Submitted:
28 January 2026
Posted:
29 January 2026
You are already at the latest version
Abstract
Death domain-associated protein 6 (DAXX) was originally identified as a key regulator of Fas receptor-mediated apoptosis. Recent studies have found that it plays a central role in many biological processes, such as cell metabolism, aging and immunity. DAXX, through its nuclear localization and epigenetic regulatory capabilities, participates in the maintenance of metabolic homeostasis, DNA damage repair, and telomere stability, and modulates immune responses by regulating the transcriptional programs of immune-related genes. This review systematically discusses the molecular mechanisms and recent advances regarding DAXX's involvement in metabolism, aging, and immunity, and explores its potential as a therapeutic target, providing a theoretical basis for the study of related diseases and clinical interventions.
Keywords:
DAXX
; metabolism
; aging
; immunity
; inflammation
1. Introduction
Metabolic homeostasis, cellular aging, and immune regulation are crucial biological processes for maintaining overall health, and their dynamic balance plays a decisive role in tissue function and internal environment stability. Disruption of these processes is closely linked to the development of various chronic diseases[1,2]. These biological processes are not independent, but are interrelated through nuclear mechanisms such as chromatin state and transcriptional regulation, collectively influencing the cellular response to metabolic changes and stress stimuli. Recent studies have shown that DAXX, a multifunctional nuclear regulatory protein, plays an integrative role in these processes through its chromatin remodeling and epigenetic regulatory capabilities[3,4]. DAXX is involved not only in the regulation of energy metabolism[5], but also in DNA damage response, telomere maintenance[6], and the maintenance of immune homeostasis[7]. This review focuses on the nuclear localization characteristics of DAXX and systematically explores its molecular mechanisms and functions in metabolism, aging, and immune regulation, aiming to elucidate its pivotal role in both physiological and pathological conditions.
2. Basic Functions of DAXX
2.1. Molecular Structure and Functional Domain of Daxx
DAXX protein is encoded by DAXX gene located in the short arm of human chromosome 6 (6p 21.3). The full length of DAXX gene is about 80 kb, including 16 exons. Its translation product is a protein composed of 740 amino acids with a molecular weight of about 81 kDa, which is highly conserved in many mammals[8]. Its primary sequence contains acidic and basic regions, which provide the basis for the interaction of diverse proteins[9]. In the overall conformation, DAXX lacks typical repeat structure, and its secondary structure is mainly composed of helical and disordered regions, in which the C-terminal is rich in intrinsic disorder (IDR), while the N-terminal forms a stable folding domain[10,11].
At the functional domain level, the N-terminus of DAXX harbors a conserved four-helix bundle (4HB) that binds to signaling molecules, including Ras association domain family member 1C (RASSF1C), p53, and mouse double minute 2 homolog (MDM2), thereby regulating apoptosis and transcriptional repression[12]. Its central region, the histone binding domain (HBD), consists of six α helices and is the core structure that binds the H3.3 – H4 dimer and performs histone chaperone functions[13]. The C-terminal acidic region is involved in chromatin remodeling and gene silencing while enhancing H3.3/H4 affinity[12,13]. In addition, two small ubiquitin-modifying (SUMO) interaction motifs (SIMs) enable DAXX to bind to SUMOylated proteins, thereby regulating subcellular localization and transcriptional regulatory activity[10,14].
In terms of subunit interaction, DAXX mainly forms complexes with H3.3/H4 dimers, which determines its specific location in heterochromatin, telomeres and centromeres, and helps to maintain the higher structure of chromatin and genome stability[13]. There is a synergistic effect between different domains of DAXX: for example, HBD cooperates with the C-terminal acidic region to improve the affinity for H3.3/H4[12].The SUMO binding of SIM may affect the enrichment of DAXX in the nuclear body of promyelocytic leukemia protein (promyelocytic leukemia protein, PML), thus regulating chromatin silencing and DNA repair[11,15].
2.2. Nuclear Localization and Function of DAXX
The diverse functions of DAXX are highly contingent upon its localization within the nucleus. In contrast to its primary role in the cytoplasm, where it mainly mediates apoptotic signaling, DAXX exerts critical functions in the nucleus by binding to chromatin, participating in transcriptional regulation, and maintaining genomic stability. This feature renders it a classic example of a "nuclear localization-dependent" regulatory factor[16,17,18].
At the chromatin level, DAXX is an important partner for histone variant H3.3 deposition. It cooperates with related complexes to load H3.3 specifically into telomeres, centromeres, and repetitive DNA regions, thereby maintaining heterochromatin stability and long-term genomic integrity[16,17,18]. If the nuclear localization is impaired, the deposition pattern of H3.3 will be disturbed, the telomere function will be impaired, and the abnormal accumulation of DNA damage signals will eventually destroy the genome homeostasis[6,19,20]. Recent studies have further revealed that Ataxia telangiectasia and Rad3-related protein(ATR)-dependent signaling is essential for the localization of DAXX to the centromere, a mechanism that guarantees the maintenance of chromatin markers and regional identity[21]. In addition, DAXX can promote the establishment of specific epigenetic modifications, such as the deposition of H3K9me3, providing a new mechanism for gene silencing and repetitive sequence suppression[19]. These findings suggest that the nuclear localization of DAXX not only guarantees the maintenance of chromatin structure, but also plays a unique role in the regulation of epigenetic modifications.
The nuclear localization of DAXX is also reflected in its aggregation in the nucleus. Previous studies have found that DAXX is highly enriched in nuclear substructures such as PML nuclear bodies, and is involved in the regulation of apoptotic signaling and transcriptional repression in this environment[22,23]. Further studies revealed that the SUMO-interacting motif (SIM) is a key element in maintaining its nuclear localization and stable existence[14,24,25]. The absence of SIM or the disruption of its SUMO binding ability will lead to DAXX can not be effectively enriched in the PML nuclear body, but diffuse in the nucleus, thus weakening its related functions. Recent studies have also shown that DAXX undergoes nuclear redistribution during viral latency and reactivation, and exhibits reversible aggregation driven by liquid-liquid phase separation, which reflects the close relationship between its nuclear localization and stress response[26]. At the same time, the level of SUMO modification directly affects the nuclear body residence and stability of DAXX, and excessive or insufficient modification will destroy its localization and function[15,27]. It has been pointed out that PML-nb, as an important non-membrane substructure in the nucleus, is a key platform for SUMO-dependent modification and chromatin regulation, and provides a necessary structural environment for the aggregation and function of DAXX[28,29].
In addition to its role in intranuclear structures such as chromatin and PML nuclear bodies, the nuclear localization of DAXX determines its functional performance in transcriptional regulation. For example, it can bind to and inhibit the activity of some transcription factors in the nucleus, thus regulating the expression of downstream genes[30]. These studies reveal that the transcriptional regulation of DAXX depends on nuclear localization rather than simple protein interactions. Recent studies have also highlighted the relationship between nuclear localization and its dynamic plasticity: DAXX can rapidly adjust its distribution under different environmental and signaling conditions, switching from a static chromatin partner to a dynamic transcriptional regulator[24,26]. This feature provides an important basis for explaining its diversified functions.
Nuclear localization abnormalities are often associated with pathological processes[31,32]. As a result of mutations found in some tumors that disrupt key domains necessary for DAXX to maintain its nuclear localization, DAXX is unable to form normal aggregated structures in the nucleus, and its functions related to chromatin loading are impaired. This impairment in function further triggers a block in the chromatin deposition process and perturbs downstream transcriptional regulation[19]. In addition, recent studies on nuclear body structure have shown that the aggregation and distribution of DAXX are regulated by a variety of signals, and its abnormalities may become the starting point of chromatin opening or genomic instability[33,34,35].
These results collectively suggest that nuclear localization is not merely a phenomenon of DAXX localization, but also a key factor determining whether its function can be normally exerted.
3. Regulatory Mechanism of DAXX in Metabolism
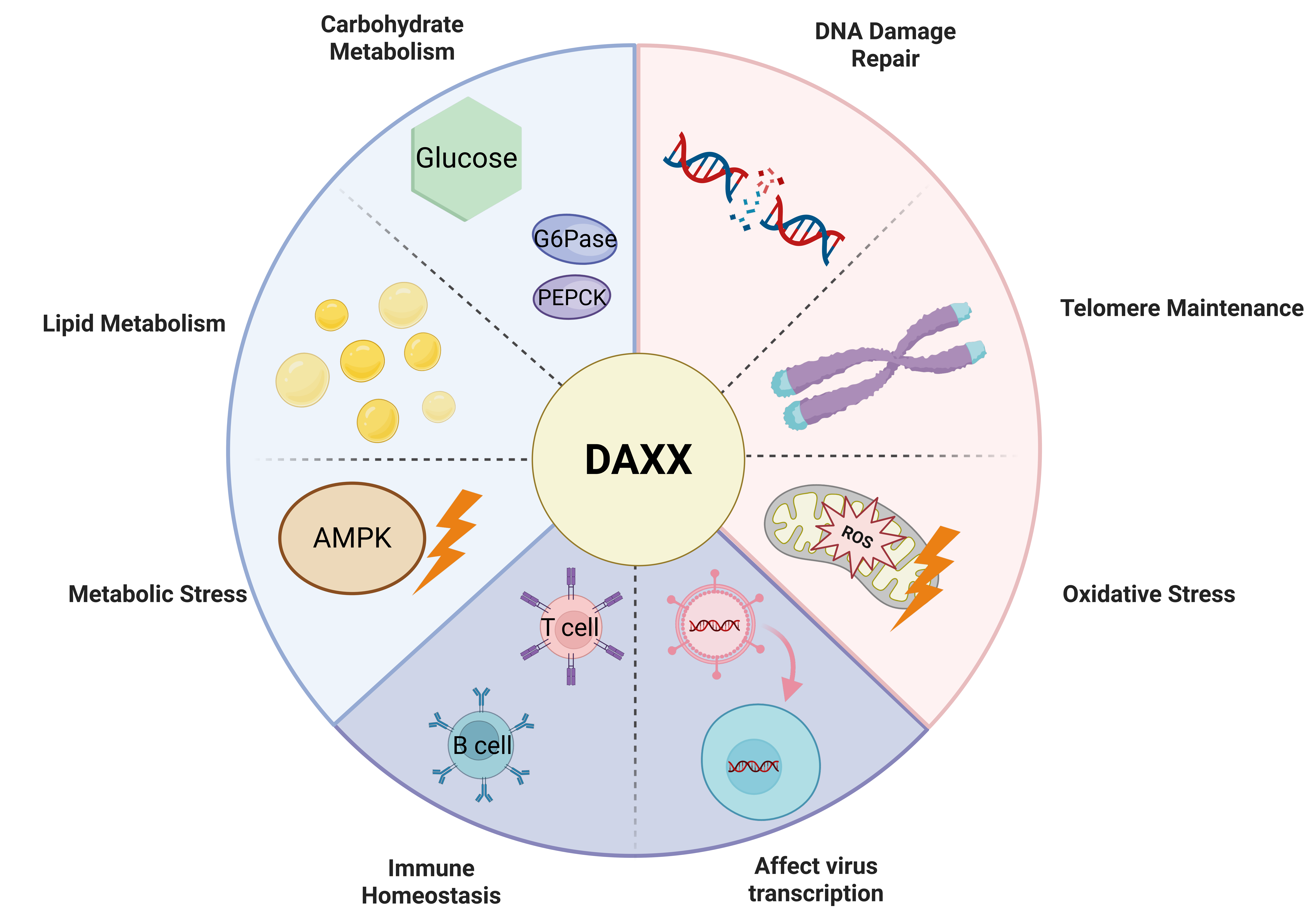
Energy metabolism is a core process for maintaining bodily life activities and tissue homeostasis, encompassing the acquisition, utilization, and storage of carbohydrates, lipids, and proteins. Its imbalance can affect cellular function, tissue homeostasis, and even overall health, and is closely related to the occurrence and development of various chronic diseases[36,37]. Recent studies have shown that DAXX is gradually found to be an important factor in the regulation of energy metabolism, and its mechanism and biological significance are attracting more and more attention. It is worth noting that the metabolic regulation of DAXX is closely related to its spatial distribution in the nucleus and epigenetic regulation, which enables it to regulate the chromatin accessibility and transcriptional activity of metabolic genes through cooperation with PML nuclear body, ATRX and a variety of transcriptional regulatory complexes, thus deeply participating in the fine regulation of glucose and lipid metabolism(Figure 1)[15,28,34].
