
Submitted:
09 January 2026
Posted:
12 January 2026
You are already at the latest version
Abstract
Background: Bipolar disorder (BD) exhibits high heritability and substantial genetic overlap with schizophrenia and major depressive disorder (MDD), yet its core pathophysiological pathways remain debated. While glutamatergic dysregulation and synaptic plasticity have been emphasized, emerging evidence from schizophrenia highlights excessive synaptic pruning. We conducted a multi-method genetic analysis to competitively test pruning versus glutamatergic/plasticity hypotheses in the latest European-ancestry BD GWAS, with parallel replication in MDD for cross-disorder comparison.Methods: Using summary statistics from a large BD GWAS (effective N ≈ 137,097) and a trans-ancestry MDD GWAS (European subsample effective N ≈ 829,250), we applied: (1) MAGMA for gene-based and competitive gene-set testing with custom pruning (shortened/expanded/specific) and glutamatergic sets (original/expanded) plus controls; (2) stratified LD score regression for partitioned heritability enrichment; (3) S-PrediXcan transcriptome-wide association in six brain tissues; and (4) two-sample Mendelian randomization with neuroplasticity proxies (e.g., educational attainment). Overlap removal enabled independence assessment; directional TWAS and MR provided functional/causal insights.Results: In BD, pruning pathways dominated across methods: expanded/specific pruning sets were Bonferroni-significant in MAGMA (p = 1.14 × 10⁻⁴/7.00 × 10⁻⁴) and showed strong LDSC enrichment (p = 4.16 × 10⁻³⁸/3.77 × 10⁻¹²), persisting independently of glutamatergic overlap. TWAS revealed activation-skewed pruning (higher predicted expression in microglial/autophagy activators) with modest glutamatergic signals. MR indicated genetically proxied higher educational attainment causally increases BD risk (IVW OR = 1.403, p = 6.69 × 10⁻⁵). In MDD, pruning was robust in heritability (LDSC p < 10⁻⁹⁰) but mediated/non-enriched in expression (TWAS p > 0.7), with glutamatergic TWAS signals (p = 0.007); education was protective (OR ≈ 0.72).Conclusion: Synaptic pruning emerges as the primary, independent pathway in BD—activation-skewed and amplified by cognitive reserve into episodic instability—distinguishing it from MDD's mediated pruning deficits with protective reserve. These findings reframe BD toward neuroimmune-pruning models, with implications for targeted therapeutics and cross-disorder nosology.
Keywords:
bipolar
; MDD
; BAD
; GWAS
; TWAS
; MR
; Mendelian
; partitioned heritability
; hypothesis
; genetics
Introduction
Bipolar disorder (BD) touches roughly 1–2 % of people worldwide. It brings repeated swings between mania and depression and, as a result, heavy personal, social, and economic costs [1,2]. Family and twin studies show a strong inherited element—around 60–80 %—yet the genes involved form a tangled, polygenic picture. Large genome-wide association studies (GWAS) have pinpointed dozens of risk spots, but together they still explain only a small slice of the total genetic risk [3,4]. Early research focused on dopamine and serotonin pathways, whereas more recent GWAS highlight genes tied to synaptic work, calcium signaling, and brain development. Many of those same pathways overlap with findings in schizophrenia (SCZ), especially in immune-related pruning processes that remove extra synapses [5,6].
Glutamate signaling has also been under the spotlight. Lithium's known action on inositol pathways and the fast-acting antidepressant effects of ketamine in bipolar depression both point toward glutamatergic imbalance [7,8]. Meanwhile, evidence from SCZ suggests that too much synaptic pruning, partly driven by complement proteins, may strip away needed brain connections [9]. Because BD shares moderate genetic links with both SCZ and major depressive disorder (MDD), teasing apart which biological routes are shared and which are unique remains a pressing question [10].
Today's very large GWAS datasets give researchers the power to explore those routes in finer detail [4,11]. Still, direct comparisons between the "extra pruning" idea and the "glutamate/plasticity" idea are rare—especially studies that combine heritability partitioning, transcriptome-wide analyses, and causal tests. The present work steps into that gap. Using the newest European-ancestry BD summary statistics, we apply four complementary tools—MAGMA gene and set tests, stratified LD score regression, S-PrediXcan across brain tissues, and two-sample Mendelian randomization. We build two custom gene lists, one for pruning and one for glutamatergic function, trimmed to avoid overlap so they can be weighed independently. Running the same pipeline in MDD lets us ask whether the leading BD pathways also matter for depression or whether they stand apart. In doing so, we hope to pin down which biological mechanisms sit at the core of BD risk and how strongly they act.
Methods
MAGMA Gene-Based and Gene-Set Analysis
We analysed publicly available summary statistics from a recent European-ancestry bipolar disorder meta-GWAS that excluded UK Biobank and 23andMe participants (file name = bip2024_eur_noUKB_no23andMe) [12]. After removing variants with missing positions or invalid p values, 6 857 849 autosomal SNPs on chromosomes 1–22 remained. Marker positions refer to the GRCh37/hg19 build. The effective sample size for downstream tests was 137 097.
All association tests were carried out with MAGMA v1.10 [13]. SNPs were assigned to protein-coding genes using NCBI37.3 coordinates with an extension of 35 kb upstream and 10 kb downstream. Linkage disequilibrium (LD) estimates came from the Phase 3 European panel of the 1000 Genomes Project. Gene statistics were calculated with the SNP-wise mean model, which accounts for LD while weighting each marker equally, and were calibrated to the effective N of 137 097.
