Submitted:
26 January 2026
Posted:
27 January 2026
You are already at the latest version
Abstract
The potential of water-soluble membrane proteins (wsMPs) has not been realized fully. The wsMPs have nearly identical functions as to their membrane-bound counterparts. In this article we improve our methodology to create water-soluble receptors to alter cellular fate. Frizzled receptors are part of the Wnt signaling pathway. Wnt-activated Frizzled receptors activate transcription factor β-catenin that affect cell growth, proliferation and epigenetic phenotypes that are well established in cellular models. Treating human embryonic kidney cells (HEK 293) with wsFrizzled receptors changes the cells fate in a fashion similar to that of chemically (CHIR99021) activated Wnt pathway. We present three examples where wsMPs are functional in dictating cellular fate, both in vitro and in vivo. We call this method in vivo deployment of recombinant viable IMPs: iDRIVE. Future work will employ wsMPs to differentiate stem cells in the production of research and clinically relevant organoids.
Keywords:
water-soluble membrane proteins
; regeneration
; stem cell
; differentiation
1. Introduction
Integral membrane proteins (IMPs) constitute 20-30% of all open reading frames (ORFs) in the genome of living organisms[1]. Despite their relative abundance and high relevance for cellular function, they remain a complex subject in research[2]. In 1935, Danielli and Harvey[3], was the first published model, based on surface tension studies, that suggested cell membranes consisted of a lipid bilayer with protein layers on both the inner and outer surfaces. These authors were the first to acknowledge the presence of proteins and their role in cellular membranes. In 1978, Glycophorin was the first integral membrane protein to be characterized by determining its primary sequence [4]. The study of membrane proteins, naturally of low abundance, is often carried out using recombinant systems [5]. Solutions to overcome typical challenges like selecting the correct host, protein purification, detergent selection for purification and maintenance of function, yields, etc. are the result of trial and error [6].
More recently, protein engineering tools have been applied to solve the overproduction of membrane proteins[7]. Making water-soluble membrane proteins is a new approach achieved by two separate groups in the last 10 years [8,9]. In 2018, Zhang and colleagues [9], published the QTY model, in which hydrophobic regions of a membrane protein are mutated to favor its interaction with water. In this seminal publication, G-protein coupled receptors (GPCRs) were used as proof-of-concept for the QTY model. The detergent-free QTY variants maintain a stable structure and retain ligand-binding activities [9]. In contrast to the QTY model, a previous expression of integral membrane proteins as water-soluble particles was achieved by using fusion proteins, the model was tested on membrane proteins of different topology and structure[8]. The method employed by Mizrachi and colleagues [8] generated a triple fusion, a soluble protein, fused to the integral membrane protein, and ultimately fused to Apolipoprotein AI (Apo AI), an amphipathic protein that provided support for the hydrophobic domain of the membrane protein, preventing destabilizing interactions with water. This technique is called SIMPLEx (solubilization of integral membrane proteins with high levels of expression) and allows the triple fusion to be expressed in the cytosol of hosting cells. In the initial 2015 report, SIMPLEx was reported to produce membrane proteins with active conformations, due to the employ of EmrE (ethidium multidrug resistance protein E) an E coli transport protein, where affinity for natural ligands was established but not its transport function [8]. More recently, SIMPLEx was demonstrated to produce fully functional membrane proteins [10].
The initial goal of the work that produces wsMPs was to improve the structural characterization of membrane proteins and aid in structure-function studies of these elusive targets. We propose that the potential of wsMPs has not been realized fully. Understanding structure-function relationships might not be always the ultimate goal, specially if wsMPs are produced as functional, and as such can be used to further our understanding of their function. In this present work we have modified the order of the triple fusion protein suggested by SIMPLEx and produced wsMPs that can be added to cells both in vitro and in vivo to alter their fate. We describe a method we named iDRIVE (in vivo deployment of recombinant viable IMPs). The technique, iDRIVE, can produce soluble functional proteins like pore-forming-proteins (PFPs), and GPCRs that can influence and dictate cellular and organismal phenotypic fates. Among other potential uses of iDRIVE, if an assay can be generated for a given IMP, mutations or truncations of proteins can be evaluated. IMPs serve as critical drug targets across various medical fields; however, they have, up to this day, been challenging. IMPs represent large obstacles in drug discovery due to the complexities involved in producing them in functional forms [11]. A significant portion of predicted disease-causal proteins have proven resistant to targeting by traditional modalities. In that sense, IMPs have largely been classified as undruggable targets [12].
2. Materials and Methods
2.1. Materials
Bacterial cells were obtained from New England Biolabs (Ipswich, MA, USA). Mammalian cells HEK 293, and C2C12 were obtained from ATCC (Manassas, VA, USA). Planaria were purchased from Carolina Biologic Supply (Burlington, NC, USA). Plasmids and DNA synthesis were obtained through TWIST Biosciences (San Francisco, CA, USA). Chemicals (CHIR99021, Retinoic Acid) were obtained through Sigma Aldrich (St. Louis, MO, USA). Media for mammalian cells and bacterial cells culture, and general salts and buffers were obtained from Genesee Scientific (El Cajon, CA, USA).
2.2. Planaria Husbandry
Planaria husbandry is continuous care for them under conditions of darkness and fresh water (Instant Ocean salt, 0.5 g/L; Spectrum Brands Inc., Middleton, WI, USA) at room temperature (in our laboratory it corresponds to 21 C). Planaria were fed beef liver once a week. Prior to experiments, Planaria are starved for 7 days. Incisions and cuts are performed with scalpels over a cold pack. Fragments are separated and kept in 6-well plates with 2 mL of Instant Ocean water. Treatments are introduced immediately and water (and treatments) are refreshed every 48 hours. For more details regarding Planaria Husbandry see Dean and Duncan 2020 review [13]. Planaria Frizzled receptor planaria Frizzled4mSx2 (https://www.uniprot.org/uniprotkb/A0A159X4Q5/entry) was used in regeneration experiments.
2.3. Core Facilities
Core facilities at Brigham Young University (Provo, Utah) provided Electron Microscopy data (https://microscopy.byu.edu/home). Core facilities at the University of Utah (https://cores.utah.edu/) (Salt Lake City, Utah) provided epigenetics analysis.
2.4. HEK 293 Cell Culture and Spheroids
Tissue culture protocols were the classical methods using DMEM and 10% BSA. Regarding spheroid formation we followed Iuchi and colleagues protocols from their publication in 2019 [14].
2.5. Protein Expression and Purification
Our protocols for protein expression and purification have been published previously [8]. Briefly, we employed Terrific broth as medium for cells to grow at 37 C. IPTG induction commenced when OD600 was 0.8-1. Temperature was then lowered to 16 C for a period of 16-18 hours. All our protein constructs contained a 6xHis tag and were purified using NiNTA [8]. For initial protein expression anti-GFP antibody (B-2), 1:1,000 (Santa Cruz Biotechnologies, California, United States of America), and anti-6xHis monoclonal antibody (MA1-135; Invitrogen, Carlsbad, CA, USA) was employed at a dilution of 1:20,000. Final steps toward developing the image was carried out using LI-COR system (Lincoln, NE, USA).
2.6. Confocal Microscopy
E. coli expressing proteins with N-terminally fused GFP were harvested and diluted 1:100 in Luria-Bertani medium. Poly-lysine (Sigma-Aldrich)-coated slide glass was used to mount the cells. A cover glass was placed over the cells and sealed in place with clear nail polish. Cells were imaged within 1 h of their preparation with a Zeiss LSM710 confocal microscope equipped with a × 100 oil immersion objective.
2.7. Negative Staining
Freshly purified OspA-TATAx3 was prepared at different concentrations (0.5, 0.25, 0.1 and 0.05 mg ml−1) for negative staining by applying a 5-μl protein drop to a carbon-coated grid (300-mesh copper grid) for 2 min and blotting with filter paper to remove excess solution. A second solution of 1.5% uranyl acetate was immediately applied for another 2 min. Dried grids were examined using a FEI Tecnai 12 Spirit Twin electron microscope. Twenty fields for each sample concentration were randomly photographed at different magnification levels and later analyzed with ImageJ software.
2.8. Biological SAXS
Biological SAXS data were collected at the Cornell High Energy Synchrotron Source (CHESS) G1 station in Ithaca, New York. Protein samples of ΔspMBP-EmrE-Apo AI* were exposed with a 250 × 250 μm beam of 9.968 keV X-ray. Sample preparation included centrifugation at 30,000g for 30 min and filtration to remove any aggregates. Samples (30 μl) were loaded and oscillated in the beam using an automated system with a plastic chip-based sample cell (2-mm path) and polystyrene X-ray transparent windows. The sample cell and X-ray flight path were placed under vacuum to reduce background scattering. Scattering patterns were captured on a Pilatus 100K-S detector (Dectris, Baden, Switzerland) at 1,504-mm distance. The exposure time was 5 s for each image and 10 images were recorded for each sample. All mathematical manipulations of the data (azimuthal integration, normalization, averaging and buffer subtraction) as well as error propagation were carried out using RAW software[15]. The range of momentum transfer was calculated to be 0.0068<q=4π sin(θ)/λ<0.28 Å−1, where 2θ is the scattering angle and λ=1.257 Å is the X-ray wavelength. Dimer and tetramer samples were run at a range of concentrations (0.3, 0.6, 1.0, 2.0, 5.0, and 10 mg ml−1) to evaluate for possible concentration effects. Molecular weight estimated from a lysozyme standard (3.5 mg ml−1, 50 mM NaOAc, 50 mM NaCl pH 4.0) agreed with our expectations within error. Radius of gyration (Rg) was calculated using both Guinier approximation and the inverse Fourier transform method as implemented in the GNOM-ATSAS 2.3 package by D. Svergun EMBL-Hamburg. The pair distance distribution function P(r) was calculated using the GNOM program [16]. The maximum dimension of the particle, Dmax, was estimated based on the goodness of the data fit and smoothness of the decaying tail. The GNOM output file for the dimer was used as input to DAMMIF [17] to perform ab initio shape reconstruction without imposing any symmetry. The 20 reconstructed bead models were superimposed and averaged using DAMAVER in the automatic mode. The mean NSD was 0.636±0.047 (n=20), where an NSD value <1 indicates close agreement between different reconstructed models.
2.9. Planar Bilayer
Planar lipid bilayer recordings were performed essentially as described by Zakharian, using painted bilayers across a Teflon aperture and symmetrical salt solutions unless otherwise indicated [18]. Protein tested was the engineered OspA-Apo AI-TATAx3-CPP at concentrations of 10-30 µM.
2.10. Epigenetics Analysis Differential Methylation
Human embryonic kidney (HEK) 293 cells were treated with either the small molecule 8 μM CHIR99021 or 5 μM constitutively active human FRZLD7 receptor. In a 6-well plate (two wells per treatment) HEK cells were treated with CHIR99021, or treated with caFRZLD7, or un-treated HEK cells were run on the Illumina Human MethylationEPIC BeadChip Version 2 array. Respectively, we will refer to these sample groups moving forward as CHEK, FHEK, and UHEK. Using the minfi package in R, beta values were produced for all 936,990 CpG sites for each HEK cell within each sample group; additionally, SWAN normalization was applied to all samples. Total QC-passed probes numbered 934,752 CpG sites. All HEK cells passed sample QC. USEQ analyses require a minimum of three samples per sample group, thus the mean beta-value between both HEK cells of an individual sample group was used for each CpG site to create a third sample that we deemed “Sample Mean.” USEQ analyses were run between CHEK versus FHEK, CHEK versus UHEK, and FHEK versus UHEK while defining our threshold Wilcoxon FDR score as 1. Bioinformatically, the top 100 differentially methylated regions (DMR) were determined for each USEQ analysis by ranking them in order of largest difference in mean beta-values per the DMR. DMRs found via USEQ were then inputted into a Stanford GREAT analysis in BED format to determine gene-region associations and their implicated biological processes.
