Submitted:
07 January 2026
Posted:
08 January 2026
You are already at the latest version
Abstract
Inflammation is a normal and essential feature of pregnancy, supporting implantation, placental development, and parturition. When dysregulated, however, inflammatory pathways contribute to major obstetric complications such as preeclampsia, fetal growth restriction (FGR), and preterm birth, which account for substantial maternal and perinatal morbidity and mortality. This review synthesizes current understanding of the maternal–fetal immune interface, examines how inflammation contributes to pregnancy disorders, and explores therapeutic strategies that link pathogenic mechanisms to targeted interventions. The placenta functions as an active immunological hub, coordinating innate and adaptive immune responses to maintain tolerance while protecting against infection. In preeclampsia and FGR, excessive activation—driven by inflammasome signaling, Th1/Th17 polarization, and altered natural killer and macrophage function—impairs placental perfusion, promotes antiangiogenic pathways, and induces systemic endothelial dysfunction. Established therapies, including low-dose aspirin, low-molecular-weight heparin, and antenatal corticosteroids, aim to mitigate inflammation and optimize fetal outcomes, while adjunctive strategies target oxidative stress, nutritional deficits, and the maternal microbiome. Emerging approaches such as cytokine-targeted biologics, inflammasome inhibitors, and mesenchymal stem cell therapies show promise but require rigorous evaluation of safety and efficacy. A deeper understanding of placental immunology and inflammatory signaling is essential to develop precision therapies. Future research should prioritize biomarker validation, pathway-specific interventions, and equitable implementation to reduce inflammation-driven pregnancy complications.
Keywords:
preeclampsia
; fetal growth restriction
; placenta
; maternal–fetal interface
; inflammation
; pregnancy complications
; targeted therapy
1. Introduction
Inflammation is a fundamental physiological process in pregnancy, supporting implantation, placental development, and the onset of labor [1,2,3,4]. Through immune modulation, the maternal immune system maintains a paradoxical balance: tolerating a semi-allogeneic fetus while maintaining intact immune defense. The success of this process hinges on precise regulation of immune activity: an early proinflammatory environment supports implantation and trophoblast invasion; an anti-inflammatory state during mid-gestation promotes fetal growth; and a resurgence of inflammation late in pregnancy triggers labor [4,5].
Evidence suggests that placental inflammation isn’t just a secondary effect but a driving force behind obstetric diseases, linking preeclampsia, fetal growth restriction (FGR), and preterm birth within a shared immunopathogenic spectrum [6,7]. Hypertensive disorders of pregnancy (HDP), particularly preeclampsia, remain among the top causes of maternal mortality worldwide [8,9]. According to the World Health Organization, preeclampsia affects around 3–5% of pregnancies and is responsible for roughly 42,000 maternal deaths and over 500,000 perinatal deaths annually, including close to 200,000 stillbirths [10,11]. The burden disproportionately hits low- and middle-income countries—due to late diagnoses, limited prenatal care, and structural inequalities—widening global disparities in maternal and neonatal outcomes [9,12]. Beyond the immediate complications, preeclampsia frequently co-occurs with preterm birth, contributing to long-term cardiovascular, metabolic, and neurodevelopmental issues for both mother and child [13,14]. Together, these conditions form a continuum of immune-mediated placental dysfunction, marked by endothelial activation, antiangiogenic signaling, and systemic inflammation [13,15,16,17].
The placenta is increasingly recognized as an immunologically dynamic organ where trophoblasts, decidual stromal cells, and immune subsets—including uterine natural killer (uNK) cells, macrophages, and regulatory T cells—coordinate tolerance and defense [3,18]. This balance is maintained by molecules such as HLA-G, PD-L1, and Galectin-1, which modulate maternal cytotoxic responses and maintain fetomaternal harmony [19]. However, placental hypoxia, oxidative stress, infection, or sterile danger signals can reprogram this immune environment, tipping it toward pathological inflammation [5,19,20,21]. The result is poor vascular remodeling, increased secretion of soluble fms-like tyrosine kinase-1 (sFlt-1), and widespread endothelial dysfunction, all of which contribute to preeclampsia and FGR [22,23,24,25,26]. Recent findings demonstrate that activation of the NLRP3 inflammasome (an intracellular protein complex] acts as a central mediator linking trophoblast stress to maternal inflammation [21,27]. NLRP3-dependent maturation of interleukins (IL)-1β and IL-18 amplifies Th1/Th17 polarization, disrupts uNK cell cytotoxicity, and perpetuates oxidative damage [28,29,30]. Cytokines such as tumor necrosis factor-α (TNF-α), IL-6, and IL-17 further boost endothelial activation by increasing vascular permeability, suppressing nitric oxide bioavailability, and promoting oxidative stress [31,32,33]. Spatial and single-cell transcriptomic studies reveal how these inflammatory circuits are organized within the placenta, shedding light on fetomaternal immune communication [18,34].
Scientific advances have begun to translate into preventive and therapeutic strategies. Aspirin, started before 16 weeks’ gestation, lowers the incidence of early preeclampsia by inhibiting cyclooxygenase-1 and modulating inflammatory signaling [35]. Complementary approaches—such as optimizing maternal nutrition, managing metabolic inflammation, and modulating the microbiome—are gaining support as regulators of immune homeostasis during pregnancy [36,37,38,39]. Meanwhile, emerging therapies—such as inflammasome inhibitors (e.g., MCC950), cytokine-targeted biologics, and mesenchymal stem cell treatments—represent the cutting edge of therapeutics [40]. Still, their clinical application demands thorough evaluation of maternal-fetal safety, dose optimization, and long-term developmental effects.
Despite years of research, immune communication between the placenta and maternal endothelium remains incompletely understood. This review aims to synthesize current evidence on how dysregulated inflammation at the maternal-fetal interface drives placental dysfunction, with particular emphasis on preeclampsia and FGR as models of inflammation-mediated obstetric disease. We integrate biomarkers, mechanisms, and therapeutic prospects to pinpoint new intervention targets that can improve pregnancy outcomes and reduce global maternal health disparities. Ultimately, our goal is to provide a practical framework that links pathogenic pathways to targeted interventions that enhance maternal and neonatal health.
2. Immune Development in the Placenta
The placenta plays a central role in pregnancy, mediating the delicate balance between maternal immune tolerance toward the fetus—a semi-allograft genetically distinct from the mother—and protection against infection [41,42]. The maternal–fetal interface, formed by the decidua and fetal trophoblast-derived tissues, constitutes a highly specialized immunological environment in which the maternal immune system adapts to sustain fetal growth while maintaining antimicrobial defense [43,44]. Successful pregnancy, therefore, requires down-regulation of cytotoxic adaptive responses and preservation of robust innate immunity to protect against pathogens [41,42,45] (Figure 1).
2.1. Immune Cells at the Maternal–Fetal Interface
Immune cell populations at this interface are recruited and regulated by cytokines secreted by trophoblasts and decidual stromal cells [46,47]. In early gestation, decidual natural killer (dNK) cells account for approximately 70% of immune cells, macrophages for 20%, and regulatory T cells (Tregs) for 10% [3,4,18,48]. Smaller populations include B lymphocytes, mast cells, and dendritic cells [3,4].
2.1.1. Natural Killer Cells
dNK cells, located around invading extravillous trophoblasts, display a pregnancy-specific phenotype (CD56^bright^CD16^–^) shaped by interleukin-15 (IL-15) and transforming growth factor-β (TGF-β) [3,49,50]. They facilitate implantation, trophoblast invasion, and spiral artery remodeling by secreting IL-8, angiogenic mediators, and interferon-inducible protein-10 (IP-10) [49]. Their numbers decline during gestation, with Tregs assuming a more prominent immunoregulatory role [21,42,50]. Ultimately, this finely tuned balance of immune activation and tolerance at the maternal-fetal interface is fundamental to successful gestation.
2.1.2. Decidual Macrophages
Macrophages in the decidua exhibit marked plasticity, responding dynamically to local microenvironmental cues [49]. They serve as primary antigen-presenting cells, recruited through trophoblast-derived IL-10 and macrophage colony-stimulating factor [51]. Two functional phenotypes predominate: pro-inflammatory M1 macrophages—most abundant during implantation and near term—and anti-inflammatory M2 macrophages, which dominate during established placentation [51]. Both subsets contribute to spiral artery remodeling, clearance of apoptotic cells, and antimicrobial defense through secretion of vascular endothelial growth factor (VEGF) and matrix metalloproteinases (MMPs) [51,52]. Through this coordinated functional versatility, decidual macrophages play an essential role in sustaining tissue homeostasis and supporting a successful pregnancy.
2.1.3. Regulatory T Cells
Regulatory T cells (Tregs) play a central role in maintaining immune tolerance during pregnancy, helping the mother’s immune system accept the semi-allogeneic fetus while still protecting against infection. Tregs secrete anti-inflammatory cytokines, including TGF-β and IL-10, promoting tolerance to fetal antigens, supporting implantation, and limiting excessive inflammation [53]. At the maternal–fetal interface, Tregs suppress excessive inflammatory responses, promote vascular remodeling, and support healthy placental development through cytokines such as IL-10 and TGF-β. When Treg number or function is reduced—or pro-inflammatory T-cell subsets dominate—immune imbalance can drive endothelial dysfunction, placental malperfusion, and tissue injury. This dysregulation has been implicated in complications including preeclampsia, fetal growth restriction, recurrent pregnancy loss, and preterm birth [41,42]. Their population expands throughout gestation, and direct interactions between trophoblasts and Tregs appear critical for their differentiation and function [53,54]. Although therapeutic strategies that boost Treg activity (e.g., targeted immunomodulation, microbiome-influenced metabolites, or cell-based approaches) are under investigation, clinical translation remains early; restoring immune tolerance without compromising host defense is the key challenge.
2.2. Mechanisms of Immune Adaptation and Tolerance
Several molecular pathways sustain maternal tolerance while preserving host defense:
- Human Leukocyte Antigen (HLA): Extravillous trophoblasts express non-classical HLA molecules (HLA-C, HLA-E, HLA-G, HLA-F) that inhibit cytotoxic immune responses and facilitate placentation. HLA-G promotes NK-cell secretion of fetal growth factors [55].
- B7 Protein Family: High expression of costimulatory molecules such as B7-H1 on trophoblasts interacts with PD-1 receptors on maternal T cells, suppressing Th17 responses and enhancing Treg activity [56].
- TNF Superfamily Members: Placental Fas ligand (FasL) and TNF-related apoptosis-inducing ligand (TRAIL) induce apoptosis in activated maternal T cells [55], preventing immune-mediated rejection.
- Pattern Recognition Receptors: Toll-like receptors (TLRs) and RIG-I like receptors on trophoblasts detect pathogen-associated molecular patterns, initiating controlled inflammatory cascades that preserve defense without disrupting tolerance [57].
- Maternal–Fetal IgG Transfer: Immunoglobulin G is actively transported across the placenta during the second and third trimesters, conferring passive immunity to the neonate and infant [58].
- Interferon-γ (IFN-γ): Produced by syncytiotrophoblast, IFN-γ plays a crucial role in antiviral protection and local immune signaling [59].
- Other Immunomodulators: Galectin-1, sex hormones such as progesterone and estrogen, and complement regulatory proteins (CD46, CD55, CD59) collectively maintain immune equilibrium and protect the fetus from complement-mediated injury [19].
Collectively, these molecular pathways orchestrate a finely tuned immunological state in which tolerance toward fetal antigens is actively maintained while essential antimicrobial defenses are preserved. This dynamic equilibrium ensures proper placental development, protects the fetus from immune-mediated damage, and supports a successful pregnancy despite the unique immunological challenges of gestation. Beyond these classical immunoregulatory pathways, emerging evidence highlights an additional layer of immune modulation involving the placental and maternal microbiomes, which may influence both immune development and susceptibility to inflammation-driven obstetric disorders.
3. The Maternal Microbiome: A Hidden Regulator of Pregnancy
Throughout pregnancy, the maternal microbiome undergoes profound physiological shifts driven by hormonal, metabolic, and immunological changes. These alterations influence maternal health and fetal development, shaping susceptibility to conditions such as gestational diabetes mellitus and other noncommunicable diseases [60,61,62,63]. In preeclampsia, studies have demonstrated consistent alterations in gut microbiota composition, including increased abundance of Proteobacteria and Actinobacteria, accompanied by reduced levels of genera such as Prevotella, Varibaculum, Lactobacillus, and Porphyromonas [64]. These changes reflect a shift toward a proinflammatory microbial profile, supporting the hypothesis that maternal gut dysbiosis may contribute to systemic inflammation, endothelial dysfunction, and ultimately placental disease. Together, these findings underscore the maternal microbiome as a dynamic regulator of immune and metabolic homeostasis during pregnancy. Understanding how microbial dysbiosis contributes to disorders such as preeclampsia offers a promising avenue for early risk stratification and targeted interventions to improve maternal and neonatal outcomes.
3.1. Placental Microbiome
For decades, the placenta was considered a sterile environment. However, recent advances in sequencing technologies have challenged this assumption, suggesting the presence of a unique placental microbiome [65]. Despite these findings, essential gaps remain regarding its origin – whether derived from the maternal oral cavity, genital tract, or hematogenous spread – and its relationship to pregnancy outcomes, both physiological and pathological [66,67,68].
