Submitted:
29 January 2026
Posted:
30 January 2026
You are already at the latest version
Abstract
Chronic obstructive pulmonary disease (COPD) and bronchial asthma (BA) are very common pathologies, both of them being characterized by a chronic bronchopulmonary inflammation. This paper aims to present the mechanisms of the two pathologies in comparison, starting from the classical approach, entering the cellular level (effector cells), then the molecular one (lipid mediators, cytokines, chemokines, reactive oxygen species, proteases, ATP, cellular senescence markers), and finally addressing the mechanisms at the quantum level. It will be explained that electron transfer through the interfacial water is essential for all cellular energy metabolism associated events. It will also be presented how biochemical reactions do not occur instantaneously and randomly, but depend on exceeding a threshold of free energy of activation and satisfying steric requirements. Another important topic addressed will be the electron-accepting property of ionic reactive oxygen species (ROS) and how cellular metabolism is regulated by the formation and decomposition of collective ROS states (this is a quantum regulated phenomenon involving a large number of entangled ROS molecules simultaneously). Finally, it will be presented how these mechanisms are altered in COPD and BA as well as the consequences of pulmonary fibrosis at the quantum level. We believe it is important for physicians to understand how the principles of quantum physics applied to experimental biology deepen the understanding of the normality and disease origin at the level of subatomic particles, molecules, and their associated electromagnetic fields.
Keywords:
chronic obstructive pulmonary disease
; bronchial asthma
; inflammation
; interfacial water
; ionic free radicals
; electromagnetic field
Introduction. Chronic obstructive pulmonary disease (COPD) and bronchial asthma (BA) are very common pathologies in the Western world, both of them being characterized by a chronic bronchopulmonary inflammation. However, the nature of the inflammatory reaction differs in each case, with a large number of clinical phenotypes. In asthma, inflammation usually responds to low doses of corticosteroids, whereas most patients with COPD are corticosteroid-resistant and require other anti-inflammatory therapies [1,2].
However, the above-mentioned reasons are not the only ones that lie at the basis of approaching the two entities. In addition, it is the fact that the assimilation of the pathophysiological mechanisms involved in each of them ensures the logical and scientific necessary data for understanding the mechanisms involved in any other bronchopulmonary pathology.
To this end, the classical mechanisms will be presented first, then the cells involved and the molecular mechanisms, so that finally, we can try to approach them at the quantum level. For many years, quantum physics has been providing us with profound information about the world of small-scale structures. While initially quantum principles were considered valid only at the level of subatomic particles, discoveries in recent years have experimentally confirmed the simultaneous dual particle-wave behavior for molecules with molecular weights up to 25 kDa. That is, for many structures existing in the living world, including our body.
Then isn't it important for a physician to understand that health and disease do not begin and end with the molecular approach?
1. Classical Pathophysiology of COPD
Chronic obstructive pulmonary disease is a pathological condition in which chronic obstructive disorders of pulmonary ventilation occur, with a progressive and irreversible character, generated by pulmonary pathological processes of complex etiology. The most common cause is long-term exposure to irritants [3].
The conditions that meet this definition are chronic obstructive bronchitis and pulmonary emphysema.
The definition of COPD excludes acute obstructive disorders, bronchial asthma and obstructive pathology of known etiology (pneumoconiosis, tuberculosis, bronchopulmonary neoplasm etc.). Chronic obstructive bronchitis and pulmonary emphysema therefore coexist in COPD, but there are multiple forms in which one of these pathologies is predominant [3,4].
Mechanisms of pulmonary hypoventilation in COPD:
a. Intrinsic narrowing of affected airways;
b. Partial or total obliteration of the small bronchi by mucus hypersecretion;
c. Decreased lung elastic recoil;
d. Bronchi and bronchioles smooth muscle spasm;
e. Extrinsic compression of small airways by hyperinflated neighboring alveoli.
- Intrinsic narrowing of affected airways
The unknown for sure etiological factor (but considered to be, most frequently, smoking) triggers an inflammatory reaction that evolves according to the classical pattern. It is not known whether it is a chronic inflammation from the beginning or it becomes chronic at some point. However, it involves bronchial mucosa edema and the release of cytokines in successive waves. Among these cytokines are the growth factors FGF (fibroblast growth factor) and TGF β (transforming growth factor beta), which induce bronchial wall hypertrophy and fibroblast activation, with collagen synthesis stimulation and the emergence of bronchial and peribronchial fibrosis [3,4,5]. Through both edema and hypertrophy followed by fibrosis, the diameter of the bronchial lumen decreases.
- b. Partial or total obliteration of the small bronchi by mucus hypersecretion
Inflammatory mediators strongly stimulate goblet cells, which secrete large amounts of mucus. After prolonged stimulation, they hypertrophy, mucus secretion becoming autonomous. The mucus clogs the small bronchi, but by gravitational effect it also falls into the alveoli, where it displaces the surfactant. Under these conditions, the alveolar collapse in exhalation can no longer be prevented [3,6].
- c. Decreased lung elastic recoil
The distension of the small airways in inhalation causes the accumulation in their wall of a force that will act as elastic recoil in exhalation. When the emphysematous component is present in COPD, the elastic fibers in the wall of the small bronchi are broken, so the elastic recoil force decreases, and the terminal bronchioles can no longer remain open [3,7,8].
- d. Bronchi and bronchioles smooth muscle spasm
It occurs following both humoral and reflex pathways.
The humoral pathway entails the diffusion of bronchospastic inflammatory mediators (histamine, prostaglandins, leukotrienes that constitute SRS-A/slow-reacting substance of anaphylaxis) into the local circulation and interstitium [3,4].
The reflex mechanism is based on the hypersensitivity of bronchial receptors, following repeated stimulation with inflammatory mediators, so that bronchospasm occurs following minor stimuli (cold air, smoke, dust etc.) [3].
- e. Extrinsic compression of small airways by hyperinflated neighboring alveoli
Partially or totally obstructed bronchioles, along with the decrease in elastic recoil through the emphysematous component, cause an increase in residual alveolar volume, with the emergence of hyperinflation and, later, emphysema bullae. The hyperinflated alveoli act as an extrinsic obstructive factor on the neighboring bronchioles [7].
The mentioned pathophysiological mechanisms do not affect all pulmonary morphofunctional units (acini), and those affected present an uneven distribution of the ventilation/perfusion ratio (V/Q) (so the first major pathophysiological mechanism of type 1 pulmonary respiratory failure/PRF is present) [3,4,9].
Through mechanisms a → e the uneven distribution of ventilation is explained.
On the other hand, the uneven distribution of perfusion is explained by:
- rupture of the pulmonary capillaries, secondary to the rupture of the alveolar septa, in emphysema;
- compression of the lung capillaries by the neighboring hyperinflated alveoli;
- uneven reflex arteriolar vasoconstriction, generated by the decrease in ventilation.
Over time, the pathological processes expand more and more, so that units with a small V/Q ratio (poorly ventilated) will predominate, a phenomenon that can be equivalent to a generalized alveolar hypoventilation (the second major pathophysiological mechanism in PRF) and which generates hypoxemia and hypercapnia, i.e. global PRF.
- Decreased diffusion through alveolar-capillary membrane (ACM). It occurs through indirect mechanisms: the decrease in the lung surface area for gas exchange and the decrease in the lung contact time (increases circulation speed by decreasing the pulmonary capillary bed) [4,10];
- -
- Intrapulmonary vascular shunt. It is generated at a certain point in COPD, when pulmonary hypertension (PH) sets in, meaning the pressure in the lung arterioles exceeds a certain limit. The consequence is the opening of arteriovenous anastomoses in different areas, with short-circuiting of the capillaries [10,11].
Pulmonary hypertension is a major pathological change, so, regardless of the presence or absence of COPD, its mechanisms must be mentioned [11,12,13].
- reducing the size of the pulmonary vascular bed, by destroying the capillaries, so that the same ventilation must be done in a smaller space;
- reflex pulmonary vasoconstriction caused by hypoventilation becomes permanent, at some point. The first result is the arterioles intraparietal tension increase, followed by hypertrophy of the vascular muscular tunic which determines a large increase in vascular resistance to blood flow;
- polycythemia secondary to chronic hypoxia causes an increase in blood viscosity, equivalent to an increase in blood flow resistance, which is attempted to be counteracted by compensatory hypertrophy of both the pulmonary arterioles and the right ventricle (RV). When RV hypertrophy can no longer compensate for cardiac function, RV failure (chronic cor pulmonale) occurs. Hypertrophy also affects the left ventricle.
2. Classical Pathophysiology of Bronchial Asthma (BA)
Bronchial asthma (BA) is a chronic inflammatory disorder of the intrapulmonary airways, determined by the participation of several types of pro-inflammatory cells, especially mast cells. In people predisposed to this condition, diffuse bronchial obstructive phenomena occur and vary in intensity, reversible spontaneously or following treatment. Bronchial inflammatory phenomena are also associated with increased bronchial reactivity to different stimuli (International Consensus Report definition) [14].
Using the traditional classification (by cause), two categories of BA are distinguished [15]:
- -
- Extrinsic (allergic) BA: determined by the inhalation of allergens from outside the body (dust, pollen, human or animal hair, flakes etc.);
- -
- Intrinsic (non-allergic/infectious) BA: determined by various bronchopulmonary infectious processes, inhalation of irritants (smoke, exhaust or industrial gases), physical exertion, inhalation of cold or humid air, drugs, emotional stress.
Clinically, we very rarely encounter pure forms of BA, allergic or infectious. Usually, the forms are intricate, with an initial allergic or non-allergic cause accompanied over time by the other form.
In all these situations, the pathophysiology of BA involves:
A. Immunological and pro-inflammatory mediators discharge abnormalities.
B. Abnormalities of the autonomic nervous system (ANS) that innervates the bronchial territory.
- A.
- Immunological and pro-inflammatory mediators discharge abnormalities
They occur mainly in allergic BA.
In principle, and without going into molecular details, BA differs from COPD by:
• increase in the activity of bronchial mast cells;
• infiltration of bronchial walls with eosinophils;
• structure alterations and desquamation of the bronchial and bronchiolar epithelium [16].
Mechanisms:
The first contact with the allergen does not cause the crisis, it is only a sensitization contact/the early sensitization phase.
Inhalation of an allergen is followed by its capture on endocytic pattern recognition receptors (PRRs) on the membrane of the antigen-presenting cell (APCs) represented by bronchial dendritic cells and macrophages. After endocytosis and in parallel with the transport to the T-dependent zones in the regional lymph nodes (paracortex), APCs processes the allergen into epitopes, coupling them with MHC II molecules and presenting them to Th2 lymphocytes, a special variety of TH2 cells, involved in allergic reactions [17,18,19]. Th2 fixes the MHC II – epitope couple on the TCR-CD3 receptor complex, a phenomenon followed by the transduction pathway triggering. Thus, following this biological information exchange, the Th2 cell is partially activated and begins to secrete small amounts of type 2 cytokines (especially IL-4, IL-5 and IL-13) [25]. At the same time, fragments of the allergen also reach the B-dependent zones in the regional ganglia (cortex) via the bloodstream and activate B cells through BCR (B cell receptors). As with Th2 cells, it is an incomplete activation.
Both types of partially activated lymphocytes move away from the antigenic contact site and meet at the border between the T- and B-dependent zones of the lymph node. Bidirectional cooperation between the two types of partially activated lymphocytes occurs. The role of APC is played by B cells, which present allergen epitopes on MHC II molecules. The partially activated Th2 lymphocyte triggers the first activation signal via the TCR-CD3 pathway. However, activation of the second signaling pathway, the one via CD40-CD40L, is also mandatory [20]. CD40L appears on the Th2 membrane only when they are at least partially activated and has an essential role in stimulating Ig E synthesis. CD40 is a constitutive receptor on the membrane of B cells, and the formation of the CD40-CD40L couple plays a major role in the full activation of the two lymphocytes [20,21]. Fully activated Th2 cells have the maximum capacity to secrete IL-4, IL-5 and IL-13, through which they paracrinely activate various cell types, mainly B cells. Fully activated B cells become plasma cells that begin to synthesize Ig E.
Thus, under the influence of paracrine stimulation, instead of secreting Ig M, possibly Ig G/Ig A, plasma cells begin to secrete Ig E, specific for allergic responses, a phenomenon called isotype switching (mechanism of type I hypersensitivity) [22,23].
Synthesized immunoglobulins E have the particularity of being cytophilic antibodies, i.e. they stay in the blood and the interstitium for a very short period of time, and they are easily attached to the cell membranes that have specific receptors (FcƐR) [24]. Bronchial mast cells are among such membranes.
All the presented phenomena occur only in people with a genetic predisposition [26,27,28], i.e. people who have:
- very large number of Th2 cells;
- very large number of B cells capable of secreting IgE;
- reduced number of Treg lymphocytes (regulators or suppressors) that inhibit the humoral immune response (HIR) producing IgE;
- particular configurations of MHC I and II molecules on the membrane of bronchial dendritic cells and macrophages. The coupling of allergen-derived epitopes and the special MHCs induces a very intense stimulation of Th2 cells. The result will be the disruption of the functional balance between Th2 and Treg cells and the production of an exaggerated amount of Ig E.
Conclusion: the first contact with the allergen (sensitization) is manifested by the production of Ig E and has no clinical expression.
The second...nth contact with the same allergen causes clinical manifestations/the effector phase. The allergen is quickly attached to Igs E, which, in turn, are attached to mast cell FcƐRI receptors, with the receptors change in configuration and in the oscillation of constituent atoms (and subatomic components). The consequence is the activation of the cytoskeleton (actin and microtubule networks), with degranulation and release of very large amounts of pro-inflammatory mediators [29].
Bronchial mast cells are arranged in two layers: a superficial one, among the epithelial cells (actually in direct contact with the allergen), and a deep layer, under the bronchial epithelium basement membrane [30].
Upon degranulation, superficial mast cells release proteases that break the junctions between epithelial cells, allowing the allergen to penetrate deeply and induce mast cell degranulation at this level as well. Thus, pre- and neo-formed/de novo synthesized mediators are released from both superficial and deep mast cells [31].
