Submitted:
26 December 2025
Posted:
29 December 2025
You are already at the latest version
Abstract
Metabolic dysfunction–associated steatotic liver disease (MASLD) is the leading cause of chronic liver disease worldwide, distinguished by pronounced clinical heterogeneity and a frequent dissociation between metabolic risk factors and the degree of hepatic injury. These observations, together with the limited contribution of genetic heritability, have prompted a re-evaluation of the traditional conceptual framework of the disease. In this context, the question has emerged as to whether MASLD could be, at least in part, transmissible condition. While there is no evidence to suggest that MASLD is contagious in humans, as no data support person-to-person transmission, gnotobiotic animal studies demonstrate that human gut microbiota can transfer susceptibility to steatosis, inflammation, and systemic metabolic disturbances through immunometabolic mechanisms, independent of host genetics. In parallel, human studies involving microbiota-targeted interventions support the concept that the gut ecosystem is a modifiable determinant of metabolic and hepatic phenotypes. Crucially, these findings do not imply natural transmission of disease, but rather underscore the functional plasticity of microbiota-host interactions. This narrative review integrates epidemiological, experimental, and clinical data to explore the hypothesis that MASLD may be functionally transmissible. MASLD is increasingly recognized as an eco-biological disease, where liver disease risk is not only shaped by host genetics and environment, but also by the ecological configuration and functional outputs of the gut microbiome. This perspective redefines disease susceptibility as, in part, context-dependent and microbiota-mediated, without implying infectiousness in the traditional sense.
Keywords:
1. Introduction
2. MASLD Is Not Only About the Liver, but the Environment
3. Experimental Evidence Supporting How Hepatic Risk Becomes Transferable
4. Clinical Evidence in Humans Demonstrate Modulability Without Contagiousness
5. Sharing Microbiota Is Not Equivalent to Sharing Disease
6. From Clinical Modulability to Functional Transmissibility of Metabolic–Hepatic Risk
7. Implications for Public Health: From the Non-Contagious Individual to Population-Level Ecosystem Risk
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Younossi, ZM; Koenig, AB; Abdelatif, D; Fazel, Y; Henry, L; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64(1), 73–84. [Google Scholar] [CrossRef]
- Estes, C; Anstee, QM; Arias-Loste, MT; Bantel, H; Bellentani, S; Caballeria, J; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol. 2018, 69(4), 896–904. [Google Scholar] [CrossRef]
- Younossi, ZM; de Avila, L; Petta, S; Hagström, H; Kim, SU; Nakajima, A; et al. Predictors of fibrosis, clinical events and mortality in MASLD: Data from the Global-MASLD study. Hepatology 2025. [Google Scholar] [CrossRef]
- Younossi, ZM; Paik, JM; Lazarus, JV; Burra, P; Eguchi, Y; Tacke, F; et al. Projected Global Clinical, Humanistic, and Economic Impact of Metabolic Dysfunction-Associated Steatohepatitis (MASH): The Cost of Inaction Based on Data From Nine Countries. Clin Gastroenterol Hepatol 2025. [Google Scholar] [CrossRef]