Recent research by Saadaoui et al. using molecular biology and 16S rRNA sequencing, discovered different placental microbiomes according to the various outcomes, showing differences between the oral microbiome, with information about specific bacteria (Treponema maltophilum) and the outcome observed in these pregnancies in specific moments (potential biomarker), and reporting information about some bacteria as Ureaplasma urealyticum more abundant in PTB placenta [67,69]. Some studies have identified four major phyla in the placental microbiome (Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria), and changes in the placental microbiome may be associated with adverse pregnancy outcomes and comorbidities, including preterm birth (PTB), gestational diabetes mellitus, and severe chorioamnionitis. In some cases, contamination from ascending vaginal bacteria might influence the changes in the placental microbiome [70].
Based on the information to date, the placental microbiome is unique but resembles that of an oral origin [65,71]. It’s well established that the placenta contributes to early immune development, and maternal antigens may be associated with the initial formation of immune cell populations and clinical outcomes. If antibiotics are administered to the mother during the prenatal period, dysbiosis occurs, resulting in altered infant immune development and increased susceptibility to viral infections [72]. Although many questions remain open, it is well established that numerous adjustments occur in women during pregnancy (hormonal, immunological, physiological), which increase the risk of other clinical conditions, such as infections distant from the placenta (vaginal, oral), and are associated with PTB and LBW (low birth weight) [54,73].
4. Evidence of Altered Inflammation in Preeclampsia
Normal pregnancy is characterized by a controlled, low-grade systemic inflammatory state that is essential for implantation, placental development, and preparation for parturition [74,75]. Compared with the non-pregnant state, pregnancy is associated with leukocytosis [74], increased complement activation, and dynamic shifts in peripheral leukocyte populations [74]. Distinct immunological phases have been described, reflecting changes in both innate and adaptive immunity [4,42,73,74]. This physiological inflammatory environment represents a balance between proinflammatory mediators—such as TNF-α, IL-2, IL-4, IL-6, IL-8, IL-10, interferon-γ (IFN-γ), and monocyte chemotactic protein-1 (MCP-1) [4,41,42]—and anti-inflammatory/regulatory mechanisms (Figure 2).
Preeclampsia shares some features with the inflammatory state of normal pregnancy, but the intensity of the response is markedly increased [13,76,77]. This exaggerated inflammation is thought to originate from syncytiotrophoblast stress due to hypoxia, oxidative stress, or both [5,75,76,78], leading to the release of proinflammatory cytokines and antiangiogenic factors [5,16,75]. The inflammatory response may be triggered by both exogenous danger signals (e.g., pathogens) and endogenous signals (products of trauma, ischemia, necrosis, or oxidative stress) [79,80,81]. It involves inflammatory leukocytes (granulocytes, macrophages, and NK cells) in concert with endothelial cells, platelets, and soluble mediators, such as complement [42,77].
Placental-derived antiangiogenic factors—such as increased sFlt-1 and soluble endoglin (sEng) and decreased placental growth factor (PlGF)—are distinctive features of preeclampsia and contribute to widespread endothelial injury [83,84]. In summary, preeclampsia represents the convergence of dysregulated inflammation, imbalanced angiogenic signaling, and heightened long-term cardiovascular vulnerability [77,78,84]. Excess maternal inflammatory activation injures the endothelium and amplifies oxidative stress, whereas anti-angiogenic factors, such as sFlt-1 and sEng, disrupt placental perfusion and propagate systemic vascular dysfunction. These acute pathophysiologic pathways not only drive the clinical syndrome of hypertension, proteinuria, and multi-organ involvement but also leave a persistent imprint on maternal cardiovascular health, explaining the markedly increased risk of later hypertension, ischemic heart disease, and stroke [22,29,85]. Understanding how immune, vascular, and metabolic networks intersect in preeclampsia is essential for developing therapies that protect both immediate pregnancy outcomes and lifelong cardiovascular health.
4.1. Immune Dysregulation and Cytokine Imbalance
Since Medawar’s 1953 hypothesis that the fetus is an “allograft” [44,86], early theories posited that maternal immune privilege at the maternal–fetal interface was necessary to maintain pregnancy [55,74]. These models emphasized maternal immune suppression, reduced expression of major histocompatibility complex (MHC) antigens in fetal tissue, and a shift from Th1 to Th2 cytokine dominance [57,81,86,87]. Contemporary evidence, however, indicates that a balanced interplay between pro- and anti-inflammatory responses is essential for successful pregnancy [81,86]. In early gestation, implantation and placental development are inherently proinflammatory but are tightly regulated by anti-inflammatory mediators, such as IL-10 and Tregs [1,54].
In preeclampsia, this balance is disrupted [77,88]. There is an overactivation of Th1 and Th17 responses, reduced Treg activity, and increased activation of cytotoxic NK cells and autoreactive B cells [77,87]. This leads to increased innate immune activation in both the maternal circulation and the uteroplacental unit [75,76,81]. The result is superficial trophoblast invasion, poor spiral artery remodeling, and worsening placental ischemia with escalating oxidative stress [21,87,89,90].
4.2. Cytokines and Endothelial Dysfunction in Preeclampsia
Chronic immune activation in preeclampsia is characterized by elevated levels of proinflammatory cytokines, particularly TNF-α, IL-6, and IL-17 [31,32,77,87,91]. These act locally and systemically, perpetuating oxidative stress, vascular inflammation, and endothelial damage.
- TNF-α and IL-6: Promote endothelial adhesion molecule expression, increase vascular permeability, and impair endothelial nitric oxide synthase (eNOS) activity, reducing nitric oxide (NO) bioavailability and vasodilation [91,92]. Experimental models show that a two-fold increase in TNF-α elevates mean arterial pressure (MAP) and reduces renal plasma flow and glomerular filtration rate (GFR) [92,93]. IL-6 exerts similar effects and has been linked to increased plasma renin activity [92,94].
- IL-17: In animal models, IL-17 increases MAP, induces placental oxidative stress, and stimulates B cells to produce angiotensin II type 1 receptor autoantibodies (AT1-AA), exacerbating hypertension and vascular injury [95].
Together, these inflammatory and immunological disturbances integrate placental dysfunction with maternal endothelial injury, forming the central pathogenic axis that underlies the clinical manifestations of preeclampsia (Figure 3).
5. Inflammation and Fetal Growth Restriction
FGR is a complex clinical condition associated with increased perinatal morbidity and mortality [96,97]. It is primarily a consequence of placental insufficiency and alterations in the intrauterine inflammatory environment, both of which can have lasting effects on perinatal and long-term health [98,99,100]. FGR is defined as the failure of a fetus to achieve its genetically determined growth potential [97]. Clinically, the most widely accepted criterion is an estimated fetal weight below the 10th percentile for gestational age, as defined by standardized growth charts [101]. Multiple etiological factors contribute to FGR, with inflammation playing a pivotal role in its pathogenesis [102]. Excessive placental inflammation can be triggered by maternal stress, hypoxia, and endogenous danger signals such as uric acid crystals [6,28,79]. These stimuli activate placental inflammasomes—, particularly NLRP3, through IL-1–dependent pathways, impairing trophoblast function and contributing to placental insufficiency [27,28,29] (Figure 4).
The NLRP3 inflammasome, a multiprotein complex, mediates the maturation of proinflammatory cytokines such as IL-1β and IL-18, which are critical in amplifying inflammatory responses [28,32]. Experimental models demonstrate that sustained maternal inflammation can disrupt fetal metabolic programming, affecting skeletal muscle glucose metabolism and β-cell function, with potential implications for future metabolic disease risk [103]. Placental inflammation is also associated with increased macrophage infiltration and a proinflammatory cytokine profile in both placental tissue and maternal serum [6,72,88]. Furthermore, inflammation in FGR may extend beyond the placenta, affecting other systems such as the central nervous system—where neuroinflammation can impair brain development [104,105,106]—and the skin, where inflammatory changes may predispose to childhood atopic diseases [62].
Assessment of inflammatory involvement in FGR includes evaluation of both systemic and local biomarkers. Elevated maternal serum levels of TNF-α, IL-6, and high-sensitivity C-reactive protein (hs-CRP) have been reported in pregnancies complicated by FGR, with correlations to adverse neonatal outcomes such as low birth weight and reduced Apgar scores [79,103,107]. Histological and immunohistochemical analyses of placental tissue reveal increased macrophage density and a predominance of proinflammatory markers [108,109]. Additionally, hypoxia-induced factors such as hypoxia-inducible factor 1-alpha (HIF-1α) accumulate in placentas of pregnancies complicated with FGR, linking placental hypoxia to sterile inflammation and dysfunction [110]. The inflammatory milieu associated with FGR may have lasting effects on both the fetus and the mother. Children with a history of poor postnatal growth demonstrate elevated circulating inflammatory markers—such as C-reactive protein, hepatocyte growth factor, IL-8, and TNF-α—compared with age-matched controls [111]. This low-grade inflammation may increase the risk of future metabolic and cardiovascular disease. Animal studies indicate that FGR can blunt the neonatal proinflammatory response to infectious stimuli, mediated by reduced nuclear factor kappa B (NF-κB) activation [112]. This suggests impaired innate immunity and increased susceptibility to infection. On the maternal side, pregnancies complicated by FGR often show elevated proinflammatory cytokines, reinforcing the concept of a shared maternal–fetal inflammatory environment [6].
6. Immune Dysregulation in Placental Insufficiency: Clinical Implications and Translational Perspectives
There is consistent evidence that preeclampsia and FGR are associated with increased circulating inflammatory markers, including TNF-α, IL-6, and IL-17 [33,77,81,95]. These cytokines amplify oxidative stress in placental tissue, aggravating endothelial dysfunction and contributing to the most acute and clinically severe stages of the disease. However, interpreting these biomarkers in clinical practice remains challenging, as inflammation can also be influenced by maternal characteristics such as advanced age, prolonged interpregnancy interval, obesity, and comorbidities, including chronic hypertension, diabetes, renal disease, and autoimmune conditions (Figure 5).
Beyond heightened inflammation, preeclampsia is also characterized by impaired anti-inflammatory regulation [77]. A consistent finding across studies is the reduction in Tregs and decreased IL-10 production—both essential for maintaining immunological tolerance at the maternal–fetal interface. Several reports indicate that Treg cell depletion is more pronounced in early-onset, severe preeclampsia compared with late-onset or milder forms [82,86,113]. Reduced Treg function promotes increased trophoblast apoptosis, shallow trophoblast invasion, and inadequate spiral artery remodeling [53,86], highlighting a mechanistic link between immune dysregulation and the placental malperfusion that precedes clinical disease [77].
IL-10 plays a central role in this process. As a key anti-inflammatory cytokine that restrains Th1-driven inflammation and supports Treg differentiation, its reduction in preeclampsia further skews the maternal immune milieu toward a cytotoxic, proinflammatory state. This imbalance contributes to hypertension, endothelial activation, and ultimately impaired fetal growth [5,75]. Together, these findings underscore the importance of immune homeostasis for optimal pregnancy outcomes. Clinically, they highlight potential avenues for intervention: therapies that enhance Treg function, restore IL-10 signaling, or attenuate excessive cytokine activity could help rebalance inflammatory pathways and improve maternal and perinatal outcomes. Although still experimental, strategies such as Treg cell transfer, IL-10–based therapies, and selective immunomodulators hold promise and warrant further investigation in well-designed translational studies [77].
Strengthening the bridge between immunopathology and clinical care is essential. By identifying immune signatures that distinguish pathological inflammation from physiological adaptation, future diagnostic tools and targeted therapies may allow earlier detection, personalized risk stratification, and more effective prevention of the maternal and neonatal complications associated with preeclampsia.
7. Therapy Development
Therapeutic strategies for pregnancy-associated inflammatory disorders must achieve a delicate balance between modulating the maternal immune response and ensuring fetal safety. The primary objective is to attenuate excessive inflammation—implicated in adverse outcomes such as preeclampsia, FGR, and preterm birth—while preserving adequate host defense against infections. Current management combines evidence-based interventions from established clinical guidelines with emerging approaches that target specific inflammatory pathways (Table 1). Understanding how these interventions modulate the maternal–fetal immune landscape is essential for developing more precise and effective therapies.
7.1. Established Therapies
7.1.1. Low-Dose Aspirin
Low-dose aspirin (LDA), initiated before 16 weeks of gestation, reduces the incidence of preeclampsia and other placental-mediated complications. Its mechanism involves inhibition of platelet aggregation and modulation of inflammatory responses via selective cyclooxygenase-1 (COX-1) blockade [35,114]. Emerging clinical guidelines increasingly support the use of LDA in women identified as high risk for preeclampsia following first-trimester screening. Evidence from randomized trials and meta-analyses demonstrates that initiating LDA—ideally before 16 weeks—significantly reduces the risk of preeclampsia, particularly early-onset disease, and associated outcomes, including preterm birth and fetal growth restriction [35,114]. Screening-based strategies enable targeted treatment of women most likely to benefit, thereby optimizing the risk–benefit balance. Collectively, these data underpin current recommendations endorsing prophylactic LDA as a cornerstone of prevention in appropriately selected pregnancies.
