Submitted:
27 January 2026
Posted:
29 January 2026
You are already at the latest version
Abstract
The pharmaceutical industry is undergoing a transformative revolution driven by artificial intelligence, fundamentally reshaping drug discovery and early development processes. This comprehensive review examines how AI technologies from machine learning to deep neural networks are enhancing predictive accuracy and operational efficiency across the entire development pipeline. By analyzing complex biological data, these computational approaches enable unprecedented precision in target identification, lead optimization, and preclinical assessment, significantly accelerating therapeutic development. However, substantial challenges persist in implementation, including data harmonization issues, model interpretability constraints, and integration barriers within existing regulatory frameworks. This analysis critically evaluates both the transformative potential and practical limitations of AI applications, highlighting their capacity to not only streamline development pipelines but also pioneer innovative approaches in personalized medicine and novel therapeutic solutions for complex diseases, while addressing the critical hurdles that must be overcome for successful integration into pharmaceutical research and development.
Keywords:
machine learning
; deep learning
; drug discovery
; de novo design
; preclinical
1. Introduction
Despite being in charge of providing essential treatments, the traditional drug discovery paradigm is plagued by severe inefficiencies. With over 90% of candidates failing during clinical development, bringing a new medication to market is a ten-year process that costs billions of dollars and has an alarming attrition rate [1,2]. A large percentage of these failures are caused by inadequate efficacy or unanticipated safety concerns, highlighting the urgent need for more reliable and predictive preclinical models for compound selection and target validation [3].
Artificial Intelligence (AI) has become a revolutionary force in this difficult environment. Advanced machine learning (ML), deep learning (DL), and the availability of large biological datasets have come together to create previously unheard-of possibilities for complicated information analysis that go beyond the purview of conventional statistics [4]. AI has evolved from a futuristic idea to a vital instrument that is changing the pharmaceutical sector. It promises to speed up the entire process by providing completely new, data-driven methods and streamlining existing workflows.
The use of AI in drug discovery and early development will be thoroughly examined in this paper. We will examine the ways in which technologies like natural language processing, graph neural networks, and generative models are transforming important phases, from target discovery and de novo drug creation to clinical trial optimization. We will also critically analyze the important obstacles that come with this change, such as ethical issues, model interpretability, and data quality. This article attempts to shed light on AI's significant influence and its promise to provide patients with more effective treatments more quickly and effectively by combining case stories and recent research.
2. Artificial Intelligence (AI) Technologies
Artificial intelligence (AI) is transforming drug research and development primarily through machine learning (ML) and deep learning (DL) techniques. By training models with optimization techniques like gradient descent and backpropagation, these technologies may learn from complex biological and chemical data. In particular, DL techniques have shown a lot of promise for a variety of clinical applications, including patient monitoring, medical image analysis, sickness identification, and therapy prediction [5,6].
2.1. Machine Learning
The three main categories of machine learning (ML) are reinforcement learning, unsupervised learning, and supervised learning (Figure 1). With machine learning (ML), models learn from data to either identify underlying patterns or make predictions. By mapping inputs to known outputs using algorithms trained on labeled datasets, supervised learning enables precise prediction on new, unseen data, such as the identification of bioactive chemical compounds in drug discovery [7]. Unsupervised learning, on the other hand, examines unlabeled data to find hidden structures or organic groupings [8,9]. This is particularly helpful for applications like illness subtype classification and gene expression clustering. By interacting with its surroundings and providing feedback in the form of rewards, reinforcement learning focuses on teaching an agent to make the best choices [7].
Traditional machine learning (ML) techniques that are most frequently employed in the drug discovery pipeline include Artificial Neural Networks (ANNs), Random Forest (RF), Support Vector Machine (SVM), k-Nearest Neighbors (kNN), and Naïve Bayesian (NB) Classifier. In data analysis for biological activity and molecular property prediction, each technique has a specific function. The majority category of the 'k' nearest data points is used by the kNN technique to categorize a sample. Weighted kNN (WkNN) is a methodology that can be modified for particular uses, including enhancing drug-disease association matrices for repositioning studies [10]. The NB classifier, which is used to differentiate between activators and non-activators of the pregnane X receptor (PXR) to increase screening efficiency, is well-known for its ease of use and speed. It trains on known categories to predict unknown data [11]. As demonstrated by the PredMS model for predicting the metabolic stability of small compounds, the RF algorithm uses an ensemble of regression trees with bootstrap aggregation to achieve excellent predictive accuracy [12]. SVM is a powerful two-class classification model that finds the optimal hyperplane to maximize separation between categories, making it crucial for predicting molecular interactions and traits like neurotoxicity. Its accuracy, sensitivity, and specificity all exceed 80% [10,13]. Finally, computer programs known as artificial neural networks (ANNs) may learn complex relationships by simulating biological neural networks. ANNs are crucial for drug screening and design because they mimic the brain's experience-based learning capabilities [13].
2.2. Deep Learning
Deep learning algorithms, a potent subset of machine learning, have gained enormous popularity in the drug research sector because of their capacity to replicate intricate patterns in large datasets. Among the most popular designs are convolutional neural networks (CNNs), generative adversarial networks (GANs), and recurrent neural networks (RNNs). CNNs are very good at processing spatial data because they use convolutional and pooling layers to extract and compress characteristics. For example, the CAMP framework collects both local and global information using CNNs to predict binary peptide-protein interactions [14]. The adversarial process that GANs employ, which consists of a discriminator that evaluates the legitimacy of the generated data samples and a generator that creates new data samples, has been enhanced by dense networks to boost the efficiency of molecular sequence synthesis [15]. Last but not least, RNNs have proven effective in challenging tasks like significantly improving the extraction of drug-drug interactions. They understand forward and backward correlations by using their internal memory and are specialized for sequential data [16].
