Submitted:
25 December 2025
Posted:
29 December 2025
You are already at the latest version
Abstract
Hypoxic pulmonary vasoconstriction (HPV) is a rapid and reversible constrictor response of the pulmonary vasculature, and especially its small muscular precapillary arteries, which is initiated by episodes of local alveolar hypoxia. Acting as a protective homeostatic vasomotor mechanism, HPV enables maximal gas exchange by diverting blood from poorly ventilated alveoli in to those rich in oxygen, thereby optimizing oxygen uptake and the ventilation-perfusion (V/Q) ratio so as to maintain the arterial oxygen partial pressure (PaO2) within the physiological range. HPV is an intrinsic mechanism of pulmonary artery smooth muscle cells (PASMC), and requires an O2 sensor which acts through mediator(s) to trigger effector mechanisms within these cells to evoke constriction. Whereas HPV effector mechanisms are reasonably well-defined, the nature of the O2 sensor and mediators remain in dispute. The three most comprehensive models of O2 sensing in HPV share a focus on the concept that hypoxia activates effector mechanisms by inducing a change in the PASMC cytoplasmic redox state. According to the Redox Theory, first proposed by Kenneth Weir and Stephen Archer in 1995, hypoxia inhibits mitochondrial production of reactive oxygen species (ROS), thereby causing the cytoplasm to become more reduced. This inhibits ongoing vasorelaxation maintained by the opening of voltage-gated K+ channels. In contrast, according to the Mitochondrial ROS hypothesis, introduced by Paul Schumacker and Naveen Chandel in 2001, hypoxia increases mitochondrial ROS production, causing an oxidizing shift in the cytoplasmic redox poise which activate several vasoconstricting pathways. In a third scenario, developed by Michael Wolin and Sachin Gupte, hypoxia evokes contraction by causing a fall in H2O2 production by NADPH oxidase, and by activating the pentose phosphate pathway. These effects inhibit basal vasorelaxation maintained by the guanylate cyclase and protein kinase G and also stimulate vasoconstricting mechanisms. In this comprehensive review, we summarize the key studies contributing to the development of these proposals and then subject the evidence supporting them to critical appraisal, based in part on how well they accord with the wider literature and recent developments in our understanding of how cells shape and deploy redox mechanisms in order to regulate cell function.
Keywords:
O2 sensing
; hypoxia
; hypoxic pulmonary vasoconstriction
; KV channel
; mitochondria
; NADPH oxidase
; reactive oxygen species
; redox
; hydrogen peroxide
; NADH
Section 1: Introduction; Definition and Description of HPV
1.1. Basic Properties of HPV
Hypoxic pulmonary vasoconstriction (HPV) is a rapid and reversible constrictor response of the pulmonary vasculature, and especially its small muscular precapillary arteries, to episodes of local alveolar hypoxia [1,2,3]. Acting as a protective vasomotor mechanism which enables maximal gas exchange by diverting blood from poorly ventilated alveoli in to those rich in oxygen content on a breath-to-breath basis, HPV optimizes oxygen uptake and the ventilation to perfusion (V/Q) ratio to maintain the arterial oxygen partial pressure (PO2) within the physiological range [4]. In parallel with HPV, acute hypoxia provokes systemic arterial vasodilation, thereby promoting oxygen delivery to body tissues by increasing blood flow [5].
Studies in isolated perfused whole lungs or lung lobes from a range of species indicate that HPV begins to develop when PO2 falls below ~80 Torr, becomes half-maximal at ~50 Torr, and reaches its maximum amplitude at ~20 Torr [3]. Depending on the experimental model and conditions used, HPV generally manifests as a rapidly developing and sustained increase in pulmonary artery pressure (PAP) or pulmonary vascular constriction which in many studies exhibits a biphasic profile in which an initial transient development of force (Phase 1) is superimposed upon or followed by a slow and progressive increase in tension (Phase 2). The biphasic time course is typically observed when more severe levels of hypoxia are used to evoke the response [3]. In humans, HPV evoked when end-tidal PO2 (a measure of the alveolar PO2) was halved demonstrated an initial rapid increase in pulmonary vascular resistance which reached a steady state within minutes. This was followed by a second slower increase that began after 30 – 60 minutes and plateaued after ~3 hours [6,7]. HPV is a function of the PO2 in both the alveoli and the blood perfusing the pulmonary arteries (PA), with the former being of greater importance [8]. It does not require, but is modulated by, the autonomic nervous system and diverse vasoactive factors [9] [3].
The mechanisms responsible for the two phases of HPV appear to be, at least to some extent, different [10,11]. Phase 1 is mediated by an increase in the cytoplasmic Ca2+ concentration ([Ca2+]cyt) due to a rapid decrease in K+ channel activity leading to membrane depolarization and opening of L-type voltage-gated Ca2+ channels, [10,12], as well as release of Ca2+ from the sarcoplasmic reticulum and the resulting stimulation of store operated Ca2+ entry (SOCE) [10,13,14] in pulmonary artery smooth muscle cells (PASMC). Most studies of HPV in isolated perfused lungs or PA have used short periods of hypoxia (≤ 15 minutes), suggesting that their conclusions apply mainly to Phase 1. Studies of O2 sensing in isolated or cultured PASMC have also typically employed short hypoxic challenges, but the extent to which their results are representative of one or the other phase of HPV in more intact systems is unclear.
Phase 2 HPV is less well characterized but probably involves multiple pathways contributing to an increase in [Ca2+]cyt as well as rho kinase-mediated Ca2+ sensitization [15]. Unlike phase 1, sustained HPV has generally (but not always) been found to be endothelium-dependent [16]. The evidence for an involvement of membrane depolarization in Phase 2 HPV is mixed [10,14,17,18]. Both phases are reversible, but if global hypoxia lasts longer than 24 hours, persistent HPV of the entire pulmonary circulation causes a sustained increase in PAP. This, together with phenotypical, biochemical and functional changes in each pulmonary vascular cell type, gradually leads to pulmonary vascular remodeling, which can be irreversible.
1.2. Proposed Models of O2 Sensing in HPV
HPV is conceptualized as requiring an O2 sensor which is coupled through mediator(s) to effector mechanisms that evoke constriction by raising PASMC [Ca2+]cyt and/or causing Ca2+ sensitization [19] . It is generally (but not universally [20]) thought that all of these components of the response reside within the PASMC, although extrinsic influences (e.g. endothelial factors) can modulate HPV [3].
This review focuses on describing current theories which propose that the mitochondria and/or NADPH oxidase (Nox) are O2 sensors which, acting through effects on reactive oxygen species (ROS) or cellular redox couples as mediators, modify the function of contractile effectors to cause HPV. We do not cover either the extensive set of known or putative HPV effectors, which have been described in detail elsewhere (e.g. [3] [21] [22] (except to discuss those which are proposed to be regulated by redox mechanisms where appropriate), or the role of ROS/redox mechanisms in pulmonary hypertension resulting from chronic pulmonary hypoxia (see e.g. [23,24,25,26,27,28,29,30,31] for reviews of this subject).
The term ‘reactive oxygen species’ has been criticized in light of evidence that the various types of ROS exert their own specific actions, typically on different sets of biomolecules, with ‘oxidants’ suggested to be a better alternative when the relevant oxidizing species has not been identified [32]. However, to be consistent with other reviews of O2 sensing in HPV [21,33,34,35], and because the term ‘oxidants’ also encompasses reactive sulfur species, we prefer to refer to changes in superoxide/H2O2 production as increases or decreases in ROS, rather than oxidants.
There are currently three well-established proposals for O2 sensing in PASMC in which ROS play pivotal roles. The bulk of this review is devoted to a description of the investigations which contributed to the development of these hypotheses, and an evaluation of what we feel are their strengths and weaknesses.
- According to the Redox theory, introduced by Kenneth Weir and Stephen Archer in 1995 [36], hypoxia decreases the production of ROS by the mitochondrial electron transport chain (ETC), leading to a fall in [ROS] and/or the reduction of redox couples in the cytoplasm. This evokes the closure of voltage gated K+ (KV) channels which are activated by basal ROS production under normoxic conditions, thus causing membrane depolarization, the opening of voltage-gated Ca2+ channels (VGCC), and therefore contraction. Although the Redox Theory stresses the importance of KV channels, there are also other effectors which could potentially respond to a fall in ROS or a reduction of cytoplasmic redox couples in such a way as to cause HPV. For example, there is evidence that the reduction of Cys42 on PKG1a (protein kinase G a1), which may exert a tonic vasorelaxing influence on PASMC, diminishes its activity [37].
- A proposal which we will refer to as the Mitochondrial ROS hypothesis, developed by Paul Schumacker, Naveen Chandal and colleagues and first described in 2001 [38], proposes that hypoxia causes an increase in mitochondrial ROS production, leading to a higher [ROS], and/or oxidation of redox couples, in the cytoplasm. This hypoxia-induced rise in cytoplasmic ROS may be supplemented by a PKCϵ-mediated stimulation of Nox which is triggered by the mitochondrial ROS [39]. The rise in cytoplasmic [ROS] might evoke contraction through multiple effector pathways, potentially including Ca2+ release from the sarcoplasmic reticulum, an increase in store operated Ca2+ influx, and RhoA/ Rho kinase-mediated Ca2+ sensitization [13,14,15].
- The processes responsible for O2 sensing and HPV have also been the subject of an extensive series of papers by the laboratories of Michael Wolin and Sachin Gupte (see [40] for a review ). In agreement with the Redox theory, these authors propose that hypoxia causes contraction by removing a normoxic vasodilating influence, although this is seen as being maintained largely by oxidation-induced activation of soluble guanylate cyclase and protein kinase G (PKGa1) rather than by the opening of KV channels. In addition, they have presented evidence that hypoxia activates the pentose phosphate pathway, thereby increasing the production of NADPH, and that this contributes to the inhibition of PKGa1 and also activates other Ca2+-dependent and -independent contractile mechanisms.
Two additional O2 sensing mechanisms have been suggested to bring about HPV. According to the AMP kinase (AMPK) hypothesis, introduced by Mark Evans and co-workers[41] [42], HPV is caused by a small but meaningful inhibition of mitochondrial respiration which results in an increase in the cellular [AMP]/[ATP] ratio. This activates AMPK, which causes PASMC contraction through several mechanisms, the best defined of which is phosphorylation -induced inhibition of KV1.5 channels, leading to membrane depolarization and Ca2+ influx [43,44]. Furthermore, Kenneth Olson’s laboratory [45,46,47,48] has suggested that O2-dependent metabolism of hydrogen sulfide (H2S; aka sulfide) constitutes an O2 sensor in many tissues, including pulmonary arteries and carotid body chemoreceptor cells (CBCC). According to this proposal, hypoxia suppresses the oxidation of sulfide, causing increased cellular concentrations of sulfide and/or reactive sulfur species. These activate effector mechanisms which cause, for example, HPV. Neither of these hypotheses as proposed by its originators incorporates the involvement of ROS. However, a possible role for ROS in both schemes is possible, as discussed in Sections 6.2 and 6.3.
1.3. Normoxia, Physoxia and Hypoxia
The O2 concentrations (18-21 % O2) which are used almost invariably in physiological experiments to simulate ‘normoxia’ are substantially higher than ‘physoxic’ concentrations (i.e. those experienced under baseline conditions by cells in vivo) [49]. There is a growing literature [50,51,52] attesting as to how this can distort many aspects of cell phenotype. Additional problems inherent in the routine use of effectively hyperoxic conditions in studies of acute O2 sensing have been discussed in excellent reviews by Alva et al [53] and Olson [47], the former of which also presents an authoritative and perceptive discussion of many of the other issues around the role of ROS in O2 sensing which we cover in this paper. The intra-alveolar PO2 is ~13.3%, and that in arterial blood averages 12.3%. Measurements of lung tissue PO2 in anesthetized animals breathing room air have yielded lower values. For example, O2 electrodes placed in peri-bronchial lymph nodes in dogs reported PO2 levels of ~53Torr, [54] and ~38 Torr [55], and an electron paramagnetic resonance sensor inserted into lung tissue of rats also detected a PO2 of ~38 Torr, which fell to ~26 Torr in animals breathing 10% O2 [56]. It is not clear why the PO2 in lung tissue is so much lower than it is in the alveoli, but one can speculate that this could be due to the influence of the PO2 in PA, which enter the lung containing blood with a mixed venous PO2 of ~40 Torr. Importantly, it was shown in anesthetized dogs that the mixed venous PO2 exerts an marked influence on HPV [8], presumably because the blood within a substantial fraction of the PA responsible for HPV has not yet been fully oxygenated and is affecting the PO2 within the vascular wall. The effective PO2 stimulus for HPV (PSO2) in this study was calculated to be determined by the alveolar and mixed venous O2 concentrations according to the equation: PSO2 = (PAO20.59 x PmvO2 0.41). A similar weighting was observed in isolated perfused rat lung [57]. In this case, the PSO2 in vivo when breathing room air would be roughly 70 Torr, suggesting that this might be an appropriate PO2 to use in experiments to simulate physoxia under normoxic conditions when conducting studies of HPV. The mitochondria and Noxs, which are likely to be the two main sources of superoxide/hyperoxide in PASMC, produce ROS in a manner which is linearly (mitochondria[58]) or hyperbolically (Nox [53,59]) dependent on the PO2. For example, Nox4, which is thought to be constitutively active and therefore could be an important source of H2O2 in PASMC under basal conditions [60], has a KmO2 of ~136 Torr [59]. Thus, PASMC and other cells in preparations used to study O2 sensing in HPV are likely to have been undergoing a degree of oxidative stress under experimental ‘normoxic’ conditions.
A related issue is that the O2 concentration experienced by cultured cells in standard O2-controlled incubators is usually supra-physoxic when the PO2 is set to 18%. Even so, under certain conditions (e.g. high cell density) the PO2 in the medium can be much lower than the physoxic level due to cellular O2 consumption [61]. Cellular respiration can have an especially marked effect on the pericellular PO2 in cells in culture [62], an effect which has been shown to be inhibited by block of the electron transport chain (ETC] [63]. This is a concern because experimental interventions used to study hypoxia often decrease the activity of the ETC, in which case they may also increase the pericellular PO2 and therefore diminish the degree of hypoxia experienced by cells. Unfortunately, the extent to which these factors might have had an impact on the results of the investigations we will discuss is unclear.
The question of the range of levels of hypoxia which are appropriate for studying HPV has probably not been taken as seriously as it should have been in some papers. In many cases, for example, the level of hypoxia is defined as the % O2 content in the gas mixture used to bubble the solution or in the incubator housing the cell chamber, with no information provided about the PO2 in the solution. In an influential review of HPV, Moudgil et al [19] stressed the necessity of using physiological levels of hypoxia in studies of HPV, suggesting that the level of hypoxia at the summit of Mount Everest (8488 meters; alveolar and arterial PO2 of ~43Torr recorded during the Operation Everest II expedition [64] might be an appropriate guideline, since the PO2 in the small PA which are the main site of HPV should be very close to that in the alveoli.
On the other hand, if one takes into account the influence of the mixed venous PO2 on HPV as described above, and factors in the resting PmvO2 (22 Torr) observed in another study by the Everest II expedition team [65] at a simulated altitude of 8488 meters, the PSO2 for HPV at this height would be ~33 Torr. Moreover, other studies have indicated that elevation has a larger effect on the arterial and alveolar PO2; for example, PAO2 and PmvO2 values obtained at 8488 meters during an earlier expedition to Everest were 35 and 21 Torr, respectively [66], and Grocott et al [67] obtained arterial PO2 values of 48, 42, 34 and 24 Torr at elevations of 5300, 6400, 7100 and 8400 meters, with an alveolar PO2 of 30 Torr at 8400 meters, although the corresponding PmvO2 levels were not reported. Taking into account also that even moderate levels of activity further lower the PmvO2 at altitude [65], it can be argued that lower levels of hypoxia, perhaps down those approximating the PO2 range at which HPV reaches its maximum amplitude (~20 Torr) are also suitable for studying physiologically relevant O2 sensing mechanisms in HPV. Nonetheless, since the mechanisms responsible for O2 sensing in PA may well vary according to the level of hypoxia, as is thought to occur in chemoreceptor cells of the carotid body (CBCC) [68], ideally the mechanisms of O2 sensing in HPV should be assessed over a range of low PO2 levels. This has not been done in any organized way.
Section 2: Reactive Oxygen Species as Signaling Molecules
2.1. ROS Definition and Function
ROS include oxygen radicals such as superoxide (O2-), hydroxyl (·OH) and peroxyl (RO2·), as well as oxidants such as hydrogen peroxide (H2O2) and hypochlorous acid (HClO). These are formed by cell metabolism and can oxidize biological molecules, although their reactivity varies between species [69,70]. O2-, the first species formed by single- or multi-enzyme reactions through the single-electron reduction of water, rapidly dismutates to H2O2, a process which occurs spontaneously and is also catalyzed by superoxide dismutase (SOD). ·OH , which can be formed by the reaction of H2O2 with Fe2+ (Fenton reaction) or of water with Fe3+ [71], is extremely reactive and is scavenged immediately upon its production. H2O2 is relatively stable and is able to cross membranes via aquaporins [72], properties which allow it to act as a signaling molecule. O2· can also react with nitric oxide (NO) to form the reactive nitrogen species peroxynitrite (ONOO-), which has a signaling function [73]. The two cellular sources of ROS which have received the most attention with regard to O2 sensing and HPV are the mitochondrial electron transport chain (ETC) and Nox.
ROS can cause oxidative modifications of nucleic acids, carbohydrates, lipids and protein. Their excessive production therefore results in deleterious effects on cells, a situation referred to as oxidative stress. However, cells use smaller spatiotemporally controlled increases in H2O2 to regulate their normal function and initiate adaptive responses (oxidative eustress) [74], with HPV being a possible example.
2.2. H2O2 Signaling
H2O2 signaling depends on its ability to react with thiolates (S-), the anionic form of cysteine sulfhydryl (-SH groups), leading to modifications of protein structure and therefore function (Figure 1). Since the pKa value of the thiol in free cysteine is ~8.5, the fraction of cysteines in the reactive thiolate form should be small at the physiological cytoplasmic pH (e.g. ~7.3 in PASMC, [75]). However, the pKa of cysteine thiols can be lowered by their proximity to positively charged amino acids [76] and by other factors related to their local environment within the protein structure [77,78]. Thus, a fraction of cysteine residues is able to react with H2O2 and other oxidizing species including reactive nitrogen or sulfur species, which cause S-nitrosation and S-sulfhydration, respectively. Reactive cysteines can also undergo S-thiolation, most importantly by oxidized glutathione (GSSG), causing s-glutathionylation. It is thought that disulfide bond formation and s-glutathionylation are the most important oxidative modifications for H2O2 signaling [79]. These modifications can alter three-dimensional protein structure, leading to protein translocation [80,81] or substrate targeting [80,82], ultimately causing rapid functional changes that can induce or fine-tune cell signaling during acute responses.
In order to minimize its potential damaging effects on cells, basal cytoplasmic concentrations of H2O2 are maintained in the low nanomolar range by the thioredoxin (Trx) and glutaredoxin (Grx) antioxidant systems, acting in concert with peroxiredoxins (Prxs), and Grx-dependent glutathione peroxidases (Gpx) (Figure 2) [85,86]. These systems, which are also largely responsible for terminating redox signaling by reversing oxidative thiol modifications, ultimately require the oxidation of NADPH, which is regenerated from NADP+ by the pentose phosphate pathway and other enzymatic mechanisms [87]. The Prx family, comprising six isoforms which are differentially expressed in the various cell compartments, is generally thought to be particularly important for the removal of H2O2; it was estimated, for example, that 78% and 21% of H2O2 scavenging in cells is dependent on Prx and Gpx, respectively [88]. Intriguingly, however, a recent study using the novel NADPH sensor NAPStar3b has shown that that the glutathione system plays the predominant role in antioxidant defence in HEK293 cells [89]. H2O2 is also scavenged by the enzyme catalase, which is located mainly but not exclusively in peroxisomes. It is believed that the antioxidant effect of catalase, which does not require cofactors, is more important at higher H2O2 concentrations, at which the activities of Prx and Gpx may be limited by the necessity for their recycling by Trx and Grx [90].
The extent to which the direct reactions between H2O2 and protein thiolates described above can account for redox signaling has been questioned on kinetic grounds. Scavenging of H2O2 by Prx is rapid, with a 2nd order rate constant inthe range of 105–108 M-1 [88,91], whereas H2O2 reacts much more slowly with cysteine thiolates (k ≈ 101 - 102 M-1).
Despite these constraints, there is evidence that cells can use H2O2 as a highly specific signaling molecule if it is formed very close to its target protein (e.g. [92]). Longer range specific targeting can also be achieved by way of the formation of thiol peroxidase-based redox relays (Figure 3) in which Prx (or Gpx ) is used to convey oxidizing equivalents from H2O2 to a target protein, rather than sending them down an antioxidant pathway to be consumed by NADPH [93,94]. In a one-step relay, once oxidized by H2O2, Prx oxidizes the target protein rather than Trx. In a two-step relay, Prx oxidizes Trx, which then oxidizes the target protein rather than oxidizing Trx reductase (TrxR). The fact that Prx is able to act in both antioxidant and peroxidase capacities can reconcile the apparent paradox that H2O2, despite being subject to swift and efficient scavenging by cellular antioxidant pathways, can initiate the rapid and reversible oxidization of relatively unreactive thiols. The initial reaction between H2O2 and Prxs is very fast, with rate constants in the range of 107-108 M−1 s−1 for all five isoforms; similar rate constants are also seen for the reaction of Prxs and peroxynitrite [95,96]. The factors which determine the balance between the antioxidant and oxidant/signaling activities of Prxs are still being worked out. One possibility described in an elegant study by Portillo-Ledesma et al [95] is that the antioxidant capacity of Prx becomes saturated at a certain threshold concentration of oxidant (e.g. H2O2), above which it can oxidize target proteins. They showed that this threshold is particularly low (4 nM H2O2) for the cytoplasmic isoform Prx2, suggesting that it can oxidize target protein at relatively low H2O2 concentrations while the antioxidant activity of Prx isoforms with higher thresholds (e.g. Prx1) continues. The activity of Prx is also regulated by several post-translational modifications [97].
The specificity of Prx dependent redox relays is fostered by the rapid association of Prxs, once they are oxidized, with discrete sets of target proteins. This occurs through the formation of disulfide bonds at its active site, which happens during target protein oxidation. For example, in experiments carried out in HEK293T cells, Px1 and Prx2, which are both expressed in the cytoplasm, were shown to associate with 735 and 165 proteins, respectively [98]. Of these, only 53 bound to both isoforms. This highlights the specificity of these interactions, which was shown to depend on the local amino acid sequence around the target cysteine. Of the 1233 proteins found to bind to one or more of the five Prx isoforms expressed in these cells, ~80% had previously been identified [76] as harboring redox-sensitive cysteines, suggesting that Prx-dependent oxidative modifications of protein thiols probably play a widespread role in H2O2-signaling. It has also been proposed that redox relays which utilize Gpx and GSSG to cause S-glutathionylation of target cysteines are important in the redox regulation of many proteins, especially within mitochondria [99].
Section 3: Cellular Mechanisms of ROS Production
Mitochondrial complexes together with a microsomal ETC, the Noxs, and other oxidases (e.g., glucose oxidase, xanthine oxidase), peroxisomes, and cyclooxygenases comprise the main biological machinery that directly produces or is causally involved in oxidant formation. Of these various sources of ROS, research on HPV has focused only on the potential roles of mitochondria and Nox.
3.1. Mitochondrial Regulation of Cytoplasmic H2O2
Although mitochondria can release H2O2 into the cytoplasm, e.g. during ischemia-reperfusion [100] and inflammation [101], they can also act as an H2O2 sink [58,102,103,104]. Thus, their net effect on cytoplasmic H2O2 levels depends on the balance between these two processes.
3.1.1. Mitochondrial ROS Production
The mechanisms responsible for mitochondrial ROS production have been described by numerous excellent reviews (e.g. [58,105,106,107,108,109]. Here, we will focus mainly on aspects relevant to the possible effects of hypoxia.
3.1.1.1. Oxidative Phosphorylation
Within mitochondria, the energy released by the metabolic oxidation of glucose, fatty acids and amino acids is captured in the form of a highly negative redox potential by the transfer of electrons to NAD+ and, via succinate, to FAD, producing NADH and FADH2, respectively. These electrons are transferred from NADH/FADH2 into the electron transport chain (ETC), consisting of 4 protein complexes with successively more positive redox potentials (Figure 4), and are then used to reduce O2 to H2O. The energy released as the electrons flow down this redox potential gradient is used by complexes I,III and IV to pump protons outwardly across the inner mitochondrial membrane (IMM). The energy stored in the resulting transmembrane electrochemical gradient (DP or protonmotive force) is used by ATP synthase to phosphorylate ADP, generating ATP which provides energy for cellular processes. DP has two components: DΨm, the membrane potential across the IMM, and DpH, the pH gradient across the IMM. It has recently been proposed that DΨm is generated, not only by the proton gradient across the IMM, but also by a Na+ gradient which also exists across this membrane due to the operation of Na+/H+ exchange, which is mediated by complex I [110]. The process by which the energy lost by electrons as they are transferred from metabolic substrates to O2 through the ETC is coupled via DP to the production of ATP is termed oxidative phosphorylation.
3.1.1.2. Electron Flow Through the ETC
Electrons from NADH and FADH2 enter the ETC at complexes I and II, respectively, which use them to reduce coenzyme Q (CoQ; ubiquinone), a small-molecule constituent of the IMM, yielding CoQH2 (ubiquinol). CoQ can also be reduced to CoQH2 by electrons transferred during the oxidation of a number of substrates, including dihydroorotate, proline, fatty acids, glycerol 3- phosphate and H2S. Each substrate is oxidized by a specific dehydrogenase (e.g. proline dehydrogenase reduces CoQ by oxidizing proline to pyrroline-5-carboxylate).
CoQH2 binds to complex III (aka Q-cytochrome c oxidoreductase or cytochrome bc1 complex), at the Qo site, which is close to the outer side of the IMM. One electron from CoQH2 bound at this site is transferred to the nearby Reiske iron-sulfur cluster, (and thence, in succession, to cytochrome c1, cytochrome c, and complex IV, which utilizes pairs of electrons to reduce O2 to water). This releases two protons to the intermembrane space (IMS). The other electron is transferred to the bL (b566) heme group, which is also on the outer side of the IMM. From there, it moves to the bH (b562) heme group, which is closer to the matrix side of the IMM. This movement is driven by the more positive redox potential of bH compared to bL, but is opposed by DΨm; thus a high DΨm will retard this electron transfer. A second ubiquinone binding site, Qi, is close to bH, and the electron on bH is transferred to UQ bound to this site, forming ubisemiquinone (CoQ.-). When the next molecule of CoQH2 is oxidized at Qo, the electron that it donates to bL and bH reduces the ubisemiquinone at Qi to CoQH2, which returns to the CoQH2 pool in the IMM. This causes the uptake of two protons from the matrix. The net result of this process, referred to as the Q cycle, is that for each CoQH2 oxidized, two electrons flow via cytochrome c to complex IV, two protons are taken up from the matrix, and four protons enter the IMS, helping to generate DP [111,112].
3.1.1.3. Factors Governing Mitochondrial ROS Production
There are more than a dozen sites within the mitochondria at which electrons flowing into or through the ETC can react with O2 to form superoxide; some of the sites can also produce H2O2 (see Figure 5, and the legend to Figure for a list of the sites and a definition of their abbreviations) Many of these sites are locations at which dehydrogenases transfer electrons from metabolic substrates to NAD+ or CoQ. Superoxide produced is rapidly reduced to H2O2 by MnSOD (SOD2) in the mitochondrial matrix or Cu/ZnSOD (SOD1) in the IMS. Nevertheless, superoxide evidently exists long enough to exert local effects, for example oxidizing the Fe-S cluster of mitochondrial aconitase in the matrix, leading to its loss of catalytic function [113]. The ratio of mitochondrial superoxide + H2O2 production to O2 consumption is in the range of 0.1 to 0.5% [114].
The rate of superoxide/H202 production at each site is a function of the O2 concentration ([O2]), the concentration of the site in the reduced state (X*¯), and a second order rate constant (k):
where X*¯ is the product of the concentration of the site and the fraction of the site which is in the reduced state [58,106]. Overall mitochondrial ROS production is the sum of that produced at each site. Whereas some sites are incorporated into proteins as prosthetic groups (e.g. flavin mononucleotide in the IF site on complex I), IQ in complex I and IIIQ in complex III serve as sites within the complexes at which ubisemiquinone is formed and can donate electrons to O2 to form superoxide or H2O2. The mitochondrial ROS-producing sites fall into two isopotential groups defined by the redox couple involved in ROS production. The NADH-linked sites (the a-keto ketoacid dehydrogenase complexes and Complex IF) operate at a redox potential of ~ -280 mV, whereas the coenzyme-Q linked sites (the CoQ linked dehydrogenases, complex ICoQ, complex IIF, and complex IIICoQ) [115] operate at a potential of ~+20 mV [109]. The relative amounts of ROS produced by each site vary between cell types and under different conditions [109,114,116]. However, complex ICoQ, complex IIF , KGDH, PDH, OADH and IIICoQ are thought to be important mitochondrial ROS sources [58,117], with IIICoQ having the highest capacity for ROS production, at least in skeletal muscle [109].
(dROS/dt) = ([O2])(X*¯)(k)
Figure 5.
Mitochondrial ROS production. There are more than a dozen sites (e.g. IQ, GCoQ) at which electrons can ‘leak out’ of the ETC and react with O2 to form superoxide and/or H2O2. In addition to the classical sites of ROS production at complex I (IF and IQ), complex II (IIF) and complex III (IICoQ), ROS are also produced by dehydrogenases associated with the inner mitochondrial membrane during the oxidation of metabolites. Several of these sites (IF and also PF, KF, BF, and OF which are at the ROS-producing loci of a-ketoacid dehydrogenase complexes) are in redox equilibrium with the mitochondrial NAD+: NADH couple. The other sites, which include IQ (also termed ICoQ), IIF, IIICoQ , GCoQ, EF, DCoQ, PCoQ and the ROS producing site on sulfide quinone oxidoreductase (SQOR) are in redox equilibrium with CoQ: CoQH2 couple in the inner mitochondrial membrane. The GCoQ, EF, DCoQ, PCoQ sites are on located on the dehydrogenases sn-glycerol-3-phosphate (sn-GPD), flavoprotein electron-transferring co-enzyme Q oxidoreductase (ETFQO), dihydroororate dehydrogenase (DHODH), and proline dehydrogenase (PRODH). The content of the figure and the names of the cites are based on references [118] and [58].
Figure 5.
Mitochondrial ROS production. There are more than a dozen sites (e.g. IQ, GCoQ) at which electrons can ‘leak out’ of the ETC and react with O2 to form superoxide and/or H2O2. In addition to the classical sites of ROS production at complex I (IF and IQ), complex II (IIF) and complex III (IICoQ), ROS are also produced by dehydrogenases associated with the inner mitochondrial membrane during the oxidation of metabolites. Several of these sites (IF and also PF, KF, BF, and OF which are at the ROS-producing loci of a-ketoacid dehydrogenase complexes) are in redox equilibrium with the mitochondrial NAD+: NADH couple. The other sites, which include IQ (also termed ICoQ), IIF, IIICoQ , GCoQ, EF, DCoQ, PCoQ and the ROS producing site on sulfide quinone oxidoreductase (SQOR) are in redox equilibrium with CoQ: CoQH2 couple in the inner mitochondrial membrane. The GCoQ, EF, DCoQ, PCoQ sites are on located on the dehydrogenases sn-glycerol-3-phosphate (sn-GPD), flavoprotein electron-transferring co-enzyme Q oxidoreductase (ETFQO), dihydroororate dehydrogenase (DHODH), and proline dehydrogenase (PRODH). The content of the figure and the names of the cites are based on references [118] and [58].
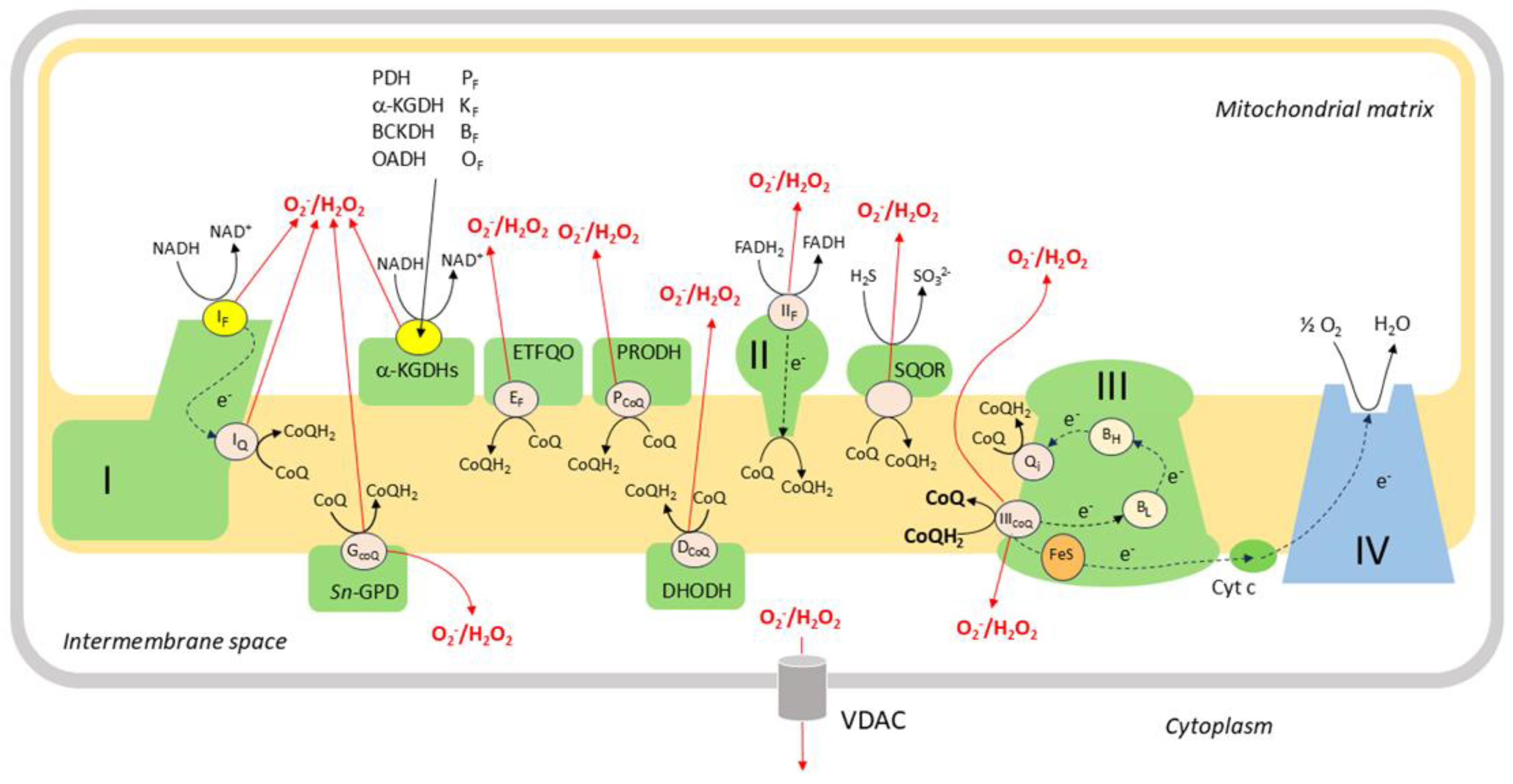
Whereas all of these sites release ROS into the mitochondrial matrix, a substantial fraction of ROS produced by Complex IIICoQ and GCoQ is also released into the IMS [109], from which H2O2 is able to diffuse into the cytoplasm through voltage-dependent anion channels (VDAC) in the outer mitochondrial membrane (OMM) [119,120]. H2O2 released into the matrix can also access the cytoplasm, as evidenced by observations that ROS released into the matrix by complex I can be detected in the cytoplasm [121] and in the medium around isolated mitochondria [115] and intact cells [122], but is to some extent consumed by intramitochondrial antioxidant mechanisms before it can do so. The ability of Complex IIICoQ to release ROS into the IMS, and also its position as the ‘bottleneck’ through which all electrons from the CoQ pool pass on their way to O2 [58], has led to suggestions that it is the main site of mitochondrial ROS production involved in regulating the redox balance of the cytoplasm [58,120,123].
