1. Introduction
Hemoglobin is the main protein responsible for oxygen transportation in the human body and the major component of red blood cells. The adult HbA (α2β2) is a tetrameric protein whose coding genes are located in two separate globin gene cluster families located on different chromosomes. During fetal life, the fetal hemoglobin (α2γ2) is the predominant hemoglobin and at birth HbF is progressively replaced by HbA. When compared to HbA, HbF has a higher affinity for oxygen. The P50 of HbF lower than P50 of HbA; this higher affinity of HbF for oxygen is important to obtain oxygen from maternal circulation. This property of HbF is important and was exploited in some studies of gene therapy.
Hemoglobinopathies, the genetic diseases caused by mutations of globin genes, represent the most common monogenic disorders worldwide. The genetic causes of hemoglobinopathies are represented by DNA mutations in or near the globin genes. These DNA variants may cause a defect in globin synthesis (thalassemias) or the synthesis of a mutant, defective hemoglobin, causing diseases such as sickle cell disease (SCD).
The most frequent hemoglobinopathies are β-thalassemia and SCD.
Beta-thalassemias are a group of recessively inherited hemoglobin disorders characterized by reduced or no production of hemoglobin and chronic anemia. The evolutionary association between the thalassemia carrier condition and resistance to malaria explains the geographic distribution of the disease involving areas such as the Mediterranean basin, Middle east, Southeast Asia and sub-Saharian Africa. More than 350 mutations are known for β-thalassemia; however, only 20 β-thalassemia mutations account for more than 80% of the β-thalassemia mutations across the globe. Thalassemia affected over 1,300,000 people worldwide in 2021, and the WHO declared it a major health concern [
1]. Globally, approximately 60,000 newborns are born with β-thalassemia major per year, with the majority liing in developing countries [
2]. β-thalassemia carriers account for approximately 3% of the global population. The disease is characterized by absent/low β-globin synthesis, with consequent large excess of free α-chains that are unstable and precipitate in developing erythroid cells causing oxidative damage and premature cell death in bone marrow (dyserythropoiesis) [
3]. This pathologic mechanism determines a condition of severe anemia, requiring continuous transfusion support [
3]. Advances in therapeutic support have consistently improved the life expectancy of individuals with β-thalassemia. Curative approaches to β-thalassemia are represented either by allogeneic hematopoietic stem cell transplantation (HSCT) from compatible healthy donors or by gene therapy.
SCD is a recessive hereditary disorder caused by a point mutation responsible for the substitution of Glu6 with Val in the beta globin gene, leading to the production of the abnormal hemoglobin (HbS) that has the tendency to polymerize, with consequent erythrocyte sickling, vaso-occlusion and multi-organ damage [
4,
5]. The incidence of SCD is estimated to be 515,000 neonates each year, the majority in sub-Sharan Africa, which accounts for nearly 80% 0f global cases [
6]. In 2021, an estimated 7.74 million people were living with SCD globally [
6]. SCD is characterized by a considerable phenotypic complexity. At clinical level, several acute complications, such as acute chest syndromes, stroke and acute pain events are commonly observed; chronic complications can damage all organs. In spite consistent progresses in the supportive care to SCD patients, the prognosis remains poor, and the only curative approaches are represented by allo-HSCT or gene therapy. Studies carried out in the last two decades have shown very significant improvement in curative approaches to β-thalassemia and SCD based on allo-HSCT and gene therapy. This review analyzes these studies and discusses the problem of therapeutic choices for β-thalassemic and SCD patients.
2. Hematopoietic Stem Cell Transplantation in SCD
Several challenges have limited the widespread adoption of HSCT for patients with SCD, mainly represented by the scarce availability of fully matched bone marrow donors and by the transplant-related morbidity and mortality [
7]. The greatest success is achieved when HSCT is made using an HLA-matched related donor (MRD) and a myeloablative conditioning regimen in young SCD patients. Various factors limit this approach, represented not only by the scarce donor availability but also by age at HSCT, associated organ dysfunction, and resource accessibility [
8]. HLA-matched HSCT using an HLA identical sibling donor (8/8 match) is the gold standard and is associated with overall survival rates ranging from 88% to 97% and event-free survival rates ranging from 81% to 92%.
2.1. HSCT with HLA-Matched Donors in SCD Patients
However, finding a matched donor is a significant obstacle, as fewer than 15%-20% of patients have an HLA-identical sibling donor. In a recent study on the evaluation of 128 SCD patients undergoing allo-HSCT, a matched HLA sibling donor was observed in about 50% of cases; in this study, the number of siblings per patient was 2 [
9]. Allo-HSCT requires an adequate conditioning to ablate recipient HSCs in the bone marrow and to replace these cells by transplantation of donor WT-HSCs that have to engraft and to repopulate bone marrow, and an immunosuppression sufficient to prevent graft rejection. Allo-HSCT with a myeloablative regimen offers the advantage to be associated with a higher frequency of full-donor chimerism compared to allo-HSCT with non-myeloablative conditioning. In SCD patients with severe disease and with an age comprised between 2 and 20 years, MRD myeloablative allo-HSCT resulted in a high rate of EFS and OS (>95%) with a low GVHD and a low rate of transplantation-related mortality (due to GVHD) [
10].
Thus, HLA-identical sibling HSCT after myeloablative conditioning with anti-thymocyte globulin should be considered as a standard of care for SCD children who are at high risk for stroke. [
10]
.
In a study of 736 SCD patients who underwent HSCT from an HLA-matched sibling donor between 1986 and 2017, OS and EFS were better in younger patients [
11]
. Particularly, SCD patients have been subdivided into the age groups: 0-5 years; 6-15 years; >15 years [
11]
. The 4-year OS and EFS rates in these three age groups were 100%, 95% and 88%, respectively for OS and 93%, 89% and 81%, respectively for EFS [
11]
. Furthermore, in patients with ages >1515 years, aGVHD and cGVHD incidence increased up to 17% and 20%, respectively [
11]
. These findings support an early referral of SCD patients to HSCT [
11]
.
A large retrospective international survey based on the analysis of 872 SCD patients (760 children and 113 adults) who have undergone MRD allo-HSCT after myeloablative conditioning showed a 5-year EFS and OS of 91.4% and 92.9%, respectively [
11]
. EFS was lower with increasing age, the rate of graft failure was low (2.3%) and death rate was 7% (mostly related to infectious events) [
12]
. It is of interest to note that, despite the excellent 5-year EFS and OS, in 10% of patients who died, death occurred beyond 5 years, an event seemingly related to both end organ damage from SCD and transplant [
12]
. Bernaudin and coworkers reported the long-term outcomes of 234 SCD patients under 30 years of age who have been transplanted after myeloablative conditioning; all patients received matched-sibling donor stem cell transplantation [
13]
. The 5-year EFS was 97.9% [
12]
. At day 100, acute GVHD was observed in 20.1% of cases; chronic GVHD was observed in 24 patients but was extensive in only six cases [
13]
. At 1 year, 44% of patients displayed mixed chimerism but not SCD-related events occurred in these patients with mixed chimerism, even those with 15-20% donor cells [
13]
. The major long-term concern with SCD patients undergoing myeloablative allo-HSCT was infertility risk in both genders [
1]
Walters et al. compared outcomes prospectively through 2 years after biologic assignment to a donor or no donor (standard of care) arm based on the availability of a donor either an HLA-matched sibling or an unrelated donor [
14]
. The study compared the survival of these two groups of patients 2 years after biologic assignment [
14]
. The study enrolled a total of 113 participants, 28 in the donor arm and 85 in the no donor arm; the 2-year probabilities of 2-year survival were 89% and 93%, in the donor vs no-donor arms [
14]
. Assignment to the donor arm led to improvement in vascular occlusive events, fatigue and social function [
14]
. These observations showed that short-term mortality after HSCT was not excessive when compared with standard of care for SCD; at long-term, it is expected an advantage in survival, as well as in quality of life in the donor arm compared to the no-donor arm [
14]
.
2.2. HSCT in SCD Patients Using Nonmyeloablative Conditioning Regimens
It was shown that a minimum state of peripheral blood chimerism of 20% is sufficient to reverse the sickle phenotype [
15]
. The relationship between chimerism, HbS levels, and symptomatic disease was correlated retrospectively in 95 SCD patients who had chimerism reports available at 1 and 2 years after transplantation [
15]
. Among the patients with mixed chimerism, the lowest documented chimerism without symptomatic disease was 26% [
16]
. These observations suggest that stable donor chimerism greater than 25% is associated with resolution of SCD-related symptoms [
16]
.
In line with these observations, non-myeloablative (NMA) conditioning was developed to enlarge transplantation opportunities to SCD patients unable to tolerate the toxicities induced by myeloablative regimens, inducing a condition of stable mixed donor chimerism and reversing the pathologic effect induced by SCD [
17,
18,
19]
.
Achieving long-term donor engraftment and minimizing transplantation-related complications in NMA allo-HSCT requires and adequate regimen of immunosuppression. In this context, the National Heart, Lung, and Blood Institute (NHLBI) NMA regimen with Alemtuzumab, low-dose radiation, and sirolimus in individuals with severe SCD undergoing MRD allo-HSCT showed a high and sustained engraftment, favorable overall and disease-free survival, low regimen-related toxicity, no-transplantation-related mortality, no acute or chronic GVHD and secondary graft failure in 13% of cases [
17,
19]
. In this regimen, SCD patients received Alemtuzumab at increasing doses at day -7 to -3 before infusion of allo-PBSCs from G-CSF-mobilized HLA-identical sibling donor, total body irradiation of 300 rad two days before transplantation, and sirolimus administration starting from day 1 before transplantation to two years after transplantation [
17,
18,
19]
. Inaman and coworkers have reported the outcomes observed in 91 adult SCD patients with severe disease who were transplanted with MRDs after the NHLBI NMA regimen: a part of patients received the standard conditioning regimen and another part of patients the same conditioning regimen, preceded by a preconditioning immunodepletion [
20]
. At a median follow-up of 7.3 years, OS was 90%, SCD-free survival 85%, mixed myeloid chimerism 43%; outcomes were comparable in the two conditioning regimen protocols. [
20]
. The graft failure rate observed in this protocol can be reduced by adding a preconditioning immunodepletion based on Pentostatin and oral Cyclophosphamide administration [
20]
. Hematologic malignancy or abnormal cytogenetics developed in 7 out 91 patients [
20]
. Interestingly, G-CSF administration post-HSCT to adult SCD patients
For adult SCD patients, myeloablative regimens are not feasible due to existing organ damage [
21]
. Thus, several reduced-intensity nonmyeloablative regimens were used, combining myelosuppressive agents (such as busulfan, threosulfan and thiotepa) and immunosuppressive agents (such as cyclophosphamide and fludarabine) with or without low-dose total-body-irradiation (TBI) [
21]
.
Alzahrani et al. reported the results of a clinical study carries out in 122 SCD patients, predominantly adult patients (median age 29 years, with only 9% of patients with <18 years) undergoing MRD allo-HSCT with G-CSF mobilized PB HSCs, after nonmyeloablative conditioning based on TBI (300 cGy) and alemtuzumab [
22]
. Overall survival at 1 year and at 5 year was 98% and 93%, respectively [
20]
. 16 patients (13%) experienced graft failure: 13 primary and 3 secondary failures [
22]
. Median neutrophil and platelet engraftment occurred on day 22, and 19, respectively [
22]
. Mean donor myeloid and lymphoid (CD3) chimerisms at 5 year post-transplant were 88% and 53%, respectively [
22]
. Two patients developed acute-GVHD of grade 1 and 2, with no chronic GVHD [
22]
. No patient developed solid cancer; two patients developed a myelodysplastic syndrome and one patient a chronic myeloid leukemia [
21]
. Hematological parameters markedly improved post-HSCT; SCD-related organ damage stopped to progress [
22]
. Interestingly, G-CSF administration post-HSCT to adult SCD patients may improve myeloid chimerism and may mitigate the risk of secondary graft loss with this non-myeloablative approach [
23]
.
More recently, Damlaj et al. reported the results observed in 200 SCD patients with severe SCD, mean age of 26 years, undergoing NMA, MRD allo-HSCT, treated between 2015 and 2021 at the Division of Hematology of Rivadh, South Arabia [
24]
. A part of patients received G-CSF post-transplant to accelerate neutrophil recovery. 17 patients (8.5%) experienced graft failure: 3 as primary and 14 as a secondary event [
24]
. 76% of patients discontinued sirolimus at the latest follow-up. 3-year EFS and OS were 88% and 95%, respectively [
24]
. At multivariate analysis resulted that non-ABO incompatibility, as evidenced by the presence of alloantibodies against donor non-ABO antigens, was associated with increased risk of graft failure [
25]
. Patients with graft failure had inferior survival and strategies to reduce this event are required in this setting of allo-HSCT [
25]
. The same authors recently reported the study of 115 SCD patients undergoing an alternative reduced-intensity conditioning regimen based The same authors recently reported the study of 115 SCD patients undergoing an alternative reduced-intensity conditioning regimen based on an initial 4-wk administration of prednisone at alternate days (to attenuate inflammation, to promote endothelial stabilization and to mitigate intravascular hemolysis) and then on the administration of intravenous busulfan, fludarabine and ATG [
25]
. GVHD prophylaxis utilized either Cyclosporine A/Methotrexate or Tacrolimus/Methotrexate. At 1 yr the OS was 96.5%; the incidence of acute GVHD and chronic GVHD was 19% and 10%, respectively [
26]
.
2.3. Haploidentical HSCT in SCD Patients
When an HLA identical sibling is not available, donor from a stem cell registry can be used, either identifying a matched unrelated donor or an haploidentical-related donor (frequently a parent) can be used. The main studies involving HLA-haploidentical HSCT in SCD patients are reported in Table II. Initial studies in SCD patients provided evidence that haploidentical HSCT was an unacceptable strategy for these patients due to high rates of graft failure and of death [
27]
.
In this context, emblematic was the study carried out by Shenoy and coworkers [
28]
. These authors have evaluated in a group of 30 children with severe SCD in the context of a phase II clinical study (BMT CTN 0601) of HLA-unrelated (URD) bone marrow transplantation: a regimen of Alemtuzumab, Fludarabine and Melphalan was used using Alemtuzumab early to provide recipient immune suppression to overcome the consistent risk of graft rejection in SCD patients, while laso mitigating toxicities associated with myeloablative agents [
28]
. GVHD prophylaxis included calcineurin inhibitor, short-corse methotrexate and methylprednisolone [
28]
. The 1 and 2 years EFS and OS were 76% and 69% respectively and 86% and 79%, respectively [
26]
. The day-100 incidence rate of acute GVHD II-IV was 20%, and the 1-year incidence rate of chronic GVHD was 62% (in 38% of cases, chronic GVHD was extensive) [
28]
. There were 7 GVHD-related deaths [
28]
. The 5- and 8-year probability of OS were 68% and of EFS 61% and 57%, respectively [
29]
. These observations indicate that engraftment and cure were achievable with a reduced-intensity regimen following unrelated donor HSCT; however, GVHD prophylaxis regimen was inadequate, particularly in patients over 13 years of age, compromising outcomes and increasing mortality [
29]
.
The introduction of techniques of T-cell depletion and of treatment preventing GVHD development, such as post-transplant cyclophosphamide improved the outcomes of haplo-HSCT in SCD patients [
27]
. A notable example of the significant effect of post-transplantation Cyclophosphamide in preventing GVHD is given by the study of Fitz hough et al. in patients with SCD and severe organ damage who undergo haploidentical HSCT [
30]
. These authors evaluated, in the context of a phase 1-2 trial, a non-myeloablative HLA-haploidentical PBSCT approach based on Alemtuzumab, 400 cGy total body irradiation and increasing doses of post-transplantation Cyclophosphamide: 0mg/kg (cohort 1), 50mg/kg (cohort 2), 100 mg/kg (cohort 3) [
30]
. No transplant-related mortality was observed and 90% of patients remained disease-free in cohort 3 compared to 0% in cohort 1 [
30]
. There was no grade 2 to 4 aGVHD or extensive cGVHD [
30]
. Furthermore, de la Fuente and coworkers explored haplo-HSCT in a group of 15 SCD patients with an age included from 7 to 40 years who received a conditioning regimen based on Fludarabine+Cyclophopshamide+Thiotepa+ATG+TBI and a GVHD prophylaxis based on post-transplantation Cyclophosphamide+Sirolimus+MMF [
31]
. At a median follow-up of 13 months, 95% of patients achieveda stable engraftment with an OS of 100%; two patients had grade III-IV aGVHD and 1 patient had mild cGVHD [
31]
.
Three different strategies of haplo-HSCT have been explored in SCD patients: (i) a nonmyeloablative, T-cell replete, bone marrow transplant with thiotepa and post-transplant cyclophosphamide, with the aim of achieving complete donor chimerism; (ii) a nonmyeloablative, in vivo T-cell depletion, using peripheral blood stem cells with the aim of achieving a stable mixed donor-recipient chimerism; and (iii) a myeloablative, ex vivo T-cell depletion using PBSCs and advanced technology graft manipulation, with the aim of achieving complete donor chimerism [
27]
.