2.3. Applications of AI in Drug Discovery and Development
3.3.1. Target Identification and Validation
Artificial intelligence is fundamentally transforming early-stage cancer drug discovery by enhancing the identification and validation of novel therapeutic targets. For target identification, AI employs network-based algorithms to analyze complex biological data, modeling cellular pathways to predict new targets through concepts like "guilt-by-association" [17,18]. A pivotal application is in synthetic lethality, where AI models, particularly those integrating knowledge graphs with graph neural networks, pinpoint gene pairs whose simultaneous inactivation selectively kills cancer cells with specific mutations, sparing healthy tissues [19,20]. Furthermore, machine learning models assess a target's "druggability." The DrugnomeAI model, for instance, uses a semi-supervised framework to classify genes as potential drug targets, though its potential bias towards known target classes remains a consideration . This AI-driven focus on molecular mechanisms improves therapeutic efficacy and reduces costly trial-and-error [21].
In the subsequent target validation phase, AI accelerates methods like high-throughput screening (HTS) and virtual screening (VS). AI-driven HTS has successfully identified novel compounds, such as kinase inhibitors against STK33 and DDR1, that induce cancer cell death [22]. In VS, AI platforms use deep learning to predict drug-target interactions, rapidly identifying potent inhibitors . AI also streamlines drug repurposing by finding new anticancer uses for existing drugs, saving significant time and resources [19,20]. Many previous studies have applied these approaches to evaluate the inhibitory potential of plant-derived compounds against various cancer types [23,24,25,26,27].
3.3.2. De Novo Drug Design and Optimization
De novo drug design utilizes AI to create novel therapeutic compounds from scratch, moving beyond traditional methods like pharmacophore modeling which struggle with molecular flexibility and computational complexity [28]. By employing algorithms such as deep reinforcement learning, AI efficiently explores chemical space, leveraging large datasets of bioactivity and protein structures to generate molecules with optimized properties and reduce human bias [29,30]. Tools like REINVENT 2.0 exemplify this approach, though their effectiveness depends on training data quality, and a common limitation is that generated molecules can be challenging to synthesize [31]. Other platforms, including DeepChem, have successfully created novel inhibitors for targets like protein-protein interactions in breast cancer and selective cyclin-dependent kinase (CDK) inhibitors, demonstrating AI's practical impact in developing targeted therapies [32]. A list of AI-based tools used for drug discovery are described in Table 1.
In the subsequent lead optimization phase, initial hits are refined into drug candidates with improved efficacy, safety, and synthetic accessibility [33]. AI augments this process through in silico methods like QSAR models and fragment-based drug discovery, predicting how structural changes will affect a compound's properties and thus prioritizing the most promising candidates for testing [34]. The AI tool DeepFrag, for instance, uses deep learning to analyze 3D receptor-ligand structures and suggest chemical fragments that enhance binding affinity [29,30]. Practical applications include IBM Watson proposing modifications for breast cancer treatments and deep learning models generating superior CDK inhibitors, significantly accelerating development and reducing costs [35,36]. A critical consideration, however, is the potential for algorithmic bias from training data, a challenge that requires ongoing attention.
3.3.3. Preclinical Development
Preclinical drug development is changing as a result of artificial intelligence's introduction of sophisticated predictive tools that enhance the evaluation of pharmacokinetic behavior, safety, and efficacy. Largely responsible for this advancement is AI's ability to decipher complicated and sizable datasets, which allows for precise prediction of important chemical and biological parameters and lessens reliance on time-consuming and resource-intensive experimental processes. The prediction of pharmacokinetics (PK) and ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) is significantly reduced.
Algorithms for machine learning (ML) are able to assess molecular structures and forecast important characteristics including drug-drug interaction potential, metabolic degradation, and membrane permeability. From large molecular datasets, supervised learning methods like as Support Vector Machines (SVM) and Artificial Neural Networks (ANNs) have shown excellent accuracy in predicting intestinal absorption in humans [37,38]. These techniques are operationalized by tools like pkCSM and admetSAR, which enable quick in silico screening and early prioritizing of compounds with desired pharmacokinetic properties. Deep learning, which can identify minute structure–toxicity correlations, is another way AI is improving toxicity prediction.
High-throughput toxicity screening is supported by programs like the Tox21 challenge and systems like DeepTox, which allow for the early removal of hazardous substances [39]. Additionally, by analyzing imaging and multi-omics datasets to find markers linked to treatment response and disease prognosis, AI improves biomarker discovery. Clinically significant biomarkers like CA19-9 and CA125 have been identified by platforms like PandaOmics, enabling patient stratification and individualized treatment plans [40,41].
3.3.4. Clinical Development
Artificial intelligence (AI) is revolutionizing clinical drug research by optimizing efficacy, safety, and predicting accuracy throughout the trial lifecycle. In order to increase enrollment and diversity the cohort, it begins by revolutionizing patient recruitment and identification. AI technologies like Deep 6 AI swiftly identify eligible candidates by examining genetic databases and electronic health records (EHRs) [70]. In order to lead more intelligent trial design and produce more successful and efficient research, AI also employs predictive analytics on historical data to forecast potential outcomes, the ideal dosage, and adverse occurrences [71,72]. Through the analysis of real-time data from wearables and clinical reports, artificial intelligence (AI) enhances patient monitoring and safety during the course of a trial, ensuring protocol adherence and aiding early side effect detection [73]. Finally, AI streamlines data management and regulatory compliance by automating reporting tasks and unlocking hidden insights from massive datasets. Artificial intelligence-derived real-world evidence is welcomed by the USFDA and other agencies [73]. With these integrated operations, which include everything from intelligent recruiting and predictive design to real-time monitoring and automated analysis, AI significantly lowers the risk of clinical development and increases the likelihood of providing effective innovative medications to patients [74,75].
4. Critical View: Limitations and Challenges of AI in Drug Discovery
Artificial intelligence has the potential to revolutionize drug discovery and early development, but there are also serious obstacles that need to be recognized and overcome. These restrictions fall under four general categories: ethical governance, operational integration, model openness, and data integrity.