The sites proposed to be responsible for hypoxia-induced changes in ROS production in PASMC (and CBCC, the other extensively-studied cell type which responds acutely to physiological hypoxia) include ICoQ [124,125]; IIF [126], and IIICoQ [38,127,128,129,130]. IQ can produce ROS during either forward electron transport (when the electron is coming from NADH via IF) or during reverse electron transport (RET), which occurs when the Q pool is highly reduced, causing a backflux of electrons from CoQH2 into complex I [131]. The mechanism by which the semiquinone is formed and donates an electron to O2 at IIICoQ is controversial. It is thought that superoxide/H2O2 is produced at this site via the reduction of O2 by ubisemiquinone, which exists transiently at IIICoQ after CoQH2 has lost one electron to the Reiske complex (referred to as the ‘semi-forward reaction’), or due to the backflow of an electron from bL (the ‘semi-reverse reaction’) [132,133,134]. Thus, superoxide production by IIICoQ is promoted by an increase in DΨm, which slows the loss of electrons from bL, [135] and by the drug antimycin, which causes an accumulation of electrons at bL and bH by preventing the binding of CoQ to Qi [133]. Note that IIICoQ is located at the site where CoQH2 binds to complex III, which is generally termed Qo.
Since ROS production by ICoQ and IIICoQ is linked to co-enzyme Q, the extent to which the CoQ pool is reduced is a key determinant of mitochondrial ROS production. Importantly, it is thought that ROS production at IIICoQ is maximal when the CoQ pool is partially but not fully reduced. [134,136,137].
A number of compounds which affect mitochondrial ROS production by binding to specific sites within the ETC are available. Of these, rotenone, myxothiazol, antimycin, cyanide and sodium azide have been used most widely to study the role of the mitochondria in HPV. Rotenone binds to IQ, thereby blocking the ETC at this point. Myxothiazol blocks the ETC at complex III by binding to IIICoQ. Antimycin binds to Qi in complex III, stopping electron flow by interrupting the Q cycle. Cyanide and azide, which binds to the Fea3- CuB binuclear center in complex IV [138], block the ETC by preventing the reduction of O2 to H2O2.
3.1.1.4. Mechanisms by Which Hypoxia May Increase Mitochondrial ROS Production
The ‘metabolic hypothesis’, initially developed to explain O2 sensing in CBCC [139], proposes that hypoxia, by causing a perturbation of the state of the ETC, generates a signal which engages with effector pathways to initiate appropriate cellular responses. According to the mitochondrial ROS hypothesis, a variant of this proposal, one way in which hypoxia can affect the ETC is that a decreased availability of O2 tends to slow the rate of its reaction with electrons at complex IV (aka cytochrome aa3, cytochrome c oxidase or Cox). This reaction, the rate-limiting process in mitochondrial electron transport, comprises a series of steps in which two pairs of electrons transferred from cytochrome c are used to reduce an O2 molecule to two H2O molecules. This reaction is irreversible, whereas the upstream series of redox reactions which couple the oxidation of NADH and FADH2 to the reduction of cytochrome c, are at near-equilibrium with each other. As a result, a decrease of the activity of Cox will cause all parts of the ETC upstream of complex IV, and also the NAD+:NADH and FAD:FADH2 redox couples, to become more reduced [140]. The consequent reduction of the CoQ pool leads to higher ROS production by Complex IIICoQ [132].
In accordance with this concept, levels of physiological hypoxia which initiate HPV in PASMC (and transmitter release by CBCC) cause an immediate increase in cellular NAD(P)H fluorescence which is probably due mainly to a rise in the mitochondrial NADH/NAD ratio. For example, half maximal increases in NAD(P)H fluorescence were observed at PO2 levels of 21 and 15 Torr in PASMC and CBCC, respectively [141] [142]. This implies that hypoxia is impeding the flow of electrons though the ETC (although as described below, this might not result in a commensurate inhibition of O2 consumption). Similarly, moderate hypoxia was shown to cause the reduction of cytochrome c in CBCC [143] and of CoQ and cytochromes c and aa3 in PASMC [129,144]. Hypoxia also alters the magnitude of DΨm in both types of cells (although, puzzlingly, in opposite directions [130,145,146]).
Notably, however, the reduction of O2 at Cox has an apparent P50 (i.e. the PO2 at which the activity of the complex, as reflected by cellular O2 consumption, is half maximal) of ~0.8 Torr in PASMC [144]. This is ~60 times lower than the PO2 level which half-maximally stimulates HPV. This raises the question of how mild to moderate levels of hypoxia which cause HPV could perturb the ETC, thereby causing a signaling effect (e.g. reduction of the CoQ pool and increased ROS production by Complex IIICoQ), even though they are too high to affect its rate-limiting step.
Several explanations have been advanced to explain this apparent discrepancy [3] [34] [141]. One is that the basal mitochondrial PO2 may be lower than that in the extracellular space (ECS) [147,148] and can therefore fall during moderate hypoxia to a level small enough to significantly limit the activity of Cox. However, the ECS to mitochondria PO2 gradient in PASMC would have to be very substantial for a fall in PO2 associated with moderate hypoxia to exert a meaningful effect on the activity of Cox [141].
Gnaiger and colleagues proposed another explanation for this conundrum, based on their finding that the apparent P50 for Cox is proportional to its activity [149]. According to their model, oxidase activity is controlled by the rate at which electrons are being fed into it by cytochrome c. This in turn depends on the relative expression of Cox compared to that of the upstream complexes which provide cytochrome c with electrons; in effect, the individual cytochrome c enzymes compete with each other for the pool of reduced cytochrome c. Thus, if the expression of Cox relative to that of complexes 1-3 is low, the supply to electrons to each cytochrome c, and therefore its activity and the apparent P50, will be higher. Thus, cells with a relatively low expression of Cox will be more sensitive to moderate hypoxia.
The P50 is also increased by nitric oxide (NO) which competes with O2 for binding to Cox [150], especially under hypoxic conditions. The presence of endogenous NOS was shown to be associated with increases in the reduction of cytochromes BH, c1, and aa3 and also ROS production under hypoxic conditions In RAW246.7 cells [151]. In line with a role for NO-mediated block of Cox in O2 sensing, the eNOS antagonist N(ω)-nitro-L-arginine methyl ester (L-NAME) blocked hypoxia-induced ROS production and O2 sensing in endothelial cells [152,153,154].
Determining whether an interaction between NO and Cox in PASMC is involved in O2 sensing during HPV is not straightforward, since NO production by the pulmonary vascular endothelium exerts a powerful vasodilating influence which inhibits HPV by activating the guanylate cyclase/cyclic GMP/protein kinase G pathway. This has been demonstrated by studies in which knockout of eNOS and application of non-selective NOS antagonists increased the amplitude of HPV in studies carried out in perfused lung [155] [16,156] [157,158], and in humans in vivo [159]. The fact that HPV persisted [160] or was increased [158] when the synthesis of NO by NOS in PA was prevented seems to militate against the possibility that NO could be playing a role in O2 sensing in HPV. Even so, there is evidence that at least some types of cells possess NOS-independent pathways for synthesizing NO, and that these are stimulated by hypoxia. For example, hypoxia was shown to induce mitochondria from rat liver to produce NO through a Cox-mediated reduction of nitrite (NO2-) [161] . Although the importance of this mechanism has been disputed [162], hypoxia is also thought to activate other heme-dependent pathways within cells which can also generate NO from nitrite [163]. Furthermore, it has been shown that Cox plays an important role in metabolizing NO and that this process is inhibited by hypoxia [162]. This would also help to maintain NO levels during hypoxia, in which case there might be enough present in PASMC during hypoxia to compete with O2 for binding to Cox. Therefore, although there is currently no evidence that NO is involved in O2 sensing in HPV, it remains an intriguing possibility.
The mitochondrial concentration of H2S has also been proposed to increase during hypoxia [164], and since it also inhibits Cox, with a submicromolar Ki for the isolated enzyme [165], it might have a similar effect to that proposed for NO. However, much higher concentrations of H2S appear to be required to block Cox in intact cells and tissues [166,167]. These concentrations are unlikely to exist under physiological conditions, although this cannot be ruled out [168,169].
Another mechanism by which moderate levels of hypoxia could affect oxidative phosphorylation is described in a study carried out in isolated mitochondria from rat liver by Wilson et al [170], and further supported by work using mitochondria from cardiac myocytes [171]. Wilson and colleagues presented evidence that the actual P50 for Cox is much higher than its apparent value, which is typically derived by plotting the rate of respiration (measured as mitochondrial O2 consumption) vs PO2. According to this scheme, even relatively moderate levels of hypoxia (<40 Torr in this study) decrease the activity of Cox. This would tend to slow the flow of electrons from cytochrome c to Cox, causing cytochrome c to become more reduced. However, this reduction of cytochrome c increases its tendency to ‘push’ electrons onto Cox. These two tendencies, which have opposing effects on electron flux, cancel each other out. Thus, hypoxia does not alter the rate at which electrons flow into Cox, or on the rate at which it reduces O2 to water (i.e. the rate of O2 consumption, which is equivalent to respiration). Instead, the inhibitory effect of moderate hypoxia on Cox is reflected by an increased reduction of cytochrome c rather than by a decrease in respiration. Wilson and colleagues observed a reduction of cytochrome c which was half-maximal at a PO2 of 12 Torr, a value which they proposed was a reflection of the actual P50 for the activity of Cox. This compensatory mechanism would fail as the PO2 falls towards zero because cytochrome c becomes maximally reduced. Thus, with very severe hypoxia, O2 consumption falls steeply, and the PO2 - dependency of this decline would give rise to the very low apparent P50 for the reaction between Cox and O2 which is observed when measuring O2 consumption as a function of PO2. Because the reactions of oxidative phosphorylation upstream of Cox are at near- equilibrium [140,172], the increased reduction of cytochrome c at PO2 levels well above the apparent P50 for Cox would ‘back up’ into complexes 1 - 3 and the co-enzyme Q pool, causing these to become more reduced, and would also raise the NADH/NAD+ ratio. This would elevate the degree of reduction for both CoQ- and NADH- linked sites of ROS production, tending to increase H2O2 production.
This mechanism, which would be further promoted if hypoxia also decreased the cell energy state [173], is not necessarily restricted to any particular type of cell. However, it has also been suggested that Cox in PASMC and CBCC is unusually sensitive to hypoxia (i.e. has a higher P50) because they both express high levels of Cox4i2, an atypical subunit of Cox which has been shown to increase its sensitivity to O2 [174] and is negligibly expressed in other types of cell [175]. If the P50 for Cox was raised sufficiently, this would enable physiological levels of hypoxia to decrease the activity of COX and therefore slow the flux of electrons through the ETC and increase the degree of reduction at CoQ- and NADH-linked ROS-producing sites. Supporting this possibility, the P50 for the hypoxia-induced rise in NAD(P)H in sympathetic neurons was 0.3 Torr, as compared to 15 Torr in CBCC (n.b. the rise in NAD(P)H reflects the inhibition of Cox, since this carries out the rate limiting step of respiration and therefore also of the oxidation of NADH to NAD) [176].
Another factor which may cause a hypoxia-induced increase in mitochondrial ROS production, in this case independently of any effect on Cox, is a rise in the concentration of the Krebs cycle intermediate succinate. Couchani et al [100] showed that ischemia led to the reversal of the reaction carried out by succinate dehydrogenase (complex II), causing an accumulation of succinate in myocardial cells. Cardiac reperfusion then evoked a burst of ROS production due to the oxidation of the accumulated succinate by complex II, and this was blocked by the drug rotenone, which prevents the binding of quinones to complex I. This suggested that the ROS burst was mediated by complex I and was due to RET, which causes a high level of ROS production by complex I, probably at the 1CoQ site [109,177] [178].
The role of succinate and RET in O2 sensing during HPV is currently unknown. However, based in part on the observation that the complex II blocker dimethyl malonate depressed the response of CBCC to hypoxia, López-Barneo’s laboratory [179,180] initially suggested that succinate-linked RET contributes to acute O2-sensing in these cells by causing ROS production by complex I; a similar finding was reported by Swiderska et al [181] who also used dimethyl malonate. Nevertheless, it has more recently been reported that O2 sensing in CBCC was not obviously affected in mice in which RET was strongly depressed due to the introduction of a mutation into the ancillary complex I subunit ND6[182]. The involvement of RET in O2 sensing in CBCC can also be questioned because, whereas ROS generated at complex I due to RET are released into the mitochondrial matrix [109], the matrix redox balance in CBCC was reduced, rather than oxidized, by hypoxia [180]. In addition, the use of dimethyl malonate is problematic because it could potentially depress ROS production at complex III by decreasing succinate-mediated reduction of the CoQ pool [132].
Hernansanz-Agustin and colleagues [183,184] have proposed a mechanism by which hypoxia can increase complex III-mediated ROS production which is based on evidence that complex I has an intrinsic Na+/H+ antiport activity (pumping Na+ from the IMS into the matrix in exchange for protons) which is greatly increased when it undergoes a hypoxia-induced transition from its normal active state to a ‘de-active’ conformation in which it loses its usual NADH-ubiquinone oxidoreductase/proton-pumping activity [185] [186]. A deficit of O2 is proposed to promote this ‘A/D transition’ by causing a net reduction of the CoQ pool, thus inhibiting electron flow though complex I because there is less oxidized CoQ (ubiquinone) available to accept electrons in quinone binding site [187]. Interestingly, the active/de-active ratio varies over a broad PO2 range, and deactivation can occur quickly, rendering it a feasible target for moderate hypoxia [188,189].
According to this scheme, A/D transition induced by hypoxia causes matrix acidification by promoting H+ influx via the complex I Na+/H+ antiport and by inhibiting the normal Complex I mediated pumping of H+ from matrix to IMS [183]. This acidification causes the partial solubilization of the calcium phosphate complex which is present at a very high concentration in the [190]. The resulting increase in free [Ca2+]mito promotes Na+ entry into the matrix via the mitochondrial Na+/Ca2+/Li+ antiporter (NCLX). This causes a rise in matrix [Na+] which decreases the fluidity of the IMM, especially in the leaflet of the membrane adjacent to the matrix. The authors presented evidence that whereas an increase in [Na+] did not affect the function of the individual respiratory complexes when they were studied in isolation (i.e. in a mitochondrial membrane preparation), it did decrease the flow of electrons from complex II and G3PD to cytochrome in intact mitochondria. They proposed that the decreased fluidity of the IMM due to the rise in matrix Na+ induced by hypoxia would retard the mobility of CoQH2 in this membrane, thereby slowing electron flow from complex II and G3PD to complex III. This would slow the Q cycle, thereby increasing ROS production by prolonging the lifetime of ubisemiquinone at the Qo site. This mechanism is suggested to contribute to O2 sensing in HPV [183] (see section 5.2.3).
There is also evidence that an increased Ca2+ concentration in the mitochondrial matrix, which has been shown to occur in PASMC during hypoxia (see Section 5.2.1) stimulates pyruvate dehydrogenase, a-ketoglutarate dehydrogenase and complex II [191]. This increases the flux of electrons into the CoQ pool, thereby stimulating oxidative phosphorylation and increasing ROS production [192,193]. It has alternatively been proposed that an increased [Ca2+]mito elevates DΨm by reversing a brake on Cox exerted by ongoing ATP-dependent phosphorylation of this complex [194]. Regardless of the mechanism by which [Ca2+]mito or other factors elevate DΨm, there is abundant evidence that an increased (i.e. hyperpolarized) mitochondrial membrane potential powerfully stimulates ROS production at complexes 1 and 3 [132,195,135]. Ramzan et al [196] suggested that an increase in [Ca2+]cyto, which raises [Ca2+]mito via influx through the mitochondrial Ca2+ uniporter [192,197], accounts for the rise in mitochondrial ROS production evoked by a range of stressors, including hypoxia, in a variety of cell types. However, in this case, increased mitochondrial ROS production could not be the initial O2 sensing signal (which would be needed to generate the rise in [Ca2+]cyto), although it could act to amplify this signal.
Hajnóczky and colleagues have presented an intriguing proposal that an increase in the Ca2+ concentration in the mitochondrial matrix may cause it to swell due to the increased influx of K+ (and therefore water) into the matrix via mitoKCa channels in the IMM [92]. This then causes a compression of the mitochondrial cristae, which are continuous with the IMS. This would decrease the volume of the cristae/IMS compartment, increasing its [H2O2] and thereby promoting the leakage of H2O2 into the cytoplasm.
Notably, the concept that an increase in [Ca2+]mito raises mitochondrial ROS production has been vigorously contested [198].
3.1.1.5. Possible Mechanisms by Which Hypoxia Could Decrease Mitochondrial ROS Production
According to Eqn 1, a fall in PO2 would tend to decrease mitochondrial ROS production by the law of mass action, and most papers which have described hypoxia-induced falls in mitochondrial ROS production in cells or isolated mitochondria seem to have implicitly assumed that they occurred for this reason. This idea is consistent with evidence that ROS production by isolated mitochondria is linearly related to the O2 concentration over a wide PO2 range [199,200], although others have reported a hyperbolic relationship between the PO2 and ROS production [115,201], this does not necessarily conflict with the idea that the reactions between O2 with electrons leaving ROS-producing sites in the ETC are linearly dependent on the [O2] [137].
Conversely, Archer and colleagues have suggested that a fall in PO2 also diminishes ROS production by decreasing the rate of electron flow though the ETC, thereby limiting the availability of electrons at the sites in the proximal ETC at which they can react with O2 [36,202], see also [203]. This would appear to imply that the location at which hypoxia is slowing electron flow must be either at or upstream of the site where the ROS which regulate HPV are being produced, since upstream block of the ETC would be predicted to increase electron occupancy at this site and therefore promote its ROS production. This is consistent with evidence from this laboratory (but not others, see [3]) that the effects of hypoxia on ROS production are mimicked by the IQ-blocker rotenone [204]. Similarly, these effects have been reported to require the complex I subunit Ndufs2 [125], which forms part of ICoQ and interacts with the N2 Fe-S cluster that is believed to mediate the formation of semiquinone at this site [205] [206] [207] (see Section 5.1.1) . Nevertheless, the nature of the interactions between hypoxia, Ndufs2, and a slowing of electron flow which could result in decreased ROS production by complex I remain to be defined.
3.1.2. Mitochondrial ROS Consumption
Mitochondrial matrix consumption of H2O2 depends on the GSH and Trx2 redoxin systems (see Section 2.2), and on peroxiredoxins and catalase [58,104,208]. ROS released into the IMS are subject to degradation by SOD1, GSH and cytochrome C [209]. The antioxidant activities of GSH, TRX2 and peroxiredoxins require that they be returned to the reduced state by NADPH after having been oxidized due to the scavenging of H2O2. The maintenance of sufficient matrix NADPH to perform this function depends on the activity of the Krebs cycle enzymes malic enzyme (ME3) and isocitrate dehydrogenase (IDH2), which reduce NADP+, and also on nicotinamide nucleotide transhydrogenase (NNT), an enzyme in the IMM which couples the oxidation of NADH to the reduction of NADP+ within the matrix.
These antioxidant intra-mitochondrial mechanisms can affect the redox balance of the cytoplasm in two ways. Firstly, they limit the extent to which H2O2 generated within the matrix reaches the cytoplasm [103,208]. Secondly, the mitochondria can act as a sink for cytoplasmic H2O2. A study [104] which examined the effect of knocking out and overexpressing mitochondrial anti-oxidant enzymes on the redox state of the cytoplasm in a H9c2 cells exposed to the organic peroxide tert-butyl hydroperoxide (t-BOOH)(1 mM) reported that knockout of NNT, ME3 or IDH2, and also the mitochondrial anti-oxidant enzyme thioredoxin2, increased the extent to which t-BOOH oxidized the cytoplasm. In contrast, knockout of cytoplasmic antioxidant enzymes did not ameliorate the effects of t-BOOH on the mitochondria. The authors suggested that the mitochondria may function to degrade H2O2 which leaks into cells from the ECS. Interestingly, another paper reported that application of 25 and 500 μM extracellular H2O2 to EA.hy926 cells caused a rise in cellular H2O2 which was larger and occurred more rapidly in the mitochondria compared to the cytoplasm and nucleus [210]. These reports suggest that the mitochondria play a crucial role in the ability of cells to scavenge H2O2 leaking in from the extracellular space, although whether this occurs under physiological conditions can be questioned given the antioxidant prowess of cytoplasmic Prxs [211] (see Section 2.2).
Oxidation of the matrix leads to s-glutathionylation of complex I and other proteins associated with ETC ROS production, including pyruvate dehydrogenase (PDH) and a-ketoglutarate dehydrogenase (KGDH) [79]. This depresses oxidative phosphorylation and ROS production, thus stabilizing the redox state of the matrix. Another factor promoting mitochondrial redox homeostasis is that the activity of NNT is crucially dependent on DP, meaning that both the production and NADPH-dependent scavenging of ROS within the matrix are enhanced by increases in [NADH] and DP. Nevertheless, Treberg et al [208] demonstrated that the effect of DΨm on mitochondrial ROS production was greater than that on peroxiredoxin-mediated ROS scavenging, suggesting that increasing DΨm would produce a net rise in matrix [H2O2].
3.2. ROS Production by NADPH Oxidases
The seven members of the Nox family of oxidoreductases (Noxs 1-5 and Duox 1 and 2) function as adjustable sources of superoxide and/or H2O2 which regulate the activities of nearby target proteins. Four Nox isoforms (Nox 1, 2, 4 and 5) are expressed in vascular smooth muscle cells [60]. Nox2 has been shown not to be required for HPV, at least in mice [212], and the role in HPV of Nox 5, which is expressed in primates but not rodents, is unknown. However, Noxes 1 and 4 have been proposed to be involved in HPV (see Section 5.2.5 and Section 8). Both of these Nox isoforms are heterodimers comprising a membrane-spanning oxidoreductase subunit, which transfers electrons from NADPH to O2 via flavin adenine dinucleotide (FAD) and two heme groups, and p22phox, a smaller transmembrane scaffolding protein. Nevertheless, they differ in important ways.
3.2.1. Nox1
Nox1 is present in the plasmalemma, localized to the caveoli in vascular smooth muscle cells (VSMC) [213]. It releases superoxide into the ECS, from where it can enter cells either via plasmalemmal anion channels, or, after being converted to H2O2 by EC-SOD, through aquaporins [214]. Nox1 can also be endocytosed upon its activation by certain stimuli, potentially leading to redox signaling by superoxide and/or H2O2 both within the endosome and the cytoplasm [215]. Activation of Nox1 is triggered by the binding of the cytoplasmic proteins p47phox, p67phox, and p40Phox. Its recruitment of these proteins is triggered by the phosphorylation of p47phox by protein kinase C (PKC) and/or a Src family tyrosine kinase [60,216]. Alternatively, p47phox and p67phox may be replaced by their homologues NoxO1 and NoxA1, respectively. In this case, NoxO1 is constitutively tethered to p22phox, and superoxide production is stimulated by the binding of NoxA1 to NoxO1 [217], which is promoted by PKC [218], and also requires the phosphorylation of threonine 429 on Nox1 (e.g. by protein kinase C β1; [219]. The combination of p47phox and NoxA1 has also been shown to stimulate Nox in VSMC [220].
Activation of PKC and Src kinases can be initiated by diverse stimuli, including lipid mediators produced by phospholipases A2, C and D following their activation by the binding of agonists to multiple types of G-protein coupled receptors. PKC and Src family kinases can also be activated by each other, and by ROS [216], thereby enabling positive feedback. Full activation of Nox1 occurs with the subsequent binding of activated monomeric G protein Rac1. This in turn necessitates the stimulation of Rac guanine exchange factors following their tyrosine phosphorylation and binding to phosphatidyl inositol triphosphate, which is generated by transactivation of the epidermal growth factor receptor and the stimulation of phosphatidyl inositol-3 kinase [60].
3.2.2. Nox4
Nox4 is thought to be the most highly expressed isoform in VSMC, in which it is localized to the plasma membrane in association with focal adhesions [213]. It is also expressed in the sarcoplasmic reticulum [221], mitochondria [222], and nucleus [223]. Nox4 primarily produces H2O2, which presumably gains access to the cytoplasm via aquaporins in the plasmalemma and intracellular membranes. The Nox4/p22phox dimer is constitutively active, suggesting that it can produce appreciable levels of H2O2 under basal conditions, and that its ability to do so may regulated by its level of expression [224], and, acutely, by the availability of NADPH and also O2 [59]. H2O2 production by plasmalemmal Nox4 is increased by its association with polymerase delta interacting protein 2 (Poldip2) [225] and in VSMC can be enhanced acutely by G protein coupled vasoconstrictors through mechanisms that are not well understood. Little is known about the regulation of intracellular Nox4 [60].
One important property of Nox4 which makes it well suited to act as an sensor for acute changes in the PO2 is that its Km (O2) (the PO2 at which its production of H2O2 is half maximal) has been reported to be ~136 Torr, such that its activity would be decreased by even mild hypoxia. The Km (O2) for Nox1 seems not to have been measured, although that for Nox2 is ~25 Torr [59].
4. Intra- and Extracellular H2O2 Concentrations
Recent reviews state that the basal intracellular [H2O2] is in the range of 1-5 nM [32,226,227], citing as evidence an elegant study by Oshino et al [228] which took advantage of the fact that the enzyme catalase reacts with H2O2 to form the intermediate Compound 1 (which can be monitored spectrophotometrically) to calculate that the intracellular cellular concentration of H2O2 in rat liver under resting conditions was on the order of 10-9 M [228,229] , see also [230].
A similar estimate was reported by Lyublinskaya & Antunes [231], who, using an approach based on measuring the kinetics of oxidation of the H2O2 sensor HyPer , determined that the basal cytoplasmic [H2O2]cyt in K562 cells is in the low nanomolar range [231]. Their study was designed to determine the involvement of extracellular H2O2 in setting the basal cytoplasmic [H2O2]. The concept that extracellular [H2O2] ([H2O2]EC) is an important determinant of the cytoplasmic [H2O2] had emerged from studies showing that a bolus of H2O2 added to the solution bathing cells in culture is taken up (via aquaporins [72] [232]) and then rapidly scavenged by intracellular antioxidant systems [233,234,235]. Because the removal of H2O2 by cellular antioxidants is much faster than its uptake, a large H2O2 concentration gradient across the cell membrane develops immediately upon its extracellular application [236]. The magnitude of this gradient is maintained at a constant level as the H2O2 in the bath is taken up by the cells, such that intra-and extracellular [H2O2] undergo a parallel exponential decline [237].
The steady-state transmembrane gradient can be assessed by keeping the cell number low enough to ensure that the uptake of H2O2 has a negligible effect on its concentration in the bath, or by using an H2O2 - regulating system such as glucose/glucose oxidase/catalase to stabilize the [H2O2] in the bathing solution [238,239]. Using the former approach, Lyublinskaya & Antunes reported that the cytoplasmic [H2O2], which was 2.2 nM when no H2O2 was added to the bath, rose to 3.5 and 8.9 nM, respectively, when 1 and 2.5 μM H2O2 were applied to the solution. The authors calculated that an average trans-plasmalemmal H2O2 concentration gradient of ~390-fold was present over this range of extracellular [H2O2].
The existence of even larger (~1000-fold) [H2O2] gradients between extracellular and cytoplasmic [H2O2] had previously been predicted by mathematical models developed by Adimora et al [237] and Lim et al [240], which took into account information about the rates of transmembrane H2O2 flux, cellular production, and scavenging e.g. [235,241]. Importantly, whereas cellular H2O2 was initially thought to be scavenged by catalase and glutathione peroxidase [234], yielding calculated transmembrane gradients of which were small (e.g. ~7 in Jurkat T cells [233]), these models took into account more recent evidence that Prxs, which are present in micromolar concentrations in cells and can have a very high affinity for H2O2 (e.g. Prx2), are predominantly responsible for removing it from the cytoplasm [88]. Importantly, Lim et al [240] went on to show that the scavenging of H2O2 following its entry into the cell would also set up a significant peroxide concentration gradient within the cytoplasm which would develop within milliseconds and remain stable if the [H2O2]EC was constant. Thus, they calculated that [H2O2] at the inner face of the plasmalemma in a spherical cell with a radius of 10 microns would be ~0.3% of that in the extracellular medium, falling to ~0.08% and 0.02% at 2 and 4 microns into the cell, respectively. These studies imply that the basal intracellular [H2O2] in the subplasmalemmal region is strongly influenced by the [H2O2]EC.
The physiological implications of these findings are difficult to parse, firstly because the [H2O2] in solutions bathing biological preparations in vitro is typically not controlled or measured during investigations of its intracellular dynamics or effects, and secondly because little is known about the [H2O2]EC in vivo. Although basal [H2O2]EC has been suggested to be 1-5 μM [32,226,227], this seems to have been measured only by one laboratory, which reported mean in vivo values of 3 and 5 μM for [H2O2]EC in rat ventrolateral medulla and hippocampus, respectively [242,243,244]. Additional indirect evidence supporting the possibility that cells experience [H2O2]EC in this range came from an investigation in which the [H2O2]EC immediately adjacent to the extracellular surface of the plasmalemma in A549 (tumor-derived) cells in culture was measured using a membrane-tethered sensor [245]. This showed that average [H2O2]EC close to the membrane was ~2 μM, and also revealed the presence of discrete H2O2 hotspots, presumably associated with the presence of a higher local expression of Nox in that area of the membrane, with concentrations of up to ~12 μM. [246]. The authors suggested that these areas were likely to give rise to corresponding H2O2 hotspots in the subplasmalemmal cytoplasm, although they did not confirm this.
Parenthetically, basal plasma [H2O2] is also generally thought to be 1-5 μM [247]. However, Sousa et al [248] contend that the basal plasma [H2O2] has been overestimated, and that due to its uptake by erythrocytes, is in the range of 10 nM. They note, for example, that taking into account measurements of the permeability of erythrocytes to H2O2, Prx2, which should be heavily oxidized when the plasma [H2O2] is ≥ 80 nM, is instead found to be almost completely reduced under basal conditions [249].
Although the evidence is very limited, the agreement between the measured values of intra- and extracellular [H2O2] and the ~1000-fold [H2O2] gradient predicted by mathematical models makes a neat story. Nevertheless, it has also been suggested that the basal cytoplasmic [H2O2] may be in the sub-nanomolar range [250]. Lim et al [211] concluded on theoretical grounds that, absent H2O2 influx from the extracellular space, cellular H2O2 production and scavenging should set the basal cytoplasmic [H2O2] at ~80 pM. Since according to their later model [240] the influence of [H2O2]EC decreases steeply moving away from the cell membrane, [H2O2] would be predicted to approach this level in the core of large cells. Moreover, using HEK293-MSR cells expressing the novel high-sensitivity H2O2 indicator Apex2 [251], Eid et al [252] detected a sustained increase in intracellular [H2O2] upon the application of [H2O2]EC down to 25 nM in HEK293-MSR cells. Sommer et al [130] found that application of 124 nM H2O2 caused membrane depolarization and inhibited the KV current in PASMC, and this laboratory has recently reported that the applying as little as 10 nM H2O2 inhibited the current through KV1.5 channels co-expressed with KVb1.4 ancillary subunits in oocytes [253]. This implies that the basal [H2O2] even adjacent to the cell membrane could have been in the subnanomolar range, although whether this applies to cells in vivo would presumably depend on the balance between the rates of H2O2 influx from the ECS and its intracellular scavenging.
Another implication of the presence of powerful antioxidant mechanisms present in the cytoplasm is that increases in [H2O2] emanating from intracellular sites are highly localized. This was demonstrated by Mishina et al [254], using cells expressing HyPer fused to growth factor receptors in the plasmalemma and protein tyrosine phosphatase 1B (PTP1B) in the endoplasmic reticulum (ER) membrane. They found, for example, that applying epidermal growth factor (EGF) to HeLa cells expressing an EGF receptor – HyPer fusion construct led to the internalization of a fraction of the EGF receptor into endosomes, while the rest remained in the plasmalemma. The endosomal fraction reported an increase in [H2O2] due to Nox activity associated with receptor activation, while the portion in the plasmalemma did not. This demonstrated that only internalized EGF receptor was activated, and also that H2O2 was not able to diffuse from far enough from the endosomes to reach the plasmalemma. Likewise, H2O2 generated by Nox in response to platelet derived growth factor (PDGF) was sensed by PTP1B-HyPer in the ER membrane but not by PDGF receptor-HyPer in the endosomal membrane. In a subsequent study in HeLa cells, this group demonstrated the presence of a steep cytoplasmic H2O2 concentration gradient when a H2O2 generating system (D-amino acid oxidase/alanine) was localized to the nucleus [255].
However, an investigation utilizing the highly sensitive indicator HyPer7 failed to detect cytoplasmic H2O2 gradients, or indeed any rise in the global cytoplasmic [H2O2], associated with an increase in mitochondrial ROS production induced by D-amino acid oxidase/alanine in K562 cells [256]. On the other hand, by applying antimycin to cells fed with galactose, which enhances mitochondrial ROS production, Hoehne et al [121] were able to demonstrate using HyPer7 that an increase in mitochondrial ROS production at complex III did cause a small rise in [H2O2]cyt in HEK293 cells, although this was not observed in cells fed with glucose. Both papers concluded that H2O2 was being released by the mitochondria, but that it (and any HyPer7 molecules it may have oxidized [257]) were being reduced before they could diffuse far enough to generate a measurable signal in the overall cytoplasm. Notably, recent experiments using HyPer7 also detected an increase in cytoplasmic H2O2 elicited by mitochondrial D-amino acid oxidase/alanine in HeLa cells, leading the authors to suggest that these cells may have less effective antioxidant mechanisms compared to K562 cells. Interestingly, using the novel H2O2 indicator HyPerFLEX, which is more sensitive than HyPer7, they also observed an accompanying increase in H2O2 in the nucleus and endoplasmic reticulum [258].
Notably, studies by Hajnoczky and colleagues have shown that the close apposition of mitochondria to the endo/sarcoplasmic reticulum (ER) means that mitochondrial ROS release into the nanodomain between these organelles can oxidize a group of cysteines in the cytoplasmic-facing suppressor domain of the IP3R, inducing Ca2+ release [92,259,260]. Using a compartmentally-targeted redox sensor (Grx1-Ro-GFP2), they demonstrated in HEPG2 cells [259] that the oxidative signal due to mitochondrial ROS release was restricted to the mitochondria/ER nanodomain, and did not result in a general cytoplasmic oxidation. Intriguingly, they also presented evidence that the ER-mitochondrial interface nanodomain was more oxidized than the bulk cytoplasm even under basal conditions, suggesting the existence of a standing intracellular redox gradient which may function to enable ER Ca2+ release.
