Submitted:
26 January 2026
Posted:
27 January 2026
You are already at the latest version
Abstract
Background: Natural killer (NK) cells are essential innate immune effectors with significant therapeutic potential in cancer immunotherapy. However, strategies to enhance NK cell maturation and effector function remain limited. Metadichol, a nano-emulsion of policosanol capable of modulating 49 nuclear receptors, has demonstrated immunomodulatory properties, yet its effects on NK cell developmental programming have not been systematically characterized. Understanding how Metadichol regulates NK cell maturation, particularly through the critical IL-15/CD122 signaling axis, could reveal novel immunotherapeutic approaches.Methods: Human peripheral blood mononuclear cells (PBMCs) were treated with Metadichol at concentrations ranging from 0.1 pg/ml to 100 ng/ml. Expression of 19 genes critical for NK cell development, including surface markers, transcription factors, and cytokine receptors, was analyzed by qRT-PCR. Regulatory network analysis was performed to elucidate mechanistic pathways, with particular focus on IL-15 signaling and NF-κB-independent regulatory mechanisms.Results: Metadichol induced a coordinated "push-pull" developmental program characterized by dramatic upregulation of mature NK cell markers concurrent with suppression of early progenitor markers. CD122 (IL-2Rβ), the essential receptor for IL-15 responsiveness, showed robust 9.19-fold upregulation (p<0.05), while NKG2D increased 8.59-fold (p<0.001) and NKp80 increased 4.91-fold (p<0.05). Conversely, early developmental markers CD127, CD7, and CD45RA were suppressed to 0.08, 0.09, and 0.02-fold of control levels, respectively (all p<0.001). Mechanistic analysis revealed that Metadichol enhances IL-15 signaling through the mTORC1→E4BP4→EOMES→CD122 positive feedback loop. Critically, despite NF-κB inhibition, Metadichol maintains IL-15 and IRF-1 regulation through alternative pathways including TLR-TRIF signaling, nuclear receptor activation (VDR, RARs, PPARs), sirtuin-mediated STAT modulation, and TP53-IRF-1 cross-regulation.Conclusions: Metadichol drives NK cell maturation through a novel push-pull mechanism that simultaneously accelerates developmental progression while suppressing progenitor phenotypes. The pronounced enhancement of CD122 expression amplifies IL-15 responsiveness through NF-κB-independent pathways, achieving immunomodulation that promotes NK cell cytotoxic capacity without excessive inflammation. These findings establish Metadichol as a promising agent for NK cell-based cancer immunotherapy and infectious disease treatment, offering a unique multi-pathway approach to enhancing innate immune function.
Keywords:
Metadichol
; natural killer cells
; CD122
; IL-15
; NK cell development
; push-pull mechanism
; nuclear receptors
; immunotherapy
; NF-κB-independent signaling
Introduction
Natural Killer Cell Biology and Development
Natural killer (NK) cells represent a critical component of the innate immune system, serving as first-line defenders against viral infections and malignant transformation. Arachchige et al. provide a comprehensive review of human NK cells from development to effector functions. [1] and the role of natural killer cells in autoimmune diseases. [2] and also the molecular mechanism of NK cell function and its importance in cancer immunotherapy. [3] These lymphocytes, comprising approximately 5-15% of circulating lymphocytes, possess the unique ability to recognize and eliminate aberrant cells without prior sensitization.
Natural killer (NK) cell development occurs in several distinct stages. Reviews have summarized NK cell development, maturation, and clinical applications, [4,5] as well as explored whether human NK cells develop through a single pathway or multiple routes. [6,7] Studies have identified early stages of human NK cell development in both primary and secondary lymphoid tissues.[8] The process involves NK cells gaining and losing specific surface markers in sequence.
Key Surface Markers in NK Cell Development
NKG2D (CD314/KLRK1): NKG2D is a type II transmembrane C-type lectin-like receptor that serves as a major activating receptor on NK cells and their ligands in cancer immunotherapy. [9] NKG2D is a master regulator of immune cell responsiveness. [10] and recognizes stress-induced ligands, including MICA (MHC Class I Chain-related protein A), MICB (MHC Class I Chain-related protein B), and ULBPs (UL16-Binding Proteins) on transformed or infected cells.
CD122 (IL-2Rβ): The β chain of the IL-2 receptor, CD122, pairs with the common γ chain to form the functional receptor for IL-15. and it has been shown how IL-2 interacts with IL-2Rβ. [11] CD8+CD122+ T-cells have been identified as a newly emerging regulator with immunosuppressive function. [12] Recent research has shown that the abundance and availability of CD122 constrains lymphopenia-induced homeostatic proliferation. [13] Work on developmental and functional control of NK cells by cytokines. [14]. demonstrated that CD122 signaling in CD8+ memory T cells drives co-stimulation-independent alloreactivity. [15] and showed that targeting CD122 enhances antitumor immunity. [16]
CD127 (IL-7Rα): CD127 expression characterizes early lymphoid progenitors, soluble IL-7Rα/sCD127 in health and disease, and its therapeutic potential. [17] CD127 expression inversely correlates with FoxP3 and identifies regulatory T cells. [18] and IL-7 signaling and CD127 receptor regulation. [19] leads to human epistatic interaction that controls IL7R splicing. [20]
CD56 (NCAM): Neural cell adhesion molecule serves as the defining marker of human NK cells. CD56 as a biomarker and therapeutic target in multiple myeloma, [21] and in the immune system it is more than a marker for cytotoxicity. [22] Recent work discussed the significance of NK cell CD56 brightness. [23] and demonstrated that CD56 regulates human NK cell cytotoxicity through Pyk2. [24] It has been demonstrated that CD56/NCAM mediates cell migration of human NK cells. [25] and established CD56 as a pathogen recognition receptor on human NK cells. [26]
CD7: CD7 is an early T and NK cell marker and is characterized by the expression and function of CD7 on human NK cells.[27] and CD7 is a differentiation marker identifying multiple CD8 T cell effector subsets.[28]
CD38: This ecto-enzyme marks activated lymphocytes and is an immunomodulatory molecule in inflammation and autoimmunity.[29] CD38 has an important role in the immune response to infection.[30] and is a significant regulator of macrophage function.[31] CD38 plays a role in regulation of immune response in cancer.[32] and CD38/CD31 interactions activate genetic pathways in Chronic Lymphocytic Leukemia.[33] CD38 regulates B cell antigen receptor signaling, [34] and contributes to human NK cell responses to multiple myeloma.[35] and enhanced anti-tumor activity of CD38-CAR NK cells.[36] CD38 is a target therapy in multiple myeloma, [37] and CD38 antibodies in multiple myeloma.[38]
CD45RA: CD45RA is an isoform of protein tyrosine phosphatase expressed on naive lymphocytes.[39] CD45 isoform profile identifies NK subsets with differential activity.[40] and properties of end-stage human T cells are defined by CD45RA re-expression.[41] CD45RA+ naive T cells are implicated in pancreatic cancer.[42] Unique phenotypes of CD4 effector memory T cells have been shown to be re-expressing CD45RA.[43,44]
CD117 (c-Kit): CD117 is the stem cell factor receptor marking hematopoietic progenitors and is expressed in normal and neoplastic tissues.[45] Foster characterized CD117/c-kit in cancer stem cell-mediated progression, [46] in testis and other tissues.[47] Signal transduction via stem cell factor receptor/c-Kit.[48] demonstrated selective hematopoietic stem cell ablation using CD117-targeting.[49]
CD34: CD34 is a cell surface glycoprotein marking hematopoietic stem cells and there is evidence for CD34 as a common marker for diverse progenitors.[50] CD34 structure, functions have a marked relationship with cancer stem cells.[51] and CD34 is not just a marker on hematopoietic stem/progenitor cells.[52] CD34 positive cells are used as endothelial progenitor cells in regenerative medicine.[53] and this puts CD34- cells at the apex of the human hematopoietic stem cell hierarchy.[54] There is an association of CD34 cell dose with survival after allogeneic transplantation.[55]
NKp80 (KLRF1): NKp80 marks a critical developmental checkpoint in NK cell maturation. NKp80 defines a critical step during human NK cell development.[56] Mutual activation of NK cells and monocytes mediated by NKp80-AICL interaction.[57] and researchers have developed a novel NKp80-based strategy for universal identification of NK cells.[58]
1.3. Natural Cytotoxicity Receptors
CD336 (NKp44/NCR2): NKp44 is a natural cytotoxicity receptor expressed on activated NK cells as a damage-associated molecular pattern recognition receptor.[64] In NKp44-ligand interactions in NK cell regulation.[65]) is expressed on the majority of porcine NK cells ex vivo.[66] Multiple tumors were targeted using NCR2-based chimeric receptor T-cells.[67] and there is a developed NKp44-based chimeric antigen receptor for synovial sarcoma.[68]
CD337 (NKp30/NCR3): NKp30 is a key membrane molecule in cancer and infection defense, and its expression serves as a prognostic immune biomarker in AML.[69] NKp30 is a key membrane molecule.[70] Expression of NKp46 (CD335) in human NK and T-cell neoplasia.[71] NKp46 is a diagnostic biomarker in gastrointestinal T-cell lymphoproliferative diseases,[72] and accentuated NK lymphocyte CD335 (NKp46) expression predicts pregnancy failures.[73]
IL1R1 (Interleukin-1 Receptor Type 1): IL1R1 mediates inflammatory cytokine signaling. IL-1R1/MyD88 signaling and inflammasome are essential for pulmonary fibrosis development.[74] IL-1β selectively expands immature human NK cells in secondary lymphoid tissue.[75] and it raises therapeutic prospects of targeting IL-1R1 for neuroinflammation.[76,77]
Metadichol: A Novel Nuclear Receptor Modulator
Metadichol is a nano-emulsion formulation of long-chain aliphatic alcohols (policosanols). [78,79,80] Metadichol is a nano lipid emulsion capable of expressing all 49 nuclear receptors in stem and somatic cells,[81] providing a mechanistic basis for its broad regulatory effects. Metadichol acts as a natural ligand for expression of Yamanaka reprogramming factors.[82]
Materials and Methods
A commercial service provider (Skanda Life Sciences, Bangalore, India) performed the quantitative q-RT‒PCR, Western blot analysis, and cell culture work. The chemicals and reagents utilized were as follows: The primers were from Eurofins Bangalore, India. Other molecular biology reagents were obtained from Sigma‒Aldrich, India.
PBMC Isolation Protocol
Fresh human blood was collected into EDTA-containing tubes and diluted 1:1 with PBS. The diluted blood was carefully layered onto Histopaque-1077 and centrifuged at 400 × g for 30 minutes at room temperature with the brake disengaged. The mononuclear cell layer at the interface was carefully collected, washed twice with PBS (250 × g, 10 minutes), and resuspended in RPMI 1640 medium supplemented with 10% FBS. Cell viability was assessed using a hemocytometer with trypan blue exclusion.
RNA Isolation
Following treatment, cells were harvested, washed with ice-cold PBS, and lysed in TRIzol reagent. Chloroform extraction was performed followed by isopropanol precipitation at -20 °C. The RNA pellet was washed with 70% ethanol, air-dried, and resuspended in DEPC-treated water. Total RNA concentration and purity were assessed using a SpectraDrop spectrophotometer (SpectraMax i3x, Molecular Devices).
cDNA Synthesis
Complementary DNA was synthesized from 500 ng of total RNA using the PrimeScript RT Reagent Kit with oligo-dT primers according to the manufacturer’s protocol. The reaction (20 µL total volume) was incubated at 50 °C for 30 minutes followed by reverse transcriptase inactivation at 85 °C for 5 minutes using an Applied Biosystems Veriti thermal cycler.
Quantitative Real-Time PCR Analysis
Quantitative PCR was performed in 20 µL reactions containing 1.4 µL cDNA template, 10 µL SYBR Green Master Mix, and 1 µM each of gene-specific forward and reverse primers. Thermal cycling parameters consisted of initial enzyme activation at 95 °C for 2 minutes, followed by 39 cycles of denaturation at 95 °C for 5 seconds and annealing/extension at the primer-specific temperature for 30 seconds. Melt curve analysis was performed from 65 °C to 95 °C to verify amplicon specificity. Relative gene expression was calculated using the 2-ΔΔCq method with GAPDH as the reference gene.
Primer Sequences
Results
Metadichol Modulates NK Cell Surface Marker Expression
Results of treatment with Metadichol are illustrated in summarized Figure 1, Figure 2, Figure 3, Figure 4, Figure 5 and Figure 6. The most striking observation (Figure 1) was dramatic upregulation of NKG2D at 1 pg/ml (8.59±3.74 fold, p<0.001). CD122 showed robust induction at 1 pg/ml (9.19±7.68 fold) and 100 ng/ml (9.14±6.42 fold, p<0.05). NKP80 demonstrated significant upregulation at 1 pg/ml (4.91±2.94 fold, p<0.05).
Heatmap of NK cell surface marker expression (Figure 1) following Metadichol treatment. Expression of 20 NK cell-associated genes was analyzed by qRT-PCR across four Metadichol concentrations (1 pg, 100 pg, 1 ng, 100 ng/ml) normalized to untreated controls. Color intensity represents fold change relative to control (scale: 0-9).
Notable findings include robust CD122 upregulation at 1 pg/ml (9.19-fold) and 100 ng/ml (9.14-fold), dramatic
NKG2D induction at 1 pg/ml (8.59-fold), and consistent suppression of early progenitor markers CD127, CD45RA, and CD7 across all concentrations.
EOMES shows progressive increase from 0.90 at 1 pg/ml to 1.50-fold at 100 ng/ml.
IL-15 peaks at 1 pg/ml (1.60-fold). TBET demonstrates concentration-dependent modulation with highest expression at 100 pg/ml (1.85-fold) and 100 ng/ml (1.70-fold).
Bar graphs (Figure 2) shows fold changes (mean ± SEM) for six representative markers: NKG2D (activating receptor), CD122 (IL-15 receptor β chain), CD56 (NCAM), CD127 (IL-7Rα), NKP80 (maturation marker), and CD7 (early marker). Red dashed lines indicate baseline (fold change = 1). Statistical significance: *p<0.05, ***p<0.001 vs. control. NKG2D shows peak response at 1 pg/ml (8.59-fold), while CD122 demonstrates sustained upregulation across concentrations. CD127 and CD7 show consistent suppression indicative of accelerated maturation.
Figure 3 shows the correlation matrix between the expressed genes The Pearson correlation coefficients were calculated based on expression patterns across the four Metadichol concentrations.
Notable findings:
Strong positive correlations (co-expressed markers):
CD10 ↔ IL1R1 (r = 0.98)
CD34 ↔ CD122 (r = 0.96)
CD117 ↔ NKP80 (r = 0.95)
CD117 ↔ NKG2A (r = 0.94)
EOMES ↔ TBET (r = 0.94) — both are T-box transcription factors involved in NK cell maturation
Strong negative correlations (inverse expression patterns):
CD338 ↔ CD122 (r = -0.99)
CD45RA ↔ CD122 (r = -0.97)
CD7 ↔ CD122 (r = -0.96)
CD337 ↔ NKG2D (r = -0.93)
These correlations suggest coordinated transcriptional programs in response to Metadichol treatment. The strong co-expression of CD122 (IL-2Rβ) with CD117 and NKP80, alongside its inverse relationship with CD338 and CD45RA, may indicate distinct NK cell subpopulation responses or differentiation states being modulated by the treatment.
Figure 4 shows the expression of key transcription factors TBET, EOMES, and IL-15.
Bar graphs showing fold change across five Metadichol concentrations (0.1 pg/ml to 100 ng/ml). TBET shows biphasic response with peak at 0.1 pg/ml (~2.8-fold) and significant upregulation at 100 ng/ml (*p<0.05). EOMES demonstrates progressive increase from suppression at 0.1 pg/ml (*p<0.05) to 1.5-fold elevation at 100 ng/ml. IL-15 peaks at 1 pg/ml (~1.6-fold, *p<0.05), supporting enhanced cytokine production capacity. Gray dashed lines indicate baseline expression (fold change = 1).
Individual Dose-Response Curves
This panel displays (Figure 5) individual dose-response curves for each of the nine highly responsive NK cell markers in separate subplots. Each graph plots fold change (y-axis) against Metadichol concentration on a logarithmic scale (x-axis). A horizontal dashed line at fold change = 1 represents the control baseline. Maximum expression values are annotated on each curve.
Key Observations
- CD122 (IL-2Rβ) exhibits the highest overall response with a maximum fold change of 9.19 at 1 pg, demonstrating an inverse dose-response relationship with high expression at both 1 pg and 100 ng but reduced expression at intermediate concentrations.
- NKG2D shows a striking hormetic response pattern with peak expression (8.59-fold) at the lowest dose tested (1 pg), declining sharply at higher concentrations.
- NKP80 displays a U-shaped response curve with elevated expression at both low (1 pg: 4.91-fold) and high (100 ng: 6.28-fold) concentrations.
- CD338 uniquely peaks at 100 pg (3.10-fold), suggesting optimal activation at intermediate concentrations.
