Submitted:
11 January 2026
Posted:
27 January 2026
You are already at the latest version
Abstract
Cardiovascular disease (CVD) continues to be the leading non-communicable disease and the main cause of death worldwide, with atherosclerosis being the primary underlying condition factor. Emerging evidence highlights the crucial role of the host microbiota in regulating immune responses and metabolic pathways involved in atherogenesis. Gut and oral microbiota imbalance has been associated with atherosclerosis development, marked by a decrease in anti-inflammatory, butyrate-producing taxa such as Faecalibacterium prausnitzii, Roseburia intestinalis, and Blautia, along with an increase in pro-inflammatory bacteria like Enterobacteriaceae, Streptococcus spp., and Chlamydia pneumoniae. The detection of oral microbes within vascular and atherosclerotic lesions further connects oral health with cardiovascular outcomes. Microbial metabolites, such as trimethylamine-N-oxide (TMAO) and microbial-associated molecular patterns, have been shown to increase the risk of coronary heart disease. Conversely, certain gut bacteria, notably Akkermansia muciniphila, Bacteroides dorei, and Bacteroides vulgatus, exhibit protective effects by reducing atherosclerotic lesions and systemic inflammation. Additionally, gut microbiota-derived metabolites, particularly short-chain fatty acids (SCFAs) and bile acid derivatives, regulate immune homeostasis and lipid metabolism via GPR41 and GPR43 receptors. Although microbial signatures associated with atherosclerosis remain debated, growing evidence supports the microbiota and its metabolites as integral to the immune-metabolic axis influencing CVD. Understanding these dual roles may guide the development of microbiome-targeted interventions, including dietary modifications, prebiotics, probiotics, and receptor agonists, to prevent and manage atherosclerosis.
Keywords:
Atherosclerosis
; Trimethylamine-oxide
; short chain fatty acids
; gut microbiota
; Dysbiosis
1. Introduction
Cardiovascular diseases (CVDs) consist of various conditions that affect the heart and blood vessels, including coronary artery disease, cerebrovascular disease, rheumatic heart disease, and many more. The World Health Organization (WHO) reports approximately 17.9 million deaths worldwide each year due to CVDs. Myocardial infarctions and strokes account for 80% of total CVD deaths, with one-third of deaths in individuals under the age of 70. In the current 21st century, the primary cause of mortality in India is CVDs. CVDs affect the Indian population 10 years earlier than their European counterparts [1]. In Western countries, a quarter of deaths due to CVDs happen before 70 years of age, whereas half of CVD-related deaths occur before 70 years in India [2]. Moreover, countries with lower incomes, like India, have significantly higher death rates due to CVD compared to middle and high-income nations [3]. The mortality rate from CVDs doubled to 2.8 million in 2016, up from 1.3 million deaths in 1990, with a 34.3% increase in the crude death rate. Most CVD deaths occur in people over 70, but there is a significant number of deaths in younger age groups as well. In 2016, India, which accounts for 17.8% of the world’s population, was responsible for 23.1% of the global Disability-Adjusted Life Years (DALYs) caused by ischemic heart disease, 14.0% due to stroke, and a significant 37.6% due to rheumatic heart disease. CVDs represent nearly 30% of all non-communicable diseases (NCDs) in India, with atherosclerosis being the leading cause. The development of atherosclerosis is associated with traditional risk factors, altered lifestyle, and hereditary risk factors. Atherosclerosis often leads to ischemic heart disease (IHD), which is increasingly affecting younger individuals in India with an age of less than or equal to 45 years. In some cases, sudden cardiac death (SCD) can be a symptom of IHD in younger individuals [3]. Atherosclerosis is characterized by a chronic inflammatory process that begins with endothelial injury, leading to lipid accumulation and plaque formation within the intima. The term 'atherosclerosis' is derived from 'atherosis', referring to the accumulation of lipid-laden macrophages, and 'sclerosis', referring to the fibrosis of the arterial wall with smooth muscle cells, leukocytes, and connective tissue [3]. This process involves a complex interaction among dysregulated lipid metabolism, acute inflammation, and vascular cell dysfunction. Additionally, endothelial cells, smooth muscle cells (SMCs), and immune cells, including innate effector cells such as macrophages and adaptive lymphocytes such as T cells and B cells, are involved [4]. In recent years, the involvement of gut microbiota has added another layer of complexity to an already complex system. Microbiota affects host metabolism, immunity, and vascular function through various bioactive metabolites, including trimethylamine-N-oxide (TMAO), bile acid derivatives, short-chain fatty acids (SCFAs), and tryptophan metabolites. Specifically, they can influence immune cell polarization, endothelial function, and oxidative stress pathways [6]. Additionally, microbiota dysbiosis can trigger systemic inflammation by compromising the intestinal permeability and lipopolysaccharide (LPS) translocation, linking gut-derived signals to vascular health injury.
A more comprehensive and detailed mechanistic framework for atherosclerosis can be created by integrating the mechanisms of oxidative stress, immune activation, and microbiota-derived metabolites. This perspective highlights new therapeutic opportunities targeting the microbiota–metabolite–immunity axis and bridges the gap between traditional and emerging risk factors.
2. Conventional and Emerging Cardiovascular Risk Determinants
2.1. Traditional Risk Factor: The Chronic Driver of Vascular Injuries
The growing prevalence of atherosclerotic cardiovascular disease (ASCVD) can be attributed to risk factors such as dyslipidemia, hypertension, smoking, diabetes mellitus, dietary habits, and a sedentary lifestyle. These risk factors can activate common pathogenic mechanisms, such as endothelial dysfunction, oxidative stress, and inflammatory signalling. For example, dyslipidemia is characterized by elevated low-density lipoprotein (LDL) cholesterol, which can lead to the formation of oxidized LDL (oxLDL). These oxLDL particles are potent pro-inflammatory antigens that contribute significantly to vascular damage [5]. Obesity is strongly associated with cardiovascular issues and is a major global health concern. Meanwhile, some studies point to an "obesity paradox" that challenges the idea of obesity as a health risk. More research is necessary to clarify these findings [8]. Hypertension worsens this condition by increasing shear stress on blood vessels, which can result in endothelial injury. Research indicates that high blood pressure can cause significant oxidative stress, further weakening vascular integrity [9]. Moreover, elevated glucose levels from diabetes accelerate the formation of advanced glycation end-products (AGEs), intensifying oxidative stress within blood vessels [6]. Dietary habits significantly influence cardiovascular health. The American Heart Association (AHA) recommends a Mediterranean diet (MD) rich in unprocessed foods, dietary fibre, and healthy fats, with less dairy products, which are linked to lower rates of heart disease [11]. Psychological factors also influence cardiovascular health, where depression increases the risk of CVD, and stress plays a role in the development of atherosclerosis. [7]. Together, these factors form a vicious cycle that aggravates the risk of cardiovascular disease. Evidence from both population studies and mechanistic research shows that these traditional risk factors closely interact with the body's immune system and gut microbial metabolism, collectively affecting the inflammatory state of blood vessels.
2.2. Emerging and Non-Classical Risk Factor:
The human gastrointestinal (GI) tract contains a diverse and active microbiota, supported by its nutrient-rich environment [8]. The gut microbiota consists of over 10 trillion microbial cells, including various symbiotic microorganisms. These microorganisms play a vital role in modulating immune responses and metabolic processes through their metabolites [14]. Due to their broad metabolic capabilities, they have been described as a "metabolic organ". [9]. Metagenomic sequencing of microbial DNA has revealed the vast diversity of the gut microbiota [10]. Microbiota dysbiosis has emerged as a significant contributor to the pathophysiology of ASCVD [11]. Changes in gut microbial composition can lead to increased production of pro-atherogenic metabolites, TMAO and secondary bile acids. At the same time, it reduces the synthesis of anti-inflammatory SCFAs. This dysbiotic shift maintains a systemic pro-inflammatory environment, increases oxidative stress, and disrupts endothelial homeostasis. For example, a metagenome-wide association study (MWAS) of individuals with ACVD showed differences from healthy subjects, identifying higher levels of taxa from the Enterobacteriaceae family, such as Escherichia coli, Klebsiella spp., Enterobacter aerogenes, and bacteria from the oral cavity, including Streptococcus spp., Lactobacillus salivarius, Solobacterium moorei, and Atopobium parvulum in ACVD patients [18]. In contrast, ACVD patients showed lower levels of beneficial bacteria such as Roseburia intestinalis and Faecalibacterium prausnitzii, which are known butyrate producers, as well as other common gut microbiota members like Bacteroides spp., Prevotella copri, and Alistipes shahii [12]. A study using terminal restriction fragment length polymorphism (T-RFLP) revealed a higher Firmicutes/Bacteroidetes (F/B) ratio in individuals with coronary artery disease (CAD) than in healthy individuals. The Firmicutes phylum mainly comprises Lactobacillales and Clostridium, and the Bacteroidetes phylum, including Bacteroides and Prevotella [13]. Research has also underscored the diet-microbiota interaction concerning cardiovascular disease [14]. Metagenomic studies have revealed a higher abundance of Collinsella, Enterobacteriaceae, and Streptococcus spp. in patients with atherosclerosis, whereas Roseburia and Eubacterium are more commonly found in healthy individuals [21].
Further investigations have identified Chryseomonas, Veillonella, and Streptococcus in atherosclerotic plaques and changes in cholesterol levels [15]. Furthermore, Helicobacter pylori, known for causing gastric ulcers, has been associated with atherosclerosis [16]. A reduction in genera such as Blautia, Collinsella, and some unclassified Erysipelotrichaceae and Ruminococcaceae has been observed in patients with heart failure compared to healthy controls. Notably, Blautia is associated with anti-inflammatory effects [24], and Akkermansia muciniphila has been shown to enhance gut barrier integrity and protect against atherosclerosis [17].
Several bacterial species, such as Chlamydia pneumoniae, Staphylococcus spp., Streptococcus spp., Klebsiella pneumoniae, Proteus vulgaris, Burkholderia spp., and Pseudomonas aeruginosa, have been associated with the accelerated progression of cardiovascular disease [18]. Virome studies in patients with cardiovascular disease have reported a higher prevalence of bacteriophages, especially those associated with Propionibacterium, Pseudomonas, and Rhizobium. This contradicts the virome of healthy individuals, which is more enriched in eukaryotic viruses such as Lymphocystis virus (LCV) and Torque teno viruses (TTV). The findings suggest that the presence of bacterial and viral DNA elements in the bloodstream could be associated with increased circulating DNA in individuals with cardiovascular disease, a connection that warrants further investigation [19].
Earlier studies and investigations underscored the connection between atherosclerosis development and infections, such as those in the oral cavity, respiratory system (including Chlamydia pneumoniae), and those caused by Helicobacter pylori [20]. Furthermore, the previous study showed the presence of periodontal bacteria in atherosclerotic plaques [21]. Various other studies further confirmed the presence of bacteria like P. gingivalis and A. actinomycetemcomitans in these plaques [22,23]. The origin of microbes and their association with CVDs are listed in Table 1. Recent investigations have continued to link the oral microbiota and atherosclerosis, underscoring the significance of oral bacteria in the pathogenesis of the disease [24].
3. Lipid Peroxidation in Vascular Inflammation
Lipid peroxidation is a crucial internal chain reaction that significantly contributes to lipid oxidation. When lipids with carbon-carbon double bonds are attacked by free radicals or non-radical species, they lose a hydrogen atom from a carbon and gain an oxygen atom. This process results in the formation of lipid peroxyl radicals and hydroperoxides [25].
Lipids play a significant role in the onset of cardiovascular diseases, mainly due to their predisposition to oxidation. These lipids, present in the bloodstream and various tissues, may bind with lipoproteins or albumin. Lipoproteins have a core filled with cholesterol esters and triglycerides, surrounded by free cholesterol, phospholipids, and apolipoproteins, which are the main transporters of lipids that do not dissolve in water. Lipoproteins are categorized into various types based on their size, lipid makeup, and apolipoproteins, including chylomicrons, chylomicron remnants, very low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), low-density lipoprotein (LDL), high-density lipoprotein (HDL), and lipoprotein (a) (Lp(a)). These lipoproteins play a crucial role in processing and transporting dietary lipids from the intestines and lipids from the liver. HDL plays a crucial role in reverse cholesterol transport (RCT), facilitating the transport of cholesterol from the body's tissues back to the liver and intestines. On the other hand, LDL mainly transports cholesterol to cells, which raises the risk of heart disease. Oxidative mechanisms in the development of atherosclerosis are complex and influenced by multiple factors. Initially, LDL particles in the arterial wall can undergo varying degrees of oxidation, which contributes to the progression of atherosclerosis. OxLDL can lead to endothelial damage, promote the adhesion of inflammatory cells, and induce macrophage transformation [26]. This process contributes to endothelial dysfunction by suppressing endothelial nitric oxide synthase (eNOS), stimulating inflammatory cytokine release, and promoting platelet aggregation. Oxidized LDL induces the activation of pro-inflammatory genes, including (tumor necrosis factor α) TNFα, (interleukin 1β)IL-1β, and (interleukin 6) IL-6, through the stimulation of peroxisome proliferator-activated receptor gamma (PPARγ) [27]. Furthermore, lipid peroxidation byproducts elevated the expression of monocyte chemotactic protein-1 (MCP-1), interferon-γ-inducible protein-10, and cyclooxygenase-2 (COX-2) and facilitated the transcriptional activation of nuclear factor (NF)-κB following the uptake of modified LDL. These substances further exacerbate atherosclerosis by producing leukotrienes, acrolein, and other inflammatory agents that activate endothelial and foam cells and cause mast cell degranulation, intensifying inflammatory damage [28,29]. The endothelium plays a vital role in regulating vascular tone, maintaining homeostasis, facilitating molecular exchange between the blood and tissues, and signaling immune responses and inflammation [30].
Endothelial dysfunction and damage cause LDL cholesterol to seep into the vessel wall, especially the intima, where it undergoes oxidation. The buildup of cholesterol oxidation products, such as oxysterols and aldehydes, is prominent in atherosclerotic lesions. Scavenger receptors on endothelial cells, like the lectin-like oxidized LDL receptor 1 (LOX-1), facilitate the uptake of oxLDL and contribute to foam cell formation, thus speeding up atherosclerosis. LOX-1 is implicated in endothelial cell apoptosis and is associated with elevated levels of MCP-1 and transforming growth factor β1 (TGF-β1). Furthermore, LOX-1 downregulates the activity of eNOS and stimulates the synthesis of matrix metalloproteinases in vascular smooth muscle cells (VSMCs) [31].
Although HDL is considered protective due to its role in reverse cholesterol transport, there is evidence that HDL can be oxidized, altering its beneficial properties into harmful ones [32]. HDLs represent a diverse and functionally complex family of lipoproteins, characterized by multiple subclasses that vary in their physical dimensions, structural morphology, and lipid and apolipoprotein composition. This inherent heterogeneity renders HDLs vulnerable to various oxidative modifications induced by reactive oxygen species, including peroxyl and hydroxyl radicals, aldehyde compounds, and oxidants generated by myeloperoxidase (MPO) [33]. Such changes lead to dysfunctional HDL, as evidenced by reduced sphingosine-1-phosphate levels in patients with coronary artery disease [34]. The oxidation of lipids, driven by free radicals, generates biologically relevant aldehydes, including advanced lipoxidation end products (ALEs), malondialdehyde (MDA), 4-hydroxynonenal (4-HNE), and acrolein [35,36]. These oxidation byproducts trigger inflammation, extracellular matrix deposition, and arterial wall remodeling, all of which are vital to the progression of atherosclerosis [37]. Endothelial cells are highly susceptible to 4-HNE-induced damage. A rise in 4-HNE levels decreases NO availability by lowering dimethylarginine dimethylaminohydrolase (DDAH) activity, which can elevate methylarginine levels, diminish eNOS-derived NO production, and consequently, endothelial dysfunction [38]. ALEs also cause monocyte activation and vascular complications by activating inflammatory pathways [39].
In addition, 4-HNE prompts macrophages to produce matrix metalloproteinase-9 (MMP-9) [40] and MMP-2 in vascular smooth muscle cells [41]. These enzymes are responsible for the degradation of collagen fibers and may destabilize atherosclerotic plaques and promote thrombus formation. 4-HNE activates matrix metalloproteinases-1 and -2 in vascular smooth muscle cells. These enzymes not only degrade the extracellular matrix [42], but it also activates protein kinase B (Akt) [41], p38 mitogen-activated protein kinase (MAPK)[43]. Its phosphorylation of c-jun N-terminal kinase (JNK) is also noteworthy [44].
Recent research has highlighted the buildup of 4-HNE-adducts, especially in the smooth muscle cells of the human aorta. This buildup tends to increase with age and is common in people with advanced atherosclerosis [45]. Additionally, 4-HNE levels are elevated in the adventitia, which is associated with the vasa vasorum and microcapillaries found in atherosclerotic lesions [46]. While 4-HNE does not directly affect aortic elastin, it plays a role in its breakdown and decreases its ability to regenerate [47]. Moreover, other oxidant sources within the vessel wall, such as myeloperoxidase, lipoxygenases, mitochondria, and uncoupled eNOS, are often found in human atherosclerotic lesions [48].
4. Immune Cells and Immune Response in Atherosclerosis
Atherosclerosis begins with the retention of apolipoprotein-B-containing lipoproteins, such as LDL, and their oxidative or enzymatic modification within the arterial intima. These modified lipoproteins act as damage-associated molecular patterns that trigger local innate and adaptive immune responses. This chronic condition typically starts with dysfunctional endothelial cells, which are characterized by the upregulation of surface cell adhesion molecules such as vascular cell adhesion molecule 1 (VCAM-1), intracellular cell adhesion molecule 1 (ICAM-1), and E-selectin [49]. This leads to the recruitment of circulating monocytes and other leukocytes to these regions [50]. The monocytes migrate to the site and transform into macrophages that internalize lipoproteins via scavenger receptors such as SR-A, (cluster of differentiation 36) CD36, and LOX-1, and via pinocytosis. These lipid-laden macrophages transformed themselves into foam cells that secrete proinflammatory cytokines and matrix remodelling enzymes [51]. A significant fraction of CD4+ T cells belonging to the T helper type 1 (Th1) group infiltrate the arterial damage in response to LDL. These Th1 cells produce proinflammatory cytokines, including interferon-γ (IFN-γ) and TNF-α [52]. IL-6 produced by arterial walls during atherosclerosis can prompt the liver to initiate an acute-phase response. Activation of the acute-phase response in the liver causes the release of acute-phase reactants, such as (C-reactive protein) CRP, into the bloodstream [53]. The association between elevated CRP levels and myocardial infarction highlights the role of inflammation in myocardial ischemia [54]. CRP concentrations have been identified as an independent marker for forecasting cardiovascular incidents [55]. Although CRP does not directly cause atherosclerosis [55], its role as an indicator of vascular inflammation makes it a valuable marker for assessing statin treatment in patients with intermediate risk levels [56]. The invading monocytes differentiate into macrophages under the influence of granulocyte-macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF), which are secreted by endothelial cells, among others. Macrophages derived from monocytes constitute a significant portion of the cell population in atherosclerotic plaques due to ongoing recruitment, differentiation, and proliferation at the site [57].
Research involving in vitro and in vivo models has shown that macrophages can transform into either an inflammatory M1 type or a less inflammatory M2 type, guided by the type of activation signals they receive [58]. IL-4 or IL-13 can stimulate the development of M2, whereas IFN-γ and lipopolysaccharide trigger classical activation of M1 macrophages. It has been observed that M1 macrophages are laden with lipids, in contrast to M2 macrophages, which hold fewer lipids and are located away from the central lipid area [59]. Identifying these macrophage subsets in live organisms presents difficulties, as over a third of macrophages extracted from the aortas of atherosclerotic mice do not conform to the standard M1 or M2 categories. A study by Kadi et al. [60] transformed macrophage into a subset after treatment with oxidised phospholipid. This particular macrophage is characterized by a novel phenotype, referred to as Mox macrophages. This phenotype exhibits a significant upregulation of Nuclear factor erythroid 2-related factor 2 (NRF2) mediated gene expression involved in redox reactions. Their findings showed that Mox macrophages accounted for approximately 30% of the total macrophage population in the advanced atherosclerotic lesions in Ldl-/- mice.
Macrophages are known for their adaptability and interactions with other cell types, notably T cells, which can influence their polarization in their immediate surroundings. Interestingly, vascular smooth muscle cells can transform into macrophage-like cells in response to lipid accumulation under controlled conditions [61]. In vivo experiments tracking cell fate in murine model have revealed that roughly 30% of smooth muscle cells within atherosclerotic lesions display attributes typically associated with macrophages. This observation is consistent with findings in human lesions [62,63,64]. A significant portion of these transformed smooth muscle cells show similarities to the Mox cell type in the plaques of Apoe−/− mice [62].
4.1. Innate Immunity in Atherosclerosis
In atherosclerosis, oxLDL plays a significant role in triggering macrophage activation (Figure 1), where the NF-κB moves into the nucleus [65]. Within atherosclerotic lesions, these macrophages are known to express multiple Toll-like receptors (TLRs). These receptors can detect pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). TLR2 and TLR4 are activated by modified LDL and its by-products [66]. Furthermore, elements such as heat shock proteins, bacterial toxins, and viral glycoproteins present in the lesions also serve as both internal and external triggers for TLRs [52]. The interaction of these TLRs, via the (Myeloid Differentiation Primary Response Gene88) MyD88 adaptor protein, leads to the activation of signalling pathways that result in the secretion of pro-atherosclerotic cytokines such as IL-1β and IL-18 [67]. Moreover, the innate immune system is further activated by products of LDL oxidation, such as lysophosphatidylcholine and oxidized non-esterified fatty acids, which are generated by the enzyme lipoprotein-associated phospholipase A2 (Lp-PLA2) [68].
Cholesterol crystals in foam cells are known to activate the (NOD-like receptor 3) NLRP3 inflammasome, which converts pro-caspase-1 into its active form, caspase-1. This activation results in the production and release of IL-18 and IL-1β, leading to inflammation and immune responses [69]. Inhibiting IL-1β as a strategy to prevent cardiovascular incidents has been explored in clinical trials [70]. The release of IL-1β from foam cells also prompts SMCs to produce IL-6, which, in turn, stimulates the liver to initiate an acute-phase response, including the production of CRP [71].