Section 5: Models of O2 Sensing in HPV
Although they have opposed views on the effects of hypoxia on cytoplasmic oxidant signaling in PASMC, the Redox and Mitochondrial ROS theories both propose that the mitochondria are the primary O2 sensors responsible for causing HPV. This concept is supported by evidence that 1. blockers of the proximal ETC abolish HPV (see Section 7.1), 2. cultured PASMC depleted of their mitochondria by ethidium bromide treatment [261] do not demonstrate hypoxia-induced rises in [Ca2+]cyt [38], and 3. mitochondria in PASMC are distributed more peripherally compared to those in mesenteric artery SMC (MASMC), so are better placed to influence effectors located in the plasmalemma [262].
We will first describe the experiments leading to development of each theory separately in Section 5.1 and Section 5.2 Because these proposals share a focus on mitochondrial ROS production, the studies carried out by their proponents have often used similar approaches (e.g. assessing the effects of ETC blockers or pro- and anti-oxidants, measurement of ROS), and have therefore yielded information relevant to both. We will therefore consider this evidence as a whole and discuss its implications in Section 7.
5.1. The Redox Theory
The Redox theory of hypoxia of HPV, proposed by Kenneth Weir and Stephen Archer in the mid 1990’s [36,263], is based on the concept that HPV is due to the removal by hypoxia of a tonic PASMC vasorelaxation [264] which is maintained under normoxic conditions by a level of cytoplasmic ROS which is higher than that present in systemic artery VSMC. Recent work by Archer’s laboratory ascribes this difference to the higher expression of the complex I subunit Ndufs2 in PASMC [125].
According to this hypothesis (Figure 6), the relatively high ongoing PASMC production of H2O2 by mitochondria under normoxic conditions leads to a basal activation of voltage-gated K+ (KV) channels which maintains the cells in a relaxed state by ensuring that the membrane potential (Em) is negative enough to suppress the activity of L-type voltage-gated Ca2+ channels, thereby minimizing Ca2+ influx. By decreasing the flux of electrons through the ETC, hypoxia diminishes the production of mitochondrial superoxide/H2O2 [33]. The resulting fall in the cytoplasmic [H2O2] depresses KV channel activity, causing membrane depolarization and an increased Ca2+ influx which evokes HPV. The mechanism by which ROS activate the KV channels is unknown but could involve oxidation of KV channel protein thiol-containing residues. ROS are seen as exerting a similar effect on KV channels in systemic artery VSMC, although this influence is smaller under normoxic conditions because mitochondrial ROS production is lower. In contrast to PA, systemic arteries dilate to hypoxia, and this is proposed to be due, at least in part, to increased mitochondrial ROS production and the activation of KV channels [145]. Thus, the opposite responses of pulmonary and systemic arteries to hypoxia are seen as being due to its contrary effect on mitochondrial ROS production, rather than to fundamental differences between the hypoxia-sensitive effector pathways controlling VSMC force development in these arteries and/or their responsiveness to ROS [33].
Although the proponents of the Redox theory have generally stressed the effect of hypoxia on ROS, they also view hypoxia-induced reduction of cellular redox couples (e.g. NAD+/NADH, NADP/NADPH, GSSG/GSH) as potentially contributing to HPV [36,125]. This could occur, for example, through an action of NAD(P)H on KV channel b-subunits, which can modulate channel gating by virtue of their activity as aldehyde dehydrogenases [265]. However, taking into account that the cytoplasmic redox potential of the NADP+/NADPH couple in the cytoplasm is approximately -390 mV [266], this couple is already almost entirely reduced under normoxic conditions. This suggests that a fall in [ROS] in and of itself is unlikely to increase the free [NADPH], making it a poor candidate for mediating hypoxia-induced changes in KV channel activity associated with decreased ROS production. On the other hand, the NAD+/NADH couple, which has a cytoplasmic redox potential of ~-240, is highly oxidized [252,266]. This results in a low free cytoplasmic [NADH] [266,267] which is in the appropriate range for regulating KV channels [268]. Given its involvement in controlling the oxidation of cellular thiol switches, the GSSG/GSH redox couple is of particular interest with regard to signaling. Importantly, the GSSG/GSH ratio has been shown to be altered by the application of low concentrations of extracellular [H2O2], and also by hypoxia [269,270] (see Section 6.4 for a further discussion of the role of these redox couples in HPV).
Similarly, although they have emphasized the role of KV channels as the crucial ROS-sensitive effectors, the involvement of other redox-sensitive downstream pathways which could also respond to a fall in ROS by to promote contraction (or inhibit relaxation) are not ruled out [271]. There is, for example, evidence from Michael Wolin’s laboratory that a decrease in H2O2 production contributes to HPV by suppressing basal vasodilation due to activity of soluble guanylate cyclase (sGC) and protein kinase G (PKGa1). However, the model for HPV developed by Wolin and colleagues differs from the Redox theory in that it is Nox rather than the mitochondria which is seen as being responsible for basal ROS production, and, unlike the Redox theory, it also proposes an important role for the pentose phosphate pathway in HPV (see Section 8).
5.1.1. Evidence for a Fall in Mitochondrial ROS Production and PASMC Reduction as the O2 Sensor in HPV
A series of studies in the 1980’s, carried out mainly by Weir, Archer and colleagues, investigated the effects of oxidizing and reducing agents on HPV. Weir et al [272] examined the effect of IV injection of the oxidizing agent diamide on HPV recorded in anesthetized dogs when the inspired O2 level was reduced to 12%, and on the pressor response to PGF2a. Diamide markedly diminished the increases in PAP and PVR to elicited by both hypoxia and PGF2a without affecting cardiac output or systemic vascular resistance. Burghuber et al [273] examined the effect of H2O2, generated by adding glucose/glucose oxidase to the perfusate, on HPV in isolated perfused rat lung, finding that it did not alter PAP under normoxic conditions, but abolished HPV and halved the amplitude of the pressor response to angiotensin 2. These responses were prevented if catalase was added to the solution and were mimicked by perfusing the lungs with physiological saline solution (PSS) containing ~10-6 M H2O2. Although glucose/glucose oxidase caused the lungs to release PGF2a and TXA2, these were unlikely to have mediated its effects on pulmonary vasoactivity, which persisted when their synthesis was blocked using indomethacin.
Subsequent studies from Weir and colleagues were also performed in isolated perfused rat lungs in which HPV was evoked by reducing the PO2 in the perfusate to ~40 Torr. Weir et al [274] found that diamide applied during hypoxia also reversed HPV in this preparation, and that adding xanthine/xanthine oxidase (X/XO) to the solution to generate ROS had a similar effect which was prevented if the X/XO blocker allopurinol was present. They suggested that X/XO might be acting by oxidizing cellular sulfhydryl groups, and soon afterwards proposed that hypoxia was inducing PA constriction by causing a similar effect [275]. Archer et al [276] confirmed this effect of X/XO on HPV and found that it was attenuated if SOD and catalase were added to the perfusate. Application of SOD and CAT which had been incorporated into liposomes to facilitate their entry into PA cells similarly suppressed the effect of X/XO. If added in the absence of X/XO, these liposomes increased baseline PAP and enhanced both HPV and the pressor response to angiotensin 2. These results supported the concept that reduction of redox couples in PASMC tended to promote PA constriction, both under normoxic and hypoxic conditions. Both diamide and X/XO suppressed HPV, although it is difficult to interpret this effect because the response to angiotensin 2, which was used as a pre-tone agent to amplify HPV [277] was similarly inhibited by X/XO, and the effect of diamide on the angiotensin 2 response was not described.
Archer et al [278] performed simultaneous measurements of PAP and pulmonary ROS production in isolated and ventilated rat lungs while changing the PO2 in the inspired gas. Luminol or lucigenin were included in the perfusion solution as ROS indicators, and the PO2 was varied from the normoxic baseline (20%) to 0, 1, 2.5, 10, or 95%. The key result of this paper was that lowering the PO2 to ≤ 2.5% evoked a fall in luminescence with both indicators that paralleled the decrease in PO2 and preceded the onset of HPV, which was sustained with 1% and 2.5% O2, but transient with 0% O2. Including catalase in the perfusate did not affect lucigenin luminescence under normoxic conditions, whereas this was strongly diminished by SOD. SOD similarly depressed luminol luminescence, which was however somewhat increased by catalase. SOD slightly raised basal PAP, and strongly enhanced HPV and the pressor response to angiotensin 2, whereas catalase had no effect on PAP under any condition. Based on their observations, the authors suggested that hypoxia was decreasing pulmonary superoxide levels. The ability of SOD to increase basal and stimulated vascular tone echoed earlier findings and was seen to support the idea that the oxidation of cell redox couples exerts a tonic vasodilating influence on PA. However, from a contemporary perspective, which views H2O2 rather than superoxide as mediating most ROS-dependent intracellular signaling [32], the observations that SOD caused contraction and enhanced HPV appear to be inconsistent with the Redox theory.
A crucial step in the development of the Redox theory came with the observation, which emerged from a collaboration between the Stephen Archer and Joseph Hume’s laboratory [279], that hypoxia inhibited the K+ current in pulmonary (but not renal) artery SMC. This raised the question of whether hypoxia was inhibiting K+ channels by virtue of its effects on cell redox. Based on the observation that blockers of the ETC cause PA contraction [280] and would be predicted to mimic hypoxia by suppressing mitochondrial ROS production and reducing cell redox couples such as NADH/NAD+ and GSH/GSSG, Archer et al [263] investigated the effects of several ETC blockers on PAP and ROS production in their rat lung model, and on the K+ current in freshly isolated rat PASMC. Rotenone, antimycin or cyanide, which act at complexes 1, 3 and 4, respectively, were applied as a bolus injection into the perfusate inflow, and at the doses used all produced an increase in PAP similar to that evoked by hypoxia (ventilation with 2.5% O2). Pulmonary ROS production, assessed using luminol and lucigenin, was decreased by both rotenone and antimycin. Both blockers also abolished HPV. The fall in lung ROS production caused by hypoxia was strongly attenuated by pre-treatment with antimycin, but was unaffected by rotenone (n.b. this appears to be inconsistent with later evidence from this laboratory [125] that HPV is due to decreased ROS production by complex I). Cyanide caused a dose-dependent increase in PAP comparable to that caused by rotenone and antimycin, but unlike these blockers had no effect on HPV and caused a transient increase in lucigenin luminescence. Both of these responses were depressed by SOD, suggesting that they were superoxide-dependent. The K+ current they recorded in PASMC was strongly blocked by both TEA and 4-AP and was attenuated by hypoxia. The current was also reversibly suppressed by rotenone and antimycin but was not significantly affected by this concentration of cyanide.
In discussing their findings, the authors noted that whereas Rounds & McMurtry [280] had previously highlighted the resemblance between the effects of hypoxia and ETC blockers on pulmonary artery contraction and had speculated that hypoxia might causing a pressor response in PA by depleting ATP, this seemed unlikely in light of evidence that high energy phosphate levels in PASMC are little changed by hypoxia [281,282,283]. They proposed instead that rotenone and antimycin were causing PA contraction by suppressing mitochondrial ROS production and pointed out that effects of these drugs on PAP, ROS production, and the PASMC K+ current resembled those of hypoxia. The ability of both ETC blockers to abolish HPV also suggested that they were acting via the same mechanism as hypoxia. The observation that cyanide, which did not diminish ROS, had no effect on either HPV or the K+ current, also aligned with the idea that a fall in ROS was linked to K+ channel inhibition and contraction. They therefore concluded that that hypoxia was causing HPV by decreasing mitochondrial ROS production (or causing a reducing shift in cell redox couples) which led to a decreased opening of voltage-dependent K+ channels. These papers established the basic outline of the Redox theory, which was then set forth in detail in a review by Weir and Archer in 1995 [36].
Observations that PA constrict in response to hypoxia, whereas systemic arteries dilate (e.g. [160]) could potentially be explained by differences in either the O2 sensor or the mediators/effectors to which it is coupled. Michelakis et al [145] therefore examined whether the hypoxia-induced fall in ROS which had been observed in PA also occurred in small renal arteries, and whether mitochondria from the SMC of these responded differently to hypoxia.
They observed that both hypoxia (pO2 ~40Torr) and rotenone inhibited the whole cell K+ current in rat PASMC, whereas they increased its amplitude in renal artery smooth muscle cells (RASMC). Likewise, both stimuli increased PAP in isolated lungs, but reduced the arterial pressure in perfused kidneys. Lucigenin-enhanced luminescence was >20 times higher in isolated PA compared to RA under normoxic conditions. Hypoxia decreased luminescence by ~80% in PA but increased it about 5-fold in RA, and rotenone had a similar effect. Utilizing Amplex Red or 2',7'-dichlorodihydrofluorescein (DCFH) as indicators, they found that the level of H2O2 under normoxic conditions was higher in PA than in RA. The H2O2 signal was decreased in PA by hypoxia and antimycin, although it was not increased by either hypoxia or rotenone in RA. Cyanide had no effect on the DCFH signal in either artery.
In further experiments, they characterized mitochondria isolated from homogenates of whole lung (LM) and kidney (KM), using glutamate and succinate to support respiration. In line with their observations in the arteries, ROS production, assessed using lucigenin, was higher in LM vs KM. ROS production was inhibited by antimycin but not cyanide in LM, and neither drug had an effect in the KM.
Using Western blotting, they found higher expression of complexes I and 3 in RA compared to PA, and pointed out that this was consistent with their additional observations that O2 consumption, indicative of respiration, was greater in KM compared to LM, and that the mitochondrial membrane potential (Δψm), assessed using JC-1 and TMRM, was more hyperpolarized in RASMC than in PASMC. Moreover, hypoxia increased Dψm in PASMC but had the opposite effect in RASMC. Based on their findings that glutathione levels and Mn SOD protein expression were substantially higher in the PA compared to RA, which was consistent with an adaptation of PASMC to the relatively O2-rich pulmonary milieu, the authors also proposed that PASMC are more oxidized than RASMC, although this conclusion is questionable because the concentrations of oxidized and reduced glutathione, which reflect the cytoplasmic redox potential, were not reported.
The differential effect of hypoxia on the lucigenin vs DCFH/Amplex Red signals in RASMC led the authors to suggest that hypoxia increases superoxide, but not H2O2, in these cells, possibly due to the low expression of MnSOD in RASMC. However, using the genetically encoded H2O2 indicator HyPer this laboratory subsequently observed that hypoxia increased cytoplasmic H2O2 in these cells [125], suggesting perhaps that Amplex Red and DCFH were sensing ROS other than H2O2 in their experiments.
Notably, the physiological significance of these results is uncertain, since whereas the physoxic PO2 for mesenteric arteries is much lower than it is for pulmonary arteries, there is no indication in the paper that this was taken into account in designing the experiments.
Wu et al [284] used dihydroethidium (DHE) in the experiments (although the ‘normoxic’ PO2 was not stated, a hypoxic PO2 of 40 Torr was used in preparations from both arteries) to compare the effect of hypoxia (pO2 ~30 Torr) on ROS production by cultured human smooth muscle cells from PA and coronary artery (CASMC). In accord with the Redox theory, they found that hypoxia decreased superoxide levels in PASMC. However, hypoxia also caused a similar fall in DHE oxidation in CASMC. The authors pointed out that coronary arteries dilate to hypoxia, and that the fall in superoxide in both types of cells took several minutes to occur whereas HPV in lungs develops within seconds. This led them to speculate that ROS may not be involved in HPV, or that the opposite contractile response of pulmonary and coronary arteries to hypoxia might be due to differences in the effector mechanisms coupled to ROS. Similar observations were made by Mehta et al [285], who found that moderate hypoxia (5% O2) significantly decreased ROS production in both primary cultured human PASMC and coronary artery SMC. This effect was seen with three indicators (DCFH, DHE and Amplex Red). The fall in ROS showed a (non-significant) trend to be more substantial in the PASMC with each indicator.
In 2019, Dunham-Snary and colleagues [125] published a study of the role of ROS in HPV in rats and mice which included a further comparison of the properties of pulmonary and renal artery mitochondria. This paper presents the most recent and comprehensive evidence currently available to support the Redox theory and its results relating to the acute effects of hypoxia will therefore be considered in detail. The authors incubated isolated LM and KM at a concentration of ~2.5 mg protein/ml in physiological medium for 15 minutes under normoxic or hypoxic conditions. Using Amplex Red, they measured the H2O2 concentration in the conditioned media, finding that under normoxic conditions this was much higher in medium conditioned with LM (~1 μΜ) compared to KM (<0.2 μΜ). These results were consistent with their earlier findings [145].
Micropolarimetry experiments were carried out to compare the O2 consumption rate in cultured PASMC and RASMC, finding that it was about twice as high in the former. Since mitochondria contribute the vast bulk of cellular O2 consumption, this appears to contradict their earlier observation that the rate of respiration in isolated LM was about a third of that in KM [145]. The authors did not discuss possible reasons for these divergent results, but it is possible that they may have been due to cellular influences on mitochondrial function which would be absent in isolated mitochondria. If so, this suggests that effects of hypoxia on isolated mitochondria cannot simply be extrapolated to more intact preparations [53,286].
Isolated PSS- perfused rat lung was used to examine the effect of oxidants on HPV evoked using perfusate gassed with 2.5% O2. Injection of 0.1 mL of LM-conditioned medium into the perfusion inflow caused a rapid and marked reversal of HPV (by 56%), whereas injection of KM-conditioned medium, or medium conditioned with LM in the presence of catalase, had no effect on HPV. Perfusion with PSS containing 100 μM t-BOOH also reversed HPV (by 75%), although 10 μΜ t-BOOH had a negligible effect.
Cultured rat PASMC and RASMC were transfected with HyPer-dMito or HyPer-dCyto to monitor H2O2 levels in the mitochondria and cytoplasm, respectively [287]. In PASMC, hypoxia (pO2 ~20 Torr) decreased H2O2 levels in both the mitochondria and cytoplasm. In contrast, hypoxia had no effect on mitochondrial ROS and increased cytoplasmic ROS in RASMC. Hypoxia also increased the [Ca2+]cyt in PASMC. This rise in [Ca2+] cyt was completely reversed by 1 mM t-BOOH and subsequently restored by application of catalase. The effects of hypoxia on [Ca2+]cyt and mitochondrial [ROS] were mimicked by rotenone, which also diminished both the concentration of H2O2 in PASMC- conditioned medium and the reversal of HPV induced by this medium. Taken together, these results support the idea that mitochondria in PASMC produce more ROS than those in RASMC under normoxic conditions, that hypoxia causes a fall in ROS production which increases PASMC [Ca2+]i, and that reducing and oxidizing stimuli cause changes in [Ca2+]cyt in the cells which are consistent with the Redox theory, as does block of the proximal ETC with rotenone.
Ndufs2 is a subunit of complex I which plays an important role in the binding and reduction of ubiquinone by complex I [288,289,290,291]. It had been demonstrated [179] that conditional knockout of Ndufs2 in CBCC prevented activation of the carotid body by hypoxia, leading Dunham-Snary et al [125] to investigate its involvement in HPV. They found that the mRNA expression of Ndufs2 was higher in PASMC than in RASMC, as was its protein expression (normalized to TOMM20, another mitochondrial protein). Moreover, complex I in PASMC was enriched in Nduf2s compared to RASMC.
siRNA knockdown of Ndufs2 in PASMC inhibited respiration and increased cellular levels of NADH, consistent with its role in the enabling of electron flow in the ETC. Ndufs2 knockout was not associated with a fall in the quantity of complex I, but its activity, assessed using protein immunocaptured from cell homogenates, was depressed. Knockdown of Ndufs2 also abolished the hypoxia-induced rise in [Ca2+]cyt and decreased the release of H2O2 by LM. Importantly, the increase in both mitochondrial and cytoplasmic ROS evoked in PASMC by hypoxia was also decreased. In contrast, siRNA knockdown of Uqcrfs1, the protein containing the Rieske Fe-S cluster (RISP) in complex III, or of COX4i2, a Cox subunit proposed to contribute to O2 sensing [130] did not prevent these effects. Knockdown of Ndufs1, a protein in the N-domain of complex I, was also without effect.
In further experiments, siRNA directed against Ndufs2 or control siRNA was administered once to rats via airway nebulization. After 48 hours, compared to the controls, the si-Ndufs2 rats demonstrated diminished levels of Ndufs2 mRNA in lung homogenates and exhibited a profound fall in Ndufs2 levels co-localizing with smooth muscle actin, as revealed by immunofluorescence studies of small PA in lung sections. The animals were anesthetized and PAP pressure was measured in vivo under normoxic and hypoxic (10% O2) conditions. PAP under normoxic conditions was not different in the two groups, but hypoxia caused a much smaller increase in PAP in the si-Ndufs2 rats compared to the controls. Similarly, treatment of mice with Ndufs2 siRNA over a period of more than a week had no effect on basal PAP, but profoundly depressed HPV, compared to animals treated with control siRNA. Likewise, the increase in PAP evoked by rotenone in the controls was almost absent in the si-Ndufs2 rats, whereas the response to phenylephrine was enhanced in the latter group. This may have been due to the down-regulation of KV1.5 which also occurred in the si-Ndufs2 animals. They also found that exposing mouse lungs in vitro to hypoxia for 30 minutes increased the ratio of reduced to total Ndufs2. 289Hypoxia also inhibited complex I activity, an effect mimicked by treatment with the reducing agent dithiothreitol (DTT). They speculated that this might be due to the reduction of the mitochondrial milieu caused by hypoxia.
Based on the specific inhibitory effect of effect of Nduf2s knockdown on H2O2 production and the rise in [Ca2+]cyt evoked by hypoxia, as well as their evidence that hypoxia caused a fall in cytoplasmic H2O2 and that HPV was reversed by oxidants, the authors proposed that HPV was triggered by a fall in ROS production which they suggested was due to inhibition of the production of uncoupled electrons (i.e. electrons leaking from the ETC) at Ndufs2.
In accordance with the concept that Ndufs2 is involved in producing ROS in an PO2-dependent manner, a subsequent paper from this laboratory [292] reported that the IQ site, in which Ndufs2 plays a vital role in the transfer of electrons from complex I to ubiquinone [289], is responsible for the increase in ROS production associated with a rise in PO2 in the ductus arteriosus.
A letter by Hüttemann et al [293] criticized the conclusion that COX4i2 was not involved in HPV on the basis that its knockdown in the Dunham-Snary study, which amounted to ~55%, was insufficient to significantly affect its role in O2 sensing. Their contention was based on their unpublished finding that HPV was largely intact in COX4i2+/- mice, whereas it was abolished in homozygous knockouts [130].
In a response letter, Dunham-Snary & Archer [294] expressed their belief in the validity of their findings, and criticized the study by Hüttemann and colleagues [130] based on its use of a very low pO2 to elicit HPV and on the small amplitude of the HPV they obtained. They also noted that their conclusion that Ndufs2 was crucial in O2 sensing in PASMC was consistent with the earlier demonstration by Fernandez-Aguera et al [179] that it was the O2 sensor in CBCC, supporting the concept that common O2 sensing mechanisms are operative in various oxygen sensing tissues [271]. However, they failed to mention that the proposed role of Ndufs2 in O2 sensing in CBCC [179] was opposite to theirs, since its knockdown prevented a hypoxia-induced increase in mitochondrial ROS production, and the resulting suppression the K+ current, in these cells. Also, K+ currents were inhibited by oxidants (H2O2 and diamide) and increased by the putative antioxidant N-acetyl cysteine. Based on subsequent work [180], it was proposed that hypoxia increases ROS production in CBCC by causing reduction of the ubiquinone pool, thereby lengthening the dwell-time of electrons at the Ndufs2 quinone binding site and increasing the probability that they will react with O2 at this point. It has also been reported that an increase in ROS production dependent on NDUF2.1, the C. elegans ortholog of Ndufs2, is responsible for the locomotor response to hypoxia in these organisms [295]. Similarly, Hernansanz- Agustin et al [184] presented evidence that whereas hypoxia caused a marked increase in cytoplasmic [H2O2], which they measured using HyPer, in bovine aortic endothelial cells transfected with scrambled siRNA, the opposite effect occurred if Ndufs2 was knocked out. They also found that Ndufs2 knockout increased the basal (normoxic) cytoplasmic [H2O2].
In considering the results reported by Dunham-Snary et al [294], it is perplexing that reversal of HPV by the LM-conditioned medium was of similar in magnitude to that exerted by much higher concentrations of the membrane permeable H2O2 t-BOOH. In these experiments. 0.1 mL of the conditioned medium, which had a H2O2 concentration of ~1 μM, was injected into the lung inflow line of the perfusion system, which had a volume of 40 ml and was being continually recirculated at a flow rate of ~0.25 mL/s. The conditioned medium would have been diluted rapidly upon injection and the concentration of H2O2 in the perfusate would have very quickly fallen into the low nM range during the 5 minute period over which HPV was progressively reversed. In comparison, application of concentrations of the membrane permeable H2O2 analog t-BOOH which were ≥4 orders of magnitude higher were required to cause a comparable reversal of HPV (EC50 > 50 μΜ). In contrast, in a study the which directly compared the vasorelaxing effects of H2O2 and t-BOOH (carried out using PGF2a – constricted rat aorta) these oxidants evoked vasorelaxation with very similar concentration-dependencies; with an EC50 for both of ≈ 20 μΜ, [296]. Similarly, 1 μM t-BOOH and H2O2 caused identical levels of oxidation when applied to BY4742 yeast cells [297].
The results and implications of the experiments in which protein components of the mitochondrial complexes were knocked down can also be questioned. Ndufs2 knockdown depressed respiration, indicative of a decrease in the activity of the ETC, and also has been shown to cause additional and widespread effects on mitochondrial function, including the depolarization of DΨm [298]. This may occur because the reduction of CoQ, which is thought to initiate proton translocation by complex I, requires electrons donated by an aspartate-histidine pair in the b1-b2 loop of Ndufs2, [299]. A change in the configuration of Ndufs2 may also play a role in the active to de-active transition of complex I which was proposed [183] to mediate the increase in ROS responsible for HPV [207]. It is therefore difficult to interpret the effect of Ndufs2 knockdown on O2 sensing in PASMC.
It is also curious that the knockdown of Ndufs1 and Uqcrfs1 apparently had no effect on the response to hypoxia in PASMC. Ndufs 1 is one of the 14 catalytic-core subunits required for complex I to function, and contains three (N1b, N4, N5) of the seven Fe-S clusters which form a ‘wire’ carrying electrons from the NADH binding pocket to the ubiquinone binding site [300]. Since Ndufs1 is upstream of N2, the Fe-S cluster with which Ndufs2 interacts, the lack of effect of its knockdown on the response to hypoxia would also appear to rule out a role for Ndufs2 in regulating ROS production at either IQ or IF. More generally, knockout of either Ndufs1 or Uqcrfs1, which is required for electron flow from Qo to cytochrome c and thence to Cox and O2, should suppress mitochondrial respiration and depolarize DΨm, thereby presumably causing the dysfunction of any redox-mediated mitochondrial O2 sensing mechanism [301,302].
5.1.2. The Involvement of ROS in PASMC K+ Channel Inhibition During Hypoxia
The Redox theory posits that membrane depolarization due to the hypoxia-induced closure of KV channels, particularly those incorporating the KV1.5 subunit, is a primary effector mechanism for HPV, and that this inhibition is mediated by a fall in [H2O2] and/or an increase in [NAD(P)H] in the cytoplasm.
The concept that KV channel closure contributes to HPV is supported by abundant evidence [17,279,303,304,305,306,307,308,309,310,311,312,313], although other processes, including intracellular Ca2+ release, store-operated Ca2+ entry (SOCE), and Ca2+ sensitization are also important [3,10,12,14,305], and additional depolarizing mechanisms (e.g. TASK-1; non-selective cation channels) [314,315,316] have been identified.
On the other hand, the proposal that this inhibition of KV channels during HPV requires a reduction of the cytoplasmic redox balance remains controversial. In keeping with the focus of this review on the O2 sensing rather than effector mechanisms of HPV, and because the question of how KV channels in PASMC are regulated by redox mechanisms has been critiqued extensively [3,317,318,319], but seems not to have moved any closer to being resolved over the past decade, we will consider this issue only briefly.
A number of laboratories have investigated the redox regulation of KV channels in PASMC by monitoring whole cell or membrane patch currents while applying oxidants, reductants, or ETC blockers, which were used to decrease mitochondrial ROS production. Both rotenone and antimycin (10mM) were shown to block the K+ current in PASMC [145,263]. The effect of rotenone is in accordance with Redox theory, although that of antimycin is difficult to interpret, since it is generally thought to increase mitochondrial ROS production [109]. A more detailed study [320] showed that although antimycin, myxothiazol, and rotenone depressed the KV current in PASMC at positive potentials, they caused a hyperpolarizing shift in its activation threshold, implying that these blockers should open rather than shut KV channels in the physiological range of membrane potentials. The authors provided evidence that the ETC blockers were exerting these effects on the KV current by increasing the cytoplasmic cellular [Mg2+]. This could be due to mitochondrial depolarization, an effect exerted by all ETC inhibitors, which could explain why both rotenone and antimycin inhibited the current at positive potentials. To complicate matters further, both rotenone and antimycin inhibited the K+ current with IC50 values of ~1mM in H146 (small cell lung carcinoma) cells by a mechanism which was independent of the mitochondria [321].
The effects of reductants and oxidants on K+ currents are summarized in Table 1 and Table 2, respectively. The majority of studies produced evidence supporting the viewpoint of the Redox theory with regard to the redox regulation of PASMC KV channels by hypoxia. However, other investigations have reported observations inconsistent with this view [130,320,322,323] and there is also evidence that hypoxia-induced KV channel inhibition is mediated by a rise in [Ca2+]cyt due to Ca2+ release from the sarcoplasmic reticulum [324,325]. Moreover, LY83583, which causes intracellular superoxide production, increased the KV current in the physiological range of membrane potentials in PASMC, as predicted by the Redox theory, but caused PA constriction rather than relaxation [326]. This suggests that the contractile response of these cells to alterations in the cytoplasmic redox state might not depend only on changes in KV channel activity.
Yandjue et al [253] recently showed that the effect of H2O2 on the KV1.5 current expressed in oocytes is strongly dependent, not only on its concentration, but on the potential at which the current is evoked. Application of low concentrations of extracellular [H2O2] (0.01 – 100 μM), a range which is likely to cause physiologically relevant variationsin its cytoplasmic concentration (see Section 4), tended to not affect or inhibit the current at -20 mV, whereas it was increased by concentrations above 1 mM. Although the implications of these results for the redox regulation of KV1.5 during HPV remain unclear, the observations made in this paper highlight the need to study the effect of redox mechanisms on channel activity during using conditions which more realistically simulate the effects of hypoxia on specific redox couples and oxidant concentrations. More generally, little is known at the molecular level about the redox regulation of the KV1.x and KV2.X isoforms thought to play a role in HPV, and the results of the few studies which have examined this in more depth [327,267,328] suggest that an accurate determination of how redox mechanisms (and other factors such as [Ca2+]cyt or channel phosphorylation [44,324,329] contribute to KV channel closure during HPV will require a more refined and concerted approach to unravelling the effects of hypoxia and redox agents on these channels than has previously been attempted. It will be particularly important to characterize the effects of hypoxia and perturbations on specific redox couples on KVb-subunit function, the oxidation of cytoplasmic cysteine residues on the KV1.5α subunits, and the membrane trafficking of KV channels (which can change rapidly [327,330]) in native PASMC.

Table 1a.
Effects of oxidants on K+ currents in PASMC.
Table 1b.
Effects of reductants on K+ currents in PASMC.
n.b. Except where noted, PASMC for these studies were freshly isolated on the day that currents were recorded.
5.2. The Mitochondrial ROS Theory of HPV
The proposal that HPV is caused by a fall in cellular [ROS] was challenged by Marshall et al [339], who reported that hypoxia increased ROS in cultured PASMC from fetal calves, and by Killilea et al [340], who made a similar observation in cultured PASMC from rats. Abdalla & Will [341] had also observed that HPV in isolated guinea pig PA was enhanced by two SOD inhibitors (DETCA and TETA), and suggested that HPV was due to a rise in cellular superoxide levels.
Whereas Marshall et al ascribed their results to an activation of Nox, Paul Schumacker and colleagues proposed in 2001 that hypoxia promotes ROS production by PASMC mitochondria, leading to a rise in cytoplasmic [ROS] that results in the stimulation of redox-sensitive effector pathways which cause HPV [38]. This concept grew out of observations by Schumacker’s laboratory that hypoxia increased mitochondrial ROS production in cardiac myocytes, leading to stabilization of HIF1α [342], and in HEP3b cells, stimulating the expression- of erythropoietin, glycolytic enzymes, and vascular endothelial growth factor [343].
This ‘Mitochondrial ROS hypothesis’ was supported and extended in subsequent papers from this and other laboratories, several of which have presented evidence that the site of ROS production is complex III. It has also been proposed that the increase in mitochondrial ROS leads to additional ROS production by Nox which enhances the pressor response to hypoxia (see Section 5.2.5). A schematic of the mitochondrial ROS hypothesis which has grown out of the investigations described in Section 5.2 is shown in Figure 7.
Waypa et al [38] studied HPV recorded in isolated ventilated rat lung and microvascular PASMC in primary culture. Hypoxia was imposed by reducing the [O2] in the ventilating gas from 21 to 2%, and angiotensin 2 was used to create pretone. HPV was abolished by rotenone, and myxothiazol, which blocks electron transport in complex III at the ubiquinol oxidation site (Qo), had a similar effect. Conversely, I ng/mL antimycin, which binds to complex III more distally at the Qi site where cytochrome b562 is oxidized, had no effect on HPV, and cyanide augmented the amplitude of HPV. All of the ETC blockers except myxothiazol caused a transient contraction.
Pyrrolidinedithiolcarbamate (PDTC), an antioxidant said to enhance H2O2 clearance by reducing oxidized glutathione, reversibly attenuated HPV by 65% and blocked hypoxic contraction of PASMC. The glutathione peroxidase mimetic ebselen caused a profound and irreversible block of HPV, and diethyldithiocarbamate (DDC), a cytosolic Cu,Zn SOD blocker, inhibited HPV by 68%. Application of DIDS, an anion channel blocker used here to prevent mitochondrial superoxide from entering the cytoplasm through the mitochondrial inner membrane anion channel (IMAC), inhibited HPV to a similar extent, and also blunted the contraction caused by antimycin. At the concentrations used in these experiments, none of these blockers affected the pressor response to the TXA2 agonist U46619, which was used as a negative control to detect non-specific effects on contraction. In contrast, 3μM apocynin abolished both HPV and the U46619 contraction, whereas it had no effect on either response at a lower concentration (0.3μΜ). Waypa et al [38] also found that hypoxia caused a largely reversible increase in the release of ROS from PASMC (monitored using DCHF-DA), which was attenuated by myxothiazol. In addition, incubation of PASMC in ethidium bromide for two weeks to deplete cellular mitochondria [261] diminished HPV by 70%.
