Submitted:
09 January 2026
Posted:
13 January 2026
You are already at the latest version
Abstract
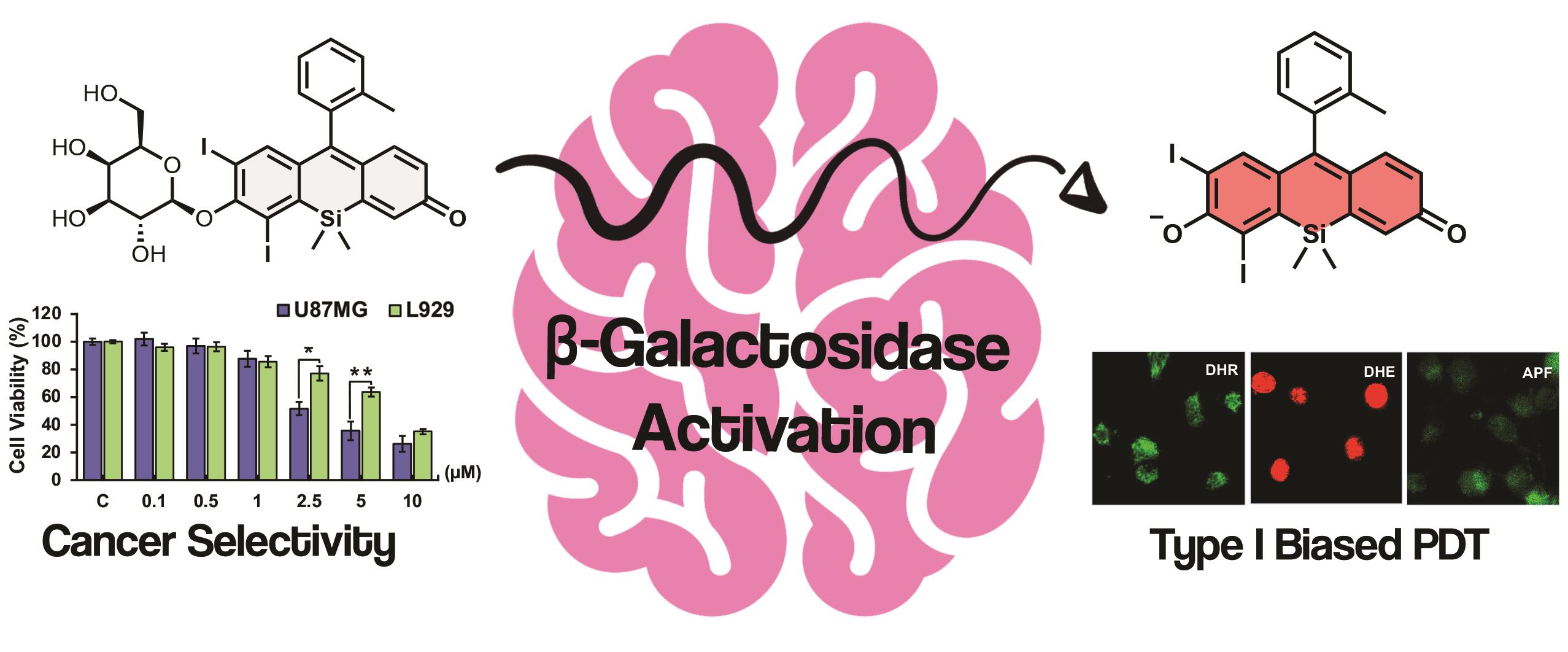
Photodynamic therapy (PDT) offers a promising complementary strategy for the treatment of glioblastoma multiforme (GBM); however, achieving selective activation in tumor tissue and maintaining efficacy under hypoxic conditions remain significant limitations. In this study, we present the synthesis and functional evaluation of Gal-SiX, an enzymatically activatable Si-xanthene photosensitizer designed to address these challenges. Prepared through an improved 10-step synthetic route, Gal-SiX displays a clear turn-on fluorescence and absorbance response upon β-galactosidase activation and generates reactive oxygen species efficiently in aqueous media. Mechanistic studies revealed that Gal-SiX enables both Type I and Type II PDT pathways, an advantageous feature for GBM, where oxygen availability is restricted. In vitro assays conducted on U87MG glioblastoma cells and L929 healthy fibroblasts demonstrated meaningful selectivity, with IC50 values of 3.30 μM and 7.19 μM, respectively. Gal-SiX also showed minimal dark toxicity (>80 μM) and potent light-induced cytotoxicity, yielding a phototoxicity index of 24.8 in glioblastoma cells. Confocal imaging and MTT assays consistently demonstrated its activation and PDT efficacy. Overall, this work introduces the first activatable Si-xanthene–based PDT agent for glioblastoma and provides the first evidence that the Si-xanthene scaffold can support dual Type I/II phototoxicity. These results underscore Gal-SiX’s potential as a selective PDT platform for addressing the unique constraints of GBM biology.
Keywords:
photodynamic therapy
; glioblastoma
; β-galactosidase
; activatable PDT agents
1. Introduction
Cancer remains one of the foremost global health concerns, since effective treatment modalities for a range of cancer types are still elusive.[1,2] While significant progress has been made in cancer prevention, early detection, and treatment across various types of cancers, brain tumors persist as an unresolved clinical challenge.[3,4] Despite their lower incidence compared to many other types of cancer, brain tumors are among the most feared due to their high mortality rates.[5] Among the pediatric solid tumors, brain cancers are also the most prevalent and most fatal type of cancer. Furthermore, several adverse effects of medical operations such as surgery, chemotherapy, and/or radiotherapy are often observed in people who have suffered from a brain tumor and have survived into adulthood.[6]
Among all types of brain tumors, glioblastoma is the most aggressive one, with a two-year survival rate below 35%.[7] In recent years, significant progress has been made in molecular profiling and genomic characterization, enhancing our understanding of the interdependence of glioblastoma proliferation and invasion. This has opened avenues for discoveries that may aid in differential diagnosis and novel treatment strategies.[8] Although several FDA-approved drugs have been developed for the treatment of glioblastoma, only three are currently used: Carmustine, Temozolomide, and Bevacizumab. Unfortunately, none of them has shown superiority over conventional chemotherapy and radiation in terms of extending survival.[9] The inadequacy of current treatment modalities for brain tumors emphasizes the urgent need for further research to develop more effective alternatives.
PDT is an alternative treatment method for diverse cancer types. It has gained attention due to its minimally invasive nature and fewer side effects compared to existing treatment methods[10,11] and holds potential for overcoming the barriers of brain cancer treatment. Nevertheless, its applicability remains limited to a small group of cancers due to the poor penetration of the requisite light through human tissue needed to trigger singlet oxygen generation and the lack of selective targeting of cancer cells.[12] Another obstacle to PDT is the hypoxic nature of certain tumors, particularly glioblastomas. This characteristic feature of these tumors hinders the therapeutic efficacy of PDT.[13] To enhance PDT performance, numerous efforts have been made to increase the O2 concentration in the tumor area, including direct delivery of exogenous O2 to the tumor,[14,15] in situ O2 generation,[16,17] reduction of tumor O2 consumption by inhibiting respiration,[18,19] regulation of the tumor microenvironment,[20] and inhibition of hypoxia-inducible factor 1α (HIF-1α) signaling.[21,22] Furthermore, O2-independent or Type I-biased PDT, which relies more on radical formation than on classical singlet oxygen, is being actively explored as a way to bypass hypoxia-driven resistance.[23] Several PDT agents, including organic photosensitizers and metal complexes, have demonstrated potent photocytotoxicity against various cancer types. [24,25,26,27,28,29,30,31,32,33,34,35,36,37] However, studies focusing on hypoxic glioblastomas using Type I-based PDT agents remain scarce, and although targeted PDT approaches have been reported, no examples of activity-based PDT have been described to the best of our knowledge.[38]
Furthermore, designing an ideal photosensitizer presents another challenge for the effective use of PDT. To achieve deep tissue penetration, PS should absorb light within the therapeutic window (600-850 nm).[39,40,41] Despite the development of numerous fluorophores in recent decades, only a small number have been modified into PSs due to the absence of absorption maxima within the therapeutic window, low aqueous solubility, and difficulties with structural modification.[42,43,44,45] The promising properties of xanthene–based dyes, such as water solubility, photostability, and ease of modification[46,47,48,49,50,51,52,53] motivated us to investigate improvements on classical xanthene dyes such as fluorescein and rhodamine towards the realization of red/near infrared (NIR) absorbing/emitting dyes with highly cancer-selective activation for imaging and treatment opportunities. Previously, extending conjugation within the molecule has been a favored methodology to shift absorption toward the NIR region.[54,55] This ultimately results in higher molecular weight and renders them ineffective for brain cancer treatment due to the highly selective nature of the blood-brain barrier (BBB).[56,57] On the other hand, modification of certain parts of the xanthene core was shown to be highly effective in shifting the absorption maximum toward the NIR region without significantly affecting molecular weight.[54,55,58] In 2011, Nagano and colleagues introduced a red-shifted analogue of fluorescein by replacing the oxygen atom at the 10th position of the xanthene core with a silicon atom, resulting in a 100 nm red shift while preserving all the promising properties of classical fluorescein analogues.[54] To date, Si-Xanthenes has been used in numerous imaging studies; however, its applicability in PDT was demonstrated only recently by our group in our pursuit of viable PDT agents for brain cancer treatment.[43,59,60,61,62] Tetra-iodinated Si-Xanthene (SF-I) proved to be a theranostic agent, exhibiting significant toxicity toward various peripheral cancer cell lines. Nonetheless, the lack of an effective targeting moiety hinders its potential as a potent PDT agent.[63]
β-Galactosidase (β-gal) is a lysosomal glycoside hydrolase that catalyzes the cleavage of β-D-galactosides into monosaccharides, playing a fundamental role in cellular catabolism and turnover of glycoconjugates and complex carbohydrates.[64,65,66] Under physiological conditions, β-gal activity is ubiquitous in lysosomes, but its expression and activity become significantly altered in various pathological states, particularly in the context of cancer biology and cellular senescence. One of the most widely studied phenomena involving β-gal in mammalian cells is senescence-associated β-galactosidase (SA-β-gal) activity, which is used as a canonical biomarker for senescent cells both in vitro and in tumor tissues.[67] Elevated β-gal activity has been observed in various malignancies, including ovarian, gastric, and other epithelial cancers[68,69] and this overexpression has been leveraged for selective imaging and detection of tumor and metastatic tissue using β-gal-activated probes.[64,65] Although abnormal β-gal levels are generally attributed to metastatic ovarian cancer,[66] significantly elevated β-gal levels in gliomas compared to healthy cells have also been reported.[70,71] Due to its distinct cancer selectivity, the β-gal enzyme is utilized in fluorescent diagnostic studies.[69,72,73] However, β-gal enzyme-activatable phototherapeutic drugs remain surprisingly rare, with the first β-gal activatable PDT agent for brain cancers, using an iodinated resorufin photosensitizer, having been demonstrated by our group.[74]
Herein, we report the development of iodinated Si-Xanthene, modified with a β-gal-responsive β-D-galactopyranose moiety, as an activatable PDT agent (Gal-SiX). Gal-SiX demonstrated selectivity toward glioma (U87MG) cells over healthy (L929) cells in vitro. Gal-SiX remains in an OFF state until activation by β-gal and exhibits significantly low toxicity. Upon cleavage of the glycosidic linkage between the β-D-galactopyranose moiety and the photosensitizer (PS) in the presence of high β-gal levels in glioblastoma, the highly cytotoxic and moderately emissive iodinated Si-Xanthene core (SiX) is revealed (Figure 1). Thus, the first β-gal activatable Si-Xanthene-based PS for theranostic PDT applications has been demonstrated and tested in glioblastoma in vitro. It is essential to note that SiX was not separately synthesized in this work; SiX refers to the released species after β-gal treatment of Gal-SiX.
2. Materials and Methods
2.1. Chemistry
2.1.1. General
The starting materials and solvents were purchased from Sigma-Aldrich, ABCR, TCI, and Merck. Solvents used for column chromatography, Hexane, EtOAc, and DCM, were distilled prior to use over CaCl2. All reactions were conducted under a nitrogen atmosphere, unless otherwise specified. Reaction solvents (Diethyl ether, THF, DCM, Toluene, and DMF) were directly used from the MBraun MBSPS5 solvent drying system. The reactions were monitored by thin-layer chromatography (TLC) (Merck Silica Gel 60 F254) and visualized by UV light at 254 nm and 366 nm. Column chromatography of all products was performed using Merck Silica Gel (particle size: 0.040-0.063 mm, 230-400 mesh ASTM).
The structural analysis of the synthesized compounds was conducted using NMR and HRMS. 1H and 13C nuclear magnetic resonance spectra of the compounds were recorded in deuterated solvents with Bruker Avance III Ultrashield 400 Hz NMR spectrometer. The chemical shifts were reported in parts per million (ppm) relative to tetramethylsilane (TMS) as an internal reference. High-Resolution Mass Spectra (HRMS) were processed for the novel compounds using Time of Flight mass analyzer with Water Synapt MS System. The purity of the final compound was confirmed by reverse-phase HPLC to be>95%.
The photophysical analyses were performed through absorbance and fluorescence measurements. Absorption spectra were collected using a double-beam Jasco V-730 (Easton, MD, USA) UV-vis spectrophotometer. A Cary Eclipse fluorescence spectrophotometer (Santa Clara, CA, USA) equipped with a Cary single-cell Peltier temperature controller (Agilent Technologies) was used to record the fluorescence emission spectra. All measurements were performed at room temperature using 10 mm quartz (3.5 mL, 111-QS, Hellma).
2.1.2. Synthesis
Compound 1: In a 250 mL Schlenk tube, commercially available 3-bromoaniline (8.10 g, 47.1 mmol) and K2CO3 (18.88 g, 136.6 mmol, 2.9 eq.) were suspended with dry MeCN (110 mL) under nitrogen. BnBr (18.8 mL, 160.1 mmol, 3.4 eq.) was added slowly and then the mixture was heated to 80°C for 16 hours. After completion of reaction, MeCN evaporated and the crude product was treated with distilled water (80 mL) and extracted with EtOAc (2 x 80 mL). Collected organic phases were dried over Na2SO4, filtered and the solvent was evaporated. The crude product redissolved with EtOAc (5 mL) and added to cold hexane (20 mL) dropwise to precipitate the product. After the addition was complete, the hexane/ethyl acetate mixture was removed the precipitate was dried under vacuum. The target product, compound 1, was obtained as a white solid (13.6 g, 82%). 1H NMR (400 MHz, CDCl3) δ 7.35 (t, J = 7.3 Hz, 4H), 7.29 (d, J = 7.1 Hz, 2H), 7.23 (d, J = 7.3 175 Hz, 4H), 7.01 (t, J = 8.1 Hz, 1H), 6.90 (s, 1H), 6.82 (d, J = 7.7 Hz, 1H), 6.64 (dd, J = 8.3, 2.0 Hz, 1H), 4.63 (s, 4H). 13C NMR (100 MHz, CDCl3) δ 150.6, 137.9, 130.6, 128.9, 127.2, 126.7, 123.6, 119.7, 115.15, 111.2, 54.1.