4.1. Foundational Data Challenges
The caliber of the data used to train AI models is inextricably connected to their performance. Data bias is a major issue, as models trained on non-representative datasets (e.g., over-representing particular chemical spaces or demographics) provide skewed predictions that do not apply to larger populations or new classes of compounds [76,77]. Moreover, the generation of unified training datasets is a difficult and time-consuming process because to the significant challenges posed by data heterogeneity from various sources (such as chemical assays, clinical trials, and omics data) [78].
4.2. The "Black Box" Problem
The lack of interpretability and transparency in many complex AI models, especially Deep Neural Networks (DNNs), is a major obstacle to their mainstream adoption. When these technologies operate as "black boxes," it is difficult to understand the reasoning behind their forecasts. In an area where knowing the "why" behind a decision is crucial for clinical application and regulatory approval, this opacity undermines confidence, makes accountability more difficult, and raises safety concerns [79,80].
3. Operational and Ethical Hurdles
Concerns about intellectual property, data privacy, and worker displacement make it difficult to integrate AI into traditional, frequently inflexible pharmaceutical workflows, which calls for significant adjustments to infrastructure and knowledge. The sector faces ethical challenges in protecting patient privacy, obtaining informed consent for data use, and creating precise legal frameworks for responsibility. Professional surveys underscore the need for clear ethical standards by highlighting serious worries about cybersecurity, job displacement, and a lack of strong legal control [81,82,83].
5. Perspectives and Future Directions
The future of artificial intelligence (AI) in drug discovery depends on creating complex plans to get around these restrictions and take use of new technical synergies.
5.1. Mitigating Data Limitations with Synthetic Data and Advanced Models
Synthetic data (SD) creation with Generative Adversarial Networks (GANs) and Variational Autoencoders (VAEs) is a viable option to address data shortage and bias. By producing actual biological data (such as single-cell RNA sequencing) and supplementing underrepresented patient groups in datasets, SD can increase the generalizability and robustness of models [77,84]. Additionally, new architectures that push the limits of sequence modeling, such as State Space Models (SSMs), offer a more computationally efficient option for evaluating lengthy biological sequences than Transformers [85].
5.2. Toward Explainable AI and Robust Integration
In order to promote regulatory acceptance, boost confidence, and demystify model judgments, future projects must prioritize the development of explainable AI. AI may be integrated with the Internet of Things (IoT) to enable real-time monitoring in clinical settings and with other technologies, like blockchain, to enhance data security and integrity, resulting in a more safe and efficient data ecosystem [86,87].
5.3. The Push for Personalized Medicine and Multi-Omics Integration
The combination of multi-omics techniques and artificial intelligence is poised to transform precision medicine. Artificial intelligence (AI) can identify new therapeutic targets and provide biomarkers for patient stratification by combining data from genomes, proteomics, and metabolomics, allowing for genuinely customized treatment interventions [88,89]. The development of treatments for intricate illnesses like cancer and neurological disorders will depend heavily on this comprehensive, systems-level understanding of illness.
5.4. Evolving Regulatory and Global Health Frameworks
Regulatory frameworks must change in concert with AI if it is to realize its full potential. To guarantee safety and effectiveness without limiting innovation, this entails developing precise criteria for AI model validation, deployment, and ongoing monitoring [90]. Last but not least, artificial intelligence (AI) has enormous potential to improve healthcare outcomes and access for underprivileged populations around the world by significantly speeding up the development of medicines for uncommon and neglected diseases [91].
6. Conclusion
Artificial intelligence's incorporation into drug research and early development is a paradigm shift that is radically altering the pharmaceutical industry. AI shows great promise in accelerating target identification, producing novel chemical structures, predicting ADMET characteristics, and optimizing clinical trials through the use of machine learning and deep learning algorithms. It is anticipated that this technological revolution would improve success rates and drastically cut down on development expenses and timetables. Data quality problems, algorithmic bias, model interpretability issues, and ethical concerns about data privacy and regulatory compliance are just a few of the significant obstacles that still exist. Creating strong validation frameworks, improving explainable AI techniques, and encouraging interdisciplinary cooperation are key to the future of AI-driven drug development. AI has the potential to open up previously unheard-of possibilities for personalized medicine and novel cancer treatments as these technologies develop alongside cutting-edge strategies like synthetic biology and multi-omics integration. In the end, this could revolutionize patient care by enabling the development of more accurate, effective, and efficient treatments.
Author Contributions
Cromwel Tepap Zemnou: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Supervision, Resources, Writing – original draft, Writing – review & editing. Gabriel Tchuente KAMSU: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. Ramelle Ngakam: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. Etienne Junior Tcheumeni: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
This work was not supported by any funding or grant.