3.1. Lipid Metabolism
Lipid metabolism is the core component of energy metabolism and signal transduction, which covers cholesterol metabolism, fatty acid synthesis, fatty acid oxidation, triglyceride synthesis and other links[38]. These metabolic pathways are highly coordinated under physiological conditions and work together to maintain cellular homeostasis[39].However, under pathological conditions, the imbalance of any link may lead to lipid accumulation and abnormal energy metabolism, and then lead to obesity, diabetes, non-alcoholic fatty liver disease (MASLD/MASH) and cancer[40,41]. In this process, DAXX precisely couples epigenetic regulation with metabolic gene transcription activity through its spatial localization in the nucleus and chromatin regulation ability. DAXX can affect the activation and inhibition of genes related to lipid metabolism by cooperating with PML nuclear body, ATRX and various transcription complexes, and regulate lipid homeostasis in both directions according to the changes of metabolic environment[12,21].
Research indicates that DAXX plays a pivotal role in cholesterol and fatty acid synthesis. The crucial steps of these two synthesis processes occur during the stage where sterol regulatory element-binding proteins (SREBP) precursors are cleaved and enter the cell nucleus[5,42]. DAXX relies on its nuclear localization ability to synergize with SREBP1/2 in regulating the expression of metabolic genes. Its activation state directly determines the transcription level of synthesis-related genes and the metabolic direction[5]. In the cholesterol synthesis pathway, SREBP exists as a precursor in the endoplasmic reticulum. When the cholesterol level of cells decreases, the chaperone protein SREBP cleavage-activating protein(SCAP) transports it to the Golgi apparatus and releases its N-terminal fragment into the nucleus after cleavage by site 1 protease and site 2 protease, thus driving the transcription of key genes such as HMGCR, HMGCS1 and LDLR[43,44,45]. Promotes cholesterol production. Its nuclear transcriptional activity is influenced by chromatin accessibility and nuclear body microenvironment (such as PML nuclear body)[15,35]. In this process, DAXX depends on its SUMO-binding domain to interact with SREBP1/2, stabilize its binding on the promoter of lipid metabolism-related genes, significantly up-regulate the expression of HMGCR and HMGCS1, and promote cholesterol accumulation[5].This mechanism is particularly evident in tumor cells such as breast cancer[5]. In addition, SREBP1/2 also plays a key role in fatty acid synthesis. The activated fragment formed after SREBP1/2 cleavage can up-regulate ACC, FASN, SCD1, ACLY and other genes, promote the supply of acetyl-Coa to malonyl-Coa, and the synthesis and desaturation of long-chain fatty acid chains[46]. DAXX enhances the transcriptional activity of SREBP1/2 by virtue of its SIM2 domain binding to SUMO, thereby promoting fatty acid synthesis[5,47].This mechanism is particularly prominent in tumor cells, and when the DAXX key domain is damaged, the pathway is blocked, resulting in a decline in synthesis and inhibition of tumor cell growth[5]. At the same time, the effect of DAXX is also closely related to androgen receptor (AR). In HepG2 and LNCaP cells, DAXX weakened the ability of AR to activate SREBP2 by interacting with the N-terminal and DNA-binding domain of AR. Especially in LNCaP prostate cancer cells, SUMO modification of AR enhances its binding to DAXX, thereby enhancing their synergistic inhibition on the nuclear chromatin platform, inhibiting SREBP2 activation and reducing the expression of downstream synthetic genes[30,48,49].
This bidirectional regulation may be due to the different epigenetic status of DAXX in different cellular environments. The level of SUMOylation and its spatial distribution in the nuclear body of PML may determine the accessibility and interaction stability of DAXX to different transcriptional programs (such as SREBP or AR-related pathways)[15,50].In addition, the position of DAXX in the metabolic signaling pathway may be dynamically regulated by energy sensing mechanisms such as AMP-activated protein kinase(AMPK) and mechanistic target of rapamycin(mTOR), so that it tends to promote synthesis under high metabolic pressure and to inhibit feedback under steady-state conditions[51]. It is speculated that the bidirectional effect of DAXX essentially reflects its selective redirection of metabolic gene expression programs in different energy and signaling backgrounds.
The DAXX – SREBP 1/2 axis plays a key regulatory role in the upstream synthesis of cholesterol and fatty acid metabolism, and its influence may extend to the lipid storage proces[52]. The synthesis of triacylglycerol (TAG) is dependent on diacylglycerol acyltransferase 1/2 (DGAT1/2), and the transcription of DGAT1/2 is indirectly regulated by SREBP1[46,53,54]. Based on DAXX can enhance the transcriptional activity of SREBP1[5].It can be speculated that it may indirectly regulate the expression of DGAT1/2 to affect the production of TAG and the accumulation of lipid droplets. In contrast, when the DAXX – SREBP axis is damaged, the supply of fatty acids and cholesterol is reduced, and the power of TAG production is also reduced. This indirect regulation through substrate accumulation and coordinated transmission of metabolic pathways suggests that DAXX – SREBP mechanism may be involved in lipid storage and long-term metabolic adaptation, and its specific function still needs further experimental confirmation.
Cholesterol homeostasis also depends on the regulation of efflux and transport pathways[55]. DAXX has been shown to upregulate caveolin-1 expression and enhance cholesterol transport to high density lipoproteins or plasma membranes, thereby promoting cholesterol efflux and preventing lipid deposition[56,57,58]. This mechanism suggests that DAXX plays a coordinating role in the import and export of cholesterol metabolism through nuclear location-dependent transcriptional regulation.
Fatty acid oxidation is the core metabolic process to maintain energy homeostasis, which is mainly regulated by AMPK and its downstream target carnitine palmitoyltransferase 1A[59]. During energy deprivation, AMPK is activated, which inhibits lipid synthesis and promotes fatty acid oxidation into mitochondria, thereby preventing lipid accumulation[60]. In a mouse model of high-fat diet-induced obesity, overexpression of DAXX activates the AMPK-related kinase MPK38, whose N-terminus (1 – 440aa) binds to the C-terminus (270 – 643aa) of MPK38 to enhance its stability and promote activation. Activated MPK38 exerts AMPK-like effects through ASK1/TGF-β/p53 signaling pathway, significantly increasing the level of fatty acid oxidation[61]. This mechanism not only reduces the number of lipid droplets in hepatocytes, but also reduces serum triglyceride and total cholesterol[61], thus playing a protective role in the maintenance of global metabolic homeostasis, which provides a new perspective for understanding the mechanism of DAXX in metabolic diseases such as obesity and fatty liver.
3.2. Glycometabolism
Imbalance of glucose metabolism is an important pathological basis for many diseases, such as insulin resistance, obesity, type II diabetes, cardiovascular disease and metabolic reprogramming of cancer[53].Glucose uptake, glycolysis, and gluconeogenesis act in a coordinated manner to maintain systemic glucose homeostasis and energy supply, and they participate in the regulation of disease progression through interactions with transcription factors and signaling pathways[62,63].At these glycometabolic nodes, DAXX influences the homeostasis of metabolic pathways by regulating the chromatin accessibility of key glycometabolic enzymes and transcription factors. This epigenetic regulatory mechanism enables DAXX to deeply participate in the transcriptional regulation of the glycometabolic network and play an important role in metabolic adaptation and pathological progression.
Glucose transport into cells is mainly mediated by glucose transporters (GLUTs)[64] GLUT4 is the most insulin-sensitive transporter in skeletal muscle and adipose tissue[65].Intracellular recycling of GLUT4 and membrane translocation depend on microtubule network regulated by kinesin 5B and JNK1[66]. It is worth noting that DAXX can directly interact with GLUT4, and they can be modified by SUMOylation. In the transport system related to insulin stimulation, DAXX, as a scaffold protein, cooperates with JNK1 and KIF5B to assemble a complex, participates in the transport process of GLUT4 vesicles, and synergistically affects the membrane translocation efficiency of GLUT4. Thereby further regulating glucose intake. Loss of DAXX leads to the retention of GLUT4 in endosomal compartments, thereby impairing transmembrane glucose transport and ultimately disrupting energy acquisition and utilization in tissues[67,68,69].
Glycolysis is a major pathway for rapid energy supply in cells, and its key rate-limiting step is controlled by hexokinase (HK), phosphofructokinase 1 (PFK1) and pyruvate kinase (PK[70] HK2 and PKM2 are particularly important in tumor metabolic reprogramming[71]. Studies have shown that in small cell lung cancer, DAXX binds to long non-coding RNA CALML3-AS1, enhances GLUT4-mediated glucose uptake, upregulates HK2 and PKM2 levels, accelerates glycolysis and lactate production, provides sufficient ATP and metabolic intermediates for tumor cells, and supports their rapid proliferation and survival[67]. Although studies have described DAXX as a tumor suppressor[18].However, this difference may be due to the different cellular environment and binding partners, suggesting that the role of DAXX in glucose metabolism is not single, and whether it has anti-tumor potential still needs further study. The role of DAXX in the regulation of glucose metabolism depends not only on its interaction with metabolism-related molecules and complexes, but also on its nuclear localization and regulatory function at the chromatin level. In the mother-infant model of type I diabetes mellitus, the abnormal expression of DAXX may be significantly related to H3K4me3 modification in the promoter region of glycolytic genes[72]. H3K4me3 is a typical transcriptional activation marker, which occurs in the promoter region of the nucleus, and its state directly affects the expression of metabolism-related genes[73]. H3K4me3 has been reported to undergo alterations in hyperglycemic conditions, which are accompanied by remodeling of chromatin accessibility and transcriptional activity programs in promoter regions, thus driving changes in metabolism-related gene expression profiles[74,75]. Given that DAXX possesses histone chaperone and chromatin regulatory functions[6], whether it influences the transcriptional activity of glucose metabolism genes by participating in or regulating H3K4me3-related chromatin states remains to be further systematically studied.
Gluconeogenesis occurs predominantly in the liver, where the rate of conversion of substrates such as lactate, glycerol, and amino acids into glucose is tightly regulated by phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) [76]. In DAXX knockout mice, the expression levels of PEPCK and G6Pas in the liver were significantly increased, resulting in enhanced gluconeogenesis. Restoring the expression of DAXX can inhibit the expression of PEPCK and G6Pase, reduce the level of liver gluconeogenesis and improve the level of blood glucose[61].
It can be seen that DAXX has many regulatory functions in glucose metabolism. It interacts with GLUT4 and related complexes to affect glucose uptake and regulate the expression of key enzymes in glycolysis, thus changing the activity of downstream metabolic pathways. In the liver, DAXX inhibits gluconeogenesis by negatively regulating PEPCK and G6Pase. The association of DAXX with H3K4me3 modification in the promoter region of glycolytic genes suggests that its role is not only dependent on complex assembly and signal regulation at the cytoplasmic level, but also closely related to nuclear localization and epigenetic regulation. Therefore, DAXX may play an important role in maintaining glucose homeostasis as well as in metabolic reprogramming under pathological conditions, and may represent a potential avenue for research into glucose metabolism related diseases.
3.3. Antioxidant Response
Antioxidant response is an important defense process for cells to cope with metabolic disorders and environmental stresses, and plays a key role in many diseases[77]. It is usually induced by factors such as nutrient loading, hypoxia or excessive reactive oxygen species (ROS), thus breaking the metabolic homeostasis[78,79]. Under this condition, cells initiate defense mechanisms, such as removing damaged cells, activating antioxidant defense and energy redistribution, to maintain survival and function[80]. It was found that the expression of DAXX was often induced in this process, accompanied by translocation from nucleus to cytoplasm, which provided a spatial basis for its regulation of multiple signaling pathways[81].
