Submitted:
01 January 2026
Posted:
05 January 2026
You are already at the latest version
Abstract
Extracellular vesicles (EVs) reflect their spatiotemporal cellular origins and, when released into the peripheral bloodstream, represent sources of biomarkers for processes occurring in inaccessible tissues, such as the brain. Positive selection by immunoaffinity isolation targeting a specific EV surface marker can be used to preferentially enrich for EVs from a particular cell type over other EVs in circulation. However, the case of L1CAM immunocapture for enrichment of brain-derived neuronal EVs has exemplified the biological and technical influences affecting the outcomes of positive selection, which are discussed here. These include findings from the wider (non-EV) literature showing that soluble L1CAM is a binding partner of the common EV marker CD9. Additional studies show that CD9 is not expressed by the vast majority of neurons in the brain, but it is highly expressed by neurons of the peripheral nervous system, which could affect the interpretation of studies demonstrating the co-localisation of L1CAM and CD9 on EVs to assert isolation of EVs derived from neurons in the brain. Next, emerging negative selection strategies are discussed which, when combined with positive selection, could overcome some of the challenges associated with enriching for EV-based biomarkers. These strategies include depleting EVs on a cell-by-cell type basis, as well as targeting common EV markers to simultaneously deplete diverse, undesired EV populations whilst selecting for the EVs of interest. An example is given of how CD9 depletion could enrich and select for brain-derived neuronal EVs. Additionally, studies that have used negative selection alone to select for EVs, and the advantages of this approach for biomarker detection, are highlighted. Finally, based on these insights, considerations for the choice of future positive and negative selection targets are discussed, including how understanding the nuances of EV heterogeneity could facilitate the process of EV-based biomarker discovery and their translation to the clinic.
Keywords:
extracellular vesicle
; biomarker
; neuron
; positive selection
; negative selection
; immunodepletion
; L1CAM
; CD9
Plain language summary
Extracellular vesicles are miniscule parcels released by cells with functions that include communication. These parcels contain molecular messages in the form of proteins and RNA which can be read by other cells that receive the parcels. Since the messages reflect the status of the sending cell, they can also warn other cells about a disease affecting the sending cell.
The parcels are transported between cells by the movement of fluids in the body, including the bloodstream. Because of this, researchers in the lab can take a blood sample then use techniques to separate the parcels from the blood and read the molecular messages contained within. This is a useful approach for understanding diseases in inaccessible tissues such as the brain because the disease processes can be indirectly studied via the parcels that were released into the blood by brain cells.
A barrier to this approach is that most of the parcels found in the blood are derived from blood cells and these mask disease-specific signals coming from the brain. To overcome this, researchers can use an approach called positive selection to target a brain cell marker on the parcels’ surface and enrich brain-specific signals. However, in a large, complex structure such as the human body, many cells release parcels with overlapping markers which can again obscure brain-specific messages. This article looks at how issues such as these have been raised and addressed when targeting a protein called L1CAM to enrich parcels coming from neuronal cells in the brain. Negative selection is also discussed as an alternative approach, where the unwanted parcels are targeted for removal to enrich the desired parcels in the sample. Together, these approaches could clarify messages coming from the brain so that we can identify early warning signals (biomarkers) in disease diagnostics and design more effective treatments.
Main body
Introduction
Neuropathological conditions cause major disease burden and mortality, but there are few effective therapies available. Moreover, differential diagnosis remains challenging due to overlapping clinical symptoms and a lack of molecular markers which can be used to distinguish different neurological disorders in the clinic (Dutta et al., 2023). The massive financial and health costs for individuals and society caused by these conditions necessitates a greater understanding of the underlying neuropathophysiological mechanisms to develop drugs to prevent their onset and identify biomarkers for risk stratification, diagnosis, and monitoring disease progression and response to therapy. However, the discovery of such biomarkers is hindered by the inaccessibility of the brain, and central nervous system (CNS) as a whole, and direct access to brain tissue is too invasive to be used in routine clinical tests. Therefore, much of our understanding of neuropathological disorders is based on animal studies (which may not translate to humans) or brain imaging techniques, which yield little information about molecular and cellular neuropathophysiological events (Trivedi and Bhat, 2023).
Liquid biopsies of peripheral biofluids (e.g. blood plasma) offer a less invasive way to monitor neuropathophysiological molecular processes in humans in real-time through analysis of brain-derived molecules in the bloodstream, such as proteins, RNA, and DNA (Trivedi and Bhat, 2023). However, currently there is a lack of biomarkers for neurological disorders that can be accessed in peripheral biofluids. Biomarker identification by analysing soluble molecules in the peripheral circulation has been hampered by the low concentration of molecules derived from the central nervous system (CNS) in the periphery, difficulties in tracing individual molecules back to their cell type of origin, and degradation of potential biomarkers by circulating enzymes (Ciferri et al., 2021; Zhou et al., 2021).
There are some cases where biomarker assays for single soluble molecules have been developed into clinical tests, such as for neurofilament light chain (NFL), which is a neuron-enriched cytoskeletal protein. The concentration of NFL in peripheral biofluids has been developed into a biomarker assay because NFL is released following axonal damage or neuronal degeneration and levels of NFL are therefore elevated in traumatic brain injury, multiple sclerosis, and other neurodegenerative diseases (Ashrafzadeh-Kian et al., 2024). However, whilst elevated NFL levels do serve as a general biomarker of neuronal damage, they are unable to distinguish neurological conditions such as Alzheimer’s disease and frontotemporal dementia nor indicate the molecular events that led to the release of NFL (Truffi et al., 2023). Moreover, NFL levels can be a result of co-morbidities and lifestyle habits unrelated to the neurological disorder of interest, limiting its biomarker potential (Ladang et al., 2022). Therefore, there is increasing interest in multicomponent extracellular vesicles (EVs) in liquid biopsies as a source of biomarkers for neuropathological conditions (Zanirati et al., 2024).
Extracellular vesicles as sources of biomarkers
EVs are heterogenous spherical packages of bioactive molecules (proteins, lipids, nucleic acids, glycans, and metabolites) protected by a lipid bilayer which are released by viable cells (Welsh et al., 2024). Because EVs are stable in biofluids, they can carry molecular messages to distant cells and act in intercellular communication. The composition of EVs is determined by their mode of biogenesis and cellular origins; thus, EVs represent a molecular signature of their parent cell at that moment in time and their presence in biofluids makes them attractive for use as biomarkers (van Niel et al., 2018). The biomarker component of EVs could be represented by a single molecular biomarker in the EV cargo, or a signature of several different biomolecules within the EV unit (Foroni et al., 2020; Tutrone et al., 2023).
EVs from blood have already been characterised to identify proteomic patterns associated with (and distinguishing) different cancers in large patient cohorts (Hoshino et al., 2020) and EV-based biomarkers have been shown to outperform circulating nucleic acids in terms of sensitivity and selectivity for tumour detection (Foroni et al., 2020). Furthermore, EV-based biomarkers have already entered the clinic: the commercial ExoDxTM Prostate test is a gene expression array for three EV-encapsulated RNA biomarkers in urine that provides risk stratification of prostate cancer and guides the clinical need for biopsy (Tutrone et al., 2023). With this precedent, Box 1 highlights some of the areas in which EV-based biomarkers could lead to improved clinical outcomes for patients.
Box 1. Areas in which isolating EV-based biomarkers from inaccessible tissues in peripheral biofluids could lead to improved clinical outcomes for patients.
|
The advantages EV-based biomarkers over single soluble molecules relate to 1) the multicomponent nature of EVs and 2) the protection offered by the EV lipid bilayer (Ciferri et al., 2021; Zhou et al., 2021). Firstly, being multicomponent enables EVs to concentrate multiple copies of the biomarker in a single EV unit, enhancing the signal-to-noise ratio of the biomarker (Tutrone et al., 2023). Moreover, analysis of a combination of different biomarkers (e.g. proteins and nucleic acids) within one EV can be performed to increase the specificity of the biomarker signature, which further increases the signal-to-noise ratio compared to potential biomarkers in freely circulating single molecules (Foroni et al., 2020). EVs can also be enriched from biofluids using various techniques (e.g. size-exclusion chromatography, ultracentrifugation, etc.), which further enhances the biomarker signal-to-noise ratio by removing non-EV molecules to reduce background noise. Additionally, the composition of EVs reflects their cell type of origin, so cell type-specific EVs can be isolated by targeting a cell type-specific protein exposed on the EV surface (for example, by antibody-based immunoaffinity isolation). This yields a population of EVs which can be analysed to provide insight into cell type-specific processes during disease, which also amplifies the biomarker signal-to-noise ratio by removing the background noise caused by EVs from other cells (Amin et al., 2024).
Secondly, the EV lipid bilayer protects the biomarker cargo from degradation by circulating enzymes (e.g. proteases and RNases). This can prolong the amount of time an EV-enclosed biomarker remains in circulation, as well as enabling molecular biomarkers to transfer between distal tissues via the bloodstream whilst contained within an EV (Zhao et al., 2019). The protection conferred by EVs extends to the ex vivo setting (e.g. after a blood sample has been taken in the clinic) and, so long as the EV membrane remains intact, cargo molecules are protected during sample processing (e.g. centrifuging blood to acquire plasma) and storage. Indeed, EV biomarker cargo can remain stable within biofluids stored for multiple years (Jeyaram et al., 2017). Therefore, the stability of EVs and their biomarkers during sample processing and storage makes them clinically amenable because clinical assays do not need to be performed immediately.
Extracellular vesicle-based biomarkers of neuropathological conditions
Because brain-derived (BD)EVs enter the peripheral circulation (Banks et al., 2020; Rufino-Ramos et al, 2022; Rufino-Ramos et al, 2023), the isolation and analysis of BDEVs from patient blood samples could offer an unprecedented source of biomarkers for cell type-specific neuropathological molecular processes. However, although EVs are relatively abundant in blood plasma (approximately 1010 EVs/ml; Johnsen et al., 2019), most of these originate from blood cells and platelets, whilst tissue-derived EVs make up a relatively small proportion of the total EVs in circulation (Li et al., 2020). For example, EVs derived from brain tissue have been estimated to constitute <1% of the total blood plasma EV population (Li et al, 2020), or <108 EVs/ml. Therefore, because EVs released by peripheral cells make up the majority of EVs in circulation, molecular patterns that reflect cell type-specific changes in the brain are masked by peripheral EVs, obscuring potential biomarkers of neurological disorders.
To enable analysis of brain-derived EV-based biomarkers in peripheral biofluids, immunoaffinity isolation can be performed to positively select for EVs on a cell-by-cell type basis (Figure 1A) and unmask biomarkers of neurological disorders by reducing the background noise of peripheral EVs (Ganesh et al., 2023). However, this approach relies on the identification of specific cell markers, while the use of a single protein to definitively trace EVs back to their cellular source is complicated in large multicellular organism where most proteins show expression in multiple cell types and in multiple tissues (Uhlén et al., 2015). Moreover, the isolation of CNS neuronal EVs is further complicated by EVs derived from neurons of the peripheral nervous system (PNS; e.g. enteric neurons that innervate the gastrointestinal tract), which share the expression of many neuronal markers (Uhlén et al., 2015; Zeisel et al., 2018). This hinders the identification of EV proteins unique to CNS neurons that can be detected in EVs in the periphery and used as targets for selective immunoisolation.
The case of L1CAM for the immunocapture of brain-derived neuronal extracellular vesicles
The most widely targeted protein for immunocapture of CNS neuronal EVs from peripheral biofluids has been L1 cell adhesion molecule (L1CAM) (Gomes et al., 2022), which is a single-pass type 1 membrane protein (https://www.uniprot.org/uniprotkb/P32004/entry; UniProt Consortium, 2025). Whilst L1CAM-associated biomarker signatures have been identified that correlate with neurological disease (Dunlop et al., 2023; Dutta et al., 2023) and treatment response (Athauda et al., 2019; Mustapic et al., 2019), the validity of L1CAM as a target to capture CNS neuronal EVs from the peripheral circulation is contested (Gomes et al., 2022; Dutta et al., 2023). Firstly, L1CAM is not exclusively expressed by CNS neurons and transcriptomic and proteomic datasets show that L1CAM is expressed at higher levels in enteric neurons and in the kidneys and skin, as well as at lower levels in other tissues (http://mousebrain.org/genes/L1cam.html, Zeisel et al., 2018; v25.0 proteinatlas.org/ENSG00000198910-L1CAM, Uhlén et al., 2015). This indicates that other tissues may also release L1CAM-positive EVs into circulation, which could be enriched following L1CAM immunocapture. This is supported by estimations that 5-10% of EVs in plasma are L1CAM-positive (Mustapic et al., 2017; Kumar et al., 2022), whilst less than 1% of EVs in plasma originate from the brain (Li et al., 2020).
Secondly, Norman et al. (2021) reported that L1CAM in circulation is present as a soluble isoform and not integral or exclusive to CNS neuronal EVs. Through fractionation of blood plasma by density gradient centrifugation, as well as size-exclusion chromatography, L1CAM eluted with albumin (a proxy for soluble protein), but not in the fractions containing the EV markers CD9, CD63, and CD81 when detected using a digital ELISA (Simoa®) (Norman et al., 2021), which can detect fg/ml levels of proteins (Rissin et al., 2010). At the same time, the authors revealed the existence of a soluble, alternatively spliced isoform of L1CAM in plasma, containing both the internal and external domains of the protein, but lacking its transmembrane region. Since this isoform was indistinguishable from the integral membrane protein by Western blotting, the authors indicated that the L1CAM detected after L1CAM immunocapture from plasma could be this isoform, and that other proteins detected in the immunoprecipitate were co-isolated through direct interactions with the L1CAM antibody/beads. This was demonstrated with soluble α-synuclein, which preferentially bound the UJ127 L1CAM antibody clone compared to control IgG (Norman et al., 2021).