Other innate immune cells, such as neutrophils, mast cells, natural killer (NK) cells, and natural killer T (NKT) cells, are also present in plaques but in smaller numbers than macrophages. In hypercholesterolemic mice, neutrophils are recruited during the early stages of atherosclerosis but are absent in later stages [72]. NK and NKT cells can exacerbate atherosclerosis, likely through the release of IFN-γ [73,74]. However, NKT cells might also limit disease progression by producing the anti-inflammatory cytokine IL-10 [75]. γδ T cells appear to have no significant effect on atherosclerosis [76]. Although several small populations of innate immune cells are involved at different stages of the disease, macrophages are the main effector cells in the plaque.
4.2. Adaptive Immunity in Atherosclerosis
The adaptive immune system is essential in coordinating the immune response to atherosclerosis. T cells, B cells, and their effector molecules are key players in this condition (Figure 2). Atherosclerosis may not be classified as an autoimmune disease, but compelling evidence from both clinical and preclinical studies showed that autoimmune responses can be observed in individuals with atherosclerosis and in animal models. Two key subsets of T cells, CD4+ and CD8+, play a significant role in this process and highlight the adaptive immune system’s multifaceted nature. Antigen-presenting cells (APCs), particularly macrophages within the atherosclerotic plaque, adventitial B cells, and dendritic cells (DCs), are central to modulating adaptive immunity [77]. These APCs migrate to the lymph nodes, where they present the plague-derived peptides on their MHC (major histocompatibility complex) class I or MHC class II molecules to the naïve CD4+ or CD8+ T cells, respectively [78]. The interaction between co-stimulatory molecules such as CD28 and CD40L on APCs with T cell receptor (TCR) is essential for inducing clonal proliferation [79] and differentiation of T cells into various effector subsets such as T helper 1 (Th1), T helper 2 (Th2), T helper 17 (Th17), CD4+ regulatory T cells (Treg), and CD8+ cytotoxic T cells [80]. These specialized effector cells, with immunological memory, migrate from draining lymph nodes to the systemic circulation, where they accumulate in the atherosclerotic plaque [81]. Effector cells expressed receptors such as CCR5 [82], CXCR6 [83], and CXCR3 [84] as homing receptors for the atherosclerotic lesion. For instance, Th1 cells, which are predominant dominant in atherosclerotic lesions, are characterized by the expression of T-box transcription factor (T-bet)[85]. These cells secrete pro-inflammatory cytokines like IFN-γ and TNF, which polarize macrophages towards M1 effector cells and T cells, amplifying the atherogenic inflammation [86]. IFN-γ has a dual role; while it restricts the VSMCs' proliferation [91], it also accelerates the degradation of collagen [92], leading to plaque instability. On the other hand, TNF-α increased the lesion size by promoting leukocyte infiltration [87] and elevating the LDL levels [88]. Meanwhile, Th2 T helper cells, which express transcription factor GATA-binding protein 3 (GATA3), are essential for establishing humoral immunity [89]. In addition, Th17 cells, expressing the transcription factor RARA-related orphan receptor gamma (RORγ), represent a significant subset of CD4+ T cells in atherosclerosis [90]. The actions of Th2 and Th17 cells can swing either toward pro- or anti-atherogenic effects depending on the cytokines they release [91]. For instance, IL-4 from Th2 cells promotes atherogenic [92], while IL-5 and IL-13 can inhibit the Th1 response, presenting a protective mechanism against atherosclerosis [93]. In addition, IL-5 promotes the production of antibodies against the oxLDL by inducing B1 cell maturation. Similarly, cytokines such as IL-17, IFN-γ, IL-6, and GM-CSF produced by Th17 not only accelerate atherosclerosis progression but also worsen leukocyte accumulation in plaques [94,95]. Treg cells play a vital role in maintaining immune balance by secreting essential anti-inflammatory cytokines such as IL-10 and TNF-β. They are instrumental in preventing atherosclerosis by inhibiting the proliferation and functional activity of CD4+, CD8+, B cells, NKT cells, and APCs, as well as cytokine production. Furthermore, Treg cells enhance macrophage efferocytosis, a process critical for cellular cleanup and tissue health [96]. However, in the presence of ApoB100, these beneficial Treg cells can lose CD25 and FoxP3 expression and exhibit a pro-inflammatory Th1/Th17 phenotype with cytotoxic properties [97]. A fraction of Treg cells can convert into T follicle helper cells, thus losing their ability to suppress immune responses and prevent atherosclerosis [98]. MHC class I molecules play a crucial role in presenting apolipoprotein B-derived epitopes to CD8+ T cells, which subsequently differentiate into CD8+ cytotoxic T lymphocytes (CTLs) 97. CD8+ T cells can either contribute to the progression of atherosclerosis or protect against it. For example, CD8+ cytotoxic T cells can promote necrotic core formation by releasing cytotoxic granules, especially perforin-1 and granzymes, to induce apoptosis in macrophages, VSMCs, and endothelial cells. Moreover, they aggravate inflammation and monocytosis in the bone marrow by producing pro-inflammatory cytokines like TNF-α and IFN-γ [99]. Additionally, a specific subset of CD8+ T cells with regulatory functions can actively reduce atherosclerosis by limiting the accumulation of macrophages and Th1 cells [108], as well as decreasing B cell-mediated atherogenic antibody production [109].
B cells, like T cells, detect antigens and other foreign molecules with the B cell receptors (BCRs). These receptors play a significant role in the activation, maturation, and differentiation of B cells into antibody-producing plasma cells. B cells are broadly categorized into two types, B1 cells and B2 cells [100]. B1 cells have a unique ability to mature independently into B1 plasma cells, which secrete IgM antibodies that specifically target oxidized low-density lipoprotein (oxLDL), while also producing the anti-inflammatory cytokine IL-10. This dual action is crucial, as IgM antibodies directly counteract oxLDL, thereby reducing foam cell formation and inhibiting their activation, while IL-10 effectively modulates the immune response and promotes homeostasis [101]. On the other hand, B2 cells rely on T follicular helper (Tfh) cells for their activation and subsequent maturation into B2 plasma cells [102]. These B2 plasma cells release IgG antibodies and pro-inflammatory cytokines like TNF-α, which can, unfortunately, drive the progression of atherosclerosis [103].
5. Gut-Derived Metabolites and Atherosclerosis
The gut microbiome breaks down both diet-derived and host-produced substances, producing metabolites that significantly influence microbial communities and host cells. It is believed that more than half of the metabolites in feces and urine originate from or are altered by the gut microbiota [104]. These metabolites fall into three main categories (1) metabolites that come from daily diet and modified by the gut microbiota, like compound K [105]; (2) metabolites originated from the host and transformed by the gut microbiota, for instance, secondary bile acids [106]; and (3) metabolites that are produce by the gut microbiota, including short-chain fatty acids [107].
These metabolites do not localize in the gastrointestinal tract but instead travel throughout the body, mainly through the small intestine [108]. Depending on the type of cell they interact with, these metabolites have varying effects on the host cells. For instance, it affects vital functions such as intestinal epithelial regeneration, maintenance of the intestine, and the balance of the mucosal immune system [109].
5.1. Trimethylamine N-Oxide and Its Role in Cardiovascular Disease
Trimethylamine (TMA) is a compound produced in the gut microbiome from dietary components such as betaine, L-carnitine, γ-butyrobetaine (GBB), choline, and various choline-rich compounds [110]. The transformation of choline into TMA is catalyzed by enzymes encoded by the cutC/D gene cluster, where CutC represents a glycyl radical enzyme with choline trimethylamine-lyase activity and CutD signifies a glycyl radical-activating protein. These enzymes, together with proteins responsible for the construction of microcompartments that isolate acetaldehyde, a TMA by-product, are present across several bacterial phyla, including Firmicutes (e.g., Clostridium XIVa strains) [111], Actinobacteria, Proteobacteria (e.g., Desulfovibrio desulfuricans, Desulfovibrio alaskensis) [112], and Eubacterium sp. strain AB3007 [111]. Nonetheless, the Bacteroidetes phylum lacks these Cut genes and the TMA-lyase functionality [110].
The gene duo cnt A/B is crucial for synthesizing a two-component oxidoreductase enzyme that converts L-carnitine into TMA, initially producing γ-butyrobetaine in the ileum, which is later converted to TMA in the cecum and colon [113]. Similarly, the genes yeaW/X encode an oxygenase and an oxidoreductase, which play a role in the generation of TMA from substrates such as γ-butyrobetaine, L-carnitine, choline, and betaine [114]. A variety of gut microbiota, including Gamma-proteobacteria (e.g., Klebsiella pneumoniae, E. coli, Citrobacter, Providencia, and Shigella), Betaproteobacteria (e.g., Achromobacter), Firmicutes (e.g., Sporosarcina), and Actinobacteria, possess orthologs and homologs of the CntA/CntB and YeaW/YeaX genes. However, it is noteworthy that bacteria from the Bacteroidetes phylum do not contribute to TMA production [115]. Romano et al.'s research identified eight species within the primary phyla of Firmicutes and Proteobacteria that can metabolize choline to TMA, including Anaerococcus hydrogenalis, Clostridium asparagiforme, Clostridium hathewayi, Clostridium sporogenes, Escherichia fergusonii, Proteus penneri, Providencia rettgeri, and strains of Edwardsiella tarda, indicating a potential acquisition through horizontal gene transfer [110].
Dietary choices, especially fish and seafood, are high in TMAO and TMA. About 50% of the TMAO consumed is directly absorbed and excreted in urine, while the remaining portion is converted into TMA by TMAO reductase in the gut [116]. Certain gut microorganisms can change TMA into TMAO using TMA monooxygenase [117]. Additionally, TMA and TMAO serve as substrates for gut bacteria to produce dimethylamine (DMA) and formaldehyde, mediated by trimethylamine dehydrogenase (TMADH) and TMAO demethylase [117]. Most TMA formed or ingested in the gut is quickly absorbed into the portal circulation. It is oxidized to TMAO by liver flavin-containing monooxygenases (FMOs), with FMO3 being especially important due to its much higher specificity compared to FMO1 [114].
Plasma TMAO concentrations vary significantly among individuals based on age, diet, and other factors. TMAO levels tend to increase with age, and cholic acid, a bile salt, can boost plasma TMAO levels by promoting FMO3 expression through the bile acid–activated nuclear receptor FXR. TMAO levels are also affected by sex hormones, with testosterone decreasing FMO3 expression and estrogen increasing it. Trimethylaminuria may occur due to the notable decrease in TMAO levels that happens during menstruation [118].
Dietary choices significantly impact TMAO synthesis, with vegetarians usually producing less TMA than omnivores. Diets high in fat or typical Western diets correlate with higher plasma TMAO concentrations. Consumption of foods rich in non-digestible carbohydrates could reduce the production of TMAO by changing the composition of gut bacteria, even though there is evidence of a short-term rise in TMAO levels in the blood after a boost in the consumption of non-digestible starch [118].
TMAO is implicated in the development of CVD, especially atherosclerosis (Figure 3). TMAO levels could be indicative of the likelihood of CVD occurrences and major adverse cardiovascular events (MACE), such as myocardial infarction, stroke, and death [119]. Research on mice fed with Western diet found a connection between elevated TMAO levels in the blood and several adverse cardiac outcomes. These included cardiac dysfunction, heart fibrosis, increased levels of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), alongside a decrease in anti-inflammatory cytokines like IL-10 [120].
TMAO interferes with normal endothelial cell function and contributes to CVD. Application of TMAO to human monocytic THP-1 cells and human umbilical vein endothelial cells (HUVECs) enhances monocyte adhesion via the PKC/NF-κB pathway, resulting in elevated VCAM-1 levels [121]. In a similar mechanism, exposure of human aortic endothelial cells (HAECs) and vascular smooth muscle cells to TMAO results in greater leukocyte adhesion by activating the MAPK/NF-κB pathway [122]. TMAO further triggers the release of pro-inflammatory proteins such as cyclooxygenase 2, E-selectin, IL-6, and intracellular adhesion molecule 1 by stimulating NF-κB [122]. Moreover, TMAO boosts the expression and functionality of tissue factor (TF) in primary human coronary artery endothelial cells (HCAECs) via the NF-κB pathway, which may lead to atherothrombosis [123].
The cytokines IL-23 and IL-22 can influence atherosclerosis by affecting the composition of pro-atherogenic microbiota and their byproducts, such as TMAO. Inhibiting the IL-23 and IL-22 pathway results in elevated systemic concentrations of TMAO, demonstrating their importance in regulating the microbial generation of TMAO [124]. IL-22 is notably produced by Th17 cells, with IL-23 being essential for the function of these cells [125,126]. The specific roles of IL-22 and IL-23 in atherosclerosis remain to be fully elucidated.
Individuals suffering from metabolic disorders such as type 2 diabetes and chronic kidney disease exhibit a higher concentration of microbes that produce TMA. In these patients, increased levels of TMAO are associated with signs of inflammation and endothelial dysfunction [127]. TMAO affects cholesterol and sterol metabolism across biological systems, including the bile acid pool, thereby affecting lipid balance [113]. TMAO decreases the activity of the CYP7A1 enzyme, which is essential for bile acid production and cholesterol breakdown. Certain human CYP7A1 gene variations have been linked to lower CYP7A1 activity and an increased risk of atherosclerosis [128].
Intake of TMAO precursors such as choline and carnitine, or direct supplementation with TMAO, can inhibit reverse cholesterol transport. This reduction in activity can be attributed to the lowered levels of specific intestinal cholesterol transporters. Notably, ABCG5/8 promotes cholesterol efflux, and the Niemann-Pick C1-like 1 (NPC1L1) protein aids in the absorption of cholesterol into enterocytes [129]. Additionally, TMAO increases macrophage expression of scavenger receptors CD36 and SR-A1, which cause lipid accumulation and foam cell formation in atherosclerotic lesions [130,131]. OxLDL activates these receptors, and the MAPK/JNK signalling pathway might play a key role in TMAO-induced atherosclerosis [132].
Elevated levels of TMAO trigger NLRP3 inflammasome activation, increasing caspase-1 activity and IL-1β secretion. Additionally, this process increases the endothelial cells' permeability, aiding in the onset of endothelial damage and the development of atherosclerosis. This phenomenon has been observed in carotid artery endothelial cells (CAECs) and in mice that have undergone partial ligation of the carotid arteries [133]. Furthermore, TMAO is capable of inducing oxidative stress and triggering the activation of the TXNIP-NLRP3 inflammasome, which leads to the release of the inflammatory cytokines IL-18 and IL-1β in a manner dependent on both concentration and time [134]. In addition, Yue et al. found that TMAO activates the NLRP3 inflammasome and stimulates the production of reactive oxygen species (ROS) in fetal human colon cells (FHCs) [135].
In individuals suffering from ischemic stroke, there is a significant correlation between the levels of TMAO and the proportion of pro-inflammatory intermediate monocytes, specifically CD14++CD16+ [136]. Levels of TMAO are associated with markers of monocyte activation and inflammation, such as sCD14 and sCD163, in individuals with HIV who also have carotid atherosclerosis [137]. Moreover, sCD14 levels have been shown to independently correlate with TMAO levels in untreated HIV-infected patients [138]. Additionally, mice on a choline-rich diet, which increases TMAO synthesis, show higher levels of pro-inflammatory Ly6C high monocytes than those on a standard diet [136].
A study of 271 German adults found a significant association between high plasma TMAO levels and low-grade inflammation. Higher TMAO levels were associated with elevated TNF-α and soluble TNF receptors (sTNF-R p75 and sTNF-R p55). However, no changes were noted in plasma IL-6 and C-reactive protein levels [139]. On the other hand, Chou et al. reported a positive correlation between TMAO levels and both IL-1β and high-sensitivity CRP (hsCRP) in patients with stable angina [140]. CRP is recognized as an inflammation marker used to assess the risk of atherosclerosis and cardiovascular events [139,141]. In vitro studies with endothelial progenitor cells (EPCs) have demonstrated that TMAO induces cellular inflammation and oxidative stress [140].
5.2. Short-Chain Fatty Acids and Their Role in Metabolism
Short-chain fatty acids, which contain 1-6 carbon atoms, are produced through the anaerobic fermentation of non-absorbed nutrients, including resistant starch, dietary fiber, and complex carbohydrates [142]. Acetate, propionate, and butyrate are important SCFAs that are abundant in the cecum and have concentrations of about 130 mmol/kg, which is comparable to what is found in the portal vein [143]. The composition of the gut microbiota, gut transit time, and diet all affect SCFA synthesis [144]. The breakdown of dietary fiber by the gut microbiota follows three main metabolic routes: hydrolysis, glycolysis, and the pentose-phosphate pathway [145]. The phyla Firmicutes and Bacteroidetes, making up roughly 70% of total SCFAs [145], with Bacteroidetes mainly producing acetate and propionate, and Firmicutes being the primary sources of butyrate [146].
Acetate, a two-carbon SCFA, is synthesized through the Wood-Ljungdahl and acetyl-CoA pathways by various bacteria, including Lactobacillus spp., Bifidobacterium spp., Akkermansia muciniphila, Bacteroides spp., Prevotella spp., Ruminococcus spp., and Streptococcus spp. [146]. Propionate is produced by species such as Phascolarctobacterium spp., Bacteroides spp., Dialister spp., Veillonella spp., Salmonella spp., Roseburia inulinivorans, Ruminococcus obeum, Megasphaera elsdenii, and Coprococcus catus through the succinate, acrylate, and propanediol pathways [146]. Butyrate is generated by Roseburia spp., Clostridium leptum, Eubacterium hallii, Coprococcus eutactus, Faecalibacterium prausnitzii, Eubacterium rectale, and Anaerostipes caccae via the butyryl-CoA: acetate CoA-transferase and phosphotransbutyrylase/butyrate kinase pathways [147].
About 5–10% of the host's energy comes from SCFAs, which are absorbed through the colonic epithelium and enter the hepatic portal circulation [148]. Besides providing energy to the host, SCFAs function as signaling molecules that influence inflammatory responses, systemic blood pressure, and autonomic systems. They inhibit histone deacetylases, alter phagocytosis and chemotaxis, produce reactive oxygen species, promote cell division, and maintain the integrity of the intestinal barrier [148]. For instance, n-butyrate, a four-carbon SCFA, supports aerobic fatty acid oxidation in mitochondria, producing acetyl-CoA for the Krebs cycle [149]. SCFAs also contribute to the synthesis of cholesterol, long-chain fatty acids, and gluconeogenesis [145]. Butyrate influences the expression of claudin 1 (CLDN1), mucin genes, and other tight junction proteins vital for intestinal barrier strength [160], is essential for immune balance, reduces endotoxemia and inflammation risks, and lessens adipose tissue activation [150]. An overview of various effects of SCFAs on several organs is shown in Figure 4.
SCFAs are recognized by various free fatty acid receptors (FFARs), including FFAR2 (GPR43), FFAR3 (GPR41), hydroxycarboxylic acid receptor 2 (HCA2, also known as GPR109a), and other G protein-coupled receptors like Olfr-87 in mice and OR51E2 in humans [148,151,152], expressed in different tissues such as white adipose tissue and pancreatic β and α cells. These receptors mediate inflammatory responses and species-specific effects [153]. FFAR signalling is pivotal in modulating immune and inflammatory responses. SCFAs affect cellular functions through receptor activation, intracellular metabolism modulation, and histone deacetylase (HDAC) inhibition [154].
Peptide YY (PYY) and glucagon-like peptide-1 (GLP-1), anorectic hormones secreted by enteroendocrine cells, influence satiety, feeding behaviour, and glucose homeostasis, generally aiding in weight loss and reducing diabetes risk [155,156]. SCFAs prompt the secretion of these hormones [157], enhancing glucose tolerance, boosting intestinal gluconeogenesis, and curbing weight gain [158]. GPR41 activation has been associated with increased production of gut hormones, leptin, and PYY in humans and mice [159], while GPR43 activation in adipocytes reduces insulin-dependent fat accumulation, leading to leaner mice compared to their wild-type counterparts [160]. Furthermore, through its interactions with SCFAs, GPR41 regulates the sympathetic nervous system (SNS), influences a number of physiological processes, and may help explain how diet, probiotics, and prebiotics affect body's homeostasis [161].
SCFAs play a crucial role in reducing blood pressure by inhibiting renin release in the juxtaglomerular apparatus and facilitating relaxation of vascular smooth muscle cells via GPCRs, including olfactory receptor 78 [162]. In models of Dextran sulfate sodium-induced colitis, dietary fiber and SCFAs have been observed to activate GPR43 on colonic epithelial cells. This activation triggers the NLRP3 inflammasome, thereby improving colitis by mediating gut homeostasis through IL-18 [163]. Moreover, SCFAs play a critical role in promoting IL-22 secretion by CD4+ T cells and innate lymphoid cells (ILCs) via GPR41 activation and HDAC inhibition. This process enhances the aryl hydrocarbon receptor (AhR) levels and hypoxia-inducible factor 1α (HIF-1α). This process helps suppress intestinal inflammation and supports intestinal homeostasis [164]. Neutrophils that do not express GPR43 show an increased movement towards standard chemoattractants and lipopolysaccharides, indicating that a deficiency in SCFA levels could aggravate systemic inflammation by diminishing GPR43-mediated signalling in immune cells [165].
It is well known that SCFAs influence gene expression by directly inhibiting HDACs, which causes histone hyperacetylation [166]. This modification stimulates gene expression by increasing chromatin accessibility [167]. Butyrate and propionate specifically affect both local and systemic immune responses by preserving immunological homeostasis and preventing allergies and autoimmunity through their effects on the intestinal mucosa and lumen [168,169]. SCFAs promote a tolerogenic immune response by inhibiting the expression of co-stimulatory molecules on antigen-presenting cells by blocking HDAC activity and signalling via GPR109A [170]. Additionally, SCFAs support hematopoiesis by promoting the development of dendritic cell precursors that have phagocytic properties but reduced ability to support Th2 cell effector functions, resulting in lower allergic reactions [171]. Butyrate and propionate also promote the acetylation of histones at the Foxp3 locus [172], which supports the thymic production and peripheral conversion of T cells into FoxP3+ regulatory T cells [173].
