
Submitted:
26 January 2026
Posted:
27 January 2026
You are already at the latest version
Abstract
Non-small cell lung cancer (NSCLC) is the leading cause of lung cancer deaths, with re-sistance to targeted therapies posing a major clinical challenge. Drug-tolerant persister (DTP) cells are key contributors to resistance and targeting them offers new strategies to enhance existing treatments. MicroRNAs (miRNAs), particularly the tumour-suppressive miR-15/107 family, offer promise due to their ability to target multiple oncogenic path-ways. This study evaluated a synthetic consensus miRNA mimic, conmiR-15/107, in NSCLC cell line models. Dose–response assays showed robust, dose-dependent growth inhibition in both EGFR mutant (PC9) and KRAS mutant (H358 and A549) lung adeno-carcinoma cells, but not in the human bronchial epithelial cell line BEAS-2B. When combined with EGFR inhibitors (osimertinib and gefitinib) in PC9 cells, the mimics showed a higher rate of growth inhibition as compared to the controls and reduced IC50 values. Similarly, conmiR-15/107 enhanced growth inhibition by the KRAS inhibitors sotorasib and adagrasib in H358 cells. RT-qPCR confirmed downregulation of conmiR-15/107 targets including MEK1, BCL2, and BRCA1, suggesting a multi-target mechanism of action. Long-term assays showed that the mimics reduced the survival of DTPs in osimertinib-treated PC9 cells as well as sotorasib-treated H358 cells. These findings support conmiR-15/107 as a potential adjunct to targeted therapy, capable of enhancing treatment efficacy and delaying resistance in lung adenocarcinoma.
Keywords:
microRNA
; targeted therapy
; lung adenocarcinoma
; drug tolerance
1. Introduction
Lung cancer is the predominant cause of cancer-associated mortality globally, highlighting the critical demand for novel and more durable therapeutic approaches [1]. Although targeted therapies have significantly improved outcomes in non-small cell lung cancer (NSCLC), their long-term efficacy is often hindered by the inevitable development of drug resistance [2]. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), such as the first generation gefitinib, and the third-generation agent osimertinib, have improved progression-free survival when compared to chemotherapy in patients with EGFR-mutant NSCLC [3,4]. Despite frequent responses, resistance inevitably emerges through mechanisms such as secondary mutations in EGFR (e.g., T790M, C797S), activation of bypass signalling pathways (e.g., MET or HER2 amplification), or phenotypic changes such as transdifferentiation into small cell lung cancer (SCLC) or epithelial-to-mesenchymal transition (EMT) [5]. Similarly, the recent discovery and clinical development of KRASG12C inhibitors, such as sotorasib and adagrasib [6,7,8,9], has provided a novel approach for this previously “undruggable” target, but adaptive resistance limits the durability of responses for both of these new drugs [10,11].
A growing body of evidence indicates that drug-tolerant persister (DTP) cells play a central role in the emergence of resistance to targeted therapies [12,13,14]. These rare, slow-cycling subpopulations survive initial drug exposure through reversible, non-genetic adaptations, such as chromatin remodelling [15], altered metabolic states [16], or activation of survival pathways [11,17]. While DTPs typically represent only a small fraction of the tumour, they can persist under prolonged therapeutic pressure and eventually give rise to genetically resistant clones [18,19,20], posing a major challenge to the long-term efficacy of EGFR and KRAS inhibitors [11,17]. The lack of mutational alterations in DTPs makes them difficult to target, and while vulnerabilities in several pathways altered in DTPs have been explored, to date none have been broadly effective (reviewed in [12,13]).
MicroRNAs (miRNAs) may be particularly well suited to the challenge of targeting DTPs due to their ability to post-transcriptionally regulate multiple genes and pathways simultaneously [21]. MiRNAs are short (17–24 nucleotide) non-coding RNAs that associate with the RNA-induced silencing complex (RISC) and bind complementary recognition sequences in target mRNAs to inhibit their translation [22]. Among tumour-suppressive miRNAs, the miR-15/107 family has garnered particular interest due to its widespread dysregulation across cancer types, including NSCLC, and its involvement in resistance-related pathways [23]. Members of this family play key roles in regulating key oncogenic processes, including apoptosis, cell proliferation, cell cycle progression, and EMT [24]. Targets of this family include MEK1, MAPK3 and CRAF, which are all components of MAPK signalling [25,26]. Notably, many of these targets are downstream of receptor tyrosine kinase signalling pathways, including EGFR, suggesting that miR-15/107-based therapies may offer benefit when combined with EGFR inhibitors.
Multiple preclinical investigations have examined the restoration of miRNA expression using synthetic mimics as a therapeutic approach to improve the effectiveness of existing targeted therapies [27]. In preclinical models, restoration of miR-34a using a mimic has been reported to re-sensitize cancer cells to EGFR inhibitors, including erlotinib [28] and osimertinib [29]. In KRAS-mutant NSCLC cell lines, Fanini et al. reported that miR-16 mimic transfection re-sensitized cells to EGFR inhibition with erlotinib and surpassed the therapeutic effect of dual EGFR/MEK blockade using selumetinib–erlotinib. [25]. Our lab developed a series of synthetic consensus sequence mimics, “conmiR-15/107,” based on members of the highly conserved miR-15/107 superfamily [30], most of which share the 5’-AGCAGC-3’ seed sequence [23]. These engineered constructs were designed to replicate the tumour-suppressive functions of the entire family by targeting oncogenic effectors from multiple pathways. In vitro, conmiR-15/107 constructs induced robust growth inhibition across several cancer cell lines, attributed to downregulation of genes such as BCL2 and CCND1 [30]. In vivo studies in xenograft models of PM and NSCLC demonstrated significant tumour growth suppression, further supporting its therapeutic potential.
Therapeutic applications of miRNA mimics and inhibitors are increasingly being explored, with several candidates advancing to clinical trials. For instance, MRX34, a liposomal miR-34a mimic, was among the first to be tested clinically in advanced solid tumours, where it showed early signs of antitumour efficacy; however, the study was prematurely terminated as a result of severe immune-mediated toxicities [31]. Following this, the phase I TARGOmiR study assessed the delivery of a miR-16 mimic via EGFR-targeted minicells (EnGeneIC Dream Vectors) in pleural mesothelioma (PM) patients, providing evidence of feasibility and potential clinical benefit [32]. This trial was open to NSCLC patients but none were enrolled and thus this avenue of investigation remains unexplored.
Based on the known targets of members of the miR-15/107 family, the goal of this study was to investigate the therapeutic potential of the conmiR-15/107 consensus mimics in oncogene-driven NSCLC models. Specifically, we aimed to evaluate their ability of these constructs to enhance the efficacy of targeted therapies and inhibit DTPs.
2. Results
conmiR-15/107 shows dose dependent growth inhibitory effect
In a previous study we showed that the conmiR-15/107 constructs have a growth inhibitory effect in multiple cancer types including NSCLC cells [32]. Here the effects of conmiR-15/107 constructs on cell proliferation were tested in the oncogene-driven adenocarcinoma cell lines PC9 (EGFR exon 19 deletion), H358 (KRASG12C) and A549 (KRASG12S), and compared with effects on the BEAS-2B cell line as a normal lung epithelial control. In PC9 cells, conmiR-15/107.2 and conmiR-15/107.4 induced a dose-dependent inhibition of growth, with approximately 50% growth inhibition at the highest concentration (Figure 1A). A miR-15a mimic showed a similar, though slightly less potent, effect. In H358 cells, both constructs again caused a dose-dependent growth inhibition, with a maximum of approximately 60% inhibition at 20 nM, with similar results observed with miR-15a (Figure 1B). In A549 (Figure 1C) conmiR-15/107.2 induced 70% inhibition and conmiR-15/107.4 40% inhibition at 20 nM. In BEAS-2B cells, miR-15a, conmiR-15/107.2 and conmiR-15/107.4 had a lesser effect on cell growth except at the highest concentration (20 nM), where it had a 25% inhibition (Figure 1D).
miR-15/107 sensitises lung adenocarcinoma cells to targeted therapy
Individual members of the miR-15/107 family have been shown to sensitise NSCLC to targeted therapy [25], and previously we showed that the conmiR-15/107 constructs could sensitize PM and NSCLC cells to gemcitabine treatment [30]. To investigate whether conmiR-15/107 constructs sensitized EGFR- and KRASG12C-mutant lung adenocarcinoma cells to targeted therapy, PC9 and H358 cells were transfected with 2 nM of conmiR-15/107.2, conmiR-15/107.4, or control, followed by treatment with increasing concentrations of the EGFR inhibitors osimertinib and gefitinib, or the KRAS inhibitors sotorasib and adagrasib.
