Submitted:
30 December 2025
Posted:
31 December 2025
You are already at the latest version
Abstract
Neurodegenerative research has long hypothesized that aggregated proteins such as amyloid‑β (Aβ), tau, and α‑synuclein (αSyn) are intrinsically toxic and are directly associated with the etiologies of Alzheimer’s disease (AD) and Parkinson’s disease (PD). However, emerging scientific evidence challenges this view. Plasma p‑tau217 shows weak correlation with cognitive severity, αSyn seed amplification assays provide only binary diagnostic support, and anti‑amyloid monoclonal antibodies yield modest short-term benefit while increasing amyloid-related imaging abnormality (ARIA) risk. Postmortem pathology and fluid biomarkers explain only a limited amount of variance in clinical outcomes, undermining their role as surrogate endpoints. We propose a biophysical framework in which aggregation reflects a supersaturation-driven phase transition that signals depletion of soluble, functional monomers rather than the emergence of toxic species. Within this paradigm, amyloid plaques, neurofibrillary tangles, and Lewy bodies represent tombstones of lost protein function, and neurodegeneration occurs when monomer supply falls below neuronal demand. This shift has practical implications for biomarker interpretation, staging, and therapeutic design. Future directions include quantifying monomer flux using stable-isotope labeling kinetics (SILK), integrating supply and demand ratios, and prioritizing mechanism-testing trials that restore protein homeostasis rather than indiscriminately clear aggregates. By reframing pathology as a marker of stress rather than a maker of disease, this approach may enable more effective precision therapeutics based on human biology.
Keywords:
1. Introduction
2. Biomarker–Clinical Mismatch in AD
2.1. The p-tau217 “Mismatch”: When Clinical Severity Diverges from Pathology
2.2. Resilience and Compensation: Pathology as Tombstone of Function
3. Reinterpreting Aβ Biology: From Toxic Waste to Essential Peptide
3.1. The Aβ42 Depletion Hypothesis and γ-Secretase Function
3.2. OLE Illusions and Survivor Bias Under Anti-Aβ mAbs
3.3. Aβ as Protector: Evidence Against Intrinsic Neurotoxicity
4. αSyn: What Seed Amplification Assays Can and Cannot Tell Us
4.1. Binary Diagnostic Support, Not a Quantitative Severity Readout
4.2. Protective Role and the Paradox of Pathology-Based Severity
4.3. Pathology Without Toxicity: Signals from Peripheral Autonomic Nerves
5. Prion Paradigms, Strains, and the Physics of Aggregation
5.1. Replication vs. Precipitation: Why Prionization Analogies Mislead
5.2. Kinetic vs Thermodynamic Control in Amyloid Formation
6. Infection, Surface Catalysis, and Protein Depletion
7. Precision Medicine in PD and AD: From Epidemiology to Hypothesis-Testing Trials
7.1. The Pharmaco-Epidemiology Disconnect
7.2. Three Guiding Principles for PD Research
8. Clinical Staging and the Risks of Surrogate Determinism
9. Therapeutic Lessons Across Neurodegenerations
9.1. Anti-Aβ mAbs: Mechanism of Modest Benefit and Safety Trade-Offs
9.2. PD Anti-αSyn Strategies and Bayesian Re-Analyses
9.3. Lessons From Other Neurodegenerations
10. Counter-Arguments and Open Questions
11. Practical Implications and Future Directions
11.1. Biomarker Use
11.2. Quantifying Monomer Flux in Humans
11.3. Mapping Neuronal Demand Relative to Supply
11.4. Therapeutic-Strategy Trials
11.4.1. CSF Aβ42 Augmentation
11.4.2. Neutralizing Surface Catalysis
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ADAD | Autosomal dominant Alzheimer's Disease |
| Anti-Aβ mAbs | Anti-amyloid-β monoclonal antibodies |
| Aβ | Amyloid-β |
| ARIA | Amyloid-related imaging abnormalities |
| CBS | Corticobasal syndrome |
| CDR-SB | Clinical dementia rating–sum of boxes |
| CSF | Cerebrospinal fluid |
| DSAD | Down syndrome-associated Alzheimer's Disease |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MCI | Mild cognitive impairment |
| NDI | Neuronal demand index |
| NfL | Neurofilament light chain |
| OLE | Open-label extension |
| PD | Parkinson’s disease |
| PSEN1 | Presenilin-1 |
| SAA | Seed amplification assays |
| SILK | Stable isotope labelling kinetics |
| TDP-43 | TAR DNA-binding protein 43 |
| α-Synuclein | αSyn |
Appendix A

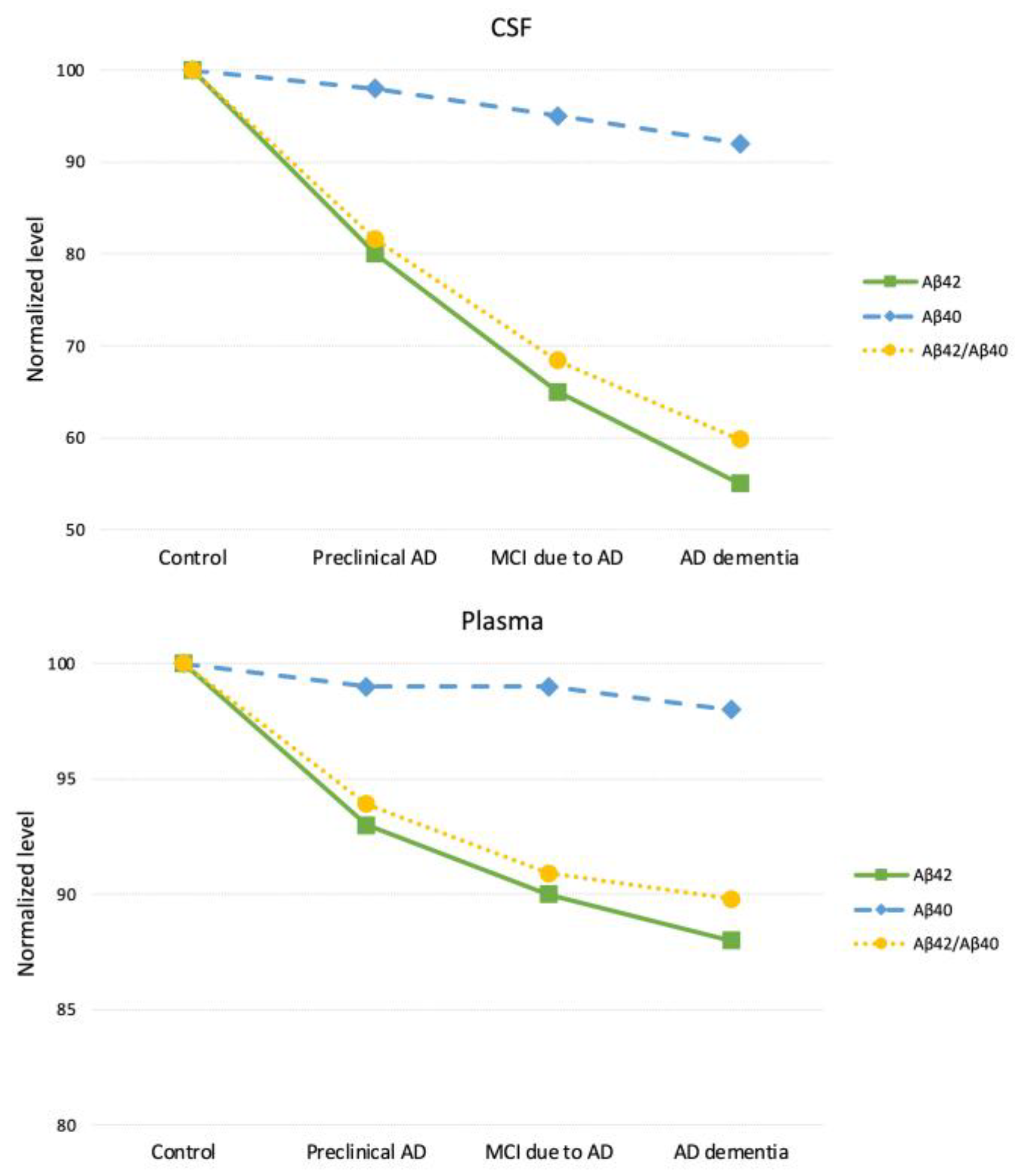
| Matrix | CSF Aβ42/Aβ40 | CSF Aβ42/Aβ40 (normalized) | Plasma Aβ42/Aβ40 | Plasma Aβ42/Aβ40 (normalized) |
|---|---|---|---|---|
| Control | 0.093 | 100 | 0.132 | 100 |
| Preclinical AD | 0.076 | 81.6 | 0.124 | 93.9 |
| MCI due to AD | 0.064 | 68.4 | 0.120 | 90.9 |
| AD Dementia | 0.056 | 59.8 | 0.119 | 89.8 |
| Assay | Electrochemiluminescence immunoassay | Immunoprecipitation-Mass Spectrometry | ||
| Reference | Hansson et al. (2018) [42] Wojdała et al. (2023) [70] |
Nakamura et al. (2018) [69] Wojdała et al. (2023) [70] |
||
References
- Bigi, A.; Lombardo, E.; Cascella, R.; Cecchi, C. The Toxicity of Protein Aggregates: New Insights into the Mechanisms. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Lovestone, S.; McLoughlin, D.M. Protein Aggregates and Dementia: Is There a Common Toxicity? J Neurol Neurosurg Psychiatry 2002, 72, 152–161. [Google Scholar] [CrossRef]
- Tsoi, P.S.; Quan, M.D.; Ferreon, J.C.; Ferreon, A.C.M. Aggregation of Disordered Proteins Associated with Neurodegeneration. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Chopra, A.; Lang, A.E.; Höglinger, G.; Outeiro, T.F. Towards a Biological Diagnosis of PD. Parkinsonism Relat Disord 2024, 122, 106078. [Google Scholar] [CrossRef]
- Pitton Rissardo, J.; Fornari Caprara, A. Disease-Modifying Therapies for Parkinson’s Disease: Biological Mechanisms, Pharmacological Strategies, and Clinical Pipeline. Preprints 2025. [Google Scholar] [CrossRef]
- Keefe, R.S.E.; Kraemer, H.C.; Epstein, R.S.; Frank, E.; Haynes, G.; Laughren, T.P.; McNulty, J.; Reed, S.D.; Sanchez, J.; Leon, A.C. Defining a Clinically Meaningful Effect for the Design and Interpretation of Randomized Controlled Trials. Innov Clin Neurosci 2013, 10, 4S–19S. [Google Scholar] [PubMed]
- Abanto, J.; Dwivedi, A.K.; Imbimbo, B.P.; Espay, A.J. Increases in Amyloid-Β42 Slow Cognitive and Clinical Decline in Alzheimer’s Disease Trials. Brain 2024, 147, 3513–3521. [Google Scholar] [CrossRef] [PubMed]