Seven a-priori gene sets were tested competitively:
- 23 focused glutamatergic-receptor target genes.
- An expanded glutamatergic/synaptic-plasticity list (130 genes).
- A core synaptic pruning list (38 genes).
- A broad pruning list (262 genes).
- A "specific pruning" list (225 genes) created by removing the 37 glutamatergic/plasticity genes that overlap with the broad pruning set.
- A monoaminergic control list (104 genes).
- A housekeeping-gene control list (184 genes).
Set membership was covering glutamatergic signalling, microglia-mediated pruning, classical monoaminergic pathways, and constitutively expressed housekeeping functions. For each gene we converted the MAGMA p value to a Z score and compared the mean Z of every set with the genome-wide mean using one-sided one-sample t tests. Genome-wide Bonferroni correction across 18 348 genes set the gene-level threshold at p < 2.73 × 10⁻⁶. Gene-set p values were adjusted for the seven planned tests by Bonferroni (p < 0.0071) and by the false-discovery rate (FDR).
Partitioned Heritability with Stratified LD-Score Regression
To examine whether the polygenic signal observed in MAGMA is mirrored at the level of SNP-based heritability, we applied stratified LD-score regression (S-LDSC) [14]. Seven binary annotations were built, one for each a-priori gene set analysed earlier. Every gene was padded by 10 kb upstream and downstream, limited to autosomal coordinates, and SNPs falling within those intervals were flagged. European reference LD scores from the 1000 Genomes Phase 3 baseline-LD model (v2.2) were used throughout.
Summary statistics from the bipolar disorder GWAS were "munged" to Z-scores, and the effective sample size (Neff ≈ 137 097) supplied to S-LDSC. Enrichment was defined as the proportion of SNP-heritability captured by an annotation divided by its share of SNPs; an LD-corrected estimate was obtained by regressing χ² values on LD scores. Significance was evaluated with the one-sided Mann-Whitney U test that S-LDSC uses to compare χ² distributions of annotated versus background SNPs. Bonferroni adjustment across the seven planned tests set the experiment-wide threshold at p < 0.0071; false-discovery-rate (FDR) q-values were also computed.
Transcriptome-Wide Association Analyses with S-PrediXcan
To test whether genetically regulated expression influences the liability to bipolar disorder we ran S-PrediXcan, the summary-data implementation of PrediXcan [15,16]. GWAS summary statistics were first harmonised: Z-scores were calculated from reported odds ratios and standard errors, or from p values when standard errors were unavailable; the effective sample size was 137 097.
Expression prediction weights were taken from GTEx release v8 and were those produced with the MASHR/elastic-net procedure described by [17]. Six CNS tissues were evaluated—frontal cortex (BA9), anterior cingulate cortex (BA24), hippocampus, amygdala, caudate and nucleus accumbens.
The seven a-priori gene sets interrogated in earlier MAGMA and S-LDSC analyses were re-analysed here. For each gene–tissue pair we recorded nominal significance (p < 0.05), Benjamini–Hochberg FDR < 0.05 across all pairs, and Bonferroni significance within tissue. Competitive enrichment was tested by comparing absolute TWAS Z-scores for genes inside each set with those for all other expressed genes by one-sided Mann-Whitney U tests.
Two-Sample Mendelian Randomization
We evaluated potential causal effects of neuroplasticity-related traits on bipolar disorder by performing two-sample Mendelian randomization (MR) following the framework outlined by [18]. Independent instruments (r² < 0.001, 10 Mb window) were extracted from published genome-wide association studies (GWAS) for seven exposures that have been linked to cognitive reserve or neural plasticity: years of schooling [19], general cognitive performance, intelligence quotient (IQ) scores [20], fluid intelligence, reaction time, hippocampal volume, and major depressive disorder (included as a benchmark phenotype with recognised bidirectional overlap with bipolar disorder). Genome-wide significance (p < 5 × 10⁻⁸) defined the primary variant list; for phenotypes yielding fewer than three instruments, the threshold was relaxed to p < 5 × 10⁻⁶.
Outcome data were GWAS summary statistics for bipolar disorder (effective N ≈ 137 097). Variants were harmonised so that alleles were aligned across exposure and outcome datasets; palindromic markers with ambiguous minor-allele frequencies (0.42–0.58) were omitted. The principal estimator was the inverse-variance weighted (IVW) model with multiplicative random effects. Robustness was assessed with the weighted median, weighted mode, and MR-Egger procedures [21,22]. Instrument strength was gauged with mean F statistics (> 10 interpreted as adequate). Horizontal pleiotropy was probed through the MR-Egger intercept and Cochran's Q heterogeneity index. All analyses were scripted in R; code calculated odds ratios (ORs) per genetically predicted one-SD increase in each exposure.
Comparison with the MDD GWAS
To evaluate whether the observed genetic signals were specific to bipolar disorder or shared with related mood disorders, we replicated the full analytical pipeline—encompassing MAGMA gene and gene-set testing, stratified LD score regression for partitioned heritability, S-PrediXcan TWAS across brain tissues, and two-sample Mendelian randomization with neuroplasticity proxies—using summary statistics from a large trans-ancestry genome-wide association study of major depressive disorder [11].