In this context two recent studies were particularly relevant. Kassim et al reported the results of a prospective phase II study of haploidentical HSCT in SCD patients; 70 children and young SCD patients without an HLA-matched sibling donor or an available first- or second-degree haploidentical relative were enrolled; patients with a history of stroke or end-organ damage were included [
31]
. Conditioning was achieved with a regimen with multiple agents, including anti-thymocyte globulin, fludarabine, thiotepa, cyclophosphamide and TBI (200 centigray); GVHGD prophylaxis was achieved with post-transplant cyclophosphamide [
32]
. At two years, EFS was 82.6% and OS 94.1%; all the five deaths observed in the study were related to infectious complications [
32]
. Graft failure was observed in 11.4% of patients, who received a second haploidentical transplant; GVHD was observed in 10% of patients [
32]
.
A second study (in the context of the phase II BMT-CTN1507 clinical trial) involving 54 adult SCD patients undergoing haploidentical bone marrow transplantation after a conditioning similar to that reported in the other study, showed a 2-year EFS of 88% and an OS of 95%; two patients had graft failure (one primary and one secondary graft failure); the rate of grade 3 acute GVHD was 4.8% and of chronic GVHD was 22.4% (with 7% of patients with moderate/severe chronic GVHD) [
33]
. The median time to neutrophil and platelet recovery was 25.5 days and 34.5 days, respectively; 95% of patients achieved a condition of full donor chimerism; all patients with full donor engraftment displayed a significant improvement of hematological parameters [
33]
.
A third study, always in the context of the BMT-CTN 1507 trial evaluated 39 pediatric SCD patients (median age 12.5 years); the conditioning regimen was similar to that reported in the study on adult patients [
34]
. The results obtained in this study showed: a 2-year EFS and OS post-transplantation of 79% and 94.5%, respectively; graft failure occurred in 15.4% of patients; the median time to neutrophil and platelet engraftment was 22 and 31 days, respectively; cGVHD was observed in 30% of patients; two deaths occurred for infectious complications [
34]
.
Another recent study confirmed the good outcomes of haplo-HSCT in adult SCD patients [
34]
. In this study, 22 adult SCD patients underwent haplo-HSCT after a conditioning regimen based on anti-thymocyte globulin, fludarabine, cyclophosphamide to prevent GVHD [
35]
. Engraftment was observed in 20/22 patients with full donor chimerism, while 2 patients had secondary graft failure; acute GVHD and chronic GVHD were observed in 2 and 3 patients, respectively; organ damage remained stable or improved, and no vaso-occlusive events were observed post-transplant [
35]
.
Other studies of haplo-HSCT in SCD patients were based on the use of donor cells depleted of T-lymphocytes either by a selective T-cell depletion or by purification of CD34+ cell population, enriched in HSCs and deprived in T-cells.
Foell and coworkers performed a phase II clinical trial to evaluate α/β T-cell depleted HSCT in children and adults. SCD patients lacking sibling HLA-identical donors who have failed prior hydroxyurea therapy [
36]
. These investigators used a myeloablative conditioning regimen based on Fludarabine+Thiotepa+Treosulfan+ATG and a GVHD prophylaxis based on Tacrolimus and MMF [
36]
. The patients received CD3+/CD19+ or TCRαβ+/CD19+ PBSC grafts. After a median follow-up of 22 months, OS and EFS were both 88%, transplantation-related mortality was 12%, grade I-II and grade III-IV aGVHD were 28% and 0%, respectively, moderate or severe cGVHD were observed in 16% and 0% of cases, respectively [
36]
. An updated analysis of this study extended to 29 patients confirmed these findings [
37]
.
Gibson reported the outcomes observed in 11 SCD patients who have received unrelated peripheral blood donor stem cell transplantation with partial T-cell depletion ]. Conditioning included Hydroxyurea, ATG, Fludarabine, Thiotepa, and Busulfan; graft manipulation included CD3+/CD19+ depletion with 1x105 cells/kg addback or TCRα/β+/CD19+ depletion [
38]
. 1-yr and 3-yr OS was 93.8% and 81.3%, respectively [
38]
. No patient developed grade III-IV aGVHD or moderate to severe cGVHD [
38]
. All patients receiving 10/10 HLA-compatible donors had stable engraftment, while 60% of patients receiving grafts from 9/10 HLA compatible donors had graft failure [
38]
.
Gilman and coworkers have reported the results of a phase II study in children and young adults with severe SCD using a reduced-intensity conditioning regimen with CD34+-selected, T-cell depleted PBSC grafts. 9/10 patients survived 49 months after transplantation with no sickle cell-related complications. Two patients had grade II-IV GVHD and 1 cGVHD. Surviving patients had stable chimerism. Epstein Barr virus-related posttransplant lymphoproliferative disorder (PTLD) occurred in 3 patients, and 1 patient died as a consequence of treatment of PTLD [
39]
.
Cairo et at explored another strategy of haplo-HSCT based on the use of the purified CD34+ cell population, enriched in HSCs and deprived of lymphoid cells [
40]
. Using CD34+ cells, they reported the results observed in 19 familial haploidentical HSCT in children and adolescent SCD patients lacking sibling HLA identical donors and have failed prior hydroxyurea therapy [
40]
. These investigators used a myeloablative conditioning regimen based on Fludarabine+Thiotepa+Treosulfan+TAG and a GVHD prophylaxis based on Tacrolimus and MMF [
40]
. The patients received CD3+/CD19+ or TCRα/β+/CD19+ PBSC grafts [
40]
. After a median follow-up of 22 months, OS and EFS were both 88%, transplantation-related mortality was 12%, grade I-II or II-IV aGVHD 28% and 0%, respectively, moderate or sevre cGVHD was observed in 16% and 0% of cases, respectively [
40]
.
The initial criteria of eligibility of SCD patients for haploidentical transplantation studies involved the enrollment of patients with overt stroke, subsequently enlarged to patients with patients with silent cerebral infarcts. More recently, it was proposed incremental eligibility criteria, including also patients with life threatening acute chest syndrome, pulmonary hypertension, persistent systemic hypertension, acute untreatable pain and repeated episodes of major priapism [
41]
.
Shrestha et al. reported the development of a novel HLA-specific flow cytometry-based assay (HFA) as a tool to evaluate lymphocyte donor chimerism as a potential predictive indicator of engraftment outcomes. This assay utilizes cell surface markers (CD3, CD56 and HLA antibodies targeting a donor or patient-specific HLA for each case) for analysis of lymphocyte donor chimerism. This assay confirmed the strong donor chimerism for most of adult SCD patients enrolled in the BMT CTN 1507 study [
42]
. HFA testing identified the two patients with scarce donor engraftment before they were diagnosed as patients with a secondary graft failure [
42]
.
Eapen et al. reported the results of a retrospective study aiming to explore the impact of donor type and conditioning regimen intensity on allo-HSCT outcomes in a group of 910 SCD patients. The study reported the data of 90 US centers referring their results to the Center for International Blood and marrow transplant research (CIBMTR). These patients underwent allo-HSCT from MRD, haploidentical-related (HAPLO), matched unrelated (MUD) and mismatched unrelated (MMUD); different conditioning regimens were used in these patients, either myeloablative or reduced intensity regimens or nonmyeloablative regimens [
43]
. Conditioning regimens were considered myeloablative when busulfan was administered at concentrations greater than 150 mg/m2; regimens using lower doses of busulfan or melphalan were considered reduced intensity; TBI regimens (200-400 cGy) were considered non-myeloablative ; myeloablative conditioning is the regimen with the highest intensity and non-myeloablative with lowest intensity, whereas the reduced intensity regimens fall in an intermediate intensity category [
43]
. Overall survival was lower in patients aged 13 years or older than in those younger than 13 years; in those who received myeloablative and RIC than in those who received non-myeloablative regimens and after transplantation of grafts from donors who were not HLA-matched siblings [
43]
. EFS was lower in recipients aged ≥13 years, those transplanted with grafts from HAPLO, MUD and MMUD compared to MRD and those conditioned with reduced-intensity regimens compared to non-myeloablative regimens [
43]
. EFS did not differ between myeloablative and non-myeloablative regimens [
43]
. Mortality risks and graft failure were higher after any alternative donor transplantation (HAPLO, MUD or MMUD) compared to HLA-matched sibling transplantation (MRD) and resulted in lower EFS [
43]
. Donor type was associated with graft failure: compared with MRD, the proportion of patients with graft failure increased among recipients of HAPLO, MUD and MMUD transplants; graft failure rate did not change between HAPLO and MUD patients [
43]
. Myeloablative and reduced-intensity regimens were associated with higher mortality, and higher incidence of acute and chronic GVHD. GVHD incidence increased in recipients of transplants from HAPLO, MUD and MMUD compared to MRD donors [
43]
. The most relevant conclusions of this study are that EFS and OS are improved in SCD patients who received an allo-HSCT at age 12 years or lower and those with an MRD; for those without a MRD, different types of alternative donors seem to be equivalent.
Prior et al. compared the outcomes of allogeneic matched related donor HSCT for SCD. [
44]
Particularly, they compared four different conditioning regimens: non-myeloablative regimen-based on TBI (300-400 cGy) + Sirolimus + ATG/Alemtuzumab [
44]
; Busulfan/Fludarabine + CNI/MTX + ATG/Alemtuzumab as described by Krishnamurti et al. [
45]
; non-myeloablative conditioning regimen based on Fludarabine/Melphalan + CNI/MTX + ATG/Alemtuzab as described by King et al. [
46]
: myeloablative conditioning regimen based on Busulfan/Cyclophosphamide + CNI/MTX/Prednisone + ATG/Alemtuzumab as represented by Walters et al. [
47]
. For pediatric patients, highest EFS was observed with the conditioning regimens reported by Krishnamurti et al (3-year EFS 94%) [
45]
and lowest EFS with the nonmyeloablative regimen of Hsieh et al (3-year EFS 57%) [
17]
]. Rates of GVHD were lowest in the patients reported by Hsieh et al (3-year rate of 0%) [
17]
and highest in the cohort of patients of King et al (3-year rate of 20.4%) [
40]
. For adult patients, EFS rates were similar between the Hsieh et al cohort [
17]
and Krishnamurti et al cohort [
45]
, while cGVHD was lower in the Hsieh et al cohort [
26]
. These findings suggest that pediatric patients had the highest EFS rates using myeloablative conditioning regimen, while non-myeloablative conditioning resulted in the lowest EFS rates; pediatric patients had the highest EFS rates using myeloablative conditioning regimens, while non-myeloablative conditioning regimens resulted in the lowest EFS rates [
44]
.
Krishinamurti et al. have retrospectively analyzed a group of 763 SCD patients who have undergone allo-HSCT from MRD, HAPLO, MUD and MMUD donors after myeloablative, RIC or non-myeloablative conditioning and have analyzed in these patients the incidence and risk factors of pain crisis after HSCT [
48]
. The results of this study showed that increased risk of pain crisis after HSCT was observed in patients aged >10 years at HSCT, in age ≥18 years, in those undergoing HSCT with alternate donors either HAPLO, MUD or MMUD, and in those with graft failure [
48]
.
Stenger et al. analyzed the long-term survival and late effects of allo-HSCT in a group of 1013 SCD patients undergoing allo-HSCT at 112 centers (51% MRD, 21% mismatch related donors MMRD, 11% matched unrelated donors and myeloablative in 66% of cases); GVHD prophylaxis was variable, involving tacrolimus, cyclosporine or post-transplant cyclophosphamide [
49]
. After a median follow-up pf 60 months, 86% of transplanted patients remained cured with no SCD symptoms; most of patients had no SCD-related complications post-transplant; 70% of patients remained free of acute GVHD and 74% of chronic GVHD [
49]
. At 1 and 7 years post-HSCT, OS and EFS were 95% and 90%, 89% and 83%, respectively [
49]
. Older age at HSCT, alternative donor and grade III-IV GVHD were significantly associated with shorter survival [
49]
. Therefore, outcomes are superior in patients who undergo HSCT at younger age, using BM MRD donors and in those who remain GVHD-free [
49]
.
2.4. Long-Term Effects of HSCT for SCD
The large number of HSCTs carried out in SCD patients allowed to perform an analysis of the major determinants of the rate of success and of the complications (early and late) of this procedure. HSCT is a highly intensive treatment associated with many toxicities which may impact patient-reported outcomes and the quality of life.
Recent studies including increasing numbers of HSCTs with alternative donors, the introduction of reduced-intensity conditioning and non-myeloablative conditioning regimens and efficient GVHD prophylaxis, such as post-transplant Cyclophosphamide, have provided evidence that most SCD patients who underwent HSCT were alive, cured of SCD, and free of late events post-HSCT [
50]
.The mata-analysis of 58 studies of HSCT in SCD patients showed an OS rate of 94% and an EFS rate of 86% [
50]
. Clinical outcomes in these patients varied according to the type opf donors, conditioning procedures/regimens, GVHD prophylaxis regimens and stem cell sources [
49,
50]
. Older age at HSCT, alternative donors and grade III-IV aGVHD are determinants significantly associated with inferior survival [
49,
50]
.
Limited data defined long-term health outcomes after allo-HSCT for SCD and rarely the HSCT outcomes were compared with chronic standard SCD therapies. In this context, some retrospective studies have analyzed long-term organ function after HSCT for SCD.
In SCD patients who received myeloablative HSCT from HLA-identic sibling donors with successful engraftment there was the resolution of SCD-related events after transplantation, such as pain, stroke or acute chest pain [
51]
. Vaso-occlusive events represent key pathological determinants in the genesis of tissue damage. Vaso-occlusive events requiring medical care are significantly reduced in patients with SCD after allo-HSCT; this reduction improves over time and is particularly evident beyond 1 year post-HSCT [
52]
.
SCD causes various central nervous system (CNS) abnormalities and cerebral vasculopathy was the main indication for HSCT in SCD patients; usually CNS abnormalities are stabilized or improved after HSCT, with normal or stable brain magnetic resonance imaging [
53]
. Cases of stroke after HSCT occurring in patients with preexisting CNS disease and graft failure or rejection have been reported [
51]
. In adult SCD patients with CNS involvement with an history of depression, a high incidence of suicidal ideation was observed [
54]
.
Cardiopulmonary complications are frequently observed in SCD patients and seem to stabilize or to improve 1-2 years after HSCT. However, more post-HSCT patients had low cardiac ejection fraction than pre-HSCT patients [
53]
.
At baseline, patients with SCD have to face some infertility risk factors and this condition may be considerably impacted by gonadotoxic treatments associated with HSCT, caused by alkylating agents and TBI [
49,
50]
. Conditioning with myeloablative Busulfan and Cyclophosphamide causes serious gonadotoxicity particularly among post-pubertal females; reduced intensity conditioning and non-myeloablative conditioning are less gonadotoxic, but their effects on fertility need to be assessed at long-term [
49]
. Female patients with SCD who undergo HSCT have high rates of diminished ovarian reserve, while the effects on premature ovarian insufficiency are variable [
56]
. These observations support the view that patients need to be counseled about the risk of infertility and informed on the strategies that can be adopted to favor fertility preservation.
An improvement in quality of life of SCD patients who received allo-HASCT is a finding largely expected given the curative effect of transplantation in these patients. However, it must be considered that HSCT is a highly intensive treatment associated with many toxicities which might impact patient-reported outcomes and the quality of life. A prospective evaluation of patients-reported outcomes in 17 adolescents and adults with SCD undergoing HSCT showed a significant improvement of physical function and pain interference after 1 year [
45]
. Other three small retrospective studies have explored the effects of HSCT on quality of life showing that patients with successful transplants are usually able to pursue their objectives, but fatigues and avascular necrosis can negatively affect the quality of life of some patients [
57,
58,
59]
.
Two recent studies more carefully assessed quality of life in SCD patients who received allo-HSCT. Nirmal et al. reported the assessment of patient-parent reported quality of life involving 62 SCD patients who underwent HSCT with a follow-up of 59 months post-transplant [
60]
. These patients experienced, post-HSCT, improved physical health, improved scholastic performance, decreased hospital visits and were able to pursue their personal life goals [
60]
. Dovern and coworkers explored changes in quality of life of adult SCD patients with SCD following allo-HSCT, through assessment of physical, mental and social health before HSCT and 6-, 12- and 18-months post-transplant [
61]
. The results of this assessment showed an improvement of quality of life at physical, mental and social health level after HSCT; however, these effects on mental health are complex and support the need for a psychosocial support early during the process of curative therapies [
61]
.
3. Hematopoietic Stem Cell Transplantation in Beta-Thalassemia
HSCT is an established procedure in β-thalassemic patients with a consistent definition of the clinical and biologic parameters that affect transplantation outcomes.
3.1. HSCT in β-Thalassemic Patients with HLA-Identical Donors After a Myeloablative Conditioning Regimen
Studies carried out in the last 3-4 decades showed that HSCT is an important therapeutic option for β-thalassemic patients. The most consistent experience occurred in Pesaro, Italy where a standard pre-transplantation risk assessment was developed. This system classifies β-thalassemic patients into 3 risk classes based on liver size assessed by physical examination, the presence or absence of liver fibrosis as assessed by histological analysis of liver biopsy, and adherence to regular iron chelation: class I, no or minimal risk factors; class II, presence of one or two risk factors; class III, presence of 3 risk factors [
62,
63]. For patients undergoing HSCT, assessment of liver pathology by liver biopsy plays a crucial role in risk stratification and clinical decision-making, particularly for patients displaying an abnormal liver elastography [
64]. These studies showed that after HLA-matched sibling HSCT, the thalassemia-free survival probabilities for Pesaro class I or II and class III recipients were 87%, 85% and 80%, respectively [
65,
66]. Other studies outside Italy showed 5-yr probabilities of OS and EFS for patients with Pesaro risk class II corresponding to 91% and 88%, respectively and 64% and 62%, respectively for Pesaro risk class III [
67]. Mortality risks were higher for patients higher than 7 years of age anf those with hepatomegaly before HSCT [
67].