- Fabrizi, E.; Ancidoni, A.; Locuratolo, N.; Piscopo, P.; Della Gatta, F.; Salemme, S.; Pani, S.M.; Marconi, D.; Vignatelli, L.; Sagliocca, L.; et al. The Italian Guideline on Diagnosis and Treatment of Dementia and Mild Cognitive Impairment. Age Ageing 2024, 53. [Google Scholar] [CrossRef]
- Ashton, N.J.; Brum, W.S.; Di Molfetta, G.; Benedet, A.L.; Arslan, B.; Jonaitis, E.; Langhough, R.E.; Cody, K.; Wilson, R.; Carlsson, C.M.; et al. Diagnostic Accuracy of a Plasma Phosphorylated Tau 217 Immunoassay for Alzheimer Disease Pathology. JAMA Neurol 2024, 81, 255–263. [Google Scholar] [CrossRef]
- Benedet, A.L.; Arslan, B.; Tan, K.; Huber, H.; Pola, I.; Di Molfetta, G.; Kvartsberg, H.; Dolado, A.O.; Janelidze, S.; Blennow, K.; et al. Diagnostic Value of Serum P-Tau217 in Alzheimer Disease: Equal to Plasma in Levels and Clinical Utility? Clin Chem 2025, hvaf162. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; Cardoso, F.; Frucht, S.J.; Imarisio, A.; Halliday, G.M.; Lees, A.J. Refutation of the αSyn-SAA-Based Staging for Parkinson’s Progression (Neuronal α-Synuclein Disease-Integrated Staging System [NSD-ISS]). Mov Disord 2025, 40, 1744–1745. [Google Scholar] [CrossRef]
- Alkhalifa, A.E.; Al Mokhlf, A.; Ali, H.; Al-Ghraiybah, N.F.; Syropoulou, V. Anti-Amyloid Monoclonal Antibodies for Alzheimer’s Disease: Evidence, ARIA Risk, and Precision Patient Selection. J Pers Med 2025, 15. [Google Scholar] [CrossRef]
- Espay, A.J.; Kepp, K.P.; Herrup, K. Lecanemab and Donanemab as Therapies for Alzheimer’s Disease: An Illustrated Perspective on the Data. eNeuro 2024, 11. [Google Scholar] [CrossRef]
- Pitton Rissardo, J.; Fornari Caprara, A.L. Disease-Modifying Trials in Parkinson’s Disease: Challenges, Lessons, and Future Directions. Preprints 2025. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prions. Proc Natl Acad Sci U S A 1998, 95, 13363–13383. [Google Scholar] [CrossRef]
- Condello, C.; Westaway, D.; Prusiner, S.B. Expanding the Prion Paradigm to Include Alzheimer and Parkinson Diseases. JAMA Neurol 2024, 81, 1023–1024. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Melki, R. Prying into the Prion Hypothesis for Parkinson’s Disease. J Neurosci 2017, 37, 9808–9818. [Google Scholar] [CrossRef]
- Dill, K.A.; MacCallum, J.L. The Protein-Folding Problem, 50 Years On. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.J.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.A.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An Analytical Solution to the Kinetics of Breakable Filament Assembly. Science 2009, 326, 1533–1537. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Höglinger, G.; Lang, A.E.; Vieira, T.C.R.G. Protein Misfolding: Understanding Biology to Classify and Treat Synucleinopathies. J Neural Transm (Vienna) 2025, 132, 1645–1654. [Google Scholar] [CrossRef]
- Christ, W.; Kapell, S.; Sobkowiak, M.J.; Mermelekas, G.; Evertsson, B.; Sork, H.; Saher, O.; Bazaz, S.; Gustafsson, O.; Cardenas, E.I.; et al. SARS-CoV-2 and HSV-1 Induce Amyloid Aggregation in Human CSF Resulting in Drastic Soluble Protein Depletion. ACS Chem Neurosci 2024, 15, 4095–4104. [Google Scholar] [CrossRef]
- Duncan, G.B.; Dickson, S.P.; Kaplan, J.M.; Johnson, S.B.; Duke, T.M.; Dayley, C.W.; Hendrix, S.B.; Altstiel, L.D.; Mallinckrodt, C.H. Leveraging Recent Advances in Plasma Biomarkers to Optimize Early Proof of Concept Trials in Alzheimer’s Disease. Alzheimers Dement (N Y) 2025, 11, e70183. [Google Scholar] [CrossRef]
- Feizpour, A.; Doré, V.; Doecke, J.D.; Saad, Z.S.; Triana-Baltzer, G.; Slemmon, R.; Maruff, P.; Krishnadas, N.; Bourgeat, P.; Huang, K.; et al. Two-Year Prognostic Utility of Plasma P217+tau across the Alzheimer’s Continuum. J Prev Alzheimers Dis 2023, 10, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.J.; Pascoal, T.A.; Karikari, T.K.; Benedet, A.L.; Lantero-Rodriguez, J.; Brinkmalm, G.; Snellman, A.; Schöll, M.; Troakes, C.; Hye, A.; et al. Plasma P-Tau231: A New Biomarker for Incipient Alzheimer’s Disease Pathology. Acta Neuropathol 2021, 141, 709–724. [Google Scholar] [CrossRef]
- Brown, C.A.; Mundada, N.S.; Cousins, K.A.Q.; Sadeghpour, N.; Lyu, X.; McGrew, E.; Korecka, M.; Chen-Plotkin, A.; Xie, L.; Wisse, L.E.M.; et al. Tau-Clinical Mismatch Identifies Individuals with Co-Pathology and Predicts Clinical Trajectory. medRxiv : the preprint server for health sciences 2025. [Google Scholar]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative Accuracy of Plasma Phospho-Tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA 2020, 324, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Tideman, P.; Mattsson-Carlgren, N.; Schindler, S.E.; Smith, R.; Ossenkoppele, R.; Calling, S.; West, T.; Monane, M.; Verghese, P.B.; et al. Blood Biomarkers to Detect Alzheimer Disease in Primary Care and Secondary Care. JAMA 2024, 332, 1245–1257. [Google Scholar] [CrossRef]
- Arslan, B.; Gobom, J.; Andreasson, U.; Gonzalez-Ortiz, F.; Ashton, N.J.; Zetterberg, H.; Kvartsberg, H. Comparative Analysis of Plasma P-Tau217 Immunoassays: Challenges for Standardization and Harmonization. Clin Chem Lab Med 2025. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, Y.; Yu, J. Blood-Based Biomarkers of Alzheimer’s Disease: Standardization and Comprehensiveness. Neuroprotection 2024, 2, 246–254. [Google Scholar] [CrossRef]
- Anyaiwe, O.; Nataraj, N.; Gudikandula, B.S. Computational Risk Stratification of Preclinical Alzheimer’s in Younger Adults. Diagnostics (Basel) 2025, 15. [Google Scholar] [CrossRef]
- Pitton Rissardo, J.; McGarry, A.; Shi, Y.; Fornari Caprara, A.L.; Kannarkat, G.T. Alpha-Synuclein Neurobiology in Parkinson’s Disease: A Comprehensive Review of Its Role, Mechanisms, and Therapeutic Perspectives. Brain Sciences 2025, 15, 1260. [Google Scholar] [CrossRef]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-Synuclein Strains in Parkinson’s Disease and Multiple System Atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, G.; Stoops, E.; Vanbrabant, J.; Demeyer, L.; Francois, C.; Vanhooren, M.; Ma, Y.; Farris, C.M.; Concha-Marambio, L.; Paolini Paoletti, F.; et al. Phosphorylated α-Synuclein in CSF and Plasma Does Not Reflect Synucleinopathy. NPJ Parkinsons Dis 2025, 11, 232. [Google Scholar] [CrossRef]