- Arrese, M; Arab, JP; Barrera, F; Kaufmann, B; Valenti, L; Feldstein, AE. Insights into Nonalcoholic Fatty-Liver Disease Heterogeneity. Semin Liver Dis. 2021, 41(4), 421–34. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S; Pirola, CJ. Genetics of Nonalcoholic Fatty Liver Disease: From Pathogenesis to Therapeutics. Semin Liver Dis. 2019, 39(2), 124–40. [Google Scholar] [CrossRef]
- Martínez-Arranz, I; Bruzzone, C; Noureddin, M; Gil-Redondo, R; Mincholé, I; Bizkarguenaga, M; et al. Metabolic subtypes of patients with NAFLD exhibit distinctive cardiovascular risk profiles. Hepatology 2022, 76(4), 1121–34. [Google Scholar] [CrossRef]
- Kolodziejczyk, AA; Zheng, D; Shibolet, O; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol Med. 2019, 11(2). [Google Scholar] [CrossRef] [PubMed]
- Abdelhameed, F; Mustafa, A; Kite, C; Lagojda, L; Dallaway, A; Than, NN; et al. Gut Microbiota and Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD): Emerging Pathogenic Mechanisms and Therapeutic Implications. Livers 2025, 5(1), 11. [Google Scholar] [CrossRef]
- Nistal, E; Sáenz de Miera, LE; Ballesteros Pomar, M; Sánchez-Campos, S; García-Mediavilla, MV; Álvarez-Cuenllas, B; et al. An altered fecal microbiota profile in patients with non-alcoholic fatty liver disease (NAFLD) associated with obesity. Rev Esp Enferm Dig. 2019, 111(4), 275–82. [Google Scholar] [CrossRef]
- Francque, SM; Marchesini, G; Kautz, A; Walmsley, M; Dorner, R; Lazarus, JV; et al. Non-alcoholic fatty liver disease: A patient guideline. JHEP Rep. 2021, 3(5), 100322. [Google Scholar] [CrossRef]
- Bahitham, W; Banoun, Y; Aljahdali, M; Almuaiqly, G; Bahshwan, SM; Aljahdali, L; et al. “Trust your gut”: exploring the connection between gut microbiome dysbiosis and the advancement of Metabolic Associated Steatosis Liver Disease (MASLD)/Metabolic Associated Steatohepatitis (MASH): a systematic review of animal and human studies. Front Nutr. 2025, 12, 1637071. [Google Scholar] [CrossRef] [PubMed]
- Lanthier, N; Delzenne, N. Targeting the Gut Microbiome to Treat Metabolic Dysfunction-Associated Fatty Liver Disease: Ready for Prime Time? Cells 2022, 11(17). [Google Scholar] [CrossRef]
- Tilg, H; Zmora, N; Adolph, TE; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol. 2020, 20(1), 40–54. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H; Adolph, TE. Liver microbes controlling immunity: Facts and pitfalls. Cell Metab. 2022, 34(4), 510–2. [Google Scholar] [CrossRef]
- Yuan, J; Chen, C; Cui, J; Lu, J; Yan, C; Wei, X; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30(4), 675–88.e7. [Google Scholar] [CrossRef]
- Iruzubieta, P; Medina, JM; Fernández-López, R; Crespo, J; de la Cruz, F. A Role for Gut Microbiome Fermentative Pathways in Fatty Liver Disease Progression. J Clin Med. 2020, 9(5). [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, A; Robles-Vera, I; Mañanes, D; Galán, M; Femenía-Muiña, M; Redondo-Urzainqui, A; et al. Imidazole propionate is a driver and therapeutic target in atherosclerosis. Nature 2025, 645(8079), 254–61. [Google Scholar] [CrossRef]
- Hu, Y; Li, J; Wang, B; Zhu, L; Li, Y; Ivey, KL; et al. Interplay between diet, circulating indolepropionate concentrations and cardiometabolic health in US populations. Gut 2023, 72(12), 2260–71. [Google Scholar] [CrossRef]