In a large analysis of 1061 cases of matched sibling donor transplants from the EBMT registry involving β-thal patients with a median age of 7 years showed a long-term OS and thalassemia-free survival (TFS) of 91% and 83%, respectively [
68]. A statistically significant difference in OS was reported in the group of patients with an age comprised between 10 and <14 years compared to those in the group of 14 to <18 years of age (96% and 82%, respectively); in the group of patients with <18 years of age (96% vs 82%, respectively); in the group of patients with >18years OS and TFS were 80% and 77%, respectively [
68]. A subsequent EBMT study showed that a HSCT with fully matched unrelated donor is associated with outcomes identical to those obtained with an MSD [
69,
70]. (
Figure 1).
HSCT in β-thalassemia continuously expanded and the current worldwide experience showed OS rates around 90%, with disease-free survival around 70-80%; furthermore, advanced transplantation techniques, such as modified conditioning regimens, have reduced the risk for high-risk β-thalassemic patients, making outcomes more similar across all Pesaro classes; despite these improvements, factors like iron overload from pretreatment exposure negatively impact outcomes, particularly in older patients.
Recent retrospective analyses have provided a long-term evaluation of survival and of late effects of HSCT in β-thalassemia patients who have undergone allo-HSCT with a HLA-identical-related donor sibling after a myeloablative conditioning regimen [
71]. In a group of 137 β-thalassemic patients who received allo-HSCT the cumulative incidence of nonrelapse mortality and thalassemia recurrence was 9.5% at 1 years and 10.2% at 39 years; the 39-years cumulative incidence of overall survival and disease-free survival were 81.4% and 74.5%, respectively; early death and late death transplant-related causes were observed in 9.5% and 0.7% of patients, respectively; full chimerism was observed in 94.5% of cured patients [
71]. OS was related to the age at the moment of HSCT: 84.7% in patients <10 years; 80.9% in patients 10-17 years; 74.3% in patients >18 years [
71]. Patients cured had normal hematological parameters [
71]. 15 cases of secondary solid cancers were observed for a 39-year cumulative incidence of 16.4% [
71]. The conclusion of this long-term evaluation of β-thalassemic patients who have received allo-HSCT was that allo-HSCT represents a curative option for most of these patients at the price of a nonnegligible early and late mortality [
71]. Similar conclusions were reached by Caocci et al. in a long-term analysis on 258 β-thalassemic patients, including 97 adults (age ≥16 years) [
71]. After 30-year follow-up, OS among all patients was 82.6%, compared to 71.2% among adult patients [
72].
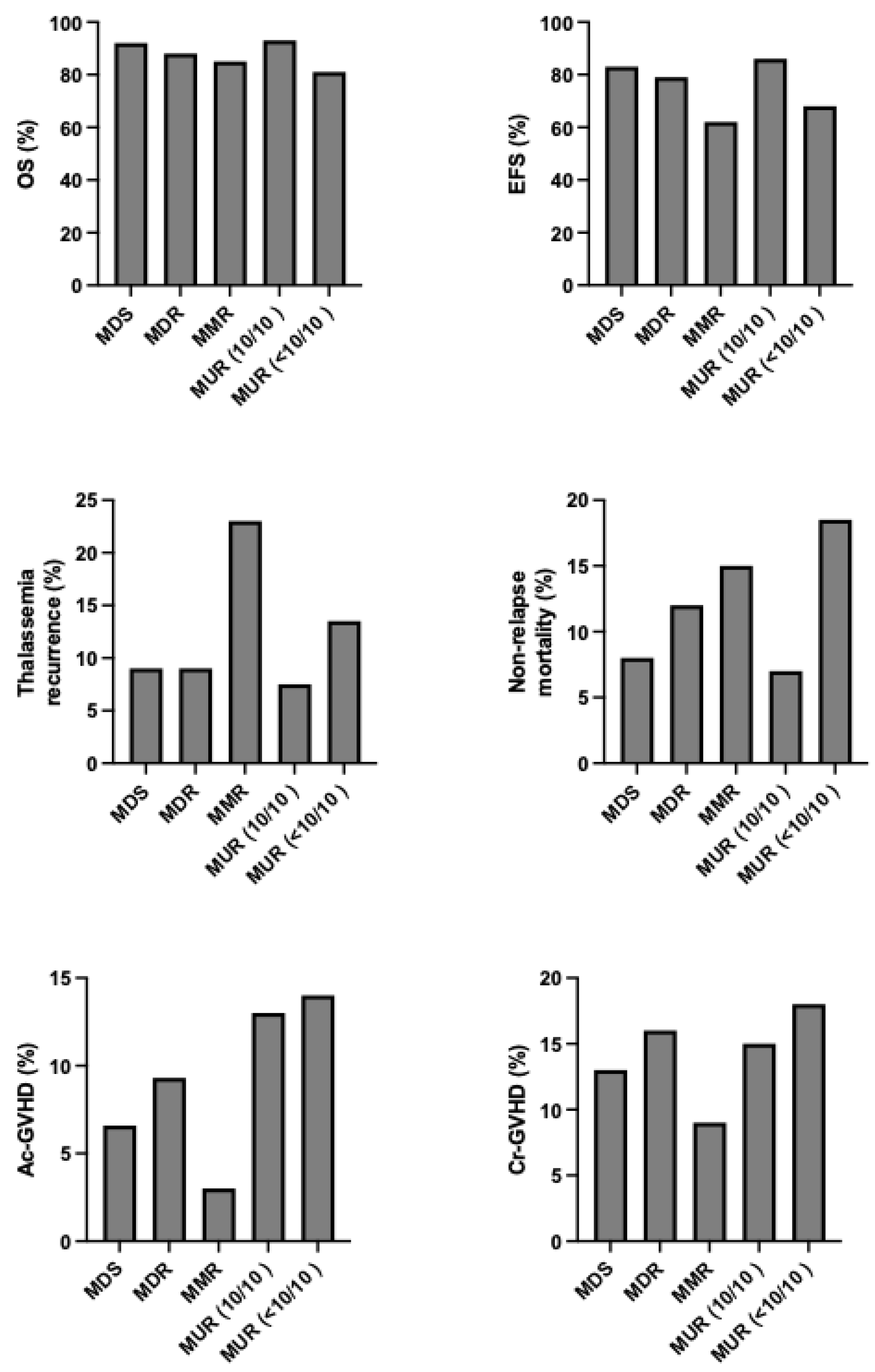
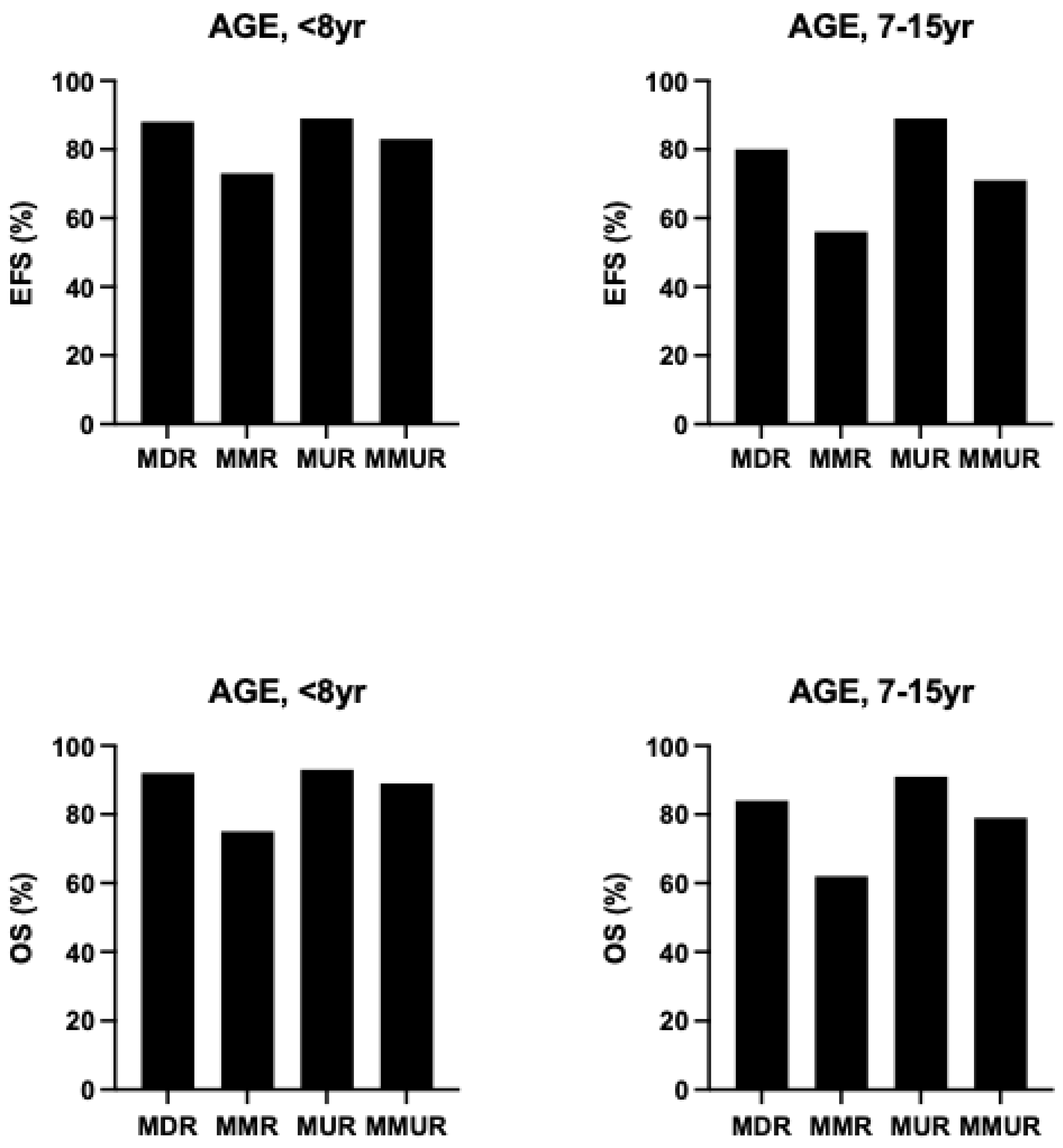
As above discussed, among the clinical determinants of transplantation outcomes, the most relevant is represented by the risk class of the patients, mainly defined the absence or presence of hepatomegaly and portal hepatic fibrosis. An international survey no more than 1000 β-thalassemia patients undergoing HSCT showed that outcomes are strongly affected by the age of patients at the transplantation: overall survival for patients ≤6 years, 7 to 15 years, and 16 to 25 years, the 5-year OS was 90%, 84% and 63%, respectively and the EFS was 86%, 80% and 63%, respectively [
73]. (
Figure 2) Among the biologic factors, the most relevant determinant was HLA-matching: overall and event-free survival were similar for HLA-matched relative and HLA-matched unrelated donor transplantation (OS 89% vs 87% and EFS 86% vs 82%, respectively); overall and event-free survival were lower for HLA mismatched than HLA matched transplants [
73]. The graft failure for patients ≤6 years, 7 to 15 years and 16 to 25 years were 8%, 10% and 22%, respectively. A high rate of graft failure was observed after HLA-mismatched transplants (HLA-matched relative 85% and HLA-mismatched relative 22%) [
73]. aGVHD and cGVHD were higher in HLA-mismatched relative than in HLA-matched relative 35% vs 12% and 20% vs 8%, respectively. [
73].
These findings were confirmed in a study of Xu et al on 281 Chinese β-thalassemic patients who received HSCT from HLA-identical sibling donors; the patients were subdivided into 3 groups: group 1 <4 years; group 2 5-8 years; group 3 >8 years. OS in group 1, 2 and 3 were 96.2%, 95% and 85.2%, respectively [
67]. These findings support the view that an age >8 years at HSCT negatively affects transplantation outcomes [
74].
In this context, it is important to note that HLA-typing techniques have led to define five key human leukocyte antigen (HLA) loci (HLA-A,-B, -C, -DRB1 and -DQB1) crucial for immune recognition, whose matching between donor and recipient allows to define a condition of good compatibility (10/10 compatibility). Thus, although HLA-matched sibling transplantation represents the gold standard, an 8/8 allele matched donor (at HLA-A, -B, and -C, and -DRB1) is the minimum requirement, but transplantation from 10/10 donors represents a good choice for patients without HLA-matched donor. However, the achievement of 12/12 match across all six HLA loci, including HLA-DP1, is considered the gold standard for optimal outcomes, since mismatches are associated with higher risk of GVHD and graft failure, particularly in HSCTs in patients with non-malignant conditions. In fact, HLA-DP1 mismatches in otherwise matched transplants associate with an increased incidence of GVHD [
75] and post-transplant Cyclophosphamide improves survival in HLA-DP1 mismatched unrelated donor allo-HSCT [
76].
The availability of a matched donor sibling for HSCT is limited so that probability of finding a suitable fully compatible donor is below 25%. Due to the paucity of matched donor availability, haploidentical HSCT (Haplo-HSCT) is a reasonable alternative. Two different regimens for haplo-HSCT were used: ex vivo t-cell depletion via TCRαβ/CD19+ depletion and in vivo T-cell depletion with post-transplantation cyclophosphamide.
3.2. Haplo-HSCT in β-Thalassemia Using Post-Transplant Cyclophosphamide as GVHD Prophylaxis
Post-transplant Cyclophosphamide is typically used in association with other immunosuppressive drugs, usually administered at days +3 and +4 post-transplant.
Wen et al. reported the study of 20 β-thalassemic patients (median age 8 years) undergoing mismatched HSCT (15 unrelated and 5 related donors). The patients received a conditioning regimen containing thymoglobulin, Busulfan, Fludarabine and Thiotepa; GVHD prophylaxis consisted of Cyclophosphamide, Mycophenolate Mofetil and Thiotepa administered post-transplantation [
68]. All transplanted patients had a successful engraftment with >95% of donor-derived cells at day 30 post-transplantation; after a median follow-up time of 38 months, OS and TFS were 100% [
77]. The cumulative incidence of grade II-III acute GVHD was 10% (no patient developed grade IV GVHD); no patient developed either limited or extensive chronic GVHD [
77]. The rates of OS, TFS and the incidence of acute and chronic GVHD compare favorably with those previously observed by the same authors in mismatched HSCT in β-thalassemic patients using a different strategy of GVHD prophylaxis [
77]. Lu et al. reported the results observed in 40 β-thalassemic patients receiving allo-HSCT from 10 haploid-related donors (Haplo-RD) and from 30 unrelated donors, 15 with HLA-matched (MUD) and 15 with haploid HLA compatibility (Haplo-UD); these patients received HSCT after a conditioning regimen based on Fludarabine, Busulfan, Cyclophosphamide and Thiotepa and with a GVHD prophylaxis including post-transplantation Cyclophosphamide, ATG and Cyclosporine [
78]. Importantly, in HSCT involving Haplo-RD and Haplo-UD the GVHD regimen was based on a low-dose ATG (1.mg/Kg/3d) and a semi-dose of Cyclophosphamide (25mg/Kg/2d) [
69]. The incidence of grade III-IV acute GVHD was 7.5% and of chronic GVHD was 10% [
69]. The OS and TFS were both 92.5%. In the group of Haplo-RD patients OS and TFS were both 100% [
78]. Anurathapan and coworkers developed a reduced-intensity conditioning regimen (Fludarabine and Busulfan) and initially explored the regimen in HLA-matched allo-HSCT of β-thalassemic patients with age >10 years and hepatomegaly and observed that this regimen was comparable to a conventional myeloablative regimen in terms of OS and EFS [
79]. In a subsequent study this approach was extended to 83 β-thalassemic patients undergoing haplo-HSCT [
71]. In this study, the patients were first submitted to a pharmacologic pretransplant immune suppression phase and two courses of dexamethasone and Fludarabine, followed by posttransplant conditioning with Fludarabine, i.v. Busulfan and post-transplant GVHD prophylaxis with Cyclophosphamide, Tacrolimus and mycophenolate mofetil [
80]. The 3-year OS and EFS were both 96% and there were no secondary graft failures [
80]. 7% of treated patients developed severe GVHD [
80].
Hu et al. reported the results of a clinical study on 54 pediatric β-thalassemic patients who have undergone allo-HSCT from haploidentical donors after a myeloablative conditioning regimen, with post-transplant cyclophosphamide and low-dose methotrexate as a GVHD prophylaxis regimen [
81]. After a median follow-up of 520 days, OS and EFS rates were 98.1% and 90.7%, respectively [
81]. The cumulative incidence of grade II-III acute GVDH was 13.8% and the frequency of chronic GVHD was 28.5% [
81]. Importantly, in this study it was shown that the inclusion of a high-dose cyclophosphamide dose (200mg/Kg) compared to a low-dose cyclophosphamide dose (120 mg/Kg) resulted in a better OS and EFS and a lower incidence of chronic GVHD [
81]. More recently, the same authors have reported the results of a retrospective analysis on 160 β-thalassemic patients undergoing allo-HSCT using PBSCs from haplo-identical donors or matched unrelated donors using the above-reported GVHD prophylaxis regimen using post-transplant Cyclophosphamide [
82]. The results of this study showed that OS, thalassemia-free survival, aGVHD and cGVHD were similar in HSCT from haploidentical and matched donors [
82].