Conflict of Interests
The authors report there are no competing interests to declare.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
References
- DiMasi, J. A.; Grabowski, H. G.; Hansen, R. W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J Health Econ 2016, vol. 47, 20–33. [Google Scholar] [CrossRef]
- Wouters, O. J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. JAMA 2020, vol. 323(no. 9), 844–853. [Google Scholar] [CrossRef]
- S. M. Paul et al., How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov 2010, vol. 9(no. 3), 203–214. [CrossRef]
- Murmu, A.; Győrffy, B. Artificial intelligence methods available for cancer research. Front Med 2024, vol. 18(no. 5), 778–797. [Google Scholar] [CrossRef]
- Lac, L.; Leung, C. K.; Hu, P. Computational frameworks integrating deep learning and statistical models in mining multimodal omics data. Journal of Biomedical Informatics 2024, vol. 152, 104629. [Google Scholar] [CrossRef]
- Y. Wang et al., XGraphCDS: An explainable deep learning model for predicting drug sensitivity from gene pathways and chemical structures. Computers in Biology and Medicine 2024, vol. 168, 107746. [CrossRef]
- Md. A. Islam, Md. Z. Hasan Majumder, Md. A. Hussein, K. M. Hossain, and Md. S. Miah, A review of machine learning and deep learning algorithms for Parkinson’s disease detection using handwriting and voice datasets. Heliyon 2024, vol. 10(no. 3), e25469. [CrossRef]
- Mak, K.-K.; Pichika, M. R. Artificial intelligence in drug development: present status and future prospects. Drug Discovery Today 2019, vol. 24(no. 3), 773–780. [Google Scholar] [CrossRef] [PubMed]
- Chen, R. R. Chen et al., Transcriptome sequencing and functional characterization of new sesquiterpene synthases from Curcuma wenyujin. Archives of Biochemistry and Biophysics 2021, vol. 709, 108986. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yang, B.; Duan, G.; Wang, J. ITRPCA: a new model for computational drug repositioning based on improved tensor robust principal component analysis. Front. Genet. 2023, vol. 14. [Google Scholar] [CrossRef] [PubMed]
- H. Shi et al., Absorption, Distribution, Metabolism, Excretion, and Toxicity Evaluation in Drug Discovery. 14. Prediction of Human Pregnane X Receptor Activators by Using Naive Bayesian Classification Technique. Chem. Res. Toxicol. 2015, vol. 28(no. 1), 116–125. [CrossRef] [PubMed]
- Ryu, J. Y.; Lee, J. H.; Lee, B. H.; Song, J. S.; Ahn, S.; Oh, K.-S. PredMS: a random forest model for predicting metabolic stability of drug candidates in human liver microsomes. Bioinformatics 2022, vol. 38(no. 2), 364–368. [Google Scholar] [CrossRef] [PubMed]
- P. N. Shiammala et al., Exploring the artificial intelligence and machine learning models in the context of drug design difficulties and future potential for the pharmaceutical sectors. Methods 2023, vol. 219, 82–94. [CrossRef]
- Y. Lei et al., A deep-learning framework for multi-level peptide–protein interaction prediction. Nat Commun 2021, vol. 12(no. 1), 5465. [CrossRef]
- F. Wang et al., Generating new protein sequences by using dense network and attention mechanism. MBE 2023, vol. 20(no. 2), 4178–4197. [CrossRef]
- Lim, S.; Lee, K.; Kang, J. Drug drug interaction extraction from the literature using a recursive neural network. PLOS ONE 2018, vol. 13(no. 1), e0190926. [Google Scholar] [CrossRef]
- Galindez, G.; Sadegh, S.; Baumbach, J.; Kacprowski, T.; List, M. Network-based approaches for modeling disease regulation and progression. Computational and Structural Biotechnology Journal 2023, vol. 21, 780–795. [Google Scholar] [CrossRef]
- Y. You et al., Artificial intelligence in cancer target identification and drug discovery. Sig Transduct Target Ther 2022, vol. 7(no. 1), 156. [CrossRef] [PubMed]
- V. Previtali et al., New Horizons of Synthetic Lethality in Cancer: Current Development and Future Perspectives. J. Med. Chem. 2024, vol. 67(no. 14), 11488–11521. [CrossRef]
- Wang, L.; Yan, Z.; Xiang, P.; Yan, L.; Zhang, Z. MR microneurography of human peripheral fascicles using a clinical 3T MR scanner. Journal of Neuroradiology 2023, vol. 50(no. 2), 253–257. [Google Scholar] [CrossRef]
- K. N, K. K, K. N, and K. K, Target identification and validation in research. World Journal of Biology Pharmacy and Health Sciences 2024, vol. 17(no. 3), 107–117. [CrossRef]
- A. Zhavoronkov et al., Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol 2019, vol. 37(no. 9), 1038–1040. [CrossRef]
- Zemnou, C. T.; Karim, E. M.; Chtita, S.; Zanchi, F. B. Impact of mutations on KAT6A enzyme and inhibitory potential of compounds from Withania somnifera using computational approaches. Computers in Biology and Medicine 2025, vol. 190, 110041. [Google Scholar] [CrossRef]
- Tepap Zemnou, C.; Anissi, J.; Bounou, S.; Zanchi, F. In Silico Approach for Assessment of the Anti-Tumor Potential of Cannabinoid Compounds by Targeting Glucose-6-Phosphate Dehydrogenase Enzyme. Chemistry & Biodiversity 2024, vol. 21, e202401338. [Google Scholar] [CrossRef]
- C. Tepap Zemnou, In silico Comparative Study of the Anti-Cancer Potential of Inhibitors of Glucose-6-Phosphate Dehydrogenase Enzyme Using ADMET Analysis, Molecular Docking, and Molecular Dynamic Simulation. Advanced Theory and Simulations vol. 8(no. 5), 2400757, 2025. [CrossRef]