7.1.2. Anticoagulants
Low molecular weight heparin (LMWH) is indicated in selected high-risk pregnancies, particularly in the context of thrombophilia or recurrent pregnancy loss. Beyond its anticoagulant properties, LMWH exhibits anti-inflammatory effects by modulating complement activation and leukocyte trafficking [85]. A growing body of evidence suggests that adjunctive LMWH may confer benefit in women at high risk of preeclampsia who do not have thrombophilia. In a meta-analysis of 10 randomized trials (1,758 participants), LMWH significantly reduced the incidence of preeclampsia, with risk reduction confined mainly to studies in which low-dose aspirin was the primary intervention. The combination of LMWH and LDA was also associated with fewer preterm births and fewer cases of fetal growth restriction, without affecting placental abruption rates. Overall, these findings support the selective use of LMWH as an adjunct to LDA in carefully defined high-risk populations [115].
7.1.3. Corticosteroids
Antenatal corticosteroids remain a cornerstone when preterm delivery is anticipated in pregnancies complicated by fetal growth restriction or preeclampsia, primarily because they accelerate fetal lung maturation and reduce neonatal respiratory distress, intraventricular hemorrhage, and death [116,117]. Although they do not directly treat placental insufficiency or maternal disease, their use can safely extend the window for optimizing delivery timing and adjunctive care. In these high-risk contexts, clinicians must balance benefits against potential maternal effects—including transient hyperglycemia, fluid retention, and a small increase in infectious risk—particularly in women with diabetes or severe hypertension. Overall, a single standard course before early preterm birth is strongly supported, while repeat dosing requires individualized consideration [116,117].
These established therapies delineate the current therapeutic landscape—effective in specific contexts yet fundamentally constrained by the need to safeguard the maternal–fetal unit. This limitation underscores an urgent scientific priority: to design next-generation immunomodulatory approaches that selectively suppress pathological inflammation while preserving fetal development. As insights into placental immunobiology expand, the development of truly targeted, precision therapies in pregnancy are shifting from possibility to near inevitability.
7.2. Adjunctive and Supportive Interventions
7.2.1. Antioxidants and Nutritional Modulation
Oxidative stress is a key contributor to placental inflammation [118,119]. Although antioxidants such as vitamins C and E have been extensively evaluated, large clinical trials have not demonstrated consistent benefit in preventing preeclampsia [81]. Melatonin, with potent antioxidant and vasoprotective properties, has shown promising results in small-scale studies [120,121], suggesting a potential role for targeted antioxidant therapy. Experimental data demonstrate that melatonin reduces placental oxidative stress and enhances antioxidant defenses without altering key anti-angiogenic mediators, and that it partially protects endothelial function in vitro. In a phase I clinical study involving women with established preeclampsia, melatonin administration was safe for both mother and fetus [120]. It was associated with a meaningful prolongation of pregnancy, along with reduced requirements for escalation of antihypertensive therapy [120]. Although significant biochemical markers of disease severity remained unchanged, these findings suggest that melatonin may attenuate maternal endothelial injury and help stabilize the disease long enough to improve perinatal outcomes, warranting larger confirmatory trials.
Recent high-quality evidence challenges earlier assumptions regarding routine calcium supplementation for the prevention of preeclampsia [122]. Across large, prospectively registered randomized trials, calcium showed little to no effect on preeclampsia, preterm birth, or major maternal and perinatal outcomes, regardless of baseline intake or dose. Overall, current data suggest that dietary calcium alone is unlikely to modify preeclampsia risk meaningfully and should not replace proven preventive strategies in high-risk pregnancies [122].
7.2.3. Lifestyle and Microbiome Modulation
Optimizing maternal diet, engaging in regular moderate physical activity, and using probiotic supplementation are under investigation as strategies to modulate systemic inflammation and enhance immune tolerance during pregnancy [36,123]. Lifestyle and microbiome modulation are emerging areas of interest in strategies to reduce the risk of preeclampsia, although the evidence remains preliminary. Observational and mechanistic studies suggest that healthy dietary patterns, appropriate weight gain, physical activity, and smoking avoidance may attenuate systemic inflammation, improve metabolic function, and support vascular health—pathways central to preeclampsia [124]. Parallel research indicates that alterations in the gut, vaginal, and placental microbiomes may influence immune tolerance, endothelial function, and oxidative stress, raising the possibility that probiotics, prebiotics, or diet-driven microbial shifts could play a preventive role. However, high-quality randomized trials are scarce, and standardized interventions have not yet been defined. For now, lifestyle optimization should be encouraged as part of comprehensive antenatal care, while microbiome-targeted therapies remain promising but investigational.
7.3. Emerging and Experimental Therapies
7.3.1. Cytokine-Targeted Biologics
Monoclonal antibodies that neutralize pro-inflammatory cytokines (e.g., anti–TNF-α, anti–IL-6) have been successful in treating autoimmune diseases and are now under consideration for the treatment of pregnancy-related inflammatory disorders [125]. Their capacity to exert highly specific immunomodulatory effects makes them an appealing, although still experimental, therapeutic avenue. Experimental models suggest these cytokines contribute to endothelial dysfunction, placental malperfusion, and systemic oxidative stress, and targeted blockade can partially restore vascular homeostasis. Clinically, experience in pregnant patients largely comes from women treated for autoimmune diseases, where selected agents (e.g., certain anti–TNF drugs) have shown acceptable safety profiles when carefully monitored; however, robust randomized data specific to hypertensive or placental disorders are lacking. At present, cytokine-neutralizing antibodies remain investigational for pregnancy-specific indications, and their future role will depend on trials that balance maternal benefit with fetal immune and developmental safety.
7.3.2. Inflammasome Inhibitors
Given the pivotal role of NLRP3 inflammasome activation in placental inflammation and FGR, small-molecule inhibitors such as MCC950 are being tested in preclinical models [126]. By targeting upstream inflammatory signaling, these agents may help mitigate placental injury. Inflammasome inhibitors, particularly those targeting NLRP3, offer a novel approach for pregnancy disorders in which excessive IL-1β and IL-18 signaling drives endothelial and placental injury, such as preeclampsia and fetal growth restriction [127]. Preclinical models suggest that pharmacologic blockade of inflammasome activation can attenuate oxidative stress, vascular dysfunction, and placental inflammation, thereby improving maternal hemodynamics and fetal growth [127]. However, clinical experience in pregnancy is virtually absent, and concerns remain regarding host defense and fetal immune development. At present, inflammasome inhibition is a promising but purely experimental strategy that requires rigorous safety and efficacy trials before translation to obstetric care.
7.3.3. Cell-Based Therapies
Mesenchymal stem cell (MSC) therapy represents a novel approach aimed at promoting immune tolerance and facilitating tissue repair at the maternal–fetal interface [128]. Early data indicate that MSCs possess strong immunomodulatory and regenerative potential, positioning them as a promising frontier in perinatal therapeutics. Cell-based therapies are emerging as an innovative strategy for preeclampsia, aiming to repair rather than merely temporize placental and endothelial injury [128]. Mesenchymal stromal cells, endothelial progenitor cells, and regulatory T-cell–based approaches have been shown in preclinical models to improve uteroplacental perfusion, dampen inflammation, and restore endothelial function, primarily through paracrine and immunomodulatory mechanisms. Early-phase studies outside pregnancy suggest acceptable safety profiles for some platforms, but robust clinical data in pregnant populations are lacking. For now, cell-based therapies remain experimental, offering a compelling future avenue for disease modification in severe or early-onset preeclampsia [128].
7.3.4. Safety Considerations
The administration of anti-inflammatory agents during pregnancy requires rigorous assessment of their teratogenic potential, effects on fetal immune development, and potential long-term effects on offspring. Current guidelines advocate for individualized risk–benefit evaluation and preferential use of therapies with well-established safety profiles [129,130].
7.4. Future Therapies
While current and emerging therapeutic approaches offer promising avenues for managing pregnancy-associated inflammatory disorders, ongoing research is advancing toward even more targeted and biologically precise interventions. Among these, exosome-based strategies are among the most innovative and rapidly evolving frontiers.
8. Exosomal Signaling at the Maternal–Fetal Interface: Orchestrating Immune and Vascular Adaptation
Exosomes are nanoscale extracellular vesicles that transport proteins, DNA, mRNA, and microRNAs (miRNAs) and function as key mediators of intercellular communication across multiple tissues [131]. They are secreted by diverse cell types—including trophoblasts, endothelial cells, and immune cells—and participate actively in both physiological and pathological immune modulation [82,131,132,133]. Through selective cargo delivery, exosomes influence inflammatory pathways by transporting miRNAs that regulate interferons (IFN-α/γ), TNF-α, and various interleukins central to immune activation [134].
During pregnancy, placental exosomes contribute significantly to homeostasis, inflammation control, and maternal–fetal tolerance [133,135,136,137]. Specific miRNAs such as miR-146a and miR-21 play central roles in regulating immune responses, controlling macrophage activity, and modulating hypoxia-driven inflammation [82,138,139]. Placental exosomes also help maintain tolerance to the semi-allogeneic fetus by shaping maternal immune cell phenotypes and restraining cytotoxic responses that might otherwise threaten placental integrity [138].
Alterations in exosomal composition have been closely linked to complications such as preeclampsia. Several studies have demonstrated that preeclamptic placentas release exosomes enriched with inflammatory and antiangiogenic cargo—including dysregulated miRNAs and increased sFlt-1—which antagonizes PlGF and VEGF, contributing to impaired trophoblast invasion and endothelial dysfunction [140]. These findings position exosomes as both mechanistic contributors and potential early biomarkers of disease [141]. Given their profound immunomodulatory properties, exosomes have emerged as promising candidates for next-generation therapies. Preclinical models indicate that mesenchymal stromal cell-derived extracellular vesicles can prevent preeclampsia-like physiology by restoring uterine immune balance, reducing inflammation, and improving placental vascular remodeling [142]. Such evidence suggests that exosome-based interventions may one day enable targeted, noninvasive modulation of the uteroplacental environment to prevent hypertensive disorders and their associated maternal–fetal complications.
Taken together, these insights highlight exosomes as a compelling therapeutic and diagnostic frontier in reproductive immunology. Their dual potential—as biomarkers and as immunomodulatory agents—suggests that they may play a transformative role in the future management of inflammation-mediated pregnancy disorders [139,141]. Collectively, these adjunctive and experimental interventions reflect a rapidly advancing field that seeks to refine immune modulation without compromising fetal development. As understanding of placental immunobiology deepens, the transition from broadly acting therapies to targeted, mechanism-based strategies is becoming increasingly feasible, offering a path toward safer and more effective treatments for pregnancy-associated inflammatory disorders.
9. Future Perspectives
Advances in immunology and molecular biology are rapidly reshaping our understanding of pregnancy-related inflammatory disorders. In the next decade, integration of multi-omics approaches—including transcriptomics, proteomics, and metabolomics—may allow the identification of precise molecular signatures for early risk stratification and targeted intervention. Artificial intelligence-driven predictive models could enable real-time clinical decision support by combining biomarker data with maternal and fetal parameters [102]. Furthermore, global health strategies must address disparities in access to diagnostic tools and therapeutics, ensuring that innovations benefit populations in low- and middle-income countries where the burden of preeclampsia, FGR, and preterm birth remains highest. Collaborative translational research networks will be essential to accelerate the validation of novel therapies, clarify their safety profiles, and implement them within equitable, sustainable healthcare frameworks.
10. Conclusions
Pregnancy-associated inflammatory disorders represent a significant challenge in maternal–fetal medicine due to their complex pathophysiology, heterogeneous clinical presentations, and potential for long-term consequences in both mother and child. Evidence reviewed here highlights the central role of immune and inflammatory mechanisms in conditions such as preeclampsia and FGR and underscores the placenta as a pivotal site for immune regulation. Advances in our understanding of the maternal–fetal immune interface have opened new opportunities for targeted interventions, ranging from established pharmacologic strategies to emerging biologic and cell-based therapies. Future research should focus on refining diagnostic biomarkers, elucidating precise inflammatory pathways, and ensuring the safety and efficacy of novel interventions, to improve pregnancy outcomes and reduce intergenerational health risks.
Author Contributions
Natalia Maestre conceived the initial outline, conducted the literature search, synthesized the evidence, and drafted the first version of the manuscript. Roberto Zapata contributed to data interpretation, critically revised the manuscript for clinical relevance, and assisted in structuring the figures and tables. Mariana Devia supported literature screening, prepared summary sections, and contributed to manuscript editing. Margarita M. Ochoa-Díaz provided conceptual input on immunological mechanisms, reviewed all drafts, and refined the discussion. Walter Anichiarico contributed to methodological oversight, verified sources, and critically reviewed the final version for accuracy and coherence. Jezid Miranda (senior author) supervised the project, contributed to conceptual design, integrated clinical and translational perspectives, provided major revisions across multiple drafts, and approved the final manuscript for submission. All authors read and approved the final version of the manuscript.
