with tumor malignancy [73]. Based on in silico binding predictions, chromatin immunoprecipitation (ChIP) assays demonstrated binding of SP1 to the NLRP6 promoter region, and knockdown of SP1 resulted in reduced NLRP6 expression. However, the functional relevance of the predicted binding sites was not validated using reporter assays. In addition, as the sources of human and mouse CD8⁺ T cells used in this study were not clearly described, the interpretation of the immunological data warrants caution.
Concluding Remarks
Over the past decade, NLRP6 has emerged as a multifaceted regulator of host defense and tissue homeostasis, whose functions extend far beyond its originally proposed role as a canonical inflammasome sensor. Accumulating evidence from studies in the intestine, liver, lung, and immune system has revealed that NLRP6 can exert both protective and detrimental effects depending on tissue context, cell type, and the nature and severity of the challenge. This context dependency appears to be a defining feature of NLRP6 biology and likely reflects its ability to engage multiple downstream pathways.
In the intestine, NLRP6 has been most consistently linked to host protection, where it contributes to antiviral defense, epithelial integrity, and the maintenance of microbial and metabolic homeostasis. Notably, NLRP6 can function not only through classical inflammasome-dependent pathways but also through inflammasome-independent mechanisms, such as the activation of antiviral signaling cascades leading to the induction of interferon-stimulated genes. In the liver, a large body of work using conventional Nlrp6-deficient mice has highlighted the importance of the gut–liver axis, demonstrating that NLRP6-dependent control of the intestinal microbiota and its metabolites indirectly shapes susceptibility to metabolic and inflammatory liver diseases. More recent studies using hepatocyte-specific deletion models further suggest that NLRP6 can also act in a cell-intrinsic manner, for example by restraining NF-κB signaling to protect hepatocytes from excessive inflammatory damage. Together, these findings strongly argue that NLRP6 is deeply involved in sensing or responding to infection-associated or microbiota-derived metabolic cues, as well as to viral infections, at barrier and metabolic organs.
Beyond these organ-level functions, NLRP6 has also been implicated in diverse cellular processes, including the regulation of ubiquitination, autophagy, and multiple forms of cell death such as pyroptosis and necrosis. These observations raise the intriguing possibility that NLRP6 should be viewed not merely as a pathogen sensor, but rather as a broader signaling hub that integrates stress signals, metabolic states, and infection-derived cues to fine-tune cellular and tissue responses. Such a perspective may help explain why NLRP6 can either limit or exacerbate pathology depending on the experimental setting, for instance by dampening inflammatory signaling in some contexts while promoting cytokine release or cell death in others.
In immune cells, however, several fundamental questions remain unresolved. Although NLRP6 expression and function have been reported in macrophages, neutrophils, and other immune cell types, the upstream signals in these cells are still poorly defined. Given the strong links between NLRP6, metabolites, and antiviral responses observed in the gut and liver, it is tempting to speculate that similar principles may apply in immune cells. NLRP6 may participate in the direct sensing of metabolic alterations associated with infection or tissue damage, or in the integration of metabolic and antiviral signaling pathways that shape immune cell function. If so, NLRP6 could represent an important molecular bridge between immunometabolism and innate immune signaling.
Looking forward, several key issues need to be addressed. These include the identification of bona fide upstream ligands or activating pathways for NLRP6 in different cell types, the clarification of how its inflammasome-dependent and -independent functions are coordinated, and the dissection of its cell type–specific roles using conditional and temporally controlled genetic models. A deeper mechanistic understanding of these aspects will help reconcile seemingly divergent observations and clarify why NLRP6 can act as either a protector or a promoter of disease. Ultimately, such insights may open new avenues for therapeutic strategies aimed at modulating NLRP6 signaling in infections, inflammatory disorders, metabolic diseases, and cancer.
Author Contributions
Conceptualization, T.Y.; writing—original draft preparation, T.Y.; writing—review and editing, V.J. and S.G.G.; funding acquisition, T.Y. and S.G.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Ichiro Kanehara Foundation and the Gout and uric acid foundation of Japan (to T.Y.), National Institutes on Aging (NIA) grant U19AG060909 (to S.G.G.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zheng, D.; Kern, L.; Elinav, E. The NLRP6 inflammasome. Immunology 2021, 162, 281–289. [Google Scholar] [CrossRef]