3.3. Haplo-HSCT in β-Thalassemia with TCRαβ+-Depleted Donor Cells
Ex vivo T and B-cell-depleted haplo-matched transplants represent another strategy for HSCT from haploidentical donors. T-cell and B-cell depleted haplo-HSCT are associated with a low rate of acute and chronic GVHD and a low rate of transplant-related mortality but with a high rate of graft failure; patients who experienced graft failure may be re-transplanted.
Several studies have explored the use of haploidentical TCR and B-cell-depleted donors in allo-HSCT in children with nonmalignant disorders, including β-thalassemia. In this context, Bertaina and coworkers have explored in a clinical trial 23 children with nonmalignant disorders who received HLA-haploidentical HSCT after
ex vivo elimination of αβ
+ T-cells and CD19
+ B-cells; no patient received any post-transplantation pharmacologic prophylaxis of GVDH; all but 4 patients engrafted, but rescued after a second transplant; transplantation-related mortality was 9.3% and after 2 years OS and DFS were both 91.1% [
83].
Gaziev and coworkers have examined the outcomes of haplo-HSCT using TCRαβ
+-depleted grafts in 14 children with hemoglobinopathies (TCR group) compared with the outcomes observed in a historical group of 40 patients with hemoglobinopathies transplanted with CD34
+ selected grafts [
84]. Patients received a conditioning regimen consisting of Busulfan, Thiotepa, Cyclophosphamide and ATG, preceded by Fludarabine, Hydroxyurea and Azathioprine. In the TCR group, the 5-year OS and DFS were 84% and 69%, respectively, with 14% of primary graft failure; in the CD34 group, the 5-year OS and DFS were 78% and 39%, respectively, with a graft failure of 45% [
84]. The incidence of acute GVHD and chronic GVHD were 28% and 21% in the TCR group and 29% and 10% in the CD34 group [
84]. Suboptimal recovery of CD4
+ during the first six months post-transplantation was observed in both groups of patients [
84].
Another clinical study reported the results observed in 70 pediatric patients with nonmalignant disorders undergoing TCRαβ/CD19-depleted haploidentical HSCT from HLA-partially matched relative donors [
85]. The median age at transplantation was 3.5 years; primary engraftment was observed in 51 patients, while 19 and 2 patients experience primary or secondary graft failure, respectively; most graft failures were observed in children with aplastic anemia or β-thalassemia; all but 5 patients exhibiting graft failure were retransplanted; six patients (8.5%) died of infectious complications ; grade 1 or 2 GVHD was observed in 14.4% of patients; the 5-year probability of overall and disease-free survival were 91.4% and 86.8%, respectively [
85].
Since a complete
ex vivo T cell depletion while mitigating the risk of developing GVHD, exposes to a higher risk of graft failure and of severe infections due to delayed immune reconstitution, a strategy was developed for CD3
+/CD19
+ depletion with a targeted CD3
+ addback of 1x10
5 CD3
+ T cells/Kg to the graft before infusion [
86]. All 12 patients displayed rapid engraftment, were alive with >92% of peripheral blood chimerism [
86]. No patient developed grade III-IV GVHD [
86]. Another study confirmed the safety and the efficacy of unrelated donor stem cell transplantation with partial T-cell depletion in a group of 16 pediatric patients with hemoglobinopathies [
87].
Liao and coworkers have reported a study on 266 β-thalassemic patients undergoing haplo-HSCT with TCRαβ
+-depleted donor cells with the specific aim of defining an optimal conditioning regimen for HSC engraftment [
88]. The patients were subdivided into two groups depending on the timing of ATG and Cyclophosphamide administration. In
TDH-A group ATG was administered on days -8 to -7 without Cyclophosphamide on days -2 and -1; in TDH-B group, ATG was administered on days -21 to -19, with 50mg/Kg cyclophosphamide on day -2 and 25 mg/Kg cyclophosphamide on day -1 [
89].The rest of the conditioning regimen was identical for the two groups of patients and was based on cyclophosphamide, Busulfan, Fludarabine and Thiotepa; the conditioning regimen included also the infusion of a low number of donor lymphocytes [
89]. Remarkable differences were observed for groups TDH-A and TDH-B. In the group TDH-A 30 patients were enrolled, 18 (60%) achieved a successful engraftment, while 12 (40%) had primary graft failure (all underwent salvage HSCT with alternative donors); one death occurred and with a follow-up of 1523 days, OS, TFS, TRM (transplant-related mortality) were 96.7%, 56.7% and 40%, respectively [
89]. In the group TDH-B, 236 β-thalassemic patients were enrolled, 231 achieving a successful engraftment (97.9%), while (2.1%) showing primary graft failure; 10 deaths occurred and with a follow-up of 717 days, OS. TFS and TRM were 95.4%, 94.1% and 4.6%, respectively [
89]. Grade III-IV GVHD was 10.5% among TDH-A and 4.7% among TDH-B patients; chronic GVHD was 16.6% in TDH-A group and 8.6% in the group TDH-B [
89]. These observations showed that advancing ATG administration to day -21 (group TDH-B) significantly promoted engraftment; pretransplant (day-2/-1) administration of Cyclophosphamide clears recipient lymphocytes without compromising donor stem cell engraftment with low rates of acute and chronic GVHD [
89].
Giardino and coworkers have retrospectively analyzed 20 children with congenital non-malignant disease who underwent haploidentical stem cell transplantation after TCRαβ
+ and CD19
+ depletion; no GVHD prophylaxis was given when TCRαβ
+ cells in the graft were <1x10
5 cells/Kg [
90]. Engraftment was observed in 85% of patients; 15% of patients displayed primary graft failure; 10% of patients exhibited secondary graft failure; all these patients received a second transplant [
90]. The cumulative incidence of acute and chronic GVHD was 15% [
90]. After a median follow-up of 4 years, 18 patients (90%) are alive [
90].
Liao et al have analyzed in a group of 196 β-thalassemic patients who have underwent allo-HSCT the effect of different stem cells used for transplantation on the immune reconstitution, GVHD and viral infections; three groups of HSCTs were analyzed: MUD-HSCT, Haplo-HSCT and TCRαβ
+ T-cell depleted HSCT (TD-HSCT) [
91]. TD-HSCTs exhibited a faster immune reconstitution compared to the other two groups; TDH and MUD-HSCTs were associated with a lower incidence of cGVHD; the occurrence of viral infections was higher in the TDH group than in the other two groups [
91].
Meissner et al. have retrospectively analyzed 124 transfusion-dependent β-thalassemic patients, Pesaro 1-2 risk class, who have undergone allo-HSCT. OS and TFS were 94.5% and 88% after Threosulfan-Fludarabine-Thiotepa and 96.9% and 96.9% after Busulfan-Fludarabine-based conditioning; mixed chimerism below 75% occurred predominantly in Treosulfan-based regimens; OS and TFS did not differ significantly between various types of matched donors; mismatched UD-HSCTs displayed a lower OS and TFS rate [
92]. The main risk-reducing factors were represented by high CD3
+ cell count in the graft, Busulfan-conditioning, pre-conditioning therapy [
92].
3.4. Allo-HSCT in β-Thalassemic Patients with Reduced-Intensity Conditioning Regimens
Patient characteristics, such as age, Pesaro risk class, and availability of suitable donors are key determinant factors for a successful HSCT in β-thalassemia patients. Over the last years conditioning regimens for allo-HSCT of β-thalassemic patients have evolved to provide an adaptation to overcome patient-related limitations, to increase the opportunity of transplantation for more patients including unrelated donors and to reduce adverse outcomes related to toxicity events or to graft failure or GVHD [
93].
Several studies have explored the use of cytotoxic agents less toxic than Busulfan in the conditioning regimen. Treosulfan (dihydroxybusulfan), structurally similar to Busulfan, is a prodrug alternative to Busulfan conditioning prior to HSCT. Treosulfan is administered as a prodrug and is converted nonenzymatically and generates various active metabolites that are responsible of its cytotoxic effects.
The standard myeloablative conditioning used for allo-HSCT of β-thalassemic patients consisted of Busulfan (14-16 mg/Kg), Cyclophosphamide (160-200 mg/Kg) with or without ATG. In high-risk β-thalassemic patients is associated with a significant toxicity and a significant rate of graft rejection. The introduction of a toxicity-reduced conditioning regimen containing Treosulfan, Fludabarine and Thiotepa and a peripheral blood stem cell graft in older β-thalassemic patients with high-risk disease (Pesaro class III) resulted in improved transplant outcomes (5-year OS 71.9% with Treosulfan compared to 54.5% with Busulfan) and a reduction of early transplant-related mortality (46% with Busulfan and 13% with Treosulfan [
94,
95].
Dose exposure-response studies suggest a consistent interindividual variation in Treosulfan exposure resulting in variable HSCT outcomes, supporting the importance of drug monitoring of Treosulfan [
96]. Measuring pharmacokinetics of Treosulfan at the first dose and individualizing the third dose may represent an optimal strategy in nonmalignant diseases [
87]. Van der Stoep et al reported the study of 110 pediatric patients undergoing allo-HSCT after a Treosulfan-based conditioning regimen; the analysis of the exposure to Treosulfan of these patients showed a relationship with early and long-term clinical outcomes [
97]. A similar study in β-thalassemia patients who underwent allo-HSCT after a Treosulfan-based conditioning regimen showed that exposure to this drug predicts thalassemia-free survival [
98]. Importantly, lower Treosulfan exposure resulted in an increased risk of graft rejection and early transplant-related mortality [
98].
A large retrospective study (772 β-thalassemic patients, 410 treated with a Busulfan regimen and 362 with Treosulfan regimen) showed that the use of Busulfan or Treosulfan as the backbone of myeloablative conditioning for patients with transfusion-dependent thalassemia undergoing HSCT resulted in comparable high cure rates (2-year OS of 92.7% after Busulfan and Fludarabine conditioning and 94.7% after Treosulfan and Fludarabine conditioning) [
99]. Sykora et al have reported the results of a prospective randomized, phase 2 trial comparing the safety and the efficacy of Busulfan plus Fludarabine with Treosulfan plus Fludarabine preparative regimens in patients with nonmalignant disease undergoing allo-HSCT [
100]. Treosulfan-based regimen resulted to be less toxic than Busulfan, showing a lower transplantation-related mortality than Busulfan (3.9% vs 12%, respectively) [
100]. Overall survival favored Treosulfan (96%) compared to Busulfan (88%) [
100]. The rate of graft failure, particularly of secondary graft failure, was higher in transplantations conditioned with Treosulfan compared to Busulfan [
91]. Acute GVHD of at least grade I/II was observed in 8% in the Busulfan arm as compared to 13.7% in the Treosulfan arm. Moderate/severe chronic GVHD was more frequent among patients treated with Busulfan (14%) compared to Treosulfan (2%) [
100].
Kleinschmidt and coworkers have explored the safety and the efficacy of T-cell-depleted haplo-HSCT for pediatric and young adult β-thalassemic patients [
101]. Twenty patients with transfusion-dependent thalassemia (median age 10 years) received either a matched sibling donor or a haplo-HSCT after a conditioning regimen based of ATG, Treosulfan, Thiotepa and Fludarabine; immunosuppression consisted of calcineurin inhibitor and mycophenolate mofetil [
92]. At a median fellow-up of 37 months, OS and DFS were both 100%% for MDS and 92% in haplo-HSCT; two patients in the haplo-HSCT had graft failure; no acute GVHD >grade III or severe chronic GVHD were observed [
101].
Treosulfan is generally associated with a favorable long-term safety profile and fewer toxicities compared to Busulfan. Long-term follow-up studies in children with β-thalassemia post-HSCT show no significant reduction in growth potential, with up to 80% of patients achieving optimal growth rate [
102]. Treosulfan-based regimens are associated with low endothelial toxicity and less passage across the blood-brain barrier compared to Busulfan [
103]. Although most of acute toxicities and long-term adverse effects are less frequent in SCD and β-thalassemia patients who underwent allo-HSCT with a Treosulfan-based regimen compared to a Busulfan-based regimens, it was noted that higher Treosulfan exposure increases the risk of skin toxicity [
104].
4. Different Sources of HSCs for HSCT in SCD and β-Thalassemic Patients
Bone marrow (BM), peripheral blood (PB) and cord blood (CB) represent a suitable source of HSCs for HSCT in SCD and β-thalassemic patients. Bone marrow transplantation (BMT), peripheral blood stem cell transplantation (PBSCT) and cord blood stem cell transplantation (CBSCT) represent effective, potentially curative options for SCD and β-thalassemia, usually associated with comparable survival outcomes.
Few studies have directly compared in β-thalassemic patients the outcomes of PBSCT and BMT. Studies carried out using these two different sources of HSCs showed that both are safe for donors being associated with a low rate of adverse events, PBSCs induced a faster platelet and neutrophil recovery after HSCT probably related to a higher content of CD34
+ cells in mobilized PB than in BM and PBSCT is usually associated with a higher risk of developing GVHD than BMT. In a first study, Ghavamzadeh et al. compared class I-II Pesaro-risk
β-thalassemic children who underwent HLA-matched PBSCT or BMT after a myeloablative conditioning regimen based on Busulfan and Cyclophosphamide, followed by Cyclosporine +/- Methotrexate for GVHD prophylaxis [
105]. The comparative analysis of the outcomes observed in 87 PBSCT patients and 96 BMT patients showed that: the median time to neutrophil and platelet recovery in PBSCT was significant shorter than in BMT patients; grade II-IV GVHD and cGVHD were more frequent in PBSCT than in BMT group; the 2-year OS was similar after PBSCT and BMT (83% vs 89%, respectively) [
105]. A second more recent study by the same authors based on the long-term outcomes of 567 β-thalassemic patients who received allo-HSCT between 1998 and 2015 showed a similar OS after BMT and PBSCT; aGVHD and cGVHD were more frequent after PBSCT compared to after BMT, but the difference was not statistically significant [
106].
Locatelli et al. analyzed the outcomes of 485 patients with β-thalassemia major or SCD who received HLA-identical CBSCT or BMT [
98]. In comparison with patients receiving BMT, those receiving CBSCT had slower neutrophil recovery, less aGVHD and none, extensive GVHD [
107]. The 6-year OS after BMT and CBSCT was 95% and 97%; the 6-year DFS was 86% and 80% in β-thalassemia patients after BMT and CBSCT, respectively and 92% and 90% in SCD patients after BMT and CBSCT, respectively [
107].
The analysis of 44 patients with β-thalassemia or SCD who underwent related CBSCT with a GVHD prophylaxis based on Cyclosporin A alone or in association with methotrexate showed that 36 patients displayed engraftment and 8 patients a graft failure (6primary and 2 secondary graft failure); 3 of these 8 patients had sustained donor engraftment after BMT from the same donor [
108]. The occurrence of GVHD was low, with 4 patients experiencing grade 2 aGVHD and 2 limited cGVHD [
99].
Zwolsman and coworkers have retrospectively evaluated 44 SCD patients undergoing haploidentical HSCT: 26 patients received BMT and 15 received PBSCT [
109]. All the patients received a non-myeloablative conditioning regimen consisting of ATG, Thiotepa, Fludarabine, Cyclophosphamide, TBI (2Gy) and post-transplantation Cyclophosphamide. 1-year EFS was 100% in the PBSCT group and 85% in the BM group [
100]. Initially, all patients engrafted; one BM recipient displayed secondary growth failure with autologous reconstitution; three patients in the BMT group died [
109]. Future prospective studies are required to compare haplo-HSCT in SCD patients using BM or PBSC [
109].
Two recent studies explored the safety and the efficacy of PBSCs for haplo-HSCT in adult patients with severe SCD [
35,
110]. These studies reported the outcomes observed in 22 consecutive adults with severe SCD who underwent haplo-HSCT with PBSCs after a conditioning regimen based on ATG, Fludarabine, Cyclophosphamide, TBI (3Gy) and post-transplantation Cyclophosphamide [
35,
110]. The patients received an infusion of G-CSF-mobilized haploidentical-PBSCs [
35,
110]. After a follow-up of 4.8 years, 95% of patients were alive, 86% alive without GVHD, 90% remained free of vascular occlusive events, Hb levels increased from 8.2 g/dL pre-transplantation to 14.3 g/dL post-transplantation; a GVHD ≥2 grade was observed in 2 patients and moderate to severe cGVHD occurred in 3 patients [
35]. Organ damage/dysfunction remained stable or improved [
35,
110].
4. Complications Associated with Allo-HSCT in Patients with Hemoglobinopathies
Allo-HSCT in β-thalassemic and SCD patients is frequently associated with some complications that can impair the outcomes of transplantation in these patients.
4.1. Acute and Chronic GVHD
GVHD represents one of the most frequent complications after allo-HSCT, particularly in patients undergoing mismatched HSCT. In a recent systematic review and meta-analysis of allo-HSCT studies in SCD patients it was estimated a global incidence of 20% of acute-GVHD (aGVHD) and 14% of chronic GVHD (cGVHD) [
111]. In a recent analysis of outcomes and complications observed in β-thalassemia patients who received allo-HSCT in the United States reported a frequency of aGVHD of 42% and of cGVHD of 24% [
112]. The analysis of the EBMT registry including 2807 consecutive β-thalassemia patients who underwent allo-HSCT (mostly from MRDs) showed that the rate of aGVHD grade III/IV and cGVHD extensive occurred in 8.4% and 4.4% of children and 9.5% and 7.2% of adults, respectively [
113].