Subsequent studies have corroborated that the majority of L1CAM in circulation is not an integral EV component using size-exclusion chromatography, but these studies have also indicated that there is a small amount of L1CAM that elutes with the EV fraction (Dutta et al., 2021; Yan et al., 2023). Additionally, Blommer et al. (2023) and Kumar et al. (2022) reported that L1CAM could be detected on single EVs through co-localisation with canonical EV markers, as demonstrated by immunoelectron microscopy with CD63 (Kumar et al., 2022); high-resolution fluorescent confocal microscopy with ALIX (Blommer et al. 2023); and single particle interferometric imaging sensing (SP-IRIS) with CD9, CD63, and CD81 (Blommer et al. 2023). For this, both studies used a used a commercial precipitation reagent (ExoQuick®) to precipitate particles from plasma before performing L1CAM immunocapture (Blommer et al. 2023; Kumar et al., 2022).
However, the reliability and reproducibility of biomarker studies using precipitation agents to concentrate EVs before L1CAM capture has been questioned due to the associated aggregation of soluble proteins with EVs, and subsequent non-specific capture during immunoisolation, leading to high variability in inter-study results (Taha, 2023; Yan et al., 2023). For example, in the case of α-synuclein derived from the blood of healthy volunteers, Yan et al. (2023) highlighted that the 3099.9 (±2986) pg/ml concentration of α-synuclein obtained by Blommer et al. (2023) using precipitation followed by L1CAM immunocapture was approximately 233-fold higher than the 13.3 (±4.5) pg/ml concentration obtained when size-exclusion chromatography was used to separate EVs and soluble proteins (Yan et al., 2023). This difference was attributed to the tendency of α-synuclein, which is present in blood plasma at a concentration of 42 ng/ml (v25.0 proteinatlas.org/ENSG00000145335-SNCA/blood#blood_conc_ms, Uhlén et al., 2015), to precipitate and aggregate in the presence of the precipitation reagent, as well as bind to surfaces including immunocapture beads, polymers in the precipitation reagent, and EVs (Yan et al., 2023).
To try to definitively show the association of L1CAM with EVs in plasma, Nogueras-Ortiz et al. (2024) employed multiple EV isolation techniques and single-EV analysis methods to localise the ectodomain of L1CAM (as the intended target for L1CAM immunocapture) to the outer surface of EVs. This included differential ultracentrifugation followed by high-resolution confocal fluorescent microscopy to show that the L1CAM ectodomain co-localised with EV markers CD63, CD81, and ALIX on EVs derived from neuronal cells in vitro (specifically, primary rat hippocampal neurons and human induced pluripotent stem cell-derived neurons). Additionally, nanoscale multiplex flow cytometry analysis (FCA) showed that 97% of L1CAM-positive events were double positive for ‘pan-tetraspanins’ (CD9, CD63, and CD81) and that a higher number of L1CAM/pan-tetraspanin(++) events occurred in larger EVs that pelleted at 10,000 g compared with smaller EVs that pelleted 120,000 g (Nogueras-Ortiz et al., 2024).
Using FCA to analyse total EVs from plasma (pelleted by a single ultracentrifugation step at 120,000 g), Nogueras-Ortiz et al. (2024) also reported that 3.8% were L1CAM-positive, and 67.4% of L1CAM-positive events were co-labelled with at least one of CD9, CD63, or CD81. To show the neuronal origin of plasma-derived EVs by FCA, it was demonstrated that 33.6% of L1CAM-positive events were co-labelled with VAMP2 and 54% with β-III-tubulin (two proteins enriched in neurons), whilst only 5.0% (VAMP2) or 4.1% (β-III-tubulin) of EVs from the total plasma pool were labelled with these proteins (Nogueras-Ortiz et al., 2024). Moreover, only 2.4% L1CAM-negative events were immunopositive for VAMP2, and 1.3% for β-III-tubulin, indicating the specificity of L1CAM co-labelling with neuron-enriched proteins. Out of the total EVs in plasma, 1.3% were double-positive for L1CAM/VAMP2, 2.1% for L1CAM/β-III-tubulin, and 2.5% L1CAM/pan-tetraspanin (CD9, CD63, and CD81). Furthermore, following L1CAM immunocapture, an EV multiplexing assay was used to show that the isolated pan-tetraspanin(+) population carried GAP43, another neuron-enriched protein (Nogueras-Ortiz et al., 2024). Based on these quantitative findings, it was estimated that two-thirds of L1CAM-positive EVs were from neuronal cells, whilst one-third had a non-neuronal origin (Nogueras-Ortiz et al., 2024).
Additionally, in contrast to the results of Norman et al. (2021), Nogueras-Ortiz et al. (2024) did detect the L1CAM ectodomain using a Simoa® digital ELISA in the fractions of plasma containing CD9, CD63, or CD81 following size-exclusion chromatography (although notably both studies employed different chromatography columns and resins). The latter study also sought to address whether free, soluble L1CAM in blood could bind non-specifically to the surface of EVs (Nogueras-Ortiz et al., 2024). For this, the authors incubated EVs from HEK-293 cells (an embryonic kidney cell line that do not express L1CAM) with particle-depleted plasma for 3.5 h at 37 °C before assessing the adsorption of L1CAM or APOA1 to the surface of EVs (APOA1 is a recognised component of the non-integral, surface corona coating EVs in plasma; Toth et al., 2021). After the incubation, FCA showed that APOA1, but not L1CAM, was detected on the surface of HEK-293 EVs, demonstrating that free, soluble L1CAM in particle-depleted plasma does not bind HEK-293 EVs (Nogueras-Ortiz et al., 2024).
Unanswered questions regarding the use of L1CAM for the immunocapture of brain-derived neuronal extracellular vesicles
Together, the abovementioned studies make substantial contributions to understanding the complexities of targeting EVs from a single cellular source (CNS neurons) using immunoaffinity isolation. However, they also highlight some of the limitations of this approach and there remains unresolved questions regarding the use of L1CAM immunocapture to obtain brain-derived neuronal biomarkers. It is recognised that the methodology of L1CAM immunocapture is unable to distinguish between EVs derived from neurons of the CNS and PNS, thereby limiting the recovery of biomarkers specific for brain disorders (Nogueras-Ortiz et al., 2024). The relative contribution of EVs to the peripheral bloodstream from these two sources remains unknown (Nogueras-Ortiz et al., 2024); however, in comparison with the blood-brain barrier, the blood-nerve barrier (between peripheral neurons and the bloodstream) represents a relatively ‘leaky’ interface (Malong et al., 2023) by which peripheral neuron-derived EVs may enter circulation.
Notably, transcriptomic data shows that L1CAM, β-III-tubulin, and GAP43 are expressed at higher levels in peripheral neurons than the CNS, whilst VAMP2 is expressed at similar levels in CNS and PNS neurons (Table 1). Additionally, L1CAM, VAMP2, β-III-tubulin, and GAP43 have widely different protein concentrations in plasma (proteinatlas.org; Uhlén et al., 2015), ranging from 0.029 ng/ml for GAP43 to 71 ng/ml for β-III-tubulin (Table 1). The concentration of β-III-tubulin in plasma is also 17-fold the concentration of CD9 (Table 1), with CD9 representing the most abundant tetraspanin in plasma, found on the majority (approximately 62-72%) of plasma EVs (Hoshino et al., 2020; Karimi et al., 2022; Salmond et al., 2021; Tordoff et al., 2024). This indicates that some of these neuron-enriched proteins are either particularly abundant components of EVs, or they have non-neuronal sources, or they are secreted in a soluble form. Therefore, this highlights that further markers are required to delineate the origins of CNS neuronal EVs from peripheral sources.
Furthermore, it remains unknown how biomarker studies using immunocapture antibodies targeting the internal (intravesicular) domain of L1CAM apparently isolate L1CAM-positive EVs (Chen et al., 2024; Dagur et al., 2020; Kluge et al., 2022; Schaeffer et al., 2024). Since the L1CAM ectodomain is on the EV surface (Nogueras-Ortiz et al., 2024) and L1CAM is a single-pass type 1 membrane protein (https://www.uniprot.org/uniprotkb/P32004/entry; UniProt Consortium, 2025), it would be anticipated that the internal domain and C-terminus of the protein are located within the EV lumen (as illustrated in Figure 2A). Therefore, with this transmembrane model, the internal domain of L1CAM should be inaccessible to antibody binding (Figure 2A). Notably, antibodies targeting the L1CAM internal domain, which have been experimentally mapped to binding the protein’s C-terminus (Chen et al., 2010; Girbes Minguez et al. 2020), would also not bind the soluble L1CAM ectodomain (released as a result of ectodomain shedding; Maretzky et al., 2005) adsorbed onto the EV surface because this cleavage product lacks the internal domain of L1CAM.
However, studies using internal domain antibodies for L1CAM immunocapture (Chen et al., 2024; Dagur et al., 2020; Kluge et al., 2022; Schaeffer et al., 2024) reported the isolation of particles with the size profile of EVs by transmission electron microscopy (Kluge et al., 2022) and nanoparticle tracking analysis (Dagur et al., 2020). Similarly, L1CAM internal domain immunocapture isolated full-length L1CAM, EV-enriched proteins (CD9, CD63, CD81, TSG101) and neuron-enriched proteins (β-III-tubulin, synaptophysin, enolase 2) (Dagur et al., 2020; Kluge et al., 2022). Since these studies detected L1CAM with a molecular weight >32 kDa (the size of the internal domain and transmembrane region alone; Maretzky et al., 2005), it indicates that both the internal domain and ectodomain of the protein were captured by the internal domain antibody (Dagur et al., 2020; Kluge et al., 2022). To explain this, Gomes and Witwer (2022) postulated that the L1CAM internal domain could be exposed to antibodies on damaged EVs or particles with an inverted membrane topology formed from cell membrane fragments, enabling immunocapture of full-length L1CAM and other proteins.
An alternative explanation is that the L1CAM internal domain antibodies capture the soluble, alternatively spliced isoform of L1CAM (described in plasma by Norman et al., 2021) bound to the surface of EVs (illustrated in Figure 2B). Although it was shown that soluble L1CAM did not bind exogenous EVs in particle-depleted plasma when compared with APOA1 (Nogueras-Ortiz et al., 2024), the binding of soluble L1CAM to EVs may depend on context and specific protein-protein interactions since the binding of EVs and soluble plasma proteins is specific and not universal (Su et al., 2024). Notably, the concentration of APOA1 in plasma is ~160,000 ng/ml (v25.0 proteinatlas.org/ENSG00000118137-APOA1/blood#blood_conc_ms, Uhlén et al., 2015), approximately 12,308-fold higher than L1CAM (13 ng/ml; v25.0 proteinatlas.org/ENSG00000198910-L1CAM/blood#blood_conc_ms). Whilst interactions between EVs and free L1CAM molecules in plasma would be comparatively rare, L1CAM is one of the most abundant soluble proteins secreted by neurons (Tüshaus et al., 2020). Therefore, it remains to be explored if full-length soluble L1CAM could be adsorbed onto the surface of EVs in the neuronal microenvironment, which could explain how studies performing L1CAM immunocapture using the internal domain antibody show the presence of neuron-enriched proteins in the immunoprecipitate (Dagur et al., 2020; Kluge et al., 2022). This could be tested experimentally by incubating L1CAM-negative EVs with conditioned media from in vitro cultures of neurons. Further controls are also required to show that the observed EV isolation is not the result of off-target binding of the immunocapture antibody, as observed with α-synuclein for the UJ127 clone against the L1CAM ectodomain (Norman et al., 2021).
It is also noteworthy that soluble L1CAM is a CD9 binding protein via its ectodomain (Schmidt et al., 1996; Jouannet et al., 2016). This was demonstrated in immunoprecipitation experiments with soluble cell lysates derived from N2A neuroblastoma (Schmidt et al., 1996) or U2 osteosarcoma cells (Jouannet et al., 2016), with L1CAM and CD9 found to form a stable and specific molecular complex that was concentration-dependent and saturable (Schmidt et al., 1996). Thus, this interaction could provide a mechanism by which soluble full-length L1CAM binds the EV surface, leaving its internal domain exposed for immunocapture. Relevant to this interaction is that CD9 inhibits ADAM10 and ADAM17 (Arduise et al., 2008; Gutiérrez-López et al., 2011), two metalloproteases that cleave L1CAM to release the ectodomain (Maretzky et al., 2005), and it is unknown how this negative regulation could impact the presence of full-length L1CAM on CD9-positive EVs.
Notably, the vast majority of neurons in the adult CNS do not express CD9 (Figure 3), as demonstrated by proteomic and transcriptomic datasets (proteinatlas.org, Uhlén et al., 2015; mousebrain.org, Zeisel et al., 2018), as well as in studies investigating the expression of CD9 in the nervous system (e.g. Doh-Uhra et al., 2000; Kagawa et al., 1997; Table 2). Instead, these same datasets show that CD9 is highly expressed by PNS neurons (Figure 3, Table 1, Table 2), providing a physiological context for which soluble L1CAM could bind CD9-positive EVs secreted by PNS neurons. Together, context-dependent CD9 expression and CD9-L1CAM interactions need to be taken into account when assessing the presence of L1CAM on EVs. This is especially important because 1) several EV analysis platforms using CD9-L1CAM binding have been used to demonstrate the presence of EV-integral L1CAM (when soluble L1CAM is a CD9 binding protein) and 2) these platforms have also been used to assert the presence of brain-derived neuronal EVs through the presence of CD9 and L1CAM (Bravo-Miana et al., 2024; Hotta et al., 2022; Nogueras-Ortiz et al., 2024), even though the vast majority of neurons in the brain do not express CD9 (Figure 3, Table 2).