- Schnabl, B; Damman, CJ; Carr, RM. Metabolic dysfunction-associated steatotic liver disease and the gut microbiome: pathogenic insights and therapeutic innovations. J Clin Invest. 2025, 135(7). [Google Scholar] [CrossRef]
- Gómez-Hurtado, I; Gallego-Durán, R; Zapater, P; Ampuero, J; Aller, R; Crespo, J; et al. Bacterial antigen translocation and age as BMI-independent contributing factors on systemic inflammation in NAFLD patients. Liver Int. 2020, 40(9), 2182–93. [Google Scholar] [CrossRef] [PubMed]
- Martino, C; Zaramela, LS; Gao, B; Embree, M; Tarasova, J; Parker, SJ; et al. Acetate reprograms gut microbiota during alcohol consumption. Nat Commun. 2022, 13(1), 4630. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J; Vigliotti, C; Witjes, J; Le, P; Holleboom, AG; Verheij, J; et al. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol 2020, 17(5), 279–97. [Google Scholar] [CrossRef] [PubMed]
- Santos-Laso, A; Gutiérrez-Larrañaga, M; Alonso-Peña, M; Medina, JM; Iruzubieta, P; Arias-Loste, MT; et al. Pathophysiological Mechanisms in Non-Alcoholic Fatty Liver Disease: From Drivers to Targets. Biomedicines 2021, 10(1). [Google Scholar] [CrossRef]
- Lau, HC; Zhang, X; Yu, J. Gut microbiome in metabolic dysfunction-associated steatotic liver disease and associated hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2025, 22(9), 619–38. [Google Scholar] [CrossRef]
- El-Sayed, AM; Galea, S. Epidemiology and the People’s Health: By Nancy Krieger. American Journal of Epidemiology 2012, 175(8), 854–6. [Google Scholar] [CrossRef]
- Wild, CP. Complementing the genome with an “exposome”: the outstanding challenge of environmental exposure measurement in molecular epidemiology. In Cancer Epidemiol Biomarkers; Prev. 14. United States; 2005; pp. 1847–1850. [Google Scholar]
- Hood, L; Friend, SH. Predictive, personalized, preventive, participatory (P4) cancer medicine. Nat Rev Clin Oncol. 2011, 8(3), 184–7. [Google Scholar] [CrossRef]
- Heddes, M; Altaha, B; Niu, Y; Reitmeier, S; Kleigrewe, K; Haller, D; et al. The intestinal clock drives the microbiome to maintain gastrointestinal homeostasis. Nat Commun. 2022, 13(1), 6068. [Google Scholar] [CrossRef]
- Medina-Méndez, JM; Iruzubieta, P; Fernández-López, R; Crespo, J; de la Cruz, F. Bacterial metabolic signatures in MASLD predicted through gene-centric studies in stool metagenomes. BMC Microbiol 2025. [Google Scholar] [CrossRef]
- Le Roy, T; Llopis, M; Lepage, P; Bruneau, A; Rabot, S; Bevilacqua, C; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62(12), 1787–94. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J; Elinav, E; Jin, C; Hao, L; Mehal, WZ; Strowig, T; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482(7384), 179–85. [Google Scholar] [CrossRef] [PubMed]
- Leinwand, JC; Paul, B; Chen, R; Xu, F; Sierra, MA; Paluru, MM; et al. Intrahepatic microbes govern liver immunity by programming NKT cells. J Clin Invest. 2022, 132(8). [Google Scholar] [CrossRef]
- Ridaura, VK; Faith, JJ; Rey, FE; Cheng, J; Duncan, AE; Kau, AL; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341(6150), 1241214. [Google Scholar] [CrossRef]