2.11. ATP Assay
ATPlite ATP Luminescence Assay System (Revvity 6016736, Shelton, CT, USA), is a sensitive method to measure cell viability, proliferation, or cytotoxicity by detecting ATP concentration intracellularly, the cell's energy indicator, using firefly luciferase. The analysis was conducted using manufacturer’s instructions .
2.12. Hypertrophy Assay
C2C12 cells were seeded into a 24-well flask. We used DMEM (Gen Clone MS014S) media with 10% FBS. We had four replicates for the negative control, IGF-1, and GPR56. After cells grew to 90-100% confluency, cells were differentiated to myotubes with a 2% Horse FBS (…) DMEM media for four days. We treated the differentiated C2C12 myotubes with IGF-1 (Sino Biological 10598-HNAE) 100 ng/ml for 72 hours and recombinant GPR56 ng/ml every 24 hours until 72 hours is complete. The cells were then fixed using PFA 4% for 10 mins, incubated with permeabilization buffer for 5 minutes, and incubated at 4 degrees Celsius overnight with primary Anti-Myosin (Developmental Studies MF20) 1:50 dilution. The secondary antibody of Alexa Fluor 488 goat anti-mouse IgG (Invitrogen #A11059) followed at 1:100 dilution in PBS for 1 hour in dark. The cells were then stained with a 2.5 mg/ml DAPI diluted solution in 5 mls of PBS. Before imaging, 3 drops of Fluormount G were added to each well. Imaging was performed using a Revolve ECHO microscope at 10x focus. Each well was split into 4 pictures. We measured each myotube 5 times through its diameter and averaged the measurements to one myotube diameter length. We performed over 150 measurements per image leading to 30 myotubes measured per image. We ended up with a total of 120 myotubes measured per well. We had four replicates each per negative Control, IGF-1 and GPR56 treatments. Statistical analysis was performed for ANOVA (https://astatsa.com/OneWay_Anova_with_TukeyHSD/ ) and the bar graph was created using R studio.
2.13. Statistical Analysis
Statistical comparisons between multiple assays were performed using one-way analysis of variance (ANOVA (https://astatsa.com/OneWay_Anova_with_TukeyHSD/)). Unpaired Student’s t-tests were used for comparisons between two groups. A p-value less than 0.05 was considered statistically significant. Bar graphs were created using R studio.
3. Results and Discussion
3.1. Design of iDRIVE (In Vivo Deployment of Recombinant Viable IMPs)
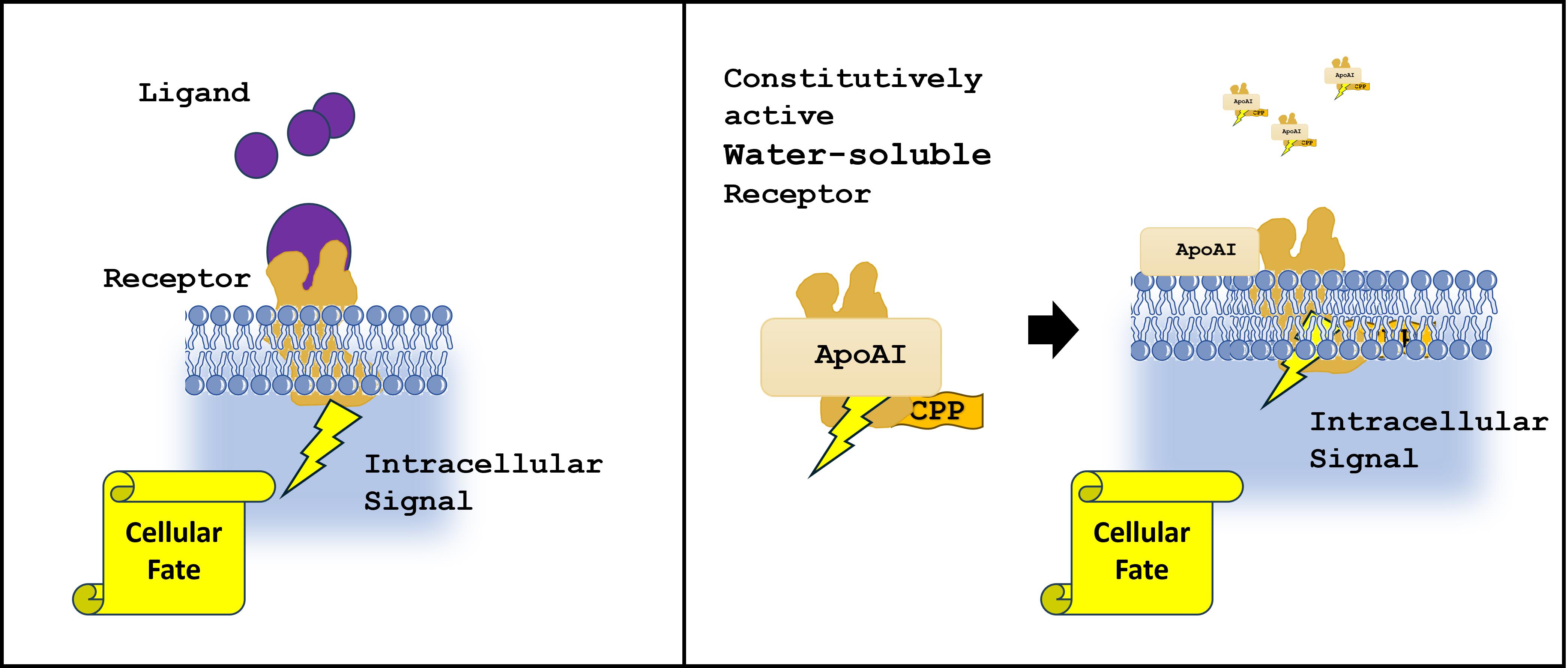
Initially we have hypothesized that wsMPs can be a tool for research in areas like structural biology, and we have demonstrated the power of our first method, SIMPLEx, beyond production of active IMPs, but also functional [8,10,19]. In the present publication, our hypothesis is in part based on working with SIMPLEx and pore-forming proteins (PFPs), where the presence of SIMPLEx PFP seemed to be affecting negatively the growth of mammalian cells in culture (unpublished data, until now). Apolipoprotein AI (Apo AI) is one of the proteins employed in the SIMPLEx particles to solubilize IMPs [8,10]. Apo AI is well documented to bind and solubilize lipids in lipoprotein particles (chylomicrons) and to interact directly with the plasma membrane to extract phospholipids and cholesterol [20]. Our hypothesis then is further modified to include the following statement: reorganizing the triple fusion of proteins from SIMPLEx will result in a wsMP particle that can be further enticed to interact with the plasma membrane by including a cell-penetrating peptide (CPP). The sequence of the triple fusion will be constrained by the presence of an IMP. In 2015, Mizrachi and colleagues [8], demonstrate the need for a decoy protein, a soluble first protein that will divert the ribosome from translocating the IMP to the plasma membrane before it can be folded and protected from interacting with water by the Apo AI protein. Thus, the only possible permutation is as follows: decoy protein (OspA), Apo AI, IMP, and finally CPP.
Figure 1A reminds us the native location of a membrane protein. Water-soluble membrane proteins (wsMPs) were first described as tools for structural and molecular biology of membrane proteins [8] (Figue 1B, top panel). The strategy proposed by those authors used a decoy soluble protein fused to the N-terminus of the membrane protein, while Apo AI was used as a shield protein, fused to the C-terminal of the membrane protein. Currently we propose a re-arrangement of the components of the triple fusion (Figure 1B, bottom panel), the decoy remains as the most N-terminal protein, followed by Apo AI, and the membrane protein fused C-terminal of it. This new triple fusion resulted in a soluble particle that hosted EmrE, the same membrane protein used in the 2015 Nat Comm article [8]. The final modification comes in the form of a CPP [21] that is placed C-terminal of the membrane protein. Figure 1D-F demonstrates qualitatively the solubility of the membrane proteins hosted by SIMPLEx [8] or our current design, iDRIVE.
As demonstrated by Western Blot (Figure 1C) or the confocal images of E coli hosting GFP-Apo AI or SIMPLEx-EmrE or iDRIVE-EmrE. Our new design can be described as an alternative method to create wsMPs. Our hypothesis is supported by these results. We employed EmrE as our IMP since data already exists in the literature, and thus creates a strong argument for our evaluation of the data.
3.2. iDRIVE Interacts with Quiescent and Proliferating Cells
Following the success of creating water-soluble EmrE using iDRIVE we set out to test, qualitatively, if iDRIVE-EmrE can be deployed to cells. We initially tested the protocol using cells that are more vulnerable or actively dividing. With that goal in mind, we tested deploying iDRIVE-EmrE to Planaria that has been cut transversally and are in the process of regeneration [23]. Planaria are flat worms that are capable of regeneration [24]. In planaria, the stem cells are called neoblasts, a population of adult somatic stem cells that supports continuous tissue turnover and whole-body regeneration and includes clonogenic cells that are individually pluripotent [23,25]. Figure 2 illustrates the shape of Brown Planaria (A) and a transversal cut performed to one individual (B). Figure 2C demonstrates the effects of deploying a fusion of GFP-Apo AI (10 µM for 48 hours). Light microscopy identifies the fragment of Planaria that will be then examined with Fluorescent microscopy. Fluorescent microscopy of said fragment seems to indicate that green fluorescence is not observed. Thus, we infer that GFP- Apo AI does not transfer to cells in the fragment of Planaria exposed to the protein for 48 h. On the other hand, Figure 2D, green fluorescence can be detected on a fragment of Planaria subject to 10 µM concentration of iDRIVE-EmrE for 48 h. The fluorescent microscope image seems to indicate that green fluorescence is transferred to the severed fragment of Planaria. Although the initial thought was to provide actively dividing cells with iDRIVE-EmrE which are the cells exposed after the cut, fluorescence can be observed in many different structures of the Planaria fragment. Thus far, iDRIVE seems to apply to both, quiescent and proliferating cells.