- Carlos, A.F.; Tosakulwong, N.; Weigand, S.D.; Senjem, M.L.; Schwarz, C.G.; Knopman, D.S.; Boeve, B.F.; Petersen, R.C.; Nguyen, A.T.; Reichard, R.R.; et al. TDP-43 Pathology Effect on Volume and Flortaucipir Uptake in Alzheimer’s Disease. Alzheimers Dement 2023, 19, 2343–2354. [Google Scholar] [CrossRef]
- Espay, A.J.; Sturchio, A.; Imarisio, A.; Hill, E.J.; Williamson, B.; Montemagno, K.; Hoffmann, C.; Roy, H.L.; Milovanovic, D.; Manfredsson, F.P. Physics of Protein Aggregation in Normal and Accelerated Brain Aging. Bioessays 2025, 47, e70030. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Sturchio, A.; Espay, A.J. The Shift to a Proteinopenia Paradigm in Neurodegeneration. Handb Clin Neurol 2023, 193, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.S.; Yu, L.; Lamar, M.; Schneider, J.A.; Boyle, P.A.; Bennett, D.A. Education and Cognitive Reserve in Old Age. Neurology 2019, 92, e1041–e1050. [Google Scholar] [CrossRef]
- Gonzalez-Ortiz, F.; Vávra, J.; Payne, E.; Kirsebom, B.-E.; Sjöbom, U.; Santos, C.; Júlvez, J.; Kramer, K.; Zalcberg, D.; Montoliu-Gaya, L.; et al. The Potential Dual Role of Tau Phosphorylation: Plasma Phosphorylated-Tau217 in Newborns and Alzheimer’s Disease. Brain Commun 2025, 7, fcaf221. [Google Scholar] [CrossRef]
- van der Harg, J.M.; Nölle, A.; Zwart, R.; Boerema, A.S.; van Haastert, E.S.; Strijkstra, A.M.; Hoozemans, J.J.; Scheper, W. The Unfolded Protein Response Mediates Reversible Tau Phosphorylation Induced by Metabolic Stress. Cell Death Dis 2014, 5, e1393. [Google Scholar] [CrossRef]
- Siderowf, A.; Concha-Marambio, L.; Lafontant, D.-E.; Farris, C.M.; Ma, Y.; Urenia, P.A.; Nguyen, H.; Alcalay, R.N.; Chahine, L.M.; Foroud, T.; et al. Assessment of Heterogeneity among Participants in the Parkinson’s Progression Markers Initiative Cohort Using α-Synuclein Seed Amplification: A Cross-Sectional Study. Lancet Neurol 2023, 22, 407–417. [Google Scholar] [CrossRef]
- Espay, A.J.; Lees, A.J.; Cardoso, F.; Frucht, S.J.; Erskine, D.; Sandoval, I.M.; Bernal-Conde, L.D.; Sturchio, A.; Imarisio, A.; Hoffmann, C.; et al. The α-Synuclein Seed Amplification Assay: Interpreting a Test of Parkinson’s Pathology. Parkinsonism Relat Disord 2025, 131, 107256. [Google Scholar] [CrossRef]
- Hansson, O.; Seibyl, J.; Stomrud, E.; Zetterberg, H.; Trojanowski, J.Q.; Bittner, T.; Lifke, V.; Corradini, V.; Eichenlaub, U.; Batrla, R.; et al. CSF Biomarkers of Alzheimer’s Disease Concord with Amyloid-β PET and Predict Clinical Progression: A Study of Fully Automated Immunoassays in BioFINDER and ADNI Cohorts. Alzheimers Dement 2018, 14, 1470–1481. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in Physiology and Pathology. Nat Rev Neurosci 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and Tau--a Toxic Pas de Deux in Alzheimer’s Disease. Nat Rev Neurosci 2011, 12, 65–72. [Google Scholar] [CrossRef]
- Puzzo, D.; Privitera, L.; Fa’, M.; Staniszewski, A.; Hashimoto, G.; Aziz, F.; Sakurai, M.; Ribe, E.M.; Troy, C.M.; Mercken, M.; et al. Endogenous Amyloid-β Is Necessary for Hippocampal Synaptic Plasticity and Memory. Ann Neurol 2011, 69, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Bocancea, D.I.; Svenningsson, A.L.; van Loenhoud, A.C.; Groot, C.; Barkhof, F.; Strandberg, O.; Smith, R.; La Joie, R.; Rosen, H.J.; Pontecorvo, M.J.; et al. Determinants of Cognitive and Brain Resilience to Tau Pathology: A Longitudinal Analysis. Brain 2023, 146, 3719–3734. [Google Scholar] [CrossRef] [PubMed]
- Ziontz, J.; Bilgel, M.; Shafer, A.T.; Moghekar, A.; Elkins, W.; Helphrey, J.; Gomez, G.; June, D.; McDonald, M.A.; Dannals, R.F.; et al. Tau Pathology in Cognitively Normal Older Adults. Alzheimers Dement (Amst) 2019, 11, 637–645. [Google Scholar] [CrossRef]
- St-Onge, F.; Chapleau, M.; Breitner, J.C.S.; Villeneuve, S.; Pichet Binette, A. Tau Accumulation and Its Spatial Progression across the Alzheimer’s Disease Spectrum. Brain Commun 2024, 6, fcae031. [Google Scholar] [CrossRef]
- Guerrero-Muñoz, M.J.; Gerson, J.; Castillo-Carranza, D.L. Tau Oligomers: The Toxic Player at Synapses in Alzheimer’s Disease. Front Cell Neurosci 2015, 9, 464. [Google Scholar] [CrossRef]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The Many Faces of Tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Schonhaut, D.R.; Schöll, M.; Lockhart, S.N.; Ayakta, N.; Baker, S.L.; O’Neil, J.P.; Janabi, M.; Lazaris, A.; Cantwell, A.; et al. Tau PET Patterns Mirror Clinical and Neuroanatomical Variability in Alzheimer’s Disease. Brain 2016, 139, 1551–1567. [Google Scholar] [CrossRef]
- Jack, C.R.J.; Wiste, H.J.; Weigand, S.D.; Therneau, T.M.; Knopman, D.S.; Lowe, V.; Vemuri, P.; Mielke, M.M.; Roberts, R.O.; Machulda, M.M.; et al. Age-Specific and Sex-Specific Prevalence of Cerebral β-Amyloidosis, Tauopathy, and Neurodegeneration in Cognitively Unimpaired Individuals Aged 50-95 Years: A Cross-Sectional Study. Lancet Neurol 2017, 16, 435–444. [Google Scholar] [CrossRef]
- Ottoy, J.; Owsicki, N.; Bilgel, M.; Binette, A.P.; Salvadó, G.; Kang, M.S.; Cash, D.M.; Ewers, M.; La Joie, R.; Wisse, L.E.M.; et al. Recent Advances in Neuroimaging of Alzheimer’s Disease and Related Dementias. Alzheimers Dement 2025, 21, e70648. [Google Scholar] [CrossRef] [PubMed]
- Barthélemy, N.R.; Li, Y.; Joseph-Mathurin, N.; Gordon, B.A.; Hassenstab, J.; Benzinger, T.L.S.; Buckles, V.; Fagan, A.M.; Perrin, R.J.; Goate, A.M.; et al. A Soluble Phosphorylated Tau Signature Links Tau, Amyloid and the Evolution of Stages of Dominantly Inherited Alzheimer’s Disease. Nat Med 2020, 26, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Sgourakis, N.G.; Yan, Y.; McCallum, S.A.; Wang, C.; Garcia, A.E. The Alzheimer’s Peptides Abeta40 and 42 Adopt Distinct Conformations in Water: A Combined MD / NMR Study. J Mol Biol 2007, 368, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; Herrup, K.; Kepp, K.P.; Daly, T. The Proteinopenia Hypothesis: Loss of Aβ(42) and the Onset of Alzheimer’s Disease. Ageing Res Rev 2023, 92, 102112. [Google Scholar] [CrossRef]
- Okochi, M.; Tagami, S.; Yanagida, K.; Takami, M.; Kodama, T.S.; Mori, K.; Nakayama, T.; Ihara, Y.; Takeda, M. γ-Secretase Modulators and Presenilin 1 Mutants Act Differently on Presenilin/γ-Secretase Function to Cleave Aβ42 and Aβ43. Cell Rep 2013, 3, 42–51. [Google Scholar] [CrossRef]