On the one hand, ROS are able to activate the ASK1 – JNK signaling axis, thereby inducing apoptosis to clear damaged cells[82]. In the cytosolic environment, DAXX can act as a binding scaffold for ASK1, promote the activation of JNK pathway, promote apoptosis, and help the body to remove severely damaged cells in time and maintain tissue homeostasis[9].On the other hand, cells also enhance stress tolerance through antioxidant pathways[83]. The selective autophagy receptor SQSTM1 (sequestosome 1, SQSTM1)/p62 can mediate the degradation of kelch-like ECH-associated protein 1 (KEAP1), thereby releasing the transcription factor Nrf2. Promote the expression of antioxidant genes[84].During this process, the C-terminal domain of DAXX can interact with SQSTM1/p62, inducing its oligomerization and enhancing its liquid–liquid phase separation (LLPS) capacity, thereby promoting the formation of larger SQSTM1 condensates[85]. These condensates sequester KEAP1 in the cytoplasm, thereby preventing KEAP1-mediated ubiquitination and degradation of Nrf2, allowing Nrf2 to stabilize and translocate into the nucleus, where it subsequently activates the transcription of downstream antioxidant genes[85].Further studies have shown that this process does not depend on the PB1 domain of SQSTM1, but on the specific site of DAXX to enhance protein interaction[86,87]. Notably, during the regulation of reactive oxygen species (ROS) clearance, DAXX not only influences metabolic homeostasis but also exerts profound effects on the process of cellular senescence, suggesting a dual role that bridges metabolism and aging.
4. The Role of DAXX in the Aging Process
With the increase of age, the tissues and cells of the body will gradually enter the aging state. Aging is a process of functional degeneration of the body over time, with the gradual accumulation of DNA damage, the decline of repair efficiency and the change of chromatin structure[88].Telomeres keep shortening, making cells more susceptible to senescence[89]. At the same time, elevated levels of reactive oxygen species can damage proteins and lipids[90].In addition, the long-term effects of chronic low-grade inflammation further disturb tissue homeostasis[91]. These mechanisms are intertwined, making aging highly complex and heterogeneous, and providing a fundamental perspective for revealing key regulatory factors and potential intervention strategies. At the same time, DAXX, as a chromatin and stress-related regulatory protein, is gradually being concerned about its potential role in the aging process(Figure 2).
4.1. DNA Damage Repair
DNA damage is an important factor in promoting cell aging[92]. In the process of aging, cells are more prone to double strand breaks, base modification and replication stress, which will destroy genome stability and accelerate aging if not repaired for a long time[88].DNA damage response mainly includes recognition, signal transduction and repair[88].However, it is often disturbed in senescent cells, resulting in the accumulation of abnormal signals[93]. In recent years, in addition to classical repair factors, DAXX has been regarded as a potential key molecule linking DNA damage and aging because of its regulatory role in DNA damage response[7].
The first is the damage sensing stage. When the double-strand break occurs, the histone variant γH2AX will rapidly deposit in the damaged area, and further recruit and activate checkpoint kinase 2 (CHK2), thus amplifying the initial signal[94]. Among them, DAXX is involved in damage recognition by co-localization with PML nuclear bodies in the nucleus and limits the fragmentation signal[95]. PML nuclear body is an important DNA damage sensing structure in the nucleus[96].Therefore, this localization indicates that the role of DAXX in this phase depends on its nuclear distribution. If DAXX is absent, the levels of γH2ax and p-CHK2 are significantly increased, and DNA damage signals are amplified, accompanied by the accumulation of tumor suppressor protein p53 and the overexpression of its downstream cyclin-dependent kinase inhibitor p21, which promotes cells to enter the senescence state in advance[95].
Second is the signal transduction stage. During this phase, PML nuclear bodies act as key signaling platforms and cooperate with ATM/ATR to ensure the transmission of DNA damage response signals[97]. Meanwhile, HDACs are recruited to induce transient chromatin compaction, facilitating the assembly of repair factors. DNA methyltransferase 1 (DNMT1) is rapidly and transiently recruited to DNA double-strand breaks, where it exerts a significant influence on the regulation of the DNA damage response and the rate of DSB repair[98]. In this step, DAXX binds to the PML nuclear body via its C-terminal SIM and is enriched in the nucleus by phosphorylation by the tyrosine kinase casein kinase 2(CK2)[15]. This intranuclear enrichment is a prerequisite for its regulation of injury signaling. It has been shown that DAXX can form complexes with HDAC and DNMT1 in the nucleus[99].It is suggested that it may assist the amplification of ATM/ATR signals by regulating the nuclear chromatin environment.
Finally, the stage of damage repair. At this stage, ATRX, a chromatin remodeling factor, is responsible for depositing H3.3 in heterochromatin and repetitive sequence regions to maintain the stability of nucleosome structure and provide the necessary platform for repair factors[100]. DAXX acts as a histone chaperone in the nucleus and cooperates with ATRX to efficiently deliver H3.3 to specific genomic regions, thereby ensuring the accuracy of repair[101]. This nuclear location-dependent histone loading process is a critical step in maintaining genome stability. If this mechanism is impaired, aberrant structural G-quadruplexes in DNA may gradually accumulate, leading to a decrease in repair accuracy and further amplification of the damage signal[102,103,104].
In conclusion, the localization of DAXX in the nucleus is the basis of its involvement in various aspects of DNA damage response. In the process of perception, transmission and repair, DAXX participates in signal recognition and transmission, and maintains chromatin homeostasis as a histone chaperone, relying on its distribution in nuclear structures such as nuclear bodies, chromatin and heterochromatin. In addition, its interaction with regulatory factors such as HDACs and DNMT1 in the nucleus helps to regulate the strength and timing of repair signals, thus playing a key role in the maintenance of genome stability and the regulation of cell senescence.
4.2. Maintenance of Telomeres
Telomeres, located at the ends of chromosomes, are composed of repetitive sequences and specific binding proteins, which can maintain the integrity of chromosomes[105]. With cell division, telomeres gradually shorten, and when they exceed the critical threshold, they will trigger DNA damage response and push cells into the aging state[89]. The stability of telomeres depends on factors such as telomere length, structural protection, and chromatin modification[89]. This also provides an entry point for DAXX to link telomere maintenance to the process of cell senescence.
In the process of telomere elongation, telomeric repeat-containing RNA (TERRA) can form telR.-Loops, promote homologous recombination repair mechanisms that drive alternative telomere elongation (alternative lengthening of telomeres, ALT)[106]. Decreased levels of H3K9me3 at telomeres have been shown to significantly enhance ALT activity[107],suggesting a negative correlation between heterochromatin modifications and ALT[108,109]. DAXX forms a complex with ATRX in the telomeric chromatin environment, limiting aberrant ALT activation by inhibiting TERRA expression[110]. The stability of telomere structure depends on TIN2 (TRF1-interacting nuclear factor 2)[111].When TIN2 is absent, the DAXX/ATRX complex accumulates abnormally at telomere ends and detaches from telomeres during cell differentiation[112],Which in turn induces DNA damage and impedes differentiation. Because the DAXX/ATRX complex is capable of both depositing H3.3 and promoting H3K9me3 modification[19], its localization within telomeric chromatin is not only a prerequisite for the execution of these functions but also suggests that, in the absence of TIN2, DAXX may partially compensate for defective telomere protection by enhancing heterochromatin stability.
4.3. Oxidative Stress and Inflammatory Aging
Oxidative stress is considered to be an important driving force after DNA damage and telomere dysfunction during aging, and its core mechanism is that intracellular ROS levels continue to rise to break the homeostasis[92]. Excessive ROS can damage proteins, lipids and nucleic acids, and accelerate aging[90]. In this process, DAXX can activate p62 – Nrf2 pathway through LLPS and enhance ROS scavenging capacity[85]. This mechanism echoes the mode of action in metabolic stress, suggesting that DAXX closely links metabolic stress to the driving force of aging by regulating the Nrf2 – ROS network.
Under the background of continuous oxidative stress, the aging process is often accompanied by "inflammatory aging", which is characterized by low but persistent chronic inflammation[113]. It is mainly driven by the long-term release of senescence-associated secretory phenotype (SASP) factors (such as IL-6, IL-8, IL-1β, etc.)[91], which can amplify damage signals between cells and accelerate functional degradation. In this process, sustained activation of the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) signaling pathway is considered a key molecular basis for maintaining chronic low-grade inflammation and amplifying aging-associated tissue damage[114]. Studies have shown that DAXX can regulate both the canonical and non-canonical NF-κB pathways: on the one hand, DAXX can bind to NF-κB p65 (RelA) and inhibit its acetylation level and DNA binding activity, thereby blocking the transcription of pro-inflammatory genes such as IL-8 and IκBα[115]; On the other hand, DAXX can also inhibit the expression of its downstream target gene cIAP2 by interacting with RelB, thus limiting the continuous activation and diffusion of inflammatory signals[116]. During aging, chronic inflammatory activation of microglia is considered to be the main manifestation of inflammatory aging in the central nervous system[117]. Against this background, studies on neuroinflammation-related models have further revealed its specific mode of action: in Pdcd4-deficient microglia, DAXX is more likely to localize in the nucleus and enhance its interaction with PPARγ, thus promoting the transcription of anti-inflammatory factor IL-10; Under LPS-stimulated conditions, this regulatory axis reduces inflammatory cell infiltration in neural tissue and alleviates neuroinflammation-related phenotypes[118]. In addition, at the SASP molecular level, DAXX can also be enriched in the IL-6 promoter region and recruit the deacetylase HDAC1 to reduce the level of local histone H3 acetylation, thereby selectively inhibiting the transcription of IL-6[119], suggesting that it may limit the sustained expression of key amplification factors during inflammatory aging through epigenetic means. These findings suggest that DAXX may affect the formation of inflammatory microenvironment during aging by regulating inflammation-related transcriptional programs, and play a potential regulatory role in the intersection of inflammation and aging.
5. The Role of DAXX in Inflammation and Immune Homeostasis
Immune homeostasis is an important basis for the body to resist infection, eliminate abnormal cells and maintain homeostasis, which depends on the synergy of innate and adaptive immunity, as well as the fine regulation of immune tolerance and immune surveillance mechanisms[120,121]. The maintenance of immune homeostasis depends not only on the development and functional status of immune cells, but also on the strict control of transcriptional regulation and chromatin status on the expression threshold of immune-related genes[122]. When immune regulation is out of balance, abnormal activation or inhibition of immune response may weaken the body's defense capability and promote the occurrence and development of chronic diseases and tumors[120]. Recent studies have shown that DAXX, as a nuclear location-dependent chromatin regulatory protein, plays an important role in the multi-level regulation of immune homeostasis. By participating in interferon-associated immune responses[123,124], maintaining epigenetic silencing of endogenous retroviruses(ERV)[7,125], and regulating the activation threshold of key immune signaling pathways such as cGAS – STING [126,127], It affects the intensity, persistence and immune tolerance of innate and adaptive immune responses. These processes indicate that the maintenance of immune homeostasis is highly dependent on the cooperation of epigenetic and signal regulation in the nucleus, and DAXX is at the key intersection of this regulatory network.
Innate immunity, as the primary line of defense against exogenous pathogens and endogenous danger signals, relies on IFN-I signaling pathways to initiate defense responses rapidly in the early stage[128]. The PML nuclear body acts as a limiting factor in this process and is able to inhibit transcriptional activation of the viral genome[129]. DAXX has been shown to be an important component of this mechanism by binding to the PML nuclear body and acting as a histone chaperone to deposit H3.3 onto the viral genome, prompting chromatization and heterochromatization, thereby inhibiting early viral gene transcription[130]. DAXX also exhibits extensive restriction in RNA virus infections. For example, in the HIV-1 model, DAXX inhibits reverse transcription and decapsidation through a SUMO-dependent mechanism, thereby enhancing interferon-associated antiviral responses[123].In SARS-CoV-2 infection, CRISPR/Cas9 screening confirmed that DAXX is a key host restriction factor, and its deletion will significantly improve the efficiency of viral replication[126]. A similar phenomenon was seen in a pancreas-specific DAXX knockout mouse model, in which the ERV pathway is significantly activated, and virus-like transcripts accumulate and stimulate innate immune receptors, leaving the body chronically overactivated[7].Thus, DAXX prevents the occurrence of immune hyperresponsiveness by maintaining ERV silencing and limiting viral replication.