Based on these results, the authors proposed that hypoxia induces an increase in mitochondrial ROS production at a site upstream of where antimycin binds to complex III, and that this was leads to an increase in the cytoplasmic concentration of H2O2 which acts through several effector mechanisms to cause HPV. The authors pointed out that drugs acting distal to ubisemiquinone, including antimycin, tend to increase ROS during normoxia by increasing the reduction of the ubiquinone pool [344], whereas more proximal block would suppress ROS production by oxidizing this pool. The lack of HPV in po cells, and the inability of apocynin to exert a selective block of HPV supported the idea that ROS produced by the mitochondria rather than Nox were regulating HPV (n.b. however, apocynin is not a dependable blocker of Nox; [345]). Based on the block of HPV by DIDS, they also suggested that hypoxia might alternatively increase the entry of superoxide into the cytoplasm by opening IMAC, although in subsequent papers this idea was not pursued.
Leach et al [346] employed a pharmacological approach to evaluate the role of the mitochondrial complexes in HPV using isolated rat intrapulmonary arteries (IPA). They observed that the ETC blockers rotenone, myxothiazol, cyanide and the complex II substrate succinate had no effect on basal tone under normoxic conditions. Rotenone caused an increase in NAD(P)H (assessed by measuring tissue autofluorescence) similar in amplitude to that induced by anoxia. This was not reversed by application of 5 mM succinate, consistent with a complete block of the ETC at complex I by rotenone. Hypoxia (PO2 of 15-18 Torr) applied to IPA which were slightly pre-constricted with PGF2a caused a biphasic HPV, as typically observed in this preparation under these conditions. Application of either rotenone or myxothiazol abolished both phases of HPV. Importantly, rotenone did not block either phase of HPV if it was co-applied with succinate, whereas succinate did not reverse the inhibition of HPV by myxothiazol. These results indicated that HPV in these arteries required the provision of electrons to the myxothiazol binding site (i.e. Qo) of complex III, which succinate would be able to restore in the presence of rotenone but not myxothiazol. The authors pointed out that whereas this effect of succinate was consistent with the concept that HPV was caused by an increase in ROS production by the ETC distal to this site, it was difficult to explain if hypoxia was causing HPV by mimicking the effect of rotenone and inducing a decrease in mitochondrial ROS production, as proposed by the Redox theory. The lack of effect of rotenone on basal tension was also inconsistent with the Redox model. Subsequent studies have shown that succinate can also increase ROS production by causing reversed electron transport (RET) through complex I [100,179]. Although this would have been favored by the combination of succinate and myxothiazol, which should highly reduce the ubiquinone pool [177], it seems unlikely that ROS production by RET could explain the restoration of HPV by succinate in this study, since this would have been prevented by rotenone [177,347].
Waypa et al [348] investigated the effect of ETC blockers and other agents on the hypoxia-induced rise in [Ca2+]cyt in PASMC using fura 2. Exposure to medium equilibrated with 1.5% O2 caused a sustained rise in [Ca2+]cyt recorded over 9 minutes. This was attenuated by DPI (here used to block complex I), rotenone and myxothiazol, but was unaffected by antimycin and cyanide; none of these drugs significantly affected the increases in [Ca2+]cyt evoked by angiotensin 2 or 50 μM H2O2. In PASMC in which catalase had been overexpressed by recombinant adenovirus transduction, the responses to hypoxia and exogenous H2O2, but not angiotensin 2, were diminished compared to those in control cells overexpressing LacZ. Application of cyanide caused a rise in [Ca2+]cyt which was antagonized by myxothiazol and was smaller in PASMC overexpressing catalase, but not lacZ. These results provided further support for the model illustrated in Figure 7. The contrasting effects on HPV of the proximal (rotenone, DPI and myxothiazol) as compared to distal (antimycin and cyanide) ETC blockers led them to propose that mitochondrial ROS production at the ubisemiquinone site in complex III was crucial for triggering the rise in [Ca2+]cyt during HPV. The observation that HPV was blocked by catalase overexpression suggested that H2O2 rather than superoxide was the important ROS for HPV, consistent with the idea that H2O2 is generated from superoxide by MnSOD in the intermembrane space of the mitochondria, and then crosses the outer membrane to access the cytoplasm.
Weissmann et al [349] carried out a comprehensive study of the effects on HPV of blockers of complexes 1-3, as well as mitochondrial uncouplers which should decrease ROS production. The aim of the investigation was to test the predictions of the Mitochondrial ROS model as set out by Waypa et al [38]. In this and other investigations carried out by this laboratory [144,350,351,352], HPV was recorded in isolated perfused rabbit lungs which were ventilated via the trachea and the PO2 in the ventilating gas mixture was lowered from 21 to 3% for 10 minutes to reduce the alveolar PO2 from ~160 to ~23 Torr and evoke the initial transient component of HPV (phase 1). The thromboxane A2 agonist U46619 was used as a negative control, based on the assumption that the contraction it evokes should be resistant to ETC block, since it was not known to require mitochondrial ROS production. Experiments were repeated in the presence of L-NMMA to determine whether there were any confounding effects of the mitochondrial blockers which were being exerted due to interference with NO release. Whereas some of the results supported the Mitochondrial ROS theory (e.g. blocking complexes 1 and 2 antagonized HPV), others did not (e.g. antimycin did not cause a sustained contraction), allowing the authors to conclude only that mitochondria are somehow involved in phase 1 HPV during a brief hypoxic exposure. Importantly, they also observed that most of the blockers they studied strongly attenuated the response to U46619, and/or were interacting with NO system, suggesting that these drugs have off-target actions on contraction which could obscure the effects they exert on HPV by altering mitochondrial ROS production.
Liu et al [353] used perfusion myography to evaluate the role of ROS in HPV (evoked by exposing the arteries to pO2 = 29 Torr) in 7th generation branches of porcine PA. HPV was abolished when the solution contained 150 U/ml SOD or SOD + 200 U/ml CAT. They used lucigenin and also EPR spectroscopy with the spin probe DMPO in an attempt to determine the effect of hypoxia on cellular ROS in the arteries, but although both approaches yielded results suggestive of an increase, in neither case were the results significant (n.b. the use of DMPO may also have been problematic, [354]). However, hypoxia (pO2 34 Torr) did cause a rapid and largely reversible increase in DCFH florescence in primary cultured PASMC. This was strongly suppressed by SOD and completely prevented by both CAT and SOD + CAT, leading the authors to conclude that it was due to a rise in cellular oxidative stress rather than hypoxia-induced changes in DCFH leakage or metabolism. The authors confirmed, using FITC labelled SOD and CAT, that both enzymes were entering the cells. Notably, the observation that SOD and SOD + CAT similarly suppressed HPV suggested it was mainly an increase in cellular [superoxide] rather than [H2O2] which was causing contraction, a finding which appears to be inconsistent with the current view [32] that H2O2 rather than superoxide is the crucial ROS for cellular signaling.
Weissmann et al [352] compared the effects of ETC blockers on the 1st and 2nd phases of HPV in rabbit lungs. The complex I blocker MPP+, applied at a concentration which slightly blocked phase 1, strongly blocked phase 2. Similar semi-selective effects on phase 2 were exerted by rotenone and cyanide. In contrast, 3-NPA (3-nitroproprionic acid; blocks complex III) and antimycin at the concentrations used caused a similar and marked suppression of both phases.
In view of the shortcomings of ROS indicators such as lucigenin, DCFH and luminol (see section 7.4), Guzy et al [355] developed a novel fluorescence resonance energy transfer probe, HSP-FRET, in order to evaluate the role of mitochondrial ROS in hypoxia-induced stabilization of HIF-1a. Their study, carried out in 143B cells, showed that application of exogenous H2O2 and hypoxia, but not NO, increased the FRET signal, and that stigmatellin (blocks the ETC at the Qo site in complex III), suppressed the response to hypoxia. Their results suggested that HSP-FRET was able to detect H2O2, and that, at least in these cells, hypoxia increases the production of mitochondrial ROS, which then enters the cytoplasm.
This laboratory [356] then used HSP-FRET to determine whether hypoxia caused an increase in ROS in PASMC. They described HSP-FRET as being predominantly expressed in the cytoplasm, although specific evidence for this was not provided. In PASMC transfected with HSP-FRET, hypoxia (1.5% O2) caused a gradual increase in ROS which started after ~3 minutes. A simultaneous increase in [Ca2+]cyt, assessed using fura 2 also occurred, and similar results were obtained in experiments using YC2.3, a FRET-based Ca2+ sensor. Hypoxia also decreased the GSH/GSSG ratio, considered to reflect the general cell redox state [357], from ~50 to ~30. The effects of hypoxia on oxidant signaling and [Ca2+]cyt were suppressed by the antioxidants PDTC and N-acetyl-L-cysteine (NAC), neither of which affected the signal from either probe under basal conditions. Overexpression of glutathione peroxidise in order to deplete cytoplasmic H2O2 suppressed the effects of hypoxia on both [Ca2+]cyt and oxidant signaling, as did overexpression of catalase in either the mitochondria or the cytoplasm. Conversely, overexpression of cytosolic Cu, Zn SOD (SOD 1) had no significant effect on the responses of HSP-FRET or YC2.3 to hypoxia, while overexpression of mitochondrial Mn SOD (SOD 2) similarly did not affect oxidant signaling and enhanced the [Ca2+]cyt rise induced by hypoxia. In a subsequent paper from this group, overexpression of Prx5 in the mitochondrial IMS also was found to diminish the rise in [Ca2+]cyt evoked by hypoxia (1.5% O2 applied for 15 minutes) in these cells [358].
These results supported the concept that hypoxia causes an increase in cytoplasmic H2O2 and an oxidizing shift in the redox balance of PASMC resulting from mitochondrial ROS production. The observation that PDTC and NAC blocked the effects of hypoxia but did not themselves cause any response argued against the proposal of the Redox theory that cytoplasmic oxidation under basal conditions maintains vasorelaxation by causing an ongoing stimulation of KV channels.
A study by Wang et al [359] which examined the role of mitochondrial ROS in the hypoxia-induced rise in [Ca2+]cyt in freshly isolated mouse PASMC yielded results similar to those reported by Waypa and colleagues. They found that hypoxia (1% O2 for 5 minutes) increased ROS levels (assessed using DCFH and lucigenin) and [Ca2+]cyt. Pharmacological blockade of complexes 1, 2 and the Qo site of complex III did not affect the basal ROS or [Ca2+]cyt, signals, but strongly antagonized the effects of hypoxia. In contrast, block of complex III at the Qi site with antimycin or at complex 4 with NaN3 had no effect on ROS levels or [Ca2+]cyt under either normoxic or hypoxic conditions. The effects of hypoxia on ROS levels and [Ca2+]cyt in PASMC from mice overexpressing glutathione peroxidase or catalase were smaller than those observed in control mice, whereas they were enhanced in PASMC from glutathione peroxidase knockout mice. They also found that application of 51 μM H2O2 caused an increase in the DCFH signal similar to that evoked by hypoxia but resulted in a much smaller increase in [Ca2+]cyt. This led them to propose that mitochondrial H2O2 production was necessary but not sufficient to evoke the full hypoxia-induced increase in [Ca2+]cyt, or alternatively that this response might depend on the mitochondrial compartmentalization of ROS, which would not occur when peroxide was applied exogenously. A later investigation [360] similarly reported that hypoxia-induced increases in [Ca2+]cyt in cultured human PASMC were prevented by blocking complexes 1, 2 and Qo, but were unaffected by block of Qi or complex IV, and that HPV in small PA from humans showed the same pattern of responsiveness to these drugs.
In order to determine whether hypoxia-induced redox signaling in PASMC was compartmentalized, Waypa et al [361] transfected PASMC with Ro-GFP, another genetically encoded ratiometric redox indicator. Ro-GFP expression was directed to the mitochondrial matrix (Mito-Ro-GFP) or the mitochondrial intermembrane space (IMS-Ro-GFP) by adding polypeptide targeting sequences to its structure; non-targeted Ro-GFP was used to assess the cytoplasmic redox state. Rather than providing a readout of [H2O2], Ro-GFP fluorescence is sensitive to the redox state of the GSH/GSSG couple [362]. Because the Ro-GFP signal is ratiometric, it can be calibrated, allowing the degree of its oxidation to be calculated.
Hypoxia (1.5% O2) caused a progressive increase in Cyto (non-targeted)-RoGTP oxidation from 19 to 35% over 30 minutes. IMS-Ro-GFP had a much higher level of oxidation under normoxic conditions (48%) but was still significantly oxidized (to 62%) by 30 min of hypoxia. In contrast, Mito-Ro-GFP, which was even more highly oxidized under normoxic conditions (63%), became more reduced during hypoxia (to 44%). Overexpression of catalase in the cytoplasm had no effect on the normoxic oxidation level of Ro-GFP but prevented its hypoxia-induced oxidation. Overexpression of catalase in the mitochondrial matrix had no effect on the oxidation of Mito-Ro-GFP under either normoxic or hypoxic conditions. These results imply that changes in H2O2 production during hypoxia influence the redox potential in the cytoplasm but not in the mitochondrial matrix. Hypoxia caused the same pattern of compartmentalized changes in Ro-GFP oxidation in renal artery SMC. However, experiments with YC2.3 showed that hypoxia caused an increase in [Ca2+]cyt in PASMC, but not in the renal arteriolar cells. In this case, if mitochondrial ROS are involved in O2 sensing in both types of SMC, their coupling to effector pathways must differ.
These investigations can be criticized because they were carried out in cultured SMC which may have undergone phenotypic changes. In order to characterize oxidant signaling under more physiological conditions, Desireddi et al. [363] transfected mouse lung slices with Cyto-Ro-GFP, a process which required their incubation in culture medium for two-days. The vascular endothelium was labelled with Cell Tracker Red so the vascular lumen could be imaged and used to measure contraction. The effect of hypoxia on [Ca2+]cyt was assessed in parallel experiments using Fura 2, although this was done after only one day of organ culture, as Ca2+ signaling was lost thereafter.
Small PA in lung slices contracted to angiotensin 2, high K+ PSS and hypoxia after the two day period in organ culture. Hypoxia (1.5% O2) caused a rapid and sustained increase in Ro-GFP oxidation from 11 to 17%, which reversed upon re-oxygenation. Overexpression of catalase in lung slices had no effect on basal Ro-GFP oxidation but almost abolished the effect of hypoxia. Hypoxia caused an immediate increase in [Ca2+]cyt which was diminished by catalase overexpression, and was abolished by EUK-134, a SOD-catalase mimic [364]. The increase in [Ca2+]cyt was absent in slices superfused with Ca2+- free PSS containing 2.5 mM EGTA, suggesting that it was due to the influx of extracellular Ca2+. However, this concentration of EGTA can deplete releasable intracellular Ca2+ stores [365], so these might also have been involved.
These results demonstrated that hypoxia caused a reversible increase in Ro-GFP oxidation (i.e. oxidation of the GSH/GSSG redox couple) in small PA which, although exposed to culture conditions for two days, retained the ability to contract. It is noteworthy, however, that the size of the contractions was small compared to those previously described in freshly prepared mouse lung slices [366,367], and the proportion of arteries demonstrating a contraction to hypoxia was not stated. Also, the loss of Ca2+ signaling caused by the two-day incubation period suggests that changes in the PASMC phenotype had occurred.
5.2.1. Role of Mitochondrial Ca2+ and the Rieske Iron-Sulfur Protein in Hypoxia-Induced ROS Production
The Rieske iron-sulfur protein (RISP; also termed ubiquinol-cytochrome c reductase, Rieske iron-sulfur protein 1 and Uqcrfs1), is a subunit of complex III. RISP contains a 2Fe-2S cluster which accepts electrons from the Qo site and passes them on to cytochrome c1, and has been proposed to play a pivotal role in mediating a hypoxia-induced increase in ROS production by complex III [368].
Korde et al [127] examined the effect of RISP knockdown on the response to hypoxia in isolated mouse PA, cultured PASMC, mitochondria isolated from PA homogenates, and complex III obtained from these using immunocapture. PASMC were transfected with the genetically encoded ratiometric H2O2 indicator pHyPer-dMito [287] which the authors described as being expressed in the mitochondrial IMS. Hypoxia (PO2 = 13 Torr) markedly increased the pHyPer-dMito signal and enhanced ROS production (assessed using DCFH) by isolated mitochondria and complex III. Hypoxia increased the activity of isolated complex III but had no effect on O2 consumption or ATP production by the isolated mitochondria. Mitochondrial H2O2 under hypoxic conditions was greatly diminished in PASMC in which RISP expression was strongly depressed by siRNA treatment, as was hypoxia-induced ROS production in isolated mitochondria and Complex III obtained from these cells. RISP knockdown also strongly depressed HPV and the associated rise in [Ca2+] in isolated PA. Overexpression of RISP had opposite effects. Based on these results, the authors proposed that complex III in PASMC is intrinsically sensitive to hypoxia and speculated that this might involve an effect on the transfer of electrons from ubisemiquinone to cytochrome c1 by RISP.
Based on their observation [369] that hypoxia induces Ca2+ release from the SR in PASMC which is dependent on ROS released from the mitochondria, and evidence that Ca2+ causes increased ROS production by mitochondria (Section 3.1.1.4), this laboratory investigated whether a hypoxia induces a positive feedback process involving Ca2+ and ROS release in PASMC [370]. They found that that SR Ca2+ release induced by caffeine (0.2 mM to 2M) or norepinephrine (1 μM – 10 mM) caused increases in mitochondrial [Ca2+] and cytoplasmic [ROS], which was monitored using several ROS probes, including the cytoplasmic H2O2 indicator pHyPer-cyto. In addition, raising the [Ca2+] in solution bathing mitochondria isolated from these cells also stimulated their ROS production. Hypoxia also increased mitochondrial [Ca2+] and ROS production by PASMC and isolated mitochondria. These effects were antagonised by RU360, a blocker of the mitochondrial Ca2+ uniporter (MCU). RISP knockdown attenuated basal as well as Ca2+- and hypoxia-induced ROS production by PASMC and mitochondria isolated from these cells. These results suggested that hypoxia stimulates complex III-dependent ROS production by increasing mitochondrial [Ca2+], and that this involves Ca2+ release from the SR and the opening of the MCU. The authors also reported that SMC cultured from mesenteric arteries had a much lower expression of RISP compared to PASMC and did not increase their ROS production in response to hypoxia, supporting the concept that RISP is important in ROS production and O2 sensing.
Evidence for a role of RISP in O2 sensing was also provided by Waypa et al [128], who used compartmentally-targeted Ro-GFP to compare the effects of hypoxia (30 minutes: 1.5% O2) on oxidant signaling in pulmonary and renal artery SMC from conditional knockout mice in which RISP expression was either normal or decreased by ~60%. In control PASMC, hypoxia caused a pattern of compartmental redox alterations resembling that which had been seen in rat PASMC: the cytoplasm and IMS became more oxidized and the mitochondrial matrix became more reduced [361]. In contrast, Cyto-Ro-GFP in RISP-depleted cells was more oxidized under normoxic conditions and hypoxia caused the cytoplasm to become more reduced. Hypoxia did not alter the redox state of IMS-Ro-GFP in RISP-depleted cells, and the redox state of the mitochondrial matrix, which was more reduced under normoxic conditions compared to controls, was also unaffected by hypoxia. Echoing their earlier observation in rat [361], they found that the pattern of compartmental changes in Ro-GFP oxidation in renal artery SMC induced by hypoxia was similar to that in PASMC, as were the effects of RISP-deletion. In additional in vivo experiments, injection of SMC-MHC-Cre/RISPflox/flox mice with tamoxifen was used to knock down smooth muscle RISP expression by ~60%. Measurements of PA diameter and [Ca2+]cyt in lung slices revealed that HPV and the associated rise in PASMC [Ca2+]cyt was suppressed in tamoxifen- vs vehicle-treated animals. Moreover, whereas PA wall thickness, RV mass, Fulton index, cardiac function and baseline normoxic PAP were similar in tamoxifen- vs vehicle-treated animals, the increase in right ventricular systolic pressure caused by ventilating the animals with a hypoxic gas mixture (5% O2/95% N2) was strongly reduced in the tamoxifen group. The authors proposed that RISP ablation acted at site Qo to inhibit the formation of ubisemiquinone, which can donate an electron to O2 to form superoxide. Based on the effects of RISP deletion on the differential compartmental oxidation of Ro-GFP, they suggested that hypoxia might change the configuration of complex III, such that the ROS it produced were directed into the IMS rather than the mitochondrial matrix.
Importantly, however, the depletion of RISP would be predicted to disrupt the Q cycle and the redox state of Qo, which governs the production of ROS by complex III. Because the flow of electrons through RISP is necessary for the reduction of cytochrome C, its removal has been shown to strongly inhibit respiration [371,372,373], and can cause mitochondrial depolarization [374,375], and the upregulation of glycolysis, an increase in the NADH/NAD ratio [371,373] and the decreased expression of multiple isoforms of NADPH oxidase [372]. Although there is also countervailing evidence that cells can maintain a normal mitochondrial membrane potential and rate of glycolysis following RISP knockdown [193], its potential for causing disruption of mitochondrial function is such that it the results of these studies should be interpreted with caution.
5.2.2. Mitochondrial Hyperpolarization, Reduction of the Quinone Pool, and Cox4i2
Several studies by Sommer and colleagues [129,130,144] presented evidence that hyperpolarization of the mitochondrial membrane potential (ΔΨm) and reduction of components of the ETC promote hypoxia-induced mitochondrial ROS production in PASMC.
Sommer et al [144] evaluated the effect of hypoxia on the redox state of cytochromes bL/H, c and aa3 in isolated perfused lung and PASMC from rabbit using remission spectrophotometry, a technique in which the redox state of multiple cytochromes is inferred from the wavelength-dependency of light absorbance by cells. They found that HPV was evoked when the pO2 was lowered to ≤ 75Torr and increased progressively as the level of hypoxia was deepened. A significant change in absorbance was observed at pO2 ≤ 53Torr, and analysis of the spectra indicated that reduction of cytochromes c, aa3 and bL/H occurred at pO2 values of 38, 23 and 8 Torr respectively in the lungs. Based on measurements of the P50 for oxygen consumption, they calculated (see [149]) that respiration was significantly inhibited at ambient PO2 levels of <100 Torr (e.g. by ~2 and ~10% at pO2 of 38 and 8 Torr, respectively). Experiments in primary cultured microvascular PASMC showed that both [Ca2+]cyt and ΔΨm were increased at a pO2 of 23 and 8 Torr. Mitochondrial ROS production (monitored using MitoSox) was also increased at a pO2 of 8 Torr. Application of either the complex IV blocker cyanide or the complex III/Qi site blocker HQNQ to isolated lungs caused a reduction of all of the cytochromes and an inhibition of HPV, with both effects developing over the same concentration ranges of each blocker.
The authors concluded that HPV was due to an increase in mitochondrial ROS production by complex III, which was linked to an increased reduction of components of the ETC associated with a subtle but significant inhibition of respiration evoked by hypoxia. They highlighted their observation that cytochrome bL/H remained more oxidized than the other cytochromes, speculating that this might reflect its involvement in ROS production, as suggested previously in a report showing that superoxide production was associated with a similar pattern of hypoxia-induced cytochrome reduction in a monocyte cell line [151].
Cox4 is one of the 10 regulatory subunits of complex IV encoded by nuclear DNA. When phosphorylated, Cox4 functions to inhibit complex IV at a high ATP/ADP ratio [376]. Cox4 exists as two isoforms, Cox4i1 and Cox4i2. The expression of Cox4i1 strongly predominates in most tissues, but Cox4i2 is also highly expressed in lung [175] and CBCC [377,378]. Cox4i2 has been shown to promote respiration [353] and therefore potentially increase ΔΨm, a key determinant of mitochondrial ROS production.
Sommer et al [130] therefore explored the relationship between ΔΨm, ROS production and downstream mechanisms causing HPV in Cox4i2 knockout and wildtype mice. They found that hypoxia (1% O2 for 5 or 10 minutes) caused a hyperpolarization of ΔΨm in primary cultured PASMC from wildtype mice. This was associated with increased mitochondrial superoxide production, a rise in cellular [H2O2], the inhibition of plasmalemmal KV channels, membrane depolarization, and Ca2+ influx. All of these effects of hypoxia were greatly diminished in PASMC from Cox4i2-/- mice. Hypoxia-induced membrane depolarization was also inhibited in human PASMC treated with anti-Cox4i2 siRNA. The results presented in this paper are too extensive to describe in detail; however, the most important evidence is listed in Figure 8.
In essence, both phases of HPV, which this laboratory had first observed in isolated perfused lung from wild type mice [352], were virtually absent in Cox4i2-/- mice. Notably, however, since their studies in PASMC utilized only brief exposures to hypoxia, the specific effector mechanisms shown in Figure 8 are not necessarily involved in phase 2 HPV, although it too was Cox4i2-dependent.
In accordance with their model, the authors found that hypoxia-induced superoxide production was restored by overexpressing wildtype Cox4i2 in PASMC from Cox4i2-/- mice. However, this was not case when Cox4i2 in which cysteine 109 had been replaced by a serine or alanine was overexpressed in these cells. Additional experiments suggested that cysteines 41 and 45 on Cox4i2 were also required for superoxide production. These results suggested that these cysteines might act as thiol switches which would allow Cox4i2 to be redox-regulated during hypoxia in such a way as to increase ΔΨm. However, their original hypothesis that Cox4i2 might be increasing ΔΨm by enhancing respiration during hypoxia was not borne out by their experiments, and they also found that mitoTEMPO did not prevent hypoxia from increasing ΔΨm, which was not consistent with redox regulation of Cox4i2 being responsible for this effect. Thus, whereas the study presents evidence that the hypoxia-induced increase in ΔΨm in PASMC was dependent on the presence of Cox4i2, it didn’t show that a specific effect of hypoxia on Cox4i2 was causing the rise in ΔΨm. . It is particularly noteworthy that HPV, recorded as an increase in PAP in isolated perfused mouse lung, was inhibited in a concentration dependent-manner by S3QEL2, which blocks ROS production by Complex III without affecting respiration.
A subsequent paper from this group [379], again employed ESR to show that hypoxia (1% O2) increased superoxide levels in mouse PASMC, and also recorded a rapid increase in cytoplasmic H2O2 using Hypercyto. In contrast, hypoxia strongly decreased superoxide levels in whole lung homogenates. A similar effect occurred in cultured lung fibroblasts, leading them to speculate that this might explain the hypoxia-induced fall in ROS in lung homogenate, which had also previously been seen in perfused lungs [278,380]. Using MitoSox, they confirmed their earlier finding [144] that hypoxia increased ROS in the mitochondrial matrix, a finding which however runs counter to measurements made using genetically encoded ROS indicators [125,361]. The authors did not discuss this discrepancy, which could conceivably reflect drawbacks of MitoSox [381].
To further investigate mitochondrial ROS production by the distal ETC, this laboratory [129] utilized AOX mice, which express an alternative oxidase derived from the marine invertebrate Ciona intestinalis. AOX becomes active if the flux of electrons through the distal ETC is slowed enough to cause substantial reduction of the quinone pool [382], creating an alternative pathway for electrons to flow from QH2 to O2 which bypasses complexes 3 and 4. This prevents over-reduction of the quinone pool while maintaining oxidative phosphorylation, with no apparent deleterious effects on mice [383]. As initially described in tobacco cells, AOX suppresses basal ROS production [384].
They observed that the pressor response to cyanide and both phases of HPV evoked by hypoxia (1% O2 in the ventilating gas mixture) in isolated perfused lungs from WT mice were almost abolished in by AOX expression but were restored by applying the AOX inhibitor n-propyl gallate. Hypoxia-induced PASMC depolarization was decreased by >50% in AOX compared to wildtype mice, and n-propyl gallate caused further depolarization in the PASMC from AOX mice, but not from controls. HPV in isolated PA recorded in control PSS was depressed in the AOX mice, but if arteries were incubated in PSS containing 20 mm K+ to evoke a degree of resting membrane depolarization, HPV in the WT and AOX mice was of similar amplitude. Perfusion of isolated lungs from AOX with high K+ PSS also produced some restoration of HPV, although it remained far smaller than that seen in wildtype mouse lung perfused with this solution. They also used ESR to show that the hypoxia-induced increase in superoxide seen in PASMC from wildtype mice was absent in those from AOX mice. Similarly, hypoxia hyperpolarized ΔΨm in PASMC from WT but not AOX mice.
In further experiments, they transfected rat PASMC with either an AOX-containing or empty expression vector and saw that the presence of AOX slightly enhanced respiration, at very low PO2. This observation that AOX increased O2 consumption under hypoxic conditions supported the notion that hypoxia was causing reduction of the quinone pool, since AOX should not be able to donate electrons to O2 unless this pool is substantially reduced [382]. In accordance with this possibility, and with their earlier observations [144], they found using Raman spectroscopy that hypoxia reduced the quinone pool and cytochrome c, but not cytochrome b, in WT PASMC. The quinone pool was also reduced in the AOX PASMC, but the redox state of cytochromes b and c was unchanged, which supported the idea that electron flow though AOX was bypassing complexes 3 and 4.
These investigations by Sommer and colleagues suggest a model in which hypoxia increases ROS production by complex III, possibly via the escape of electrons from cytochrome bH/L, and that increased reduction of the quinone pool due to a slight inhibition of respiration, as well as mitochondrial hyperpolarization, which had previously been observed by other laboratories [43,145] contribute to this response.
It is noteworthy that Cox4i2A has also been implicated in O2 sensing in the carotid body [385], reported that the normal hypoxic ventilatory response, which is dependent on O2 sensing by CBCC, was virtually abolished in Cox4i2 null mice, as was the rise in [Ca2+]cyt in CBCC which underlies the secretory response of these cells to a fall in PO2. The rise in NADH evoked by hypoxia in CBCC from wildtype controls was also absent in the knockouts. Similar effects were seen when the expression of Cox4i2 was reduced by knocking out EPas1, the gene coding for Hif1a. EPas1 knockout also diminished the expression of two other mitochondrial proteins, COX8b, another regulatory subunit of complex IV, and NDUFA4L2, which interacts with complex IV [376]. These two proteins, together with Cox4i2, have been proposed to contribute to the unusually high Km[O2] of CBCC that increases their sensitivity to hypoxia [377,378], although how they would do so is unclear.
The observation that hypoxia had no effect on [NADH] in CBCC from Cox4i2 knockouts, which suggested that it was not inhibiting respiration, is consistent with evidence from a study by Pajuelo-Riguera and colleagues which compared the PO2 sensitivity of Cox in HEK293 cells exclusively expressing either Cox4i1 or Cox4i2 [174]. Their key finding was that the Km[O]2 for mitochondrial respiration was almost ~2 fold higher in the cells expressing Cox4i2, indicating that respiration would be more depressed in these cells at low PO2 levels. The authors pointed out that this should promote reduction of the proximal ETC and of the NAD+/NADH couple, both factors known to increase mitochondrial ROS production, and was consistent with the loss of HPV seen in PA from Cox4i2 knockout mice.
Notably, however, the effect of Cox4i2 on the Km[O2] described in this paper appears to be insufficient to itself cause a meaningful depression of respiration except during severe hypoxia (see Eqn 1 in [149]), implying that its expression in O2 sensing cells might be necessary but in itself insufficient to promote mitochondrial ROS production at moderate levels of hypoxia. The authors suggested that the expression of COX8b and NDUFA4L2 might also be important for the hypoxia-induced ROS production, possibly through their interaction with Cox4i2. It would therefore be of interest to see if these proteins are expressed in PASMC, and if combined knockout of all three proteins affects HPV at moderate levels of hypoxia.
5.2.2. HPV and Mitochondrial NCLX
As described in Section 3.1.1.4, hypoxia is proposed by Hernansanz-Agustin and colleagues [183,184] to cause complex I deactivation, leading to acidification of the mitochondrial matrix. Since mitochondria contain a very substantial amount of Ca2+ bound to phosphate as a Ca3(PO4)2 complex, the dissociation of which is highly pH-sensitive [190], acidification causes an increase in free [Ca2+]mito. According to their model, this promotes Na+ entry into the matrix through the mitochondrial NCLX. The resulting rise in [Na+]mito decreases the fluidity of the IMM, impeding electron transfer from complex II and G3PD to complex III and thereby increasing ROS production at Complex IIICoQ by slowing electron flow within the Q cycle.
Although this scheme was based on data obtained from bovine aortic endothelial cells and mouse embryonic fibroblasts, they presented evidence that this mechanism is involved in O2 sensing during HPV by showing that hypoxia (1% O2) strongly increased superoxide production by mouse PASMCs, and that this response was absent in cells in which the mitochondrial NCLX was knocked down using siRNA [183]. Furthermore, the NCLXmito inhibitor CGP-37157 (30 μM) suppressed sustained HPV in isolated rat PA, although the initial peak of the response was unaffected.
However, CGP-37157 at this concentration also blocks L-type VGCC and KV channels [386,387], both of which contribute importantly to HPV. Also arguing against a role for NCXmito in HPV, Becker and colleagues found that replacement of extracellular Na+ with Li+, which should profoundly reduce intracellular [Na+] [388] and thereby prevent Na+ influx into the mitochondria through NCLXmito, did not inhibit HPV in rat PA. Similarly, treatment of PA with the Na,K – ATPase inhibitor ouabain, which should greatly increase the [Na+] in the cytoplasm and therefore in the mitochondrial matrix [389] did not inhibit HPV. Thus, HPV persisted in spite of significant perturbations of the intracellular Na+ concentration which would be predicted to disrupt the Na+ dependent regulation of the fluidity of the mitochondrial inner membrane proposed to be responsible for the hypoxia – induced rise in mitochondrial ROS production. Nevertheless, the Becker et al study was not designed to evaluate the role of NCLX in HPV and the evidence presented by Hernansanz-Agustin et al that their scheme is involved in O2 sensing is extensive; thus, a more thorough investigation focusing on the involvement of this pathway in HPV would be desirable.
5.2.3. Hypoxia and Increased ROS Production by Complex II
Paddenberg et al [126] used DCFH-DA to characterize the effect of hypoxia (1% O2) on ROS levels in PA from mouse lung slices and cultured rabbit PASMC. They observed that three hours of hypoxia increased the number of PASMC and PAECs in PA from the lung slices which exhibited an increase in the DCFH signal above the threshold level they used to indicate ROS production (DCFH-positive cells), and that one hour of hypoxia also significantly increased DCFH fluorescence in rabbit PASMC. The hypoxia-induced increase in DCFH-positive cells was abolished by DPI, rotenone, and the complex II blockers 3-NPA and TTFA. Rotenone decreased the number of DCFH-positive cells under normoxic conditions, as did antimycin, but 3-NPA did not, leading them to propose that hypoxia was acting at complex II to increase ROS production, and also that complex III was contributing to ROS production during both normoxia and hypoxia. Using a histochemical assay, they showed that hypoxia decreased the activity of succinate dehydrogenase in the lung slices. Furthermore, application of exogenous succinate prevented the hypoxia-induced increase in ROS. Based on these results they proposed that hypoxia causes complex II, which normally accepts electrons from succinate to generate fumarate, to switch to using electrons it receives from NADH via complex I and the ubiquinol pool to reduce fumarate to succinate, as a result generating ROS.