SCFAs modulate T cells, DCs, ILCs, and macrophages. They enhance the production of anti-inflammatory cytokines such as TGF-β and IL-10, while reducing pro-inflammatory cytokines like IL-6, IL-12, IL-17a, IFN-γ, and TNF-α [174]. SCFAs also play a role in decreasing eosinophil recruitment and allergic cellular infiltration in the airways, thus mitigating inflammation and the IgE antibody response [171].
5.3. Secondary Bile Acids: Metabolism, Signaling, and Immune Modulation
Bile acids (BAs) play a critical role in human health and metabolism. They are initially produced from cholesterol as primary bile acids in the liver, and they circulate between the liver and the colon as part of the enterohepatic system circulation. In the colon, gut microbiota convert primary BAs into secondary BAs via various enzymatic activities, including deconjugation, oxidation, epimerization, dehydroxylation, esterification, and desulfation [175]. The process of deconjugation involves the splitting of glycine and taurine from bile acids by the action of bile salt hydrolase (BSH), an enzyme found in a wide array of bacterial species, including both Gram-positive (e.g., Bifidobacterium, Lactobacillus, Clostridium, Enterococcus) and Gram-negative (Bacteroides) bacteria[176]. Even certain archaea like Methanobrevibacter smithii and Methanosphera stadtmanae possess BSH enzymes that can break down both taurine- and glycine-conjugated bile acids [177].
Bacterial hydroxysteroid dehydrogenase (HSDH) enzymes play a key role in converting primary bile acids, such as cholic acid (CA) and chenodeoxycholic acid (CDCA), into secondary bile acids, including lithocholic acid (LCA) and deoxycholic acid (DCA). This process mainly involves dehydroxylation at the 7α position. This specific conversion is carried out by a small fraction of anaerobic bacteria belonging to genera like Bacteroides, Clostridium, Eubacterium, Lactobacillus, and Escherichia, which constitute a minor portion of the gut and colonic microbiota [178]. These anaerobic species represent less than 0.025% of the total gut microbiome and 0.0001% of the colonic microbiota [179]. The majority of these bacteria belong to the Clostridium genus, such as C. hiranonis, C. hylemonae, C. sordelli, and C. scindens [180]. Furthermore, a fraction of obligate anaerobes belonging to phyla Actinobacteria, Proteobacteria, Firmicutes, and Bacteroidetes can epimerize bile acids to produce ursodeoxycholic acid (UDCA) in the large intestine [181].
Secondary BAs, especially DCA and LCA, can re-enter the enterohepatic circulation and influence host physiology and disease by activating the nuclear FXR and the G protein-coupled receptor 5 (TGR5) on cell membranes [182]. Besides assisting in nutrient digestion and absorption, bile acids also regulate inflammation, fibrosis, and intestinal barrier function. For instance, FXR activation can maintain intestinal structure and permeability by preventing intestinal ischemia-reperfusion damage [183]. The discovery of BAs' receptors in endothelial cells, fibroblasts, and cardiomyocytes suggested a broader role for bile acids in cardiovascular health, and in treating heart failure caused by gut dysbiosis [184].
The EGFR/Src/ERK signaling pathway can be suppressed by DCA's interaction with FXR at normal levels, lowering the intestinal epithelial cell (IEC) proliferation [185]. Bile acids have been shown to activate the AP-1 and c-Myc signaling pathways, which may also assist in the development and metastasis of colon cancer cells [186]. Furthermore, bile acids can influence enteroendocrine L-cells by activating FXR and TGR5; TGR5 activation increases intracellular cAMP and calcium levels, promoting GLP-1 secretion [187].
TGR5 and FXR, receptors expressed in various immune cells, are notably activated by microbial bile acid derivatives, such as DCA and LCA. Bile acids play an crucial role in regulating the cytokine levels, as evidenced by transcriptomic studies, which show they can lower pro-inflammatory cytokines and chemokines, improve anti-inflammatory responses, promote wound healing, and transform pro-inflammatory macrophages into anti-inflammatory cells [188]. Specifically, LCA is a strong inhibitor of the NLRP3 inflammasome in bone marrow-derived macrophages (BMDMs), blocking caspase-1 maturation and the secretion of IL-1β and IL-18 via the TGR5–cAMP–PKA pathway. Phosphorylation and ubiquitination of the NLRP3 inflammasome prevent the caspase-1-dependent processing and release of pro-inflammatory cytokines [189]. Moreover, secondary BAs may affect adaptive immunity. For instance, LCA inhibits Th1 cell activation and decreases ERK1/2 phosphorylation by interacting with vitamin D receptor. In both human and mouse T cells, treatment with secondary BAs decreased STAT1α/β phosphorylation and Th1-specific cytokine production [190].
5.4. Metabolic Endotoxins and Their Implications in Cardiometabolic Diseases
A well-balanced gut microbiota is vital in maintaining immune and metabolic equilibrium, offering protection against harmful pathogens. Metabolic endotoxins or lipopolysaccharides (LPS) derived from the outer membranes of Gram-negative bacteria are prevalent in the gut [191]. The gut microbiota hosts approximately one trillion bacterial cells, which contribute nearly a gram of LPS within the intestinal lumen. In a dysfunctional gut barrier, gut integrity is compromised and can facilitate the leakage of LPS and other endotoxins into the bloodstream, thereby leading to a range of metabolic issues [192]. An imbalance in the gut microbiota, known as dysbiosis, is associated with an increased risk of cardiometabolic disorders, including hypertension, high cholesterol, and insulin resistance, all of which are significant risk factors for CVD [193]. The difference between a normal gut and a dysbiotic gut is described in Figure 5.
Characterized by elevated LPS levels in the circulation, which can be two to three times the normal [150], metabolic endotoxemia triggers oxidative stress, harms mitochondrial DNA and proteins in the heart, increases endothelial cell adhesion molecule expression, and encourages atherosclerosis-promoting activities [194].
Once endotoxins enter the systemic circulation, they bind to carrier molecules such as lipopolysaccharide-binding protein (LBP), bactericidal/permeability-increasing protein (BPI), soluble CD14, or serum lipoproteins, such as HDL and VLDL. LBP aids in extracting LPS from bacterial membranes and prevents the free endotoxins from aggregating in the bloodstream [195]. Complexes of LPS-LBP then bind to CD14 receptors on the surface of innate immune cells such as macrophages, neutrophils, and dendritic cells [196]. This complex transfers the endotoxin to the TLR4/MD2 complex, with the lipid A segment of LPS specifically binding to Toll-like receptor 4 (TLR4) [197]. This interaction activates adapter proteins and kinases, including MyD88, IRAK, TRAF6, NIK, and IκB, activating NF-κB [198]. NF-κB translocates to the nucleus to bind to various gene promoters, initiating the transcription of genes involved in inflammation, including clotting factors, complement proteins, acute phase reactants, cytokines, chemokines, and nitric oxide synthase[199].
Adipocytes expressing TLR4/MD2 complexes rely on soluble CD14 for LPS binding because they lack CD14 receptors on their membranes [150]. Interestingly, even cells lacking TLR4 can respond to LPS [200]. In patients with chronic heart failure (CHF), NF-κB activity is heightened in leukocytes, with increased expression of TLR2 and TLR4 on circulating monocytes compared to healthy individuals [201,202,203]. Higher levels of TNF-α, regulated by TLR4, correlate with more severe functional impairments in the Studies of Left Ventricular Dysfunction (SOLVD) trials [204] underscoring the roles of LPS, NF-κB, and TLR4 in chronic heart failure and positioning endotoxins as potential initiators of inflammation.
It is interesting to note that exposure to LPS can elevate blood insulin and glucose levels and induce weight gain in mice fed a high-fat diet [150]. Research indicates that LPS can cause oxidation of low-density lipoproteins [205]. This condition is implicated by an increase in permeability of the gut and, consequently, elevated circulating LPS levels, which are frequently observed in cardiovascular diseases [206]. Additionally, metabolic endotoxemia is associated with hypertriglyceridemia and the progression of fatty liver disease [207]. Moreover, studies have shown that LPS can increase the activity of endothelial lipase, which lowers the HDL levels [208], and it has been implicated in the development of plaque, progression of atherosclerotic lesions, and the secretion of pro-inflammatory molecules by endothelial cells [209].
6. Therapeutic Strategies Targeting Gut Microbiota-Derived Metabolites for Atherosclerosis
Gut microbiota-derived metabolites, including TMAO, SCFAs, secondary bile acids, and microbial LPS, have been recognized as significant modulators of atherogenesis. These metabolites affect lipid metabolism, vascular inflammation, and immune cell function. As a result, focusing on these metabolites and the related microbial pathways offers a viable supplement to traditional risk-factor management techniques. Broad dietary and lifestyle changes, precise microbiome modulation, and the use of small-molecule inhibitors that target microbial enzymes are examples of therapeutic interventions. The main therapeutic strategies are outlined in this review, along with the suggested mechanisms to reduce atherosclerotic risk and the available evidence supporting their development and clinical assessment.
6.1. Diet and Lifestyle:
Making dietary adjustments is a practical and economical way to affect the gut microbiota's makeup and the metabolites it produces. Diets high in fiber, low in L-carnitine and choline, and high in some polyphenols may improve cardiometabolic health and reduce toxic metabolites. Mechanistically, diet influences the substrates available to microbes, shifts the gut microbial community towards beneficial taxa, and improves the integrity of the intestinal barrier. A diet rich in fresh fruits and vegetables, dietary fibers with fewer animal products, and processed foods can lower the risk of CVD [210]. Several studies have highlighted the Mediterranean diet (MD) as a potential approach for preventing and reducing the risk of cardiovascular and metabolic diseases [211,212,213]. The CORDIOPREV trial showed that MD decreased the progression of atherosclerosis in patients with coronary heart disease [214]. Although this diet is rich in nutrients that can generate TMAO, evidence suggests that MD does not affect plasma TMAO concentrations in healthy people [215] or may even reduce them [216,217,218]. For instance, dietary apigenin has been shown to counteract several pro-atherosclerotic mechanisms associated with TMAO, such as inflammasome activation, low-density lipoprotein uptake, and leukocyte adhesion [219]. The PREDIMED trial revealed that the Mediterranean diet supplemented with extra-virgin olive oil or nuts was associated with a lower incidence of cardiovascular events than the reduced-fat diet. The trial also demonstrated that the Mediterranean diet can reshape the gut microbiota and alter microbial metabolite production, reducing cardiovascular disease risk [220]. Similarly, a study in Australia showed notable differences in the relative abundance of bacterial taxa between individuals following a Mediterranean diet and those following a low-fat diet. An increase in Butyrococcus was noted, showing an inverse correlation with changes in systolic blood pressure and significantly contributing to cardiovascular disease outcomes [221].
A cohort study of 307 healthy men using shotgun metagenome and meta-transcriptome analysis of gut microbiota by Ma et al. revealed that higher dietary intake shifts the Clostridiales compositions and their ability to utilize the carbohydrate. They also found a significant reduction in C-reactive protein, accompanied by a low or absent Prevotella copri population in the gut [222]. Murga-Garrido et al. demonstrated that various dietary fibers, including cellulose, inulin, and pectin, modulate the gut microbiota and, in turn, reduce body fat and liver triglyceride levels [223]. In a preclinical study on ApoE-/- knock-out mice, Catry et al. reported that a 15-day supplementation with inulin-type fructans (ITFs) improved endothelial function in the arteries of n-3 PUFA-deficient conditions [224]. The results demonstrate the potential of ITFs to promote cardiovascular health, especially in n-3 PUFA-deficient conditions. Likewise, a thorough examination of a randomized controlled clinical trial showed that consumption of isolated triterpene fraction lowers LDL cholesterol levels [225]. Apart from ITFs, beta-glucan supplements have also been shown to lower total LDL cholesterol levels while enhancing endothelial vascular reactivity in healthy individuals [226].
A pre-clinical study on a mouse model has shown that regular exercise increases the concentration of SCFAs [227]. A combination of aerobic and resistance training significantly lowers the prevalence of chronic metabolic disorders. Exercise also enhances gut microbial diversity and nutrient utilization, improves gut barrier function, and modulates neural and hormonal pathways [228]. It has been demonstrated that high-endurance training lowers plasma LPS levels. This implies that exercise might help lower inflammation linked to cardiovascular disease and other metabolic disorders [229]. It is important to note that although exercise benefits the gut microbiota, its effects are short-lived. This emphasizes the necessity for consistent and regular exercise to sustain a constructive impact on the gut microbiota and the associated health benefits [230].
6.2. Natural Product-Based Therapies Targeting Gut Microbiota
Natural products have gained considerable attention in research and are increasingly acknowledged as beneficial complementary therapies in clinical environments. Their diverse chemical compositions and favorable bioavailability contribute to their extensive therapeutic possibilities. Research indicates that natural products can target multiple pathways, making them promising options for preventing and treating atherosclerosis [231]. The natural products therapy of atherosclerosis showed that resveratrol, barberine, curcumin, quercetin, naringin, ginsenoside RC, paeonal, millet shell, polyphenols, flexseed oil, capsaicin, gastrodia extract, gastrodin, and gypenoside XLIX can delay the progression of atherosclerosis by targeting the gut microbiota and immune cell crosstalk; detailed mechanisms are available elsewhere [232]. Preclinical data reveal that consumption of Ligustrum robustum reduces serum TMA and TMAO levels by moderating gut microbiota, increasing fecal cholesterol excretion, decreasing serum and liver cholesterol levels, and ultimately attenuating diet-induced atherosclerosis [233]. Numerous ongoing and planned clinical trials seek to clarify these compounds' effectiveness and mechanisms. Due to their widespread availability, low toxicity, and extensive immunomodulatory effects, natural products are emerging as promising candidates for treating atherosclerosis. Future pharmaceutical development could benefit from investigating how active natural compounds influence gut microbiota metabolic pathways and assessing whether the resulting metabolites have complementary or opposing effects on atherosclerosis progression. Discovering beneficial metabolites derived from gut microbiota originating from natural products could lead to new strategies for creating innovative therapies to combat atherosclerosis.
6.3. Probiotics
Probiotics are live microorganisms that, when consumed in adequate amounts, provide several health benefits. They are highly regarded in nutraceutical science for their potential health benefit [234]. They have gained considerable attention as potential therapeutics for restoring gut microbial composition and improving cardiovascular health. The abundance of Lactobacilli in the gastrointestinal tract can significantly impact the overall well-being of the organism [235]. Probiotics colonized in the gut delay the progression of atherosclerosis by modulating the host’s micro-ecology [236]. Qiu et al. showed that L. plantarum ZDY04 may be a promising strategy for lowering plasma TMAO concentrations and mitigating TMAO-induced atherosclerosis in mice [237]. A randomized, double-blind clinical trial involving 44 patients with coronary artery disease demonstrated that 12 weeks of supplementation with Lactobacillus rhamnosus combined with caloric restriction resulted in anti-inflammatory effects and significant weight reduction [238]. Administration of a probiotic containing Limnosilactobacillus fermentum on a high-fat diet to Wistar rats restored gut microbial balance associated with improvements in metabolic disturbances and blood pressure abnormalities [239]. Probiotic Bifidobacterium animalis subsp. Lactis F1-3-2 intervention can cause a decrease in TMA levels in the cecum of mice and an improvement of lipid metabolism by acting on farnesoid X receptors and cholesterol 7-alpha hydroxylase [240]. Likewise, Bifidobacterium breve Bb4 and Bifidobacterium longum strains BL1 and BL7 were found to decrease plasma TMAO and plasma and cecal TMA levels. However, no change in the expression of FMO, FMO3, or FXR proteins, nor in TMAO excretion, was observed [241]. Supplementation with Lactobacillus fermentum ZJUIDS06 was shown to elevate colonic SCFA levels and increase the relative abundance of Parabacteroides in the cecum of hyperlipidemic hamsters, a key indicator of enhanced SCFA production and improved serum cholesterol profiles [242]. Similarly, Lactobacillus reuteri CCFM8631 enhanced acetate, propionate, and butyrate concentrations while reducing fecal TMAO levels in atherosclerosis model mice. This probiotic treatment also modulates the gut microbial environment by elevating the abundance of Deferribacteres, Lachnospiraceae NK4A136 group, Lactobacillus, and Dubosiella, while decreasing Erysipelatoclostridium and Romboutsia populations [243]. Pham and his team developed a probiotic cocktail using the commercially available E. coli Nissle (1917) strain, EcN_TL. They engineered it to continuously produce SCFAs by incorporating the biosynthetic pathways for propionate and butyrate. They observed that the daily intake of EcN_TL markedly reduced myocardial injury and cardiac function. Further analysis elucidated that EcN_TL lessens the neutrophil infiltration into the infarct site and promotes polarization of wound healing macrophages. They also revealed that plasma SCFAs protected the cardiomyocyte from inflammation by suppressing the NF-κB activation pathway [244]. Akkermansia muciniphila, a SCFAs producer, is well known for its probiotic properties and has been associated with regulating glucose, insulin, and leptin [245]. Research into the 16S rRNA gene has identified a reduced presence of Bacteroides vulgatus and Bacteroides dorei in individuals suffering from CAD. Oral administration of these bacteria into mice prone to atherosclerosis diminished atherosclerotic lesions and mitigated endotoxemia. This is achieved by reducing the production of gut microbial LPS and inhibiting inflammatory immune responses [246]. Although LPS generally has harmful effects, the LPS derived from Pantoea agglomerans (LPSp) has demonstrated a capability to decrease LDL cholesterol levels in people with hyperlipidemia. In ApoE−/− mice on a high-fat diet, LPSp treatment reduced body weight, plasma LDL, and oxidized LDL concentrations, and decreased proinflammatory mediators [247]. Modifying the gut microbiota with probiotic supplements offers a new approach for treating conditions such as coronary artery disease and hyperlipidemia. Ongoing research highlights the potential of probiotics as supportive therapies for cardiovascular health, emphasizing the need for deeper investigation and clinical validation.
6.4. Pharmacological Modulation of the Gut Microbiome in Cardiovascular Disease
Pharmacological interventions can modulate the gut microbiota for cardiovascular health benefits. It is an emerging and promising approach to tackle the atherosclerotic burden. Several drugs and bioactive compounds have shown potential in modulating the composition and functionality of gut microbes, which in turn can influence cardiovascular health. These strategies involve using agents that directly or indirectly target the gut microbiota, including antibiotics, bioactive compounds, and other microbiome-targeted pharmaceuticals.
Metformin, an anti-diabetic drug, minimized the risk of myocardial infarction onset in type 2 diabetes patients in a study to evaluate the risk of all-cause mortality and MACE [258]. Metformin can upregulate glucose uptake in peripheral tissues and regulate the pathway by increasing both plasma and pancreatic levels of GLP-1, the central molecule responsible for SCFA-associated cardioprotective function. It can also reduce the BA absorption by inhibiting the apical sodium-dependent bile acid transporter (ASBT), thereby disrupting the primary BA-mediated FXR activation [259]. A cohort study reveals a significant positive association between metformin and higher abundance of Akkermensia muciniphila and SCFAs producers like Butyrivibrio, Bifidobacterium bifidum, Megasphaera [260]. This suggests that metformin not only regulates blood glucose but also promotes a healthier gut microbiome. Acarbose, an alpha-glycosidase inhibitor, is also an alternative to metformin that can upregulate BSH, increase the Lactobacillus and Bifidobacterium populations, and downregulate the Bacteroides, Alistipes, and Clostridium populations [261]. A preclinical study on the cutC/D inhibitor showed that a single dose of Iodomethylcholine (IMC) and Fluoromethylcholine (FMC) significantly reduced TMAO levels for up to 3 days, while minimizing the risk of thrombosis and other significant complications associated with CVD [262]. Meldonium, another TMAO-inhibitory compound, reduces TMAO level by inhibiting L-carnitine metabolism intermediate gamma-butyrobetaine, significantly lowering the K. pneumoniae TMAO production [248].
Although antibiotic therapy demonstrated beneficial effects in preclinical studies, several clinical trials reported no significant impact on the disease progression [249]. Conversely, a long-term longitudinal study involving approximately 36,000 adult women revealed that prolonged antibiotic use was linked to an increased risk of cardiovascular disease, likely due to persistent microbiome disturbances, including the loss of beneficial probiotic bacteria [250]. While antibiotics may exhibit short-term benefits in a controlled experimental setup, long-term clinical use could disturb the gut microbiota ecology and expedite cardiovascular risk. This underscores the importance of developing a targeted, narrow therapeutic approach that modulates the gut microbiota without compromising its beneficial functions.
Despite several preclinical and translational studies, the clinical effectiveness and safety of gut microbiota-targeted pharmacotherapies require further validation through large-scale human trials. Future research must integrate multi-omics approaches and precision medicine frameworks to elucidate the interactions among hosts, microbes, and drugs. This understanding will pave the way for personalized microbiome-based therapeutics for the management of cardiovascular diseases.
6.5. Fecal Microbiome Transplantation
Since the US Food and Drug Administration approved fecal microbiome transplantation (FMT) for managing Clostridioides difficile infection, it has been used to treat inflammatory bowel disease, irritable bowel syndrome, constipation, metabolic syndrome, and other diseases. Transfer of gut microbiota from lean donors to individuals with metabolic syndrome has been shown to enhance insulin sensitivity, accompanied by a rise in butyrate-producing bacterial populations, particularly Roseburia intestinalis and Eubacterium hallii [251]. The therapeutic effect of FMT can be associated with a broader variety of bacteria, viruses, fungi, and archaea that successfully adapt within the recipient’s gut, resulting in enhanced functional diversity of the microbiota. FMT can be used to reduce the level of circulating TMAO and the risk of atherosclerosis [252]. In the present scenario, there is a lack of substantial data to establish the effectiveness of fecal microbiota transplantation as a therapeutic for CVD. With the advances in new technology and research, a deeper understanding of the gut microbiota interaction could pave the way for FMT application in promoting cardiovascular health and other systemic conditions.