Compound 2: In a 250 mL Schlenk tube, compound 1 (4.85 g, 13.8 mmol, 2 eq.) and formaldehyde solution (2.07 g, 6.90 mmol, 30%) were placed and dissolved with AcOH (40 mL). Then the mixture was heated to 80°C and stirred for 16 hours at that temperature. After that, mixture cooled to room temperature and neutralized with satd. NaHCO3 and satd. NaOH solutions and then extracted with CHCl3 (2 x 50 mL). Collected organic phases were dried over Na2SO4, filtered and the solvent was evaporated. The crude product purified with column chromatography (silica, hexane:EtOAc 6:1). The target product, compound 2, was obtained as a white solid (4.05 g, 82%). 1H NMR (400 MHz, CDCl3) δ 7.24 (t, J = 7.1 Hz, 8H), 7.18 (d, J = 6.5 Hz, 4H), 7.13 (d, J = 7.4 Hz, 8H), 6.88 (d, J = 2.0 Hz, 2H), 6.70 (d, J = 8.5 Hz, 2H), 6.47 (dd, J = 8.4, 2.2 Hz, 2H), 4.50 (s, 8H), 3.86 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 148.8, 138.1, 131.1, 128.8, 127.4, 127.1, 126.7, 125.8, 125.8, 116.0, 111.8, 54.1, 39.9
Compound 3: In a 100 mL Schlenk tube, compound 2 (2.00 g, 2.79 mmol) was added and was dissolved with dry THF (12 mL) under nitrogen and was cooled to -78°C. Then, sec-BuLi (6.50 mL, 7.95 mmol, 1.4 M, 2.85 eq.) was added dropwise and stirred at that temperature for 1 h. 1.9 mL SiMe2Cl2 from stock solution ((stock solution: 1.9 mL SiMe2Cl2 + 3.8 mL THF), 5.30 mmol, 1.9 eq.) was added dropwise then reaction mixture warmed to room temperature slowly. After reaching the room temperature, mixture stirred at that temperature for 3 h. After that, reaction mixture was treated with 1 N HCl (1 mL) then neutralized with NaHCO3 (25 mL) and extracted with CHCl3 (2 x 50 mL). Collected organic phases were dried over Na2SO4, filtered and the solvent was evaporated. The crude product was placed in 100 mL two necked round-bottomed balloon and dissolved with acetone (26 mL) and cooled to 0°C. To reaction mixture KMnO4 (1.22 g, 7.67 mmol, 2.75 eq.) in portions over 1.5 hours and stirred for 2.5 hours more at that temperature. After that, reaction mixture filtered through celite and washed with CHCl3. The crude product was purified with column chromatography (neutral alumina hexane:CHCl3 1:2). The target product, compound 3, was obtained as a yellow solid (0.99 g, 57%). 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 9.0 Hz, 2H), 7.34 – 7.28 (m, 8H), 7.23 (d, J = 7.3 Hz, 12H), 6.86 (dd, J = 9.0, 2.7 Hz, 2H), 6.80 (d, J = 2.7 Hz, 2H), 4.71 (s, 8H), 0.14 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 185.1, 150.7, 140.6, 137.8, 131.8, 130.4, 128.9, 127.3, 126.7, 115.2, 113.7, 54.2, -1.4.
Compound 4: In a parr reactor, compound 3 (0.60 g, 1.0 mmol) was placed and dissolved with MeOH:CHCl3 mixture (30 mL, 4:1). To this solution Pd-C (0.12 g, 20% by mass) was added then air removed by vacuum. Reactor was refilled with H2 gas (3.6 bar) and stirred for 3 days. After completion of reaction, the mixture was filtered through celite and washed with MeOH. The crude product was purified with column chromatography (silica, MeOH:DCM, 1:9). The target product, compound 4, obtained as a white solid (0.20 g, 77%). 1H NMR (400 MHz, MeOD) δ 8.13 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 2.4 Hz, 2H), 6.75 (dd, J = 8.7, 2.4 Hz, 2H), 0.40 (s, 6H). 13C NMR (100 MHz, MeOD) δ 187.6, 153.1, 142.8, 132.9, 131.1, 118.6, 116.7, -1.2.
Compound 5: In a 250 mL two-necked round bottomed flask, compound 4 (0.10 g, 0.4 mmol) was added and dissolved with MeOH : 6 N H2SO4 (92 mL, 1:1) under nitrogen and was cooled to 0 °C. Solution of NaNO2 (0.17 g, 2.5 mmol, 6.7 eq.) in H2O (4 mL) was added dropwise and stirred for 1 hour at that temperature. After that, mixture was added dropwise to the boiling 1 N H2SO4 (135 °C, 100 mL) over 1 hour. Then stirred 10 more minutes at that temperature before cooling to room temperature. After completion of reaction, mixture extracted with CHCl3 (2 x 50 mL). Collected organic phases were washed with brine, dried over Na2SO4, filtered and the solvent was evaporated. The crude product was purified with column chromatography (silica, MeOH:DCM 5:95). The target product, compound 5, obtained as a white solid (61 mg, 61%). 1H NMR (400 MHz, MeOD) δ 8.22 (d, J = 8.8 Hz, 2H), 7.03 (d, J = 2.6 Hz, 2H), 6.91 (dd, J = 8.8, 2.6 Hz, 2H), 0.39 (s, 6H). 13C NMR (100 MHz, MeOD) δ 187.8, 162.3, 143.2, 133.9, 133.5, 120.2, 118.5, -1.4.
Compound 6: In a 250 mL two-necked round bottomed flask, compound 5 (100 mg, 0.37 mmol) and imidazole (260 mg, 3.83 mmol, 10.4 eq.) were added and dissolved with dry DCM (55 mL) under nitrogen. To this solution, solution of TBDMSCl (562 mg, 3.74 mmol, 10.1 eq.) in dry DCM (13 mL) was added slowly and stirred for 16 hours at room temperature. After completion of reaction, the mixture was treated with distilled water (50 mL) and extracted. Collected organic phases were washed with brine, dried over Na2SO4, filtered and the solvent was evaporated. The crude product was purified with column chromatography (silica, hexane:DCM 1:8). The target product, compound 6, obtained as a white solid (153 mg, 83%). 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 8.7 Hz, 2H), 7.05 (d, J = 2.2 Hz, 2H), 6.99 (dd, J = 8.7, 2.5 Hz, 2H), 1.01 (s, 18H), 0.47 (s, 6H), 0.27 (s, 12H). 13C NMR (100 MHz, CDCl3) δ 186.1, 158.9, 141.3, 134.7, 132.4, 123.8, 121.9, 25.8, 18.4, -1.4, -4.2.
Compound 7: In a 100 mL two necked round bottom flask, commercially available o-bromotoluene (343 mg, 2.00 mmol, 5 eq.) was added and dissolved with dry THF (30 mL) under nitrogen and cooled to -78°C. After that, n-BuLi (0.80 mL, 2.0 mmol, 2.5 M, 5 eq.) was added dropwise and stirred for 30 minutes at that temperature. Then, compound 6 (200 mg, 0.40 mmol) in dry THF (5 mL) was added to the reaction mixture dropwise and the mixture was allowed to warm room temperature slowly and stirred 1.5 hours more at that temperature. After completion of reaction, mixture was treated with 1 M HCl (12 mL) and extracted with DCM (50 mL). Collected organic phases were washed with brine, dried over Na2SO4, filtered and the solvent was evaporated. The crude product was purified with column chromatography (silica, MeOH:CHCl3 1:18). The target product, compound 7, obtained as a dark orange solid (86 mg, 63%). 1H NMR (400 MHz, CDCl3) δ 7.38 – 7.34 (m, J = 7.4 Hz, 1H), 7.29 (d, J = 8.2 Hz, 2H), 7.09 – 7.04 (m, 3H), 6.94 (d, J = 9.4 Hz, 2H), 6.56 (dd, J = 9.4, 2.1 Hz, 2H), 3.49 (s, 1H), 2.03 (s, 3H), 0.43 (s, 3H), 0.41 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.6, 161.5, 146.0, 140.5, 139.3, 135.9, 130.2, 129.9, 129.2, 129.0, 128.5, 125.7, 122.1, 19.6, -1.2, -1.5.
Compound 8: In a 100 mL two necked round bottom flask, compound 7 (69 mg, 0.20 mmol) and CsCO3 (326 mg, 1.00 mmol, 5 eq.) were added and dissolved with dry MeCN (45 mL) under nitrogen. After that, acetobromo-α-D-galactose (348 mg, 1.00 mmol, 5 eq.) was added and stirred for 16 hours. After completion of reaction, MeCN was removed from reaction mixture with reduced pressure and mixture was dissolved in CHCl3 (50 mL) and extracted with distilled water (50 mL) and washed with brine (50 mL). Collected organic phases were dried over Na2SO4, filtered and the solvent was evaporated. The crude product was purified with column chromatography (silica, MeOH:CHCl3 1:18). The target product, compound 8, obtained as an orange solid (75 mg, 56%). 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.33 (m, 1H), 7.33 – 7.27 (m, 2H), 7.24 (d, J = 2.1 Hz, 1H), 7.06 (t, J = 6.5 Hz, 1H), 6.94 (d, J = 10.1 Hz, 1H), 6.85 – 6.76 (m, J = 9.2, 5.6 Hz, 3H), 6.22 (dd, J = 10.1, 1.9 Hz, 1H), 5.53 – 5.42 (m, 2H), 5.16 – 5.08 (m, 2H), 4.20 – 4.04 (m, 3H), 2.16 (s, 3H), 2.04 (dd, J = 6.7, 1.1 Hz, 6H), 2.00 – 1.97 (m, 6H), 0.46 (t, J = 6.0 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 184.4, 170.3, 170.2, 170.1, 169.4, 157.0, 157.0, 154.9, 146.8, 141.6, 140.6, 139.1, 137.2, 136.1, 136.0, 135.7, 135.0, 130.3, 130.2, 129.2, 128.4, 128.0, 125.9, 125.9, 122.9, 116.6, 98.2, 71.2, 70.7, 68.4, 66.7, 61.3, 61.3, 53.5, 20.8, 20.6, 20.6, 19.5, -1.2, -1.2, -1.4, -1.5.
Compound 9: In a 50 mL Schlenk tube, compound 8 (64 mg, 0.10 mmol) was added, and dissolved with EtOH (10 mL) under nitrogen. After that, I2 (48 mg, 0.19 mmol, 2 eq.) was added to the mixture and stirred for 15 minutes at 60 °C. Then, HIO3 (33.4 mg, 0.19 mmol, 2 eq.) solution in H2O (1.0 mL) was added dropwise to the reaction mixture and temperature raised to 78 °C stirred for 2 hours. After consumption of all starting material, EtOH was evaporated, and mixture treated with 10% Na2S2O3 solution (10 mL) and extracted with EtOAc (50 mL). Collected organic phases were dried over Na2SO4, filtered and the solvent was evaporated. The crude product was purified with column chromatography (silica, MeOH:DCM 1:18). The target product, compound 9, obtained as a dark orange solid (75 mg, 85%). 1H NMR (400 MHz, CDCl3) δ 7.74 (s, 1H), 7.41 (t, J = 7.2 Hz, 1H), 7.36 – 7.29 (m, 2H), 7.24 (s, 1H), 7.06 (t, J = 6.7 Hz, 1H), 6.80 (s, 2H), 5.50 (dd, J = 10.3, 8.0 Hz, 1H), 5.45 (d, J = 3.2 Hz, 1H), 5.18 (d, J = 7.7 Hz, 1H), 5.13 (dd, J = 10.4, 3.2 Hz, 1H), 4.21 – 4.08 (m, 3H), 2.17 (s, 3H), 2.06 (s, 3H), 2.04 (s, 3H), 2.02 – 1.98 (m, J = 4.8 Hz, 6H), 0.78 (t, J = 5.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 171.9, 170.1, 170.0, 169.9, 169.2, 158.1, 157.3, 151.4, 151.3, 143.0, 138.9, 135.6, 135.5, 134.2, 131.9, 130.3, 128.8, 128.8, 125.9, 123.0, 118.2, 116.6, 98.0, 98.0, 97.7, 71.2, 70.5, 68.3, 66.6, 61.2, 61.2, 20.6, 20.5, 20.4, 19.5, 19.5, -1.2, -1.2, -1.3, -1.3.
Gal-SiX: In a 25 mL Schlenk tube, compound 9 (75 mg, 0.1 mmol) was added, and dissolved with MeOH (7 mL) under nitrogen and cooled to 0°C. To this solution, NaOMe (10 mg, 0.4, 4.4 eq.) in MeOH (1.1 mL) solution added dropwise and stirred for 1 hour at that temperature. After completion of reaction, mixture was neutralized with amberlyst, then filtered. Collected filtrate was evaporated and the crude product was purified with preparative HPLC. The target product, Gal-SiX, obtained as a dark orange solid (57 mg, 93%). 1H NMR (400 MHz, MeOD) δ 7.82 (s, 1H), 7.55 – 7.48 (m, J = 7.5, 5.1 Hz, 2H), 7.47 – 7.38 (m, J = 14.3, 7.1 Hz, 2H), 7.17 (d, J = 7.4 Hz, 1H), 7.05 – 6.99 (m, 1H), 6.90 (d, J = 9.1 Hz, 1H), 5.03 (dd, J = 7.7, 5.1 Hz, 2H), 3.91 (d, J = 3.3 Hz, 1H), 3.85 (dd, J = 9.5, 7.9 Hz, 1H), 3.79 – 3.74 (m, 2H), 3.62 (dd, J = 9.7, 3.2 Hz, 2H), 2.07 (s, 3H), 0.84 (dd, J = 5.3, 3.5 Hz, 6H). 13C NMR (100 MHz, MeOD) δ 172., 160.2, 160., 159.9, 152.7, 151.9, 143.0, 139.0, 136.0, 135.4, 132.7, 131.1, 130.0, 128.6, 125.6, 122.7, 122.6, 117.5, 117.4, 115.8, 100.4, 100.4, 99.9, 95.3, 75.9, 73.2, 70.5, 68.7, 61.0, 18.0, -2.6, -2.7, -2.7, -2.8. HRMS (ESI/MS) m/z: [M+H]+ Calcd. for C22H18I2O2Si+ 596.9200; Found 596.9240
2.1.3. HPLC Analysis
Reverse phase HPLC analyses were conducted using Agilent Technologies 1260 Infinity II series HPLC systems with DAD detector. All the analyses were performed by using gradient elution with different Milli Q (0.1% TFA) and acetonitrile (0.08% TFA) systems as mobile phase and the column compartment temperature was 40°C. HPLC purifications were performed using a Phenomenex Kinetex 5 μm C18 100 A semi-preparative column with a flow rate of 1.5 mL/min. And for the purities, an Agilent Technologies Poroshell 120 EC-C18 analytical column was used with a flow rate of 0.5 ml/min.
2.2. Photophysical Characterization
2.2.1. Enzymatic Activation
To demonstrate the enzymatic cleavage of the handle group of Gal-SiX in the presence of β-galactosidase, Gal-SiX (10 μM) in PBS buffer (pH 7.4, 1% DMSO) was incubated with β-galactosidase (5 U), and absorbance spectra were recorded at 0, 10, 20, 30, 40, and 75 minutes. Time-dependent activation was observed, with complete activation achieved after 40 minutes of incubation.