- CD10, CD117, and NKG2A demonstrate classical dose-dependent increases with maximum expression at the highest concentration (100 ng)
Figure 6: Dose-Response Overlay
This composite figure superimposes all nine dose-response curves on a single plot, enabling direct comparison of response magnitudes and kinetics across markers. Each gene is represented by a distinct color, with concentrations displayed on a logarithmic x-axis.
Key Observations
- Magnitude stratification: CD122 and NKG2D clearly separate from other markers as the most responsive genes, with fold changes reaching 8–10 at their peak concentrations.
- Divergent trajectories: The overlay reveals that genes do not respond uniformly—while some markers increase monotonically with dose (CD10, CD117, NKG2A), others show inverse (NKG2D) or non-monotonic (CD122, NKP80) relationships.
- Concentration-dependent crossover: Several curves intersect at intermediate doses (100 pg–1 ng), indicating that the relative expression hierarchy among markers shifts depending on Metadichol concentration.
- Therapeutic windows: The overlay suggests that ultra-low doses (1 pg) preferentially activate cytotoxicity-associated receptors (NKG2D, CD122), while higher doses (100 ng) favor maturation markers (NKG2A, CD117).
Figure 7 categorizes the nine responsive genes into three panels based on their dose-response pattern type: (1) Inverse/Hormetic Response, (2) Bell-Shaped/Biphasic Response, and (3) Dose-Dependent Increase. This classification provides mechanistic insight into how different NK cell pathways respond to Metadichol.
Pattern Classification
| Pattern | Genes | Biological Interpretation |
| Inverse/Hormetic (Peak at 1 pg) | CD122, NKG2D | Ultra-sensitive signaling; possible receptor saturation or negative feedback at higher doses; suggests potent activation at picomolar concentrations |
| Bell-Shaped (Peak at 100 pg–1 ng) | CD338, CD7, CD336 | Optimal therapeutic window at intermediate concentrations; balanced receptor engagement; CD336 (NKp44) activation indicates enhanced NK cell activation state |
| Monotonic Increase (Peak at 100 ng) | NKP80, CD10, NKG2A, CD117 | Classical pharmacological response; cumulative transcriptional activation; NKG2A and CD117 upregulation suggests enhanced NK cell maturation and licensing |
Functional Implications
- Activating receptors: NKG2D (hormetic) and NKP80/CD336 (biphasic/monotonic) are activating receptors involved in tumor cell recognition. Their upregulation suggests enhanced cytotoxic potential.
- Inhibitory receptor: NKG2A (monotonic increase) is an inhibitory receptor that recognizes HLA-E. Its upregulation alongside activating receptors indicates balanced immunomodulation rather than uncontrolled activation.
- Cytokine signaling: CD122 (IL-2Rβ) is essential for IL-2 and IL-15 signaling, which drive NK cell proliferation and survival. Its hormetic response suggests maximal proliferative signaling at ultra-low Metadichol doses.
Key points
The dose-response analysis reveals that Metadichol exerts complex, non-linear effects on NK cell surface marker expression. The three visualizations collectively demonstrate:
- Ultra-low dose efficacy: The hormetic responses of CD122 and NKG2D indicate that picomolar concentrations may be pharmacologically active, a finding with significant implications for therapeutic dosing.
- Pathway-specific responses: Different NK cell signaling pathways exhibit distinct concentration thresholds, suggesting engagement of multiple molecular targets.
- Balanced immunomodulation: Coordinate regulation of activating and inhibitory receptors indicates physiologically relevant immune enhancement rather than pathological hyperactivation.
- Concentration-dependent phenotypes: The overlay analysis reveals that optimal concentrations differ by target gene, necessitating careful dose selection based on desired immunological endpoints.
These findings support further investigation of Metadichol as an NK cell immunomodulator, with particular attention to the observed hormetic effects at ultra-low concentrations.
Figure 8 Four-panel summary
Comprehensive analysis of gene expression changes following Metadichol treatment
Panel description; (A) Gene Expression Changes Overview showing distribution of upregulated (10 genes, >1.5-fold), unchanged (6 genes, 0.5-1.5-fold), and downregulated (4 genes, <0.5-fold) markers; (B) Statistical Significance Distribution pie chart showing proportion of significantly altered genes (p<0.05); (C) Top Responsive NK Cell Markers ranked by maximum fold change, with CD122, NKG2D, and NKP80 showing highest responses; (D) Dose-Dependent Expression Pattern demonstrating the characteristic biphasic response curve of Metadichol treatment with peaks at low and high concentrations.
Discussion
Metadichol’s Multi-Pathway Regulatory Network
Metadichol functions as a pleiotropic immunomodulator through its capacity to engage multiple regulatory pathways (Figure 9). [81,82,83] NK Cell Gene Regulatory Flywheel depicting the interconnected network of surface markers, transcription factors, and cytokines modulated by Metadichol. Red arrows indicate activation/upregulation pathways; green arrows indicate inhibition/downregulation. Central regulatory nodes include master transcription factors T-BET and EOMES. The network illustrates how changes in early markers (CD34, CD127, CD7) propagate through intermediate regulators to affect mature NK cell markers (NKG2D, NKP80, CD122). This flywheel model explains the coordinated “push-pull” developmental acceleration mechanism whereby simultaneous activation and inhibition pathways drive maturation.
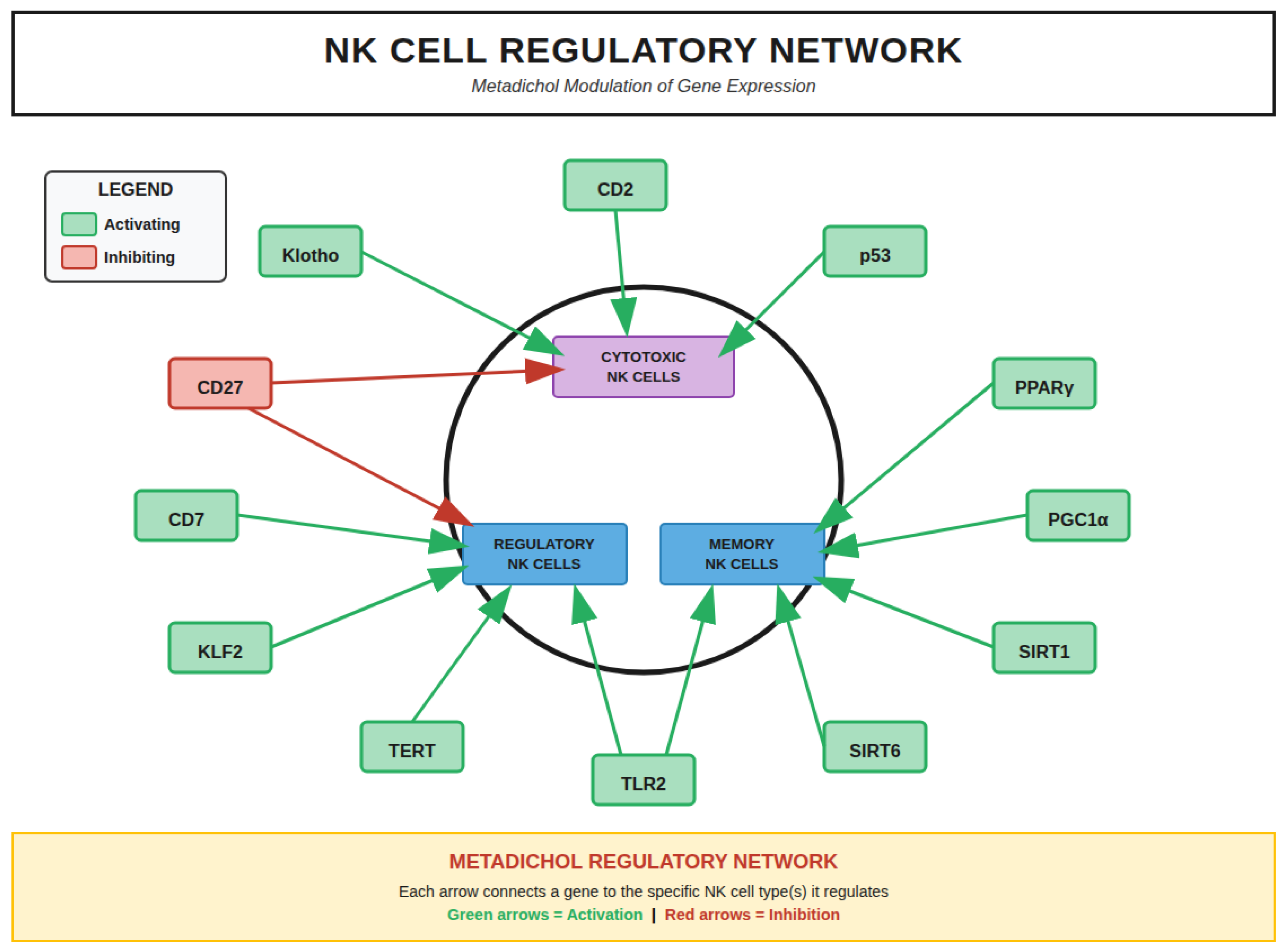
NK Cell Gene Regulatory Networks
NK Cell Gene Regulatory Network (Figure 11) showing comprehensive interactions between NK cell genes (outer ring, blue nodes) and regulatory factors (inner ring). The network integrates multiple regulatory pathways: Toll-like Receptors
(TLR2, TLR3, TLR4, TLR7, TLR9; purple), Nuclear Receptors (VDR, RAR, RXR; orange), Circadian genes (BMAL1, CLOCK, PER2; yellow), FOX transcription factors (FOXO1, FOXO3), and KLF transcription factors (KLF2, KLF4). Red arrows indicate activation (54 interactions); green arrows indicate downregulation (2 interactions). Gray nodes (CD7, CD10, CD45RA, NKG2A) represent markers with no documented regulation by these factors.
NK Cell Development Pathway
Human NK cell development proceeds through discrete stages characterized by sequential acquisition and loss of specific surface markers and transcription factors (Figure 11), shows six developmental stages: HSC (Hematopoietic Stem Cell) → CLP (Common Lymphoid Progenitor) → NKP (NK Progenitor) → iNK (Immature NK) → Mature NK (CD56bright/CD56dim) → Activated NK (Effector). Each stage displays characteristic surface markers, transcription factors, and key regulators. Metadichol’s non-viral cellular reprogramming effects are shown at the top, including induction of Yamanaka factors [82] (OCT4, SOX2, KLF4, c-MYC, NANOG) and modulation of DDIT4/REDD1 pathway. [83] Key regulatory events and Metadichol effects are detailed for each developmental transition.
Metadichol Effects on NK Cell Developmental Stages
Analysis of the 19 genes measured in our RT-PCR study reveals stage-specific effects of Metadichol treatment across the six recognized stages of NK cell development (Figure 11). The data demonstrate a clear pattern of accelerated developmental progression, with downregulation of early-stage markers and upregulation of late-stage maturation markers.
Stage 1 (Pro-NK/Hematopoietic Stem Cell): At this earliest stage, Metadichol treatment results in downregulation of stem cell markers CD34 (↓0.82), CD45RA (↓0.21), and CD244 (↓0.71). This pattern indicates accelerated exit from the stem cell state, pushing cells toward lineage commitment.
Stage 2 (Pre-NK/Common Lymphoid Progenitor): Continued downregulation of CD34 (↓0.82), CD38 (↓0.46), CD127 (↓0.30), CD45RA (↓0.21), CD244 (↓0.71), and CD7 (↓0.79) is observed. Importantly, upregulation of CD10 (↑2.29) and CD117 (↑1.26) marks the initiation of NK lineage commitment. This stage represents the critical transition point where cells become responsive to NK-differentiating signals.
Stage 3 (Immature NK/NK Progenitor): A pivotal shift occurs at this stage with the dramatic upregulation of CD122 (↑16.97-fold), the IL-15 receptor β-chain. This represents the switch from IL-7 dependence (CD127↓0.30) to IL-15 dependence, which is essential for NK cell survival and proliferation. CD117 (↑1.26) remains elevated while early markers continue to decline (CD45RA↓0.21, IL1R1↓0.06).
Stage 4 (CD56bright/Immature NK): This cytokine-producing stage shows sustained CD122 upregulation (↑16.97) with the emergence of NKG2D (↑3.39) and CD117 (↑1.26). The downregulation of IL1R1 (↓0.06) and CD45RA (↓0.21) continues, while CD336 (↑1.38) and CD337 (↓0.84) show differential NCR expression. This stage is characterized by enhanced cytokine production capacity.
Stage 5 (Transitional/Maturing NK): The acquisition of cytotoxic function is marked by sustained upregulation of CD122 (↑16.97), NKG2D (↑3.39), and the critical appearance of NKP80 (↑3.89). CD56 downregulation (↓0.68) indicates transition from CD56bright to CD56dim phenotype. NKG2A shows modest downregulation (↓0.79), reducing inhibitory signaling. CD244 (↓0.71) and CD337 (↓0.84) continue to decline.
Stage 6 (CD56dim/Mature Cytotoxic NK): The terminal cytotoxic effector phenotype is characterized by maximal expression of activating receptors: CD122 (↑16.97), NKG2D (↑3.39), and NKP80 (↑3.89). T-bet upregulation (↑1.30) confirms terminal maturation. The continued downregulation of CD56 (↓0.68) and NKG2A (↓0.79) with stable
Push-Pull Developmental Acceleration Mechanism
The pattern of gene expression changes induced by Metadichol reveals a coordinated “push-pull” mechanism that accelerates NK cell developmental progression (Figure 12). This mechanism operates through three complementary processes.
PUSH Mechanism - Forcing Exit from Immature Stages: The downregulation of early markers including CD34 (↓0.82), CD127 (↓0.30), CD45RA (↓0.21), and IL1R1 (↓0.06) creates a “push” force that drives cells out progenitor markers removes the molecular anchors that maintain cells in early developmental states, forcing progression toward maturity.
PULL Mechanism - Attracting Cells Toward Mature Phenotype: Simultaneously, the dramatic upregulation of late-stage markers including CD122 (↑16.97), NKG2D (↑3.39), and NKP80 (↑3.89) creates a “pull” force that attracts cells toward the mature cytotoxic phenotype. These markers define functional NK cells capable of tumor recognition and killing, and their premature expression accelerates acquisition of effector function.
BALANCE Mechanism - Ensuring Survival During Transition: The critical balance element is the 16.97-fold upregulation of CD122, which ensures that IL-15 survival signaling compensates for the loss of early survival signals (IL-7/CD127). Without this compensation, the downregulation of CD127 (IL-7Rα) could The massive CD122 upregulation provides a survival bridge that allows cells to safely traverse the developmental stages.
Impact of Metadichol-Induced Gene Regulation on NK Cell Development: Push-Pull Mechanism.
Stage-Specific Impacts, Risks, and Compensatory Mechanisms
Push-Pull Mechanism. Comprehensive six-stage analysis showing downregulated markers (red boxes), upregulated markers (green boxes), developmental impact (yellow boxes), potential risks (orange boxes), and compensatory mechanisms (blue boxes) at each stage.. KEY MECHANISM: PUSH = Downregulation of early markers (CD34, CD127, CD45RA, IL1R1) forces cells to exit immature stages; PULL = Upregulation of late markers (CD122, NKG2D, NKP80) attracts cells toward mature cytotoxic phenotype; BALANCE = CD122↑ (6.97x) ensures IL-15 survival signaling compensates for loss of early survival signals (IL-7/CD127). Each stage includes specific impacts, risks, and compensatory mechanisms.
Stage-Specific Impacts, Risks, and Compensatory Mechanisms
NK Cell Developmental Stages with Metadichol Effects on Measured Markers. Six-stage developmental scheme
Stage 1-2 Transition (Pro-NK to Pre-NK): The primary impact is accelerated exit from stem state and rapid NK lineage commitment. The potential risk is premature differentiation and loss of IL-7 survival signal. Compensation is provided by CD117 upregulation (↑1.26), which maintains SCF responsiveness, and CD122 upregulation (↑16.97), which enables IL-15 responsiveness before complete loss of IL-7 signaling.
Stage 3 Transition (Immature NK): The critical switch to IL-15 dependence occurs here. The impact is enhanced survival through IL-15 signaling. The risk is inflammatory vulnerability during the transition period. Compensation is provided by IL1R1 downregulation (↓0.06), which protects cells from inflammatory exhaustion while CD122 upregulation ensures robust IL-15 responsiveness.
Stage 4 Transition (CD56bright): Enhanced cytokine production capacity is the primary impact. The risk is reduced proliferation as cells transition toward terminal differentiation. Compensation occurs through NKG2D upregulation (↑3.39), which enhances activation signals that maintain cellular responses even as proliferative capacity decreases.
Stage 5 Transition (Transitional): Acquisition of cytotoxic function defines this stage. The potential risk is over-activation leading to inappropriate responses. This is balanced by NKG2A expression (↓0.79 but not absent), which maintains inhibitory checkpoints while allowing enhanced activation through upregulated NKG2D and NKP80.
Stage 6 (CD56dim Mature): The terminal cytotoxic effector phenotype is achieved with maximum NKG2D (↑3.39), NKP80 (↑3.89), and T-bet (↑1.30) expression. The risk is reduced self-renewal capacity in terminally differentiated cells. Compensation is provided by T-bet upregulation, which stabilizes the mature phenotype and maintains effector function even without further proliferative potential.