7. Conclusion
In conclusion, the host microbiota has a complex and significant role in human health. The development of next-generation sequencing (NGS) technologies has further highlighted this fact. These technologies have revealed a wide variety of microorganisms previously impossible to cultivate, underscoring the significant impact of the microbiota on illnesses such as diabetes, Parkinson's disease, Alzheimer's disease, and cardiovascular diseases. The gut microbiota has a multifaceted impact on cardiovascular disease and is crucial for maintaining cardiovascular health. Signalling molecules that control host gut hormones and aid in the liver's synthesis of cholesterol are produced by microbial metabolism in the gut. Some microbial strains produce complex exopolysaccharides and enzymes that may indirectly affect bile acid cleavage or reabsorption, thereby reducing cholesterol levels. However, some gut microbiota components can partake in harmful pathways, transforming beneficial dietary micronutrients into detrimental compounds. Nowadays, artificial intelligence and machine learning (AI-ML) are being used to analyse big data generated from the NGS. Researchers are integrating AI-ML in clinical settings to predict and develop models in response to dietary interventions or probiotic treatments. These prediction models might help clinicians make informed decisions, leading to an optimized cardiovascular health management regime.
Future research must focus on integrating AI-ML and multi-omics data to elucidate the precise mechanisms by which the gut microbiome contributes to health maintenance and CVD development. It should also continue exploring novel strategies, emphasizing the value of combining microbiome data with state-of-the-art technologies to develop tailored, efficient treatments for CVD and other illnesses.
Author Contributions
R.W. conceptualized and designed the study. M.G.S. searched the literature, prepared the first draft, figures, and table. R.W. oversaw the drafting process and critically reviewed the manuscript. All authors reviewed and edited the final draft of the manuscript and collectively approved the final manuscript.
Funding
This study received funding from the Indian Council of Medical Research (ICMR), New Delhi, India, under its Junior Research Fellowship program, awarded to M.G.S. (award grant no.: 3/1/3/JRF-2019/HRD-070(31035)).
Data Availability Statement
No datasets were generated or analysed during the current study.
Acknowledgments
The Council of Scientific and Industrial Research (CSIR), New Delhi, and the CSIR-NEIST, Jorhat, Assam, are gratefully acknowledged for providing the necessary facilities, thereby facilitating the smooth progression of the study. The author would also like to thank the Publication & Intellectual Property Rights Committee, CSIR-NEIST for reviewing and approving this study (manuscript approval No. CSIR-NEIST/PUB/2024/077 Dated: 18-10-2024).
Competing Interests
The authors declare no competing interests.
Abbreviations
| CVDs | cardiovascular diseases |
| WHO | World Health Organization |
| DALYs | Disability Adjusted Life Years |
| NCD | non-communicable disease |
| IHD | ischemic heart disease |
| SCD | sudden cardiac death |
| SMC | smooth muscle cell |
| TMAO | trimethylamine-n-oxide |
| SCFA | short-chain fatty acid |
| LPS | lipopolysaccharides |
| ASCVD | Atherosclerotic cardiovascular disease |
| LDL | Low-density lipoprotein |
| oxLDL | Oxidised Low-density lipoprotein |
| AGEs | advanced glycation-end product |
| AHA | American Heart Association |
| GI | gastrointestinal |
| MWAS | metagenome-wide association |
| T-RFLP | terminal restriction fragment length polymorphism |
| CAD | coronary artery disease |
| LCV | Lymphocystis Virus |
| TTV | Torque teno virus |
| VLDL | very low-density lipoprotein |
| HDL | high-density lipoprotein |
| Lp(a) | lipoprotein (a) |
| RCT | Reverse Cholesterol Transport |
| eNOS | endothelial nitric oxide synthase |
| TNF-α | tumor necrosis factor-α |
| IL-1β | interleukin-1β |
| IL-6 | interleukin-6 |
| PPARγ | peroxisome proliferator activator receptor γ |
| MCP-1 | monocyte chemoattractant protein-1 |
| COX-2 | cyclooxygenase-2 |
| NF-κB | nuclear factor κB |
| LOX-1 | lectin-like oxidised LDL receptor 1 |
| TGF-β1 | transforming growth factor-β1 |
| VSMCs | vascular smooth muscle cells |
| MPO | myeloperoxidase |
| ALEs | advanced lipoxidation end product |
| MDA | malondialdehyde |
| 4HNE | 4-hydroxynonenal |
| DDAH | dimethyl aminohydrolase |
| MMP | matrix metalloproteinase |
| MAPK | mitogen-activated protein kinase |
| JNK | c-jun N-terminal kinase |
| VCAM-1 | vascular cell adhesion molecule |
| ICAM | intracellular cell adhesion molecule |
| Th1 | T helper type 1 |
| INF-γ | interferon-γ |
| GM-CSF | granulocyte macrophage colony-stimulating factor |
| M-CSF | macrophage colony-stimulating factor |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| TLRs | Toll-like receptors |
| PAMPs | pathogen-associated molecular pattern |
| DAMPs | damage-associated molecular pattern |
| MyD88 | Myeloid Differentiation Primary Response Gene88 |
| Lp-PLA2 | Lipoprotein-associated phospholipase A2 |
| NLRP3 | NOD-like receptor 3 |
| NK | natural killer |
| NKT | natural killer T cells |
| APC | antigen-presenting cells |
| DC | dendritic cells |
| MHC | major histocompatibility complex |
| T-bet | T-box transcription factor |
| GATA3 | GATA binding protein 3 |
| RORγ | RARA-related orphan receptor γ |
| CTLs | cytotoxic T lymphocytes |
| BCR | B cell recptor |
| Tfh | T follicular helper |
| IgG | immunoglobulin G |
| IgM | immunoglobulin M |
| GBB | gamma butyrobetaine |
| TMADH | trimethylamine dehydrogenase |
| FMO | flavin monooxygenase |
| FXR | farnesoid X receptor |
| MACE | major adverse cardiovascular event |
| HAECs | human aortic endothelial cells |
| HCAECs | human coronary aortic endothelial cells |
| CYP7a1 | Cholesterol 7 alpha-hydrolase |
| ABCG5/8 | ATP-binding cassette G5/8 |
| NPC1L1 | Neimann-Pick C1-like 1 |
| ROS | reactive oxygen species |
| FHCs | fetal human colon cells |
| HIV | human immunodeficiency virus |
| EPCs | endothelial progenitor cells |
| CLDN1 | claudin1 |
| FFARs | free fatty acid receptors |
| HCA2 | hydroxycarboxylic acid receptor 2 |
| HDAC | histone deacetylase |
| PYY | peptide YY |
| GLP-1 | glucagon-like peptide 1 |
| SNS | sympathetic nervous system |
| ILCs | innate lymphoid cells |
| AhR | aryl hydrocarbon receptor |
| HIF-1α | hypoxia-inducible factor 1α |
| HSDH | hydroxysteroid dehydrogenase |
| CA | cholic acid |
| CDCA | chenodeoxycholic acid |
| LCA | lithocholic acid |
| DCA | deoxycholic acid |
| UDCA | ursodeoxycholic acid |
| IEC | intestinal epithelial cells |
| cAMP | cyclic adenosine monophosphate |
| BMDMs | bone marrow-derived macrophages |
| LBP | lipopolysaccharide-binding protein |
| BPI | bactericidal/permeability-increasing protein |
| CHF | chronic heart failure |
| SOLVD | Studies of Left Ventricular Dysfunction |
| CORDIOPREV | Coronary Diet Intervention with Olive Oil and Cardiovascular Prevention |
| PREDIMED | Prevención con dieta Mediterránea |
| ITF | inulin-type fructans |
| ASBT | apical sodium-dependent bile acid transporter |
| IMC | Iodomethylcholine |
| FMC | Fluoromethylcholine |
| FMT | fecal microbiome transplantation |
References
- Xavier, D.; Pais, P.; Devereaux, P.J.; Xie, C.; Prabhakaran, D.; Reddy, K.S.; Gupta, R.; Joshi, P.; Kerkar, P.; Thanikachalam, S.; et al. Treatment and Outcomes of Acute Coronary Syndromes in India (CREATE): A Prospective Analysis of Registry Data. Lancet 2008, 371, 1435–1442. [Google Scholar] [CrossRef]
- Sivadasanpillai, H.; Leeder, S.; M, H.; Jeemon, P.; Dorairaj, P. A Race against Time: The Challenge of Cardiovascular Diseases in Developing Economies; 2015; ISBN 978-81-930819-0-7. [Google Scholar]
- Z, R. ACE Insertion/Deletion (I/D) Polymorphism and Diabetic Nephropathy. Journal of nephropathology 2012, 1. [Google Scholar] [CrossRef]
- Azar, P.; Bochaton-Piallat, M.-L. Deciphering Metabolic Reprogramming in Atherosclerosis through a Multi-Omics Approach. Cardiovasc Res 2025, 121, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr Atheroscler Rep 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.; Petroianu, G.; Adem, A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules 2022, 12, 542. [Google Scholar] [CrossRef]
- Dar, T.; Radfar, A.; Abohashem, S.; Pitman, R.K.; Tawakol, A.; Osborne, M.T. Psychosocial Stress and Cardiovascular Disease. Curr Treat Options Cardiovasc Med 2019, 21, 23. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Kitai, T.; Hazen, S.L. Gut Microbiota in Cardiovascular Health and Disease. Circulation research 2017, 120, 1183. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; Shanahan, F. The Gut Flora as a Forgotten Organ. EMBO Rep 2006, 7, 688–693. [Google Scholar] [CrossRef]
- F, T.; C, M.; S, D.; Ga, L.; S, B.; A, M.; D.P., F; van S., D; M, V. The Infant Gut Microbiome as a Microbial Organ Influencing Host Well-Being. Italian journal of pediatrics 2020, 46. [Google Scholar] [CrossRef]
- Yang, L.; Yang, J.; Zhang, T.; Xie, X.; Wu, Q. Gut Microbiota: A Novel Strategy Affecting Atherosclerosis. Microbiology Spectrum 2025, 13, e00482-24. [Google Scholar] [CrossRef]
- Jie, Z.; Xia, H.; Zhong, S.-L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The Gut Microbiome in Atherosclerotic Cardiovascular Disease. Nat Commun 2017, 8, 845. [Google Scholar] [CrossRef]
- Emoto, T.; Yamashita, T.; Sasaki, N.; Hirota, Y.; Hayashi, T.; So, A.; Kasahara, K.; Yodoi, K.; Matsumoto, T.; Mizoguchi, T.; et al. Analysis of Gut Microbiota in Coronary Artery Disease Patients: A Possible Link between Gut Microbiota and Coronary Artery Disease. J Atheroscler Thromb 2016, 23, 908–921. [Google Scholar] [CrossRef]
- Kasahara, K.; Krautkramer, K.A.; Org, E.; Romano, K.A.; Kerby, R.L.; Vivas, E.I.; Mehrabian, M.; Denu, J.M.; Bäckhed, F.; Lusis, A.J.; et al. Interactions between Roseburia Intestinalis and Diet Modulate Atherogenesis in a Murine Model. Nat Microbiol 2018, 3, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- O, K.; A, S.; J, F.; F, F.; J, S.; V, T.; Cj, B.; R, K.; B, F.; Re, L.; et al. Human Oral, Gut, and Plaque Microbiota in Patients with Atherosclerosis. Proceedings of the National Academy of Sciences of the United States of America 2011, 108 Suppl 1. [Google Scholar] [CrossRef]
- Kim, T.J.; Lee, H.; Kang, M.; Kim, J.E.; Choi, Y.-H.; Min, Y.W.; Min, B.-H.; Lee, J.H.; Son, H.J.; Rhee, P.-L.; et al. Helicobacter Pylori Is Associated with Dyslipidemia but Not with Other Risk Factors of Cardiovascular Disease. Sci Rep 2016, 6, 38015. [Google Scholar] [CrossRef] [PubMed]
- J, L.; S, L.; Pm, V.; Cw, W.; A, X. Akkermansia Muciniphila Protects Against Atherosclerosis by Preventing Metabolic Endotoxemia-Induced Inflammation in Apoe-/- Mice. Circulation 2016, 133. [Google Scholar] [CrossRef]
- Ott, S.J.; El Mokhtari, N.E.; Musfeldt, M.; Hellmig, S.; Freitag, S.; Rehman, A.; Kühbacher, T.; Nikolaus, S.; Namsolleck, P.; Blaut, M.; et al. Detection of Diverse Bacterial Signatures in Atherosclerotic Lesions of Patients with Coronary Heart Disease. Circulation 2006, 113, 929–937. [Google Scholar] [CrossRef]
- Dinakaran, V.; Rathinavel, A.; Pushpanathan, M.; Sivakumar, R.; Gunasekaran, P.; Rajendhran, J. Elevated Levels of Circulating DNA in Cardiovascular Disease Patients: Metagenomic Profiling of Microbiome in the Circulation. PLoS One 2014, 9, e105221. [Google Scholar] [CrossRef]
- Pothineni, N.V.K.; Subramany, S.; Kuriakose, K.; Shirazi, L.F.; Romeo, F.; Shah, P.K.; Mehta, J.L. Infections, Atherosclerosis, and Coronary Heart Disease. Eur Heart J 2017, 38, 3195–3201. [Google Scholar] [CrossRef]
- Haraszthy, V.I.; Zambon, J.J.; Trevisan, M.; Zeid, M.; Genco, R.J. Identification of Periodontal Pathogens in Atheromatous Plaques. J Periodontol 2000, 71, 1554–1560. [Google Scholar] [CrossRef]
- Kozarov, E.V.; Dorn, B.R.; Shelburne, C.E.; Dunn, W.A.; Progulske-Fox, A. Human Atherosclerotic Plaque Contains Viable Invasive Actinobacillus Actinomycetemcomitans and Porphyromonas Gingivalis. Arterioscler Thromb Vasc Biol 2005, 25, e17-18. [Google Scholar] [CrossRef]
- Padilla, C.; Lobos, O.; Hubert, E.; González, C.; Matus, S.; Pereira, M.; Hasbun, S.; Descouvieres, C. Periodontal Pathogens in Atheromatous Plaques Isolated from Patients with Chronic Periodontitis. J Periodontal Res 2006, 41, 350–353. [Google Scholar] [CrossRef]
- F, F.; V, T.; G, B.; F, B. Oral Microbiota in Patients with Atherosclerosis. Atherosclerosis 2015, 243. [Google Scholar] [CrossRef]
- Shibata, T.; Shimizu, K.; Hirano, K.; Nakashima, F.; Kikuchi, R.; Matsushita, T.; Uchida, K. Adductome-Based Identification of Biomarkers for Lipid Peroxidation. J Biol Chem 2017, 292, 8223–8235. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Raghavamenon, A.; Garelnabi, M.O.; Santanam, N. Oxidized Low-Density Lipoprotein. Methods Mol Biol 2010, 610, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Lj, van T.; R, S.; Pl, van L.; Mg, N.; La, J.; Af, S. Oxidized LDL Enhances Pro-Inflammatory Responses of Alternatively Activated M2 Macrophages: A Crucial Role for Krüppel-like Factor 2. Atherosclerosis 2011, 214. [Google Scholar] [CrossRef]
- Kim, C.E.; Lee, S.J.; Seo, K.W.; Park, H.M.; Yun, J.W.; Bae, J.U.; Bae, S.S.; Kim, C.D. Acrolein Increases 5-Lipoxygenase Expression in Murine Macrophages through Activation of ERK Pathway. Toxicol Appl Pharmacol 2010, 245, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Moghe, A.; Ghare, S.; Lamoreau, B.; Mohammad, M.; Barve, S.; McClain, C.; Joshi-Barve, S. Molecular Mechanisms of Acrolein Toxicity: Relevance to Human Disease. Toxicol Sci 2015, 143, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Sandoo, A.; van Zanten, J.J.C.S.V.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The Endothelium and Its Role in Regulating Vascular Tone. Open Cardiovasc Med J 2010, 4, 302–312. [Google Scholar] [CrossRef]
- Yoshimoto, R.; Fujita, Y.; Kakino, A.; Iwamoto, S.; Takaya, T.; Sawamura, T. The Discovery of LOX-1, Its Ligands and Clinical Significance. Cardiovasc Drugs Ther 2011, 25, 379–391. [Google Scholar] [CrossRef]
- Murphy, A.J.; Akhtari, M.; Tolani, S.; Pagler, T.; Bijl, N.; Kuo, C.-L.; Wang, M.; Sanson, M.; Abramowicz, S.; Welch, C.; et al. ApoE Regulates Hematopoietic Stem Cell Proliferation, Monocytosis, and Monocyte Accumulation in Atherosclerotic Lesions in Mice. J Clin Invest 2011, 121, 4138–4149. [Google Scholar] [CrossRef]
- Riwanto, M.; Rohrer, L.; von Eckardstein, A.; Landmesser, U. Dysfunctional HDL: From Structure-Function-Relationships to Biomarkers. Handbook of Experimental Pharmacology 2015, 224, 337–366. [Google Scholar] [CrossRef]
- Sattler, K.; Lehmann, I.; Gräler, M.; Bröcker-Preuss, M.; Erbel, R.; Heusch, G.; Levkau, B. HDL-Bound Sphingosine 1-Phosphate (S1P) Predicts the Severity of Coronary Artery Atherosclerosis. Cellular Physiology and Biochemistry 2014, 34, 172–184. [Google Scholar] [CrossRef]
- Niki, E.; Yoshida, Y.; Saito, Y.; Noguchi, N. Lipid Peroxidation: Mechanisms, Inhibition, and Biological Effects. Biochemical and Biophysical Research Communications 2005, 338, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Fritz, K.S.; Petersen, D.R. An Overview of the Chemistry and Biology of Reactive Aldehydes. Free Radic Biol Med 2013, 59, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Leonarduzzi, G.; Gamba, P.; Gargiulo, S.; Biasi, F.; Poli, G. Inflammation-Related Gene Expression by Lipid Oxidation-Derived Products in the Progression of Atherosclerosis. Free Radic Biol Med 2012, 52, 19–34. [Google Scholar] [CrossRef]
- Sp, F.; Lj, D.; Je, G.; N, P.; L, Z.; Kb, G.-C.; Aj, C. Mechanism of 4-HNE Mediated Inhibition of hDDAH-1: Implications in No Regulation. Biochemistry 2008, 47. [Google Scholar] [CrossRef] [PubMed]
- N, S.; Jl, F.; Y, L.; Pm, S.; S, R.; R, N. Proinflammatory Effects of Advanced Lipoxidation End Products in Monocytes. Diabetes 2008, 57. [Google Scholar] [CrossRef]
- Schrimpe-Rutledge, A.C.; Fong, K.Y.; Wright, D.W. Impact of 4-Hydroxynonenal on Matrix Metalloproteinase-9 Regulation in Lipopolysaccharide-Stimulated RAW 264.7 Cells. Cell Biochem Funct 2015, 33, 59–66. [Google Scholar] [CrossRef]
- Lee, S.J.; Seo, K.W.; Yun, M.R.; Bae, S.S.; Lee, W.S.; Hong, K.W.; Kim, C.D. 4-Hydroxynonenal Enhances MMP-2 Production in Vascular Smooth Muscle Cells via Mitochondrial ROS-Mediated Activation of the Akt/NF-kappaB Signaling Pathways. Free Radic Biol Med 2008, 45, 1487–1492. [Google Scholar] [CrossRef]
- S, X.; Ac, L.; Ai, G. Common Pathogenic Features of Atherosclerosis and Calcific Aortic Stenosis: Role of Transforming Growth Factor-Beta. Cardiovascular pathology: the official journal of the Society for Cardiovascular Pathology 2010, 19. [Google Scholar] [CrossRef]
- Vatsyayan, R.; Kothari, H.; Pendurthi, U.R.; Rao, L.V.M. 4-Hydroxy-2-Nonenal Enhances Tissue Factor Activity in Human Monocytic Cells via P38 Mitogen-Activated Protein Kinase Activation-Dependent Phosphatidylserine Exposure. Arterioscler Thromb Vasc Biol 2013, 33, 1601–1611. [Google Scholar] [CrossRef] [PubMed]