PC9 cells transfected with conmiR-15/107.2 and conmiR-15/107.4 were significantly more sensitive to both osimertinib and gefitinib treatment at all concentrations tested, with all combinations leading to >50% growth inhibition (Figure 2A-B), precluding comparison of IC50 values. Both conmiR-15/107.2 and conmiR-15/107.4 also sensitised PC9 cells to erlotinib, although to a lesser extent (Figure S1). Control experiments confirmed that 2 nM mimic alone had minimal impact on cell viability and that transfection efficiency was consistent across conditions (Figure 2G-I). To determine whether conmiR-15/107.2 and conmiR-15/107.4 could increase sensitivity to EGFR inhibitors in EGFR wild-type cells, we transfected A549 cells with the constructs and treated with osimertinib and gefitinib. As expected, A549 cells were considerably less sensitive than PC9 cells to osimertinib (Figure 2E) and essentially insensitive to erlotinib (Figure 2F). While the combination with conmiR-15/107.2 and conmiR-15/107.4 increased sensitivity to both drugs, these changes were not statistically significant. The mimics also failed to significantly increase sensitivity to the MEK inhibitor trametinib (Figure S1).
Next we evaluated whether conmiR-15/107 constructs sensitize KRAS-mutant NSCLC cells to KRASG12C inhibitors. H358 cells were transfected with 2 nM of conmiR-15/107.2, conmiR-15/107.4, or control, followed by treatment with increasing concentrations of sotorasib or adagrasib. Both constructs enhanced drug sensitivity to KRAS inhibitors, with reduced cell viability (Figure 2C-D) and lowered estimated IC50 values. H358 cells transfected with conmiR-15/107.2 and conmiR-15/107.4 were significantly more sensitive to both sotorasib and adagrasib treatment at all concentrations of targeted therapy tested.
miR-15/107 consensus mimics regulate multiple targets in PC9 cells
Given the clear growth inhibitory effect of both consensus mimics, we next investigated possible mechanistic effectors of the constructs. To investigate the regulatory impact of conmiR-15/107 constructs on known miR-15/107 family targets, PC9 cells were transfected with 2 nM of C81, conmiR-15/107.2, or conmiR-15/107.4 and treated with DMSO, 2 μM osimertinib, or 5 μM gefitinib. Western blot results indicated no reduction in phosphorylated ERK following transfection with conmiR-15/107.2 or conmiR-15/107.4 in PC9 and H358 cells (Figure 3A–B), therefore subsequent experiments focused on identifying alternative targets. As both BCL2 and CCND1 were confirmed as targets of the conmiR-15/107 constructs in PM cell lines [32], we investigated whether this was also the case in NSCLC models. BCL2, an anti-apoptotic gene, was consistently downregulated across all treatment conditions (Figure 3C). In DMSO-treated cells, BCL2 expression was reduced ~3-fold in both conmiR-transfected conditions compared to C81. In osimertinib-treated cells, reductions reached ~5-fold, while gefitinib treatment resulted in ~3-fold (conmiR-15/107.2) and ~6-fold (conmiR-15/107.4) downregulation. In contrast, neither conmiR-15/107.2 nor conmiR-15/107.4 significantly altered CCND1 mRNA levels under any condition (Figure 3D).
As MEK1, a component of the MAPK/ERK pathway and involved in EGFR signalling, has been shown to be downregulated by miR-15/107 family members [27] we investigated whether the consensus mimic constructs affected its expression. Following transfection with both constructs MEK1 was found to be significantly downregulated under all conditions (Figure 3E). In DMSO-treated cells, MEK1 expression decreased ~1.4-fold. Under osimertinib, MEK1 was reduced ~3-fold in both construct conditions. Gefitinib treatment led to ~2-fold (conmiR-15/107.2) and ~3-fold (conmiR-15/107.4) downregulation. Finally, BRCA1, involved in DNA damage repair and another potential target [38], was strongly suppressed following conmiR-15/107 transfection under drug treatment (Figure 3F). Under osimertinib, BRCA1 expression was reduced ~3.7-fold (107.2) and ~6-fold (107.4). Under gefitinib treatment, BRCA1 decreased ~3-fold (107.2, non-significant) and ~5.5-fold (107.4) relative to control.
miR-15/107 inhibits the survival of DTPs
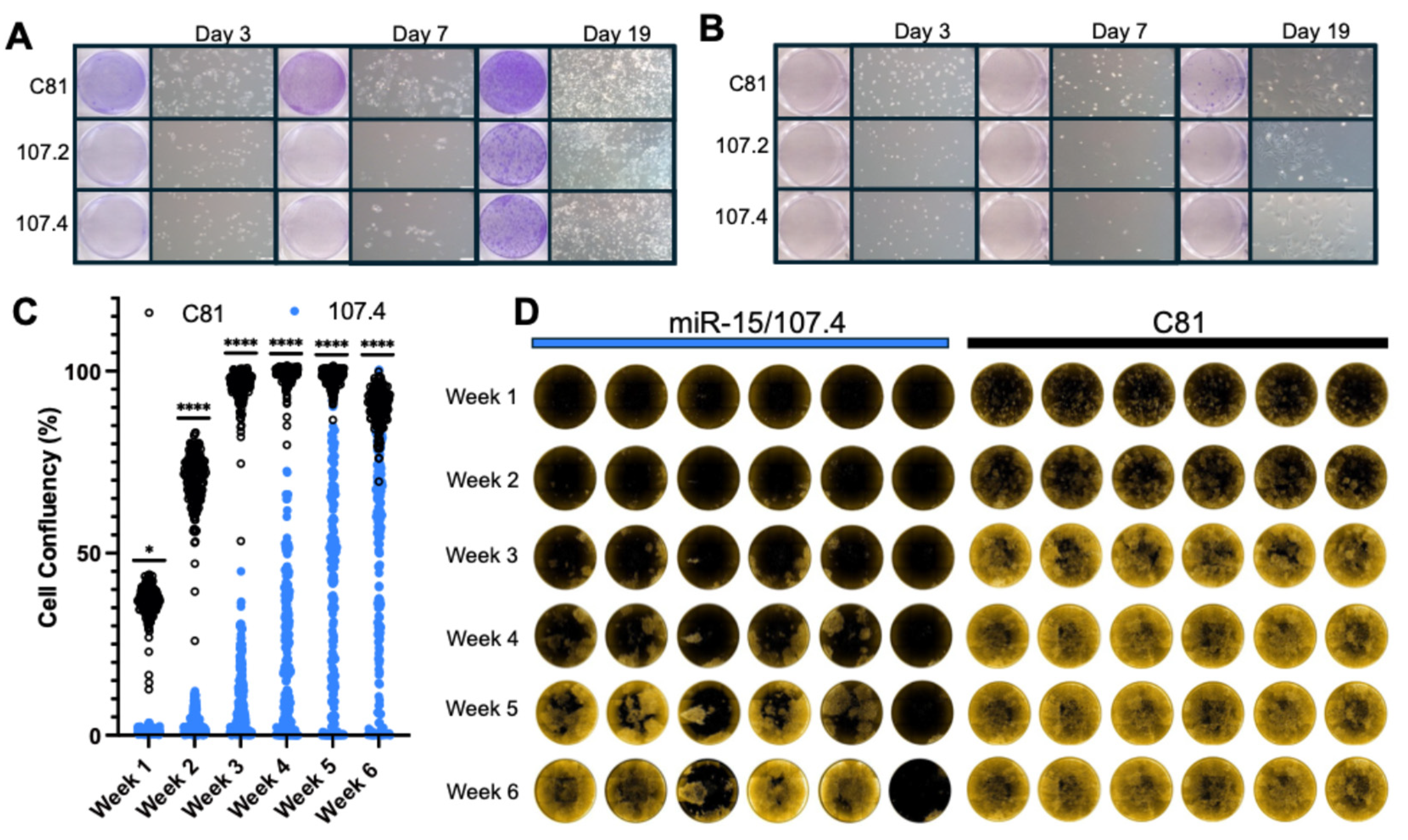
As conmiR-15/107 transfection enhanced sensitivity of PC9 cells to osimertinib and H358 to sotorasib, we next assessed the impact of these constructs on the survival of DTPs resulting from extended drug treatment. PC9 cells are an established model of drug tolerance [15,18,36], and we recently showed that H358 cells also exhibit transcriptional features consistent with this state [Davis]. PC9 cells were transfected with conmiR-15/107.2, conmiR-15/107.4 or control mimics (2 nM) and then treated with osimertinib (2 µM) or DMSO. Cell survival was monitored over 19 days, and it was found that the constructs greatly reduced the number of DTPs in both osimertinib-treated PC9 cells (Figure 4A) and sotorasib-treated H358 cells (Figure 4B). The effect was more pronounced in osimertinib-treated PC9 cells, with clear decreases in DTPs surviving combination treatment at all time points when compared with control mimic transfected cells (Figure 4A). In sotorasib-treated H358 cells, there were fewer surviving DTPs, and this fraction was further inhibited by transfection with mimics (Figure 4B).