Preformed mediators: histamine, acid hydrolases, proteolytic enzymes (tryptase, chymase), proteoglycans (heparin. serglycin), and certain cytokines (like TNF-α) [31,32].
Neo-formed mediators: PAF (platelet activating factor), TXA2 (thromboxane A2), LTB4 (leukotriene B4), 5-HETE (5 hydroxyeicosatetraenoic acid), SRS-A (slow-reacting substance of anaphylaxis, consisting of leukotrienes LTC4, D4 and E4), cytokines (TNF, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-11, IL-12, IL-13, IL-16, IL-33, IFNγ), chemokines and chemokine ligands (CCL1–CCL4, CCL8, CCL9, CXCL2, CXCL8, and XCL1) [31,32,33,34,35]
Depending on their actions, pre- and neo-formed mediators can be grouped into bronchospastic and chemoattractant mediators [36].
a. Bronchospastic mediators: histamine, SRS-A, PAF, TXA2, PG E2, PG F2. They attach to GPCR-type receptors (G protein-coupled receptors), to the membranes of bronchial smooth muscle cells, and use cGMP as a second messenger [37].
• For eosinophils: histamine, PAF, LT B4, 5 HETE, IL 5, PG E2 and PG F2.
Eosinophils attracted to the bronchial wall release, in turn:
- acid hydrolases and ROS, which amplify the inflammatory process;
-MBP (major basic protein), which exerts two unfavorable effects: it blocks the movements of bronchial cilia (mucus accumulates and forms plugs) and favors desquamation of bronchial epithelial cells, with the amplification of obstruction.
• For neutrophils: LT B4, PG D2, PG E2. Attracted neutrophils activate and release lytic enzymes, reactive oxygen and nitrogen species (ROS, RNS) in the bronchial wall, thereby amplifying inflammatory phenomena.
• For platelets: PAF, TXA2. Locally attracted platelets meet the endothelium modified by inflammatory cytokines and get activated, triggering hemostasis and partial obstruction of bronchial vessels (micronecrosis).
In addition, the growth factors TGF-β (transforming growth factor beta) and FGF (fibroblast growth factor) are also released from activated resident macrophages, with stimulation of fibroblasts and the onset of bronchial fibrosis.
Conclusions:
1. The mechanisms of bronchial obstruction have approximately the same pattern as in COPD, but, unlike COPD, they disappear between attacks (reversible bronchial obstruction).
2. As in COPD, both mast cells and neutrophils as well as macrophages are activated, but the activation of the latter does not have fully elucidated mechanisms in BA.
3. Both mast cells and activated macrophages release various cytokines, including IL-3 and IL-5, which further stimulate mast cell degranulation, but also complement activation, with intrabronchial discharge of anaphylatoxins C3a, C5a and C4a (with a secondary role), which trigger inflammation and bronchospasm by direct mechanism.
- B. Abnormalities of the autonomic nervous system (ANS) that innervates the bronchial territory.
They entail:
- hereditary decrease in the number of β adrenergic receptors (through which bronchodilation is induced);
- hereditary increase in the number of α adrenergic receptors (bronchoconstriction);
- hereditary predominance of bronchial parasympathetic activity;
- increased reactivity of irritation receptors, in connection with cholinergic fibers;
- abnormalities in the activity of non-adrenergic non-cholinergic bronchial nerve fibers (NANC). Under normal conditions, these fibers release both bronchodilator compounds (active vascular-intestinal peptide, VIP, and nitric oxide, NO) and bronchoconstrictors (tachykinins, such as neurokinins A and B, substance P, calcitonin).
In non-allergic BA, people with a hereditary predisposition show an excess of bronchoconstrictor activity. Moreover, in these people, the hypothesis of the existence of a pathological, exacerbated, variant of the inflammatory response is also advanced [41,42].
Involvement of PRF pathophysiological mechanisms in bronchial asthma
● V/Q mismatch occurs in mild and moderate BA.
The uneven ventilation distribution is explained by the fact that the inflammatory reaction is not so strong that all the bronchioles are obstructed by mucus, and the rupture of the alveolar septa does not uniformly affect the entire lung.
The uneven perfusion distribution is explained by the reflex vasoconstriction in poorly ventilated areas.
V/Q ratio mismatch is the primary mechanism of PRF onset in early or mild and moderate BA.
• In severe BA and in the elderly, generalized alveolar hypoventilation is present. In addition, bronchial hypertrophy and fibrosis occur, through increased and prolonged releases of FGF and TGFβ. Hypoxemia and hypercapnia, i.e. global PRF, also occurs.
A secondary role is played by the decrease in O2 diffusion through ACM (indirect mechanism: decrease in the exchange surface and lung contact time) and the intrapulmonary vascular shunt, secondary to the increase in pressure in the pulmonary arterioles (mechanisms identical to COPD).
3. Pathophysiological Details in COPD and BA—Similarities and Differences Regarding:
- EFFECTOR CELLS
- MOLECULAR MECHANISMS
- MORPHO-FUNCTIONAL CONSEQUENCES OF INFLAMMATION
- QUANTUM IMPLICATIONS OF MOLECULAR MECHANISMS
From a clinical point of view, there is a wide phenotypic variety of COPD cases, some of which present predominantly small airway pathology, while others are characterized mainly by the alveolar space enlargement and the emphysematous component.
In contrast to asthma, in COPD, inflammation is mainly located in the small airways and lung parenchyma, being associated with systemic inflammation. The main cause of the airways obstruction is their narrowing, first by inflammation, then by the fibrosis and collapse caused by the rupture of elastic fibers. The reversible cholinergic contraction of small airways is added. The obstruction is also maintained by mucus hypersecretion, amplified by ciliary dysfunction [43,44].
The airway obstruction in COPD is usually progressive, unlike in asthma, where it basically does not progress. Even in mild forms, there is bronchial obstruction and atelectasis, but the obstruction precedes the onset of emphysema. Longitudinal studies on COPD demonstrate that 50% of cases show an accelerated decline in lung function, while the rest evolve within age limits, but in the latter, the onset of the pathology is either in the intrauterine period or in childhood [43,44].
In COPD, the number of macrophages, T and B cells increases in the bronchial wall and lung parenchyma, so there are both innate and adaptive immunity mechanisms [43,44]. The same pattern of response is observed in smokers without the airway limitation, but it is amplified in COPD, without the mechanism to be fully understood [45].
On the other hand, most patients with bronchial asthma have an atopic terrain and present an inflammatory pattern of an allergic type, extended from the trachea to the peripheral airways. This allergic type of inflammation is induced by Th2 cells, which secretes IL-4, IL-5 and IL-13. That is why it is called T2-high asthma [46]. Other asthmatic patients do not have this type of inflammation, being called non-T2/low-T2, these forms being more severe [47]. The mentioned heterogeneity of asthma is now widespread, but it is very difficult to establish a clear link between molecular mechanisms (endotype) and clinical phenotypes [48].
In asthma, airway stenosis occurs primarily through contraction of bronchial smooth muscle and, secondarily, through edema generated by increased permeability and bronchial vasodilation. In addition, there are structural abnormalities, such as the smooth muscle hypertrophy and fibrosis, which contribute to the irreversibility of changes in the bronchial wall [44,48]. Plugs of viscous mucus, rich in glycoproteins and plasma proteins, can completely obstruct the airway. Inflammation causes not only the narrowing of the bronchial lumen, but also the hyperreactivity of the wall, the defining asthma phenomenon [49]. The hyperreactivity molecular mechanisms are not fully elucidated, but certainly incriminate: the release of mediators from inflammatory cells (mainly mast cells), increased smooth muscle contractility and sensitivity of bronchial nerve endings to pre-existing airway narrowing, through geometric effects [50].
Even though asthma is relatively easy to control with appropriate medication, approximately 5% of patients do not respond to treatment, being considered to have severe forms [51]. In these situations, several inflammatory response subtypes have been identified, which can benefit from personalized medication.
- -
- EFFECTOR CELLS IN COPD AND BA
In both pathologies, not only innate immunity cells (neutrophils, macrophages, eosinophils, mast cells, NK lymphocytes, γδ T lymphocytes, ILC – innate lymphoid cells, dendritic cells), but also adaptive immunity cells (T and B cells) participate. To them, epithelial cells, endothelial cells and fibroblasts are added, which are also sources of inflammatory mediators.
Mast cells
- They are the most important effector cells in BA, through the release of bronchoconstrictor mediators: histamine, SRS-A (LT C4, D4 and E4, also known as cysteinyl-leukotrienes) and PG D2 [52]. In allergic asthma, mast cells degranulate after the allergen is bound to IgE, which in turn binds to membrane receptors FcεR1. Another cause of degranulation is osmolarity changes secondary to hyperventilation. Apart from the classical chemoattractants that have already been presented, mast cells are directly recruited to the surface of the airways by the stem cell factor (SCF), released by the epithelial cells. SCF binds to mast cell c-Kit receptors [53,54]. Upon activation, mast cells release IL-4, IL-5, and IL-13, as well as neurotrophins, which appear to play an important role in the late response to allergens [55]. Mast cell-specific proteases, tryptase and chymase, are also released. Tryptase has a strong pro-inflammatory action too, contributing to the onset of bronchial hyperreactivity, while chymase has a profibrotic role, activating TGFβ [56].
- On the other hand, mast cells do not seem to have a significant role in COPD, which could explain the absence of variable airway narrowing, in stark contrast to BA [57]. An increased number of mast cells has been described in patients with centrilobular emphysema, being linked to bronchial hyperresponsiveness and possibly airway fibrosis [58,59].
Macrophages
As in other tissues, lung macrophages are very heterogeneous, some being pro-inflammatory and others anti-inflammatory, having a regenerative role.
• In BA, the role of macrophages is not very clear, as it is not established whether the pro-inflammatory or anti-inflammatory phenotype acts. They can be also activated by Ig E-bound allergens, which, in turn, attach to low-affinity FcεRII (CD23) receptors on the macrophage membrane [60]. Moreover, in patients with severe forms of BA, it has been observed a decrease in the macrophage secretion of IL-10, which is an anti-inflammatory cytokine [61].
• In COPD, macrophages play the main role in inflammation, being shown to increase their number in the airways, lung parenchyma and bronchoalveolar lavage fluid. They originate in circulating monocytes, chemoattracted by the chemokines CCL2 and CXCL1 (CXC chemokine ligand 1), highly present in sputum and bronchoalveolar lavage fluid. Furthermore, the macrophages in COPD patients show a greater response to CXCL1 than the macrophages in smokers or non-smokers without COPD. It is explained by the increase in the number and activity of CXCR2 receptors, which bind to CXCL1 [57,62].
In COPD, the M1 profile of pro-inflammatory macrophages predominates, although an increase in the number of M2 macrophages, which participate in the defective remodeling of the airways and lung parenchyma, has also been identified [57,62,63]. M1 macrophages in COPD patients release the inflammatory mediators IL-1β, IL-6, TNFα, CXCL1, CXCL8, CCL2, LTB4 and ROS in significantly higher amounts compared to macrophages in normal patients. They also show increased secretion of matrix metalloproteinases MMP-2, MMP-9, MMP-12 and cathepsins K, L and S [62,63,64]. Macrophages have shown the mentioned differences even when maintained for several days in culture, being clearly different from the macrophages from normal individuals, smokers and non-smokers.
Macrophages also release CXCL9, CXCL10, and CXCL11, chemoattractants for T CD8+ cytotoxic cells and for TH1 CD4+ lymphocytes that display CXC3 membrane receptors for these chemokines [64,65,66].
Both alveolar-resident macrophages and those derived from blood monocytes have shown a reduced capacity to phagocytose bacteria, a phenomenon that could explain why approximately 50% of COPD patients have lower airways chronically colonized with Haemophilus influenzae or Streptococcus pneumonia [67,68]. This pattern has also been described in patients with severe BA [69]. Macrophages have also demonstrated a reduced ability to phagocytose apoptotic bodies (efferocytosis) [70,71].
Broncho-pulmonary dendritic cells
They are phagocytes that act as antigen-presenting cells, capturing allergens, processing them in lysosomes, obtaining epitopes, which will then be presented on MHC II molecules to TH cells. They stimulate the differentiation of TH cells into the TH2 profile, thus activating the humoral immune response (HIR). BA patients respiratory epithelial cells release the thymic stromal lymphopoietin (TSLP), a cytokine which stimulates at this level the dendritic cells to release chemoattractant cytokines for blood TH2 cells [72]. Moreover, the number of dendritic cells is increased in small airways of COPD patients, especially in severe forms [73].
Eosinophils
• They are the main effector cells of inflammation in BA, being attracted from the bloodstream by the endothelial cell adhesion molecules. Then, will be directed into the submucosa by the chemokines CCL11 (eotaxin) and CCL5 (RANTES), secreted by respiratory epithelial cells [74,75]. Once extravascular, eosinophils become activated and increase their survival time in the airways. Th2 cells and the innate lymphoid cell population ILCs (ILC2) secrete IL-5, which stimulates medullary production of eosinophils and their survival in the airways [76]. ILC2 are relatively recently discovered innate immune cells, being derived from lymphoid progenitor cells. These cells secrete signal molecules (IL-5, IL-13), through which they participate in the control of both innate and acquired immunity [77]. ILC2 are resident cells in different tissues, predominantly in the mucous membranes. They participate in the maintenance of tissue homeostasis, morphogenesis, metabolism, repair and regeneration. These lymphoid cells are considered to play an important role in intrinsic, non-allergic BA [77].
Experimental blockage of IL-5 and specific receptors, IL-5R, has caused a massive and prolonged decrease in the number of eosinophils in blood and sputum, without diminishing bronchial hyperreactivity and symptoms of BA [78,79].
Eosinophils also release growth factors, such as TGFβ, involved in bronchial remodeling [80].
• If in BA eosinophils are the main leukocytes, their role in COPD is much less clear. However, in certain stable COPD cases, their increase in airways and bronchoalveolar lavage fluid has been shown. The presence of eosinophils in such patients indicates a better therapeutic response to bronchodilators and corticosteroids, as well as the possibility of COPD-BA coexistence. Also, the eosinophils increase in COPD is attributed to ILC2, stimulated by IL-33, a cytokine released by attacked epithelial cells [81,82].