Clarification over the association of L1CAM with EVs could come through proteinase K assays, which have already been used to demonstrate that the L1CAM ectodomain exists on the EV surface, or free in solution, in brain-derived EV preparations (You et al., 2023). Such assays involve proteinase K digesting the exposed proteins in the EV preparation, whilst the EV lipid bilayer protects proteins within the EV lumen (Théry et al., 2018). Subsequent analysis (e.g. Western blotting) then reveals which proteins had been digested (i.e. those proteins on the EV surface or free in solution) (Théry et al., 2018). Thus, this approach could also be used to resolve the association of the L1CAM internal domain with EVs, where protection of the internal domain following proteinase K treatment would confirm that L1CAM exists on EVs as a transmembrane protein with the internal domain located within the EV lumen (Figure 2C). Notably, in such a case, following digestion L1CAM would be detected at ~32 kDa by Western blotting using an internal domain antibody because the ectodomain would have been digested by proteinase K (Maretzky et al., 2005). Conversely, if both the internal domain and ectodomain were exposed to proteinase K digestion (either because L1CAM exists as the isoform free in solution or bound to the EV surface), the internal domain would not be detected following proteinase K treatment (Figure 2D). Of note, related to this, You et al. (2023) targeted ATP1A3 to enrich for neuronal EVs from plasma and used the XVIF9-G10 clone antibody that has also been shown to bind the cytoplasmic (and anticipated intravesicular) domain of the ATP1A3 protein at the protein’s N-terminus (Arystarkhova and Sweadner, 1996). However, when You et al. (2023) performed a proteinase K assay with EVs derived from human brain tissue (enriched using a protocol of enzymatic digestion and density gradient centrifugation) followed by Western blotting for ATP1A3 with the XVIF9-G10 clone antibody, proteinase K was shown to completely digest the epitope, indicating that the internal domain of ATP1A3 was exposed to proteinase K, not protected by the EV membrane, and thus external to the EV (You et al., 2023). Further clarity is therefore required on how these proteins are located in relation to EVs and the EV lipid bilayer.
Negative selection by immunodepletion: a key step for enrichment and specificity of rare extracellular vesicle-based biomarkers?
Because of concerns over the specificity of L1CAM to brain-derived neuronal EVs, other targets have been investigated for CNS neuronal EV isolation from the periphery. These include ATP1A3, GluR2, GAP43, NLGN3, and NCAM (Eitan et al., 2023; Fiandaca et al., 2015; Ko et al., 2016; You et al., 2023). However, these all show expression in peripheral neurons, and some are expressed at high levels in other non-neuronal cell types in the body (proteinatlas.org, Uhlén et al., 2015; mousebrain.org, Zeisel et al., 2018), indicating that they may be unable to distinguish between EVs from neurons of the CNS and PNS, or EVs released by other cell types that express these proteins. In this regard, it is notable that studies targeting L1CAM (for example, Dunlop et al., 2023; Hotta et al., 2022; Kumar et al., 2022), GAP43 and NLGN3 (Eitan et al., 2023), and ATP1A3 (You et al., 2023) from plasma show that the immunocaptured EVs are enriched in CD9, which is highly expressed by PNS neurons but not expressed by the vast majority of CNS neurons (Figure 3, Table 1, Table 2). This suggests that a sizeable proportion of these EVs are actually originating from PNS neurons/other non-neuronal sources, rather than from neurons in the brain.
Together, this indicates that the single-step immunocapture approach of targeting enriched neuronal proteins for CNS neuronal EV isolation is unable to distinguish CNS-derived neuronal EVs from PNS-derived EVs in circulation; thereby, obscuring potentially critical insights into mechanisms underlying neuropathophysiological processes and masking brain-derived biomarkers. Moreover, the molecular overlap of PNS and CNS neurons remains an obstacle to identifying new targets for positive selection of CNS neuronal EVs that are 1) only expressed in CNS neurons; 2) released in CNS neuronal EVs; and 3) released into peripheral biofluids in sufficient number for biomarker discovery and detection. The need for more selective and specific isolation of CNS neuronal EVs (and associated biomarkers) has directed attention towards the strategy of combining positive selection of CNS neuronal EVs with negative selection by depleting contaminating EV populations (Nogueras-Ortiz et al., 2024). Figure 1B shows an illustration of this approach, where positive and negative selection are combined to select for a rare EV population, thereby amplifying the signal-to-noise ratio of cell type-specific biomarkers.
Such an approach has not been widely applied in EV research but, in cases where negative selection has been used, this has mostly been to remove EVs on a cell-by-cell type basis by targeting cell type-enriched markers to deplete abundant EV populations. In blood plasma, the majority of circulating EVs are blood cell-derived (Li et al., 2020) and the most abundant EVs by cell type originate from platelets (constituting between 30-51% of all plasma EVs; Auber et al., 2022; Li et al., 2020). Therefore, depletion of platelet EVs, as well as EVs derived from other blood cell types, has been used to successfully enrich for rare EV populations in plasma. Notarangelo et al. (2019) combined CD41 depletion with CD235 depletion to deplete platelet and erythrocyte EVs, which together have been estimated to constitute <55% of the total EVs in plasma (Auber et al., 2022; Li et al., 2020). This achieved a 4-5 fold enrichment of rare EVs (and associated biomarkers) derived from breast cancer cells in plasma EV samples, thereby demonstrating that the inclusion of a negative selection step increased the amount of material for biomarker detection in rare EV populations (Notarangelo et al., 2019).
Ko et al. (2016) also used negative selection by depleting CD45 (a leukocyte EV marker) and CD61 (a platelet EV marker) to enrich for and detect neuronal EVs in serum using a microfluidic device. For this, serum loaded into the microfluidics chamber was incubated with CD45/CD61 negative selection beads for 30 minutes, as well as positive selection beads targeting CD81. Using negative pressure to draw the serum through the device, the larger (7 µm) negative selection beads were then separated from the sample on a 5 µm porous membrane before the smaller (2.2 µm) positive selection beads were trapped on a 1 µm porous membrane. Following washes, a custom-made ELISA was then performed to detect the neuron-enriched protein, GluR2, in the positively selected EV population (Ko et al., 2016).
Using this device, Ko et al. (2016) reported that the inclusion CD45/CD61 depletion removed background EVs to enable the detection of GluR2 in mouse serum (that had been spiked with cortical neuron cultured media), when GluR2 could not be detected without the negative selection step. Similarly, the immunodepletion step enhanced the detection of changes in the level of GluR2-positive EVs in mouse serum following mild traumatic brain injury, showing that depletion of abundant blood EVs can be used to amplify the biomarker signal originating from neuronal EV populations bearing GluR2 (Ko et al., 2016). In another example combining positive and negative selection, Ganesh et al. (2023) found that preceding neuronal EV capture from plasma with either depletion of CD235-positive EVs derived from erythrocytes (constituting 4-5% of total plasma EVs; Auber et al., 2022; Li et al., 2020), abundant free plasma proteins (albumin and immunoglobulin), or combined CD235/albumin/immunoglobulin depletion, increased the number of proteins that were identified in neuronal EVs isolated by positive selection using a proprietary platform and analysed by mass spectrometry-based proteomics, thereby increasing the number of potential biomarker proteins that were detected.
Collectively, these examples demonstrate that immunodepletion is an effective strategy to enrich for rare EV populations, and their associated biomarkers, when used in combination with positive selection. However, strategies targeting cellular markers to deplete abundant EVs in plasma have the limitation that they only deplete EVs released by specific cell types. Similarly, solely focussing on depletion of abundant EVs released by particular cells may be unable to separate two EV populations from closely related cell types (e.g. CNS and PNS neurons) which both release EVs bearing the desired target for positive selection. Concerns have also been raised that depleting abundant plasma proteins could compromise EV recovery due to interactions between plasma proteins and specific EV populations (Su et al., 2024).
Taking advantage of the heterogeneity of common extracellular vesicle markers for immunodepletion
An alternative negative selection approach is targeting a common EV marker not found on the EV population of interest to simultaneously deplete multiple contaminating EV populations from different cell types. Almousa et al. (2025) developed a method of negative selection through depletion of the common EV marker CD63 to enable the enrichment of bacterial EVs from mouse faecal samples. Since no universal surface marker existed to isolate EVs from diverse bacteria by positive selection, performing CD63 depletion instead maximised the removal of heterogenous murine EVs from the sample (faecal EV isolated by ultracentrifugation) by targeting a single protein common to eukaryotic EVs but absent from the bacterial EVs (Almousa et al., 2025).
To examine the selectivity of CD63 depletion in enriching for bacterial EVs, unbiased metaproteomic analysis was then performed. Of the proteins identified in total faecal EVs from control mice, 64.2% (4133 proteins) were of bacterial origin, whilst 35.8% (2308 proteins) were derived from the mouse. However, following CD63 depletion, the percentage of proteins identified as bacterial rose to 90.3% (6692 proteins), with 9.7% (723) derived from the murine host. Correspondingly, the CD63-positive fraction contained 98.4% (4707) proteins of eukaryotic origin, and 1.6% (79) bacterial origin (Almousa et al., 2025). Immunogold electron microscopy and Western blotting for the eukaryotic EV markers CD63 and syntenin, as well as the bacterial markers LPS and outer membrane protein C, were also used to confirm the separation of bacterial EVs from those derived from the murine host. Thus, the role of bacterial EVs in pain sensitivity in a mouse model of diet induced obesity could be analysed by limiting the confounding presence of murine EVs, thereby enabling the observed biological effects to be attributed to the bacterial-enriched faecal EV population (Almousa et al., 2025).
This approach could also be used in human biomarker studies due to the heterogeneity of markers on EVs derived from different cellular and subcellular origins (Kugeratski et al., 2021; Spitzberg et al., 2023; Su et al., 2024; Zarovni et al., 2025). Thus, rather than depleting contaminating EVs on a cell-by-cell type basis, this approach would target a single, common marker for negative selection. This could enable the separation of the desired rare EV population from a multitude of EVs released by diverse cell types in a variety of biofluids (Almousa et al., 2025).
For example, the common EV marker CD9 is not expressed by the vast majority of neurons in the brain (Figure 3, Table 1, Table 2) and brain-derived neuronal EVs are a desired source of biomarkers for neurological disorders (Nogueras-Ortiz et al., 2024). Conversely, CD9 is the most abundant tetraspanin in blood plasma (Tordoff et al., 2024), found on 62-72% of plasma EVs (Hoshino et al., 2020; Tordoff et al., 2024). CD9 is also relatively common in EVs derived from different cell types and biofluids (Hoshino et al., 2020; Ko et al., 2021; Spitzberg et al., 2023). Therefore, CD9 depletion could be used to enrich for CNS-derived neuronal EVs in a variety of biological samples, for example by removing 62-72% of contaminating plasma EVs. In contrast, a depletion strategy targeting a cell marker for platelet EVs (constituting ~30-51% of EVs in plasma; Auber et al., 2022; Li et al., 2020) would be of limited use and could only be employed in certain biofluids. While there may be scope for combining the depletion of common EV markers with the depletion of markers of abundant cell type-specific EVs to further enrich for rare EV populations, the case for each additional depletion target needs to be considered. For example, combining CD9 depletion with CD41 depletion to remove platelet EVs (as the most abundant EVs by cell type in plasma) would have a negligible effect since 98% of CD41-positive EVs are CD9-positive (Khanna et al., 2023).
Moreover, depleting common EV markers could enable the isolation of relatively pure cell type-specific EV populations compared to using positive selection alone by removing contaminating EVs that also display the positive selection target (and so are co-isolated during immunoisolation). For example, CD9 is highly expressed by PNS neurons but is not expressed by neurons in the brain (Figure 2, Table 1, Table 2), indicating that negative selection to deplete CD9 will deplete CD9-positive PNS neuronal EVs and select for CNS neuronal EVs for biomarker analysis. Thus, CD9 depletion could be used to overcome the molecular overlap of positive selection targets shared between neurons of the CNS and PNS (Table 1). Accordingly, CD9 has been used as a marker to distinguish cells of the CNS and PNS by immunofluorescence microscopy and single-cell RNA sequencing (Ishibashi et al., 2004; Zeisel et al., 2018). This suggests that a strategy of CD9 depletion in combination with positive selection of neuron-enriched proteins could be used to separate brain-derived neuronal EVs (and associated biomarkers) from EVs released by PNS neurons and other cell types, which could not be achieved using a negative selection approach targeting e.g. platelet EVs for depletion.
Untouched isolation to select for tissue-derived extracellular vesicles
Whilst a combination of positive and negative selection is required for isolation of specific EV populations from a single cellular source, negative selection by itself has also been employed to enrich for rare EVs derived from tissue (as depicted in Figure 1C). Yu et al. (2022) developed an ‘untouched’ isolation strategy by using immunodepletion to separate tumour cell-derived EVs from heterogenous tissue-derived EVs without the need for a positive isolation step. After enriching for EVs from tissue interstitial fluid using an enzymatic digestion and ultracentrifugation protocol, non-tumour cell EVs were depleted by targeting CD45, CD144, CD41, and CD235 (removing leukocyte, endothelial, platelet, and erythrocyte EVs, respectively). By simultaneously depleting EVs from these four different cell types, which had previously constituted 62.4% of the total EV pool (52.4% leukocyte EVs, 5.1% endothelial EVs, 3.4% platelet EVs, 1.5% erythrocyte EVs), the proportion of tumour-derived EVs in the EV preparation increased from 37.6% (pre-negative selection) to 92.5% (post-negative selection) (Yu et al., 2022).
There were multiple reasons for omitting positive immunoaffinity isolation of the desired EV population. Firstly, it eliminated the need to identify a cell type-specific EV marker for positive selection of the tumour EV population and enabled enrichment of the entire heterogenous assortment of tumour cell-derived EVs. This overcame a limitation of positive selection strategies that only select for EVs bearing the positive selection target, whilst disregarding EV subpopulations released by the cell of interest that do not carry that marker, potentially missing EVs with biomarker cargo that may inform about the disease state (Ramirez et al., 2018).