5.2.4. NAD(P)H Oxidase as a Source of Increased ROS During HPV
In 1996, Marshall et al [339] presented evidence that hypoxia increases Nox-mediated superoxide production in PAs. They employed immunohistochemistry and spectrophotometry, respectively, to detect the presence of gp91-phox and the unique Nox cytochrome (b-245) in cultured PASMC from fetal calves, and immunoblotting to demonstrate its expression in both PA and systemic arteries. Using lucigenin as a nominal superoxide indicator, they examined the effect of 20 min of hypoxia (~40 Torr) on ROS production by primary cultured SMC from small and large PA, as well as aorta and ear arteries. Hypoxia greatly increased superoxide production in each type of SMC, particularly those from small PA. This effect was absent if SOD (163 units/ml) was present, leading the authors to conclude that lucigenin was detecting an increase in extracellular superoxide. Hypoxia-induced superoxide production was virtually abolished by the Nox antagonist DPI (diphenyleneiodonium) which however has many other effects, including block of complex I[390]) but was unaffected by myxothiazol. HPV in isolated PA from cats was blocked dose-dependently by DPI, with an ID50 of ~0.81 μM, whereas the pressor response to norepinephrine was ~15-fold less sensitive to DPI. The authors concluded that hypoxia was causing an activation of Nox, rather than the inhibition which had been described previously [391] (see Section 8.1), and suggested that the Km of the oxidase for O2 (10 μM or ~7 Torr) was low enough to ensure that superoxide production would not be limited by the fall in pO2 associated with moderate hypoxia.
The role of Nox in HPV was subsequently challenged by the observation that HPV was intact in lungs from mice with chronic granulomatous disease, which lack the phagocytic form of gp91-phox (Nox2) [198]. Lungs from these mice also exhibited a profound suppression of normoxic ROS production (detected using lucigenin), and in contrast to the observations of Marshall et al [339], ROS production in isolated PA from control mice was strongly depressed by moderate (~40 Torr) hypoxia.
However, Weissmann et al [350] thereafter reported the expression of Mox-1, a different isoform of Nox (now called Nox1), in PASMC from rabbit lung. In order to study its role in HPV while avoiding problems associated with the non-selectivity of the NADPH oxidase blockers diphenyleneiodonium (DPI) and apocynin [392], they used AEBSF, a serine protease inhibitor which had been shown to block NADPH oxidase in phagocytes [393] and inhibited the response to hypoxia in carotid body [394]. AEBSF was also an flawed blocker, as it caused a substantial increase in PAP in isolated perfused rabbit lung. However, this effect was transient and when the PAP had returned to baseline level the pressor response to 10 minutes of hypoxia (O2 of ~23 Torr in the ventilating gas mixture) was depressed by AESBF in a concentration-dependent manner. The authors also demonstrated that HPV, but not the pressor response to U46619, was attenuated by the SOD inhibitor TETA, which itself did not affect PAP. These results led them to conclude that ROS production by Nox was activated by hypoxia and contributed to HPV.
Weissmann et al [351] reported that HPV in isolated perfused rabbit lung is biphasic, observing that PAP rose to reach a peak ~5 minutes after the imposition of hypoxia (phase 1), then fell by ~50% to reach a nadir at ~15 minutes, and thereafter rose gradually to reach a plateau after 2-3 hours (phase 2). The antioxidant nitro blue tetrazolium (NBT), applied at an initial concentration of 0.9 μM, strongly depressed both phases, as did TETA, applied at an initial concentration of 25 mM. DPI (initial concentration 1.5 μM), applied in the presence of L-NMMA, diminished phase 1 by ~50%, whereas phase 2 was almost abolished. DPI itself evoked a minor increase in baseline PAP, whereas TETA and NBT caused a small fall. However, Weissmann et al 2006 [352] observed that AEBSF (initial concentration 500μM) abolished phase 1 HPV in the rabbit lung, without significantly affecting phase 2. Like Archer and colleagues [212], they found that phase 1 was of similar amplitude in gp91-phox knockout mice and wildtype controls. However, phase 1 was significantly depressed in p47-phox knockouts, in which ROS production by both Nox1 and Nox2 would be depressed. In contrast, the pressor response to U46619 was similar in all three groups of mice. Baseline normoxic PAP was also not different in the p47-phox knockouts, suggesting that basal ROS production by NOX1 or 2 was not contributing to ongoing normoxic vasodilation. These data suggested that ROS production by Nox1 was partly responsible for phase 1 HPV.
In 2008, Rathore et al [39] found that protein for Nox1, Nox4 and the p22-, p47- and p67-phox components of Nox was similarly expressed in small endothelium-denuded pulmonary and mesenteric arteries from mice, but saw no expression of Nox2 in either artery. Normoxic arteries or those subjected to 5 minutes of hypoxia (1% O2) were homogenized and centrifuged to isolate a plasma membrane-containing fraction and the activity of Nox was monitored by adding NADPH and measuring the SOD-inhibitable (i.e. superoxide-dependent) reduction of cytochrome C. Nox activity was increased in PA but not MA, presumably reflecting an effect caused by hypoxia which had persisted during the isolation process. This was associated with an increased plasmalemmal expression of p47-phox, consistent with Nox activation.
Nox activity during hypoxia was strongly suppressed by the nominal Nox blocker apocynin (1μM) and was smaller in PA from p47-phox knockouts compared to wildtype controls. Similar effects were also observed in PA from PKCe knockout mice comparted to wildtype controls and in PA treated with a peptide inhibitor of PKCe translocation (10 μΜ) but not Gö6796 (0.1 μM), a blocker of conventional PKC isoforms. Although it’s not clear that these interventions diminished the effect of hypoxia per se, since their effects on Nox activity under normoxic conditions were not described, parallel experiments utilizing DCFH as an indicator showed that in each case hypoxia-induced ROS production was attenuated in freshly isolated PASMC with no effect apparent during normoxia. Cell shortening and rises in [Ca2+]cyt in response to hypoxia were also decreased in PASMC treated with apocynin and in those from p47-phox -/- mice, whereas [Ca2+]cyt and the contractile response to caffeine were not affected under normoxic conditions.
The authors also observed that Nox activity during hypoxia was strongly suppressed by rotenone and myxothiazol. They had previously found that hypoxia increased the activity of PKCe in PASMC, that this effect was also sensitive to these ETC blockers, and that the expression of PKCe in PA was much higher than in MA [395]. They therefore proposed that increased mitochondrial ROS production in PASMC due to hypoxia was activating PKCe, causing a stimulation of Nox1 that generated additional superoxide which acted via effector pathways to cause HPV. They suggested that the Nox2 expression which had previously been reported in mouse lung might [212] might occur mainly in non-SMC or in pulmonary veins. They also pointed out that the lack of effect of blocking or knocking out p47-phox on basal [Ca2+]cyt or the caffeine-induced contraction was not consistent with the concept that that ongoing Nox-mediated ROS production maintains a tonic normoxic vasodilation which can be reversed by hypoxia (see Section 8).
In 2011, Frazziano et al [323] similarly detected a role for both mitochondrial and Nox-derived ROS production in HPV evoked in small rat PA by 10 minutes of hypoxia (22-26 Torr). This group had earlier proposed that HPV is due to the activation of neutral sphingomyelinase 2 (nSMase-2), this causing an increased production of ceramide and a consequent stimulation of PKCζ which acts to depress the KV current [396,397]. They observed that the increase in cellular ceramide caused by hypoxia was abolished by rotenone, suggesting its dependency on mitochondrial ROS production. Application of exogenous ceramide caused the production of ROS (monitored using DCFH and DHE), and this was blocked by a PKCζ – inhibitory peptide. Hypoxia also caused the phosphorylation of p47-phox and its association with caveolin-1, and this was blocked by the PKCζ – inhibitory peptide [323]. They also found that 300 μM apocynin strongly antagonized HPV. Based on these results, they proposed that nSMase-2 is stimulated by mitochondrial ROS, generating ceramide which activates Nox via a PKCζ -mediated phosphorylation of p47phox, leading to the production of ROS which inhibit KV channels to cause HPV. The evidence that hypoxia induced a PKCζ – dependent phosphorylation of p47phox which was blocked by rotenone supports the model developed by Rathore et al (2008) in that mitochondrial ROS would act through PKC to promote further oxidant signaling by Nox, although the two schemes differ with regard to which PKC isoform causes p47phox activation.
It is notable, however, that the effects of apocynin described in these papers are uninterpretable, since Heumüller et al [345] showed that it blocks Nox only after being converted by myeloperoxidase into a dimer and that this does not occur in VSMC, which lack this enzyme. Apocynin was also found to act as an antioxidant which interacts with both biomolecules and with ROS indicators such as lucigenin.
In contrast to the findings of these papers, Connolly et al (2013) [18], who studied HPV in rat IPA in the absence of pre-tone (under which condition Phase 1 HPV asismall), found that the Nox inhibitor VAS2870 (10 μM ) had no effect on HPV, although the broad spectrum PKC antagonist Gö6983 (3 μΜ), which would be expected to block both PKCϵ and PKCζ [398], reduced HPV by ~30%.
Veith and colleagues [399] compared the effect of hypoxia (1% O2) in perfused lungs from wild-type and Nox4-/- mice, finding that neither phase of HPV was different in these two groups. The basal right ventricular systolic pressure (RSVP), an indicator of the PAP, was also not significantly affected by Nox4 knockout. Murtaza et al [400] subsequently compared the effect of hypoxia on pre-acinar and smaller intra-acinar arterioles in lung slices from these two groups, and saw that wildtype intra-acinar arteries demonstrated a substantial sustained constriction which was halved in the Nox4 knockouts, whereas the pre-acinar arteries showed a smaller response which was similar in the two groups. In view of the results from Veith et al, they suggested that constriction of intra-acinar arteries does not contribute to the overall hypoxia-induced rise in PAP [401] but might be involved in the redistribution of blood flow responsible for ventilation-perfusion matching.
Nagaraj et al [402] recorded HPV (evoked by 1% O2) in perfused lungs from wild type and p22phox-/- mice, reporting that whereas the basal normoxic PAP and initial transient phase of HPV were similar the two groups, the sustained phase was decreased by ~40%. They also showed that hypoxia increased the activity of RhoK in lung slices from wildtype mice, and that this effect was absent in the p22phox knockouts. Since Noxs 1 and 4 are the isoforms most likely to be involved in HPV, and both require p22phox for their activity, these results suggest that ROS production by Nox1 may contribute to sustained HPV by stimulating Rho kinase-mediated Ca2+ sensitization.
The Role of NADPH Oxidase in HPV: Summary and Conclusions
Although Nox2 is not involved in HPV [212], at least in mice, the studies in rodents described above generally support the possibility that HPV depends to some extent on a hypoxia-induced activation of ROS production by Nox1 which is dependent on PKC, a well-characterized activator of Nox1/2 [60]. The possibility that activation of Nox1 by 5 minutes of hypoxia is dependent on mitochondrial ROS production acting via PKCe, as evidenced by Rathore and colleagues [39,395], is also is consistent with observations that the amplitude of phase 1 HPV in mice was decreased by ~1/3 when p47-phox was knocked out (Weissmann et al, 2006)[352], but was abolished in Cox4i2-/- mice, along with superoxide production [130]. The involvement of a mitochondrial ROS-PKCe -Nox1 axis in the initial phase of HPV is also supported by evidence that the pressor response to a 5 minute hypoxic challenge in isolated perfused rat lung was decreased by ~50% [403] in PKCe knockout mice, and that the rapid increase in [Ca2+]cyt evoked by hypoxia was strongly depressed in PASMC from these knockouts and from wildtype mice treated with PKCe inhibitor peptide [395]. Moreover, the finding that Ro-31-8220, which blocks multiple PKC isoforms including PKCe depressed phase 1 HPV in rat intrapulmonary arteries but had no effect on phase 2 [404] supports the lack of involvement of this pathway in sustained HPV. The investigation by Nagaraj et al [402] also implicates Nox1 in HPV. However, they observed that it was phase 2 rather than phase 1 HPV which was Nox-dependent. The reason for this discrepancy is unknown.
Importantly, with the exception of the initial paper by Marshall et al [339], the investigations described above ignored the question of whether the Km (O2) for Nox has an impact on ROS production during hypoxia. The Km(O2) for Nox1 has not been reported, but if it is similar to that of Nox2 (~25 torr) [59], its ability to generate ROS would be diminished at the levels of hypoxia used by most laboratories to study its involvement in HPV. This would call into question the plausibility of the findings that ROS production by Nox1 was increased at this level of hypoxia, but at the same time would imply that if these observations are valid, these studies may have greatly underestimated the ability of Nox1 to produce ROS and therefore contribute to HPV at more moderate levels of hypoxia.
This issue is even more salient with regard to the Km(O2) for Nox4, which has been shown to be ~136 Torr [59,405]. This implies that using very low levels of PO2 to impose hypoxia is unsuitable for examining whether increased ROS production by Nox4 contributes to HPV evoked by moderate hypoxia, since severe hypoxia should essentially shut the oxidase down. On the other hand, its high Km(O2) means that Nox4 is well suited to act as an O2 sensor which responds to acute hypoxia by decreasing rather than increasing the production of H2O2 [59,405]. Indeed, as described in Section 8, it has been suggested that a fall in H2O2 production by Nox4 is instrumental in causing HPV in bovine pulmonary arteries.
5.2.5. HPV Effector Mechanisms Coupled to an Increase in PASMC [ROS]
The Redox theory from the outset incorporated KV channel inhibition as the essential HPV effector mechanism. In contrast, Schumacker’s laboratory did not include any specific effector mechanisms in their initial presentation of the Mitochondrial ROS theory [38], although they later suggested [348] that increases in cytoplasmic ROS might be working though activation of phospholipase C [406], which would cause an increase in IP3 production leading to SR Ca2+ release mediated by the IP3R (see also [407]). Subsequently, it has been demonstrated that SR Ca2+ release mediated by the RYR [408] and SOCE mediated by STIM1/TRPC1/Orai1 [409] are both activated in a ROS-dependent manner during HPV. H2O2 was also shown to cause an increase in [Ca2+]cyt in PASMC mediated by SR Ca2+ release through IP3R and RyR, as well as Ca2+ influx through Ni2+- sensitive cation channels [410].
Other pro-contractile mechanisms mobilized by acute hypoxia in PASMC, including Ca2+ influx via TRPC6 channels [11,411,412] and the activation of several protein kinases [413], such as PKC [25,323], Src-family kinases [414,415] [416] and RhoK [416], have also been shown to be stimulated by ROS. It has also been demonstrated that the IP3R, which may contribute to SR Ca2+ release during HPV [3] is activated by ROS released from the mitochondria via oxidation of a group of cysteines located in its N-terminal suppressor domain [92,259,260]. Furthermore, as described in Section 5.2.4, Cogolludo and colleagues have provided evidence that an increase in ROS production by the mitochondria and Nox inhibits the opening of KV1.5 channels in PASMCs though a mechanism involving nSMase-2), ceramide and PKCζ [323,396].
6.3. H2S, ROS and HPV
Olson and colleagues proposed that hypoxia, by inhibiting the O2-dependent metabolism of H2S, causes a rise in its cellular concentration which constitutes the PO2 -sensitive signal triggering HPV. This was based on observations that 1. applying exogenous sulfide caused constricting or dilating effects mimicking those of hypoxia in multiple blood vessels from diverse species 2. blockers of sulfide-producing enzymes inhibit HPV, and 3. sulfide production by lung homogenates and pulmonary vascular preparations is increased at low PO2 [46]. However, the contention that sulfide as an O2 sensor in HPV has been challenged [434,435]. Subsequently, Olson suggested that hypoxia might also increase cellular levels of polysulfides and other reactive sulfur species (RSS), which exist in equilibrium with cellular sulfide, but at much higher concentrations [47]. Like ROS, these are feasible HPV mediators since they can regulate protein function by causing oxidative modification of reactive thiols via sulfuration [436]. However, evidence that hypoxia can elevate sulfide or polysulfide levels in PASMC quickly enough to trigger HPV is lacking. Indeed, production of sulfide and polysulfides in bovine pulmonary artery SMC was not potentiated during the first 5 hours of a hypoxic (5% O2) challenge [437].
Nevertheless, the mechanisms of the sustained contractions of rat PA induced by applying exogenous sulfide and hypoxia demonstrate many similarities [18,166]. In both cases, contraction was insensitive to the VGCC antagonist nifedipine, but was inhibited by the antioxidant TEMPOL, and by blockers of the RyR (ryanodine), RhoK (Y-27632), PKC (broad spectrum inhibitor Gö6983), and complex IIICoQ (myxothiazol). Moreover, sulfide mimicked hypoxia by causing mitochondrial hyperpolarization [130,144]. Sulfide also increased ROS production by PASMC, an effect that was abolished by the knockdown of sulfide quinone oxidoreductase (SQOR), a protein located in the IMM. SQOR forms part of the ‘sulfide oxidation unit’ which metabolizes sulfide and in doing so feeds electrons into the CoQ pool and stimulates respiration [167,438]. The contraction of trout gill to sulfide was also antagonized by an antioxidant (DETCA) and blockers of mitochondrial complexes 1, 3 and 4 [439]. In light of the proposal that sulfide stored within the mitochondria as thiosulfide can be mobilized by hypoxia [164], as well as the high affinity of SQOR for sulfide [167], it is conceivable that a small hypoxia-induced rise in intramitochondrial [sulfide] could act through SQOR to increase mitochondrial ROS production at complex IIICoQ, thereby causing HPV.
6.4. Effects of Hypoxia on Redox Couples
Cells contain several important redox couples, each of which regulates different aspects of cellular function. Although the three main redox couples (GSSG/GSH, NADP+/NADPH, NAD+/NADH) interact in important ways, the flow of electrons between them is regulated enzymatically and by membrane transport systems, so that they are not in thermodynamic equilibrium [266]. The redox potential of each couple also differs between cell compartments.
The GSH/GSSG couple functions as a conduit to transfer electrons from NADPH to cell proteins, causing their reduction, but GSSG is also used for protein glutathionylation, thereby contributing to oxidative redox signaling. Although its involvement in maintaining overall cellular antioxidant defence has been unclear, and has been overshadowed by that of the thioredoxin/peroxiredoxin pathway, a recent investigation suggests that it may it may be more important than has been previously thought, at least in some type of cells [89]. As shown in Table 5, Connolly et al (2013)[18] did not detect a difference between the normoxic redox potentials of the GSH/GSSG couple in rat PA and aorta, which are exposed to a similar normoxic plasma PO2 in vivo, [49]. They also observed that 45 minutes of hypoxia caused the oxidation of this couple in PASMC. Similarly, the GSH/GSSG ratio in cultured rat PASMC was decreased by 2 hours of hypoxia [356]. These observations are predicted by the mitochondrial ROS model and do not support the Redox theory, which posits that the cytoplasm of PASMC is relatively oxidized under normoxic conditions and becomes more reduced during hypoxia, whereas in comparison systemic SMC are more reduced in normoxia and become oxidized during hypoxia. However, the lengthy exposures to hypoxia used in both studies means that their results may not reflect the immediate response to hypoxia.
Measurements of pyridine nucleotide (i.e. NAD(P)H) fluorescence in rat and bovine PA suggest that the NAD+/NADH and/or the NADP+/NADPH redox couples may become more reduced during hypoxia [141,346,440,441]. However, these fluorescence measurements did not distinguish between NADH and NADPH, or between their free and bound forms. Since it is the concentrations of the free forms of both pyridine nucleotides which determines their redox potentials, and these constitute only a small fraction of their total contents [442], the effects of hypoxia on the individual redox potentials of both couples remain unclear.
The NADPH/NADP+ couple is the ultimate source of electrons which are used by redox relays to reduce oxidized proteins but also provides the electrons which are used by Nox to produce superoxide/H2O2. The role of NADPH in determining pulmonary vascular tone during HPV has been investigated in bovine pulmonary artery by Michael Wolin, Sachin Gupte, and colleagues. This work, which is described in Section 8, has shown, somewhat paradoxically, that a hypoxia-induced increase in NADPH may contribute to HPV both by causing the reduction of PKGa1 via Trx-1/TrxR-1, and by increasing ROS production via Nox, presumably leading to the oxidation of other as yet unidentified effector proteins.
The NADH/NAD+ couple is mainly involved in controlling respiration by providing reducing equivalents to the ETC, and also acts to reduce mitochondrial NADP+ via the NNT, thereby helping to maintain the mitochondrial matrix in a highly reduced state [58,87]. Because its redox potential is much less negative than that of NADP+: NADPH, the NAD+:NADH couple is not directly involved in regulating thiol oxidation [442]. However, via the NNT, it exerts an important indirect influence on the NADP+/NADPH ratio, and therefore thiol signaling, in the mitochondrial matrix. An increase in mitochondrial [NADH] also promotes superoxide production by Complex I, both directly, and indirectly through its effects on KGDH and PDH [87] (Section 3.1.1.3 and Section 3.1.1.4).
Although an increase in NADH has been suggested to cause HPV through a ROS-independent pathway (inhibition of cyclic ADPR hydrolase) [443], the impact of changes in the cytoplasmic or mitochondrial NAD+/NADH ratio on redox signaling during HPV remain unexplored. However, Lopez-Barneo’s laboratory has proposed that a rise in cytoplasmic [NADH] due to a direct effect of hypoxia makes an important contribution to O2 sensing in femoral artery myocytes [444] by suppressing voltage-gated Ca2+ currents, and in CBCC by inhibiting KV channel opening [179,445]. A role for NADH in O2 sensing is supported by evidence that hypoxia can increase the cytoplasmic free [NADH] in coronary artery myocytes [267], and that NADH binds to KV channel β subunits with a high affinity, thereby affecting channel activity [446]. However, the hypoxia-induced increase in the cytoplasmic [NADH]/[NAD+] ratio, at least in coronary myocytes, was quite small, and the concentration of NADH used to characterize its effect on the KV current in CBCC (200 μM) was almost certainly much higher than those present in cells, since the NAD+/NADH couple in the cytoplasm is almost entirely oxidized (i.e. NAD+ >> NADH). This is potentially problematic since 1. NADPH also binds to KV channel β subunits, 2. the cytoplasmic NADP+/NADPH redox couple is almost completely reduced so that NADPH >> NADP+ [266], and 3. the affinity of NADPH for the β subunit is even higher than that of NADH. As a result, it has been questioned whether physiological levels of cytoplasmic free NADH can regulate KV channel activity, since they may not be sufficient to compete with NADPH for binding to KV b-subunits [268].
It is noteworthy, however, that because the redox state of the cytoplasmic NAD+/NADH couple is strongly influenced by the lactate/pyruvate ratio, increases in cellular lactate which can occur under conditions such as systemic hypoxia can lead to a significant rise in cellular [NADH], which may contribute to O2 sensing in CBCC [447].
7. Critique of the Redox and Mitochondrial ROS Theories
As described in Section 5.1 and Section 5.2, the Redox and Mitochondrial ROS theories propose opposite effects of hypoxia on mitochondrial ROS production in PASMCs. The Redox model is based mainly on work by Stephen Archer, Kenneth Weir, Evangelos Michelakis and colleagues, whereas the most salient findings supporting the Mitochondrial ROS theory have come from the labs of Paul Schumacker, Norbert Weissmann and Yong-Xiao Wang. Although the papers from the proponents of these two theories present apparently irreconcilable bodies of evidence, other authors have also published results which can be used to assess the extent to which the literature as a whole supports the predictions of either theory.
Predictions of the Redox theory: The key tenet of the Redox theory is that hypoxia causes contraction by decreasing mitochondrial ROS production. In setting forth the Redox theory [19,33,204], Archer and colleagues have invoked observations in the literature which are in line with the following predictions:
- 4.
- Blockers of the ETC which induce a fall in mitochondrial ROS production should mimic hypoxia by causing inhibition of PASMC K+ currents and contraction and should also prevent HPV.
- 5.
- Similarly, anti-oxidants should cause a sustained contraction. Their effect on HPV is more difficult to predict, since a small anti-oxidant effect might add to that of hypoxia to enhance HPV, while a large anti-oxidant effect could abolish any further response to hypoxia.
- 6.
- Pro-oxidants should prevent and reverse HPV.
Most importantly, hypoxia should diminish mitochondrial production of ROS in PASMC, thus causing a fall in cytoplasmic [ROS] and/or a reduction of cytoplasmic redox couples involved in regulating reactive thiols, particularly on KV channels.
The Redox theory also predicts that hypoxia, reductants, and blockers of the ETC which decrease mitochondrial ROS production should inhibit PASMC KV currents, whereas oxidants should have the opposite effects. Whereas it is well established that these currents are indeed suppressed by hypoxia in PASMCs, as described in Section 5.1.2 there is no direct evidence (e.g. changes in reactive thiol oxidation or interactions between KV a and b subunits in native PASMCs) that this due to redox mechanisms.
Predictions of the mitochondrial ROS model: The Mitochondrial ROS theory is predicated on the concept that hypoxia increases mitochondrial ROS production in PASMC, causing a rise in [ROS] in the mitochondrial IMS and the cytoplasm which induces the oxidation of cytoplasmic redox couples which regulate reactive thiols. This effect of hypoxia is seen as not being unique to PASMC, and indeed the Mitochondrial ROS theory grew out of observations that hypoxia increased ROS production in cardiomyocytes [342]. In their reviews, Schumacker and colleagues have focused mainly on evidence that hypoxia increases ROS in PASMC and in other types of cell rather than emphasizing the effects of ETC blockers, anti-oxidants and pro-oxidants on HPV. However, the responses of PASMC to these agents predicted by this hypothesis are reasonably clear:
- 7.
- Blockers of the ETC which diminish mitochondrial ROS production should not cause contraction under normoxic conditions but should inhibit HPV.
- 8.
- ETC blockers like myxothiazol or rotenone which act at or upstream of the Qo site of complex III should inhibit HPV. Antimycin, shown by investigators to increase complex III-mediated ROS production in many types of cells, would be expected to cause PASMC to contract during normoxia, but its effects on HPV are difficult to predict.
- 9.
- Anti-oxidants should have no effect on PASMC tension in normoxia, but should block HPV.
In accordance with this theory, there is evidence that multiple contractile effector pathways in PASMC are activated by oxidation, although again identification of specific reactive cysteines oxidized in the constituent effector proteins of these pathways in these cells is lacking.
In the next several sections, we critically assess these theories by examining the results from the substantial number of papers which have investigated the effects of ETC blockers, and anti- and pro-oxidants on PASMC contraction and cytoplasmic Ca2+ levels under normoxic and hypoxic conditions. We also review the effects of hypoxia on PASMC [H2O2], superoxide and the ambient cytoplasmic thiol/disulfide equilibrium which have been obtained from studies employing genetically encoded ROS indicators or ESR. Finally, having considered this evidence, as well as the literature relating to the effects of ETC blockers and pro- and anti-oxidants on PASMC KV currents, we describe what we feel are the strengths and weaknesses of the Redox and Mitochondrial ROS theories.
7.1. Pulmonary Effects of ETC Blockers
Observations that blockers of the proximal ETC decrease PASMC [ROS] production [145,263], increase PAP in perfused lung or cause PA constriction [145,263,280], block HPV [263], and mimic the effects of hypoxia on K+ currents in SMC from PA [145,263], renal arteries [145] and ductus arteriosus [448] have consistently been invoked to support the Redox theory [19,33,36,125,263]. It is noteworthy, however, that the meaning of the term ‘proximal ETC’ used in these papers varied; in some cases it referred to the portion of the ETC upstream of ubisemiquinone [19], whereas in others it included the Qi site in complex III, since antimycin is described as a blocker of the proximal ETC [33,125]. This ambiguity is worth keeping in mind, since, as described below, it is unlikely that antimycin would produce the same effect on mitochondrial ROS production as drugs such as rotenone which act upstream of ubisemiquinone.
Results of studies of the effects of ETC blockers on PASMC, isolated PA, or lung preparations are summarized in Table 2. Rotenone has been used more than any other ETC blocker to examine the role of mitochondrial ROS production in HPV and was seen to decrease basal ROS levels in most studies where this was examined. Rotenone generally increased basal tension in normoxia and was almost always observed to strongly inhibit HPV or reverse hypoxia-induced rises in [Ca2+]cyt. If rotenone was truly decreasing ROS production (which is however questionable [109,117,136,256,445,449,450]) these effects are predicted by the Redox theory. However, as noted previously [3], the generally observed transience of the rotenone-induced contraction does not accord with this model. The ability of rotenone to block HPV is also consistent with the Mitochondrial ROS theory (since it would oxidize the CoQ pool and thereby diminish ROS production by complex III), although the rise in basal tension it caused argues against this scheme.
Table 2.
Effects of ETC blockers on PASMC, PA or perfused lung preparations.
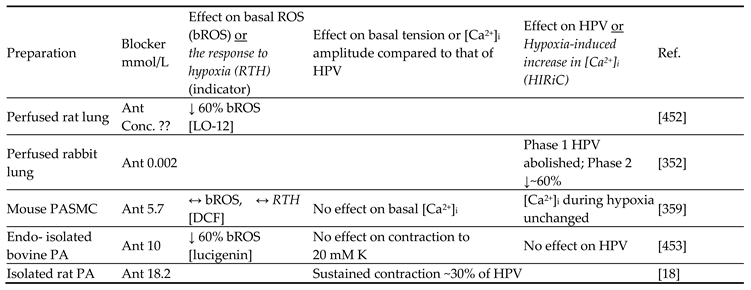
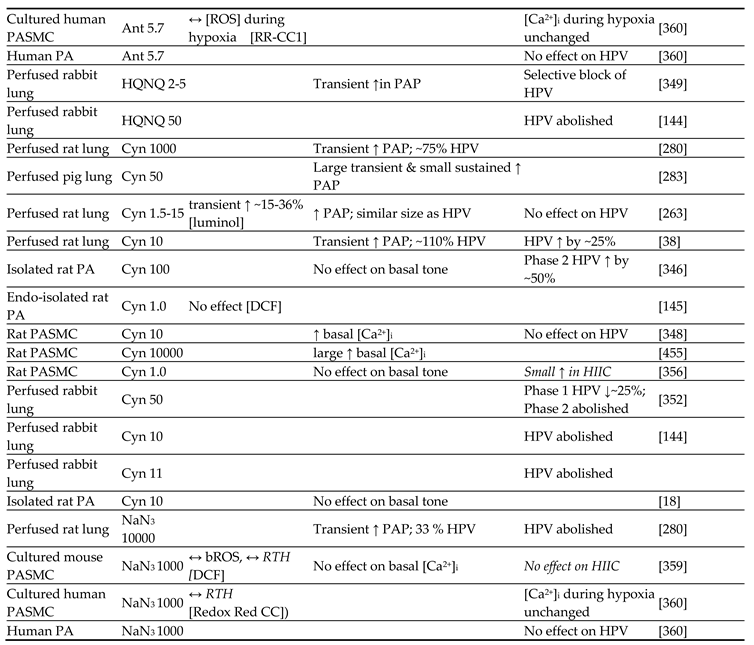
Complex I blockers: Rotenone (ROT), 1-methyl-4-phenylpyridinium (MPP+); Complex II blockers: 3-nitropropionic acid (NPA), 2-tenoyltrifluoroacetone (TTFA); Complex III blockers: myxothiazol (Myx; blocks at Qo), antimycin (Ant; blocks at Qi), 2-n-heptyl-4-hydroxyquinoline-N-oxide (HQNO; blocks at Qi). Complex IV blockers: cyanide (Cyn), sodium azide (NaN3) 1 Endo - = endothelium-denuded. Abbreviations: RTH = Response of ROS signal to hypoxia; DCF = dichlorofluorescein; HIIC = Hypoxia-induced increased in {Ca2+] signal.
Unlike rotenone, myxothiazol was not observed to alter basal ROS production in any of the three studies in which this was studied. Studies of mitochondria from various tissues show that myothiazol generally has no effect on, or increases, basal mitochondrial ROS production, depending on the cell type and substrates used to fuel oxidative phosphorylation [114,120,344,456,457]. Myxothiazol strongly antagonized the hypoxia-induced increase in ROS recorded in several studies and was consistently seen to depress HPV without itself altering basal tone or [Ca2+]cyt. Since myxothiazol blocks ROS production by complex III (although it can increase ROS release upstream of complex III), this combination of effects is generally consistent with the Mitochondrial ROS theory. In light of the observations that myxothiazol did not change basal [ROS], its lack of effect on basal tone also fits with the Redox Theory. However, since the potential effects of myxothiazol on any hypoxia-induced decrease in ROS production by complex I in PASMC are uncertain, the implications for the Redox theory of the block of HPV it exerted are unclear.
Antimycin was seen to lower (6 studies) or not affect (2 studies) ROS levels. These results are perplexing, since there is a widespread consensus, emerging from numerous studies in multiple types of cells, that antimycin increases ROS production by complex III, and can also do so at more proximal sites in the ETC by causing reduction of the CoQ pool [114,121]. If antimycin was indeed causing a paradoxical fall in basal ROS production, the increase in basal contraction it usually (in 4 of 5 studies) evoked was in accord with the Redox theory. However, although antimycin blocked HPV in 3 studies, as predicted by the Redox theory, it more often (6 studies) had no effect on or increased the response to hypoxia, which does not conform to this model. On the other hand, the normoxic contraction it often caused is consistent with the Mitochondrial ROS theory [38,342], although the apparent fall in ROS production is not. In any case, since no single investigation of antimycin in PA or PASMC evaluated its effects on all of the key parameters, (basal [ROS] and tension development or [Ca2+]cyt in normoxia and hypoxia), it is difficult to draw any meaningful conclusions from its observed effects.
The effect on HPV of the complex IV blockers cyanide and NaN2, is difficult to predict from either the Redox or Mitochondrial ROS hypotheses. However, since these drugs would be expected to mimic the inhibition of complex IV caused by hypoxia, they can induce reduction of the CoQ pool which might increase ROS production by complex III. Thus, evocation of contraction by these drugs would be consistent with the Mitochondrial ROS hypothesis [348], although this would probably depend on the whether the degree of the block was similar to that caused by hypoxia. Cyanide was observed to cause contraction in most studies in which it was used, this was never seen with azide. The reasons for this difference are not obvious.
In summary, the use of conventional ETC blockers to study HPV has yielded inconsistent results which, when taken together, do not provide convincing support for either the Redox or Mitochondrial ROS models. More importantly, although these drugs, if employed intelligently, can provide important information about how hypoxia influences mitochondrial ROS production, their use has a number of drawbacks which need to be taken into account when interpreting their effects. Firstly, the effects of ETC blockers on overall mitochondrial ROS production are difficult to predict. For example, rotenone inhibits ROS production at complex ICoQ and downstream sites of the ETC, but tends to have the opposite effect on complex IF, OF and PF [109]. Secondly inhibition of respiration caused by ETC blockers has manifold impacts on mitochondrial function which go beyond their direct effects on ROS production [458]. One of these which is likely to have immediate consequences for vascular tone is that ETC blockers depolarize DΨm in isolated mitochondria and intact cells from many cells/tissues, including pulmonary artery and other SMC [459,460,461]. This inhibits mitochondrial Ca2+ uptake, leading to complex effects on Ca2+ signaling in SMC which generally tend to inhibit contraction by interfering with the generation of Ca2+ waves [461,462,463] and by causing the opening of BKCa channels, even though global [Ca2+]cyt may not change [463] or even increase [461]. Also, by depressing O2 consumption, inhibition of respiration can increase the intracellular O2 concentration [464,465,466]. This could potentially decrease the effect of hypoxia on extramitochondrial O2 sensors, thereby attenuating HPV. In addition, there is evidence from Hernansanz-Agustin et al [183] that rotenone can both mimic and prevent hypoxia-induced complex I deactivation. In addition, rotenone at concentrations of > 0.5 μM as used in many but not all HPV studies, can block K+ channels [321]. Finally, it is apparent from the comprehensive investigation by Weissmann and colleagues that the observed effects of ETC blockers on PAP in perfused lungs are greatly distorted by the effects of NO release [349].