Enhancement of the IL-15/CD122 Signaling Axis
One of the most striking findings of this study was the marked upregulation of CD122 (IL-2Rβ), with expression reaching 9.19-fold at 1 pg/ml and 9.14-fold at 100 ng/ml (p<0.05). CD122 forms a critical component of the IL-15 receptor complex, and its upregulation has profound implications for NK cell biology. IL-15 signaling through a receptor complex containing CD122 and the common γ-chain (CD132) activates JAK1/JAK3 and STAT5 signaling pathways, which are essential for NK cell survival and proliferation. [84,85] and demonstrated reversible defects are seen in NK cell lineages in IL-15-deficient mice, [86] and IL-15-mediated survival of NK cells is determined by interactions among Bim, Noxa, and Mcl-1, with CD122 expression being rate-limiting for this survival signaling . [87]
The strong positive correlation observed between CD122 and CD34 (r=0.960) in our dataset reflects the developmental relationship between these markers during early NK cell ontogeny. It is established that human NK cell development in secondary lymphoid tissues proceeds through discrete stages marked by sequential acquisition of CD122 expression. [88,89] Further characterized the location and cellular stages of NK cell development, demonstrating that CD34+CD117+ cells acquire CD122 expression as they commit to the NK lineage. [90] The inverse correlation between CD122 and early progenitor markers including CD45RA (r=-0.972) and CD7 (r=-0.957) observed in our study suggests that Metadichol-induced CD122 upregulation is associated with progression beyond the early progenitor stage toward a more mature phenotype.
Integrated Gene-Gene Interactions and Signaling Pathways:
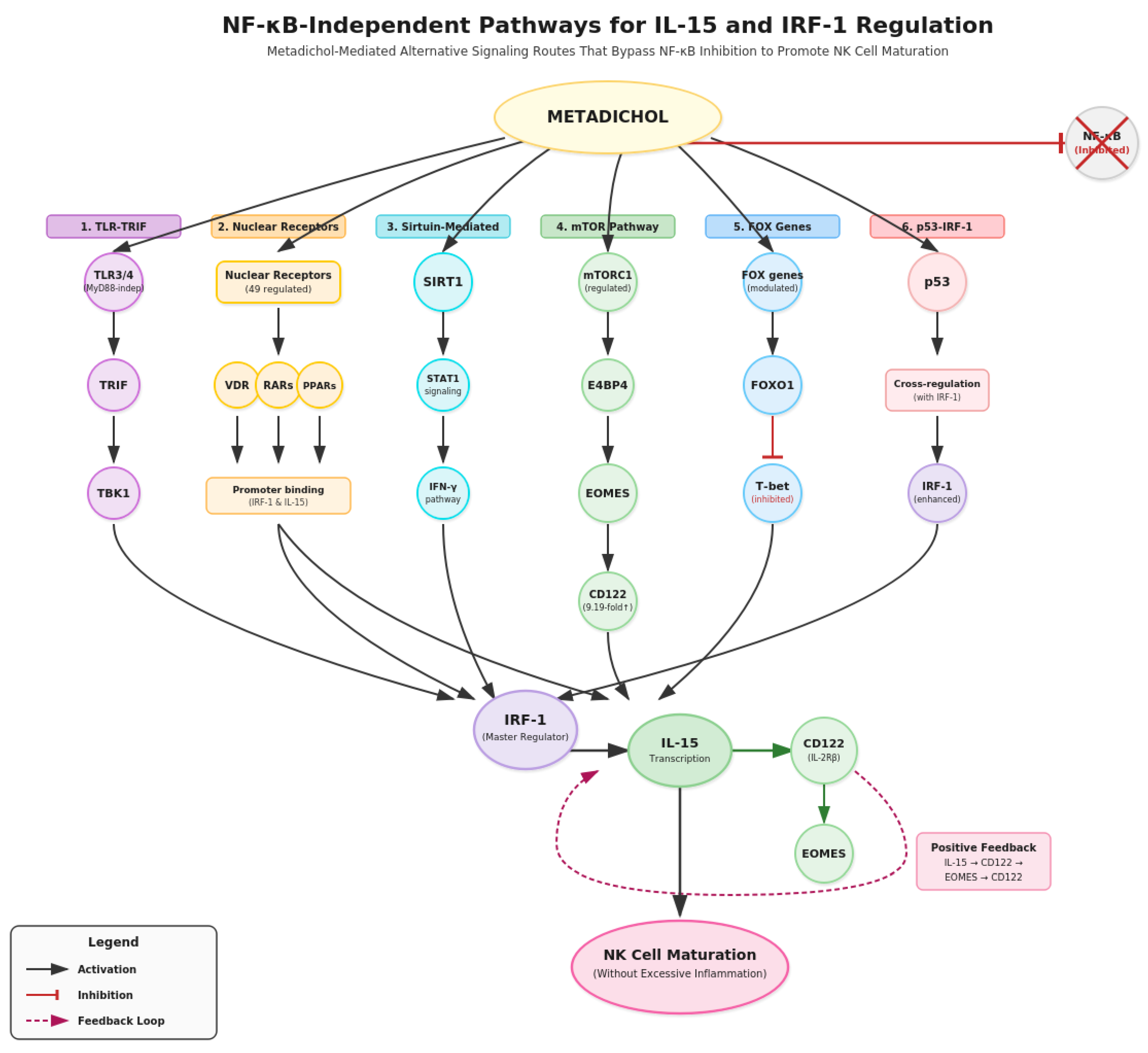
NK Cell Gene Regulatory Network (Figure 14) with NF-κB Independent Routes.
Key Metadichol Pathways (NF-κB Independent Routes): TLRs → TRIF → IRF-1 → IL-15; VDR/RAR/PPAR → IRF-1; SIRT1 → STAT1 → IRF-1; mTORC1 → E4BP4 → EOMES.
The integrated network visualization (Figure 14) reveals the complex interplay between cytokines, transcription factors, receptors, and signaling molecules that regulate NK cell function. Central to this network is IL-15, which connects multiple pathways including mTORC1 signaling, STAT activation, and transcription factor induction. The Metadichol pathways operate through NF-κB independent routes, including TLRs → TRIF → IRF-1 → IL-15, VDR/RAR/PPAR → IRF-1, SIRT1 → STAT1 → IRF-1, and mTORC1 → E4BP4 → EOMES. These alternative pathways may explain how Metadichol achieves immune activation without excessive inflammation. The network demonstrates 54 activation pathways and 2 inhibitory pathways that coordinate NK cell responses.
mTOR Signaling and Metabolic Control of NK Cell Function
The metabolic checkpoint kinase mTOR represents a central node in Metadichol’s regulatory network with direct implications for NK cell biology. mTOR is essential for IL-15 signaling during NK cell development and activation. [91] IL-15 binding to CD122 activates mTORC1 and mTORC2 complexes, which regulate NK cell metabolism, proliferation, and cytotoxic capacity. mTORC1 and mTORC2 differentially promote NK cell development, with mTORC1 controlling cell growth and mTORC2 regulating cytokine production. [92,93]
Metadichol’s capacity to modulate mTOR signaling provides a mechanism for optimizing NK cell metabolic fitness. The upregulation of CD122 observed in our study would enhance IL-15-mTOR signaling, promoting the metabolic reprogramming required for NK cell effector function. Importantly, Metadichol also influences DDIT4/REDD1, a negative regulator of mTOR. [83] REDD1 regulates mTOR hyperactivation. [94]
Figure 13.
Upregulation of Activating Receptors and Cytotoxic Potential
The 8.59-fold upregulation of NKG2D at 1 pg/ml (p<0.001) represents a particularly significant finding given the central role of this receptor in tumor immunosurveillance. NKG2D (KLRK1) recognizes stress-induced ligands including MICA, MICB, and ULBPs on transformed or infected cells, providing a dominant activation signal for NK cell-mediated cytotoxicity.[95] Lanier emphasized that NKG2D functions as a master regulator of NK cell activation, capable of overcoming inhibitory signals when sufficiently engaged.
The relationship between NKG2D upregulation and tumor recognition is further enhanced by Metadichol’s effects on p53 signaling. Textor et al. demonstrated that human NK cells are alerted to induction of p53 in cancer cells through upregulation of the NKG2D ligands ULBP1 and ULBP2. [96] Pharmacological activation of p53 triggers anticancer innate immune response through induction of ULBP2 . Metadichol’s capacity to modulate TP53 expression in tumor cells, combined with NKG2D upregulation on NK cells, creates a synergistic mechanism for enhanced tumor recognition and elimination.
NKp80 (KLRF1), which showed 4.91-fold upregulation at 1 pg/ml (p<0.05), marks a critical developmental checkpoint in NK cell maturation. NKp80 is a triggering receptor that cooperates with natural cytotoxicity receptors (NCRs) to provide optimal NK cell activation.[97] NK cell receptors are tools to analyze NK cell development, subsets, and function, establishing NKp80 as a marker of the Stage 2 to Stage 3 transition.[98] The strong correlation between NKp80 and CD117 (r=0.945) observed in our dataset is consistent with this temporal expression pattern during NK development.
Toll-Like Receptor Modulation and NK Cell Priming
Metadichol’s regulatory network includes modulation of all Toll-like receptors (TLRs), which has profound effects on NK cell activation and function. The integration of TLR, NCR, and KIR signaling in NK cells demonstrates that TLR engagement primes NK cells for enhanced cytotoxicity. [99] TLR3, TLR7, and NKG2D regulate IFN-γ secretion and cytotoxicity in human NK cells stimulated with IL-12. [100] TLR7/8-mediated activation of human NK cells results in IFN-γ production. [101]
The capacity of Metadichol to modulate TLR signaling provides an additional mechanism for NK cell activation that complements the observed upregulation of activating receptors. TLR-mediated priming enhances NK cell responsiveness to subsequent activating receptor engagement, lowering the threshold for cytotoxic degranulation. This is particularly relevant in the context of tumor immunosurveillance, where danger-associated molecular patterns (DAMPs) released by dying tumor cells can engage TLRs on NK cells.
Transcription Factor Coordination: FOXO1, T-bet, and EOMES
The near-perfect correlation between T-bet and EOMES expression (r=0.957) observed in our correlation analysis aligns with their established roles as cooperative master regulators of NK cell development and function. T-bet and EOMES control key checkpoints of NK cell maturation, with both transcription factors required for acquisition of full effector function. [102,103] These T-box transcription factors share target genes including CD122, cytotoxic effector molecules (perforin, granzymes), and cytokine genes (IFN-γ).
FOXO1 serves as an important negative regulator in this transcriptional network. It has been demonstrated that transcription factor FOXO1 is a negative regulator of NK cell maturation and function.[104] CD226 regulates NK cell antitumor responses via phosphorylation-mediated inactivation of FOXO1. [105] FOXO1 represses T-bet-mediated effector functions. [106] The interplay between FOXO1, T-bet, and EOMES creates a regulatory circuit that controls the balance between NK cell quiescence and activation.
Nuclear Receptor Signaling in NK Cell Development
Metadichol’s capacity to activate all 48 nuclear receptors, has significant implications for NK cell biology. Rhee et al. reviewed the regulation of NK cell development and function by nuclear receptor signaling, demonstrating that the vitamin D receptor (VDR), retinoic acid receptors (RARs), and other nuclear receptors influence NK cell differentiation and effector function.[107] A recent publication has characterized vitamin D receptor biology and signaling in immune function. [108] A-trans retinoic acid enhances effector functions of human NK cells. [109]
Yamanaka Factors and Cellular Reprogramming Potential
A unique aspect of Metadichol’s mechanism involves its capacity to induce expression of Yamanaka reprogramming factors (OCT4, SOX2, KLF4, c-MYC, NANOG). These defined factors could induce pluripotent stem cells from somatic cells. [110] Metadichol acts as a natural ligand for expression of Yamanaka reprogramming factors in human cardiac, fibroblast, and cancer cell lines. [82] representing a non-viral approach to cellular reprogramming. The KLF family of transcription factors, in particular, regulates T cell and NK cell activation, differentiation, and prevention of exhaustion.
Modulation of Inhibitory Receptor NKG2A
NKG2A, the major inhibitory receptor recognizing HLA-E on target cells, showed significant downregulation at 100 pg/ml (p<0.01). This finding has important implications for NK cell functional capacity. Anti-NKG2A monoclonal antibodies function as checkpoint inhibitors that promote anti-tumor immunity by unleashing both T and NK cells. [111] NKG2A engagement recruits SHP-1 and SHP-2 phosphatases that dephosphorylate signaling molecules downstream of activating receptors, effectively blocking NKG2D-mediated and NCR-mediated activation. [112,113] The downregulation of NKG2A at specific concentrations, combined with upregulation of activating receptors (NKG2D, NKp80), shifts the balance toward NK cell activation.
Anti-Cancer Therapeutic Implications
The gene expression profile induced by Metadichol has significant implications for NK cell and T cell regulation in anti-cancer therapy (Figure 15). The multi-pathway approach suggests several therapeutic applications:
Enhancement of Immune Checkpoint Therapy: The combination of NKG2D upregulation (↑3.39) and NKG2A downregulation (↓0.79) mimics the functional effect of NKG2A checkpoint inhibitors currently in clinical development. [114]
Improvement of Adoptive Cell Therapy: The dramatic enhancement of IL-15 responsiveness through CD122 upregulation (↑16.97-fold) could significantly augment NK cell-based immunotherapies, including CAR-NK and adoptive cell transfer approaches.
Metabolic Optimization: Metadichol’s modulation of the mTOR pathway provides metabolic support for sustained anti-tumor activity while the DDIT4/REDD1 pathway prevents exhaustion.
Push-Pull Developmental Acceleration: The coordinated downregulation of early markers with upregulation of mature effector markers accelerates the production of cytotoxic NK cells capable of tumor cell recognition and killing.
Figure 14.
Metadichol’s regulatory effects on NK cell and T cell function in the context of anti-cancer immunity, showing enhanced activation, metabolic support, and anti-exhaustion effects.
Integrated Mechanism of Metadichol Action
Integrating the observed expression changes with established NK cell regulatory networks and Metadichol’s multi-pathway effects (Figure 15)
Figure 15.
Gene regulatory network model depicting Metadichol-mediated modulatio.

Conclusions
Natural killer cells are indispensable sentinels of the innate immune system, providing critical first-line defense against malignancies and viral infections. In cancer, NK cells perform essential immunosurveillance functions, directly recognizing and eliminating tumor cells without prior sensitization—a unique capability that has propelled NK cell-based therapies to the forefront of cancer immunotherapy. Clinical applications including CAR-NK cells, adoptive NK cell transfer, and NK cell engagers have demonstrated promising efficacy against hematological malignancies and solid tumors, yet optimizing NK cell maturation and cytotoxic function remains a critical challenge.
The clinical success of these immunotherapies depends fundamentally on generating mature, cytotoxically competent effector cells—a process critically dependent on IL-15 signaling. IL-15, acting through its receptor containing CD122 (IL-2Rβ), governs NK cell survival, proliferation, and acquisition of effector function, making the IL-15/CD122 axis the central regulatory node in NK cell biology.
This study demonstrates that Metadichol potently enhances this critical developmental pathway. The 9.19-fold upregulation of CD122 dramatically amplifies IL-15 responsiveness, while concurrent upregulation of NKG2D (8.59-fold) and NKp80 (4.91-fold) confirms accelerated maturation toward functional cytotoxic phenotypes. Simultaneously, suppression of early progenitor markers (CD127, CD45RA, CD7) indicates developmental progression beyond IL-7-dependent stages toward IL-15-dependent maturation.
Mechanistically, Metadichol achieves this through NF-κB-independent pathways, including TLR-TRIF signaling, nuclear receptor activation, and the mTORC1→E4BP4→EOMES→CD122 positive feedback loop—enhancing IL-15 signaling without excessive inflammation.
The coordinated “push-pull” mechanism identified here represents a novel paradigm for NK cell developmental manipulation, positioning Metadichol as a promising agent for enhancing NK cell-based cancer immunotherapy and infectious disease treatment given its non toxic nature with a LD50 greater than 5000 mg per kilo..[115,116]
Supplementary Materials
Raw data is available on request. The author is the founder of Nanorx Inc. and is a major shareholder in the company. This study was conducted independently by a third-party external laboratory on commercial terms to any eliminate bias in our results.