- Ghimire, L.; Paudel, S.; Jin, L.; Jeyaseelan, S. The NLRP6 inflammasome in health and disease. Mucosal Immunol 2020, 13, 388–398. [Google Scholar] [CrossRef]
- Sundaram, B.; Tweedell, R.E.; Prasanth Kumar, S.; Kanneganti, T.D. The NLR family of innate immune and cell death sensors. Immunity 2024, 57, 674–699. [Google Scholar] [CrossRef]
- Li, R.; Zan, Y.; Wang, D.; Chen, X.; Wang, A.; Tan, H.; Zhang, G.; Ding, S.; Shen, C.; Wu, H.; et al. A mouse model to distinguish NLRP6-mediated inflammasome-dependent and -independent functions. Proc Natl Acad Sci U S A 2024, 121, e2321419121. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalova, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Nunez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol 2011, 186, 7187–7194. [Google Scholar] [CrossRef] [PubMed]
- Normand, S.; Delanoye-Crespin, A.; Bressenot, A.; Huot, L.; Grandjean, T.; Peyrin-Biroulet, L.; Lemoine, Y.; Hot, D.; Chamaillard, M. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci U S A 2011, 108, 9601–9606. [Google Scholar] [CrossRef]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef]
- Ghimire, L.; Paudel, S.; Jin, L.; Baral, P.; Cai, S.; Jeyaseelan, S. NLRP6 negatively regulates pulmonary host defense in Gram-positive bacterial infection through modulating neutrophil recruitment and function. PLoS Pathog 2018, 14, e1007308. [Google Scholar] [CrossRef]
- Li, S.; Fu, Y.; Jia, X.; Liu, Z.; Qian, Z.; Zha, H.; Lei, G.; Yu, L.; Zhang, X.; Zhang, T.; et al. NLRP6 deficiency enhances macrophage-mediated phagocytosis via E-Syt1 to inhibit hepatocellular carcinoma progression. Gut 2025, 74, 1883–1895. [Google Scholar] [CrossRef]
- Valino-Rivas, L.; Cuarental, L.; Nunez, G.; Sanz, A.B.; Ortiz, A.; Sanchez-Nino, M.D. Loss of NLRP6 expression increases the severity of acute kidney injury. Nephrol Dial Transplant 2020, 35, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.F.; Li, Z.G.; Zhang, Y.; Ju, X.H.; Liu, X.W.; Zhou, A.M.; Chen, J. NLRP6 Inflammasome Ameliorates Brain Injury after Intracerebral Hemorrhage. Front Cell Neurosci 2017, 11, 206. [Google Scholar] [CrossRef]
- Anand, P.K.; Malireddi, R.K.; Lukens, J.R.; Vogel, P.; Bertin, J.; Lamkanfi, M.; Kanneganti, T.D. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 2012, 488, 389–393. [Google Scholar] [CrossRef]
- Chenuet, P.; Marquant, Q.; Fauconnier, L.; Youness, A.; Mellier, M.; Marchiol, T.; Rouxel, N.; Messaoud-Nacer, Y.; Maillet, I.; Ledru, A.; et al. NLRP6 negatively regulates type 2 immune responses in mice. Allergy 2022, 77, 3320–3336. [Google Scholar] [CrossRef]
- Wang, P.; Zhu, S.; Yang, L.; Cui, S.; Pan, W.; Jackson, R.; Zheng, Y.; Rongvaux, A.; Sun, Q.; Yang, G.; et al. Nlrp6 regulates intestinal antiviral innate immunity. Science 2015, 350, 826–830. [Google Scholar] [CrossRef]
- Hara, H.; Seregin, S.S.; Yang, D.; Fukase, K.; Chamaillard, M.; Alnemri, E.S.; Inohara, N.; Chen, G.Y.; Nunez, G. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 2018, 175, 1651–1664 e1614. [Google Scholar] [CrossRef]
- Shen, C.; Li, R.; Negro, R.; Cheng, J.; Vora, S.M.; Fu, T.M.; Wang, A.; He, K.; Andreeva, L.; Gao, P.; et al. Phase separation drives RNA virus-induced activation of the NLRP6 inflammasome. Cell 2021, 184, 5759–5774 e5720. [Google Scholar] [CrossRef]
- Hu, B.; Elinav, E.; Huber, S.; Strowig, T.; Hao, L.; Hafemann, A.; Jin, C.; Wunderlich, C.; Wunderlich, T.; Eisenbarth, S.C.; et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci U S A 2013, 110, 9862–9867. [Google Scholar] [CrossRef]
- Gremel, G.; Wanders, A.; Cedernaes, J.; Fagerberg, L.; Hallstrom, B.; Edlund, K.; Sjostedt, E.; Uhlen, M.; Ponten, F. The human gastrointestinal tract-specific transcriptome and proteome as defined by RNA sequencing and antibody-based profiling. J Gastroenterol 2015, 50, 46–57. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Zheng, J.; Hu, G.; Wang, J.; Huang, C.; Lou, L.; Wang, X.; Zeng, Y. Mucosal adherent bacterial dysbiosis in patients with colorectal adenomas. Sci Rep 2016, 6, 26337. [Google Scholar] [CrossRef]
- Seregin, S.S.; Golovchenko, N.; Schaf, B.; Chen, J.; Eaton, K.A.; Chen, G.Y. NLRP6 function in inflammatory monocytes reduces susceptibility to chemically induced intestinal injury. Mucosal Immunol 2017, 10, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol 2001, 19, 423–474. [Google Scholar] [CrossRef]