Results
MAGMA Gene-Based and Gene-Set Analysis
The gene-based scan yielded 218 genome-wide significant genes and 4 594 additional genes with nominal evidence (p < 0.05). Leading signals included FEN1 (p = 1.66 × 10⁻¹³), TMEM258 (p = 8.62 × 10⁻¹³), the FADS1/FADS2 locus (≈ 10⁻¹²), and MSRA (p = 4.28 × 10⁻¹²), echoing earlier work that implicates lipid metabolism and DNA repair in bipolar disorder.
Competitive testing highlighted synaptic pruning pathways (Table 1). The broad pruning set showed the strongest enrichment (raw p = 1.14 × 10⁻⁴; Bonferroni p = 7.99 × 10⁻⁴), anchored by CACNA1C (p = 6.96 × 10⁻¹¹), SHANK2 (p = 2.70 × 10⁻⁹), and several complement and MHC class I genes. When glutamatergic-overlapping genes were removed, the specific pruning set remained significant (raw p = 7.00 × 10⁻⁴; Bonferroni p = 0.0049), confirming that the signal is not driven solely by glutamate-related genes.
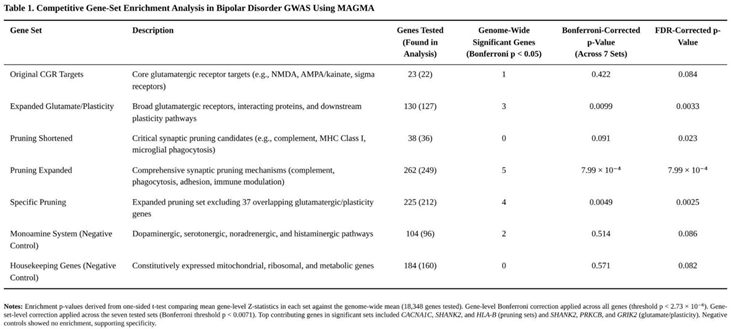
The expanded glutamatergic/plasticity set also passed correction (raw p = 0.0014; Bonferroni p = 0.0099), with SHANK2, PRKCB, and GRIK2 contributing prominently. The core pruning list was FDR-significant (raw p = 0.013; FDR p = 0.023) but fell short of Bonferroni adjustment.
Neither the original 23-gene CGR set (p = 0.060) nor the negative-control monoaminergic (p = 0.073) and housekeeping (p = 0.082) lists showed evidence of enrichment after multiple-testing correction, supporting the selectivity of the pruning and glutamatergic findings.
Partitioned Heritability with Stratified LD-Score Regression
Partitioned heritability analysis corroborated the gene-set signals detected by MAGMA (Table 2). Despite comprising only 0.15 % of SNPs, the 23-gene core glutamatergic receptor (CGR) annotation explained 1.44-fold more heritability than expected; after LD correction this rose to 1.67-fold (p = 1.16 × 10⁻¹¹²), easily surpassing the Bonferroni boundary. The broader 130-gene glutamatergic/synaptic-plasticity list covered 0.73 % of SNPs and displayed a raw enrichment of 1.14-fold that climbed to 1.39-fold after LD adjustment (p = 8.04 × 10⁻⁷⁰).
Synaptic pruning annotations were likewise enriched. The 38-gene "short" pruning set (0.18 % of SNPs) produced 1.16-fold raw enrichment (p = 1.23 × 10⁻³⁷). Expanding the list to 262 genes diluted the raw estimate to 1.10-fold but the signal remained robust (p = 4.16 × 10⁻³⁸). Removing the 37 genes that overlap with glutamatergic pathways still left a significant 1.07-fold enrichment (p = 3.77 × 10⁻¹²), indicating that pruning contributes to heritability independently of glutamatergic genes.
Control annotations behaved as anticipated. Housekeeping genes, which span 0.24 % of SNPs, showed modest but significant enrichment (raw = 1.12-fold, LD-adjusted = 1.22-fold, p = 2.31 × 10⁻¹⁹), likely because of their broad genomic footprint. By contrast, the monoaminergic list (0.34 % of SNPs) exhibited no enrichment (raw = 0.98-fold, p ≈ 1), underscoring the specificity of the glutamatergic and pruning findings. After Bonferroni correction, all target-based annotations except the monoaminergic control remained significant.
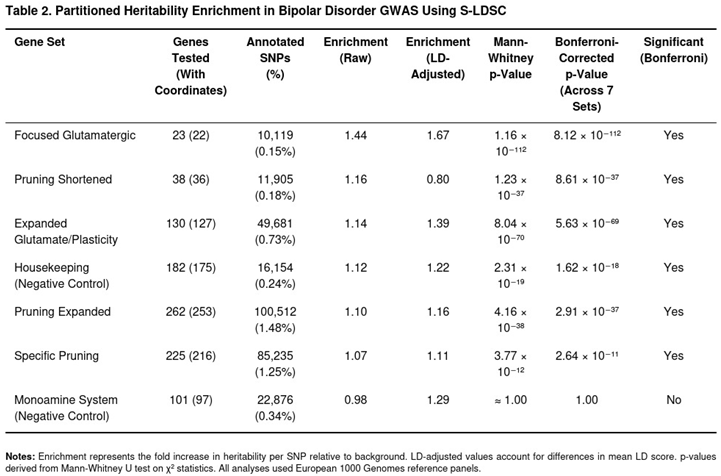
Taken together, the S-LDSC results reinforce the MAGMA evidence: genes involved in glutamatergic signalling and, independently, in microglia-mediated synaptic pruning capture a disproportionate share of bipolar disorder heritability in Europeans.