- Tj, L.; Jt, L.; Sk, M.; Ch, K.; Jw, P.; Tk, K. Age-Related Differential Growth Rate and Response to 4-Hydroxynonenal in Mouse Aortic Smooth Muscle Cells. International journal of molecular medicine 2006, 17. [Google Scholar]
- Zarkovic, K.; Larroque-Cardoso, P.; Pucelle, M.; Salvayre, R.; Waeg, G.; Nègre-Salvayre, A.; Zarkovic, N. Elastin Aging and Lipid Oxidation Products in Human Aorta. Redox Biol 2014, 4, 109–117. [Google Scholar] [CrossRef]
- Subbotin, V.M. Neovascularization of Coronary Tunica Intima (DIT) Is the Cause of Coronary Atherosclerosis. Lipoproteins Invade Coronary Intima via Neovascularization from Adventitial Vasa Vasorum, but Not from the Arterial Lumen: A Hypothesis. Theor Biol Med Model 2012, 9, 11. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, C.E.; Yun, M.R.; Seo, K.W.; Park, H.M.; Yun, J.W.; Shin, H.K.; Bae, S.S.; Kim, C.D. 4-Hydroxynonenal Enhances MMP-9 Production in Murine Macrophages via 5-Lipoxygenase-Mediated Activation of ERK and P38 MAPK. Toxicol Appl Pharmacol 2010, 242, 191–198. [Google Scholar] [CrossRef]
- Stocker, R.; Keaney, J.F., Jr. New Insights on Oxidative Stress in the Artery Wall. Journal of Thrombosis and Haemostasis 2005, 3, 1825–1834. [Google Scholar] [CrossRef]
- Björkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent Developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef]
- Nakashima, Y.; Raines, E.W.; Plump, A.S.; Breslow, J.L.; Ross, R. Upregulation of VCAM-1 and ICAM-1 at Atherosclerosis-Prone Sites on the Endothelium in the ApoE-Deficient Mouse. Arterioscler Thromb Vasc Biol 1998, 18, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and Challenges in Translating the Biology of Atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Am, L.; Gk, H. Innate Immune Signals in Atherosclerosis. Clinical immunology (Orlando, Fla.) 2010, 134. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, Atherosclerosis, and Coronary Artery Disease. N Engl J Med 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- de Beer, F.C.; Hind, C.R.; Fox, K.M.; Allan, R.M.; Maseri, A.; Pepys, M.B. Measurement of Serum C-Reactive Protein Concentration in Myocardial Ischaemia and Infarction. Br Heart J 1982, 47, 239–243. [Google Scholar] [CrossRef]
- Tennent, G.A.; Hutchinson, W.L.; Kahan, M.C.; Hirschfield, G.M.; Gallimore, J.R.; Lewin, J.; Sabin, C.A.; Dhillon, A.P.; Pepys, M.B. Transgenic Human CRP Is Not Pro-Atherogenic, pro-Atherothrombotic or pro-Inflammatory in apoE-/- Mice. Atherosclerosis 2008, 196, 248–255. [Google Scholar] [CrossRef]
- Hoefer, I.E.; Steffens, S.; Ala-Korpela, M.; Bäck, M.; Badimon, L.; Bochaton-Piallat, M.-L.; Boulanger, C.M.; Caligiuri, G.; Dimmeler, S.; Egido, J.; et al. Novel Methodologies for Biomarker Discovery in Atherosclerosis. Eur Heart J 2015, 36, 2635–2642. [Google Scholar] [CrossRef]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.-L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.S.; Smyth, D.; et al. Local Proliferation Dominates Lesional Macrophage Accumulation in Atherosclerosis. Nat Med 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J Immunol 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Baron, M.; Bouhlel, M.A.; Vanhoutte, J.; Copin, C.; Sebti, Y.; Derudas, B.; Mayi, T.; Bories, G.; Tailleux, A.; et al. Human Atherosclerotic Plaque Alternative Macrophages Display Low Cholesterol Handling but High Phagocytosis Because of Distinct Activities of the PPARγ and LXRα Pathways. Circ Res 2011, 108, 985–995. [Google Scholar] [CrossRef]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a Novel Macrophage Phenotype That Develops in Response to Atherogenic Phospholipids via Nrf2. Circ Res 2010, 107, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of Mouse Aortic Smooth Muscle Cells to a Macrophage-like State after Cholesterol Loading. Proc Natl Acad Sci U S A 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of Vascular Smooth Muscle Cells to Macrophage-like Cells during Atherogenesis. Circ Res 2014, 115, 662–667. [Google Scholar] [CrossRef]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.C.; Greene, E.S.; Straub, A.C.; et al. KLF4-Dependent Phenotypic Modulation of Smooth Muscle Cells Has a Key Role in Atherosclerotic Plaque Pathogenesis. Nat Med 2015, 21, 628–637. [Google Scholar] [CrossRef]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-like Cells in Human Atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef]
- Edfeldt, K.; Swedenborg, J.; Hansson, G.K.; Yan, Z. Expression of Toll-like Receptors in Human Atherosclerotic Lesions: A Possible Pathway for Plaque Activation. Circulation 2002, 105, 1158–1161. [Google Scholar] [CrossRef]
- West, X.Z.; Malinin, N.L.; Merkulova, A.A.; Tischenko, M.; Kerr, B.A.; Borden, E.C.; Podrez, E.A.; Salomon, R.G.; Byzova, T.V. Oxidative Stress Induces Angiogenesis by Activating TLR2 with Novel Endogenous Ligands. Nature 2010, 467, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Björkbacka, H.; Kunjathoor, V.V.; Moore, K.J.; Koehn, S.; Ordija, C.M.; Lee, M.A.; Means, T.; Halmen, K.; Luster, A.D.; Golenbock, D.T.; et al. Reduced Atherosclerosis in MyD88-Null Mice Links Elevated Serum Cholesterol Levels to Activation of Innate Immunity Signaling Pathways. Nat Med 2004, 10, 416–421. [Google Scholar] [CrossRef] [PubMed]
- H.-C., E; G, C.; H, P.; K, O.; P, K. Phospholipase A(2) in Vascular Disease. Circulation research 2001, 89. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci 2019, 20, 3328. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.H.; Genest, J.; Gotto, A.M.; Kastelein, J.J.P.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to Prevent Vascular Events in Men and Women with Elevated C-Reactive Protein. N Engl J Med 2008, 359, 2195–2207. [Google Scholar] [CrossRef]
- Maier, W.; Altwegg, L.A.; Corti, R.; Gay, S.; Hersberger, M.; Maly, F.E.; Sütsch, G.; Roffi, M.; Neidhart, M.; Eberli, F.R.; et al. Inflammatory Markers at the Site of Ruptured Plaque in Acute Myocardial Infarction: Locally Increased Interleukin-6 and Serum Amyloid A but Decreased C-Reactive Protein. Circulation 2005, 111, 1355–1361. [Google Scholar] [CrossRef]
- M, D.; Rt, M.; van Z., M; C, W.; O, S. Hyperlipidemia-Triggered Neutrophilia Promotes Early Atherosclerosis. Circulation 2010, 122. [Google Scholar] [CrossRef]
- Tupin, E.; Nicoletti, A.; Elhage, R.; Rudling, M.; Ljunggren, H.-G.; Hansson, G.K.; Berne, G.P. CD1d-Dependent Activation of NKT Cells Aggravates Atherosclerosis. J Exp Med 2004, 199, 417–422. [Google Scholar] [CrossRef]
- Whitman, S.C.; Rateri, D.L.; Szilvassy, S.J.; Yokoyama, W.; Daugherty, A. Depletion of Natural Killer Cell Function Decreases Atherosclerosis in Low-Density Lipoprotein Receptor Null Mice. Arteriosclerosis, Thrombosis, and Vascular Biology 2004, 24, 1049–1054. [Google Scholar] [CrossRef]
- Gh, van P.; Ej, van W.; Ad, H.; de V., P; Tj, van B.; J, K. Effect of Natural Killer T Cell Activation on the Initiation of Atherosclerosis. Thrombosis and haemostasis 2009, 102. [Google Scholar] [CrossRef]
- Hy, C.; R, W.; Cc, H. Gammadelta (Γδ) T Lymphocytes Do Not Impact the Development of Early Atherosclerosis. Atherosclerosis 2014, 234. [Google Scholar] [CrossRef]
- Bobryshev, Y.V.; Lord, R.S.A. Mapping of Vascular Dendritic Cells in Atherosclerotic Arteries Suggests Their Involvement in Local Immune-Inflammatory Reactions. Cardiovasc Res 1998, 37, 799–810. [Google Scholar] [CrossRef]
- Saigusa, R.; Winkels, H.; Ley, K. T Cell Subsets and Functions in Atherosclerosis. Nat Rev Cardiol 2020, 17, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. Decisions about Dendritic Cells: Past, Present, and Future. Annu Rev Immunol 2012, 30, 1–22. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ Res 2019, 124, 315–327. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Kim, H.I.; Park, J.; Guo, J.; Huang, W. The Role of Immune Cells in Different Stages of Atherosclerosis. Int J Med Sci 2024, 21, 1129–1143. [Google Scholar] [CrossRef]
- Li, J.; McArdle, S.; Gholami, A.; Kimura, T.; Wolf, D.; Gerhardt, T.; Miller, J.; Weber, C.; Ley, K. CCR5+T-bet+FoxP3+ Effector CD4 T Cells Drive Atherosclerosis. Circ Res 2016, 118, 1540–1552. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Harry, B.L.; Ludwig, A.; Liehn, E.A.; Sanders, J.M.; Bruce, A.; Weber, C.; Ley, K. CXCR6 Promotes Atherosclerosis by Supporting T-Cell Homing, Interferon-Gamma Production, and Macrophage Accumulation in the Aortic Wall. Circulation 2007, 116, 1801–1811. [Google Scholar] [CrossRef]
- Brinkman, C.C.; Peske, J.D.; Engelhard, V.H. Peripheral Tissue Homing Receptor Control of Naïve, Effector, and Memory CD8 T Cell Localization in Lymphoid and Non-Lymphoid Tissues. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A Novel Transcription Factor, T-Bet, Directs Th1 Lineage Commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J Immunol 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Edamitsu, S.; Matsukawa, A.; Ohkawara, S.; Takagi, K.; Nariuchi, H.; Yoshinaga, M. Role of TNF Alpha, IL-1, and IL-1ra in the Mediation of Leukocyte Infiltration and Increased Vascular Permeability in Rabbits with LPS-Induced Pleurisy. Clin Immunol Immunopathol 1995, 75, 68–74. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Bian, F.; Wu, P.; Xing, S.; Xu, G.; Li, W.; Chi, J.; Ouyang, C.; Zheng, T.; et al. TNF-α Promotes Early Atherosclerosis by Increasing Transcytosis of LDL across Endothelial Cells: Crosstalk between NF-κB and PPAR-γ. J Mol Cell Cardiol 2014, 72, 85–94. [Google Scholar] [CrossRef]
- Nakayama, T.; Hirahara, K.; Onodera, A.; Endo, Y.; Hosokawa, H.; Shinoda, K.; Tumes, D.J.; Okamoto, Y. Th2 Cells in Health and Disease. Annual Review of Immunology 2017, 35, 53–84. [Google Scholar] [CrossRef]
- Marks, B.R.; Nowyhed, H.N.; Choi, J.-Y.; Poholek, A.C.; Odegard, J.M.; Flavell, R.A.; Craft, J. Thymic Self-Reactivity Selects Natural Interleukin 17-Producing T Cells That Can Regulate Peripheral Inflammation. Nat Immunol 2009, 10, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Snijckers, R.P.M.; Foks, A.C. Adaptive Immunity and Atherosclerosis: Aging at Its Crossroads. Front. Immunol. 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Davenport, P.; Tipping, P.G. The Role of Interleukin-4 and Interleukin-12 in the Progression of Atherosclerosis in Apolipoprotein E-Deficient Mice. Am J Pathol 2003, 163, 1117–1125. [Google Scholar] [CrossRef]
- Fernández-Gallego, N.; Castillo-González, R.; Méndez-Barbero, N.; López-Sanz, C.; Obeso, D.; Villaseñor, A.; Escribese, M.M.; López-Melgar, B.; Salamanca, J.; Benedicto-Buendía, A.; et al. The Impact of Type 2 Immunity and Allergic Diseases in Atherosclerosis. Allergy 2022, 77, 3249–3266. [Google Scholar] [CrossRef]
- Smith, E.; Prasad, K.-M.R.; Butcher, M.; Dobrian, A.; Kolls, J.K.; Ley, K.; Galkina, E. Blockade of Interleukin-17A Results in Reduced Atherosclerosis in Apolipoprotein E-Deficient Mice. Circulation 2010, 121, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, C.E.; Mele, F.; Aschenbrenner, D.; Jarrossay, D.; Ronchi, F.; Gattorno, M.; Monticelli, S.; Lanzavecchia, A.; Sallusto, F. Pathogen-Induced Human TH17 Cells Produce IFN-γ or IL-10 and Are Regulated by IL-1β. Nature 2012, 484, 514–518. [Google Scholar] [CrossRef]
- Foks, A.C.; Lichtman, A.H.; Kuiper, J. Treating Atherosclerosis with Regulatory T Cells. Arterioscler Thromb Vasc Biol 2015, 35, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Freuchet, A.; Roy, P.; Armstrong, S.S.; Oliaeimotlagh, M.; Kumar, S.; Orecchioni, M.; Ali, A.J.; Khan, A.; Makings, J.; Lyu, Q.; et al. Identification of Human exTreg Cells as CD16+CD56+ Cytotoxic CD4+ T Cells. Nat Immunol 2023, 24, 1748–1761. [Google Scholar] [CrossRef]
- Gaddis, D.E.; Padgett, L.E.; Wu, R.; McSkimming, C.; Romines, V.; Taylor, A.M.; McNamara, C.A.; Kronenberg, M.; Crotty, S.; Thomas, M.J.; et al. Apolipoprotein AI Prevents Regulatory to Follicular Helper T Cell Switching during Atherosclerosis. Nat Commun 2018, 9, 1095. [Google Scholar] [CrossRef]
- Kyaw, T.; Winship, A.; Tay, C.; Kanellakis, P.; Hosseini, H.; Cao, A.; Li, P.; Tipping, P.; Bobik, A.; Toh, B.-H. Cytotoxic and Proinflammatory CD8+ T Lymphocytes Promote Development of Vulnerable Atherosclerotic Plaques in apoE-Deficient Mice. Circulation 2013, 127, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Tsiantoulas, D.; Binder, C.J.; Mallat, Z. The Role of B Cells in Atherosclerosis. Nat Rev Cardiol 2019, 16, 180–196. [Google Scholar] [CrossRef]
- Kyaw, T.; Tipping, P.; Bobik, A.; Toh, B.-H. Protective Role of Natural IgM-Producing B1a Cells in Atherosclerosis. Trends Cardiovasc Med 2012, 22, 48–53. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ Res 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- Zheng, X.; Xie, G.; Zhao, A.; Zhao, L.; Yao, C.; Chiu, N.H.L.; Zhou, Z.; Bao, Y.; Jia, W.; Nicholson, J.K.; et al. The Footprints of Gut Microbial-Mammalian Co-Metabolism. J Proteome Res 2011, 10, 5512–5522. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-H.; Chen, Y.; Smith, T.C.; Karna, S.L.R.; Seshu, J. Short-Chain Fatty Acids Alter Metabolic and Virulence Attributes of Borrelia Burgdorferi. Infect Immun 2018, 86, e00217-18. [Google Scholar] [CrossRef] [PubMed]
- Y, M.; Ja, G.; C.-T., L; Bc, O.; E, J.; Ej, S.; W, L.; Sl, P.; Ar, H.; Is, O. Association between Dietary Fiber and Serum C-Reactive Protein. The American journal of clinical nutrition 2006, 83. [Google Scholar] [CrossRef]
- Postler, T.S.; Ghosh, S. Understanding the Holobiont: How Microbial Metabolites Affect Human Health and Shape the Immune System. Cell Metab 2017, 26, 110–130. [Google Scholar] [CrossRef]
- Uchimura, Y.; Fuhrer, T.; Li, H.; Lawson, M.A.; Zimmermann, M.; Yilmaz, B.; Zindel, J.; Ronchi, F.; Sorribas, M.; Hapfelmeier, S.; et al. Antibodies Set Boundaries Limiting Microbial Metabolite Penetration and the Resultant Mammalian Host Response. Immunity 2018, 49, 545–559.e5. [Google Scholar] [CrossRef]
- Parker, A.; Fonseca, S.; Carding, S.R. Gut Microbes and Metabolites as Modulators of Blood-Brain Barrier Integrity and Brain Health. Gut Microbes 2020, 11, 135–157. [Google Scholar] [CrossRef]
- Romano, K.A.; Vivas, E.I.; Amador-Noguez, D.; Rey, F.E. Intestinal Microbiota Composition Modulates Choline Bioavailability from Diet and Accumulation of the Proatherogenic Metabolite Trimethylamine-N-Oxide. mBio 2015, 6, e02481. [Google Scholar] [CrossRef]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the Trimethylamine-Producing Bacteria of the Human Gut Microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef]
- Craciun, S.; Balskus, E.P. Microbial Conversion of Choline to Trimethylamine Requires a Glycyl Radical Enzyme. Proc Natl Acad Sci U S A 2012, 109, 21307–21312. [Google Scholar] [CrossRef]
- Koeth, R.A.; Levison, B.S.; Culley, M.K.; Buffa, J.A.; Wang, Z.; Gregory, J.C.; Org, E.; Wu, Y.; Li, L.; Smith, J.D.; et al. γ-Butyrobetaine Is a Proatherogenic Intermediate in Gut Microbial Metabolism of L-Carnitine to TMAO. Cell Metab 2014, 20, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Warrier, M. Trimethylamine N-Oxide, the Microbiome, and Heart and Kidney Disease. Annu Rev Nutr 2017, 37, 157–181. [Google Scholar] [CrossRef]
- G, F.; V.-S., S; J, R. Microbiology Meets Big Data: The Case of Gut Microbiota-Derived Trimethylamine. Annual review of microbiology 2015, 69. [Google Scholar] [CrossRef]
- Fennema, D.; Phillips, I.R.; Shephard, E.A. Trimethylamine and Trimethylamine N-Oxide, a Flavin-Containing Monooxygenase 3 (FMO3)-Mediated Host-Microbiome Metabolic Axis Implicated in Health and Disease. Drug Metab Dispos 2016, 44, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- C.-G., J; A, G.; V, S.; N, P.; B, B.; A, S. The Complex Metabolism of Trimethylamine in Humans: Endogenous and Exogenous Sources. Expert reviews in molecular medicine 2016, 18. [Google Scholar] [CrossRef]
- Janeiro, M.H.; Ramírez, M.J.; Milagro, F.I.; Martínez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10, 1398. [Google Scholar] [CrossRef]
- Trøseid, M.; Ueland, T.; Hov, J.R.; Svardal, A.; Gregersen, I.; Dahl, C.P.; Aakhus, S.; Gude, E.; Bjørndal, B.; Halvorsen, B.; et al. Microbiota-Dependent Metabolite Trimethylamine-N-Oxide Is Associated with Disease Severity and Survival of Patients with Chronic Heart Failure. J Intern Med 2015, 277, 717–726. [Google Scholar] [CrossRef]
- K, C.; X, Z.; M, F.; D, L.; H, Z. Gut Microbiota-Dependent Metabolite Trimethylamine N-Oxide Contributes to Cardiac Dysfunction in Western Diet-Induced Obese Mice. Frontiers in physiology 2017, 8. [Google Scholar] [CrossRef]
- Ma, G.; Pan, B.; Chen, Y.; Guo, C.; Zhao, M.; Zheng, L.; Chen, B. Trimethylamine N-Oxide in Atherogenesis: Impairing Endothelial Self-Repair Capacity and Enhancing Monocyte Adhesion. Biosci Rep 2017, 37, BSR20160244. [Google Scholar] [CrossRef]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB. J Am Heart Assoc 2016, 5, e002767. [Google Scholar] [CrossRef]
- X, C.; X, Q.; Y, L.; C, Y.; X, Y. Trimethylamine N-Oxide Promotes Tissue Factor Expression and Activity in Vascular Endothelial Cells: A New Link between Trimethylamine N-Oxide and Atherosclerotic Thrombosis. Thrombosis research 2019, 177. [Google Scholar] [CrossRef]