To further investigate the impact of construct transfection in the context of continued osimertinib pressure, cell growth and viability were tracked for six weeks using a high-content imaging assay. PC9 cells transduced with mCherry were transfected with conmiR-15/107.4 or C81 control mimics (2 nM) and then treated with osimertinib. Monitoring confluency over time revealed that conmiR-15/107.4 significantly reduced the number of DTP cells compared with control-transfected cells at early time points (Figure 4C). The delay in growth was maintained throughout the observation period, although the response in each replicate well was heterogeneous (Figure 4D; additional wells in Figure S2). From the comparison of individual wells, it is clear that despite the transfection method used resulting in >95% transfection efficiency (data not shown), the response to osimertinib of cells in parallel conmiR-15/107.4-transfected wells ranged from complete inhibition of cell survival, through colony formation to full confluency by week 6 (Figure 4D and Figure S2). This is in contrast to the effects of control-transfected cells, for which cell growth in the presence of osimertinib was more uniform, with almost all wells reaching confluency by 3 weeks of continued treatment (Figure 4C-D).
3. Discussion
In recent years, miRNA mimics have emerged as a promising therapeutic strategy due to their ability to regulate multiple oncogenic pathways simultaneously [39]. A notable example is the first-in-human phase 1 clinical trial of a miR-15/107–based mimic in PM, which demonstrated both objective clinical responses and good tolerability [24]. Building on these encouraging clinical findings, the present study extends the evaluation of this therapeutic approach to NSCLC model. Here, we report follow-up preclinical data showing the activity of a synthetic miR-15/107 consensus mimic in NSCLC and explore its potential to be used in combination with existing targeted therapies as an adjuvant strategy to enhance efficacy and delay resistance.
The conmiR-15/107 construct inhibited proliferation in both PC9 and H358 cells in a dose-dependent manner, and its efficacy was comparable to that of miR-15a, a native family member. This suggests that the consensus design effectively preserves the tumour-suppressive properties of the family, though it does not appear to confer substantially greater potency. Previous reports have described stronger activity relative to miR-16 [32], but such discrepancies may reflect differences in comparator choice, cellular context, or overlapping target repertoires. Importantly, the level of growth inhibition observed here is consistent with earlier studies of miR-15/107 mimics in adenocarcinoma models and aligns with the efficacy reported for other therapeutic miRNA mimics, including miR-34a and let-7 [40,41]. Together, these findings indicate that consensus-based constructs can match the performance of natural family members while potentially broadening applicability across tumour types by converging on shared oncogenic effectors.
The introduction of conmiR-15/107 constructs enhanced the activity of multiple targeted therapies in NSCLC models, encompassing both EGFR-mutant and KRAS-mutant cell lines. ConmiR-15/107 constructs increased the effectiveness of EGFR inhibition in PC9 cells, enhancing responses to both gefitinib and osimertinib. This effect may result from complementary inhibition of the ERK/MAPK pathway, whereby EGFR inhibitors attenuate upstream signaling, while conmiR-15/107 is predicted to suppress downstream effectors such as MEK1. These observations are consistent with prior reports. Fanini et al. [27] showed that a miR-16 mimic increased A549 sensitivity to erlotinib by targeting key ERK/MAPK components, including MAPK3 (ERK1), MAP2K1 (MEK1), and CRAF. Multiple previous studies have found that a variety of miRNA mimics targeting EGFR or the EGFR-signalling pathway can improve the efficacy of EGFR-TKIs for example gefitinib [42,43,44]. This sensitisation effect extended to gefitinib resistant cell lines in many cases. More broadly, tumour-suppressive miRNAs such as miR-34a and let-7 have been paired with first- and third-generation EGFR-TKIs, yielding potentiated—and in some studies synergistic—growth inhibition [30,31,45].
In KRASG12C mutant H358 cells, the conmiR-15/107 constructs enhanced responses to the KRASG12C-specific inhibitors sotorasib and adagrasib. This indicates that the constructs may influence downstream pathways in a manner that enhances cellular responsiveness to KRAS blockade. While previous studies have shown that miR-16 can target KRAS [46] and senstise lung adenocarcinoma cells to targeted therapies [27], to our knowledge the effect of miR-15/107 family members on response to KRASG12C inhibitors has not been tested, suggesting that this may represent a novel therapeutic approach. This is supported by other RNA-based strategies targeting KRAS that have entered the clinic. For example, the iExoKRASG12D Phase I trial (NCT03608631) tested engineered exosomes loaded with KRASG12D-specific siRNA in pancreatic cancer patients [47]. The therapy was well tolerated and showed molecular evidence of target engagement, including reduced KRAS signalling and increased CD8+ T-cell infiltration. Similarly, an ASCO-reported Phase I trial evaluated KRASG12D siRNA-loaded exosomes, also demonstrating safety and signs of disease stabilization in advanced cases of Pancreatic ductal adenocarcinoma [48].
There is also growing evidence that combining KRAS inhibition with MAPK pathway targeting can improve therapeutic efficacy [49,50]. Further support for RNA-based combination approaches comes from Xue et al., who showed that siRNA-mediated KRAS knockdown, when paired with a miR-34a mimic delivered via lung-targeting nanoparticles, produced marked tumour growth inhibition in NSCLC mouse models [51]. Additional support for RNA-based strategies targeting KRAS comes from the work of Acunzo et al., who designed artificial miRNAs (amiRs) that selectively target point-mutated KRASG12S and KRASG12D transcripts while sparing the wild-type allele [52]. While their study focused on direct allele-specific silencing rather than pathway modulation, it reinforces the broader principle that engineered miRNA-based therapeutics can be harnessed to overcome the limitations of current targeted therapies in the treatment of KRAS-mutant NSCLC.
The addition of conmiR-15/107 constructs significantly suppressed DTP outgrowth under osimertinib pressure, a finding observed in both the 19-day assay and corroborated by 6-week live-cell imaging. Multiple factors may contribute to the early suppression of DTP emergence. Among them, the targeting of downstream effectors of the EGFR pathway—such as MEK1—by the miRNA constructs likely reinforces the blockade achieved by EGFR inhibitors. Despite the encouraging early effects, DTPs eventually re-emerged, reflecting the short-lived activity of miRNA mimics [53]. Their efficacy is confined to the immediate post-transfection window, as dilution during cell division quickly reduces activity, limiting sustained suppression. The re-emergence of DTP populations, and the variability in their delay, were consistently observed. This variability reflects a key limitation of miRNA-based therapy—one shared with conventional targeted treatments—namely the inherent heterogeneity of DTPs [54,55]. Within otherwise sensitive populations, rare cells express higher levels of proteins that confer tolerance, influenced by stochastic fluctuations or epigenetic variability [56,57]. The abundance and state of these pre-DTP subpopulations at the time of drug exposure critically shape both the initial response and timing of regrowth.
Despite limited evidence on the role of the miR-15/107 family in DTPs, several other miRNAs have been directly implicated in persister cell biology. In EGFR-mutant NSCLC models, miR-147b was upregulated in osimertinib-tolerant cells and promotes a reversible tolerant state through TCA cycle disruption and pseudohypoxia [58], while miR-21-5p/3p drove tolerance by repressing ADSL and rewiring purine metabolism [59]; inhibition of either pathway restored drug sensitivity. Broader screening approaches also support a central role for miRNAs in tolerance. In a large-scale analysis of PC9-derived DTP cells, multiple miRNAs were found to be downregulated, among them miR-371-3p, which normally acts to suppress persister formation by targeting PRDX6 and reducing redox buffering [60]. The loss of tumour-suppressive miRNAs in DTPs highlights how changes in the miRNA landscape actively shape tolerance phenotypes.
The miR-15/107 family remains a promising therapeutic axis, yet our findings underscore that durable efficacy will require strategies capable of overcoming both delivery challenges and the heterogeneity of persister populations. Novel platforms such as Selective Organ Targeting (SORT) lipid nanoparticles and tumour-targeted minicells, exemplified by the TARGOmiR trial, offer potential solutions [24,61,62]. SORT LNPs incorporate charge-modifying components that enable organ-selective distribution, and encouragingly, preclinical studies have demonstrated their ability to deliver functional small RNAs, including siRNAs, to the lung [63,64]. However, these systems remain untested in humans, and important limitations persist, including lack of tumour specificity, inefficient endosomal escape [65], and the risk of systemic toxicity [23,66]. To try and address these concerns, chemically modified miRNAs combined with targeted ligand conjugation [67] and endosomal escape agents have been developed [68]. For example the DUPA-nigericin-miR-34a have demonstrated selective uptake by PSMA-positive cells and a corresponding decrease in proliferation in vitro and in vivo [68].