Abbreviations
Surface Markers & Receptors
| Abbreviation | Full Name |
| CD7 | Cluster of Differentiation 7 |
| CD10 | Neprilysin/Membrane Metalloendopeptidase (MME) |
| CD34 | Hematopoietic Progenitor Cell Antigen CD34 |
| CD38 | Cyclic ADP Ribose Hydrolase |
| CD45RA | Protein Tyrosine Phosphatase Receptor Type C (naive isoform) |
| CD56 | Neural Cell Adhesion Molecule (NCAM) |
| CD117 | c-Kit; Stem Cell Factor Receptor |
| CD122 | Interleukin-2 Receptor Beta (IL-2Rβ) |
| CD127 | Interleukin-7 Receptor Alpha (IL-7Rα) |
| CD132 | Common Gamma Chain (γc) |
| CD244 | 2B4/SLAMF4 |
| CD314 | NKG2D/KLRK1 |
| CD336 | NKp44/NCR2 |
| CD337 | NKp30/NCR3 |
| IL1R1 | Interleukin-1 Receptor Type 1 |
| NCR | Natural Cytotoxicity Receptor |
| NKG2A | Natural Killer Group 2 Member A (CD159a/KLRC1) |
| NKG2D | Natural Killer Group 2 Member D (CD314/KLRK1) |
| NKp30 | Natural Killer Cell p30-Related Protein (NCR3/CD337) |
| NKp44 | Natural Killer Cell p44-Related Protein (NCR2/CD336) |
| NKp46 | Natural Killer Cell p46-Related Protein (NCR1/CD335) |
| NKp80 | Natural Killer Cell p80-Related Protein (KLRF1) |
NKG2D Ligands
| Abbreviation | Full Name |
| MICA | MHC Class I Chain-Related Protein A |
| MICB | MHC Class I Chain-Related Protein B |
| ULBPs | UL16-Binding Proteins (ULBP1-6) |
Cytokines & Growth Factors
| Abbreviation | Full Name |
| IFN-γ | Interferon-Gamma |
| IL-2 | Interleukin-2 |
| IL-7 | Interleukin-7 |
| IL-15 | Interleukin-15 |
| SCF | Stem Cell Factor |
| TNF-α | Tumor Necrosis Factor-Alpha |
Transcription Factors
| Abbreviation | Full Name |
| ATF4 | Activating Transcription Factor 4 |
| E4BP4 | E4 Promoter-Binding Protein 4 (NFIL3) |
| EOMES | Eomesodermin |
| FOXO1 | Forkhead Box O1 |
| FOXO3 | Forkhead Box O3 |
| IRF-1 | Interferon Regulatory Factor 1 |
| KLF2 | Krüppel-Like Factor 2 |
| KLF4 | Krüppel-Like Factor 4 |
| NF-κB | Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells |
| OCT4 | Octamer-Binding Transcription Factor 4 |
| SOX2 | SRY-Box Transcription Factor 2 |
| STAT5 | Signal Transducer and Activator of Transcription 5 |
| T-bet | T-Box Transcription Factor 21 (TBX21) |
| TP53 | Tumor Protein P53 |
Signaling Molecules & Kinases
| Abbreviation | Full Name |
| JAK1 | Janus Kinase 1 |
| JAK3 | Janus Kinase 3 |
| mTOR | Mechanistic Target of Rapamycin |
| mTORC1 | Mechanistic Target of Rapamycin Complex 1 |
| mTORC2 | Mechanistic Target of Rapamycin Complex 2 |
| REDD1 | Regulated in Development and DNA Damage Responses 1 (DDIT4) |
| TBK1 | TANK-Binding Kinase 1 |
| TRIF | TIR-Domain-Containing Adapter-Inducing Interferon-β |
Nuclear Receptors
| Abbreviation | Full Name |
| PPAR | Peroxisome Proliferator-Activated Receptor |
| PPARγ | Peroxisome Proliferator-Activated Receptor Gamma |
| RAR | Retinoic Acid Receptor |
| VDR | Vitamin D Receptor |
Toll-Like Receptors
| Abbreviation | Full Name |
| TLR | Toll-Like Receptor |
| TLR2 | Toll-Like Receptor 2 |
| TLR3 | Toll-Like Receptor 3 |
| TLR4 | Toll-Like Receptor 4 |
Housekeeping & Reference Genes
| Abbreviation | Full Name |
| ACTB | Actin Beta |
| B2M | Beta-2-Microglobulin |
| GAPDH | Glyceraldehyde-3-Phosphate Dehydrogenase |
| HPRT1 | Hypoxanthine Phosphoribosyltransferase 1 |
Cell Types & Developmental Stages
| Abbreviation | Full Name |
| CAR-NK | Chimeric Antigen Receptor Natural Killer Cell |
| CLP | Common Lymphoid Progenitor |
| HSC | Hematopoietic Stem Cell |
| iNK | Immature Natural Killer Cell |
| NK | Natural Killer |
| NKP | Natural Killer Progenitor |
| PBMC | Peripheral Blood Mononuclear Cell |
Techniques & Methods
| Abbreviation | Full Name |
| cDNA | Complementary DNA |
| EDTA | Ethylenediaminetetraacetic Acid |
| FBS | Fetal Bovine Serum |
| PBS | Phosphate-Buffered Saline |
| qRT-PCR | Quantitative Real-Time Polymerase Chain Reaction |
| RNA | Ribonucleic Acid |
| SEM | Standard Error of the Mean |
Clinical & Disease Terms
| Abbreviation | Full Name |
| AML | Acute Myeloid Leukemia |
| DAMP | Damage-Associated Molecular Pattern |
| HLA-E | Human Leukocyte Antigen-E |
| MHC | Major Histocompatibility Complex |
References
- Arachchige, A.S.P.M. Human NK cells: From development to effector functions. J. Endotoxin Res. 2021, 27, 212–229. [Google Scholar] [CrossRef]
- Kucuksezer, U.C.; Cetin, E.A.; Esen, F.; Tahrali, I.; Akdeniz, N.; Gelmez, M.Y.; Deniz, G. The Role of Natural Killer Cells in Autoimmune Diseases. Front. Immunol. 2021, 12, 622306. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef]
- Cichocki, F.; Grzywacz, B.; Miller, J.S. Human NK Cell Development: One Road or Many? Front. Immunol. 2019, 10, 2078. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; Grzywacz, B.; Miller, J.S. Human NK Cell Development: One Road or Many? Front. Immunol. 2019, 10, 2078. [Google Scholar] [CrossRef]
- Eissens, D.N.; Spanholtz, J.; Van Der Meer, A.; Van Cranenbroek, B.; Dolstra, H.; Kwekkeboom, J.; Preijers, F.W.M.B.; Joosten, I. Defining Early Human NK Cell Developmental Stages in Primary and Secondary Lymphoid Tissues. PLOS ONE 2012, 7, e30930. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, S.; Xin, J.; Wang, J.; Yao, C.; Zhang, Z. Role of NKG2D and its ligands in cancer immunotherapy. 2019, 9, 2064–2078. [Google Scholar]
- Wensveen, F.M.; Jelenčić, V.; Polić, B. NKG2D: A Master Regulator of Immune Cell Responsiveness. Front. Immunol. 2018, 9, 441. [Google Scholar] [CrossRef]
- Zhou, X.; Hao, Q.; Zhang, Q.; et al. Interleukin-2 (IL-2) interacts with IL-2 receptor beta (IL-2Rβ). Front Immunol. 2020, 11, 584983. [Google Scholar] [CrossRef]
- Liu, J.; Chen, D.; Nie, G.D.; et al. CD8+CD122+ T-cells: a newly emerging regulator with immunosuppressive function. Mediators Inflamm. 2015, 2015, 690190. [Google Scholar] [CrossRef]
- Keller, H.R.; Srivastava, S.; Hombach, A.; Spranger, S.; Cendales, L.; Bhavsar, J.D. The abundance and availability of cytokine receptor IL-2Rβ (CD122) constrain the lymphopenia-induced homeostatic proliferation. J Immunol. 2020, 204, 3227–3235. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tian, Z.; Wei, H. Developmental and Functional Control of Natural Killer Cells by Cytokines. Front. Immunol. 2017, 8, 930. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.V.; Dong, Y.; Higginbotham, L.B.; Kim, S.C.; Breeden, C.P.; Stobert, E.A.; Jenkins, J.; Tso, J.Y.; Larsen, C.P.; Adams, A.B. CD122 signaling in CD8+ memory T cells drives costimulation-independent rejection. J. Clin. Investig. 2018, 128, 4557–4572. [Google Scholar] [CrossRef]
- Villarreal, D.O.; L’Huillier, A.; Bhardwaj, N.; et al. Targeting of CD122 enhances antitumor immunity by modulating the adaptive immune response. J Clin Invest. 2017, 127, 4307–4319. [Google Scholar] [CrossRef]
- Barros, P.O.; Berthoud, T.K.; Aloufi, N.; Angel, J.B. Soluble IL-7Rα/sCD127 in health, disease, and its potential role as a therapeutic agent. Immunotargets Ther. 2021, 10, 47–62. [Google Scholar] [CrossRef]
- Liu, W.; Putnam, A.L.; Xu-Yu, Z.; Szot, G.L.; Lee, M.R.; Zhu, S.; Gottlieb, P.A.; Kapranov, P.; Gingeras, T.R.; de St Groth, B.F.; et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 2006, 203, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Carrette, F.; Surh, C.D. IL-7 signaling and CD127 receptor regulation in the control of T cell homeostasis. Semin. Immunol. 2012, 24, 209–217. [Google Scholar] [CrossRef]
- Galarza-Muñoz, G.; Briggs, F.B.; Evsyukova, I.; Schott-Lerner, G.; Kennedy, E.M.; Nyanhete, T.; Wang, L.; Bergamaschi, L.; Widen, S.G.; Tomaras, G.D.; et al. Human Epistatic Interaction Controls IL7R Splicing and Increases Multiple Sclerosis Risk. Cell 2017, 169, 72–84.e13. [Google Scholar] [CrossRef]
- Cottini, F.; Rodriguez, J.; Hughes, T.; Sharma, N.; Guo, L.; Lozanski, G.; Liu, B.; Cocucci, E.; Yang, Y.; Benson, D. Redefining CD56 as a Biomarker and Therapeutic Target in Multiple Myeloma. Mol. Cancer Res. 2022, 20, 1083–1095. [Google Scholar] [CrossRef]
- Van Acker, H.H.; Capsomidis, A.; Smits, E.L.; Van Tendeloo, V.F. CD56 in the Immune System: More Than a Marker for Cytotoxicity? Front. Immunol. 2017, 8, 892–892. [Google Scholar] [CrossRef]
- Poznanski, S.M.; Ashkar, A.A. Shining light on the significance of NK cell CD56 brightness. Cell. Mol. Immunol. 2018, 15, 1071–1073. [Google Scholar] [CrossRef] [PubMed]
- Gunesch, J.T.; Dixon, A.L.; Ebrahim, T.A.; Berrien-Elliott, M.M.; Tatineni, S.; Kumar, T.; Hegewisch-Solloa, E.; A Fehniger, T.; Mace, E.M. CD56 regulates human NK cell cytotoxicity through Pyk2. eLife 2020, 9, e57346. [Google Scholar] [CrossRef]
- Gunesch, J.T.; Orange, J.S. CD56/NCAM mediates cell migration of human NK cells by regulating cell polarity and actin flow. Mol Biol Cell. 2024, 35, ar45. [Google Scholar] [CrossRef]
- Ziegler, S.; Weiss, E.; Schmitt, A.-L.; Schlegel, J.; Burgert, A.; Terpitz, U.; Sauer, M.; Moretta, L.; Sivori, S.; Leonhardt, I.; et al. CD56 Is a Pathogen Recognition Receptor on Human Natural Killer Cells. Sci. Rep. 2017, 7, 5874. [Google Scholar] [CrossRef] [PubMed]
- Rabinowich, H.; Pricop, L.; Herberman, R.B.; Whiteside, T.L. Expression and function of CD7 molecule on human natural killer cells. J. Immunol. 1994, 152, 517–526. [Google Scholar] [CrossRef]
- Aandahl, E.M.; Sandberg, J.K.; Beckerman, K.P.; Taskén, K.; Moretto, W.J.; Nixon, D.F. CD7 Is a Differentiation Marker That Identifies Multiple CD8 T Cell Effector Subsets. J. Immunol. 2003, 170, 2349–2355. [Google Scholar] [CrossRef]
- Piedra-Quintero, Z.L.; Wilson, Z.; Nava, P.; Guerau-De-Arellano, M. CD38: An Immunomodulatory Molecule in Inflammation and Autoimmunity. Front. Immunol. 2020, 11, 597959. [Google Scholar] [CrossRef]