Neutrophils
- Some patients with severe BA have shown an increase in the activated neutrophils number in the airways and sputum, a phenomenon also observed in some mild or moderate BA forms [83,84]. The mechanisms of neutrophil-induced inflammation in BA are not clear, but they appear to be related to the treatment with corticosteroids, which prolong neutrophils lifespan and decrease antibacterial resistance. Another mechanism involves TH17 lymphocytes, known to have a strong pro-inflammatory action by activating neutrophils. They secrete the A and F forms of IL-17 and have been identified in severe asthma. [85].
• On the other hand, the involvement of neutrophils in COPD inflammation is well known. The clearest evidence is the increase in the number of activated neutrophils in sputum and bronchoalveolar lavage fluid and the correlation of this number with the severity of the disease [86]. Neutrophils have even been identified in the bronchial lumen and in the lung parenchyma [87].
In smokers, tobacco has been shown to stimulate medullary granulocyte production and their survival in the respiratory tract. The mechanism appears to be initiated by the aggression exerted by smoking on bronchial epithelial cells and pulmonary macrophages, which release the granulocyte and granulocyte-macrophage colony-stimulating factors, G-CSF and GM-CSF. The use of GM-CSF blocking antibodies inhibits neutrophil-induced inflammation [88].
In patients with COPD, peribronchial endothelial cells expose an increased number of E-selectin molecules on the luminal membrane to which blood neutrophils adhere. Epithelial cells, local macrophages and activated T cells also secrete the chemoattractant factors LTB4, CXCL1, CXCL5 (ENA-78) and CXCL8, through which they attract and target neutrophils [89]. Even neutrophils already in the bronchial wall are an important source of CXCL8, attracting other neutrophils from the blood [89]. So, COPD patients neutrophils show aberrant, greatly exacerbated responses to chemoattractant factors [89].
Once inside the inflammatory foci, neutrophils are activated and secrete myeloperoxidase, serine proteases (elastase, cathepsin G, proteinase 3, matrix metalloproteinases MMP-8 and MMP-9), which contribute to alveolar destruction [87,88,89]. Cathepsin G and proteinase 3 strongly stimulate goblet cells and submucosal glands to secrete mucus [91]. Moreover, activated neutrophils secrete lipocalins, proteins involved in immune responses, transport of pheromones and retinoids, synthesis of prostaglandins and biological information exchange between malignant cells [90].
T Lymphocytes
• In patients with BA, the TH2 profile predominates (especially Th2), in contrast to normal individuals, whose airways contain mostly TH1 [92]. The cytokines secreted by Th2 cells (predominantly IL-5) recruit eosinophils and mast cells and help them survive in the airways. They also stimulate Ig E synthesis (IL-4 and IL-13) [55]. T reg cells (regulatory/suppressor T lymphocytes) suppress Th2 effects, but they are low in BA [92,93].
Innate lymphoid cells (ILC2) are controlled by the epithelial cytokines IL-25, IL-33 and TSLP (thymic stromal lymphopoietin) and appear to have an important function in intrinsic and severe BA [77,94].
TH17 cells have already been mentioned, being a lymphocyte population that increases in number in patients with severe asthma and stimulates the CXCL8 release from epithelial cells. This chemokine, in turn, attracts neutrophils to the airways and lung parenchyma [85,89].
A distinct population of TH is that of TH9 cells, (IL-9-secreting lymphocytes), which are present in great number in BA and stimulate T cells survival in the airways [95,96]. At this level, they also activate T cells, B cells and macrophages [105].
• In COPD, there is clear evidence of increased T cells number in the airways and lung parenchyma, predominantly CD8+ cytotoxic lymphocytes (TcCD8+) [97]. The likely explanation is the transformation of bacterially colonized epithelial cells in the lower respiratory tract into target cells for cytotoxic lymphocytes [97,98]. A clear argument is also the increase in the number of TH1, which activates TcCD8+, and of TH17 (secretors of IL-17A and IL-22), which activates neutrophils [99]. T cells number is correlated with the level of alveolar destruction and the severity of airway obstruction. Both TH CD4+ and Tc CD8+ lymphocytes strongly express the CXCR3 receptor for the chemokines CXCL9, CXCL10, and CXCL11, which are highly present in COPD [99,100].
In patients with emphysema, alveolar cell apoptosis is induced by mediators secreted by TcCD8+ cells (perforins, granzymes and TNFα) [97,101]. Another interesting phenomenon discovered in COPD patients is the immunosenescence of some Tc cells, which no longer express the stimulatory co-receptor CD28 (CD28 null T cells), but show an increased capacity to release perforins and granzymes [102,103].
Lymphoid cells are also increased in COPD, especially ILC3, which are innate immunity cells functionally equivalent to TH17. They too secrete IL-17 and IL-22, activating neutrophilic inflammation [104,105].
B Lymphocytes
They are the Ig E-secreting cells, following stimulation with IL-4, IL-5 and IL-13 and have been identified in both allergic and non-allergic BA [107,108]. Moreover, patients with severe forms of COPD show an increased number of B lymphocytes in the peripheral airways and in the lung parenchyma [109].
Autoimmunity mechanisms are present in both BA and COPD, triggered by the lung tissue damage and deficient T reg cells function [110,111]. Thus, autoantibodies have been identified in non-allergic BA, but their function is unclear [112]. In smokers, cellular and interstitial lung lesions occur that antigenically alter structures at this level. Furthermore, oxidative stress generates carbonylated and citrullinated proteins, against which autoantibodies are synthesized. They have been identified together with anti-endothelial antibodies in the serum of patients with COPD, especially in severe forms [109]. In all these situations, immune complexes activate complement via the classical pathway, as evidenced by the increased intrapulmonary presence of complement factors in patients with COPD [113].
Structural cells
In both AB and COPD, epithelial and endothelial cells, fibroblasts, and bronchial smooth muscle cells can secrete inflammatory mediators and cytokines when they are attacked and/or activated in an inflammatory focus [70,71].
- -
- MOLECULAR MECHANISMS
All the mentioned cell types release a great diversity of inflammatory mediators, many of them having the role of signaling molecules too. In order to remain oriented in this molecular variety, it is useful to understand that molecular actions are redundant (induce the same response at various sites of action), “tune in”, becoming coherent through compatible electromagnetic emission, in terms of both local agglomeration and their effects. This statement is scientifically correct because it has long been known that any molecule emits an electromagnetic field [114]. It is also essential to know that all these molecules associate the interfacial water layer, which is in fact the ordering and functional coherence factor, through the generated electron and proton fields [115,116,117]. If in a short-term inflammation, the cellular and molecular agglomeration can be functionally coherent for the purpose of effective local and general defense of the body, in chronic airway pathology and especially in COPD, these new functional balances, obtained for “damage conditions”, are short-lived, break easily, becoming disease generators themselves, through all kinds of vicious signaling circles.
Lipid mediators
Prostaglandins and leukotrienes released in the two pathologies trigger bronchoconstriction, increased vascular permeability and chemoattractant effect, as it has already been presented. Currently, their differentiated actions in COPD, compared to BA, are not clear or systematized.
• In BA, TXA2 and SRS-A (LTC4, D4, E4) predominate. LTD4 has a strong bronchoconstrictor effect, but the administration of receptor antagonists is less effective than β sympathomimetics, which indicates the existence of the additive action of other bronchoconstrictors [118,119]. Prostaglandin D2 (PGD2) has a strong bronchoconstrictor effect too, being also chemoattractant for Th2 lymphocytes, eosinophils and mast cells by binding to DP2R receptors [120].
• In COPD, lipid mediators have a different profile than in asthma. A significant increase in PGE2, PGF2α and LTB4 has been identified [121]. LTB4 has a strong neutrophil chemoattractant effect and, more recently, T cells chemoattraction has also been proved [122].
Cytokines
They have an essential role in the synergistic unfolding of inflammation mechanisms, in both COPD and BA.
• In BA, the cytokines released by activated Th2 cells mediate allergic inflammation, while the “classical” pro-inflammatory cytokines TNFα and IL-1β increase significantly in more severe forms [123]. The use of anti-IL-5 or anti-IL-13 blocking antibodies has been associated with clinical benefit. TLSP is a cytokine released in asthma by epithelial cells and in turn stimulates LTh2 chemokine release [94]. Th17 lymphocytes releases IL-17A/F and IL-22, which stimulate neutrophilic inflammation in more severe forms of BA [124]. On the other hand, the anti-inflammatory cytokine IL-10 is deficient in asthma. TH9 lymphocytes produce IL-9, who stimulates T cells, B cells and macrophages and may promote inflammation by activating Th2 cells and by increasing mast cell accumulation [125]. This interleukin also activates Arg1+ interstitial macrophages, which secrete the chemokine CCL5. This attracts eosinophils, T cells, and blood monocytes to the lungs, triggering type 2 inflammation [106]
• In COPD, the TNFα level increases, mainly through its production in macrophages. Through the initiated transduction cascade, TNFα strongly activates the transcription factor NF-κB, which, among actions, stimulates gene transcription for other proinflammatory cytokines, including IL-6 [126]. It is also responsible for the occurrence of cachexia in some severe and advanced forms, inducing apoptosis in striated muscle cells, by stimulating TNFR death receptors. Anti-TNF therapies have not proven to be very effective in COPD because of their strong side effects [127,128].
Furthermore, the other prototypical inflammatory cytokines, IL-1β and IL-6, have demonstrated significant elevation in the serum and sputum of COPD patients. IL-17A and IL-22, produced by LTh17 and ILC3 innate lymphoid cells, also show increase in the airways. The alarmins IL-33 and TLSP are produced in increased concentrations by epithelial cells and stimulate ILCs [128].
Chemokines
They play an important role in both COPD and BA, signaling through G protein-coupled receptors (GPCRs).
• In BA, epithelial cells secrete CCL11, a chemoattractant for eosinophils, which exerts its actions through CCR3. In parallel, dendritic cells secrete CCL17 and CCL22, chemoattractants for Th2 cells, to whose membranes they attach via CCR4 receptors [15,19,66,129].
• In COPD, macrophages, neutrophils, T cells that have already entered the airways, as well as epithelial cells secrete the chemokine CCL8, a chemoattractant for neutrophils, by binding with high affinity to their CXCR2 receptors. CCL1 and CCL5 also act on these receptors, inducing the neutrophils and monocytes chemoattraction. Moreover, in patients with COPD, monocyte chemoattraction determined by CCL1 is significantly stronger than in normal smokers (without COPD), a phenomenon explained by the increase in the number of CCR2 receptors on these patients monocytes membranes. Also in COPD, CCL5 activates CCR5 receptors on T cells and CCR3 receptors on eosinophils, causing chemoattraction of these cells to the airway wall. In addition, the chemokines CXCL9, CXCL10 and CXCL11 secreted in COPD cause chemoattraction of monocytes and TH1 and Tc lymphocytes, acting on their CXCR3 receptors [15,19,66,129].
Reactive oxygen species
They are involved in important signaling mechanisms in COPD and BA, their local and systemic concentration being increased in these pathologies. They come from both an external source (cigarette smoke and polluted air) and from endogenous production, secondary to the activation of macrophages, neutrophils and eosinophils. In patients with BA, increases in 8-isoprostane (arachidonic acid oxidation compound) and ethane (lipid peroxidation product) levels have been recorded in exhaled air, both compounds resulting from ROS attack on membrane lipids [130]. Moreover, a direct correlation has been established between ROS levels and disease severity, both in COPD and BA, by amplifying the inflammatory reaction (secondary to the transcription factor NF-kB activation) and reducing the response to anti-inflammatory therapy with corticosteroids [131].
In patients with COPD, a decrease in antioxidant levels and antiprotease activity (such as that of α1-antitrypsin) has also been associated, a phenomenon that accelerates elastin degradation in the lung parenchyma [132]. ROS also reduce the expression and activity of sirtuin-1, a key reparative molecule with an anti-aging role, the cellular aging phenomenon being accelerated in COPD [133].
Proteases
They are present in any inflammatory reaction, therefore in COPD and BA too. Among the first to be released is mast cell tryptase, which is also thought to play a role in bronchial hyperreactivity in asthma. In COPD, elastases are major players in the development of emphysema. By degrading elastin, elastases generate intermediate peptide compounds, called matrikines, with a chemoattractant effect for neutrophils by binding to their CXCR2 receptors [134]. In a vicious circle, activated neutrophils release matrix metalloproteinases, such as MMP-9, which additionally generate matrikines and degrade elastin. MMP-9 is the predominant elastase in COPD, being secreted by macrophages, neutrophils and epithelial cells [134]. Neutrophil elastase has also been identified as a potent stimulator of bronchial mucus secretion, by binding to epidermal growth factor receptors, EGFR [135].
ATP
Adenosine triphosphate (ATP) exerts both intracellular and extracellular effects. If in the cell the ATP molecule plays an essential role in energy metabolism, in the extracellular environment it attaches to purinergic receptors and aggravates airway inflammation. One of the identified mechanisms is binding to mast cell P2Y2 receptors, followed by degranulation [136]. It is also a potent activator of afferent nerve fibers in the airways through the use of P2X3 receptors. Blockers of these receptors have a very effective antitussive action [137].
Secretory phenotype associated with cellular senescence
Relatively recently, markers of cellular senescence have been identified in the airway smooth muscle cells of elderly patients with BA: the presence of oxidative stress and chronic inflammation, telomere shortening, autophagy/mitophagy [138,139].
There is also an increasing evidence that a lung aging process occurs in COPD, manifested by the accumulation of senescent alveolar epithelial, endothelial and fibroblast cells. These cells release inflammatory compounds having a particular profile: TNF-α, IL-β, IL-6, CCL2, CXCL1, CXCL8, TGF-β, MMP-9 and ROS, known as the senescence-associated secretory phenotype, whereby cellular senescence is amplified and disseminated [138]. In patients with COPD, these compounds have increased levels, both pulmonary and systemic, considered to be involved in the occurrence of comorbidities (cardiovascular disease, type 2 diabetes and chronic kidney disease), which accelerate the process of cellular aging. The senescence-associated secretory phenotype is transmitted from one cell to another via extracellular vesicles [139,140].