To move toward clinical application, future studies will need to evaluate the activity of conmiR-15/107 in vivo, where tumour complexity, immune interactions, and delivery barriers will determine efficacy. Ultimately, rational combinations with targeted therapies and adaptive approaches that address DTP heterogeneity will be required to translate these encouraging findings into durable clinical benefit. In conclusion, conmiR-15/107 enhances the initial activity of EGFR and KRAS inhibitors and postpones the emergence of persister cells, demonstrating clear potential as an adjuvant strategy in NSCLC. However, the transient activity of miRNA mimics and the heterogeneous nature of persister populations remain significant hurdles. Overcoming these limitations through improved delivery technologies and strategic combination regimens will be critical to advancing consensus-based miRNA therapeutics toward clinical application.
4. Materials and Methods
Cell Culture
A549, BEAS-2B, PC9 and H358 cell lines were obtained from ATCC (American Type Culture Collection). Cells were grown in RPMI 1640 medium with GlutaMax (Thermo Fisher Scientific) supplemented with 10 % fetal bovine serum (Moregate). All the cell lines were regularly tested for Mycoplasma (Mycoalert Mycoplasma Detection Kit, Lonza). The cell line identity was confirmed using STR profiling carried out by Cell Bank Australia (Sydney, Australia) using the Promega PP16HS kit.
Transfections
Reverse transfections were performed in all experiments using miRNA mimics or siRNAs (outlined in Table S1) and Lipofectamine RNAiMax (Thermo Fisher Scientific). All transfections were performed using the manufacturers recommendations for Lipofectamine RNAiMax. The conmiR-15/107 constructs consisted of a consensus mimic as described previously [30]. The mimic of endogenous miRNA miR-15a consisted of the mature miRNA sequence and passenger strand. RRM1 siRNA was employed as a positive control for transfection efficiency, as its successful delivery is known to induce robust cell death [33,34]. C81, a non-targeting siRNA [33,34], was employed as a negative control.
Growth Inhibition assays
To determine the half-maximal inhibitory concentration (IC₅₀) of candidate compounds, serial dilutions were prepared and applied to cells seeded in 96-well plates. Cells were transfected with miRNA mimics at the indicated concentrations, and after 24 hours, treated with the indicated targeted therapy drug under the respective experimental conditions. Cells were then allowed to grow until controls reached confluency, at which point proliferation was assessed using a SYBR Green-based assay (ThermoFisher Scientific) as previously described [33,34]. Non-linear regression analysis was used to determine an estimated IC50 values of the resulting growth curve.
RT-qPCR
To determine the expression of target mRNA, transfections were performed in 6-well plates with miRNA mimics (2 nM). After 24 hours, cells were treated with respective targeted therapy or vehicle control (DMSO). Following incubation (48 hours), RNA was extracted using the PureLink™ RNA Mini Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. RNA concentration and quality was determined by NanoPhotometer N60 (Implen Inc). Isolated RNA (1000 ng) was used to make cDNA. qScript cDNA Synthesis Master Mix (Quantabio) was used according to manufacturer’s instructions. Thermal cycling conditions were as follows: annealing for 5 min at 25 °C; cDNA synthesis for 30 min at 42 °C; and denaturing for 5 min at 85 °C. The resulting cDNA was diluted (10 ng/µL) and RT-qPCR reactions were performed with TB Green® Premix Ex Taq™ II (Tli RNaseH Plus, Takara) using 10 ng cDNA per reaction and forward and reverse primers (4 nM; Table S2). A ROX reference dye (Takara) was included as an internal control. Ubiquitin C (UBC) was used as a reference gene for normalization, with relative expression calculated using the 2-ΔΔCT method [35].
Target Protein expression
To assess target protein expression, cells were transfected with miRNA mimics (2 nM) in 10-cm dishes. After 24 hr, the media was replaced, and following an additional 48 hours, the cells were treated with the respective targeted therapy or vehicle control (DMSO). After a 4-hour incubation, the media was removed and washed with ice cold PBS. Proteins were extracted using 1× RIPA buffer (NaCl 150 mM, NP40 1% v/v, sodium deoxycholate 0.5% v/v, SDS 0.1% v/v, Tris (pH 7.4) 25 mM) supplemented with protease/phosphatase inhibitors (Roche). Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Equal amounts of protein (50 µg) were mixed with 1 × Laemmli buffer and 5% v/v 2-mercaptoethanol (Sigma-Aldrich), then denatured at 98 °C for 5 minutes. Samples were separated on a Mini-PROTEAN TGX 10-well precast gel (4-20% gradient, Bio-Rad) alongside Precision Plus Protein Dual Color Standards (Bio-Rad) and subsequently transferred to a nitrocellulose, membrane (0.2 μm pore size) for antibody staining. The following primary antibodies were used at 1:1000 dilution in blocking buffer (5% trim milk in TBS-Tween [0.1% Tween]): anti-p44/42 MAPK (ERK1/2) (#9102 Cell Signalling Technology) and anti-phospho-p44/42 MAPK (pERK1/2) (#9101 Cell Signalling Technology). The secondary antibody (IRDye® 680RD, Goat anti-Rabbit, LI-COR) was used in a 1:20000 dilution in blocking buffer. Western blots were imaged using the LI-COR ODESSEY CLx imaging system. The software package Image Studio Software Version 6.0 (LI-CoR) was used to analyse the images.
Long-term proliferation assays
To investigate the impact of conmiR mimics on drug tolerance, growth inhibition assays were performed in either 6- or 96-well plates. PC9 or H358 cells were transfected in 6-well plates with miRNA mimics (2 nM) and one day later treated with respective targeted therapy. Drug treatments were renewed every 4 to 5 days, and cells were maintained under these conditions for 3, 7, or 19 days to generate DTPs [36]. At each timepoint, wells were photographed and stained with crystal violet (Sigma-Aldrich) to assess cell proliferation and confluence. Longer-term growth assays were performed to assess the growth of transfected cells over six weeks. PC9 cells stably expressing pSin-mCherry-PuroR [37] were transfected in Greiner µClear glass-bottom 96-well plates. Twenty-four hours post-transfection, cells were treated with 2 µM osimertinib, with drug-containing media refreshed weekly, one day prior to imaging. Plates were imaged weekly using the Opera Phenix High Content Screening System (Perkin Elmer). Brightfield and mCherry fluorescence images were acquired with a 5× air objective (NA 0.16, non-confocal), using the following settings: Brightfield (10 ms exposure, 5% laser power and 90.0 µm focal height) and mCherry (250 ms exposure, 100% laser power, –88.0 µm focal height). Image acquisition and pre-processing were carried out using Harmony Software (v5.1, Revvity), and data were subsequently analysed in Signals Image Artist (v1.0, Revvity).
Statistical Analysis:
Growth inhibition assays and RT-qPCR were an average of three biological replicates. The statistical difference for the IC50 proliferation assays were calculated between the transfection conditions at each concentration of the targeted therapy using a two-tailed, t-test. Statistical differences between RT-qPCR experimental groups and the confluency estimates in long-term proliferation assays were evaluated using an unpaired, non-parametric Mann-Whitney test in GraphPad Prism. A p-value < 0.05 was considered significant in these experiments.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: conmiR-15/107 constructs in combination with targeted therapy efficacy, Figure S2: High-content imaging of NSCLC cells transfected with control (C81) or miR-107.4 constructs under targeted treatment, over a period of 6 weeks; Table S1: siRNA and microRNA mimic sequences; Table S2: Primer sequences used for RT-qPCR; Table S3. Confluency measurements for long-term assays.
Author Contributions
Conceptualization, G.R. and C.D.; methodology, C.C, M.T, W.D., C.D and G.R.; formal analysis, X.X.; investigation, C.C., N.S., M.T and W.D.; writing—original draft preparation, C.C.; writing—review and editing, all authors; supervision, C.D. and G.R.; funding acquisition, G.R. and C.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by funding from Otago Innovation Limited (University of Otago).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Informed Consent Statement
Not applicable.