- Glaría, E.; Valledor, A.F. Roles of CD38 in the Immune Response to Infection. Cells 2020, 9, 228. [Google Scholar] [CrossRef]
- Li, W.; Li, Y.; Jin, X.; Liao, Q.; Chen, Z.; Peng, H.; Zhou, Y. CD38: A Significant Regulator of Macrophage Function. Front. Oncol. 2022, 12, 775649. [Google Scholar] [CrossRef] [PubMed]
- Polano, M.; Fabbiani, E.; Andreuzzi, E.; Di Cintio, F.; Bedon, L.; Gentilini, D.; Mongiat, M.; Ius, T.; Arcicasa, M.; Skrap, M.; et al. A New Epigenetic Model to Stratify Glioma Patients According to Their Immunosuppressive State. Cells 2021, 10, 576. [Google Scholar] [CrossRef]
- Shah, K.G.; Wu, R.; Jacob, A.; Blau, S.A.; Ji, Y.; Dong, W.; Marini, C.P.; Ravikumar, T.S.; Coppa, G.F.; Wang, P. Human Ghrelin Ameliorates Organ Injury and Improves Survival after Radiation Injury Combined with Severe Sepsis. Mol. Med. 2009, 15, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Camponeschi, A.; Kläsener, K.; Sundell, T.; Lundqvist, C.; Manna, P.T.; Ayoubzadeh, N.; Sundqvist, M.; Thorarinsdottir, K.; Gatto, M.; Visentini, M.; et al. Human CD38 regulates B cell antigen receptor dynamic organization in normal and malignant B cells. J. Exp. Med. 2022, 219, e20220201. [Google Scholar] [CrossRef]
- Gurney, M.; Gornalusse, G.G.; Wang, X.; et al. CD38 contributes to human natural killer cell responses to multiple myeloma. bioRxiv. 2019. [Google Scholar] [CrossRef]
- Pulliam, T.; Jani, S.; Jing, L.; Ryu, H.; Jojic, A.; Shasha, C.; Zhang, J.; Kulikauskas, R.; Church, C.; Garnett-Benson, C.; et al. Circulating cancer-specific CD8 T cell frequency is associated with response to PD-1 blockade in Merkel cell carcinoma. Cell Rep. Med. 2024, 5, 101412. [Google Scholar] [CrossRef]
- Jiao, Y.; Yi, M.; Xu, L.; Chu, Q.; Yan, Y.; Luo, S.; Wu, K. CD38: targeted therapy in multiple myeloma and therapeutic potential for solid cancers. Expert Opin. Investig. Drugs 2020, 29, 1295–1308. [Google Scholar] [CrossRef] [PubMed]
- van de Donk, N.W.C.J.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef]
- Al Barashdi, M.A.; Ali, A.; McMullin, M.F.; Mills, K. Protein tyrosine phosphatase receptor type C (PTPRC or CD45). J. Clin. Pathol. 2021, 74, 548–552. [Google Scholar] [CrossRef]
- Krzywinska, E.; Cornillon, A.; Allende-Vega, N.; Vo, D.-N.; Rene, C.; Lu, Z.-Y.; Pasero, C.; Olive, D.; Fegueux, N.; Ceballos, P.; et al. CD45 Isoform Profile Identifies Natural Killer (NK) Subsets with Differential Activity. PLOS ONE 2016, 11, e0150434. [Google Scholar] [CrossRef]
- Henson, S.M.; E Riddell, N.; Akbar, A.N. Properties of end-stage human T cells defined by CD45RA re-expression. Curr. Opin. Immunol. 2012, 24, 476–481. [Google Scholar] [CrossRef]
- Hang, J.; Liu, Y.; Zhang, Y.; et al. The clinical implication of CD45RA+ naïve T cells in advanced pancreatic cancer. J Transl Med. 2019, 17, 97. [Google Scholar] [CrossRef]
- Tian, Y.; Babor, M.; Lane, J.; Schulten, V.; Patil, V.S.; Seumois, G.; Rosales, S.L.; Fu, Z.; Picarda, G.; Burel, J.; et al. Unique phenotypes and clonal expansions of human CD4 effector memory T cells re-expressing CD45RA. Nat. Commun. 2017, 8, 1703. [Google Scholar] [CrossRef] [PubMed]
- McGuire, D.J.; Akondy, R.S.; Yang, S.; Edupuganti, S.; Nagar, S.; Michael, G.; De Rosa, S.C.; Newell, E.W.; Farber, D.L.; Kissick, H.T.; et al. Regulation of CD45 isoforms during human effector and memory CD8 T cell differentiation: Implications for T cell nomenclature. Proc. Natl. Acad. Sci. 2025, 122, e2322982122. [Google Scholar] [CrossRef]
- Miettinen, M.; Lasota, J. KIT (CD117): A Review on Expression in Normal and Neoplastic Tissues, and Mutations and Their Clinicopathologic Correlation. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.M.; Zaidi, D.; Young, T.R.; Mobley, M.E.; Kerr, B.A. CD117/c-kit in Cancer Stem Cell-Mediated Progression and Therapeutic Resistance. Biomedicines 2018, 6, 31. [Google Scholar] [CrossRef]
- Cardoso, H.J.; Figueira, M.I.; Socorro, S. The stem cell factor (SCF)/c-KIT signalling in testis and prostate cancer. J. Cell Commun. Signal. 2017, 11, 297–307. [Google Scholar] [CrossRef]
- Rönnstrand, L. Signal transduction via the stem cell factor receptor/c-Kit. Cell. Mol. Life Sci. 2004, 61, 2535–2548. [Google Scholar] [CrossRef]
- Czechowicz, A.; Palchaudhuri, R.; Sber, M.; et al. Selective hematopoietic stem cell ablation using CD117-targeting. Nat Biotechnol. 2019, 37, 1192–1201. [Google Scholar] [CrossRef]
- Sidney, L.E.; Branch, M.J.; Dunphy, S.E.; Dua, H.S.; Hopkinson, A. Concise Review: Evidence for CD34 as a Common Marker for Diverse Progenitors. Stem Cells 2014, 32, 1380–1389. [Google Scholar] [CrossRef]
- Radu, P.; Zurzu, M.; Paic, V.; Bratucu, M.; Garofil, D.; Tigora, A.; Georgescu, V.; Prunoiu, V.; Pasnicu, C.; Popa, F.; et al. CD34—Structure, Functions and Relationship with Cancer Stem Cells. Medicina 2023, 59, 938. [Google Scholar] [CrossRef]
- AbuSamra, D.B.; Aleely, F.; Wu, Y.; et al. Not just a marker: CD34 on human hematopoietic stem/progenitor cells. Blood. 2017, 129, 1568–1578. [Google Scholar] [CrossRef]
- Hassanpour, M.; Salybekov, A.A.; Kobayashi, S.; Asahara, T. CD34 positive cells as endothelial progenitor cells in biology and medicine. Front. Cell Dev. Biol. 2023, 11, 1128134. [Google Scholar] [CrossRef]
- Merkle, F.T.; Eggan, K. Modeling Human Disease with Pluripotent Stem Cells: from Genome Association to Function. Cell Stem Cell 2013, 12, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.B.J.-W.; Chan, L.-L.; Tan, E.-K. Perceived Cognitive Changes following Chimeric Antigen Receptor T Cell Therapy in Lymphoma: Perceptual Anticipation? Biol. Blood Marrow Transplant. 2022, 29, 64. [Google Scholar] [CrossRef]
- Freud, A.G.; Keller, K.A.; Scoville, S.D.; Mundy-Bosse, B.L.; Cheng, S.; Youssef, Y.; Hughes, T.; Zhang, X.; Mo, X.; Porcu, P.; et al. NKp80 Defines a Critical Step during Human Natural Killer Cell Development. Cell Rep. 2016, 16, 379–391. [Google Scholar] [CrossRef]
- Welte, S.; Kuttruff, S.; Waldhauer, I.; Steinle, A. Mutual activation of natural killer cells and monocytes mediated by NKp80-AICL interaction. Nat. Immunol. 2006, 7, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Morán-Plata, F.J.; Muñoz-García, N.; González-González, M.; Pozo, J.; Carretero-Domínguez, S.; Mateos, S.; Barrena, S.; Belhassen-García, M.; Lau, C.; Teixeira, M.D.A.; et al. A novel NKp80-based strategy for universal identification of normal, reactive and tumor/clonal natural killer-cells in blood. Front. Immunol. 2024, 15, 1423689. [Google Scholar] [CrossRef]
- Sun, L.; Gang, X.; Li, Z.; Zhao, X.; Zhou, T.; Zhang, S.; Wang, G. Advances in Understanding the Roles of CD244 (SLAMF4) in Immune Regulation and Associated Diseases. Front. Immunol. 2021, 12, 648182. [Google Scholar] [CrossRef]
- Agresta, L.; Hoebe, K.H.N.; Janssen, E.M. The Emerging Role of CD244 Signaling in Immune Cells of the Tumor Microenvironment. Front. Immunol. 2018, 9, 2809. [Google Scholar] [CrossRef]
- Deng, Z.; Liu, Y.; Zhou, H. Distinct roles of CD244 expression in cancer diagnosis and prognosis: A pan-cancer analysis. Heliyon 2024, 10, e28928. [Google Scholar] [CrossRef]
- Wang, X.; Xiong, H.; Ning, Z. Implications of NKG2A in immunity and immune-mediated diseases. Front. Immunol. 2022, 13, 960852. [Google Scholar] [CrossRef]
- Chen, D.G.; Xie, J.; Choi, J.; Ng, R.H.; Zhang, R.; Li, S.; Edmark, R.; Zheng, H.; Solomon, B.; Campbell, K.M.; et al. Integrative systems biology reveals NKG2A-biased immune responses correlate with protection in infectious disease, autoimmune disease, and cancer. Cell Rep. 2024, 43, 113872. [Google Scholar] [CrossRef]
- Horton, N.C.; Mathew, P.A. NKp44 and Natural Cytotoxicity Receptors as Damage-Associated Molecular Pattern Recognition Receptors. Front. Immunol. 2015, 6, 31. [Google Scholar] [CrossRef]
- Parodi, M.; Favoreel, H.; Candiano, G.; Gaggero, S.; Sivori, S.; Mingari, M.C.; Moretta, L.; Vitale, M.; Cantoni, C. NKp44-NKp44 Ligand Interactions in the Regulation of Natural Killer Cells and Other Innate Lymphoid Cells in Humans. Front. Immunol. 2019, 10, 719. [Google Scholar] [CrossRef]
- Mair, K.H.; Crossman, A.J.; Wagner, B.; Babasyan, S.; Noronha, L.; Boyd, P.; Zarlenga, D.; Stadler, M.; van Dongen, K.A.; Gerner, W.; et al. The Natural Cytotoxicity Receptor NKp44 (NCR2, CD336) Is Expressed on the Majority of Porcine NK Cells Ex Vivo Without Stimulation. Front. Immunol. 2022, 13, 767530. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, V.; Shamalov, K.; Meir, S.; Hoogi, S.; Sarkar, R.; Pinker, S.; Markel, G.; Porgador, A.; Cohen, C.J. Targeting Multiple Tumors Using T-Cells Engineered to Express a Natural Cytotoxicity Receptor 2-Based Chimeric Receptor. Front. Immunol. 2017, 8, 1212. [Google Scholar] [CrossRef]
- Murayama, Y.; Hirayama, A.V.; Houchins, J.P.; et al. NKp44-based chimeric antigen receptor effectively targets synovial sarcomas. J Immunother Cancer. 2022, 10, e004947. [Google Scholar] [CrossRef]
- Sarfraz, M.; Afzal, A.; Raza, S.M.; Bashir, S.; Madni, A.; Khan, M.W.; Ma, X.; Xiang, G. Liposomal co-delivered oleanolic acid attenuates doxorubicin-induced multi-organ toxicity in hepatocellular carcinoma. Oncotarget 2017, 8, 47136–47153. [Google Scholar] [CrossRef]
- Cui, Z.J. Deep-sea photodynamic vision at low light level — Which is more important, prosthetic retinal or apo-rhodopsin moiety? FASEB J. 2025, 39, e70470. [Google Scholar] [CrossRef] [PubMed]