Transcriptome-Wide Association Analyses with S-PrediXcan
Across ~65 600 gene–tissue pairs, S-PrediXcan produced 349 Bonferroni-significant associations and 2 113 FDR-significant ones. Prominent findings included reduced predicted expression of HTR6 in frontal cortex and increased C4A expression in anterior cingulate cortex; several mitochondrial and ribosomal transcripts in the housekeeping reference set were also significant.
At the pathway level (Table 3) glutamatergic gene sets showed little evidence of enrichment. The original 23-gene CGR panel contained one FDR-level hit (CYP2D6 in nucleus accumbens) and no enrichment relative to the genomic background (|Z| ratio 1.03, p = 0.72). The expanded glutamate/synaptic-plasticity set (130 genes) had seven FDR-significant members but was similarly un-enriched (ratio 0.99, p = 0.79).
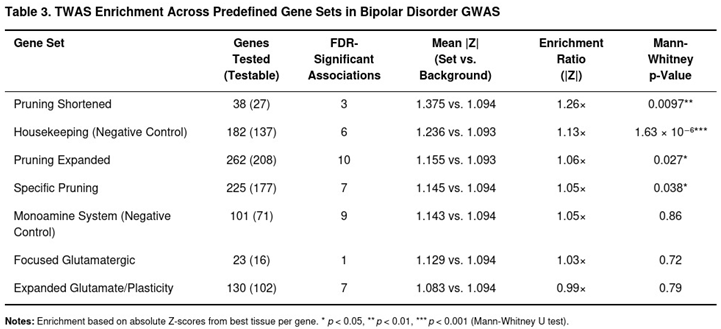
By contrast, pruning-related annotations yielded stronger signals. The 38-gene shortened pruning list produced three FDR-significant associations (C4A, HLA-B, HLA-C) and a 26 % increase in average |Z| values over background (p = 0.0097). The larger 262-gene pruning panel generated ten FDR associations and a modest but significant enrichment (ratio 1.06, p = 0.027). Removing glutamatergic overlap (225-gene specific pruning list) preserved seven FDR hits and comparable enrichment (ratio 1.05, p = 0.038).
Control sets behaved as expected. The monoaminergic list yielded nine FDR-significant genes yet no enrichment (ratio 1.05, p = 0.86). In contrast, the broadly expressed housekeeping set showed clear enrichment (ratio 1.13, p = 1.6 × 10⁻⁶), driven by mitochondrial and ribosomal components.
Collectively, brain-based TWAS supports a contribution of complement/MHC-linked pruning genes to bipolar disorder risk, while offering scant evidence that glutamatergic receptor or plasticity genes are broadly dysregulated at the expression level.
Two-Sample Mendelian Randomization
Educational attainment showed a consistent positive association with bipolar disorder liability (Table 4). The IVW model yielded β = 0.338, corresponding to OR = 1.40 (95 % CI 1.19–1.66, p = 6.7 × 10⁻⁵). This result was corroborated by the weighted median (OR = 1.47, p = 1.3 × 10⁻⁵) and weighted mode estimators. Although Q statistics indicated heterogeneity (I² = 70.6 %), the MR-Egger intercept was null (p = 0.864), and MR-PRESSO adjustment for three outlier variants left the point estimate materially unchanged.
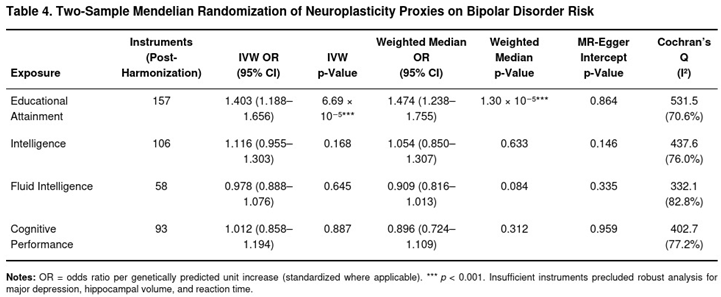
For intelligence, fluid intelligence, and cognitive performance the IVW estimates were non-significant (all p > 0.15) and concordant across sensitivity tests. Mean F values exceeded 25 for these instruments, arguing against weak-instrument bias. Reaction time, hippocampal volume, and major depression produced fewer than three valid instruments after clumping, precluding reliable MR; exploratory IVW runs were therefore not interpreted.
Collectively, the analyses suggest that genetically proxied years of schooling—but not intelligence or other cognitive metrics—may increase bipolar disorder risk. While pleiotropic pathways cannot be entirely ruled out, the robustness across MR estimators supports a directional effect from educational attainment to disease susceptibility rather than the reverse.