- C. Tepap Zemnou and R. Ibrahim, Network Pharmacology-based Approach to Investigate the Mechanism of Folium Artemisiae Argyi in the Treatment of Hepatocellular Carcinoma. International Journal of Advancement in Life Sciences Research 2025, vol. 08, 74–94. [CrossRef]
- C. Tepap Zemnou, Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation Revealed the Molecular Targets and Potential Mechanism of Nauclea Latifolia in the Treatment of Breast Cancer. Chemistry & Biodiversity 2024, vol. 22, e202402423. [CrossRef]
- Lewis, R. A.; Sirockin, F. 2/3D Pharmacophore Definitions and Their Application. In in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier, 2025. [Google Scholar] [CrossRef]
- V. D. Mouchlis et al., Advances in De Novo Drug Design: From Conventional to Machine Learning Methods. International Journal of Molecular Sciences 2021, vol. 22(no. 4), 1676. [CrossRef]
- Singh, S.; Kaur, N.; Gehlot, A. Application of artificial intelligence in drug design: A review. Computers in Biology and Medicine 2024, vol. 179, 108810. [Google Scholar] [CrossRef]
- T. Blaschke et al., REINVENT 2.0: An AI Tool for De Novo Drug Design. J. Chem. Inf. Model. 2020, vol. 60(no. 12), 5918–5922. [CrossRef]
- Bhisetti, G.; Fang, C. Artificial Intelligence–Enabled De Novo DesignDe novo designof Novel Compounds that Are Synthesizable. In Artificial Intelligence in Drug Design; Heifetz, A., Ed.; Springer US: New York, NY, 2022; pp. 409–419. [Google Scholar] [CrossRef]
- Wang, S.; Dong, G.; Sheng, C. Structural simplification: an efficient strategy in lead optimization. Acta Pharmaceutica Sinica B 2019, vol. 9(no. 5), 880–901. [Google Scholar] [CrossRef] [PubMed]
- L. R. de Souza Neto et al., In silico Strategies to Support Fragment-to-Lead Optimization in Drug Discovery. Front. Chem. 2020, vol. 8. [CrossRef]
- Crucitti, D.; Pérez Míguez, C.; Díaz Arias, J. Á.; Fernandez Prada, D. B.; A. Mosquera Orgueira. De novo drug design through artificial intelligence: an introduction. Front. Hematol. 2024, vol. 3. [Google Scholar] [CrossRef]
- Sarvepalli, S.; Vadarevu, S. Role of artificial intelligence in cancer drug discovery and development. Cancer Letters 2025, vol. 627, 217821. [Google Scholar] [CrossRef]
- Pires, D. E. V.; Blundell, T. L.; Ascher, D. B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, vol. 58(no. 9), 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- H. Yang et al., admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, vol. 35(no. 6), 1067–1069. [CrossRef]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity prediction using deep learning. Front. Environ. Sci. 2016, vol. 3, no. FEB. [Google Scholar] [CrossRef]
- P. Kamya et al., PandaOmics: An AI-Driven Platform for Therapeutic Target and Biomarker Discovery. J Chem Inf Model 2024, vol. 64(no. 10), 3961–3969. [CrossRef]
- Z. Zhang et al., Identifying tumor markers-stratified subtypes (CA-125/CA19-9/carcinoembryonic antigen) in cervical adenocarcinoma. Int J Biol Markers 2023, vol. 38(no. 3–4), 223–232. [CrossRef]
- M. Varadi et al., AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 2022, vol. 50(no. D1), D439–D444. [CrossRef]
- Boiko, D. A.; MacKnight, R.; Kline, B.; Gomes, G. Autonomous chemical research with large language models. Nature 2023, vol. 624(no. 7992), 570–578. [Google Scholar] [CrossRef]
- Wójcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open Drug Discovery Toolkit (ODDT): a new open-source player in the drug discovery field. J Cheminform 2015, vol. 7(no. 1), 26. [Google Scholar] [CrossRef]
- Sanchez-Lengeling, B.; Outeiral, C.; Guimaraes, G. L.; Aspuru-Guzik, A. Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC). In ChemRxiv; 17 Aug 2017. [Google Scholar] [CrossRef]
- Jin, W.; Barzilay, R.; Jaakkola, T. Junction Tree Variational Autoencoder for Molecular Graph Generation. In in Proceedings of the 35th International Conference on Machine Learning, PMLR, July 2018; pp. 2323–2332. Available online: https://proceedings.mlr.press/v80/jin18a.html.
- Maryasin, B.; Marquetand, P.; Maulide, N. Machine Learning for Organic Synthesis: Are Robots Replacing Chemists? Angewandte Chemie International Edition 2018, vol. 57(no. 24), 6978–6980. [Google Scholar] [CrossRef] [PubMed]
- Altae-Tran, H.; Ramsundar, B.; Pappu, A. S.; Pande, V. Low Data Drug Discovery with One-Shot Learning. ACS Cent. Sci. 2017, vol. 3(no. 4), 283–293. [Google Scholar] [CrossRef]
- Coley, C. W.; Barzilay, R.; Green, W. H.; Jaakkola, T. S.; Jensen, K. F. Convolutional Embedding of Attributed Molecular Graphs for Physical Property Prediction. J. Chem. Inf. Model. 2017, vol. 57(no. 8), 1757–1772. [Google Scholar] [CrossRef]
- Xu, Y.; Ma, J.; Liaw, A.; Sheridan, R. P.; Svetnik, V. Demystifying Multitask Deep Neural Networks for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model. 2017, vol. 57(no. 10), 2490–2504. [Google Scholar] [CrossRef] [PubMed]
- D. K. Duvenaud et al., “Convolutional Networks on Graphs for Learning Molecular Fingerprints,” in Advances in Neural Information Processing Systems. Curran Associates, Inc., 2015. 23 Oct 2025. Available online: https://proceedings.neurips.cc/paper_files/paper/2015/hash/f9be311e65d81a9ad8150a60844bb94c-Abstract.html.