2.2.2. Fluorescence Quantum Yield
Fluorescence quantum yields of the samples were investigated using a fluorescence spectrometer (FLS 1000, Edinburgh Instruments) equipped with an integrating sphere accessory. A continuous-wave xenon lamp served as the excitation source, and the emitted fluorescence was detected with a standard photomultiplier (PMT-900) covering a wavelength range of 200-800 nm. During measurements, the PMT was cooled to -20 °C using a built-in housing to reduce undesired dark current noise.
For quantum yield measurement, an integrating sphere (Edinburgh Instruments) was placed inside the spectrometer's sample compartment. The internal cavity of the sphere was coated with a PTFE-like material, enabling a reflectance of approximately >99% (>95%) over the wavelength ranges of 400-1500 nm and 250-2500 nm, respectively. The sphere had two ports positioned 90° apart. The excitation beam was directed to the sample through the excitation port, and the fluorescence was collected from the emission port. The excitation port of the sphere included a lens to effectively focus the beam on the sample, while the emission port was an open aperture.
Prior to the experiments with the PSs, blank spectra were measured using the reference solvents (PBS, pH 7.4, 1% DMSO). For both measurements (blank and sample), two identical quartz cuvettes with equal volumes were used. Initially, the reference sample was placed inside the sphere, and the emission/excitation slits were adjusted to the excitation wavelength to ensure the PMT's response remained linear during measurements. To cover a scattering range, the emission scans started 20 nm below the actual excitation wavelengths and ended at 900 nm. Additionally, the step size and integration time of the measurements were set to 1 nm and 0.2 seconds, respectively.
Upon completing all emission measurements of the samples and references, the quantum yields of the samples were determined using the Fluoracle software. The built-in analysis tool calculates the quantum yield (QY) as:
where Es (EB) and Ss (SB) are the selected areas for the emitted and scattered signals of the sample (blank).
𝑄𝑌=(𝐸𝑠−𝐸𝐵)/(𝑆𝐵−𝑆𝑠),
2.2.3. Singlet Oxygen Trap Experiment
Singlet oxygen quantum yields were calculated using the following equation and methylene blue as the reference fluorophore according to our latest experimental procedure [58].
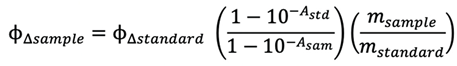
2.2.4. General Procedure for Detection of ROS Type
To identify the type of reactive oxygen species (ROS) generated, several ROS-specific fluorescent probes were employed: Singlet Oxygen Sensor Green (SOSG) for the detection of singlet oxygen (1O2), Dihydrorhodamine 123 (DHR123) for superoxide anion (O2•-), and 2-[6-(4′-amino)phenoxy-3H-xanthen-3-on-9-yl]benzoic acid (APF) for hydroxyl radical (·OH). The interaction between the probes and the generated ROS was assessed following photoirradiation of Gal-SiX in PBS buffer (pH 7.4) containing 1% DMSO, after incubation with β-galactosidase enzyme for 40 minutes at 37 °C.
Probe concentrations were maintained at 5 μM, and the activated Gal-SiX concentration was set at 10 μM. Fluorescence measurements were performed using a spectrofluorometer with emission wavelengths set at 525 nm for SOSG, 526 nm for DHR123, and 517 nm for APF. The cuvette containing the photosensitizer and the respective probe was irradiated using a 595 nm LED light source placed 10 cm away. Each irradiation lasted for 10 seconds, and the fluorescence intensity was monitored for up to 100 seconds.
2.2.5. Interference Studies
To evaluate potential interference from biologically relevant molecules, various ions and small molecules-including sodium iodide (NaI), potassium chloride (KCl), lithium fluoride (LiF), calcium chloride (CaCl₂), sodium nitrite (NaNO₂), sodium thiosulfate (Na₂S₂O₃), thiourea, hydrogen peroxide (H₂O₂), glucose, and homocysteine were tested. Each was prepared at a concentration of 1 mM in PBS and added to a solution of Gal-SiX (10 μM) in PBS buffer (pH 7.4, 1% DMSO), which was incubated in the presence or absence of β-galactosidase (5 U). The resulting solutions absorbance were recorded.
2.3. In vitro Experiments
2.3.1. Cell Culture
Human glioblastoma cells (U87MG) and healthy cells (L929, mouse fibroblast cells) were used in this study. L929 mouse fibroblast cells were selected as the healthy control line because they are non-transformed, robust, and widely recommended for in vitro cytotoxicity testing by ISO 10993-5:2009 (Annex C: MTT cytotoxicity test) owing to their reproducible growth and sensitivity.[75] Their use provides a standardized benchmark for comparative phototoxicity studies[76] and ensures methodological consistency with U87MG cells under identical culture and irradiation conditions. Cells were cultured under standard conditions (in high-glucose DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, 0.5% amphotericin B, and 2 mM glutamine at 37 °C with 5% CO₂). The cells were passaged every 3-4 days when they reached 80-90% confluency. Gal-SiX was dissolved in cell culture-grade DMSO and further diluted in complete medium for experimental use.
2.3.2. Photodynamic Therapy
For photodynamic therapy studies, cells were incubated with increasing concentrations of Gal-SiX (0-10 μM) for 0.5-2 hours in the dark, followed by LED light irradiation (595 nm, 8.12 mW/cm2) for 2 hours. The incubation medium was not replaced before irradiation to maintain continuous β-galactosidase-mediated activation of the Gal-SiX during the entire incubation period. This approach preserves both intracellular and extracellular activation kinetics, ensuring accurate assessment of the agent’s full photodynamic potential under enzyme-active conditions and reflecting the in vitro microenvironment of solid tumors, where limited diffusion and sustained enzymatic activity allow local accumulation of the activated PS.[77,78,79,80] After the irradiation period, cells were further incubated in the dark for 24 hours to evaluate phototoxic effects. In parallel experiments for index determinations, U87MG and L929 cells were treated with increasing concentrations of Gal-SiX (0.1-160 μM) for 24 hours under dark conditions, followed by viability assessment using the MTT assay. Phototoxicity index (PI), selectivity index (SI), and in vitro therapeutic index (TI) values were calculated as follows:
PI=IC50, dark /IC50, light
SI= IC50, light (L929)/IC50, light (U87MG)
TI= IC50, dark (L929) /IC50, light (U87MG)
2.3.3. Cell Viability Analysis
Cells were seeded at 5x104 cells/well on a flat-bottom 96-well plate in complete medium for 24 h. After incubation, cells were treated with Gal-SiXas described above. Following experimental protocols, cells were treated with 0.5 mg/ml MTT in a fresh medium for 2-4 h at 37 °C. The formazan crystals were dissolved with 10% SDS in PBS (0.01 N HCl) and incubated at 37 °C overnight. Absorbance values of each well were measured at 490 nm and 570 nm wavelengths using a MultiskanSky Microplate Reader (Thermo Scientific, USA). The results were expressed as percentages relative to the control samples treated with DMSO (0.2%), (n=6-8). IC50 values were determined using concentration-normalized response curves derived from non-linear regression analysis (GraphPad Prism 9.02, GraphPad Software Inc.).
2.3.4. Cellular Internalization and Activation
For cellular internalization and activation studies, U87MG and L929 cells were seeded in black 24-well plates (2× 104 cells/well) and incubated overnight. Cells were treated with Gal-SiX(1 µM) for 0.5, 1, 2, and 4 h, then washed twice with 1X PBS. Cells were stained with Hoechst 33342 (0.5 µg/ml) for 15 minutes in serum-free medium at 37 °C. After washing steps, cells were fixed with 4% paraformaldehyde for 15-20 min at RT. Confocal images were captured using a Zeiss LSM 900 CLSM at a 40X objective with constant exposure parameters. The fluorescence intensities of each time point were normalized relative to those of the 0.5 h L929 cells to evaluate the time-dependent activation and internalization states. Each experimental condition was performed in triplicate (n = 3). D-galactose (competitive inhibitor for β-galactosidase) was used to evaluate the cage-cleavage of Gal-SiX. In brief, cells were pre-treated with the increasing concentrations (5-50 mM) of D-galactose (in PBS) for 4 h, followed byGal-SiX (2.5 µM, 1 h) post-treatment and PDT application. After 24 hours of dark incubation, MTT analysis was performed (n = 6).
2.3.5. Subcellular co-localization experiments
Cells were seeded in 35 mm glass-bottom confocal dishes (2 × 10⁴ cells/well) and treated with Gal-SiX (1 µM and 2.5 µM) for 1 h. After incubation periods, cells were washed with PBS and incubated with the following organelle-specific trackers: MitoTracker™ Green FM (100 nM, 45 min), LysoTracker™ Yellow HCK-123 (75 nM, 45 min), ER-Tracker™ Green (BODIPY™ FL Glibenclamide) (1 µM, 30 min) and Hoechst 33342 (0.5 µg/ml, 15 min). Images were captures under the conditions with specified excitation/emission wavelengths for Hoechst, 361/497 nm; MitoTracker, 488/516 nm; LysoTracker, 465/535 nm; ER-Tracker, 504/511 nm; Gal-SiX, (Alexa 594 filter; 570-620 nm). The fluorescence intensities of the photosensitizers were maintained consistently by adjusting the image processing parameters uniformly. Co-localization was evaluated by calculating Pearson’s correlation coefficients (PCCs) using the co-localization analysis tool in Zeiss Zen Blue software, comparing red channel intensity with organelle-specific green signals. All experimental conditions were conducted in triplicate (40X and 63X, n = 3).
2.3.6. Scavenger assays
Reactive oxygen species (ROS) generation was evaluated using the DCFH-DA (2′,7′-dichlorofluorescin diacetate) probe via confocal microscopy. Briefly, U87MG glioblastoma cells were seeded in 96-well plates at a density of 3 × 105 cells/well and allowed to adhere overnight under standard culture conditions. The cells were then treated with Gal-SiX at its IC50 concentration for 1 hour in the dark, followed by LED light irradiation (595 nm, 8.12 mW/cm²) for 2 hours. To investigate the contribution of different ROS types, parallel groups were co-treated with specific ROS scavengers added 1 hour prior to photosensitizer administration. The scavengers and their working concentrations were as follows: N-acetylcysteine (NAC, 5 mM) thiol based ROS scavenger; sodium azide (NaN₃, 5 mM) and histidine (5 mM) for singlet oxygen (1O₂); Tiron (100 μM) for superoxide anion (O2• –); mannitol (25 mM) for hydroxyl radicals (HO·); and Trolox (25 μM) for peroxyl radicals (ROO·).
For imaging, cells were washed twice with PBS and incubated with DCFH-DA (20 μM) and Hoechst 33342 (0.5 μg/mL) in serum-free medium for 20–30 minutes at 37 °C. After staining, cells were rewashed with PBS, and fresh serum-free medium was added. Fluorescence imaging was performed using a Zeiss LSM 900 confocal laser scanning microscope at 488/535 nm (ex/em) for DCF and 361/497 nm (ex/em) for Hoechst, using a 10X objective. Representative fields from each condition were captured to assess intracellular ROS levels and the effect of scavengers. All experiments were performed in biological triplicate (n = 6). For viability assessment, cells were incubated in the dark subsequently after the scavenger assay protocol for an additional 24 hours under standard conditions, followed by MTT analysis.
2.3.7. Intracellular Type I ROS Detection
The DCF probe was used to detect intracellular general reactive oxygen species. The DHR123 probe was employed to monitor hydrogen peroxide, hypochlorous acid, and mitochondrial ROS. The DHE probe was used specifically for the detection of superoxide anion (O2•-). The APF probe was used to evaluate hydroxyl radical (HO·), hypochlorous acid (HOCl), and peroxynitrite anion (ONOO⁻). After incubation with Gal-SiX(2.5 μM) for 1 hour in the dark, the medium was removed, and cells were subsequently stained with DCF (10 μM), DHR123 (5 μM), DHE (5 μM), or APF (10 μM) in fresh medium for an additional 30 minutes at 37 °C. Cells were then irradiated with or without LED light (595 nm, 8.12 mW/cm²) to evaluate ROS production under both light-exposed and dark conditions. Following irradiation, cells were washed twice with PBS, counterstained with Hoechst 33342 (0.5 μg/mL) for 15 minutes to visualize the nuclei, and then washed again with PBS. Confocal fluorescence imaging was performed using a 40X objective with the following excitation/emission settings: 488/535 nm for DCF, 508/536 nm for DHR123, 535/610 nm for DHE, 490/515 nm for APF, and 361/497 nm for Hoechst. No background fluorescence was observed under these conditions. (n=3).
2.3.8. Acridine orange/Ethidium Bromide (AO/EtBr) Dual Staining
Acridine orange/ethidium bromide (AO/EtBr) dual staining was performed to distinguish live, apoptotic or necrotic cells based on differential membrane permeability. In brief, U87MG and L929 cells were plated on a 48-well plate at a density of 5x104 cells/well and treated with IC50 value ofGal-SiX. After the irradiation period, cells were kept at 37 °C for 20 min and stained with AO (2.5 μg/mL) /EtBr (2.5 μg/mL) in serum-free DMEM for an additional 20-30 min at dark. After washing periods with 1X PBS, confocal images were obtained at 500 nm/525 nm (ex/em) and 530 nm/617 nm (ex/em) wavelengths for AO and EtBr dyes, respectively. (10X, n=6)
2.3.9. TBARS Assay for Lipid Peroxidation
The impact of Gal-SiX on cellular lipid peroxidation was assessed using the thiobarbituric acid reactive substances (TBARS) assay, which quantifies malondialdehyde (MDA), a stable end-product of lipid oxidation.[81] Cells were seeded in 6-well plates at a density of 1 ×10⁶ cells per well and allowed to adhere overnight. Subsequently, the PS was applied at an IC50 concentration following the established PDT protocol. Lipid peroxidation was evaluated immediately after irradiation and after a 24 h post-treatment period. At each time point, cells were collected and lysed in RIPA buffer (Merck, Germany). Lysates were then processed according to the TBARS procedure. Briefly, samples were mixed with 0.8% thiobarbituric acid prepared in 20% trichloroacetic acid, incubated at 95 °C for 2-2.5 h, and the absorbance of the resulting chromogenic product was measured at 532 and 555 nm using a spectrophotometer (Thermo, Germany). Results were expressed as a percentage relative to untreated controls and normalized to protein content and cell viability (n = 3-4).
2.3.10. Determination of Free Unsaturated Lipid Content
Intracellular lipid accumulation following Gal-SiX treatment was quantified by the sulfo-phospho-vanillin assay, which primarily detects unsaturated free lipids.[82] Cell lysates were prepared as described in the lipid peroxidation experiment. Protein concentration was determined using the bicinchoninic acid (BCA) method (Thermo Scientific, Germany). For analysis, 40 µL of each lysate was combined with 200 µL of concentrated H₂SO₄, incubated at 90 °C for 10 min, and then cooled to room temperature. Subsequently, 120 µL of vanillin solution (1 mg/mL in 17% phosphoric acid) was added, producing a red chromophore that was quantified spectrophotometrically at 520-540 nm. Data were normalized to protein concentration and expressed as a percentage of the untreated control group (n=3-4).