Funding
No specific funding was received for this work.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Abbreviations
| FGR | Fetal growth restriction |
| Th1 | T-helper 1 cells |
| Th2 | T-helper 2 cells |
| HDP | Hypertensive disorders of pregnancy |
| uNK | Uterine natural killer cells |
| HLA-G | Human Leukocyte Antigen-G |
| PD-L1 | Programmed Death-Ligand 1 |
| sFlt-1 | soluble fms-like tyrosine kinase-1 |
| NLRP3 | NOD-like receptor family, pyrin domain containing 3 |
| IL | Interleukin |
| TNF-α | Tumor necrosis factor-α |
| dNK | Decidual natural killer cells |
| CD56 | Cluster of Differentiation 56 |
| IP-10 | Interferon-inducible protein-10 |
| VEGF | Vascular endothelial growth factor |
| Tregs | Regulatory T cells |
| TGF-β | Transforming Growth Factor-Beta |
| FasL | Placental Fas ligand |
| TLRs | Toll-like receptors |
| PTB | Preterm birth |
| LBW | Low birth weight |
| MCP-1 | Monocyte chemotactic protein-1 |
| PlGF | Placental growth factor |
| sEng | Soluble endoglin |
| MHC | Histocompatibility complex |
| eNOS | Endothelial nitric oxide synthase |
| NO | Nitric oxide |
| MAP | Mean arterial pressure |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| NF-κB | Nuclear factor kappa B |
| LDA | Low-dose aspirin |
| COX-1 | Cyclooxygenase-1 |
| LMWH | Low molecular weight heparin |
| MSC | Mesenchymal stem cell |
| miRNAs | microRNAs |
References
- Dekel, N.; Gnainsky, Y.; Granot, I.; Racicot, K.; Mor, G. The role of inflammation for a successful implantation. Am J Reprod Immunol N Y N 1989 2014, 72, 141–7. [Google Scholar] [CrossRef]
- Dimitriadis, E.; Rolnik, D.L.; Zhou, W.; Estrada-Gutierrez, G.; Koga, K.; Francisco, R.P.V.; et al. Pre-eclampsia. Nat Rev Dis Primer 2023, 9, 8. [Google Scholar] [CrossRef]
- Hanna, J; Goldman-Wohl, D; Hamani, Y; Avraham, I; Greenfield, C; Natanson-Yaron, S; et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med. 2006, 12, 1065–74. [Google Scholar] [CrossRef]
- Mor, G; Aldo, P; Alvero, AB. The unique immunological and microbial aspects of pregnancy. Nat Rev Immunol. 2017, 17, 469–82. [Google Scholar] [CrossRef]
- Redman, CWG; Sargent, IL. Placental Stress and Pre-eclampsia: A Revised View. Placenta 2009, 30, 38–42. [Google Scholar] [CrossRef]
- Burton, GJ; Jauniaux, E. Pathophysiology of placental-derived fetal growth restriction. Am J Obstet Gynecol. 2018, 218, S745–61. [Google Scholar] [CrossRef]
- Staff, AC. The two-stage placental model of preeclampsia: An update. J Reprod Immunol 2019, 134-135, 1–10. [Google Scholar] [CrossRef]
- UNFPA WBG; WHO; UNICEF. Trends in Maternal Mortality: 1990 to 2015; WHO Geneva, 2015. [Google Scholar]
- Abalos, E; Cuesta, C; Grosso, AL; Chou, D; Say, L. Global and regional estimates of preeclampsia and eclampsia: A systematic review. Eur J Obstet Gynecol Reprod Biol. 2013, 170, 1–7. [Google Scholar] [CrossRef]
- World Health Organization. Maternal mortality: key facts [Internet]; Geneva, Switzerland, 2023. [Google Scholar]
- World Health Organization (WHO). United Nations Children’s Fund (UNICEF). Maternal and perinatal death surveillance and response: materials to support implementation [Internet]; Who/Unicef, 2021. [Google Scholar]
- Burstein, R; Henry, NJ; Collison, ML; Marczak, LB; Sligar, A; Watson, S; et al. Mapping 123 million neonatal, infant and child deaths between 2000 and 2017. Nature 2019, 574, 353–8. [Google Scholar] [CrossRef]
- Roberts, JM; Hubel, CA. The two stage model of preeclampsia: variations on the theme. Placenta 2009, 30 Suppl A(Suppl A), S32–37. [Google Scholar] [CrossRef]
- Carr, H; Cnattingius, S; Granath, F; Ludvigsson, JF; Edstedt Bonamy, AK. Preterm Birth and Risk of Heart Failure Up to Early Adulthood. J Am Coll Cardiol. 2017, 69, 2634–42. [Google Scholar] [CrossRef]
- Crispi, F; Dominguez, C; Llurba, E; Martin-Gallan, P; Cabero, L; Gratacos, E. Placental angiogenic growth factors and uterine artery Doppler findings for characterization of different subsets in preeclampsia and in isolated intrauterine growth restriction. Am J Obstet Gynecol. 2006, 195, 201–7. [Google Scholar] [CrossRef]
- Karumanchi, SA. Angiogenic Factors in Preeclampsia: From Diagnosis to Therapy. Hypertension 2016, 67, 1072–9. [Google Scholar] [CrossRef]
- Llurba, E; Crispi, F; Verlohren, S. Update on the pathophysiological implications and clinical role of angiogenic factors in pregnancy. Fetal Diagn Ther. 2015, 37, 81–92. [Google Scholar] [CrossRef]
- Vento-Tormo, R; Efremova, M; Botting, RA; Turco, MY; Vento-Tormo, M; Meyer, KB; et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 2018, 563, 347–53. [Google Scholar] [CrossRef]
- Blois, SM; Ilarregui, JM; Tometten, M; Garcia, M; Orsal, AS; Cordo-Russo, R; et al. A pivotal role for galectin-1 in fetomaternal tolerance. Nat Med. 2007, 13, 1450–7. [Google Scholar] [CrossRef]
- Redman, CWG; Tannetta, DS; Dragovic, RA; Gardiner, C; Southcombe, JH; Collett, GP; et al. Review: Does size matter? Placental debris and the pathophysiology of pre-eclampsia. Placenta 2012. [Google Scholar] [CrossRef]
- Deer, E; Herrock, O; Campbell, N; Cornelius, D; Fitzgerald, S; Amaral, LM; et al. The role of immune cells and mediators in preeclampsia. Nat Rev Nephrol. 2023, 19, 257–70. [Google Scholar] [CrossRef]
- Hausvater, A; Giannone, T; Sandoval, YHG; Doonan, RJ; Antonopoulos, CN; Matsoukis, IL; et al. The association between preeclampsia and arterial stiffness. J Hypertens 2012. [Google Scholar] [CrossRef]
- Herraiz, I; Droge, LA; Gomez-Montes, E; Henrich, W; Galindo, A; Verlohren, S. Characterization of the soluble fms-like tyrosine kinase-1 to placental growth factor ratio in pregnancies complicated by fetal growth restriction. Obstet Gynecol. 2014, 124, 265–73. [Google Scholar] [CrossRef]
- Maynard, SE; Min, JY; Merchan, J; Lim, KH; Li, J; Mondal, S; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003, 111, 649–58. [Google Scholar] [CrossRef]
- Shibata, E; Rajakumar, A; Powers, RW; Larkin, RW; Gilmour, C; Bodnar, LM; et al. Soluble fms-like tyrosine kinase 1 is increased in preeclampsia but not in normotensive pregnancies with small-for-gestational-age neonates: relationship to circulating placental growth factor. J Clin Endocrinol Metab. 2005, 90, 4895–903. [Google Scholar] [CrossRef]
- Levine, RJ; Maynard, SE; Qian, C; Lim, KH; England, LJ; Yu, KF; et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004, 350, 672–83. [Google Scholar] [CrossRef]
- Moss, CG; Dilworth, MR; Harris, LK; Freeman, S; Heazell, AEP. Understanding a Potential Role for the NLRP3 Inflammasome in Placenta-Mediated Pregnancy Complications. Am J Reprod Immunol N Y N 1989 2025, 93, e70077. [Google Scholar] [CrossRef]
- Alfian, I; Chakraborty, A; Yong, HEJ; Saini, S; Lau, RWK; Kalionis, B; et al. The Placental NLRP3 Inflammasome and Its Downstream Targets, Caspase-1 and Interleukin-6, Are Increased in Human Fetal Growth Restriction: Implications for Aberrant Inflammation-Induced Trophoblast Dysfunction. Cells 2022, 11(9). [Google Scholar] [CrossRef]
- Garcia-Puente, LM; Fraile-Martinez, O; García-Montero, C; Bujan, J; De León-Luis, JA; Bravo, C; et al. Placentas from Women with Late-Onset Preeclampsia Exhibit Increased Expression of the NLRP3 Inflammasome Machinery. Biomolecules 2023, 13(11). [Google Scholar] [CrossRef]
- Menkhorst, E; Santos, LL; Zhou, W; Yang, G; Winship, AL; Rainczuk, KE; et al. IL11 activates the placental inflammasome to drive preeclampsia. Front Immunol. 2023, 14, 1175926. [Google Scholar] [CrossRef]
- Szarka, A; Rigó, JJ; Lázár, L; Beko, G; Molvarec, A. Circulating cytokines, chemokines and adhesion molecules in normal pregnancy and preeclampsia determined by multiplex suspension array. BMC Immunol. 2010, 11, 59. [Google Scholar] [CrossRef]
- Pinheiro, MB; Martins-Filho, OA; Mota, APL; Alpoim, PN; Godoi, LC; Silveira, ACO; et al. Severe preeclampsia goes along with a cytokine network disturbance towards a systemic inflammatory state. Cytokine 2013, 62, 165–73. [Google Scholar] [CrossRef]
- Tosun, M; Celik, H; Avci, B; Yavuz, E; Alper, T; Malatyalioğlu, E. Maternal and umbilical serum levels of interleukin-6, interleukin-8, and tumor necrosis factor-alpha in normal pregnancies and in pregnancies complicated by preeclampsia. J Matern-Fetal Neonatal Med Off J Eur Assoc Perinat Med Fed Asia Ocean Perinat Soc Int Soc Perinat Obstet 2010, 23, 880–6. [Google Scholar]
- Zheng, S; Xu, P; Shen, H; Shu, C. Immune dysregulation in preeclampsia: integrative analysis of peripheral transcriptomes and placental single-cell land-scapes. Front Immunol. 2025, 16, 1638603. [Google Scholar] [CrossRef]
- Rolnik, DL; Wright, D; Poon, LC; O’Gorman, N; Syngelaki, A; de Paco Matallana, C; et al. Aspirin versus Placebo in Pregnancies at High Risk for Preterm Preeclampsia. N Engl J Med. 2017, 377, 613–22. [Google Scholar] [CrossRef]
- García-Montero, C; Fraile-Martinez, O; De Leon-Oliva, D; Boaru, DL; Garcia-Puente, LM; De León-Luis, JA; et al. Exploring the Role of Mediterranean and Westernized Diets and Their Main Nutrients in the Modulation of Oxidative Stress in the Placenta: A Narrative Review. Antioxid Basel Switz 2023, 12(11). [Google Scholar] [CrossRef]
- Fernandes, KA; Lim, AI. Maternal-driven immune education in offspring. Immunol Rev. 2024, 323(1), 288–302. [Google Scholar] [CrossRef]
- Delaroque, C; Chassaing, B. Microbiome in heritage: how maternal microbiome transmission impacts next generation health. Microbiome 2025, 13(1), 196. [Google Scholar] [CrossRef]
- Andonotopo, W; Bachnas, MA; Dewantiningrum, J; Adi Pramono, MB; Sulistyowati, S; Hariyasa Sanjaya, IN; et al. Nutriepigenomics in perinatal medicine: maternal nutrition as a modulator of fetal gene expression and long-term health. J Perinat Med 2025. [Google Scholar] [CrossRef]
- Mangan, MSJ; Olhava, EJ; Roush, WR; Seidel, HM; Glick, GD; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018, 17, 688. [Google Scholar] [CrossRef]
- Mor, G; Cardenas, I. The immune system in pregnancy: a unique complexity. Am J Reprod Immunol N Y N 1989 2010, 63, 425–33. [Google Scholar]
- Racicot, K; Kwon, JY; Aldo, P; Silasi, M; Mor, G. Understanding the complexity of the immune system during pregnancy. Am J Reprod Immunol N Y N 1989 2014, 72, 107–16. [Google Scholar] [CrossRef]
- Riley, JK. Trophoblast immune receptors in maternal-fetal tolerance. Immunol Invest. 2008, 37(5), 395–426. [Google Scholar] [CrossRef]
- Huppertz, B; Ghosh, D; Sengupta, J. An integrative view on the physiology of human early placental villi. Prog Biophys Mol Biol. 2014, 114, 33–48. [Google Scholar] [CrossRef]
- Burton, GJ; Woods, AW; Jauniaux, E; Kingdom, JCP. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta 2009, 30, 473–82. [Google Scholar] [CrossRef]
- Iwasaki, A; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015, 16(4), 343–53. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–35. [Google Scholar] [CrossRef]
- Levenson, D; Romero, R; Miller, D; Galaz, J; Garcia-Flores, V; Neshek, B; et al. The maternal-fetal interface at single-cell resolution: uncovering the cellular anatomy of the placenta and decidua. Am J Obstet Gynecol. 2025, 232, S55–79. [Google Scholar] [CrossRef]
- Moffett, A; Colucci, F. Uterine NK cells: active regulators at the maternal-fetal interface. J Clin Invest. 2014, 124, 1872–9. [Google Scholar] [CrossRef]