Systemic inflammation and comorbidities
• In BA, inflammation is limited to the airway mucosa, with little evidence of systemic inflammation. Comorbidities are usually the other allergic manifestations (allergic rhinitis and atopic dermatitis), as well as gastroesophageal reflux, secondary to bronchodilator therapy, which relaxes the gastroesophageal sphincter [141,142]. Another comorbidity is obesity, which may decrease the therapeutic response in patients with asthma. The adipokines secreted by adipose tissue amplify the inflammatory response. In addition, obese asthmatic patients show an alteration of the gut microbiota, which produces short-chain fatty acids and other compounds that activate immune cells [142].
• Unlike in BA, systemic inflammation is present in COPD patients with severe forms or during exacerbations, being manifested by increases in the cytokines, chemokines and acute phase proteins blood levels, as well as by significant changes in the blood count. The presence of systemic inflammation is associated with a poor prognosis in COPD [143]. However, it cannot be said with certainty whether the systemic inflammatory markers originate from the pulmonary inflammatory foci, reflect a real systemic inflammation or are generated by comorbidities. In a large population-based study, the presence of systemic inflammation (increased C-reactive protein, fibrinogen, and leukocytes) has been associated with a 2- to 4-fold increased risk of COPD-associated cardiovascular disease, diabetes, pneumonia, and lung cancer. 70% of COPD patients have shown increases in serum concentrations of CRP, IL-6, CXCL8, fibrinogen, TNFα and leukocyte count, while in 16% of them inflammation has been persistent and associated with more exacerbations and increase in mortality [144,145].
- -
- INFLAMMATION MORPHO-FUNCTIONAL CONSEQUENCES
Changes in bronchial smooth muscle
- In patients with COPD, it has not been highlighted a significant increase in the bronchial wall muscle mass, probably because, among the released inflammatory mediators, there are not sufficient concentrations of the specific growth factors [57].
- On the other hand, in BA, bronchoconstriction is the main mechanism of the bronchial lumen narrowing and it is induced mainly by mast cell mediators. Bronchoconstriction is associated with the bronchial smooth muscle hypertrophy and hyperplasia, stimulated by growth factors such as PDGF and endothelin-1, released by inflammatory cells and epithelial cells. In turn, smooth muscle cells also release inflammatory mediators, which contribute to the lumen narrowing [145].
Changes in nutrient vasculature
In COPD, there is a decrease in airway vasculature, probably through reduced secretion of the vascular endothelial growth factor, VEGF [57]. On the other hand, in BA, the blood flow in the bronchial mucosa vessels is increased, inducing the lumen narrowing. The growth of the peribronchial vascular network, secondary to VEGF stimulation, also contributes to this phenomenon. Moreover, in BA, permeability is increased in the post-capillary venules, generating airway edema and inflammatory exudate [145,146].
Mucus hypersecretion
It is a pathophysiological mechanism present in both COPD and BA, which induces the airway obstruction. It is accompanied by mucociliary function alteration, submucosal glands hyperplasia in the large bronchi and an increase in the goblet epithelial cells number. The result is the presence of viscous mucus plugs that, in fatal forms of BA, can even completely obstruct the airways. IL-13 is a potent stimulator of mucus secretion and it is antagonized by corticosteroids [146]. Another pathophysiological mechanism induced in inflammation starts from excess ROS and TGFα, which stimulates the EGFR receptor, triggering signals completed by mucin gene expression [146,147].
Moreover, neutrophils release proteases that strongly stimulate mucus secretion, a phenomenon that is confirmed by infiltration of the bronchial wall with neutrophils in patients with COPD and severe forms of BA [148].
Inflammation effects on neural regulatory mechanisms
Inflammation plays an important role in the onset of bronchoconstriction and the cough reflex.
- Effects of inflammation on the cough reflex:
Sensitive nerve endings are activated for a long time by inflammatory mediators, becoming hyperreactive. Thus, prostaglandins, leukotrienes, ATP and low pH activate transient receptor potential (TRP) channels, a type of ion channels in the structure of sensory endings, triggering the cough reflex, in both BA and COPD [57,145]. An indirect mechanism is added, generated by neurotrophins, in particular by NGF (nerve growth factor), which are released from the bronchial epithelial cells and mast cells of patients with BA. This mechanism induces an increase in the number and sensitivity of the airways nerve endings [149]. The role of neurotrophins in COPD is not clear, although the studies conducted in our department on neurotrophin-3 behavior in COPD patients have shown a significant increase in serum concentrations compared to healthy individuals [150].
- Effects of inflammation on bronchoconstriction:
The cholinergic mechanism triggering, which involves the acetylcholine release, bronchial muscarinic receptors stimulation and bronchoconstriction occurrence, can be achieved neuronally or non-neuronally [151].
- Inflammatory mediators stimulate sensitive nerve endings, with the initiation of a motor reflex, manifested by bronchoconstriction, mediated by either cholinergic mechanisms or neuropeptide release. In addition, inflammatory mediators induce an increase in the number of parasympathetic ganglia in the airways. In turn, the neuropeptides (primarily, substance P) released by the sensitive nerves stimulated in bronchial inflammation also have a pro-inflammatory role, generating a self-maintenance mechanism of the inflammatory reaction. However, the role of neurogenic inflammation in COPD and BA has not yet been clearly proven (the administration of inhibitors of substance P and other neurokinins has not determined a significant clinical benefit) [152,153].
- -
- Bronchoconstriction can be also triggered by a non-neuronal mechanism, which involves the release of acetylcholine from bronchial epithelial cells and inflammatory cells, this being an important inducer of bronchial spasm in the peripheral area of the bronchial trees, where cholinergic innervation is very reduced [151].
Therefore, anticholinergic medication is effective in both neuronal and non-neuronal mechanism. In COPD, the use of this type of medication is of great importance, because bronchial cholinergic hypertonia is the only reversible mechanism of the bronchial lumen reduction. On the other hand, the β sympathomimetic medication has a much more effective bronchodilator effect in BA than the anticholinergic one, because it also antagonizes the bronchoconstrictor effects of prostaglandins and leukotrienes [152,153].
Bronchial fibrosis
It is present in both COPD and BA, representing an aberrant reparative response induced by prolonged inflammatory aggression on the bronchial epithelium.
- In all asthmatic patients, the basement membrane shows apparent thickening due to subepithelial fibrosis (deposition of type III and IV collagen fibers) and eosinophil aggregation beneath the epithelial basement membrane [153,154]. The most likely molecular substrate of these changes is the release of TGFβ from the epithelial and inflammatory cells activated by repeated bronchoconstriction-induced mechanical stress. As BA worsens, fibrosis extends beyond the space beneath the basement membrane and encompasses the entire bronchial wall, presumably through the phenotypic transformation of bronchial smooth muscle cells into myofibroblasts [153]
- In COPD, peribronchiolar fibrosis is an important factor in disease progression. The molecular mechanism is also initiated by fibrogenic inflammatory mediators, released by epithelial cells, and activated macrophages: TGFβ, connective tissue growth factor (CTGF) and endothelin. An important histopathological observation is that fibrosis of the small airways is an early change in the development of COPD, preceding the onset of emphysema. As long as the inflammatory mechanisms can be inhibited, the airway narrowing is reversible [153,154,155].
The resolution of inflammation is an active and intense process, for which the body is endowed with endogenous mechanisms, more recently discovered, initiated by lipoxins, resolvins E, protectins D and maresins [155]. All these molecules are derivatives of polyunsaturated fatty acids and act on specific phagocytic receptors, with the resolution of phagocyte-induced inflammation. The compounds listed above inhibit the recruitment of blood neutrophils into tissues and enhance the efferocytosis of apoptotic neutrophils. Maresin-1 is the most powerful known activator of efferocytosis performed by macrophages (efferocytosis is the process of removing apoptotic bodies and involves mechanisms similar to those of phagocytosis) [155]. The phagocytes deficiency in the phagocytosis and efferocytosis favors the maintenance of bronchial and peribronchial inflammation. The phenomenon is confirmed in patients with COPD [153,154,155].
- -
- QUANTUM IMPLICATIONS OF MOLECULAR MECHANISMS
- -
- A. Quantum behavior of reactive oxygen species and other inflammation mediators
The discussion in this context must necessarily be preceded by the presentation and understanding of the special properties of water, which is a polar molecule, having an electric dipole, where the positive and negative charges are separated by a "length" of the dipole [156]. This configuration makes it possible to establish bonds between several water molecules, more precisely between the positive region of one molecule (hydrogen atoms) and the negative region of another (oxygen). This process also continues with other ions, so that any positive or negative ion can attract and is attracted to the oxygen or the protons in a water molecule. The phenomenon is called hydration [156]. Water has a surface tension at any interface, including the one with atmospheric air, generated by dipolar bonds between molecules. This type of polymerization gives the formed water clusters crystal-like properties, i.e. symmetry and order. Water clusters form hydration layers around molecules. Thus, in biological systems liquid water interacts not only with small solutes but also with large hydrophilic or hydrophobic surfaces, such as proteins, nucleic acids, various organelles or cell membranes [156]. Incoherent inelastic neutron scattering studies of several types of cells and tissues suggest that 20–30% of the total water (intra and extracellular) of these systems is interfacial water, i.e. water located in a 1–4 nm (approx.) layer adjacent to the cell surface [156,157,158,159]. The remaining 70-80% is "unstructured" bulk water. At the nanoscale, the interfacial water adjacent to hydrophilic surfaces has a 2-106 times higher viscosity than bulk water, while water in the vicinity of hydrophobic surfaces has no significant changes in viscosity [156,157,158,159]. Moreover, water located more than 1 nm from hydrophobic surfaces has fewer hydrogen bonds and behaves more like a liquid-vapor interface water than a “bulk” water [156,159].
Therefore, we can state that the water around a hydrophilic surface is organized in three concentric layers: interfacial water, semi-structured water and "bulk" water [156,157,158,159].
However, biological water can be considered interfacial (structured) water, due to the fact that there is almost no point in the body placed at a distance much greater than a fraction of a micron from a surface [159].
Structured/interfacial water has various interactions with biological systems as follows [156,157,158,159]:
- it increases the biological interfaces electrical conductivity, facilitating metabolism and maintaining the differences in electrical potential at the level of cellular organelles;
- it absorbs, stores and emits electromagnetic energy, phenomena that allow the storage and transmission of energy and biological information. However, it has long been known that any molecule emits an electromagnetic field;
- it has a role in thermolysis;
- it solves the problems of intracellular crowding and molecular self-assembly, through chirality and magnetization.
The exogenous stress exerted on the interfacial water can destroy its structure, initiating a series of events in the intra- and extracellular spaces, leading to neurological dysfunctions and diseases, infectious diseases, malignancies and death. We can consider that this is the level at which the disease is triggered [156,157,158,159]. In other words, destructive changes first occur in the aqueous interface, the volume in the vicinity of the surface of a biological macromolecule [156]. At this level, the structure and properties of water are different from those of unstructured/bulk liquid water, as we have already discussed ("Interface" means the surface area between two phases, while "interphase" is the volume of the narrow region between two phases).
The studies carried out proved that the interfacial water has a glassy, semi-crystalline appearance, being considered a different phase from ordinary liquid water. That's why it was called Pollack's Fourth Phase. It is essentially characterized by the coherence of the electromagnetic field emitted by each molecule (quantum coherence) [156].
Electron transfer through the interfacial water is essential for all cellular energy metabolism associated events [157,158]. Anyone can easily imagine the material transfer of electrons in a chemical environment. However, quantum physics offers us a much more subtle and profound mechanism: the non-material transfer of electrons/particles.
The simplified diagram below (Figure 1) demonstrates the non-material transfer of electrons from interfacial water, a phenomenon called electron quantum condensation [158]. Without requiring the reader to have a deep understanding of these quantum electrochemistry reactions, it is useful to note the ongoing generation of hydroxyl and superoxide radical species, ionic radical forms of ROS:
Electron quantum condensation occurs in two stages and it is the result of the excitation of the interfacial water coherent phase, excited water that subsequently develops a phase instability.
In the first stage, the almost free electrons generated in the energetically excited interfacial water molecules emit an electromagnetic vortex field whose energy, in turn, excites the interfacial water molecules. This excitation energy generates not only molecular coherence but also an energy surplus, a phase instability [158,159,160].
In the second stage, there is quantum condensation (attraction) into the vortex of some additional electrons from the environment, which are given the excitation energy and which themselves become emitters of the energy given to other atoms, which become radical species. Following the reactions in Figure 1, we observe how the quantum condensation of electrons is associated with the formation of molecular hydrogen, hydroxyl and superoxide anions, serving as a source of ionic radical molecules in living organisms [158,159,160].
Enzymes containing metals of variable valence (i.e. all heme-containing enzymes) achieve “modulation” of the functions of their associated interfacial water. First, the stage of growth of the coherent phase occurs, i.e. the increase in the energy of the electrons in the enzyme-metal complexes and in the associated water. Then, phase instability is generated, which is accompanied by the electrons condensation in the quantum-connected enzymatic sites, with the release of their energy to free radicals formed in the associated water. In other words, the heme-proteins are molecules that catalyze radical reactions, being able to induce the balancing (coupling) of the opposite spin of different radical species [158,159,160,161,162]. This mode of action is specific to antioxidant enzymes (catalase and peroxidase) as well as to oxygen transporters (hemoglobin and myoglobin) and cytochromes in the mitochondrial respiratory chain.
How do free radicals influence chemical bonds formation?
Reactive oxygen species, being energetically excited, have a short life span and show a tendency to recombine, with the generation of radical pairs.
To form, radical pairs require for their electron spin to be correlated, meaning that molecules must have opposite spin. Under normal conditions, the thermal and enzymatic reactions that produce free radicals in biological systems involve the energetically stable triplet states of oxygen molecules. The electrons in chemical bonds that break homolytically (equally) to form free radicals have antiparallel spin, as do the free radicals themselves. Since the spin must be antiparallel for bond formation, it is expected that the newly released free radicals with antiparallel spin to immediately recombine. However, the energy released from the reaction in the environment causes their rapid separation again, so that the definitive combination of the radicals in pairs occurs rather rarely [158,159,160,161,162].