3.3. iDRIVE Allows IMPs to Re-Insert into the Plasma Membrane
To further illustrate the ability of iDRIVE to deliver membrane proteins to a living cell we resourced to Planar Lipid Bilayer (PLB) [18]. To establish the proof of concept we first engineered a synthetic pore-forming protein (PFP). Approximately one third of proteins synthesized in the bacterial cytoplasm are destined for extra-cytoplasmic locations. They pass the cytoplasmic membrane using a transport system that involve pore formation, and signal peptides to direct protein export. In bacteria, the Twin-arginine translocation (TAT) pathway, is more exclusive, with only 30 native substrates in the Gram-negative bacterium E. coli, and it is not universally conserved [26]. In 2013, Walther and colleagues [27] suggested a mechanism for the small Tat-A (https://www.uniprot.org/uniprotkb/P69428/entry ) protein to fold during the pore-forming process that enables translocation. Electrostatic “charge zippers” could be responsible for the transition of Tat-A from a single membrane protein to a multimeric unit that creates the pore. Each subunit of Tat-A consists of a transmembrane segment, an amphipathic helix (APH), and a C-terminal populated with charged amino acids (CAA) [28]. The sequence of charges in the transmembrane is complementary to the charges present on the APH, suggesting that the protein can be “zipped up” by as many as seven salt bridges [27]. The length of the resulting hairpin matches the lipid bilayer height; hence a transmembrane pore could self-assemble via intra- and intermolecular interactions [27]. These steric interactions were characterized by molecular dynamics simulations by the authors [27]. Our team subscribed to this hypothesis and followed up using protein engineering to create a synthetic construct that will reconstitute a trimer of Tat-A by fusing the monomeric Tat-A between amino acids 1-48, which excludes the CAA domain. We proceeded to create a triple fusion of truncated Tat-A in tandem (see Figure 3A and B) which we called TATAx3. We prepared the TATAx3 protein to be expressed without any other modifications besides a 6xHis tag in a pET28a plasmid. The expected result is that TATAx3 will express as a membrane protein. If TATAx3 inserts in the membrane it might not harm the cells without further assembly. Figure 3C demonstrates that when bacterial cells express TATAx3 under the direction of IPTG, cell number and proliferation decline until the population cannot overcome the process and disappears. These data indicate that TATAx3 may assemble beyond its single unit and may recreate a pore that without control causes cellular death. PFPs kill cells either by causing irreparable membrane rupture or by triggering regulated cell death pathways once pores disturb ionic and metabolic homeostasis [29]. In the literature [28], an inner diameter of about 40 Å for a Tat-A pore was measured using electron microscopy at low resolution, which may correspond to 12–14 subunits. We purified a water-soluble TATAx3 (SIMPLEx-TATAx3) and examined it using Negative Staining (Electron Microscopy) (Figure 3D). We confirmed that under the conditions of expression and purification the diameter observed an outside diameter of 75±7 nm and an inside diameter of 62±7 nm. These data seem to indicate TATAx3 may assemble as a tetramer or pentamer. Furthermore, we opted for a structural approach. SIMPLEx-TATAx3 was the result of OspA-TATAx3-Apo AI fusion. We replaced OspA, a small globular protein, with maltose binding protein (MBP) to create a bulging decoy protein that may provide more evidence of the assembly. The goal was to create a structural feature that could allow us to determine a definitive number of subunits in the pore forming structure of TATAx3. Figure 3E corresponds to the purified MBP-TATAx3-Apo AI, analyzed using small angle x-ray scattering (SAXS) [22,30]. Figure 3E depicts three different orientations of the tetramer of TATAx3 observed under these conditions.
Following our previous experimentation, we are satisfied that TATAx3 is a tool to further the proof of concept of iDRIVE. TATAx3 is a PFP that is constitutively formed. We continued our hypothesis testing design by employing Planar Lipid Bilayer (PLB) [18]. Figure 4A depicts a schematic of PBL, and the essential elements of the protocol. Once the lipid bilayer is formed, iDRIVE-TATAx3 is added to one of the chambers and changes in current are monitored (pico ampers, pA). Figure 4B seems to indicate that there are measurable changes (pA) over time (min) as iDRIVE enables TATAx3 to enter the bilayer and inserts as a constitutively formed pore, equilibrating ions on both sides of the bilayer. The asterisks correspond to new pores entering the bilayer.
To conclude our efforts for strong proof of concept, we deployed iDRIVE-TATAx3 to HEK 293 cells in culture. Here we replaced OspA with GFP to enable fluorescence monitoring of the experimental outcome. We hypothesize that insertion of TATAx3 from the soluble particle to the lipid membrane could drive changes to the cell’s morphology and perhaps be sufficient to cause cell death [29], as observed for bacterial cells (Figure 3C). Under these conditions, we tested HEK 293 cells at a density of 60% confluency in the presence of 20 µM GFP-Apo AI-CPP and iDRIVE-TATAx3 (GFP-Apo AI-TATAx3-CPP). Figure 4 C-F is a qualitative experiment that indicates that the deployment of GFP-Apo AI does not increase the level of observed fluorescence (Figure 4D), nor change the morphology of cells (Figure 4C). On the other hand, deploying iDRIVE-TATAx3 leaves sufficient fluorescence behind that within 12 hours we can observe the cells under the Confocal Microscope. Furthermore, after exposure of HEK 293 cells to iDRIVE-TATAx3 for 24 hours we observe the cells have changed morphology (rounded). Cell rounding in vitro is a well-recognized early morphological hallmark of apoptosis and therefore commonly correlates with cells undergoing programmed cell death rather than remaining viable [31]. As mammalian cells tend to double in size in a period of 24 hours, we also suggest the data indicates cellular death may occur since confluency does not seem to increase (data not included). Finally, punctae of fluorescent objects are observed in the interior of some of the cells (Figure 4E and F). In mammalian cells, the appearance of cytoplasmic puncta may indicate the formation of condensed structures such as autophagosomes or stress granules [32]. Although by itself cannot be interpreted as a specific cell fate without molecular markers or functional assays it seems to indicate the fluorescent condensates are inside of the cell.
3.4. iDRIVE Allows IMPs to Activate Signal Pathways In Vitro
Having observed iDRIVE succeed in solubilizing IMPs, delivering IMPs to Planaria cells, and delivering a functional IMP to cells in culture, we set out to investigate iDRIVE properties further: wsMPs by means of iDRIVE can activate signal pathways and direct cellular fate.
Wnt signaling is critical for proper development of the embryo and for tissue homeostasis in the adult [33]. Activation of this signaling cascade is initiated by binding of the secreted Wnt to their receptors, Frizzled (FZD) [34]. Wnt signal transduction relies on the activation of β-catenin, normally sequestered by a protein complex to prevent its migration to the nucleus, and thus avoid changes in gene expression [35]. In the laboratory, a small molecule can be used in stem cell differentiation, CHIR99021 [36], to activate the Wnt/β-catenin pathway. CHIR99021 inhibits GSK-3α and GSK-3β at low-nanomolar IC50 values [37]. CHIR99021 blocks GSK-3–dependent phosphorylation of β-catenin, preventing its ubiquitination and proteasomal degradation, this matches the main mechanism of action of Wnt activating FZD receptors [34,35,37].
More recently, in the year 2021, Xu and colleagues [38] created a constitutively active human Frizzled 7 receptor and study its structural properties using Cryo-Electron Microscopy. Constitutive activity is the ability of a GPCR to undergo agonist-independent isomerization from an inactive (R) state to an active (R*) state [39]. We followed their strategy to create a constitutively human FDZ7 receptor that can be used to activate signal transduction and allow for gene expression changes by activating the Wnt/β-catenin pathway in the absence of Wnt. HEK 293 cells naturally express various Frizzled (FZD) receptors (like FZD2, FZD7, FZD1, FZD4, FZD9) and Wnt signaling components, making them a standard model to study Wnt-FZD interactions and activation of the canonical Wnt/β-catenin pathway [40]. Furthermore, we transformed HEK 293 2D cell culture into 3D embryoid bodies (EB) [41]. Using EB is a simple form of 3D cell structures, more complex than 2D cultures [41]. EBs provide an opportunity to monitor the penetration of iDRIVE into the EB to guide cellular fate. This strategy will prepare iDRIVE for testing in vivo.
First, we tested HEK 293 EBs (seen in Figure 5A) and their metabolism by measuring ATP levels after treatments. When energy demand rises, ATP is produced and further hydrolyzed to ADP/AMP, which allosterically stimulates key metabolic enzymes and pathways to ramp up ATP production [42]. Figure 5B and C are the results of ATP levels after treatments. As observed in Figure 5B, neither CHIR99021 or iDRIVE-caFZD7 increased ATP levels. The literature provided an additional candidate to activate metabolism in HEK 293 cells. First, a review on retinoids and mitochondrial gene expression indicated that retinoic acid has been shown to regulate mitochondrial transcription and function, directly connecting retinoic acid (RA) signaling to cellular bioenergetics. Second, HEK 293 cells have functional retinoid processing machinery [43,44]. Lastly, RA signaling is tightly interconnected with the Wnt/β-catenin pathway, with RA shown to modulate Wnt ligand/receptor expression and to either suppress or enhance canonical Wnt signaling in a cell-dependent manner [43,45]. Figure 5C identifies an increase in metabolism (ATP levels) when CHIR99021 or caFDZ7 are combined with RA (5 µM). In a recent study [45], the combination of CHIR99021 and RA was successful in differentiating human embryonic stem cells (hESCs) into primordial germ cells (PGCs) via activating Wnt signaling pathway and metabolism.
The goal of the Epigenetics experiment in analyzing the DNA methylation signatures of HEK 293 cells and said methylation signature changes by treatment used was to determine if efficacy varies between the use of CHIR99021 and FZD7 treatment in cell gene expression differentiation. USEQ analysis between CHEK and UHEK yielded 7,789 DMRs (Wilcoxon FDR≥1); USEQ analysis between FHEK and UHEK yielded 6,933 DMRs (Wilcoxon FDR≥1). Wilcoxon score or –log10 FDR–like metric used for ranking DMRs. When comparing DMRs of both USEQ analyses, 5,124 DMRs were found to be in common. This supports our hypothesis of the effective, alternative use of FZD7 in the differentiation of cells or tissue. Our USEQ analysis between CHEK and FHEK only yielded 1222 DMRs (Wilcoxon FDR≥1). The lack of DMRs between treatment groups indicates epigenetic similarities between both treatment groups’ subsequent human adipose organoid differentiation, thus further supporting our hypothesis. Systematically developmental signaling pathways (Wnt/β-catenin, BMP/SMAD, FGF/ERK, LIF/STAT3, etc.) directly modulate chromatin modifiers and histone marks, altering nucleosome occupancy and gene accessibility [46]. Thus, development and differentiation can be equated in the sense that they both need these signal pathways. Figure 5D shows DMR graph, in which treatments are compared to the untreated EBs. The results seem to identify greater homology between CHIR99021 and caFZD7, while both have epigenetic profiles that are very different than the untreated EBs. Additionally, among the top 100 epigenetic modifications caused by CHIR99021 or caFZD7, 58 DMRs among them are the same. Under these conditions, iDRIVE-caFZD7 represents a tool that in vitro is capable of altering cellular fate in a fashion similar to other methods identified in stem cell research and animal models. Furthermore, treatment of intestinal tissue in vitro and in vivo with CHIR99021 had a protective effect from radiation [47]. Our data supports that caFZD7 delivered via iDRIVE could have a similar effect.
3.5. iDRIVE Allows IMPs to Activate Signal Pathways In Vitro. The Case of Myotube Hypertrophy
Endowed with the above-mentioned results, that iDRIVE can deliver active signaling molecules to the plasma membrane, and alter cellular fate we sought to solidify the evidence by testing one more system in vitro, the hypertrophy of muscle cells (murine C2C12 cells). In classical experiments of myotube hypertrophy, insulin growth factor-1 (IGF-1) is responsible for this phenotype in C2C12 cells in culture [48]. Additionally, adhesion G-protein coupled receptors (aGPCRs) like GPR56 have also been associated with myotube hypertrophy [49]. This family of GPCR contain an autoproteolysis-inducing (GAIN) domain, a unique and essential protein domain found in most aGPCRs that mediates their activation and self-cleavage into two fragments: an N-terminal fragment (NTF) and a C-terminal fragment (CTF) [50]. The GAIN domain includes a tethered agonist sequence called the Stachel, which is crucial for activating the receptor [50]. While many aGPCRs undergo this process, some are activated without cleavage or via other domains, like the SEA domain in GPR110 [51]. It is possible to generate a constitutively active GPR56 to induce myotubule hypertrophy [52]. We proposed then to prepare iDRIVE-caGPR56 and compare it to IGF-1 in an assay to trigger myotube hypertrophy in murine C2C12 cells. C2C12 myoblasts stably transfected with IGF-1 cDNA, or treated with IGF-1, form myotubes that are significantly hypertrophied when compared to the control; authors suggested that IGF-1 is both necessary and sufficient for hypertrophy C2C12 cells [48].