- Fatkhullina, A.R.; Peshkova, I.O.; Dzutsev, A.; Aghayev, T.; McCulloch, J.A.; Thovarai, V.; Badger, J.H.; Vats, R.; Sundd, P.; Tang, H.-Y.; et al. An Interleukin-23-Interleukin-22 Axis Regulates Intestinal Microbial Homeostasis to Protect from Diet-Induced Atherosclerosis. Immunity 2018, 49, 943–957.e9. [Google Scholar] [CrossRef]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate Lymphoid Cells Drive Interleukin-23-Dependent Innate Intestinal Pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef]
- Peshkova, I.O.; Schaefer, G.; Koltsova, E.K. Atherosclerosis and Aortic Aneurysm - Is Inflammation a Common Denominator? FEBS J 2016, 283, 1636–1652. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaide, M.A.I.; Singh, R.; Datta, P.; Rewers-Felkins, K.A.; Salguero, M.V.; Al-Obaidi, I.; Kottapalli, K.R.; Vasylyeva, T.L. Gut Microbiota-Dependent Trimethylamine-N-Oxide and Serum Biomarkers in Patients with T2DM and Advanced CKD. J Clin Med 2017, 6, 86. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Feskens, E.J.M.; Boer, J.M.A.; Müller, M. The Potential Influence of Genetic Variants in Genes along Bile Acid and Bile Metabolic Pathway on Blood Cholesterol Levels in the Population. Atherosclerosis 2010, 210, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Ra, K.; Z, W.; Bs, L.; Ja, B.; E, O.; Bt, S.; Eb, B.; X, F.; Y, W.; L, L.; et al. Intestinal Microbiota Metabolism of L-Carnitine, a Nutrient in Red Meat, Promotes Atherosclerosis. Nature medicine 2013, 19. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut Flora Metabolism of Phosphatidylcholine Promotes Cardiovascular Disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-Lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef]
- J, G.; C, Y.; B, W.; X, Z.; T, H.; Y, G.; J, L. Trimethylamine N-Oxide Promotes Atherosclerosis via CD36-Dependent MAPK/JNK Pathway. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2018, 97. [Google Scholar] [CrossRef]
- Liu, D.; Zeng, X.; Li, X.; Mehta, J.L.; Wang, X. Role of NLRP3 Inflammasome in the Pathogenesis of Cardiovascular Diseases. Basic Res Cardiol 2017, 113, 5. [Google Scholar] [CrossRef]
- X, S.; X, J.; Y, M.; Y, L.; L, Z.; Y, H.; Y, C. Trimethylamine N-Oxide Induces Inflammation and Endothelial Dysfunction in Human Umbilical Vein Endothelial Cells via Activating ROS-TXNIP-NLRP3 Inflammasome. Biochemical and biophysical research communications 2016, 481. [Google Scholar] [CrossRef]
- Yue, C.; Yang, X.; Li, J.; Chen, X.; Zhao, X.; Chen, Y.; Wen, Y. Trimethylamine N-Oxide Prime NLRP3 Inflammasome via Inhibiting ATG16L1-Induced Autophagy in Colonic Epithelial Cells. Biochem Biophys Res Commun 2017, 490, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Li, X.S.; Liman, T.G.; Bledau, N.; Schmidt, D.; Zimmermann, F.; Kränkel, N.; Widera, C.; Sonnenschein, K.; Haghikia, A.; et al. Gut Microbiota-Dependent Trimethylamine N-Oxide Predicts Risk of Cardiovascular Events in Patients With Stroke and Is Related to Proinflammatory Monocytes. Arterioscler Thromb Vasc Biol 2018, 38, 2225–2235. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Clish, C.B.; Hua, S.; Scott, J.M.; Hanna, D.B.; Burk, R.D.; Haberlen, S.A.; Shah, S.J.; Margolick, J.B.; Sears, C.L.; et al. Gut Microbial-Related Choline Metabolite Trimethylamine-N-Oxide Is Associated With Progression of Carotid Artery Atherosclerosis in HIV Infection. J Infect Dis 2018, 218, 1474–1479. [Google Scholar] [CrossRef]
- Haissman, J.M.; Haugaard, A.K.; Ostrowski, S.R.; Berge, R.K.; Hov, J.R.; Trøseid, M.; Nielsen, S.D. Microbiota-Dependent Metabolite and Cardiovascular Disease Marker Trimethylamine-N-Oxide (TMAO) Is Associated with Monocyte Activation but Not Platelet Function in Untreated HIV Infection. BMC Infect Dis 2017, 17, 445. [Google Scholar] [CrossRef]
- Rohrmann, S.; Linseisen, J.; Allenspach, M.; von Eckardstein, A.; Müller, D. Plasma Concentrations of Trimethylamine-N-Oxide Are Directly Associated with Dairy Food Consumption and Low-Grade Inflammation in a German Adult Population. J Nutr 2016, 146, 283–289. [Google Scholar] [CrossRef]
- Chou, R.-H.; Chen, C.-Y.; Chen, I.-C.; Huang, H.-L.; Lu, Y.-W.; Kuo, C.-S.; Chang, C.-C.; Huang, P.-H.; Chen, J.-W.; Lin, S.-J. Trimethylamine N-Oxide, Circulating Endothelial Progenitor Cells, and Endothelial Function in Patients with Stable Angina. Sci Rep 2019, 9, 4249. [Google Scholar] [CrossRef]
- Burke-Gaffney, A.; Brooks, A.V.S.; Bogle, R.G. Regulation of Chemokine Expression in Atherosclerosis. Vascul Pharmacol 2002, 38, 283–292. [Google Scholar] [CrossRef]
- B, D.; V.O., L; B, V.; K, V. The Role of Short-Chain Fatty Acids in Microbiota-Gut-Brain Communication. Nature reviews. Gastroenterology & hepatology 2019, 16. [Google Scholar] [CrossRef]
- Jh, C.; Ew, P.; Wj, B.; Cp, N.; Gt, M. Short Chain Fatty Acids in Human Large Intestine, Portal, Hepatic and Venous Blood. Gut 1987, 28. [Google Scholar] [CrossRef] [PubMed]
- Soldavini, J.; Kaunitz, J.D. Pathobiology and Potential Therapeutic Value of Intestinal Short-Chain Fatty Acids in Gut Inflammation and Obesity. Dig Dis Sci 2013, 58, 2756–2766. [Google Scholar] [CrossRef]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The Role of Short-Chain Fatty Acids in the Interplay between Diet, Gut Microbiota, and Host Energy Metabolism. J Lipid Res 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.; Redondo-Blanco, S.; Gutiérrez-del-Río, I.; Miguélez, E.M.; Villar, C.J.; Lombó, F. Colon Microbiota Fermentation of Dietary Prebiotics towards Short-Chain Fatty Acids and Their Roles as Anti-Inflammatory and Antitumour Agents: A Review. Journal of Functional Foods 2016, 25, 511–522. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are Short Chain Fatty Acids in Gut Microbiota Defensive Players for Inflammation and Atherosclerosis? J Atheroscler Thromb 2017, 24, 660–672. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The Microbiome and Butyrate Regulate Energy Metabolism and Autophagy in the Mammalian Colon. Cell Metab 2011, 13, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- D, B.; Ab, T.; G, M.; Ce, M. The Pharmacology and Function of Receptors for Short-Chain Fatty Acids. Molecular pharmacology 2016, 89. [Google Scholar] [CrossRef]
- Pluznick, J.L. Gut Microbiota in Renal Physiology: Focus on Short-Chain Fatty Acids and Their Receptors. Kidney Int 2016, 90, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Ang, Z.; Ding, J.L. GPR41 and GPR43 in Obesity and Inflammation - Protective or Causative? Front Immunol 2016, 7, 28. [Google Scholar] [CrossRef]
- L, A.; M, N.; O, B.; A, P.; L, A.; L, D.; T, M.; X, Z.; I, E.; L, M. Short Chain Fatty Acids (SCFA) Reprogram Gene Expression in Human Malignant Epithelial and Lymphoid Cells. PloS one 2016, 11. [Google Scholar] [CrossRef]
- G, T.; H, H.; Ys, L.; He, P.; Am, H.; E, D.; J, C.; J, G.; F, R.; Fm, G. Short-Chain Fatty Acids Stimulate Glucagon-like Peptide-1 Secretion via the G-Protein-Coupled Receptor FFAR2. Diabetes 2012, 61. [Google Scholar] [CrossRef]
- Caylak, E. Anorexigenic Peptides in Health and Disease. In eLS; John Wiley & Sons, Ltd, 2012; ISBN 978-0-470-01590-2. [Google Scholar]
- Puddu, A.; Sanguineti, R.; Montecucco, F.; Viviani, G.L. Evidence for the Gut Microbiota Short-Chain Fatty Acids as Key Pathophysiological Molecules Improving Diabetes. Mediators Inflamm 2014, 2014, 162021. [Google Scholar] [CrossRef] [PubMed]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-Generated Metabolites Promote Metabolic Benefits via Gut-Brain Neural Circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef]
- Bs, S.; A, S.; T, M.; Fe, R.; F, B.; Jk, M.; Re, H.; Sc, W.; J, C.; M, Y.; et al. Effects of the Gut Microbiota on Host Adiposity Are Modulated by the Short-Chain Fatty-Acid Binding G Protein-Coupled Receptor, Gpr41. Proceedings of the National Academy of Sciences of the United States of America 2008, 105. [Google Scholar] [CrossRef]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The Gut Microbiota Suppresses Insulin-Mediated Fat Accumulation via the Short-Chain Fatty Acid Receptor GPR43. Nat Commun 2013, 4, 1829. [Google Scholar] [CrossRef]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-Chain Fatty Acids and Ketones Directly Regulate Sympathetic Nervous System via G Protein-Coupled Receptor 41 (GPR41). Proceedings of the National Academy of Sciences 2011, 108, 8030–8035. [Google Scholar] [CrossRef]
- Jl, P.; Rj, P.; H, G.; Z, P.; A, S.; J, H.; I, B.; Lx, W.; F, R.; T, W.; et al. Olfactory Receptor Responding to Gut Microbiota-Derived Signals Plays a Role in Renin Secretion and Blood Pressure Regulation. Proceedings of the National Academy of Sciences of the United States of America 2013, 110. [Google Scholar] [CrossRef]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-Sensing Receptors GPR43 and GPR109A Facilitate Dietary Fibre-Induced Gut Homeostasis through Regulation of the Inflammasome. Nat Commun 2015, 6, 6734. [Google Scholar] [CrossRef]
- W, Y.; T, Y.; X, H.; Aj, B.; L, X.; Y, L.; J, S.; F, P.; J, Z.; W, Z.; et al. Intestinal Microbiota-Derived Short-Chain Fatty Acids Regulation of Immune Cell IL-22 Production and Gut Immunity. Nature communications 2020, 11. [Google Scholar] [CrossRef]
- Me, K.; R, S.; Aj, N.; Ac, O.; Lj, M.; L, B.; Cr, M.; Ch, W. G Protein-Coupled Receptor 43 Modulates Neutrophil Recruitment during Acute Inflammation. PloS one 2016, 11. [Google Scholar] [CrossRef]
- Singh, N.; Thangaraju, M.; Prasad, P.D.; Martin, P.M.; Lambert, N.A.; Boettger, T.; Offermanns, S.; Ganapathy, V. Blockade of Dendritic Cell Development by Bacterial Fermentation Products Butyrate and Propionate through a Transporter (Slc5a8)-Dependent Inhibition of Histone Deacetylases. J Biol Chem 2010, 285, 27601–27608. [Google Scholar] [CrossRef] [PubMed]
- Alenghat, T.; Artis, D. Epigenomic Regulation of Host-Microbiota Interactions. Trends Immunol 2014, 35, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The Microbiome and Innate Immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef]
- Blander, J.M.; Longman, R.S.; Iliev, I.D.; Sonnenberg, G.F.; Artis, D. Regulation of Inflammation by Microbiota Interactions with the Host. Nat Immunol 2017, 18, 851–860. [Google Scholar] [CrossRef]
- Kaisar, M.M.M.; Pelgrom, L.R.; van der Ham, A.J.; Yazdanbakhsh, M.; Everts, B. Butyrate Conditions Human Dendritic Cells to Prime Type 1 Regulatory T Cells via Both Histone Deacetylase Inhibition and G Protein-Coupled Receptor 109A Signaling. Front Immunol 2017, 8, 1429. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut Microbiota Metabolism of Dietary Fiber Influences Allergic Airway Disease and Hematopoiesis. Nat Med 2014, 20, 159–166. [Google Scholar] [CrossRef]
- N, A.; C, C.; X, F.; S, D.; van der V., J; deRoos, P; H, L.; Jr, C.; K, P.; Pj, C.; et al. Metabolites Produced by Commensal Bacteria Promote Peripheral Regulatory T-Cell Generation. Nature 2013, 504. [Google Scholar] [CrossRef]
- T, A.; G, G.; T, G.; T, M.; L, W.; Jl, R.; Ww, H. Histone/Protein Deacetylase Inhibitors Increase Suppressive Functions of Human FOXP3+ Tregs. Clinical immunology (Orlando, Fla.) 2010, 136. [Google Scholar] [CrossRef]
- Sanchez, H.N.; Moroney, J.B.; Gan, H.; Shen, T.; Im, J.L.; Li, T.; Taylor, J.R.; Zan, H.; Casali, P. B Cell-Intrinsic Epigenetic Modulation of Antibody Responses by Dietary Fiber-Derived Short-Chain Fatty Acids. Nat Commun 2020, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Busnelli, M.; Manzini, S.; Chiesa, G. The Gut Microbiota Affects Host Pathophysiology as an Endocrine Organ: A Focus on Cardiovascular Disease. Nutrients 2019, 12, 79. [Google Scholar] [CrossRef]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.M.; Marchesi, J.R. Functional and Comparative Metagenomic Analysis of Bile Salt Hydrolase Activity in the Human Gut Microbiome. Proc Natl Acad Sci U S A 2008, 105, 13580–13585. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Chiu, C.; Hung, S.; Huang, W.-C.; Lee, Y.-P.; Liu, J.-Y.; Huang, Y.-T.; Chen, T.; Chuang, H. Gnotobiotic Mice Inoculated with Firmicutes, but Not Bacteroidetes, Deteriorate Nonalcoholic Fatty Liver Disease Severity by Modulating Hepatic Lipid Metabolism. Nutrition research 2019, 69, 20–29. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Bajaj, J.S. The Human Gut Sterolbiome: Bile Acid-Microbiome Endocrine Aspects and Therapeutics. Acta Pharm Sin B 2015, 5, 99–105. [Google Scholar] [CrossRef]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision Microbiome Reconstitution Restores Bile Acid Mediated Resistance to Clostridium Difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef] [PubMed]
- N, M.; L, R.; B, S.; A, M.; S, D. Intestinal Bacteria Interplay With Bile and Cholesterol Metabolism: Implications on Host Physiology. Frontiers in physiology 2019, 10. [Google Scholar] [CrossRef]
- Branchereau, M.; Burcelin, R.; Heymes, C. The Gut Microbiome and Heart Failure: A Better Gut for a Better Heart. Rev Endocr Metab Disord 2019, 20, 407–414. [Google Scholar] [CrossRef]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of Bile Acids and Bile Acid Receptors in Metabolic Regulation. Physiol Rev 2009, 89, 147–191. [Google Scholar] [CrossRef]
- Hanafi, N.I.; Mohamed, A.S.; Sheikh Abdul Kadir, S.H.; Othman, M.H.D. Overview of Bile Acids Signaling and Perspective on the Signal of Ursodeoxycholic Acid, the Most Hydrophilic Bile Acid, in the Heart. Biomolecules 2018, 8, 159. [Google Scholar] [CrossRef]
- Dossa, A.Y.; Escobar, O.; Golden, J.; Frey, M.R.; Ford, H.R.; Gayer, C.P. Bile Acids Regulate Intestinal Cell Proliferation by Modulating EGFR and FXR Signaling. Am J Physiol Gastrointest Liver Physiol 2016, 310, G81–92. [Google Scholar] [CrossRef]
- H, Z.; S, U.; B, R.; D, L.; M, B. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. International journal of molecular sciences 2019, 20. [Google Scholar] [CrossRef]
- Parker, H.E.; Wallis, K.; le Roux, C.W.; Wong, K.Y.; Reimann, F.; Gribble, F.M. Molecular Mechanisms Underlying Bile Acid-Stimulated Glucagon-like Peptide-1 Secretion. Br J Pharmacol 2012, 165, 414–423. [Google Scholar] [CrossRef]
- Wammers, M.; Schupp, A.-K.; Bode, J.G.; Ehlting, C.; Wolf, S.; Deenen, R.; Köhrer, K.; Häussinger, D.; Graf, D. Reprogramming of Pro-Inflammatory Human Macrophages to an Anti-Inflammatory Phenotype by Bile Acids. Sci Rep 2018, 8, 255. [Google Scholar] [CrossRef]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef]
- Pols, T.W.H.; Puchner, T.; Korkmaz, H.I.; Vos, M.; Soeters, M.R.; de Vries, C.J.M. Lithocholic Acid Controls Adaptive Immune Responses by Inhibition of Th1 Activation through the Vitamin D Receptor. PLoS One 2017, 12, e0176715. [Google Scholar] [CrossRef]
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide Endotoxins. Annu Rev Biochem 2002, 71, 635–700. [Google Scholar] [CrossRef] [PubMed]
- Pj, T.; Re, L.; M, H.; Cm, F.-L.; R, K.; Ji, G. The Human Microbiome Project. Nature 2007, 449. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the Gut Microbiota in Disease. Microb Ecol Health Dis 2015, 26, 26191. [Google Scholar] [CrossRef]
- Hb, S.; Ke, W.-W.; M, C.; L, T.; Ca, P. Lipopolysaccharide Induces Oxidative Cardiac Mitochondrial Damage and Biogenesis. Cardiovascular research 2004, 64. [Google Scholar] [CrossRef]
- Ll, S.; Gm, D.; Nl, W. Potential Role of Endotoxin as a Proinflammatory Mediator of Atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology 2004, 24. [Google Scholar] [CrossRef]
- Wright, S.D.; Ramos, R.A.; Tobias, P.S.; Ulevitch, R.J.; Mathison, J.C. CD14, a Receptor for Complexes of Lipopolysaccharide (LPS) and LPS Binding Protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 Signal Transduction Pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- M.B., F; Jg, V.; N, R. Toll-like Receptor Signaling Pathways in Cardiovascular Diseases: Challenges and Opportunities. International reviews of immunology 2012, 31. [Google Scholar] [CrossRef]
- Blomkalns, A.L.; Stoll, L.L.; Shaheen, W.; Romig-Martin, S.A.; Dickson, E.W.; Weintraub, N.L.; Denning, G.M. Low Level Bacterial Endotoxin Activates Two Distinct Signaling Pathways in Human Peripheral Blood Mononuclear Cells. J Inflamm (Lond) 2011, 8, 4. [Google Scholar] [CrossRef]
- Tan, Y.; Kagan, J.C. A Cross-Disciplinary Perspective on the Innate Immune Responses to Bacterial Lipopolysaccharide. Mol Cell 2014, 54, 212–223. [Google Scholar] [CrossRef]
- Jankowska, E.A.; von Haehling, S.; Czarny, A.; Zaczynska, E.; Kus, A.; Anker, S.D.; Banasiak, W.; Ponikowski, P. Activation of the NF-kappaB System in Peripheral Blood Leukocytes from Patients with Chronic Heart Failure. Eur J Heart Fail 2005, 7, 984–990. [Google Scholar] [CrossRef]
- Földes, G.; von Haehling, S.; Okonko, D.O.; Jankowska, E.A.; Poole-Wilson, P.A.; Anker, S.D. Fluvastatin Reduces Increased Blood Monocyte Toll-like Receptor 4 Expression in Whole Blood from Patients with Chronic Heart Failure. Int J Cardiol 2008, 124, 80–85. [Google Scholar] [CrossRef]
- Kuwahata, S.; Fujita, S.; Orihara, K.; Hamasaki, S.; Oba, R.; Hirai, H.; Nagata, K.; Ishida, S.; Kataoka, T.; Oketani, N.; et al. High Expression Level of Toll-like Receptor 2 on Monocytes Is an Important Risk Factor for Arteriosclerotic Disease. Atherosclerosis 2010, 209, 248–254. [Google Scholar] [CrossRef]