- Fei, N; Zhao, L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. Isme j. 2013, 7(4), 880–4. [Google Scholar] [CrossRef]
- Juárez-Fernández, M; Goikoetxea-Usandizaga, N; Porras, D; García-Mediavilla, MV; Bravo, M; Serrano-Maciá, M; et al. Enhanced mitochondrial activity reshapes a gut microbiota profile that delays NASH progression. Hepatology 2023, 77(5), 1654–69. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y; Tang, L; Chen, Q; Wu, L; He, W; Tu, D; et al. Disulfiram ameliorates nonalcoholic steatohepatitis by modulating the gut microbiota and bile acid metabolism. Nat Commun. 2022, 13(1), 6862. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D; Pan, Q; Shen, F; Cao, HX; Ding, WJ; Chen, YW; et al. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci Rep. 2017, 7(1), 1529. [Google Scholar] [CrossRef]
- Wang, R; Li, H; Yang, X; Xue, X; Deng, L; Shen, J; et al. Genetically Obese Human Gut Microbiota Induces Liver Steatosis in Germ-Free Mice Fed on Normal Diet. Front Microbiol 2018, 9, 1602. [Google Scholar] [CrossRef]
- Sun, X; Wu, J; Liu, L; Chen, Y; Tang, Y; Liu, S; et al. Transcriptional switch of hepatocytes initiates macrophage recruitment and T-cell suppression in endotoxemia. J Hepatol. 2022, 77(2), 436–52. [Google Scholar] [CrossRef]
- Vrieze, A; Van Nood, E; Holleman, F; Salojärvi, J; Kootte, RS; Bartelsman, JF; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143(4), 913–6.e7. [Google Scholar] [CrossRef]
- Kootte, RS; Levin, E; Salojärvi, J; Smits, LP; Hartstra, AV; Udayappan, SD; et al. Improvement of Insulin Sensitivity after Lean Donor Feces in Metabolic Syndrome Is Driven by Baseline Intestinal Microbiota Composition. Cell Metab. 2017, 26(4), 611–9.e6. [Google Scholar] [CrossRef]
- Proença, IM; Allegretti, JR; Bernardo, WM; de Moura, DTH; Ponte Neto, AM; Matsubayashi, CO; et al. Fecal microbiota transplantation improves metabolic syndrome parameters: systematic review with meta-analysis based on randomized clinical trials. Nutr Res. 2020, 83, 1–14. [Google Scholar] [CrossRef]
- Bajaj, JS; Fagan, A; Gavis, EA; Kassam, Z; Sikaroodi, M; Gillevet, PM. Long-term Outcomes of Fecal Microbiota Transplantation in Patients With Cirrhosis. Gastroenterology 2019, 156(6), 1921–3.e3. [Google Scholar] [CrossRef]
- Rasmussen, DGK; Anstee, QM; Torstenson, R; Golding, B; Patterson, SD; Brass, C; et al. NAFLD and NASH biomarker qualification in the LITMUS consortium - Lessons learned. J Hepatol 2023, 78(4), 852–65. [Google Scholar] [CrossRef]
- Yang, W; Jin, Q; Xiao, D; Li, X; Huang, D. Interaction mechanism and intervention strategy between metabolic dysfunction-associated steatotic liver disease and intestinal microbiota. Front Microbiol 2025, 16, 1597995. [Google Scholar] [CrossRef]
- Groenewegen, B; Ruissen, MM; Crossette, E; Menon, R; Prince, AL; Norman, JM; et al. Consecutive fecal microbiota transplantation for metabolic dysfunction-associated steatotic liver disease: a randomized controlled trial. Gut Microbes 2025, 17(1), 2541035. [Google Scholar] [CrossRef]
- Del Barrio, M; Lavín, L; Santos-Laso, Á; Arias-Loste, MT; Odriozola, A; Rodriguez-Duque, JC; et al. Faecal Microbiota Transplantation, Paving the Way to Treat Non-Alcoholic Fatty Liver Disease. Int J Mol Sci. 2023, 24(7). [Google Scholar] [CrossRef] [PubMed]