Much more important in terms of consequences is the magnetic interaction between the electron spin of the radical pair and the spin of the nuclei of neighboring hydrogen and oxygen atoms. As a result of this interaction, the spin state of the oxygen electrons of the radical pair changes, which now acquires a singlet character (energetically excited, unstable state) [158].
The fact that chemical bonds are formed between free radicals with electrons of opposite spin does not mean that the homolytic breaking of these bonds automatically implies rapid recombination of the resulting radicals. Some free radicals do not recombine immediately and, because the energy released from the reaction diffuses into the immediate vicinity, it induces the threshold energy value to be exceeded for the formation of another chemical bond. In this way, reactive oxygen species modulate biochemical reactions in the environment [158,159,160,161,162].
Conclusions:
1. Biochemical reactions do not occur instantaneously and randomly, but depend on exceeding a threshold of free energy of activation and satisfying steric requirements (i.e. a reaction can only occur if the reactants, free radicals or other atomic and molecular species, approach each other in a certain direction) [158,159,160,161,162]. The directed approach between the reactants could be the result of the action of the electromagnetic vortex field associated with the electronic vortices of the free radicals.
2. It is now becoming clear that ionic ROS are very important electron acceptors of the body and that cellular metabolism is regulated through them. As a general mechanism, this regulation involves the formation and decay of collective states of ROS (a quantum-regulated phenomenon involving a large number of entangled ROS molecules simultaneously) [158,159,162,163,164]. Therefore, the biological activity and the functional orientation of cellular metabolism directly depend on the activity of primary electron receptors.
3. These phenomena occur physiologically, both at the alveolar-capillary level and in every cell of our body, and the formation of small concentrations of ionic ROS is an essential mechanism, indispensable for the coherent functioning of our cells.
What happens in COPD and BA?
The disorders associated with these pathologies are initiated by repetitive and/or persistent inflammation. This means the accumulation of a large number of inflammatory mediators and exudated water molecules precisely at the level of these facilitated quantum transfer structures, both of matter and energy-informational (physical/biological information). Each newly released inflammatory molecule associates a layer of interfacial water that will generate electron vortices and proton currents, according to the structure and electrical polarization of that molecule [158]. So, there will be generated electromagnetic emissions that have completely different spectra than those emitted under normal conditions. In addition, inflammatory cells, especially phagocytes, will release far above normal amounts of reactive oxygen and nitrogen species, which do nothing but pick up the energy of the environmental electron vortices and attract new electrons, to then transfer the energy of the vortices of excitation to other structures, foreign or specific to the body, degrading them [158,159,160,161,162,163].
Chronic lung inflammatory phenomena are associated with varying degrees of hypoxia, which becomes severe during BA attacks and in advanced forms of COPD. Under normal conditions, oxygen is the final electron acceptor for efficient energy production in the form of ATP. Thus, for the proper functioning of energy metabolism, mitochondria need oxygen to combine with the electrons from the cytochrome transport chain and with the protons generated by the proton gradient, constituting water.
When oxygen drops significantly, the electrons resulting from mitochondrial metabolic reactions can no longer be picked up and transformed, being released in excess and yielding their energy to the free radicals that emerge. Moreover, oxygen deficiency will also induce proton excess, with acidosis [158,159,160,161,162,163]. Under these conditions, hydrogen peroxide (H2O2) is converted into hydroxyl radical (-OH-•), in the presence of bivalent iron or other transition metals. Free radicals formed excessively in hypoxia and inflammation attack protein and lipid structures as well as nucleic acids in an attempt to lose their excitation energy. The result is an increase in their molecular size and a decrease in their availability for oxygen, aggravating in a vicious cycle mitochondrial, nuclear and cellular dysfunction as a whole [163,164].
In the presence of physiological concentrations of free radicals, the lipid compounds polymerize, with the formation of nano-sized solid structures, of rubbery gel type, while the peroxidized lipids behave like crystalline structures [164,165]. In the event of high concentrations of ROS, excessive lipid peroxidation is associated with severe permeability disorders of the outer membrane and organelle membranes. It results in the occurrence of a temporary or permanent damage of the hydro-electromagnetic properties of the tissues, at the level of the exclusion zones of the interfacial water [165,166,167].
All these conditions force the cell to extreme responses, such as necrosis or apoptosis.
Conclusions:
1. The synthesis of small concentrations of reactive oxygen and nitrogen species is essential for normal metabolic reactions and it is accompanied by a normal tissue electromagnetic emission;
2. The oxidative stress in inflammation and hypoxia is associated with the disruption of mitochondrial, nuclear and cell membrane functioning, lowering the tissue excitability threshold to electromagnetic fields.
- B. Quantum consequences of fibrosis
Another phenomenon generated by the inflammatory reaction, with essential pathological implications at a quantum level too, is the occurrence of bronchial and interstitial fibrosis.
As an essential histo-physiological element, we must remember that intrapulmonary connective tissue has as its elementary unit of form and energy-information exchange the spiral collagen fiber. At the intracellular level, the functional counterpart of collagen is the cytoskeleton, whose basic component is the microtubule network. Microtubules connect the nucleus to other cytosolic organelles and structures, being made up of tubulins (six known types, denoted α-ζ), proteins assembled in the same spiral form as extracellular collagen fibers [168]. As in a fractal progression, intracellular spirals of microtubules are connected by membrane junctional proteins to extracellular collagen spirals, which then join the basement membranes with interstitial collagen, then with the collagen of the capsules and fascia, of muscles, tendons and skin [167,168,169].
A continuum is formed that unites the intracellular space with the extracellular one and then with the macroscopic one, a continuum that has as a common structure element a spiral shape and as a common electrochemical and electrodynamic element the polar structure and the presence of a continuous layer of interfacial water, which functions as a facilitated transport environment [158,169,170]. All this astonishing structure, which connects the whole organism, could be functionally assimilated to a network of optical fibers that transfer all biological information in all directions at a speed close to the speed of light [170,171]. It is a fundamental direction of experimental research that should not be abandoned.
Arguments
The most interesting characteristic of collagen is hydrophilicity, i.e. the association with biological water. Most of the hydrogen bonds in the collagen structure are mediated by interfacial water. Thus, water molecules bind hydrogen between the carbonyl groups of the same chain or between different chains of the collagen triple helix, as well as between the hydroxyl groups of hydroxyproline and the carbonyl groups of the same or different chains. The number of water molecules involved in binding two groups varies along the helix. Water bridges are crucial for connecting adjacent collagen triple helices and maintaining intermolecular spaces. It is as if water “dictates” the spatial configuration of collagen [170,171,172].
The hydrogen bonding network is also present in the associated interfacial water. Each water molecule binds to four other molecules, forming a three-dimensional network that extends around the collagen [158,170,171,172]. As mentioned before, the structured water associated with collagen is highly ordered, as a single phase.
For 40-50 years, the transport of protons through collagen and the ability of this molecule to interact with electromagnetic fields, whose photons have frequencies from UV (ultraviolet) to IR (infrared), have been experimentally proven [172,173,174]. The result of the interaction will be the capture of photon energy and the excitation of valence electrons from different collagen groups, followed by their return to the stable energy phases, through the emission of new photons, with double energy. The frequency of these photons is doubled and the wavelength is halved [158].
Collagen is the most abundant protein in the body and, through its internal structural order and associated interfacial water, as well as its ability to interact with electromagnetic fields, has the behavior of a liquid crystal (liquid crystals are considered states of matter in the mesophase, i.e. in an intermediate phase between solid and liquid crystals) [158,174,175].
As the most abundant protein of the extracellular matrix and connective tissue, collagen transfers its liquid crystal behavior to the entire body. In physics, ordered, crystalline structures are known to facilitate collective, coherent behavior over short and long distances. In conclusion, we can state again that the structures containing collagen structurally and functionally cohere the whole body [174,175].
Coming back to the cell, the microtubules cytoskeletal structures are, in turn, polar, therefore hydrophilic. The water associated with these protein structures conducts electrons through quantum tunneling, from the negatively charged perinuclear centrosome to the positively charged outer surface of the cell membrane. These electrons are needed, among other things, to ensure actin polymerization, essential in all cellular events, and their main source is mitochondria [176,177]. Under hypoxia conditions, mitochondrial production of electrons increases significantly, so that actin polymerization becomes excessive, this intracellular disorder being transmitted to neighboring or distant cells through the spiral continuum of collagen fibers [177].
All these hydrophilic intracellular, membrane and extracellular materials are characterized by different densities of electrons and protons on their surfaces and they can get coherent when hydrophilic surfaces, water and the energy of an electromagnetic field come into contact, in space and time.
If, in the light of this information, we try to understand the consequences of pulmonary fibrosis, we will come to the following findings: the newly synthesized collagen fibers no longer respect the initial geometry of the lung, they are arranged anarchically and short-circuit or interrupt the normal transport pathways, not only in alveoli and capillaries, but also in interstitium, between alveoli, between acini, between bronchioles..., between the lung and the pleura, between the lung and the rest of the body.
Moreover, excess collagen “unbalances” the overall emission of the lung, by accentuating the emission of electron and proton currents from the associated structured water. We can imagine a “discordant” emission of the lung, difficult to ignore by the rest of the body, which is also faced with multiple areas more or less “discordant” because of the tissue hypoxia induced by pulmonary suffering. It is irrelevant whether these perturbations are initiated intracellularly, on the membrane, or in the interstitium. The continuum that joins the structures ensures the multidirectional propagation of “disagreement”.
It remains for the future research to establish whether the quantum disturbances of the oxygen transfer through the alveolar-capillary membrane simultaneously trigger, through “pathological coherence”, alterations of the cellular energy metabolism in the rest of the body or whether these alterations are strictly secondary, succeeding pulmonary suffering over time.
Conclusions
- All diseases of the body involve the appearance of disruptions in the mechanisms of structural and functional interrelationship between organs, tissues, cells and molecules. We have presented these disruptions for the two prototypical lung pathologies, COPD and asthma, but the approach is valid for any other disease.
- The molecular level is by no means the last at which physiological and pathological events in living organisms take place. The principles of quantum physics applied in experimental biology prove the origin of normality and disease at the level of subatomic particles, phenomena that must be understood by physicians.
- All stages of manifestation of normality and disease, from the organism and organs to subatomic particles and fields, involve overlapping phenomena, by no means excluding each other.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Declaration of interests
The author declares no competing interests.
Ethics, Consent to Participate, and Consent to Publish declarations
not applicable.
References
- Barnes, PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013, 131(3), 636–45. [Google Scholar] [CrossRef] [PubMed]
- Hodge, G.; Roscioli, E.; Jersmann, H.; et al. Steroid resistance in COPD is associated with impaired molecular chaperone Hsp90 expression by pro-inflammatory lymphocytes. Respir Res 2016, 17, 135. [Google Scholar] [CrossRef]
- Barnes, P.J. Cellular and molecular mechanisms of asthma and COPD. Clin Sci (Lond) 2017, 131(13), 1541–1558. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, SO; Cunha, CMCD; Soares, GMV; Silva, PL; Silva, AR; Gonçalves-de-Albuquerque, CF. Mechanisms, Pathophysiology and Currently Proposed Treatments of Chronic Obstructive Pulmonary Disease. Pharmaceuticals (Basel) 2021, 14(10), 979. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kraik, K; Tota, M; Laska, J; Łacwik, J; Paździerz, Ł; Sędek, Ł; Gomułka, K. The Role of Transforming Growth Factor-β (TGF-β) in Asthma and Chronic Obstructive Pulmonary Disease (COPD). Cells 2024, 13(15), 1271. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Agudelo, CW; Kumley, BK; Area-Gomez, E; Xu, Y; Dabo, AJ; Geraghty, P; Campos, M; Foronjy, R; Garcia-Arcos, I. Decreased surfactant lipids correlate with lung function in chronic obstructive pulmonary disease (COPD). PLoS One 2020, 15(2), e0228279. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Koopman, M; Posthuma, R; Vanfleteren, LEGW; Simons, SO; Franssen, FME. Lung Hyperinflation as Treatable Trait in Chronic Obstructive Pulmonary Disease: A Narrative Review. Int J Chron Obstruct Pulmon Dis. 2024, 19, 1561–1578. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Papandrinopoulou, D; Tzouda, V; Tsoukalas, G. Lung compliance and chronic obstructive pulmonary disease. Pulm Med. 2012, 2012, 542769. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Swenson, KE; Swenson, ER. Pathophysiology of Acute Respiratory Distress Syndrome and COVID-19 Lung Injury. Crit Care Clin. 2021, 37(4), 749–776. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sakao, S; Voelkel, NF; Tatsumi, K. The vascular bed in COPD: pulmonary hypertension and pulmonary vascular alterations. Eur Respir Rev 2014, 23(133), 350–5. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Raimondi Cominesi, D.; Forcione, M.; Pozzi, M.; et al. Pulmonary shunt in critical care: a practical approach with clinical scenarios. J Anesth Analg Crit Care 2024, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Chan, SY; Loscalzo, J. Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol. 2008, 44(1), 14–30. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guignabert, C.; Aman, J.; Bonnet, S.; et al. Pathology and pathobiology of pulmonary hypertension: current insights and future directions. European Respiratory Journal 2024, 2401095. [Google Scholar] [CrossRef] [PubMed]
- International consensus report on diagnosis and treatment of asthma. National Heart, Lung, and Blood Institute, National Institutes of Health. Bethesda, Maryland 20892. Publication no. European Respiratory Journal 1992, 1992 5(5), 601–641. [CrossRef]
- Habib, N; Pasha, MA; Tang, DD. Current Understanding of Asthma Pathogenesis and Biomarkers. Cells 2022, 11(17), 2764. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barnes, P.J. Immunology of asthma and chronic obstructive pulmonary disease. Nat. Immunol. Rev. 8 2008, 183–192. [Google Scholar] [CrossRef]
- Maggi, E. The TH1/TH2 paradigm in allergy. Immunotechnology 1998, Volume 3(Issue 4), 233–244. [Google Scholar] [CrossRef]
- Bujoreanu, I; Gupta, V. Anatomy, Lymph Nodes. In StatPearls [Internet]; StatPearls Publishing: Treasure Island (FL), 25 Jul 2023; Available online: https://www.ncbi.nlm.nih.gov/books/NBK557717/.