Figure 6A has the structure of an aGPCR, with a large extracellular domain that is typically connected with the surface of a neighboring cell [50]. Figure 6B, corresponds to data measuring the diameter of myotubes in C2C12 cells untreated or treated with IGF-1 or iDRIVE-caGPR56. Both treatments are statistically different than the untreated myotube diameters. More importantly, caGPR56 delivered through iDRIVE seems to be more impactful than IGF-1. The phenotype is the important outcome considering IGF-1 [53] and GPR56 [50] have different signal pathways that in C2C12 cells result in a similar outcome, myotube hypertrophy. In the past years, Dr. Michael Levin’s laboratory (https://drmichaellevin.org/ ) has presented evidence that supports the concept of morphospace, the high-dimensional space of all possible anatomical and morphological configurations that a biological system (e.g., cells, tissues, or organisms) can explore and navigate toward target states by differently activated pathways [54,55]. In our case the target state is hypertrophy, and the means to achieve it can be either IGF-1 or caGPR56. Morphospace is linked to bioelectricity and can be manipulated through connexins and Gap Junctions formation [54,55]. In the literature IGF-1 is related to Gap Junctions [56] but there is no connection between them and GPR56. Gap junctions (connexins) can mediate adhesion independently of channel function via hemichannel docking, but no studies link this to aGPCR expression, signaling, or regulation to them [57,58].
3.6. iDRIVE Allows IMPs to Activate Signal Pathways In Vivo. The Case of Planaria Regeneration
Our final experiment aims to demonstrate that iDRIVE can trigger signal transduction in vivo. We will resource to the Wnt/FZD/β-catenin signaling pathway as a tool to qualify the outcome and the model will be Planaria. Planaria are freshwater flatworms with remarkable regeneration, regrowing a complete body—including brain, gut, and organs—from tiny fragments via pluripotent neoblasts [59].
When a Planaria is cut transversally, two wounds form. These wounds in spite of having similar origin will develop different programs to direct the regeneration of a tail (for the segment containing the head) and a head for the tail segment. In the head segment, the wound will follow Wnt signaling in the neoblasts to regenerate the tail [60]. In the tail, Wnt signaling will be initially active, nevertheless, cells will produce an enzyme called Notum that will inhibit Wnt signaling [60]. Notum directly deacylates Wnt ligands, removing the essential palmitoleate group required for binding to Frizzled receptors, thereby inactivating Wnt proteins and suppressing downstream signaling [61]. Thus, the expression of Notum results in inactivation of Wnt signaling and ultimately the regeneration of a head. Concluding, Wnt ON results in a tail, Wnt OFF results in a head. As an example, recent studies have generated two-tailed Planaria by manipulating the Wnt signaling using RNAi [62]. Two-tailed Planaria can be obtained by altering bioelectric patterns in regenerating Planaria [63] or using RNAi [64]. Success in altering Planaria regeneration has never been achieved using proteins or wsMPs.
Figure 7A shows the important molecules in the Wnt signal pathway and that are relevant to understanding Planaria regeneration. Figure 7B corresponds to the strategy followed and the outcomes proposed upon treatment.
Following husbandry protocols, we prepared Planaria for the experiment. After transversal cuts, tails were separated and exposed to 8 µM iDRIVE-caFZD. The constitutively active FZD was the translation of the mutations performed in the human FZD7[38]. We tested two Planaria FZD receptors (pFrizzled4mSx2, pFrizzled1/2/7mSx2), similar results were observed. Data presented in this manuscript corresponds to pFrizzled4mSx2 and will be referred to as capFZD.
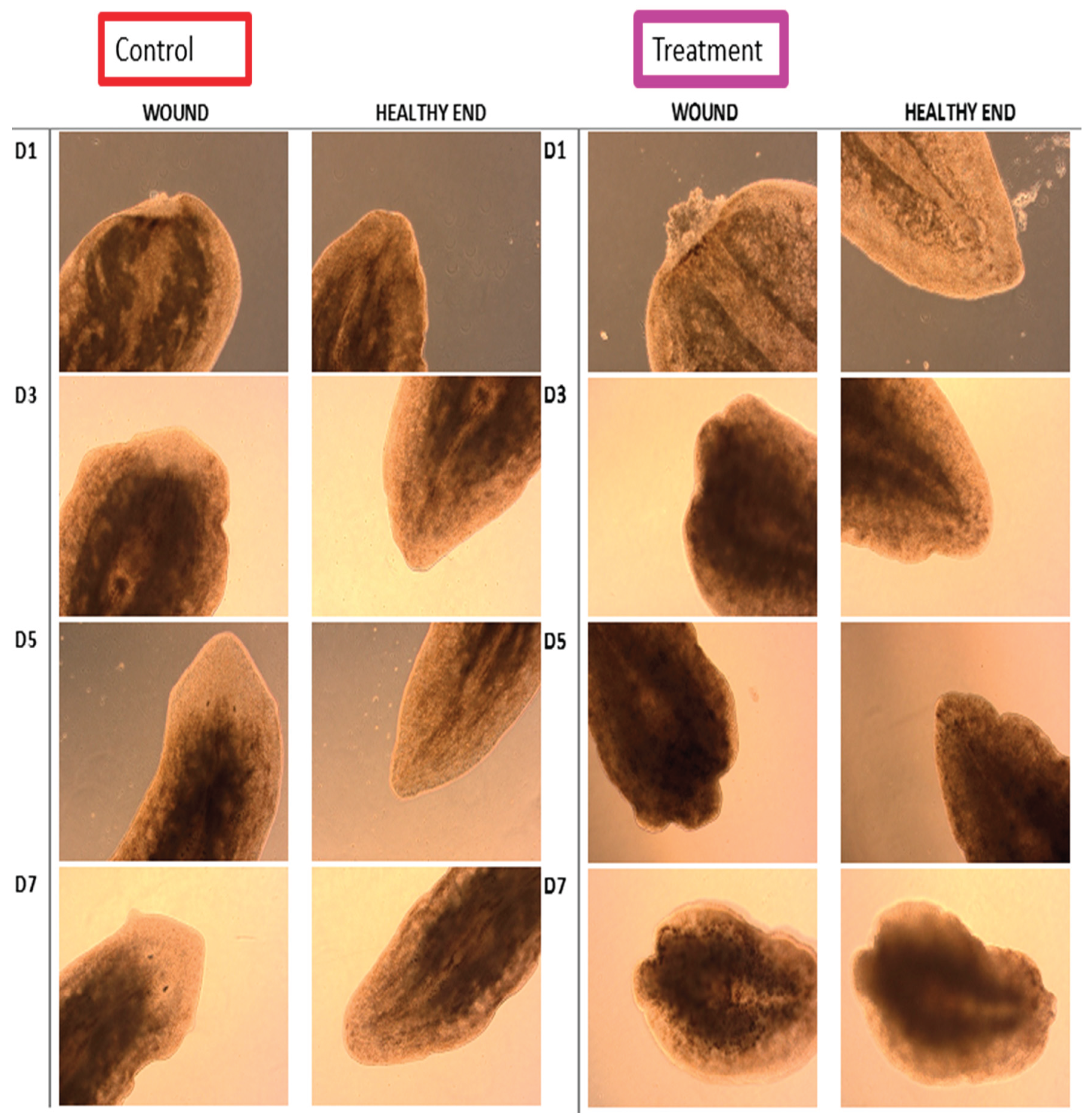
Figure 8 contains visual representation of the changes to tails during the process of regeneration. Control images correspond to Planaria tails untreated. For clarity the images of the wound and the tail were separated. The process of full regeneration is achieved between 5 and 7 days. Our results demonstrate that treated tails (iDRIVE-capFZD) do not regenerate a head, as expected, suggesting that our tools work also in vivo and are a suitable solution for altering cellular fate.
The tool presented here, iDRIVE-capFZD offers new paradigms to the study of regeneration, cell proliferation, and genetic and molecular aspects of the regeneration process. Canonical Wnt signaling is mediated by Wnt–Frizzled (Fz)–LRP5/6 complexes, inhibiting β-catenin degradation and driving TCF/LEF transcription. Non-canonical pathways include Wnt/PCP (Fzd–Dishevelled–RhoA/JNK for polarity/migration) and Wnt/Calcium (Fzd–G proteins–PLC/IP3R–Calcium/NFAT for asymmetric cell division) [65]. The study of constitutively active receptors of the GPCR class has not advanced sufficient to differentiate which pathway (canonical or non-canonical) may be activated [66], presenting with research opportunities. GPCRs form the largest family of membrane protein receptors, encoded by ~800–1,000 genes (~4–5% of the human genome) [67,68]. Approximately 35% of FDA-approved drugs (~500–700 agents) target ~108–134 GPCRs, generating $180B+ annual sales; they underpin β-blockers, opioids, antihistamines, and antipsychotics, among many others [69].
4. Conclusion
With our new strategy for protein engineering of wsMPs we have presented evidence that iDRIVE can solubilize IMPs. We present data that strongly suggests that IMPs are able to interact with the plasma membrane of cells in vitro and in vivo and re-enter the lipid bilayer. We also identify properties of the iDRIVE system that enables active signaling IMPs to actively drive instructions related to their native program. We suggest that wsMPs in the iDRIVE system can be used as tools to replace DNA/RNA manipulations of cells, tissues, or organisms. These manipulations can also replace small molecules or toxic signals in the process of stem cell differentiation. Additionally, we propose that as proteins have a finite life span in the cell, iDRIVE signaling may result in phenotypic or genotypic changes, without the permanent sequalae of genetic manipulations, for example transgenic animal models, or viral infections. Finally, if only ~15% of all human proteins are druggable and >60% of approved targets are membrane proteins, then the vast majority of membrane proteins—on the order of thousands of individual integral membrane proteins—are still undrugged and, in practice, undruggable by current approaches. Providing access to functional IMPs is an increment in solving the undruggable genome.