- Drucker, DJ. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018, 27(4), 740–56. [Google Scholar] [CrossRef]
- Dulai, AS; Min, M; Sivamani, RK. The Gut Microbiome’s Influence on Incretins and Impact on Blood Glucose Control. Biomedicines 2024, 12(12). [Google Scholar] [CrossRef] [PubMed]
- Tilg, H; Petta, S; Stefan, N; Targher, G. Metabolic Dysfunction-Associated Steatotic Liver Disease in Adults: A Review. Jama 2025. [Google Scholar] [CrossRef] [PubMed]
- Song, SJ; Lauber, C; Costello, EK; Lozupone, CA; Humphrey, G; Berg-Lyons, D; et al. Cohabiting family members share microbiota with one another and with their dogs. Elife 2013, 2, e00458. [Google Scholar] [CrossRef]
- Dill-McFarland, KA; Tang, ZZ; Kemis, JH; Kerby, RL; Chen, G; Palloni, A; et al. Close social relationships correlate with human gut microbiota composition. Sci Rep. 2019, 9(1), 703. [Google Scholar] [CrossRef]
- Rothschild, D; Weissbrod, O; Barkan, E; Kurilshikov, A; Korem, T; Zeevi, D; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555(7695), 210–5. [Google Scholar] [CrossRef]
- Lang, S; Demir, M; Martin, A; Jiang, L; Zhang, X; Duan, Y; et al. Intestinal Virome Signature Associated With Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 159(5), 1839–52. [Google Scholar] [CrossRef] [PubMed]
- Odriozola, A; Santos-Laso, A; Del Barrio, M; Cabezas, J; Iruzubieta, P; Arias-Loste, MT; et al. Fatty Liver Disease, Metabolism and Alcohol Interplay: A Comprehensive Review. Int J Mol Sci. 2023, 24(9). [Google Scholar] [CrossRef]
- Smillie, CS; Sauk, J; Gevers, D; Friedman, J; Sung, J; Youngster, I; et al. Strain Tracking Reveals the Determinants of Bacterial Engraftment in the Human Gut Following Fecal Microbiota Transplantation. Cell Host Microbe 2018, 23(2), 229–40.e5. [Google Scholar] [CrossRef]
- Chen, Q; Wu, C; Xu, J; Ye, C; Chen, X; Tian, H; et al. Donor-recipient intermicrobial interactions impact transfer of subspecies and fecal microbiota transplantation outcome. Cell Host Microbe 2024, 32(3), 349–65.e4. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, MA; Ramaswamy, R; Lin, H; Lehmann, C; Moran, A; Mullowney, MW; et al. Fecal Butyrate and Deoxycholic Acid Concentrations Correlate With Mortality in Patients With Liver Disease. Gastro Hep Adv. 2025, 4(8), 100695. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q; Stappenbeck, TS. Local barriers configure systemic communications between the host and microbiota. Science 2022, 376(6596), 950–5. [Google Scholar] [CrossRef]
- Yang, J; Hou, L; Wang, J; Xiao, L; Zhang, J; Yin, N; et al. Unfavourable intrauterine environment contributes to abnormal gut microbiome and metabolome in twins. Gut 2022, 71(12), 2451–62. [Google Scholar] [CrossRef]
- Nageswaran, V; Carreras, A; Reinshagen, L; Beck, KR; Steinfeldt, J; Henricsson, M; et al. Gut Microbial Metabolite Imidazole Propionate Impairs Endothelial Cell Function and Promotes the Development of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2025, 45(5), 823–39. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q; Wang, W; Li, Y; Liu, Y. Imidazole propionate in type 2 diabetes mellitus and cardiovascular diseases: a mini review. Front Immunol 2024, 15, 1454210. [Google Scholar] [CrossRef] [PubMed]