- Dai, S.M.; Matsuno, H.; Nakamura, H.; Nishioka, K.; Yudoh, K. Interleukin-18 enhances monocyte tumor necrosis factor alpha and interleukin-1beta production induced by direct contact with T lymphocytes: implications in rheumatoid arthritis. Arthritis Rheum 2004, 50, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Takagi, T.; Handa, O.; Ishikawa, T.; Nakagawa, S.; Yamaguchi, T.; Yoshida, N.; Minami, M.; Kita, M.; Imanishi, J.; et al. Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-alpha deficient mice. J Gastroenterol Hepatol 2003, 18, 560–569. [Google Scholar] [CrossRef]
- Noti, M.; Corazza, N.; Mueller, C.; Berger, B.; Brunner, T. TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J Exp Med 2010, 207, 1057–1066. [Google Scholar] [CrossRef]
- Schneider, K.M.; Mohs, A.; Gui, W.; Galvez, E.J.C.; Candels, L.S.; Hoenicke, L.; Muthukumarasamy, U.; Holland, C.H.; Elfers, C.; Kilic, K.; et al. Imbalanced gut microbiota fuels hepatocellular carcinoma development by shaping the hepatic inflammatory microenvironment. Nat Commun 2022, 13, 3964. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L.; Lu, W.; Farhataziz, N.; Gonzalez, A.; Xing, J.; Zhang, Z. TRIM29 controls enteric RNA virus-induced intestinal inflammation by targeting NLRP6 and NLRP9b signaling pathways. Mucosal Immunol 2025, 18, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Winsor, N.J.; Bayer, G.; Singh, O.; Chan, J.K.; Li, L.Y.; Lieng, B.Y.; Foerster, E.; Popovic, A.; Tsankov, B.K.; Maughan, H.; et al. Cross-kingdom-mediated detection of intestinal protozoa through NLRP6. Cell Host Microbe 2025, 33, 388–407 e389. [Google Scholar] [CrossRef]
- Mukherjee, S.; Kumar, R.; Tsakem Lenou, E.; Basrur, V.; Kontoyiannis, D.L.; Ioakeimidis, F.; Mosialos, G.; Theiss, A.L.; Flavell, R.A.; Venuprasad, K. Deubiquitination of NLRP6 inflammasome by Cyld critically regulates intestinal inflammation. Nat Immunol 2020, 21, 626–635. [Google Scholar] [CrossRef]
- Mao, X.; Qiu, X.; Jiao, C.; Lu, M.; Zhao, X.; Li, X.; Li, J.; Ma, J.; Zhang, H. Candida albicans SC5314 inhibits NLRP3/NLRP6 inflammasome expression and dampens human intestinal barrier activity in Caco-2 cell monolayer model. Cytokine 2020, 126, 154882. [Google Scholar] [CrossRef]
- Deng, Q.; Yang, S.; Huang, K.; Zhu, Y.; Sun, L.; Cao, Y.; Dong, K.; Li, Y.; Wu, S.; Huang, R. NLRP6 induces RIP1 kinase-dependent necroptosis via TAK1-mediated p38(MAPK)/MK2 phosphorylation in S. typhimurium infection. iScience 2024, 27, 109339. [Google Scholar] [CrossRef]
- Sun, L.; Huang, K.; Deng, Q.; Zhu, Y.; Cao, Y.; Dong, K.; Yang, S.; Li, Y.; Wu, S.; Huang, R. REV-ERBalpha negatively regulates NLRP6 transcription and reduces the severity of Salmonella infection in mice. Heliyon 2024, 10, e28432. [Google Scholar] [CrossRef]
- Pourcet, B.; Zecchin, M.; Ferri, L.; Beauchamp, J.; Sitaula, S.; Billon, C.; Delhaye, S.; Vanhoutte, J.; Mayeuf-Louchart, A.; Thorel, Q.; et al. Nuclear Receptor Subfamily 1 Group D Member 1 Regulates Circadian Activity of NLRP3 Inflammasome to Reduce the Severity of Fulminant Hepatitis in Mice. Gastroenterology 2018, 154, 1449–1464 e1420. [Google Scholar] [CrossRef]
- Lam, M.T.; Cho, H.; Lesch, H.P.; Gosselin, D.; Heinz, S.; Tanaka-Oishi, Y.; Benner, C.; Kaikkonen, M.U.; Kim, A.S.; Kosaka, M.; et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 2013, 498, 511–515. [Google Scholar] [CrossRef]
- Ghimire, L.; Paudel, S.; Le, J.; Jin, L.; Cai, S.; Bhattarai, D.; Jeyaseelan, S. NLRP6 negatively regulates host defense against polymicrobial sepsis. Front Immunol 2024, 15, 1248907. [Google Scholar] [CrossRef] [PubMed]
- Radulovic, K.; Ayata, C.K.; Mak'Anyengo, R.; Lechner, K.; Wuggenig, P.; Kaya, B.; Hruz, P.; Gomez de Aguero, M.; Broz, P.; Weigmann, B.; et al. NLRP6 Deficiency in CD4 T Cells Decreases T Cell Survival Associated with Increased Cell Death. J Immunol 2019, 203, 544–556. [Google Scholar] [CrossRef]
- Cai, S.; Paudel, S.; Jin, L.; Ghimire, L.; Taylor, C.M.; Wakamatsu, N.; Bhattarai, D.; Jeyaseelan, S. NLRP6 modulates neutrophil homeostasis in bacterial pneumonia-derived sepsis. Mucosal Immunol 2021, 14, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Chen, S.; Gui, X.; Li, Y.; Sun, Y.; Zhu, H.; Dai, Y.; Zhang, J.; Li, X.; Ju, W.; et al. Platelet NLRP6 protects against microvascular thrombosis in sepsis. Blood 2025, 146, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Huang, Y.; Ye, Z.; Chen, W.; Zhang, N.; Wen, Z.; Ge, C. Short-chain fatty acids attenuate sepsis-induced gut dysbiosis and hippocampal neuroinflammation via NLRP6 inflammasome activation in mice. Int J Surg 2026, 112, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wu, X.; Peng, L.; Chen, T.; Huang, Q.; Wang, Y.; Ye, C.; Peng, Y.; Hu, D.; Fang, R. The Critical Role of NLRP6 Inflammasome in Streptococcus pneumoniae Infection In Vitro and In Vivo. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783 e764. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Heikenwalder, M.; Ganal-Vonarburg, S.C. The Liver at the Nexus of Host-Microbial Interactions. Cell Host Microbe 2016, 20, 561–571. [Google Scholar] [CrossRef]