- Freud, A.G.; Zhao, S.; Wei, S.; Gitana, G.M.; Molina-Kirsch, H.F.; Atwater, S.K.; Natkunam, Y. Expression of the Activating Receptor, NKp46 (CD335), in Human Natural Killer and T-Cell Neoplasia. Am. J. Clin. Pathol. 2013, 140, 853–866. [Google Scholar] [CrossRef]
- Cheminant, M.; Bruneau, J.; Malamut, G.; et al. NKp46 is a diagnostic biomarker in gastrointestinal T-cell lymphoproliferative diseases. Gut. 2019, 68, 1396–1406. [Google Scholar] [CrossRef]
- Sica, M.; Piazzolla, P.; Amparore, D.; Verri, P.; De Cillis, S.; Piramide, F.; Volpi, G.; Piana, A.; Di Dio, M.; Alba, S.; et al. 3D Model Artificial Intelligence-Guided Automatic Augmented Reality Images during Robotic Partial Nephrectomy. Diagnostics 2023, 13, 3454. [Google Scholar] [CrossRef] [PubMed]
- Gasse, P.; Mary, C.; Guenon, I.; Noulin, N.; Charron, S.; Schnyder-Candrian, S.; Schnyder, B.; Akira, S.; Quesniaux, V.F.; Lagente, V.; et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J. Clin. Investig. 2007, 117, 3786–3799. [Google Scholar] [CrossRef]
- Hughes, T.; Becknell, B.; Freud, A.G.; et al. IL-1β selectively expands IL-22+ immature human NK cells in secondary lymphoid tissue. Immunity. 2010, 32, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Luís, J.P.; Simões, C.J.V.; Brito, R.M.M. The Therapeutic Prospects of Targeting IL-1R1 for the Modulation of Neuroinflammation in Central Nervous System Disorders. Int. J. Mol. Sci. 2022, 23, 1731. [Google Scholar] [CrossRef] [PubMed]
- Chen, I. Restoring order. Nat. Struct. Mol. Biol. 2012, 19, 267–267. [Google Scholar] [CrossRef]
- Raghavan, PR. Policosanol nanoparticles. US Patent 8,722,093, 2014. [Google Scholar]
- Raghavan, PR. Policosanol nanoparticles. US Patent 9,006,292, 2015. [Google Scholar]
- Raghavan, PR. Policosanol nanoparticles. US Patent 9,034,383, 2015. [Google Scholar]
- Raghavan, P.R. Metadichol® A Nano Lipid Emulsion that Expresses All 49 Nuclear Receptors in Stem and Somatic Cells. Arch. Clin. Biomed. Res. 2022, 7, 543–555. [Google Scholar] [CrossRef]
- Richards, J.; Danielson, A.; Stuart, R.; Richards, A.; Laurin, E. Reverse Remodeling of Methamphetamine-Associated Cardiomyopathy: An Update on Mechanisms for Recovery. Med Res. Arch. 2024, 12. [Google Scholar] [CrossRef]
- Raghavan, P.R. Beyond Rapamycin: Metadichol Represents a New Class of Multi-Target mTOR Modulators. Med Res. Arch. 2025, 13. [Google Scholar] [CrossRef]
- A Johnston, J.; Bacon, C.M.; Finbloom, D.S.; Rees, R.C.; Kaplan, D.; Shibuya, K.; Ortaldo, J.R.; Gupta, S.; Chen, Y.Q.; Giri, J.D. Tyrosine phosphorylation and activation of STAT5, STAT3, and Janus kinases by interleukins 2 and 15. Proc. Natl. Acad. Sci. 1995, 92, 8705–8709. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Kawahara, A.; Fujii, H.; Nakagawa, Y.; Minami, Y.; Liu, Z.-J.; Oishi, I.; Silvennoinen, O.; Witthuhn, B.A.; Ihle, J.N.; et al. Functional Activation of Jak1 and Jak3 by Selective Association with IL-2 Receptor Subunits. Science 1994, 266, 1045–1047. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.K.; Glaccum, M.; Brown, S.N.; Butz, E.A.; Viney, J.L.; Embers, M.; Matsuki, N.; Charrier, K.; Sedger, L.; Willis, C.R.; et al. Reversible Defects in Natural Killer and Memory Cd8 T Cell Lineages in Interleukin 15–Deficient Mice. J. Exp. Med. 2000, 191, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Puthalakath, H.; Gunn, P.; Naik, E.; Michalak, E.M.; Smyth, M.J.; Tabarias, H.; A Degli-Esposti, M.; Dewson, G.; Willis, S.N.; et al. Interleukin 15–mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat. Immunol. 2007, 8, 856–863. [Google Scholar] [CrossRef]
- Freud, A.G.; Caligiuri, M.A. Human natural killer cell development. Immunol. Rev. 2006, 214, 56–72. [Google Scholar] [CrossRef]
- Freud, A.G.; Yu, J.; Caligiuri, M.A. Human natural killer cell development in secondary lymphoid tissues. Semin. Immunol. 2014, 26, 132–137. [Google Scholar] [CrossRef]
- Yu, J.; Freud, A.G.; Caligiuri, M.A. Location and cellular stages of natural killer cell development. Trends Immunol. 2013, 34, 573–582. [Google Scholar] [CrossRef]
- Marçais, A.; Cherfils-Vicini, J.; Viant, C.; Degouve, S.; Viel, S.; Fenis, A.; Rabilloud, J.; Mayol, K.; Tavares, A.; Bienvenu, J.; et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 2014, 15, 749–757. [Google Scholar] [CrossRef]
- Yang, M.; Li, D.; Chang, Z.; Yang, Z.; Tian, Z.; Dong, Z. PDK1 orchestrates early NK cell development through induction of E4BP4 expression and maintenance of IL-15 responsiveness. J. Exp. Med. 2015, 212, 253–265. [Google Scholar] [CrossRef]
- Wang, F.; Meng, M.; Mo, B.; Yang, Y.; Ji, Y.; Huang, P.; Lai, W.; Pan, X.; You, T.; Luo, H.; et al. Crosstalks between mTORC1 and mTORC2 variagate cytokine signaling to control NK maturation and effector function. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK Cells and T Cells by NKG2D, a Receptor for Stress-Inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Textor, S.; Fiegler, N.; Arnold, A.; Porgador, A.; Hofmann, T.G.; Cerwenka, A. Human NK Cells Are Alerted to Induction of p53 in Cancer Cells by Upregulation of the NKG2D Ligands ULBP1 and ULBP2. Cancer Res. 2011, 71, 5998–6009. [Google Scholar] [CrossRef] [PubMed]
- Welte, S.; Kuttruff, S.; Waldhauer, I.; Steinle, A. Mutual activation of natural killer cells and monocytes mediated by NKp80-AICL interaction. Nat. Immunol. 2006, 7, 1334–1342. [Google Scholar] [CrossRef]
- Freud, A.G.; Keller, K.A.; Scoville, S.D.; Mundy-Bosse, B.L.; Cheng, S.; Youssef, Y.; Hughes, T.; Zhang, X.; Mo, X.; Porcu, P.; et al. NKp80 Defines a Critical Step during Human Natural Killer Cell Development. Cell Rep. 2016, 16, 379–391. [Google Scholar] [CrossRef]
- Sivori, S.; Falco, M.; Della Chiesa, M.; Carlomagno, S.; Vitale, M.; Moretta, L.; Moretta, A. CpG and double-stranded RNA trigger human NK cells by Toll-like receptors: Induction of cytokine release and cytotoxicity against tumors and dendritic cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10116–10121. [Google Scholar] [CrossRef]
- Girart, M.V.; Fuertes, M.B.; Domaica, C.I.; Rossi, L.E.; Zwirner, N.W. Engagement of TLR3, TLR7, and NKG2D Regulate IFN-γ Secretion but Not NKG2D-Mediated Cytotoxicity by Human NK Cells Stimulated with Suboptimal Doses of IL-12. J. Immunol. 2007, 179, 3472–3479. [Google Scholar] [CrossRef]
- Hart, O.M.; Athie-Morales, V.; O’Connor, G.M.; Gardiner, C.M. TLR7/8-Mediated Activation of Human NK Cells Results in Accessory Cell-Dependent IFN-γ Production. J. Immunol. 2005, 175, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.M.; Chaix, J.; Rupp, L.J.; Wu, J.; Madera, S.; Sun, J.C.; Lindsten, T.; Reiner, S.L. The Transcription Factors T-bet and Eomes Control Key Checkpoints of Natural Killer Cell Maturation. Immunity 2012, 36, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Simonetta, F.; Pradier, A.; Roosnek, E. T-bet and Eomesodermin in NK Cell Development, Maturation, and Function. Front. Immunol. 2016, 7, 241. [Google Scholar] [CrossRef]
- Deng, Y.; Kerdiles, Y.; Chu, J.; Yuan, S.; Wang, Y.; Chen, X.; Mao, H.; Zhang, L.; Zhang, J.; Hughes, T.; et al. Transcription Factor Foxo1 Is a Negative Regulator of Natural Killer Cell Maturation and Function. Immunity 2015, 42, 457–470. [Google Scholar] [CrossRef]
- Du, X.; De Almeida, P.; Manieri, N.; Nagata, D.D.A.; Wu, T.D.; Bowles, K.H.; Arumugam, V.; Schartner, J.; Cubas, R.; Mittman, S.; et al. CD226 regulates natural killer cell antitumor responses via phosphorylation-mediated inactivation of transcription factor FOXO1. Proc. Natl. Acad. Sci. USA 2018, 115, E11731–E11740. [Google Scholar] [CrossRef]
- Rao, R.R.; Li, Q.; Bupp, M.R.G.; Shrikant, P.A. Transcription Factor Foxo1 Represses T-bet-Mediated Effector Functions and Promotes Memory CD8+ T Cell Differentiation. Immunity 2012, 36, 374–387. [Google Scholar] [CrossRef]
- Rhee, E.J.; Kwon, H.; Lee, S. Regulation of natural killer cell development and function by nuclear receptor signaling. Arch Pharm Res. 2018, 41, 527–538. [Google Scholar] [CrossRef]
- Charoenngam, N.; Holick, M.F. Immunologic Effects of Vitamin D on Human Health and Disease. Nutrients 2020, 12, 2097. [Google Scholar] [CrossRef] [PubMed]
- Konjević, G.M.; Vuletić, A.M.; Mirjačić Martinović, K.M.; Larsen, A.K.; Jurišić, V.B. The role of cytokines in the regulation of NK cells in the tumor environment. Cytokine 2019, 117, 30–40. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef]
- Long, E.O.; Kim, H.S.; Liu, D.; Peterson, M.E.; Rajagopalan, S. Controlling Natural Killer Cell Responses: Integration of Signals for Activation and Inhibition. Annu. Rev. Immunol. 2013, 31, 227–258. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.J.; O'Callaghan, C.A.; Söderström, K.; D'Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- van Hall, T.; André, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef] [PubMed]
- Aleman, C.; Mas, R.; Hernandez, C.; Rodeiro, I.; Cerejido, E.; Noa, M.; Capote, A.; Menendez, R.; Amor, A.; Fraga, V. A 12-month study of policosanol oral toxicity in Sprague Dawley rats. Toxicol. Lett. 1994, 70, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Alemán, C.L.; Ferreiro, R.M.; Puig, M.N.; Guerra, I.R.; Ortega, C.H.; Capote, A. Carcinogenicity of policosanol in sprague dawley rats: A 24 month study. Teratog. Carcinog. Mutagen. 1994, 14, 239–249. [Google Scholar] [CrossRef] [PubMed]
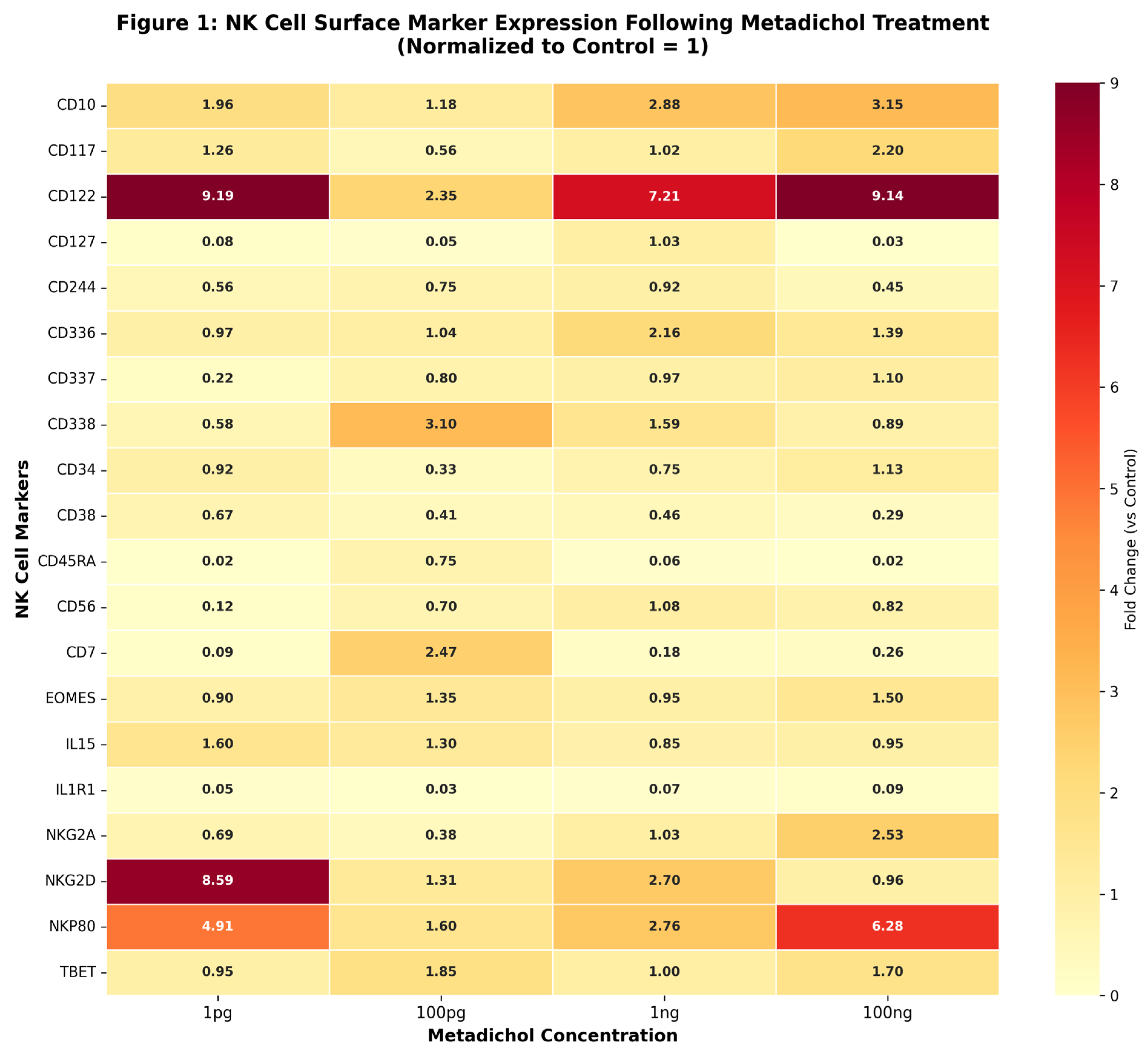
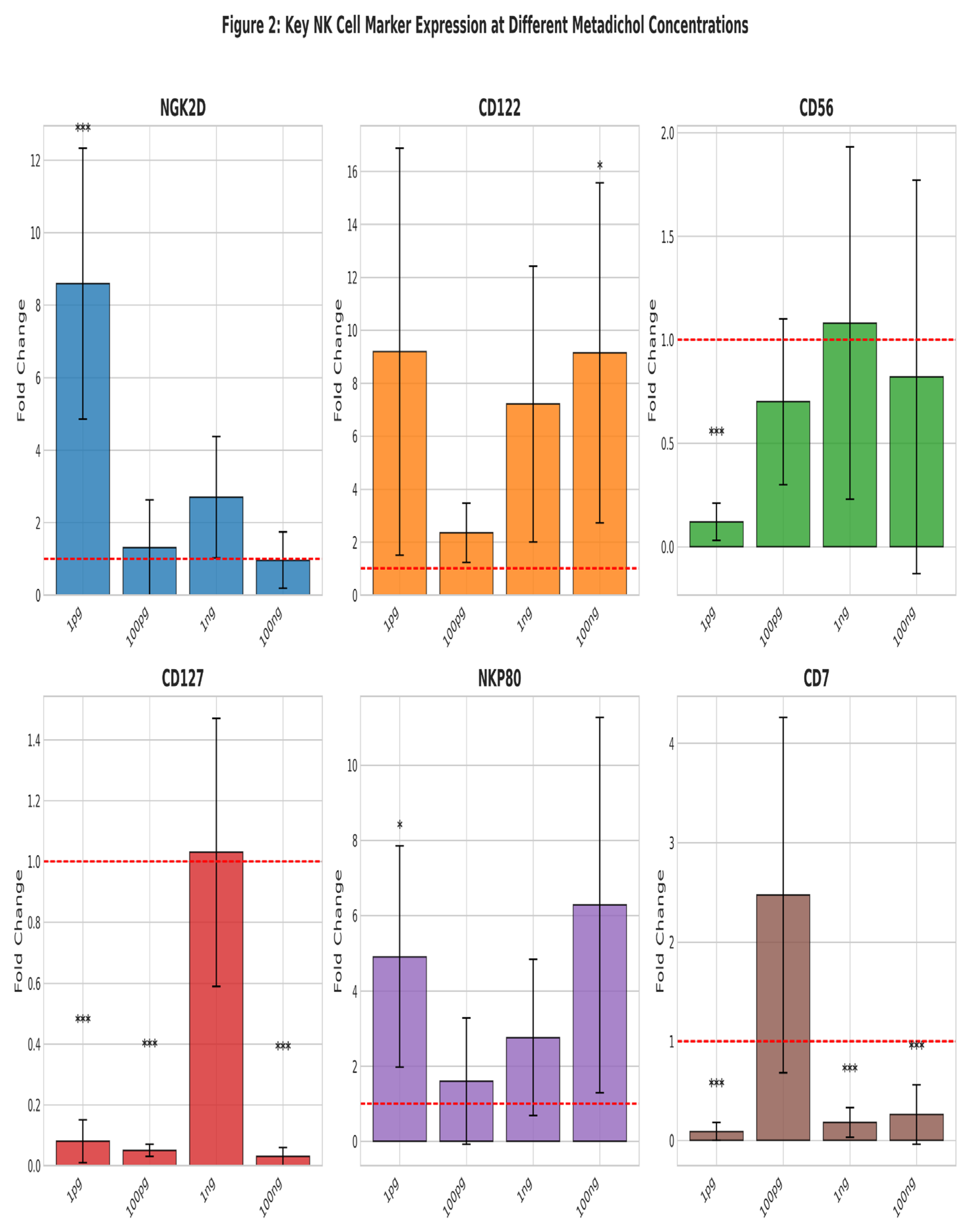
Figure 3.
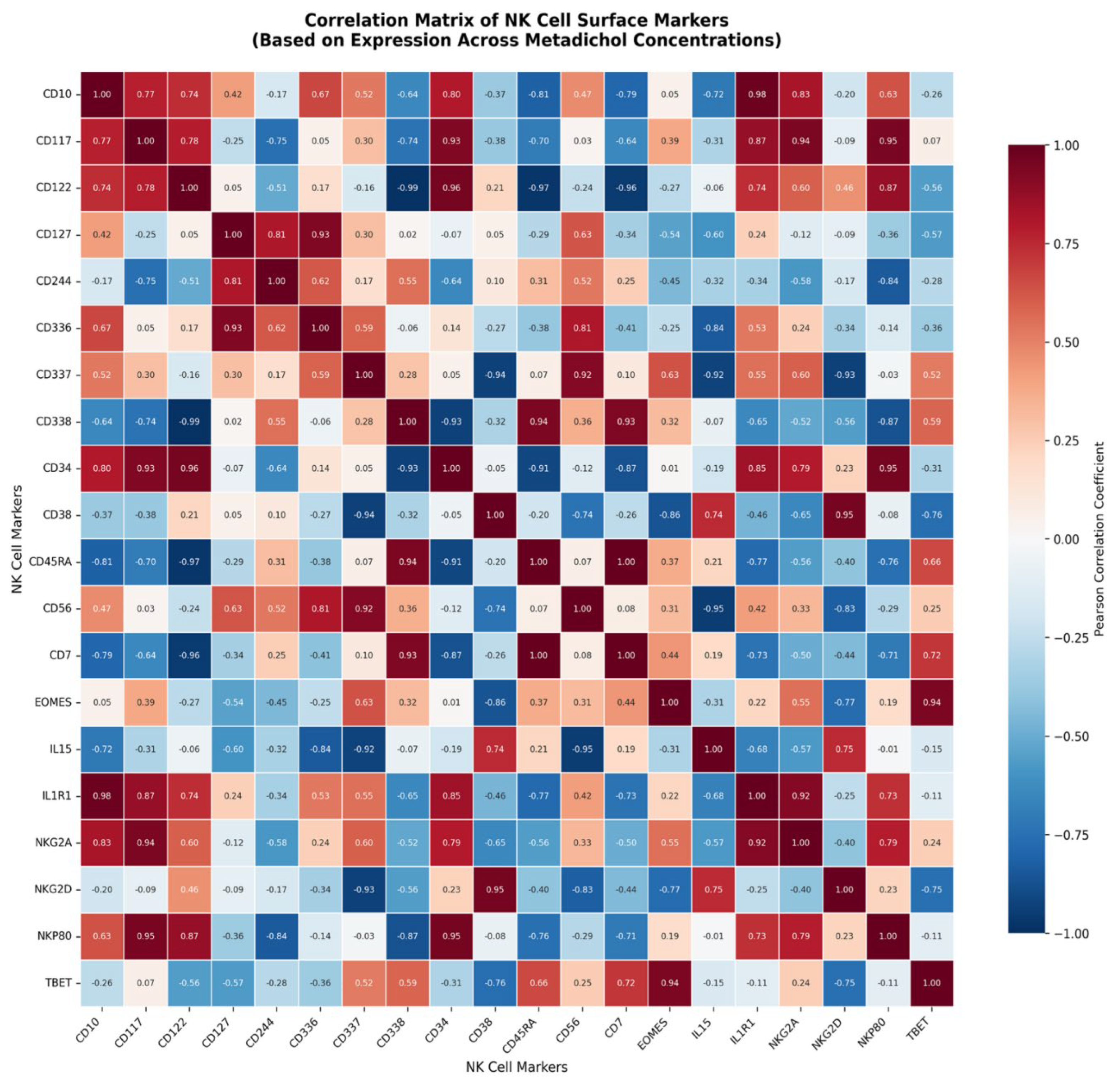
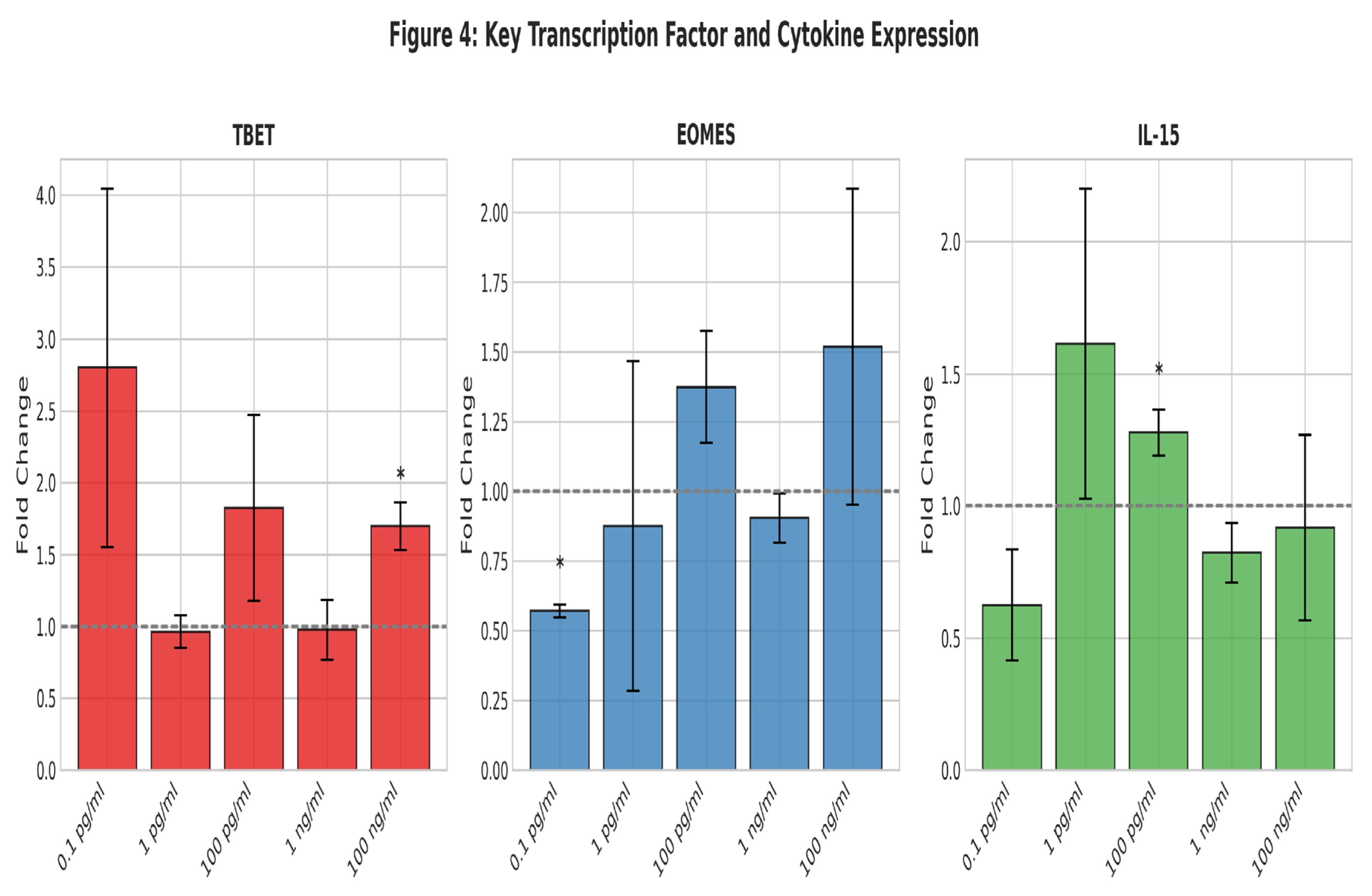
Figure 5.