Adaptive immunity depends on the differentiation and functional maintenance of T and B cells, and its core feature is to provide lasting protection through antigen-specific recognition and immune memory[131].T cell receptor (TCR) will trigger the activation and expansion of T cells after recognizing antigens, thus ensuring the strength of immune response[120]. It has been shown that T-cell specific deletion of DAXX results in a significant decrease in the number of peripheral mature T cells, and these cells are more susceptible to apoptosis after TCR activation, suggesting that DAXX plays a protective role in the survival of activated T cells and avoids premature attenuation of immune response[132].Meanwhile, B cell differentiation determines the quality and persistence of antibody response, which is regulated by multiple transcription factors, including paired box protein 5 (Pax5), which is the core factor driving B cell lineage development[133]. Studies have shown that DAXX can assist Pax5 to perform both transcriptional activation and repression functions, thus playing an intermediate role in the early stage of B cell differentiation[134].In addition, DAXX is also an essential factor in the IFN-α-mediated inhibitory pathway to protect pro-B cells from inhibition[135],thereby ensuring as to ensure the development process and functional maturity of B cells. These findings suggest that DAXX plays an important role in the maintenance of adaptive immune homeostasis by supporting the survival of T cells to maintain the persistence of immune responses and by participating in transcriptional regulation during the differentiation of B cells to promote their early development, thereby indirectly affecting antigen-specific recognition and the formation of immune memory.
Immune tolerance is a key mechanism for the body to prevent the immune system from attacking its own components, once destroyed, it may lead to autoimmune diseases. Abnormally high expression of pro-inflammatory factors such as IL-6 can drive the immune system to attack autoantigens[136]; In view of the transcriptional repression of IL-6 expression by DAXX described above[119], the functional significance of this regulatory mechanism in the maintenance of immune tolerance has also been gradually concerned, suggesting that DAXX may be involved in regulating the tolerance threshold of immune response by limiting the continuous accumulation of pro-inflammatory signals. In addition, if hematopoietic stem cells are excessively biased towards granulocytic differentiation, it will lead to an increase in the proportion of pro-inflammatory cells[137] and destroy immune homeostasis; In combination with the known role of DAXX in the inhibition of inflammatory signaling and the regulation of immune cell survival[119,132], it is suggested that it may also have the potential function of inhibiting pro-inflammatory bias and maintaining the diversity of immune cell population during hematopoietic differentiation. These findings suggest that DAXX participates in the maintenance of immune tolerance through multiple mechanisms and avoids the occurrence of autoimmune reactions.
Previous studies have demonstrated that DAXX exerts an inhibitory role in the regulation of inflammatory signaling[119].Together with its function in immune cell survival[132], this suggests that DAXX may also play a potential role during hematopoietic differentiation by restraining pro-inflammatory bias and maintaining the diversity of immune cell populations.These findings suggest that DAXX participates in the maintenance of immune tolerance through multiple mechanisms and avoids the occurrence of autoimmune reactions.
Tumor immunity depends on the effective recognition and elimination of tumor antigens by the immune system, and immune escape is a key mechanism for the long-term survival and expansion of tumor cells[138]. The cyclic GMP-AMP synthase (cGAS) – interferon gene stimulating factor (stimulator of interferon genes, STING) pathway plays a central role in tumor immunity. Its activation can induce the production of interferon and a variety of immune effector molecules, thereby promoting anti-tumor immunity[139]. It has been shown that overexpression of DAXX inhibits the activation of the cGAS – STING pathway, impairs the production of immune effector molecules, and helps tumor cells achieve immune escape[140]. In viral infection studies, herpes simplex virus type 1 (HSV-1) uses its encoded immediate early protein ICP0 to degrade PML and MORC family protein 3, thereby disrupting the integrity of the PML-NB complex and relieving restrictions including DAXX, ultimately promoting transcriptional activation of viral genes[141].Such disruption of PML-NB architecture suggests an important role for nuclear DAXX in maintaining immune-related chromatin states and gene accessibility.
Therefore, DAXX is involved in innate defense, lymphocyte homeostasis, immune tolerance and tumor surveillance. By regulating chromatin accessibility in the nucleus, participating in transcription complex assembly, and regulating signaling thresholds, DAXX establishes a dynamic balance between preventing over-activation of the immune response and maintaining an effective immune response. Its function is obviously context-dependent in different cell types and pathological environments, suggesting the complexity of this regulatory network. If future studies can further clarify the direct molecular links between DAXX and key immune pathways, it will help to reveal the relationship between DAXX and aging, infection and tumorigenesis, and may provide new ideas for the intervention of immune-related diseases(Figure 3).
6. Conclusions
In this review, we summarized the biological characteristics of DAXX from the aspects of structure and function, and revealed the core position of its nuclear localization in coordinating multiple life processes. As the junction of chromatin regulation and signal integration, DAXX can reshape the transcriptional environment, regulate epigenetic modifications and affect key signaling thresholds, thus forming a continuous and highly coupled regulatory network at multiple system levels, such as energy metabolism, cell aging and immune balance. Its role is not only limited to the regulation of a single molecular pathway, but also provides structural basis and functional support for the maintenance of cellular homeostasis and environmental adaptation through the cooperation of spatial localization, chromatin state and signal integration.
The diversity of DAXX is reflected in its high degree of "environmental dependence" and "bidirectionality". In metabolic regulation, DAXX promotes lipid synthesis by activating SREBP1/2 and fatty acid oxidation by activating MPK38, depending on the specific metabolic state of the cell. During aging, DAXX cooperates with ATRX in the nucleus to maintain the integrity of telomeres and genomes by stabilizing the deposition of H3.3, which is one of the core mechanisms to delay aging. In addition, it can also enhance the antioxidant capacity of cells by activating the Nrf2 pathway, so as to resist the oxidative stress damage caused by aging and chronic inflammation. At the level of immune regulation, DAXX affects the intensity and persistence of immune response by fine-tuning the transcription program of immune-related genes, and participates in the body's defense response to pathogen stimulation. It is this integrated function across multiple biological levels, such as metabolic homeostasis, aging regulation and immune homeostasis, that makes DAXX a key molecule in understanding the interweaving of these processes.
Therefore, further exploration of the function of DAXX will not only help us to understand its role in normal physiological and pathological conditions, but also provide new ideas for finding the common molecular roots of many chronic diseases, such as obesity, diabetes and neurodegenerative diseases. Future studies can further focus on the specific molecular mechanisms of DAXX and core signaling pathways such as NF-κB, and explore the specific differences of its functions in different tissues and pathological environments, so as to clarify its location in aging and chronic diseases, which will help to promote DAXX from molecular mechanism research to clinical application. It also provides theoretical support for precise intervention(Figure 4).
Author Contributions
Conceptualization: J.Z., L.Z., and Q.T.; first draft preparation: J.Z. and L.Z.; review and editing:Q.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China(82474115), Technology Innovation team of Hunan Province (2021RC4064) and Basic Research Center for the Prevention of Vascular Diseases with Chinese and Western Medicine Integration.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
We are grateful to the colleagues from Qinhui Tuo’s laboratory for their insightful suggestions and comments during the discussion of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 4HB | Four-helix bundle |
| ACC | Acetyl-CoA carboxylase |
| ALT | Alternative lengthening of telomeres |
| AMPK | AMP-activated protein kinase |
| AR | Androgen receptor |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| ATR | Ataxia telangiectasia and Rad3-related protein |
| ATRX | Alpha-thalassemia/mental retardation syndrome X-linked |
| CALML3-AS1 | CALML3 antisense RNA 1 |
| cGAS | Cyclic GMP-AMP synthase |
| CHK2 | Checkpoint kinase 2 |
| CK2 | Casein kinase 2 |
| CPT1A | Carnitine palmitoyltransferase 1A |
| DAXX | Death domain-associated protein 6 |
| DGAT | Diacylglycerol acyltransferase |
| DNMT1 | DNA methyltransferase 1 |
| DSB | DNA double-strand break |
| ERV | Endogenous retrovirus |
| FASN | Fatty acid synthase |
| G6Pase | Glucose-6-phosphatase |
| GLUT | Glucose transporter |
| H3.3 | Histone H3 variant 3 |
| H3K4me3 | Trimethylation of histone H3 lysine 4 |
| H3K9me3 | Trimethylation of histone H3 lysine 9 |
| HDAC | Histone deacetylase |
| HBD | Histone-binding domain |
| HK | Hexokinase |
| HMGCR | 3-hydroxy-3-methylglutaryl-CoA reductase |
| HMGCS1 | 3-hydroxy-3-methylglutaryl-CoA synthase 1 |
| IFN-I | Type I interferon |
| JNK | c-Jun N-terminal kinase |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| KIF5B | Kinesin family member 5B |
| LDLR | Low-density lipoprotein receptor |
| LLPS | Liquid–liquid phase separation |
| MASLD | Metabolic dysfunction-associated steatotic liver disease |
| MASH | Metabolic dysfunction-associated steatohepatitis |
| MDM2 | Mouse double minute 2 homolog |
| MPK38 | Maternal embryonic leucine zipper kinase (MELK) |
| mTOR | Mechanistic target of rapamycin |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| PB1 | Phox and Bem1 domain |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PFK1 | Phosphofructokinase 1 |
| PK | Pyruvate kinase |
| PML | Promyelocytic leukemia protein |
| PML-NB | Promyelocytic leukemia nuclear body |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| RASSF1C | Ras association domain family member 1C |
| ROS | Reactive oxygen species |
| SCAP | SREBP cleavage-activating protein |
| SCD1 | Stearoyl-CoA desaturase 1 |
| SIM | SUMO-interacting motif |
| SQSTM1 | Sequestosome 1 |
| SREBP | Sterol regulatory element-binding protein |
| STING | Stimulator of interferon genes |
| SUMO | Small ubiquitin-like modifier |
| TAG | Triacylglycerol |
| TERRA | Telomeric repeat-containing RNA |
| TIN2 | TRF1-interacting nuclear factor 2 |
| TCR | T cell receptor |
References
- Kim, H H; Dixit, V D. Metabolic regulation of immunological aging[J]. Nature Aging 2025, 5(8), 1425–1440. [Google Scholar] [CrossRef]
- Franceschi, C; Garagnani, P; Parini, P; et al. Inflammaging: a new immune-metabolic viewpoint for age-related diseases[J]. Nature Reviews. Endocrinology 2018, 14(10), 576–590. [Google Scholar] [CrossRef]
- Allis, C D; Jenuwein, T. The molecular hallmarks of epigenetic control[J]. Nature Reviews. Genetics 2016, 17(8), 487–500. [Google Scholar] [CrossRef]
- Choi, J; Kim, T; Cho, E J. HIRA vs. DAXX: The two axes shaping the histone H3.3 landscape[J]. Experimental & Molecular Medicine 2024, 56(2), 251–263. [Google Scholar]
- Mahmud, I; Tian, G; Wang, J; et al. DAXX drives de novo lipogenesis and contributes to tumorigenesis[J]. Nature Communications 2023, 14(1), 1927. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P W; Elsaesser, S J; Noh, K M; et al. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres[J]. Proceedings of the National Academy of Sciences of the United States of America 2010, 107(32), 14075–14080. [Google Scholar] [CrossRef]