- Urban, G.; Subrahmanya, N.; Baldi, P. Inner and Outer Recursive Neural Networks for Chemoinformatics Applications. J. Chem. Inf. Model. 2018, vol. 58(no. 2), 207–211. [Google Scholar] [CrossRef] [PubMed]
- E. N. Feinberg et al., PotentialNet for Molecular Property Prediction. ACS Cent. Sci. 2018, vol. 4(no. 11), 1520–1530. [CrossRef]
- Durrant, J. D.; McCammon, J. A. NNScore: A Neural-Network-Based Scoring Function for the Characterization of Protein−Ligand Complexes. J. Chem. Inf. Model. 2010, vol. 50(no. 10), 1865–1871. [Google Scholar] [CrossRef]
- Awale, M.; Reymond, J.-L. Polypharmacology Browser PPB2: Target Prediction Combining Nearest Neighbors with Machine Learning. J. Chem. Inf. Model. 2019, vol. 59(no. 1), 10–17. [Google Scholar] [CrossRef]
- C. W. Coley, L. Rogers, W. H. Green, and K. F. Jensen, SCScore: Synthetic Complexity Learned from a Reaction Corpus. J. Chem. Inf. Model. 2018, vol. 58(no. 2), 252–261. [CrossRef]
- Pereira, J. C.; Caffarena, E. R.; dos Santos, C. N. Boosting Docking-Based Virtual Screening with Deep Learning. J. Chem. Inf. Model. 2016, vol. 56(no. 12), 2495–2506. [Google Scholar] [CrossRef]
- Stork, C.; Chen, Y.; Šícho, M.; Kirchmair, J. Hit Dexter 2.0: Machine-Learning Models for the Prediction of Frequent Hitters. J. Chem. Inf. Model. 2019, vol. 59(no. 3), 1030–1043. [Google Scholar] [CrossRef]
- Yasuo, N.; Sekijima, M. Improved Method of Structure-Based Virtual Screening via Interaction-Energy-Based Learning. J. Chem. Inf. Model. 2019, vol. 59(no. 3), 1050–1061. [Google Scholar] [CrossRef]
- Cho, A. No room for error. Science 2020, vol. 369(no. 6500), 130–133. [Google Scholar] [CrossRef] [PubMed]
- Gromski, P. S.; Granda, J. M.; Cronin, L. Universal Chemical Synthesis and Discovery with ‘The Chemputer. Trends in Chemistry 2020, vol. 2(no. 1), 4–12. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Wang, S.; Tang, X. EQUIBIND: A geometric deep learning-based protein-ligand binding prediction method. Drug Discov Ther 2023, vol. 17(no. 5), 363–364. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Stärk, H.; Jing, B.; Barzilay, R.; Jaakkola, T. DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking. arXiv:2210.01776. 2023. [Google Scholar] [CrossRef]
- A. T. McNutt et al., GNINA 1.0: molecular docking with deep learning. J Cheminform 2021, vol. 13(no. 1), 43. [CrossRef]
- S. D. Dimitrov et al., QSAR Toolbox – workflow and major functionalities. SAR and QSAR in Environmental Research 2016, vol. 27(no. 3), 203–219. [CrossRef]
- P. Jing et al., Quantitative studies on structure–ORAC relationships of anthocyanins from eggplant and radish using 3D-QSAR. Food Chemistry 2014, vol. 145, 365–371. [CrossRef]
- Tosco, P.; Balle, T. Open3DQSAR: a new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J Mol Model 2011, vol. 17(no. 1), 201–208. [Google Scholar] [CrossRef] [PubMed]
- Ambure, P.; Halder, A. K.; González Díaz, H.; Cordeiro, M. N. D. S. QSAR-Co: An Open Source Software for Developing Robust Multitasking or Multitarget Classification-Based QSAR Models. J. Chem. Inf. Model. 2019, vol. 59(no. 6), 2538–2544. [Google Scholar] [CrossRef]
- Vainio, M. J.; Johnson, M. S. McQSAR: A Multiconformational Quantitative Structure−Activity Relationship Engine Driven by Genetic Algorithms. J. Chem. Inf. Model. 2005, vol. 45(no. 6), 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Nashwan, M. J.; Hani, S. B. Transforming cancer clinical trials: The integral role of artificial intelligence in electronic health records for efficient patient recruitment. Contemporary Clinical Trials Communications 2023, vol. 36, 101223. [Google Scholar] [CrossRef] [PubMed]
- A. Farnoud, A. J. Ohnmacht, M. Meinel, and M. P. Menden, Can artificial intelligence accelerate preclinical drug discovery and precision medicine? Expert Opinion on Drug Discovery 2022, vol. 17(no. 7), 661–665. [CrossRef]
- Wieder, R.; Adam, N. Drug repositioning for cancer in the era of AI, big omics, and real-world data. Critical Reviews in Oncology/Hematology 2022, vol. 175, 103730. [Google Scholar] [CrossRef]
- Chen, Z.; Min, M. R.; Parthasarathy, S.; Ning, X. A deep generative model for molecule optimization via one fragment modification. Nat Mach Intell 2021, vol. 3(no. 12), 1040–1049. [Google Scholar] [CrossRef]