- Huang, C.; Liu, Q.; Tang, Q.; Jing, X.; Wu, T.; Zhang, J.; Zhang, G.; Zhou, J.; Zhang, Z.; Zhao, Y.; et al. Hepatocyte-specific deletion of Nlrp6 in mice exacerbates the development of non-alcoholic steatohepatitis. Free Radic Biol Med 2021, 169, 110–121. [Google Scholar] [CrossRef]
- Ji, X.; Li, L.; Lu, P.; Li, X.; Tian, D.; Liu, M. NLRP6 exerts a protective role via NF-kB with involvement of CCL20 in a mouse model of alcoholic hepatitis. Biochem Biophys Res Commun 2020, 528, 485–492. [Google Scholar] [CrossRef]
- Mainz, R.E.; Albers, S.; Haque, M.; Sonntag, R.; Treichel, N.S.; Clavel, T.; Latz, E.; Schneider, K.M.; Trautwein, C.; Otto, T. NLRP6 Inflammasome Modulates Disease Progression in a Chronic-Plus-Binge Mouse Model of Alcoholic Liver Disease. Cells 2022, 11. [Google Scholar] [CrossRef]
- Li, M.; Chen, Y.; Shi, J.; Ju, W.; Qi, K.; Fu, C.; Li, Z.; Zhang, X.; Qiao, J.; Xu, K.; et al. NLRP6 deficiency aggravates liver injury after allogeneic hematopoietic stem cell transplantation. Int Immunopharmacol 2019, 74, 105740. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Li, C.; Lei, G.; Zhou, L.; Chen, X.; Jia, X.; Lu, Y. Intestinal Candida albicans Promotes Hepatocarcinogenesis by Up-Regulating NLRP6. Front Microbiol 2022, 13, 812771. [Google Scholar] [CrossRef]
- Chang, L.; Xu, L.; Tian, Y.; Liu, Z.; Song, M.; Li, S.; Zhang, X.; Chen, Y.; Hao, Q.; Lu, Y.; et al. NLRP6 deficiency suppresses colorectal cancer liver metastasis growth by modulating M-MDSC-induced immunosuppressive microenvironment. Biochim Biophys Acta Mol Basis Dis 2024, 1870, 167035. [Google Scholar] [CrossRef] [PubMed]
- Lucke, J.; Zhang, T.; Zazara, D.E.; Seeger, P.; Izbicki, J.R.; Hackert, T.; Huber, S.; Giannou, A.D. Protocol for generating lung and liver metastasis in mice using models that bypass intravasation. STAR Protoc 2024, 5, 102696. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Zhou, X.; Wang, G.; Jie, X.; Xing, B.; Xu, Y.; Chen, Y.; Li, J.; Zhu, K.; Wu, Z.; et al. NLRP6 is required for cancer-derived exosome-modified macrophage M2 polarization and promotes metastasis in small cell lung cancer. Cell Death Dis 2022, 13, 891. [Google Scholar] [CrossRef]
- Kempster, S.L.; Belteki, G.; Forhead, A.J.; Fowden, A.L.; Catalano, R.D.; Lam, B.Y.; McFarlane, I.; Charnock-Jones, D.S.; Smith, G.C. Developmental control of the Nlrp6 inflammasome and a substrate, IL-18, in mammalian intestine. Am J Physiol Gastrointest Liver Physiol 2011, 300, G253–263. [Google Scholar] [CrossRef]
- Wang, H.; Xu, G.; Huang, Z.; Li, W.; Cai, H.; Zhang, Y.; Xiong, D.; Liu, G.; Wang, S.; Xue, Z.; et al. LRP6 targeting suppresses gastric tumorigenesis via P14(ARF)-Mdm2-P53-dependent cellular senescence. Oncotarget 2017, 8, 111597–111607. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, C.; Chen, J. NLRP6, decreased in gastric cancer, suppresses tumorigenicity of gastric cancer cells. Cancer Manag Res 2018, 10, 6431–6444. [Google Scholar] [CrossRef] [PubMed]
- Tabassam, F.H.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori-associated regulation of forkhead transcription factors FoxO1/3a in human gastric cells. Helicobacter 2012, 17, 193–202. [Google Scholar] [CrossRef]
- Wang, X.; Wu, X.; Wang, Q.; Zhang, Y.; Wang, C.; Chen, J. NLRP6 suppresses gastric cancer growth via GRP78 ubiquitination. Exp Cell Res 2020, 395, 112177. [Google Scholar] [CrossRef]
- Lin, M.; Mo, Y.; Li, C.M.; Liu, Y.Z.; Feng, X.P. GRP78 as a potential therapeutic target in cancer treatment: an updated review of its role in chemoradiotherapy resistance of cancer cells. Med Oncol 2025, 42, 49. [Google Scholar] [CrossRef]
- Hernandez, I.; Cohen, M. Linking cell-surface GRP78 to cancer: From basic research to clinical value of GRP78 antibodies. Cancer Lett 2022, 524, 1–14. [Google Scholar] [CrossRef]
- Bai, Y.; Li, S. Long noncoding RNA OIP5-AS1 aggravates cell proliferation, migration in gastric cancer by epigenetically silencing NLRP6 expression via binding EZH2. J Cell Biochem 2020, 121, 353–362. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Dashti, S.; Farsi, M.; Hussen, B.M.; Taheri, M. A review on the role of oncogenic lncRNA OIP5-AS1 in human malignancies. Biomed Pharmacother 2021, 137, 111366. [Google Scholar] [CrossRef]
- Izquierdo, M.C.; Sanz, A.B.; Mezzano, S.; Blanco, J.; Carrasco, S.; Sanchez-Nino, M.D.; Benito-Martin, A.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. TWEAK (tumor necrosis factor-like weak inducer of apoptosis) activates CXCL16 expression during renal tubulointerstitial inflammation. Kidney Int 2012, 81, 1098–1107. [Google Scholar] [CrossRef]
- Ydens, E.; Demon, D.; Lornet, G.; De Winter, V.; Timmerman, V.; Lamkanfi, M.; Janssens, S. Nlrp6 promotes recovery after peripheral nerve injury independently of inflammasomes. J Neuroinflammation 2015, 12, 143. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Zhang, J.; Zhang, L.; Wang, Y.; Li, Z.; Zhao, J. Effects of NLRP6 in Cerebral Ischemia/Reperfusion (I/R) Injury in Rats. J Mol Neurosci 2019, 69, 411–418. [Google Scholar] [CrossRef]