2.3.11. Intracellular Thiols Detection
Cellular reduced thiol levels (-SH) were quantified using a colorimetric method according to the manufacturer's instructions, which relies on the reaction of -SH with Ellman’s reagent (5,5′-dithiobis-2-nitrobenzoic acid, DTNB) to generate a yellow-colored 2-nitro-5-thiobenzoate product.[83] Cell lysates were prepared as described previously, and Ellman’s reagent (200 μM) was freshly prepared in 1X PBS. Equal volumes of lysate (100 μL) and reagent (100 μL) were mixed and incubated for 30 min at room temperature in the dark. The absorbance of the reaction product was measured at 412 nm using a microplate reader (Thermo, Germany). Free thiol levels were calculated as percentages relative to untreated control cells and normalized to protein concentration (n = 3-4).
2.3.12. Statistical Analysis
Statistical analyses were performed using GraphPad Prism 9.02 (GraphPad Software Inc.). For multiple group comparisons, the Kruskal–Wallis test was employed, followed by Dunn’s post hoc test. All data are presented as mean ± standard deviation (SD). A p value of p<0.05 was considered statistically significant.
3. Results and Discussion
3.1. Synthesis of Gal-SiX
The synthesis of Gal-SiX was achieved via 10-step process by modifying the original synthetic route of Tokyo Magenta (TM) developed by Nagano et al.[54] (Scheme 1) This modification incorporated a greener approach by changing the protecting groups for the aniline moieties and using catalytic amounts of palladium for the deprotection, rather than stoichiometric quantities of the palladium reagent (Scheme 1, boxed reactions).
The synthetic pathway for Gal-SiX, as shown in Scheme 1, begins with the protection of commercially available 3-bromoaniline using a benzyl protective group, which results in the formation of a dibenzyl-protected aniline derivative, compound 1, with an 82% yield. Compound 1 is then condensed with formaldehyde to establish a carbon bridge within the xanthene core. After the successful synthesis of compound 2, the dimethylsilane moiety is introduced into the core through a lithium-halogen exchange reaction using sec-BuLi. Once the silicon bridge is established, the crude product is dissolved in acetone, and KMnO4 is used to oxidize the dibenzylic position, yielding compound 3. The deprotection of the benzyl groups proved challenging. Initial efforts to remove the four benzyl groups via hydrogenation at 1 atmosphere of pressure required 6 days to complete. Implementing the reaction in a Parr reactor with 3.6 bar H2 reduced the reaction time to three days while maintaining a comparable yield. Upon completing the optimization studies for the synthesis of compound 4, the Sandmeyer reaction was utilized to get compound 5. The hydroxyl groups were then protected with TBDMS to give compound 6 with high yield. Following the synthesis of the Si-xanthone core, commercially available o-bromo toluene was introduced at the 9th position of the Si-xanthone core, leading to the formation of compound 7, TM. TM has been widely used in numerous imaging studies due to its exceptional photophysical properties.[75,76,77,78] Compound 7 was further modified with a β-galactose derivative to function as an activatable photosensitizer (aPS). The incorporation of heavy atoms into the PS was achieved by iodinating compound 8 with molecular iodine in the presence of HIO3, with a high yield. Finally, the desired PS, Gal-SiX, was obtained by removing the acetyl protective groups from the β-Gal moiety. The synthesized PS, Gal-SiX, was characterized using 1H, 13C NMR, and HRMS (Figure S29-31). Before investigating its photophysical properties and conducting in vitro cell studies, Gal-SiX was purified using reverse-phase HPLC to achieve a purity greater than 95% (Figure S1).
3.2. Optical Characterization
First, the absorption and fluorescence spectra of Gal-SiX were recorded before β-galactosidase enzyme addition in PBS buffer (pH 7.4, 1% DMSO), and Gal-SiXexhibited an absorption maximum at 486 nm and showed no detectable emission. (Figure 2a)
Upon incubation of Gal-SiX with β-galactosidase (5 U) for 40 min (37 °C), a red-shifted absorption maximum centered at 598 nm, which belongs to the SiX (Scheme 1), was obtained, accompanied by fluorescence emission with a maximum at 613 nm. (Figure 2b, c) ΦF was calculated as 6.4% for SiX by employing Tokyo Magenta as a reference dye,[54] which is significantly lower compared to Tokyo Magenta, as expected, due to intersystem crossing (ISC) by the heavy atom effect (Table 1). To demonstrate the ex vivo activation of Gal-SiX upon incubation with β-galactosidase, Gal-SiX (10 μM) in PBS buffer (pH 7.4, 1% DMSO) was incubated with β-galactosidase (5 U), and absorbance spectra were recorded at various time intervals (0-75 minutes). Full activation was observed after 40 minutes of incubation (Figure S2), and subsequent photophysical measurements were performed following 40 minutes of incubation with β-galactosidase (5 U). Gal-SiXwas further treated with biologically relevant analytes in the presence or absence of β-galactosidase to test selectivity toward this enzyme. No significant change was recorded in the absorbance spectra of Gal-SiX in the presence of these analytes, indicating high selectivity toward β-galactosidase. (Figure S6)
Before conducting ROS generation measurements, the photostability of Gal-SiX and SiX was evaluated under irradiation with a 595 nm LED light source at a distance of 10 cm (3.50 mW/cm²). After 60 minutes dark incubation in cell culture medium in the presence or absence of β-galactosidase, absorbance spectra of Gal-SiX and SiX were recorded after various time intervals up to 2 hours light exposure. To our delight, no significant change in the absorption maxima was observed for Gal-SiX and the cleaved active form SiX (Figure S3a-d). Subsequently, the singlet oxygen (1O2) generation capacity of SiX was investigated using the commonly employed 1O2 trap molecule, 2,2′-(anthracene-9,10-diyl)bis(methylene)dimalonic acid (ADMDA). Upon irradiation of β-Gal-treated Gal-SiX (SiX) with an LED at 595 nm, a time-dependent decrease in the absorption peak of ADMDA at 380 nm was observed due to cycloaddition reaction between photosensitized 1O2 and the anthracene ring (Figure S4-5). In contrast, no change in the absorption signal was observed when Gal-SiX was irradiated in the absence of the enzyme, as the photosensitizer cannot absorb the excitation light under these conditions (Figure S3a). The singlet oxygen quantum yield for SiX was calculated to be 52% (Table 1) using methylene blue as a reference (ΦΔ = 0.52 in PBS buffer).[79] To further support the 1O2 generation, SiX was illuminated in the presence of singlet oxygen sensor green (SOSG) and a notable increase in SOSG emission (Figure 2d) indicated a successful 1O2 generation.
To further evaluate the ROS generation capacity of Gal-SiX, the PS was incubated with β-galactosidase (5 U) for 40 minutes at 37 °C and treated with dihydrorhodamine 123 (DHR123) and 2-[6-(4′-amino)phenoxy-3H-xanthen-3-on-9-yl]benzoic acid (APF) to investigate its superoxide and other Type I ROS (peroxyl, hydroxyl, hypochlorite anions) generation capabilities. (Figure 2d) A marked enhancement in the fluorescence (15-fold) of oxidized DHR123 supported a strong ROS production through Type I mechanism. (Figure 2e) Additionally, a slight increase in APF emission suggested a measurable hydroxyl radical (·OH) and peroxynitrite (ONOO−) generation capacity for SiX. (Figure 2f).
3.3. In vitro Studies
3.3.1. Cytotoxicity Analysis
The phototoxicity of Gal-SiX was systematically evaluated in U87MG glioblastoma and L929 healthy cells. To determine the optimal incubation and light exposure conditions, cells were first incubated with increasing concentrations (0.5–10 μM) of Gal-SiX for 0.5, 1, and 2 hours in the dark, followed by LED irradiation at 1 cm over the lid (595 nm, 8.12 mW/cm²) for 0.5, 1, or 2 hours, and subsequent incubation in the dark for 24 hours (Figure S7). In U87MG cells, a more pronounced reduction in cell viability was observed with longer light exposure times, particularly at higher concentrations and after 1- or 2-hour pre-incubation periods. In contrast, L929 cells exhibited relatively higher viability under similar conditions, indicating lower phototoxic sensitivity. Based on these optimization results, a protocol consisting of 1-hour pre-incubation followed by 2-hour LED irradiation was selected for further studies. Under these optimized conditions, U87MG cells demonstrated a concentration-dependent decrease in cell viability upon light activation, with significant effects observed at concentrations ≥1 μM (Figure 3a). In contrast, dark-treated cells maintained high viability even at the highest tested concentrations, confirming minimal dark toxicity. Similar trends were observed in L929 cells; however, the overall phototoxicity was less pronounced than in U87MG cells (Figure 3b). The calculated IC50 values (at optimized conditions) were 3.30 ± 0.11 μM for U87MG cells and 7.19 ± 0.32 μM for L929 cells. IC50 values for the other conditions were presented in Supplementary Table S2. The photodynamic performance and tumor selectivity of Gal-SiX were quantitatively evaluated using established metrics, including the phototoxicity index (PI), selectivity index (SI), and in vitro therapeutic index (TI). Gal-SiXexhibited high dark IC50 values of 81.92 ± 2.94 μM in U87MG and 116.6 ± 3.34 μM in L929 cells, confirming its minimal dark cytotoxicity. Under light irradiation, these values translated to a PI of 24.8 and an SI of 2.18, indicating that tumor cells were approximately two-fold more susceptible to Gal-SiX under light conditions than healthy fibroblast cells. Furthermore, the calculated TI (35.3) underscores the broad therapeutic window ofGal-SiX, indicating that high concentrations safely tolerated by normal cells in the dark are sufficient to induce potent, light-activated phototoxicity in U87MG cells (Table S3).
3.3.2. Cellular Uptake and Activation-induced Cell Death
To establish the correlation between internalization and the observed cytotoxicity, the intracellular uptake and activation kinetics of Gal-SiX were further investigated in U87MG and L929 cells at different dark incubation periods (0.5, 1, 2, and 4 h) with confocal microscopy (Figure S8). In U87MG cells, a progressive increase in red fluorescence intensity was observed with more extended incubation periods, indicating time-dependent internalization and activation of Gal-SiX.
Notably, a strong intracellular signal was observed after 1 hour, which further intensified at 2 and 4 hours. In contrast, L929 cells displayed comparatively lower fluorescence intensity at all time points, suggesting reduced uptake and/or activation efficiency in non-tumor cells. These findings confirm selective and gradual intracellular accumulation of Gal-SiX in glioblastoma cells, supporting its tumor-targeted activation profile after 1 h incubation (Figure 3c), which supports the statistically significant phototoxicity profile of Gal-SiX in U87MG cells compared to L929 cells (Figure 3d). To further support the β-galactosidase-dependent activation of Gal-SiX in U87MG cells were pre-incubated with increasing concentrations of D-galactose (5–50 mM) as a competitive inhibitor.[80] A concentration-dependent increase in cell viability was observed with D-galactose pre-treatment, confirming that Gal-SiX activation is specifically mediated by endogenous β-galactosidase activity (Figure 4a).
Following confirmation of cellular uptake and activation, the underlying mechanism of cell death was elucidated. For this purpose, apoptotic markers were assessed. Live/dead cell staining using acridine orange and ethidium bromide (EtBr) revealed a substantial increase in red fluorescence in U87MG cells at IC50, consistent with extensive loss of plasma membrane integrity and chromatin condensation, indicative of late apoptosis and secondary necrosis.[81,82,83] The predominance of EtBr-positive nuclei and fragmented or swollen morphologies supports activation of terminal cell death programs rather than transient stress responses.[83,84,85] In contrast, L929 cells showed predominantly green fluorescence with intact nuclear morphology, suggesting higher viability and minimal induction of apoptosis or necrosis (Figure 3e).
3.3.3. Subcellular Localization
Mitochondria are critical regulators of intrinsic apoptosis, as their photodamage can lead to mitochondrial membrane depolarization, cytochrome c release, and activation of downstream pathways. Similarly, lysosomal targeting in PDT has been shown to induce lysosomal membrane permeabilization, resulting in the release of cathepsins and subsequent activation of apoptotic or necrotic pathways. The preferential accumulation of photosensitizers within mitochondria and lysosomes is therefore considered a critical determinant of PDT efficacy, as both organelles are highly susceptible to localized oxidative stress and serve as irreversible intracellular damage checkpoints.[86]
To investigate the intracellular trafficking and to determine the subcellular localization of Gal-SiX, U87MG cells were stained with organelle-specific trackers (Figure S9). Confocal images demonstrated that Gal-SiX predominantly accumulates in mitochondria and lysosomes, as evidenced by substantial overlap with mitotracker (PCC = 0.79 ± 0.02) and lysotracker (PCC = 0.82 ± 0.03) (Figure 3f), while partial distribution was observed in the endoplasmic reticulum (PCC = 0.60 ± 0.04).[87] We believe that the accumulation of Gal-SiX in mitochondria and lysosomes plays a key role in mediating its phototoxic effects in glioblastoma cells.
3.3.4. Mechanistic Insights into Type I/II Mediated Phototoxicity
To verify whether the observed cytotoxicity resulted from Gal-SiX induced oxidative stress, the intracellular ROS generation capability was examined in U87MG cells. Robust green fluorescence was observed with DCF staining (general ROS; Figure 4c) and DHR123 (mitochondrial ROS; Figure 4d) after LED irradiation, highlighting extensive intracellular oxidative stress. Notably, strong red fluorescence from DHE (superoxide anion; Figure 4e) and partial green fluorescence from APF (hydroxyl radical and peroxynitrite; Figure 4f) were evident under light-activated conditions, supporting predominant Type I ROS production. In contrast, minimal fluorescence emission was observed in dark or control groups, confirming the strict light-dependent activation of Gal-SiX.
Moreover, based on the data obtained from cell viability assays in the presence of scavengers (Figure 4b), co-treatment with tiron (superoxide anion scavenger), mannitol (hydroxyl radical scavenger), trolox (peroxyl radical scavenger), and N-acetylcysteine (broad-spectrum thiol-based ROS scavenger) significantly rescued cell viability following photodynamic application, indicating a substantial contribution of Type I ROS pathways. A substantial but relatively lower protective effect was also observed with sodium azide and, to a lesser extent, with histidine, which primarily quench singlet oxygen (1O2), suggesting that Type II mechanisms also play an essential role in Gal-SiX-mediated phototoxicity, but to a lesser extent than Type I. Type I-biased systems show predominant inhibition of phototoxicity upon radical scavenging, consistent with your interpretation that superoxide, hydroxyl and peroxyl radicals substantially contribute to cell death.[88,89] Additionally, the effects of ROS scavengers on intracellular DCF fluorescence were further visualized using confocal microscopy and are presented in Supplementary Figure S10, which supports the viability assay results.