- Wasylishen, A R; Sun, C; Moyer, S M; et al. Daxx maintains endogenous retroviral silencing and restricts cellular plasticity in vivo[J]. Science Advances 2020, 6(32), eaba8415. [Google Scholar] [CrossRef] [PubMed]
- Kiriakidou, M; Driscoll, D A; Lopez-Guisa, J M; et al. Cloning and expression of primate daxx cDNAs and mapping of the human gene to chromosome 6p21.3 in the MHC region[J]. DNA and cell biology 1997, 16(11), 1289–1298. [Google Scholar] [CrossRef]
- Yang, X; Khosravi-Far, R; Chang, H Y; et al. Daxx, a novel fas-binding protein that activates JNK and apoptosis[J]. Cell 1997, 89(7), 1067–1076. [Google Scholar] [CrossRef]
- Escobar-Cabrera, E; Lau, D K W; Giovinazzi, S; et al. Structural characterization of the DAXX N-terminal helical bundle domain and its complex with Rassf1C[J]. Structure (London, England: 1993) 2010, 18(12), 1642–1653. [Google Scholar] [CrossRef]
- Clatterbuck Soper, S F; Walker, R L; Pineda, M A; et al. Cancer-associated DAXX mutations reveal a critical role for ATRX localization in ALT suppression[J/OL]. bioRxiv: The Preprint Server for Biology 2024, 2024.11.18.624165. [Google Scholar] [CrossRef]
- Mahmud, I; Liao, D. DAXX in cancer: Phenomena, processes, mechanisms and regulation[J]. Nucleic Acids Research 2019, 47(15), 7734–7752. [Google Scholar] [CrossRef]
- Bogolyubova, I; Bogolyubov, D. DAXX is a crucial factor for proper development of mammalian oocytes and early embryos[J]. International Journal of Molecular Sciences 2021, 22(3), 1313. [Google Scholar] [CrossRef] [PubMed]
- Lin, D Y; Huang, Y S; Jeng, J C; et al. Role of SUMO-interacting motif in daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors[J]. Molecular Cell 2006, 24(3), 341–354. [Google Scholar] [CrossRef]
- Gao, J; Liu, T; Yang, D; et al. The dynamic regulation of daxx-mediated transcriptional inhibition by SUMO and PML NBs[J]. International journal of molecular sciences 2025, 26(14), 6703. [Google Scholar] [CrossRef]
- Elsässer, S J; Huang, H; Lewis, P W; et al. DAXX envelops a histone H3.3-H4 dimer for H3.3-specific recognition[J]. Nature 2012, 491(7425), 560–565. [Google Scholar] [CrossRef] [PubMed]
- Chang, H Y; Nishitoh, H; Yang, X; et al. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein daxx[J]. Science (New York, N.Y.) 1998, 281(5384), 1860–1863. [Google Scholar] [CrossRef]
- Pan, W W; Zhou, J J; Liu, X M; et al. Death domain-associated protein DAXX promotes ovarian cancer development and chemoresistance[J]. The Journal of Biological Chemistry 2013, 288(19), 13620–13630. [Google Scholar] [CrossRef]
- Carraro, M; Hendriks, I A; Hammond, C M; et al. DAXX adds a de novo H3.3K9me3 deposition pathway to the histone chaperone network[J]. Molecular Cell 2023, 83(7), 1075–1092.e9. [Google Scholar] [CrossRef]
- Yeung, P L; Chen, L Y; Tsai, S C; et al. Daxx contains two nuclear localization signals and interacts with importin alpha3[J]. Journal of Cellular Biochemistry 2008, 103(2), 456–470. [Google Scholar] [CrossRef]
- Trier, I; Black, E M; Joo, Y K; et al. ATR protects centromere identity by promoting DAXX association with PML nuclear bodies[J]. Cell Reports 2023, 42(5), 112495. [Google Scholar] [CrossRef]
- Torii, S; Egan, D A; Evans, R A; et al. Human daxx regulates fas-induced apoptosis from nuclear PML oncogenic domains (PODs)[J]. The EMBO journal 1999, 18(21), 6037–6049. [Google Scholar] [CrossRef]
- Chang, F T M; McGhie, J D; Chan, F L; et al. PML bodies provide an important platform for the maintenance of telomeric chromatin integrity in embryonic stem cells[J]. Nucleic Acids Research 2013, 41(8), 4447–4458. [Google Scholar] [CrossRef]
- Chen, C; Sun, X; Xie, W; et al. Opposing biological functions of the cytoplasm and nucleus DAXX modified by SUMO-2/3 in gastric cancer[J]. Cell Death & Disease 2020, 11(7), 514. [Google Scholar]
- Santiago, A; Godsey, A C; Hossain, J; et al. Identification of two independent SUMO-interacting motifs in daxx: evolutionary conservation from drosophila to humans and their biochemical functions[J]. Cell Cycle (Georgetown, Tex.) 2009, 8(1), 76–87. [Google Scholar] [CrossRef]
- Vladimirova, O; De Leo, A; Deng, Z; et al. Phase separation and DAXX redistribution contribute to LANA nuclear body and KSHV genome dynamics during latency and reactivation[J]. PLoS pathogens 2021, 17(1), e1009231. [Google Scholar] [CrossRef]
- Mascle, X H; Gagnon, C; Wahba, H M; et al. Acetylation of SUMO1 alters interactions with the SIMs of PML and daxx in a protein-specific manner[J]. Structure (london, England: 1993) 2020, 28(2), 157–168.e5. [Google Scholar] [CrossRef] [PubMed]
- Dorosz, K; Majewska, L; Kijowski, J. Structure and function of PML nuclear bodies: a brief overview of key cellular roles[J]. Biomolecules 2025, 15(9), 1291. [Google Scholar] [CrossRef] [PubMed]
- Abou-Ghali, M; Lallemand-Breitenbach, V. PML nuclear bodies: the cancer connection and beyond[J]. Nucleus (austin, Tex.) 2024, 15(1), 2321265. [Google Scholar] [CrossRef] [PubMed]
- Lin, D Y; Fang, H I; Ma, A H; et al. Negative modulation of androgen receptor transcriptional activity by daxx[J]. Molecular and Cellular Biology 2004, 24(24), 10529–10541. [Google Scholar] [CrossRef]
- Jung, Y S; Kim, H Y; Kim, J; et al. Physical interactions and functional coupling between daxx and sodium hydrogen exchanger 1 in ischemic cell death[J]. Journal of Biological Chemistry 2008, 283(2), 1018–1025. [Google Scholar] [CrossRef]
- Xiong, G; Li, L; Sun, S; et al. Subcellular localization of DAXX influence ox-LDL induced apoptosis in macrophages[J]. Molecular Biology Reports 2014, 41(11), 7183–7190. [Google Scholar] [CrossRef]
- Gulve, N; Su, C; Deng, Z; et al. DAXX-ATRX regulation of p53 chromatin binding and DNA damage response[J]. Nature Communications 2022, 13(1), 5033. [Google Scholar] [CrossRef]
- Pinto, L M; Pailas, A; Bondarchenko, M; et al. DAXX promotes centromeric stability independently of ATRX by preventing the accumulation of R-loop-induced DNA double-stranded breaks[J]. Nucleic Acids Research 2023, 52(3), 1136–1155. [Google Scholar] [CrossRef] [PubMed]
- Salsman, J; Rapkin, L M; Margam, N N; et al. Myogenic differentiation triggers PML nuclear body loss and DAXX relocalization to chromocentres[J]. Cell Death and Disease 2017, 8(3), e2724. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S; Unnikrishnan, A G; Baruah, M P; et al. Metabolic and energy imbalance in dysglycemia-based chronic disease[J]. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy 2021, 14, 165–184. [Google Scholar] [CrossRef]
- H L, S W, J W, et al. Energy metabolism in health and diseases[J]. Signal Transduction and Targeted Therapy 2025, 10(1), 1–71.
- Chandel, N S. Lipid metabolism[J]. Cold Spring Harbor Perspectives in Biology 2021, 13(9), a040576. [Google Scholar] [CrossRef]
- Hotamisligil, G S. Inflammation, metaflammation and immunometabolic disorders[J]. Nature 2017, 542(7640), 177–185. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J; Lo, C H. Editorial: Lipid metabolism dysregulation in obesity-related diseases and neurodegeneration[J]. Frontiers in Endocrinology 2025, 16, 1564003. [Google Scholar] [CrossRef]
- Samuel, V T; Shulman, G I. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux[J]. Journal of Clinical Investigation 2016, 126(1), 12–22. [Google Scholar] [CrossRef]
- Brown, M S; Goldstein, J L. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor[J]. Cell 1997, 89(3), 331–340. [Google Scholar] [CrossRef] [PubMed]
- Lee, S H; Lee, J H; Im, S S. The cellular function of SCAP in metabolic signaling[J]. Experimental & Molecular Medicine 2020, 52(5), 724–729. [Google Scholar]
- Horton, J D; Goldstein, J L; Brown, M S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver[J]. Journal of Clinical Investigation 2002, 109(9), 1125–1131. [Google Scholar] [CrossRef]
- Chandrasekaran, P; Weiskirchen, R. The role of SCAP/SREBP as central regulators of lipid metabolism in hepatic steatosis[J]. International journal of molecular sciences 2024, 25(2), 1109. [Google Scholar] [CrossRef]
- Zhao, Q; Lin, X; Wang, G. Targeting SREBP-1-mediated lipogenesis as potential strategies for cancer[J]. Frontiers in Oncology 2022, 12, 952371. [Google Scholar] [CrossRef]
- Eberlé, D; Hegarty, B; Bossard, P; et al. SREBP transcription factors: master regulators of lipid homeostasis[J]. Biochimie 2004, 86(11), 839–848. [Google Scholar] [CrossRef]
- Li, T P; Sun, S W; Xiong, G Z; et al. Direct interaction of daxx and androgen receptor is required for their regulatory activity in cholesterol biosynthesis[J]. Pharmacology 2021, 106(1-2), 29–36. [Google Scholar] [CrossRef]
- Swinnen, J V; Verhoeven, G. Androgens and the control of lipid metabolism in human prostate cancer cells[J]. Journal of Steroid Biochemistry and Molecular Biology 1998, 65(1-6), 191–198. [Google Scholar] [CrossRef] [PubMed]
- Vertegaal, A C O. Signalling mechanisms and cellular functions of SUMO[J]. Nature Reviews. Molecular Cell Biology 2022, 23(11), 715–731. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T; Fan, J; Abu-Zaid, A; et al. Nuclear mTOR signaling orchestrates transcriptional programs underlying cellular growth and metabolism[J]. Cells 2024, 13(9), 781. [Google Scholar] [CrossRef]
- Hajirahimkhan, A; Brown, K A; Clare, S E; et al. SREBP1-dependent metabolism as a potential target for breast cancer risk reduction[J]. Cancers 2025, 17(10), 1664. [Google Scholar] [CrossRef]
- Anand, S; Patel, T N. Integrating the metabolic and molecular circuits in diabetes, obesity and cancer: A comprehensive review[J]. Discover Oncology 2024, 15(1), 779. [Google Scholar] [CrossRef]
- Rong, S; Xia, M; Vale, G; et al. DGAT2 inhibition blocks SREBP-1 cleavage and improves hepatic steatosis by increasing phosphatidylethanolamine in the ER[J]. Cell Metabolism 2024, 36(3), 617–629.e7. [Google Scholar] [CrossRef]
- Juhl, A D; Wüstner, D. Pathways and mechanisms of cellular cholesterol efflux-insight from imaging[J]. Frontiers in Cell and Developmental Biology 2022, 10, 834408. [Google Scholar] [CrossRef]
- Chen, L; Zhao, Z W; Zeng, P H; et al. Molecular mechanisms for ABCA1-mediated cholesterol efflux[J]. Cell Cycle (Georgetown, Tex.) 2022, 21(11), 1121–1139. [Google Scholar] [CrossRef] [PubMed]
- Parton, R G; del Pozo, M A. Caveolae as plasma membrane sensors, protectors and organizers[J]. Nature Reviews. Molecular Cell Biology 2013, 14(2), 98–112. [Google Scholar] [CrossRef]
- Tuo, Q H; Liang, L; Zhu, B Y; et al. Effect of daxx on cholesterol accumulation in hepatic cells[J]. World Journal of Gastroenterology 2008, 14(3), 435–440. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I R; Joshi, M. CPT1A-mediated fat oxidation, mechanisms, and therapeutic potential[J]. Endocrinology 2020, 161(2), bqz046. [Google Scholar] [CrossRef] [PubMed]
- Fang, C; Pan, J; Qu, N; et al. The AMPK pathway in fatty liver disease[J]. Frontiers in Physiology 2022, 13, 970292. [Google Scholar] [CrossRef]
- Seong, H A; Manoharan, R; Ha, H. DAXX ameliorates metabolic dysfunction in mice with diet-induced obesity by activating the AMP-activated protein kinase-related kinase MPK38/MELK[J]. Biochemical and Biophysical Research Communications 2021, 572, 164–170. [Google Scholar] [CrossRef]
- Ramatchandirin, B; Pearah, A; He, L. Regulation of liver glucose and lipid metabolism by transcriptional factors and coactivators[J]. Life (Basel, Switzerland) 2023, 13(2), 515. [Google Scholar] [CrossRef]
- Han, H S; Kang, G; Kim, J S; et al. Regulation of glucose metabolism from a liver-centric perspective[J]. Experimental & Molecular Medicine 2016, 48(3), e218. [Google Scholar]
- Drobiova, H; Alhamar, G; Ahmad, R; et al. GLUT4 trafficking and storage vesicles: molecular architecture, regulatory networks, and their disruption in insulin resistance[J]. International journal of molecular sciences 2025, 26(15), 7568. [Google Scholar] [CrossRef]
- Richter, E A; Bilan, P J; Klip, A. A comprehensive view of muscle glucose uptake: Regulation by insulin, contractile activity, and exercise[J]. Physiological Reviews 2025, 105(3), 1867–1945. [Google Scholar] [CrossRef]
- Knudsen, J R; Persson, K W; Henriquez-Olguin, C; et al. Microtubule-mediated GLUT4 trafficking is disrupted in insulin-resistant skeletal muscle[J]. eLife 2023, 12, e83338. [Google Scholar] [CrossRef] [PubMed]