Head-to-Head Comparison to the MDD Results
Cross-phenotype evaluation revealed overlapping yet disorder-specific enrichment patterns (Table 5). In MAGMA competitive testing, pathways linked to microglial pruning showed the strongest and most consistent signals. The expanded pruning list reached Bonferroni-corrected significance for both bipolar disorder (uncorrected p = 1.14 × 10⁻⁴; corrected p = 7.99 × 10⁻⁴) and major depression (uncorrected p = 5.70 × 10⁻⁵; corrected p = 3.99 × 10⁻⁴). Comparable support emerged for the specific pruning list (bipolar corrected p = 4.9 × 10⁻³; depression corrected p = 3.0 × 10⁻³). Glutamatergic and plasticity genes were also enriched, although the signal was weaker (bipolar corrected p = 9.9 × 10⁻³; depression corrected p = 1.42 × 10⁻²). In contrast, the original, smaller CGR target panel was only nominally suggestive in bipolar disorder (p = 0.060) and fell short of strict correction in depression. Neither monoaminergic genes nor housekeeping controls showed appreciable enrichment after correction, confirming their suitability as negative controls.
Partitioned heritability analyses with stratified LD-score regression (S-LDSC) reinforced these observations but highlighted sharper contrasts between the two disorders. Depression exhibited very large pruning signals, with Mann–Whitney p values reaching 3.75 × 10⁻¹¹³ for the expanded list and 1.24 × 10⁻⁹² for the specific list. Bipolar disorder showed the same direction of effect, yet with more modest probabilities (4.16 × 10⁻³⁸ and 3.77 × 10⁻¹², respectively). The glutamatergic annotations displayed the opposite pattern: they were strongly enriched in bipolar disorder (p = 8.04 × 10⁻⁷⁰) but entirely absent in depression (p = 0.91). Again, control categories remained null.
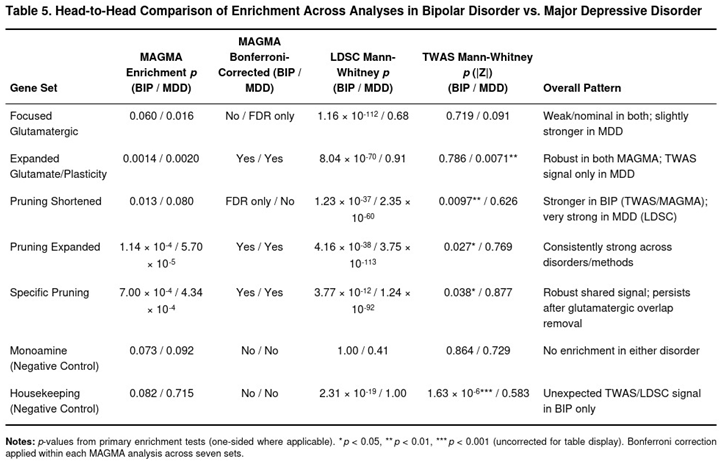
Transcriptome-wide association study (TWAS) data yielded weaker overall signals, but a subset of results aligned with the genomic findings. The shortened pruning list was enriched in bipolar disorder (p = 0.0097) whereas the same list was uninformative for depression (p = 0.626). Housekeeping genes, included solely as a technical benchmark, were unexpectedly over-represented in bipolar TWAS output (p = 1.63 × 10⁻⁶) yet remained neutral in depression. Conversely, expanded glutamatergic genes showed enrichment in depression (p = 0.0071) but not in bipolar disorder.
Mendelian randomization revealed markedly divergent causal estimates between bipolar disorder and major depression. In bipolar disorder, genetically predicted higher educational attainment showed a positive association with risk (IVW OR = 1.403, 95% CI 1.188–1.656, p = 6.69 × 10⁻⁵), consistent across sensitivity methods and robust to pleiotropy checks. No robust effects emerged for other proxies (intelligence, fluid intelligence, cognitive performance).
In major depression, the direction reversed sharply: higher educational attainment was protective (IVW OR = 0.721, 95% CI 0.661–0.785, p = 7.53 × 10⁻¹⁴), with consistent negative estimates in weighted median and mode analyses. Cognitive performance (OR = 0.903, p = 0.0077) and fluid intelligence (OR = 0.953, p = 0.0365) also suggested protective effects, though weaker. Intelligence showed no clear signal (OR = 0.943, p = 0.133). Instrument strength was adequate (mean F > 40 across analyses), with heterogeneity present but no evidence of directional pleiotropy (Egger intercepts non-significant).
Discussion
Deriving an Etiological Hypothesis for Bipolar Disorder from the Divergence
When the genetic fingerprints of bipolar disorder (BD) and major depressive disorder (MDD) are laid side by side, a picture emerges that is equal parts overlap and divergence. Signals tied to microglial pruning pop out in BD even after every glutamatergic gene is stripped away (MAGMA p = 0.0049; TWAS |Z|-enrichment p = 0.038). In MDD the very same pruning genes explain an enormous slice of SNP-based heritability (LDSC p < 10⁻⁹²) yet do not change predicted expression at all (TWAS p = 0.877). One reading is that pruning is an active driver of mood instability in BD, whereas in MDD it may sit further downstream, a consequence of more general plasticity trouble [11,22]. Glutamatergic pathways are statistically enriched in both illnesses, but only MDD shows a convincing expression signal (TWAS p = 0.0071 versus 0.786 in BD).
Figure 1.