Conflicts of Interest
GR and NvZ are inventors of an awarded patent related to the consensus sequence microRNA mimics. All other authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Tan, A.C.; Tan, D.S.W. Targeted Therapies for Lung Cancer Patients With Oncogenic Driver Molecular Alterations. J Clin Oncol 2022, 40, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Planchard, D.; Janne, P.A.; Cheng, Y.; Yang, J.C.; Yanagitani, N.; Kim, S.W.; Sugawara, S.; Yu, Y.; Fan, Y.; Geater, S.L.; et al. Osimertinib with or without Chemotherapy in EGFR-Mutated Advanced NSCLC. N Engl J Med 2023, 389, 1935–1948. [Google Scholar] [CrossRef]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRAS(G12C) Mutation. N Engl J Med 2022, 387, 120–131. [Google Scholar] [CrossRef]
- Awad, M.M.; Liu, S.; Rybkin, II; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med 2021, 384, 2382–2393. [Google Scholar] [CrossRef]
- Isermann, T.; Sers, C.; Der, C.J.; Papke, B. KRAS inhibitors: resistance drivers and combinatorial strategies. Trends Cancer 2025, 11, 91–116. [Google Scholar] [CrossRef]
- Pu, Y.; Li, L.; Peng, H.; Liu, L.; Heymann, D.; Robert, C.; Vallette, F.; Shen, S. Drug-tolerant persister cells in cancer: the cutting edges and future directions. Nature Reviews Clinical Oncology 2023, 1–15. [Google Scholar] [CrossRef]
- Russo, M.; Chen, M.; Mariella, E.; Peng, H.; Rehman, S.K.; Sancho, E.; Sogari, A.; Toh, T.S.; Balaban, N.Q.; Batlle, E.; et al. Cancer drug-tolerant persister cells: from biological questions to cli nical opportunities. Nature Reviews Cancer 2024, 24, 694–717. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, M.; Dong, B.; Wang, Y.; Ding, Z.; Shen, S. Drug-tolerant persister cells in cancer: bridging the gaps between bench and bedside. Nat Commun 2025, 16, 10048. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Criscione, S.W.; Martin, M.J.; Oien, D.B.; Gorthi, A.; Miragaia, R.J.; Zhang, J.; Chen, H.; Karl, D.L.; Mendler, K.; Markovets, A.; et al. The landscape of therapeutic vulnerabilities in EGFR inhibitor osimertinib drug tolerant persister cells. NPJ Precis Oncol 2022, 6, 95. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magri, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Russo, M.; Pompei, S.; Sogari, A.; Corigliano, M.; Crisafulli, G.; Puliafito, A.; Lamba, S.; Erriquez, J.; Bertotti, A.; Gherardi, M.; et al. A modified fluctuation-test framework characterizes the population dynamics and mutation rate of colorectal cancer persister cells. Nat Genet 2022, 54, 976–984. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct Target Ther 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.; Lee, S.; Senavirathne, G.; Lai, E.C. microRNAs in action: biogenesis, function and regulation. Nature Reviews Genetics 2023, 24, 816–833. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br J Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: a first-in-man, phase 1, open-label, dose-escalation study. The Lancet Oncology 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Finnerty, J.R.; Wang, W.X.; Hebert, S.S.; Wilfred, B.R.; Mao, G.; Nelson, P.T. The miR-15/107 group of microRNA genes: evolutionary biology, cellular functions, and roles in human diseases. J Mol Biol 2010, 402, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Turco, C.; Donzelli, S.; Fontemaggi, G. miR-15/107 microRNA Gene Group: Characteristics and Functional Implications in Cancer. Front Cell Dev Biol 2020, 8, 427. [Google Scholar] [CrossRef]
- Fanini, F.; Bandini, E.; Plousiou, M.; Carloni, S.; Wise, P.; Neviani, P.; Murtadha, M.; Foca, F.; Fabbri, F.; Vannini, I.; et al. MicroRNA-16 Restores Sensitivity to Tyrosine Kinase Inhibitors and Outperforms MEK Inhibitors in KRAS-Mutated Non-Small Cell Lung Cancer. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Chen, T.; Xiao, Q.; Wang, X.; Wang, Z.; Hu, J.; Zhang, Z.; Gong, Z.; Chen, S. miR-16 regulates proliferation and invasion of lung cancer cells via the ERK/MAPK signaling pathway by targeted inhibition of MAPK kinase 1 (MEK1). J Int Med Res 2019, 47, 5194–5204. [Google Scholar] [CrossRef] [PubMed]
- Campos-Parra, A.D.; Sánchez-Marín, D.; Acevedo-Sánchez, V. MicroRNAs as Sensitizers of Tyrosine Kinase Inhibitor Resistance in Cancer: Small Molecule Partnerships. Pharmaceuticals (Basel) 2025, 18, 492. [Google Scholar] [CrossRef]
- Zhao, J.; Kelnar, K.; Bader, A.G. In-depth analysis shows synergy between erlotinib and miR-34a. PLoS One 2014, 9, e89105. [Google Scholar] [CrossRef]
- Zhao, J.; Guerrero, A.; Kelnar, K.; Peltier, H.J.; Bader, A.G. Synergy between next generation EGFR tyrosine kinase inhibitors and miR-34a in the inhibition of non-small cell lung cancer. Lung Cancer 2017, 108, 96–102. [Google Scholar] [CrossRef]
- Reid, G.; Williams, M.; Cheng, Y.; Sarun, K.; Winata, P.; Kirschner, M.; Mugridge, N.; Weiss, J.; Molloy, M.; Brahmbhatt, H.; et al. Therapeutic potential of synthetic microRNA mimics based on the miR-15/107 consensus sequence. Cancer Gene Therapy 2025, 32, 486–496. [Google Scholar] [CrossRef]
- Reid, G.; Wallant, N.C.; Patel, R.; Antonic, A.; Saxon-Aliifaalogo, F.; Cao, H.; Webster, G.; Watson, J.D. Potent subunit-specific effects on cell growth and drug sensitivity from optimised siRNA-mediated silencing of ribonucleotide reductase. J RNAi Gene Silencing 2009, 5, 321–330. [Google Scholar] [PubMed]
- Patel, R.; t’Wallant, N.C.; Herbert, M.H.; White, D.; Murison, J.G.; Reid, G. The Potency of siRNA-Mediated Growth Inhibition Following Silencing of Essential Genes Is Dependent on siRNA Design and Varies With Target Sequence. Oligonucleotides 2009, 19, 317–328. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Maynard, A.; McCoach, C.E.; Rotow, J.K.; Harris, L.; Haderk, F.; Kerr, D.L.; Yu, E.A.; Schenk, E.L.; Tan, W.; Zee, A.; et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell 2020, 182, 1232–1251.e22. [Google Scholar] [CrossRef]
- Davis, W.J.H. The dark matter of drug tolerance: Long non-coding RNAs in drug tolerance in lung adenocarcinoma; University of Otago, 2025. [Google Scholar]
- Quann, K.; Jing, Y.; Rigoutsos, I. Post-transcriptional regulation of BRCA1 through its coding sequence by the miR-15/107 group of miRNAs. Front Genet 2015, 6, 242. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Liu, X.M.; Zhang, C.Y.; Zhou, J.B.; Shao, Y.; Liang, C.; Wang, H.M.; Hua, Z.Y.; Lu, S.D.; Ma, Z.L. MicroRNA-34a/EGFR axis plays pivotal roles in lung tumorigenesis. Oncogenesis 2017, 6, e372. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Zheng, Y.; Jiang, P.; Liu, R.; Liu, X.; Chu, Y. MicroRNA-7 inhibits the growth of human non-small cell lung cancer A549 cells through targeting BCL-2. Int J Biol Sci 2011, 7, 805–814. [Google Scholar] [CrossRef]
- Zhen, Q.; Liu, J.; Gao, L.; Liu, J.; Wang, R.; Chu, W.; Zhang, Y.; Tan, G.; Zhao, X.; Lv, B. MicroRNA-200a Targets EGFR and c-Met to Inhibit Migration, Invasion, and Gefitinib Resistance in Non-Small Cell Lung Cancer. Cytogenet Genome Res 2015, 146, 1–8. [Google Scholar] [CrossRef]
- Liu, L.; Shao, X.; Gao, W.; Zhang, Z.; Liu, P.; Wang, R.; Huang, P.; Yin, Y.; Shu, Y. MicroRNA-133b inhibits the growth of non-small-cell lung cancer by targeting the epidermal growth factor receptor. 2012, 279, 3800–3812. [Google Scholar] [CrossRef]