Secondly, since the tumour-derived EVs did not go through the immunocapture procedure, there was no need to separate the EVs from an antibody-coated surface, which can require non-physiological conditions (e.g. acid treatment/high-salt concentrations), as well as being time-consuming and inefficient in eluting intact EVs (Karimi et al., 2022). Moreover, following EV elution, antibodies remaining bound to the positive selection target on the EV surface can prevent subsequent antibody binding in detection assays, alter the protein’s structure and distribution on the membrane, and block EV internalisation by recipient cells (Chen et al., 2018; de la Torre-Escudero et al., 2019, Hoshino et al., 2015). Therefore, the untouched negative selection strategy enabled the natural properties of the EVs of interest to be preserved (Yu et al., 2022).
Thirdly, negative selection can be relatively time and labour-efficient since it does not require the washing steps performed in positive selection to separate off-target molecules adsorbed on the immunocapture surface, or an elution step (Kabe et al., 2019). For example, Sharafeldin et al. (2023) demonstrated that the majority of CD9-positive EVs could be depleted within 30 minutes from neat serum, where they constitute 75-86% of total EVs (Hoshino et al., 2020; Sharafeldin et al., 2023). This was achieved by using a magnetic field to promote interactions between EVs and the magnetic immunocapture beads within a microfluidics device (Sharafeldin et al., 2023). Therefore, the single-step negative selection approach could shorten the duration and hands-on processing time of clinical assays for EV-based biomarkers, whilst amplifying the biomarker signal-to-noise ratio by removing abundant EV populations unrelated to the disease state.
Based on these advantages of omitting the positive selection step, the ‘untouched’ tumour cell-derived EVs enriched by Yu et al. (2022) were maintained in a biocompatible form (termed ‘natural EVs’ by the authors) during the isolation procedure, which enabled direct experimentation with biological systems. It was found that these natural tumour cell-derived EVs remained detectable in circulation almost four times longer (120 minutes) than the tissue-derived EVs (pre-negative selection), which only remained in circulation for 40 minutes after being injected into the tail veins of mice (Yu et al., 2022). Additionally, the tumour cell-derived EVs displayed a specific tropism for the lymph nodes, whereas the heterogenous tissue-derived EVs did not display the same tissue distribution specificity. Moreover, EVs derived from an in vitro cell line corresponding to the tumour type (oral squamous cell carcinoma) did not interact with CD8 T cells and B cells to the same extent as the ex vivo tumour cell-derived EVs. This demonstrates the limitations of EVs isolated from in vitro cultures in replicating the biological effects of EVs produced in a physiological environment (Yu et al., 2022).
The purity of EVs derived from a single cellular source obtained by Yu et al. (2022) from tissue using negative selection alone might be difficult to achieve in more complex biofluids such as plasma due to the diversity of cells that contribute to the total EV pool. However, single-step negative selection may be sufficient to amplify the signal-to-noise ratio of pre-determined EV-based biomarkers in plasma that had previously been identified through positive selection or combined positive and negative selection strategies. For the abovementioned reasons, this would facilitate the enrichment and direct detection of EV-based biomarkers derived from a single cellular origin without the need for further sample processing (e.g. positive selection), especially if the negative selection antibodies could be integrated into a microfluidics device (Yu et al., 2022).
Alternatively, the ‘untouched’ single-step negative selection approach could be used for biomarker discovery by unveiling correlative changes in peripheral EVs that mirror disease states in tissue. Profiling total EVs from blood (without positive or negative selection) does not yield cell type-specific disease information (Fordjour et al., 2023). However, it has been used to identify biomarker signatures in blood samples through changes in peripheral EV miRNA content in Alzheimer’s disease and amyotrophic lateral sclerosis (Cheng et al., 2020; Vassileff et al., 2024). This method has the advantage that it does not require isolation of cell type-specific EVs, so the identified biomarkers could feasibly be used in clinical tests with minimal processing of the sample. Incorporating single-step negative selection could benefit this total-profiling approach by removing abundant EVs to unmask biomarker signatures that had otherwise been obscured, whilst only adding a limited amount of hands-on labour.
The use of negative EV markers to screen for contaminating EV populations in rare EV isolates has also been proposed to determine the origin of detected biomarker signals before more comprehensive analysis and clarify whether the biological signal truly comes from the EVs of interest (Newman et al., 2023). The introduction of negative markers as a screening tool could be particularly useful where positive markers alone are unable to distinguish two EV populations released by closely related cell types (for example, EVs from neurons of the CNS and PNS).
Selection of future targets to isolate biomarkers in rare extracellular vesicle populations
There are now many datasets from which candidate targets for positive and negative selection of rare EV populations can be identified. These include the Human Protein Atlas (proteinatlas.org, Uhlén et al., 2015) and the Mouse Brain Atlas (mousebrain.org; Zeisel et al., 2018), as well as EV proteomics atlases of cells, tissues, biofluids, and disease states (Hoshino et al., 2020; Huang et al., 2023; Kugeratski et al., 2021; Liu et al., 2022). However, after selecting a candidate EV target, the case of L1CAM exemplifies the biological and technological factors that could affect cell type-specific EV isolation and may influence the interpretation of biological signals in biomarker studies.
These factors include alternative (non-EV) mechanisms of secretion of the candidate immunoisolation target, such as those involving alternatively spliced forms or ectodomain shedding (Norman et al., 2021). For ectodomain shedding, there are databases (SheddomeDB; Huang et al., 2023), predictive tools (DEEPSMP; Cao et al., 2020), and tissue-specific studies (e.g. for the mouse brain; Lobete et al., 2024) that can indicate whether a membrane protein is likely to be shed through proteolytic processing. Relatedly, 71% of identified glycoproteins in the murine neuronal secretome have been shown to be shed ectodomains (Tüshaus et al., 2020) and as many as 8% of annotated human transmembrane proteins are predicted to undergo ectodomain shedding (Tien et al., 2017). Whilst alternative secretory mechanisms do not preclude the utility of a candidate target for EV immunoisolation (Nogueras-Ortiz et al., 2024), they may need to be considered as potential confounding factors during the experimental process. In that regard, proteinase K assays have been used to show that the targeted protein domains exist outside of the EV lumen (You et al., 2023) and could be used to clarify the association of transmembrane immunocapture targets with EVs (Figure 2C), as well as differentiate intravesicular biomarker cargo from free, soluble forms of the same protein (Gilboa et al., 2024).
Negative selection approaches have emerged as alternative enrichment strategies to positive selection alone, with advantages that include enhancing the biomarker signal and preserving natural properties of EVs (Ganesh et al., 2023; Ko et al., 2016; Notarangelo et al., 2019; Yu et al., 2022). Whilst ideal targets for positive selection are specific for EVs from the cell type of interest, negative selection targets benefit from being general markers of EVs to deplete diverse, non-target EV populations (Almousa et al., 2025). Therefore, shifting away from using CD9, CD63, and CD81 to define EV populations irrespective of EV cellular and subcellular origins, and taking advantage of EV marker heterogeneity (Kugeratski et al., 2021; Spitzberg et al., 2023; Su et al., 2024), could facilitate discovery of common EV markers as negative selection targets.
Incorporating negative selection into EV biomarker detection protocols could also promote future clinical translation by reducing pre-analytical variability caused by the release of EVs into blood samples ex vivo. Platelet EVs released during blood processing and storage are a major source of contamination with EVs enriched in CD41, CD42, CD62, and CD9 that are not related to the disease state (Małys et al., 2023) and the degree of contamination varies depending on methodological differences (Karimi et al., 2022). Therefore, depletion of platelet-derived EVs by targeting platelet-enriched proteins will facilitate standardisation in the detection of biomarkers in EVs by removing contaminating platelet EVs which could mask the biomarker signal to differing extents depending on ex vivo treatment of the blood sample.
Additionally, context-dependent biological factors need to be accounted for in EV immunoisolation studies since the presence of surface markers on EVs can change depending on cell activation states (Yang et al., 2018), pathological conditions, or the age and sex of the sample donor (Huang et al., 2025). Similarly, technical factors during the EV isolation procedure can influence the presence of surface markers on EVs. For example, enzymatic digestion of tissue to access EVs within the intercellular space was shown to also digest exposed domains of EV proteins, affecting the recovery of certain markers on EVs derived from brain tissue (Matamoros-Angles et al., 2024). Moreover, EV isolation methods used to separate total EVs from biofluids, and freezing of the sample, both differentially impact recovery of certain EV populations and, thus, the presence of the targeted EV population in the sample (Droste et al., 2021; Gelibter et al., 2022; Paget et al., 2022).
Finally, the utility of a candidate for immunoisolation may depend on the availability of a commercial antibody for the targeted molecule since generating and validating new antibodies can be time consuming, costly, and technically challenging. Advances in affinity agents, like the development of oligonucleotide aptamers targeting CD9, CD63, and PTK7 (An et al., 2023; Xue et al., 2021; Zhang et al., 2019), may reduce the costs associated with scalable antibody production for clinical assays of novel EV-based biomarkers (Roy et al., 2021) and enable capture and release of intact EVs under physiological conditions (Brambilla et al., 2021). Additionally, the use of emerging biosensing technologies, which enable real-time analysis of binding kinetics to EVs (Yildizhan et al., 2021), could streamline the process of optimising binding agents for immunoisolation. With the correct affinity agent, the capture efficiency for the targeted EV population can be as high as 100% (Dias et al., 2024; Fortunato et al., 2022), facilitating the recovery of low abundance EV populations for detection of their biomarker cargo in clinical assays.
Grant information
Adam P.S. Bennett was supported by Horizon Europe Marie Skłodowska-Curie Actions grant agreement number 101067960.
Data statement
No data are associated with this article.
Competing interests
Adam P.S. Bennett is the inventor on a patent application relating to methods for the isolation and analysis of central nervous system-derived neuronal extracellular vesicles (WO/2025/259235).
References
- Almousa S, Kim S, Kumar A, Su Y, Singh S, Mishra S, Fonseca MM, Rather HA, Romero-Sandoval EA, Hsu FC, Singh R, Yadav H, Mishra S, Deep G. Bacterial nanovesicles as interkingdom signaling moieties mediating pain hypersensitivity. ACS Nano. 2025 Jan 28;19(3):3210-3225. [CrossRef]
- Amin S, Massoumi H, Tewari D, Roy A, Chaudhuri M, Jazayerli C, Krishan A, Singh M, Soleimani M, Karaca EE, Mirzaei A, Guaiquil VH, Rosenblatt MI, Djalilian AR, Jalilian E. Cell type-specific extracellular vesicles and their impact on health and disease. Int J Mol Sci. 2024 Feb 27;25(5):2730. [CrossRef]
- An J, Park H, Kim J, Park H, Kim TH, Park C, Kim J, Lee MH, Lee T. Extended-gate field-effect transistor consisted of a CD9 aptamer and MXene for exosome detection in human serum. ACS Sens. 2023 Aug 25;8(8):3174-3186.
- Arduise C, Abache T, Li L, Billard M, Chabanon A, Ludwig A, Mauduit P, Boucheix C, Rubinstein E, Le Naour F. Tetraspanins regulate ADAM10-mediated cleavage of TNF-alpha and epidermal growth factor. J Immunol. 2008 Nov 15;181(10):7002-13. [CrossRef]
- Arystarkhova E, Sweadner KJ. Isoform-specific monoclonal antibodies to Na,K-ATPase alpha subunits. Evidence for a tissue-specific post-translational modification of the alpha subunit. J Biol Chem. 1996 Sep 20;271(38):23407-17.
- Ashrafzadeh-Kian S, Figdore D, Larson B, Deters R, Abou-Diwan C, Bornhorst J, Algeciras-Schimnich A. Head-to-head comparison of four plasma neurofilament light chain (NfL) immunoassays. Clin Chim Acta. 2024 Jul 15;561:119817. [CrossRef]
- Athauda D, Gulyani S, Karnati HK, Li Y, Tweedie D, Mustapic M, Chawla S, Chowdhury K, Skene SS, Greig NH, Kapogiannis D, Foltynie T. Utility of neuronal-derived exosomes to examine molecular mechanisms that affect motor function in patients with Parkinson disease: a secondary analysis of the exenatide-pd trial. JAMA Neurol. 2019 Apr 1;76(4):420-429.