The "Pruned-but-Potent" Etiological Model of Bipolar Disorder versus Major Depressive Disorder. This schematic illustrates the divergence of shared immunogenetic risks into distinct pathophysiological pathways. While both disorders share genetic roots in the MHC locus, Bipolar Disorder (left, blue) is characterized by active, activation-skewed synaptic pruning driven by neuroinflammation, resulting in efficient but unstable neural circuits. Major Depressive Disorder (right, orange) is characterized by broad plasticity deficits and neural erosion. High cognitive reserve (center, green) acts as a divergent modifier: it amplifies risk in Bipolar Disorder by over-activating thinned circuits (leading to mania), while buffering risk in MDD by compensating for neural deficits. This divergence suggests distinct therapeutic targets: complement modulation for Bipolar Disorder versus plasticity boosters for MDD.
Figure 1.
The "Pruned-but-Potent" Etiological Model of Bipolar Disorder versus Major Depressive Disorder. This schematic illustrates the divergence of shared immunogenetic risks into distinct pathophysiological pathways. While both disorders share genetic roots in the MHC locus, Bipolar Disorder (left, blue) is characterized by active, activation-skewed synaptic pruning driven by neuroinflammation, resulting in efficient but unstable neural circuits. Major Depressive Disorder (right, orange) is characterized by broad plasticity deficits and neural erosion. High cognitive reserve (center, green) acts as a divergent modifier: it amplifies risk in Bipolar Disorder by over-activating thinned circuits (leading to mania), while buffering risk in MDD by compensating for neural deficits. This divergence suggests distinct therapeutic targets: complement modulation for Bipolar Disorder versus plasticity boosters for MDD.
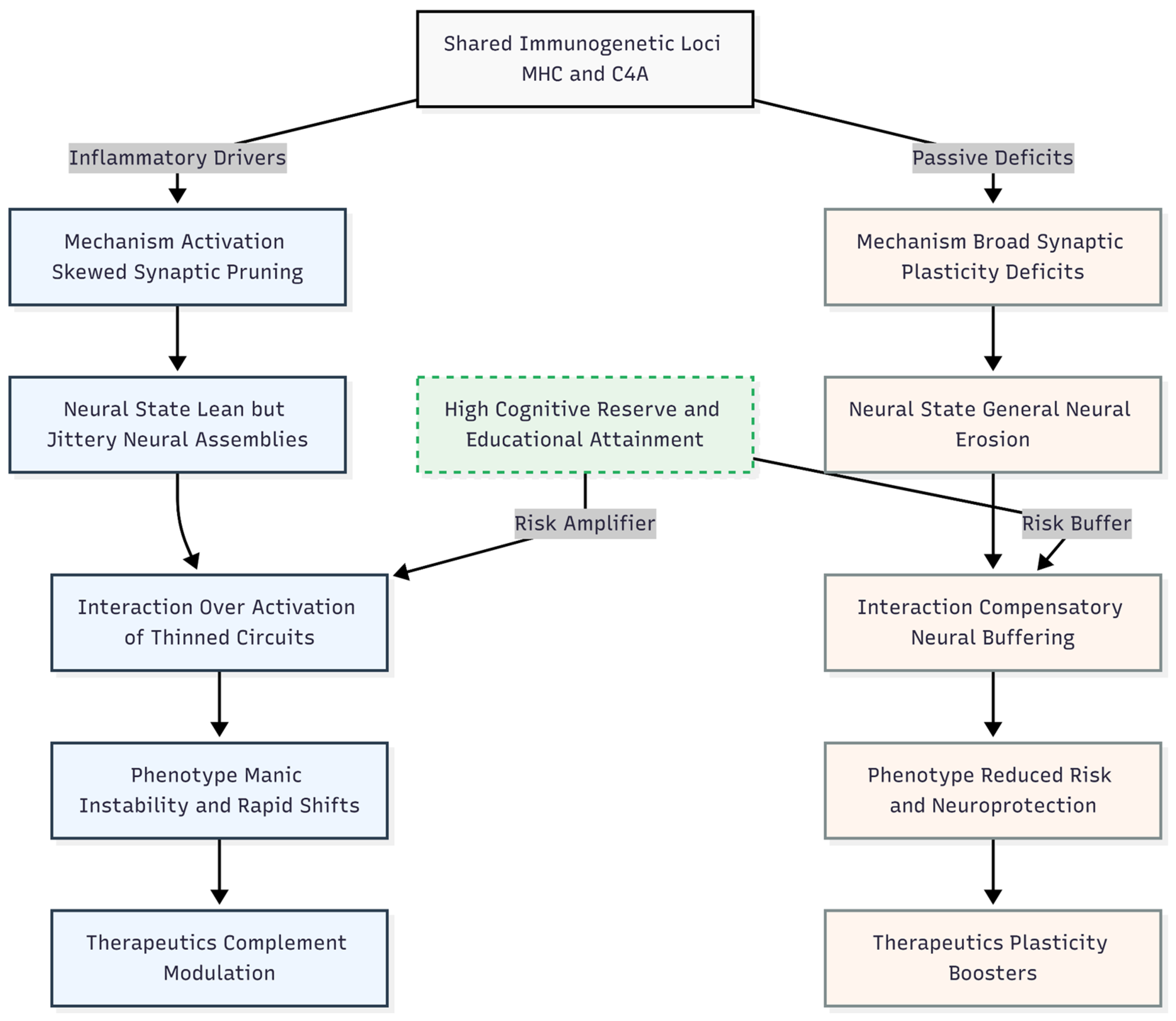
Mendelian-randomization adds another layer. A genetic tilt toward higher educational attainment—often used as a proxy for cognitive reserve—raises the odds of BD (OR = 1.403, p = 6.7 × 10⁻⁵) yet protects against MDD (OR = 0.721, p = 7.5 × 10⁻¹⁴). The same pattern holds for intelligence scores: helpful in depression, neutral in bipolar. Put together, the results suggest that a robust cognitive network can paradoxically fuel BD by over-activating circuits already thinned by pruning, whereas it cushions the broader, slower deficits typical of MDD [22].