GVHD results from immune reactions triggered by dissimilar HLA antigen molecules, inducing the immune activation of donor T-lymphocytes. Unrelated HSCT or partially HLA-mismatched HSCT are associated with an increased risk of severe GVHD, graft failure and non-relapse mortality. Strategies to prevent and to mitigate GVHD are essential to ensure the results of allo-HSCT. GVHD prophylaxis was achieved using different agents such as calcineurin inhibitor (CNI) combined with methotrexate (MTX), anti-lymphocyte antibodies polyclonal (anti-thymocyte globulin, ATG) or monoclonal (Alemtuzumab, an antibody targeting CD52, a cell surface antigen found on most lymphocytes, NK cells and monocytes) or post-transplant Cyclophosphamide. Most of these treatments showed a consistent efficacy in preventing GVHD in alloHSCT in SCD and β-thalassemia patients.
Recently, it was introduced in therapy Abatacept (a recombinant fusion protein consisting of the extracellular domain of human CTLA-4 connected to the Fc of human IgG1) that inhibits antibody-dependent, cell-mediated cytotoxicity and/or complement fixation. Experimental, preclinical studies have supported the clinical use of Abatacept in GVHD prophylaxis in SCD and β-thalassemia patients undergoing allo-HSCT.
Thus, Ngwube and coworkers showed that the addition of Abatacept to a standard GVHD prophylaxis regimen (Tacrolimus and MTX) resulted in a low frequency of GVHD in SCD patients undergoing one-antigen-mismatched unrelated donor HSCT, after reduced-intensity conditioning [
113]. Thus, the incidence of a GVHD grades II to IV and grades III to IV at day +100 was 28% and 7%, respectively [
104]. One-year incidence of cGVHD was 57% and mild/limited in all cases but 1. The 2-year OS was 100% and DFS was 92.9% [
114]. In a more recent study, the same authors evaluated the impact of Abatacept incorporated into prednisone-including GVHD prophylaxis compared to standard prophylaxis without Abatacept, in pediatric patients with hemoglobinopathies undergoing allo-HSCT from related or unrelated donors after reduced-intensity conditioning [
115]. The incidence of posterior reversible encephalopathy syndrome was 17% with standard GVHD prophylaxis and 0% with Abatacept [
115]. Acue grade 3 to 4 GVHD occurred in 28% of patients who received standard GVHD prophylaxis and 0% of patients treated with Abatacept. Among patients who received standard GVHD prophylaxis, cGVHD was observed in 2.5% (mild), 5% (moderate) and 30% (severe) of cases, compared to 25 (mild), 5% (moderate) and 5% (severe) in those treated with Abatacept [
115]. OS and EFS were 87% and 80% in patients who have received standard GV HD prophylaxis and 95% and 90%, respectively in patients treated with Abatacept [
115].
Khandelwal et al. showed that GVHD prophylaxis with Abatacept reduced severe GVHD in allo-HSCT for β-thalassemia after a myeloablative conditioned regimen with Busulfan, Fludarabine and Thiotepa. Particularly, the addition of Abatacept to a standard GVHD prophylaxis regimen with CNI and methylprednisolone reduced to 0% the occurrence of grades III-IV GVHD at day +100 compared to a frequency of 50% observed in patients who underwent standard GVHD prophylaxis without Abatacept [
116]. Thalassemia-free survival after HSCT was 100% in the Abatacept cohort compared to 62.5% in the standard cohort [
116].
Jaiswal et al. reported the development of a GVHD prophylaxis protocol in patients with non-malignant diseases undergoing haplo-HSCT from haploidentical familial donors, based on Abatacept administration on hay 0, +5, +20, +35 and every 28 days thereafter until day +180, post-transplantation Cyclophosphamide administered on day +3 and +4 and Sirolimus given from day -7 to 9 months post-HSCT [
117]. This GVHD prophylaxis protocol was explored in 40 patients with non-malignant diseases (24 with hemoglobinopathies) showing the following results: post-transplantation hemophagocytic syndrome was detected in 3 patients, leading to graft failure in 2 cases; the incidence of aGVHD was 2.6% and that of cGVHD was 14.3%; rates of non-relapse mortality, overall survival, event-free survival, and GVHD-free, event-free survival were 5%, 95%, 90% and 82%, respectively [
118]. Absence of cGVHD correlated with younger patient age [
118].
Dexter and coworkers compared the outcomes observed in 35 consecutive patients with hemoglobinopathies who underwent allo-HSCT and received Abatacept GVHD prophylaxis to those observed in a historical group of 66 patients who received allo-HSCT after an identical conditioning regimen and with a GVHD prophylaxis without Abatacept [
119]. All patients in the Abatacept group engrafted and are alive whereas there were two cases of graft failure (3%) and 3 deaths (4.5%) in the control group [
119].
4.2. Hematologic Malignancies Post-Transplantation in SCD and β-Thalassemic Patients
Population studies have reported an increased risk of hematologic malignancies in patients with SCD compared with the general population [
120,
121]. Particularly, SCD patients aged 15 to 39 years were more likely to develop leukemia compared to to those under 15 years [
121]. Furthermore, patients with more severe disease were more likely to develop leukemia compared to those with less severe disease [
121].
Various studies have explored the occurrence of secondary neoplasms after HSCT for SCD. Eapen et al explored a cohort of 1,096 transplants for SCD; the 10-year incidence of any secondary neoplasms was 2.4% and 1.7% of leukemia/MDS [
122]. The incidence of secondary neoplasms was higher with low-intensity regimens (non-myeloablative) including total-body irradiation compared to myeloablative regimens [
122]. Lawal explored 120 patients with SCD who underwent allo-HSCT and reported that 8 of these patients developed hematologic malignancies between 4 months and 9 years post-HSCT; importantly, these 8 patients displayed either a graft failure or a condition of mixed chimerism [
123]. Two fo these 8 patients had the same
TP53 mutations at baseline [
124]. These authors using a ultradeep sequencing method showed pre-existing
TP53 mutations with a VAF ranging from 0.06% to 0.34% in 4 SCD patients developing a myeloid neoplasia after allo-SCT [
125]. In line with these findings, Ali and coworkers showed that recipient cells and not donor cells are the source of hematologic malignancies after graft failure and mixed chimerism in SCD patients [
126]. Gondek et al have proposed a theory to explain why in SCD patients a curative therapy based on allo-HSCT may favor the development of leukemia. Individuals with SCD may develop clonal hematopoiesis that usually does not expand and does not progress to leukemia over time. However, after graft failure the pressure of changing from homeostatic to regenerative hematopoiesis may drive the clonal expansion and leukemic transformation of pre-existing premalignant clones, inducing in some patients the development of leukemia [
127].
Leukemia development has been associated with the acquisition of somatic mutations, which accumulate with aging in a condition defined as clonal hematopoiesis (CH). Somatic mutations in genes involved in stem cell self-renewal and differentiation are present in a minority of stem/progenitor cells and confer a survival or proliferation advantage giving rise to the development of a small clone, resulting in its significant expansion compared with non-mutant cells. Whole exome sequencing studies on 1459 patients with SCD and 6848 African-American controls have shown that SCD is associated with an increased prevalence of CH, with more frequent mutations on
DNMT3A and
TP53 genes [
127].
TP53 which is infrequently affected in the healthy population, was the second most frequently mutated gene after
DNMT3A and accounted for 13% of all CH mutations in SCD [
128]. In this study it was postulated that ineffective erythropoiesis and increased erythrocyte turnover may favor the development of a clone carrying a selective growth advantage; an additional mechanism could be related to chronic inflammation [
128].
These observations suggest that SCD itself may predispose patients to high-risk CH. Weeks et al. have screened a large cohort of SCD patients, normal individuals and of patients with other hemoglobinopathies and observed that: CH occurred earlier in SCD patients ands was more prevalent in SCD cases compared to non-SCD controls among individuals aged 0-19 years (10.6% vs 3.5%, respectively) [
129]. This difference was selectively driven by mutations of DNA damage response (DDR) driver genes (
TP53, PPM1D, CHEK2, ATM) [
129]. Prevalence of
DNMT3A and
TET2 mutations was higher in SCD among the youngest population than in normal controls age-matched (6.4% vs 2.5%, respectively) but progressively decreased with advancing age [
129]. This precocious DDR-CH is specific to SCD and is not observed in individuals with sickle cell trait [
129]. Given these observations, De Luna and coworkers have screened the occurrence of CH is 56 SCD patients candidates to transplant approaches and observed that 11 of these patients display CH mutations, the most frequent being
DNMT3A (29%),
TP53 (29%),
PPMD1 (29%) and
ASXL1 (21%) [
130]. The presence of high-risk mutations, such as
TP53, in these patients supports the inclusion of molecular profiling in the context of pre-transplant evaluation [
130].
Several studies have explored the occurrence of solid cancers and hematologic malignancies in β-thalassemic patients who received allo-HSCT. The analysis of risk of cancer in β-thalassemic patients showed a significantly increased risk of developing cancer, particularly hepatocellular carcinoma, compared to both β-thalassemic trail subjects and control general population [
131]
Santarone et al. have reported the long-term occurrence of secondary cancers in β-thalassemic patients who received allo-HSCT [
132]. With a follow-up of 39 years, the cumulative incidence of developing solid cancers in 122 transplanted β-thalassemic patients was 13.2%, compared to 3.2% and 1.3% in a population of age-matched HSC donors or in a population of β-thalassemic patients who received conventional nontransplant therapy, respectively [
132]. In another study, the same authors reported a 39-year cumulative incidence of secondary solid cancer at 16.4%, with only one case of non-Hodgkin lymphoma reported as a hematologic malignancy among secondary cancers [
61].
Solomou et al. reported a high incidence (15.8%) of clonal hematopoiesis in a cohort of 95 β-thalassemic patients [
124]. It was suggested that life-long stress in the hematopoiesis could represent a pathogenetic mechanism favoring CH development in
β-thalassemic patients [
133].
5. Cost-Effectiveness of HSCT for Hemoglobinopathies
HSCT for hemoglobinopathies is considered as a cost-effective and curative treatment, with an acceptable safety profile compared to lifelong standard care (including transfusions, iron chelation, hospitalizations, pharmaceuticals, medical support for acute events and medical treatments for organ damage), despite its high upfront cost.
HSCT offers a one-time cure, particularly for younger SCD and β-thalassemic patients who have a matched related donor. HSCT improves quality of life of both SCD and β-thalassemic patients, preventing pain crises, organ damage dependent on pathologic events related to SCD and β-thalassemia, premature death. Furthermore, HSCT improves the social participation of SCD and β-thalassemic patients to an active social life.
The evaluation of the cost of allo-HSCT for SCD and β-thalassemic patients is challenging because charges depend on many variables, including type of transplant (matched related donor HSCTs are generally less costly and more effective than Matched Unrelated Donor or Haploidentical Donor or PBSC or CB transplants), state regulations, regional health costs and insurance contracts [
134]. The cost of a HSCT for SCD and β-thalassemia varies dramatically by location ranging from
$10,000 to 50,000 in some developing countries to
$80,000 to 400,000 in uSA/Western Europe. A detailed analysis performed in the USA showed that the whole cost of allo-HSCT in SCD patients ranges from
$340,000 to
$799,000 with a median total cost of
$467,000 [
135]. Several variables affect the cost of allo-HSCT in these patients: healthcare utilization was lower among recipients of matched sibling donor grafts and in patients early transplanted (young children and this was due to a reduced post-transplant healthcare utilization [
135]. A recent evaluation showed in the USA a total median cost of
$130,000 for allo-HSCT in SCD patients encompassing a lapse of time from 6 months pre-transplant to two years post-transplant [
136]. A comparative cost analysis carried out in USA between HSCT vs supportive care showed that the former one is approximately equivalent to the cost of ten SCD hospital admissions [
136].
Recent studies have shown that gene therapy represents an alternative approach to allo-HSCT to obtain a curative effect in SCD and β-thalassemic patients. The current procedure used in gene therapy studies implies the
ex vivo gene modification of autologous stem cells and the reinfusion of genetically modified cells back to the patient. Particularly, the treatment process implies: initial evaluation involving patient selection and preparation; stem cell collection, involving the collection of HSCs from patient’s blood after HSC mobilization with G-CSF and Plerixafor; genetic modification implying genetic manipulation of autologous HSCs using either gene addition (lentiviral vectors) oe gene editing (CRISPR) to correct the genetic defect; conditioning involving the hospitalization of the patient and preparation to HSC transplantation though administration of a conditioning regimen to clear abnormal cells from the bone marrow; infusion back into the patient of genetically modified HSCs that engraft and begin to produce functional erythroid cells, leading to a long-term cure; clinical support, monitoring and follow-up [
138,
139]. Three gene therapy products have been approved for the treatment of β-hemoglobinopathies: Lyfgenia (lovotibeglogene autotemcel) and Zynteglo (betibeglogene autotemcel) that use lentiviral vectors to modify the patient’s stem cells and Casgevy (exagamglogene autotemcel) that utilizes CRISPR gene editing of the patient’s stem cells [
138,
139].
The availability of two different curative approaches for both SCD and β-thalassemic patients raises the need for a comparison between allo-HSCT and gene therapy treatments at the level of costs, efficacy, safety and patients’ eligibility. Several considerations related to the cost (the cost of allo-HSCT is five-, six-fold lower than the cost of approved gene therapies for β-hemoglobinopathies) and to the long-term effects (that are known for allo-HSCT for both SCD and β-thalassemic patients, but are only in part established for gene therapy treatments) favor allo-HSCT over gene therapy treatments [
137]. A group of experts in hemoglobinopathies and/or in HSCT and gene therapy of the EHA and EBMT has recently proposed criteria for an optimal selection of patients with hemoglobinopathies for gene therapy studies [
140]. These experts have proposed to restrict the access to gene therapy to SCD patients in good clinical conditions with no-HLA matched donor available, without irreversible severe organ impairment and able to tolerate a myeloablative conditioning regimen [
140]. Medical centers providing gene therapy must evaluate each SCD or β-thalassemic patient through a process ensuring ethical, systematic allocation of each patient either to HSCT or to gene therapy.
The existence of two curative therapies for β-hemoglobinopathies now may offer the opportunity to some patients who have experienced a failure to cure SCD or β-thalassemia with HSCT of a second curative treatment based on gene therapy [
141]. However, this opportunity must be considered with caution since exposure to inflammatory stress and conditioning chemotherapy may compromise the fitness of HSCs, reduce the hematopoietic reserve, accelerate HSC aging, and promote the accumulation of deleterious genetic alterations, all of which may contribute to negatively affect the efficacy and the safety of gene therapy [
141].
6. Conclusions
HSCT is the only proven cure for SCD and β-thalassemia and is based on the replacement of pathological HSCs with health donor cells, offering excellent outcomes in young patients with matched sibling donors but carrying a significant risk of transplant-related infections and mortality, thus requiring a careful risk-benefit assessment for eligible patients. The studies carried out in the last three decades have clearly shown that three parameters are key determinants for success of allo-HSCT in patients with β-hemoglobinopathies: donor match (with best results offered by matched sibling donors); patient’s age (with younger children <8 years-old offering the best results) and health (patients without organ damage have significantly better outcomes) and early intervention (HSCT before idevelopment of severe complications improves survival).
Since only 15-20% of patients have a sibling matched donor, to expand the opportunities of allo-HSCT for patients with β-hemoglobinopathies, transplants have been developed using matched unrelated donors or other family members (haploidentical) as donors. These HSCTs carry higher risks related to the lower incomplete immunological compatibility; however, strategies have been developed in the last two decades to reduce the risk of graft failure and to prevent aGVHD and cGVHD, with a consequent improvement of the outcomes of allo-HSCT with unrelated matched donors or with haploidentical donors. Furthermore, the development of efficient strategies of reduced-intensity conditioning and of immunosuppression have improved the outcomes of allo-HSCT in adult patients with β-hemoglobinopathies.
Gene therapy based on gene addition or gene editing strategies has led to the development of additional curative therapies for SCD and β-thalassemia, thus raising the problem of the selection of SCD and β-thalassemia patients either for allo-HSCT of for gene therapy. The adoption of gene therapy treatments of β-hemoglobinopathies should be limited to patients who do not have HLA-matched donors and is strongly limited by its high cost, five to six-fold higher than HSCT and its applicability to only few highly specialized medical centers.
Finally, it is important to emphasize that both allo-HSCT and ex vivo gene therapies require a specialized infrastructure and a considerable expertise for an HSCT, which are available only in developed countries and not in the low- and middle-income countries where SCD and β-thalassemia are mostly prevalent.
Author Contributions
G.C. and E.P. were involved in researching, writing, and editing the manu- script. U.T. was involved in conceptualization, organization, research, and editing the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this artivle.