- Gao, Y.; Fan, X.; Li, W.; Ping, W.; Deng, Y.; Fu, X. miR-138-5p reverses gefitinib resistance in non-small cell lung cancer cells via negatively regulating G protein-coupled receptor 124. Biochem Biophys Res Commun 2014, 446, 179–186. [Google Scholar] [CrossRef]
- Stahlhut, C.; Slack, F.J. Combinatorial Action of MicroRNAs let-7 and miR-34 Effectively Synergizes with Erlotinib to Suppress Non-small Cell Lung Cancer Cell Proliferation. Cell Cycle 2015, 14, 2171–2180. [Google Scholar] [CrossRef]
- You, C.; Liang, H.; Sun, W.; Li, J.; Liu, Y.; Fan, Q.; Zhang, H.; Yue, X.; Li, J.; Chen, X.; et al. Deregulation of the miR-16-KRAS axis promotes colorectal cancer. Sci Rep 2016, 6, 37459. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Smaglo, B.G.; Mahadevan, K.K.; Kirtley, M.L.; McAndrews, K.M.; Mendt, M.; Yang, S.; Maldonado, A.S.; Sugimoto, H.; Salvatierra, M.E.; et al. KRAS (G12D) -Specific Targeting with Engineered Exosomes Reprograms the Immune Microenvironment to Enable Efficacy of Immune Checkpoint Therapy in PDAC Patients. medRxiv 2025. [Google Scholar] [CrossRef]
- Surana, R.; LeBleu, V.S.; Lee, J.J.; Smaglo, B.G.; Zhao, D.; Lee, M.S.; Wolff, R.A.; Overman, M.J.; Mendt, M.C.; McAndrews, K.M.; et al. Phase I study of mesenchymal stem cell (MSC)-derived exosomes with KRASG12D siRNA in patients with metastatic pancreatic cancer harboring a KRASG12D mutation. Journal of Clinical Oncology 2022, 40, TPS633. [Google Scholar] [CrossRef]
- Yaeger, R.; Solit, D.B. Overcoming Adaptive Resistance to KRAS Inhibitors Through Vertical Pathway Targeting. Clin Cancer Res 2020, 26, 1538–1540. [Google Scholar] [CrossRef]
- Palma, G.; Khurshid, F.; Lu, K.; Woodward, B.; Husain, H. Selective KRAS G12C inhibitors in non-small cell lung cancer: chemistry, concurrent pathway alterations, and clinical outcomes. npj Precision Oncology 2021, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Dahlman, J.E.; Tammela, T.; Khan, O.F.; Sood, S.; Dave, A.; Cai, W.; Chirino, L.M.; Yang, G.R.; Bronson, R.; et al. Small RNA combination therapy for lung cancer. Proc Natl Acad Sci U S A 2014, 111, E3553–E3561. [Google Scholar] [CrossRef] [PubMed]
- Acunzo, M.; Romano, G.; Nigita, G.; Veneziano, D.; Fattore, L.; Laganà, A.; Zanesi, N.; Fadda, P.; Fassan, M.; Rizzotto, L.; et al. Selective targeting of point-mutated KRAS through artificial microRNAs. Proceedings of the National Academy of Sciences 2017, 114, E4203–E4212. [Google Scholar] [CrossRef]
- Jin, H.Y.; Gonzalez-Martin, A.; Miletic, A.V.; Lai, M.; Knight, S.; Sabouri-Ghomi, M.; Head, S.R.; Macauley, M.S.; Rickert, R.C.; Xiao, C. Transfection of microRNA Mimics Should Be Used with Caution. Front Genet 2015, 6, 340. [Google Scholar] [CrossRef]
- Goyal, Y.; Busch, G.T.; Pillai, M.; Li, J.; Boe, R.H.; Grody, E.I.; Chelvanambi, M.; Dardani, I.P.; Emert, B.; Bodkin, N.; et al. Diverse clonal fates emerge upon drug treatment of homogeneous cancer cells. Nature 2023, 1–9. [Google Scholar] [CrossRef]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hutter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef]
- Moghal, N.; Li, Q.; Stewart, E.L.; Navab, R.; Mikubo, M.; D'Arcangelo, E.; Martins-Filho, S.N.; Raghavan, V.; Pham, N.A.; Li, M.; et al. Single-Cell Analysis Reveals Transcriptomic Features of Drug-Tolerant Persisters and Stromal Adaptation in a Patient-Derived EGFR-Mutated Lung Adenocarcinoma Xenograft Model. J Thorac Oncol 2023, 18, 499–515. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.C.; Wells, J.M.; Chow, K.H.; Huang, H.; Yuan, M.; Saxena, T.; Melnick, M.A.; Politi, K.; Asara, J.M.; Costa, D.B.; et al. miR-147b-mediated TCA cycle dysfunction and pseudohypoxia initiate drug tolerance to EGFR inhibitors in lung adenocarcinoma. Nat Metab 2019, 1, 460–474. [Google Scholar] [CrossRef]
- Zhang, W.C.; Skiados, N.; Aftab, F.; Moreno, C.; Silva, L.; Corbilla, P.J.A.; Asara, J.M.; Hata, A.N.; Slack, F.J. MicroRNA-21 guide and passenger strand regulation of adenylosuccinate lyase-mediated purine metabolism promotes transition to an EGFR-TKI-tolerant persister state. Cancer Gene Therapy 2022, 29, 1878–1894. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.; Stephan, J.-P.; Cruz, D.D.; Merchant, M.; Haley, B.; Bourgon, R.; Classon, M.; Settleman, J. Functional screening implicates miR-371-3p and peroxiredoxin 6 in reversible tolerance to cancer drugs. Nature Communications 2016, 7, 12351. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nature Nanotechnology 2020, 15, 313–320. [Google Scholar] [CrossRef]
- Dilliard, S.A.; Cheng, Q.; Siegwart, D.J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proceedings of the National Academy of Sciences 2021, 118, e2109256118. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Sun, Y.; Cheng, Q.; Chatterjee, S.; Traylor, Z.; Johnson, L.T.; Coquelin, M.L.; Wang, J.; Torres, M.J.; Lian, X.; et al. Lung SORT LNPs enable precise homology-directed repair mediated CRISPR/Cas genome correction in cystic fibrosis models. Nat Commun 2023, 14, 7322. [Google Scholar] [CrossRef]
- Vaidya, A.; Moore, S.; Chatterjee, S.; Guerrero, E.; Kim, M.; Farbiak, L.; Dilliard, S.A.; Siegwart, D.J. Expanding RNAi to Kidneys, Lungs, and Spleen via Selective ORgan Targeting (SORT) siRNA Lipid Nanoparticles. Adv Mater 2024, 36, e2313791. [Google Scholar] [CrossRef]
- Dowdy, S.F.; Setten, R.L.; Cui, X.S.; Jadhav, S.G. Delivery of RNA Therapeutics: The Great Endosomal Escape! Nucleic Acid Ther 2022, 32, 361–368. [Google Scholar] [CrossRef]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol 2013, 31, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Abdelaal, A.M.; Sohal, I.S.; Iyer, S.; Sudarshan, K.; Kothandaraman, H.; Lanman, N.A.; Low, P.S.; Kasinski, A.L. A first-in-class fully modified version of miR-34a with outstanding stability, activity, and anti-tumor efficacy. Oncogene 2023, 42, 2985–2999. [Google Scholar] [CrossRef] [PubMed]
- Abdelaal, A.M.; Sohal, I.S.; Iyer, S.G.; Sudarshan, K.; Orellana, E.A.; Ozcan, K.E.; Dos Santos, A.P.; Low, P.S.; Kasinski, A.L. Selective targeting of chemically modified miR-34a to prostate cancer using a small molecule ligand and an endosomal escape agent. Mol Ther Nucleic Acids 2024, 35, 102193. [Google Scholar] [CrossRef]
Figure 1.
Cell growth inhibition by miRNA constructs in lung cancer cell lines. PC9 (A), H358 (B), A549 (C) and BEAS2B (D) cells were transfected with miRNA constructs conmiR-15/107.2, conmiR-15/107.4, or miR-15a. C81 (negative control) and RRM1 (positive control) were included as controls. Cell growth was measured across a range of construct concentrations and expressed as a percentage relative to C81-transfected cells (normalized to 100%). Data shown are representative of three independent biological replicates. Error bars indicate mean ± SD of technical repeats.
Figure 1.
Cell growth inhibition by miRNA constructs in lung cancer cell lines. PC9 (A), H358 (B), A549 (C) and BEAS2B (D) cells were transfected with miRNA constructs conmiR-15/107.2, conmiR-15/107.4, or miR-15a. C81 (negative control) and RRM1 (positive control) were included as controls. Cell growth was measured across a range of construct concentrations and expressed as a percentage relative to C81-transfected cells (normalized to 100%). Data shown are representative of three independent biological replicates. Error bars indicate mean ± SD of technical repeats.