These observations converge on a "pruned-but-potent" sketch of BD. Genes such as C4A (TWAS Z = 4.26) and RHOA (Z = 7.22) hint at heavy developmental pruning, leaving lean but jittery neural assemblies. Add a high cognitive-reserve load and those assemblies may overshoot, flipping into mania or rapid mood shifts. Epidemiology fits: many future BD patients shine academically or creatively before their first episode [23], a period when those efficient yet unstable circuits could be operating at full tilt. MDD, by contrast, shows a more even, across-the-board erosion of cognition over time [24].
Neuroinflammation may link these genomic clues to day-to-day illness. In BD, inflammatory markers spike during manic phases [25] and could push pruning into overdrive. Shared immune loci in the MHC—HLA-B, C4A and friends—tie BD and MDD together genetically, but the downstream effects appear to split by diagnosis [26]. Therapeutically, that split matters. BD might benefit more from strategies that dial back complement activity or broader inflammation—think anti-inflammatories or complement blockers—than from blanket plasticity boosters. The mixed performance of lithium across illness stages [27] may reflect exactly this tug-of-war between pruning load and neuroprogression [28,29].
Novelty and Impact
Recent work continues to show that many risk variants for bipolar disorder (BD) sit in genes that guide synaptic development, echoing overlaps with schizophrenia at complement and MHC loci [6,30]. What is new here is the layered evidence—competitive MAGMA and LDSC after removing glutamatergic overlap, brain-based TWAS with directionality, and two-sample Mendelian randomization (MR)—all pointing to synaptic pruning as a primary and independent pathway in BD. Pruning enrichment stayed significant even when glutamatergic genes were excluded, and predicted expression was higher for microglial or autophagy activators such as CD68, MAP1LC3A, and C4A.
The pattern separates BD from major depressive disorder (MDD). In MDD, pruning explains heritability but appears downstream of broader plasticity deficits in functional tests [11,22], whereas glutamatergic dysregulation drives expression signals. Borderline personality disorder shows little pruning enrichment [31,32].
A particularly novel MR finding is that genetically proxied educational attainment—often used as a marker of cognitive reserve—raises BD risk (OR = 1.403; p ≈ 6.7 × 10⁻⁵) yet protects against MDD (OR ≈ 0.72). This supports a "pruned-but-potent" model: excessive developmental pruning leaves lean, efficient, but fragile networks; high reserve can then drive these networks into manic over-activity under stress [23,33]. The model helps reconcile BD's frequent premorbid academic or creative strengths with later episodic instability and gives mechanistic weight to neuroprogression views that link cumulative inflammatory pruning to poorer outcomes [28,34]. Clinically, it highlights complement or microglial modulators as more promising than broad glutamatergic agents, which may aggravate instability [35].
Limitations
The analyses draw mainly on European-ancestry GWAS, limiting reach to other groups [11]. Use of summary statistics prevents testing gene–environment interactions central to BD, such as stress sensitization [33]. Candidate gene sets bring selection bias, and GTEx-based TWAS might overlook tissue-specific effects beyond sampled brain regions [16]. MR assumptions of no horizontal pleiotropy passed statistical checks but cannot be proven, and lifetime GWAS data cannot speak to episode timing or progression [36].
Conclusion
Multi-platform genomic evidence positions activation-skewed synaptic pruning as a core, independent mechanism in BD and suggests that greater cognitive reserve genetically heightens risk by over-engaging these pruned circuits. This framework distinguishes BD from MDD and guides attention toward neuroimmune targets and stage-specific interventions. Validation across ancestries, longitudinal cohorts, and functional studies is the next step in translating these insights into better stratification and treatment of BD.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics Declaration
Not applicable.
Conflicts of Interest
None declared.