- Pandiyan, S.; Wang, L. A comprehensive review on recent approaches for cancer drug discovery associated with artificial intelligence. Computers in Biology and Medicine 2022, vol. 150, 106140. [Google Scholar] [CrossRef]
- Zhang, B.; Shi, H.; Wang, H. Machine Learning and AI in Cancer Prognosis, Prediction, and Treatment Selection: A Critical Approach. JMDH 2023, vol. 16, 1779–1791. [Google Scholar] [CrossRef]
- K. Senthil Kumar et al., Artificial Intelligence in Clinical Oncology: From Data to Digital Pathology and Treatment. Am Soc Clin Oncol Educ Book 2023, no. 43, e390084. [CrossRef]
- Fountzilas, E.; Pearce, T.; Baysal, M. A.; Chakraborty, A.; Tsimberidou, A. M. Convergence of evolving artificial intelligence and machine learning techniques in precision oncology. NPJ Digit Med 2025, vol. 8(no. 1), 75. [Google Scholar] [CrossRef]
- A. Blanco-González et al., The Role of AI in Drug Discovery: Challenges, Opportunities, and Strategies. Pharmaceuticals (Basel) 2023, vol. 16(no. 6), 891. [CrossRef]
- Cheong, B. C. Transparency and accountability in AI systems: safeguarding wellbeing in the age of algorithmic decision-making. Front. Hum. Dyn. 2024, vol. 6. [Google Scholar] [CrossRef]
- Kiseleva, A.; Kotzinos, D.; De Hert, P. Transparency of AI in Healthcare as a Multilayered System of Accountabilities: Between Legal Requirements and Technical Limitations. Front Artif Intell 2022, vol. 5, 879603. [Google Scholar] [CrossRef]
- Y. K. Dwivedi et al., Artificial Intelligence (AI): Multidisciplinary perspectives on emerging challenges, opportunities, and agenda for research, practice and policy. International Journal of Information Management 2021, vol. 57, 101994. [CrossRef]
- Boudi, A. L.; Boudi, M.; Chan, C.; Boudi, F. B. Ethical Challenges of Artificial Intelligence in Medicine. Cureus 2024, vol. 16(no. 11), e74495. [Google Scholar] [CrossRef] [PubMed]
- Hasan, H. E.; Jaber, D.; Khabour, O. F.; Alzoubi, K. H. Ethical considerations and concerns in the implementation of AI in pharmacy practice: a cross-sectional study. BMC Med Ethics 2024, vol. 25(no. 1), 55. [Google Scholar] [CrossRef] [PubMed]
- M. Marouf et al., Realistic in silico generation and augmentation of single-cell RNA-seq data using generative adversarial networks. Nat Commun 2020, vol. 11(no. 1), 166. [CrossRef] [PubMed]
- Holmes, I.; Linder, J.; Kelley, D. Selective State Space Models Outperform Transformers at Predicting RNA-Seq Read Coverage. bioRxiv 2025, p. 2025.02.13.638190. [Google Scholar] [CrossRef]
- Toni, E.; Ayatollahi, H.; Abbaszadeh, R.; Siahpirani, A. Fotuhi. Machine Learning Techniques for Predicting Drug-Related Side Effects: A Scoping Review. Pharmaceuticals (Basel) 2024, vol. 17(no. 6), 795. [Google Scholar] [CrossRef] [PubMed]
- AlGhamdi, R.; Alassafi, M. O.; Alshdadi, A. A.; Dessouky, M. M.; Ramdan, R. A.; Aboshosha, B. W. Developing Trusted IoT Healthcare Information-Based AI and Blockchain. Processes 2023, vol. 11(no. 1), 34. [Google Scholar] [CrossRef]
- Ivanisevic, T.; Sewduth, R. N. Multi-Omics Integration for the Design of Novel Therapies and the Identification of Novel Biomarkers. Proteomes 2023, vol. 11(no. 4), 34. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform Biol Insights 2020, vol. 14, 1177932219899051. [Google Scholar] [CrossRef] [PubMed]
- Mirakhori, F.; Niazi, S. K. Harnessing the AI/ML in Drug and Biological Products Discovery and Development: The Regulatory Perspective. Pharmaceuticals (Basel) 2025, vol. 18(no. 1), 47. [Google Scholar] [CrossRef]
- Zaidan, A. M. The leading global health challenges in the artificial intelligence era. Front Public Health 2023, vol. 11, 1328918. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Overview of the core components of Artificial intelligence (AI). AI integrates machine learning (ML) and its advanced subset, deep learning (DL). ML is also divided in three main categories, including supervised, unsupervised, and reinforcement learning that incorporated many other methods.
Figure 1.
Overview of the core components of Artificial intelligence (AI). AI integrates machine learning (ML) and its advanced subset, deep learning (DL). ML is also divided in three main categories, including supervised, unsupervised, and reinforcement learning that incorporated many other methods.
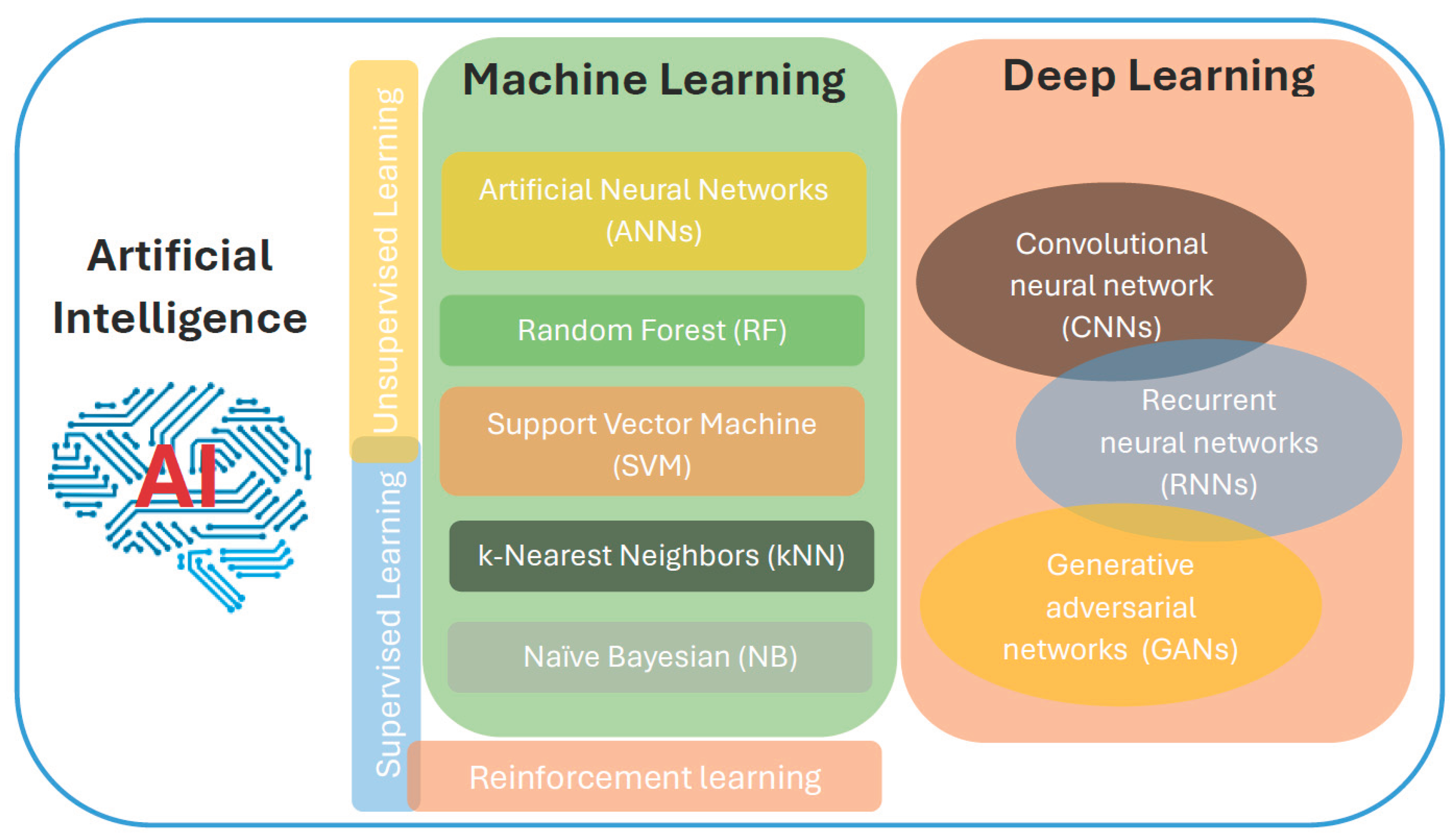

Table 1.