- Yang, Y.; Bagyinszky, E.; An, S.S.A. Presenilin-1 (PSEN1) Mutations: Clinical Phenotypes beyond Alzheimer’s Disease. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Tan, L.; Liu, J.; Yu, J.-T. The Essential Role of Soluble Aβ Oligomers in Alzheimer’s Disease. Mol Neurobiol 2016, 53, 1905–1924. [Google Scholar] [CrossRef]
- Bateman, R.J.; Aisen, P.S.; De Strooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.M.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosomal-Dominant Alzheimer’s Disease: A Review and Proposal for the Prevention of Alzheimer’s Disease. Alzheimers Res Ther 2011, 3, 1. [Google Scholar] [CrossRef]
- Chávez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The Mechanism of γ-Secretase Dysfunction in Familial Alzheimer Disease. EMBO J 2012, 31, 2261–2274. [Google Scholar] [CrossRef]
- Younkin, S.G. The Role of A Beta 42 in Alzheimer’s Disease. J Physiol Paris 1998, 92, 289–292. [Google Scholar] [CrossRef]
- Phillips, J.C. Why Aβ42 Is Much More Toxic than Aβ40. ACS Chem Neurosci 2019, 10, 2843–2847. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, D.V.; Zaretskaia, M.V.; Molkov, Y.I. Patients with Alzheimer’s Disease Have an Increased Removal Rate of Soluble Beta-Amyloid-42. PLoS One 2022, 17, e0276933. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A Phase 3 Trial of Semagacestat for Treatment of Alzheimer’s Disease. N Engl J Med 2013, 369, 341–350. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N Engl J Med 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N Engl J Med 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Fortea, J.; Pegueroles, J.; Alcolea, D.; Belbin, O.; Dols-Icardo, O.; Vaqué-Alcázar, L.; Videla, L.; Gispert, J.D.; Suárez-Calvet, M.; Johnson, S.C.; et al. APOE4 Homozygozity Represents a Distinct Genetic Form of Alzheimer’s Disease. Nat Med 2024, 30, 1284–1291. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.-X.; Martins, R.; Rowe, C.; et al. High Performance Plasma Amyloid-β Biomarkers for Alzheimer’s Disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Wojdała, A.L.; Bellomo, G.; Toja, A.; Gaetani, L.; Parnetti, L.; Chiasserini, D. CSF and Plasma Aβ42/40 across Alzheimer’s Disease Continuum: Comparison of Two Ultrasensitive Simoa(®) Assays Targeting Distinct Amyloid Regions. Clin Chem Lab Med 2024, 62, 332–340. [Google Scholar] [CrossRef]
- Parihar, M.S.; Brewer, G.J. Amyloid-β as a Modulator of Synaptic Plasticity. J Alzheimers Dis 2010, 22, 741–763. [Google Scholar] [CrossRef]
- Espay, A.J.; Ezzat, K.; Kepp, K.P.; Daly, T.; Robakis, N.K.; Meijer, L.; Imbimbo, B.P. Restoring Amyloid-Β42 and γ-Secretase Function in Alzheimer’s Disease. Brain 2025, 148, 3856–3864. [Google Scholar] [CrossRef]
- van Harten, A.C.; Visser, P.J.; Pijnenburg, Y.A.L.; Teunissen, C.E.; Blankenstein, M.A.; Scheltens, P.; van der Flier, W.M. Cerebrospinal Fluid Aβ42 Is the Best Predictor of Clinical Progression in Patients with Subjective Complaints. Alzheimers Dement 2013, 9, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.J.; Wiste, H.J.; Weigand, S.D.; Rocca, W.A.; Knopman, D.S.; Mielke, M.M.; Lowe, V.J.; Senjem, M.L.; Gunter, J.L.; Preboske, G.M.; et al. Age-Specific Population Frequencies of Cerebral β-Amyloidosis and Neurodegeneration among People with Normal Cognitive Function Aged 50-89 Years: A Cross-Sectional Study. Lancet Neurol 2014, 13, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Jansen, W.J.; Ossenkoppele, R.; Knol, D.L.; Tijms, B.M.; Scheltens, P.; Verhey, F.R.J.; Visser, P.J.; Aalten, P.; Aarsland, D.; Alcolea, D.; et al. Prevalence of Cerebral Amyloid Pathology in Persons without Dementia: A Meta-Analysis. JAMA 2015, 313, 1924–1938. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N Engl J Med 2018, 378, 1691–1703. [Google Scholar] [CrossRef]
- Kukar, T.; Murphy, M.P.; Eriksen, J.L.; Sagi, S.A.; Weggen, S.; Smith, T.E.; Ladd, T.; Khan, M.A.; Kache, R.; Beard, J.; et al. Diverse Compounds Mimic Alzheimer Disease-Causing Mutations by Augmenting Abeta42 Production. Nat Med 2005, 11, 545–550. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Clement, A.B.; Behl, C. Cholesterol-like Effects of Selective Cyclooxygenase Inhibitors and Fibrates on Cellular Membranes and Amyloid-Beta Production. Mol Pharmacol 2007, 72, 141–151. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, K.; Wang, R.; Liu, Y.; Kwak, Y.-D.; Ma, T.; Thompson, R.C.; Zhao, Y.; Smith, L.; Gasparini, L.; et al. Antidiabetic Drug Metformin (GlucophageR) Increases Biogenesis of Alzheimer’s Amyloid Peptides via up-Regulating BACE1 Transcription. Proc Natl Acad Sci U S A 2009, 106, 3907–3912. [Google Scholar] [CrossRef]
- Schoenfeld, H.A.; West, T.; Verghese, P.B.; Holubasch, M.; Shenoy, N.; Kagan, D.; Buono, C.; Zhou, W.; DeCristofaro, M.; Douville, J.; et al. The Effect of Angiotensin Receptor Neprilysin Inhibitor, Sacubitril/Valsartan, on Central Nervous System Amyloid-β Concentrations and Clearance in the Cynomolgus Monkey. Toxicol Appl Pharmacol 2017, 323, 53–65. [Google Scholar] [CrossRef]
- Rogers, J.; Kirby, L.C.; Hempelman, S.R.; Berry, D.L.; McGeer, P.L.; Kaszniak, A.W.; Zalinski, J.; Cofield, M.; Mansukhani, L.; Willson, P. Clinical Trial of Indomethacin in Alzheimer’s Disease. Neurology 1993, 43, 1609–1611. [Google Scholar] [CrossRef]
- de Jong, D.; Jansen, R.; Hoefnagels, W.; Jellesma-Eggenkamp, M.; Verbeek, M.; Borm, G.; Kremer, B. No Effect of One-Year Treatment with Indomethacin on Alzheimer’s Disease Progression: A Randomized Controlled Trial. PLoS One 2008, 3, e1475. [Google Scholar] [CrossRef]
- Sperling, R.; Henley, D.; Aisen, P.S.; Raman, R.; Donohue, M.C.; Ernstrom, K.; Rafii, M.S.; Streffer, J.; Shi, Y.; Karcher, K.; et al. Findings of Efficacy, Safety, and Biomarker Outcomes of Atabecestat in Preclinical Alzheimer Disease: A Truncated Randomized Phase 2b/3 Clinical Trial. JAMA Neurol 2021, 78, 293–301. [Google Scholar] [CrossRef]
- Ancelin, M.-L.; Carrière, I.; Barberger-Gateau, P.; Auriacombe, S.; Rouaud, O.; Fourlanos, S.; Berr, C.; Dupuy, A.-M.; Ritchie, K. Lipid Lowering Agents, Cognitive Decline, and Dementia: The Three-City Study. J Alzheimers Dis 2012, 30, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Pomilio, C.; Pérez, N.G.; Calandri, I.; Crivelli, L.; Allegri, R.; Sevlever, G.; Saravia, F. Diabetic Patients Treated with Metformin during Early Stages of Alzheimer’s Disease Show a Better Integral Performance: Data from ADNI Study. Geroscience 2022, 44, 1791–1805. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Chen, W.-M.; Wu, S.-Y.; Zhang, J. Metformin in Elderly Type 2 Diabetes Mellitus: Dose-Dependent Dementia Risk Reduction. Brain 2024, 147, 1474–1482. [Google Scholar] [CrossRef]