Conflicts of Interest
The authors declare no conflicts of interest:
References
- Kattamis, A.; Forni, G.L.; Aydinok, Y.; Viprakasit, V. Changing patterns in the epidemiology of β-thalassemia. Eur J Hematol 2020, 105, 692–703. [Google Scholar] [CrossRef]
- Tyo, Y.; Li, Y.; Ma, J.; Yang, X.; Wu, S.; Jin, J.; He, Z. Global, regional, and national burden of thalassemia, 1990-2021: a systematic analysis of the global burden of disease study 2021. eClinical Medicine 2024, 72, 102619. [Google Scholar] [CrossRef]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. β-thalassemias. N Engl J Med 2021, 384, 727–743. [Google Scholar] [CrossRef]
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle cell disease. N Engl J Med 2017, 376, 1561–1573. [Google Scholar] [CrossRef]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishinamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat Rev Dis Primers 2018, 4, 18010. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.M.; McHugh, T.A.; Oron, A.P.; Toply, C.; Lonberg, N.; Tella, V.; Wilmer, L.; Fullet, K.; Hagins, H.; Aboagye, R.; et al. Global, regional, and national prevalence and mortality burden of sickle cell disease, 200-2021: a systematic analysis from the global burden of disease study 2021. Lancet Hematol 2023, 10, e585–599. [Google Scholar] [CrossRef]
- Gluckman, E. Allogeneic transplantation strategies including haploidentical transplantation in sickle cell disease. Hematology Am Soc Hematol Educ Program 2013, 2013.370376. [Google Scholar] [CrossRef] [PubMed]
- Ball, J.; Bradley, A.; Le, A.; Tisdale, J.F.; Uchida, N. Current and future treatments for sickle cell disease: from hematopoietic stem cell transplantation to in vivo gene therapy. Mol Therapy 2025, 33, 2172–2189. [Google Scholar] [CrossRef]
- De Luna, G.; Harfouch, E.; Essaydi, A.; Redjoul, R.; Beckerich, F.; Properzi, E.; Hebert, N.; Habibi, A.; Volle, G.; Leoughre, M.P.; et al. Identification of HLA-matched related donors for hematopoietic stem cell transplant in adult sickle cell patients: experience from an adult referral center in France. Blood 2024, 144, 5361–5362. [Google Scholar] [CrossRef]
- Bernaudin, F.; Socie, G.; Kuentz, M.; Chevret, S.; Duval, M.; Bertrand, Y.; Vannier, J.P.; Yakouben, K.; Thuret, I.; Bordigoni, P.; et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood 2007, 110, 2749–2756. [Google Scholar] [CrossRef]
- Cappelli, B.; Volt, F.; Tozatto-Maio, K.; Scigliuolo, G.M.; Ferster, A.; Dupont, S.; Simões, B.P.; Al-Seraihy, A.; Aljurf, M.D.; Almohareb, F.; et al. Risk factors and outcomes according to age at transplantation with an HLA-identical sibling for sickle cell disease. Haematologica 2019, 104, e543–e546. [Google Scholar] [CrossRef]
- Gluckman, E.; Cappelli, B.; Bernaudin, F.; Labopin, M.; Volt, F.; Carreras, J.; Pinto Simoes, B.; Ferster, A.; Dupont, S.; dela Fuente, J.; et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017, 129, 1548–1556. [Google Scholar] [CrossRef]
- Bernaudin, F.; Dalle, J.H.; Borles, D.; Peffault de Latour, R.; Robin, M.; Bertrand, Y.; Pandarre, C.; Vannier, J.P.; Neven, B.; Kuentz, M.; et al. Long-term event-free survival, chimerism and fertility outcomes in 234 patients with sickle-cell anemia younger than 30 years after myeloablative conditioning and matched-sibling transplantation in France. Haematologica 2020, 105, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.C.; Eapen, M.; Liu, Y.; El Rassi, F.; Waller, E.K.; Levine, J.E.; Strousse, J.J.; Antin, J.H.; Parikh, S.H.; Bakahi, N.; et al. Hematopoietic cell transplant compared with standard care in adolescent and young adults with sickle cell disease. Blood Adv 2025, 9, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Fitzhugh, C.D.; Cordes, S.; Taylor, T.; Coles, W.; Roskom, C.; Link, M.; Hsieh, M.M.; Tisdale, J.F. At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. Blood 2017, 130, 1946–1948. [Google Scholar] [CrossRef]
- Abraham, A.; Hsieh, M.; Eapen, M.; Fitzhugh, C.; Carreras, J.; Keesler, D.; Guilcher, G.; Kamani, N.; Walters, M.C.; Boelens, J.J.; et al. Relationship between mixed donor-recipient chimerism and disease recurrence after hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant 2017, 23, 2178–2183. [Google Scholar] [CrossRef]
- Hsieh, M.M.; Fitzhugh, C.D.; Weitzel, R.P.; Link, M.E.; Coles, W.A.; Zhao, X.; Rodgers, J.P.; Powell, J.D.; Tisdale, J.F.; et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA 2014, 312, 48–56. [Google Scholar] [CrossRef]
- Iannone, R.; Casella, J.F.; Fuchs, E.J.; Chen, A.R.; Jones, R.J.; Wolfrey, A.; Amylon, M.; Sullivan, K.M.; Storb, R.F.; Walters, M.C. Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and beta-thalassemia. Biol Blood Marrow Transplant 2003, 9, 519–528. [Google Scholar] [CrossRef]
- Hsieh, M.M.; Kang, E.M.; Fitzhugh, C.D.; Link, B.; Bolan, C.D.; Kurlander, R.; Childs, R.W.; Rodgers, G.; Powell, J.D.; Tisdale, J.F. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med 2009, 361, 2309–2317. [Google Scholar] [CrossRef]
- Inam, Z.; Jeffries, N.; Link, M.; Coles, W.; Pollack, P.; Luckett, C.; Phang, O.; Harvey, E.; Martin, T.; Farrey, T.; et al. Two nonmyeloablative HLA-matched related donor allogeneic hematopoietic cell transplantation regimens in patients with severe sickle cell disease. Transplant Cell Ther 2025, 31, 305–318. [Google Scholar] [CrossRef]
- Ball, J.; Bradley, A.; Le, A.; Tisdale, J.F.; Uchida, N. Current and future treatments for sickle cell disease: from hematopoietic stem cell transplantation to in vivo gene therapy. Mol Ther 2025, 33, 2172–2191. [Google Scholar] [CrossRef]
- Alzaharani, M.; Damlaj, M.; Jeffries, N.; Alahmri, B.; Singh, A.; Rondelli, D.; Tisdale, J.F.; Saraf, S.L.; Hsieh, M.H. Non-myeloablative human leukocyte antigen-matched donor transplantation in sickle cell disease: outcomes from three independent centers. Br J Haematol 2021, 192, 761–768. [Google Scholar] [CrossRef]
- Zhu, S.; Maahs, L.; Sweiss, K.; Campbell-Lee, S.; Mahmud, N.; Gordeuk, V.; Rondelli, D.; Saraf, F. Granulocyte colony-stimulating factor may improve engraftment in adults with sickle cell disease undergoing non-myeloablative hematopoietic stem cell transplantation. Brit J Haematol 2025, 207, 1690–1693. [Google Scholar] [CrossRef] [PubMed]
- Damlaj, M.; Alahmri, B.; Alaskar, A.; Alhejazi, A.; Alsadi, H.; Ahmed, M.; Alanazi, T.; Ahmed, R.; Alharbi, A.; Shehabeddine, I.; et al. Favorable outcome of non-myeloablative allogeneic transplantation in adult patients with severe sickle cell disease: a single center experience of 200 patients. Am J Hematol 2024, 99, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Saib, I.; Alahmnari, B.; Alsadi, H.; Aljubour, Z.; Ahmed, M.; Alzahrani, M.; et al. Effect of ABO mismatch and red blood cell alloimmunization on the outcome of hematopoietic cell transplantation for sickle cell disease. Transplant Cell Ther 2025, 31, P30-E3-30. [Google Scholar] [CrossRef]
- Nasiri, A.; Samarkandi, S.; Alasbali, R.; Alahmadi, A.; Nassar, M.; Aflattah, H.; Bukhari, Y.; Fallatah, A.; Samrkandi, H.; Alzahrani, T.; et al. Steroid pre-busulfan based reduced-intensity conditioning in matched related hematopoietic stem cell transplantation: in young and adult sickle disease patients. Blood 2024, 144, 1142–1143. [Google Scholar] [CrossRef]
- Kassim, A.A.; DeBaun, M.R. The range of haploidentical transplant protocol in sickle cell disease: all haplos are not created equally. Hematology Am Soc Hematol Educ Program 2023, 2023, 532–541. [Google Scholar] [CrossRef]
- Shenoy, S.; Eapen, M.; Panepinto, J.A.; Logan, B.R.; Wu, J.; Abraham, A.; Brochstein, J.; Chaudhury, S.; Godder, K.; Haight, A.E.; et al. A trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood 2016, 128, 2561–2567. [Google Scholar] [CrossRef]
- Eapen, M.; Kou, J.; Andreanskly, M.; Bhatia, M.; Brochstein, J.; Chaudhury, S.; Haight, A.E.; Haines, H.; Jacobsohn, D.; Jaroscak, J.; et al. Long-term outcomes after unrelated donor transplantation for severe sickle cell disease on the BMT CTN 0601 trial. Am J Hematol 2024, 99, 785–788. [Google Scholar] [CrossRef]
- Fitzhugh, C.D.; Hsieh, M.; Taylor, T.; Coles, W.; Roskom, K.; Wilson, D.; Wright, E.; Jeffries, N.; Gamper, C.; Powell, J.; et al. Cyclophosphamide improved engraftment in patients with SCD and severe organ damage who undergo haploidentical PBSCT. Blood Adv 2017, 1, 652–660. [Google Scholar] [CrossRef]
- De la Fuente, J.; Dhedin, N.; Koyama, T.; Bernaudin, F.; Kuentz, M.; Karnik, L.; Socié, G.; Culos, K.A.; Brodsky, R.A.; DeBaun, M.R.; et al. Haploidentical cone marrow transplantation with post-transplantation cyclophosphamide plus thiotepa improves donor engraftment in patients with sickle cell disease anemia: results of an international learning collaborative. Biol Blood Marrow Transplant 2019, 25, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Kassim, A.A.; de la Fuente, J.; Nur, E.; Wilkerson, K.L.; Alahmari, A.D.; Seber, A.; Bonfim, C.; Pinto Simoes, B.; Alzahrani, M.; Eckrich, M.J.; et al. An international learning collaborative phase 2 trial for haploidentical bone marrow transplant in sickle cell disease. Blood 2024, 143, 2654–2663. [Google Scholar] [CrossRef]
- Kassim, A.A.; Walters, M.C.; Eapen, M.; Smith, M.; Logan, B.R.; Solh, M.; McKinney, C.; Ross, M.; Kent, M.; Abusin, G.A.; et al. Haploidentical bone marrow transplantation for sickle cell disease. N Engl J Med Evid 2025, 4. [Google Scholar] [CrossRef]
- Walters, M.; Kassim, A.; Eapen, M.; Milner, J.; Nickel, R.; Gillio, A.; Yassine, K.; Skiles, J.; McKinney, C.; Rangarajan, H.; et al. Reduced intensity haploidentical bone marrow transplantation in children with severe sickle cell disease (SCD): BMT CTN 1507. Blood 2025, 146, abs 25–11982. [Google Scholar] [CrossRef]
- Rodriguez, A.M.A.; Zucchetti, E.; Sweiss, K.; Sanchez, M.; Galvez, C.; Mahmud, N.; Koshy, M.; Saraf, S.; Rondelli, D. Haploidentical hematopoietic stem cell transplantation in adults with severe sickle cell disease: long-term outcomes using a modified conditioning regimen with peripheral blood stem cells. Blood 2025, 146, abs25–8487. [Google Scholar] [CrossRef]
- Foell, J.; Schulte, J.H.; Pfirstinger, B.; Troger, A.; Wolff, D.; Edinger, M.; Hofmann, P.; Aslandis, C.; Lang, P.; Holelr, E.; et al. Haploidentical CD3 or α/β T-dell depleted HSCT in advanced stage sickle cell disease. Bone Marrow Transplant 2019, 54, 1859–1867. [Google Scholar] [CrossRef]
- Carbacioglu, S.; Troger, A.; Kleinschmidt, K.; Hanafee-Alali, T.; Brosig, A.M.; Jakob, M.; Kramer, S.; Offner, R.; Wolff, D.; Foell, J. Haploidentical AB T cell depleted HSCT represents an alternative treatment option in pediatric and adult patients with sickle cell disease (SCD). Blood 2023, 142, 4915–4916. [Google Scholar] [CrossRef]
- Gibson, N.M.; Elgarten, C.W.; Oved, J.H.; Wray, L.; Freedman, J.; Khandros, J.; Worster, E.; Nicholas, P.; Kadauke, S.; Wang, Y.; et al. Outcomes of unrelated donor stem cell transplantation with partial T cell depletion for pediatric patients with hemoglobinopathies. Transplant Cell Therapy 2025, 31, 828.e1–828.e14. [Google Scholar] [CrossRef] [PubMed]
- Gilman, A.L.; Eckrich, M.J.; Epstein, S.; Bamhart, C.; Cannon, M.; Fukes, T.; Hyland, M.; Shah, K.; Crfochowski, D.; Champion, E.; et al. Alternative donor hematopoietic stem cell transplantation. Blood Adv 2017, 1, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Cairo, M.S.; Talamo, J.A.; Moore, T.B.; Shi, Q.; Weinberg, R.S.; Grossman, B.; Shenoy, S. Familial haploidentical stem cell transplant in children and adolescents with high-risk sickle cell disease: a phase 2 clinical trial. JAMA Pediatrics 2019, 174, 195–197. [Google Scholar] [CrossRef]
- John, T.D.; Walters, M.C.; Rangarajan, H.; Rahim, M.; McKinney, C.; Bollard, C.; Ausin, G.; Eapen, M.; Kassim, A.A.; DeBaun, M. Incremental eligibility criteria for the BMT CTN 1507 haploidentical trial for children with sickle cell disease. Blood Adv 2024, 8, 6055–6062. [Google Scholar] [CrossRef] [PubMed]
- Shestra, J.; Motta, C.; Keller, M.; Abraham, A.; Lazarski, C.; Nicke, R.; Bollard, C. Predicting donor engraftment success in adults post identical bone marrow transplant for sickle cell disease: BMT CTN 1507. Blood 2025, 146, abs25–12371. [Google Scholar]
- Eapen, M.; Brazauskas, R.; Walters, M.C.; Bernaudin, F.; Bo-Subait, K.; Fithugh, C.D.; Hankins, J.B.; Kanter, J.; Meerpohl, J.J.; Bolanos; Meade, J.; et al. Impact of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: a reprospective, cohort study. Lancet Hematol 2019, 6, e585–e596. [Google Scholar] [CrossRef] [PubMed]
- Prior, D.; Liang, J.; Deng, Y.; Shah, N.; Flagg, A.; Krishnamurti, L. Comparison of regimens used for allogenic matched related donor hematopoietic cell transplantation for sickle cell disease. Transplant Cell Ther 2025, 31, 594.e1–594.e13. [Google Scholar] [CrossRef]
- Krishinamurti, L.; Neuberg, D.; Sullivan, K; Kamani, N.; Abraham, A.; Campigotto, F.; Zhang, W.; Dahdoul, T.; De Caisro, L.; Parikh, S.; et al. Bone marrow transplantation for adolescents and young adults with sickle cell disease: results of a prospective multicenter pilot study. Am J Hematol 2019, 94, 446–454. [Google Scholar] [CrossRef]
- King, A.; Kamani, N.; Bunin, N.; Shadev, I.; Brochstein, J.; Hayashi, R.; Grimley, M.; Abraham, A.; Dioguardi, J.; Chan, K.W.; et al. Successful matched sibling donor marrow transplantation following reduced toxicity conditioning in children with hemoglobinopathies. Am J Hematol 2015, 90, 1093–1098. [Google Scholar] [CrossRef]
- Walters, N.C.; Patience, M.; Leisenring, W.; Echman, J.R.; Scott, J.P.; Mentzer; Davies, S.C.; Ohene-Frempong, K.; Bernaudin, F.; Matthews, D.C.; et al. Bone marrow transplantation for sickle cell disease. N Engl J Med 1996, 335, 369–376. [Google Scholar] [CrossRef]
- Krishinamurti, L.; Liang, J.; He, Z.; Deng, Y.; Nallagatla, Y.; Hamidi, R.; Flagg, A.; Shah, N. Incidence and risk factors of pain crisis after hematopoietic cell transplantation for sickle cell disease. Blood Adv 2024, 8, 1908–1919. [Google Scholar] [CrossRef]
- Stenger, E.; Brazauskas, R.; Peterson, A.; Bo-Subait, K.; Battiwala, M.; Buchbinder, D.; Hamilton, B.K.; Hamilton, B.K.; Savani, B.M.; Schoemans, H.; et al. Long-term survival and late effects following hematopoietic cell transplantation for sickle cell disease into a modern era: a center for international blood and marrow transplant research (CIBMTR) analysis. Blood 2025, 146, abs25–13922. [Google Scholar] [CrossRef]