- Xiao, H.; Chen, H.; Jiang, R.; Zhang, L.; Wang, L.; Gan, H.; Jiang, N.; Zhao, J.; Zhai, X.; Liang, P. NLRP6 contributes to inflammation and brain injury following intracerebral haemorrhage by activating autophagy. J Mol Med (Berl) 2020, 98, 1319–1331. [Google Scholar] [CrossRef]
- Huang, X.; Tan, J.; Ji, Y.; Luo, J.; Zhao, Y.; Zhao, J. BRCC3 mediates inflammation and pyroptosis in cerebral ischemia/reperfusion injury by activating the NLRP6 inflammasome. CNS Neurosci Ther 2024, 30, e14697. [Google Scholar] [CrossRef]
- Tang, C.; Wang, Q.; Shen, J.; Wang, C.; Ding, H.; Wen, S.; Yang, F.; Jiao, R.; Wu, X.; Li, J.; et al. Neuron stem cell NLRP6 sustains hippocampal neurogenesis to resist stress-induced depression. Acta Pharm Sin B 2023, 13, 2017–2038. [Google Scholar] [CrossRef]
- Du, Y.; He, C.; An, Y.; Huang, Y.; Zhang, H.; Fu, W.; Wang, M.; Shan, Z.; Xie, J.; Yang, Y.; et al. The Role of Short Chain Fatty Acids in Inflammation and Body Health. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Oladapo, A.; Kannan, M.; Deshetty, U.M.; Singh, S.; Buch, S.; Periyasamy, P. Methamphetamine-mediated astrocytic pyroptosis and neuroinflammation involves miR-152-NLRP6 inflammasome signaling axis. Redox Biol 2025, 80, 103517. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Kannan, M.; Oladapo, A.; Deshetty, U.M.; Ray, S.; Buch, S.; Periyasamy, P. Ethanol modulates astrocyte activation and neuroinflammation via miR-339/NLRP6 inflammasome signaling. Free Radic Biol Med 2025, 226, 1–12. [Google Scholar] [CrossRef]
- Zhi, F.; Li, B.; Zhang, C.; Xia, F.; Wang, R.; Xie, W.; Cai, S.; Zhang, D.; Kong, R.; Hu, Y.; et al. NLRP6 potentiates PI3K/AKT signalling by promoting autophagic degradation of p85alpha to drive tumorigenesis. Nat Commun 2023, 14, 6069. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cao, F.; Xiong, Y.; Zhou, H. SP1 transcriptionally activates NLRP6 inflammasome and induces immune evasion and radioresistance in glioma cells. Int Immunopharmacol 2021, 98, 107858. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Yin, H.; Qin, S.; Zhang, K.; Guan, Y.; Fang, R.; Wang, H.; Li, G.; Jiang, Z.; Sun, F.; et al. NLRP6 self-assembles into a linear molecular platform following LPS binding and ATP stimulation. Sci Rep 2020, 10, 198. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Xie, P.; Wang, J.; Yang, F.; Li, S.; Jiang, H.; Wu, X.; Zhou, F.; Li, J. Nlrp6 protects from corticosterone-induced NSPC ferroptosis by modulating RIG-1/MAVS-mediated mitophagy. Redox Biol 2024, 73, 103196. [Google Scholar] [CrossRef]
- Wang, D.; Chen, X.; Sui, K.; Wang, A.; Li, R.; Zhou, W.; Zhu, X.; Hua, Y.; Yuan, S.; Zhou, R.; et al. UBE2O-mediated monoubiquitination licenses NLRP6 inflammasome activation in the intestine. Cell Host Microbe 2026, 34, 86–102 e108. [Google Scholar] [CrossRef]
- Premadasa, L.S.; McDew-White, M.; Romero, L.; Gondo, B.; Drawec, J.A.; Ling, B.; Okeoma, C.M.; Mohan, M. Epigenetic modulation of the NLRP6 inflammasome sensor as a therapeutic modality to reduce necroptosis-driven gastrointestinal mucosal dysfunction in HIV/SIV infection. Cell Commun Signal 2025, 23, 199. [Google Scholar] [CrossRef]
Figure 1.
Structure and molecular functions of NLRP6. Like other members of the NOD-like receptor (NLR) family, NLRP6 is composed of three characteristic domains: an N-terminal pyrin domain (PYD), a central NACHT (nucleotide-binding and oligomerization) domain, and a C-terminal leucine-rich repeat (LRR) domain. The PYD functions as an effector domain that interacts with the inflammasome adaptor ASC and triggers filamentous oligomerization during inflammasome assembly. In mouse NLRP6, the amino acid residues Arg39 (R39) and Trp50 (W50) have been identified as critical for ASC binding [4] The central NACHT domain mediates ATP binding and hydrolysis and plays a central role in controlling NLRP6 self-oligomerization. The C-terminal LRR domain adopts a horseshoe-like structure and functions as a sensor that directly or indirectly recognizes pathogen-derived ligands. Binding to dsRNA is mediated by a region spanning the central NACHT and C-terminal LRR domains [19]. In particular, a lysine-rich region (K350–354 in mouse NLRP6) is essential for multivalent interactions with dsRNA and for promoting liquid–liquid phase separation (LLPS), which may serve as a hub for integrating and regulating multiple signaling pathways [19]. In addition, NLRP6 has been reported to directly recognize lipoteichoic acid (LTA), a cell wall component of Gram-positive bacteria such as Listeria, via its LRR domain [18]. Structural analyses have suggested the presence of a density patch near the LRR region of NLRP6 that may be involved in LPS binding; however, a specific amino acid–level binding pocket has not yet been identified [74].