Collectively, the obtained results demonstrate that Gal-SiX acts as a dual Type I/II agent, generating superoxide anion, hydroxyl radicals, peroxynitrite as well as singlet oxygen in tumor cells, which leads to effective and selective phototoxicity. This dual effect of Gal-SiX makes it advantageous for overcoming hypoxic tumor microenvironments where pure Type II agents are often less efficient. These findings align well with the broader literature showing that dual Type I/II photosensitizers can generate both radical species (superoxide, hydroxyl radical, peroxynitrite-derived chemistry) and singlet oxygen in tumor cells, resulting in potent and often hypoxia-tolerant phototoxicity.[90] Reviews and mechanistic studies further emphasize that such dual-mode PSs are particularly advantageous in hypoxic tumor microenvironments, where purely Type II (singlet oxygen-only) agents lose efficiency, whereas Type I radical pathways remain effective and can even be enhanced, leading to more reliable and selective PDT outcomes in solid tumors.[91,92]
3.3.5. Dual Type I/II Based Oxidative Mechanisms Underlying Cell Death
Further analyses were conducted to elucidate the mechanistic insights that contribute to the cell death mechanisms induced by Gal-SiX. Lipid peroxidation, a key endpoint of ROS-mediated membrane damage, was evaluated.[93] The results demonstrated that Gal-SiX induced a significant increase in lipid peroxidation immediately after light exposure (133.70±2.27 %) (**p<0.01 vs. control), which further enhanced during the post-irradiation period and reached to drastic levels at 24 h (304.67±2.92 %) (#p<0.001 vs. control) in U87MG cells (Figure S11a). This was further supported by the sulfo-phospho-vanillin assay,[94] which confirmed a pronounced elevation of lipid products in Gal-SiX–treated cells. A marked accumulation of free lipid content was detected at 24 h under LED irradiation (206.19±14.43 %) (#p<0.001 vs. control), consistent with the release of unsaturated lipid products upon disruption of membranous structures such as mitochondria, lysosomes, and the plasma membrane (Figure S11b). Concomitantly, a notable depletion of intracellular thiol levels was observed after LED exposure at 24 h (80.8±2.6 %) (*p<0.05 vs. control), indicating a depletion of glutathione-dependent antioxidant defenses and reinforcing the role of oxidative stress in the phototoxic response (Figure S11c).[95] Taken together, the increased lipid peroxidation, elevated lipid accumulation, and thiol depletion indicate that Gal-SiX–induced phototoxicity brings forth the possibility of ferroptosis-like oxidative cell death mechanisms, in addition to direct ROS-mediated membrane injury [96].
4. Conclusions
In summary, the first β-Galactosidase-activatable Si-Xanthene-based PS, Gal-SiX, was developed via an improved 10-step synthetic process. This PS demonstrated a remarkable turn-on response in both absorbance and fluorescence signals, with a significant capacity for ROS generation in an aqueous medium upon activation by the β-Galactosidase enzyme. The potential of Gal-SiXas an activatable PS was then tested in vitro on glioblastoma (U87MG) and healthy (L929) cell lines, where it showed no dark toxicity toward either cell type. Upon irradiation, the presence of the β-Gal moiety enabled selective treatment of glioblastoma cells (IC50 values of 3.30 μM vs 7.19 μM). Detailed MTT and confocal microscopy analysis revealed that Gal-SiXexhibits a dual Type I/II mechanism of action in PDT, which is beneficial for the treatment of hypoxic tumors. Further biochemical assays confirmed this dual mechanism, asGal-SiX induced a drastic increase in lipid peroxidation, accompanied by significant accumulation of lipid degradation products and depletion of intracellular thiols. These hallmarks of oxidative membrane damage indicate exhaustion of glutathione-dependent defenses, engagement of ferroptosis-like death pathways, and ROS-induced disruption of membranous structures. Our work on the development of Type I mechanism-biased PDT agents with NIR absorption and on their evaluation in hypoxic tumors with different non-apoptotic cell death mechanisms is ongoing.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
O.K. and Z.E. Conceptualization, methodology, validation, data curation O.K., D.K., M.F., and Z.E; formal analysis, G.G.; investigation, resources, visualization, supervision, project administration, funding acquisition O.K., Z.E., and G.G.; writing—original draft preparation, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
The research leading to these results has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. [852614]).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Acknowledgments
We thank Cevahir Ceren Akgul for the help in photophysical measurements. We also thank Safacan Kolemen for valuable discussions.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Debela, D.T.; Muzazu, S.G.Y.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New Approaches and Procedures for Cancer Treatment: Current Perspectives. SAGE Open Med 2021, 9, 20503121211034370. [CrossRef]
- Ghufran, S.; Priyanka, S.; Govinda, R.D. The Global Concern for Cancer Emergence and Its Prevention: A Systematic Unveiling of the Present Scenario. In Bioprospecting of Tropical Medicinal Plants; Springer Nature : Switzerland, 2023; pp. 1429–1455.
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to Curing Primary Brain Tumours. Nat Rev Clin Oncol 2019, 16, 509–520. [CrossRef]
- Khalighi, S.; Reddy, K.; Midya, A.; Pandav, K.B.; Madabhushi, A.; Abedalthagafi, M. Artificial Intelligence in Neuro-Oncology: Advances and Challenges in Brain Tumor Diagnosis, Prognosis, and Precision Treatment. NPJ Precis Oncol 2024, 8, 80. [CrossRef]
- Koo, Y.E.L.; Reddy, G.R.; Bhojani, M.; Schneider, R.; Philbert, M.A.; Rehemtulla, A.; Ross, B.D.; Kopelman, R. Brain Cancer Diagnosis and Therapy with Nanoplatforms. Adv Drug Deliv Rev 2006, 58, 1556–1577. [CrossRef]
- Brinkman, T.M.; Krasin, M.J.; Liu, W.; Armstrong, G.T.; Ojha, R.P.; Sadighi, Z.S.; Gupta, P.; Kimberg, C.; Srivastava, D.; Merchant, T.E.; et al. Long-Term Neurocognitive Functioning and Social Attainment in Adult Survivors of Pediatric CNS Tumors: Results from the St Jude Lifetime Cohort Study. Journal of Clinical Oncology 2016, 34, 1358–1367. [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2010-2014. Neuro Oncol 2017, 19, v1–v88. [CrossRef]
- Verdugo, E.; Puerto, I.; Medina, M.Á. An Update on the Molecular Biology of Glioblastoma, with Clinical Implications and Progress in Its Treatment. Cancer Commun 2022, 42, 1083–1111. [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. New England Journal of Medicine 2014, 370, 699–708. [CrossRef]
- Dhillon, S.K.; Porter, S.L.; Rizk, N.; Sheng, Y.; McKaig, T.; Burnett, K.; White, B.; Nesbitt, H.; Matin, R.N.; McHale, A.P.; et al. Rose Bengal-Amphiphilic Peptide Conjugate for Enhanced Photodynamic Therapy of Malignant Melanoma. J Med Chem 2020, 63, 1328–1336. [CrossRef]
- Xiao, Q.; Lin, H.; Wu, J.; Pang, X.; Zhou, Q.; Jiang, Y.; Wang, P.; Leung, W.; Lee, H.; Jiang, S.; et al. Pyridine-Embedded Phenothiazinium Dyes as Lysosome-Targeted Photosensitizers for Highly Efficient Photodynamic Antitumor Therapy. J Med Chem 2020, 63, 4896–4907. [CrossRef]
- Dolmans, D.E.J.G.J.; Fukurmura, D.; Jain, R.K. Photodynamic Therapy for Cancer. Nat Rev Cancer 2003, 3, 380–387. [CrossRef]
- Wan, Y.; Fu, L.H.; Li, C.; Lin, J.; Huang, P. Conquering the Hypoxia Limitation for Photodynamic Therapy. Advanced Materials 2021, 33.
- Wang, Y.; Luo, S.; Wu, Y.; Tang, P.; Liu, J.; Liu, Z.; Shen, S.; Ren, H.; Wu, D. Highly Penetrable and On-Demand Oxygen Release with Tumor Activity Composite Nanosystem for Photothermal/Photodynamic Synergetic Therapy. ACS Nano 2020, 14, 17046–17062. [CrossRef]
- Zhang, Y.; Bo, S.; Feng, T.; Qin, X.; Wan, Y.; Jiang, S.; Li, C.; Lin, J.; Wang, T.; Zhou, X.; et al. A Versatile Theranostic Nanoemulsion for Architecture-Dependent Multimodal Imaging and Dually Augmented Photodynamic Therapy. Advanced Materials 2019, 31. [CrossRef]
- Fu, L.H.; Wan, Y.; Li, C.; Qi, C.; He, T.; Yang, C.; Zhang, Y.; Lin, J.; Huang, P. Biodegradable Calcium Phosphate Nanotheranostics with Tumor-Specific Activatable Cascade Catalytic Reactions-Augmented Photodynamic Therapy. Adv Funct Mater 2021, 31. [CrossRef]
- Huo, M.; Wang, L.; Zhang, L.; Wei, C.; Chen, Y.; Shi, J. Photosynthetic Tumor Oxygenation by Photosensitizer-Containing Cyanobacteria for Enhanced Photodynamic Therapy. Angewandte Chemie 2020, 132, 1922–1929. [CrossRef]
- Yu, W.; Liu, T.; Zhang, M.; Wang, Z.; Ye, J.; Li, C.X.; Liu, W.; Li, R.; Feng, J.; Zhang, X.Z. O2 Economizer for Inhibiting Cell Respiration to Combat the Hypoxia Obstacle in Tumor Treatments. ACS Nano 2019, 13, 1784–1794. [CrossRef]
- Fan, Y.T.; Zhou, T.J.; Cui, P.F.; He, Y.J.; Chang, X.; Xing, L.; Jiang, H.L. Modulation of Intracellular Oxygen Pressure by Dual-Drug Nanoparticles to Enhance Photodynamic Therapy. Adv Funct Mater 2019, 29. [CrossRef]
- Gong, H.; Chao, Y.; Xiang, J.; Han, X.; Song, G.; Feng, L.; Liu, J.; Yang, G.; Chen, Q.; Liu, Z. Hyaluronidase To Enhance Nanoparticle-Based Photodynamic Tumor Therapy. Nano Lett 2016, 16, 2512–2521. [CrossRef]
- Zhu, D.; Duo, Y.; Suo, M.; Zhao, Y.; Xia, L.; Zheng, Z.; Li, Y.; Tang, B.Z. Tumor-Exocytosed Exosome/Aggregation-Induced Emission Luminogen Hybrid Nanovesicles Facilitate Efficient Tumor Penetration and Photodynamic Therapy. Angewandte Chemie - International Edition 2020, 59, 13836–13843. [CrossRef]
- Deng, Y.; Jia, F.; Chen, S.; Shen, Z.; Jin, Q.; Fu, G.; Ji, J. Nitric Oxide as an All-Rounder for Enhanced Photodynamic Therapy: Hypoxia Relief, Glutathione Depletion and Reactive Nitrogen Species Generation. Biomaterials 2018, 187, 55–65. [CrossRef]
- Krzykawska-Serda, M.; Dąbrowski, J.M.; Arnaut, L.G.; Szczygieł, M.; Urbańska, K.; Stochel, G.; Elas, M. The Role of Strong Hypoxia in Tumors after Treatment in the Outcome of Bacteriochlorin-Based Photodynamic Therapy. Free Radic Biol Med 2014, 73, 239–251. [CrossRef]
- Luo, T.; Ni, K.; Culbert, A.; Lan, G.; Li, Z.; Jiang, X.; Kaufmann, M.; Lin, W. Nanoscale Metal-Organic Frameworks Stabilize Bacteriochlorins for Type i and Type II Photodynamic Therapy. J Am Chem Soc 2020, 142, 7334–7339. [CrossRef]
- Li, M.; Xiong, T.; Du, J.; Tian, R.; Xiao, M.; Guo, L.; Long, S.; Fan, J.; Sun, W.; Shao, K.; et al. Superoxide Radical Photogenerator with Amplification Effect: Surmounting the Achilles’ Heels of Photodynamic Oncotherapy. J Am Chem Soc 2019, 141, 2695–2702. [CrossRef]
- Zhang, K.; Yu, Z.; Meng, X.; Zhao, W.; Shi, Z.; Yang, Z.; Dong, H.; Zhang, X. A Bacteriochlorin-Based Metal–Organic Framework Nanosheet Superoxide Radical Generator for Photoacoustic Imaging-Guided Highly Efficient Photodynamic Therapy. Advanced Science 2019, 6. [CrossRef]
- Li, M.; Xia, J.; Tian, R.; Wang, J.; Fan, J.; Du, J.; Long, S.; Song, X.; Foley, J.W.; Peng, X. Near-Infrared Light-Initiated Molecular Superoxide Radical Generator: Rejuvenating Photodynamic Therapy against Hypoxic Tumors. J Am Chem Soc 2018, 140, 14851–14859. [CrossRef]
- Nguyen, V.N.; Qi, S.; Kim, S.; Kwon, N.; Kim, G.; Yim, Y.; Park, S.; Yoon, J. An Emerging Molecular Design Approach to Heavy-Atom-Free Photosensitizers for Enhanced Photodynamic Therapy under Hypoxia. J Am Chem Soc 2019, 141, 16243–16248. [CrossRef]
- Lv, Z.; Wei, H.; Li, Q.; Su, X.; Liu, S.; Zhang, K.Y.; Lv, W.; Zhao, Q.; Li, X.; Huang, W. Achieving Efficient Photodynamic Therapy under Both Normoxia and Hypoxia Using Cyclometalated Ru(II) Photosensitizer through Type i Photochemical Process. Chem Sci 2018, 9, 502–512. [CrossRef]
- Ou, M.; Pan, C.; Yu, Y.; Wang, X.; Zhou, Y.; Zhang, H.; Cheng, Q.; Wu, M.; Ji, X.; Mei, L. Two-Dimensional Highly Oxidized Ilmenite Nanosheets Equipped with Z-Scheme Heterojunction for Regulating Tumor Microenvironment and Enhancing Reactive Oxygen Species Generation. Chemical Engineering Journal 2020, 390. [CrossRef]
- Pan, C.; Ou, M.; Cheng, Q.; Zhou, Y.; Yu, Y.; Li, Z.; Zhang, F.; Xia, D.; Mei, L.; Ji, X. Z-Scheme Heterojunction Functionalized Pyrite Nanosheets for Modulating Tumor Microenvironment and Strengthening Photo/Chemodynamic Therapeutic Effects. Adv Funct Mater 2020, 30. [CrossRef]
- Chen, D.; Wang, Z.; Dai, H.; Lv, X.; Ma, Q.; Yang, D.P.; Shao, J.; Xu, Z.; Dong, X. Boosting O2•− Photogeneration via Promoting Intersystem-Crossing and Electron-Donating Efficiency of Aza-BODIPY-Based Nanoplatforms for Hypoxic-Tumor Photodynamic Therapy. Small Methods 2020, 4. [CrossRef]
- Cheng, Y.; Kong, X.; Chang, Y.; Feng, Y.; Zheng, R.; Wu, X.; Xu, K.; Gao, X.; Zhang, H. Spatiotemporally Synchronous Oxygen Self-Supply and Reactive Oxygen Species Production on Z-Scheme Heterostructures for Hypoxic Tumor Therapy. Advanced Materials 2020, 32. [CrossRef]
- Cui, X.; Zhang, J.; Wan, Y.; Fang, F.; Chen, R.; Shen, D.; Huang, Z.; Tian, S.; Xiao, Y.; Li, X.; et al. Dual Fenton Catalytic Nanoreactor for Integrative Type-I and Type-II Photodynamic Therapy against Hypoxic Cancer Cells. ACS Appl Bio Mater 2019, 2, 3854–3860. [CrossRef]
- Pan, C.; Ou, M.; Cheng, Q.; Zhou, Y.; Yu, Y.; Li, Z.; Zhang, F.; Xia, D.; Mei, L.; Ji, X. Z-Scheme Heterojunction Functionalized Pyrite Nanosheets for Modulating Tumor Microenvironment and Strengthening Photo/Chemodynamic Therapeutic Effects. Adv Funct Mater 2020, 30. [CrossRef]
- Hou, Z.; Zhang, Y.; Deng, K.; Chen, Y.; Li, X.; Deng, X.; Cheng, Z.; Lian, H.; Li, C.; Lin, J. UV-Emitting Upconversion-Based TiO2 Photosensitizing Nanoplatform: Near-Infrared Light Mediated in Vivo Photodynamic Therapy via Mitochondria-Involved Apoptosis Pathway. ACS Nano 2015, 9, 2584–2599. [CrossRef]
- Zhang, C.; Zhao, K.; Bu, W.; Ni, D.; Liu, Y.; Feng, J.; Shi, J. Marriage of Scintillator and Semiconductor for Synchronous Radiotherapy and Deep Photodynamic Therapy with Diminished Oxygen Dependence. Angewandte Chemie - International Edition 2015, 54, 1770–1774. [CrossRef]
- Zhang, Y.; Zhao, M.; Cheng, D.; Zhu, J.; Cheng, B.; Miao, M.; Li, Q.; Miao, Q. Self-Assembled Type I Nanophotosensitizer with NIR-II Fluorescence Emission for Imaging-Guided Targeted Phototherapy of Glioblastoma. ACS Appl Nano Mater 2024, 7, 22117–22129. [CrossRef]
- Lismont, M.; Dreesen, L.; Wuttke, S. Metal-Organic Framework Nanoparticles in Photodynamic Therapy: Current Status and Perspectives. Adv Funct Mater 2017, 27, 1606314. [CrossRef]
- Chilakamarthi, U.; Giribabu, L. Photodynamic Therapy: Past, Presentand Future. The Chemical Record 2017, 17, 775–802.