References
- Wallin, E.; von Heijne, G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms . Protein Sci 1998, 7(4), 1029–38. [Google Scholar] [CrossRef]
- Hedin, L.E.; Illergard, K.; Elofsson, A. An introduction to membrane proteins . J Proteome Res 2011, 10(8), 3324–31. [Google Scholar] [CrossRef]
- Danielli, J.F.; Harvey, E.N. The tension at the surface of mackerel egg oil, with remarks on the nature of the cell surface . Journal of Cellular and Comparative Physiology 1935, 5(4), 483–494. [Google Scholar] [CrossRef]
- Tomita, M.; Furthmayr, H.; Marchesi, V.T. Primary structure of human erythrocyte glycophorin A. Isolation and characterization of peptides and complete amino acid sequence . Biochemistry 1978, 17(22), 4756–70. [Google Scholar] [CrossRef]
- Tate, C.G. Overexpression of mammalian integral membrane proteins for structural studies . FEBS Lett 2001, 504(3), 94–8. [Google Scholar] [CrossRef]
- Hedfalk, K. Further advances in the production of membrane proteins in Pichia pastoris . Bioengineered 2013, 4(6), 363–7. [Google Scholar] [CrossRef]
- Scott, D.J. Stabilizing membrane proteins through protein engineering . Curr Opin Chem Biol 2013, 17(3), 427–35. [Google Scholar] [CrossRef] [PubMed]
- Mizrachi, D. Making water-soluble integral membrane proteins in vivo using an amphipathic protein fusion strategy . Nat Commun 2015, 6, 6826. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. QTY code enables design of detergent-free chemokine receptors that retain ligand-binding activities . Proc Natl Acad Sci U S A 2018, 115(37), E8652–E8659. [Google Scholar] [CrossRef]
- Jaroentomeechai, T. A universal glycoenzyme biosynthesis pipeline that enables efficient cell-free remodeling of glycans . Nat Commun 2022, 13(1), 6325. [Google Scholar] [CrossRef] [PubMed]
- Gulezian, E. Membrane protein production and formulation for drug discovery . Trends Pharmacol Sci 2021, 42(8), 657–674. [Google Scholar] [CrossRef]
- Swenson, C.S. Tackling Undruggable Targets with Designer Peptidomimetics and Synthetic Biologics . Chem Rev 2024, 124(22), 13020–13093. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.R.P.; Duncan, E.M. Laboratory Maintenance and Propagation of Freshwater Planarians . Curr Protoc Microbiol 2020, 59(1), p. e120. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, K. Different morphologies of human embryonic kidney 293T cells in various types of culture dishes . Cytotechnology 2020, 72(1), 131–140. [Google Scholar] [CrossRef]
- Behrens, M.A. Structural analysis of RNA helicases with small-angle X-ray scattering . Methods Enzymol 2012, 511, 191–212. [Google Scholar] [PubMed]
- Svergun, D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria . Journal of Applied Crystallography 1992, 25(4), 495–503. [Google Scholar] [CrossRef]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering . J Appl Crystallogr 2009, 42 Pt 2, 342–346. [Google Scholar] [CrossRef]
- Zakharian, E. Recording of ion channel activity in planar lipid bilayer experiments . Methods Mol Biol 2013, 998, 109–18. [Google Scholar]
- Mizrachi, D. A water-soluble DsbB variant that catalyzes disulfide-bond formation in vivo . Nat Chem Biol 2017, 13(9), 1022–1028. [Google Scholar] [CrossRef]
- Tricerri, M.A. Interaction of apolipoprotein A-I in three different conformations with palmitoyl oleoyl phosphatidylcholine vesicles . J Lipid Res 2002, 43(2), 187–97. [Google Scholar] [CrossRef]
- Davis, H.C.; Posey, N.D.; Tew, G.N. Protein Binding and Release by Polymeric Cell-Penetrating Peptide Mimics . Biomacromolecules 2022, 23(1), 57–66. [Google Scholar] [CrossRef] [PubMed]
- Malaby, A.W. Methods for analysis of size-exclusion chromatography-small-angle X-ray scattering and reconstruction of protein scattering . J Appl Crystallogr 2015, 48 Pt 4, 1102–1113. [Google Scholar] [CrossRef]
- Scalf, S.M.; Wu, Q.; Guo, S. Molecular basis of cell fate plasticity - insights from the privileged cells . Curr Opin Genet Dev 2025. 93, 102354. [Google Scholar] [CrossRef]
- Rink, J.C. Stem cell systems and regeneration in planaria . Dev Genes Evol 2013, 223(1-2), 67–84. [Google Scholar] [CrossRef]
- Wagner, D.E.; Wang, I.E.; Reddien, P.W. Clonogenic neoblasts are pluripotent adult stem cells that underlie planarian regeneration . Science 2011, 332(6031), 811–6. [Google Scholar] [CrossRef]
- Hutcheon, G.W.; Bolhuis, A. The archaeal twin-arginine translocation pathway . Biochem Soc Trans 2003, 31 Pt 3, 686–9. [Google Scholar] [CrossRef]
- Walther, T.H. Folding and self-assembly of the TatA translocation pore based on a charge zipper mechanism . Cell 2013, 152(1-2), 316–26. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, U. The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter . Proc Natl Acad Sci U S A 2005, 102(30), 10482–6. [Google Scholar] [CrossRef] [PubMed]
- Flores-Romero, H.; Ros, U.; Garcia-Saez, A.J. Pore formation in regulated cell death . EMBO J 2020, 39(23), p. e105753. [Google Scholar] [CrossRef] [PubMed]
- Iino, H. Small-angle X-ray scattering analysis reveals the ATP-bound monomeric state of the ATPase domain from the homodimeric MutL endonuclease, a GHKL phosphotransferase superfamily protein . Extremophiles 2015, 19(3), 643–56. [Google Scholar] [CrossRef]
- Doonan, F.; Cotter, T.G. Morphological assessment of apoptosis . Methods 2008, 44(3), 200–4. [Google Scholar] [CrossRef]
- Ho, C.J. Puncta intended: connecting the dots between autophagy and cell stress networks . Autophagy 2021, 17(4), 1028–1033. [Google Scholar] [CrossRef]
- Sonavane, P.R.; Willert, K. Controlling Wnt Signaling Specificity and Implications for Targeting WNTs Pharmacologically . Handb Exp Pharmacol 2021, 269, 3–28. [Google Scholar]
- Martinez-Marin, D. Frizzled receptors: gatekeepers of Wnt signaling in development and disease . Front Cell Dev Biol 2025, 13, 1599355. [Google Scholar] [CrossRef]
- Liu, J. Wnt/beta-catenin signalling: function, biological mechanisms, and therapeutic opportunities . Signal Transduct Target Ther 2022, 7(1), p. 3. [Google Scholar] [CrossRef]
- Ai, Z. CHIR99021 enhances Klf4 Expression through beta-Catenin Signaling and miR-7a Regulation in J1 Mouse Embryonic Stem Cells . PLoS One 2016, 11(3), e0150936. [Google Scholar] [CrossRef]
- Ye, S. Pleiotropy of glycogen synthase kinase-3 inhibition by CHIR99021 promotes self-renewal of embryonic stem cells from refractory mouse strains . PLoS One 2012, 7(4), e35892. [Google Scholar] [CrossRef] [PubMed]
- Xu, L. Cryo-EM structure of constitutively active human Frizzled 7 in complex with heterotrimeric G(s) . Cell Res 2021, 31(12), 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Seifert, R.; Wenzel-Seifert, K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors . Naunyn Schmiedebergs Arch Pharmacol 2002, 366(5), 381–416. [Google Scholar] [CrossRef]
- Voss, J.H. WNT-induced association of Frizzled and LRP6 is not sufficient for the initiation of WNT/beta-catenin signaling . Nat Commun 2025, 16(1), 4848. [Google Scholar] [CrossRef] [PubMed]
- Pettinato, G.; Wen, X.; Zhang, N. Engineering Strategies for the Formation of Embryoid Bodies from Human Pluripotent Stem Cells . Stem Cells Dev 2015, 24(14), 1595–609. [Google Scholar] [CrossRef]
- Meurer, F. Standard Gibbs energy of metabolic reactions: II. Glucose-6-phosphatase reaction and ATP hydrolysis . Biophys Chem 2017, 223, 30–38. [Google Scholar] [CrossRef]
- Brueggemann, L.I.; Sullivan, J.M. HEK293S cells have functional retinoid processing machinery . J Gen Physiol 2002, 119(6), 593–612. [Google Scholar] [CrossRef]
- Moise, A.R. Activation of retinoic acid receptors by dihydroretinoids . Mol Pharmacol 2009, 76(6), 1228–37. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T. CHIR99021 combined with retinoic acid promotes the differentiation of primordial germ cells from human embryonic stem cells . Oncotarget 2017, 8(5), 7814–7826. [Google Scholar] [CrossRef] [PubMed]
- Fagnocchi, L.; Mazzoleni, S.; Zippo, A. Integration of Signaling Pathways with the Epigenetic Machinery in the Maintenance of Stem Cells . In Stem Cells Int; 2016; p. 8652748. [Google Scholar]
- Wang, X. Pharmacologically blocking p53-dependent apoptosis protects intestinal stem cells and mice from radiation . Sci Rep 2015, 5, 8566. [Google Scholar] [CrossRef]
- Semsarian, C. Insulin-like growth factor (IGF-I) induces myotube hypertrophy associated with an increase in anaerobic glycolysis in a clonal skeletal-muscle cell model . Biochem J 1999, 339 Pt 2)(Pt 2, 443–51. [Google Scholar] [CrossRef] [PubMed]
- White, J.P. G protein-coupled receptor 56 regulates mechanical overload-induced muscle hypertrophy . Proc Natl Acad Sci U S A 2014, 111(44), 15756–61. [Google Scholar] [CrossRef]
- Lala, T.; Hall, R.A. Adhesion G protein-coupled receptors: structure, signaling, physiology, and pathophysiology . Physiol Rev 2022, 102(4), 1587–1624. [Google Scholar] [CrossRef]
- Stoveken, H.M. Adhesion G protein-coupled receptors are activated by exposure of a cryptic tethered agonist . Proc Natl Acad Sci U S A 2015, 112(19), 6194–9. [Google Scholar] [CrossRef]
- Jallouli, R. G protein selectivity profile of GPR56/ADGRG1 and its effect on downstream effectors . Cell Mol Life Sci 2024, 81(1), p. 383. [Google Scholar] [CrossRef]
- Werner, H. The IGF1 Signaling Pathway: From Basic Concepts to Therapeutic Opportunities . Int J Mol Sci 2023, 24(19). [Google Scholar] [CrossRef]
- Cervera, J.; Levin, M.; Mafe, S. Morphology changes induced by intercellular gap junction blocking: A reaction-diffusion mechanism . Biosystems 2021, 209, 104511. [Google Scholar] [CrossRef]
- Cervera, J.; Levin, M.; Mafe, S. Top-down perspectives on cell membrane potential and protein transcription . Sci Rep 2025, 16(1), p. 1996. [Google Scholar] [CrossRef]
- Aberg, N.D. Insulin-like growth factor-I increases astrocyte intercellular gap junctional communication and connexin43 expression in vitro . J Neurosci Res 2003, 74(1), 12–22. [Google Scholar] [CrossRef]
- Prochnow, N.; Dermietzel, R. Connexons and cell adhesion: a romantic phase . Histochem Cell Biol 2008, 130(1), 71–7. [Google Scholar] [CrossRef]
- Langenhan, T.; Aust, G.; Hamann, J. Sticky signaling--adhesion class G protein-coupled receptors take the stage . Sci Signal 2013, 6(276), p. re3. [Google Scholar] [CrossRef]
- Karami, A. Planarians: an In Vivo Model for Regenerative Medicine . Int J Stem Cells 2015, 8(2), 128–33. [Google Scholar] [CrossRef]
- Gurley, K.A.; Rink, J.C.; Alvarado, A. Sanchez. Beta-catenin defines head versus tail identity during planarian regeneration and homeostasis . Science 2008, 319(5861), 323–7. [Google Scholar] [CrossRef]
- Kakugawa, S. Notum deacylates Wnt proteins to suppress signalling activity . Nature 2015, 519(7542), 187–192. [Google Scholar] [CrossRef]
- Pascual-Carreras, E. Wnt/beta-catenin signalling is required for pole-specific chromatin remodeling during planarian regeneration . Nat Commun 2023, 14(1), p. 298. [Google Scholar] [CrossRef]
- Beane, W.S. A chemical genetics approach reveals H,K-ATPase-mediated membrane voltage is required for planarian head regeneration . Chem Biol 2011, 18(1), 77–89. [Google Scholar]
- Adler, C.E.; Alvarado, A.S. Systemic RNA Interference in Planarians by Feeding of dsRNA Containing Bacteria . Methods Mol Biol 2018, 1774, 445–454. [Google Scholar]
- Xue, C. Wnt signaling pathways in biology and disease: mechanisms and therapeutic advances . Signal Transduct Target Ther 2025, 10(1), p. 106. [Google Scholar] [CrossRef]
- Tao, Y.X. Constitutive activation of G protein-coupled receptors and diseases: insights into mechanisms of activation and therapeutics . Pharmacol Ther 2008, 120(2), 129–48. [Google Scholar] [CrossRef]
- Zhang, M. G protein-coupled receptors (GPCRs): advances in structures, mechanisms, and drug discovery . Signal Transduct Target Ther 2024, 9(1), p. 88. [Google Scholar]
- Liu, S. G Protein-Coupled Receptors: A Century of Research and Discovery . Circ Res 2024, 135(1), 174–197. [Google Scholar] [CrossRef]
- Sriram, K. and P.A. Insel, G. Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol Pharmacol 2018, 93(4), 251–258. [CrossRef]
Figure 1.