- Alippe, Y; Hatterschide, J; Coyne, CB; Diamond, MS. Innate immune responses to pathogens at the maternal-fetal interface. Nat Rev Immunol. 2025, 25, 869–84. [Google Scholar] [CrossRef]
- Svensson-Arvelund, J; Ernerudh, J. The Role of Macrophages in Promoting and Maintaining Homeostasis at the Fetal-Maternal Interface. Am J Reprod Immunol N Y N 1989 2015, 74, 100–9. [Google Scholar] [CrossRef]
- Houser, BL. Decidual macrophages and their roles at the maternal-fetal interface. Yale J Biol Med. 2012, 85, 105–18. [Google Scholar]
- Aluvihare, VR; Kallikourdis, M; Betz, AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat Immunol. 2004, 5, 266–71. [Google Scholar] [CrossRef]
- Robertson, SA; Care, AS; Moldenhauer, LM. Regulatory T cells in embryo implantation and the immune response to pregnancy. J Clin Invest. 2018, 128, 4224–35. [Google Scholar] [CrossRef]
- Tilburgs, T; Scherjon, SA; van der Mast, BJ; Haasnoot, GW; Versteeg-V D Voort-Maarschalk, M; Roelen, DL; et al. Fetal-maternal HLA-C mismatch is associated with decidual T cell activation and induction of functional T regulatory cells. J Reprod Immunol. 2009, 82, 148–57. [Google Scholar] [CrossRef]
- Saito, S; Nakashima, A. Review: The role of autophagy in extravillous trophoblast function under hypoxia. Placenta 2013, 34 Suppl, S79–84. [Google Scholar] [CrossRef]
- Abrahams, VM; Visintin, I; Aldo, PB; Guller, S; Romero, R; Mor, G. A role for TLRs in the regulation of immune cell migration by first trimester trophoblast cells. J Immunol Baltim Md 1950 2005, 175, 8096–104. [Google Scholar] [CrossRef]
- Palmeira, P; Quinello, C; Silveira-Lessa, AL; Zago, CA; Carneiro-Sampaio, M. IgG placental transfer in healthy and pathological pregnancies. Clin Dev Immunol. 2012, 2012, 985646. [Google Scholar] [CrossRef]
- Gomez-Lopez, N; Romero, R; Xu, Y; Miller, D; Leng, Y; Panaitescu, B; et al. The immunophenotype of amniotic fluid leukocytes in normal and complicated pregnancies. Am J Reprod Immunol. 2018, 79, e12827. [Google Scholar] [CrossRef]
- Frishman, S; Nuriel-Ohayon, M; Turjeman, S; Pinto, Y; Yariv, O; Tenenbaum-Gavish, K; et al. Positive effects of diet-induced microbiome modification on GDM in mice following human faecal transfer. Gut 2024, 73, e17. [Google Scholar] [CrossRef]
- Koren, O; Konnikova, L; Brodin, P; Mysorekar, IU; Collado, MC. The maternal gut microbiome in pregnancy: implications for the developing immune system. Nat Rev Gastroenterol Hepatol 2024, 21, 35–45. [Google Scholar] [CrossRef]
- Fasano, A; Chassaing, B; Haller, D; Flores Ventura, E; Carmen-Collado, M; Pastor, N; et al. Microbiota during pregnancy and early life: role in maternal-neonatal outcomes based on human evidence. Gut Microbes 2024, 16, 2392009. [Google Scholar] [CrossRef]
- Pinto, Y; Frishman, S; Turjeman, S; Eshel, A; Nuriel-Ohayon, M; Shrossel, O; et al. Gestational diabetes is driven by microbiota-induced inflammation months before diagnosis. Gut 2023, 72, 918–28. [Google Scholar] [CrossRef]
- Huang, L; Cai, M; Li, L; Zhang, X; Xu, Y; Xiao, J; et al. Gut microbiota changes in preeclampsia, abnormal placental growth and healthy pregnant women. BMC Microbiol. 2021, 21, 265. [Google Scholar] [CrossRef]
- Aagaard, K; Ma, J; Antony, KM; Ganu, R; Petrosino, J; Versalovic, J. The placenta harbors a unique microbiome. Sci Transl Med. 2014, 6, 237ra65. [Google Scholar] [CrossRef]
- Panzer, JJ; Romero, R; Greenberg, JM; Winters, AD; Galaz, J; Gomez-Lopez, N; et al. Is there a placental microbiota? A critical review and re-analysis of published placental microbiota datasets. BMC Microbiol 2023, 23, 76. [Google Scholar] [CrossRef]
- Saadaoui, M; Djekidel, MN; Murugesan, S; Kumar, M; Elhag, D; Singh, P; et al. Exploring the composition of placental microbiome and its potential origin in preterm birth. Front Cell Infect Microbiol 2024, 14, 1486409. [Google Scholar] [CrossRef]
- de Goffau, MC; Lager, S; Sovio, U; Gaccioli, F; Cook, E; Peacock, SJ; et al. Human placenta has no microbiome but can contain potential pathogens. Nature 2019, 572, 329–34. [Google Scholar] [CrossRef]
- Arora, N; Sadovsky, Y; Dermody, TS; Coyne, CB. Microbial Vertical Transmission during Human Pregnancy. Cell Host Microbe 2017, 21, 561–7. [Google Scholar] [CrossRef]
- Romero, R; Hassan, SS; Gajer, P; Tarca, AL; Fadrosh, DW; Bieda, J; et al. The vaginal microbiota of pregnant women who subsequently have spontaneous preterm labor and delivery and those with a normal delivery at term. Microbiome 2014, 2, 18. [Google Scholar] [CrossRef]
- Zakis, DR; Paulissen, E; Kornete, L; Kaan, AMM; Nicu, EA; Zaura, E. The evidence for placental microbiome and its composition in healthy pregnancies: A systematic review. J Reprod Immunol. 2022, 149, 103455. [Google Scholar] [CrossRef]
- Reijnders, IF; Mulders, AGMGJ; Koster, MPH. Placental development and function in women with a history of placenta-related complications: a systematic review. Acta Obstet Gynecol Scand. 2018, 97(3), 248–57. [Google Scholar] [CrossRef]
- Sargent, IL; Borzychowski, AM; Redman, CWG. Immunoregulation in normal pregnancy and pre-eclampsia: an overview. Reprod Biomed Online 2006, 13, 680–6. [Google Scholar] [CrossRef]
- Tilburgs, T; Roelen, DL; van der Mast, BJ; van Schip, JJ; Kleijburg, C; de Groot-Swings, GM; et al. Differential distribution of CD4(+)CD25(bright) and CD8(+)CD28(-) T-cells in decidua and maternal blood during human pregnancy. Placenta 2006, 27, S47–53. [Google Scholar] [CrossRef]
- Redman, CWG; Sargent, IL. Preeclampsia and the systemic inflammatory response. Semin Nephrol. 2004, 24, 565–70. [Google Scholar] [CrossRef]
- Redman, CW; Sargent, IL; Staff, AC. IFPA Senior Award Lecture: making sense of pre-eclampsia - two placental causes of preeclampsia? Placenta 2014, 35, S20–25. [Google Scholar] [CrossRef]
- Boulanger, H; Bounan, S; Mahdhi, A; Drouin, D; Ahriz-Saksi, S; Guimiot, F; et al. Immunologic aspects of preeclampsia. AJOG Glob Rep. 2024, 4, 100321. [Google Scholar] [CrossRef]
- Deer, E; Vaka, VR; McMaster, KM; Wallace, K; Cornelius, DC; Amaral, LM; et al. Vascular endothelial mitochondrial oxidative stress in response to preeclampsia: a role for angiotension II type 1 autoantibodies. Am J Obstet Gynecol. 2021, 3(1). [Google Scholar] [CrossRef]
- Brien, ME; Duval, C; Palacios, J; Boufaied, I; Hudon-Thibeault, AA; Nadeau-Vallée, M; et al. Uric Acid Crystals Induce Placental Inflammation and Alter Trophoblast Function via an IL-1-Dependent Pathway: Implications for Fetal Growth Restriction. J Immunol Baltim Md 1950 2017, 198, 443–51. [Google Scholar] [CrossRef]
- Matzinger, P. The danger model: a renewed sense of self. Science 2002, 296, 301–5. [Google Scholar] [CrossRef]
- Sacks, GP; Studena, K; Sargent, K; Redman, CW. Normal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsis. Am J Obstet Gynecol. 1998, 179, 80–6. [Google Scholar] [CrossRef]
- Salomon, C; Yee, SW; Mitchell, MD; Rice, GE. The Possible Role of Extravillous Trophoblast-Derived Exosomes on the Uterine Spiral Arterial Remodeling under Both Normal and Pathological Conditions. BioMed Res Int. 2014, 2014. [Google Scholar] [CrossRef]
- Romero, R; Nien, JK; Espinoza, J; Todem, D; Fu, W; Chung, H; et al. A longitudinal study of angiogenic (placental growth factor) and anti-angiogenic (soluble endoglin and soluble vascular endothelial growth factor receptor-1) factors in normal pregnancy and patients destined to develop preeclampsia and deliver a small fo. J Matern-Fetal Neonatal Med. 2008, 21, 9–23. [Google Scholar] [CrossRef]
- Levine, RJ; Lam, C; Qian, C; Yu, KF; Maynard, SE; Sachs, BP; et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006, 355, 992–1005. [Google Scholar] [CrossRef]
- American College of Obstetricians Gynecologists’ Committee. Gestational Hypertension and Preeclampsia. Obstet Gynecol. 2020, 135, e237-60. [Google Scholar]
- La Rocca, C; Carbone, F; Longobardi, S; Matarese, G. The immunology of pregnancy: Regulatory T cells control maternal immune tolerance toward the fetus. Immunol Lett. 2014, 162, 41–8. [Google Scholar] [CrossRef]
- Dhillion, P; Wallace, K; Herse, F; Scott, J; Wallukat, G; Heath, J; et al. IL-17-mediated oxidative stress is an important stimulator of AT1-AA and hypertension during pregnancy. Am J Physiol Regul Integr Comp Physiol. 2012, 303, R353–358. [Google Scholar] [CrossRef]
- Geldenhuys, J; Rossouw, TM; Lombaard, HA; Ehlers, MM; Kock, MM. Disruption in the Regulation of Immune Responses in the Placental Subtype of Preeclampsia. Front Immunol. 2018, 9, 1659. [Google Scholar] [CrossRef]
- Kharfi, A; Giguère, Y; Sapin, V; Massé, J; Dastugue, B; Forest, JC. Trophoblastic remodeling in normal and preeclamptic pregnancies: implication of cytokines. Clin Biochem. 2003, 36, 323–31. [Google Scholar] [CrossRef]
- Lyall, F; Robson, SC; Bulmer, JN. Spiral artery remodeling and trophoblast invasion in preeclampsia and fetal growth restriction: relationship to clinical outcome. Hypertens. 2013, 62, 1046–54. [Google Scholar] [CrossRef]
- Madazli, R; Aydin, S; Uludag, S; Vildan, O; Tolun, N. Maternal plasma levels of cytokines in normal and preeclamptic pregnancies and their relationship with diastolic blood pressure and fibronectin levels. Acta Obstet Gynecol Scand. 2003, 82, 797–802. [Google Scholar] [CrossRef]
- Aggarwal, R; Jain, AK; Mittal, P; Kohli, M; Jawanjal, P; Rath, G. Association of pro- and anti-inflammatory cytokines in preeclampsia. J Clin Lab Anal. 2019, 33, e22834. [Google Scholar] [CrossRef]
- LaMarca, B; Speed, J; Fournier, L; Babcock, SA; Berry, H; Cockrell, K; et al. Hypertension in Response to Chronic Reductions in Uterine Perfusion in Pregnant Rats. Hypertension 2008, 52, 1161–7. [Google Scholar] [CrossRef]
- Gadonski, G; LaMarca, BBD; Sullivan, E; Bennett, W; Chandler, D; Granger, JP. Hypertension Produced by Reductions in Uterine Perfusion in the Pregnant Rat. Hypertension 2006, 48, 711–6. [Google Scholar] [CrossRef]
- Dhillion, P; Wallace, K; Herse, F; Scott, J; Wallukat, G; Heath, J; et al. IL-17-mediated oxidative stress is an important stimulator of AT1-AA and hypertension during pregnancy. Am J Physiol-Regul Integr Comp Physiol. 2012, 303, R353–8. [Google Scholar] [CrossRef]
- Crispi, F; Crovetto, F; Gratacos, E. Intrauterine growth restriction and later cardiovascular function. Early Hum Dev. 2018, 126, 23–7. [Google Scholar] [CrossRef]
- Figueras, F; Gratacos, E. Stage-based approach to the management of fetal growth restriction. Prenat Diagn 2014, 34, 655–9. [Google Scholar] [CrossRef]
- Crispi, F; Figueras, F; Cruz-Lemini, M; Bartrons, J; Bijnens, B; Gratacos, E. Cardiovascular programming in children born small for gestational age and relationship with prenatal signs of severity. Am J Obstet Gynecol. 2012, 207, e1-9. [Google Scholar] [CrossRef]
- Crispi, F; Bijnens, B; Figueras, F; Bartrons, J; Eixarch, E; Le Noble, F; et al. Fetal growth restriction results in remodeled and less efficient hearts in children. Circulation 2010, 121, 2427–36. [Google Scholar] [CrossRef]
- Crispi, F; Miranda, J; Gratacós, E. Long-term cardiovascular consequences of fetal growth restriction: biology, clinical implications, and opportunities for prevention of adult disease. Am J Obstet Gynecol. 2018, 218, S869–79. [Google Scholar] [CrossRef]