- Yin, H.; Stephenson, M.; Gibson, J.; Sampson, E.; Shi, G.; Sainuddin, T.; Monro, S.; McFarland, S.A. In Vitro Multiwavelength PDT with 3IL States: Teaching Old Molecules New Tricks. Inorg Chem 2014, 53, 4548–4559. [CrossRef]
- Yoon, I.; Li, J.Z.; Shim, Y.K. Advance in Photosensitizers and Light Delivery for Photodynamic Therapy. Clin Endosc 2013, 46, 7–23. [CrossRef]
- Karaman, O.; Alkan, G.A.; Kizilenis, C.; Akgul, C.C.; Gunbas, G. Xanthene Dyes for Cancer Imaging and Treatment: A Material Odyssey. Coord Chem Rev 2023, 475, 214841. [CrossRef]
- Zhao, X.; Liu, J.; Fan, J.; Chao, H.; Peng, X. Recent Progress in Photosensitizers for Overcoming the Challenges of Photodynamic Therapy: From Molecular Design to Application. Chem Soc Rev 2021, 50, 4185–4219.
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic Therapy of Cancer: An Update. CA Cancer J Clin 2011, 61, 250–281.
- Chen, X.; Pradhan, T.; Wang, F.; Kim, J.S.; Yoon, J. Fluorescent Chemosensors Based on Spiroring-Opening of Xanthenes and Related Derivatives. Chem Rev 2012, 112, 1910–1956. [CrossRef]
- Samanta, S.; Lai, K.; Wu, F.; Liu, Y.; Cai, S.; Yang, X.; Qu, J.; Yang, Z. Xanthene, Cyanine, Oxazine and BODIPY: The Four Pillars of the Fluorophore Empire for Super-Resolution Bioimaging. Chem Soc Rev 2023, 52, 7197–7261.
- Bergmann, E. V.; Cavalaro, A.P.B.; Kimura, N.M.; Zanuto, V.S.; Astrath, N.G.C.; Herculano, L.S.; Malacarne, L.C. Photophysical Characterization of Xanthene Dyes. Spectrochim Acta A Mol Biomol Spectrosc 2025, 327, 125345. [CrossRef]
- Kamino, S.; Uchiyama, M. Xanthene-Based Functional Dyes: Towards New Molecules Operating in the near-Infrared Region. Org Biomol Chem 2023, 21, 2458–2471. [CrossRef]
- Zhu, T.; Ding, J.; Zheng, F.; Fang, Y.; Huang, W.; Yin, Y.; Zeng, W. Synergistic Cancer Therapy: An NIR-Activated Methylene Blue-Nitrogen Mustard Prodrug for Combined Chemotherapy and Photodynamic Therapy. J Med Chem 2025, 68, 7630–7641. [CrossRef]
- Yao, S.; Xu, F.; Wang, Y.; Shang, J.; Li, S.; Xu, X.; Liu, Z.; He, W.; Guo, Z.; Chen, Y. Photoinduced Synergism of Ferroptosis/Pyroptosis/Oncosis by an O2-Independent Photocatalyst for Enhanced Tumor Immunotherapy. J Am Chem Soc 2025, 147, 11132–11144. [CrossRef]
- Chen, W.; Mao, X.Q.; Wang, X.Z.; Liao, Y.C.; Yin, X.Y.; Wu, H.L.; Chen, T.Y.; Liu, M.Q.; Wang, T.; Yu, R.Q. Data-Driven Discovery of near-Infrared Type I Photosensitizers for RNA-Targeted Tumor Photodynamic Therapy. Chem Sci 2025, 16, 14455–14467. [CrossRef]
- Cheng, Z.; Benson, S.; Mendive-Tapia, L.; Nestoros, E.; Lochenie, C.; Seah, D.; Chang, K.Y.; Feng, Y.; Vendrell, M. Enzyme-Activatable Near-Infrared Hemicyanines as Modular Scaffolds for in Vivo Photodynamic Therapy. Angewandte Chemie - International Edition 2024, 63. [CrossRef]
- Egawa, T.; Koide, Y.; Hanaoka, K.; Komatsu, T.; CooTeraiper, T.; Nagano, T. Development of a Fluorescein Analogue, TokyoMagenta, as a Novel Scaffold for Fluorescence Probes in Red Region. Chemical Communications 2011, 47, 4162–4164. [CrossRef]
- Fukazawa, A.; Suda, S.; Taki, M.; Yamaguchi, E.; Grzybowski, M.; Sato, Y.; Higashiyama, T.; Yamaguchi, S. Phospha-Fluorescein: A Red-Emissive Fluorescein Analogue with High Photobleaching Resistance. Chemical Communications 2016, 52, 1120–1123. [CrossRef]
- Mo, F.; Pellerino, A.; Soffietti, R.; Rudà, R. Blood-Brain Barrier in Brain Tumors: Biology and Clinical Relevance. Int J Mol Sci 2021, 22, 12654. [CrossRef]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The Blood–Brain Barrier and Blood–Tumour Barrier in Brain Tumours and Metastases. Nat Rev Cancer 2020, 20, 26–41. [CrossRef]
- Karaman, O.; Yesilcimen, E.; Forough, M.; Elmazoglu, Z.; Gunbas, G. Drastic Impact of Donor Substituents on Xanthenes in the PDT of Glioblastoma. JACS Au 2025, 5, 5346–5358. [CrossRef]
- Zhou, X.; Miao, L.; Zhou, W.; Chen, Y.; Ruan, Y.; Wang, X.; Wang, G.; Bao, P.; Qiao, Q.; Xu, Z. Enhancing the Photostability of Red Fluorescent Proteins through FRET with Si-Rhodamine for Dynamic Super-Resolution Fluorescence Imaging. Chem Sci 2025, 16, 10476–10486. [CrossRef]
- Wang, K.; Zhou, J.; Geng, Y.; Zhang, G.; Zhang, Z.; Jin, Y.; Liu, X.; Wang, Z. Lysosome-Targeted Si-Rhodamine Derivative for NO Imaging in Mice Brain with Neurological Diseases. Anal Chem 2025, 97, 12728–12735. [CrossRef]
- Xu, Q.; Jiao, C.; Liu, X.; Yang, Y.; Ren, J.; Wang, P.; Pan, X.; Wu, Y.; Yan, Z.; Zeng, J. A Highly Sensitive and Fast-Response Fluorescence Nanoprobe for in Vivo Imaging of Hypochlorous Acid. J Hazard Mater 2025, 487, 137282. [CrossRef]
- Wang, N.; Li, H.; Kong, D.; Feng, X.; Li, C.; Cui, X.; Wang, T. Dibutylated Si-Fluorescein: Enhanced Hydrophobicity for Fluorogenic Labeling in Vivo. Dyes and Pigments 2025, 240, 112841. [CrossRef]
- Cetin, S.; Elmazoglu, Z.; Karaman, O.; Gunduz, H.; Gunbas, G.; Kolemen, S. Balanced Intersystem Crossing in Iodinated Silicon-Fluoresceins Allows New Class of Red Shifted Theranostic Agents. ACS Med Chem Lett 2021, 12, 752–757. [CrossRef]
- Jiang, G.; Zeng, G.; Zhu, W.; Li, Y.; Dong, X.; Zhang, G.; Fan, X.; Wang, J.; Wu, Y.; Tang, B.Z. A Selective and Light-up Fluorescent Probe for β-Galactosidase Activity Detection and Imaging in Living Cells Based on an AIE Tetraphenylethylene Derivative. Chemical Communications 2017, 53, 4505–4508. [CrossRef]
- Wu, F.; Liu, J.; Tao, M.; Wang, M.; Ren, X.; Hai, Z. β-Galactosidase-Activatable Fluorescent and Photoacoustic Imaging of Tumor Senescence. Anal Chem 2023, 95, 10481–10485. [CrossRef]
- Chatterjee, S.K.; Bhattacharya, M.; Barlow, J.J. Glycosyltransferase and Glycosidase Activities in Ovarian Cancer Patients. Cancer Res 1979, 39, 1943–1951.
- Valieva, Y.; Ivanova, E.; Fayzullin, A.; Kurkov, A.; Igrunkova, A. Senescence-Associated β-Galactosidase Detection in Pathology. Diagnostics 2022, 12, 2309. [CrossRef]
- Kubo, H.; Murayama, Y.; Ogawa, S.; Matsumoto, T.; Yubakami, M.; Ohashi, T.; Kubota, T.; Okamoto, K.; Kamiya, M.; Urano, Y.; et al. β-Galactosidase Is a Target Enzyme for Detecting Peritoneal Metastasis of Gastric Cancer. Sci Rep 2021, 11, 10664. [CrossRef]
- Chen, J.A.; Pan, H.; Wang, Z.; Gao, J.; Tan, J.; Ouyang, Z.; Guo, W.; Gu, X. Imaging of Ovarian Cancers Using Enzyme Activatable Probes with Second Near-Infrared Window Emission. Chemical Communications 2020, 56, 2731–2734. [CrossRef]
- Wielgat, P.; Walczuk, U.; Szajda, S.; Bień, M.; Zimnoch, L.; Mariak, Z.; Zwierz, K. Activity of Lysosomal Exoglycosidases in Human Gliomas. J Neurooncol 2006, 80, 243–249. [CrossRef]
- Xu, X.; Shen, X.; Feng, W.; Yang, D.; Jin, L.; Wang, J.; Wang, M.; Ting, Z.; Xue, F.; Zhang, J.; et al. D-Galactose Induces Senescence of Glioblastoma Cells through YAP-CDK6 Pathway. Aging (Albany NY) 2020, 12, 18501.
- Chen, J.A.; Guo, W.; Wang, Z.; Sun, N.; Pan, H.; Tan, J.; Ouyang, Z.; Fu, W.; Wang, Y.; Hu, W.; et al. In Vivo Imaging of Senescent Vascular Cells in Atherosclerotic Mice Using a β-Galactosidase-Activatable Nanoprobe. Anal Chem 2020, 92, 12613–12621. [CrossRef]
- Gu, K.; Qiu, W.; Guo, Z.; Yan, C.; Zhu, S.; Yao, D.; Shi, P.; Tian, H.; Zhu, W.H. An Enzyme-Activatable Probe Liberating AIEgens: On-Site Sensing and Long-Term Tracking of β-Galactosidase in Ovarian Cancer Cells. Chem Sci 2019, 10, 398–405. [CrossRef]
- Almammadov, T.; Elmazoglu, Z.; Atakan, G.; Kepil, D.; Aykent, G.; Kolemen, S.; Gunbas, G. Locked and Loaded: SS-Galactosidase Activated Photodynamic Therapy Agent Enables Selective Imaging and Targeted Treatment of Glioblastoma Multiforme Cancer Cells. ACS Appl Bio Mater 2022, 5, 4284–4293. [CrossRef]
- International Organization for Standardization Biological Evaluation of Medical Devices—Part 5: Tests for in Vitro Cytotoxicity; ISO 10993-5:2009; Geneva, 2009;
- Onaral, F.G.; Silindir-Gunay, M.; Uluturk, S.; Ozturk, S.C.; Cakir-Aktas, C.; Esendagli, G. Development and In Vitro and In Vivo Efficacy Investigation of Multifunctional, Targeted, Theranostic Liposomes for Imaging and Photodynamic Therapy of Glioblastoma. J Drug Deliv Sci Technol 2024, 101. [CrossRef]
- Chiba, M.; Kamiya, M.; Tsuda-Sakurai, K.; Fujisawa, Y.; Kosakamoto, H.; Kojima, R.; Miura, M.; Urano, Y. Activatable Photosensitizer for Targeted Ablation of LacZ-Positive Cells with Single-Cell Resolution. ACS Cent Sci 2019, 5, 1676–1681. [CrossRef]
- Zhang, R.; Wong, K. High Performance Enzyme Kinetics of Turnover, Activation and Inhibition for Translational Drug Discovery. Expert Opin Drug Discov 2017, 12, 17–37.
- Markovic, M.; Ben-Shabat, S.; Dahan, A. Computational Simulations to Guide Enzyme-Mediated Prodrug Activation. Int J Mol Sci 2020, 21.
- Valieva, Y.; Ivanova, E.; Fayzullin, A.; Kurkov, A.; Igrunkova, A. Senescence-Associated β-Galactosidase Detection in Pathology. Diagnostics 2022, 12.