Strategic design of iDRIVE. A) Depiction of a membrane protein. The protein is embedded in the membrane, while soluble domains are intra- or extracellularly oriented. B) Graphical representation of wsMPs strategies using fusion of proteins, OspA (decoy), Apo AI (amphipathic protein), EmrE (membrane protein), cell penetrating peptide (CPP). SIMPLEx [22] and iDRIVE are different permutations of these proteins. C) Western blot, anti-GFP. Soluble extracts of cells (BL21 DE3) expressing the different proteins tested. Lane 1 (negative control, un-transfected cells), Lane 2-3 (GFP-Apo AI), Lane 4-6 (GFP-EmrE-Apo AI), Lanes 7-9 (GFP-Apo AI-EmrE-CPP). D-E) Confocal microscopy of BL21 DE3 cells expressing the proteins tested.
Figure 1.
Strategic design of iDRIVE. A) Depiction of a membrane protein. The protein is embedded in the membrane, while soluble domains are intra- or extracellularly oriented. B) Graphical representation of wsMPs strategies using fusion of proteins, OspA (decoy), Apo AI (amphipathic protein), EmrE (membrane protein), cell penetrating peptide (CPP). SIMPLEx [22] and iDRIVE are different permutations of these proteins. C) Western blot, anti-GFP. Soluble extracts of cells (BL21 DE3) expressing the different proteins tested. Lane 1 (negative control, un-transfected cells), Lane 2-3 (GFP-Apo AI), Lane 4-6 (GFP-EmrE-Apo AI), Lanes 7-9 (GFP-Apo AI-EmrE-CPP). D-E) Confocal microscopy of BL21 DE3 cells expressing the proteins tested.
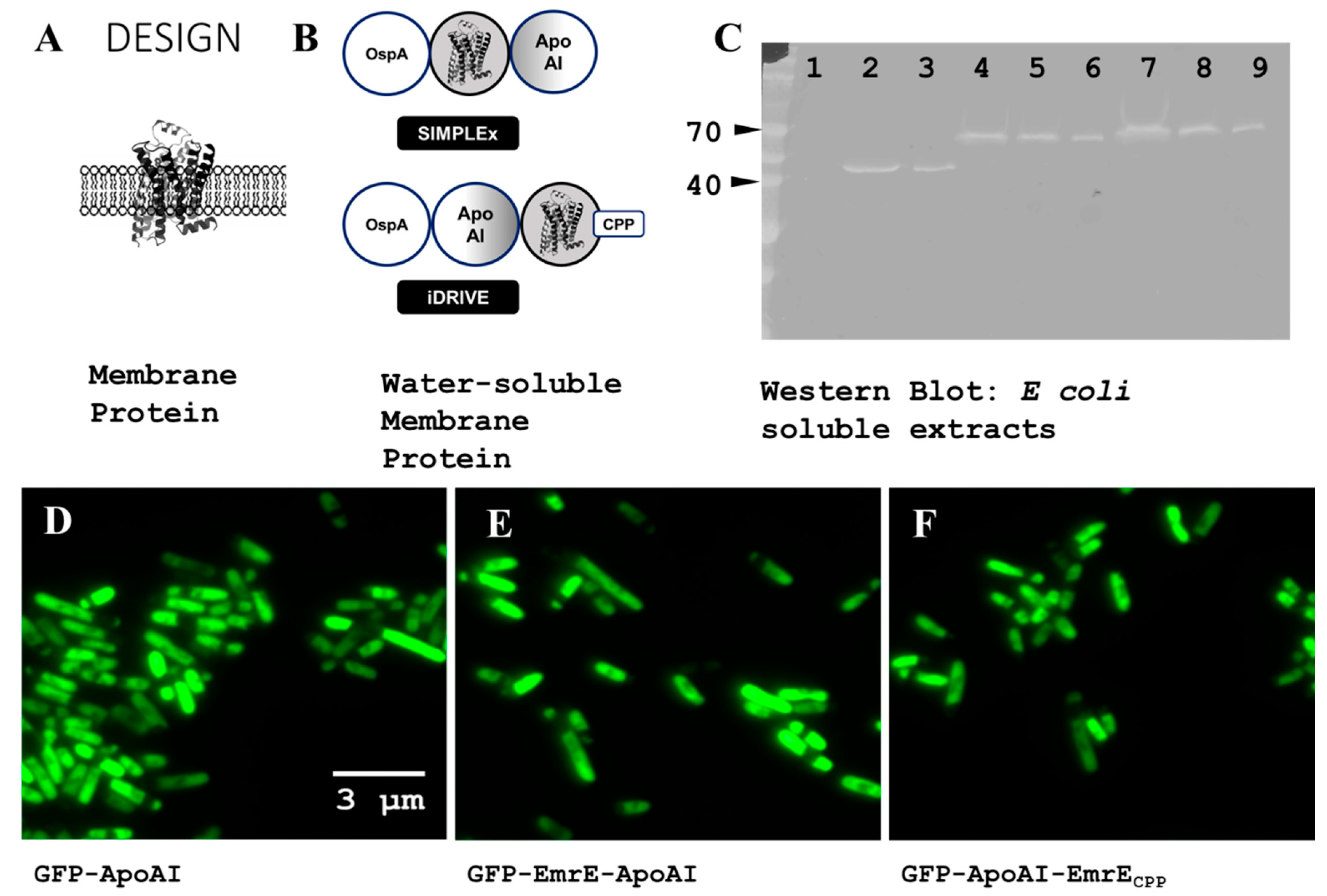
Figure 2.
Planaria interactions with iDRIVE. A) Planaria in their environment. Their size is typically visible with the naked eye, a few millimeters. B) Planaria that has been cut transversally with a scalpel. C) Fragment of Planaria exposed to GFP-Apo AI fusion proteins. Using Confocal Microscopy, fluorescence is not detected after refreshing the water in the well. D) Fragment of Planaria exposed to GFP-Apo AI-EmrE-CPP. Using Confocal Microscopy, fluorescence is detected after refreshing the water in the well. Fluorescence is observed as emanating from the body of the Planaria.
Figure 2.
Planaria interactions with iDRIVE. A) Planaria in their environment. Their size is typically visible with the naked eye, a few millimeters. B) Planaria that has been cut transversally with a scalpel. C) Fragment of Planaria exposed to GFP-Apo AI fusion proteins. Using Confocal Microscopy, fluorescence is not detected after refreshing the water in the well. D) Fragment of Planaria exposed to GFP-Apo AI-EmrE-CPP. Using Confocal Microscopy, fluorescence is detected after refreshing the water in the well. Fluorescence is observed as emanating from the body of the Planaria.
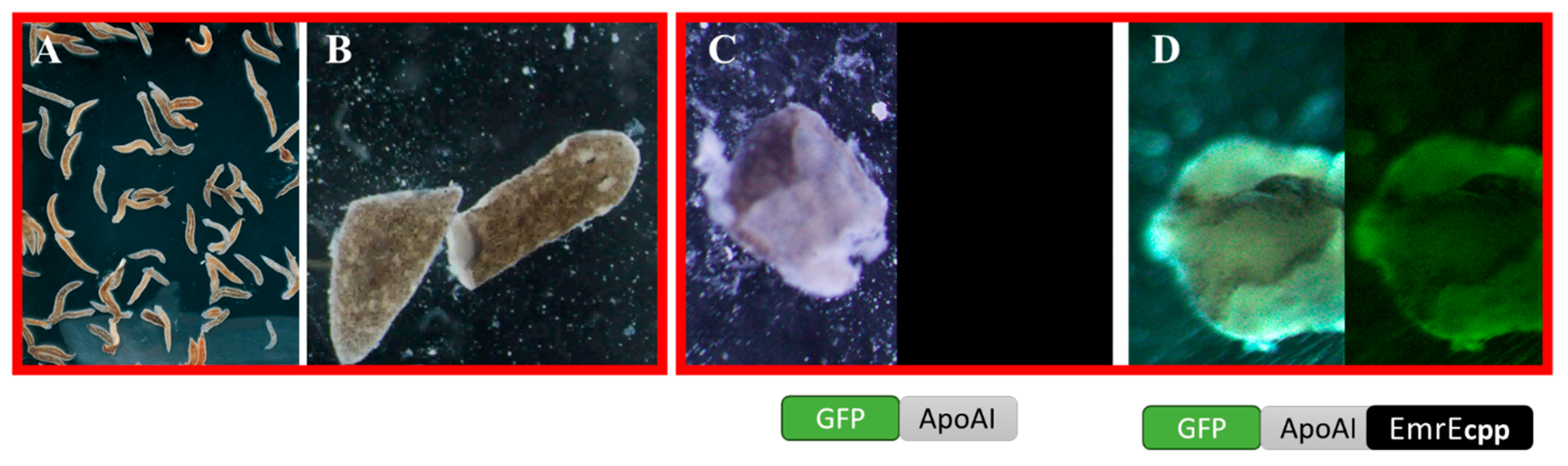
Figure 3.
design of a TATAx3 pore forming. Based on the article by Walther 2013 [27], we hypothesized that the design of a synthetic pore-forming protein can be achieved through the “charge zipper” matching the transmembrane domain and the APH domain. A) Tat A protein in the plasma membrane. B) Proposed fusion of truncated Tat A (amino acids 1-48), three units fused. C) Monitoring cellular growth (BL21 DE3) at OD600. Cells expressing TatA (blue line) or the triple fusion of TatA (TATAx3) (yellow line). The arrow indicates the addition of IPTG (1 mM). D) Negative staining image of SIMPLEx TATAx3. OspA SIMPLEx TATAx3 is purified from BL21 DE3 and subject to Negative Staining using Uranyl Acetate. As the protein forms pores, the Uranyl Acetate accumulates inside the pore. E) Small-angle x-ray scattering (SAXS) volume of MBP-TATAx3-Apo AI. A pore is observed as the soluble protein appears composed of a homo-tetramer MBP-TATAx3-Apo AI.
Figure 3.
design of a TATAx3 pore forming. Based on the article by Walther 2013 [27], we hypothesized that the design of a synthetic pore-forming protein can be achieved through the “charge zipper” matching the transmembrane domain and the APH domain. A) Tat A protein in the plasma membrane. B) Proposed fusion of truncated Tat A (amino acids 1-48), three units fused. C) Monitoring cellular growth (BL21 DE3) at OD600. Cells expressing TatA (blue line) or the triple fusion of TatA (TATAx3) (yellow line). The arrow indicates the addition of IPTG (1 mM). D) Negative staining image of SIMPLEx TATAx3. OspA SIMPLEx TATAx3 is purified from BL21 DE3 and subject to Negative Staining using Uranyl Acetate. As the protein forms pores, the Uranyl Acetate accumulates inside the pore. E) Small-angle x-ray scattering (SAXS) volume of MBP-TATAx3-Apo AI. A pore is observed as the soluble protein appears composed of a homo-tetramer MBP-TATAx3-Apo AI.