AI-based tools for drug discovery.
| Tool Name | Category | Key Feature(s) | Algorithm(s)/Architecture | Ref. |
|---|---|---|---|---|
| AlphaFold | Protein Structure Prediction | Predicts 3D protein structures with high accuracy. | Deep Neural Networks (DNN) | [42] |
| Coscientist | Chemical Reaction Planning | Autonomously plans and executes chemical experiments using literature search. | Large Language Models (LLM), Deep Neural Networks (DNN) | [43] |
| ODDT | Molecular Modeling | A comprehensive toolkit for molecular modeling and chemoinformatics. | Random Forest (RF), Neural Network Score (NNScore) | [44] |
| REINVENT | Molecular Generation | Designs nov[40,41]el molecules from scratch. | Recurrent Neural Networks (RNN), Reinforcement Learning | [31] |
| ORGANIC | Molecular Generation | Generates molecules with desired properties. | Machine Learning (ML) | [45] |
| JunctionTree VAE | Molecular Generation | Creates new, valid molecular structures. | Variational Autoencoder (VAE) | [46] |
| Chemical VAE | Molecular Generation | Generates new chemical compounds automatically. | Variational Autoencoder (VAE) | [47] |
| DeepChem | Molecular Property Prediction | A Python library for various drug discovery predictions. | Deep Learning (DL) | [48] |
| Conv_qsar_fast | Molecular Property Prediction | Predicts molecular properties from structural data. | Convolutional Neural Network (CNN) | [49] |
| DeepNeuralNetQSAR | Molecular Property Prediction | Forecasts molecular activity levels. | Deep Neural Networks (DNN) | [50] |
| Neural Graph Fingerprints | Molecular Property Prediction | Predicts properties of new molecules using their graph structure. | Convolutional Neural Network (CNN) | [51] |
| InnerOuterRNN | Molecular Property Prediction | Estimates chemical, physical, and biological properties. | Recurrent Neural Networks (RNN) | [52] |
| DeepTox | Molecular Property Prediction | Assesses the toxicity of chemical compounds. | Deep Learning (DL) | [39] |
| PotentialNet | Molecular Property Prediction | Estimates ligand-binding affinity. | Graph Convolutional Neural Network (CNN) | [53] |
| NNScore | Molecular Property Prediction | Scores protein-ligand binding affinity. | Neural Network | [54] |
| PPB2 | Molecular Property Prediction | Predicts poly-pharmacology (interaction with multiple targets). | Machine Learning (ML), Nearest Neighbor | [55] |
| SCScore | Molecular Property Prediction | Rates the synthetic complexity of a molecule. | Neural Network | [56] |
| DeltaVina | Drug Discovery | Improves the prediction of binding affinity. | Random Forest (RF), AutoDock Scoring | [57] |
| Hit Dexter | Drug Discovery | Identifies compounds that may interfere with biochemical assays. | Machine Learning (ML) | [58] |
| SIEVE-Score | Drug Discovery | An advanced scoring function for structure-based virtual screening. | Interaction-Energy-Based Learning | [59] |
| QML | Quantum Machine Learning | A Python toolkit for molecular modeling using quantum algorithms. | Machine Learning (ML) | [60] |
| Chemputer | Chemical Synthesis | A system for automating and documenting chemical synthesis procedures. | Chemical Programming Language (Not standard AI) | [61] |
| EquiBind | Molecular docking and virtual screening | Performs direct, "blind" prediction of ligand binding poses without the need for a traditional search procedure. | A deep learning model | [62] |
| DiffDock | .Molecular docking and virtual screening | Molecular docking that provides confidence estimates for its predicted ligand poses | A diffusion-based generative model | [63] |
| GNINA | Molecular docking and virtual screening | Pose prediction and scoring, offering high accuracy in structure-based virtual screening. | Convolutional neural networks (CNNs) | [64] |
| QSAR ToolBox | QSAR | An integrated platform for chemical grouping, read-across, and structural similarity analysis by combining experimental data and computational inference. | Traditional Machine Learning | [65] |
| SYBYL-X | QSAR | A comprehensive molecular modeling suite supporting structure-based drug design, lead optimization, and molecular docking. | Classical Machine Learning | [66] |
| Open3DQSAR | QSAR | Used to develop 3D-QSAR models via alignment, field-based descriptors, and regression optimization. | Classical Machine Learning | [67] |
| QSAR-Co | QSAR | Builds multi-target QSAR classification and regression models with multiple descriptor types. | Supervised Machine Learning | [68] |
| McQSAR | QSAR | Automated generation and optimization of QSAR models using evolutionary algorithms. | Machine Learning Optimisation Algorithm | [69] |
| PkCSM | Pharmacokinetics & Toxicity Prediction | Predicts key ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties using graph-based signatures as molecular descriptors. | Based on distance-based graph kernels and supervised learning. | [37] |
| AdmetSAR | Pharmacokinetics & Toxicity Prediction | A comprehensive source and prediction tool for chemical ADMET properties, featuring a large, curated database. | Various machine learning models (e.g., Random Forest, SVM). | [38] |
| DeepTox | Toxicity Prediction | Predicts the toxicity of chemical compounds by identifying toxicophores using deep learning. | Deep Learning (DL) | [39] |
| PandaOmics | Biomarker & Target Discovery | An AI-driven platform for analyzing multi-omics data to identify novel disease biomarkers and therapeutic targets. | Machine Learning (ML), including natural language processing for text mining. | [40] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