- Yang, H; Luo, K; Peters, BA; Wang, Y; Zhang, Y; Daviglus, M; et al. Diet, Gut Microbiota, and Histidine Metabolism Toward Imidazole Propionate Production in Relation to Type 2 Diabetes. Diabetes Care 2025, 48(7), 1225–32. [Google Scholar] [CrossRef]
- Crespo, J; Iruzubieta, P; Alonso-Peña, M; Jiménez- González, C; Argos Vélez, P; Martínez-Chantar, M. The Histidine Crossroad: An Integrative Theory of the Gut–Liver–Vascular Axis in MASLD. Unpublished data.
- Truong, DT; Tett, A; Pasolli, E; Huttenhower, C; Segata, N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017, 27(4), 626–38. [Google Scholar] [CrossRef] [PubMed]
- Anyansi, C; Straub, TJ; Manson, AL; Earl, AM; Abeel, T. Computational Methods for Strain-Level Microbial Detection in Colony and Metagenome Sequencing Data. Front Microbiol 2020, 11, 1925. [Google Scholar] [CrossRef]
- Vatanen, T; Jabbar, KS; Ruohtula, T; Honkanen, J; Avila-Pacheco, J; Siljander, H; et al. Mobile genetic elements from the maternal microbiome shape infant gut microbial assembly and metabolism. Cell. 2022, 185(26), 4921–36.e15. [Google Scholar] [CrossRef]
- Xiao, L; Zhao, F. Microbial transmission, colonisation and succession: from pregnancy to infancy. Gut 2023, 72(4), 772–86. [Google Scholar] [CrossRef]
- Crespo, J; Iruzubieta, P; Fernández Rodríguez, CM. The liver as a thermometer of cardiometabolic health: time to prioritize MASLD in Global Health Policy. Rev Esp Enferm Dig 2025. [Google Scholar] [CrossRef]
- Acharya, C; Bajaj, JS. Chronic Liver Diseases and the Microbiome-Translating Our Knowledge of Gut Microbiota to Management of Chronic Liver Disease. Gastroenterology 2021, 160(2), 556–72. [Google Scholar] [CrossRef]
- Philips, CA; Augustine, P. Gut Barrier and Microbiota in Cirrhosis. J Clin Exp Hepatol 2022, 12(2), 625–38. [Google Scholar] [CrossRef] [PubMed]
- Trebicka, J; Macnaughtan, J; Schnabl, B; Shawcross, DL; Bajaj, JS. The microbiota in cirrhosis and its role in hepatic decompensation. J Hepatol 2021, 75 Suppl 1(Suppl 1), S67–s81. [Google Scholar] [CrossRef]
- Rose, G. Sick individuals and sick populations. Int J Epidemiol 1985, 14(1), 32–8. [Google Scholar] [CrossRef]
- Iruzubieta, P; de Vega, T; Crespo, J. Overlooked determinants and unequal outcomes: rethinking metabolic dysfunction-associated steatotic liver disease beyond the biomedical model. Lancet Gastroenterol Hepatol 2025, 10(12), 1132–42. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-González, C; Alonso-Peña, M; Argos Vélez, P; Crespo, J; Iruzubieta, P. Unraveling MASLD: The Role of Gut Microbiota, Dietary Modulation, and AI-Driven Lifestyle Interventions. Nutrients 2025, 17(9). [Google Scholar] [CrossRef]
- Adolph, TE; Zhang, J. Diet fuelling inflammatory bowel diseases: preclinical and clinical concepts. Gut 2022, 71(12), 2574–86. [Google Scholar] [CrossRef]
- Mukherjee, A; Breselge, S; Dimidi, E; Marco, ML; Cotter, PD. Fermented foods and gastrointestinal health: underlying mechanisms. Nat Rev Gastroenterol Hepatol 2024, 21(4), 248–66. [Google Scholar] [CrossRef]
- Almeida, JI; Tenreiro, MF; Martinez-Santamaria, L; Guerrero-Aspizua, S; Gisbert, JP; Alves, PM; et al. Hallmarks of the human intestinal microbiome on liver maturation and function. J Hepatol. 2022, 76(3), 694–725. [Google Scholar] [CrossRef] [PubMed]