- Grewal, P.K.; Abboud, A.; Myserlis, E.P.; Goldschmidt, M.E.; Butler, J.; Skopicki, H.A.; Kalogeropoulos, A.P. Sacubitril/Valsartan and Cognitive Outcomes in Heart Failure With Reduced Ejection Fraction. JACC Adv 2023, 2, 100372. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J Prev Alzheimers Dis 2022, 9, 197–210. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A Randomized, Double-Blind, Phase 2b Proof-of-Concept Clinical Trial in Early Alzheimer’s Disease with Lecanemab, an Anti-Aβ Protofibril Antibody. Alzheimers Res Ther 2021, 13, 80. [Google Scholar] [CrossRef]
- Veitch, D.P.; Weiner, M.W.; Miller, M.; Aisen, P.S.; Ashford, M.A.; Beckett, L.A.; Green, R.C.; Harvey, D.; Jack, C.R.J.; Jagust, W.; et al. The Alzheimer’s Disease Neuroimaging Initiative in the Era of Alzheimer’s Disease Treatment: A Review of ADNI Studies from 2021 to 2022. Alzheimers Dement 2024, 20, 652–694. [Google Scholar] [CrossRef]
- Jutten, R.J.; Sikkes, S.A.M.; Van der Flier, W.M.; Scheltens, P.; Visser, P.J.; Tijms, B.M. Finding Treatment Effects in Alzheimer Trials in the Face of Disease Progression Heterogeneity. Neurology 2021, 96, e2673–e2684. [Google Scholar] [CrossRef]
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients With Early Alzheimer Disease. JAMA Neurol 2022, 79, 13–21. [Google Scholar] [CrossRef]
- Bobbins, A.; Davies, M.; Lynn, E.; Roy, D.; Yeomans, A.; Shakir, S.A.W. Safety and Effectiveness of the Anti-Amyloid Monoclonal Antibody (mAb) Drug Lecanemab for Early Alzheimer’s Disease: The Pharmacovigilance Perspective. Br J Clin Pharmacol 2025, 91, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Birks, J.S.; Harvey, R.J. Donepezil for Dementia Due to Alzheimer’s Disease. Cochrane Database Syst Rev 2018, 6, CD001190. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-W.; Hsu, T.-W.; Kao, Y.-C.; Lin, Y.-H.; Thompson, T.; Carvalho, A.F.; Stubbs, B.; Tseng, P.-T.; Yang, F.-C.; Tsai, C.-K.; et al. The Efficacy and Safety of Anti-Amyloid Monoclonal Antibody versus Acetylcholinesterase Inhibitor with an in-Depth Analysis across Genotypes and Disease Stages: A Systematic Review and Meta-Analysis. J Prev Alzheimers Dis 2025, 12, 100195. [Google Scholar] [CrossRef]
- Filip, P.; McCarten, J.R.; Hemmy, L.; Crocker, J.; Wolf, M.; Thotland, J.; Cayci, Z.; Kes, T.; Michaeli, S.; Terpstra, M.; et al. Clinical and MRI Correlates of β-Amyloid Load Inconsistent With Its Presumed Neurotoxicity in Cognitively Healthy Ageing. J Neurochem 2025, 169, e70241. [Google Scholar] [CrossRef]
- Smith, M.A.; Casadesus, G.; Joseph, J.A.; Perry, G. Amyloid-Beta and Tau Serve Antioxidant Functions in the Aging and Alzheimer Brain. Free Radic Biol Med 2002, 33, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Sawada, M.; Yamaguchi, K.; Hirano, M.; Noji, M.; So, M.; Otzen, D.; Kawata, Y.; Goto, Y. Amyloid Formation of α-Synuclein Based on the Solubility- and Supersaturation-Dependent Mechanism. Langmuir 2020, 36, 4671–4681. [Google Scholar] [CrossRef] [PubMed]
- Belete, D.; Mattjie, C.; Huxford, B.; Bestwick, J.P.; Noyce, A.J.; Simonet, C. Evaluating the Role of α-Synuclein Seed Amplification as a Disease Progression Marker: Evidence and Uncertainties. Mov Disord Clin Pract 2025. [Google Scholar] [CrossRef]
- Rissardo, J.P.; Fornari Caprara, A.L. Alpha-Synuclein Seed Amplification Assays in Parkinson’s Disease: A Systematic Review and Network Meta-Analysis. Clin Pract 2025, 15. [Google Scholar] [CrossRef] [PubMed]
- Giehm, L.; Lorenzen, N.; Otzen, D.E. Assays for α-Synuclein Aggregation. Methods 2011, 53, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Goetz, C.G.; Mestre, T.A.; Sampaio, C.; Adler, C.H.; Berg, D.; Bloem, B.R.; Burn, D.J.; Fitts, M.S.; Gasser, T.; et al. A Statement of the MDS on Biological Definition, Staging, and Classification of Parkinson’s Disease. Mov Disord 2024, 39, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Ticca, A.; Lerche, S.; Mastrangelo, A.; Mammana, A.; Wurster, I.; Vittoriosi, E.; Roeben, B.; Hauser, A.-K.; Deuschle, C.; et al. Quantification of Cerebrospinal Fluid α-Synuclein Seeds by Endpoint Dilution Seed Amplification Assay in Parkinson’s Disease. NPJ Parkinsons Dis 2025. [Google Scholar] [CrossRef]
- Ezzat, K.; Espay, A.J. Can Prions Carry Biological Information? ACS Omega 2025, 10, 57842–57845. [Google Scholar] [CrossRef]
- Koloteva-Levine, N.; Aubrey, L.D.; Marchante, R.; Purton, T.J.; Hiscock, J.R.; Tuite, M.F.; Xue, W.-F. Amyloid Particles Facilitate Surface-Catalyzed Cross-Seeding by Acting as Promiscuous Nanoparticles. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef]
- Candelise, N.; Schmitz, M.; Thüne, K.; Cramm, M.; Rabano, A.; Zafar, S.; Stoops, E.; Vanderstichele, H.; Villar-Pique, A.; Llorens, F.; et al. Effect of the Micro-Environment on α-Synuclein Conversion and Implication in Seeded Conversion Assays. Transl Neurodegener 2020, 9, 5. [Google Scholar] [CrossRef]
- Palleis, C.; Bernhardt, A.M.; Weidinger, E.; Fietzek, U.M.; Jäck, A.; Katzdobler, S.; Gnörich, J.; Bauer, T.; Franzmeier, N.; Perneczky, R.; et al. A Biomarker-Based Classification of Corticobasal Syndrome. Mov Disord 2025. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Burke, R.E.; Dauer, W.T.; Vonsattel, J.P.G. A Critical Evaluation of the Braak Staging Scheme for Parkinson’s Disease. Ann Neurol 2008, 64, 485–491. [Google Scholar] [CrossRef]
- Pitton Rissardo, J.; Fornari Caprara, A.L. Protein Aggregation and Proteostasis Failure in Neurodegenerative Diseases: Mechanisms, Structural Insights, and Therapeutic Strategies. Preprints 2025. [Google Scholar] [CrossRef]
- Isonaka, R.; Sullivan, P.; Goldstein, D.S. Pathophysiological Significance of α-Synuclein in Sympathetic Nerves: In Vivo Observations. Neurology 2025, 104, e210215. [Google Scholar] [CrossRef] [PubMed]
- Neupane, S.; De Cecco, E.; Aguzzi, A. The Hidden Cell-to-Cell Trail of α-Synuclein Aggregates. J Mol Biol 2023, 435, 167930. [Google Scholar] [CrossRef]