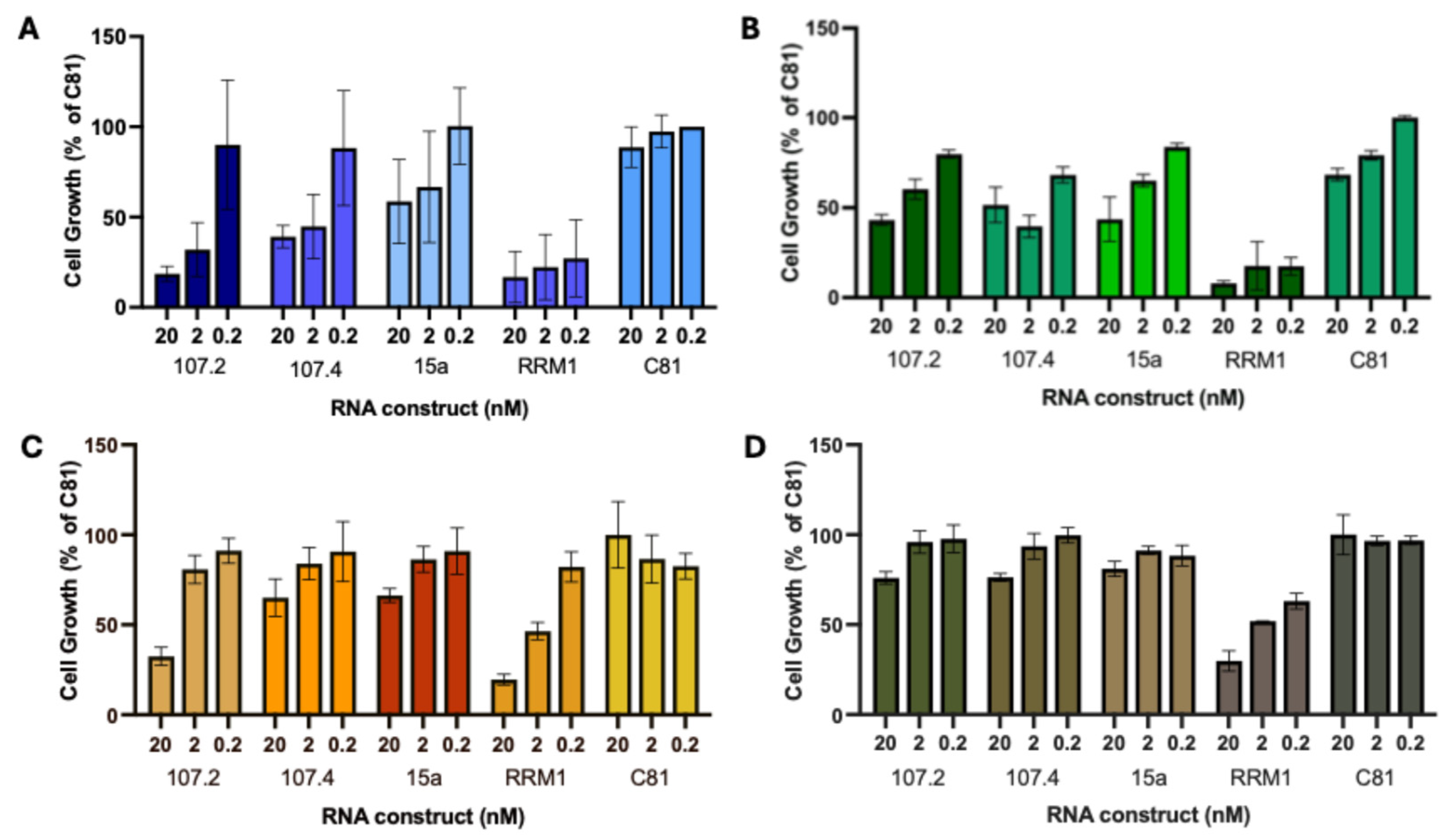
Figure 2.
conmiR-15/107 constructs increase targeted therapy efficacy. Dose-response curve showing cell growth of PC9 (A,B), H358 (C,D) and A549 (E,F) cells transfected with C81 (negative control), conmiR-15/107.2, or conmiR-15/107.4, and treated with increasing concentrations of osimertinib (A,E), gefitinib (B), erlotinib (F), sotorasib (C), and adagrasib (D). Estimated IC50 values (n=3) for sotorasib: 36 ± 4 nM for control versus 14±3 (p<0.05) and 12 ±4 nM (p<0.05) for conmiR-15/107.2 and conmiR-15/107.4, respectively; for adagrasib: 42±1 versus 17±3 (p<0.05) and 12±5 nM (p<0.01). Data shown are representative of three independent biological replicates. Error bars indicate mean ± SD of technical repeats normalised to C81 control. (G-I) Relative growth of mimic transfected cells in PC9 (G), H358 (H) and A549 (I) cells, normalized to C81 controls. Data represent mean ± SD of three biological replicates. In panels A to D, all values significantly different (p<0.05, paired t-test) between C81 control and each consensus mimic, except for the respective highest (A, B) or lowest (C, D) concentration.
Figure 2.
conmiR-15/107 constructs increase targeted therapy efficacy. Dose-response curve showing cell growth of PC9 (A,B), H358 (C,D) and A549 (E,F) cells transfected with C81 (negative control), conmiR-15/107.2, or conmiR-15/107.4, and treated with increasing concentrations of osimertinib (A,E), gefitinib (B), erlotinib (F), sotorasib (C), and adagrasib (D). Estimated IC50 values (n=3) for sotorasib: 36 ± 4 nM for control versus 14±3 (p<0.05) and 12 ±4 nM (p<0.05) for conmiR-15/107.2 and conmiR-15/107.4, respectively; for adagrasib: 42±1 versus 17±3 (p<0.05) and 12±5 nM (p<0.01). Data shown are representative of three independent biological replicates. Error bars indicate mean ± SD of technical repeats normalised to C81 control. (G-I) Relative growth of mimic transfected cells in PC9 (G), H358 (H) and A549 (I) cells, normalized to C81 controls. Data represent mean ± SD of three biological replicates. In panels A to D, all values significantly different (p<0.05, paired t-test) between C81 control and each consensus mimic, except for the respective highest (A, B) or lowest (C, D) concentration.
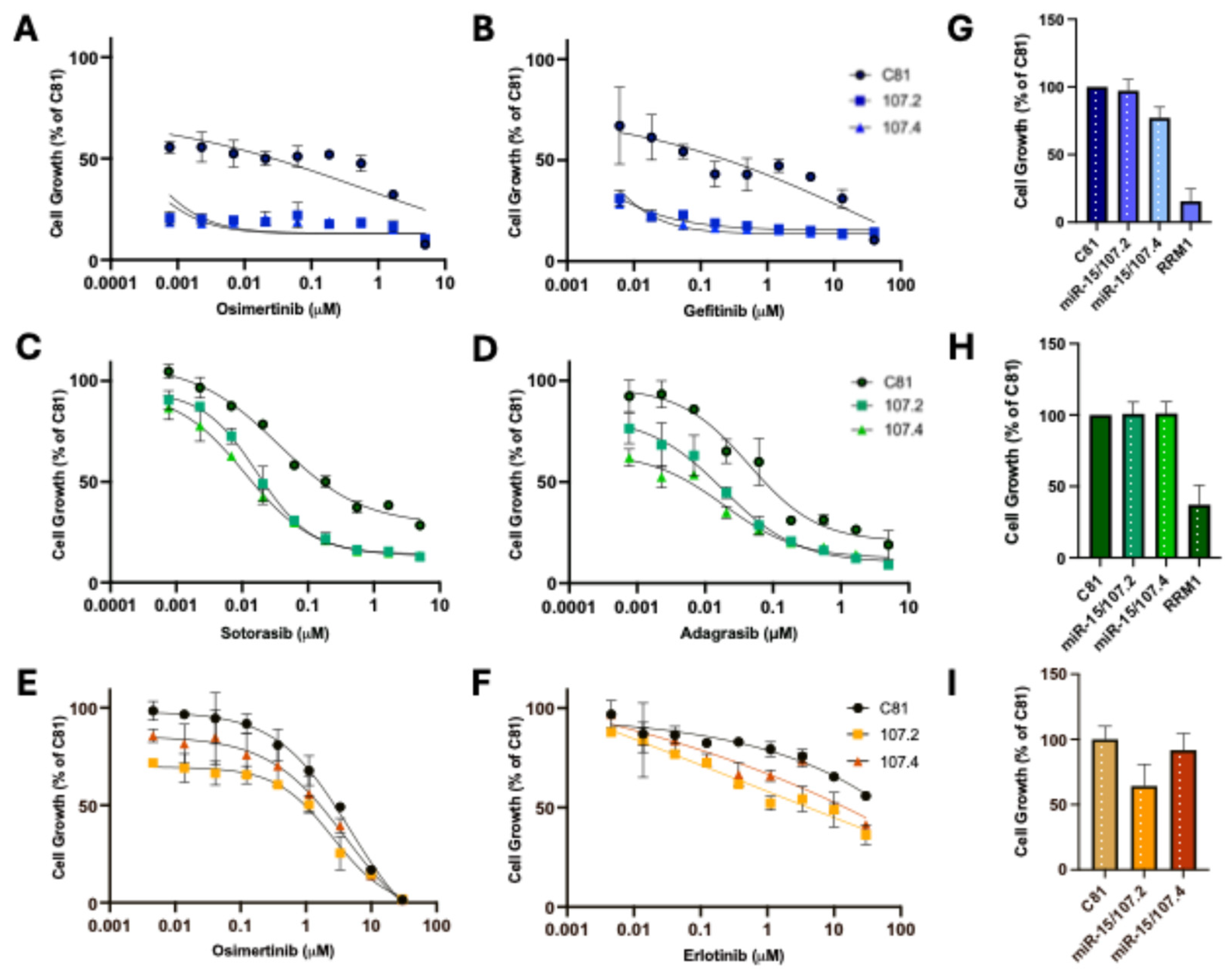
Figure 3.