- Auber M, Svenningsen P. An estimate of extracellular vesicle secretion rates of human blood cells. J Extracell Biol. 2022 Jun 2;1(6):e46. [CrossRef]
- Banerjee SA, Patterson PH. Schwann cell CD9 expression is regulated by axons. Mol Cell Neurosci. 1995 Oct;6(5):462-73. [CrossRef]
- Banks WA, Sharma P, Bullock KM, Hansen KM, Ludwig N, Whiteside TL. Transport of extracellular vesicles across the blood-brain barrier: brain pharmacokinetics and effects of inflammation. Int J Mol Sci. 2020 Jun 21;21(12):4407. [CrossRef]
- Blommer J, Pitcher T, Mustapic M, Eren E, Yao PJ, Vreones MP, Pucha KA, Dalrymple-Alford J, Shoorangiz R, Meissner WG, Anderson T, Kapogiannis D. Extracellular vesicle biomarkers for cognitive impairment in Parkinson’s disease. Brain. 2023 Jan 5;146(1):195-208. [CrossRef]
- Brambilla D, Sola L, Ferretti AM, Chiodi E, Zarovni N, Fortunato D, Criscuoli M, Dolo V, Giusti I, Murdica V, Kluszczyńska K, Czernek L, Düchler M, Vago R, Chiari M. EV Separation: Release of intact extracellular vesicles immunocaptured on magnetic particles. Anal Chem. 2021 Apr 6;93(13):5476-5483. [CrossRef]
- Bravo-Miana RDC, Arizaga-Echebarria JK, Sabas-Ortega V, Crespillo-Velasco H, Prada A, Castillo-Triviño T, Otaegui D. Tetraspanins, GLAST and L1CAM quantification in single extracellular vesicles from cerebrospinal fluid and serum of people with multiple sclerosis. Biomedicines. 2024 Oct 2;12(10):2245. [CrossRef]
- Cao Z, Du W, Li G, Cao H. DEEPSMP: A deep learning model for predicting the ectodomain shedding events of membrane proteins. J Bioinform Comput Biol. 2020 Jun;18(3):2050017. [CrossRef]
- Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, Yu Z, Yang J, Wang B, Sun H, Xia H, Man Q, Zhong W, Antelo LF, Wu B, Xiong X, Liu X, Guan L, Li T, Liu S, Yang R, Lu Y, Dong L, McGettigan S, Somasundaram R, Radhakrishnan R, Mills G, Lu Y, Kim J, Chen YH, Dong H, Zhao Y, Karakousis GC, Mitchell TC, Schuchter LM, Herlyn M, Wherry EJ, Xu X, Guo W. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018 Aug;560(7718):382-386. [CrossRef]
- Chen MM, Lee CY, Leland HA, Lin GY, Montgomery AM, Silletti S. Inside-out regulation of L1 conformation, integrin binding, proteolysis, and concomitant cell migration. Mol Biol Cell. 2010 May 15;21(10):1671-85. [CrossRef]
- Chen Z, Li W, Meng B, Xu C, Huang Y, Li G, Wen Z, Liu J, Mao Z. Neuronal-enriched small extracellular vesicles trigger a PD-L1-mediated broad suppression of T cells in Parkinson’s disease. iScience. 2024 Jun 11;27(7):110243. [CrossRef]
- Cheng L, Vella LJ, Barnham KJ, McLean C, Masters CL, Hill AF. Small RNA fingerprinting of Alzheimer’s disease frontal cortex extracellular vesicles and their comparison with peripheral extracellular vesicles. J Extracell Vesicles. 2020 Jun 4;9(1):1766822. [CrossRef]
- Ciferri MC, Quarto R, Tasso R. extracellular vesicles as biomarkers and therapeutic tools: from pre-clinical to clinical applications. Biology (Basel). 2021 Apr 23;10(5):359. [CrossRef]
- Dagur RS, Liao K, Sil S, Niu F, Sun Z, Lyubchenko YL, Peeples ES, Hu G, Buch S. Neuronal-derived extracellular vesicles are enriched in the brain and serum of HIV-1 transgenic rats. J Extracell Vesicles. 2019 Dec 20;9(1):1703249. [CrossRef]
- de la Torre-Escudero E, Gerlach JQ, Bennett APS, Cwiklinski K, Jewhurst HL, Huson KM, Joshi L, Kilcoyne M, O’Neill S, Dalton JP, Robinson MW. Surface molecules of extracellular vesicles secreted by the helminth pathogen Fasciola hepatica direct their internalisation by host cells. PLoS Negl Trop Dis. 2019 Jan 18;13(1):e0007087. [CrossRef]
- Dias T, Figueiras R, Vagueiro S, Domingues R, Hung YH, Sethi J, Persia E, Arsène P. An electro-optical platform for the ultrasensitive detection of small extracellular vesicle sub-types and their protein epitope counts. iScience. 2024 Apr 30;27(6):109866. [CrossRef]
- Doh-Ura K, Mekada E, Ogomori K, Iwaki T. Enhanced CD9 expression in the mouse and human brains infected with transmissible spongiform encephalopathies. J Neuropathol Exp Neurol. 2000 Sep;59(9):774-85. [CrossRef]
- Droste M, Tertel T, Jeruschke S, Dittrich R, Kontopoulou E, Walkenfort B, Börger V, Hoyer PF, Büscher AK, Thakur BK, Giebel B. Single extracellular vesicle analysis performed by imaging flow cytometry and nanoparticle tracking analysis evaluate the accuracy of urinary extracellular vesicle preparation techniques differently. Int J Mol Sci. 2021 Nov 18;22(22):12436. [CrossRef]
- Dunlop RA, Banack SA, Cox PA. L1CAM immunocapture generates a unique extracellular vesicle population with a reproducible miRNA fingerprint. RNA Biol. 2023 Jan;20(1):140-148. [CrossRef]
- Dutta S, Hornung S, Kruayatidee A, Maina KN, Del Rosario I, Paul KC, Wong DY, Duarte Folle A, Markovic D, Palma JA, Serrano GE, Adler CH, Perlman SL, Poon WW, Kang UJ, Alcalay RN, Sklerov M, Gylys KH, Kaufmann H, Fogel BL, Bronstein JM, Ritz B, Bitan G. α-Synuclein in blood exosomes immunoprecipitated using neuronal and oligodendroglial markers distinguishes Parkinson’s disease from multiple system atrophy. Acta Neuropathol. 2021 Sep;142(3):495-511.
- Dutta S, Hornung S, Taha HB, Bitan G. Biomarkers for parkinsonian disorders in CNS-originating EVs: promise and challenges. Acta Neuropathol. 2023 May;145(5):515-540. [CrossRef]
- Eitan E, Thornton-Wells T, Elgart K, Erden E, Gershun E, Levine A, Volpert O, Azadeh M, Smith DG, Kapogiannis D. Synaptic proteins in neuron-derived extracellular vesicles as biomarkers for Alzheimer’s disease: novel methodology and clinical proof of concept. Extracell Vesicles Circ Nucl Acids. 2023 Mar;4(1):133-150. [CrossRef]
- Fiandaca MS, Kapogiannis D, Mapstone M, Boxer A, Eitan E, Schwartz JB, Abner EL, Petersen RC, Federoff HJ, Miller BL, Goetzl EJ. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case-control study. Alzheimers Dement. 2015 Jun;11(6):600-7.e1. [CrossRef]
- Fordjour FK, Abuelreich S, Hong X, Chatterjee E, Lallai V, Ng M, Saftics A, Deng F, Carnel-Amar N, Wakimoto H, Shimizu K, Bautista M, Phu TA, Vu NK, Geiger PC, Raffai RL, Fowler CD, Das S, Christenson LK, Jovanovic-Talisman T, Gould SJ. Exomap1 mouse: a transgenic model for in vivo studies of exosome biology. Extracell Vesicle. 2023 Dec;2:100030. [CrossRef]
- Foroni C, Zarovni N, Bianciardi L, Bernardi S, Triggiani L, Zocco D, Venturella M, Chiesi A, Valcamonico F, Berruti A. When less is more: specific capture and analysis of tumor exosomes in plasma increases the sensitivity of liquid biopsy for comprehensive detection of multiple androgen receptor phenotypes in advanced prostate cancer patients. Biomedicines. 2020 May 22;8(5):131. [CrossRef]
- Fortunato D, Giannoukakos S, Giménez-Capitán A, Hackenberg M, Molina-Vila MA, Zarovni N. Selective isolation of extracellular vesicles from minimally processed human plasma as a translational strategy for liquid biopsies. Biomark Res. 2022 Aug 7;10(1):57. [CrossRef]
- Ganesh S, Lam TT, Garcia-Milian R, Cyril D’Souza D, Nairn AC, Elgert K, Eitan E, Ranganathan M. Peripheral signature of altered synaptic integrity in young onset cannabis use disorder: A proteomic study of circulating extracellular vesicles. World J Biol Psychiatry. 2023 Sep-Oct;24(7):603-613.
- Gelibter S, Marostica G, Mandelli A, Siciliani S, Podini P, Finardi A, Furlan R. The impact of storage on extracellular vesicles: A systematic study. J Extracell Vesicles. 2022 Feb;11(2):e12162.
- Gilboa T, Ter-Ovanesyan D, Wang SC, Whiteman S, Kannarkat GT, Church GM, Chen-Plotkin AS, Walt DR. Measurement of α-synuclein as protein cargo in plasma extracellular vesicles. Proc Natl Acad Sci U S A. 2024 Nov 5;121(45):e2408949121. [CrossRef]
- Girbes Minguez M, Wolters-Eisfeld G, Lutz D, Buck F, Schachner M, Kleene R. The cell adhesion molecule L1 interacts with nuclear proteins via its intracellular domain. FASEB J. 2020 Aug;34(8):9869-9883.
- Gomes DE, Witwer KW. L1CAM-associated extracellular vesicles: a systematic review of nomenclature, sources, separation, and characterization. J Extracell Biol. 2022 Mar;1(3):e35. [CrossRef]
- Gutiérrez-López MD, Gilsanz A, Yáñez-Mó M, Ovalle S, Lafuente EM, Domínguez C, Monk PN, González-Alvaro I, Sánchez-Madrid F, Cabañas C. The sheddase activity of ADAM17/TACE is regulated by the tetraspanin CD9. Cell Mol Life Sci. 2011 Oct;68(19):3275-92. [CrossRef]
- Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, Singh S, Williams C, Soplop N, Uryu K, Pharmer L, King T, Bojmar L, Davies AE, Ararso Y, Zhang T, …Lyden D. Tumour exosome integrins determine organotropic metastasis. Nature. 2015 Nov 19;527(7578):329-35. [CrossRef]
- Hoshino A, Kim HS, Bojmar L, Gyan KE, Cioffi M, Hernandez J, Zambirinis CP, Rodrigues G, Molina H, Heissel S, Mark MT, Steiner L, Benito-Martin A, Lucotti S, Di Giannatale A, Offer K, Nakajima M, Williams C, Nogués L, Pelissier Vatter FA, …Lyden D. Extracellular vesicle and particle biomarkers define multiple human cancers. Cell. 2020 Aug 20;182(4):1044-1061.e18. [CrossRef]
- Hotta N, Tadokoro T, Henry J, Koga D, Kawata K, Ishida H, Oguma Y, Hirata A, Mitsuhashi M, Yoshitani K. Monitoring of post-brain injuries by measuring plasma levels of neuron-derived extracellular vesicles. Biomark Insights. 2022 Oct 26;17:11772719221128145. [CrossRef]
- Huang WY, Wu KP. SheddomeDB 2023: A Revision of an ectodomain shedding database based on a comprehensive literature review and online resources. J Proteome Res. 2023 Aug 4;22(8):2570-2576.