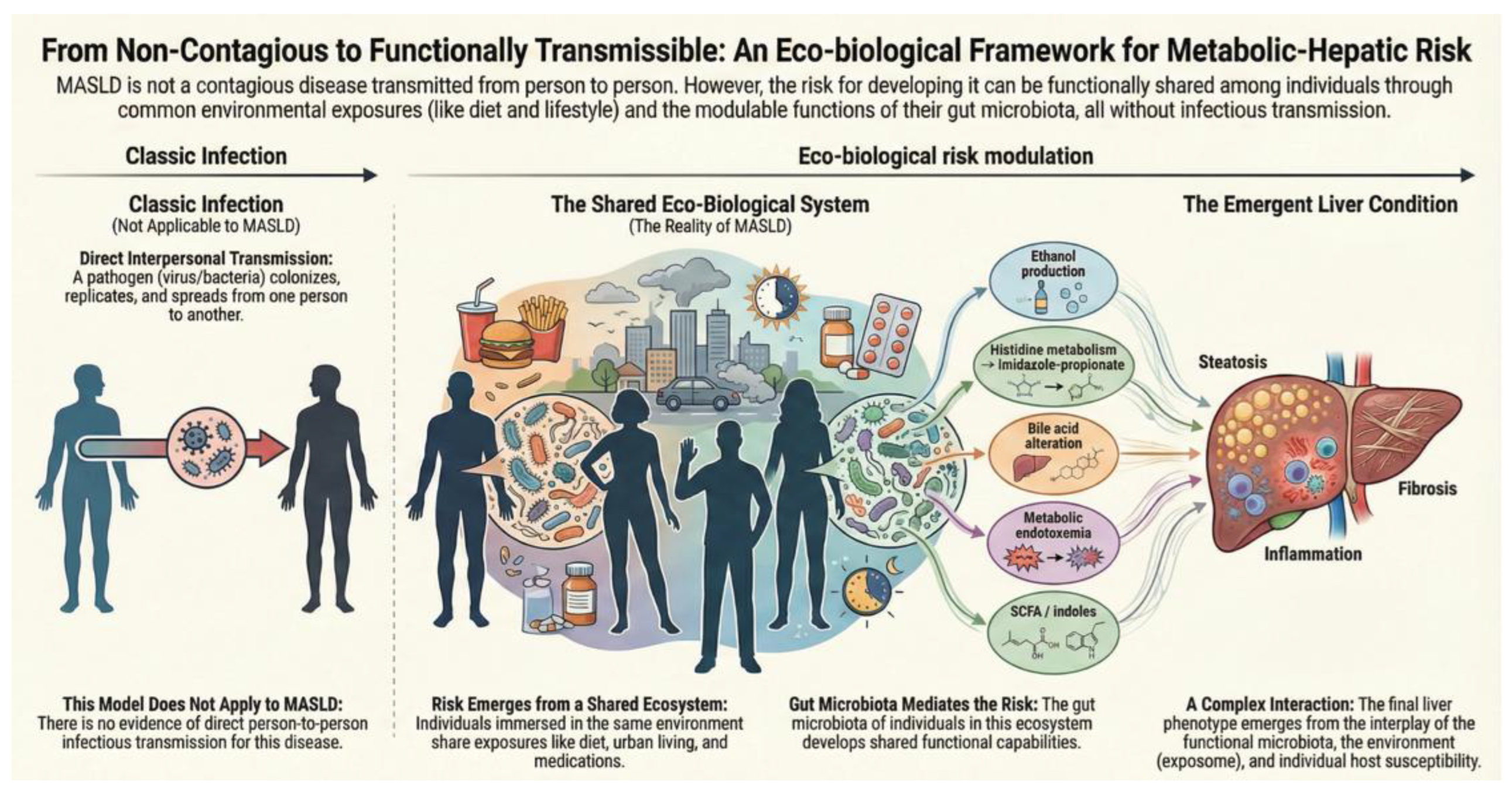


| Dimension | Infectious contagiousness | Functional transmissibility of metabolic–hepatic risk |
|---|---|---|
| Transferred biological unit | Identifiable pathogen (virus, bacterium, parasite) | Metabolic functions, microbial consortia, metabolites |
| Primary mechanism | Infection, replication, and dissemination | Immunometabolic and ecological modulation |
| Need for stable colonization | Yes, essential requirement | Not necessarily; can be transient or functional |
| Dependence on host context |
Limited | Very high (diet, exposome, genetics, metabolic state) |
| Human epidemiological evidence in MASLD |
Non-existent | Indirect, functional, and contextual |
| Experimental evidence | Not available | Robust (gnotobiotic models, FMT, microbial functions) |
| Reversibility of the phenomenon | Low | High and environment-dependent |
| Risk of interpersonal transmission | Yes | No |
| Clinical implications | Isolation, contagion control | Population prevention, ecological intervention |
| Public health relevance | Not applicable to MASLD | High |
| Dominant microbial function | Main metabolite/pathway | Metabolic–hepatic effect |
Type of evidence |
Representative references |
|---|---|---|---|---|
| Endogenous ethanol production | Ethanol | Hepatic steatosis and inflammation | Experimental | [16] |
| Dysbiotic fermentation | ↓ SCFA / ↑ alcohols | Progression to MASH | Experimental | [17] |
| Histidine metabolism |
Imidazole propionate | Insulin resistance, metabolic dysfunction | Human + experimental | [18] |
| Tryptophan metabolism | Indole-3-propionate | Cardiometabolic protection | Population-based | [19] |
| Bile acid metabolism | Secondary bile acids | Inflammation, FXR/TGR5 signaling | Mixed | [20] |
| Metabolic endotoxemia | LPS | Hepatic immune activation |
Human | [21] |
| Acetate-driven reprogramming | Systemic acetate | Alcohol-like dysbiosis | Experimental | [22] |
| Level of evidence | Study design | What it shows | What it does not show |
|---|---|---|---|
| Experimental | Gnotobiotic models, animal FMT |
Functional causality | Direct population relevance |
| Translational | Human FMT | Metabolic modifiability | Natural transmission |
| Observational | Clinical cohorts | Consistent association | Direct causality |
| Multi-omics | Metagenomics + metabolomics |
Biologically relevant function | Unique taxonomic origin |
| Epidemiological | Cohabitation, households |
Environmental convergence | Contagiousness |
| Interventional | Diet, prebiotics | Functional reversibility | Definitive cure |
| Level | Key implications |
|---|---|
| Individual | MASLD is not transmissible between people |
| Clinical | The microbiota enables risk stratification and risk modulation |
| Preventive | Early dietary and ecological interventions |
| Population | Food environments as risk modulators |
| Health-policy | Prioritize MASLD as an indicator of cardiometabolic health |
| Ethical–social | Shift focus from individual stigma to structural determinants |
| Research | Functional targets > taxonomic targets |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