- Mao, G; Liu, J. CALML3-AS1 enhances malignancies and stemness of small cell lung cancer cells through interacting with DAXX protein and promoting GLUT4-mediated aerobic glycolysis[J]. Toxicology and Applied Pharmacology 2025, 495, 117177. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, V S; Vergarajauregui, S; Tsuchiya, Y; et al. Daxx functions as a scaffold of a protein assembly constituted by GLUT4, JNK1 and KIF5B[J]. Journal of Cellular Physiology 2009, 218(2), 416–426. [Google Scholar] [CrossRef]
- van Gerwen, J; Shun-Shion, A S; Fazakerley, D J. Insulin signalling and GLUT4 trafficking in insulin resistance[J]. Biochemical Society Transactions 2023, 51(3), 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S Y; Vander Heiden, M G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation[J]. Annual Review of Cell and Developmental Biology 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Zhao, J; Jin, D; Huang, M; et al. Glycolysis in the tumor microenvironment: A driver of cancer progression and a promising therapeutic target[J]. Frontiers in Cell and Developmental Biology 2024, 12, 1416472. [Google Scholar] [CrossRef]
- Knorr, S; Skakkebæk, A; Just, J; et al. Epigenetic and transcriptomic alterations in offspring born to women with type 1 diabetes (the EPICOM study)[J]. BMC medicine 2022, 20(1), 338. [Google Scholar] [CrossRef]
- Wang, H; Fan, Z; Shliaha, P V; et al. H3K4me3 regulates RNA polymerase II promoter-proximal pause-release[J]. Nature 2023, 615(7951), 339–348. [Google Scholar] [CrossRef]
- Pandya Thakkar, N; Pereira, B M V; Katakia, Y T; et al. Elevated H3K4me3 through MLL2-WDR82 upon hyperglycemia causes jagged ligand dependent notch activation to interplay with differentiation state of endothelial cells[J]. Frontiers in Cell and Developmental Biology 2022, 10, 839109. [Google Scholar] [CrossRef]
- Zhong, W; Hong, C; Zhang, Y; et al. ASH2L-mediated H3K4me3 drives diabetic nephropathy through HIPK2 and Notch1 pathway[J]. Translational Research: the Journal of Laboratory and Clinical Medicine 2024, 264, 85–96. [Google Scholar] [CrossRef]
- Zhang, X; Yang, S; Chen, J; et al. Unraveling the Regulation of Hepatic Gluconeogenesis[J]. Frontiers in Endocrinology 2018, 9, 802. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity[J]. Annual Review of Pharmacology and Toxicology 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease[J]. Cell 2010, 140(6), 900–917. [Google Scholar] [CrossRef] [PubMed]
- Masenga, S K; Kabwe, L S; Chakulya, M; et al. Mechanisms of oxidative stress in metabolic syndrome[J]. International Journal of Molecular Sciences 2023, 24(9), 7898. [Google Scholar] [CrossRef]
- Fulda, S; Gorman, A M; Hori, O; et al. Cellular stress responses: cell survival and cell death[J]. International Journal of Cell Biology 2010, 2010, 214074. [Google Scholar] [CrossRef]
- Khelifi, A F; D’Alcontres, M S; Salomoni, P. Daxx is required for stress-induced cell death and JNK activation[J]. Cell Death & Differentiation 2005, 12(7), 724–733. [Google Scholar]
- Zhao, W; Zhuang, P; Chen, Y; et al. “double-edged sword” effect of reactive oxygen species (ROS) in tumor development and carcinogenesis[J]. Physiological Research 2023, 72(3), 301–307. [Google Scholar] [CrossRef]
- Sykiotis, G P; Bohmann, D. Stress-activated cap’n’collar transcription factors in aging and human disease[J]. Science Signaling 2010, 3(112), re3. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G P. Keap1/Nrf2 signaling pathway[J]. Antioxidants 2021, 10(6), 828. [Google Scholar] [CrossRef]
- Yang, Y; Willis, T L; Button, R W; et al. Cytoplasmic DAXX drives SQSTM1/p62 phase condensation to activate Nrf2-mediated stress response[J]. Nature Communications 2019, 10(1), 3759. [Google Scholar] [CrossRef]
- Xia, Q; Li, Y; Xu, W; et al. Enhanced liquidity of p62 droplets mediated by Smurf1 links Nrf2 activation and autophagy[J]. Cell & Bioscience 2023, 13(1), 37. [Google Scholar]
- Huang, X; Zhang, J; Yao, J; et al. Phase separation of p62: roles and regulations in autophagy[J]. Trends in Cell Biology 2025, 35(10), 854–865. [Google Scholar] [CrossRef] [PubMed]
- Shreeya, T; Ansari, M S; Kumar, P; et al. Senescence: A DNA damage response and its role in aging and neurodegenerative diseases[J]. Frontiers in Aging 2023, 4, 1292053. [Google Scholar] [CrossRef]
- Rossiello, F; Jurk, D; Passos, J F; et al. Telomere dysfunction in ageing and age-related diseases[J]. Nature Cell Biology 2022, 24(2), 135–147. [Google Scholar] [CrossRef]
- Ngo, V; Duennwald, M L. Nrf2 and oxidative stress: a general overview of mechanisms and implications in human disease[J]. Antioxidants (Basel, Switzerland) 2022, 11(12), 2345. [Google Scholar] [CrossRef]
- Olivieri, F; Prattichizzo, F; Grillari, J; et al. Cellular senescence and inflammaging in age-related diseases[J]. Mediators of Inflammation 2018, 2018, 9076485. [Google Scholar] [CrossRef]
- López-Otín, C; Blasco, M A; Partridge, L; et al. The hallmarks of aging[J]. Cell 2013, 153(6), 1194–1217. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. Living on a break: cellular senescence as a DNA-damage response[J]. Nature Reviews. Cancer 2008, 8(7), 512–522. [Google Scholar] [CrossRef]
- Gong, P; Guo, Z; Wang, S; et al. Histone phosphorylation in DNA damage response[J]. International Journal of Molecular Sciences 2025, 26(6), 2405. [Google Scholar] [CrossRef] [PubMed]
- Pan, W W; Yi, F P; Cao, L X; et al. DAXX silencing suppresses mouse ovarian surface epithelial cell growth by inducing senescence and DNA damage[J]. Gene 2013, 526(2), 287–294. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R; Pandolfi, P P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies[J]. Nature Reviews. Molecular Cell Biology 2007, 8(12), 1006–1016. [Google Scholar] [CrossRef]
- Dellaire, G; Ching, R W; Ahmed, K; et al. Promyelocytic leukemia nuclear bodies behave as DNA damage sensors whose response to DNA double-strand breaks is regulated by NBS1 and the kinases ATM, Chk2, and ATR[J]. The Journal of Cell Biology 2006, 175(1), 55–66. [Google Scholar] [CrossRef]
- Fernandez, A; O’Leary, C; O’Byrne, K J; et al. Epigenetic mechanisms in DNA double strand break repair: A clinical review[J]. Frontiers in Molecular Biosciences 2021, 8, 685440. [Google Scholar] [CrossRef] [PubMed]
- Li, H; Leo, C; Zhu, J; et al. Sequestration and inhibition of daxx-mediated transcriptional repression by PML[J]. Molecular and Cellular Biology 2000, 20(5), 1784–1796. [Google Scholar] [CrossRef]
- Juhász, S; Elbakry, A; Mathes, A; et al. ATRX promotes DNA repair synthesis and sister chromatid exchange during homologous recombination[J]. Molecular Cell 2018, 71(1), 11–24.e7. [Google Scholar] [CrossRef]
- Fan, H C; Chen, C M; Chi, C S; et al. Targeting telomerase and ATRX/DAXX inducing tumor senescence and apoptosis in the malignant glioma[J]. International Journal of Molecular Sciences 2019, 20(1), 200. [Google Scholar] [CrossRef]
- Teng, Y C; Sundaresan, A; O’Hara, R; et al. ATRX promotes heterochromatin formation to protect cells from G-quadruplex DNA-mediated stress[J]. Nature Communications 2021, 12(1), 3887. [Google Scholar] [CrossRef]
- Sato, K; Knipscheer, P. G-quadruplex resolution: from molecular mechanisms to physiological relevance[J]. DNA Repair 2023, 130, 103552. [Google Scholar] [CrossRef]
- Wulfridge, P; Sarma, K. Intertwining roles of R-loops and G-quadruplexes in DNA repair, transcription and genome organization[J]. Nature Cell Biology 2024, 26(7), 1025–1036. [Google Scholar] [CrossRef]
- Blackburn, E H. Telomeres and telomerase: the means to the end (nobel lecture)[J]. Angewandte Chemie (International Ed. in English) 2010, 49(41), 7405–7421. [Google Scholar] [CrossRef]
- Azzalin, C M. TERRA and the alternative lengthening of telomeres: A dangerous affair[J]. FEBS letters 2025, 599(2), 157–165. [Google Scholar] [CrossRef] [PubMed]
- Gauchier, M; Kan, S; Barral, A; et al. SETDB1-dependent heterochromatin stimulates alternative lengthening of telomeres[J]. Science Advances 2019, 5(5), eaav3673. [Google Scholar] [CrossRef] [PubMed]
- Carson, L M; Flynn, R L. Highlighting vulnerabilities in the alternative lengthening of telomeres pathway[J]. Current Opinion in Pharmacology 2023, 70, 102380. [Google Scholar] [CrossRef]
- Mishra, A; Patel, T N. Locking the gates of immortality: targeting alternative lengthening of telomeres (ALT) pathways[J]. Medical Oncology (Northwood, London, England) 2025, 42(3), 78. [Google Scholar] [CrossRef]
- Nera, B; Huang, H S; Lai, T; et al. Elevated levels of TRF2 induce telomeric ultrafine anaphase bridges and rapid telomere deletions[J]. Nature Communications 2015, 6(1), 10132. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P; Barnes, R; Pan, H; et al. TIN2 is an architectural protein that facilitates TRF2-mediated trans- and cis-interactions on telomeric DNA[J]. Nucleic Acids Research 2021, 49(22), 13000–13018. [Google Scholar] [CrossRef]
- S, Y; S L, F Z; et al. TIN2 deficiency leads to ALT-associated phenotypes and differentiation defects in embryonic stem cells[J]. Stem cell reports 2022, 17(5). [Google Scholar]
- Andonian, B J; Hippensteel, J A; Abuabara, K; et al. Inflammation and aging-related disease: a transdisciplinary inflammaging framework[J]. GeroScience 2025, 47(1), 515–542. [Google Scholar] [CrossRef]
- Ajoolabady, A; Pratico, D; Tang, D; et al. Immunosenescence and inflammaging: mechanisms and role in diseases[J]. Ageing Research Reviews 2024, 101, 102540. [Google Scholar] [CrossRef]
- Park, J; Lee, J H; La, M; et al. Inhibition of NF-κB acetylation and its transcriptional activity by daxx[J]. Journal of Molecular Biology 2007, 368(2), 388–397. [Google Scholar] [CrossRef] [PubMed]
- Croxton, R; Puto, L A; de Belle, I; et al. Daxx represses expression of a subset of antiapoptotic genes regulated by nuclear factor-kappaB[J]. Cancer Research 2006, 66(18), 9026–9035. [Google Scholar] [CrossRef] [PubMed]
- Singh, H; Gupta, R; Gupta, M; et al. Aging-induced alterations in microglial cells and their impact on neurodegenerative disorders[J]. Molecular Biology Reports 2025, 52(1), 515. [Google Scholar] [CrossRef]
- Li, Y; Zhan, B; Zhuang, X; et al. Microglial Pdcd4 deficiency mitigates neuroinflammation-associated depression via facilitating daxx mediated PPARγ/IL-10 signaling[J]. Journal of Neuroinflammation 2024, 21(1), 143. [Google Scholar] [CrossRef]
- Yao, Z; Zhang, Q; Li, X; et al. Death domain-associated protein 6 (daxx) selectively represses IL-6 transcription through histone deacetylase 1 (HDAC1)-mediated histone deacetylation in macrophages[J]. The Journal of Biological Chemistry 2014, 289(13), 9372–9379. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z; Liang, Q; Ren, Y; et al. Immunosenescence: Molecular mechanisms and diseases[J]. Signal Transduction and Targeted Therapy 2023, 8(1), 200. [Google Scholar] [CrossRef]
- Chaplin, D D. Overview of the immune response[J]. Journal of Allergy and Clinical Immunology 2010, 125((2) Suppl 2, S3–23. [Google Scholar] [CrossRef]
- Xu, Y; He, Z; Du, J; et al. Epigenetic modulations of immune cells: from normal development to tumor progression[J]. International Journal of Biological Sciences 2023, 19(16), 5120–5144. [Google Scholar] [CrossRef] [PubMed]
- Maillet, S; Fernandez, J; Decourcelle, M; et al. Daxx inhibits HIV-1 reverse transcription and uncoating in a SUMO-dependent manner[J]. Viruses 2020, 12(6), 636. [Google Scholar] [CrossRef]
- Boehmer, D; Zanoni, I. Interferons in health and disease[J]. Cell 2025, 188(17), 4480–4504. [Google Scholar] [CrossRef] [PubMed]
- Dopkins, N; Nixon, D F. Activation of human endogenous retroviruses and its physiological consequences[J]. Nature Reviews. Molecular Cell Biology 2024, 25(3), 212–222. [Google Scholar] [CrossRef] [PubMed]
- Mac Kain, A; Maarifi, G; Aicher, S M; et al. Identification of DAXX as a restriction factor of SARS-CoV-2 through a CRISPR/Cas9 screen[J]. Nature Communications 2022, 13(1), 2442. [Google Scholar] [CrossRef]
- Sood, A. The cGAS-STING axis: a comprehensive review from immune defense to disease pathogenesis[J]. Immunologic Research 2025, 73(1), 91. [Google Scholar] [CrossRef]
- Ivashkiv, L B; Donlin, L T. Regulation of type I interferon responses[J]. Nature Reviews. Immunology 2014, 14(1), 36–49. [Google Scholar] [CrossRef]
- Scherer, M; Read, C; Neusser, G; et al. Dual signaling via interferon and DNA damage response elicits entrapment by giant PML nuclear bodies[J]. eLife 2022, 11, e73006. [Google Scholar] [CrossRef]
- Daxx and HIRA go viral – How chromatin remodeling complexes affect DNA virus infection[Z].