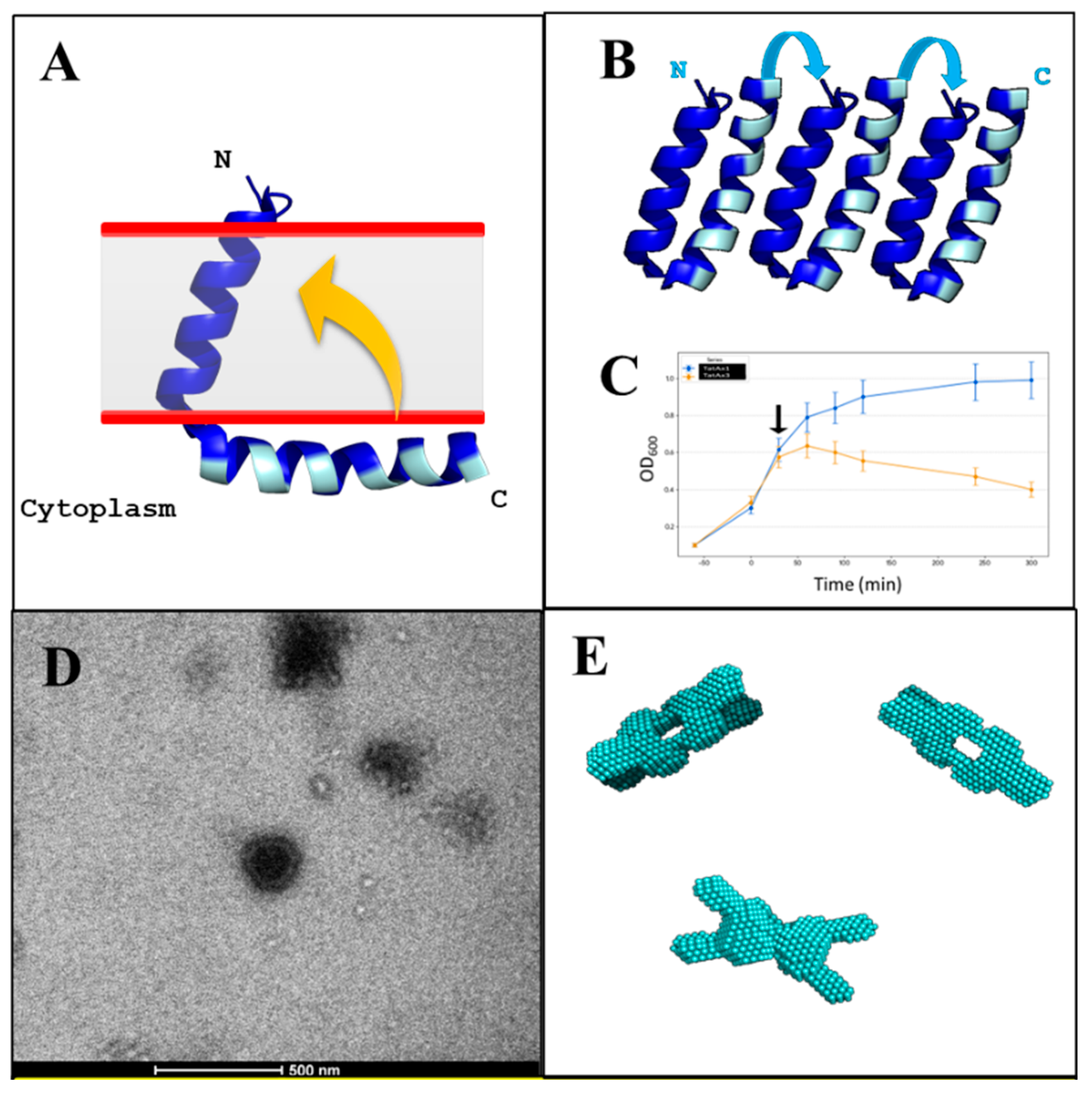
Figure 4.
iDRIVE TATAx3 re-inserts in membranes. A) Schematic representation of Planar Lipid Bilayer (PLB) and experimental approach. B) PBL Recordings after one of the chambers is exposed to iDRIVE TATAx3. C and D) HEK 293 cells after 12 h of exposure to GFP-Apo AI. Panel D is panel C where image is captured by Confocal Microscopy. E and F) HEK 293 cells after 12 h (Panel E) or 24 h (Panel F) of exposure to GFP-Apo AI-TATAx3-CPP. Both images are taken using Confocal Microscopy. Light Microscopy of panels E and F is not shown.
Figure 4.
iDRIVE TATAx3 re-inserts in membranes. A) Schematic representation of Planar Lipid Bilayer (PLB) and experimental approach. B) PBL Recordings after one of the chambers is exposed to iDRIVE TATAx3. C and D) HEK 293 cells after 12 h of exposure to GFP-Apo AI. Panel D is panel C where image is captured by Confocal Microscopy. E and F) HEK 293 cells after 12 h (Panel E) or 24 h (Panel F) of exposure to GFP-Apo AI-TATAx3-CPP. Both images are taken using Confocal Microscopy. Light Microscopy of panels E and F is not shown.
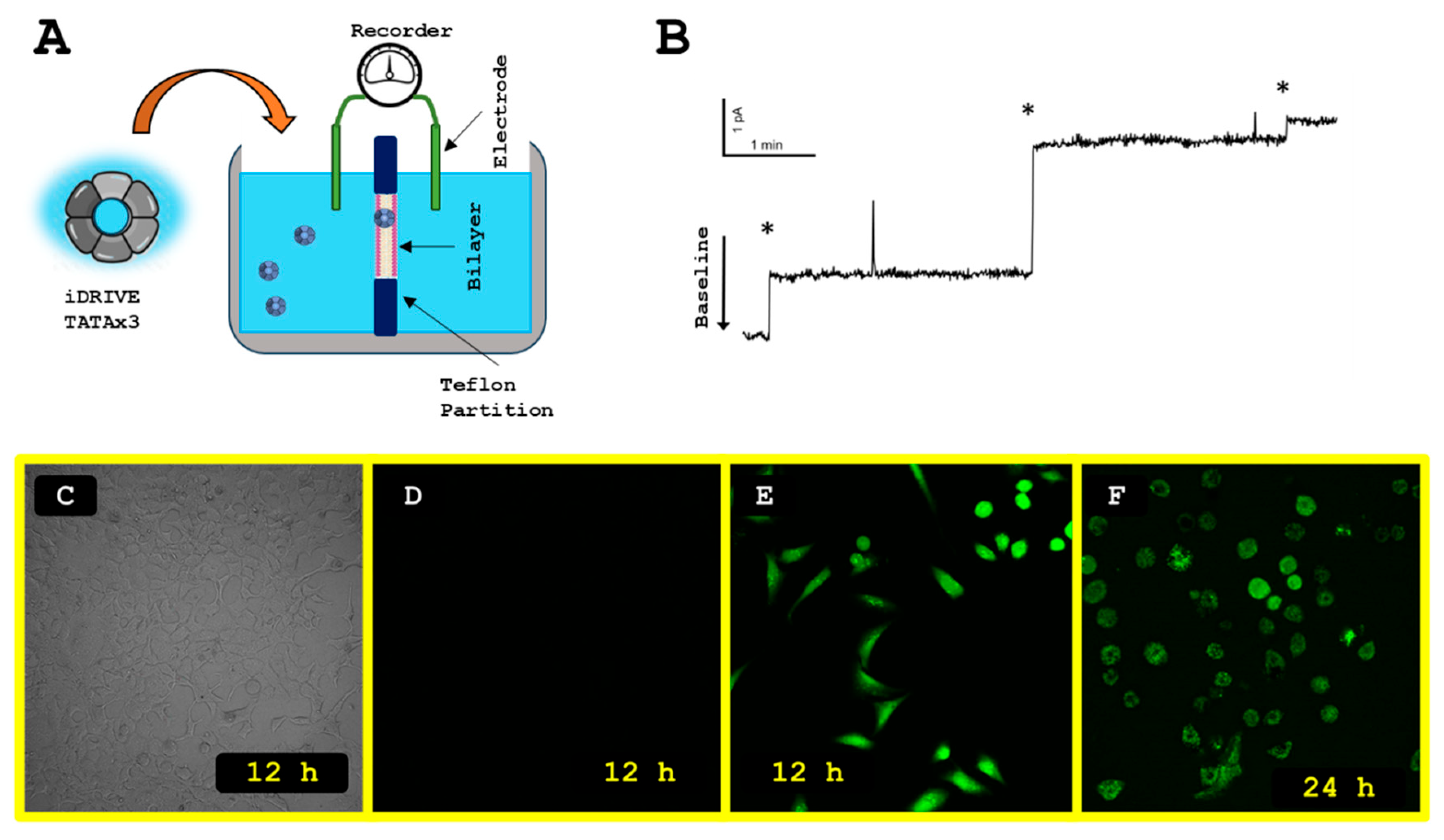
Figure 5.
HEK 293 demonstrates the power of iDRIVE. A) Imaging HEK 293 cells cultured as Embryoid Bodies (EBs). B) ATP assay of HEK 293 cells in culture. Cells are exposed to CHIR99021, iDRIVE (caFZD7), or Retinoic Acid (RA). Dark blue is used for None (no treatment) and is the control over which ATP levels are considered. C) Considering only RA stimulates metabolism (increased ATP) in HEK 293 cells, combinations of CHIR99021 and iDRIVE with RA were tested. Dark blue is used for RA only and is the control over which ATP levels are considered. D) Graphical representation of the distribution of β-values for each sample group positioned at chr1_245248281_245248411, the number one ranked differential methylated regions (DMRs) (being defined as the largest change in β-values between control and treatment groups). As shown, DMRs indicate a significant hypermethylation of the HEK 293 cells, possibly explaining similarities in the subsequent differentiation of the HEK 293 spheroids. Asterisks represent statistically significant differences against the control (p<0.04).
Figure 5.
HEK 293 demonstrates the power of iDRIVE. A) Imaging HEK 293 cells cultured as Embryoid Bodies (EBs). B) ATP assay of HEK 293 cells in culture. Cells are exposed to CHIR99021, iDRIVE (caFZD7), or Retinoic Acid (RA). Dark blue is used for None (no treatment) and is the control over which ATP levels are considered. C) Considering only RA stimulates metabolism (increased ATP) in HEK 293 cells, combinations of CHIR99021 and iDRIVE with RA were tested. Dark blue is used for RA only and is the control over which ATP levels are considered. D) Graphical representation of the distribution of β-values for each sample group positioned at chr1_245248281_245248411, the number one ranked differential methylated regions (DMRs) (being defined as the largest change in β-values between control and treatment groups). As shown, DMRs indicate a significant hypermethylation of the HEK 293 cells, possibly explaining similarities in the subsequent differentiation of the HEK 293 spheroids. Asterisks represent statistically significant differences against the control (p<0.04).