Figure 1.
Structure and molecular functions of NLRP6. Like other members of the NOD-like receptor (NLR) family, NLRP6 is composed of three characteristic domains: an N-terminal pyrin domain (PYD), a central NACHT (nucleotide-binding and oligomerization) domain, and a C-terminal leucine-rich repeat (LRR) domain. The PYD functions as an effector domain that interacts with the inflammasome adaptor ASC and triggers filamentous oligomerization during inflammasome assembly. In mouse NLRP6, the amino acid residues Arg39 (R39) and Trp50 (W50) have been identified as critical for ASC binding [4] The central NACHT domain mediates ATP binding and hydrolysis and plays a central role in controlling NLRP6 self-oligomerization. The C-terminal LRR domain adopts a horseshoe-like structure and functions as a sensor that directly or indirectly recognizes pathogen-derived ligands. Binding to dsRNA is mediated by a region spanning the central NACHT and C-terminal LRR domains [19]. In particular, a lysine-rich region (K350–354 in mouse NLRP6) is essential for multivalent interactions with dsRNA and for promoting liquid–liquid phase separation (LLPS), which may serve as a hub for integrating and regulating multiple signaling pathways [19]. In addition, NLRP6 has been reported to directly recognize lipoteichoic acid (LTA), a cell wall component of Gram-positive bacteria such as Listeria, via its LRR domain [18]. Structural analyses have suggested the presence of a density patch near the LRR region of NLRP6 that may be involved in LPS binding; however, a specific amino acid–level binding pocket has not yet been identified [74].
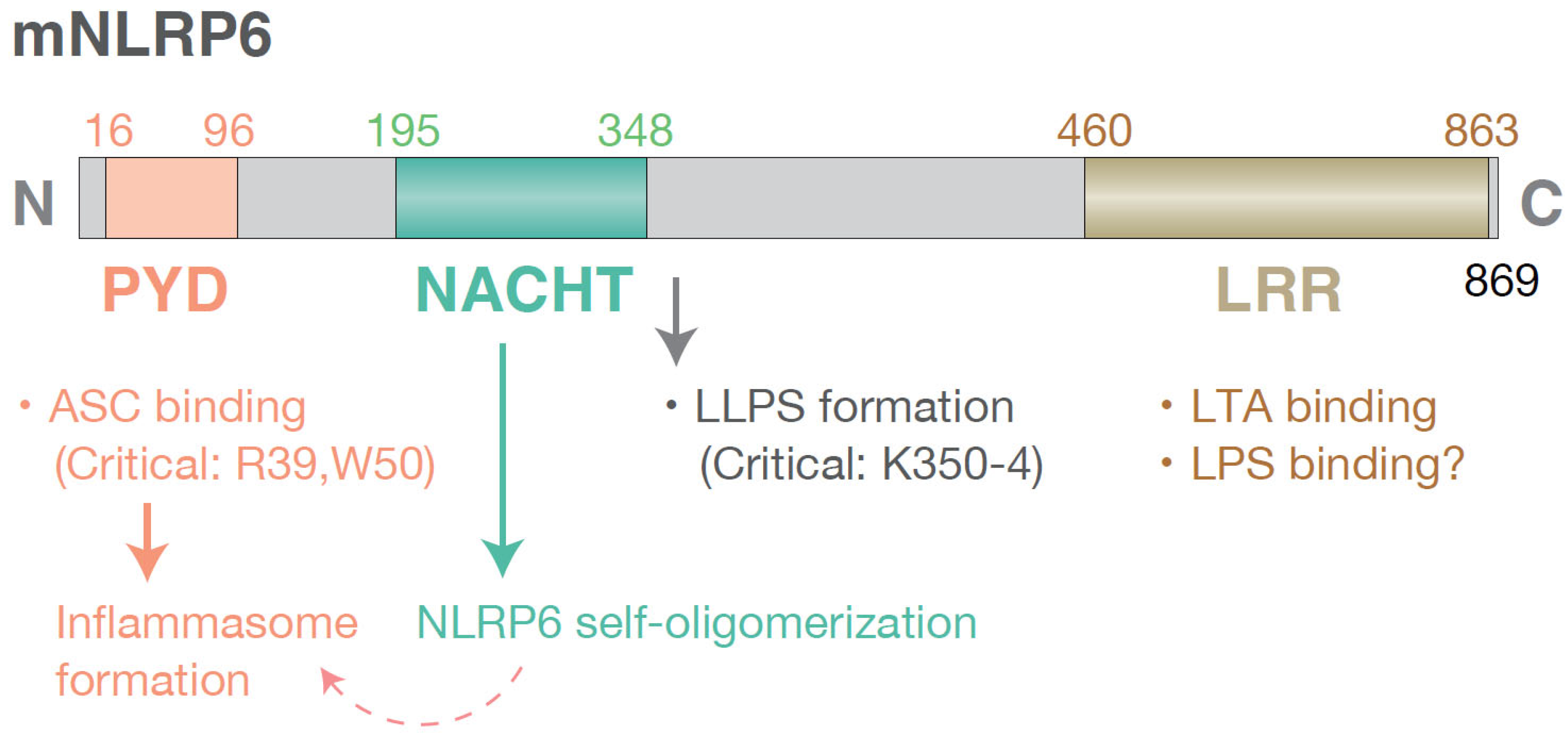
Figure 2.