- Chi, H; Pepper, M; Thomas, P G. Principles and therapeutic applications of adaptive immunity[J]. Cell 2024, 187(9), 2052–2078. [Google Scholar] [CrossRef]
- Li, J; Qian, L; Dowling, J P; et al. Daxx plays a novel role in T cell survival but is dispensable in fas-induced apoptosis[J]. PloS One 2017, 12(3), e0174011. [Google Scholar] [CrossRef] [PubMed]
- Medvedovic, J; Ebert, A; Tagoh, H; et al. Pax5: A master regulator of B cell development and leukemogenesis[J]. Advances in Immunology 2011, 111, 179–206. [Google Scholar]
- Emelyanov, A V; Kovac, C R; Sepulveda, M A; et al. The interaction of Pax5 (BSAP) with daxx can result in transcriptional activation in B cells[J]. The Journal of Biological Chemistry 2002, 277(13), 11156–11164. [Google Scholar] [CrossRef]
- Gongora, R; Stephan, R P; Zhang, Z; et al. An Essential Role for Daxx in the Inhibition of B Lymphopoiesis by Type I Interferons[J]. Immunity 2001, 14(6), 727–737. [Google Scholar] [CrossRef]
- HIRANO, T. Interleukin 6 in autoimmune and inflammatory diseases: A personal memoir[J]. Proceedings of the Japan Academy. Series B, Physical and Biological Sciences 2010, 86(7), 717–730. [Google Scholar] [CrossRef]
- Ross, J B; Myers, L M; Noh, J J; et al. Depleting myeloid-biased haematopoietic stem cells rejuvenates aged immunity[J]. Nature 2024, 628(8006), 162–170. [Google Scholar] [CrossRef] [PubMed]
- Chen, D S; Mellman, I. Elements of cancer immunity and the cancer-immune set point[J]. Nature 2017, 541(7637), 321–330. [Google Scholar] [CrossRef]
- Wang, Y; Zhu, Y; Cao, Y; et al. The activation of cGAS-STING pathway offers novel therapeutic opportunities in cancers[J]. Frontiers in Immunology 2025, 16, 1579832. [Google Scholar] [CrossRef]
- ZHU, X; HUANG, K; KAO, X; et al. Death domain-associated protein (daxx) impairs colon cancer chemotherapy by inhibiting the cGAS-STING pathway[J]. Oncology Research 2025, 33(5), 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Alandijany, T. Host intrinsic and innate intracellular immunity during herpes simplex virus type 1 (HSV-1) infection[J]. Frontiers in Microbiology 2019, 10, 2611. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
DAXX regulates metabolic pathways through nuclear and cytoplasmic mechanisms. In the nucleus, DAXX cooperates with SREBP1/2 to enhance transcription of cholesterol and fatty acid synthesis enzymes, including HMGCR and HMGCS1. DAXX also modulates lipid metabolism in an androgen receptor–dependent context. In the cytoplasm, DAXX activates the AMPK-related kinase MPK38, promoting fatty acid oxidation. DAXX further participates in glucose metabolism by facilitating GLUT4 trafficking via the JNK1–KIF5B complex and suppressing hepatic gluconeogenesis through regulation of PEPCK and G6Pase. Under metabolic stress, cytoplasmic DAXX promotes p62-mediated Nrf2 activation by facilitating KEAP1 sequestration.
Figure 1.
DAXX regulates metabolic pathways through nuclear and cytoplasmic mechanisms. In the nucleus, DAXX cooperates with SREBP1/2 to enhance transcription of cholesterol and fatty acid synthesis enzymes, including HMGCR and HMGCS1. DAXX also modulates lipid metabolism in an androgen receptor–dependent context. In the cytoplasm, DAXX activates the AMPK-related kinase MPK38, promoting fatty acid oxidation. DAXX further participates in glucose metabolism by facilitating GLUT4 trafficking via the JNK1–KIF5B complex and suppressing hepatic gluconeogenesis through regulation of PEPCK and G6Pase. Under metabolic stress, cytoplasmic DAXX promotes p62-mediated Nrf2 activation by facilitating KEAP1 sequestration.
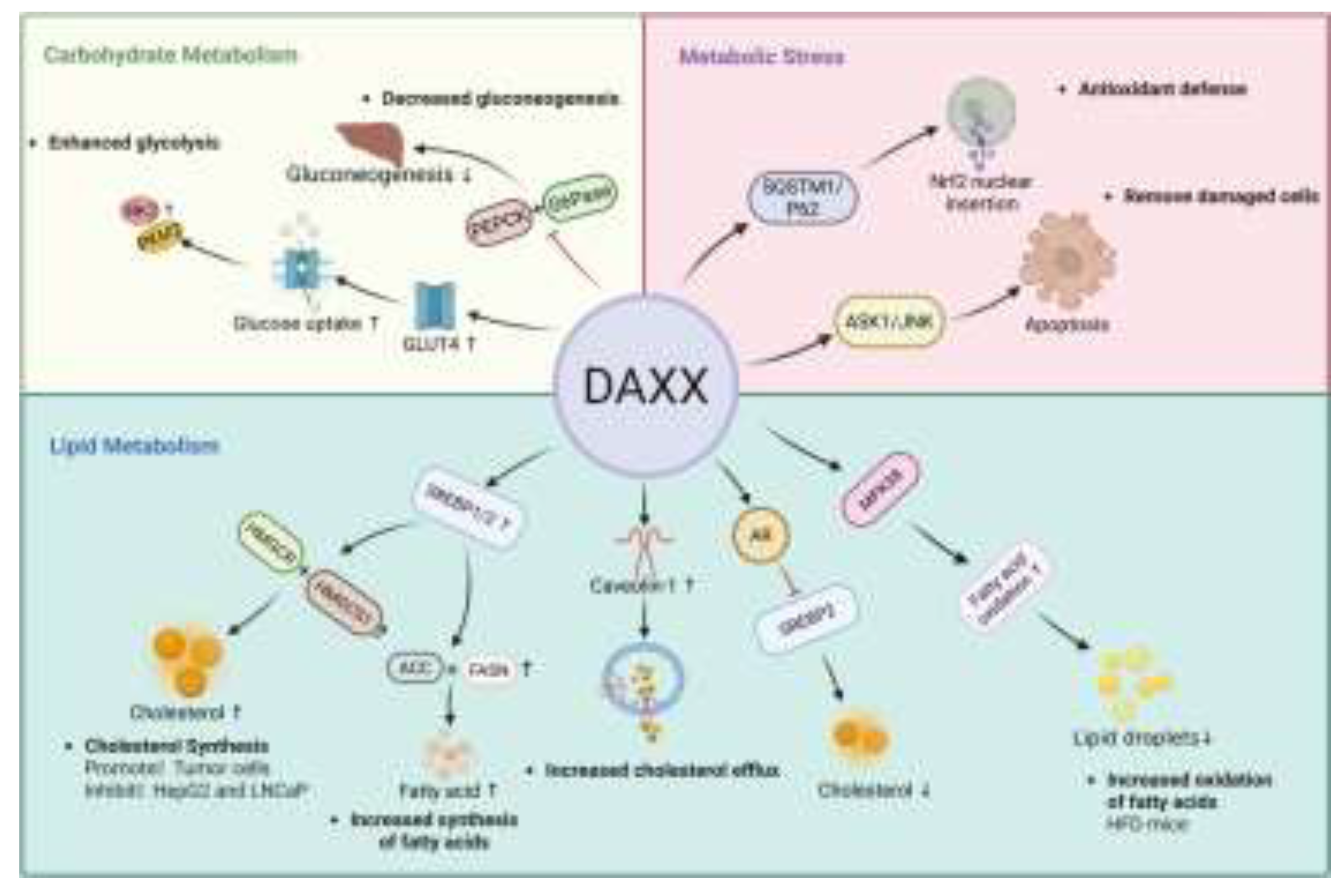
Figure 2.
DAXX contributes to aging-related processes by regulating genome stability, telomere maintenance, oxidative stress, and inflammatory signaling. In the nucleus, DAXX associates with PML nuclear bodies and cooperates with ATRX to facilitate H3.3 deposition during DNA damage response and repair. At telomeres, the DAXX–ATRX complex restrains aberrant alternative lengthening of telomeres by limiting TERRA expression. Under oxidative stress, DAXX enhances antioxidant defense through activation of the p62–Nrf2 pathway. DAXX also suppresses aging-associated inflammatory signaling by modulating NF-κB activity and limiting IL-6 transcription.
Figure 2.
DAXX contributes to aging-related processes by regulating genome stability, telomere maintenance, oxidative stress, and inflammatory signaling. In the nucleus, DAXX associates with PML nuclear bodies and cooperates with ATRX to facilitate H3.3 deposition during DNA damage response and repair. At telomeres, the DAXX–ATRX complex restrains aberrant alternative lengthening of telomeres by limiting TERRA expression. Under oxidative stress, DAXX enhances antioxidant defense through activation of the p62–Nrf2 pathway. DAXX also suppresses aging-associated inflammatory signaling by modulating NF-κB activity and limiting IL-6 transcription.