7.2. Pulmonary Vascular Effects of Antioxidants
As shown in Table 3, antioxidants have often been used to characterize how ROS affect pulmonary vascular tone under normoxic and hypoxic conditions.
The enzymes SOD and catalase have been used more frequently than any other antioxidant agents to study HPV. In most investigations, these enzymes, used either alone or in combination, had no effect on basal tone or HPV. It is unclear whether this reflected a genuine lack of response or was due to their poor entry into cells. However, application of high concentrations of SOD and catalase together to isolated perfused rat lung greatly increased HPV with no effect on basal PAP, and treatment with liposomes containing a low concentration of both enzymes, a procedure designed to increase their entry into cells, increased basal tone and caused a smaller enhancement of HPV [276]. These responses are generally in accord with the Redox theory. Conversely, HPV in pig PA was inhibited by the combination of SOD and catalase [353], which supports the mitochondrial ROS hypothesis. Interestingly, in both preparations SOD on its own exerted an effect similar to that of SOD and catalase in combination, which implies that superoxide itself must have been acting as a mediator in HPV.
Most of the studies which employed chemical antioxidants found that they inhibited HPV without affecting basal tone. Nevertheless, an antioxidant – induced increase in basal tension, which would be predicted by the Redox theory, was seen by some investigators. This was observed most often in bovine pulmonary arteries (BPA), in which ebselen, DTT and DHLA raised basal tone (although NBT and Tiron did not). HPV in BPA was inhibited by NBT and ebselen, not affected by Tiron, and increased by DTT and DHLA. It is difficult to fit these results into a coherent pattern, although Wolin and colleagues, who carried out most of these investigations, concluded that a fall in [ROS] contributes to HPV (see Section 8). However, they also found that HPV in BPA is not blocked by rotenone [453], implying that O2 sensing in BPA is independent of mitochondrial ROS production. Likewise, Olson’s laboratory, which reported the effects of DTT and DHLA, ascribed these to an increase in cellular [H2S] rather than to an effect on mitochondrial [ROS] [164].
Apart from the experiments carried out in BPA, Table 3 shows that studies employing antioxidants have mostly reported that they inhibit HPV but do not affect basal PA tone or [Ca2+]cyt tends to support the Mitochondrial ROS theory.
Nevertheless, although if employed correctly antioxidants can provide information about how ROS regulate cellular processes, their use for this purpose, especially if not supported by parallel measurements of cellular superoxide and H2O2, has come under increasing criticism [381]. Each antioxidant has a unique mechanism of action, which is often complex and/or not fully understood (e.g. TEMPOL [467]; TIRON [468] N-acetylcysteine [469]; ebselen [470]). Also, some antioxidants (e.g. N-acetylcysteine, manganese(III)tetrakis(1-methyl-4-pyridyl)porphyrin-- MnTMPyP) can increase the formation of reactive sulfur species [469,471] which could potentially mimic the effect of H2O2 on reactive cysteines [48]. In addition, SOD-mimetics such as MnTMPYP would be predicted to cause an increase in cellular H2O2 along with the fall in superoxide [472] although this may not be the case for TEMPOL and Tiron, which, although often described as SOD-mimetics, also apparently suppress the production or effect of H2O2 [467,473]. This is an important consideration when considering the effects of antioxidants on HPV, as most studies, particularly those using perfused lungs, did not include measurements of ROS (and those that did used ROS indicators which have since been largely discredited).
The overexpression or knockdown of cellular anti-oxidant enzymes allows the manipulation of redox tone in specific cellular compartments, and avoids some of the uncertainties inherent in the use of chemical anti-oxidants or proteins such as SOD and catalase [381]. Investigations using cultured PASMC from mice in which catalase was overexpressed by transfection [348,363] found that this blunted the rise in [Ca2+]cyt evoked by hypoxia in PASMC. Freshly isolated PASMC from mice which had been genetically modified to overexpress catalase also demonstrated a decreased hypoxia-induced increase in [Ca2+]cyt compared to cells from wild-type controls [359]. A similar effect was seen in PASMC from mice in which glutathione peroxidase had been overexpressed in order to enhance the metabolism of H2O2, whereas cells from glutathione peroxidase knockouts demonstrated an augmented response of [Ca2+]cyt to hypoxia. It could be argued, in line with the Redox theory, that catalase or glutathione peroxidase overexpression was causing a basal increase in [Ca2+]cyt by reducing cytoplasmic redox couples, thereby preventing a further similar effect of hypoxia. This possibility, however, is not supported by the observation [359] that the basal level of [Ca2+]cyt was not altered in PASMC from any of the genetically altered mice. Therefore, these results consistently agree with the predictions of the Mitochondrial ROS theory.
More recently, Sommer et al [129] reported that HPV was absent in isolated perfused lungs from mice expressing AOX, which has an anti-oxidant effect since it diminishes ROS production by Complex III [383]. Basal superoxide production was similar in the PASMC from AOX mice and wildtype controls, as was the resting membrane potential, indicating that baseline ROS production by Complex III was not influencing the cytosolic redox state or K+ channel activity under normoxic conditions. These results provide strong evidence favoring the Mitochondrial ROS theory.
Table 3.
Effects of antioxidants on HPV.
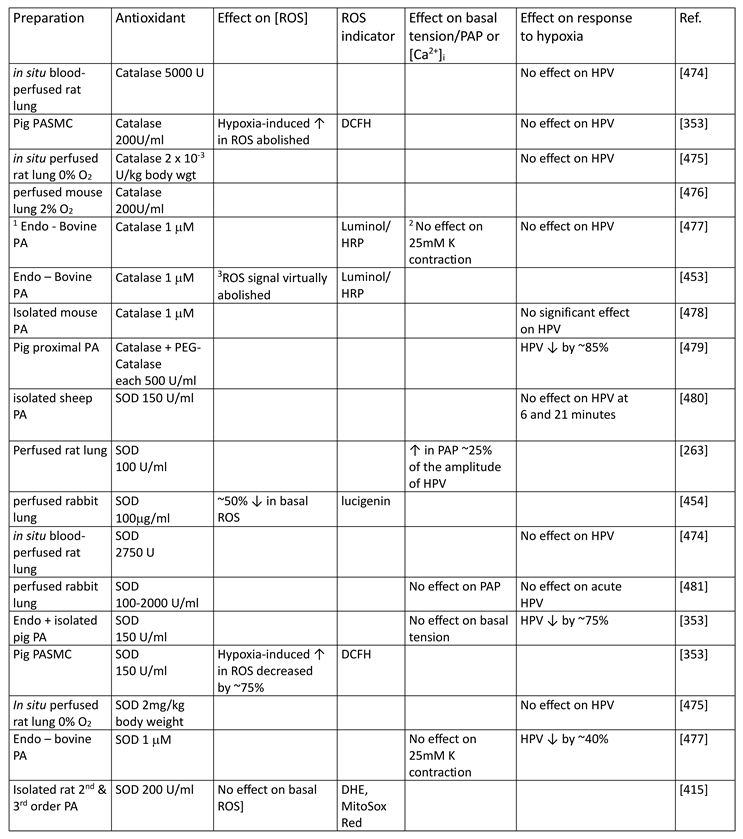
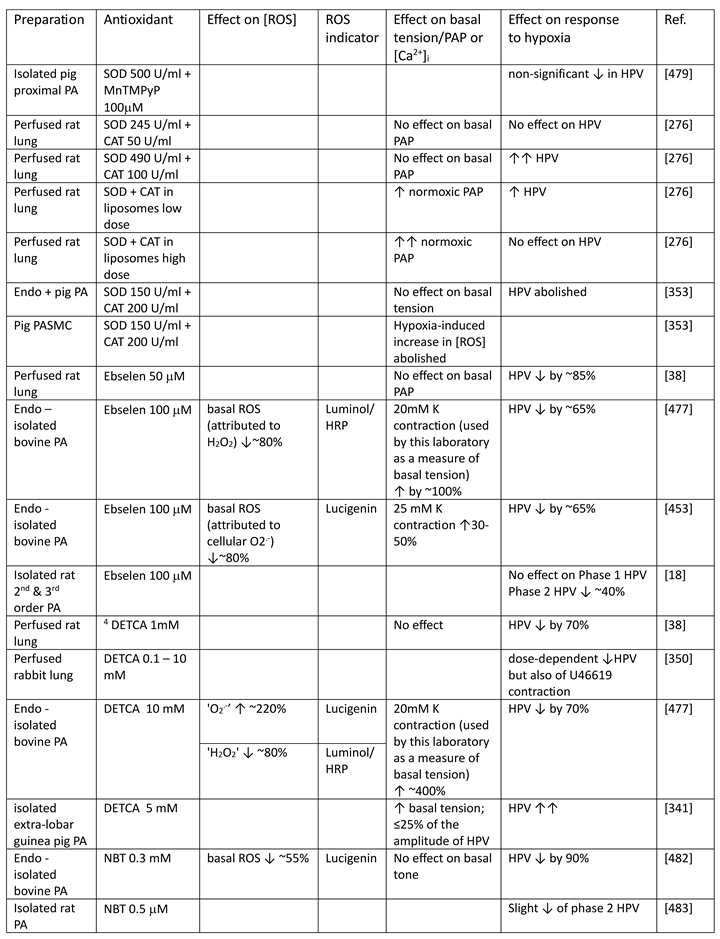
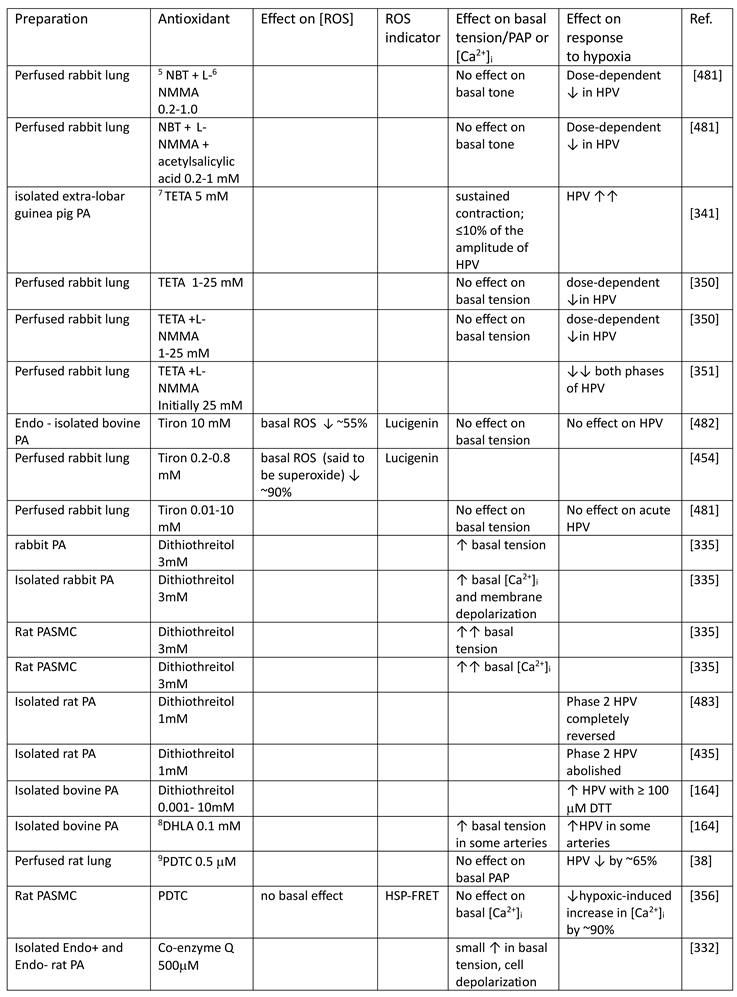
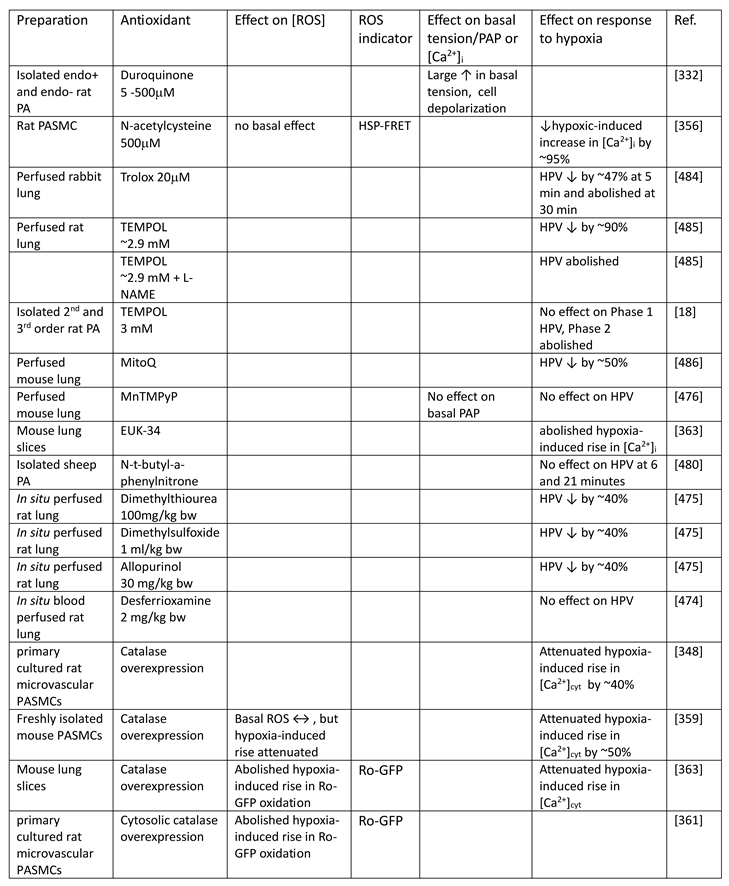
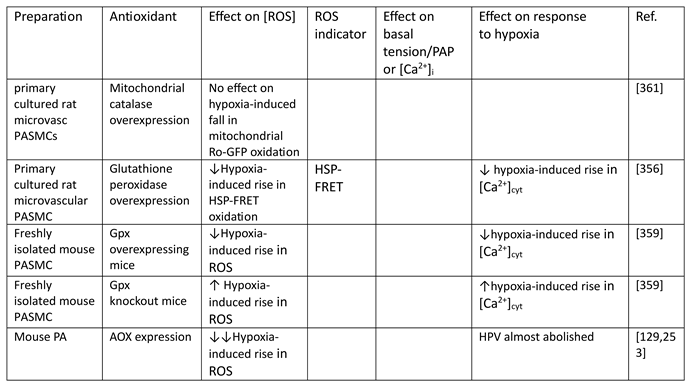
1Endo +/- refer to endothelium-intact and -denuded arteries, respectively. 2 The contraction induced by 25 mM K in this study was used by the authors as an index of basal reactivity. 3 The ROS signal provided by the lucigenin/HRP assay used in this study was considered by the authors to reflect extracellular ROS. 4 diethyldithiocarbamic acid; 5 nitro blue tetrazolium; 6 (N(G)-monomethyl L-arginine); antagonist of endothelial nitric oxide synthase; 7 triethylenetetramine; 8 dihydrolipoic acid; 9 pyrrloidinedithiocarbamate.
7.3. Effects of Oxidants on Basal Tone and HPV
Table 4a,b list the effects of H2O2 and other oxidants on pulmonary vascular tone in normoxia and also on HPV and contractions caused by various vasoconstrictors. The most widely studied oxidant, H2O2, was used over a wide concentration range (0.5 mM - 10 mM), raising the question of which concentrations are likely to produce physiologically relevant increases in intracellular [H2O2].
As discussed in Section 4, the extracellular [H2O2] in vivo is thought to be 1-5 μM [74], whereas the average cytoplasmic [H2O2] is generally said to be in the low nM range [32,226,487] but may be higher in the subplasmalemmal region. According to the mathematical model [240] developed by Sykes and colleagues, and assuming that PASMC have a width of roughly four microns [488], in the presence of an [H2O2]ECS of 2mM the basal subplasmalemmal and core [H2O2] would be ~6 and ~2 nM, respectively, and applying 10 μM H2O2 would increase these H2O2 concentrations by ~30 and ~10 nM. It has also been reported [269] [270] that the half-maximal oxidation of the cytoplasmic GSSG/GSH redox couple in HeLa cells and cardiac myocytes was induced by extracellular application of ~10 and ~20 μM H2O2, respectively. In both types of cells, the threshold for oxidation of GSH was near ~5 μΜ [H2O2] and oxidation became maximal at ~50 μM [H2O2]. Interestingly, diamide oxidized GSH over a similar range of concentrations [270]. Since the cellular GSSG/GSH ratio is a key factor in setting the cytoplasmic redox tone [357] and is crucial for maintaining cellular thiol redox balance [104] these considerations suggest that that application of exogenous H2O2 or diamide in the concentration range below ~50 mM may be suitable for mimicking physiologically relevant levels of oxidant signaling.
As shown in the tables the results from almost all studies, including those that applied H2O2 within or near this concentration range(Table 4a), form a consistent pattern, which was also observed when other oxidants were tested (Table 4b): H2O2 caused a sustained contraction/rise in [Ca2+]cyt in PA/PASMC if applied under baseline normoxic conditions, but induced relaxation or reversed rises in [Ca2+]cyt in PA/PASMC which had been constricted/stimulated by hypoxia or vasoconstrictors.
Table 4a.
Effects of H2O2 on basal tension/Ca2+, and contractile responses to hypoxia and vasoconstrictors in PASMC, pulmonary artery, and perfused lung.
Table 4a.
Effects of H2O2 on basal tension/Ca2+, and contractile responses to hypoxia and vasoconstrictors in PASMC, pulmonary artery, and perfused lung.
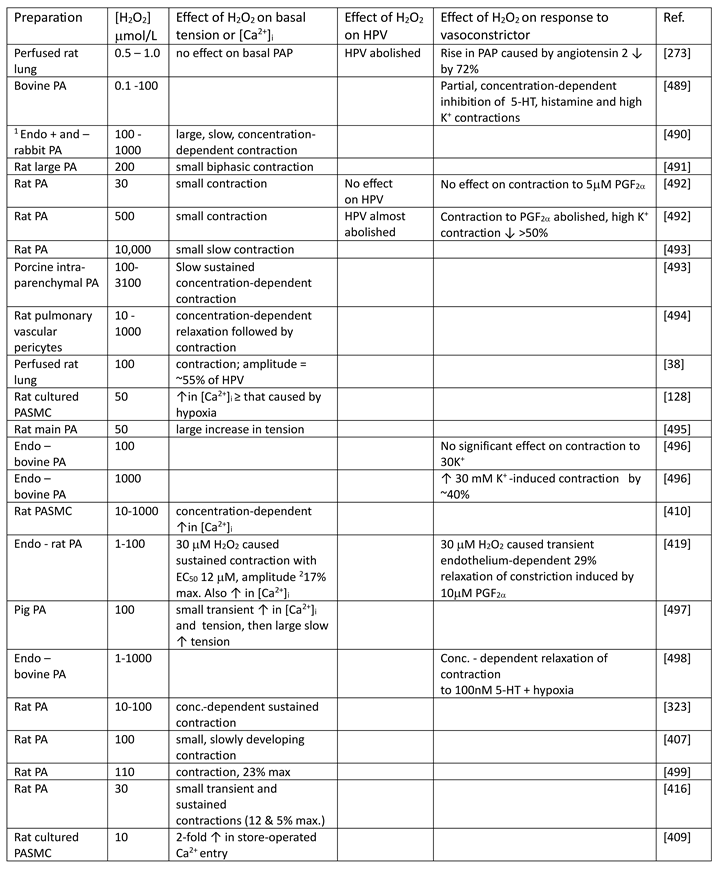
1 E- and E+ refer to endothelium-denuded and -intact arteries. 2 ‘max’ refers to the amplitude of the high K+ solution.
Table 4b.
Effects of other oxidants on basal tension/Ca2+, and contractile responses to hypoxia and vasoconstrictors in PASMC, pulmonary artery, and perfused lung.
Table 4b.
Effects of other oxidants on basal tension/Ca2+, and contractile responses to hypoxia and vasoconstrictors in PASMC, pulmonary artery, and perfused lung.
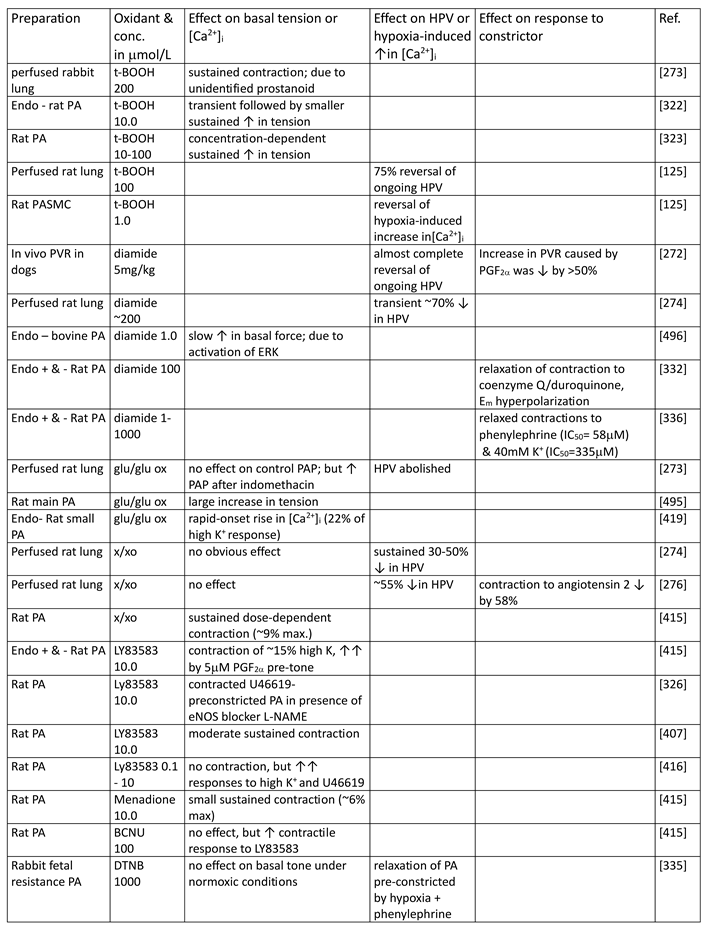
Abbreviations: t-BOOH = tert-butyl hydroperoxide; glu/glu ox = glucose/glucose oxidase; x/xo = xanthine/xanthine oxidase; BCNU = 1,3-bis(2-dichloroethyl)-1-nitrosourea; DTNB = 5,5-dithio-bis-2-nitrobenzoic acid. .
These opposing actions of H2O2 were not necessarily due to the use of different preparations or experimental set-ups by various investigators, since both were recorded when all conditions except the presence or absence of pre-constriction were identical [419]. Notably, a similar pattern has been observed upon application of H2O2 to systemic arteries [500,501].
An investigation of the constricting effects of H2O2 in PA [419] found that the EC50 for H2O2-induced contraction in rat PA was 12μM. 30 μM H2O2 caused a rapidly-developing and sustained contraction of small rat PA which was somewhat larger than that evoked by hypoxia (~22-25 Torr) under similar conditions [435]. The H2O2-induced contraction was endothelium-independent and associated with a rise in [Ca2+]i which was reversed by ryanodine but insensitive to the removal of extracellular Ca2+, indicating that it was due to Ca2+ release from the SR (see also [410]. The initial phase of the contraction (5-10 minutes) was partly blocked by the RhoK antagonist Y-27632 and was only slightly depressed by the membrane permeable Ca2+ chelator BAPTA-AM, suggesting a role for Ca2+ sensitization. The sustained contraction was BAPTA-sensitive but not affected by Y-27632. H2O2 contracted a-toxin-permeabilized PA; this effect was abolished by blockers of conventional PKC isoforms, which also strongly depressed the contraction in intact arteries, suggesting that it was also partially due to PKC-dependent Ca2+ sensitization. It was also reported that application of 10 μM H2O2 increased store-operated Ca2+ influx in cultured rat PASMC by stimulating the association of STIM1 with both Orai1 and TRPC1 [409].
Although there have been relatively few investigations of how H2O2 and other oxidants cause vasorelaxation in PA, the mechanisms which have been characterized in these arteries (opening of KV channels and activation of protein kinase G [332,336,498,502]) are in line with the evidence available from studies carried out in systemic arteries [37,503,504,505,506]. Schach et al [336] also reported that 100 μM diamide inhibited store operated Ca2+ entry in rat PA, although a later and more detailed investigation reported that 10 μM H2O2 had the opposite effect in rat PASMC [409].
Conclusions drawn on the basis of the effects extracellular H2O2 application on PASMC contraction or [Ca2+]cyt should interpreted cautiously, since cellular H2O2 signalling is highly localized [92,254,297] whereas external application of H2O2 to PASMC increases its global intracellular concentration [255]. A further complication is that adding exogenous H2O2 (and diamide) causes oxidation of the mitochondrial matrix [210,270], whereas hypoxia apparently has the opposite effect in PASMC (Section 7.4). Taken together with the evidence that H2O2 and other oxidants can activate both constricting and dilating mechanisms in PA, and that which of these dominates depends on the pre-existing level (or type [500]) of contraction, these considerations imply that whereas responses to exogenous oxidants can provide meaningful information about the redox regulation of specific contractile mechanisms if these are studied in isolation, the effect of adding exogenous oxidants on vascular tone or [Ca2+]cyt in PASMC under normoxic or hypoxic conditions may not necessarily reflect the role of endogenous ROS production in HPV.
7.4. Effects of Hypoxia on PASMC ROS Levels
Numerous investigations, many of which are described in Section 5.1 and Section 5.2, examined the effects of hypoxia on ROS in PASMC, PA, and lung tissue. As set forth in previous reviews [3,318], studies carried out prior to 2006, which utilized chemical ROS probes such as lucigenin and DCF, presented mixed results: some laboratories found that hypoxia decreased ROS and others found the opposite effect. Possible explanations for these discordant findings include the use of different preparations, levels of hypoxia, and ROS indicators. Indicator signaling arising from extracellular ROS, which could far outweigh that emanating from cells [32], may have obscured effects on cellular ROS in studies carried out in perfused lungs and isolated arteries [3]. Evidence that hypoxia causes a fall in ROS production by PAECs [507,508] and isolated lung fibroblasts [379] also suggests that the effects of hypoxia on overall [ROS] production by lung tissue may not accurately reflect what is happening in PASMC.
The chemical indicators used in these studies, most of which were said to detect superoxide levels, have also been criticized on the basis that they are in reality non-specific [509,510], although with the proper controls they can provide useful qualitative information about cellular oxidant levels [88,381]. However, there are additional difficulties in interpreting the signals arising from these probes which may arise specifically when they are used to measure the effects of changes in PO2 [88]. For example, DCFH-DA, lucigenin, luminol and L-012 are converted by cellular oxidants (DCFH-DA, luminol, L-012) or enzyme reductases (lucigenin) to intermediates which generate a signal when they react with superoxide. Unfortunately, these intermediates also react with O2 to form superoxide, a process termed ‘redox cycling’ [510,511,512]. Lucigenin has been reported to accumulate in the mitochondria and primarily detect intra-mitochondrial superoxide [513,514]. An additional confounding factor with lucigenin is that its redox cycling is promoted by NADH [515], the concentration of which is increased by hypoxia [346]. Apart from the lack of selectivity of both dihydroethidium (DHE) and MitoSox for superoxide due to their non-specific oxidation to ethidium [511], their fluorescence is also affected by their distribution into the mitochondria, which is sensitive to Δψm [381,516].
Several of these investigations also utilized the the Amplex Red/horseradish peroxidase (HRP) assay, which measures extracellular [H2O2] and can be used to detect changes in cellular [H2O2], since these lead to its altered leakage out of cells [517,518]. Amplex Red is considered to provide a high quality, specific and very sensitive measure of overall cellular H2O2 production [354]. It is therefore noteworthy that three studies using this assay reported that hypoxia decreased ROS production by PASMC [145,285,452]. However, it is not clear whether this was due to an effect on mitochondrial ROS production, since the activity of Nox is probably curtailed at the PO2 levels used in most studies of HPV [59], and has been shown to make a substantial contribution to the overall release of H2O2 from a range of cell types [519].
In view of the shortcomings of the conventional intracellular ROS indicators , several laboratories have examined the effect of hypoxia in PASMC using genetically-encoded ROS or redox indicators such as Ro-GFP [520], which responds to the ambient thiol/ disulfide equilibrium, and HyPer [287], which reacts with H2O2. In addition to responding to specific ROS or redox changes, these can be targeted to specified cell compartments and do not undergo redox cycling, so are therefore seen as greatly superior to chemical ROS indicators [362,381,521,522]. The results of these investigations are summarized in Table 5.
Table 5.
Effects of hypoxia on cell H2O2, indices of redox balance, and mitochondrial membrane potential.
Table 5.
Effects of hypoxia on cell H2O2, indices of redox balance, and mitochondrial membrane potential.
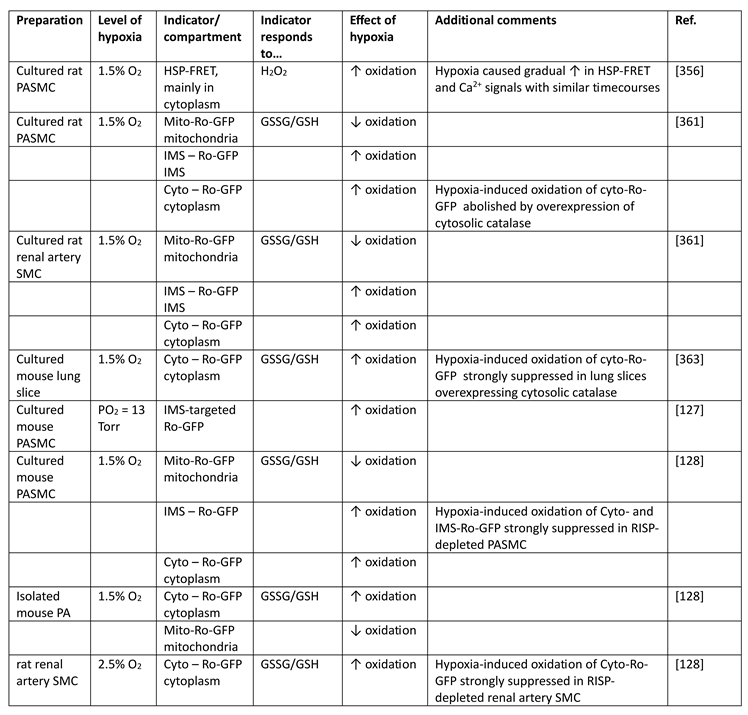
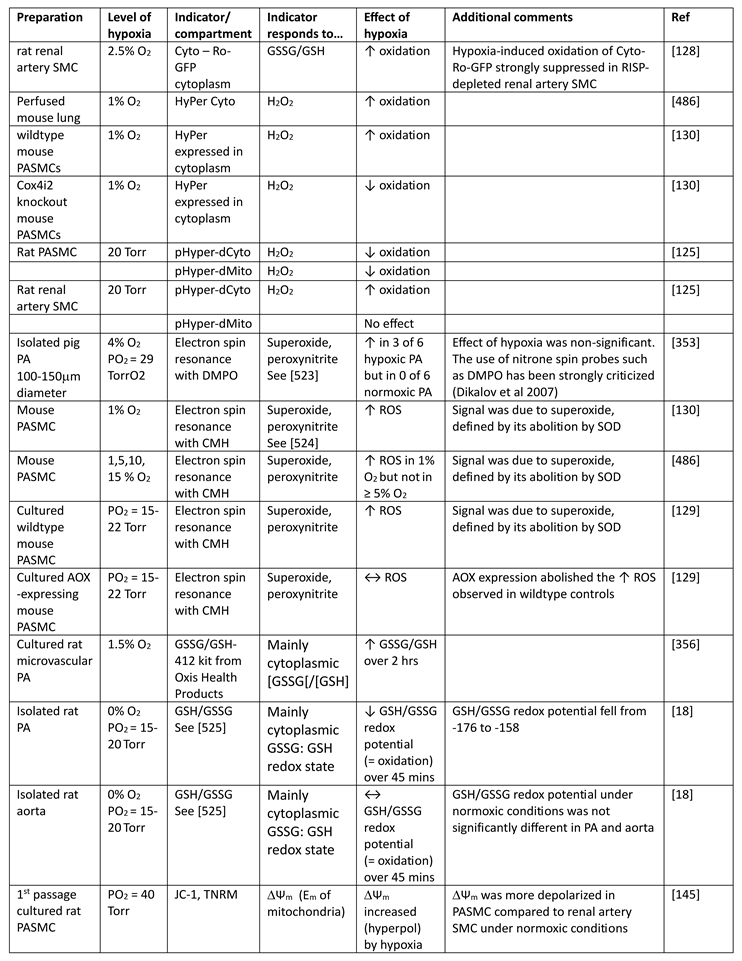
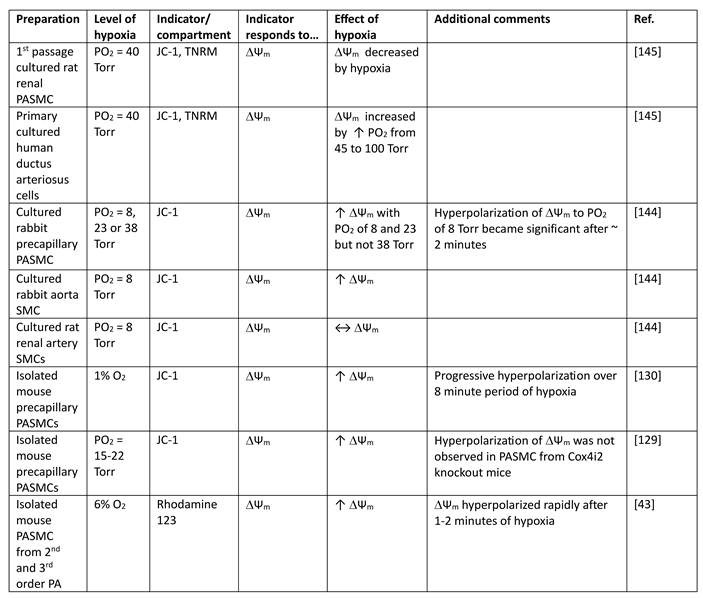
Dunham-Snary et al [125], using HyPer in cultured rat PASMC, found that hypoxia decreased [H2O2] in both the mitochondrial matrix and the cytoplasm. They suggested that since a redox mediator produced in the mitochondria must enter the cytoplasm to modulate contractile effector proteins, the fact that they detected similar effects of hypoxia on [H2O2] in both compartments supported the validity of their findings. They also speculated that the hypoxia-induced reduction of Ndufs2 thiols which they had observed (Section 5.1) was responsible for inhibiting its production of ROS, implying that the reduction of the mitochondrial matrix might be a key factor in causing HPV.
In contrast, all of the other studies using genetically encoded indicators, including one which used Hyper [130] in mouse PASMC, reported that hypoxia increased [H2O2] or oxidized the thiol/disulfide equilibrium in the cytoplasm of mouse and rat PASMC, although, in agreement with Dunham-Snary and colleagues, the mitochondrial matrix became more reduced. Several papers from Schumacker’s laboratory also reported that hypoxia increased redox tone in the mitochondrial IMS, consistent with their hypothesis that the increased ROS production by Complex III triggered by hypoxia is directed into the IMS, from which H2O2 can enter the cytoplasm. Korde et al [127] reported a similar finding.