- Komlósi, Zsolt I.; van de Veen, Willem; Kovács, Nóra; Szűcs, Gergő; Sokolowska, Milena; O'Mahony, Liam; Akdis, Mübeccel; Akdis, Cezmi A. Cellular and molecular mechanisms of allergic asthma. Molecular Aspects of Medicine 2022, Volume 85, 100995. [Google Scholar] [CrossRef]
- Mehlhop, PD; van de Rijn, M; Brewer, JP; Kisselgof, AB; Geha, RS; Oettgen, HC; Martin, TR. CD40L, but not CD40, is required for allergen-induced bronchial hyperresponsiveness in mice. In Am J Respir Cell Mol Biol; DOI] [PubMed; Google Scholar, 2000; Volume 23, pp. 646–651. [Google Scholar] [CrossRef]
- Zietkowski; Skiepko, R.; Tomasiak, M. M.; Bodzenta-Lukaszyk, A. Soluble CD40 ligand and soluble P-selectin in allergic asthma patients during exercise-induced bronchoconstriction. Journal of Investigational Allergology & Clinical Immunology 2008, vol. 18(no. 4), 272–278. [Google Scholar]
- (PDF) The Role of CD40 in Allergic Rhinitis and Airway Remodelling. Available online: https://www.researchgate.net/publication/351561816_The_Role_of_CD40_in_Allergic_Rhinitis_and_Airway_Remodelling (accessed on 11 Sep 2025).
- Eckl-Dorna, J; Villazala-Merino, S; Campion, NJ; Byazrova, M; Filatov, A; Kudlay, D; Karsonova, A; Riabova, K; Khaitov, M; Karaulov, A; Niederberger-Leppin, V; Valenta, R. Tracing IgE-Producing Cells in Allergic Patients. Cells 2019, 8(9), 994. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stanworth, D.R. The role of non-antigen receptors in mast cell signalling processes. Molecular Immunology 1984, Volume 21(Issue 12), Pages 1183–1190. [Google Scholar] [CrossRef]
- Lambrecht, Bart N. The Cytokines of Asthma. Immunity, Volume 2019, Volume 50(Issue 4), 975–991. [Google Scholar] [CrossRef] [PubMed]
- Falcon, RMG; Caoili, SEC. Immunologic, genetic, and ecological interplay of factors involved in allergic diseases. Front Allergy 2023, 4, 1215616. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mukherjee, AB; Zhang, Z. Allergic asthma: influence of genetic and environmental factors. J Biol Chem. 2011, 286(38), 32883–9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Miteva, D.; Lazova, S.; Velikova, T. Genetic and Epigenetic Factors in Risk and Susceptibility for Childhood Asthma. Allergies 2023, 3, 115–133. [Google Scholar] [CrossRef]
- Dráber P., Sulimenko V. , Dráberová E., Cytoskeleton in Mast Cell Signaling, Frontiers in Immunology, Volume 3 – 2012, 2012, https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2012.00130n DOI=10.3389/fimmu.2012.00130, ISSN=1664-3224.
- Erjefält, J. S, Mast cells in human airways: the culprit? European Respiratory Review 2014, 23(133), 299–307. [Google Scholar] [CrossRef]
- Kawa A., The role of mast cells in allergic inflammation, Respiratory Medicine, Volume 106, Issue 1, 2012, Pages 9-14, (https :// www.science direct.com /science/article/pii/S0954611111003325). ISSN 0954-6111. [CrossRef]
- Barnes, P.J. Pathophysiology of allergic inflammation. Immunol. Rev. 2011, 242(1), 31–50. [Google Scholar] [CrossRef]
- Moon, TC; Befus, AD; Kulka, M. Mast cell mediators: their differential release and the secretory pathways involved. Front Immunol 2014, 5, 569. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Halova, I; Draberova, L; Draber, P. Mast cell chemotaxis - chemoattractants and signaling pathways. Front Immunol 2012, 3, 119. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mukai, K; Tsai, M; Saito, H; Galli, SJ. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol Rev. 2018, 282(1), 121–150. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hall, S; Agrawal, DK. Key mediators in the immunopathogenesis of allergic asthma. Int Immunopharmacol 2014, 23(1), 316–29. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Pei. Inflammatory mediators mediate airway smooth muscle contraction through a G protein-coupled receptor–transmembrane protein 16A–voltage-dependent Ca2+ channel axis and contribute to bronchial hyperresponsiveness in asthma. Journal of Allergy and Clinical Immunology 2018, Volume 141(Issue 4), Pages 1259–1268.e11. [Google Scholar] [CrossRef] [PubMed]
- Luna-Gomes, T.; Bozza, T. P.; Bandeira-Melo, C. Eosinophil recruitment and activation: the role of lipid mediators. In Front. Pharmacol., Sec. Experimental Pharmacology and Drug Discovery; 2013; Volume 4. [Google Scholar] [CrossRef]
- van der Velden, VH; Hulsmann, AR. Autonomic innervation of human airways: structure, function, and pathophysiology in asthma. Neuroimmunomodulation 1999, 6(3), 145–59. [Google Scholar] [CrossRef] [PubMed]
- Canning, Brendan J.; Woo, Ariel; Mazzone, Stuart B. Neuronal Modulation of Airway and Vascular Tone and Their Influence on Nonspecific Airways Responsiveness in Asthma. Journal of Allergy 2012, 2012(108149), 7 pages. [Google Scholar] [CrossRef]
- Kistemaker, LEM; Prakash, YS. Airway Innervation and Plasticity in Asthma. Physiology (Bethesda) 2019, 34(4), 283–298. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barnes, Peter J. Neuroeffector mechanisms: The interface between inflammation and neuronal responses. Journal of Allergy and Clinical Immunology 1996, Volume 98(Issue 5), S73–S83. [Google Scholar] [CrossRef]
- Barnes, P.J. Cellular and molecular mechanisms of Chronic Obstructive Pulmonary Disease. Clin. Chest Med 2014, 35(1), 71–86. [Google Scholar] [CrossRef]
- Barnes, P.J. Asthma-COPD Overlap. Chest 2016, 149(1), 7–8. [Google Scholar] [CrossRef]
- Castaldi, P.J.; Dy, J.; Ross, J.; Chang, Y.; Washko, G.R.; Curran-Everett, D.; et al. Cluster analysis in the COPD Gene study identifies subtypes of smokers with distinct patterns of airway disease and emphysema. Thorax 2014, 69(5), 415–422. [Google Scholar] [CrossRef]
- Ricciardolo, FLM; Sprio, AE; Baroso, A; Gallo, F; Riccardi, E; Bertolini, F; Carriero, V; Arrigo, E; Ciprandi, G. Characterization of T2-Low and T2-High Asthma Phenotypes in Real-Life. Biomedicines 2021, 9(11), 1684. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Duffus, Emily K.a; Holguin, Fernandob; Rastogi, Deepac. Non-T2 asthma. Current Opinion in Pulmonary Medicine 2025, 31(3), p 287–293. [Google Scholar] [CrossRef] [PubMed]
- Lotvall, J.; Akdis, C.A.; Bacharier, L.B.; Bjermer, L.; Casale, T.B.; Custovic, A.; et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J. Allergy Clin. Immunol. 127 2011, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Cefaloni, F.; Adiletta, V.; Monteleone, G.; Cotugno Depalma, D.; Fiorenzo, E.; Boccabella, Cristina; Bonini, M. Bronchial hyperreactivity (BHR): An old but gold hallmark of asthma. HSOA Journal of Allergy Disorders & Therapy 2023. [Google Scholar] [CrossRef]
- Chapman, DG; Irvin, CG. Mechanisms of airway hyper-responsiveness in asthma: the past, present and yet to come. Clin Exp Allergy 2015, 45(4), 706–19. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hekking, P.P.; Wener, R.R.; Amelink, M.; Zwinderman, A.H.; Bouvy, M.L.; Bel, E.H. The prevalence of severe refractory asthma. J. Allergy Clin. Immunol. 2015, 135(4), 896–902. [Google Scholar] [CrossRef]
- Murphy, RC; Hallstrand, TS. Exploring the origin and regulatory role of mast cells in asthma. Curr Opin Allergy Clin Immunol 2021, 21(1), 71–78. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Campbell, E; Hogaboam, C; Lincoln, P; Lukacs, NW. Stem cell factor-induced airway hyperreactivity in allergic and normal mice. Am J Pathol 1999, 154(4), 1259–65. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, H; Chen, X; Focia, PJ; He, X. Structural basis for stem cell factor-KIT signaling and activation of class III receptor tyrosine kinases. EMBO J 2007, 26(3), 891–901. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Voisin, T; Bouvier, A; Chiu, IM. Neuro-immune interactions in allergic diseases: novel targets for therapeutics. Int Immunol 2017, 29(6), 247–261. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Caughey, GH. Mast cell tryptases and chymases in inflammation and host defense. Immunol Rev. 2007, 217, 141–54. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barnes, P.J. Inflammatory mechanisms in COPD. J. Allergy Clin. Immunol. 2016, 138(1), 16–27. [Google Scholar] [CrossRef]
- Ballarin, A.; Bazzan, E.; Zenteno, R.H.; Turato, G.; Baraldo, S.; Zanovello, D.; Mutti, E.; Hogg, J.C.; Saetta, M.; Cosio, M.G. Mast cell infiltration discriminates between histopathological phenotypes of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2012, 186, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y; Xu, J; Meng, Y; Adcock, IM; Yao, X. Role of inflammatory cells in airway remodeling in COPD. Int J Chron Obstruct Pulmon Dis 2018, 13, 3341–3348. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ezeamuzie, C. I.; Al-Attiyah, R.; Shihab, P. K.; Al-Radwan, R. Low-affinity IgE receptor (FcεRII)-mediated activation of human monocytes by both monomeric IgE and IgE/anti-IgE immune complex. International Immunopharmacology 2009, Volume 9(Issue 9), Pages 1110–1114. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y; Duru, EA; Ameredes, BT. Role of IL-10 in the resolution of airway inflammation. Curr Mol Med. 2008, 8(5), 437–45. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barnes, P.J.; Burney, P.G.J.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; et al. Chronic obstructive pulmonary disease. Nat. Rev. Primers 1 2015, 1–21. [Google Scholar] [CrossRef]
- Yamasaki, K; Eeden, SFV. Lung Macrophage Phenotypes and Functional Responses: Role in the Pathogenesis of COPD. Int J Mol Sci 2018, 19(2), 582. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Christopoulou, ME; Papakonstantinou, E; Stolz, D. Matrix Metalloproteinases in Chronic Obstructive Pulmonary Disease. Int J Mol Sci. 2023, 24(4), 3786. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tokunaga, R; Zhang, W; Naseem, M; Puccini, A; Berger, MD; Soni, S; McSkane, M; Baba, H; Lenz, HJ. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev 2018, 63, 40–47. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Müller, M; Carter, S; Hofer, MJ; Campbell, IL. Review: The chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity--a tale of conflict and conundrum. Neuropathol Appl Neurobiol 2010, 36(5), 368–87. [Google Scholar] [CrossRef] [PubMed]
- Finney, LJ; Ritchie, A; Pollard, E; Johnston, SL; Mallia, P. Lower airway colonization and inflammatory response in COPD: a focus on Haemophilus influenzae. Int J Chron Obstruct Pulmon Dis 2014, 9, 1119–32. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Love, ME; Proud, D. Respiratory Viral and Bacterial Exacerbations of COPD-The Role of the Airway Epithelium. Cells 2022, 11(9), 1416. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jabeen, MF; Sanderson, ND; Foster, D; Crook, DW; Cane, JL; Borg, C; Connolly, C; Thulborn, S; Pavord, ID; Klenerman, P; Street, TL; Hinks, TSC. Identifying Bacterial Airways Infection in Stable Severe Asthma Using Oxford Nanopore Sequencing Technologies. Microbiol Spectr 2022, 10(2), e0227921. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vandivier, RW; Henson, PM; Douglas, IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest 2006, 129(6), 1673–82. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W; Zhou, Z; Guo, X; Zuo, X; Zhang, J; An, Y; Zheng, H; Yue, Y; Wang, G; Wang, F. Efferocytosis and Respiratory Disease. Int J Mol Sci. 2023, 24(19), 14871. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Smolinska, S; Antolín-Amérigo, D; Popescu, FD; Jutel, M. Thymic Stromal Lymphopoietin (TSLP), Its Isoforms and the Interplay with the Epithelium in Allergy and Asthma. Int J Mol Sci. 2023, 24(16), 12725. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tsoumakidou, M; Demedts, IK; Brusselle, GG; Jeffery, PK. Dendritic cells in chronic obstructive pulmonary disease: new players in an old game. In Am J Respir Crit Care Med; DOI] [PubMed; Google Scholar, 2008; Volume 177, 11, pp. 1180–6. [Google Scholar] [CrossRef]
- Bao, Y; Zhu, X. Role of Chemokines and Inflammatory Cells in Respiratory Allergy. J Asthma Allergy 2022, 15, 1805–1822. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Davoine, F; Lacy, P. Eosinophil cytokines, chemokines, and growth factors: emerging roles in immunity. Front Immunol 2014, 5, 570. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Karta, MR; Broide, DH; Doherty, TA. Insights into Group 2 Innate Lymphoid Cells in Human Airway Disease. Curr Allergy Asthma Rep. 2016, 16(1), 8. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- van Rijt, L; von Richthofen, H; van Ree, R. Type 2 innate lymphoid cells: at the cross-roads in allergic asthma. Semin Immunopathol 2016, 38(4), 483–96. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bakakos, A.; Rovina, N.; Bakakos, P. Treatment Challenges in Severe Eosinophilic Asthma: Differential Response to Anti-IL-5 and Anti-IL-5R Therapy. Int. J. Mol. Sci. 2021, 22, 3969. [Google Scholar] [CrossRef] [PubMed]
- Luo, J; Chen, W; Liu, W; Jiang, S; Ye, Y; Shrimanker, R; Hynes, G; Klenerman, P; Pavord, ID; Xue, L. IL-5 antagonism reverses priming and activation of eosinophils in severe eosinophilic asthma. Mucosal Immunol 2024, 17(4), 524–536. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McBrien, CN; Menzies-Gow, A. The Biology of Eosinophils and Their Role in Asthma. Front Med (Lausanne) 2017, 4, 93. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tashkin, DP; Wechsler, ME. Role of eosinophils in airway inflammation of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2018, 13, 335–349. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nakamura, R.; Kabata, H.; Sasahara, K.; Kurihara, M.; Kagatani, J.; Sehmi, R.; Fukunaga, K. Multifaceted roles of group 2 innate lymphoid cells in respiratory diseases. Respiratory Investigation, Volume Volume 63(Issue 5, 2025), Pages 918–927. [CrossRef]
- Ray, A; Kolls, JK. Neutrophilic Inflammation in Asthma and Association with Disease Severity. Trends Immunol 2017, 38(12), 942–954. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ray, A.; Raundhal, M.; Oriss, T.B.; Ray, P.; Wenzel, S.E. Current concepts of severe asthma. J. Clin. Invest. 2016, 126(7), 2394–2403. [Google Scholar] [CrossRef]
- Chaoyu, I. Increased frequency of dual-positive TH2/TH17 cells in bronchoalveolar lavage fluid characterizes a population of patients with severe asthma. Journal of Allergy and Clinical Immunology Volume 134(Issue 5), 1175–1186.e7.