- Hoppe, S.O.; Uzunoğlu, G.; Nussbaum-Krammer, C. α-Synuclein Strains: Does Amyloid Conformation Explain the Heterogeneity of Synucleinopathies? Biomolecules 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.B.; Yagi, H.; Manno, M.; Martorana, V.; Ban, T.; Christiansen, G.; Otzen, D.E.; Goto, Y.; Rischel, C. Branching in Amyloid Fibril Growth. Biophys J 2009, 96, 1529–1536. [Google Scholar] [CrossRef]
- Törnquist, M.; Michaels, T.C.T.; Sanagavarapu, K.; Yang, X.; Meisl, G.; Cohen, S.I.A.; Knowles, T.P.J.; Linse, S. Secondary Nucleation in Amyloid Formation. Chem Commun (Camb) 2018, 54, 8667–8684. [Google Scholar] [CrossRef]
- Subedi, S.; Sasidharan, S.; Nag, N.; Saudagar, P.; Tripathi, T. Amyloid Cross-Seeding: Mechanism, Implication, and Inhibition. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Portugal Barron, D.; Guo, Z. The Supersaturation Perspective on the Amyloid Hypothesis. Chem Sci 2023, 15, 46–54. [Google Scholar] [CrossRef]
- Ezzat, K.; Sturchio, A.; Espay, A.J. Proteins Do Not Replicate, They Precipitate: Phase Transition and Loss of Function Toxicity in Amyloid Pathologies. Biology (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Ziaunys, M.; Sakalauskas, A.; Mikalauskaite, K.; Smirnovas, V. Polymorphism of Alpha-Synuclein Amyloid Fibrils Depends on Ionic Strength and Protein Concentration. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Frey, L.; Ghosh, D.; Qureshi, B.M.; Rhyner, D.; Guerrero-Ferreira, R.; Pokharna, A.; Kwiatkowski, W.; Serdiuk, T.; Picotti, P.; Riek, R.; et al. On the pH-Dependence of α-Synuclein Amyloid Polymorphism and the Role of Secondary Nucleation in Seed-Based Amyloid Propagation. Elife 2024, 12. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Pittman, J.M.; Zerweck, J.; Venkata, B.S.; Moore, P.C.; Sachleben, J.R.; Meredith, S.C. β-Amyloid Aggregation and Heterogeneous Nucleation. Protein Sci 2019, 28, 1567–1581. [Google Scholar] [CrossRef]
- Peduzzo, A.; Linse, S.; Buell, A.K. The Properties of α-Synuclein Secondary Nuclei Are Dominated by the Solution Conditions Rather than the Seed Fibril Strain. ACS Chem Neurosci 2020, 11, 909–918. [Google Scholar] [CrossRef]
- Lövestam, S.; Koh, F.A.; van Knippenberg, B.; Kotecha, A.; Murzin, A.G.; Goedert, M.; Scheres, S.H.W. Assembly of Recombinant Tau into Filaments Identical to Those of Alzheimer’s Disease and Chronic Traumatic Encephalopathy. Elife 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; So, M.; Sakurai, K.; Kardos, J.; Goto, Y. Supersaturation-Limited and Unlimited Phase Transitions Compete to Produce the Pathway Complexity in Amyloid Fibrillation. J Biol Chem 2015, 290, 18134–18145. [Google Scholar] [CrossRef]
- Goto, Y.; Nakajima, K.; Yamamoto, S.; Yamaguchi, K. Supersaturation, a Critical Factor Underlying Proteostasis of Amyloid Fibril Formation. J Mol Biol 2024, 436, 168475. [Google Scholar] [CrossRef]
- Kulkarni, P.; Leite, V.B.P.; Roy, S.; Bhattacharyya, S.; Mohanty, A.; Achuthan, S.; Singh, D.; Appadurai, R.; Rangarajan, G.; Weninger, K.; et al. Intrinsically Disordered Proteins: Ensembles at the Limits of Anfinsen’s Dogma. Biophys Rev (Melville) 2022, 3, 011306. [Google Scholar] [CrossRef]
- Tycko, R. Amyloid Polymorphism: Structural Basis and Neurobiological Relevance. Neuron 2015, 86, 632–645. [Google Scholar] [CrossRef]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril Structure of Amyloid-β(1-42) by Cryo-Electron Microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM Structures of Tau Filaments from Alzheimer’s Disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Mehra, S.; Gadhe, L.; Bera, R.; Sawner, A.S.; Maji, S.K. Structural and Functional Insights into α-Synuclein Fibril Polymorphism. Biomolecules 2021, 11. [Google Scholar] [CrossRef]
- Berry, J.; Brangwynne, C.P.; Haataja, M. Physical Principles of Intracellular Organization via Active and Passive Phase Transitions. Rep Prog Phys 2018, 81, 046601. [Google Scholar] [CrossRef]
- Vandenberghe, R. The Relationship between Amyloid Deposition, Neurodegeneration, and Cognitive Decline in Dementia. Curr Neurol Neurosci Rep 2014, 14, 498. [Google Scholar] [CrossRef]
- Rasmussen, J.; Mahler, J.; Beschorner, N.; Kaeser, S.A.; Häsler, L.M.; Baumann, F.; Nyström, S.; Portelius, E.; Blennow, K.; Lashley, T.; et al. Amyloid Polymorphisms Constitute Distinct Clouds of Conformational Variants in Different Etiological Subtypes of Alzheimer’s Disease. Proc Natl Acad Sci U S A 2017, 114, 13018–13023. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-Propagation of Pathogenic Protein Aggregates in Neurodegenerative Diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Jones, E.M.; Surewicz, W.K. Fibril Conformation as the Basis of Species- and Strain-Dependent Seeding Specificity of Mammalian Prion Amyloids. Cell 2005, 121, 63–72. [Google Scholar] [CrossRef]
- Gao, H.-M.; Hong, J.-S. Why Neurodegenerative Diseases Are Progressive: Uncontrolled Inflammation Drives Disease Progression. Trends Immunol 2008, 29, 357–365. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, C.; Morozova-Roche, L.; Zhang, J.Z.H.; Mu, Y. pH-Dependent Conformational Ensemble and Polymorphism of Amyloid-β Core Fragment. J Phys Chem B 2013, 117, 8392–8399. [Google Scholar] [CrossRef]
- Sakalauskas, A.; Ziaunys, M.; Smirnovas, V. Concentration-Dependent Polymorphism of Insulin Amyloid Fibrils. PeerJ 2019, 7, e8208. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N Engl J Med 2012, 367, 795–804. [Google Scholar] [CrossRef]
- Linse, S.; Cabaleiro-Lago, C.; Xue, W.-F.; Lynch, I.; Lindman, S.; Thulin, E.; Radford, S.E.; Dawson, K.A. Nucleation of Protein Fibrillation by Nanoparticles. Proc Natl Acad Sci U S A 2007, 104, 8691–8696. [Google Scholar] [CrossRef]
- Kuczyńska-Wiśnik, D.; Stojowska-Swędrzyńska, K.; Laskowska, E. Liquid-Liquid Phase Separation and Protective Protein Aggregates in Bacteria. Molecules 2023, 28. [Google Scholar] [CrossRef]
- Ezzat, K.; Pernemalm, M.; Pålsson, S.; Roberts, T.C.; Järver, P.; Dondalska, A.; Bestas, B.; Sobkowiak, M.J.; Levänen, B.; Sköld, M.; et al. The Viral Protein Corona Directs Viral Pathogenesis and Amyloid Aggregation. Nat Commun 2019, 10, 2331. [Google Scholar] [CrossRef]
- Grigolato, F.; Arosio, P. The Role of Surfaces on Amyloid Formation. Biophys Chem 2021, 270, 106533. [Google Scholar] [CrossRef]
- Devanand, D.P.; Wisniewski, T.; Razlighi, Q.; Qian, M.; Wei, R.; Andrews, H.F.; Acosta, E.P.; Bell, K.L.; Pelton, G.H.; Deliyannides, D.; et al. Valacyclovir Treatment of Early Symptomatic Alzheimer Disease: The VALAD Randomized Clinical Trial. JAMA 2025. [Google Scholar] [CrossRef]