Representative mechanisms by which NLRP6-mediated immune responses exert protective or detrimental effects in the host. In the intestine, NLRP6 has been consistently reported to play predominantly protective roles. In viral infections, NLRP6 activates an inflammasome-independent dsRNA–MAVS–ISG axis, thereby promoting antiviral responses [17,29], whereas in viral and other microbial infections it can also engage the inflammasome pathway [4,30]. In systemic infection models induced via the lung, intestine, or by intravenous or intraperitoneal administration, NLRP6-mediated inflammasome activation and subsequent recruitment of neutrophils and other innate immune cells contribute to host protection under relatively mild and controllable infection conditions [39]. In contrast, under high or lethal infectious burdens, NLRP6 signaling is frequently detrimental, and genetic ablation of Nlrp6 often results in improved disease outcomes. In many such contexts, NLRP6 contributes to pathology through: (i) inflammasome-independent suppression of NF-κB signaling, leading to reduced cytokine production and dampened immune responses [15,42]; (ii) IL-18–dependent induction of lymphocyte death downstream of the inflammasome [37]; and (iii) promotion of necrosis and/or pyroptosis in multiple cell types [33]. Notably, analysis using platelet-specific Nlrp6-deficient mice revealed a protective role of NLRP6 in restraining aberrant platelet activation [40], highlighting its cell type–specific functions. In the liver, many studies have relied on conventional Nlrp6-deficient mice, in which alterations in the gut microbiota and its metabolites indirectly exacerbate metabolic liver diseases, indicating that intestinal NLRP6 indirectly protects the liver via the gut–liver axis [7,28]. More recently, analyses using hepatocyte-specific Nlrp6-deficient mice have demonstrated a cell-intrinsic protective role of hepatic NLRP6, mediated at least in part through suppression of the NF-κB signaling pathway [46].
Figure 2.
Representative mechanisms by which NLRP6-mediated immune responses exert protective or detrimental effects in the host. In the intestine, NLRP6 has been consistently reported to play predominantly protective roles. In viral infections, NLRP6 activates an inflammasome-independent dsRNA–MAVS–ISG axis, thereby promoting antiviral responses [17,29], whereas in viral and other microbial infections it can also engage the inflammasome pathway [4,30]. In systemic infection models induced via the lung, intestine, or by intravenous or intraperitoneal administration, NLRP6-mediated inflammasome activation and subsequent recruitment of neutrophils and other innate immune cells contribute to host protection under relatively mild and controllable infection conditions [39]. In contrast, under high or lethal infectious burdens, NLRP6 signaling is frequently detrimental, and genetic ablation of Nlrp6 often results in improved disease outcomes. In many such contexts, NLRP6 contributes to pathology through: (i) inflammasome-independent suppression of NF-κB signaling, leading to reduced cytokine production and dampened immune responses [15,42]; (ii) IL-18–dependent induction of lymphocyte death downstream of the inflammasome [37]; and (iii) promotion of necrosis and/or pyroptosis in multiple cell types [33]. Notably, analysis using platelet-specific Nlrp6-deficient mice revealed a protective role of NLRP6 in restraining aberrant platelet activation [40], highlighting its cell type–specific functions. In the liver, many studies have relied on conventional Nlrp6-deficient mice, in which alterations in the gut microbiota and its metabolites indirectly exacerbate metabolic liver diseases, indicating that intestinal NLRP6 indirectly protects the liver via the gut–liver axis [7,28]. More recently, analyses using hepatocyte-specific Nlrp6-deficient mice have demonstrated a cell-intrinsic protective role of hepatic NLRP6, mediated at least in part through suppression of the NF-κB signaling pathway [46].
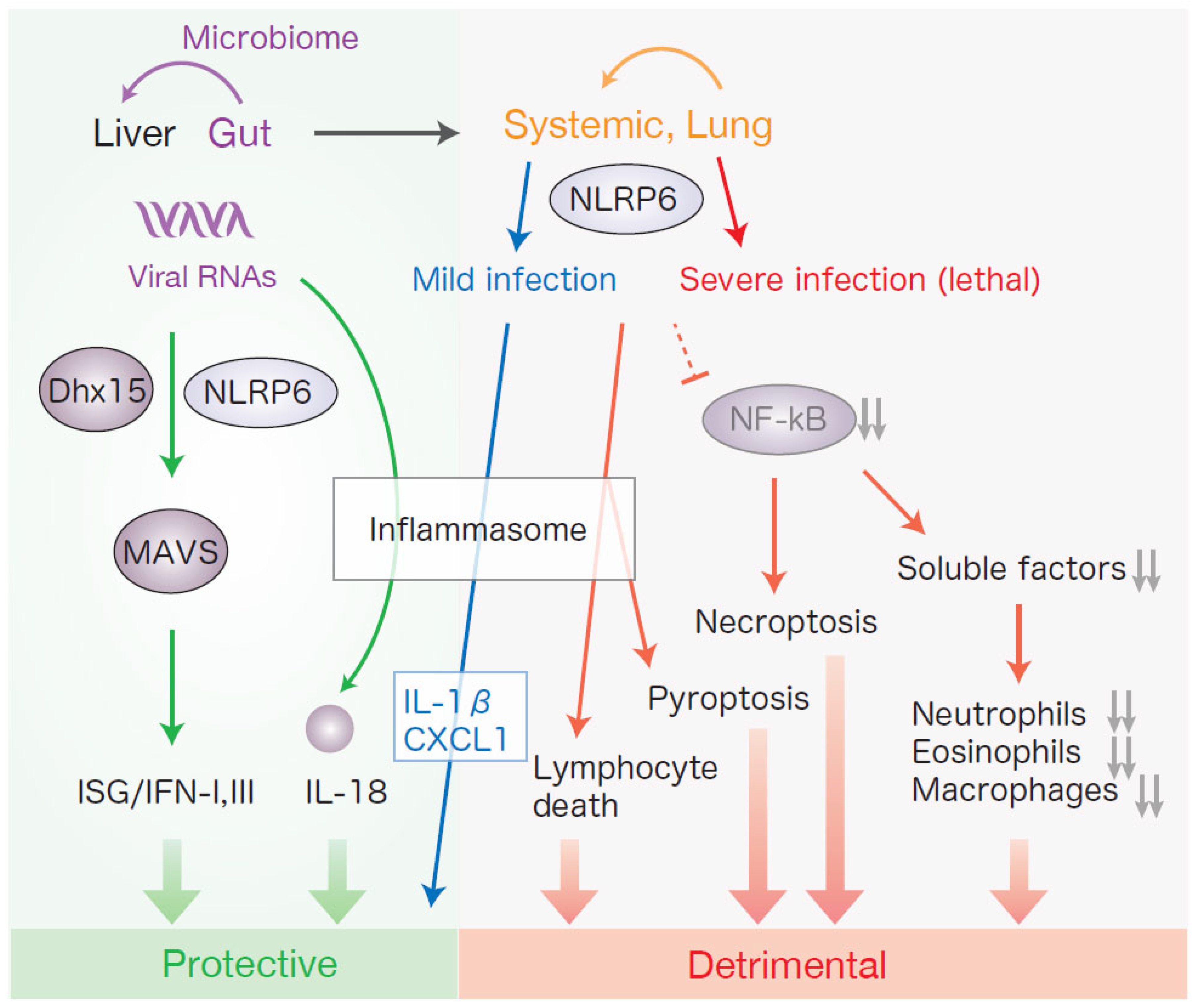

Table 1.