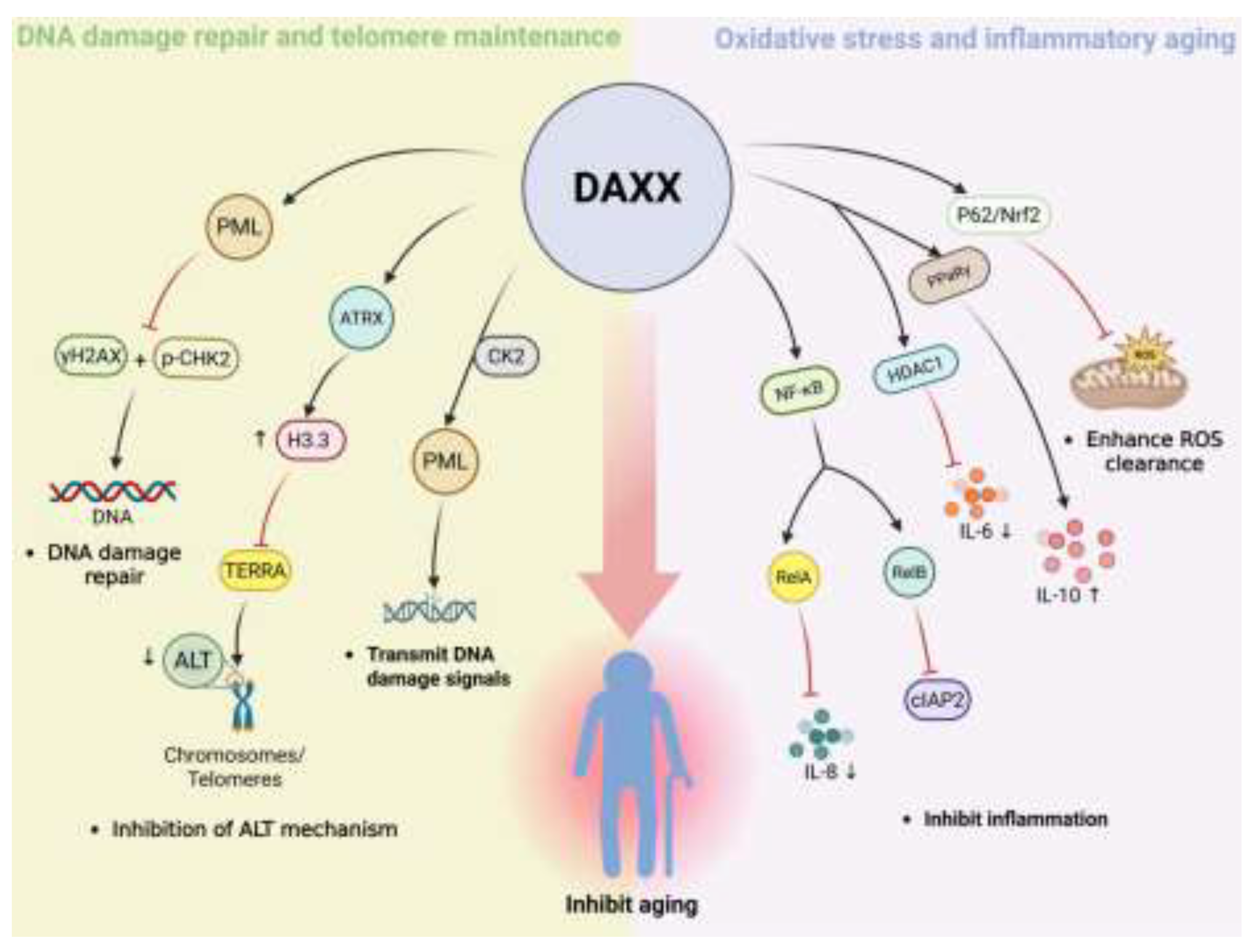
Figure 3.
DAXX regulates immune homeostasis through chromatin-based and signaling-dependent mechanisms. In innate immunity, DAXX associates with PML nuclear bodies to promote chromatinization and silencing of viral genomes and endogenous retroviruses, thereby limiting excessive interferon responses. In adaptive immunity, DAXX supports T cell survival following TCR activation and cooperates with Pax5 during early B cell differentiation. DAXX also contributes to immune tolerance by repressing pro-inflammatory gene expression, including IL-6, and modulates tumor immunity by inhibiting activation of the cGAS–STING pathway.
Figure 3.
DAXX regulates immune homeostasis through chromatin-based and signaling-dependent mechanisms. In innate immunity, DAXX associates with PML nuclear bodies to promote chromatinization and silencing of viral genomes and endogenous retroviruses, thereby limiting excessive interferon responses. In adaptive immunity, DAXX supports T cell survival following TCR activation and cooperates with Pax5 during early B cell differentiation. DAXX also contributes to immune tolerance by repressing pro-inflammatory gene expression, including IL-6, and modulates tumor immunity by inhibiting activation of the cGAS–STING pathway.
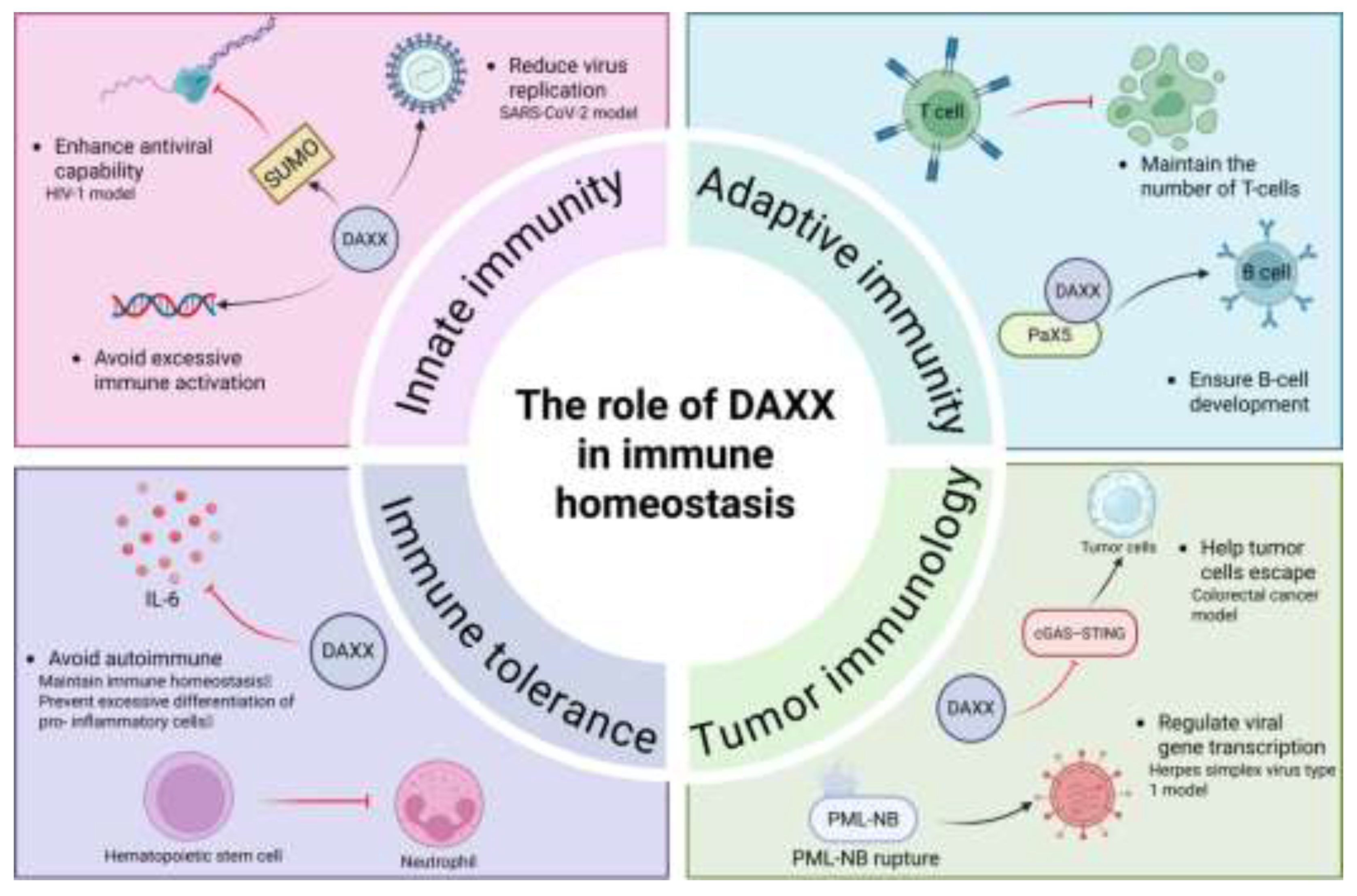
Figure 4.
Schematic overview illustrating DAXX as an integrative regulator linking metabolic control, aging-associated stress responses, and immune homeostasis. Through context-dependent nuclear localization and protein interactions, DAXX coordinates transcriptional and signaling pathways involved in energy metabolism, genome stability, oxidative stress regulation, and immune signaling.
Figure 4.
Schematic overview illustrating DAXX as an integrative regulator linking metabolic control, aging-associated stress responses, and immune homeostasis. Through context-dependent nuclear localization and protein interactions, DAXX coordinates transcriptional and signaling pathways involved in energy metabolism, genome stability, oxidative stress regulation, and immune signaling.
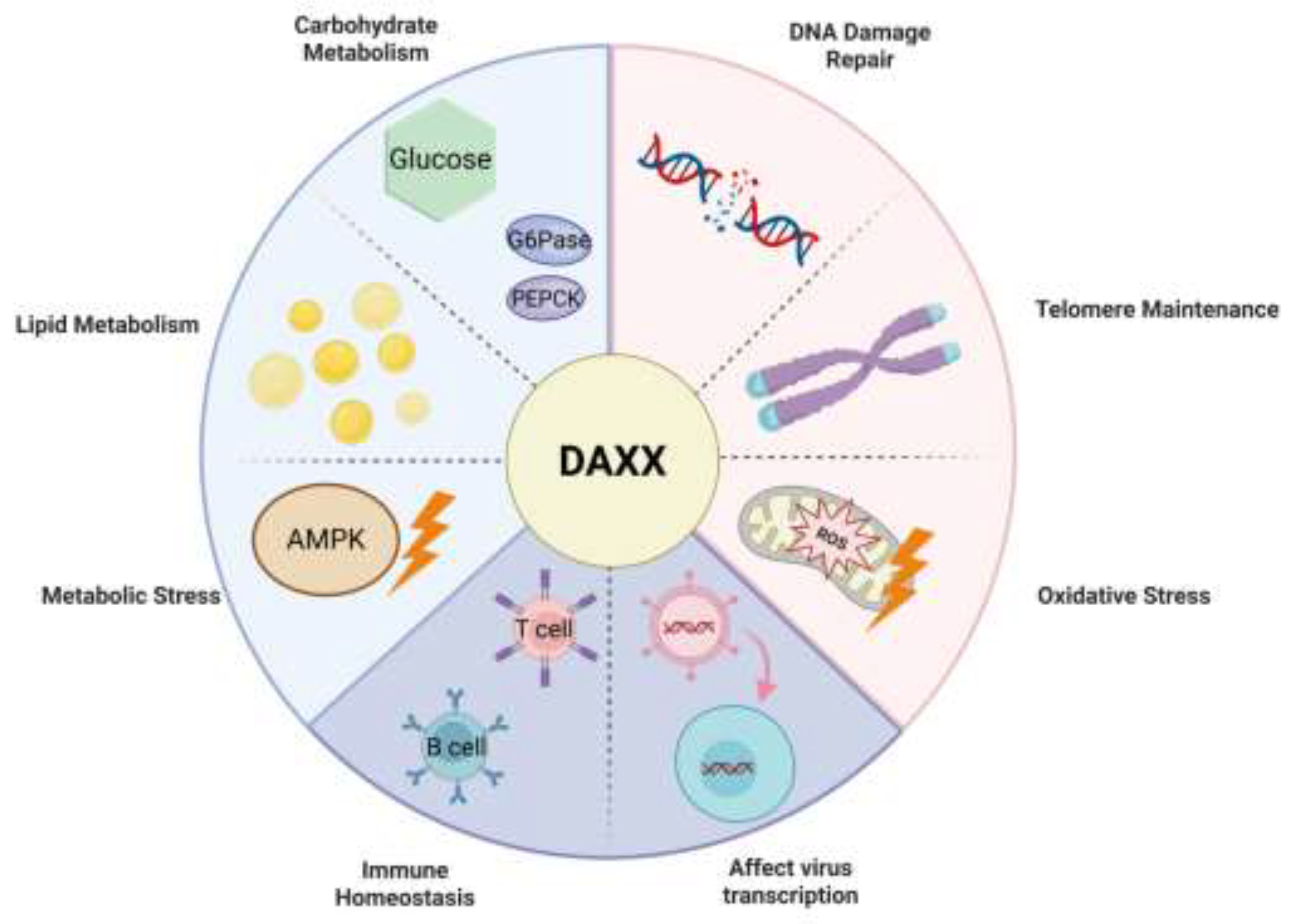
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