- Lees, CC; Stampalija, T; Baschat, AA; da Silva Costa, F; Ferrazzi, E; Figueras, F; et al. ISUOG Practice Guidelines: diagnosis and management of small-for-gestational-age fetus and fetal growth restriction. Ultrasound Obstet Gynecol. 2020, 56, 298–312. [Google Scholar] [CrossRef]
- Miranda, J; Paules, C; Noell, G; Youssef, L; Paternina-Caicedo, A; Crovetto, F; et al. Similarity network fusion to identify phenotypes of small-for-gestational-age fetuses. iScience 2023, 26, 107620. [Google Scholar] [CrossRef]
- Zarate, MA; De Dios, RK; Balasubramaniyan, D; Zheng, L; Sherlock, LG; Rozance, PJ; et al. The Acute Hepatic NF-κB-Mediated Proinflammatory Response to Endotoxemia Is Attenuated in Intrauterine Growth-Restricted Newborn Mice. Front Immunol. 2021, 12, 706774. [Google Scholar] [CrossRef]
- Arcangeli, T; Thilaganathan, B; Hooper, R; Khan, KS; Bhide, A. Neurodevelopmental delay in small babies at term: a systematic review. Ultrasound Obstet Gynecol. 2012, 40, 267–75. [Google Scholar] [CrossRef]
- Baschat, AA. Neurodevelopment after fetal growth restriction. Fetal Diagn Ther. 2014, 36, 136–42. [Google Scholar] [CrossRef]
- Eixarch, E; Meler, E; Iraola, A; Illa, M; Crispi, F; Hernandez-Andrade, E; et al. Neurodevelopmental outcome in 2-year-old infants who were small-for-gestational age term fetuses with cerebral blood flow redistribution. Ultrasound Obstet Gynecol. 2008, 32, 894–9. [Google Scholar] [CrossRef]
- Azizieh, F; Dingle, K; Raghupathy, R; Johnson, K; VanderPlas, J; Ansari, A. Multivariate analysis of cytokine profiles in pregnancy complications. Am J Reprod Immunol. 2018, 79, e12818. [Google Scholar] [CrossRef]
- Salafia, CM; Minior, VK; Pezzullo, JC; Popek, EJ; Rosenkrantz, TS; Vintzileos, AM. Intrauterine growth restriction in infants of less than thirty-two weeks’ gestation: Associated placental pathologic features. Am J Obstet Gynecol. 1995, 173, 1049–57. [Google Scholar] [CrossRef]
- Salafia, CM; Rukat, C; Dygulska, B; Miller, RK; Misra, DP. Placental chronic inflammatory histopathology and fetal growth in a cohort with universal placental examination. Placenta 2024, 154, 193–200. [Google Scholar] [CrossRef]
- Tan, CX; Yeoh, HX; Tazilan, NA; Tan, JW; Alfian, N; Zakaria, H; et al. HIF-1A Expression in Placenta of Pregnancies Complicated with Preeclampsia and Fetal Growth Restriction. Diagnostics 2025, 15, 1843. [Google Scholar] [CrossRef]
- Ordóñez-Díaz, MD; Gil-Campos, M; Flores-Rojas, K; Muñoz-Villanueva, MC; Aguilera-García, CM; Torre-Aguilar, MJ; et al. Plasma Adipokines Profile in Prepubertal Children with a History of Prematurity or Extrauterine Growth Restriction. Nutrients 2020, 12, 1201. [Google Scholar] [CrossRef]
- Zarate, MA; De Dios, RK; Balasubramaniyan, D; Zheng, L; Sherlock, LG; Rozance, PJ; et al. The Acute Hepatic NF-κB-Mediated Proinflammatory Response to Endotoxemia Is Attenuated in Intrauterine Growth-Restricted Newborn Mice. Front Immunol. 2021, 12, 706774. [Google Scholar] [CrossRef]
- Sasaki, Y; Darmochwal-Kolarz, D; Suzuki, D; Sakai, M; Ito, M; Shima, T; et al. Proportion of peripheral blood and decidual CD4+ CD25bright regulatory T cells in pre-eclampsia. Clin Exp Immunol. 2007, 149, 139–45. [Google Scholar] [CrossRef]
- ting, Xu T; Zhou, F; C yan, Deng; G qiong, Huang; ke, Li J; dong, Wang X. Low-Dose Aspirin for Preventing Preeclampsia and Its Complications: A Meta-Analysis. J Clin Hypertens. 2015, (20), 1–7. [Google Scholar]
- Chen, J; Huai, J; Yang, H. Low-molecular-weight heparin for the prevention of preeclampsia in high-risk pregnancies without thrombophilia: a systematic review and meta-analysis. BMC Pregnancy Childbirth 2024, 24, 68. [Google Scholar] [CrossRef]
- Martins, JG; Biggio, JR; Abuhamad, A. Society for Maternal-Fetal Medicine Consult Series #52: Diagnosis and management of fetal growth restriction: (Replaces Clinical Guideline Number 3, April 2012). Am J Obstet Gynecol. 2020, 223, B2–17. [Google Scholar]
- WHO recommendations on: Antenatal corticosteroids for improving preterm birth outcomes. Geneva, 2022 .
- Liabsuetrakul, T; Yamamoto, Y; Kongkamol, C; Ota, E; Mori, R; Noma, H. Medications for preventing hypertensive disorders in high-risk pregnant women: a systematic review and network meta-analysis. Syst Rev. 2022, 11, 135. [Google Scholar] [CrossRef]
- Alves, PRMM; Fragoso, MBT; Tenório, MCS; Bueno, NB; Goulart, MOF; Oliveira, ACM. The role played by oral antioxidant therapies in preventing and treating preeclampsia: An updated meta-analysis. Nutr Metab Cardiovasc Dis NMCD 2023, 33, 1277–92. [Google Scholar] [CrossRef]
- Hobson, SR; Gurusinghe, S; Lim, R; Alers, NO; Miller, SL; Kingdom, JC; et al. Melatonin improves endothelial function in vitro and prolongs pregnancy in women with early-onset preeclampsia. J Pineal Res. 2018, 65, e12508. [Google Scholar] [CrossRef]
- Dou, Y; Lin, B; Cheng, H; Wang, C; Zhao, M; Zhang, J; et al. The reduction of melatonin levels is associated with the development of preeclampsia: a meta-analysis. Hypertens Pregnancy 2019, 38, 65–72. [Google Scholar] [CrossRef]
- Cluver, CA; Rohwer, C; Rohwer, AC. Calcium supplementation during pregnancy for preventing hypertensive disorders and related problems. Cochrane Database Syst Rev. 2025, 12, CD001059. [Google Scholar]
- Gregorio, BM; Souza-Mello, V; Mandarim-de-Lacerda, CA; Aguila, MB. Maternal fish oil supplementation benefits programmed offspring from rat dams fed low-protein diet. Am J Obstet Gynecol. 2008, 199, 82.e1–7. [Google Scholar] [CrossRef]
- Crovetto, F; Crispi, F; Borras, R; Paules, C; Casas, R; Martín-Asuero, A; et al. Mediterranean diet, Mindfulness-Based Stress Reduction and usual care during pregnancy for reducing fetal growth restriction and adverse perinatal outcomes: IMPACT BCN (Improving Mothers for a better PrenAtal Care Trial BarCeloNa): a study protocol for a randomized controlled trial. Trials 2021, 22, 362. [Google Scholar]
- Zhou, J; Yan, P; Ma, W; Li, J. Cytokine modulation and immunoregulation of uterine NK cells in pregnancy disorders. Cytokine Growth Factor Rev. 2025, 81, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Santos, KN; Bizzotto, JQ; Bueno-Pereira, TO; Romao-Veiga, M; Ribeiro-Vasques, VR; Oliveira, LRC; et al. Preeclamptic plasma disrupts endothelial function and promotes inflammation in endothelial cells: beneficial effects of glibenclamide and MCC950 in this scenario. J Recept Signal Transduct Res. 2025, 45, 237–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, X; Travis, OK; Shields, CA; Tardo, GA; Giachelli, C; Nutter, CW; et al. NLRP3 inhibition improves maternal hypertension, inflammation, and vascular dysfunction in response to placental ischemia. Am J Physiol-Regul Integr Comp Physiol. 2023, 324, R556–67. [Google Scholar] [CrossRef] [PubMed]
- Grimes, S; Bombay, K; Lanes, A; Walker, M; Corsi, DJ. Potential biological therapies for severe preeclampsia: a systematic review and meta-analysis. BMC Pregnancy Childbirth 2019, 19, 163. [Google Scholar] [CrossRef]
- Magee, LA; Brown, MA; Hall, DR; Gupte, S; Hennessy, A; Karumanchi, SA; et al. The 2021 International Society for the Study of Hypertension in Pregnancy classification, diagnosis & management recommendations for international practice. Pregnancy Hypertens. 2022, 27, 148–69. [Google Scholar] [CrossRef]
- Magee, LA; Smith, GN; Bloch, C; Côté, AM; Jain, V; Nerenberg, K; et al. Guideline No. 426: Hypertensive Disorders of Pregnancy: Diagnosis, Prediction, Prevention, and Management. J Obstet Gynaecol Can JOGC J Obstet Gynecol Can JOGC 2022, 44, 547–571.e1. [Google Scholar] [CrossRef]
- Aryani, A; Denecke, B. Exosomes as a Nanodelivery System: a Key to the Future of Neuromedicine? Mol Neurobiol 2016, 53, 818–34. [Google Scholar] [CrossRef]
- An, T; Qin, S; Xu, Y; Tang, Y; Huang, Y; Situ, B; et al. Exosomes serve as tumour markers for personalized diagnostics owing to their important role in cancer metastasis. J Extracell Vesicles 2015, 4(1). [Google Scholar] [CrossRef]
- Atay, S; Gercel-Taylor, C; Kesimer, M; Taylor, DD. Morphologic and proteomic characterization of exosomes released by cultured extravillous trophoblast cells. Exp Cell Res. 2011, 317, 1192–202. [Google Scholar] [CrossRef]
- Colombo, M; Raposo, G; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu Rev Cell Dev Biol. 2014, 30, 255–89. [Google Scholar] [CrossRef]
- Bullerdiek, J; Flor, I. Exosome-delivered microRNAs of "chromosome 19 microRNA cluster" as immunomodulators in pregnancy and tumorigenesis. Mol Cytogenet. 2012, 5, 27. [Google Scholar] [CrossRef]
- Holder, B; Jones, T; Sancho Shimizu, V; Rice, TF; Donaldson, B; Bouqueau, M; et al. Macrophage Exosomes Induce Placental Inflammatory Cytokines: A Novel Mode of Maternal-Placental Messaging. Traffic 2016, 17, 168–78. [Google Scholar] [CrossRef]
- Mincheva-Nilsson, L; Baranov, V. Placenta-derived exosomes and syncytiotrophoblast microparticles and their role in human reproduction: immune modulation for pregnancy success. Am J Reprod Immunol. 2014, 72, 440–57. [Google Scholar] [CrossRef]
- Salomon, C; Kobayashi, M; Ashman, K; Sobrevia, L; Mitchell, MD; Rice, GE. Hypoxia-induced changes in the bioactivity of cytotrophoblast-derived exosomes. PloS One 2013, 8, e79636. [Google Scholar] [CrossRef]
- Salomon, C; Guanzon, D; Scholz-Romero, K; Longo, S; Correa, P; Illanes, SE; et al. Placental Exosomes as Early Biomarker of Preeclampsia: Potential Role of Exosomal MicroRNAs Across Gestation. J Clin Endocrinol Metab. 2017, 102, 3182–94. [Google Scholar] [CrossRef]
- Lykoudi, A; Kolialexi, A; Lambrou, GI; Braoudaki, M; Siristatidis, C; Papaioanou, GK; et al. Dysregulated placental microRNAs in Early and Late onset Preeclampsia. Placenta 2018, 61, 24–32. [Google Scholar] [CrossRef]
- Miranda, J; Paules, C; Nair, S; Lai, A; Palma, C; Scholz-Romero, K; et al. Placental exosomes profile in maternal and fetal circulation in intrauterine growth restriction - Liquid biopsies to monitoring fetal growth. Placenta 2018, 64, 34–43. [Google Scholar] [CrossRef]
- Taglauer, ES; Fernandez-Gonzalez, A; Willis, GR; Reis, M; Yeung, V; Liu, X; et al. Mesenchymal stromal cell-derived extracellular vesicle therapy prevents preeclamptic physiology through intrauterine immunomodulation†. Biol Reprod. 2021, 104, 457–67. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Immune development of the placenta. Left panel: Within the maternal decidua, pregnancy-specific immune remodeling is illustrated by the distribution of decidual NK (dNK) cells, macrophages, and regulatory T cells (Tregs). dNK cells adopt a tolerant phenotype that promotes spiral artery remodeling; decidual macrophages show M1 (pro-inflammatory) and M2 (pro-repair/tolerogenic) states that dynamically shift across gestation; and Tregs suppress excessive maternal immune activation through IL-10/TGF-β, fostering vascular adaptation and placental growth. Right panel: Fetal trophoblast–derived tissues express a restricted repertoire of HLA molecules (e.g., HLA-G, HLA-C, HLA-E) that interact with inhibitory and activating receptors on maternal NK and T cells, modulating immune tolerance. Co-stimulatory and checkpoint pathways (including B7 family proteins) fine-tune maternal T-cell responses, together establishing a balanced immune environment that supports placental development while preserving maternal defense.