Likewise, several papers from Sommer and colleagues reported that hypoxia increased superoxide production in cultured mouse PASMC. Although the spin probe they used (1-hydroxy-3-methoxycarbonyl-2,2,5, 5-tetramethylpyrrolidine; CMH) detects both mitochondrial and extra-mitochondrial superoxide [526], since they (and others) consistently found that the redox poise in the mitochondrial matrix tone was reduced during hypoxia, it seems likely that the increase in superoxide detected by ESR was cytoplasmic. Importantly, Sommer et al [129] also demonstrated using ESR that the hypoxia- induced increase in superoxide production was absent in PASMC from mice expressing AOX, consistent with evidence that mitochondrial ROS production associated with an increased reduction of the ubiquinone pool is depressed in these animals [383,527].
The generation of ROS by both complexes 1 and 3 is strongly influenced by Δψm [58,347,528]. As shown in Table X, hypoxia has been observed to increase Δψm in PASMC in each of the three studies in which it has been measured. This is consistent with an increase in mitochondrial ROS production.
In summary, experiments utilizing genetically-encoded ROS indicators and ESR have, with only one exception [125], supported the notion that hypoxia increases ROS production and cytoplasmic oxidant levels in PASMC. The hypoxia induced hyperpolarization of Δψm which has been reported consistently also provides indirect support for this idea.
7.5. Concluding Remarks: The Redox and Mitochondrial Models
Overall, compared to the mitochondrial ROS model, the evidence supporting the Redox model is less extensive. It is also generally older, and partly as a consequence of this, is based to a larger extent on results arising from experimental approaches (the use of ETC blockers as hypoxia surrogates, the knockout of mitochondrial complex subunits to characterize their involvement in O2 sensing) which have been criticized as being unreliable [518,529], or supportive but not definitive (the use of isolated mitochondria, pro- and anti-oxidants, and conventional small molecule ROS indicators[32]).
These considerations also apply to much of the evidence supporting the mitochondrial ROS model and may well account for some of the discrepancies between the results reported by different laboratories. However, placing this type of evidence to the side, the observations that remain predominantly favor the Mitochondrial ROS model. These include, most notably, the inhibition of HPV by S3QEL2 [130] and by the expression of AOX [129], as well of the effects of hypoxia on redox state of the cytoplasm using newer genetically coded indicators and electron spin resonance measurements shown in Table 5.
The Mitochondrial ROS model is also indirectly supported by evidence that Cox4i2, proposed to be crucial for increased hypoxia-induced ROS production during HPV (Section 5.2.2) [130], plays a similar role in the carotid body. In contrast, although the implications of Cox4i2 for the Redox model have not been discussed by its proponents, the evidence that its high expression promotes O2 sensing in both PASMC and CBCC seems at variance with the idea that hypoxia decreases ROS production, since it is widely accepted that reduction of the CoQ pool is a key stimulus for ROS production. However, whilst the comparatively high expression of Cox4i2 in the two most well-characterized types of cells seen to respond acutely to hypoxia suggests that it could be playing an important role O2-sensing, possibly by promoting mitochondrial ROS production, it is also the case that hypoxia, at levels similar to those generally used by experimenters to evoke HPV and CBCC activation, induces rapid increases in ROS in many other types of native cells/tissues or cell lines which are not known to express high levels of Cox4i2 [145,179,342,343,355,530,531,532,533,534,535,536,537,538,539]. Therefore, it remains to be established that the level of expression of Cox4i2 is a pivotal determinant of hypoxia-induced ROS production.
Despite the evidence for the mitochondrial ROS hypothesis being more compelling at this point, questions remain. For example, the observation that HPV recorded in isolated PA from AOX mice was restored if the arteries were incubated in PSS containing 20 mm K+ [129] appears to sit uneasily with evidence that an increase in ROS production by complex III is causing HPV by causing SR Ca2+ release and Ca2+ sensitization (see Section 5.2.1 and Section 5.2.5). Since 20 mM K+ , which promotes VSMC contraction by causing depolarization, was able to replace the effect of ROS production by complex III which was absent in the AOX mice, the implication is that ROS produced by complex III contribute to HPV by causing membrane depolarization. If so, the contractile effects over and above those caused by depolarization (i.e. Ca2+ release and sensitization) which are evoked by hypoxia are not due to ROS production by complex III. If nothing else, these apparently contradictory observations highlight the fact that the HPV effector mechanisms linked to an increase in ROS remain imperfectly understood.
Another unresolved aspect of the mitochondrial ROS hypothesis is that the nature of oxidizing signal responsible for HPV remains to be convincingly identified. Although H2O2 is generally seen as being the most important oxidizing species responsible for cell signaling, the evidence that this is the case for HPV is decidedly mixed. Its specific involvement in HPV is supported by evidence from three investigations showing that overexpression of catalase [356,359,363] prevented the hypoxia-induced rise in [Ca2+]cyt in PASMC, and one showing that hypoxia oxidized HyPer [130]. On the other hand, hypoxia was seen to cause a fall in the cytoplasmic concentration or cellular production of H2O2 in four other studies using AmplexRed/HRP or HyPer [145,285,294,452], and the mechanisms by which H2O2 caused contraction when applied at a concentration which probably caused a quasi-physiological intracellular signal did not closely match those thought to be responsible for HPV[419]. Also arguing against H2O2 as the HPV mediator is the carefully conducted study by Liu and colleagues showing that the application of SOD, which they verified led to its penetration into PASMC, strongly suppressed HPV in pig PA [353].
The question of the redox mediator responsible for HPV is particularly relevant in light of recent experiments carried out in HEK 293 cells by Sen et al which suggest that hypoxia may exerting opposite effects on two types of cell oxidant species [518]. This investigation aimed to determine whether the opposing effects of hypoxia on ROS production observed in PASMC and many other types of cells were genuine or might, for example, be due to the use of different indicators with non-identical responses to particular oxidant species.
The effects of hypoxia (1% O2) in HEK293 cells were recorded using the Amplex UltraRed + SOD assay, both in the presence of HRP to record H2O2, and in the absence of HRP, in which case the assay detects other oxidant species instead of H2O2. They also used DCF to measure general cell oxidation. They found that hypoxia decreased H2O2 release. Importantly, this was associated with a fall in H2O2 production by complexes 1 and 3, and also by Nox, plus other sources which were not characterized. This supports the idea that the decrease in H2O2 production was caused by a deficiency of O2 rather than a decrease in the availability of electrons at any specific site. Hypoxia also decreased DCF oxidation. On the other hand, hypoxia increased the release from the cells of (an)other oxidants(s), as sensed by Amplex UltraRed in the absence of HRP. Whereas they did not identify these other oxidant(s), work by Kalyanaranam’s laboratory shows that the oxidizing species involved is probably peroxynitrite [511], or, more likely, products of its decomposition [540]. Notably, peroxynitrite (or its products) can also increase the fluorescence of AmplexRed + HRP [540]. Even so, this reaction occurs much more slowly than that with H2O2 suggesting that the two assays were genuinely picking up opposing effects of hypoxia on H2O2 vs (an)other oxidant species.
These results therefore imply that hypoxia alters the balance between cellular production of H2O2 and other oxidizing species, thereby providing a potential explanation for the apparent indicator-dependency of the effect of hypoxia reported in both the HPV literature and by studies carried out in other types of cells and tissues.
If the unidentified oxidant was peroxynitrite, the observations of Sen et al could potentially be explained by the hypothesis, developed by Poyton and colleagues [421], that hypoxia increases the production of both NO and superoxide by the mitochondria, generating a cytoplasmic peroxynitrite signal capable of orchestrating a cellular response. One implication of this model is that since the rate at which NO and SOD react with superoxide is similar [541], they would compete for superoxide released into the IMS by complex III. Thus, an increase in [NO] evoked by hypoxia, whether originating in the mitochondria or elsewhere in the cell [163], could enhance the production of peroxynitrite while at the same time diminishing that of hydrogen peroxide. Furthermore, since Ro-GFP, ESR and HSP-FRET can sense peroxynitrite directly or indirectly through its effects on the ambient redox state, whereas HyPer responds specifically to H2O2, some of the apparently contradictory observations which set the Mitochondrial ROS and Redox models apart could in fact represent two facets of the same process.
In any case, we suggest that the role of mitochondrial ROS production in O2 sensing during HPV remains sufficiently uncertain so as to warrant further studies. For example, the observation by Sommer et al [130] that S3QEL2 inhibits HPV, which we believe is currently the most convincing evidence that it requires ROS production at complex III, has not been confirmed independently, and would be strengthened by corresponding evidence that S3QEL2 also suppresses hypoxia-induced cytoplasmic oxidation. It also would be useful if the current evidence for cytoplasmic oxidation could be broadened, for example by using newer and more sensitive indicators such as Ro-GFP2-Tsa2DeltaCRm, which is based on a modified form of peroxiredoxin, [121,297,542]. The possible involvement of NO and peroxynitrite in HPV, would appear to be another issue worth investigation.
In addition, an analysis of the effects of hypoxia on reactive cysteines in putative ROS targets such as IP3R, RyR and KV1.5 would confirm that any alteration in cytoplasmic redox state is not merely an epiphenomenon. This approach could be implemented using isolated PA or perfused lung, circumventing the use of cultured cells and avoiding attendant complications arising from their altered phenotype and isolation from their normal environment.
8. The Role of the Pentose Phosphate Pathway and the Withdrawal of Normoxic Vasodilation Maintained by Nox4, H2O2 and Protein Kinase G in HPV
Michael Wolin and colleagues developed a complex scheme for O2 sensing in HPV which resembles the Redox theory in that HPV is proposed to be due, at least in part, to hypoxia-induced withdrawal of ongoing ROS-dependent normoxic vasodilation. However, whereas according to the Redox theory this vasodilation is caused by mitochondrial ROS production and a resulting activation of KV channels, the model developed by Wolin’s laboratory envisions that normoxic vasodilation is maintained mainly by H2O2-mediated activation of the soluble guanylate cyclase (sGC)/protein kinase G (PKG) pathway, and that Nox4 rather than the mitochondrial ETC is responsible for H2O2 production.
Additional studies by Sachin Gupte, mostly carried out in collaboration with Wolin, presented evidence that hypoxia also regulates arterial tone by altering the activity of the pentose phosphate pathway (PPP), thereby leading to changes in the cellular [NADPH]/[NADP+] ratio. This would enable H2O2-independent regulation of redox networks, leading to effects on thiol oxidation which alter the function of proteins involved in controlling vascular tone. In PA, for example, hypoxia was shown to stimulate the PPP, thereby increasing the [NADPH]/[NADP+] ratio and as a result decreasing the activity of PKG1a, which is redox-sensitive [37]. Somewhat paradoxically, several of these investigations found that activation of the PPP by hypoxia is due to a rise in cytoplasmic [H2O2], which acts through PKCd to stimulate glucose-6-phosphate dehydrogenase (G-6-PD), the initial and rate limiting step in the PPP. This rise in H2O2 was also proposed to stimulate contraction through both Ca2+- dependent and -independent pathways.
Although several of these studies investigated HPV in isolated perfused lungs or pulmonary artery rings from rats [441,543] or mice [478], most of this work was carried out using isolated bovine pulmonary artery (BPA) or lung homogenates, sometimes with comparative experiments in bovine coronary arteries (BCA), which relax to hypoxia. Importantly, rotenone, which has universally been found to block HPV in other species (Table 2), had no effect on HPV in BPA [453]. This would imply that mitochondrial ROS production plays no direct role in O2 sensing in BPA, at least under the conditions used in these studies.
In most of these studies, hypoxia was imposed using PSS gassed with 5%CO2/95%N2, resulting in a pO2 of 8-10 Torr. For studies of HPV, BPA rings were denuded of their endothelium to prevent hypoxic effects which might be mediated by NO and other EDRFs, and were slightly pre-constricted, generally with 20-30 mM K+ but in some studies with U46619 or 5-HT, in order to enhance HPV. The authors generally adjusted the degree of pre-tone stimulus to maintain a similar amplitude of pre-constriction in the presence and absence of the interventions (e.g. blockers) which were being used to characterize mechanisms engaged by hypoxia or ROS.
8.1. Early Studies: HPV as the Loss of Tonic H2O2 and sGC-Mediated Vasorelaxation
A series of papers by Wolin’s laboratory published over an approximately 10-year period starting in 1987 developed the concept that PA normally generate a basal level of superoxide (and therefore H2O2) due to the activity of an NAD(P)H oxidase [391,454,482,489,544,545,546,547,548]. This causes an ongoing stimulation of soluble guanylate cyclase (sGC) and therefore protein kinase G, creating a tonic vasodilating influence. The stimulation of sGC by H2O2 was proposed to be mediated by compound 1, a form of catalase which exists during its metabolism of H2O2 [489,544]. Vasodilation due to this mechanism is suppressed by hypoxia because there is less O2 available for the formation of superoxide/H2O2, resulting in contraction (i.e. HPV). The development of this scheme is described in a review by Wolin et al [549]. The oxidase responsible for superoxide production in PA was initially seen as utilizing NADH. However, Mohazzab & Wolin [547] pointed out that its properties resembled those of the NADPH-dependent oxidoreductase which had been detected in neutrophils [550] and had been suggested to play a role in O2 sensing in the carotid body [551]. It was eventually concluded [549] that it corresponded to the phagocytic NADPH oxidase and might be the same as the vascular oxidoreductase which others had shown utilized both NADH and NADPH to generate superoxide [552]. It soon became evident that there was a group of such oxidoreductases, christened the Nox family [553], and that they preferred to use NADPH rather than NAD as a substrate for superoxide production. In later work Wolin and colleagues concentrated on defining the role of NADPH rather than NADH in mediating the effects of hypoxia, stating that the [NADPH] in cells is likely to be 10-20 μM whereas [NADH] is ~1 μM [554].
Most of the subsequent investigations by this group relevant to HPV focused on two interrelated themes. As described in Section 8.2, a series of papers published between 1999 and 2010, spearheaded by Sachin Gupte, examined how hypoxia-induced regulation of the PPP influences the redox state of the NADPH/NADP+ couple in such a way as to cause constriction of PA but relaxation of coronary arteries. Other work, published between 2010 and 2014, further examined how hypoxia, H2O2 and NADPH, acting though cellular redox networks, modulate PA tone by regulating the activity of the sGC/PKG axis (Section 8.3). In addition, Wolin’s laboratory carried out pioneering experiments to examine the important possibility that HPV is influenced by the presence of extracellular H2O2 [477,478]
8.2. PPP Activity as a Determinant of the Effects of Hypoxia on Vascular Tone
An initial investigation of the role of the PPP in regulating vascular contraction [555] was based on the hypothesis that oxidation of the heme iron on sGC would inhibit its activity by preventing the binding of NO, and that NAD(P)H, by reducing the heme iron, might restore the responsiveness of sGC to NO, thereby promoting vasorelaxation. The results of the study provided support for this hypothesis, implying that that the activity of the PPP, which generates cellular NADPH, could potentially regulate vascular tone of BPA via this mechanism.
A subsequent paper then considered two possible scenarios by which the activity of the PPP might influence HPV [543]. If NAD(P)H was indeed acting to augment the stimulation of sGC by NO, PPP activation should suppress HPV. Alternatively, based on evidence that NADP+ and GSSG activated the K+ current in PASMC [331], PPP-mediated reduction of NADP+ and/or GSSG might promote HPV by closing K+ channels and causing depolarization-induced Ca2+ influx. Their experiments showed that antagonists of glucose-6-phosphase dehydrogenase (G-6-PD), which mediates the rate-limiting step of the PPP, suppressed HPV in isolated perfused rat lungs and relaxed contractions evoked by 30 mM K+ in both PA and aorta. These results supported the second hypothesis, implying that hypoxia-induced activation of the PPP could promote HPV by reducing the cytoplasmic NADPH: NAD(P)+ couple and thereby inhibiting KV channel opening.
Gupte et al [556] similarly observed that pharmacological blockade of G-6-PD caused relaxation of bovine coronary arteries (BCA) and that this was accompanied by a fall in the tissue contents of NADPH and GSH, whereas total NAD(P)+ and GSSG increased. The reducing agent DTT attenuated this relaxation, suggesting that it was due to an oxidizing effect of some kind. However, they also found that both superoxide and H2O2 generation by BCA fell during PPP blockade, implying that this oxidation was not due to an increase in cellular [ROS]. Moreover ebselen, which would be expected to diminish the concentrations of both ROS, did not suppress relaxation. They proposed that the fall in ROS production caused by PPP blockade occurred because there was less NADPH available to be used to produce superoxide. Although this study was in coronary rather than pulmonary arteries and didn’t look at the effects of hypoxia, it was important for their subsequent studies of HPV because their observations showed that the PPP, presumably by producing NADPH, was able to influence vascular tone through ROS- and sGC-independent mechanisms, and that regulation of Ca2+ influx and release was more important in this respect than that of KV channels.
Wolin’s laboratory had previously shown [557] that hypoxia also relaxed BCA via a mechanism which appeared to be independent of ROS, since it was not affected by NBT (an antioxidant) and DPI (blocks Nox and the ETC). This suggested that the inhibition of the PPP might also be causing relaxation through this mechanism, and in accordance with this possibility Gupte & Wolin [558] reported that hypoxia increased the [GSSG]/[GSH] and [NADP+]/[NADPH] ratios in these arteries. These effects were associated with a decreased level of glucose-6-phosphate, which is produced by the first step of the PPP, suggesting that hypoxia was inhibiting this pathway. As with PPP blockade, the relaxation to hypoxia was attenuated by DTT pre-treatment. Interestingly, application to BCA of pyruvate (10 mM), which was used to promote the flux of glucose through the PPP by inhibiting phosphofructokinase [559], increased the arterial content of gluscose-6-phosphate and NAD(P)H under both normoxic and hypoxic conditions. Pyruvate also prevented the increase in arterial GSSG and the vasorelaxation evoked by hypoxia. Based on these findings, the authors proposed that, due to a relative lack of G-6-PD in BCA, hypoxia causes a ‘metabolic stress’ leading to a reduced flux of glucose through the PPP. This causes a rise in the cellular [NADP+]/[NADPH] and [GSSG]/[GSH] ratios which acts through redox networks to cause vasorelaxation by oxidizing thiol switches on a number of proteins controlling contraction (e.g. SERCA) [560].
Gupte et al [554] examined the relationship between G-6-PD and cellular superoxide levels (assessed using 5 μM lucigenin) in BPA and BCA. They reported that mRNA expression of Nox1 was not present in either artery. On the other hand, Nox2 and Nox4 mRNA and protein were similarly expressed in the two types of arteries, although superoxide levels were 40-80% higher in BPA. Both the G-6-PD blocker 6-aminonicotinamide (6-AN) and the drug apocynin, which they used as a Nox antagonist, decreased the lucigenin signal more in BPA than in BCA, suggesting that the higher level of superoxide production in BPA resulted from a higher level of Nox activity due to greater NADPH formation by the PPP. This idea was supported by their observation that the level of NADPH and the expression and activity of G-6-PD was higher in BPA than in BCA. We suggest, however, that this conclusion should be interpreted with caution, since the [NADPH]/[NADP+] ratio was higher in BCA, suggesting that the PPP might be more active in these arteries.
Gupte et al [441] investigated the involvement of the PPP and NADPH in HPV in rats. They found that 5 minutes of hypoxia caused marked increases in the amounts of glucose-6-phosphate and NADPH in the lung. The hypoxia-induced rise in NADPH was abolished by 6-AN and another G-6-PD blocker, epiandrosterone. Both drugs also greatly reduced the amplitude of HPV recorded in isolated perfused lung. Similarly, hypoxia increased the [NADPH]/[NADP+] ratio by about 75% in isolated lobar PA, and this effect, along with HPV, was suppressed by the G-6-PD antagonists. In additional experiments, sGC activity was measured in homogenates prepared from isolated PA which had been subjected to hypoxia in the presence or absence of G-6-PD blockers or the SGC antagonist 1H-[1,2,4]oxadiazolo[4,3-a] quinoxalin-1-one (ODQ). It was found that hypoxia strongly increased the activity of sGC. This effect was also prevented by G-6-PD blockade, suggesting it was due to activation of the PPP. ODQ also prevented the increased sGC activity, but, in contrast to PPP blockade, enhanced HPV.
Based on these results, the authors proposed that hypoxia activates the PPP in PA, leading to an increased synthesis of NADPH which is responsible for HPV. At the same time, the increased [NADPH] also stimulates sGC (see also [555] and [561], creating a countervailing vasodilating influence which explained why HPV was potentiated by ODQ. However, this was outweighed by the pro-contractile response, which they speculated was due to KV channel inhibition.
In summary, the results of these studies suggested that hypoxia increases and decreases NADPH production by the PPP in BPA and BCA, respectively, and that this difference is responsible for its opposite effects on vascular tone in these arteries. According to the model developed by the authors to explain their observations [562,563], hypoxia inhibits the PPP in coronary arteries, leading to vasorelaxation consequent on a fall in cellular NADPH and a resulting oxidation of cellular thiol switches (e.g. on KV channels and Ca2+ channels or pumps). Although ROS production decreases due to the fall in pO2 and/or the suppression of the PPP, their evidence suggested that this does not exert an important effect on vascular tone. In contrast, PPP activity in PA is higher under basal conditions, and is increased by hypoxia. This causes hypoxia-induced contraction (HPV) through two mechanisms. Firstly, the PPP generates sufficient NADPH under normoxic conditions to create an ongoing vasodilating influence dependent on superoxide/H2O2 produced by an NADPH oxidoreductase. This vasodilating influence is greater in BPA than in BCA because the former has a higher expression and activity of G-6-PD and therefore has higher basal NADPH levels. This ongoing vasodilation is removed by hypoxia because it attenuates ROS production, causing contraction (i.e. HPV). Secondly, PPP activation by hypoxia, leading to increased NADPH production, promotes force development by causing the reduction of GSSG and/or protein thiol switches in such a way as to suppress relaxation or enhance contraction.
Gupte et al [440] further investigated the role of the PPP in triggering HPV in 4th – 5th order BPA. Contraction mechanisms were examined in intact endothelium-denuded arteries and those which the plasma membrane had been permeabilized with a-toxin from staphylococcus aureus to enable the study of Ca2+ sensitization. They evaluated the role of G-6-PD in HPV by using PA which had been cultured with anti-G-6-PD or scrambled siRNA for two days. They also compared the responses to hypoxia in a mouse model in which expression of G-6-PD was halved due to a mutation in the 5’ promoter region of its gene, and in mice that were heterozygous for this mutation and had normal levels of G-6-PD expression. They measured G-6-PD activity and the levels of pyridine nucleotides and other metabolic intermediates in homogenates made from frozen BPA.
The authors observed that the activation of the PPP by hypoxia previously detected in rat lung also occurred in BPA. This was shown by their observations that hypoxia increased the activity of G-6-PD as well as the [NADPH]/[NADP+] ratio and the NADPH content of these arteries. They also examined the effect of hypoxia on cellular ROS levels using MitoSox and DHE in arteries which were pre-constricted with PSS containing 30 mM K+, presumably to mimic the conditions used to study HPV. Hypoxia decreased the MitoSox signal, suggesting a fall in mitochondrial ROS levels, but increased the DHE signal, which they ascribed to cytoplasmic ROS. In light of previous evidence that the activity of G-6-PD is stimulated by H2O2, [564] they speculated that an increase in cytoplasmic ROS levels caused by hypoxia was responsible for activating the PPP, a concept which was subsequently supported by the subsequent observation that H2O2 could stimulate G-6-PD in BCA through PKCδ [565]. In addition, they found that hypoxia decreased the ATP/ADP ratio and increased the lactate/pyruvate ratio, indicating that glycolysis and oxidative phosphorylation were inhibited. These effects, plus a hypoxia-induced increase in glucose uptake [346], would be expected to increase the intracellular glucose concentration and shunt glucose into the PPP, further increasing its activity.
Curiously, however, they also found that hypoxia strongly increased the NAD+/NADH ratio. This effect is the opposite to what would be expected when NADH consumption by the ETC is inhibited, and runs counter to the effects of hypoxia on this ratio observed by others in PASMC [346] and CBCC [176]. The implications of this apparent discrepancy were not discussed.
Importantly, they observed that decreasing the expression of G-6-PD strongly suppressed HPV. Furthermore, hypoxia increased contraction by stimulating both Ca2+-dependent mechanisms and Ca2+ sensitization, and both responses were almost abolished in arteries treated with anti-G-6-PD siRNA. The former effect was studied by measuring hypoxia-induced increases in contractions of intact PA evoked by 30 mM K+ PSS and 5-HT in the presence of extracellular Ca2+. The latter effect manifested as a leftward shift of the Ca2+ vs contraction curve in permeabilized PA and was also suggested by the increase in MLC20 phosphorylation which they detected in intact PA under Ca2+- free conditions. Hypoxia also increased the phosphorylation of CPI-17. The hypoxia-induced phosphorylation of MLC20 but not CPI-17 was antagonized by 6-AN, suggesting that PPP-dependent Ca2+ sensitization was not due to CPI-17. Instead, the authors suggested that hypoxia was causing Ca2+ sensitization via rho kinase, although no direct evidence for this was presented. The involvement of the PPP in O2 sensing in BPA proposed by the authors based on these results and those of earlier studies is illustrated in Figure 9.
8.2. Loss of Basal H2O2-Induced Stimulation of sGC and PKG as a Mechanism of HPV
8.2.1. Basal H2O2 Production and HPV
Ahmad et al [477] tested the hypothesis that the removal by hypoxia of an ongoing H2O2-dependent vasorelaxation contributes to HPV by using ebselen (100 μM) to mimic a hypoxia-induced fall in H2O2. They found that the contraction of BPA to 20 mM K+, which they viewed as reflecting basal normoxic contractility, was enhanced by ebselen, while HPV was strongly depressed. Treatment of BPA with the CuZn SOD blocker diethyldithiocarbamic acid (DETCA) exerted similar effects. Both ebselen and DETCA greatly decreased normoxic H2O2 levels (detected using a luminol/HRP assay). HPV was also markedly depressed in BPA which had been treated for 24 hours with CoCl2, which increased tissue levels of H2O2, putatively by enhancing the expression of extracellular SOD. This suggested that if produced extracellularly in sufficient quantities, H2O2 entry into PASMC would increase to the point where hypoxia was no longer able to depress its intracellular concentration enough to evoke HPV. This idea was supported by the observation that applying 1 mM catalase to remove extracellular H2O2 restored HPV in the CoCl2 -treated arteries.
They then [453] examined the involvement of Nox2 and Nox4 in generating the H2O2 responsible for maintaining normoxic vasorelaxation in BPA. They found that pharmacological block or siRNA knockdown of Nox2 attenuated superoxide production but did not affect HPV or the contraction to 25mM K+ under normoxic conditions. Similarly, ETC block using rotenone (10 μM) or antimycin (10 μM) decreased superoxide production but had no effect on either type of contraction. In contrast, knockdown of Nox4, which also depressed superoxide production, halved the amplitude of HPV although it too had no effect on the 25 mM K+ contraction. Based on these results, the authors concluded that Nox4 was responsible for generating H2O2 and maintaining a vasodilating influence which was removed by hypoxia, thereby causing or contributing to HPV. However, this scheme would predict that interventions which diminish intracellular [H2O2] even under normoxic conditions should increase contractility, and this conflicted with their finding that siRNA knockdown of Nox4 had no effect on the contraction to 25 mM K+. To explain this, they suggested that H2O2 produced by Nox4 was having both dilating and constricting effects, and that following H2O2 depletion the dilating effect was lost immediately whereas the constricting effect wore off more slowly. The immediate loss of the dilating effect would give rise to a rise in tension (e.g. HPV or the contraction caused by the antioxidant ebselen), whereas the eventual loss of the contractile effect would account for the fact that very prolonged H2O2 depletion associated with Nox4 knockout did not affect the response to 25 mM K+. They tested this concept by applying 25 mM K+ before and after either a short- (35 min) or long-term (several hour) treatment with ebselen (again used to mimic the H2O2-lowering effect of hypoxia) and found that short-term ebselen treatment produced the predicted enhancement of the high K+ response, whereas long-term treatment did not. The authors suggested that the pro-contractile effect of ROS produced by Nox4 might be due to the stimulation of rho kinase.
Together, the results of these two papers supported the hypothesis that HPV is due to the loss of vasorelaxation caused by H2O2 produced by Nox4.
8.2.2. HPV and Regulation of sGC and PKG by H2O2
The proposal by Wolin’s laboratory that a fall in H2O2 production caused by hypoxia evokes or promotes HPV by inhibiting the activity of the sGC/PKG pathway grew out of their initial observations suggesting that H2O2 stimulates sGC though the action of compound C, a catalase intermediate which forms while it is metabolizing H2O2 [489,544,566,567]. The link between this mechanism and HPV was based on evidence [544] that aminotriazole, which inactivates catalase by binding to compound C, caused a contraction and inhibited HPV in BPA. However, it is not clear from the results presented in this paper that the latter effect was statistically significant, and whereas aminotriazole raised basal PAP in perfused rat lung, it did not block HPV [548]. Moreover, in perfused rabbit lung, where aminotriazole did block HPV, it caused an equivalent inhibition of the contractile responses to U46619 and angiotensin 2 [481]. The ability of compound C to block sGC appears not to have been confirmed by any other laboratories.
Wolin’s laboratory subsequently explored other mechanisms by which hypoxia could regulate the sGC/PKG axis in BPA, reporting that hypoxia greatly enhanced the stimulation of cGMP production evoked by NO donors [568]. Nevertheless, sGC-mediated vasorelaxation was strongly depressed by hypoxia, although the NO-induced vasorelaxation was maintained by an sGC-independent mechanism, which their experiments suggested was the activation of SERC A. This possibility was later supported by the demonstration that NO can stimulate SERCA in arteries via peroxynitrite-mediated S-glutathiolation of Cys674 [569]. The authors did not investigate or discuss how hypoxia was apparently diminishing the ability of cGMP to cause vasodilation. However, since PKG is the only known target of cGMP in vascular smooth muscle, its inhibition by hypoxia is a likely explanation.
They then showed that diamide, an oxidant which mimics the effect of H2O2 on thiols, inhibited the activation of sGC by NO donors [561]. Blocking the PPP, which caused a fall in the level of NADPH and should therefore also lead to oxidation of thiols, had the same effect as diamide. This would predict that a fall in H2O2 and activation of the PPP caused by hypoxia should increase NO-induced stimulation of sGC, which was consistent with their earlier observation.
PKG exists as two isoforms, PKG1 and PKG2, with the former being predominantly expressed in the vasculature. Two splice variants of PKG1, PKG1α and PKG1β, are expressed in vascular smooth muscle. Both are homodimers, with the subunits held together by a leucine zipper interaction and are activated by cGMP. In 2007 Burgoyne et al [37] discovered that PKG1α can also be activated in a cGMP-independent manner by the oxidative formation of a disulfide bond between the Cys42 residues of its two subunits. Although the inactive form of PKG1α is often referred to as a monomer which dimerizes upon the formation of the disulfide bridge [503], this is incorrect; both active and inactive PKG1α are dimers [570]. The monomeric form only exists under the denaturing conditions used for immunoblotting, which cause the dimers without the disulfide bond to come apart to form monomers, whereas dimers with a disulfide bridge remain intact; this allows the proportion of active and inactive forms of PKG1α to be measured. We will use the term ‘disulfide dimer complex’ (DDC) ’ [570] to refer to the form of PKG1a activated by oxidation.
The finding that PKG1α could be directly activated by DDC formation opened up the possibility that decreases in [H2O2] caused by hypoxia could act on PKGa1 in PASMC to inhibit its vasodilating effect. This led Wolin’s laboratory [498] to investigate the effect of exogenous H2O2 on DDC formation and the activity of PKG in BPA. They also examined whether H2O2 was still able to cause vasorelaxation in arteries in which the expression of sGC had been greatly diminished by a 48 hour exposure to ODQ, an approach adapted from a protocol described in [571]. They found that application of 100mM H2 O2 to BPA significantly increased DDC formation and PKG activity (measured as the phosphorylation of the PKG1α substrate VASP- vasodilator-stimulated phosphoprotein). These effects were suppressed by pre-treating arteries with DTT, suggesting that they were due to PKG1α oxidation. Depletion of sGC by prolonged ODQ treatment, which almost eliminated the dilating response induced by a NO donor, diminished the vasorelaxation evoked by exogenous H2O2 by ~50%, indicating that H2O2 was activating PKGα1 both directly and indirectly, via sGC. Acute ODQ treatment, designed to prevent any effect of NO on sGC, did not block the response to H2O2, indicating that this was independent of NO.
Neo et al [502] studied the relative contributions of sGC and PKG1α to HPV. They found that, like H2O2, hypoxia decreased the DDC formation and activity of PKGα1. Strikingly, siRNA knockdown of PKG1α mimicked the acute effects of ebselen observed previously [453], increasing the amplitude of the contraction induced by 25 mM K+ under normoxic conditions but decreasing the amplitude of HPV by ~50%. Additionally, treating BPA with DTT mimicked hypoxia by increasing the contraction to 25mM K+ and diminishing both DDC formation and VASP phosphorylation. DTT also abolished HPV.
Depletion of sGC by prolonged ODQ treatment also potentiated the contraction to 25mM K+ under normoxic conditions while reducing the amplitude of HPV by ~60%. siRNA knockdown of the b-subunit of sGC in BPA, which reduced its expression by ~60%, caused similar effects and decreased H2O2-induced vasorelaxation and PKG activity. siRNA knock down of PKG1α also suppressed H2O2-induced relaxation and VASP phosphorylation of BPA, and attenuated HPV.
These findings supported the hypothesis that in BPA, hypoxia, by diminishing cellular H2O2 levels, causes a reduction of regulatory cysteine thiols on both sGC and PKG1a which suppresses their activities. This would attenuate ongoing normoxic vasodilation, thereby causing or augmenting HPV. In contrast to what was observed in BPA, hypoxia caused vasorelaxation in BCA, associated with increases in DDC formation and VASP phosphorylation. This presumably reflected an oxidizing shift in the NADP+/NADPH redox couple which acted through redox networks to oxidize PKG1α, rather than being due to H2O2 [556].
Neo et al [572] investigated whether PPP-induced reduction of the NADP+/NADPH couple was influencing the activity of PKG1α by regulating the redox state of antioxidant enzymes such as Trx-1 or Prx1 which could suppress its activation by reducing Cys42. In this case, it would be predicted that experimental interventions which interfered with the function of the PPP or of these enzymes would increase PKG1α DDC formation, leading to its activation and a decrease in vascular contraction.
The authors described several observations which supported the idea that the PPP creates a pro-contractile influence in BPA which is exerted via NADPH-dependent reduction (i.e. inhibition) of PKG1α. Hypoxia caused an increase in the NADPH/NADP+ ratio, which was antagonised by the PPP blocker 6-AN. PPP blockade acutely relaxed high K+-pre-constricted PA, and increased PKG1α DDC formation and VASP phosphorylation. In addition, the high K+ contraction under both normoxic and hypoxia conditions was smaller, and levels of PKG1α DDC formation and VASP phosphorylation were higher, in PA in which G-6-PD had been knocked down by siRNA, compared to controls treated with scrambled siRNA. Similar effects were seen following siRNA knockdown of Trx and TrxR-1. Knockdown of thioredoxin reductase also suppressed 6-AN-induced relaxation, DDC formation and VASP phosphorylation.