- Huang Y, Arab T, Russell AE, Mallick ER, Nagaraj R, Gizzie E, Redding-Ochoa J, Troncoso JC, Pletnikova O, Turchinovich A, Routenberg DA, Witwer KW. Toward a human brain extracellular vesicle atlas: characteristics of extracellular vesicles from different brain regions, including small RNA and protein profiles. Interdiscip Med. 2023 Oct;1(4):e20230016. [CrossRef]
- Huang Y, Feng J, Xu J, Dong L, Su W, Li B, Witwer KW, Zheng L. Associations of age and sex with characteristics of extracellular vesicles and protein-enriched fractions of blood plasma. Aging Cell. 2025 Jan;24(1):e14356. [CrossRef]
- Ishibashi T, Ding L, Ikenaka K, Inoue Y, Miyado K, Mekada E, Baba H. Tetraspanin protein CD9 is a novel paranodal component regulating paranodal junctional formation. J Neurosci. 2004 Jan 7;24(1):96-102. [CrossRef]
- Jeyaram A, Jay SM. Preservation and storage stability of extracellular vesicles for therapeutic applications. AAPS J. 2017 Nov 27;20(1):1. [CrossRef]
- Johnsen KB, Gudbergsson JM, Andresen TL, Simonsen JB. What is the blood concentration of extracellular vesicles? Implications for the use of extracellular vesicles as blood-borne biomarkers of cancer. Biochim Biophys Acta Rev Cancer. 2019 Jan;1871(1):109-116. [CrossRef]
- Jouannet S, Saint-Pol J, Fernandez L, Nguyen V, Charrin S, Boucheix C, Brou C, Milhiet PE, Rubinstein E. TspanC8 tetraspanins differentially regulate the cleavage of ADAM10 substrates, Notch activation and ADAM10 membrane compartmentalization. Cell Mol Life Sci. 2016 May;73(9):1895-915. [CrossRef]
- Kabe Y, Sakamoto S, Hatakeyama M, Yamaguchi Y, Suematsu M, Itonaga M, Handa H. Application of high-performance magnetic nanobeads to biological sensing devices. Anal Bioanal Chem. 2019 Mar;411(9):1825-1837. [CrossRef]
- Kagawa T, Mekada E, Shishido Y, Ikenaka K. Immune system-related CD9 is expressed in mouse central nervous system myelin at a very late stage of myelination. J Neurosci Res. 1997 Oct 15;50(2):312-20. [CrossRef]
- Kaprielian Z, Cho KO, Hadjiargyrou M, Patterson PH. CD9, a major platelet cell surface glycoprotein, is a ROCA antigen and is expressed in the nervous system. J Neurosci. 1995 Jan;15(1 Pt 2):562-73. [CrossRef]
- Karimi N, Dalirfardouei R, Dias T, Lötvall J, Lässer C. Tetraspanins distinguish separate extracellular vesicle subpopulations in human serum and plasma - Contributions of platelet extracellular vesicles in plasma samples. J Extracell Vesicles. 2022 May;11(5):e12213. [CrossRef]
- Khanna K, Salmond N, Halvaei S, Johnson A, Williams KC. Separation and isolation of CD9-positive extracellular vesicles from plasma using flow cytometry. Nanoscale Adv. 2023 Jul 13;5(17):4435-4446. [CrossRef]
- Kluge A, Bunk J, Schaeffer E, Drobny A, Xiang W, Knacke H, Bub S, Lückstädt W, Arnold P, Lucius R, Berg D, Zunke F. Detection of neuron-derived pathological α-synuclein in blood. Brain. 2022 Sep 14;145(9):3058-3071. [CrossRef]
- Ko J, Hemphill MA, Gabrieli D, Wu L, Yelleswarapu V, Lawrence G, Pennycooke W, Singh A, Meaney DF, Issadore D. Smartphone-enabled optofluidic exosome diagnostic for concussion recovery. Sci Rep. 2016 Aug 8;6:31215. [CrossRef]
- Ko J, Wang Y, Sheng K, Weitz DA, Weissleder R. Sequencing-based protein analysis of single extracellular vesicles. ACS Nano. 2021 Mar 23;15(3):5631-5638. [CrossRef]
- Kugeratski FG, Hodge K, Lilla S, McAndrews KM, Zhou X, Hwang RF, Zanivan S, Kalluri R. Quantitative proteomics identifies the core proteome of exosomes with syntenin-1 as the highest abundant protein and a putative universal biomarker. Nat Cell Biol. 2021 Jun;23(6):631-641. [CrossRef]
- Kuhn PH, Koroniak K, Hogl S, Colombo A, Zeitschel U, Willem M, Volbracht C, Schepers U, Imhof A, Hoffmeister A, Haass C, Roßner S, Bräse S, Lichtenthaler SF. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012 Jun 22;31(14):3157-68. [CrossRef]
- Kumar A, Sharma M, Su Y, Singh S, Hsu FC, Neth BJ, Register TC, Blennow K, Zetterberg H, Craft S, Deep G. Small extracellular vesicles in plasma reveal molecular effects of modified Mediterranean-ketogenic diet in participants with mild cognitive impairment. Brain Commun. 2022 Oct 19;4(6):fcac262. [CrossRef]
- Ladang A, Kovacs S, Lengelé L, Locquet M, Reginster JY, Bruyère O, Cavalier E. Neurofilament light chain concentration in an aging population. Aging Clin Exp Res. 2022 Feb;34(2):331-339. [CrossRef]
- Li Y, He X, Li Q, Lai H, Zhang H, Hu Z, Li Y, Huang S. EV-origin: Enumerating the tissue-cellular origin of circulating extracellular vesicles using exLR profile. Comput Struct Biotechnol J. 2020 Oct 14;18:2851-2859. [CrossRef]
- Liu CJ, Xie GY, Miao YR, Xia M, Wang Y, Lei Q, Zhang Q, Guo AY. EVAtlas: a comprehensive database for ncRNA expression in human extracellular vesicles. Nucleic Acids Res. 2022 Jan 7;50(D1):D111-D117. [CrossRef]
- Llorens-Bobadilla E, Zhao S, Baser A, Saiz-Castro G, Zwadlo K, Martin-Villalba A. Single-cell transcriptomics reveals a population of dormant neural stem cells that become activated upon brain injury. Cell Stem Cell. 2015 Sep 3;17(3):329-40. [CrossRef]
- Lobete M, Salinas T, Izquierdo-Bermejo S, Socas S, Oset-Gasque MJ, Martín-de-Saavedra MD. A methodology to globally assess ectodomain shedding using soluble fractions from the mouse brain. Front Psychiatry. 2024 Jun 19;15:1367526. [CrossRef]
- Malong L, Napoli I, Casal G, White IJ, Stierli S, Vaughan A, Cattin AL, Burden JJ, Hng KI, Bossio A, Flanagan A, Zhao HT, Lloyd AC. Characterization of the structure and control of the blood-nerve barrier identifies avenues for therapeutic delivery. Dev Cell. 2023 Feb 6;58(3):174-191.e8. [CrossRef]
- Małys MS, Köller MC, Papp K, Aigner C, Dioso D, Mucher P, Schachner H, Bonelli M, Haslacher H, Rees AJ, Kain R. Small extracellular vesicles are released ex vivo from platelets into serum and from residual blood cells into stored plasma. J Extracell Biol. 2023 May 12;2(5):e88. [CrossRef]
- Maretzky T, Schulte M, Ludwig A, Rose-John S, Blobel C, Hartmann D, Altevogt P, Saftig P, Reiss K. L1 is sequentially processed by two differently activated metalloproteases and presenilin/gamma-secretase and regulates neural cell adhesion, cell migration, and neurite outgrowth. Mol Cell Biol. 2005 Oct;25(20):9040-53. [CrossRef]
- Matamoros-Angles A, Karadjuzovic E, Mohammadi B, Song F, Brenna S, Meister SC, Siebels B, Voß H, Seuring C, Ferrer I, Schlüter H, Kneussel M, Altmeppen HC, Schweizer M, Puig B, Shafiq M, Glatzel M. Efficient enzyme-free isolation of brain-derived extracellular vesicles. J Extracell Vesicles. 2024 Nov;13(11):e70011.
- Mondal SK, Whiteside TL. Immunoaffinity-based isolation of melanoma cell-derived and T cell-derived exosomes from plasma of melanoma patients. Methods Mol Biol. 2021; 2265:305-321.
- Morton MC, Neckles VN, Seluzicki CM, Holmberg JC, Feliciano DM. Neonatal subventricular zone neural stem cells release extracellular vesicles that act as a microglial morphogen. Cell Rep. 2018 Apr 3;23(1):78-89. [CrossRef]
- Mustapic M, Eitan E, Werner JK Jr, Berkowitz ST, Lazaropoulos MP, Tran J, Goetzl EJ, Kapogiannis D. Plasma extracellular vesicles enriched for neuronal origin: a potential window into brain pathologic processes. Front Neurosci. 2017 May 22;11:278. [CrossRef]
- Mustapic M, Tran J, Craft S, Kapogiannis D. Extracellular vesicle biomarkers track cognitive changes following intranasal insulin in Alzheimer’s disease. J Alzheimers Dis. 2019;69(2):489-498. [CrossRef]
- Nakamura Y, Iwamoto R, Mekada E. Expression and distribution of CD9 in myelin of the central and peripheral nervous systems. Am J Pathol. 1996 Aug;149(2):575-83.
- Newman LA, Useckaite Z, Wu T, Sorich MJ, Rowland A. Analysis of extracellular vesicle and contaminant markers in blood derivatives using multiple reaction monitoring. Methods Mol Biol. 2023;2628:301-320.
- Nogueras-Ortiz CJ, Eren E, Yao P, Calzada E, Dunn C, Volpert O, Delgado-Peraza F, Mustapic M, Lyashkov A, Rubio FJ, Vreones M, Cheng L, You Y, Hill AF, Ikezu T, Eitan E, Goetzl EJ, Kapogiannis D. Single-extracellular vesicle (EV) analyses validate the use of L1 Cell Adhesion Molecule (L1CAM) as a reliable biomarker of neuron-derived EVs. J Extracell Vesicles. 2024 Jun;13(6):e12459. [CrossRef]
- Norman M, Ter-Ovanesyan D, Trieu W, Lazarovits R, Kowal EJK, Lee JH, Chen-Plotkin AS, Regev A, Church GM, Walt DR. L1CAM is not associated with extracellular vesicles in human cerebrospinal fluid or plasma. Nat Methods. 2021 Jun;18(6):631-634. [CrossRef]
- Notarangelo M, Zucal C, Modelska A, Pesce I, Scarduelli G, Potrich C, Lunelli L, Pederzolli C, Pavan P, la Marca G, Pasini L, Ulivi P, Beltran H, Demichelis F, Provenzani A, Quattrone A, D’Agostino VG. Ultrasensitive detection of cancer biomarkers by nickel-based isolation of polydisperse extracellular vesicles from blood. EBioMedicine. 2019 May;43:114-126. [CrossRef]
- Paget D, Checa A, Zöhrer B, Heilig R, Shanmuganathan M, Dhaliwal R, Johnson E, Jørgensen MM, Bæk R; Oxford Acute Myocardial Infarction Study (OxAMI); Wheelock CE, Channon KM, Fischer R, Anthony DC, Choudhury RP, Akbar N. Comparative and integrated analysis of plasma extracellular vesicle isolation methods in healthy volunteers and patients following myocardial infarction. J Extracell Biol. 2022 Nov 23;1(11):e66. [CrossRef]
- Polanco JC, Li C, Durisic N, Sullivan R, Götz J. Exosomes taken up by neurons hijack the endosomal pathway to spread to interconnected neurons. Acta Neuropathol Commun. 2018 Feb 15;6(1):10. [CrossRef]
- Ramirez MI, Amorim MG, Gadelha C, Milic I, Welsh JA, Freitas VM, Nawaz M, Akbar N, Couch Y, Makin L, Cooke F, Vettore AL, Batista PX, Freezor R, Pezuk JA, Rosa-Fernandes L, Carreira ACO, Devitt A, Jacobs L, Silva IT, Coakley G, Nunes DN, Carter D, Palmisano G, Dias-Neto E. Technical challenges of working with extracellular vesicles. Nanoscale. 2018 Jan 18;10(3):881-906. [CrossRef]
- Reyes R, Cardeñes B, Machado-Pineda Y, Cabañas C. Tetraspanin CD9: A key regulator of cell adhesion in the immune system. Front Immunol. 2018 Apr 30;9:863. [CrossRef]
- Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, Piech T, Patel PP, Chang L, Rivnak AJ, Ferrell EP, Randall JD, Provuncher GK, Walt DR, Duffy DC. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol. 2010 Jun;28(6):595-9. [CrossRef]
- Roy D, Pascher A, Juratli MA, Sporn JC. The potential of aptamer-mediated liquid biopsy for early detection of cancer. Int J Mol Sci. 2021 May 25;22(11):5601. [CrossRef]
- Rufino-Ramos D, Lule S, Mahjoum S, Ughetto S, Cristopher Bragg D, Pereira de Almeida L, Breakefield XO, Breyne K. Using genetically modified extracellular vesicles as a non-invasive strategy to evaluate brain-specific cargo. Biomaterials. 2022 Feb;281:121366. [CrossRef]
- Rufino-Ramos D, Leandro K, Perdigão PRL, O’Brien K, Pinto MM, Santana MM, van Solinge TS, Mahjoum S, Breakefield XO, Breyne K, Pereira de Almeida L. Extracellular communication between brain cells through functional transfer of Cre mRNA mediated by extracellular vesicles. Mol Ther. 2023 Jul 5;31(7):2220-2239. [CrossRef]
- Salmond N, Khanna K, Owen GR, Williams KC. Nanoscale flow cytometry for immunophenotyping and quantitating extracellular vesicles in blood plasma. Nanoscale. 2021 Jan 28;13(3):2012-2025. [CrossRef]
- Sanchez-Arias JC, Candlish RC, van der Slagt E, Swayne LA. Pannexin 1 regulates dendritic protrusion dynamics in immature cortical neurons. eNeuro. 2020 Aug 26;7(4):ENEURO.0079-20.2020. [CrossRef]
- Schaeffer E, Kluge A, Schulte C, Deuschle C, Bunk J, Welzel J, Maetzler W, Berg D. association of misfolded α-synuclein derived from neuronal exosomes in blood with parkinson’s disease diagnosis and duration. J Parkinsons Dis. 2024;14(4):667-679. [CrossRef]
- Schmidt C, Künemund V, Wintergerst ES, Schmitz B, Schachner M. CD9 of mouse brain is implicated in neurite outgrowth and cell migration in vitro and is associated with the alpha 6/beta 1 integrin and the neural adhesion molecule L1. J Neurosci Res. 1996 Jan 1;43(1):12-31. [CrossRef]
- Sharafeldin M, Yan S, Jiang C, Tofaris GK, Davis JJ. Alternating magnetic field-promoted nanoparticle mixing: the on-chip immunocapture of serum neuronal exosomes for Parkinson’s disease diagnostics. Anal Chem. 2023 May 23;95(20):7906-7913. [CrossRef]
- Spitzberg JD, Ferguson S, Yang KS, Peterson HM, Carlson JCT, Weissleder R. Multiplexed analysis of EV reveals specific biomarker composition with diagnostic impact. Nat Commun. 2023 Mar 4;14(1):1239. [CrossRef]
- Su X, Júnior GPO, Marie AL, Gregus M, Figueroa-Navedo A, Ghiran IC, Ivanov AR. Enhanced proteomic profiling of human plasma-derived extracellular vesicles through charge-based fractionation to advance biomarker discovery potential. J Extracell Vesicles. 2024 Dec;13(12):e70024. [CrossRef]
- Taha HB. Rethinking the reliability and accuracy of biomarkers in CNS-originating EVs for Parkinson’s disease and multiple system atrophy. Front Neurol. 2023 Sep 5;14:1192115. [CrossRef]
- Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin-Smith GK, Ayre DC, Bach JM, Bachurski D, Baharvand H, Balaj L, Baldacchino S, Bauer NN, Baxter AA, Bebawy M, Beckham C, …Zuba-Surma EK. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018 Nov 23;7(1):1535750. [CrossRef]
- Tien WS, Chen JH, Wu KP. SheddomeDB: the ectodomain shedding database for membrane-bound shed markers. BMC Bioinformatics. 2017 Mar 14;18(Suppl 3):42. [CrossRef]
- Tole S, Patterson PH. Distribution of CD9 in the developing and mature rat nervous system. Dev Dyn. 1993 Jun;197(2):94-106. [CrossRef]
- Tordoff E, Allen J, Elgart K, Elsherbini A, Kalia V, Wu H, Eren E, Kapogiannis D, Gololobova O, Witwer K, Volpert O, Eitan E. A novel multiplexed immunoassay for surface-exposed proteins in plasma extracellular vesicles. J Extracell Vesicles. 2024 Nov;13(11):e70007. [CrossRef]
- Tóth EÁ, Turiák L, Visnovitz T, Cserép C, Mázló A, Sódar BW, Försönits AI, Petővári G, Sebestyén A, Komlósi Z, Drahos L, Kittel Á, Nagy G, Bácsi A, Dénes Á, Gho YS, Szabó-Taylor KÉ, Buzás EI. Formation of a protein corona on the surface of extracellular vesicles in blood plasma. J Extracell Vesicles. 2021 Sep;10(11):e12140. [CrossRef]
- Trivedi R, Bhat KP. Liquid biopsy: creating opportunities in brain space. Br J Cancer. 2023 Nov;129(11):1727-1746. [CrossRef]
- Truffi M, Garofalo M, Ricciardi A, Cotta Ramusino M, Perini G, Scaranzin S, Gastaldi M, Albasini S, Costa A, Chiavetta V, Corsi F, Morasso C, Gagliardi S. Neurofilament-light chain quantification by Simoa and Ella in plasma from patients with dementia: a comparative study. Sci Rep. 2023 Mar 10;13(1):4041. [CrossRef]
- Tüshaus J, Müller SA, Kataka ES, Zaucha J, Sebastian Monasor L, Su M, Güner G, Jocher G, Tahirovic S, Frishman D, Simons M, Lichtenthaler SF. An optimized quantitative proteomics method establishes the cell type-resolved mouse brain secretome. EMBO J. 2020 Oct 15;39(20):e105693. [CrossRef]
- Tutrone R, Lowentritt B, Neuman B, Donovan MJ, Hallmark E, Cole TJ, Yao Y, Biesecker C, Kumar S, Verma V, Sant GR, Alter J, Skog J. ExoDx prostate test as a predictor of outcomes of high-grade prostate cancer - an interim analysis. Prostate Cancer Prostatic Dis. 2023 Sep;26(3):596-601. [CrossRef]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, …Pontén F. Proteomics. Tissue-based map of the human proteome. Science. 2015 Jan 23;347(6220):1260419. [CrossRef]
- UniProt Consortium. UniProt: the universal protein knowledgebase in 2025. Nucleic Acids Res. 2025 Jan 6;53(D1):D609-D617.