- Treusch, S.; Cyr, D.M.; Lindquist, S. Amyloid Deposits: Protection against Toxic Protein Species? Cell Cycle 2009, 8, 1668–1674. [Google Scholar] [CrossRef]
- Fulop, T.; Witkowski, J.M.; Bourgade, K.; Khalil, A.; Zerif, E.; Larbi, A.; Hirokawa, K.; Pawelec, G.; Bocti, C.; Lacombe, G.; et al. Can an Infection Hypothesis Explain the Beta Amyloid Hypothesis of Alzheimer’s Disease? Front Aging Neurosci 2018, 10, 224. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Corroboration of a Major Role for Herpes Simplex Virus Type 1 in Alzheimer’s Disease. Front Aging Neurosci 2018, 10, 324. [Google Scholar] [CrossRef]
- Muscolino, E.; Luoto, L.-M.; Brune, W. Viral Induced Protein Aggregation: A Mechanism of Immune Evasion. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s Disease-Associated Amyloid Beta-Protein Is an Antimicrobial Peptide. PLoS One 2010, 5, e9505. [Google Scholar] [CrossRef]
- Kumar, D.K.V.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β Peptide Protects against Microbial Infection in Mouse and Worm Models of Alzheimer’s Disease. Sci Transl Med 2016, 8, 340ra72. [Google Scholar] [CrossRef]
- Schwarzschild, M.A.; Ascherio, A.; Beal, M.F.; Cudkowicz, M.E.; Curhan, G.C.; Hare, J.M.; Hooper, D.C.; Kieburtz, K.D.; Macklin, E.A.; Oakes, D.; et al. Inosine to Increase Serum and Cerebrospinal Fluid Urate in Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol 2014, 71, 141–150. [Google Scholar] [CrossRef]
- Espay, A.J.; Piccinin, C.C.; Schwarzschild, M. Hope vs Hype II: We Should Stop Embarking on Epidemiology-Based Disease Modifying Clinical Trials. Parkinsonism Relat Disord 2025, 139, 107904. [Google Scholar] [CrossRef]
- Arias-Carrión, O.; Guerra-Crespo, M.; Padilla-Godínez, F.J.; Soto-Rojas, L.O.; Manjarrez, E. α-Synuclein Pathology in Synucleinopathies: Mechanisms, Biomarkers, and Therapeutic Challenges. Int J Mol Sci 2025, 26. [Google Scholar] [CrossRef]
- Aumont, E.; Therriault, J.; Kwan, A.T.H.; Carello-Collar, G.; Arfaie, S.; Hall, B.J.; Ferrari-Souza, J.-P.; Woo, M.S.; Macedo, A.C.; Tissot, C.; et al. The Evolution of Randomized Clinical Trial Designs to Assess Therapeutics in Alzheimer Disease. JAMA Netw Open 2025, 8, e2529665. [Google Scholar] [CrossRef]
- Gatto, E.M.; Espay, A.J.; Espindola, M.; Da Prat, G.; Kauffman, M.A. Protective or Pathogenic? Kinase Activity and the Neurodevelopmental Origins of G2019S LRRK2-Associated Parkinson’s Disease. Parkinsonism Relat Disord 2025, 108133. [Google Scholar] [CrossRef]
- Gates, A.J.; Brattig Correia, R.; Wang, X.; Rocha, L.M. The Effective Graph Reveals Redundancy, Canalization, and Control Pathways in Biochemical Regulation and Signaling. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef]
- Fleming, T.R.; Powers, J.H. Biomarkers and Surrogate Endpoints in Clinical Trials. Stat Med 2012, 31, 2973–2984. [Google Scholar] [CrossRef]
- Simuni, T.; Chahine, L.M.; Poston, K.; Brumm, M.; Buracchio, T.; Campbell, M.; Chowdhury, S.; Coffey, C.; Concha-Marambio, L.; Dam, T.; et al. A Biological Definition of Neuronal α-Synuclein Disease: Towards an Integrated Staging System for Research. Lancet Neurol 2024, 23, 178–190. [Google Scholar] [CrossRef]
- Dam, T.; Pagano, G.; Brumm, M.C.; Gochanour, C.; Poston, K.L.; Weintraub, D.; Chahine, L.M.; Coffey, C.; Tanner, C.M.; Kopil, C.M.; et al. Neuronal Alpha-Synuclein Disease Integrated Staging System Performance in PPMI, PASADENA, and SPARK Baseline Cohorts. NPJ Parkinsons Dis 2024, 10, 178. [Google Scholar] [CrossRef]
- Jack, C.R.J.; Andrews, J.S.; Beach, T.G.; Buracchio, T.; Dunn, B.; Graf, A.; Hansson, O.; Ho, C.; Jagust, W.; McDade, E.; et al. Revised Criteria for Diagnosis and Staging of Alzheimer’s Disease: Alzheimer’s Association Workgroup. Alzheimers Dement 2024, 20, 5143–5169. [Google Scholar] [CrossRef]
- Cummings, J.; Kinney, J. Biomarkers for Alzheimer’s Disease: Context of Use, Qualification, and Roadmap for Clinical Implementation. Medicina (Kaunas) 2022, 58. [Google Scholar] [CrossRef]
- Aiello Bowles, E.J.; Crane, P.K.; Walker, R.L.; Chubak, J.; LaCroix, A.Z.; Anderson, M.L.; Rosenberg, D.; Keene, C.D.; Larson, E.B. Cognitive Resilience to Alzheimer’s Disease Pathology in the Human Brain. J Alzheimers Dis 2019, 68, 1071–1083. [Google Scholar] [CrossRef]
- Rissardo, J.P.; Caprara, A.L. A Narrative Review on Biochemical Markers and Emerging Treatments in Prodromal Synucleinopathies. Clinics and Practice 2025, 15. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N Engl J Med 2019, 380, 2307–2316. [Google Scholar] [CrossRef]
- Russo, M.; Costa, T.; Calisi, D.; Sensi, S.L. Prasinezumab: A Bayesian Perspective on Its Efficacy. Mov Disord 2025, 40, 619–624. [Google Scholar] [CrossRef]
- Gutierrez, B.A.; Limon, A. Synaptic Disruption by Soluble Oligomers in Patients with Alzheimer’s and Parkinson’s Disease. Biomedicines 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat Neurosci 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, S.; Ikai, A.; Takashima, A. Increased Levels of Granular Tau Oligomers: An Early Sign of Brain Aging and Alzheimer’s Disease. Neurosci Res 2006, 54, 197–201. [Google Scholar] [CrossRef]
- Gelpi, E.; Höftberger, R.; Graus, F.; Ling, H.; Holton, J.L.; Dawson, T.; Popovic, M.; Pretnar-Oblak, J.; Högl, B.; Schmutzhard, E.; et al. Neuropathological Criteria of Anti-IgLON5-Related Tauopathy. Acta Neuropathol 2016, 132, 531–543. [Google Scholar] [CrossRef]
- Ashton, N.J.; Leuzy, A.; Karikari, T.K.; Mattsson-Carlgren, N.; Dodich, A.; Boccardi, M.; Corre, J.; Drzezga, A.; Nordberg, A.; Ossenkoppele, R.; et al. The Validation Status of Blood Biomarkers of Amyloid and Phospho-Tau Assessed with the 5-Phase Development Framework for AD Biomarkers. Eur J Nucl Med Mol Imaging 2021, 48, 2140–2156. [Google Scholar] [CrossRef]
- Lewczuk, P.; Wiltfang, J.; Kornhuber, J.; Verhasselt, A. Distributions of Aβ42 and Aβ42/40 in the Cerebrospinal Fluid in View of the Probability Theory. Diagnostics (Basel) 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Holland, C.; Numata, K.; Rnjak-Kovacina, J.; Seib, F.P. The Biomedical Use of Silk: Past, Present, Future. Adv Healthc Mater 2019, 8, e1800465. [Google Scholar] [CrossRef] [PubMed]