- Bahramikia, S.; Shirzadi, N.; Akbari, V. Protective Effects of Pyrogallol and Caffeic Acid against Fe2+ -Ascorbate-Induced Oxidative Stress in the Wistar Rats Liver: An in Vitro Study. Heliyon 2025, 11. [CrossRef]
- Bodnár, B.R.; Ghosal, S.; Kestecher, B.M.; Királyhidi, P.; Försönits, A.; Fekete, N.; Bugyik, E.; Komlósi, Z.I.; Pállinger, É.; Nagy, G.; et al. Isolation and Characterization of Tissue-Derived Extracellular Vesicles from Mouse Lymph Nodes. Int J Mol Sci 2025, 26, 6092. [CrossRef]
- Ginet, S.R.; Gonzalez, F.; Marano, M.L.; Salecha, M.D.; Reiner, J.E.; Caputo, G.A. Evaluation of the Ellman’s Reagent Protocol for Free Sulfhydryls Under Protein Denaturing Conditions. Analytica 2025, 6, 18. [CrossRef]
- Smaga, L.P.; Pino, N.W.; Ibarra, G.E.; Krishnamurthy, V.; Chan, J. A Photoactivatable Formaldehyde Donor with Fluorescence Monitoring Reveals Threshold to Arrest Cell Migration. J Am Chem Soc 2020, 142, 680–684. [CrossRef]
- Hirabayashi, K.; Hanaoka, K.; Takayanagi, T.; Toki, Y.; Egawa, T.; Kamiya, M.; Komatsu, T.; Ueno, T.; Terai, T.; Yoshida, K.; et al. Analysis of Chemical Equilibrium of Silicon-Substituted Fluorescein and Its Application to Develop a Scaffold for Red Fluorescent Probes. Anal Chem 2015, 87, 9061–9069. [CrossRef]
- Crovetto, L.; Orte, A.; Paredes, J.M.; Resa, S.; Valverde, J.; Castello, F.; Miguel, D.; Cuerva, J.M.; Talavera, E.M.; Alvarez-Pez, J.M. Photophysics of a Live-Cell-Marker, Red Silicon-Substituted Xanthene Dye. Journal of Physical Chemistry A 2015, 119, 10854–10862. [CrossRef]
- Best, Q.A.; Sattenapally, N.; Dyer, D.J.; Scott, C.N.; McCarroll, M.E. PH-Dependent Si-Fluorescein Hypochlorous Acid Fluorescent Probe: Spirocycle Ring-Opening and Excess Hypochlorous Acid-Induced Chlorination. J Am Chem Soc 2013, 135, 13365–13370. [CrossRef]
- Wilkinson, F.; Helman, W.P.; Ross, A.B. Quantum Yields for the Photosensitized Formation of the Lowest Electronically Excited Singlet State of Molecular Oxygen in Solution. J Phys Chem Ref Data 1993, 22, 113–262. [CrossRef]
- Chen, S.; Wang, L.; Ma, X.; Wu, Y.; Hou, S. Kill Two Birds with One Stone: A near-Infrared Ratiometric Fluorescent Probe for Simultaneous Detection of β-Galactosidase in Senescent and Cancer Cells. Sens Actuators B Chem 2022, 367, 132061–132072. [CrossRef]
- Banti, C.N.; Papatriantafyllopoulou, C.; Papachristodoulou, C.; Hatzidimitriou, A.G.; Hadjikakou, S.K. New Apoptosis Inducers Containing Anti-Inflammatory Drugs and Pnictogen Derivatives: A New Strategy in the Development of Mitochondrial Targeting Chemotherapeutics. J Med Chem 2023, 66, 4131–4149. [CrossRef]
- Kot, Y.; Klochkov, V.; Prokopiuk, V.; Sedyh, O.; Tryfonyuk, L.; Grygorova, G.; Karpenko, N.; Tomchuk, O.; Kot, K.; Onishchenko, A.; et al. GdVO4:Eu3+ and LaVO4:Eu3+ Nanoparticles Exacerbate Oxidative Stress in L929 Cells: Potential Implications for Cancer Therapy. Int J Mol Sci 2024, 25, 11687. [CrossRef]
- Baeza-Morales, A.; Pascual-García, S.; Martínez-Peinado, P.; Navarro-Sempere, A.; Segovia, Y.; Medina-García, M.; Pujalte-Satorre, C.; García, M.M.; Martínez-Espinosa, R.M.; Sempere-Ortells, J.M. Bacterioruberin Extract from Haloferax Mediterranei Induces Apoptosis and Cell Cycle Arrest in Myeloid Leukaemia Cell Lines. Sci Rep 2025, 15, 23485. [CrossRef]
- Cardoso, R.; Pereira, P.; Freitas, C.; De Freitas Silva, A.; Midlej, V.; Conte-Júnior, C.; Paschoalin, V. Nano-Encapsulated Taro Lectin Can Cross an in Vitro Blood-Brain Barrier, Induce Apoptosis and Autophagy and Inhibit the Migration of Human U-87 MG Glioblastoma Cells. Int J Nanomedicine 2025, Volume 20, 5573–5591. [CrossRef]
- Rana, N.; Chaudhary, P.K.; Prasad, R.; Sankar, M. Photodynamic Evaluation of A2BC Aminoporphyrins: Synthesis, Characterization, and Cellular Impact. ACS Appl Bio Mater 2025, 8, 5098–5108. [CrossRef]
- Doan, V.T.H.; Komatsu, Y.; Matsui, H.; Kawazoe, N.; Chen, G.; Yoshitomi, T. Singlet Oxygen-Generating Cell-Adhesive Glass Surfaces for the Fundamental Investigation of Plasma Membrane-Targeted Photodynamic Therapy. Free Radic Biol Med 2023, 207, 239–246. [CrossRef]
- Borghei, Y.S.; Hamidieh, A.A.; Lu, Y.; Hosseinkhani, S. Organic–Inorganic Hybrid Nanoflowers as a New Biomimetic Platform for ROS-Induced Apoptosis by Photodynamic Therapy. European Journal of Pharmaceutical Sciences 2023, 191, 106569. [CrossRef]
- Saczuk, K.; Kassem, A.; Dudek, M.; Sánchez, D.P.; Khrouz, L.; Allain, M.; Welch, G.C.; Sabouri, N.; Monnereau, C.; Josse, P.; et al. Organelle-Specific Thiochromenocarbazole Imide Derivative as a Heavy-Atom-Free Type I Photosensitizer for Biomolecule-Triggered Image-Guided Photodynamic Therapy. J Phys Chem Lett 2025, 16, 2273–2282. [CrossRef]
- Xu, Y.; Xie, Y.; Wan, Q.; Tian, J.; Liang, J.; Zhou, J.; Song, M.; Zhou, X.; Teng, M. Mechanism Research of Type I Reactive Oxygen Species Conversion Based on Molecular and Aggregate Levels for Tumor Photodynamic Therapy. Aggregate 2024, 5. [CrossRef]
- Liu, J.-Q.; Tse, A.K.-W.; Koncošová, M.; Ruml, T.; Tse, Y.-C.; Liu, C.-J.; Zelenka, J.; Kirakci, K.; Lang, K.; Lee, C.-S.; et al. Fluorescein-Functionalized Iridium(III) Complexes as Dual-Mode Type I Photosensitizers for Hypoxia-Tolerant Photodynamic and X-Ray-Induced Therapy. Inorg Chem 2025, 64, 10894–10905. [CrossRef]
- Xiong, T.; Chen, Y.; Li, M.; Chen, X.; Peng, X. Recent Progress of Molecular Design in Organic Type I Photosensitizers. Small 2025, 21. [CrossRef]
- Wang, H.; Qin, T.; Wang, W.; Zhou, X.; Lin, F.; Liang, G.; Yang, Z.; Chi, Z.; Tang, B.Z. Selenium-Containing Type-I Organic Photosensitizers with Dual Reactive Oxygen Species of Superoxide and Hydroxyl Radicals as Switch-Hitter for Photodynamic Therapy. Advanced Science 2023, 10. [CrossRef]
- Zhou, W.; Lim, A.; Elmadbouh, O.H.M.; Edderkaoui, M.; Osipov, A.; Mathison, A.J.; Urrutia, R.; Liu, T.; Wang, Q.; Pandol, S.J. Verteporfin Induces Lipid Peroxidation and Ferroptosis in Pancreatic Cancer Cells. Free Radic Biol Med 2024, 212, 493–504. [CrossRef]
Figure 1.
Structure of Gal-SiXand its conversion to SiX upon treatment with the enzyme.
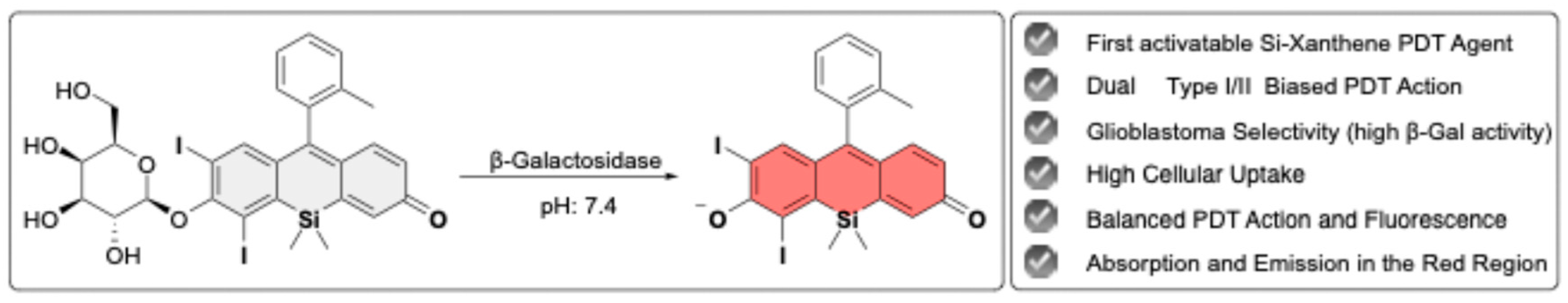
Scheme 1.
Synthetic route for Gal-SiX. a. K2CO3, BnBr, MeCN, 80 °C,16 h 82%. b. Formaldehyde, AcOH, 80 °C,16 h, 82%. c. 1-sec-BuLi, Et2O, -78 °C, 1 h, SiMe2Cl2, -78 °C 2- KMnO4 Acetone 0 °C, 4 h 57%. d. H2, Pd-C, 3.6 bar CHCl3-MeOH (1:4) 3 d, 77%. e. 1- MeOH-6 N H2SO4, NaNO2 in H2O, 0 °C, 1 h, 2-1 N H2SO4, 135 °C, 1 h, 61%. f. imidazole, TBDMSCl, DCM, 1 d, rt, 83%. g. o-bromotoluene, n-BuLi, THF, -78 °C, then 7, 63%. h. acetobromo-α-D-galactose, Cs2CO3, MeCN, 24 h, 56%. i. I2, HIO3, EtOH, 78 °C, 2 h, 85%. j. NaOMe, MeOH, 0 °C, 1 h, 90%. Structure of β-Gal cleaved Gal-SiX, SiX.
Scheme 1.
Synthetic route for Gal-SiX. a. K2CO3, BnBr, MeCN, 80 °C,16 h 82%. b. Formaldehyde, AcOH, 80 °C,16 h, 82%. c. 1-sec-BuLi, Et2O, -78 °C, 1 h, SiMe2Cl2, -78 °C 2- KMnO4 Acetone 0 °C, 4 h 57%. d. H2, Pd-C, 3.6 bar CHCl3-MeOH (1:4) 3 d, 77%. e. 1- MeOH-6 N H2SO4, NaNO2 in H2O, 0 °C, 1 h, 2-1 N H2SO4, 135 °C, 1 h, 61%. f. imidazole, TBDMSCl, DCM, 1 d, rt, 83%. g. o-bromotoluene, n-BuLi, THF, -78 °C, then 7, 63%. h. acetobromo-α-D-galactose, Cs2CO3, MeCN, 24 h, 56%. i. I2, HIO3, EtOH, 78 °C, 2 h, 85%. j. NaOMe, MeOH, 0 °C, 1 h, 90%. Structure of β-Gal cleaved Gal-SiX, SiX.
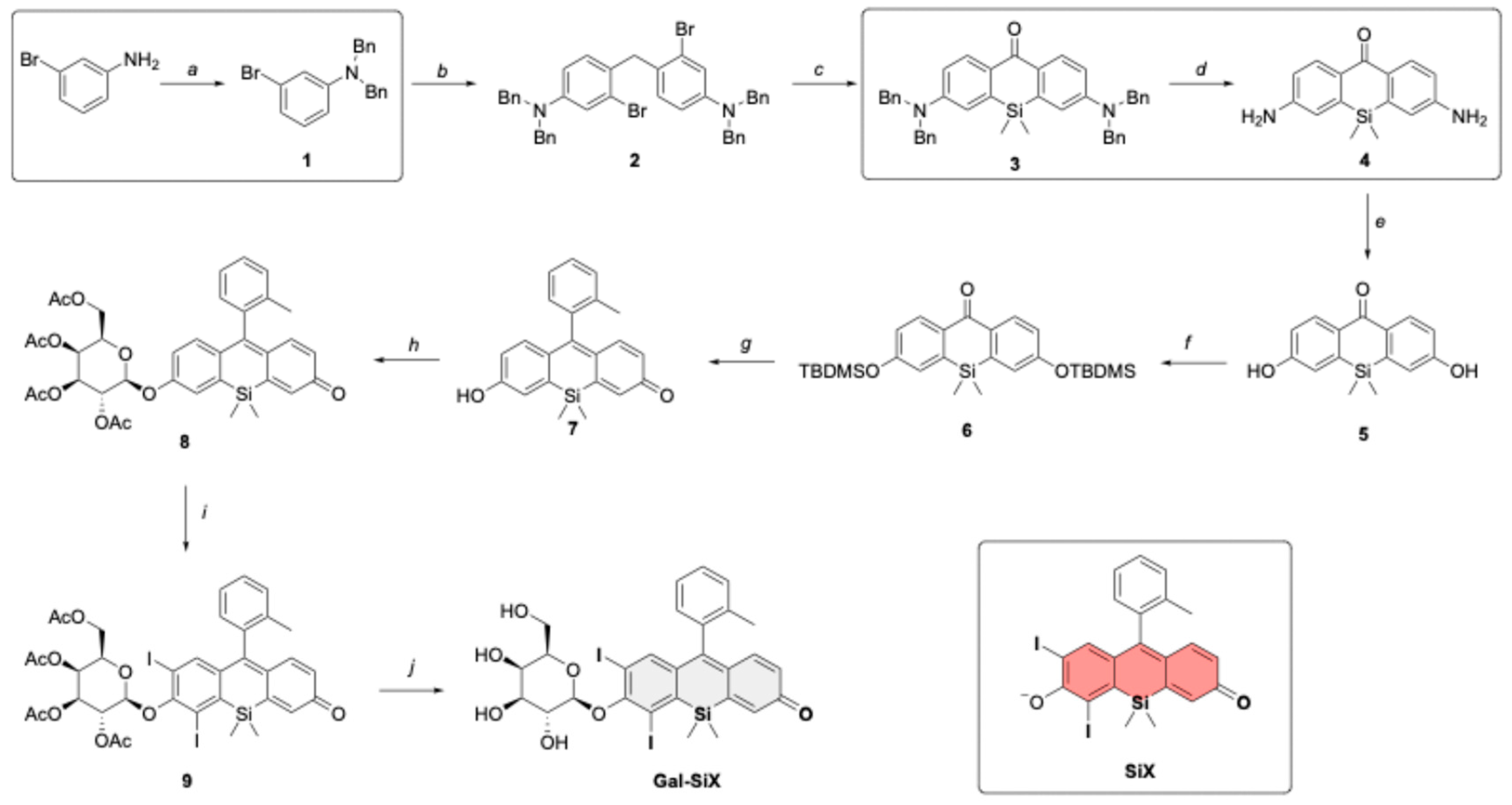
Figure 2.