- Torre-Amione, G.; Kapadia, S.; Benedict, C.; Oral, H.; Young, J.B.; Mann, D.L. Proinflammatory Cytokine Levels in Patients with Depressed Left Ventricular Ejection Fraction: A Report from the Studies of Left Ventricular Dysfunction (SOLVD). J Am Coll Cardiol 1996, 27, 1201–1206. [Google Scholar] [CrossRef]
- Ohlsson, B.G.; Englund, M.C.; Karlsson, A.L.; Knutsen, E.; Erixon, C.; Skribeck, H.; Liu, Y.; Bondjers, G.; Wiklund, O. Oxidized Low Density Lipoprotein Inhibits Lipopolysaccharide-Induced Binding of Nuclear Factor-kappaB to DNA and the Subsequent Expression of Tumor Necrosis Factor-Alpha and Interleukin-1beta in Macrophages. J Clin Invest 1996, 98, 78–89. [Google Scholar] [CrossRef]
- B, J.; Ys, B.; D, P.; Cs, S.; K, G.; Bs, L.; V, M.; M, B. Increased Circulatory Levels of Lipopolysaccharide (LPS) and Zonulin Signify Novel Biomarkers of Proinflammation in Patients with Type 2 Diabetes. Molecular and cellular biochemistry 2014, 388. [Google Scholar] [CrossRef]
- Sanz, Y.; Santacruz, A.; Palma, G.D. Insights into the Roles of Gut Microbes in Obesity. Interdisciplinary Perspectives on Infectious Diseases 2008. [Google Scholar] [CrossRef]
- Am, B.; Hw, H. Triglyceride-Rich Lipoproteins as Agents of Innate Immunity. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 2005, 41 Suppl 7. [Google Scholar] [CrossRef]
- Larsen, C.G.; Anderson, A.O.; Appella, E.; Oppenheim, J.J.; Matsushima, K. The Neutrophil-Activating Protein (NAP-1) Is Also Chemotactic for T Lymphocytes. Science 1989, 243, 1464–1466. [Google Scholar] [CrossRef]
- Cena, H.; Calder, P.C. Defining a Healthy Diet: Evidence for the Role of Contemporary Dietary Patterns in Health and Disease. Nutrients 2020, 12, 334. [Google Scholar] [CrossRef]
- Widmer, R.J.; Flammer, A.J.; Lerman, L.O.; Lerman, A. The Mediterranean Diet, Its Components, and Cardiovascular Disease. Am J Med 2015, 128, 229–238. [Google Scholar] [CrossRef]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N Engl J Med 2018, 378, e34. [Google Scholar] [CrossRef]
- Tørris, C.; Molin, M.; Cvancarova Småstuen, M. Fish Consumption and Its Possible Preventive Role on the Development and Prevalence of Metabolic Syndrome - a Systematic Review. Diabetol Metab Syndr 2014, 6, 112. [Google Scholar] [CrossRef]
- Jimenez-Torres, J.; Alcalá-Diaz, J.F.; Torres-Peña, J.D.; Gutierrez-Mariscal, F.M.; Leon-Acuña, A.; Gómez-Luna, P.; Fernández-Gandara, C.; Quintana-Navarro, G.M.; Fernandez-Garcia, J.C.; Perez-Martinez, P.; et al. Mediterranean Diet Reduces Atherosclerosis Progression in Coronary Heart Disease: An Analysis of the CORDIOPREV Randomized Controlled Trial. Stroke 2021, 52, 3440–3449. [Google Scholar] [CrossRef]
- Griffin, L.E.; Djuric, Z.; Angiletta, C.J.; Mitchell, C.M.; Baugh, M.E.; Davy, K.P.; Neilson, A.P. A Mediterranean Diet Does Not Alter Plasma Trimethylamine N-Oxide Concentrations in Healthy Adults at Risk for Colon Cancer. Food Funct 2019, 10, 2138–2147. [Google Scholar] [CrossRef]
- Barrea, L.; Annunziata, G.; Muscogiuri, G.; Laudisio, D.; Di Somma, C.; Maisto, M.; Tenore, G.C.; Colao, A.; Savastano, S. Trimethylamine N-Oxide, Mediterranean Diet, and Nutrition in Healthy, Normal-Weight Adults: Also a Matter of Sex? Nutrition 2019, 62, 7–17. [Google Scholar] [CrossRef]
- Barrea, L.; Muscogiuri, G.; Pugliese, G.; Graziadio, C.; Maisto, M.; Pivari, F.; Falco, A.; Tenore, G.C.; Colao, A.; Savastano, S. Association of the Chronotype Score with Circulating Trimethylamine N-Oxide (TMAO) Concentrations. Nutrients 2021, 13, 1671. [Google Scholar] [CrossRef]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazanetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-Level Adherence to a Mediterranean Diet Beneficially Impacts the Gut Microbiota and Associated Metabolome. Gut 2016, 65, 1812–1821. [Google Scholar] [CrossRef]
- Yamagata, K.; Hashiguchi, K.; Yamamoto, H.; Tagami, M. Dietary Apigenin Reduces Induction of LOX-1 and NLRP3 Expression, Leukocyte Adhesion, and Acetylated Low-Density Lipoprotein Uptake in Human Endothelial Cells Exposed to Trimethylamine-N-Oxide. J Cardiovasc Pharmacol 2019, 74, 558–565. [Google Scholar] [CrossRef]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N Engl J Med 2018, 378, e34. [Google Scholar] [CrossRef]
- Choo, J.M.; Murphy, K.J.; Wade, A.T.; Wang, Y.; Bracci, E.L.; Davis, C.R.; Dyer, K.A.; Woodman, R.J.; Hodgson, J.M.; Rogers, G.B. Interactions between Mediterranean Diet Supplemented with Dairy Foods and the Gut Microbiota Influence Cardiovascular Health in an Australian Population. Nutrients 2023, 15, 3645. [Google Scholar] [CrossRef]
- Ma, W.; Nguyen, L.H.; Song, M.; Wang, D.D.; Franzosa, E.A.; Cao, Y.; Joshi, A.; Drew, D.A.; Mehta, R.; Ivey, K.L.; et al. Dietary Fiber Intake, the Gut Microbiome, and Chronic Systemic Inflammation in a Cohort of Adult Men. Genome Med 2021, 13, 102. [Google Scholar] [CrossRef]
- Murga-Garrido, S.M.; Hong, Q.; Cross, T.-W.L.; Hutchison, E.R.; Han, J.; Thomas, S.P.; Vivas, E.I.; Denu, J.; Ceschin, D.G.; Tang, Z.-Z.; et al. Gut Microbiome Variation Modulates the Effects of Dietary Fiber on Host Metabolism. Microbiome 2021, 9, 117. [Google Scholar] [CrossRef]
- Catry, E.; Bindels, L.B.; Tailleux, A.; Lestavel, S.; Neyrinck, A.M.; Goossens, J.-F.; Lobysheva, I.; Plovier, H.; Essaghir, A.; Demoulin, J.-B.; et al. Targeting the Gut Microbiota with Inulin-Type Fructans: Preclinical Demonstration of a Novel Approach in the Management of Endothelial Dysfunction. Gut 2018, 67, 271–283. [Google Scholar] [CrossRef]
- Liu, F.; Prabhakar, M.; Ju, J.; Long, H.; Zhou, H.-W. Effect of Inulin-Type Fructans on Blood Lipid Profile and Glucose Level: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Eur J Clin Nutr 2017, 71, 9–20. [Google Scholar] [CrossRef]
- Cosola, C.; De Angelis, M.; Rocchetti, M.T.; Montemurno, E.; Maranzano, V.; Dalfino, G.; Manno, C.; Zito, A.; Gesualdo, M.; Ciccone, M.M.; et al. Beta-Glucans Supplementation Associates with Reduction in P-Cresyl Sulfate Levels and Improved Endothelial Vascular Reactivity in Healthy Individuals. PLoS One 2017, 12, e0169635. [Google Scholar] [CrossRef]
- Matsumoto, M.; Inoue, R.; Tsukahara, T.; Ushida, K.; Chiji, H.; Matsubara, N.; Hara, H. Voluntary Running Exercise Alters Microbiota Composition and Increases N-Butyrate Concentration in the Rat Cecum. Biosci Biotechnol Biochem 2008, 72, 572–576. [Google Scholar] [CrossRef]
- Varghese, S.; Rao, S.; Khattak, A.; Zamir, F.; Chaari, A. Physical Exercise and the Gut Microbiome: A Bidirectional Relationship Influencing Health and Performance. Nutrients 2024, 16, 3663. [Google Scholar] [CrossRef]
- Lira, F.S.; Rosa, J.C.; Pimentel, G.D.; Souza, H.A.; Caperuto, E.C.; Carnevali, L.C.; Seelaender, M.; Damaso, A.R.; Oyama, L.M.; de Mello, M.T.; et al. Endotoxin Levels Correlate Positively with a Sedentary Lifestyle and Negatively with Highly Trained Subjects. Lipids Health Dis 2010, 9, 82. [Google Scholar] [CrossRef]
- Allen, J.M.; Mailing, L.J.; Niemiro, G.M.; Moore, R.; Cook, M.D.; White, B.A.; Holscher, H.D.; Woods, J.A. Exercise Alters Gut Microbiota Composition and Function in Lean and Obese Humans. Med Sci Sports Exerc 2018, 50, 747–757. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, L.; Chen, L.; Li, L. Therapeutic Potential of Traditional Chinese Medicine in Atherosclerosis: A Review. Phytother Res 2022, 36, 4080–4100. [Google Scholar] [CrossRef]
- Jing, J.; Guo, J.; Dai, R.; Zhu, C.; Zhang, Z. Targeting Gut Microbiota and Immune Crosstalk: Potential Mechanisms of Natural Products in the Treatment of Atherosclerosis. Front Pharmacol 2023, 14, 1252907. [Google Scholar] [CrossRef]
- Liu, S.; He, F.; Zheng, T.; Wan, S.; Chen, J.; Yang, F.; Xu, X.; Pei, X. Ligustrum Robustum Alleviates Atherosclerosis by Decreasing Serum TMAO, Modulating Gut Microbiota, and Decreasing Bile Acid and Cholesterol Absorption in Mice. Mol Nutr Food Res 2021, 65, e2100014. [Google Scholar] [CrossRef]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert Consensus Document. The International Scientific Association for Probiotics and Prebiotics Consensus Statement on the Scope and Appropriate Use of the Term Probiotic. Nat Rev Gastroenterol Hepatol 2014, 11, 506–514. [Google Scholar] [CrossRef]
- Ghanbari, F.; Hasani, S.; Aghili, Z.S.; Asgary, S. The Potential Preventive Effect of Probiotics, Prebiotics, and Synbiotics on Cardiovascular Risk Factors through Modulation of Gut Microbiota: A Review. Food Sci Nutr 2024, 12, 4569–4580. [Google Scholar] [CrossRef]
- O’Morain, V.L.; Ramji, D.P. The Potential of Probiotics in the Prevention and Treatment of Atherosclerosis. Mol Nutr Food Res 2020, 64, e1900797. [Google Scholar] [CrossRef]
- Qiu, L.; Tao, X.; Xiong, H.; Yu, J.; Wei, H. Lactobacillus Plantarum ZDY04 Exhibits a Strain-Specific Property of Lowering TMAO via the Modulation of Gut Microbiota in Mice. Food Funct 2018, 9, 4299–4309. [Google Scholar] [CrossRef]
- Moludi, J.; Kafil, H.S.; Qaisar, S.A.; Gholizadeh, P.; Alizadeh, M.; Vayghyan, H.J. Effect of Probiotic Supplementation along with Calorie Restriction on Metabolic Endotoxemia, and Inflammation Markers in Coronary Artery Disease Patients: A Double Blind Placebo Controlled Randomized Clinical Trial. Nutr J 2021, 20, 47. [Google Scholar] [CrossRef]
- de Araújo Henriques Ferreira, G.; Magnani, M.; Cabral, L.; Brandão, L.R.; Noronha, M.F.; de Campos Cruz, J.; de Souza, E.L.; de Brito Alves, J.L. Potentially Probiotic Limosilactobacillus Fermentum Fruit-Derived Strains Alleviate Cardiometabolic Disorders and Gut Microbiota Impairment in Male Rats Fed a High-Fat Diet. Probiotics Antimicrob Proteins 2022, 14, 349–359. [Google Scholar] [CrossRef]
- Tong, L.; Zhang, X.; Hao, H.; Liu, Q.; Zhou, Z.; Liang, X.; Liu, T.; Gong, P.; Zhang, L.; Zhai, Z.; et al. Lactobacillus Rhamnosus GG Derived Extracellular Vesicles Modulate Gut Microbiota and Attenuate Inflammatory in DSS-Induced Colitis Mice. Nutrients 2021, 13, 3319. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, M.; Liu, Y.; Xu, M.; Shi, L.; Li, X.; Zhao, J.; Zhang, H.; Wang, G.; Chen, W. Bifidobacterium Breve and Bifidobacterium Longum Attenuate Choline-Induced Plasma Trimethylamine N-Oxide Production by Modulating Gut Microbiota in Mice. Nutrients 2022, 14, 1222. [Google Scholar] [CrossRef]
- Yang, D.; Lyu, W.; Hu, Z.; Gao, J.; Zheng, Z.; Wang, W.; Firrman, J.; Ren, D. Probiotic Effects of Lactobacillus Fermentum ZJUIDS06 and Lactobacillus Plantarum ZY08 on Hypercholesteremic Golden Hamsters. Front Nutr 2021, 8, 705763. [Google Scholar] [CrossRef]
- Wang, Q.; He, Y.; Li, X.; Zhang, T.; Liang, M.; Wang, G.; Zhao, J.; Zhang, H.; Chen, W. Lactobacillus Reuteri CCFM8631 Alleviates Hypercholesterolaemia Caused by the Paigen Atherogenic Diet by Regulating the Gut Microbiota. Nutrients 2022, 14, 1272. [Google Scholar] [CrossRef]
- Pham, Q.H.; Bui, T.V.A.; Sim, W.-S.; Lim, K.H.; Law, C.O.K.; Tan, W.; Kim, R.Y.; Chow, K.T.; Park, H.-J.; Ban, K.; et al. Daily Oral Administration of Probiotics Engineered to Constantly Secrete Short-Chain Fatty Acids Effectively Prevents Myocardial Injury from Subsequent Ischaemic Heart Disease. Cardiovasc Res 2024, 120, 1737–1751. [Google Scholar] [CrossRef]
- Schneeberger, M.; Everard, A.; Gómez-Valadés, A.G.; Matamoros, S.; Ramírez, S.; Delzenne, N.M.; Gomis, R.; Claret, M.; Cani, P.D. Akkermansia Muciniphila Inversely Correlates with the Onset of Inflammation, Altered Adipose Tissue Metabolism and Metabolic Disorders during Obesity in Mice. Sci Rep 2015, 5, 16643. [Google Scholar] [CrossRef]
- Yoshida, N.; Emoto, T.; Yamashita, T.; Watanabe, H.; Hayashi, T.; Tabata, T.; Hoshi, N.; Hatano, N.; Ozawa, G.; Sasaki, N.; et al. Bacteroides Vulgatus and Bacteroides Dorei Reduce Gut Microbial Lipopolysaccharide Production and Inhibit Atherosclerosis. Circulation 2018, 138, 2486–2498. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Inagawa, H.; Kohchi, C.; Kazumura, K.; Tsuchiya, H.; Miwa, T.; Okazaki, K.; Soma, G.-I. Oral Administration of Pantoea Agglomerans-Derived Lipopolysaccharide Prevents Development of Atherosclerosis in High-Fat Diet-Fed apoE-Deficient Mice via Ameliorating Hyperlipidemia, pro-Inflammatory Mediators and Oxidative Responses. PLoS One 2018, 13, e0195008. [Google Scholar] [CrossRef]
- Kuka, J.; Liepinsh, E.; Makrecka-Kuka, M.; Liepins, J.; Cirule, H.; Gustina, D.; Loza, E.; Zharkova-Malkova, O.; Grinberga, S.; Pugovics, O.; et al. Suppression of Intestinal Microbiota-Dependent Production of pro-Atherogenic Trimethylamine N-Oxide by Shifting L-Carnitine Microbial Degradation. Life Sciences 2014, 117, 84–92. [Google Scholar] [CrossRef]
- Sethi, N.J.; Safi, S.; Korang, S.K.; Hróbjartsson, A.; Skoog, M.; Gluud, C.; Jakobsen, J.C. Antibiotics for Secondary Prevention of Coronary Heart Disease. Cochrane Database Syst Rev 2021, 2, CD003610. [Google Scholar] [CrossRef]
- Heianza, Y.; Zheng, Y.; Ma, W.; Rimm, E.B.; Albert, C.M.; Hu, F.B.; Rexrode, K.M.; Manson, J.E.; Qi, L. Duration and Life-Stage of Antibiotic Use and Risk of Cardiovascular Events in Women. Eur Heart J 2019, 40, 3838–3845. [Google Scholar] [CrossRef]
- Vrieze, A.; Nood, E.V.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.W.M.; Dallinga–Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of Intestinal Microbiota From Lean Donors Increases Insulin Sensitivity in Individuals With Metabolic Syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef]
- Jing, L.; Zhang, H.; Xiang, Q.; Shen, L.; Guo, X.; Zhai, C.; Hu, H. Targeting Trimethylamine N-Oxide: A New Therapeutic Strategy for Alleviating Atherosclerosis. Front Cardiovasc Med 2022, 9, 864600. [Google Scholar] [CrossRef]
- Lindskog Jonsson, A.; Hållenius, F.F.; Akrami, R.; Johansson, E.; Wester, P.; Arnerlöv, C.; Bäckhed, F.; Bergström, G. Bacterial Profile in Human Atherosclerotic Plaques. Atherosclerosis 2017, 263, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Fåk, F.; Tremaroli, V.; Bergström, G.; Bäckhed, F. Oral Microbiota in Patients with Atherosclerosis. Atherosclerosis 2015, 243, 573–578. [Google Scholar] [CrossRef]
- Koren, O.; Spor, A.; Felin, J.; Fåk, F.; Stombaugh, J.; Tremaroli, V.; Behre, C.J.; Knight, R.; Fagerberg, B.; Ley, R.E.; et al. Human Oral, Gut, and Plaque Microbiota in Patients with Atherosclerosis. Proceedings of the National Academy of Sciences 2011, 108, 4592–4598. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Innate immune response in atherosclerosis. The schematic illustrates the monocyte recruitment, differentiation, and foam cell formation during atherogenesis. Circulating monocytes adhere to activated endothelium via ICAM1 and VCAM1, migrate into the intima, and differentiate into macrophages that uptake oxidized LDL, forming foam cells and releasing proinflammatory cytokines contributing to plaque progression. The Figure illustration was prepared using Servier Medical Art, licensed under a creative commons’ attribution 4.0 international license
Figure 1.
Innate immune response in atherosclerosis. The schematic illustrates the monocyte recruitment, differentiation, and foam cell formation during atherogenesis. Circulating monocytes adhere to activated endothelium via ICAM1 and VCAM1, migrate into the intima, and differentiate into macrophages that uptake oxidized LDL, forming foam cells and releasing proinflammatory cytokines contributing to plaque progression. The Figure illustration was prepared using Servier Medical Art, licensed under a creative commons’ attribution 4.0 international license
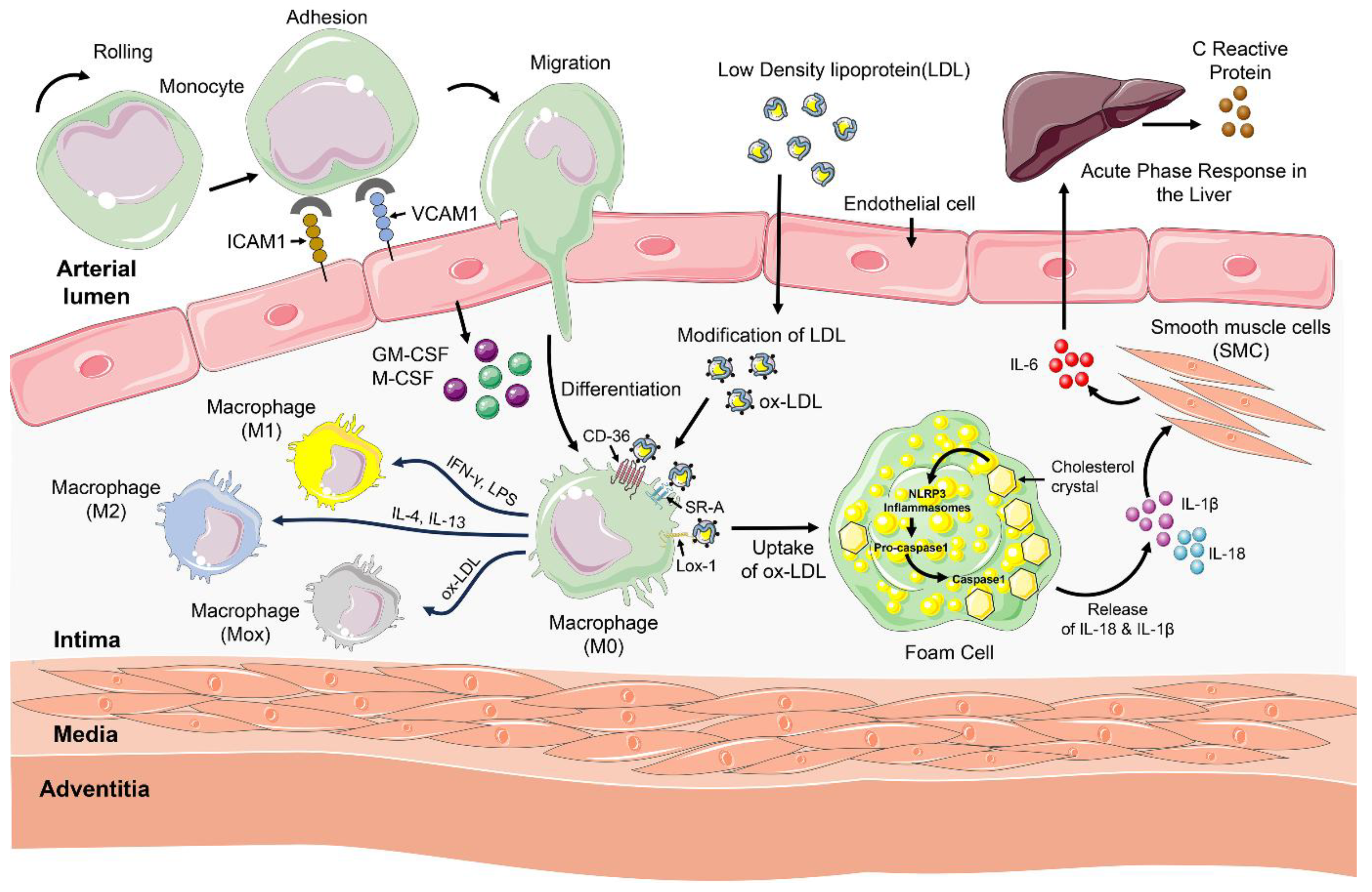
Figure 2.
Adaptive immune responses in atherosclerosis. Antigen-presenting cells activate naïve CD4⁺ and CD8⁺ T cells, leading to the differentiation of Th1, Th2, Th17, Treg, and cytotoxic subsets that modulate vascular inflammation. Th1 and Th17 cells promote plaque instability via cytokine secretion, whereas Treg cells exert anti-inflammatory effects through IL-10 and TGF-β. Activated B1 cells generate protective IgM antibodies targeting oxidized LDL, while B2 cells and Tfh cells contribute to pro-atherogenic immune activation. Effector T cells expressing CCR5 and CXCR6 migrate to atherosclerotic lesions, sustaining chronic inflammation and plaque progression. The Figure illustration was prepared using Servier Medical Art, licensed under a Creative Commons’ Attribution 4.0 International license.
Figure 2.
Adaptive immune responses in atherosclerosis. Antigen-presenting cells activate naïve CD4⁺ and CD8⁺ T cells, leading to the differentiation of Th1, Th2, Th17, Treg, and cytotoxic subsets that modulate vascular inflammation. Th1 and Th17 cells promote plaque instability via cytokine secretion, whereas Treg cells exert anti-inflammatory effects through IL-10 and TGF-β. Activated B1 cells generate protective IgM antibodies targeting oxidized LDL, while B2 cells and Tfh cells contribute to pro-atherogenic immune activation. Effector T cells expressing CCR5 and CXCR6 migrate to atherosclerotic lesions, sustaining chronic inflammation and plaque progression. The Figure illustration was prepared using Servier Medical Art, licensed under a Creative Commons’ Attribution 4.0 International license.
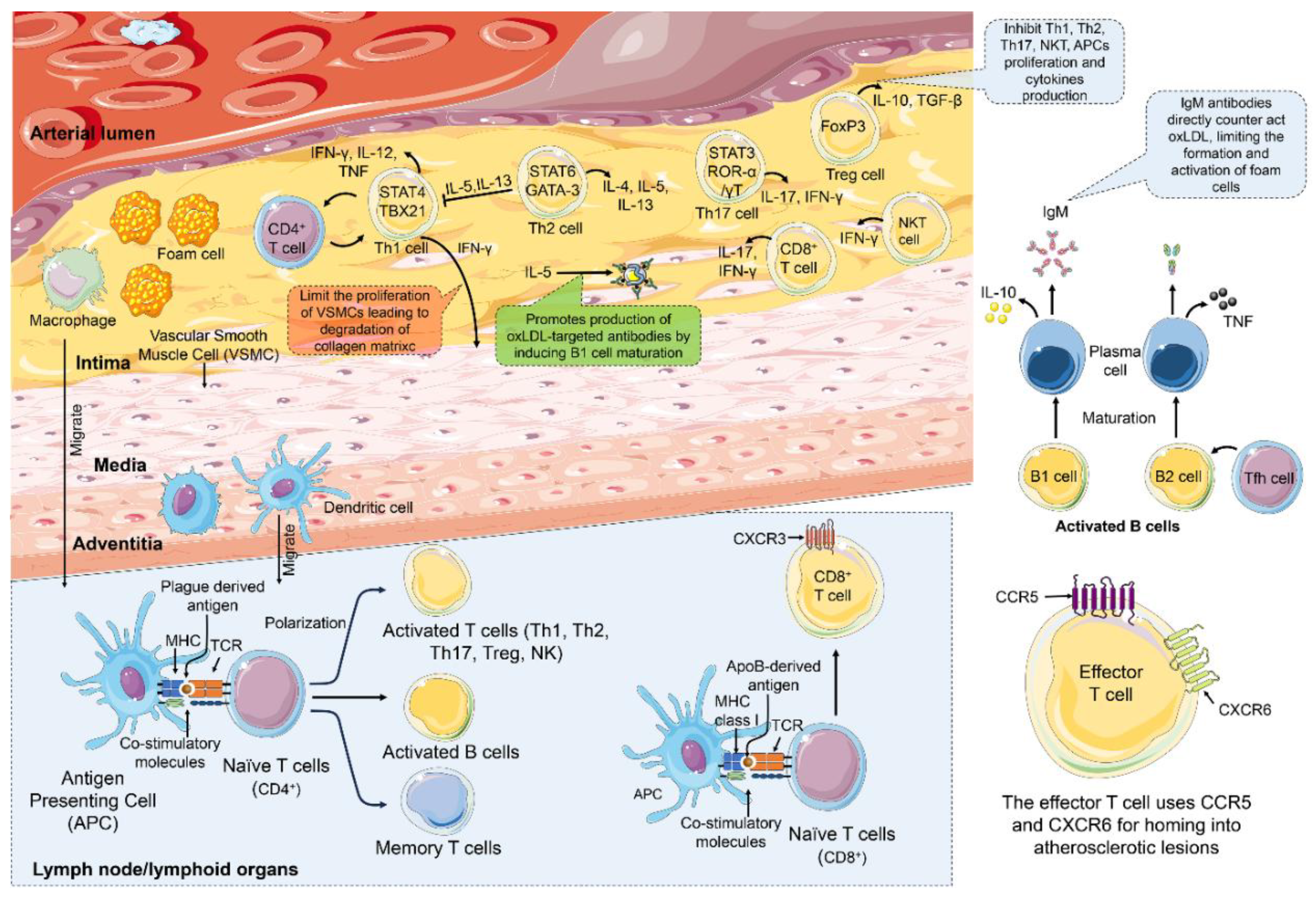
Figure 3.