- Jasper, AE; McIver, WJ; Sapey, E; Walton, GM. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Res 2019, 8, F1000 Faculty Rev–557. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hoenderdos, K; Condliffe, A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2013, 48, 531–9. [Google Scholar] [CrossRef]
- Strzelak, A; Ratajczak, A; Adamiec, A; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int J Environ Res Public Health 2018, 15(5), 1033. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rovina, N; Koutsoukou, A; Koulouris, NG. Inflammation and immune response in COPD: where do we stand? Mediators Inflamm. 2013, 2013, 413735. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, XR; Li, YP; Gao, S; Xia, W; Gao, K; Kong, QH; Qi, H; Wu, L; Zhang, J; Qu, JM; Bai, CX. Increased serum levels of lipocalin-1 and -2 in patients with stable chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2014, 9, 543–9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stæhr Gudmann, N.; Manon-Jensen, T.; Bülow Sand, J. M.; Diefenbach, C.; Sun, S.; Danielsen, A.; Karsdal, M. A.; Leeming, D. J. Lung tissue destruction by proteinase 3 and cathepsin G mediated elastin degradation is elevated in chronic obstructive pulmonary disease. Biochemical and Biophysical Research Communications 2018, Volume 503(Issue 3), Pages 1284–1290. [Google Scholar] [CrossRef]
- Harker, JA; Lloyd, CM. T helper 2 cells in asthma. J Exp Med. 2023, 220(6), e20221094. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Olsthoorn, E. M.S.; van Krimpen, A.; Hendriks, W. R.; Stadhouder, R. Chronic Inflammation in Asthma: Looking Beyond the Th2 Cell. Immunological Reviews 2025, Volume 330, e70010. [Google Scholar] [CrossRef] [PubMed]
- Nejman-Gryz, P.; Górska, K.; Paplińska-Goryca, M. Epithelial derived cytokines IL-25, IL-33 and TSLP in obstructive lung dise. European Respiratory Journal 2017, 50 (suppl 61), PA571. [Google Scholar] [CrossRef]
- Koch, S; Sopel, N; Finotto, S. Th9 and other IL-9-producing cells in allergic asthma. Semin Immunopathol 2017, 39(1), 55–68. [Google Scholar] [CrossRef] [PubMed]
- Soroosh, P; Doherty, TA. Th9 and allergic disease. Immunology 2009, 127(4), 450–8. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Williams, M; Todd, I; Fairclough, LC. The role of CD8 + T lymphocytes in chronic obstructive pulmonary disease: a systematic review. Inflamm Res 2021, 70(1), 11–18. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Janeway, CA, Jr.; Travers, P; Walport, M. Immunobiology: The Immune System in Health and Disease. T cell-mediated cytotoxicity, 5th edition; Garland Science: New York, 2001; Available online: https://www.ncbi.nlm.nih.gov/books/NBK27101/.
- Xu, J; Zeng, Q; Li, S; Su, Q; Fan, H. Inflammation mechanism and research progress of COPD. Front Immunol 2024, 15, 1404615. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Groom, JR; Luster, AD. CXCR3 in T cell function. Exp Cell Res 2011, 317(5), 620–31. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Demedts, IK; Demoor, T; Bracke, KR; Joos, GF; Brusselle, GG. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res. 2006, 7(1), 53. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lambers, C; Hacker, S; Posch, M; Hoetzenecker, K; Pollreisz, A; Lichtenauer, M; Klepetko, W; Ankersmit, HJ. T cell senescence and contraction of T cell repertoire diversity in patients with chronic obstructive pulmonary disease. Clin Exp Immunol 2009, 155(3), 466–75. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Weng, NP; Akbar, AN; Goronzy, J. CD28(-) T cells: their role in the age-associated decline of immune function. Trends Immunol 2009, 30(7), 306–12. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hsu, AT; Gottschalk, TA; Tsantikos, E; Hibbs, ML. The Role of Innate Lymphoid Cells in Chronic Respiratory Diseases. Front. Immunol. 2021, 12, 733324. [Google Scholar] [CrossRef]
- Koch, S.; Sopel, N.; Finotto, S. Th9 and other IL-9-producing cells in allergic asthma. Semin. Immunopathol. 2017, 39, 55–68. [Google Scholar] [CrossRef]
- Doherty, T.A.; Broide, D.H. Insights into the biology of IL-9 in asthma. J. Allergy Clin. Immunol. CrossRef. Google Scholar, 2022.
- Kliem, C.V.; Schaub, B. The role of regulatory B cells in immune regulation and childhood allergic asthma. Mol Cell Pediatr 2024, 11, 1. [Google Scholar] [CrossRef]
- Hammad, H.; Lambrecht, N. B. The basic immunology of asthma. Cell 2021, Volume 184(Issue 6), Pages 1469–1485. [Google Scholar] [CrossRef]
- Polverino, F; Seys, LJ; Bracke, KR; Owen, CA. B cells in chronic obstructive pulmonary disease: moving to center stage. Am J Physiol Lung Cell Mol Physiol. 2016, 311(4), L687–L695. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dong, LL; Liu, ZY; Chen, KJ; Li, ZY; Zhou, JS; Shen, HH; Chen, ZH. The persistent inflammation in COPD: is autoimmunity the core mechanism? Eur Respir Rev. 2024, 33(171), 230137. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mukherjee, M; Nair, P. Autoimmune Responses in Severe Asthma. Allergy Asthma Immunol Res 2018, 10(5), 428–447. [Google Scholar] [CrossRef] [PubMed]
- Ye, YM; Nahm, DH; Kim, SH; Kim, SH; Choi, JH; Suh, CH; Park, HS. Circulating Autoantibodies in Patients with Aspirin-intolerant Asthma: An Epiphenomenon Related to Airway Inflammation. J Korean Med Sci. 2006, 21(3), 412–417. [Google Scholar] [CrossRef] [PubMed]
- Westwood, JP; Mackay, AJ; Donaldson, G; Machin, SJ; Wedzicha, JA; Scully, M. The role of complement activation in COPD exacerbation recovery. ERJ Open Res 2016, 2(4), 00027–2016. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Panagopoulos, D.; Johansson, O.; Carlo, G. Polarization: A Key Difference between Man-made and Natural Electromagnetic Fields, in regard to Biological Activity. Sci Rep 2015, 5, 14914. [Google Scholar] [CrossRef]
- Messori, C. Deep into the Water: Exploring the Hydro-Electromagnetic and Quantum-Electrodynamic Properties of Interfacial Water in Living Systems. Open Access Library Journal 2019, 6, 1–62. [Google Scholar] [CrossRef]
- Cai, J; Griffin, E; Guarochico-Moreira, VH; Barry, D; Xin, B; Yagmurcukardes, M; Zhang, S; Geim, AK; Peeters, FM; Lozada-Hidalgo, M. Wien effect in interfacial water dissociation through proton-permeable graphene electrodes. Nat Commun 2022, 13(1), 5776. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shi, G; Lu, T; Zhang, L. Understanding the interfacial water structure in electrocatalysis. Natl Sci Rev. 2024, 11(12), nwae241. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dahlén, SE; Hansson, G; Hedqvist, P; Björck, T; Granström, E; Dahlén, B. Allergen challenge of lung tissue from asthmatics elicits bronchial contraction that correlates with the release of leukotrienes C4, D4, and E4. Proc Natl Acad Sci U S A 1983, 80(6), 1712–6. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kapoor, Y.; Kumar, K. Structural and clinical impact of anti-allergy agents: An overview. Bioorganic Chemistry, Volume 2020, Volume 94, 103351. [Google Scholar] [CrossRef]
- Arima, M; Fukuda, T. Prostaglandin D₂ and T(H)2 inflammation in the pathogenesis of bronchial asthma. Korean J Intern Med Erratum in: Korean J Intern Med. 2011 Jun;26(2):253. PMID: 21437156; PMCID: PMC3056260. 2011, 26(1), 8–18. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J. P. Mechanisms in COPD: Differences From Asthma. Chest 2000, Volume 117, Pages 10S–14S. [Google Scholar] [CrossRef] [PubMed]
- He, Rui; Chen, Yu; Cai, Qian. The role of the LTB4-BLT1 axis in health and disease. Pharmacological Research 2020, Volume 158, 104857. [Google Scholar] [CrossRef] [PubMed]
- Durrant, DM; Metzger, DW. Emerging roles of T helper subsets in the pathogenesis of asthma. Immunol Invest. 2010, 39(4-5), 526–49. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cosmi, L; Liotta, F; Maggi, E; Romagnani, S; Annunziato, F. Th17 cells: new players in asthma pathogenesis. Allergy 2011, 66, 989–998. [Google Scholar] [CrossRef]
- Wang, C; Zhou, J; Wang, J; Li, S; Fukunaga, A; Yodoi, J; Tian, H. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct Target Ther 2020, 5(1), 248. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Malaviya, R; Laskin, JD; Laskin, DL. Anti-TNFα therapy in inflammatory lung diseases. Pharmacol Ther 2017, 180, 90–98. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, J; Li, X; Huang, C; Lin, Y; Dai, Q. Change of Serum Inflammatory Cytokines Levels in Patients With Chronic Obstructive Pulmonary Disease, Pneumonia and Lung Cancer. Technol Cancer Res Treat 2020, 19, 1533033820951807. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barnes, PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008, 118(11), 3546–56. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Montuschi, P.; Collins, J.V.; Ciabattoni, G.; Lazzeri, N.; Corradi, M.; Kharitonov, S.A.; Barnes, P.J. Exhaled 8-isoprostane as anin vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am. J. Respir. Crit. Care Med. CrossRef. 2000, 162 Pt 1, 1175–1177. [Google Scholar] [CrossRef]
- Victoni, T; Barreto, E; Lagente, V; Carvalho, VF. Oxidative Imbalance as a Crucial Factor in Inflammatory Lung Diseases: Could Antioxidant Treatment Constitute a New Therapeutic Strategy? Oxid Med Cell Longev 2021, 2021, 6646923. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Orriols, G; Aljama, C; Rodriguez-Frias, F; Medina, PG; Ferrer-Costa, R; Granados, G; Nuñez, A; López-González, A; Ruiz-Satelinas, G; Miravitlles, M; Barrecheguren, M; Esquinas, C. Quantification of serum elastase inhibitory activity in patients with pulmonary emphysema with and without alpha-1 antitrypsin deficiency. PLoS One 2025, 20(6), e0324237. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Conti, V; Corbi, G; Manzo, V; Pelaia, G; Filippelli, A; Vatrella, A. Sirtuin 1 and aging theory for chronic obstructive pulmonary disease. Anal Cell Pathol (Amst) 2015, 2015, 897327. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abdul Roda, M; Fernstrand, AM; Redegeld, FA; Blalock, JE; Gaggar, A; Folkerts, G. The matrikine PGP as a potential biomarker in COPD. Am J Physiol Lung Cell Mol Physiol. 2015, 308(11), L1095–101. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, R; Wu, X; Gounni, AS; Xie, J. Mucus hypersecretion in chronic obstructive pulmonary disease: From molecular mechanisms to treatment. J Transl Int Med. 2023, 11(4), 312–315. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schulman, ES; Nishi, H; Pelleg, A. Degranulation of human mast cells: modulation by P2 receptors' agonists. Front Immunol 2023, 14, 1216580. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ramadan, A.; El-Samahy, M.; Elrosasy, A.; Al-Tawil, M.; Abdelaziz, A.; Soliman, M. A; Abouzid, M. Safety and efficacy of P2X3 receptor antagonist for the treatment of refractory or unexplained chronic cough: A systematic review and meta-analysis of 11 randomized controlled trials. Pulmonary Pharmacology & Therapeutics, Volume 2023, Volume 83, 102252. [Google Scholar] [CrossRef]
- Rivas, M; Gupta, G; Costanzo, L; Ahmed, H; Wyman, AE; Geraghty, P. Senescence: Pathogenic Driver in Chronic Obstructive Pulmonary Disease. Medicina (Kaunas) 2022, 58(6), 817. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Misawa, T; Tanaka, Y; Okada, R; Takahashi, A. Biology of extracellular vesicles secreted from senescent cells as senescence-associated secretory phenotype factors. Geriatr Gerontol Int. 2020, 20(6), 539–546. [Google Scholar] [CrossRef] [PubMed]
- Wan, R.; Srikaram, P.; Guntupalli, V.; Hu, C.; Chen, Q.; Gao, P. Cellular senescence in asthma: from pathogenesis to therapeutic challenges. eBioMedicine, Volume Volume 94(2023), 104717. [CrossRef]
- Sriprasart, T; Saiphoklang, N; Kawamatawong, T; Boonsawat, W; Mitthamsiri, W; Chirakalwasan, N; Chiewchalermsri, C; Athipongarporn, A; Kamalaporn, H; Kornthatchapong, K; Kulpraneet, M; Sompornrattanaphan, M; Oer-Areemitr, N; Rerkpattanapipat, T; Silairatana, S; Thawanaphong, S; Gaensan, T; Jirakran, K; Poachanukoon, O. Allergic rhinitis and other comorbidities associated with asthma control in Thailand. Front Med (Lausanne) 2024, 10, 1308390. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zysk, W.; Mesjasz, A.; Trzeciak, M.; Horvath, A.; Plata-Nazar, K. Gastrointestinal Comorbidities Associated with Atopic Dermatitis—A Narrative Review. Int. J. Mol. Sci. 2024, 25, 1194. [Google Scholar] [CrossRef] [PubMed]
- Agustí, A; Edwards, LD; Rennard, SI; MacNee, W; Tal-Singer, R; Miller, BE; Vestbo, J; Lomas, DA; Calverley, PM; Wouters, E; Crim, C; Yates, JC; Silverman, EK; Coxson, HO; Bakke, P; Mayer, RJ; Celli, B. Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One 2012, 7(5), e37483. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Qian, Y; Cai, C; Sun, M; Lv, D; Zhao, Y. Analyses of Factors Associated with Acute Exacerbations of Chronic Obstructive Pulmonary Disease: A Review. Int J Chron Obstruct Pulmon Dis. 2023, 18, 2707–2723. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.C.; Lutchen, K.R. Factors determining airway caliber in asthma. Critic. Rev. Biomed. Eng. 2013, 41(6), 515–532. [Google Scholar] [CrossRef] [PubMed]
- Hirota, N.; Martin, J.G. Mechanisms of airway remodeling. Chest 2013, 144(3), 1026–1032. [Google Scholar] [CrossRef]
- Shah, BK; Singh, B; Wang, Y; Xie, S; Wang, C. Mucus Hypersecretion in Chronic Obstructive Pulmonary Disease and Its Treatment. Mediators Inflamm 2023, 2023, 8840594. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gao, H; Ying, S; Dai, Y. Pathological Roles of Neutrophil-Mediated Inflammation in Asthma and Its Potential for Therapy as a Target. J Immunol Res. 2017, 2017, 3743048. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, YG; Tian, WM; Zhang, H; Li, M; Shang, YX. Nerve growth factor exacerbates allergic lung inflammation and airway remodeling in a rat model of chronic asthma. Exp Ther Med. 2013, 6(5), 1251–1258. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gheorghiu, M.; Trandafir, M.F.; Pasarica, D. Neurotrophin 3 as a new bidirectional signaling molecule between lung inflammation and the diffuse neuroendocrine system. the 43rd FEBS Congress, Prague, Czech Republic, 7-12 july 2018. [Google Scholar]
- Carlson AB, Kraus GP. Physiology, Cholinergic Receptors. [Updated 2023 Aug 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK526134/.