Target mRNA and protein expression in PC9 and H358 cells following transfection and drug treatment. PC9 (A) and H358 (B) cells were transfected with either C81, conmiR-15/107.2 or conmiR-15/107.4 and then treated with osimertinib (A) or sotorasib (B). Western Blots were performed to determine protein expression of ERK and p-ERK. (B-E) PC9 cells transfected with C81, conmiR-15/107.2 or conmiR-15/107.4 were treated with DMSO, osimertinib (2 μM), or gefitinib (5 μM). RT-qPCR measuring mRNA expression of (B) BCL2, (C) CCND1, (D) MEK1 and (E) BRCA1. Data represent the mean ± SD of three biological replicates (n=3). p < 0.05 (*), p < 0.01 (**), p < 0.001(***), p < 0.0001 (****); ns = not significant.
Figure 3.
Target mRNA and protein expression in PC9 and H358 cells following transfection and drug treatment. PC9 (A) and H358 (B) cells were transfected with either C81, conmiR-15/107.2 or conmiR-15/107.4 and then treated with osimertinib (A) or sotorasib (B). Western Blots were performed to determine protein expression of ERK and p-ERK. (B-E) PC9 cells transfected with C81, conmiR-15/107.2 or conmiR-15/107.4 were treated with DMSO, osimertinib (2 μM), or gefitinib (5 μM). RT-qPCR measuring mRNA expression of (B) BCL2, (C) CCND1, (D) MEK1 and (E) BRCA1. Data represent the mean ± SD of three biological replicates (n=3). p < 0.05 (*), p < 0.01 (**), p < 0.001(***), p < 0.0001 (****); ns = not significant.
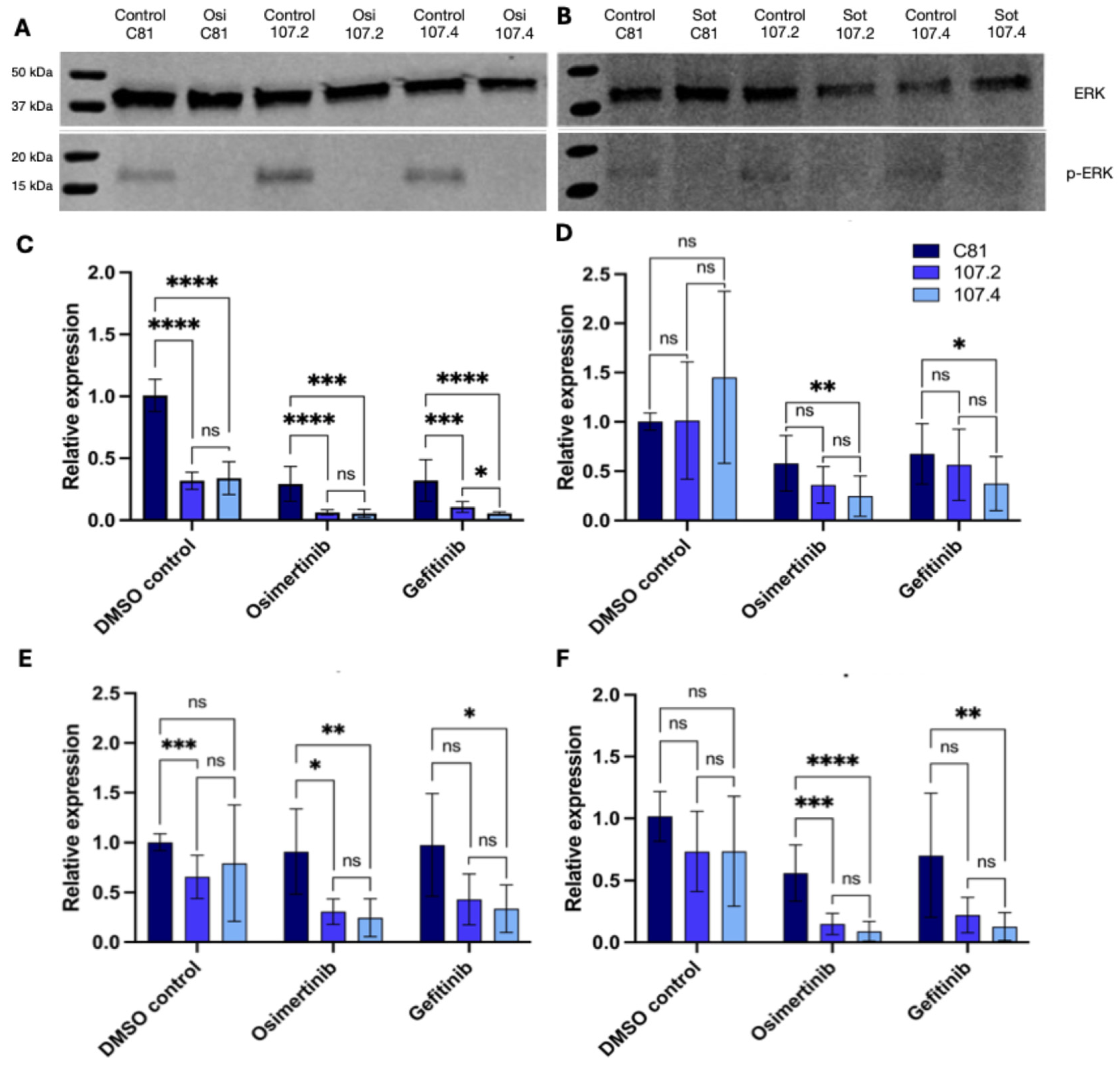
Figure 4.
Effect of consensus mimics on DTP survival following long-term drug treatment. High-content imaging and drug-tolerant persister assays of NSCLC cells transfected with control (C81) or miR-107.4 constructs under targeted treatment. Crystal violet staining and phase-contrast images of PC9 (A) and H358 (B) cells at day 3, day 7, and day 19 of treatment. Left: scanned images of 6-well plates stained with crystal violet, showing osimertinib (A) or sotorasib (B) treated cells transfected with C81, conmiR-107.2, or conmiR-107.4 constructs. Right: corresponding phase-contrast microscopy images (200 µm scale) depicting representative cell growth of transfected cells. The experiment was independently repeated three times with consistent results (n = 3). (C) Dot plot showing estimated cell confluency (%) per well across multiple time points, derived from Phenix imaging software. Data represent three plates (180 wells) per condition, with Mann-Whitney tests performed at each time point between C81 and 107.4-transfected conditions. Significance: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****). (D) Representative wells from three independent plates per condition are shown. Each row corresponds to a different time point, with images taken weekly to monitor cell growth and persistence. Images were acquired using the Phenix high-content imaging system (see also Figure S2 for images from additional wells and Table S3 for all confluency data).
Figure 4.
Effect of consensus mimics on DTP survival following long-term drug treatment. High-content imaging and drug-tolerant persister assays of NSCLC cells transfected with control (C81) or miR-107.4 constructs under targeted treatment. Crystal violet staining and phase-contrast images of PC9 (A) and H358 (B) cells at day 3, day 7, and day 19 of treatment. Left: scanned images of 6-well plates stained with crystal violet, showing osimertinib (A) or sotorasib (B) treated cells transfected with C81, conmiR-107.2, or conmiR-107.4 constructs. Right: corresponding phase-contrast microscopy images (200 µm scale) depicting representative cell growth of transfected cells. The experiment was independently repeated three times with consistent results (n = 3). (C) Dot plot showing estimated cell confluency (%) per well across multiple time points, derived from Phenix imaging software. Data represent three plates (180 wells) per condition, with Mann-Whitney tests performed at each time point between C81 and 107.4-transfected conditions. Significance: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****). (D) Representative wells from three independent plates per condition are shown. Each row corresponds to a different time point, with images taken weekly to monitor cell growth and persistence. Images were acquired using the Phenix high-content imaging system (see also Figure S2 for images from additional wells and Table S3 for all confluency data).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