- Vassileff N, Spiers JG, Lee JD, Woodruff TM, Ebrahimie E, Mohammadi Dehcheshmeh M, Hill AF, Cheng L. A panel of mirna biomarkers common to serum and brain-derived extracellular vesicles identified in mouse model of amyotrophic lateral sclerosis. Mol Neurobiol. 2024 Aug;61(8):5901-5915. [CrossRef]
- van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018 Apr;19(4):213-228. [CrossRef]
- Welsh JA, Goberdhan DCI, O’Driscoll L, Buzas EI, Blenkiron C, Bussolati B, Cai H, Di Vizio D, Driedonks TAP, Erdbrügger U, Falcon-Perez JM, Fu QL, Hill AF, Lenassi M, Lim SK, Mahoney MG, Mohanty S, Möller A, Nieuwland R, Ochiya T, …Witwer KW. Minimal information for studies of extracellular vesicles (MISEV2023): from basic to advanced approaches. J Extracell Vesicles. 2024 Feb;13(2):e12404. doi: 10.1002/jev2.12404. Erratum in: J Extracell Vesicles. 2024 May;13(5):e12451. [CrossRef]
- Xue F, Chen Y, Wen Y, Abhange K, Zhang W, Cheng G, Quinn Z, Mao W, Wan Y. Isolation of extracellular vesicles with multivalent aptamers. Analyst. 2021 Jan 4;146(1):253-261.
- Yan S, Jiang C, Davis JJ, Tofaris GK. Methodological considerations in neuronal extracellular vesicle isolation for α-synuclein biomarkers. Brain. 2023 Nov 2;146(11):e95-e97. [CrossRef]
- Yang Y, Boza-Serrano A, Dunning CJR, Clausen BH, Lambertsen KL, Deierborg T. Inflammation leads to distinct populations of extracellular vesicles from microglia. J Neuroinflammation. 2018 May 28;15(1):168. [CrossRef]
- Yildizhan Y, Vajrala VS, Geeurickx E, Declerck C, Duskunovic N, De Sutter D, Noppen S, Delport F, Schols D, Swinnen JV, Eyckerman S, Hendrix A, Lammertyn J, Spasic D. FO-SPR biosensor calibrated with recombinant extracellular vesicles enables specific and sensitive detection directly in complex matrices. J Extracell Vesicles. 2021 Feb;10(4):e12059. [CrossRef]
- You Y, Zhang Z, Sultana N, Ericsson M, Martens YA, Sun M, Kanekiyo T, Ikezu S, Shaffer SA, Ikezu T. ATP1A3 as a target for isolating neuron-specific extracellular vesicles from human brain and biofluids. Sci Adv. 2023 Sep 15;9(37):eadi3647.
- Yu ZL, Liu XC, Wu M, Shi S, Fu QY, Jia J, Chen G. Untouched isolation enables targeted functional analysis of tumour-cell-derived extracellular vesicles from tumour tissues. J Extracell Vesicles. 2022 Apr;11(4):e12214. [CrossRef]
- Zarovni N, Mladenović D, Brambilla D, Panico F, Chiari M. Stoichiometric constraints for detection of EV-borne biomarkers in blood. J Extracell Vesicles. 2025 Feb;14(2):e70034. [CrossRef]
- Zanirati G, Dos Santos PG, Alcará AM, Bruzzo F, Ghilardi IM, Wietholter V, Xavier FAC, Gonçalves JIB, Marinowic D, Shetty AK, da Costa JC. Extracellular vesicles: the next generation of biomarkers and treatment for central nervous system diseases. Int J Mol Sci. 2024 Jul 5;25(13):7371. [CrossRef]
- Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, Häring M, Braun E, Borm LE, La Manno G, Codeluppi S, Furlan A, Lee K, Skene N, Harris KD, Hjerling-Leffler J, Arenas E, Ernfors P, Marklund U, Linnarsson S. Molecular architecture of the mouse nervous system. Cell. 2018 Aug 9;174(4):999-1014.e22. [CrossRef]
- Zhang K, Yue Y, Wu S, Liu W, Shi J, Zhang Z. Rapid capture and nondestructive release of extracellular vesicles using aptamer-based magnetic isolation. ACS Sens. 2019 May 24;4(5):1245-1251. [CrossRef]
- Zhao Z, Fan J, Hsu YS, Lyon CJ, Ning B, Hu TY. Extracellular vesicles as cancer liquid biopsies: from discovery, validation, to clinical application. Lab Chip. 2019 Mar 27;19(7):1114-1140. [CrossRef]
- Zhou E, Li Y, Wu F, Guo M, Xu J, Wang S, Tan Q, Ma P, Song S, Jin Y. Circulating extracellular vesicles are effective biomarkers for predicting response to cancer therapy. EBioMedicine. 2021 May;67:103365. [CrossRef]
Figure 1.
A simplified illustration of positive and negative selection strategies to select for rare extracellular vesicle (EV) populations for biomarker discovery and detection. (A) Single-step positive selection enriches for the targeted EV population but co-isolates non-target EVs displaying the marker for positive selection. (B) A combination of positive and negative selection, where non-target EVs (including those displaying the positive selection marker) are removed by negative selection, enabling the isolation of a relatively homogeneous EV population. Negative selection also enriches the proportion of the targeted rare EV population (and associated biomarkers) by removing the majority of contaminating EVs. (C) Negative selection alone to enrich for a rare EV population for biomarker detection or discovery.
Figure 1.
A simplified illustration of positive and negative selection strategies to select for rare extracellular vesicle (EV) populations for biomarker discovery and detection. (A) Single-step positive selection enriches for the targeted EV population but co-isolates non-target EVs displaying the marker for positive selection. (B) A combination of positive and negative selection, where non-target EVs (including those displaying the positive selection marker) are removed by negative selection, enabling the isolation of a relatively homogeneous EV population. Negative selection also enriches the proportion of the targeted rare EV population (and associated biomarkers) by removing the majority of contaminating EVs. (C) Negative selection alone to enrich for a rare EV population for biomarker detection or discovery.
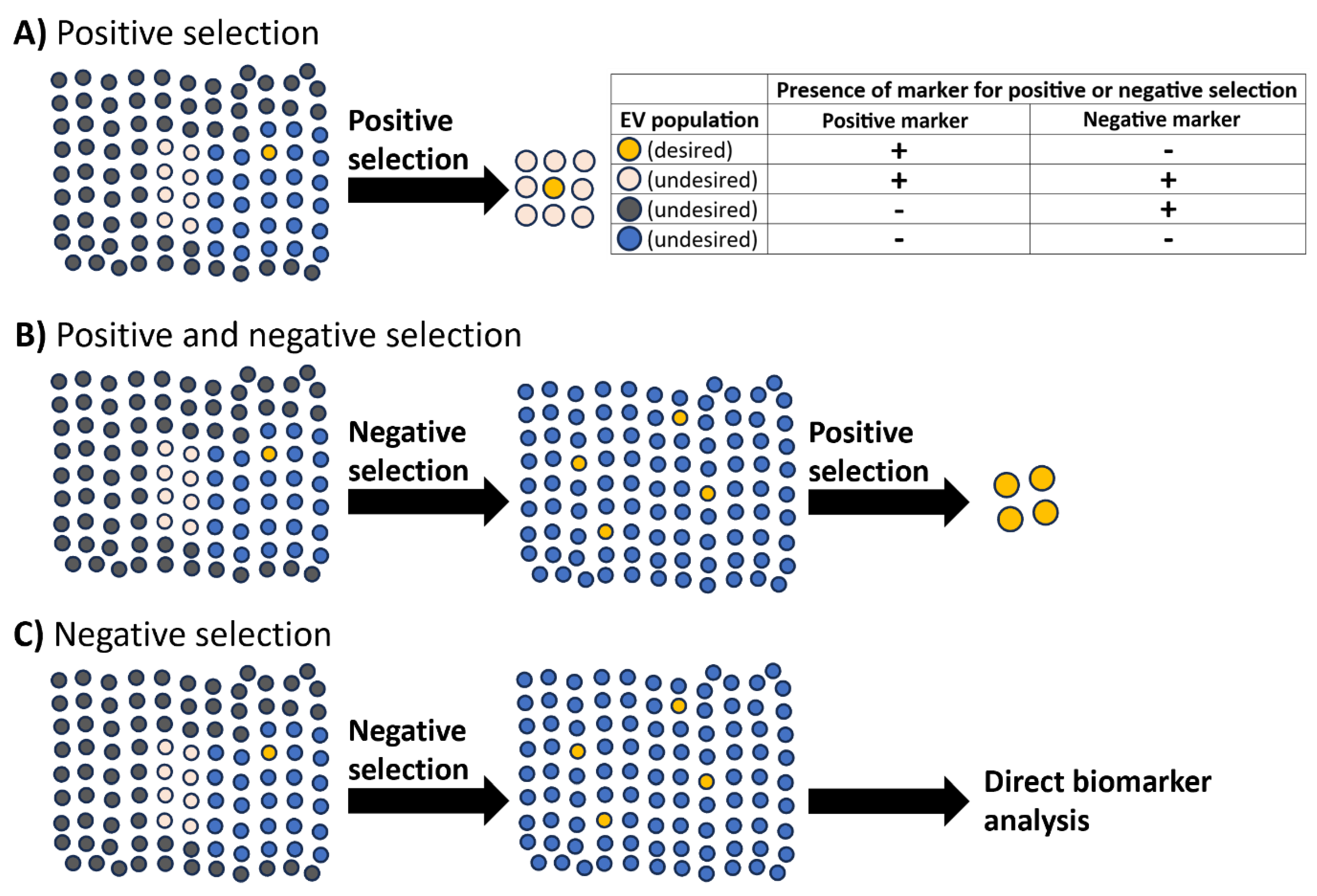
Figure 2.
Simplified illustrations of L1CAM at the extracellular vesicle (EV) surface and the effects of proteinase K treatment. (A) The anticipated single-pass transmembrane form of L1CAM in the extracellular vesicle (EV) membrane, where the intraluminal location of the internal domain does not permit binding by antibodies targeting the L1CAM internal domain. (B) An alternative model where the soluble isoform of L1CAM (lacking the transmembrane domain) is adhered to the EV surface, enabling binding by the L1CAM internal domain antibody. (C) With the transmembrane form of L1CAM seen in (A), proteinase K digests the exposed external domain of L1CAM, but the internal domain remains protected by the EV lipid bilayer, enabling the subsequent detection of the internal domain in downstream assays. (D) Proteinase K digests both the internal and external domains of L1CAM when the soluble isoform of L1CAM is adhered to the EV surface (or free in solution), as shown in (B). Consequently, the internal domain of L1CAM would not be detected in downstream assays. Of note, both forms of L1CAM seen in (A) and (B) may exist on the same EV or within the same EV preparation.
Figure 2.
Simplified illustrations of L1CAM at the extracellular vesicle (EV) surface and the effects of proteinase K treatment. (A) The anticipated single-pass transmembrane form of L1CAM in the extracellular vesicle (EV) membrane, where the intraluminal location of the internal domain does not permit binding by antibodies targeting the L1CAM internal domain. (B) An alternative model where the soluble isoform of L1CAM (lacking the transmembrane domain) is adhered to the EV surface, enabling binding by the L1CAM internal domain antibody. (C) With the transmembrane form of L1CAM seen in (A), proteinase K digests the exposed external domain of L1CAM, but the internal domain remains protected by the EV lipid bilayer, enabling the subsequent detection of the internal domain in downstream assays. (D) Proteinase K digests both the internal and external domains of L1CAM when the soluble isoform of L1CAM is adhered to the EV surface (or free in solution), as shown in (B). Consequently, the internal domain of L1CAM would not be detected in downstream assays. Of note, both forms of L1CAM seen in (A) and (B) may exist on the same EV or within the same EV preparation.