- Folarin, M.; al-Zubaidi, H.; Moore, E.; Eze, U.; Palanisamy, N. Efficacy and safety of allogeneic hematopoietic stem cell transplantation in curing sickle cell disease: a systematic review and meta-analysis of single-arm studies. Transpl Cell Therapy 2025, in press. [Google Scholar] [CrossRef] [PubMed]
- Rostami, T.; Rad, S.; Rostami, M.R.; Mirhosseini, S.A.; Alemi, H.; Khavandgar, N.; Janbabai, G.; Kiumarsi, A.; Kasaeian, A.; Mousavi, S.A. Hematopoietic stem cell transplantation in sickle cell disease: a multidimensional review. Cell Transplant 2024, 33, 1–13. [Google Scholar] [CrossRef]
- Leonard, A.; Furstenau, D.; Abraham, A.; Danbarfi, D.S.; Nickel, R.S.; Limerick, E.; Fitzhugh, C.; Hsieh, M.; Tisdale, J.F. Reduction in vaso-occlusive events following stem cell transplantation in patients with sickle cell disease. Blood Adv 2023, 7, 227–235. [Google Scholar] [CrossRef]
- Stenger, E.; Xiang, Y.; Wetzel, M.; Gillespie, S.; Chellapandian, D.; Shah, R.; Arnold, S.D.; Bhatia, M.; Chaudhury, S.; Eckrich, M.J.; et al. Long-term organ function after HCT for SCD: a report from the sickle cell transplant advocacy and research alliance. Transplant Cell Therapy 2023, 47, 47.e1–47.e10. [Google Scholar] [CrossRef]
- Mishkin, A.D.; Cheung, S.; Hoffmann, A.; Leimbach, E.; Dosopvitz, S.; Mapara, M. High incidence of suicidal ideation in a series of patients with sickle cell disease after hematopoietic stem cell transplantation. Blood Adv 2022, 6, 5542–5548. [Google Scholar] [CrossRef] [PubMed]
- Nickel, R.S.; Maher, J.; Hsieh, M.; Davis, M.; Hsieh, M.; Pecker, L. Fertility and curative therapy for sickle cell disease: a comprehensive review to guide care. J Clin Med 2022, 11, 2318. [Google Scholar] [CrossRef]
- Meacham, L.; George, S.; Veludhandi, A.; Pruett, M.; Haight, A.; Arnold, S.; Elcuri, S.; Stenger, E.; Krishnamurti, L. Female reproductive health outcomes after hematopoietic cell transplantation for sickle cell disease: is reduced intensity better than myeloablative conditioning? Tranpl Cell Therapy 2023, 29, 531.e1–531.e4. [Google Scholar] [CrossRef]
- Hastings, B.; Ptil, C.; Gallo, A.M. The experience of health-related quality of life after haploidentical stem cell transplantation for adultrs with sickle cell disease. West J Nurs Res 2019, 41, 1829–1844. [Google Scholar] [CrossRef]
- Gallo, A.M.; Patil, C.; Adenyi, T.; Hsu, L.L.; Rondelli, D.; Saraf, S. Heath-related quality of life and personal life goals of adults with sickle cell disease after hematopoietic stem cell transplantation. West J Nurs Res 2019, 41, 555–575. [Google Scholar] [CrossRef]
- Dovern, E.; Nijiland, S.; van Muilekom, M.M. Physical, mental, and social health of adult patients with sickle cell disease after allogeneic hematopoietic stem cell transplantation: a mixed-methods study. Transplant Cell Ther 2023, 29, 283.e1–283.e9. [Google Scholar] [CrossRef] [PubMed]
- Nirmal, G.; Singh, G.; Chada, V.; Gupta, N.; Srivastava, N.; Nair; Yadav, H.; Kharya, G. Patient-parent reported quality of life assessment post hematopoietic stem cell transplant for sickle cell disease: validation and clinical application of sickle cell disease burden index. Blood 2024, 144, 4927. [Google Scholar] [CrossRef]
- Dovern, E.; Nijiland, S.; Braasme, A.; van Muilekom, M.; Suijk, E.; Hoogendoom, G.; van Tuijn, C.; DeBaun, M.; Biemond, B.; Haverman, L.; et al. Changes in the quality of life of adults with sickle cell disease following allogeneic stem cell transplantation: a mixed-mehtods, prospective cohort study. Hemasphere 2025, 9, e70100. [Google Scholar] [CrossRef]
- Angelucci, E.; Baronciani, D. Allogeneic stem cell transplantation for thalassemia major. Haematologica 2008, 93, 1780–1784. [Google Scholar] [CrossRef]
- Gazlev, J.; Sodani, P.; Lucarelli, G. Hematopoietic stem cell transplantation in thalassemia. Bone Marrow Transpl 2008, 42, S41. [Google Scholar]
- Deshani, V.; Sadasivam, N.; Anjum, F.A.; Shah, R.; Patanwala, I.; Pointing, D.; Foster, L. Targeted liver assessment and the role of biopsy prior to gene therapy and HSCT in transfusion-dependent β-thalassemia: addressing an unmet need in the absence of standardized protocols. Blood 2025, 146, 7840. [Google Scholar] [CrossRef]
- Lucarelli, G.; Galimberti, M.; Polchi, P.; Angelucci, E.; Baronciani, D.; Giardini, C.; Politi, P.; Durazzi, S.M.; Muretto, P.; Albertini, P. Bone marrow transplantation in patients with thalassemia. N Engl Med 1990, 322, 417–421. [Google Scholar] [CrossRef]
- Lucarelli, G.; Galimberti, M.; Polchi, P.; Angelucci, E.; Baronciani, D.; Giardini, C.; Andreani, M.; Agostinelli, F.; Albertini, F.; Clift, R.A. Marrow transplantation in patients with thalassemia responsive to iron chelation therapy. N Engl J Med 2013, 329, 840–844. [Google Scholar] [CrossRef]
- Sabloff, M.; Chandy, M.; Wang, Z.; Logan, B.R.; Ghavamzadeh, A.; Li, C.K.; Irfan, S.M.; Bredeson, C.N.; Cowan, M.J.; Gale, R.P.; et al. HLA-matched sibling bone marrow transplantation for β-thalassemia major. Blood 2011, 117, 1745–1750. [Google Scholar] [CrossRef]
- Baronciani, D.; Angelucci, E.; Potschger, U.; Gaziev, J.; Yesilipek, A.; Zecca, M.; Orofino, M.G.; Giardini, C.; Al-Ahmari, A.; Marktel, S.; et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000-2010. Bone Marrow Transplant 2016, 51, 536–541. [Google Scholar] [CrossRef]
- Baronciani, D.; Boumendil, A.; Dalissier, A.; Gaziev, J.; Ghavamzadeh, A.; de la Fuente, D.J.; Zecca, M.; Seraihy, A.; Kupesiz, O.; Socié, G.; et al. Hemtopoietic cell transplantation in beta thalassemia and sickle cell disease: report from the European Society fo Blood and Bone Marrow Transplantation Hemoglobinopathy Registry: 2000-2017. Blood 2018, 132, 168. [Google Scholar] [CrossRef]
- Pinto, V.M.; Gambella, M.; Sodani, P.; Oevermann, L.; Angelucci, E. Hematopoietic cell transplantation in Guidelines for the management of transfusion-dependent β-thalassemia (TDT), Chapter 14 [Internet], 5th ed.; Taher, A.T., Farmakis, D., Porter, J.B., Eds.; Thalassemia International Federation: Nicosia, Cyprus, 2025. [Google Scholar]
- Santarone, S.; Angelini, S.; Natale, A.; Vaddinelli, D.; Spadano, R.; Casciani, P.; Papola, F.; DiLembo, E.; Iannetti, S.; Di Bartolomeo, P. Survival and late effects of hematopoietic cell transplantation in patients with thalassemia major. Bone marrow Transplant 2022, 57, 1989–1697. [Google Scholar] [CrossRef] [PubMed]
- Caocci, G.; Orofino, M.G.; Vacca, A.; Piroddi, A.; Piras, E.; Addari, M.C.; Carla, R.; Pilia, M.P.; Origa, R.; Mol, P.; et al. Long-term survival of beta thalassemia major patients treated with hematopoietic stem cell transplantation compared with survival with conventional treatment. Am J Hematol 2017, 92, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Mathews, V.; Kim, S.; George, B.; Hebnert, K.; Jiang, H.; Li, C.; Zhu, Y.; Keesler, D.A.; Boelens, J.J.; et al. Related and unrelated transplantation for β-thalassemia major: results of an international survey. Blood Adv 2019, 3, 2562–2570. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ruan, Y.; He, Y.; Wen, J.; Feng, X.; Liu, X.; Guo, H.; Li, C.; Haque, Q.; Wu, X. Effect of age on survival and complications after hematopoietic stem cell transplantation in children with β-thalassemia major. Blood 2019, 134, 4591. [Google Scholar] [CrossRef]
- Petersdorf, E.W.; Bentsson, M.; De Santis, D.; Dubois, V.; Felishhauer, K.; Gooley, T.; Horowitz, M.; Madrigel, A.; Malikki, M.; McKallor, C.; et al. Role of HLA-DP expression in graft-versus-host disease after unrelated donor transplantation. J Clin Oncol 2020, 38, 2712–2718. [Google Scholar] [CrossRef]
- McCurdy, S.R.; Solomon, S.R.; Shaffer, B.C.; He, M.; Bolon, Y.T.; Blouin, A.G.; Keyzner, A.; Socola, F.A.; Ibrahim, U.; Zou, J.; et al. Post-transplant cyclophosphamide improves survival in HLA-DP1 mismatched unrelated donor allogeneic transplantation. Transplant Cell Ther 2025, in press. [Google Scholar] [CrossRef]
- Wen, J.; He, Y.; Xiu, X.; Chen, L.; Wu, X.; Feng, X.; Li, C.; Liu, X.; Pei, F. Post-transplant cyclophosphamide improved outcomes of HLA mismatched hematopoietic stem cell transplantation in children with β-thalassemia major. Blood 2019, 134, 801. [Google Scholar]
- Lu, J.; Zhuang, Y.; Hong, X.; Chen, J.; Jiang, E.; Liu, Q. Combining low-dose post-transplant cyclophosphamide (PTCY) with anti-thymocyte globulin (ATG) for prophylaxis of graft-versus-host disease in alternative-donor hematopoietic stem cell transplantation for β-thalassemia major. Blood 2023, 142, 3568–3570. [Google Scholar] [CrossRef]
- Anurathapan, U.; Pakakasama, S.; Mejaruskul, P.; Sirachainan, N.; Sondgej, D.; Chuansumrit, A.; Charoenkwan, P.; Jetsrisuparb, A.; Sanpakit, K.; Pongtanakul, B.; et al. Outcomes of thalassemia patients undergoing hematopoietic stem cell transplant by using a standard myeloablative versus a novel reduced toxicity conditioning versus a novel reduced toxicity conditioning regimen according to new risk stratification. Biol Blood Marrow Transplant 2014, 20, 2066–2072. [Google Scholar]
- Anurathapan, U.; Hongeng, S.; Pakakasama, S.; Songdej, D.; Sirachainan, N.; Pongphitcha, P.; Chuansumrit, A.; Charoenkwan, P.; Jetsrisuparb, A.; Sanpakit, K.; et al. Hematopoietic stem cell transplantation for severe thalassemia patients from haploidentical donors using a novel conditioning regimen. Biol Blood Marrow Transplant 2020, 26, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Gong, S.; Chen, K.; Yang, R.; Wang, L.; Yang, K.; Nie, L.; Zou, L.; Su, T.; Chen, C.; et al. Haploidentical transplant for pediatric patients with severe thalassemia using post-transplant cyclophosphamide and methotrexate: a prospectively registered multicentre trial from the bone marrow failure working group of Hunan province, China. Brit J Haematol 2023, 200, 329–337. [Google Scholar]
- Gong, S.; Tian, X.; Yang, R.; Yang, L.; Wang, Z.; Yang, K.; Chen, K.; He, X.; Deng, W.; Yang, X.; et al. Uniform graft-versus-host disease prophylaxis using post-transplantationa cyclophosphamide, methotrexate, and cyclosporine following peripheral blood hematopoiertic stem cell transplantation from matched and haploidentical donors for transfusion-dependent thalassemia: a retrospective report from the bone marrow failure working group of Hunan province, China. Transpl Cell Therapy 2014, 30, 1213.e1–1213.e12. [Google Scholar]
- Beertaina, A.; Merli, P.; Rutella, S.; Pagliara, D.; Bernardo, M.E.; Masetti, R.; Pende, D.; Falco, M.; Handgretinger, R.; Moretta, F.; et al. HLA-haploidentical stem cell transplantation after removal of αβ+ T and B cells in children with nonmalignant disorders. Blood 2014, 124, 822–826. [Google Scholar] [CrossRef]
- Gaziev, J.; Isgrò, A.; Sodani, P.; Paciaroni, K.; De Angelis, G.; Marziali, M.; Ribersani, M.; Alfieri, C.; Lanti, A.; Galluccio, T.; Adorno, G.; et al. Haploidentical HSCT for hemoglobinopathies: improved outcomes with TCRαβ+/CD19+ -depleted grafts. Blood Adv 2018, 2, 263–270. [Google Scholar] [CrossRef]
- Merli, P.; Pagliare, D.; Galaverna, F.; Li Pira, G.; Andraeni, M.; Leone, G.; Amodio, D.; Pinto, R.M.; Bertaina, A.; Mastronuzzi, A.; et al. TCRαβ19 depleted HSCT from an HLA-haploidentical relative to treat children with different nonmalignant disorders. Blood Adv 2022, 6, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Oved, J.H.; Wang, Y.; Barrett, D.M.; Levy, E.M.; Huang, Y.; Monos, D.S.; Grupp, S.A.; Bunin, N.J.; Olson, T.S. CD3+/CD19+ depleted matched and mismatched unrelated donor hematopoietic cell transplant with targeted T cell addback is associated with excellent outcomes in pediatric patients with nonmalignant hematologic disorders. Biol Blood Marrow Transplant 2019, 25, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Gibson, N.M.; Elgarten, C.W.; Oved, J.H.; Wray, L.; Freedman, J.; Wang, Y.; Kadauke, S.; Hankins, P.; Nicholas, P.; Worster, E.; et al. Outcomes of unrelated partial T cell depleted stem cell transplantation for patients with hemoglobinopathies. Transplant Cell Therapy 2024, 30, S3091–S302. [Google Scholar] [CrossRef]
- Liao, J.; Bu, C.; He, L.; Zhang, W.; Zhou, Y.; Shi, W.; Xia, Y.; He, Y.; Li, C. Promotion of engraftment by triad of pre-HCT donor lymphocyte infusion, distant thymoglobulin and approaching cyclophosphamide in TCRαβ+-cell depleted HCT for patients with thalassemia major. Blood 2024, 144, 7315. [Google Scholar] [CrossRef]
- Liao, J.; Bu, C.; He, L.; Zhang, W.; Zhou, Y.; Shi, W.; Xia, Y.; He, Y.; Li, C. Optimized conditioning regimen enhances TCRαβ+ cell depleted hematopoietic stem cell transplantation engraftment in patients with thalassemia major. Eur Hemato Association 2025, abstr. PF108. [Google Scholar]
- Giardino, S.; Bagnasco, F.; Falco, M.; Miano, M.; Pierri, F.; Risso, M.; Terranova, P.; Di Martino, D.; Massaccesi, E.; Ricci, M.; et al. Haploidentical stem cell transplantation after TCR-αβ+ and CD19+ cells depletion in children with congenital non-malignant disease. Transplant Cell Therapy 2022, 28, 394e1–394e9. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Bu, C.; Liu, H.; Peng, Z.; Zhang, W.; Tang, H.; Wang, J.; Lu, Z.; Zhou, Y.; Xia, Y.; et al. Different sources of hematopoietic stem cell transplantation in patients with thalassemia major: relationship between immune reconstitution, graft-versus-host disease, and viral infections. Blood 2023, 142, 7093. [Google Scholar] [CrossRef]
- Meissner, B.; Lang, P.; Bader, P.; Hoenig, M.; Muller, I.; Meisel, R.; Greil, J.; Sauer, M.G.; Metzler, M.; Carbacioglu, S.; et al. Finding a balance in reduced toxicity hemaotloietic tsem cell transplantation for thalassemia: role of inf[54used CD3+ cell count and immunosuprression. Bane Marrow Transplant 2024, 59, 587–596. [Google Scholar] [CrossRef]
- Mulas, O.; Mola, B.; Caucci, G.; La Nasa, G. Conditioning regimens in patients with β-thalassemia who underwent hematopoietic stem cell transplantation: a scoping review. J Clin Medicine 2002, 11, 907. [Google Scholar] [CrossRef]
- Mathews, V.; George, B.; Viswabandya, A.; Abraham, A.; Ahmed, R.; Ganapule, A.; Sindhuvi, E.; Lakshmi, K.M.; Srivastatava, A. Improved clinical outcomes of high ris β thalassemia major patients undergoing HLA matched related allogeneic stem cell transplant with a treosulfan based conditioning regimen and peripheral blood stem cell grafts. PLoS One 2013, 8, e61637. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Abraham, A.; Lakshmi, K.; Korula, A.; Devasia, A.; Kulkarni, U.; Lionel, S.; Abubacker, F.; Srivastava, A.; Mathews, V. Treosulfan based conditioning regimen is associated with better overall survival compared to busulfan based regimens in older children with thalassemia major. Bio Blood Marrow Transplant 2019, 25, S290. [Google Scholar] [CrossRef]