(a) Absorption spectrum of Gal-SiX (10 μM) in PBS buffer (pH 7.4, 1% DMSO). (b) Absorption spectrum of Gal-SiX (10 μM) in PBS buffer (pH 7.4, 1% DMSO) upon incubation with β-galactosidase (5 U) for 40 min (37 °C). (c) Fluorescence spectrum of Gal-SiX (10 μM) in PBS buffer (pH 7.4, 1% DMSO) upon incubation with β-Galactosidase (5 U) for 30 min (37 °C). (d) Fluorescence spectra of SOSG (5 μM) containing Gal-SiX (10 μM), e. Fluorescence spectra of DHR 123 (5 μM) containing Gal-SiX (10 μM) and f. Fluorescence spectra of APF (5 μM) containing Gal-SiX (10 μM) in PBS buffer (pH: 7.4, 1% DMSO) upon incubation with β-galactosidase (5 U) for 40 min (37 °C) irradiated with LED light (595 nm, 100 s total irradiation time, 3.50 mW/cm2) to detect 1O2 generation efficiency (d), to detect O2• – generation efficiency €, to detect •OH generation efficiency (f).
Figure 2.
(a) Absorption spectrum of Gal-SiX (10 μM) in PBS buffer (pH 7.4, 1% DMSO). (b) Absorption spectrum of Gal-SiX (10 μM) in PBS buffer (pH 7.4, 1% DMSO) upon incubation with β-galactosidase (5 U) for 40 min (37 °C). (c) Fluorescence spectrum of Gal-SiX (10 μM) in PBS buffer (pH 7.4, 1% DMSO) upon incubation with β-Galactosidase (5 U) for 30 min (37 °C). (d) Fluorescence spectra of SOSG (5 μM) containing Gal-SiX (10 μM), e. Fluorescence spectra of DHR 123 (5 μM) containing Gal-SiX (10 μM) and f. Fluorescence spectra of APF (5 μM) containing Gal-SiX (10 μM) in PBS buffer (pH: 7.4, 1% DMSO) upon incubation with β-galactosidase (5 U) for 40 min (37 °C) irradiated with LED light (595 nm, 100 s total irradiation time, 3.50 mW/cm2) to detect 1O2 generation efficiency (d), to detect O2• – generation efficiency €, to detect •OH generation efficiency (f).

Figure 3.
In vitro evaluation of phototoxicity, cellular localization, and cell death mechanisms of Gal-SiX. Cell viabilities of U87MG (a) and L929 (b) cells after 1 h incubation with Gal-SiX (0.1–10 μM), followed by 2-hour LED irradiation (595 nm, 8.12 mW/cm²) and 24 h rest in the dark. (c) Confocal images depicting cellular internalization and activation state of Gal-SiX (1 μM, 1h) in U87MG and L929 cells. (d) Comparative cell viability of U87MG and L929 cells after Gal-SiX (0.1–10 μM, 1h) treatment under 2 h LED irradiation, and 24-hour dark incubation (n=6). (e) Representative confocal images showing AO/EtBr dual staining in U87MG and L929 treated with Gal-SiX (IC50, 1 h), followed by 2 h LED irradiation. Green: Acridine orange (AO), live cells; Red: Ethidium bromide (EtBr), late apoptotic or necrotic cells (n=6, Scale bar: 20 µm). (f) Subcellular localization of Gal-SiX (1 μM, 1h) (Red) with mitochondria (Green: MitoTracker) and lysosomes (Green: LysoTracker) in U87MG cells. Nuclei stained with Hoechst 33342 (blue). (n=3) Scale bar: 5 μm. *p<0.05, **p<0.01, #p<0.001 vs untreated control (C).
Figure 3.
In vitro evaluation of phototoxicity, cellular localization, and cell death mechanisms of Gal-SiX. Cell viabilities of U87MG (a) and L929 (b) cells after 1 h incubation with Gal-SiX (0.1–10 μM), followed by 2-hour LED irradiation (595 nm, 8.12 mW/cm²) and 24 h rest in the dark. (c) Confocal images depicting cellular internalization and activation state of Gal-SiX (1 μM, 1h) in U87MG and L929 cells. (d) Comparative cell viability of U87MG and L929 cells after Gal-SiX (0.1–10 μM, 1h) treatment under 2 h LED irradiation, and 24-hour dark incubation (n=6). (e) Representative confocal images showing AO/EtBr dual staining in U87MG and L929 treated with Gal-SiX (IC50, 1 h), followed by 2 h LED irradiation. Green: Acridine orange (AO), live cells; Red: Ethidium bromide (EtBr), late apoptotic or necrotic cells (n=6, Scale bar: 20 µm). (f) Subcellular localization of Gal-SiX (1 μM, 1h) (Red) with mitochondria (Green: MitoTracker) and lysosomes (Green: LysoTracker) in U87MG cells. Nuclei stained with Hoechst 33342 (blue). (n=3) Scale bar: 5 μm. *p<0.05, **p<0.01, #p<0.001 vs untreated control (C).
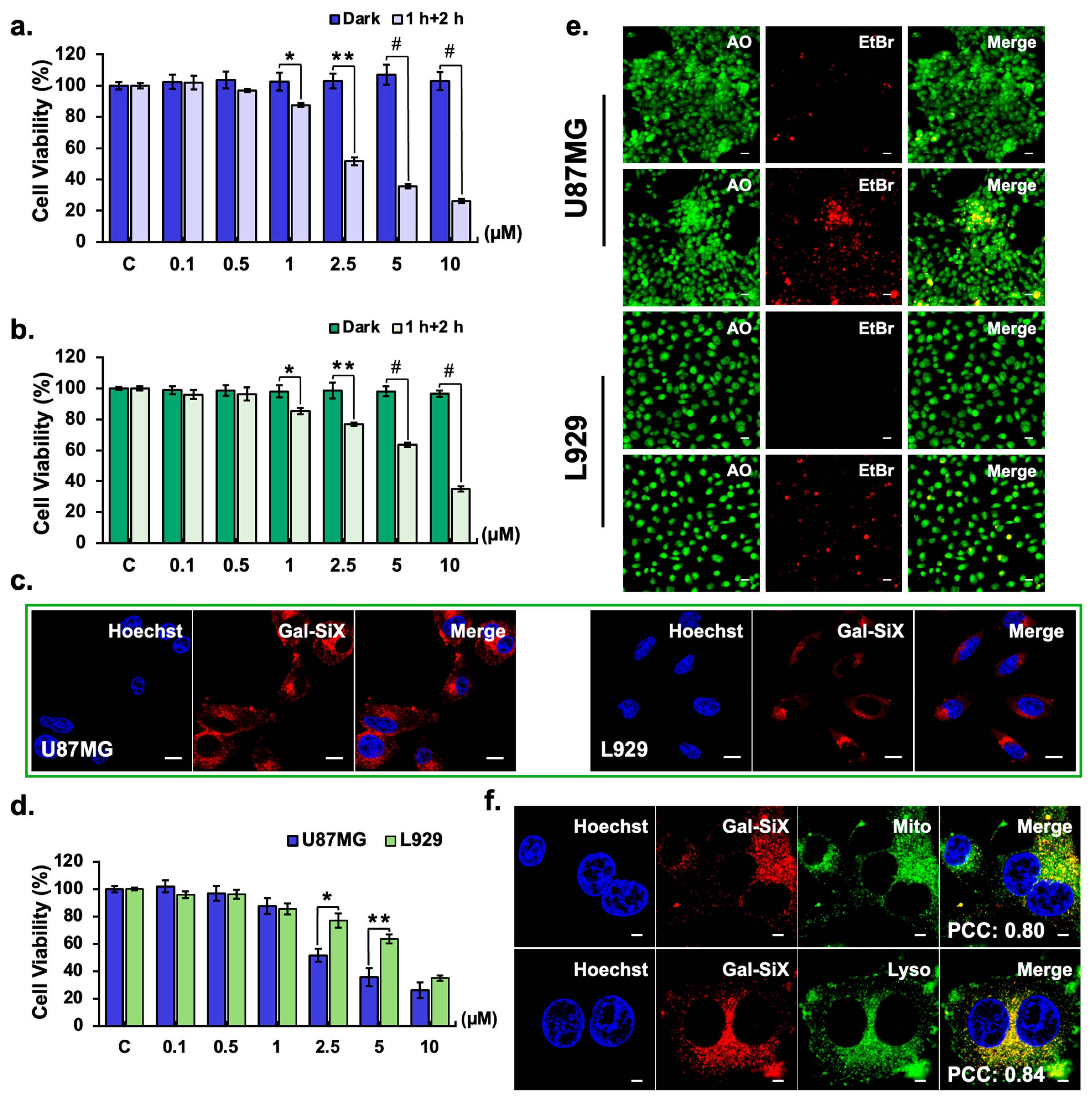
Figure 4.
Mechanistic insight for the photodynamic application of Gal-SiX in U87MG cells. (a) Cell viability analysis after pre-treatment with increasing concentrations of D-galactose (5–50 mM, 4h), then post-treated with Gal-SiX (2.5 μM, 1 h), followed by LED irradiation (2 h) and 24-hour dark incubation (n=6). (b) Cell viability analysis after co-treatment with various ROS scavengers, histidine (HIS, 5 mM), sodium azide (5 mM), Tiron (TIR, 100 µM), mannitol (MAN, 25 mM), Trolox (TRO, 25 µM), and N-acetylcysteine (NAC, 5 mM) prior to Gal-SiX treatment and PDT (n=6). Intracellular ROS detection after PDT application with Gal-SiX using different fluorescent probes: (c) DCF (Green) for general ROS, (d) DHR123 (Green) for hydrogen peroxide and mitochondrial ROS, (e) DHE (Red) for superoxide anion, and (f) APF (Green) for hydroxyl radical and peroxynitrite. Control, dark (without irradiation), and light (irradiated) conditions are presented for each probe. Nuclei stained with Hoechst 33342 (blue). Scale bars: 10 μm. *p < 0.05, **p < 0.01, #p < 0.001 vs untreated cells.
Figure 4.
Mechanistic insight for the photodynamic application of Gal-SiX in U87MG cells. (a) Cell viability analysis after pre-treatment with increasing concentrations of D-galactose (5–50 mM, 4h), then post-treated with Gal-SiX (2.5 μM, 1 h), followed by LED irradiation (2 h) and 24-hour dark incubation (n=6). (b) Cell viability analysis after co-treatment with various ROS scavengers, histidine (HIS, 5 mM), sodium azide (5 mM), Tiron (TIR, 100 µM), mannitol (MAN, 25 mM), Trolox (TRO, 25 µM), and N-acetylcysteine (NAC, 5 mM) prior to Gal-SiX treatment and PDT (n=6). Intracellular ROS detection after PDT application with Gal-SiX using different fluorescent probes: (c) DCF (Green) for general ROS, (d) DHR123 (Green) for hydrogen peroxide and mitochondrial ROS, (e) DHE (Red) for superoxide anion, and (f) APF (Green) for hydroxyl radical and peroxynitrite. Control, dark (without irradiation), and light (irradiated) conditions are presented for each probe. Nuclei stained with Hoechst 33342 (blue). Scale bars: 10 μm. *p < 0.05, **p < 0.01, #p < 0.001 vs untreated cells.
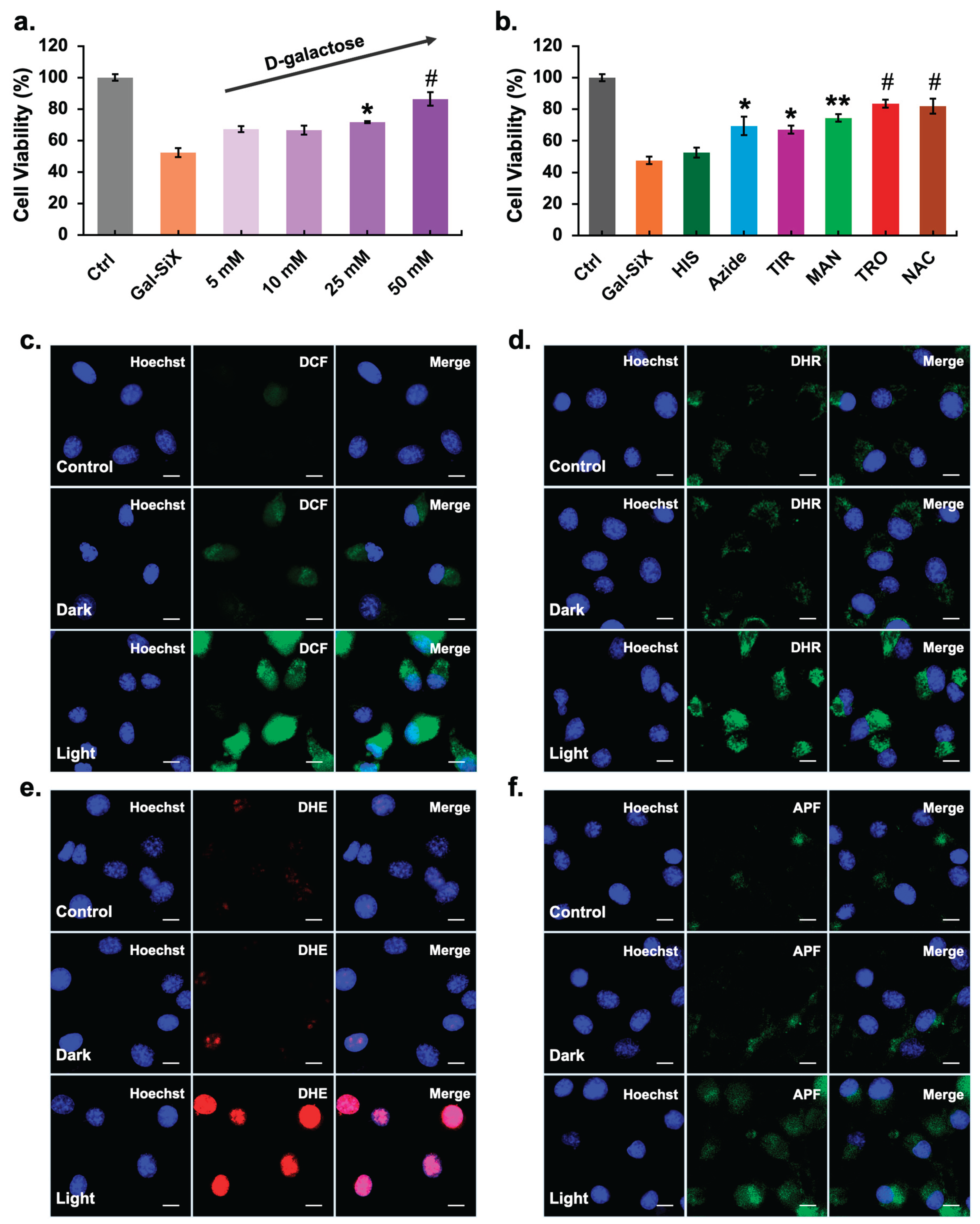

Table 1.
Photophysical properties of Gal-SiX and SiX.
| PS | λabs (nm)1 | λems (nm)1 | φF (%)1,2 | ΦΔ(%)1,3 |
|---|---|---|---|---|
| Gal-SiX | 486 | Not detectable | Not detectable | n.d.d |
| SiX | 598 | 613 | 6.4 | 52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