Figure 1.
Immune development of the placenta. Left panel: Within the maternal decidua, pregnancy-specific immune remodeling is illustrated by the distribution of decidual NK (dNK) cells, macrophages, and regulatory T cells (Tregs). dNK cells adopt a tolerant phenotype that promotes spiral artery remodeling; decidual macrophages show M1 (pro-inflammatory) and M2 (pro-repair/tolerogenic) states that dynamically shift across gestation; and Tregs suppress excessive maternal immune activation through IL-10/TGF-β, fostering vascular adaptation and placental growth. Right panel: Fetal trophoblast–derived tissues express a restricted repertoire of HLA molecules (e.g., HLA-G, HLA-C, HLA-E) that interact with inhibitory and activating receptors on maternal NK and T cells, modulating immune tolerance. Co-stimulatory and checkpoint pathways (including B7 family proteins) fine-tune maternal T-cell responses, together establishing a balanced immune environment that supports placental development while preserving maternal defense.
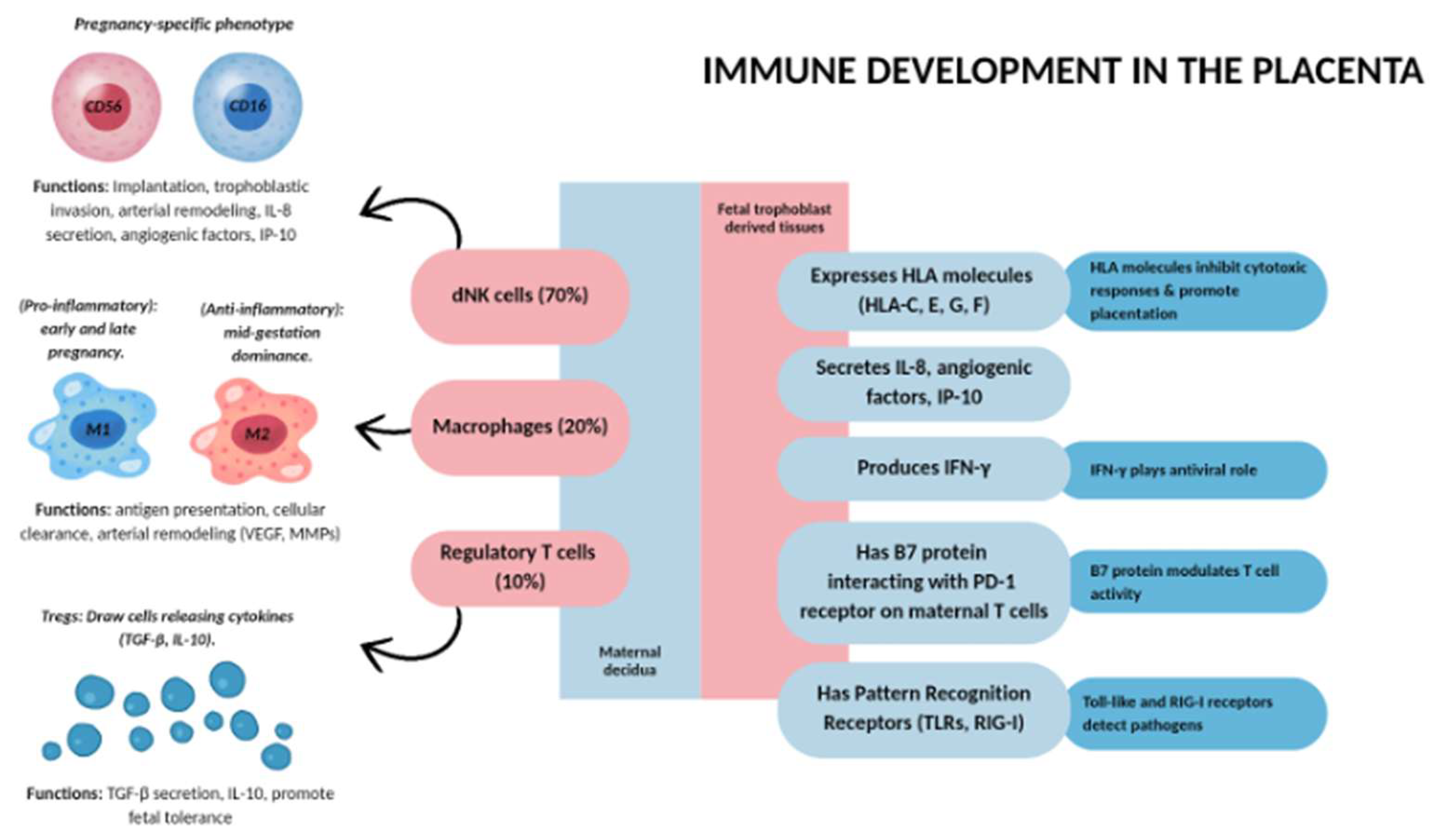
Figure 2.
Schematic overview of altered inflammation in preeclampsia. This diagram contrasts immune balance in normal pregnancy with the exaggerated inflammatory state characteristic of preeclampsia. Under physiologic conditions, a controlled, low-grade inflammatory milieu supports placental development and vascular adaptation. In preeclampsia, this equilibrium is disrupted: pro-inflammatory cytokines are elevated, anti-angiogenic factors (e.g., sFlt-1, sEng) increase, and maternal immune profiles shift toward heightened activation. These changes converge to drive endothelial dysfunction, oxidative stress, and impaired placental perfusion, culminating clinically in hypertension and multi-organ involvement.
Figure 2.
Schematic overview of altered inflammation in preeclampsia. This diagram contrasts immune balance in normal pregnancy with the exaggerated inflammatory state characteristic of preeclampsia. Under physiologic conditions, a controlled, low-grade inflammatory milieu supports placental development and vascular adaptation. In preeclampsia, this equilibrium is disrupted: pro-inflammatory cytokines are elevated, anti-angiogenic factors (e.g., sFlt-1, sEng) increase, and maternal immune profiles shift toward heightened activation. These changes converge to drive endothelial dysfunction, oxidative stress, and impaired placental perfusion, culminating clinically in hypertension and multi-organ involvement.
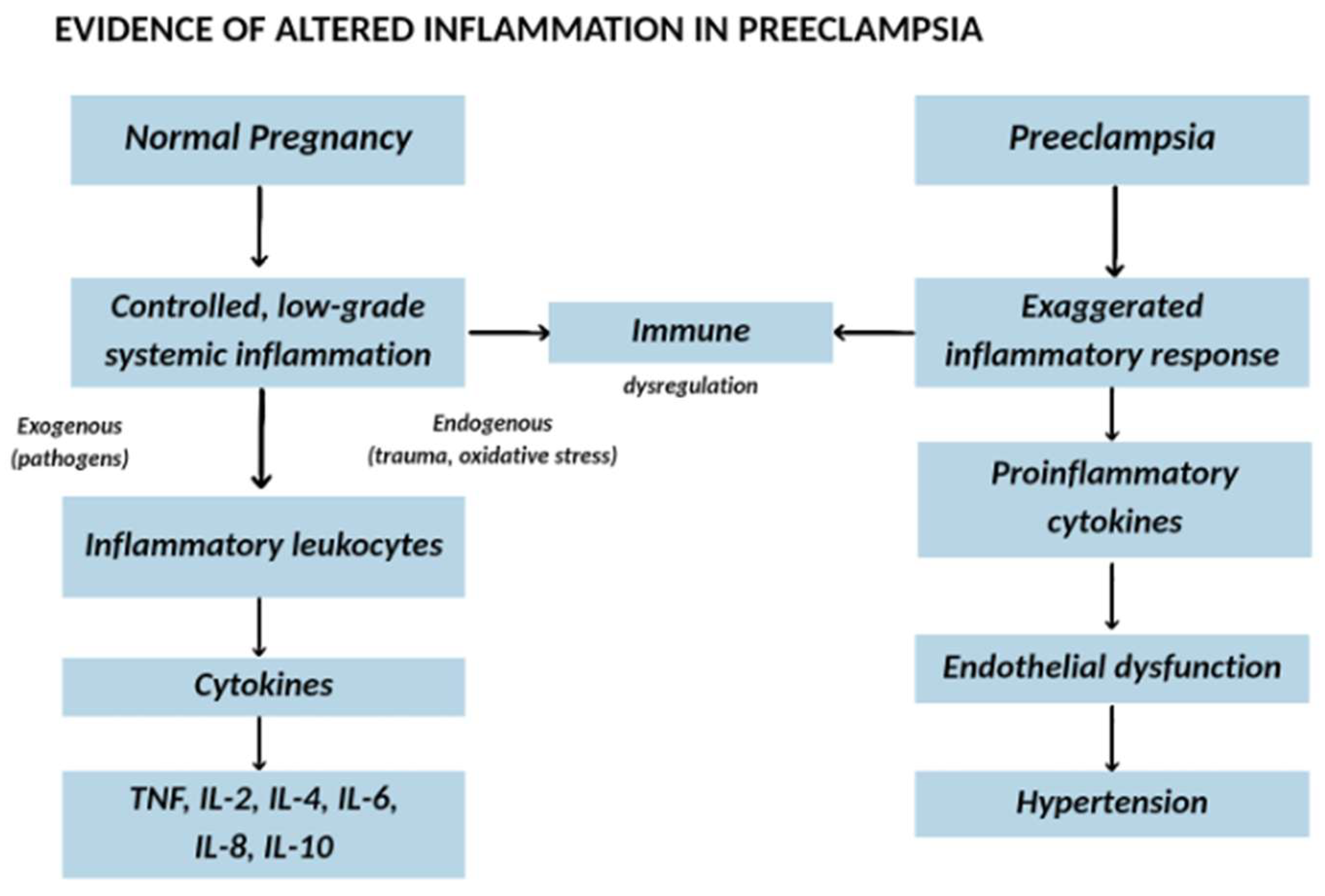
Figure 3.
Placental immune inflammation linking maternal inflammation with preeclampsia and fetal growth. This schematic illustrates how maternal immune activation shapes the maternal–fetal interface and influences placental and fetal outcomes. Maternal macrophages, regulatory T cells, and circulating cytokines modulate the decidual milieu, whereas trophoblast-derived exosomes that carry microRNAs act as key effectors coordinating crosstalk between inflammatory and angiogenic pathways. The resulting balance—or imbalance—between pro-inflammatory signals and pro-angiogenic support determines endothelial health and uteroplacental perfusion. When maternal susceptibility and heightened inflammation prevail, endothelial dysfunction and impaired placental vascularization ensue, contributing to preeclampsia and restricted fetal growth.
Figure 3.
Placental immune inflammation linking maternal inflammation with preeclampsia and fetal growth. This schematic illustrates how maternal immune activation shapes the maternal–fetal interface and influences placental and fetal outcomes. Maternal macrophages, regulatory T cells, and circulating cytokines modulate the decidual milieu, whereas trophoblast-derived exosomes that carry microRNAs act as key effectors coordinating crosstalk between inflammatory and angiogenic pathways. The resulting balance—or imbalance—between pro-inflammatory signals and pro-angiogenic support determines endothelial health and uteroplacental perfusion. When maternal susceptibility and heightened inflammation prevail, endothelial dysfunction and impaired placental vascularization ensue, contributing to preeclampsia and restricted fetal growth.