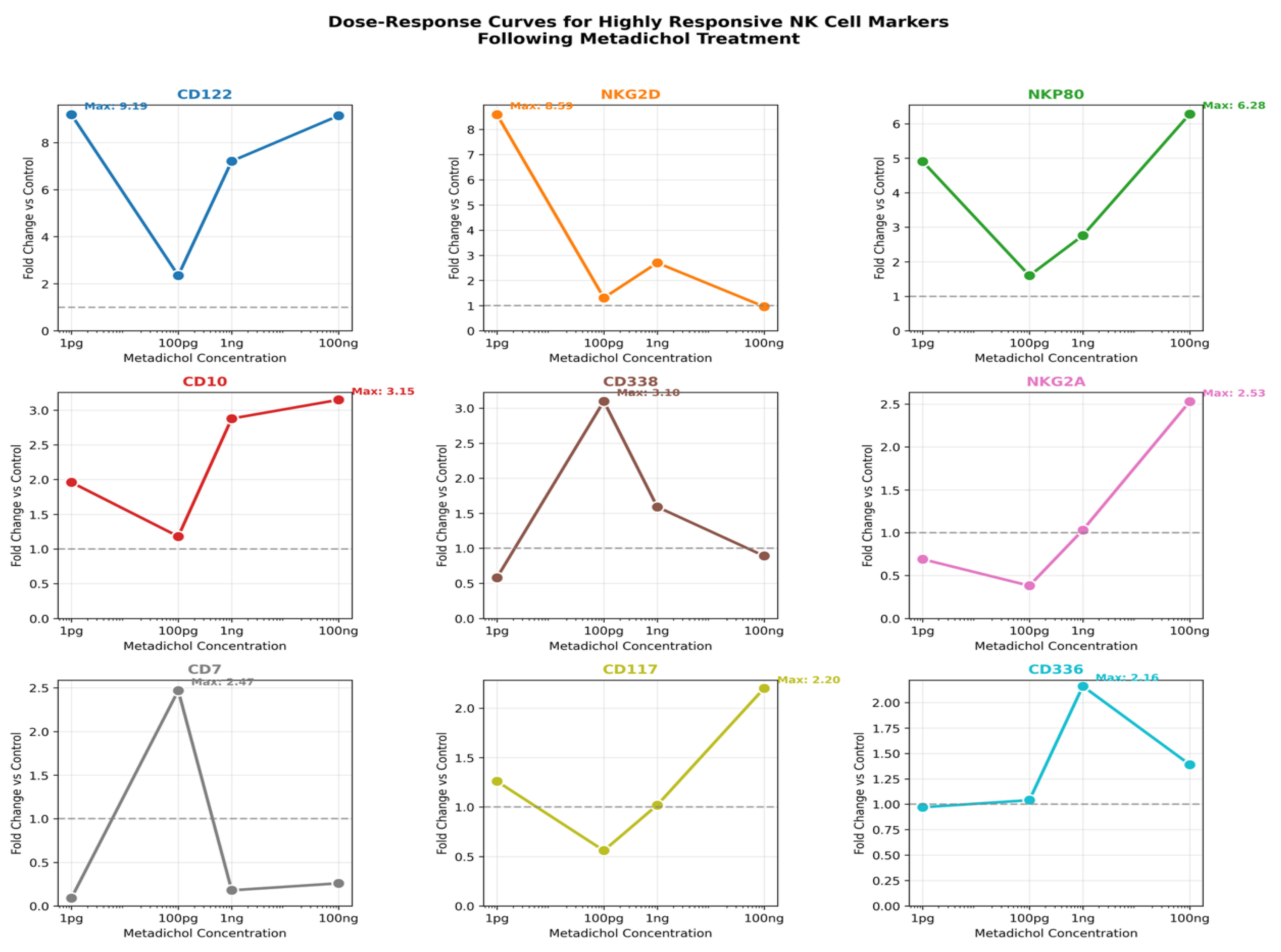
Figure 6.
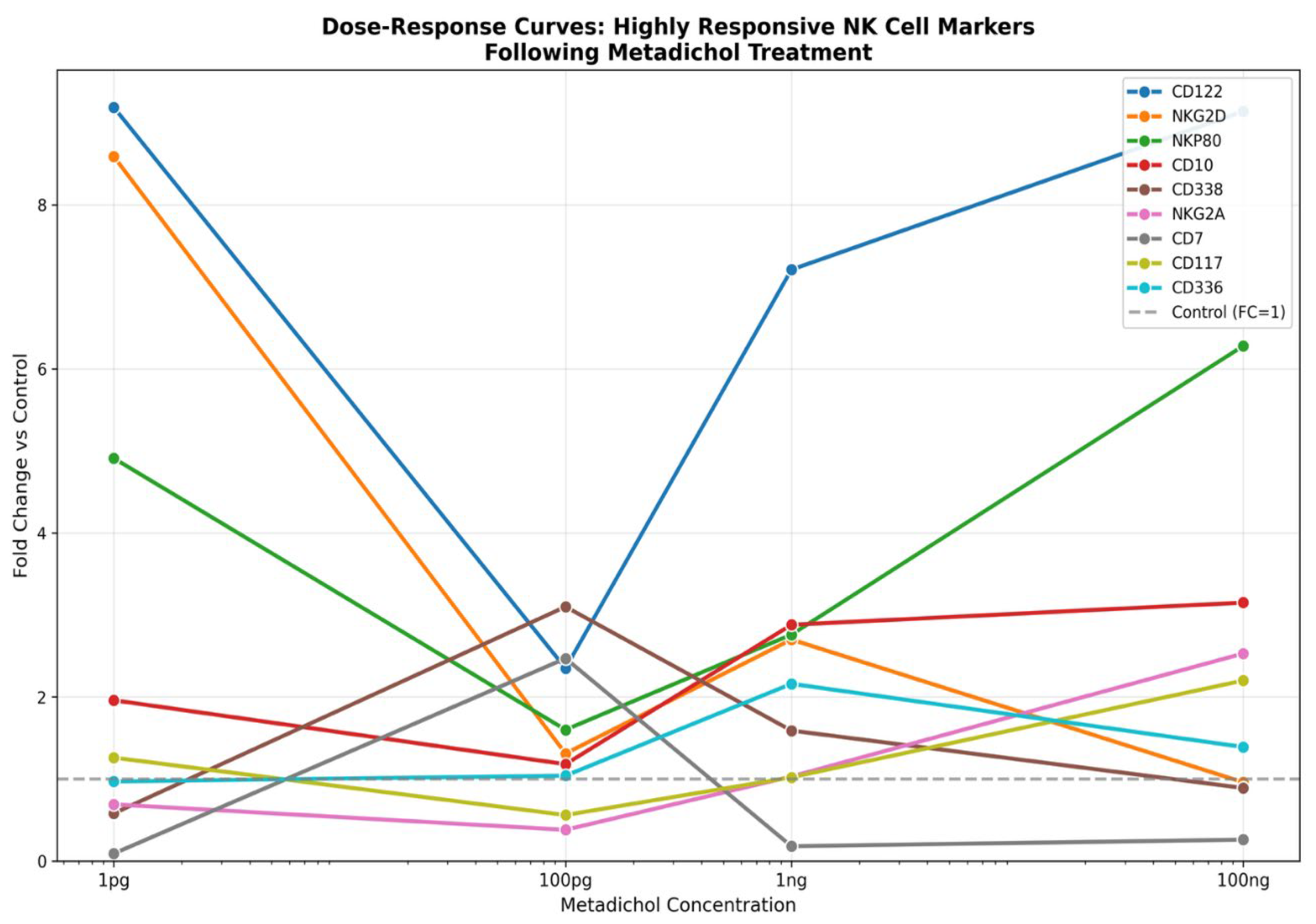
Figure 7.
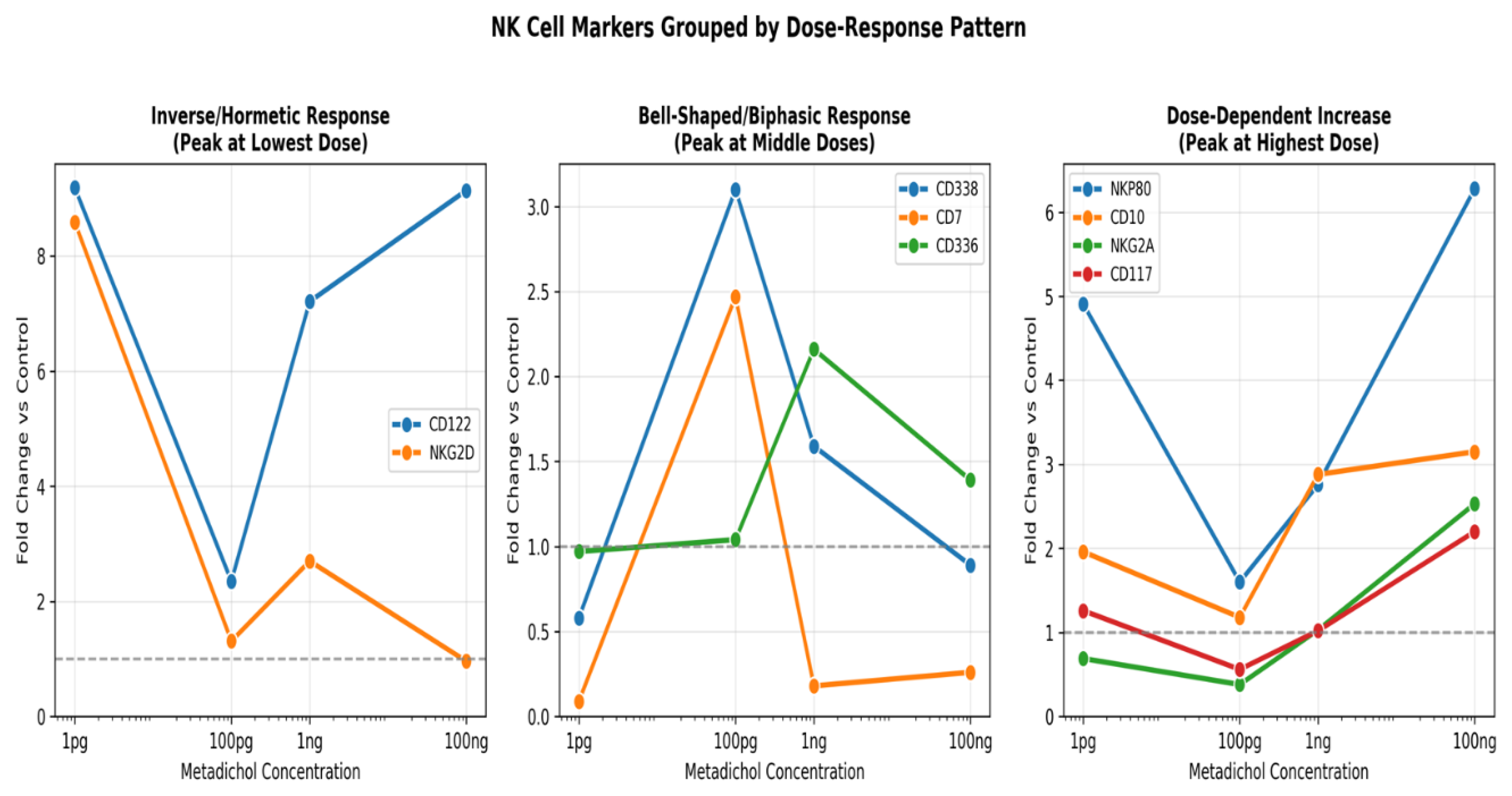
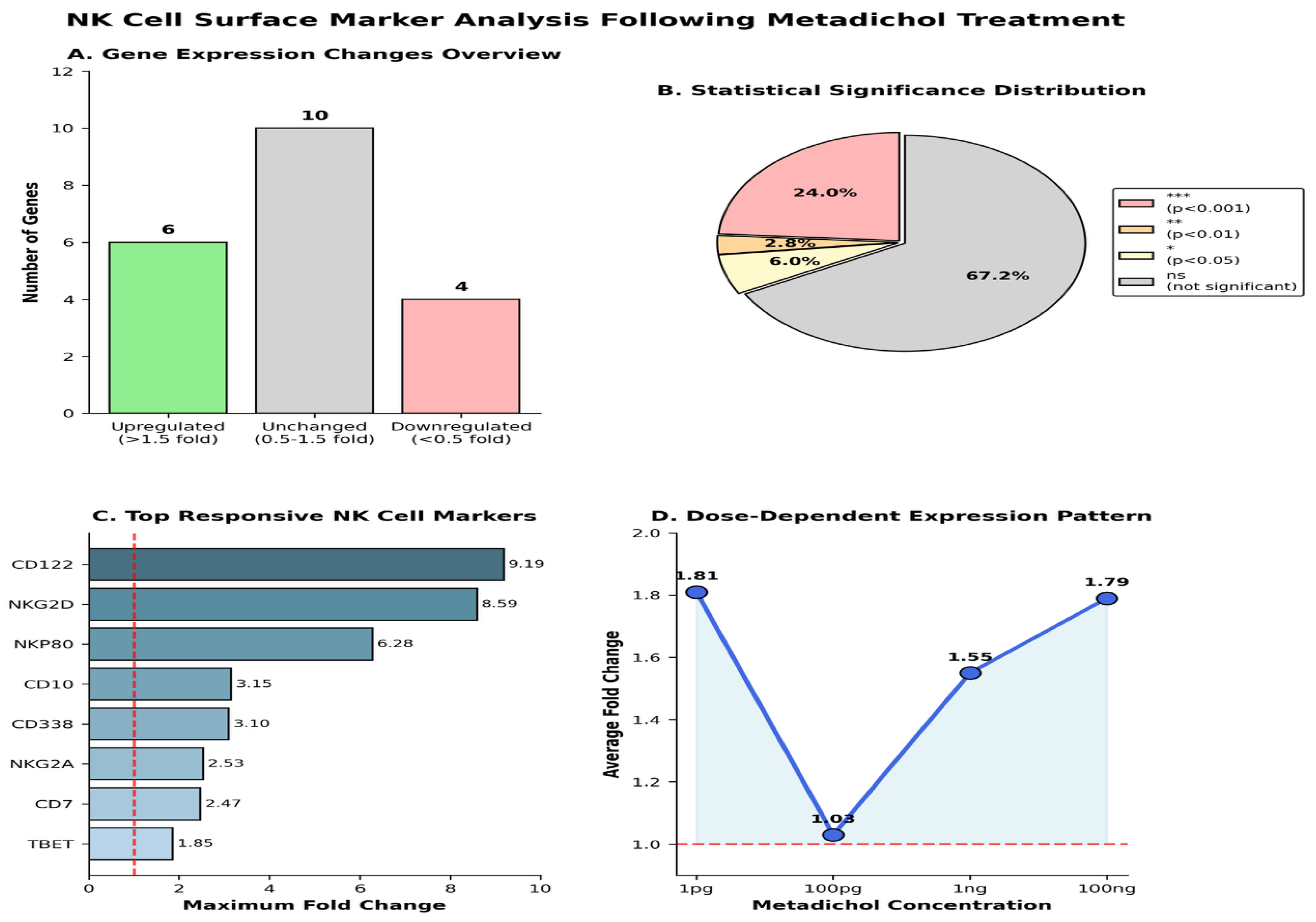
Figure 9.
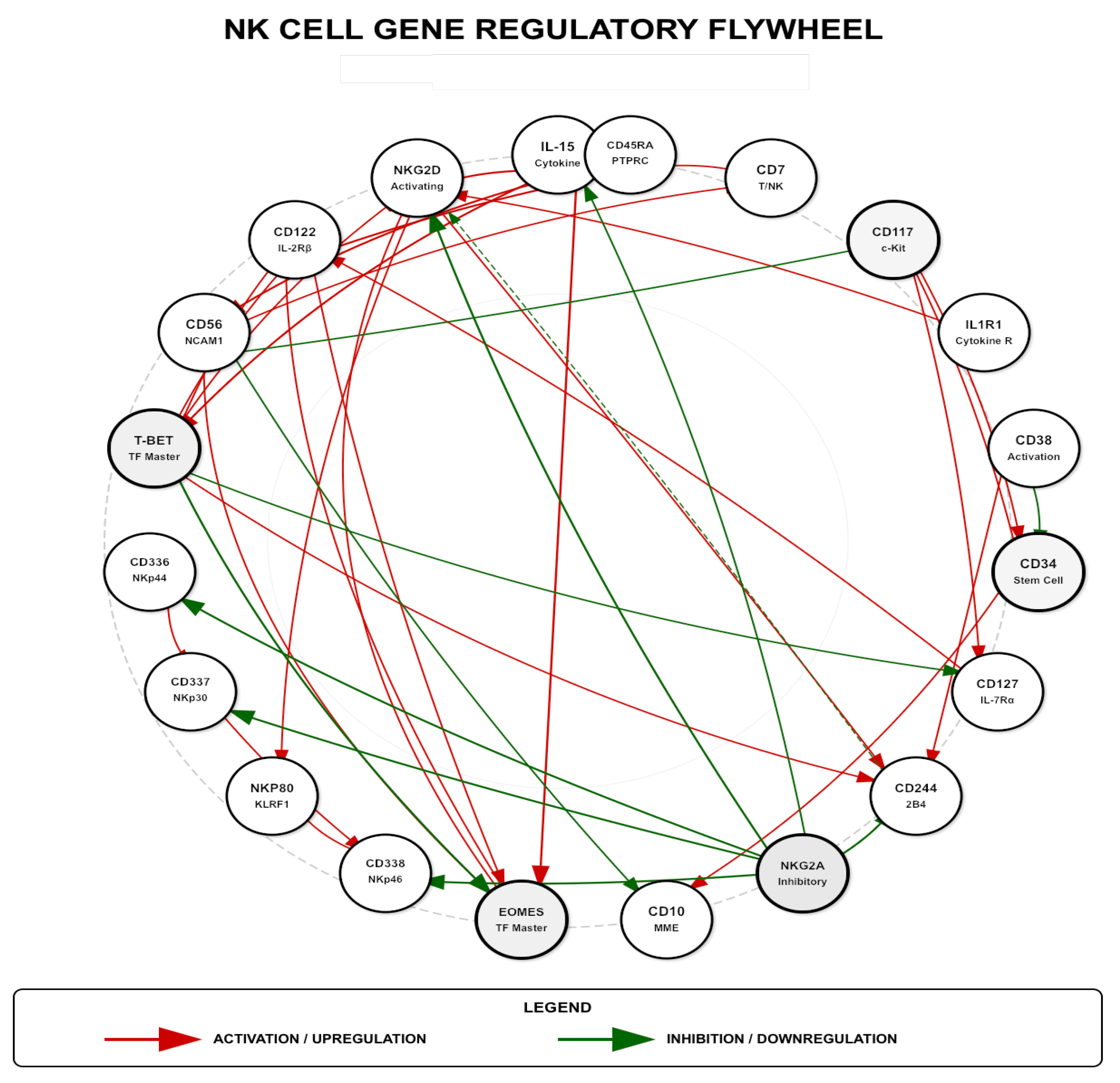
Figure 10.