Upstream regulators of NLRP6.
| Upstream regulators | Mechanisms | Locations |
| Taurine, Histamine, Spermine | Induction (taurine) or reduction (spermine and histamine) of NLRP6 inflammasome assembly | Colon [5] |
| Sphingolipids | Induction of NLRP6 inflammasome assembly | Colon [30] |
| ssRNA(EMCV) | MAVS-mediated IFN/ISG induction (together with Dhx15) | Intestinal tract [17] |
| dsRNA | Induction of LLPS of NLRP6 | MEF [19] |
| Lipoteichoic acid (LTA) | Recruitment of caspase-1 and caspase-11 to the NLRP6 inflammasome | BMDM [18] |
| α-hemolysin (hla) | Activation of the NLRP6 inflammasome | BMDM [11] |
| PPAR-γ | Transcriptional upregulation of NLRP6 | Human colon cancer cell line (Caco-2) [54] |
| Short-chain fatty acid (SCFA) | Transcriptional upregulation of Nlrp6 | Mouse neural stem cell line (NE-4C) [75] |
| SP1 | Transcriptional upregulation of NLRP6 | Human glioblastoma cell line (U87) [73] |
| REV-ERBα | Transcriptional repression of Nlrp6 | Intestinal epithelial cells [34] |
| Polyunsaturated fat (PUFA) diet, Methionine-choline deficient (MCD) diet | Transcriptional repression of Nlrp6 | Liver [46] |
| H. pylori/FOXO3 | Transcriptional repression of NLRP6 | Human gastric cancer cell lines (BGC-823, HGC-27) [56] |
| miR-152, miR-339 | Transcriptional repression of Nlrp6 (NLRP6) | Mouse primary astrocytes [56], Primary human astrocytes [71] |
| OIP5-AS1 (lncRNA)/ EZH2 | Increasing the H3K27me3 enrichment of NLRP6 promoter to represses NLRP6 expression | Human gastric cancer cell lines (AGS, SGC7901) [61] |
| Cyld (deubiquitinase) | Deubiquitination inhibited the NLRP6–ASC inflammasome complex | Colon [31] |
| BRCC3 (deubiquitinase) | Reduction of NLRP6 ubiquitination for NLRP6 stabilization | Mouse Hippocampal Nneuron Cell Line (HT22), HEK293T cells [67] |
| UBE2O* (ubiquitin conjugating enzyme) | NLRP6 monoubiquitination promotes oligomerization and cytoplasmic localization. | Intestinal epithelial cells [76] |
| TRIM29 (E3 ubiquitin ligase) | Promotes ubiquitination (degradation-inducing) of NLRP6 | Mouse intestinal organoids [29] |
| Exosomes from small cell lung cancer (SCLC) | Induction of NLRP6 expression | BMDM [53] |
Recent studies have revealed multiple upstream regulatory mechanisms controlling NLRP6 expression and activation. *UBE2O has been identified as an ubiquitin conjugating enzyme that catalyzes two distinct monoubiquitination events on NLRP6 that are essential for NLRP6-mediated intestinal immunity [76]. Specifically, monoubiquitination at the K680–687 region promotes NLRP6 oligomerization, whereas monoubiquitination at K115/130 within the nuclear localization signal facilitates its cytoplasmic localization. Although not included in this table, a study investigating mechanisms of intestinal barrier dysfunction in simian immunodeficiency virus (SIV)–induced enteropathy demonstrated hypomethylation of CpG islands in the promoters of NLRP6 and other genes in the colonic epithelium of SIV-infected rhesus macaques [77]. Conversely, long-term low-dose administration of delta-9-tetrahydrocannabinol (THC) effectively induced hypermethylation of the NLRP6 CpG island and suppressed NLRP6 expression, although the specificity of this effect for NLRP6 requires further investigation. Abbreviations: ssRNA, single-stranded RNA; EMCV, encephalomyocarditis virus; MAVS, mitochondrial antiviral signaling protein; ISG, interferon and interferon-stimulated genes; dsRNA, double-stranded RNA; LLPS, liquid-liquid phase separation; MEF, mouse embryonic fibroblast; BMDM, bone marrow derived macrophage; lncRNA, long non-coding RNA.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