- Yu, Kytikova O.; Novgorodtseva, TP; Antonyuk, MV; Gvozdenko, TA. The role of regulatory neuropeptides and neurotrophic factors in asthma pathophysiology. Russian Open Medical Journal 2019, 8, e0402. [Google Scholar] [CrossRef]
- Camp, B; Stegemann-Koniszewski, S; Schreiber, J. Infection-Associated Mechanisms of Neuro-Inflammation and Neuro-Immune Crosstalk in Chronic Respiratory Diseases. Int J Mol Sci. 2021, 22(11), 5699. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liesker, JJ; Ten Hacken, NH; Zeinstra-Smith, M; Rutgers, SR; Postma, DS; Timens, W. Reticular basement membrane in asthma and COPD: similar thickness, yet different composition. Int J Chron Obstruct Pulmon Dis. 2009, 4, 127–35. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Singh, D. Small Airway Disease in Patients with Chronic Obstructive Pulmonary Disease. Tuberc Respir Dis (Seoul) 2017, 80(4), 317–324. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barnig, C; Lutzweiler, G; Giannini, M; Lejay, A; Charles, AL; Meyer, A; Geny, B. Resolution of Inflammation after Skeletal Muscle Ischemia-Reperfusion Injury: A Focus on the Lipid Mediators Lipoxins, Resolvins, Protectins and Maresins. Antioxidants (Basel) 2022, 11(6), 1213. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gheorghiu, M. Viul de dincolo de materie: revelatii cuantice in Fiziopatologie; Ed. Scoala Ardeleana, 2021; pp. 61–70. ISBN 978-606-797-706-6. [Google Scholar]
- Ahmad M, Wolberg A, Kahwaji CI. Biochemistry, Electron Transport Chain. [Updated 2023 Sep 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK526105/.
- Stekhin, A.; Yakovleva, G.; Pronko, K.; Zemskov, V. Regulatory function of macroscopic quantum states of electrons in cell metabolism. Research Article - Clinical Practice 2018, 15(3), 707–715. [Google Scholar] [CrossRef]
- Bono, Ivan; Del Giudice, Emilio; Gamberale, Luca; Henry, Marc. Emergence of the Coherent Structure of Liquid Water. Water 2012, 4, 510–532. [Google Scholar] [CrossRef]
- Del Giudice, E.; Tedeschi, A.; Vitiello, G.; Voeikov, V. Coherent structures in liquid water close to hydrophilic surfaces 2013. J. Phys.: Conf. Ser. 442 012028. [CrossRef]
- Gardner, P.R. Resistance of Nitric Oxide Dioxygenase and Cytochrome c Oxidase to Inhibition by Nitric Oxide and Other Indications of the Spintronic Control of Electron Transfer. Biophysica 2025, 5, 41. [Google Scholar] [CrossRef]
- Villalpando-Rodriguez, GE; Gibson, SB. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxid Med Cell Longev 2021, 2021, 9912436. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Usselman, RJ; Chavarriaga, C; Castello, PR; Procopio, M; Ritz, T; Dratz, EA; Singel, DJ; Martino, CF. The Quantum Biology of Reactive Oxygen Species Partitioning Impacts Cellular Bioenergetics. Sci Rep. 2016, 6, 38543. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lüdtke, F.L.; Silva, T.J.; da Silva, M.G.; Hashimoto, J.C.; Ribeiro, A.P.B. Lipid Nanoparticles: Formulation, Production Methods and Characterization Protocols. Foods 2025, 14, 973. [Google Scholar] [CrossRef] [PubMed]
- Catalá, A; Díaz, M. Editorial: Impact of Lipid Peroxidation on the Physiology and Pathophysiology of Cell Membranes. Front Physiol. 2016, 7, 423. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schuermann, D.; Mevissen, M. Manmade Electromagnetic Fields and Oxidative Stress—Biological Effects and Consequences for Health. Int. J. Mol. Sci. 2021, 22, 3772. [Google Scholar] [CrossRef] [PubMed]
- Khan, YS; Farhana, A. Histology, Cell. In StatPearls [Internet]; StatPearls Publishing: Treasure Island (FL), 27 Mar 2025; Available online: https://www.ncbi.nlm.nih.gov/books/NBK554382/.
- Roig-Rosello, E.; Rousselle, P. The Human Epidermal Basement Membrane: A Shaped and Cell Instructive Platform That Aging Slowly Alters. Biomolecules 2020, 10, 1607. [Google Scholar] [CrossRef]
- Masic, A.; Bertinetti, L.; Schuetz, R.; et al. Osmotic pressure induced tensile forces in tendon collagen. Nat Commun 2015, 6, 5942. [Google Scholar] [CrossRef]
- Iannucci Leanne, E.; Dranoff Charles, S.; David Michael, A.; Lake Spencer, P. Optical Imaging of Dynamic Collagen Processes in Health and Disease. In Frontiers in Mechanical Engineering; 2022; Volume 8, Available online: https://www.frontiersin.org/journals/mechanical-engineering/articles/10.3389/fmech.2022.855271ISSN =2297-3079.
- Biswal, S; Agmon, N. Collagen Structured Hydration. Biomolecules 2023, 13(12), 1744. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Majewski, Alexander; Sanzari, Martin; Torzilli, Peter. Effects of ultraviolet radiation on the type-I collagen protein triple helical structure: A method for measuring structural changes through optical activity. Physical review. E, Statistical, nonlinear, and soft matter physics 2002, 65. 031920. [Google Scholar] [CrossRef]
- Gromkowska-Kępka, KJ; Puścion-Jakubik, A; Markiewicz-Żukowska, R; Socha, K. The impact of ultraviolet radiation on skin photoaging - review of in vitro studies. J Cosmet Dermatol 2021, 20(11), 3427–3431. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Giannetti, G; Matsumura, F; Caporaletti, F; Micha, D; Koenderink, GH; Ilie, IM; Bonn, M; Woutersen, S; Giubertoni, G. Water and Collagen: A Mystery Yet to Unfold. Biomacromolecules 2025, 26(5), 2784–2799. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Salvatore, L; Gallo, N; Natali, ML; Terzi, A; Sannino, A; Madaghiele, M. Mimicking the Hierarchical Organization of Natural Collagen: Toward the Development of Ideal Scaffolding Material for Tissue Regeneration. Front Bioeng Biotechnol. 2021, 9, 644595. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kalra, Aarat; Patel, Sahil; Bhuiyan, Asadullah; Preto, Jordane; Scheuer, Kyle; Mohammed, Usman; Lewis, John; Rezania, Vahid; Shankar, Karthik; Tuszynski, Jack. Investigation of the Electrical Properties of Microtubule Ensembles under Cell-Like Conditions. Nanomaterials 2020. [Google Scholar] [CrossRef]
- Chakrabarti, Rajarshi; Fung, Tak Shun; Kang, Taewook; Elonkirjo, Pieti W.; Suomalainen, Anu; Usherwood, Edward J.; Higgs, Henry N. Mitochondrial dysfunction triggers actin polymerization necessary for rapid glycolytic activation. J Cell Biol 2022, 221(11), e202201160. [Google Scholar] [CrossRef]
Figure 1.
Electron quantum condensation as a result of the coherent phase excitation of associated water during phase instability development is a two-stepped process [158 - Stekhin, A., Yakovleva, G., Pronko, K., & Zemskov, V. (2018). Regulatory function of macroscopic quantum states of electrons in cell metabolism. Research Article - Clinical Practice, 15(3), 707-715)].
Figure 1.
Electron quantum condensation as a result of the coherent phase excitation of associated water during phase instability development is a two-stepped process [158 - Stekhin, A., Yakovleva, G., Pronko, K., & Zemskov, V. (2018). Regulatory function of macroscopic quantum states of electrons in cell metabolism. Research Article - Clinical Practice, 15(3), 707-715)].
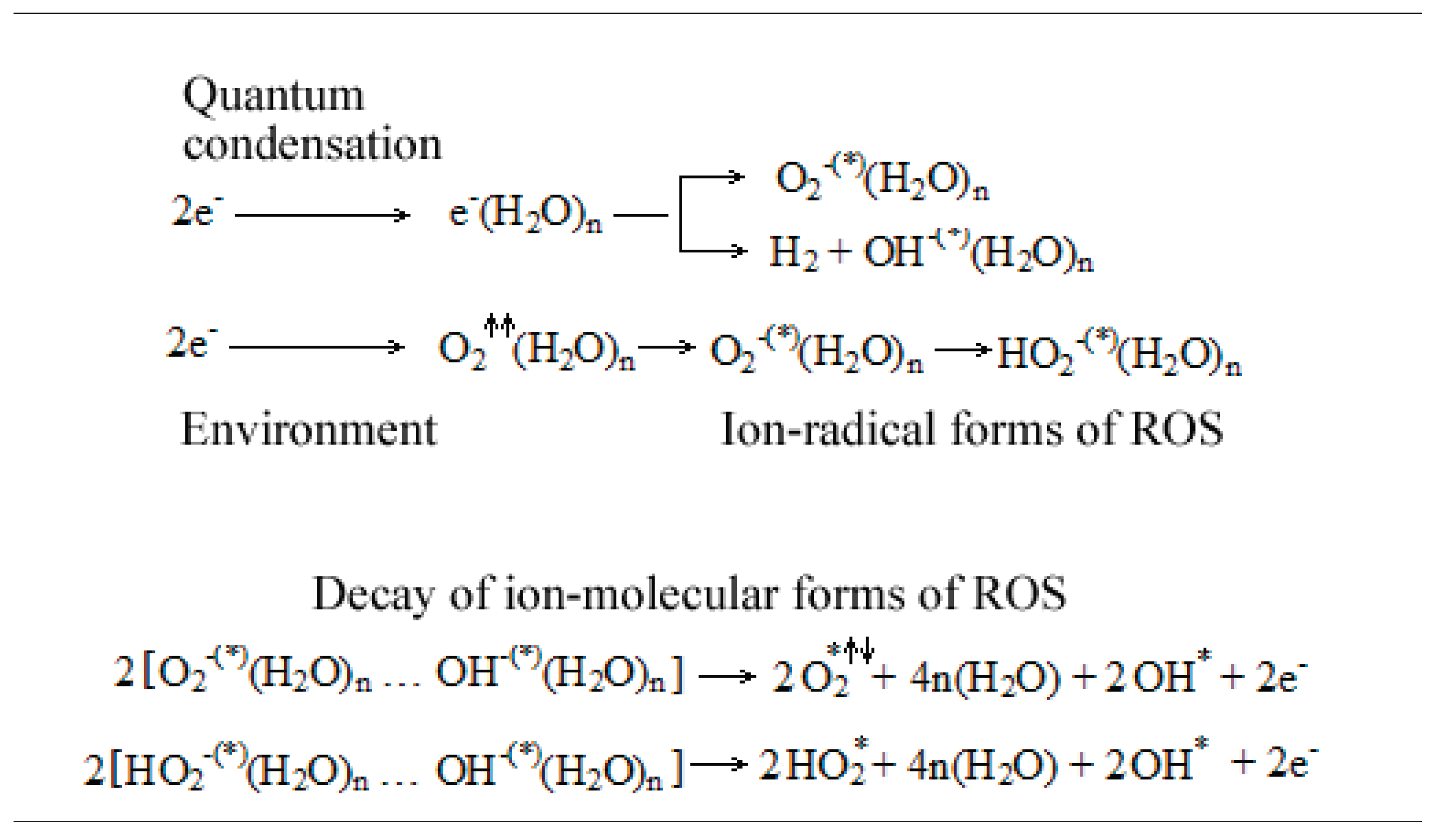
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