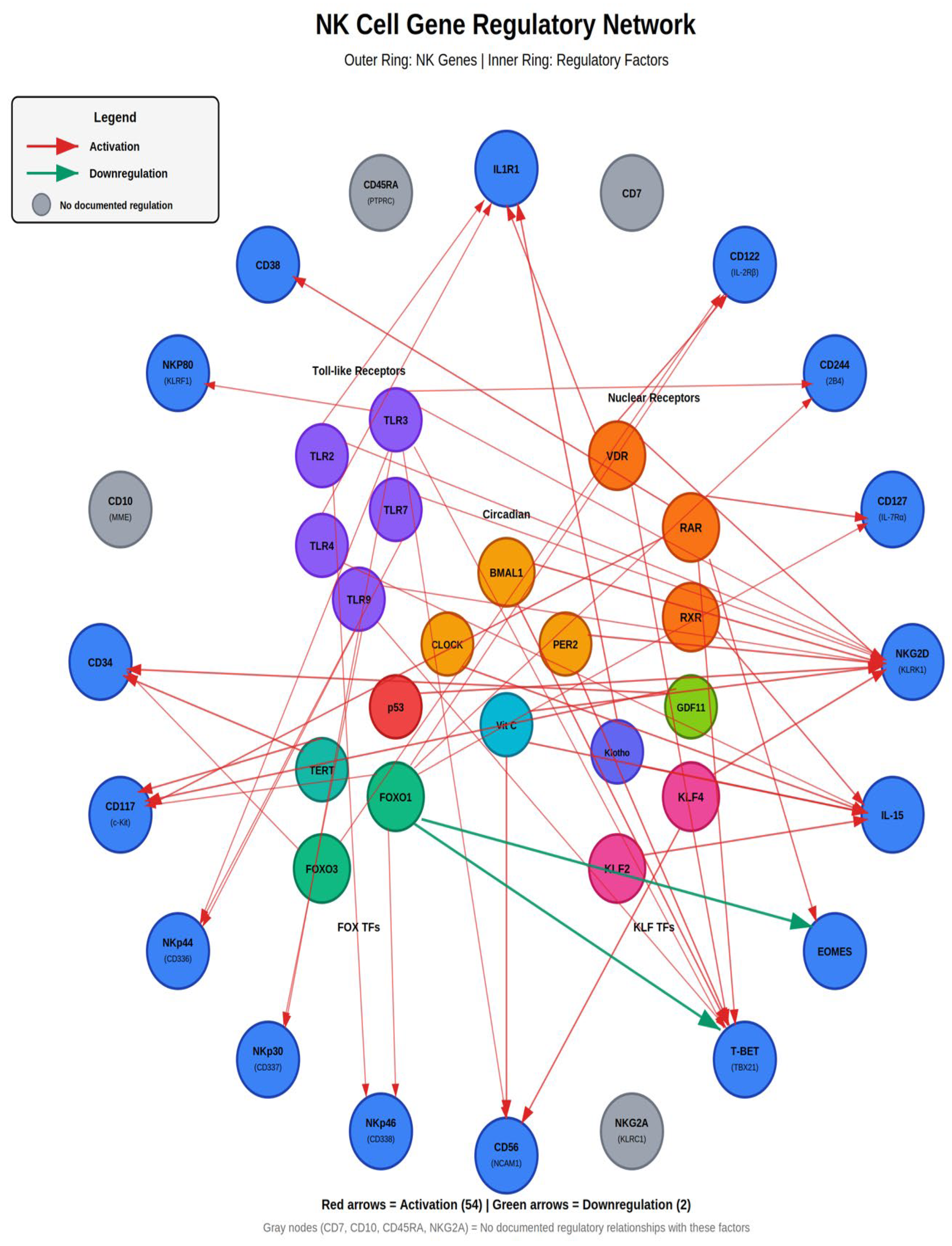
Figure 11.
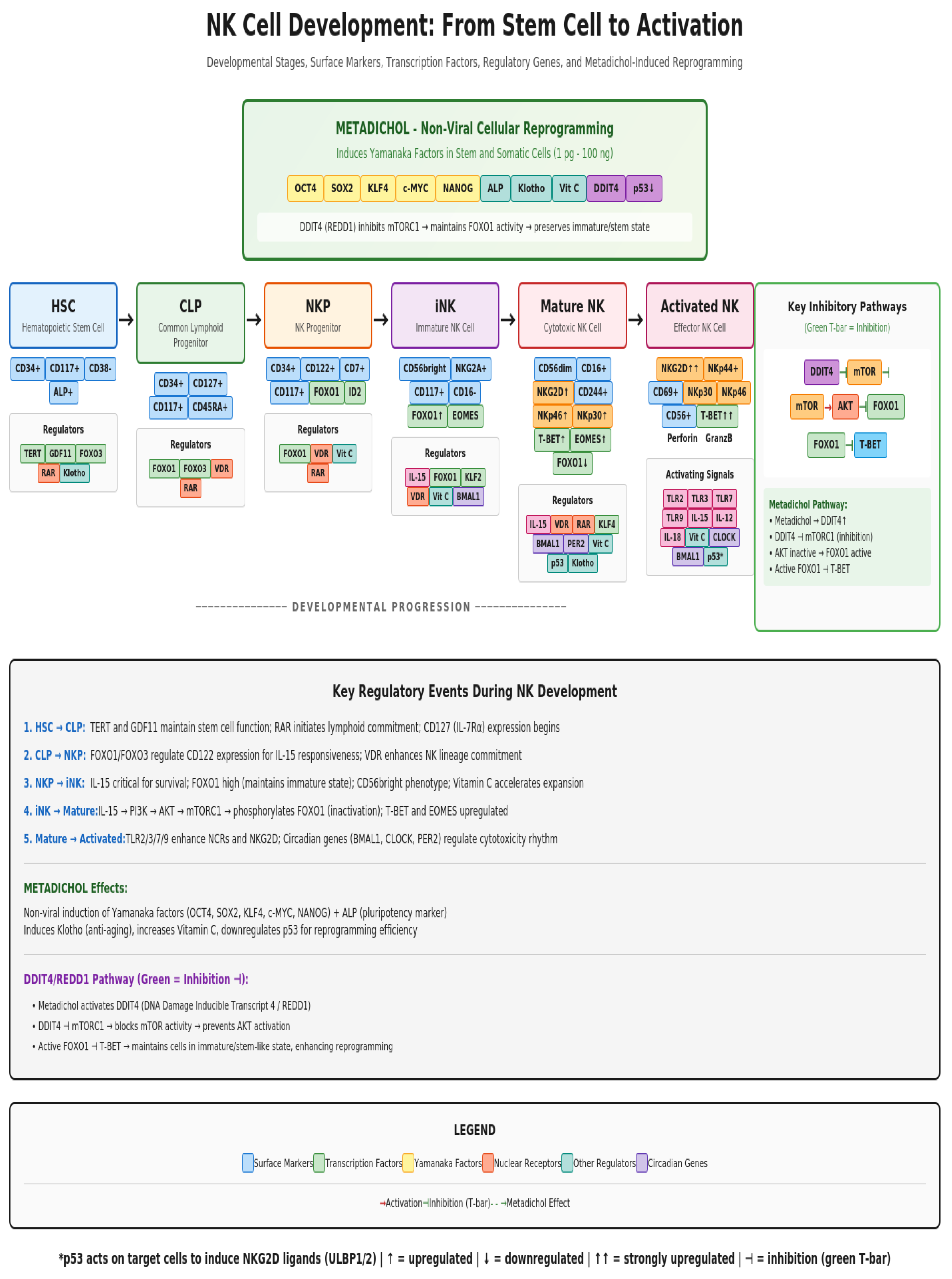
Figure 12.
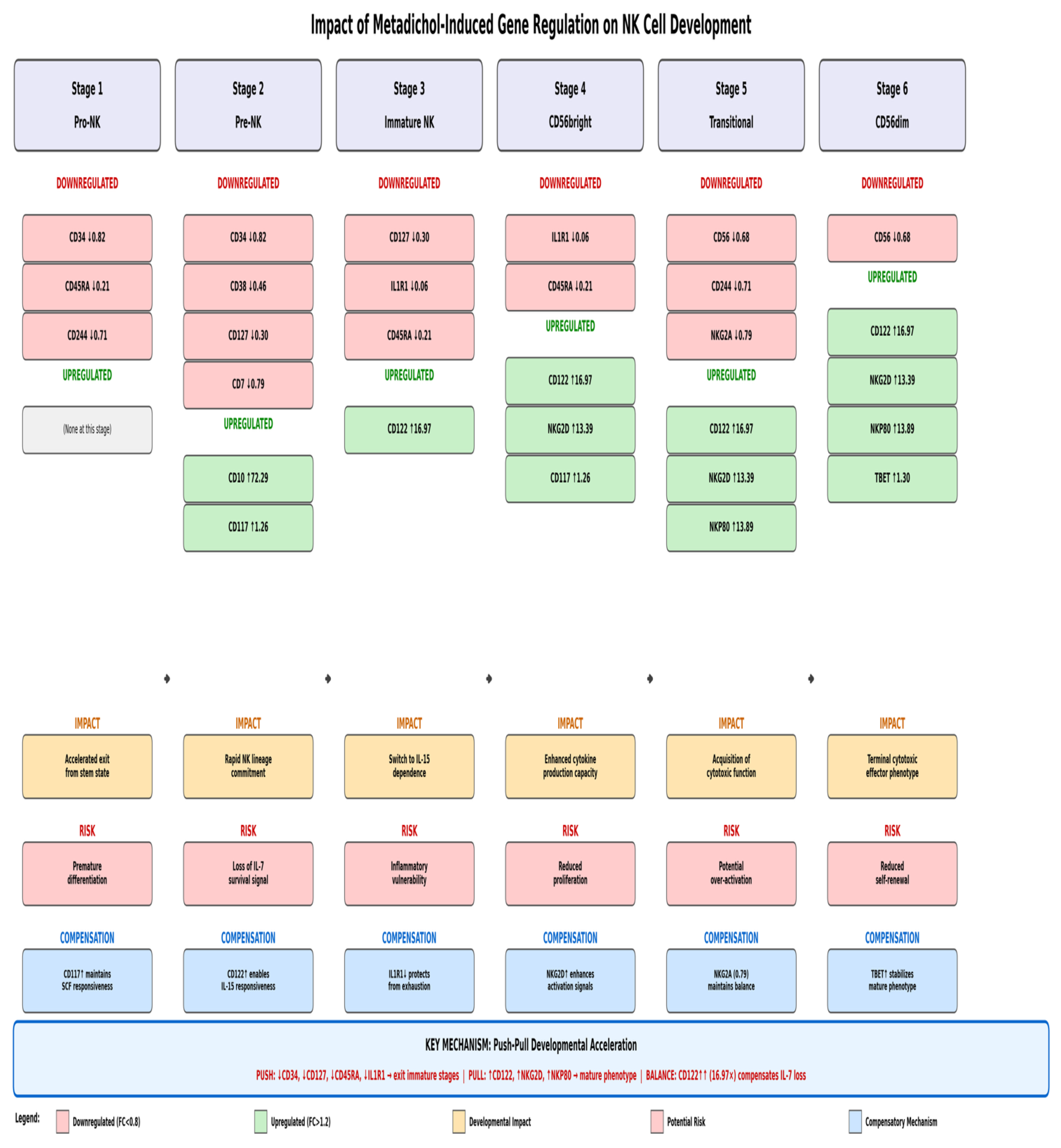

Table 1.
Metadichol Treatment Concentrations.
| Group | Cell Type | Treatment | Concentration |
|---|---|---|---|
| 1 |
Human PBMC |
Vehicle Control | — |
| 2 |
Metadichol |
0.1pg/mL | |
| 3 | 1 pg/mL | ||
| 4 | 100 pg/mL | ||
| 5 | 1 ng/mL | ||
| 6 | 100 ng/mL |
Table 2.
Total RNA yield.
| Test concentrations | ||||||
| RNA yield (ng/µl) | 0 | 0.1 pg/ ml | 1 pg/ ml | 100 pg/ ml | 1 ng/ ml | 100 ng/ ml |
| Human PBMC’s | 425.2 | 410.4 | 380.9 | 412.8 | 438.6 | 446.2 |
Table 3.
Primer details.
| Gene | Primers | Amplicon size | Annealing temperature | ||
| NGK2A | F | GCCTCTGTGGTAACGATAGTTGT | 118 | 65°C | |
| R | ATCCACTCCTCAGGACAATGGC | ||||
| CD38 | F | TCTTGCCCAGACTGGAGAAAGG | 100 | 65°C | |
| R | TGGACCACATCACAGGCAGCTT | ||||
| CD117 | F | CACCGAAGGAGGCACTTACACA | 121 | 50°C | |
| R | TGCCATTCACGAGCCTGTCGTA | ||||
| CD122 | F | CTGGAGAGATGGCCACGGT | 182 | 53°C | |
| R | GATGCCCAAGAGGTAGCCAG | ||||
| CD336 | F | CTGAGTCTCCATCTACCATCCC | 116 | 50°C | |
| R | TCTTGGCTACGAGGAGTCCACA | ||||
| CD337 | F | CCAGCATCTACGTGTGCAGAGT | 135 | 65°C | |
| R | GCATAGAATCCAGCCCGAAGGA | ||||
| CD338 | F | GTTCTCAGCAGCTCTTCGGCTT | 144 | 65°C | |
| R | TCCTCCAGACACACCACGGATA | ||||
| NKP80 | F | CGAGATCTGCAGACCAGACA | 274 | 49°C | |
| R | CGAGATCTGCAGACCAGACA | ||||
| CD34 | F | CCACAGGAGAAAGGCTGGAG | 184 | 62°C | |
| R | ATCTGGTAAGCAGGGCTGTG | ||||
| CD10 | F | CTTTAGTGCCCAGCAGTCCAAC | 128 | 56°C | |
| R | CACCAGTCAACGAGGTCTCCAT | ||||
| GAPDH | F | GTCTCCTCTGACTTCAACAGCG | 186 | 65°C | |
| R | ACCACCCTGTTGCTGTAGCCAA | ||||
| ILIR1 | F | GTGCTTTGGTACAGGGATTCCTG | 120 | 65°C | |
| R | CACAGTCAGAGGTAGACCCTTC | ||||
| CD127 | F | ATCGCAGCACTCACTGACCTGT | 100 | 50°C | |
| R | TCAGGCACTTTACCTCCACGAG | ||||
| CD244 | F | TCTACTGCCTGGAGGTCACCAG | 151 | 53°C | |
| R | GACCAAGCAAGACAGAGCCACT | ||||
| CD45RA | F | CATGCAGCTAGCAAGTGGTT | 264 | 50°C | |
| R | GAAGGGCTCAGAGTGGTTGT | ||||
| CD56 | F | CAGTCCATAGCCCTCCTCCA | 483 | 50°C | |
| R | CGGCTTTTCCACACAGGTTG | ||||
| NKG2D | F | AGATCTTCCCTCTCTGAGCA | 114 | 67°C | |
| R | GAGACCTCCGACCACGAATC | ||||
| CD7 | F | TGTCGGACACTGGCACCTACAC | 114 | 57°C | |
| R | TCCGAGCATCTGTGCCATCCTT | ||||
| EOMES | F | AAATGGGTGACCTGTGGCAAAGC | 101 | 67 | |
| R | CTCCTGTCTCATCCAGTGGGAA | ||||
| IL15 | F | AACAGAAGCCAACTGGGTGAATG | 157 | 67 | |
| R | AACAGAAGCCAACTGGGTGAATG | ||||
| TBET | F | ATTGCCGTGACTGCCTACCAGA | 150 | 67 | |
| R | ATTGCCGTGACTGCCTACCAGA | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