Schematic diagram describing the formation of TMA and TMAO from the dietary intake and its role in various organs. Diets rich in choline, betaine, and lecithin are oxidized and reduced to TMA. TMA is converted into TMAO in the liver using the FMO1/3 enzymes. In the liver, TMAO decreases the expression of CYP7A1 and increases the production of reactive oxygen species and also CRPs. In the endothelial lining of the heart, it induces monocyte and leukocyte adhesion and also increases the production of Cox2, E-selectin, IL-6, ICAM-1, and tissue factor via NF-κB. In atherosclerotic lesions, TMAO induces the expression of SR-A1and CD36 on the macrophage, increases caspase1 activity, and the expression of GPR34 and HSP70. In the gut lumen, TMA increases the abundance of Firmicutes, Actinobacteria, Gamma Proteobacteria but reduces the abundance of Bacteroidetes. In addition, it acts on the colonocyte and reduces the expression of Niemann-PickC1-like-1 and ABCG5/8. TMA increases oxidative stress, thereby releasing IL-18, IL-1β cytokines. The Figure illustration was prepared using Servier Medical Art, licensed under a Creative Commons’ Attribution 4.0 International license. Bacterial icons were taken from the FLATICONS by FREEPIK (https://www.flaticon.com/).
Figure 3.
Schematic diagram describing the formation of TMA and TMAO from the dietary intake and its role in various organs. Diets rich in choline, betaine, and lecithin are oxidized and reduced to TMA. TMA is converted into TMAO in the liver using the FMO1/3 enzymes. In the liver, TMAO decreases the expression of CYP7A1 and increases the production of reactive oxygen species and also CRPs. In the endothelial lining of the heart, it induces monocyte and leukocyte adhesion and also increases the production of Cox2, E-selectin, IL-6, ICAM-1, and tissue factor via NF-κB. In atherosclerotic lesions, TMAO induces the expression of SR-A1and CD36 on the macrophage, increases caspase1 activity, and the expression of GPR34 and HSP70. In the gut lumen, TMA increases the abundance of Firmicutes, Actinobacteria, Gamma Proteobacteria but reduces the abundance of Bacteroidetes. In addition, it acts on the colonocyte and reduces the expression of Niemann-PickC1-like-1 and ABCG5/8. TMA increases oxidative stress, thereby releasing IL-18, IL-1β cytokines. The Figure illustration was prepared using Servier Medical Art, licensed under a Creative Commons’ Attribution 4.0 International license. Bacterial icons were taken from the FLATICONS by FREEPIK (https://www.flaticon.com/).
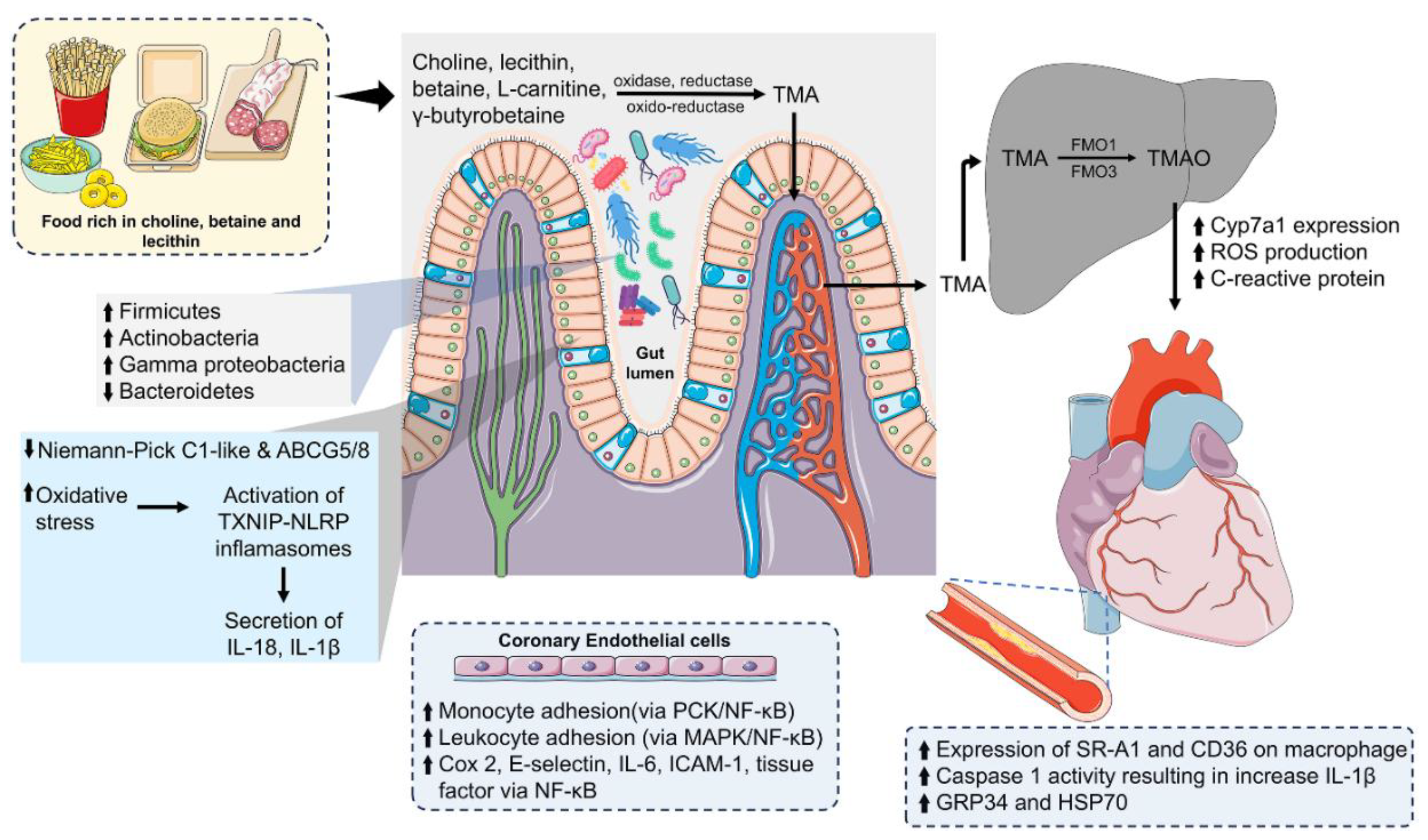
Figure 4.
Effect of SCFAs in various organs. SCFAs are derived from the anaerobic fermentation of resistant dietary fibres. In the gut SCFAs positively impact the growth of Lactobacillus, Bifidobacterium, Akkermensia muciniphia, Roseburia, Clostridium leptum, Bacteroides etc. Propionate stimulates macrophages, lymphocytes, T cells, and dendritic cells to increase the secretion of TGF-β and IL-10, while reducing the production of pro-inflammatory cytokines such as IL-6, IL-2, IL-17a, IFN-γ, and TNF-α. Notably, propionate specifically decreases renin secretion and promotes vascular dilation through Olfr78. Both propionate and acetate stimulate enteroendocrine cells to release GLP-1, GLP-2, and PYY. Additionally, n-butyrate serves as a precursor for fatty acid oxidation in the liver and skeletal muscle. Short-chain fatty acids (SCFAs) also inhibit the secretion of NF-κB, TNF-α, and IL-6 in visceral adipose tissue, while reducing insulin-dependent fat accumulation in white adipose tissue. The Figure illustration was prepared using Servier Medical Art, licensed under a Creative Commons’ Attribution 4.0 International license. Bacterial icons were taken from the FLATICONS by FREEPIK (https://www.flaticon.com/).
Figure 4.
Effect of SCFAs in various organs. SCFAs are derived from the anaerobic fermentation of resistant dietary fibres. In the gut SCFAs positively impact the growth of Lactobacillus, Bifidobacterium, Akkermensia muciniphia, Roseburia, Clostridium leptum, Bacteroides etc. Propionate stimulates macrophages, lymphocytes, T cells, and dendritic cells to increase the secretion of TGF-β and IL-10, while reducing the production of pro-inflammatory cytokines such as IL-6, IL-2, IL-17a, IFN-γ, and TNF-α. Notably, propionate specifically decreases renin secretion and promotes vascular dilation through Olfr78. Both propionate and acetate stimulate enteroendocrine cells to release GLP-1, GLP-2, and PYY. Additionally, n-butyrate serves as a precursor for fatty acid oxidation in the liver and skeletal muscle. Short-chain fatty acids (SCFAs) also inhibit the secretion of NF-κB, TNF-α, and IL-6 in visceral adipose tissue, while reducing insulin-dependent fat accumulation in white adipose tissue. The Figure illustration was prepared using Servier Medical Art, licensed under a Creative Commons’ Attribution 4.0 International license. Bacterial icons were taken from the FLATICONS by FREEPIK (https://www.flaticon.com/).
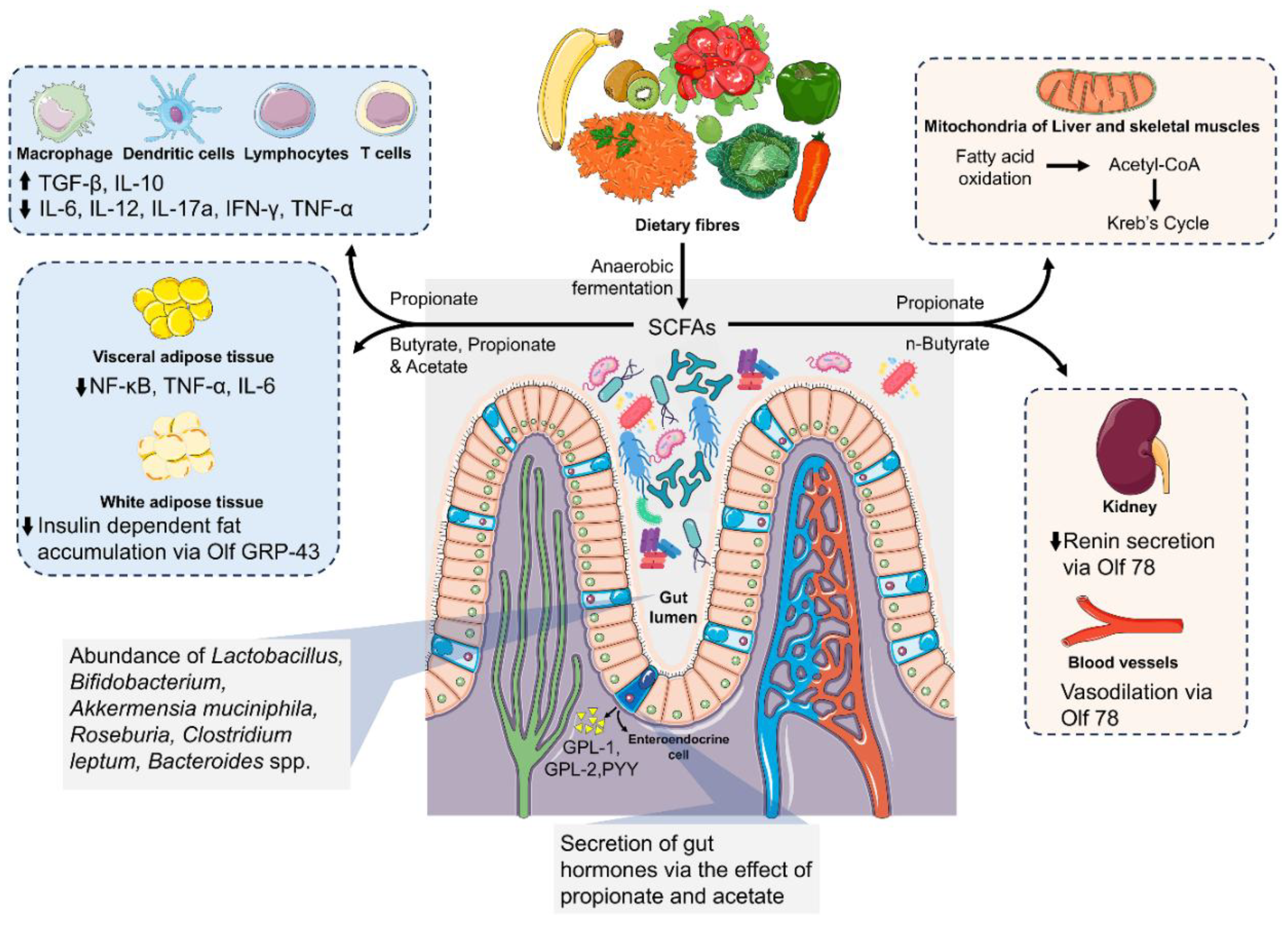
Figure 5.
Schematic image showing the difference between the normal gut and the dysbiotic atherosclerotic gut. In a healthy gut, there is an intact tight junction, with a higher abundance of beneficial bacteria, including Bacteroides, Bifidobacterium, Streptococcus, Clostridium, Lactobacillus, Ruminococcus, Enterococcus, the Akkermansia genus, and the Enterobacteriaceae family. Additionally, there is an increased presence of T regulatory cells, BSH activity, ABCG5/8 transporters, and SCFAs, along with lower levels of LPS and TMAO. In contrast, a dysbiotic atherogenic gut is characterized by a reduced abundance of Bacteroidetes and SCFA-producing Firmicutes, decreased BSH activity, ABCG5/8 transporters, T regulatory cells, and SCFAs. It also exhibits an increased abundance of Proteobacteria, higher levels of TMAO, and LPS. The Figure illustration was prepared using Servier Medical Art, licensed under a Creative Commons’ Attribution 4.0 International license. Bacterial icons were taken from the FLATICONS by FREEPIK (https://www.flaticon.com/).
Figure 5.
Schematic image showing the difference between the normal gut and the dysbiotic atherosclerotic gut. In a healthy gut, there is an intact tight junction, with a higher abundance of beneficial bacteria, including Bacteroides, Bifidobacterium, Streptococcus, Clostridium, Lactobacillus, Ruminococcus, Enterococcus, the Akkermansia genus, and the Enterobacteriaceae family. Additionally, there is an increased presence of T regulatory cells, BSH activity, ABCG5/8 transporters, and SCFAs, along with lower levels of LPS and TMAO. In contrast, a dysbiotic atherogenic gut is characterized by a reduced abundance of Bacteroidetes and SCFA-producing Firmicutes, decreased BSH activity, ABCG5/8 transporters, T regulatory cells, and SCFAs. It also exhibits an increased abundance of Proteobacteria, higher levels of TMAO, and LPS. The Figure illustration was prepared using Servier Medical Art, licensed under a Creative Commons’ Attribution 4.0 International license. Bacterial icons were taken from the FLATICONS by FREEPIK (https://www.flaticon.com/).
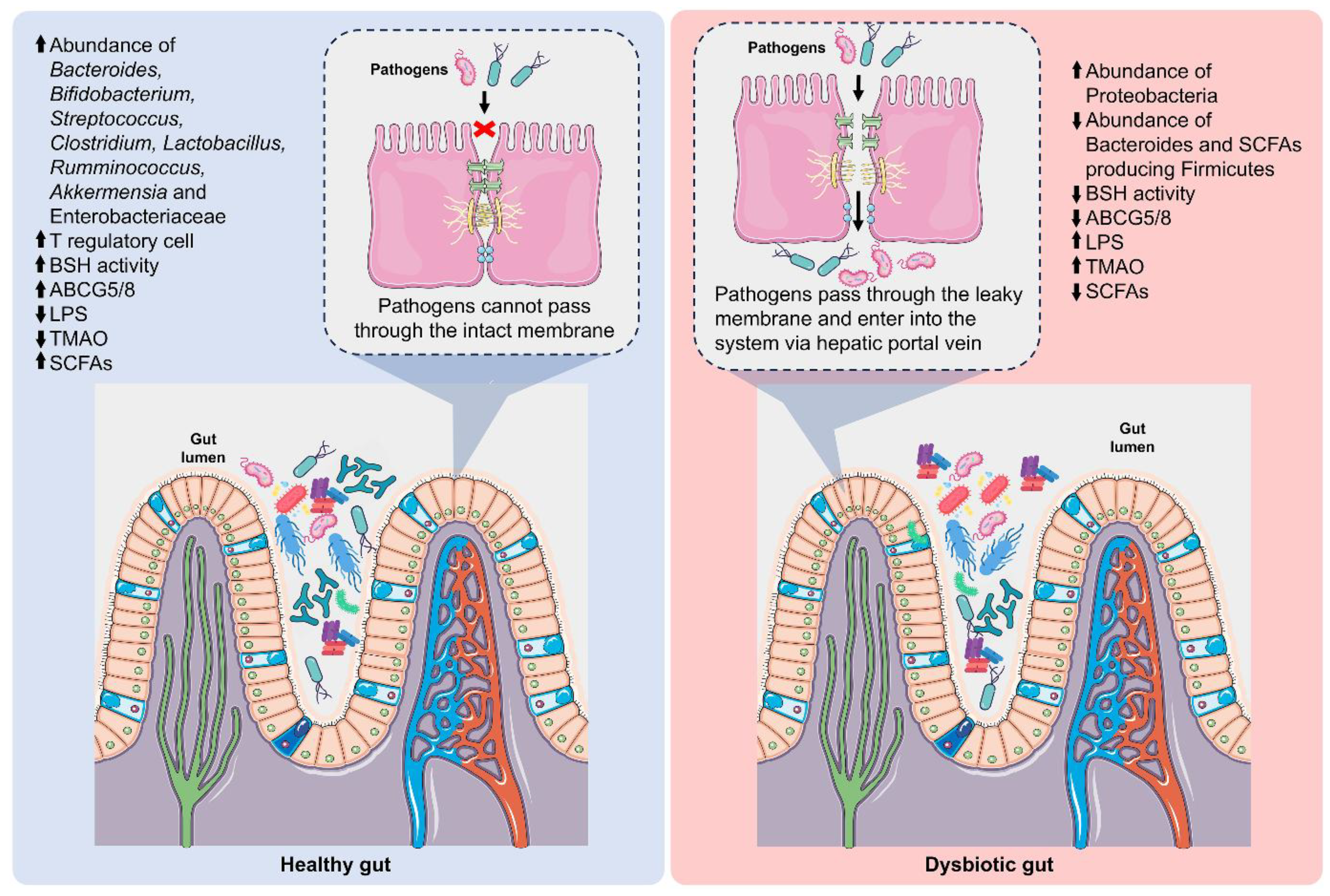

Table 1.
Oral and gut microbiota associated with CVDs.
| SL no. | Diseases | Origin of microbiota | Types of microbiota present | Inference | Reference |
|---|---|---|---|---|---|
| 1. | Coronary heart disease | Oral | Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans | Provide evidence for the presence of viable oral pathogens at the vascular sites and also at the atherosclerotic plaque. | [22] |
| 2. | Coronary Artery Disease | Gut | Phylum Bacteroidetes, Firmicutes | Decrease abundance of phylum Bacteroidetes and increase in phylum Firmicutes in patients with CAD | [13] |
| 3. | Atherosclerotic cardiovascular disease (ACVD) | Oral and Gut | Phylum Proteobacteria, Actinobacteria, Firmicutes, Cyanobacteria and Bacteroidetes. | Proteobacteria (48.3%) and Actinobacteria (40.2%), and three less dominating phyla: Firmicutes (4.0%), Cyanobacteria (3.9%) and Bacteroidetes (2.2%) in both symptomatic and asymptomatic patients |
[253] |
| 4. | Atherosclerotic cardiovascular disease (ACVD) | Oral cavity | Veillonella and Streptococcus | Bacteria from oral cavity may correlate with the disease marker of atherosclerosis. | [15] |
| 5. | Atherosclerotic cardiovascular disease (ACVD) | Gut | Enterobacteriaceae and Streptococcus spp. | Increase abundance of Enterobacteriaceae and Streptococcus spp. in ACVD patients compared to normal. | [12] |
| 6. | Atherosclerotic cardiovascular disease (ACVD) | Gut | Roseburia intestinalis | Provide protection against atherosclerosis | [14] |
| 7. | Atherosclerotic cardiovascular disease (ACVD) | Gut | Akkermansia muciniphila | Provide protection against atherosclerosis | [17] |
| 8. | Cardiovascular disease | Gut | Chlamydia pneumoniae, Staphylococcus spp., Streptococcus spp., Klebsiella pneumoniae, Proteus vulgaris, Burkholderia and Pseudomonas aeruginosa | Associated with CVD progression | [18] |
| 9. | Atherosclerotic cardiovascular disease (ACVD) | Oral and gut | Chlamydia pneumoniae and Helicobacter pylori | Associate with progression of atherosclerosis | [20] |
| 11. | Atherosclerotic cardiovascular disease (ACVD) | Oral | Anaeroglobus | Most abundant oral bacteria present in the symptomatic ACVD patient | [254] |
| 12 | Atherosclerotic cardiovascular disease (ACVD) | Oral | Chryseomonas | May contribute to the progression of Atherosclerosis | [255] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