- Chiesa, R.; Satnding, J.R.; Winter, R.; Nademi, Z.; Chu, J.; Pinner, D.; Kloprogge, F.; McLellen, S.; Amrolia, P.J.; Raok, K.; et al. Proposed therapeutic range of treosulfan in reduced toxicity pediatric allogeneic hematopoietic cell transplant conditioning: results from a prospective study. Clin Pharmacol 2020, 108, 264–273. [Google Scholar] [CrossRef]
- Van der Stoep, E.; Bertaina, A.; Moes, D.; Algeri, M.; Bredius, R.; Smiers, F.; Berguis, D.; Buddingh, E.; Mohseny, A.; Guchelaar, H.J.; et al. Impact of treosulfan exposure on early and long-term clinical outcomes in pediatric allogeneic hematopoietic stem cell transplantation recipients: a prospective multicenter study. Transplant Cell Ther 2022, 28, 99.e1–99.e7. [Google Scholar] [CrossRef] [PubMed]
- Pai, A.A.; Mohanan, E.; Panetta, J.C.; Kulkarni, U.; Illagenswan, R.S.S.; Balakrishnan, B.; Jayaraman, A.; Edison, E.S.; Lakshmi, K.; Devasia, A.; et al. Tresulfan exposure predicts thalassemia-free survival in patients with beta thalassemia major undergoing allogeneic hematopoietic cell transplantation. Clin Parmacol Therapeutics 2024, 115, 116–124. [Google Scholar] [CrossRef]
- Luftinger, R.; Zubarovskaya, N.; Gallimard, J.E.; Cseh, A.; Salzer, E.; Locatelli, F.; Algeri, M.; Yesilipek, A.; de la Fuente, J.; Isgrò, A. Busulfan-fludarabine- trosulfan-fludarabine-based myeloablative condiotingn for children with thalassemia major. Ann Hematol 2022, 101, 655–665. [Google Scholar] [CrossRef]
- Sykora, K.W.; Beler, R.; Schultz, A.; Cesaro, S.; Greil, J.; Gozdzik, J.; Sedlacek, P.; Bader, P.; Schulte, J.; Zecca, M.; et al. Treosulfan versus busulfan conditioning for allogeneic bmt in children with nonmalignant disease: a randomized phase 2 trial. Bone Marrow Transplant 2024, 59, 107–116. [Google Scholar] [CrossRef]
- Kleinschmidt, K.; Penkivech, G.; Troger, T.; Foell, J.; Hanafee-Alali, T.; Leszczak, S.; Jakob, M.; Kramer, S.; Kietz, S.; Hoffmann, P.; et al. αβ T-cell depleted haploidentical stem cell transplantation for pediatric and young adult patients with transfusion-dependent thalassemia. Bone Marrow Transplant 2025, 60, 682–689. [Google Scholar] [CrossRef]
- Ganesan, K.; Muthukumar, V.; Nair, A.; Valliyappan, S.; Uppuluri, R.; Raj, R.; et al. A 10-year follow-up study of children with thalassemia major post-transplantation using treosulfan, thiotepa, and fludarabine-based conditioning regimen and its impact on growth and puberty. Tranplant Cell Therapy 2025, 31, P926.E1–P926.E7. [Google Scholar] [CrossRef]
- Altmann, T.; Rao, K.; Hanasoge-Nataraj, R.; Patrick, K.; Carpenter, B.; Hough, R.; Ewins, A.M.; Tewari, S.; Mirci-Danicar, O.; Slatter, M.; et al. Low rates of endothelial cell dysfunction and transplant-related mortality in 537 children receiving fludarabine-treosulfan conditioning for all transplant indications: a retrospective multicentre study on behalf of the UK pediatric BMT group. Brit J Haematol 2025, 207, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Van der Stoep, E.; Bertaina, A; Moes, D.J.; Algeri, M.; Bredius, R.; Smiers, F.; Beghuis, D.; Buddingh, E.; Mohseny, A.; Guchelaar, H.J.; et al. Impact of treosulfan exposure on early and long-term clinical outcomes in pediatric allogeneic hematopoietic stem cell transplantation recipients: a prospective multicenter study. Transplant Cell Therapy 2022, 28, 99.e1–99.e7. [Google Scholar] [CrossRef]
- Ghavamzadeh, A.; Iravani, M.; Ashouri, A.; Mousavi, S.A.; Mahdavi, N.; Shamshiri, A.; Hadjibabaie, M.; Namdar, R.; Nedaeifard, L.; Ghaffari, H.; et al. Peripheral blood versus bone marrow as a source of hematopoietic stem cells for allogeneic transplantation in children with class I and II beta thalassemia major. Biol Blood Marrow Transplant 2008, 14, 301–308. [Google Scholar] [CrossRef]
- Ghavamzadeh, A.; Kasaeian, A.; Rostami, T.; Kiumarsi, A. Comparable outcomes of allogeneic peripheral blood versus bone marrow transplantation: in major thalassemia: a multivariate long-term cohort analysis. Biol Blood Marrow Transplant 2019, 25, 307–312. [Google Scholar] [CrossRef]
- Locatelli, F.; Kabbara, N.; Ruggeri, A.; Ghavamzadeh, A.; Roberts, I.; Li, C.K.; Bernaudin, F.; Vermylen, C.; Dalle, J.H.; Stein, J.; et al. Outcome of patients with hemoglobinopathies given either cord blood or bone marrow transplantation from an HLA-identical sibling. Blood 2013, 122, 1072–1078. [Google Scholar] [CrossRef]
- Locatelli, F.; Rocha, V.; Reed, W.; Bernaudin, F.; Ertem, M.; Grafakos, S.; Brichard, B.; Li, X.; Nagler, A.; Giorgiani, G.; et al. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood 2003, 101, 2137–2143. [Google Scholar] [CrossRef]
- Zwolsman, M.; Alahmri, B.; Ahmed, M.; Dovern, E.; Biemond, B.; Ahadi, H.; Almozaini, A.; Alzahrani, M.; Nur, E. Outcomes of peripheral blood stem cells versus bone marrow in adult sickle cell disease patients undergoing haploidentical allogeneic stem cell transplantation. EHA Meeting, 2025. [Google Scholar]
- Poskuriakova, E.; Zucchetti, E.; Sweiss, K.; Mahmud, N.; Koshy, M.; Rondelli, D. Haploidentical peripheral blood stem cells transplantation in 20 adult patients with sickle cell disease.
- Folarin, M.; Al-Zubaidi, H.; Moore, E.; Eze, U.; Palanisamy, N. Efficacy and safety of allogeneic hematopoietic stem cell transplantation in curing sickle cell disease: a systematic review and meta-analysis of single-arm studies. Transplant Cell Therapy 2025, in press. [Google Scholar] [CrossRef]
- Brown, D.; Li, N.; Li, J.; Imren, S.; Evans, K.; Jerry, M. Clinical outcomes and complications in patients with beta-thalassemia who received allogeneic hematopoietic stem cell transplants in the United States. 2025. Transplant Cell Therapy 2025, 31 supplement 1, S320. [Google Scholar] [CrossRef]
- Baronciani, D.; de la Fuente, J.; Galimard, J.E.; Dalissier, A.; Mousavi, A.; Yesilipek, A.; Hashem, A.; Ozturk, G.; Locatelli, F.; Alhmri, A.; et al. Age is a crucial determinant of GFRS with incidence of severe chronic GVHD reducing over time in hematopoietic cell transplantation for transfusion dependent thalassemic real world data from 2021-2021, an anlysis of the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Working Party. Blood 2024, 144, 2136–2137. [Google Scholar]
- Ngwube, A.; Shah, N.; Godder, K.; Jacobsohn, D.; Hulbert, M.L.; Shenoy, S. Abatacept is effective ad GVHD prophylaxis in unrelated donor stem cell transplantation for children with severe sickle cell disease. Blood Adv 2020, 4, 3894–3900. [Google Scholar]
- Shah, N.C.; Ngwube, A.; Suresh, T.; Sowa, A.; Abraham, A.; Anderson, E.; Andreansky, M.; Bhatia, M.; Chaudhury, S.; Cuvelier, G.; et al. Impact of abtacept inclusive graft versus-host disease prophylaxis in pediatric stem cell transplantation for hemoglobinopathy. Transplant Cell Therapy 2025, 31, 826.e1–826e.10. [Google Scholar] [CrossRef]
- Khandelwal, P.; Yah, R.F.; Yu, L.; Lane, A.; Dandoy, C.E.; El-Bietar, J.; Davies, S.M.; Grimley, M.S. Graft-versus-host disease prophylaxis with abatacept reduces severe acute graft-versus-host disease in allogeneic hematopoietic stem cell transplant for beta-thalassemia major with busulfan, fludarabine, and thiotepa. Transplantation 2021, 105, 891–896. [Google Scholar] [CrossRef]
- Jaiswal, S.R.; Agarwal, M.; Baligar, P.; Biswas, S.; Das, B.C.; Bacigalupo, A.; Chakrabarti, S. Abatacept and Pcty vased GVHD prophylaxis with CNI/Sirolimus for haploidentical HCT: donor KLRC2 genotype impacts outcome. Blood 2023, 142, 4928. [Google Scholar] [CrossRef]
- Jaiswal, S.R.; Agarwal, M.; Bhagawati, G.; Das, B.C.; Baligar, P.; Garg, M.; Biswas, S.; Chakrabarti, S. Long-term follow-up of abatacept, post-transplantation cyclophosphamide, and sirolimus-based haploidentical transplantation in younger patients with nonmalignant diseases. Transpl Cell Therapy 2024, 30, 605.e1–605.e13. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.; O’Boyle, F.; Lund, K.; Petterson, T.; Gassas, A.; Karnik, L.; de la Fuente, J.; Bremathas, S.; Hennessy, K. Abatacept abrogates the risk of acute GvHD in both matched and mismatched transplantation for hemoglobinopathies and bone marrow failure. Blood 2024, 144, 3518–3519. [Google Scholar] [CrossRef]
- Seminog, O.O.; Ogunlaja, O.I.; Yates, D.; Goldacre, M.J. Risk of individual malignant neoplasms in patients with sickle cell disease: English national record linkage study. J R Soc Med 2016, 109, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Brunson, A.; Keegan, T.H.M.; Bang, H.; Mahajan, A.; Paulukonis, S.; Wun, T. Increased risk of leukemia among sickle cell disease patients in California. Blood 2007, 130, 1597–1599. [Google Scholar] [CrossRef]
- Eapen, M.; Brazauskas, R.; Williams, D.A.; Walters, M.C.; St Martin, A.; Jacobs, B.L.; Antin, J.H.; Bonas, K.; Chaudhury, S.; Coleman-Cowger, V.H.; et al. Secondary neoplasms after hematopoietic cell transplant for sickle cell disease. J Clin Oncol 2023, 41, 2227–2237. [Google Scholar] [CrossRef]
- Lawal, R.A.; Mukherjee, D.; Limerick, E.M.; Coles, W.; Hsieh, H.M.; Dillon, L.W.; Hourigan, C.S.; Fitzhugh, C.D. Increased incidence of hematologic malignancies in SCD afte HCT in adults with graft failure and mixed chimerism. Blood 2022, 140, 2414–2418. [Google Scholar] [CrossRef] [PubMed]
- Ghannam, J.Y.; Xu, X.; Maric, I.; Dillon, L.; LI, Y.; Hsieh, M.M.; Hourigan, C.S.; Fitzhugh, C.D. Baseline TP53 mutations in adults with SCD developeing myeloid malignancy following hematopoietic cell transplantation. Blood 2020, 135, 1185–1188. [Google Scholar] [CrossRef]
- Mukhrjee, D.; Lawal, R.A.; Fitzhugh, C.D.; Hourigan, C.S.; Dillon, L.W. TP53 mutation screening for patients at risk of myeloid malignancy. Leukemia 2024, 38, 1604–1608. [Google Scholar] [CrossRef]
- Ali, M.; Limerick, E.; Hsieh, M.; Upadhyaya, K.; Xu, X.; Phang, O.; Kambala Mukendi, J.P.; Calvo, K.R.; Lopaz-Ocasio, M.; Dagur, P.; Fitzhugh, C.D. Recipeinet cells are the source of hematologic malignancies after graft failure and mixed xhimerism id adults. Am J Hematol 2025, 100, 903–905. [Google Scholar] [CrossRef]
- Gondek, V.A.; Sheenan, V.A.; Fitzhugh, C.D. Clonal hematopoiesis and the risk of hematologic malignancies after curative therapies for sickle cell disease. J Clin Med 2022, 11, 3160. [Google Scholar] [CrossRef] [PubMed]
- Pincez, T.; Lee, S.S.K.; Ilboudo, Y. Clonal hematopoiesis in sickle cell disease. Blood 2021, 138, 2148–2152. [Google Scholar] [CrossRef] [PubMed]
- Weeks, L.; Fitzhugh, C.; Pollock, S.; Osei, M.; Rickel-Young, M.; Murdoch, M.; Towsend, M.; Reilly, C.; Dinardo, C.; Sabino, E.; et al. sickle cell disease is associated with early-onset clonal hematopoiesis involving DNA damage response pathway mutations. Blood 2025, 146, abs25–7115. [Google Scholar] [CrossRef]
- De Luna, G.; Cretin, J.; Redjoul, R.; Breckerich, F.; Habibi, A.; Menouche, D.; Hebert, N.; Giovannangeli, C.; Maury, S.; Bartolucci, P.; et al. Screening for clonal hematopoiesis in patients with β-hemoglobinopathies, excluding who are candidates to transplant approaches. Blood 2025, 146, abs25–2407. [Google Scholar] [CrossRef]
- Merolle, L.; Ghirotto, L.; Schiroli, D.; Balestra, G.L.; Venturelli, F.; Bassi, M.C.; Di Bartlomeo, E.; Baricchi, R.; Marraccini, C. Risk of cancer in patients with thalassemia and sickle cell disease. Ann Medicine 2025, 57, 2522967. [Google Scholar] [CrossRef]
- Santarone, S.; Pepe, A.; Meloni, A.; Natale, A.; Pistoia, L.; Olioso, P.; Papalinetti, G.; Cuccia, L.; Spasiano, A.; Lisi, R.; et al. Secondary solid cancer following hematopoietic cell transplantation in patients with thalassemia major. Bone Marrow Transplant 2018, 53, 39–43. [Google Scholar] [CrossRef]
- Solomou, E.E.; Dalaporta, P.; Stamatia, L.; Katsika, E.V.; Cahtzielefteriou, M.; Touttoudaki, K.; Glentis, S.; Stamatopoulos, K.; Chatzidimitriou, A.; Kattamis, A. High incidence of clonal hematopoiesis in transfusion-dependent thalassemia patients. Blood 2022, 140, 8213–8214. [Google Scholar] [CrossRef]
- Jones, R.J.; Kassim, A.A.; Brodsky, R.A.; DuBaun, M.R. Is allogeneic transplantation for SCD still relevant in the era of gene therapy? Blood Adv 2025, 9, 877–882. [Google Scholar] [CrossRef]
- Arnold, S.D.; Brazauskas, R.; He, N.; Li, Y.; Aplenc, R.; Jin, Z.; Hall, M.; Atsuta, Y.; Dalai, J.; Hahn, T.; et al. Clinical risks and healthcare utilization of hematopoietic cell transplantation for sickle cell disease in the USA using merged databases. Haematologica 2017, 102, 1823–1832. [Google Scholar] [CrossRef] [PubMed]
- Gharial, J.; Guicher, G.; Spackman, E.; Sharma, A.; Arnold, S. Haelthcare costs associated with hematopoietic cell transplant for sickle cell disease in the United States and Canada: a comparative analysis. Blood 2025, 146, 6151. [Google Scholar] [CrossRef]
- Lothe, P.; Pompa, T.; Jain, M.; Patel, P; Liang, Y.; Yalamanchi, A; Ward, K.; Styler, M. Comparative cost analysis of therapy in sickle cell anemia: supportive care vs bone marrow transplant. Blood 2015, 126, 4466. [Google Scholar] [CrossRef]
- Aprile, A.; Lidonnici, M.R.; Ferrari, G. Gene therapy for hemoglobinopathies: clinical trial results and biology of hematopoietic stem cell and the bone marrow niche. Cell Rep Medicine 2025, 6, 102419. [Google Scholar] [CrossRef]
- Frangoul, H.; Stuits, A.; Bruce, K.; Domm, J.; Carroll, C.; Aide, S.; Duckworth, M.; Evasns, M.; McManus, M. Best practices in gene therapy for sickle cell disease and transfusion-dependent β-thalassemia. Transpl Cell Therapy 2025, 31, 35.2e1–35.2e10. [Google Scholar] [CrossRef]
- De Franceschi, L.; Locatelli, F.; Rees, D.; Chabannon, C.; Dalle, J.H.; Rivella, S.; Iolascon, A.; Lobitz, S.; Abboud, M.R.; de la Fuente, J.; et al. Selecting patients with sickle cell disease for gene addition or gene editing-based ther- apeutic approaches: report on behalf of a joint EHA specialized working group and EBMT hemoglobinopathies working party consensus conference. HemaSphere 2025, 9, e70089. [Google Scholar] [CrossRef]
- Sharma, A.; John, T. To pursue gene therapy or not? Is it feasible after graft failure in allogeneic hematopoietic cell transplant recipients? Blood Adv 2025, 9, 3845–3849. [Google Scholar] [CrossRef]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).