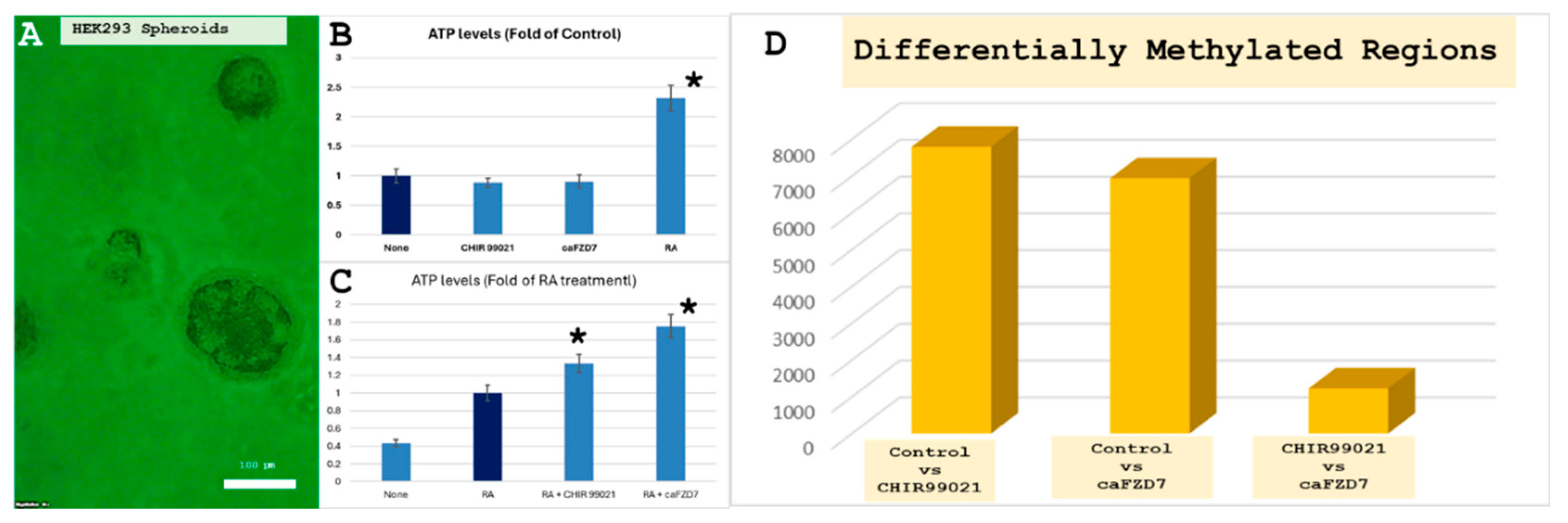
Figure 6.
C2C12 cells Myotube Hypertrophy assay. A) Representation of an adhesion GPCR (aGPCR). The N-terminus (light blue) is extracellular and is responsible for establishing contact and adhesion to other cells. B) Graphical representation of the Myotube measurement performed when C2C12 cells are exposed to no-treatment (Control) or IGF-1 or iDRIVE (caGPR56). Statistical significance between treatments and the control are represented by (**), p<0.001. C and C’) C2C12 cells, no-treatment. Green corresponds to Calcein and blue is DAPI staining. D and D’) C2C12 cells, IGF-1 treatment. E and E’) C2C12 cells, iDRIVE (GPR56) treatment. Cell size is characterized visually with the white bar, corresponding 300 µm.
Figure 6.
C2C12 cells Myotube Hypertrophy assay. A) Representation of an adhesion GPCR (aGPCR). The N-terminus (light blue) is extracellular and is responsible for establishing contact and adhesion to other cells. B) Graphical representation of the Myotube measurement performed when C2C12 cells are exposed to no-treatment (Control) or IGF-1 or iDRIVE (caGPR56). Statistical significance between treatments and the control are represented by (**), p<0.001. C and C’) C2C12 cells, no-treatment. Green corresponds to Calcein and blue is DAPI staining. D and D’) C2C12 cells, IGF-1 treatment. E and E’) C2C12 cells, iDRIVE (GPR56) treatment. Cell size is characterized visually with the white bar, corresponding 300 µm.
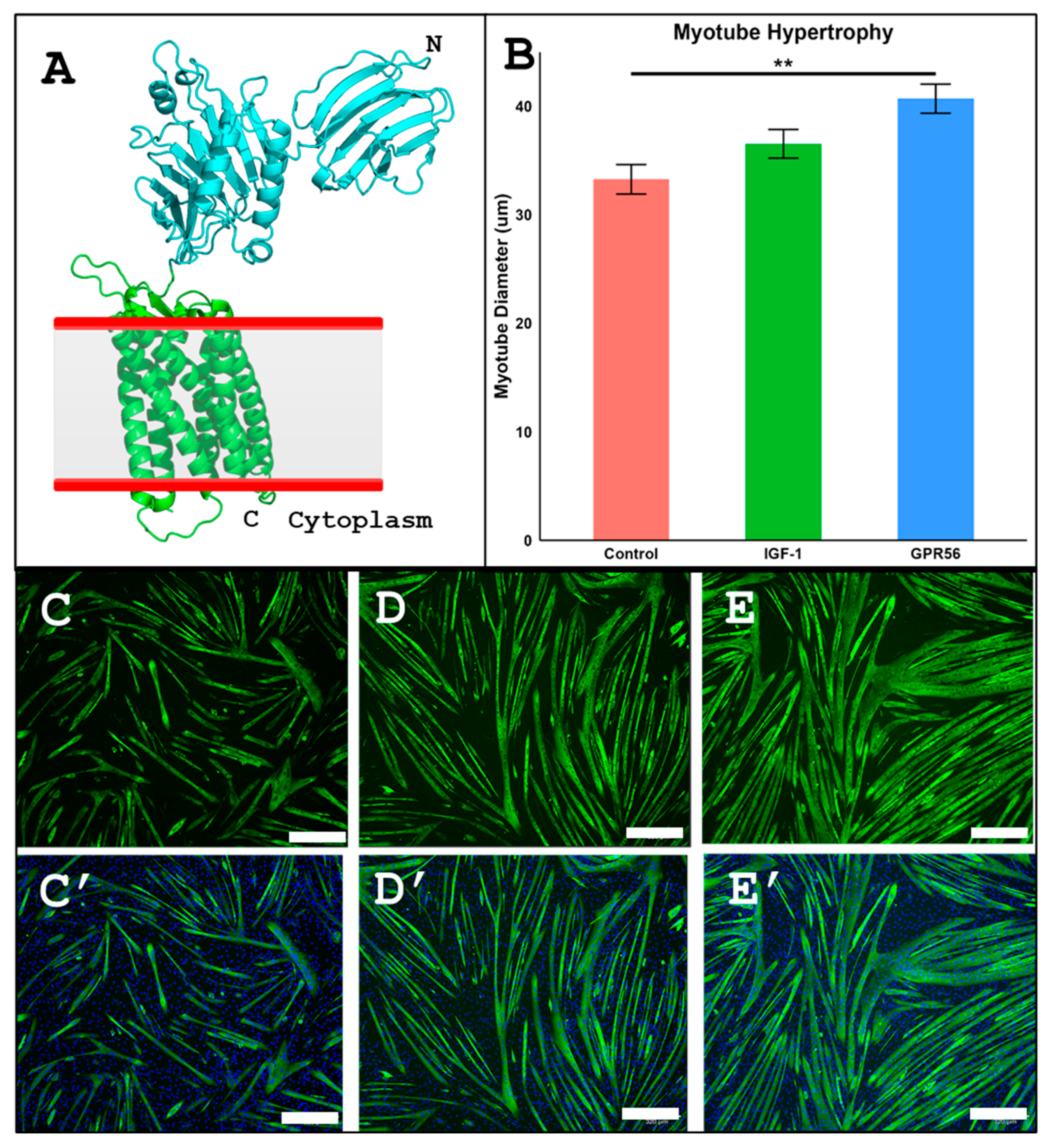
Figure 7.
basic elements of experimental design. A) Elements considered for the Wnt pathway. FLD (frizzled) receptors, LGR5 co-receptor, β-catenin is the intracellular target, Notum is the enzyme capable of inactivating Wnt signaling. B) Following our strategy, several scenarios may occur during the process of regeneration. When the Planaria are divided transversally a wound is created. On the head side, Wnt is active in the wound, Notum (its antagonist) is inactive. The tail’s wound will produce Notum, and Wnt will be inactivated as a result. If we exposed the tail to caFZD, the presence of Notum will have an effect on native extracellular components of Wnt signaling, but not on the caFZD, enabling it to signal.
Figure 7.
basic elements of experimental design. A) Elements considered for the Wnt pathway. FLD (frizzled) receptors, LGR5 co-receptor, β-catenin is the intracellular target, Notum is the enzyme capable of inactivating Wnt signaling. B) Following our strategy, several scenarios may occur during the process of regeneration. When the Planaria are divided transversally a wound is created. On the head side, Wnt is active in the wound, Notum (its antagonist) is inactive. The tail’s wound will produce Notum, and Wnt will be inactivated as a result. If we exposed the tail to caFZD, the presence of Notum will have an effect on native extracellular components of Wnt signaling, but not on the caFZD, enabling it to signal.
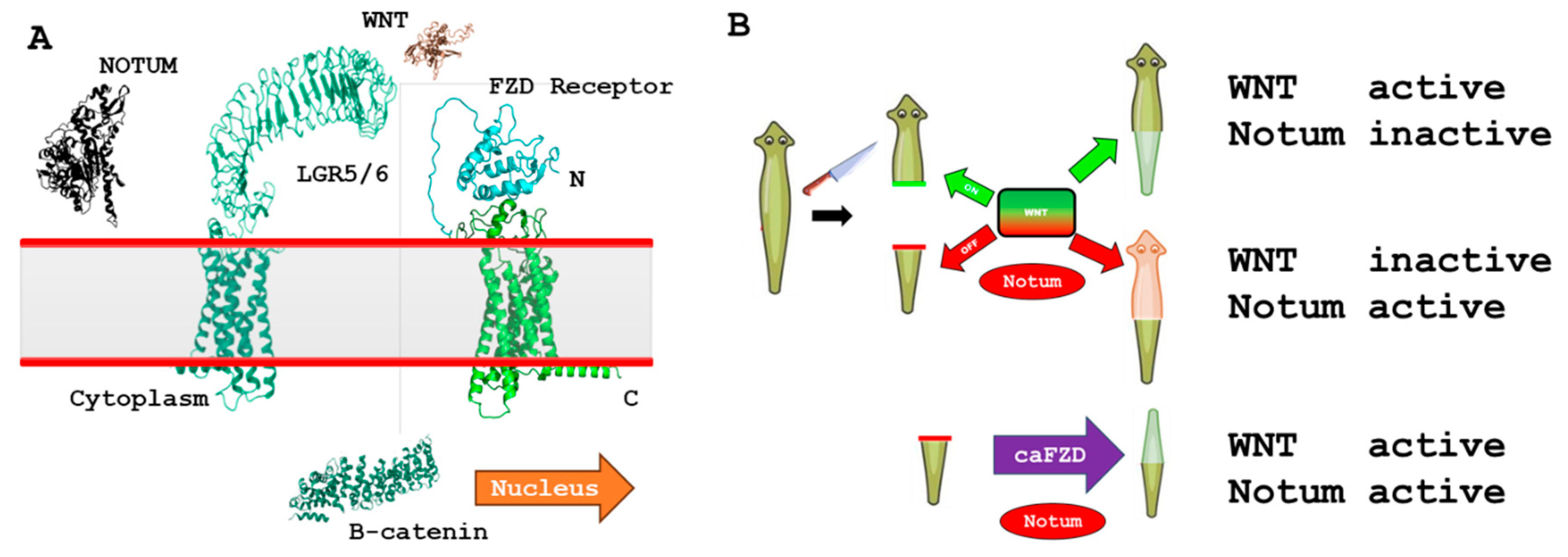
Figure 8.
Planaria regeneration. Planaria are divided into two groups, CONTROL, representing tails treated with 8 µM OspA-Apo AI, and TREATMENT, tails exposed to 8 µM iDRIVE (capFZD). The pictures indicate changes to each group daily, from day 1 (D1), the day the wounds are created, day 3 (D3), day 5 (D5), and day 7 (D7). Images of the tail are divided to capture more closely the wound side of the tail, and the tip of the tail.
Figure 8.
Planaria regeneration. Planaria are divided into two groups, CONTROL, representing tails treated with 8 µM OspA-Apo AI, and TREATMENT, tails exposed to 8 µM iDRIVE (capFZD). The pictures indicate changes to each group daily, from day 1 (D1), the day the wounds are created, day 3 (D3), day 5 (D5), and day 7 (D7). Images of the tail are divided to capture more closely the wound side of the tail, and the tip of the tail.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