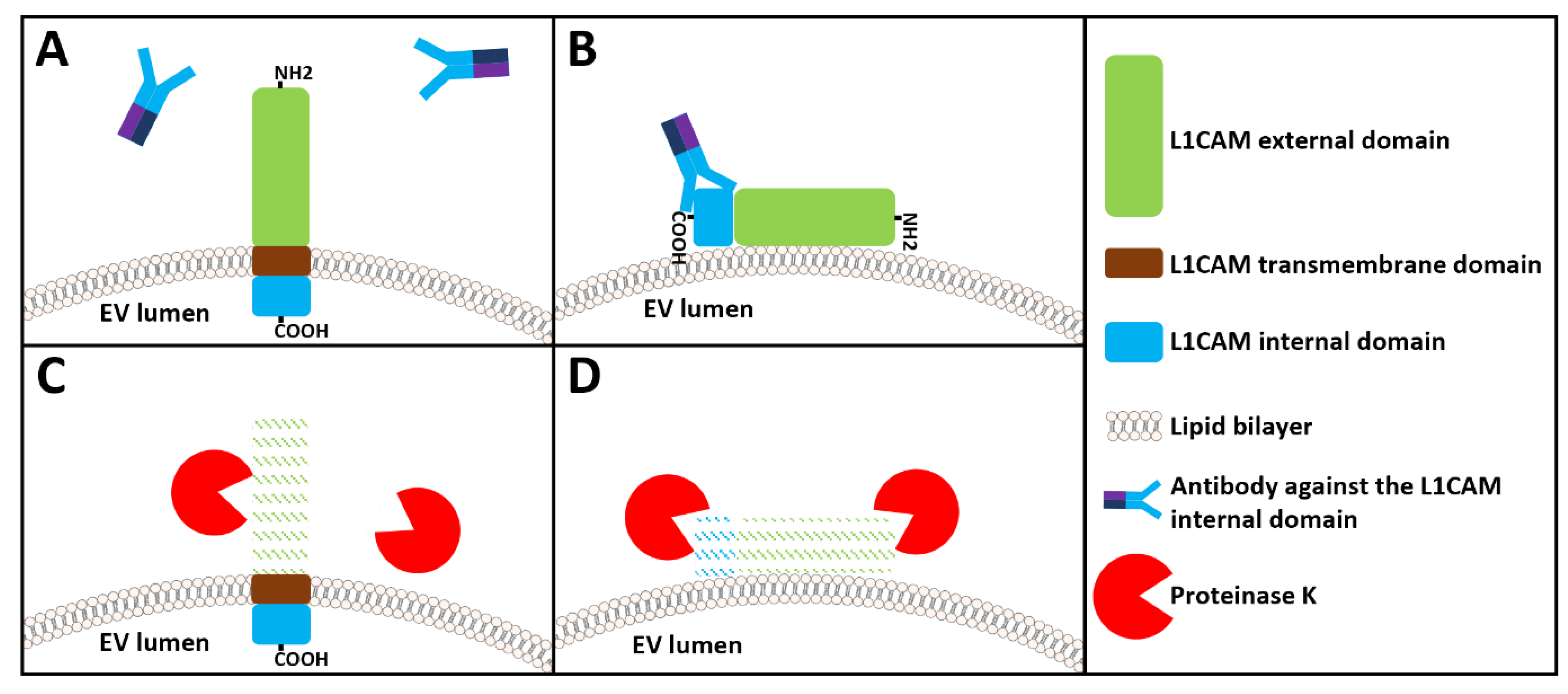
Figure 3.
Summary of CD9 expression in the nervous system. (A) The distribution of CD9 expression in different brain regions and the location summary for CD9 expression in the brain displayed in the Human Protein Atlas. (B) Summary of neuronal CD9 protein detection in the brain from the Human Protein Atlas (where neuronal CD9 detection data is available). (C) Single-cell RNA expression profile for CD9 in the brain, adapted from the Human Protein Atlas. (D) Heatmap of single-cell RNA sequencing data from the Mouse Brain Atlas showing the distribution of CD9 expression in neuronal and non-neuronal cells in the adolescent mouse nervous system; darker blue indicates higher expression. Panel A-C image credit: Human Protein Atlas (Uhlén et al., 2015) with images reproduced from v24.0 proteinatlas.org/ENSG00000010278-CD9/tissue and proteinatlas.org/ENSG00000010278-CD9/brain (A and B) and adapted from v24.0 proteinatlas.org/ENSG00000010278-CD9/single+cell (C) under the Creative Commons Attribution-ShareAlike 4.0 International License. Panel D image credit: Mouse Brain Atlas reproduced with permission (http://mousebrain.org/genes/Cd9.html; Zeisel et al., 2018).
Figure 3.
Summary of CD9 expression in the nervous system. (A) The distribution of CD9 expression in different brain regions and the location summary for CD9 expression in the brain displayed in the Human Protein Atlas. (B) Summary of neuronal CD9 protein detection in the brain from the Human Protein Atlas (where neuronal CD9 detection data is available). (C) Single-cell RNA expression profile for CD9 in the brain, adapted from the Human Protein Atlas. (D) Heatmap of single-cell RNA sequencing data from the Mouse Brain Atlas showing the distribution of CD9 expression in neuronal and non-neuronal cells in the adolescent mouse nervous system; darker blue indicates higher expression. Panel A-C image credit: Human Protein Atlas (Uhlén et al., 2015) with images reproduced from v24.0 proteinatlas.org/ENSG00000010278-CD9/tissue and proteinatlas.org/ENSG00000010278-CD9/brain (A and B) and adapted from v24.0 proteinatlas.org/ENSG00000010278-CD9/single+cell (C) under the Creative Commons Attribution-ShareAlike 4.0 International License. Panel D image credit: Mouse Brain Atlas reproduced with permission (http://mousebrain.org/genes/Cd9.html; Zeisel et al., 2018).
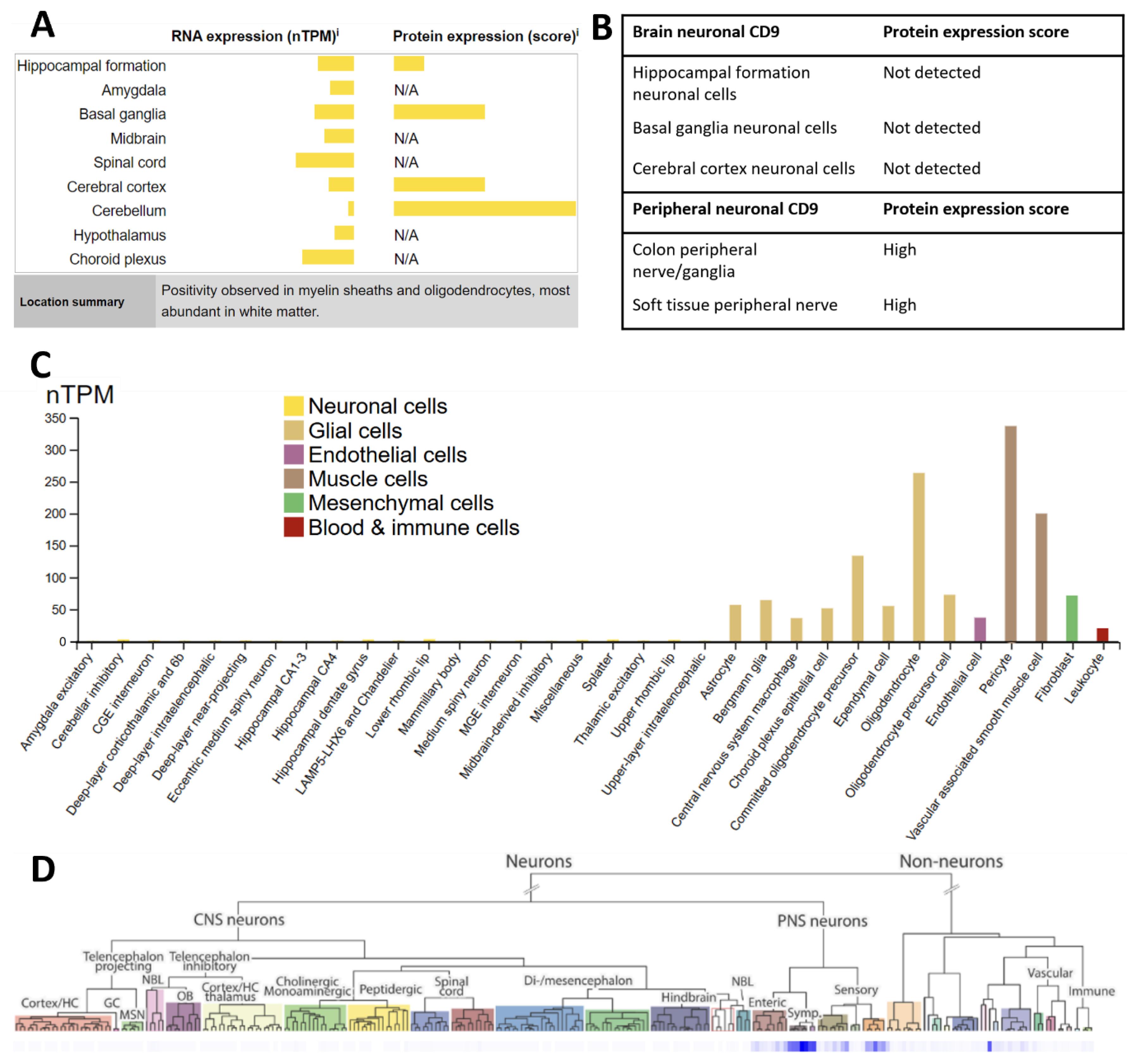

Table 1.
Expression of L1CAM, VAMP2, β-III-tubulin (TUBB3), GAP43, and CD9 in neurons of the central and peripheral nervous system (of mice), as well as their concentration in human plasma. Comparative expression levels (with arbitrary units) for neurons of the central nervous system (CNS) and peripheral nervous system (PNS) were obtained from mousebrain.org (Zeisel et al., 2018) because quantitative transcriptomic data for PNS neurons is not available in the Human Protein Atlas. N.B. CD9 expression in CNS neurons was only detected in one neuronal subtype. Protein concentrations in plasma were obtained from the Human Protein Atlas version 25.0 (Uhlén et al., 2015) at proteinatlas.org/ENSG00000198910-L1CAM, /ENSG00000220205-VAMP2, /ENSG00000258947-TUBB3, /ENSG00000172020-GAP43, and /ENSG00000010278-CD9.
Table 1.
Expression of L1CAM, VAMP2, β-III-tubulin (TUBB3), GAP43, and CD9 in neurons of the central and peripheral nervous system (of mice), as well as their concentration in human plasma. Comparative expression levels (with arbitrary units) for neurons of the central nervous system (CNS) and peripheral nervous system (PNS) were obtained from mousebrain.org (Zeisel et al., 2018) because quantitative transcriptomic data for PNS neurons is not available in the Human Protein Atlas. N.B. CD9 expression in CNS neurons was only detected in one neuronal subtype. Protein concentrations in plasma were obtained from the Human Protein Atlas version 25.0 (Uhlén et al., 2015) at proteinatlas.org/ENSG00000198910-L1CAM, /ENSG00000220205-VAMP2, /ENSG00000258947-TUBB3, /ENSG00000172020-GAP43, and /ENSG00000010278-CD9.
| Protein | Highest expression level in CNS neurons | CNS neuron subtype showing highest expression | Highest expression level in PNS neurons | PNS neuron subtype showing highest expression | Concentration in human plasma |
|---|---|---|---|---|---|
| L1CAM | 2.3 |
Cholinergic neurons, hindbrain (rhombencephalon) | 7.4 | Cholinergic enteric neurons (neural crest) | 13 ng/ml |
| VAMP2 | 15.7 | Excitatory neurons, hindbrain (rhombencephalon) | 13.0 | Noradrenergic erector muscle neurons (neural crest) | 5.2 ng/ml |
| β-III-tubulin | 18.3 | Cholinergic neurons, septal nucleus, Meissnert and diagonal band (diencephalon) | 115.1 | Noradrenergic neurons, sympathetic (neural crest) | 71 ng/ml |
| GAP43 | 42.7 | Cholinergic neurons, septal nucleus, Meissnert and diagonal band (diencephalon) | 79.7 | Noradrenergic erector muscle neurons (neural crest) | 0.029 ng/ml |
| CD9 | 0.9 | Central canal neurons, spinal cord (spinal cord) | 79.7 | Noradrenergic erector muscle neurons (neural crest) | 4.2 ng/ml |
Table 2.
Studies investigating the expression of CD9 in the nervous system and a summary of the cells in which CD9 was detected. Briefly, CD9 expression was detected in some mouse and rat embryonic and neonatal neurons in the central nervous system (CNS), but CNS neuronal CD9 expression disappeared as the animals developed. In non-neuronal cells, CD9 was detected in neural stem cells (and neural stem cell extracellular vesicles), myelinating oligodendrocytes, astrocytes, ependymal cells, and microglia. In the peripheral nervous system (PNS), CD9 was detected in peripheral neurons, satellite cells, and myelinating Schwann cells.
Table 2.
Studies investigating the expression of CD9 in the nervous system and a summary of the cells in which CD9 was detected. Briefly, CD9 expression was detected in some mouse and rat embryonic and neonatal neurons in the central nervous system (CNS), but CNS neuronal CD9 expression disappeared as the animals developed. In non-neuronal cells, CD9 was detected in neural stem cells (and neural stem cell extracellular vesicles), myelinating oligodendrocytes, astrocytes, ependymal cells, and microglia. In the peripheral nervous system (PNS), CD9 was detected in peripheral neurons, satellite cells, and myelinating Schwann cells.
| Publication | Organism | CD9 expression summary and detection method |
|---|---|---|
| Banerjee and Patterson, 1995 | Rat (neonatal and embryonic) |
|
| Doh-Uhra et al., 2000 | Mouse and human (adult) |
|
| Ishibashi et al., 2004 | Mouse (8 weeks old) |
|
| Kagawa et al., 1997 | Mouse (postnatal day 10 to adults) |
|
| Kaprielian et al., 1995 | Rat (adult) |
|
| Llorens-Bobadilla et al., 2015 | Mouse (8-12 weeks old) |
|
| Morton et al., 2018 | Mouse (neonatal) |
|
| Nakamura et al., 1996 | Human (adult) |
|
| Polanco et al., 2018 | Mouse (embryonic) |
|
| Sanchez-Arias et al., 2020 | Mouse (neonatal) |
|
| Schmidt et al., 1996 | Rats (11-day old embryos, postnatal day 4, and adult) and mouse (6 day old) |
|
| Tole and Patterson, 1993 | Rat (embryonic and adult) |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