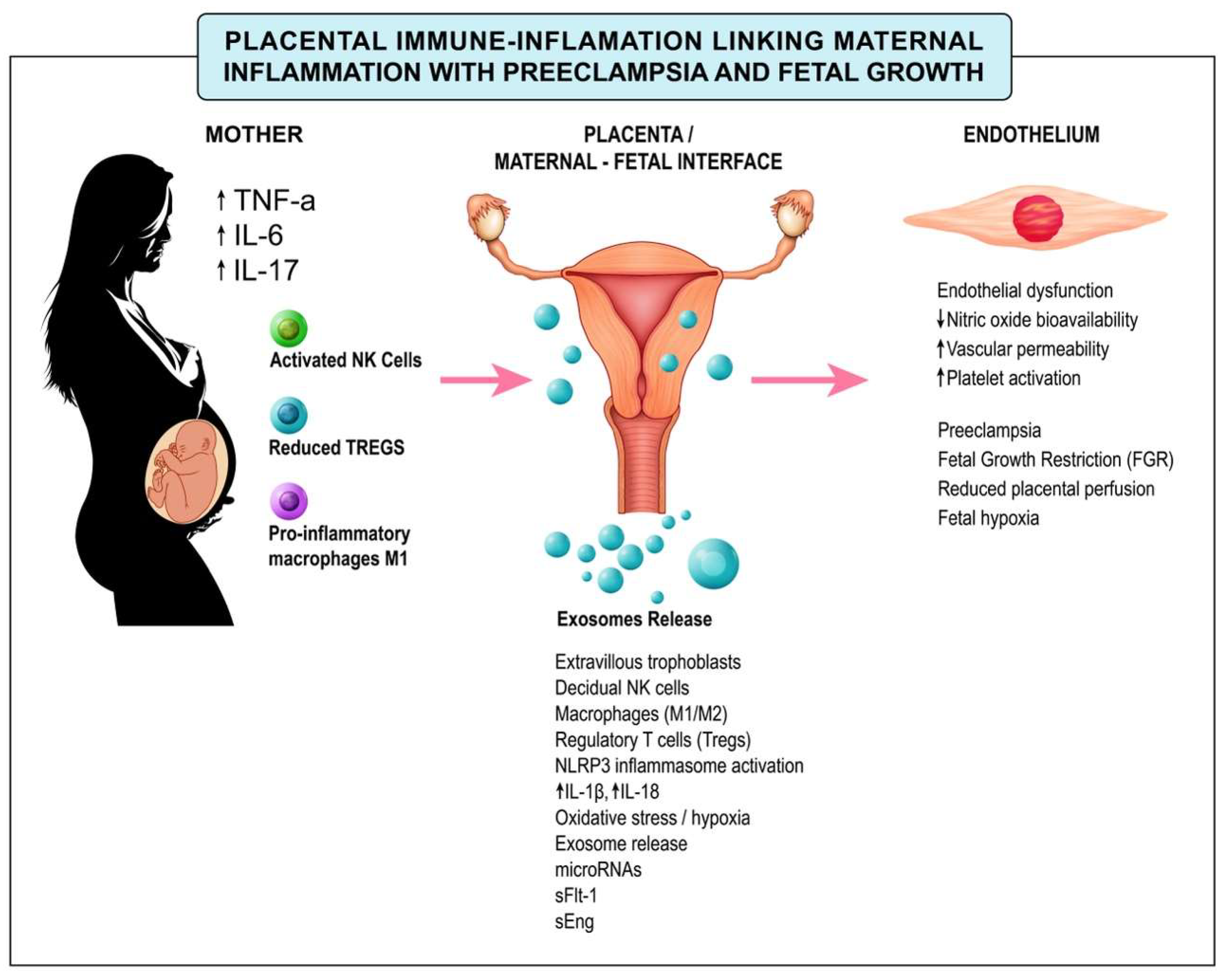
Figure 4.
Inflammatory pathways linking placental dysfunction to fetal growth restriction. Maternal hypoxia and endogenous danger signals activate the NLRP3 inflammasome, increasing IL-1β and IL-18 production and leading to trophoblast dysfunction, placental insufficiency, and maternal inflammatory biomarkers. These processes contribute to adverse fetal outcomes, including FGR, neuroinflammation, metabolic dysregulation, and long-term cardiovascular risk.
Figure 4.
Inflammatory pathways linking placental dysfunction to fetal growth restriction. Maternal hypoxia and endogenous danger signals activate the NLRP3 inflammasome, increasing IL-1β and IL-18 production and leading to trophoblast dysfunction, placental insufficiency, and maternal inflammatory biomarkers. These processes contribute to adverse fetal outcomes, including FGR, neuroinflammation, metabolic dysregulation, and long-term cardiovascular risk.
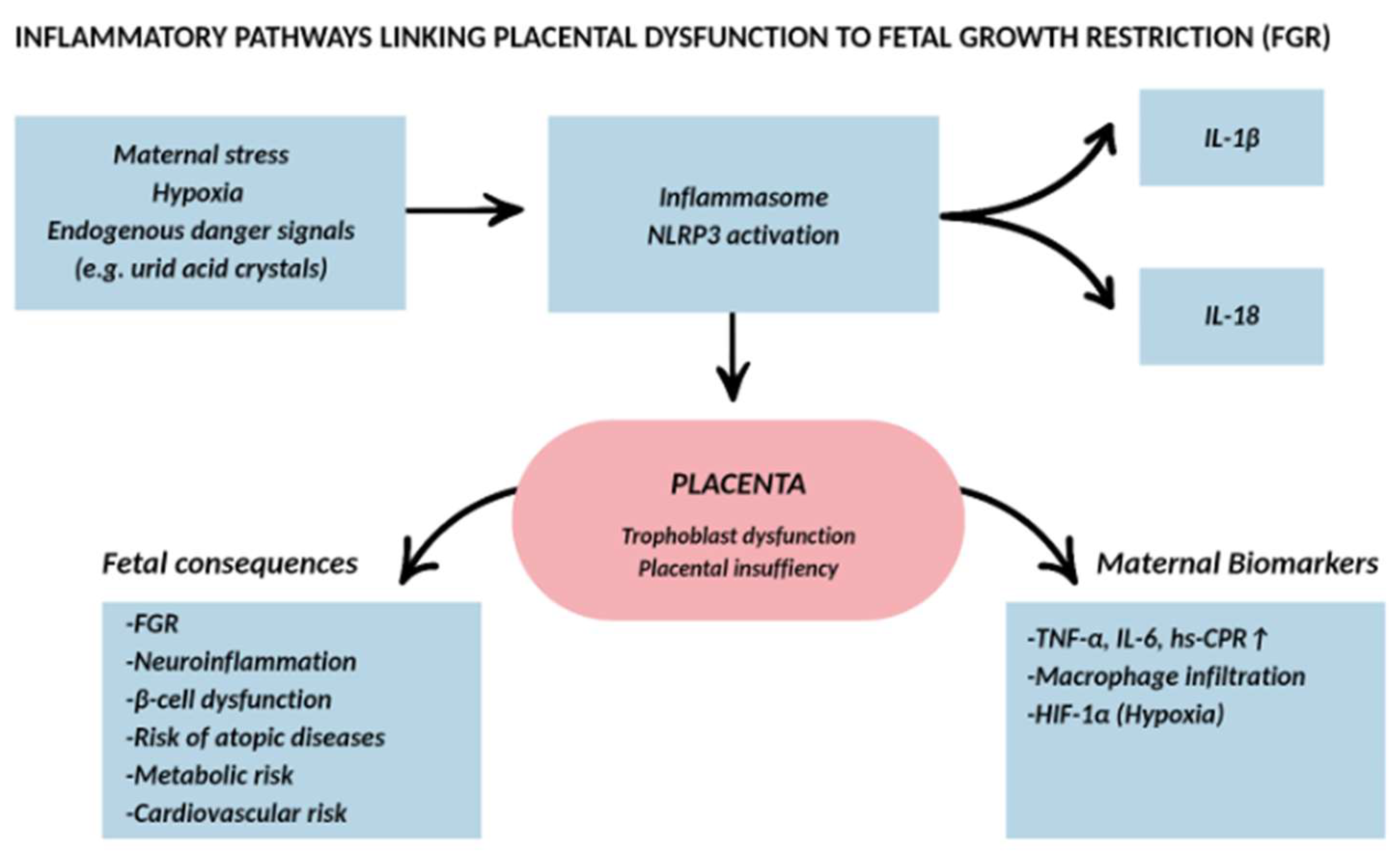
Figure 5.
Inflammatory Balance and Vascular Dysfunction as a Unifying Pathway to Preeclampsia and Fetal Growth Restriction. This model depicts pregnancy as a continuum between controlled, physiologic inflammation—necessary for implantation, placental development, and parturition—and dysregulated, exaggerated inflammation that becomes pathogenic. As inflammation intensifies, endothelial injury and angiogenic imbalance increase proportionally, leading to progressive impairment of uteroplacental perfusion. The degree of vascular involvement determines downstream fetal consequences, including altered angiogenesis and growth restriction. At the clinical level, this shared pathway manifests primarily as two outcomes: preeclampsia and fetal growth restriction, which may occur alone or together. The schematic highlights how the interplay between inflammation and vascular dysfunction provides a unified framework to understand these related pregnancy disorders.
Figure 5.
Inflammatory Balance and Vascular Dysfunction as a Unifying Pathway to Preeclampsia and Fetal Growth Restriction. This model depicts pregnancy as a continuum between controlled, physiologic inflammation—necessary for implantation, placental development, and parturition—and dysregulated, exaggerated inflammation that becomes pathogenic. As inflammation intensifies, endothelial injury and angiogenic imbalance increase proportionally, leading to progressive impairment of uteroplacental perfusion. The degree of vascular involvement determines downstream fetal consequences, including altered angiogenesis and growth restriction. At the clinical level, this shared pathway manifests primarily as two outcomes: preeclampsia and fetal growth restriction, which may occur alone or together. The schematic highlights how the interplay between inflammation and vascular dysfunction provides a unified framework to understand these related pregnancy disorders.
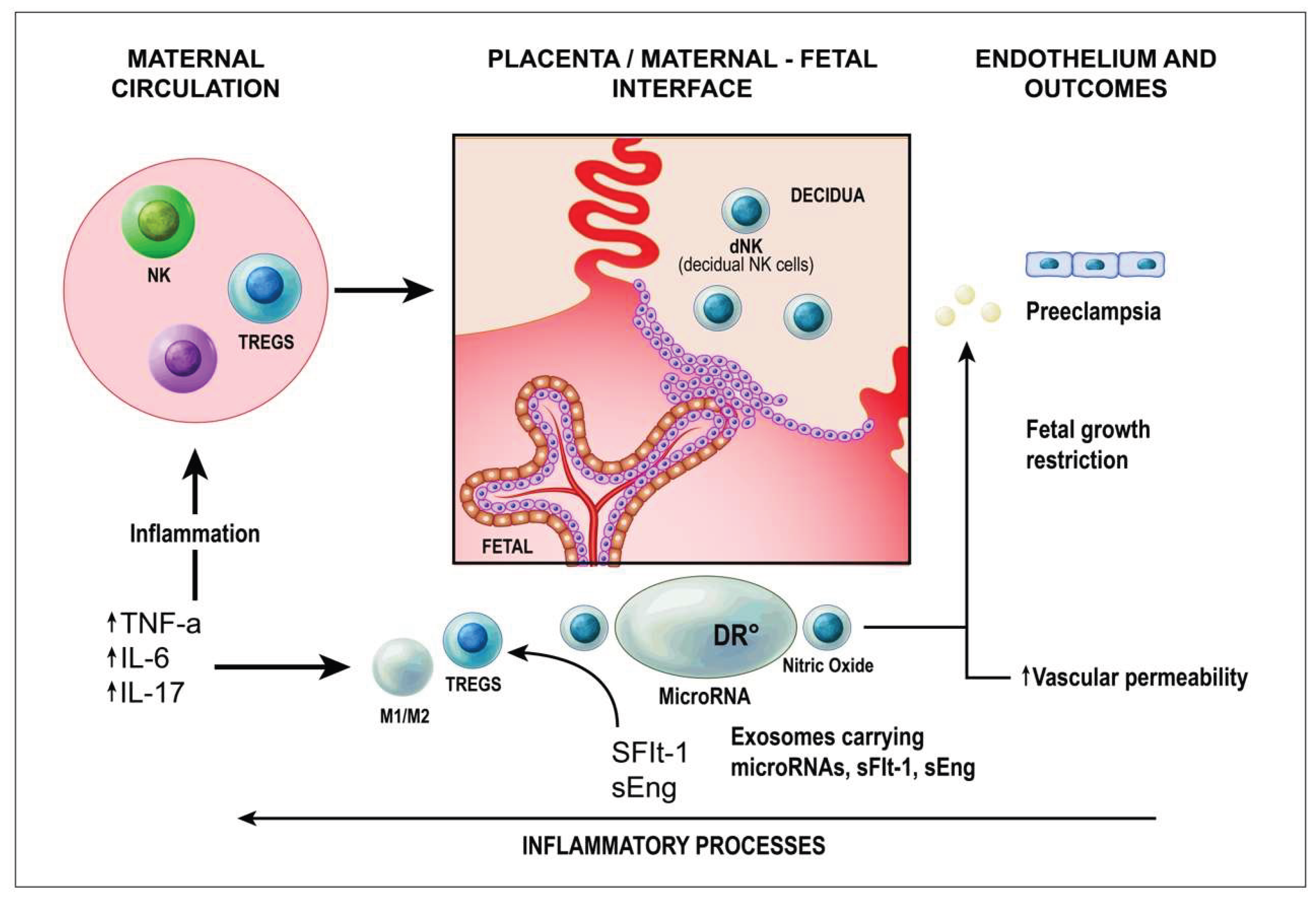

Table 1.
Comparative Therapies aimed to modulate inflammation during pregnancy.
| Category | Type | Mechanism of Action | Evidence Level |
|---|---|---|---|
| Low-Dose Aspirin | Established | COX-1 inhibition → ↓ platelet aggregation & inflammation | High – Multiple RCTs in PE prevention |
| Low Molecular Weight Heparin | Established | Anticoagulation + modulation of complement & leukocyte trafficking | Moderate – Observational studies & some RCTs |
| Antenatal Corticosteroids | Established | Promote fetal lung maturation, transient anti-inflammatory effect | High – Standard of care in threatened preterm birth |
| Antioxidants & Nutritional Modulation | Adjunctive | Reduce oxidative stress, improve endothelial function | Low to Moderate – Inconsistent RCT results |
| Lifestyle & Microbiome Modulation | Adjunctive | Improve immune tolerance & reduce systemic inflammation | Low – Ongoing trials |
| Cytokine-Targeted Biologics | Emerging | Neutralize pro-inflammatory cytokines (e.g., TNF-α, IL-6) | Experimental – Limited pregnancy-specific data |
| Inflammasome Inhibitors | Emerging | Block NLRP3 inflammasome activation | Preclinical – Animal models |
| Cell-Based Therapies | Emerging | Promote immune tolerance & tissue repair at maternal–fetal interface | Preclinical – Early-phase studies |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