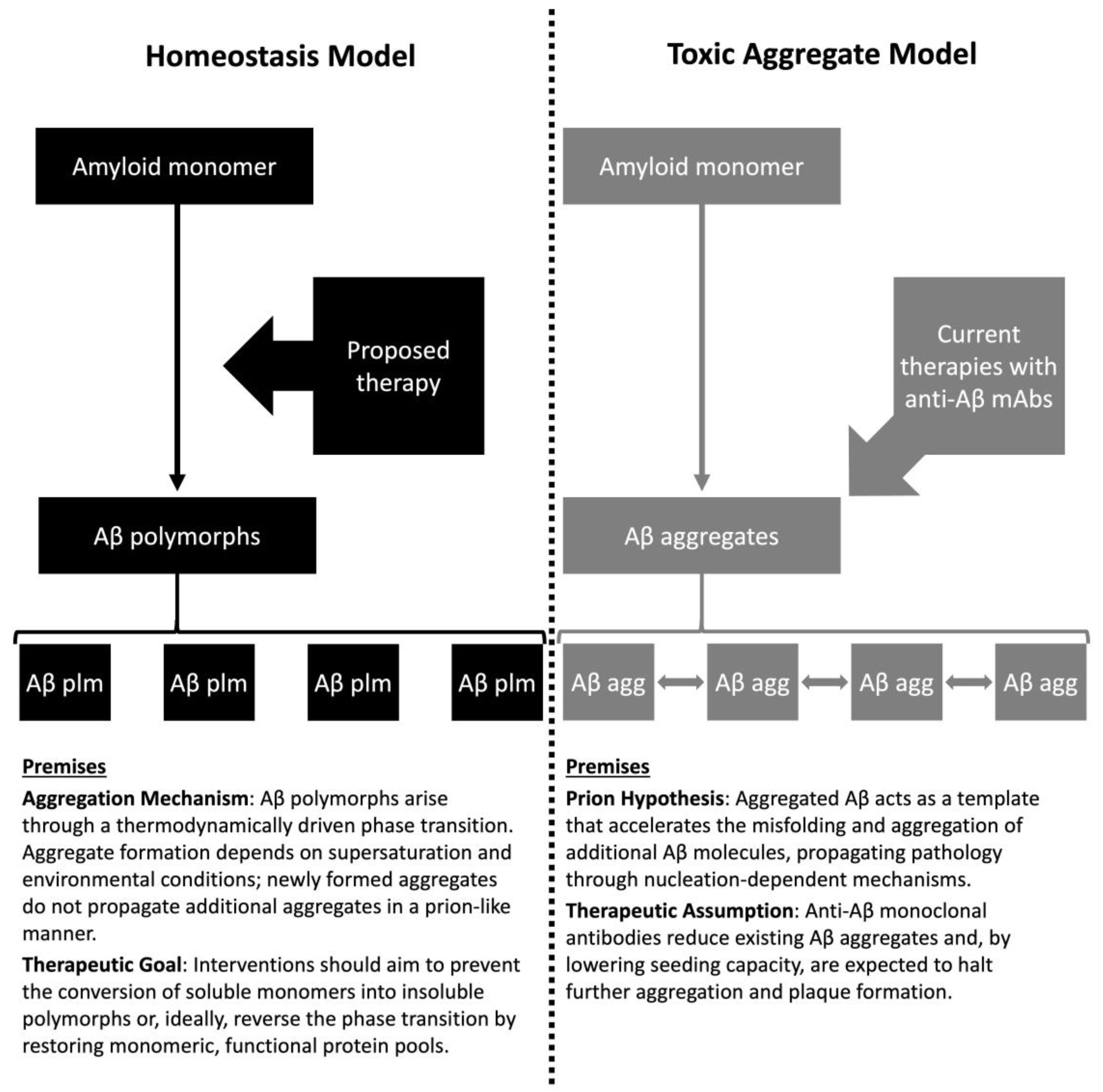
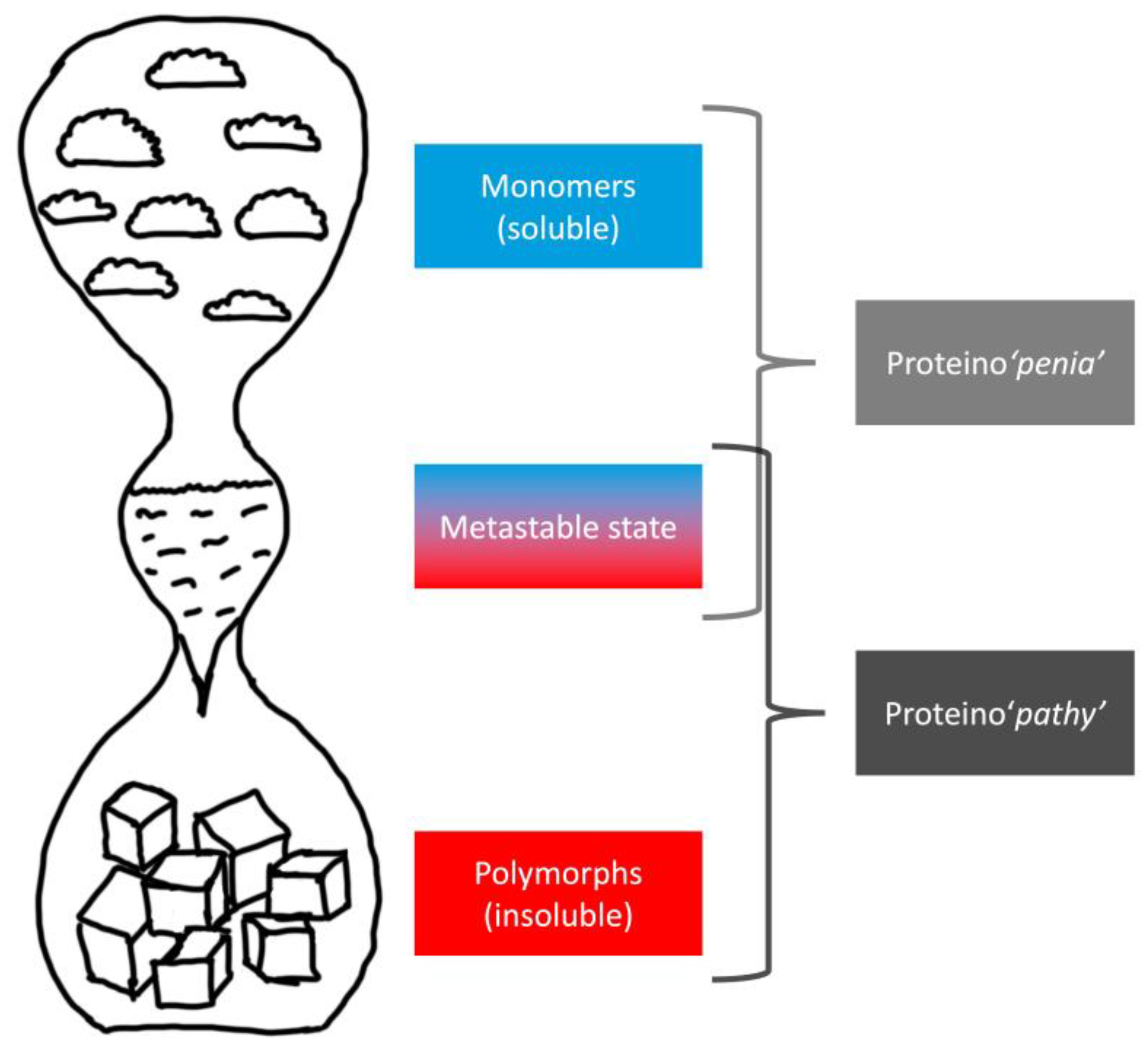
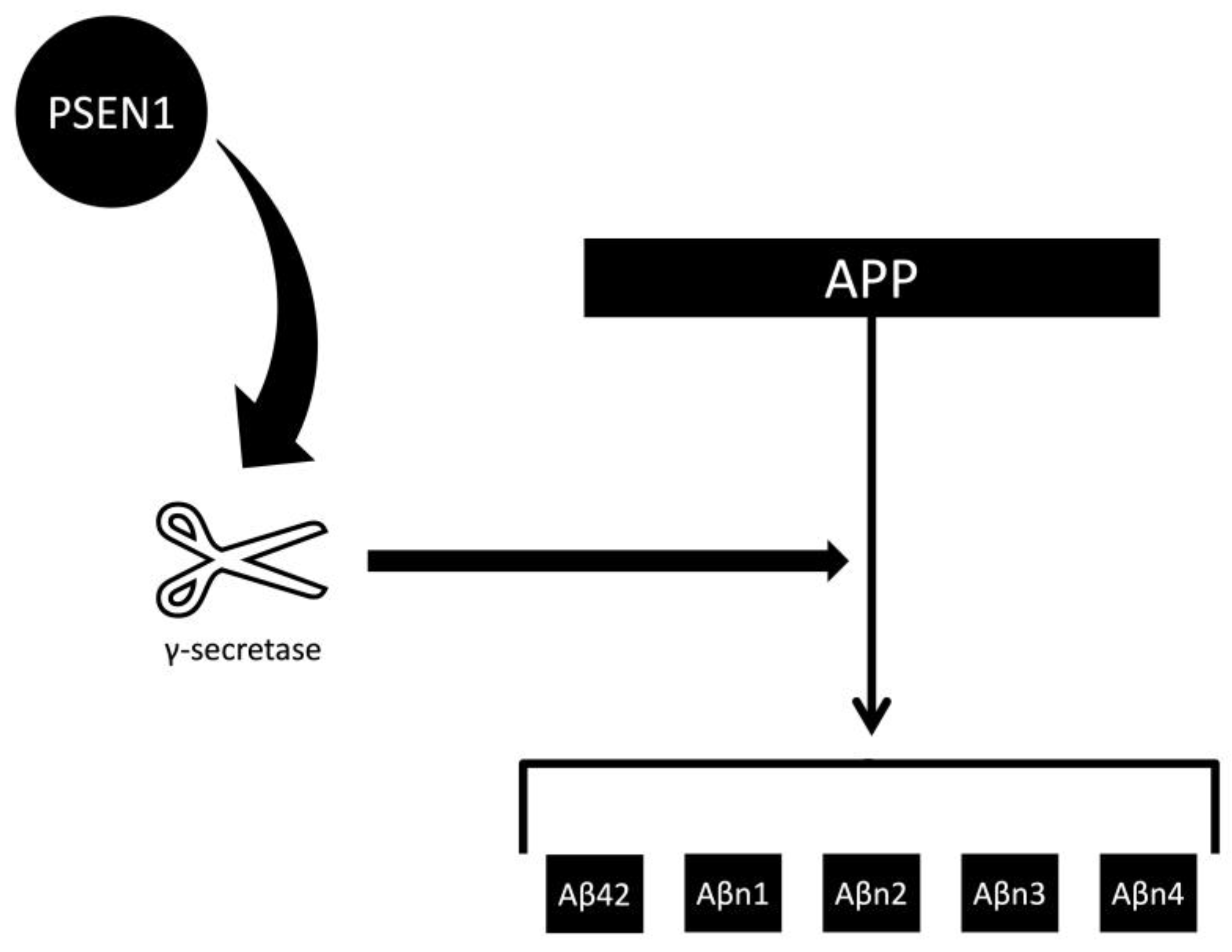
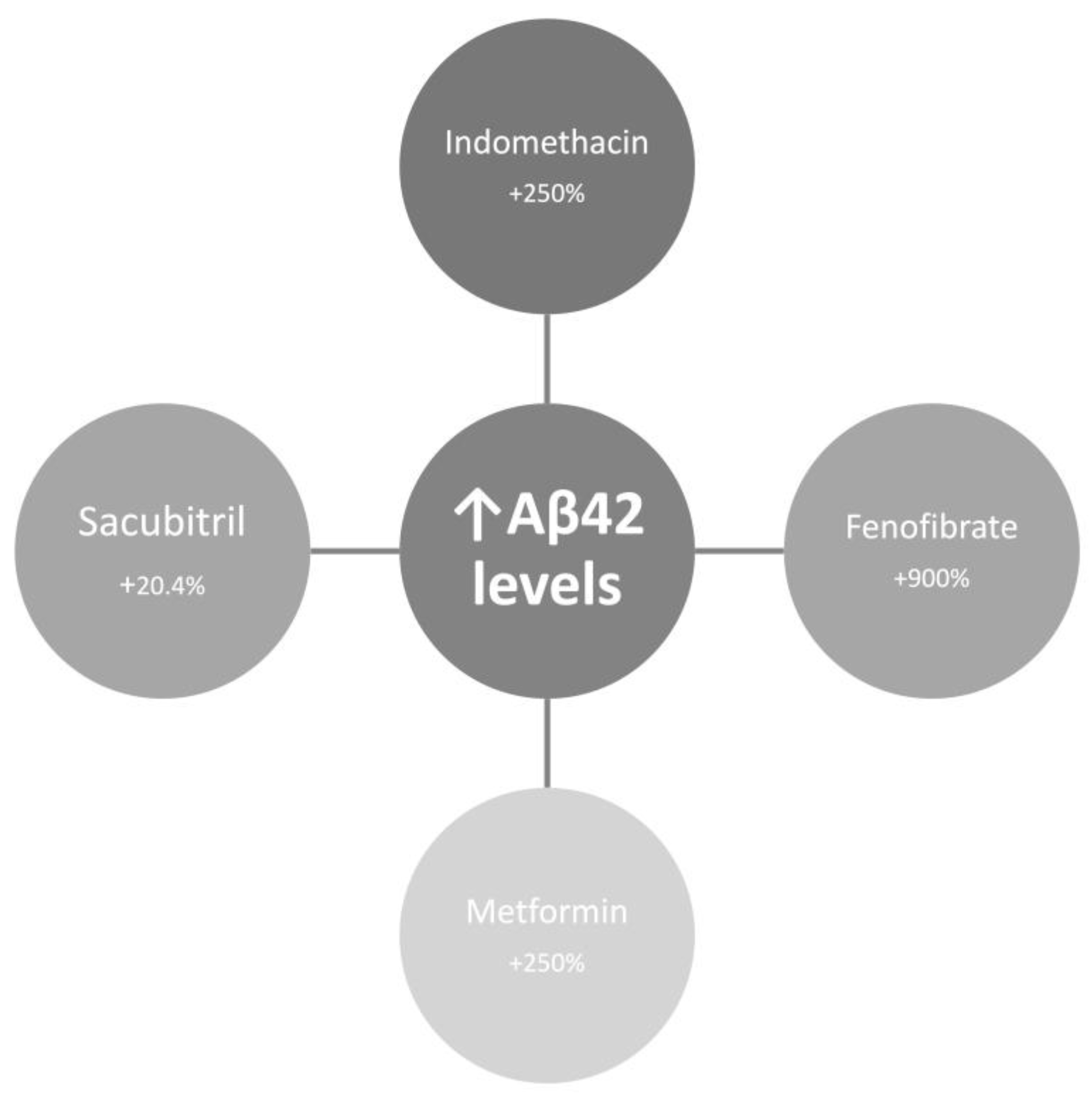
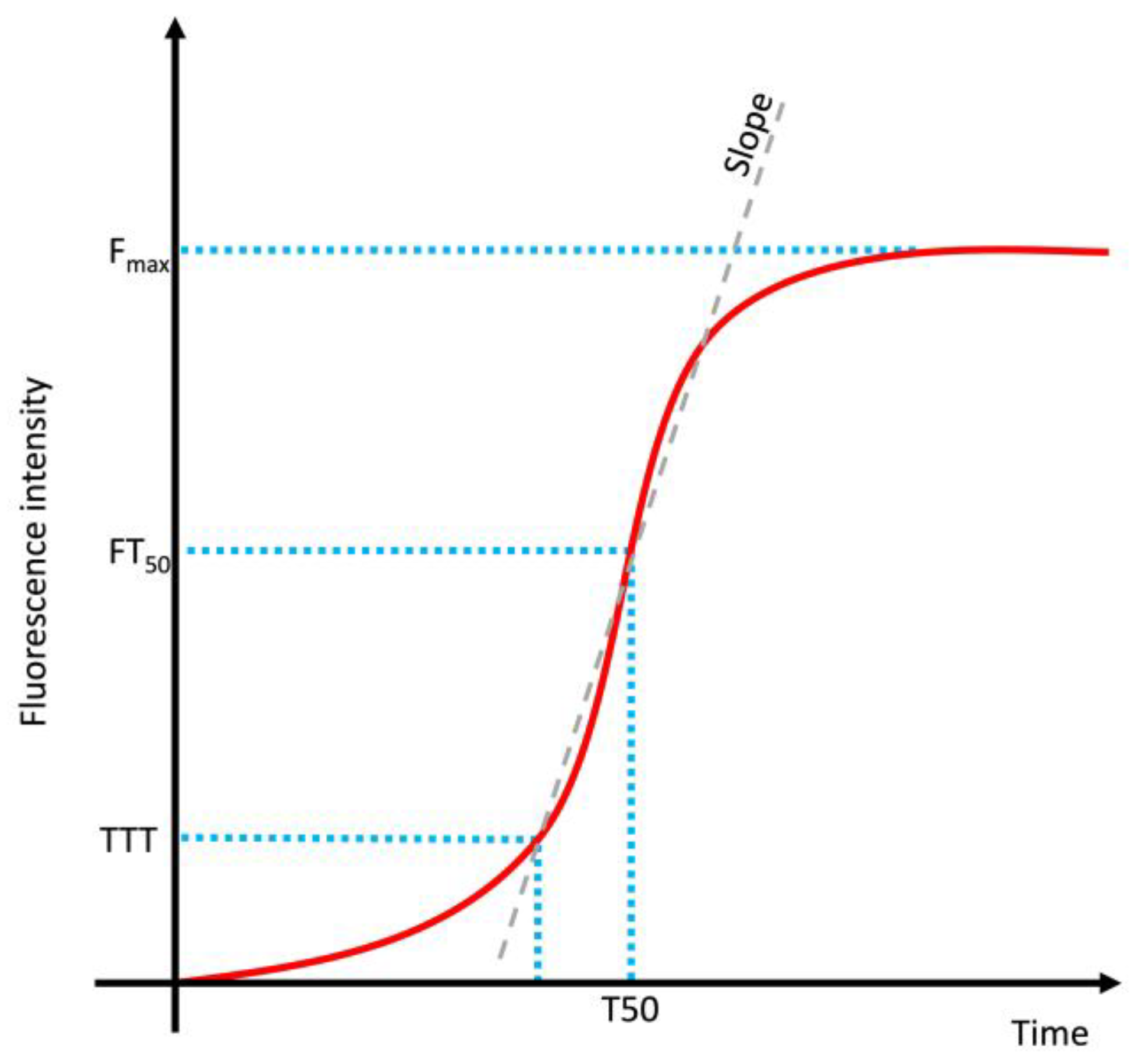
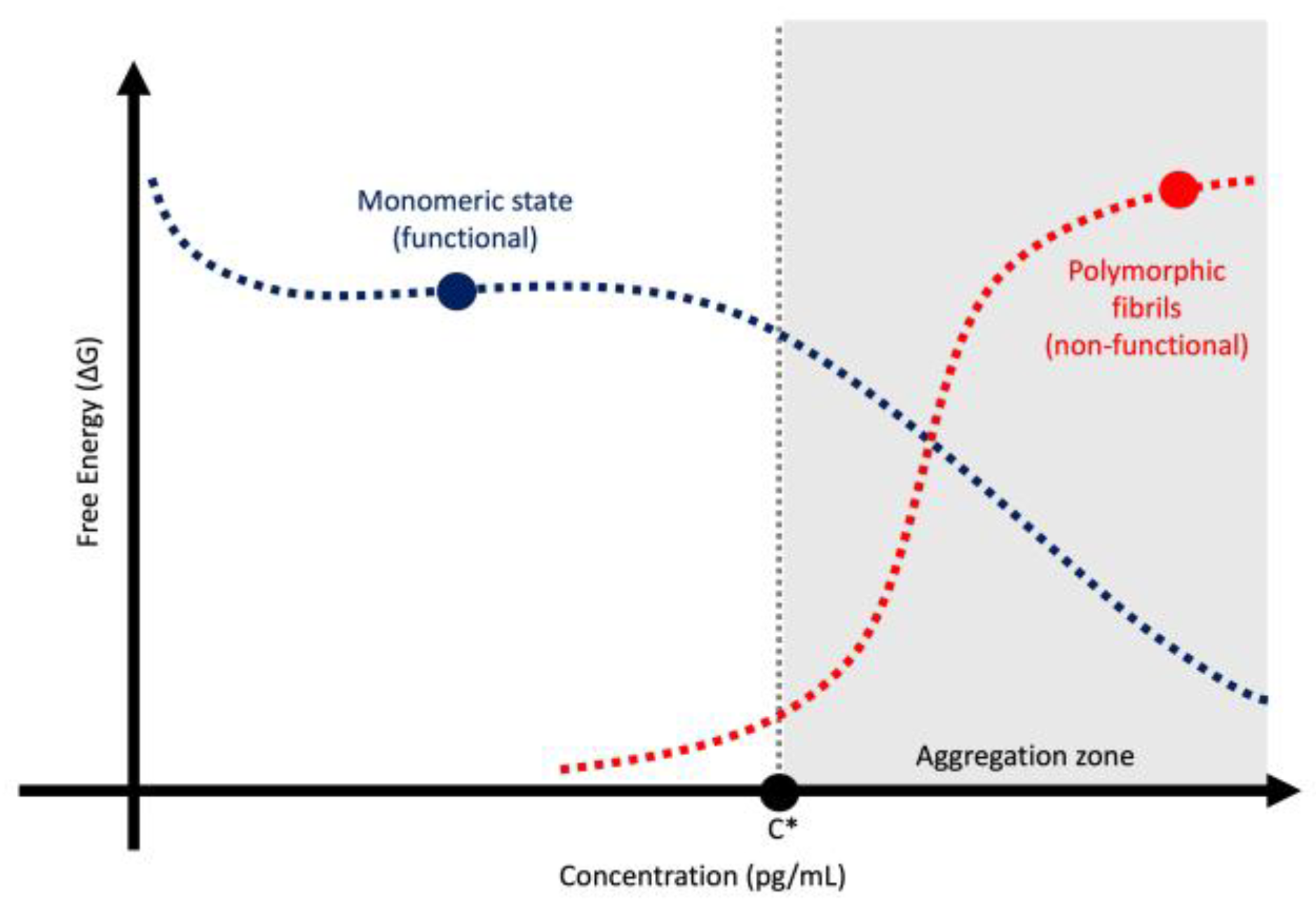
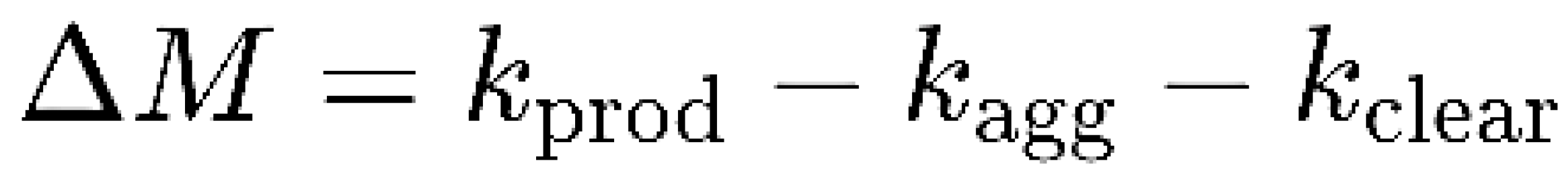
| Feature | Toxic Aggregate | Protein Homeostasis |
|---|---|---|
| Causality | Aggregates are intrinsically toxic and hierarchically causal | Aggregation reflects loss of functional monomer; pathology is reactive |
| Biomarker interpretation | Aggregate burden defines disease severity | Indicate stress response; quantify monomer and aggregate pools |
| Therapeutic goal | Clear aggregates to reduce toxicity | Restore protein homeostasis and replenish functional monomers |
| Trial design implications | Surrogate endpoints based on aggregate reduction; animal models prioritized | Human trials testing mechanistic hypotheses; endpoints focused on functional outcomes |
| Biomarker | Association with CDR-SB | Strength (Cohen's d) | Clinical Interpretation | References |
|---|---|---|---|---|
| p-tau181 | Weak | 0.01–0.12 | Tracks progression biologically, but weak link to functional severity | Duncan et al. (2025) [22] |
| p-tau217 | Weak | 0.07–0.32 | Moderate predictor of decline; best biomarker for severity among tau species | Feizpour et al. (2023) [23] |
| p-tau231 | No data | No data | Early pathology marker | Ashton et al. (2021) [24] |
| Biomarker | Expected correlation | Observed correlation | Variance explained | References |
|---|---|---|---|---|
| p-tau217 | Higher p-tau217 → worse cognition | Weak severity correlation; frequent individual mismatch | Rarely reported; clinical models show limited explanatory power | Ashton et al. (2024) [9] Palmqvist et al. (2020) [26] |
| αSyn | Faster kinetics →more advanced PD/ progression | Binary diagnostic only; misaligned with staging and progression | Severity unreported; R2 for progression unvalidated | Siderowf et al. (2023) [40] Espay et al. (2025) [41] |
| Aβ42/40 ratio | Lower ratio → worse cognition | Ratio may hide Aβ42 loss; normal ratios can occur despite severe depletion, misclassifying severity | Rarely reported; diagnostic AUC high for amyloid PET | Hansson et al. (2018) [42] Abanto et al. (2024) [7] |
| Drug | CDR-SBi | Amyloid changeii | ARIA-E & Hiii | Volume lossiv | Reference |
|---|---|---|---|---|---|
| Aducanumab (PRIME) |
-1.1 (6%) | -27% | E: 41% H: 9% |
NR | Sevigny et al. (2016) [88] |
| Aducanumab (EMERGE) |
-0.4 (2%) | -71% | E: 35% & 26% H: 20 & 16% |
Y | Budd Haeberlein et al. (2022) [89] |
| Aducanumab (ENGAGE) |
0 (0%) | -58% | E: 36% & 26% H: 19% & 16% |
Y | Budd Haeberlein et al. (2022) [89] |
| Lecanemab (BAN2401-G000–201) |
-0.4 (2%) | -31% | E: 9.9% H: 6.8% |
Y | Swanson et al. (2021) [90] |
| Lecanemab (Clarity-AD) |
-0.5 (3%) | -55% | E: 12.6% H: 17.3% |
NR | Van Dyck et al. (2023) [66] |
| Donanemab (TRAILBLAZER-ALZ) |
-0.4 (2%) | -85% | E: 26.7% H: 8.4% |
N | Mintun et al. (2021) [67] |
| Feature | Prion hypothesis | Problem with prion hypothesis | Thermodynamic model (Anfinsen dogma) | Reference |
|---|---|---|---|---|
| Core idea | Prions act as templates, imprinting their misfolded conformation on normal proteins | Branching and secondary nucleation dominate, disrupting faithful templating | Amyloid formation is a phase transition driven by supersaturation and thermodynamics | Andersen et al. (2009) [115] Törnquist et al. (2018) [116] |
| Mechanism of propagation | Elongation at fibril tips preserves cross-sectional shape | Growth occurs via branching and heterogeneous nucleation, not tip elongation | Nucleation followed by growth; branching is expected as part of phase transition | Koloteva-Levine et al. (2021) [106] |
| Sequence Requirement | Requires same protein sequence for parallel in-register stacking | Cross-seeding occurs between unrelated proteins, showing no sequence specificity | Sequence-independent; driven by packing and hydrogen bonding under high concentration | Subedi et al. (2022) [117] |
| Thermodynamic Basis | Assumes proteins leave stable native state to fit fibril tip | No thermodynamic incentive for this; amyloid formation is driven by supersaturation | Folding into cross-β structure is thermodynamically favorable at supersaturation | Portugal Barron et al. (2023) [118] Ezzat et al. (2022) [119] |
| Role of Seeds | Seeds carry conformational information | Seeds act as catalytic surfaces, not carriers of structural information | Seeds lower nucleation barrier; act as catalysts, not templates | Koloteva-Levine et al. (2021) [106] |
| Environmental Influence | Minimal role | Fibril polymorphism depends on pH, ionic strength, and other environmental conditions | Environmental conditions dictate polymorphs and ladder pairing | Ziaunys et al. (2021) [120] Frey et al. (2024) [121] |
| Spontaneous Formation | Not considered | Amyloids can form spontaneously at high concentrations (homogeneous nucleation) | Expected under supersaturation without any template | Srivastava et al. (2019) [122] |
| Cross-Sectional Shape | Preserved across generations | Shapes vary with conditions; strain concept lacks structural validation | Polymorphs arise from solution conditions, not seed structure | Peduzzo et al. (2020) [123] Lövestam et al. (2021) [124] |
| Protein | In vivo/ In vitro | Supersaturation threshold (approximation) | Note | Reference |
|---|---|---|---|---|
| Aβ42 | In vivo | 50–120 pM (200–500 pg/mL) | Healthy vs AD; depletion precedes plaque formation | Bateman et al. (2012) [140] Hansson et al. (2018) [42] |
| In vitro | >10–20 µM | Aggregation assays under physiological pH and ionic strength | Knowles et al. (2009) [19] Linse et al. (2007) [141] |
|
| Tau | In vivo | 200–600 pg/mL | Elevated in AD but aggregation depends on local concentration and phosphorylation | Barthélemy et al. (2020) [54] |
| In vitro | >2–8 µM | Phase transition observed in crowding conditions | Tsoi et al. (2023) [3] | |
| αSyn | In vivo | 0.5–1 ng/mL | PD patients show depletion of soluble αSyn; aggregation occurs intracellularly | Bellomo et al. (2025) [33] |
| In vitro | >50–70 µM | Supersaturation threshold for fibril formation under agitation | Knowles et al. (2009) [19] Frey et al. (2024) [121] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