References
- Grande, I; Berk, M; Birmaher, B; et al. Bipolar disorder. The Lancet 387(10027), 1561–1572. [CrossRef] [PubMed]
- Merikangas, KR; Jin, R; He, JP; et al. Prevalence and correlates of bipolar spectrum disorder in the World Mental Health Survey Initiative. Archives of General Psychiatry 68(3), 241–251. [CrossRef] [PubMed]
- Stahl, EA; Breen, G; Forstner, AJ; et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nature Genetics 51(5), 793–803. [CrossRef]
- Mullins, N; Forstner, AJ; O'Connell, KS; et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nature Genetics 53(6), 817–829. [CrossRef]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 179(7), 1469–1482.e11. [CrossRef]
- Trubetskoy, V; Pardiñas, AF; Qi, T; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604(7906), 502–508. [CrossRef]
- Malhi, GS; Tanious, M; Das, P; et al. Potential mechanisms of action of lithium in bipolar disorder. Current understanding. CNS drugs 27(2), 135–153. [CrossRef]
- Zarate, CA, Jr.; Singh, JB; Carlson, PJ; et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Archives of General Psychiatry 63(8), 856–864. [CrossRef] [PubMed]
- Sekar, A; Bialas, AR; de Rivera, H; et al. Schizophrenia risk from complex variation of complement component 4. Nature 530(7589), 177–183. [CrossRef]
- Coleman, JRI; Peyrot, WJ; Purves, KL; et al. Genome-wide gene-environment analyses of major depressive disorder and reported lifetime traumatic experiences in UK Biobank. Molecular psychiatry 25(7), 1430–1446. [CrossRef]
- Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Trans-ancestry genome-wide study of depression identifies 697 associations implicating cell types and pharmacotherapies. Cell 188(3), 640–652.e9. [CrossRef] [PubMed]
- O'Connell, KS; Koromina, M; van der Veen, T; et al. Genomics yields biological and phenotypic insights into bipolar disorder. Nature 639(8056), 968–975. [CrossRef]
- de Leeuw, CA; Mooij, JM; Heskes, T; et al. MAGMA: Generalized gene-set analysis of GWAS data. PLOS Computational Biology 11(4), e1004219. [CrossRef]
- Finucane, HK; Bulik-Sullivan, B; Gusev, A; et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nature Genetics 47(11), 1228–1235. [CrossRef] [PubMed]
- Barbeira, AN; Dickinson, SP; Bonazzola, R; et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nature Communications 9, 1825. [CrossRef] [PubMed]
- Gamazon, ER; Wheeler, HE; Shah, KP; et al. A gene-based association method for mapping traits using reference transcriptome data. Nature Genetics 47(9), 1091–1098. [CrossRef]
- Urbut, SM; Wang, G; Carbonetto, P; et al. Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nature Genetics 51, 187–195. [CrossRef]
- Davey Smith, G; Hemani, G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Human Molecular Genetics 23, R89–R98. [CrossRef]
- Lee, JJ; Wedow, R; Okbay, A; et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nature Genetics 50, 1112–1121. [CrossRef]
- Savage, JE; Jansen, PR; Stringer, S; et al. Genome-wide association meta-analysis in 269 867 individuals identifies new genetic and functional links to intelligence. Nature Genetics 50, 912–919. [CrossRef]
- Bowden, J; Davey Smith, G; Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. International Journal of Epidemiology 44, 512–525. [CrossRef]
- Cheung, N. From Pruning to Plasticity: Refining the Etiological Architecture of Major Depressive Disorder Through Causal and Polygenic Inference. Preprints. [CrossRef]
- MacCabe, JH; Lambe, MP; Cnattingius, S; et al. Excellent school performance at age 16 and risk of adult bipolar disorder: national cohort study. The British journal of psychiatry: the journal of mental science 196(2), 109–115. [CrossRef]
- Malhi, GS; Mann, JJ. Depression. The Lancet 392(10161), 2299–2312. [CrossRef]
- Benedetti, F; Aggio, V; Pratesi, ML; et al. Neuroinflammation in bipolar depression. Frontiers in Psychiatry 11, 71. [CrossRef] [PubMed]
- Sutantio, EH; Octaviani, ID. Neuroinflammation in the Pathogenesis of Psychiatric Disorder. Bulletin of Counseling and Psychotherapy 7(2). [CrossRef]
- Berk, M; Post, R; Ratheesh, A; et al. Staging in bipolar disorder: From theoretical framework to clinical utility. World Psychiatry 16(3), 236–244. [CrossRef]
- Berk, M; Kapczinski, F; Andreazza, AC; et al. Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neuroscience & Biobehavioral Reviews 35(3), 804–817. [CrossRef]
- Cyrino, LAR; Delwing-de Lima, D; Ullmann, OM; et al. Concepts of Neuroinflammation and Their Relationship With Impaired Mitochondrial Functions in Bipolar Disorder. Frontiers in behavioral neuroscience 15, 609487. [CrossRef]
- Coleman, JRI; Peyrot, WJ; Purves, KL; et al. Genome-wide gene-environment analyses of major depressive disorder and reported lifetime traumatic experiences in UK Biobank. Molecular psychiatry 25(7), 1430–1446. [CrossRef]
- Streit, F; Awasthi, S; Hall, AS; et al. Genome-wide association study of borderline personality disorder identifies 11 loci and highlights shared risk with mental and somatic disorders. medRxiv: the preprint server for health sciences 2024, 11.12.24316957. [Google Scholar] [CrossRef]
- Witt, SH; Streit, F; Jungkunz, M; et al. Genome-wide association study of borderline personality disorder reveals genetic overlap with bipolar disorder, major depression and schizophrenia. Translational Psychiatry 7(6), e1155. [CrossRef]
- Post, RM. The Kindling/Sensitization Model and Early Life Stress. Current topics in behavioral neurosciences 48, 255–275. [CrossRef]
- Kapczinski, F; Magalhães, PV; Balanzá-Martinez, V; et al. Staging systems in bipolar disorder: an International Society for Bipolar Disorders Task Force report. Acta Psychiatrica Scandinavica 130(5), 354–363. [CrossRef] [PubMed]
- Vieta, E; Berk, M; Schulze, TG; et al. Bipolar disorders. Nature Reviews Disease Primers 4, 18008. [CrossRef]
- Passos, IC; Mwangi, B; Vieta, E; et al. Areas of controversy in neuroprogression in bipolar disorder. Acta Psychiatrica Scandinavica 134(2), 91–103. [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