In contrast, knockdown of Prx1 had no significant effect on the amplitude of the high K+ contraction, PKG1α DDC formation, VASP phosphorylation or 6-AN-induced relaxation. They also examined the effect of TrxR-1 and Prx1 knockdown on the responses of PA to the application of 100μM H2O2, which caused PKG1α DDC formation and VASP phosphorylation. Knockdown of TrxR-1 enhanced peroxide-induced relaxation, DDC formation and VASP phosphorylation, whereas knockdown of Prx1 had the opposite effects.
The results supported the idea that the PPP exerts an inhibitory effect on the activity of PKG1α, doing so by maintaining a high level of cellular NADPH which acts through Trx-1 and TrxR-1 to reduce PKG1a, thus suppressing its vasorelaxing effect. This inhibitory effect on the underlying vasorelaxing influence exerted by PKG is enhanced during hypoxia because the PPP is activated [440], promoting HPV. This concept was in line with the observation [573] that G6P-D, Trx-1 and PKG co-precipitated, suggesting that they form a complex.
The results also suggested that Prx1 is not involved in O2 sensing in BPA. This was shown by the observation that Prx1 knockdown had no effect on the high K+ contraction under normoxic conditions, which would mean that this antioxidant enzyme is not contributing to the normoxic vasorelaxation which is removed by hypoxia. The authors also observed that depletion of sGC using chronic ODQ treatment had no effect on 6-AN mediated relaxation under hypoxic conditions, implying that the PPP and NADPH were not directly regulating cGMP production. The authors therefore concluded that regulation of PKG1a DDC formation by NADPH might contribute to hypoxic responses associated with changes in the redox state of NADPH.
Figure 10 summarizes the proposed involvement of the sGC/PKG axis and hypoxia-induced removal of a tonic normoxic H2O2-dependent vasodilation as a mechanism contributing to HPV.
8.3. Does the Presence of Extracellular H2O2 affect HPV?
As described in Section 4, it is thought that the basal extracellular H2O2 concentration is in the range of 1-5 μM, although the evidence for this is meagre. This level of extracellular H2O2 is predicted to cause an inward leak which would significantly raise its intracellular concentration, especially adjacent to the cell membrane [240], and it is therefore conceivable that this could influence the effects of changes in mitochondrial H2O2 production on redox-sensitive HPV effector targets, especially those in the plasmalemma. Wolin’s laboratory examined the possible impact of extracellular H2O2 on HPV in two studies, using catalase, which was applied at a low concentration (1μM) said to scavenge only extracellular H2O2 These investigations, carried out in isolated PA from cow [477] and mouse [478], found no effect of catalase on HPV under control conditions. However, HPV in both arteries was suppressed by manoeuvres which raised the extracellular [H2O2], an effect which was partly reversed by catalase. Based on other results in their paper, Patel et al, [478] proposed that this was due to oxidation-dependent stimulation of PKGa1, presumably exerted by an excessive inward leak of H2O2. These results should be interpreted with caution since no evidence was presented that catalase was not penetrating into the cells (which it has been shown to do [353]). However, taken at face value, they support the possibility that the presence of extracellular H2O2 might influence HPV.
8.4. Summary and Critique: Does Activation of the PPP and the Withdrawal of H2O2/PKG – Mediated Normoxic Vasodilation Cause HPV?
The picture of the involvement of ROS and cellular redox in HPV which emerges from the papers published on this subject by Wolin, Gupte and colleagues is complex, but incorporates two proposed pathways as shown in Figure 9 and Figure 10:
- Under normoxic conditions, the production of superoxide/H2O2 by Nox4 is greater in PA than in systemic arteries because the PPP, being more active, generates more NADPH. The resulting higher level of H2O2 creates an ongoing vasodilating influence by stimulating PKG1a, both directly by oxidizing Cys42 on PKG1a, and indirectly by activating sGC. Hypoxia decreases the production of superoxide/H2O2 by Nox4, thereby raising vascular tone by inhibiting this baseline vasorelaxation.
- Hypoxia also suppresses the activity of PKG1a by stimulating G-6-PD and the PPP, causing a consequent increase in the NADPH/NADP+ ratio. This acts, at least in part, through Trx-1 and TrxR-1, to reduce PKG1a, causing it to become less active, which promotes contraction. The activation of G-6-PD is proposed to result from an increase in cytoplasmic [ROS], which promotes also HPV by stimulating rho kinase and Ca2+-dependent contractile mechanisms.
Below, we look critically at several aspects of these proposals.
8.4.1. Decreased Production of H2O2 by Nox as a Cause of HPV
The high Km(O2) of Nox4 (Section 3.2.2), which allows its production of H2O2 to be sensitive to even small decreases in PO2, renders it an eminently feasible O2 sensor for HPV. It has been shown, for example, that H2O2 production by Nox4 expressed in HEK293 cells falls by ~50% when the PO2 is decreased from 150 to 80 Torr [59].
Nonetheless, the knockout of Nox4 had no effect on basal PAP or HPV [399] in mice, indicating that that it does not regulate basal vascular PA tone and is not required for the response to hypoxia, at least in this species. On the other hand, sustained HPV was diminished by the knockout of p22phox [402], suggesting that another Nox isoform, probably Nox1, plays a role in HPV. However, p22phox knockout had no effect on basal PAP. This argues against the possibility that HPV in mice is due to the withdrawal of basal Nox-induced vasodilation, suggesting instead that Nox is contributing to sustained HPV by increasing its production of ROS.
These observations do not rule out the possibility that the loss of basal ROS production by Nox4 is important for HPV in species other than the mouse. Nonetheless, the involvement of decreased ROS production by Nox4 in HPV in cow can be questioned on several fronts. Most importantly, direct evidence that hypoxia decreases ROS levels in BPA is inconsistent. A hypoxia-induced fall in ROS in intact BPA was demonstrated in only a single study [545], which measured H2O2 using a high concentration of lucigenin with HRP. Lucigenin at this concentration is subject to redox cycling making it a suboptimal indicator for examining the effect of hypoxia on ROS levels (Section 7.4). Conversely, the investigation employing the intracellular indicator DHE reported that hypoxia increases ROS in BPA [440]. Although DHE also has its drawbacks, its suitability as a detector of cell oxidation has been vigorously defended [574].
Evidence [453] that the withdrawal of normoxic vasodilation caused by Nox4-generated H2O2 contributes to HPV in BPA can also be challenged. As described in this paper, siRNA knockout of Nox4 did not affect the contraction to 25 mM K+, which the authors used an index of basal reactivity. This conflicted with their hypothesis that Nox4 exerts a tonic vasodilating effect on basal tone, which predicts that Nox4 knockout should increase the K+ contraction. The authors explained this apparent discrepancy by proposing that ROS generation by Nox4 exerts opposing vasodilating and vasoconstricting effects, and that when ROS production is acutely interrupted by hypoxia, the dilating effect is immediately removed, whereas the constricting effect only disappears after a delay of several hours.
However, down-regulation of sGC and PKG1α, the putative targets by which H2O2 produced by Nox4 was suggested to suppress basal tone, did increase the contraction to 25 mM K+ [502]. This observations seeme to undermine the authors’ explanation for the lack of an effect of Nox4 knockout on basal reactivity, which provides an important underpinning for their proposal that that Nox4-derived H2O2 causes normoxic vasodilation. As to the possibility that the attenuation of superoxide/H2O2 production by other Nox isoforms by hypoxia might contribute to HPV, this laboratory did not detect the presence of Nox1 in these arteries [554], and demonstrated that although blocking Nox2 with gp91-dsat diminished ROS levels in BPA, it had no effect on either basal reactivity or HPV [453]. We therefore believe that the proposal that a significant component of HPV in BPA is due to the loss of basal vasorelaxation associated with H2O2 production by Nox remains speculative.
8.4.2. Does a Fall in H2O2 During Hypoxia Cause HPV by Inhibiting sGC?
If HPV is partly caused by the removal of H2O2-induced basal oxidative activation of sGC and/or an increase in PPP-derived NADPH [498,502,544,545] it would be predicted that 1. block of sGC or PKG should inhibit HPV and also mimic hypoxia in causing PA constriction, 2. hypoxia should decrease the cGMP content of PA, and 3. the activity of sGC should be increased by H2O2 or equivalent oxidant stimuli.
The first prediction is supported by an in vivo study of HPV in cats [575] which showed that the sGC blocker methylene blue (MB) increased baseline PAP and abolished the response to hypoxia. MB also increased baseline normoxic PAP in pigs and sheep, although its effect on HPV was not investigated [576]. Conversely, MB [577,578] and ODQ [579] had no effect on baseline normoxic PAP, and augmented HPV in isolated perfused rat lung. In isolated rat PA, MB increased HPV, although this effect was absent in severe hypoxia [580]. Vermeersch and colleagues [581] compared HPV in wildtype mice and those in which the a1 subunit of sGCa1b1 isoform, the predominant form of sGC in the vasculature, had been rendered less active by deletion of exon 6 of the a1 subunit gene. RVSP under normoxic conditions and during a 10-minute hypoxic challenge was not different in the two groups and hypoxia had no effect on the lung cGMP content in the control mice. Also, although ODQ slightly reduced the amplitude of HPV in isolated perfused mouse lung, it did not increase normoxic PAP, going against the idea of sGC-mediated normoxic vasorelaxation [582]. Fouty et al [579] found that the PKG inhibitor Rp-8-pCPT-cGMPS had no effect on basal PAP or HPV in isolated perfused rat lung. They verified that Rp-8-pCPT-cGMPS suppressed PKG activity, by monitoring phosphorylation of the Ins(1,4,5)P3 receptor, a PKG target. The differential effect of Rp-8-pCPT-cGMPS and ODQ on HPV in this study is difficult to explain, since PKG is the only known target of cGMP in vascular smooth muscle. Nevertheless, neither drug affected basal PAP. With regard to the second prediction, contrasting effects of hypoxia on cGMP levels in PA have been reported. In BPA, hypoxia decreased cGMP in endothelium-denuded BPA [544]. In rat PA, however, hypoxia decreased the cGMP content of intact but not endothelium-denuded PA arteries [583]. This suggested that hypoxia was suppressing the activity of sGC, but that this was secondary to a fall in endothelial NO release rather than being due a direct effect of hypoxia on sGC in the smooth muscle. Taken together, the results of these studies argue against a role for sGC in causing basal PA vasorelaxation in rodents, although this might occur in cats in a manner similar to that proposed for BPA.
The proposal that H2O2 production under normoxic conditions creates a vasodilating influence in BPA which is in part due to the activation of sGC, and that this is decreased by hypoxia, rests on evidence that 1) exogenous H2O2 caused vasorelaxation and increased the cGMP content in endothelium-intact arteries [489] , 2) hypoxia decreased the cGMP content of endothelium-denuded arteries [544], 3) depletion of cellular sGCb1 using prolonged ODQ treatment increased basal reactivity (assessed as the contraction to 25mM K+ under normoxic conditions), [498], 4) depletion of sGCb1 using long term ODQ treatment or siRNA knockdown inhibited vasorelaxation and activation of PKG (assessed using VASP phosphorylation) by exogenous H2O2 under hypoxic conditions [498,502], and 5) depletion of sGC using siRNA knockdown increased basal reactivity but decreased HPV [502].
Importantly, Neo et al [498] found that the vasorelaxation of BPA induced by the H2O2 occurred in endothelium-denuded arteries, and whereas it was decreased by downregulating sGC expression, it was not affected by acute treatment with ODQ [498]. Since ODQ would block stimulation of sGC due to any remaining NO, they concluded from these results that the activation of sGC by H2O2 must be NO-independent (indeed, this laboratory had previously presented evidence that NO-dependent activation of sGC was inhibited by oxidation [561]).
In this case, their model predicts that if hypoxia causes a fall in H2O2 production it should decrease the cGMP concentration in endothelium-denuded arteries, an effect they had previously observed in BPA [544]. However, the cGMP content of endothelium-denuded rat PA was not affected by hypoxia [583], and in porcine PA, Ye et al also found that hypoxia did not affect the cyclic GMP level in the presence of either ODQ or the eNOS antagonist nitro-L-arginine [584] and that reducing agents either did not affect (DTT and GSH), or increased (L-cysteine and tris(2-carboxyethyl)phosphine), the cGMP content of nitro-L-arginine-treated arteries. Intriguingly, they also reported that sGC dimerization, which is required for its activation by NO [585], was present under basal conditions, and was decreased by both hypoxia and reductants. Accordingly, NO-dependent vasodilation was depressed by reductants. These observations contradict the evidence in BPA that oxidation depresses NO-dependent and increases NO-independent sGC activity but support the general concept that the reduction of sGC by hypoxia can cause its inhibition. However, since NO release falls during hypoxia [586], the extent to which the redox regulation of NO-dependent sGC activation could influence HPV is uncertain.
8.4.3. Does a Fall in H2O2 During Hypoxia Cause HPV by Directly Inhibiting Protein Kinase G?
The proposal that a decrease in H2O2 levels and/or an increase in NADPH production by the PPP caused by hypoxia contributes to HPV by reversing oxidation-induced PKG1α activation is consistent with extensive evidence showing that PKG exerts an important vasodilating influence in systemic arteries through its phosphorylation of multiple target proteins which control vascular tone [587]. It is also well established that PKG activity in arteries is increased by the oxidation of Cys42 leading to formation of a disulfide bond PKG1α in mice [37,570,588], and that this creates an ongoing vasodilating influence under baseline conditions, at least in systemic resistance vessels, as evidenced by the observation that blood pressure is raised in mice expressing a ‘redox-dead’ form of PKGa1 [589].
With regard to the involvement of PKG1α in HPV, this hypothesis is supported by evidence, described in Section 8.2.2, that: 1. exposure of BPA to 0.1 and 1 mM H2O2 increased PKG1α DDC formation and phosphorylation of its target VASP and these responses were reversed by DTT [498]. 2. hypoxia caused contraction and decreased PKG DDC formation and VASP phosphorylation of endothelium-denuded BPA, whereas opposite effects on PKG occurred in BCA, which relax to hypoxia, 3. DTT increased force in BPA under both aerobic and hypoxic conditions and diminished PKG1α DDC formation and VASP phosphorylation, 4. siRNA knockdown of PKG1α increased basal reactivity (i.e. the contraction to 25mM K+ under normoxic conditions) but depressed HPV [502], and 5. block of the PPP, which would reduce the supply of NADPH available to reduce and therefore inactivate PKG1α, diminished the contraction evoked by 25 mM K+ under both normoxic and hypoxia conditions [572].
However, it is not clear that the effect of the PPP on contraction exerted through the redox regulation of PKG1α [572] is specific to HPV. The authors proposed that the PPP was promoting hypoxic contraction of BPA by increasing the production of NADPH, which would transfer reducing equivalents via TrxR-1 and Trx-1 to PKG1α, thereby suppressing its vasodilating activity (Figure 10). In support of this scheme, they found that interventions used to block the influence of the PPP on contraction (i.e. siRNA knockdown of G6PD, Trx-1, TrxR-1) decreased the contraction induced by 25mM K+ under hypoxic conditions. Importantly, however, these interventions caused a comparable decrease in the high K+ contraction recorded under normoxic conditions, suggesting that the PPP similarly promotes contraction under both normoxic and hypoxic conditions rather than selectively enhancing HPV. Along similar lines, Chettimada et al, who also showed that that blocking the PPP with 6-AN increased HPV, found that this caused equivalent increases in the activity of PKG under normoxic and hypoxic conditions [573]. Rather than demonstrating that the activation of the PPP acting via an NAPDH/TrxR/Trx/ PKG1α pathway is a specific cause of HPV, these results suggest that NADPH produced by the PPP may promote contraction by inhibiting PKG1a regardless of the oxygen concentration.
There is currently no information about the role of PKG1α oxidation in acute HPV in other species, although there is evidence [590] that this is increased during chronic hypoxia and tends to ameliorate pulmonary hypertension in mice.
8.4.4. Is Activation of the PPP Important for HPV?
Although we have argued that the evidence that the PPP causes/promotes HPV by inhibiting PKG1a is unconvincing, it is possible that stimulation of the PPP by hypoxia could trigger HPV via other contractile mechanisms, e.g. activation of RhoK [440], or inhibition of KV channels by the interaction of NADPH with their b subunits [265]. Certainly, the evidence that hypoxia stimulates the PPP and causes contraction in BPA whereas it inhibits the PPP and causes relaxation in BCA offers support, albeit indirect, for the concept that it regulates the contractile response to low PO2. Unfortunately, it is difficult to draw any conclusions about the involvement of the PPP in HPV because no other laboratories seem to have investigated this. Nevertheless, in agreement with the observation that the G-6-PD antagonist 6-AN blocked HPV in BPA, we have found that dehydroepiandrosterone, which also blocks this enzyme, has a similar effect in rat PA (Figure 11).
There is also evidence that the PPP in other types of cells is rapidly stimulated by oxidative stress [591] and several mechanisms by which the oxidation of thiols in glycolytic enzymes (e.g. GAPDH, PKM2) can divert metabolic flux into the PPP have been defined (e.g. [592]. Supporting the involvement of G-6-PD and the PPP in regulating contraction, the vasoconstrictor U46619 caused an increase in G-6-PD activity in BCA which was mediated by a ROS-dependent stimulation of PKCd [565]. Also, complexes containing G-6-DP, Trx-1 and PKG can be detected in BPA homogenates [573], and L-type VGCC in BCA and human internal mammary artery SMC were shown to be activated by a direct interaction with G-6-PD in caveoli [593]. Further investigation of the role of PPP activation in HPV, focused on determining the effect of hypoxia on the redox state of the NADP+:NADPH couple [594] and on identifying the effectors through which the PPP could induce HPV, would therefore be of great interest.
9. Conclusion
The proposal that cell redox mechanisms acting on cell thiols are responsible for O2 sensing in HPV was first mooted by Kenneth Weir and colleagues in a remarkably prescient study published more than 40 years ago [274]. As we have described, experiments in the intervening time have led to the development of three main models designed to describe how hypoxia induces changes in PASMC ROS production and cytoplasmic redox state which engage with specific HPV effector mechanisms. Although many of these studies utilized techniques which were innovative and state-of-the art at the time they were carried out, the rapid pace of conceptual and technical development in the field of redox biology, especially over the past decade, has brought into question the interpretation of many the observations which underpin these models. Largely because it is supported by a larger evidence base, and one which draws more heavily on the use of newer techniques which are at present viewed as being more valid, we believe that the Mitochondrial ROS hypothesis currently provides the most convincing picture of how hypoxia acts through cell redox mechanisms to cause HPV.
Nevertheless, many interesting questions remain. The mechanisms by which hypoxia can increase mitochondrial ROS production remain uncertain, as do the identities of the HPV effectors which are linked to an increase in ROS, and the precise nature of the involvement of Cox4i2 in O2 sensing in PASMC. The possible involvement of NADH in HPV, possibly via the regulation of KV or voltage-gated Ca2+ channels, is a long-standing but unresolved concept which has been given a renewed lease of life by recent observations in CBCC. Indeed, the question as to what extent common mechanisms are used by the various homeostatic sensors of acute hypoxia present in the body remains open. Recent evidence suggesting that hypoxia may exert complex effects on the cytoplasmic redox state (due to spatial and/or temporal compartmentalization?) rather than generating a simple up/down ROS signal is also worthy of note. In addition, concerns about the use of non-physoxic conditions and regarding whether cells were being exposed to O2 concentrations which differed from those which were intended also suggest that the quest for the O2 sensing mechanism in HPV should not be regarded as finished.
Happily, progress in the development of increasingly sensitive/specific indicators for cellular H2O2 [542], peroxynitrite [595] Han, free NADH ([596]) and NADPH ([89]), as well as probes for intracellular PO2 ([597]) has made it more possible than ever to explore the involvement of cell redox mechanism as O2 sensors in PASMC and other cells. It will be interesting to see where the research in this area takes us.
Acknowledgements
We greatly appreciate the assistance of Professors Annie Beuve, Philip Eaton, Ryan Mailloux, Patrick Pagano, Armindo Salavar, Jason Treberg, David Wilson and especially Paul Schumacker, who provided very detailed and illuminating responses to our questions about many aspects of redox biology and mitochondrial function. Their inputs were invaluable in informing the writing of the manuscript. Nevertheless, the opinions we express on the many controversial issues in this field are our own.
References
- Gierhardt, M.; Pak, O.; Walmrath, D.; Seeger, W.; Grimminger, F.; Ghofrani, H.A.; Weissmann, N.; Hecker, M.; Sommer, N. Impairment of hypoxic pulmonary vasoconstriction in acute respiratory distress syndrome. Eur Respir Rev 2021, 30. [CrossRef]
- Sommer, N.; Dietrich, A.; Schermuly, R.T.; Ghofrani, H.A.; Gudermann, T.; Schulz, R.; Seeger, W.; Grimminger, F.; Weissmann, N. Regulation of hypoxic pulmonary vasoconstriction: basic mechanisms. Eur Respir J 2008, 32, 1639-1651. [CrossRef]
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol Rev 2012, 92, 367-520. [CrossRef]
- Nagendran, J.; Stewart, K.; Hoskinson, M.; Archer, S.L. An anesthesiologist's guide to hypoxic pulmonary vasoconstriction: implications for managing single-lung anesthesia and atelectasis. Curr Opin Anaesthesiol 2006, 19, 34-43. [CrossRef]
- Dinenno, F.A. Skeletal muscle vasodilation during systemic hypoxia in humans. J Appl Physiol (1985) 2016, 120, 216-225. [CrossRef]
- Dorrington, K.L.; Clar, C.; Young, J.D.; Jonas, M.; Tansley, J.G.; Robbins, P.A. Time course of the human pulmonary vascular response to 8 hours of isocapnic hypoxia. Am J Physiol 1997, 273, H1126-1134. [CrossRef]
- Talbot, N.P.; Balanos, G.M.; Dorrington, K.L.; Robbins, P.A. Two temporal components within the human pulmonary vascular response to approximately 2 h of isocapnic hypoxia. J Appl Physiol (1985) 2005, 98, 1125-1139. [CrossRef]
- Marshall, B.E.; Clarke, W.R.; Costarino, A.T.; Chen, L.; Miller, F.; Marshall, C. The dose-response relationship for hypoxic pulmonary vasoconstriction. Respir Physiol 1994, 96, 231-247. [CrossRef]
- Fishman, A.P. Acute hypoxia and pulmonary vasoconstriction in humans: uncovering the mechanism of the pressor response. Am J Physiol Lung Cell Mol Physiol 2004, 287, L893-894. [CrossRef]
- Robertson, T.P.; Hague, D.; Aaronson, P.I.; Ward, J.P. Voltage-independent calcium entry in hypoxic pulmonary vasoconstriction of intrapulmonary arteries of the rat. J Physiol 2000, 525 Pt 3, 669-680. [CrossRef]
- Weissmann, N.; Dietrich, A.; Fuchs, B.; Kalwa, H.; Ay, M.; Dumitrascu, R.; Olschewski, A.; Storch, U.; Mederos y Schnitzler, M.; Ghofrani, H.A.; et al. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci U S A 2006, 103, 19093-19098. [CrossRef]
- Jain, P.P.; Hosokawa, S.; Xiong, M.; Babicheva, A.; Zhao, T.; Rodriguez, M.; Rahimi, S.; Pourhashemi, K.; Balistrieri, F.; Lai, N.; et al. Revisiting the mechanism of hypoxic pulmonary vasoconstriction using isolated perfused/ventilated mouse lung. Pulm Circ 2020, 10, 2045894020956592. [CrossRef]
- Jabr, R.I.; Toland, H.; Gelband, C.H.; Wang, X.X.; Hume, J.R. Prominent role of intracellular Ca2+ release in hypoxic vasoconstriction of canine pulmonary artery. Br J Pharmacol 1997, 122, 21-30. [CrossRef]
- Weigand, L.; Foxson, J.; Wang, J.; Shimoda, L.A.; Sylvester, J.T. Inhibition of hypoxic pulmonary vasoconstriction by antagonists of store-operated Ca2+ and nonselective cation channels. Am J Physiol Lung Cell Mol Physiol 2005, 289, L5-L13. [CrossRef]
- Robertson, T.P.; Dipp, M.; Ward, J.P.; Aaronson, P.I.; Evans, A.M. Inhibition of sustained hypoxic vasoconstriction by Y-27632 in isolated intrapulmonary arteries and perfused lung of the rat. Br J Pharmacol 2000, 131, 5-9. [CrossRef]
- Aaronson, P.I.; Robertson, T.P.; Ward, J.P. Endothelium-derived mediators and hypoxic pulmonary vasoconstriction. Respir Physiol Neurobiol 2002, 132, 107-120. [CrossRef]
- Archer, S.L.; Wu, X.C.; Thebaud, B.; Nsair, A.; Bonnet, S.; Tyrrell, B.; McMurtry, M.S.; Hashimoto, K.; Harry, G.; Michelakis, E.D. Preferential expression and function of voltage-gated, O2-sensitive K+ channels in resistance pulmonary arteries explains regional heterogeneity in hypoxic pulmonary vasoconstriction: ionic diversity in smooth muscle cells. Circ Res 2004, 95, 308-318. [CrossRef]
- Connolly, M.J.; Prieto-Lloret, J.; Becker, S.; Ward, J.P.; Aaronson, P.I. Hypoxic pulmonary vasoconstriction in the absence of pretone: essential role for intracellular Ca2+ release. J Physiol 2013, 591, 4473-4498. [CrossRef]
- Moudgil, R.; Michelakis, E.D.; Archer, S.L. Hypoxic pulmonary vasoconstriction. J Appl Physiol (1985) 2005, 98, 390-403. [CrossRef]
- Wang, L.; Yin, J.; Nickles, H.T.; Ranke, H.; Tabuchi, A.; Hoffmann, J.; Tabeling, C.; Barbosa-Sicard, E.; Chanson, M.; Kwak, B.R.; et al. Hypoxic pulmonary vasoconstriction requires connexin 40-mediated endothelial signal conduction. J Clin Invest 2012, 122, 4218-4230. [CrossRef]
- Sommer, N.; Strielkov, I.; Pak, O.; Weissmann, N. Oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction. Eur Respir J 2016, 47, 288-303. [CrossRef]
- Dao, V.T.; Elbatreek, M.H.; Altenhofer, S.; Casas, A.I.; Pachado, M.P.; Neullens, C.T.; Knaus, U.G.; Schmidt, H. Isoform-selective NADPH oxidase inhibitor panel for pharmacological target validation. Free Radic Biol Med 2020, 148, 60-69. [CrossRef]
- Archer, S.L.; Gomberg-Maitland, M.; Maitland, M.L.; Rich, S.; Garcia, J.G.; Weir, E.K. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol 2008, 294, H570-578. [CrossRef]
- Nozik-Grayck, E.; Stenmark, K.R. Role of reactive oxygen species in chronic hypoxia-induced pulmonary hypertension and vascular remodeling. Adv Exp Med Biol 2007, 618, 101-112. [CrossRef]
- Wang, Y.X.; Zheng, Y.M. ROS-dependent signaling mechanisms for hypoxic Ca(2+) responses in pulmonary artery myocytes. Antioxid Redox Signal 2010, 12, 611-623. [CrossRef]
- Resta, T.C.; Broughton, B.R.; Jernigan, N.L. Reactive oxygen species and RhoA signaling in vascular smooth muscle: role in chronic hypoxia-induced pulmonary hypertension. Adv Exp Med Biol 2010, 661, 355-373. [CrossRef]
- Veit, F.; Pak, O.; Brandes, R.P.; Weissmann, N. Hypoxia-dependent reactive oxygen species signaling in the pulmonary circulation: focus on ion channels. Antioxid Redox Signal 2015, 22, 537-552. [CrossRef]
- Jaitovich, A.; Jourd'heuil, D. A Brief Overview of Nitric Oxide and Reactive Oxygen Species Signaling in Hypoxia-Induced Pulmonary Hypertension. Adv Exp Med Biol 2017, 967, 71-81. [CrossRef]
- Huetsch, J.C.; Suresh, K.; Shimoda, L.A. Regulation of Smooth Muscle Cell Proliferation by NADPH Oxidases in Pulmonary Hypertension. Antioxidants (Basel) 2019, 8. [CrossRef]
- Yan, S.; Resta, T.C.; Jernigan, N.L. Vasoconstrictor Mechanisms in Chronic Hypoxia-Induced Pulmonary Hypertension: Role of Oxidant Signaling. Antioxidants (Basel) 2020, 9. [CrossRef]
- Reyes-Garcia, J.; Carbajal-Garcia, A.; Di Mise, A.; Zheng, Y.M.; Wang, X.; Wang, Y.X. Important Functions and Molecular Mechanisms of Mitochondrial Redox Signaling in Pulmonary Hypertension. Antioxidants (Basel) 2022, 11. [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat Rev Mol Cell Biol 2022, 23, 499-515. [CrossRef]
- Dunham-Snary, K.J.; Hong, Z.G.; Xiong, P.Y.; Del Paggio, J.C.; Herr, J.E.; Johri, A.M.; Archer, S.L. A mitochondrial redox oxygen sensor in the pulmonary vasculature and ductus arteriosus. Pflugers Arch 2016, 468, 43-58. [CrossRef]
- Smith, K.A.; Schumacker, P.T. Sensors and signals: the role of reactive oxygen species in hypoxic pulmonary vasoconstriction. J Physiol 2019, 597, 1033-1043. [CrossRef]
- Pak, O.; Nolte, A.; Knoepp, F.; Giordano, L.; Pecina, P.; Huttemann, M.; Grossman, L.I.; Weissmann, N.; Sommer, N. Mitochondrial oxygen sensing of acute hypoxia in specialized cells - Is there a unifying mechanism? Biochim Biophys Acta Bioenerg 2022, 1863, 148911. [CrossRef]
- Weir, E.K.; Archer, S.L. The mechanism of acute hypoxic pulmonary vasoconstriction: the tale of two channels. FASEB J 1995, 9, 183-189. [CrossRef]
- Burgoyne, J.R.; Madhani, M.; Cuello, F.; Charles, R.L.; Brennan, J.P.; Schroder, E.; Browning, D.D.; Eaton, P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 2007, 317, 1393-1397. [CrossRef]
- Waypa, G.B.; Chandel, N.S.; Schumacker, P.T. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res 2001, 88, 1259-1266. [CrossRef]
- Rathore, R.; Zheng, Y.M.; Niu, C.F.; Liu, Q.H.; Korde, A.; Ho, Y.S.; Wang, Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med 2008, 45, 1223-1231. [CrossRef]
- Alruwaili, N.; Kandhi, S.; Sun, D.; Wolin, M.S. Metabolism and Redox in Pulmonary Vascular Physiology and Pathophysiology. Antioxid Redox Signal 2019, 31, 752-769. [CrossRef]
- Evans, A.M.; Mustard, K.J.; Wyatt, C.N.; Peers, C.; Dipp, M.; Kumar, P.; Kinnear, N.P.; Hardie, D.G. Does AMP-activated protein kinase couple inhibition of mitochondrial oxidative phosphorylation by hypoxia to calcium signaling in O2-sensing cells? J Biol Chem 2005, 280, 41504-41511. [CrossRef]
- Evans, A.M.; Lewis, S.A.; Ogunbayo, O.A.; Moral-Sanz, J. Modulation of the LKB1-AMPK Signalling Pathway Underpins Hypoxic Pulmonary Vasoconstriction and Pulmonary Hypertension. Adv Exp Med Biol 2015, 860, 89-99. [CrossRef]
- Moral-Sanz, J.; Lewis, S.A.; MacMillan, S.; Ross, F.A.; Thomson, A.; Viollet, B.; Foretz, M.; Moran, C.; Hardie, D.G.; Evans, A.M. The LKB1-AMPK-alpha1 signaling pathway triggers hypoxic pulmonary vasoconstriction downstream of mitochondria. Sci Signal 2018, 11. [CrossRef]
- Moral-Sanz, J.; Mahmoud, A.D.; Ross, F.A.; Eldstrom, J.; Fedida, D.; Hardie, D.G.; Evans, A.M. AMP-activated protein kinase inhibits Kv 1.5 channel currents of pulmonary arterial myocytes in response to hypoxia and inhibition of mitochondrial oxidative phosphorylation. J Physiol 2016, 594, 4901-4915. [CrossRef]
- Olson, K.R.; Dombkowski, R.A.; Russell, M.J.; Doellman, M.M.; Head, S.K.; Whitfield, N.L.; Madden, J.A. Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J Exp Biol 2006, 209, 4011-4023. [CrossRef]
- Olson, K.R.; Whitfield, N.L.; Bearden, S.E.; St Leger, J.; Nilson, E.; Gao, Y.; Madden, J.A. Hypoxic pulmonary vasodilation: a paradigm shift with a hydrogen sulfide mechanism. Am J Physiol Regul Integr Comp Physiol 2010, 298, R51-60. [CrossRef]
- Olson, K.R. Are Reactive Sulfur Species the New Reactive Oxygen Species? Antioxid Redox Signal 2020, 33, 1125-1142. [CrossRef]
- Olson, K.R. A Case for Hydrogen Sulfide Metabolism as an Oxygen Sensing Mechanism. Antioxidants (Basel) 2021, 10. [CrossRef]
- Keeley, T.P.; Mann, G.E. Defining Physiological Normoxia for Improved Translation of Cell Physiology to Animal Models and Humans. Physiol Rev 2019, 99, 161-234. [CrossRef]
- Warpsinski, G.; Smith, M.J.; Srivastava, S.; Keeley, T.P.; Siow, R.C.M.; Fraser, P.A.; Mann, G.E. Nrf2-regulated redox signaling in brain endothelial cells adapted to physiological oxygen levels: Consequences for sulforaphane mediated protection against hypoxia-reoxygenation. Redox Biol 2020, 37, 101708. [CrossRef]
- Smith, M.J.; Yang, F.; Griffiths, A.; Morrell, A.; Chapple, S.J.; Siow, R.C.M.; Stewart, T.; Maret, W.; Mann, G.E. Redox and metal profiles in human coronary endothelial and smooth muscle cells under hyperoxia, physiological normoxia and hypoxia: Effects of NRF2 signaling on intracellular zinc. Redox Biol 2023, 62, 102712. [CrossRef]
- Altun, H.Y.; Secilmis, M.; Yang, F.; Akgul Caglar, T.; Vatandaslar, E.; Toy, M.F.; Vilain, S.; Mann, G.E.; Ozturk, G.; Eroglu, E. Visualizing hydrogen peroxide and nitric oxide dynamics in endothelial cells using multispectral imaging under controlled oxygen conditions. Free Radic Biol Med 2024, 221, 89-97. [CrossRef]
- Alva, R.; Wiebe, J.E.; Stuart, J.A. Revisiting reactive oxygen species production in hypoxia. Pflugers Arch 2024, 476, 1423-1444. [CrossRef]
- Kamler, M.; Nowak, K.; Bock, M.; Herold, U.; Motsch, J.; Hagl, S.; Gebhard, M.M.; Jakob, H. Bronchial artery revascularization restores peribronchial tissue oxygenation after lung transplantation. J Heart Lung Transplant 2004, 23, 763-766. [CrossRef]
- Herold, U.; Jakob, H.; Kamler, M.; Thiele, R.; Tochtermann, U.; Weinmann, J.; Motsch, J.; Gebhard, M.M.; Hagl, S. Interruption of bronchial circulation leads to a severe decrease in peribronchial oxygen tension in standard lung transplantation technique. Eur J Cardiothorac Surg 1998, 13, 176-183. [CrossRef]
- Rivera, B.K.; Naidu, S.K.; Subramanian, K.; Joseph, M.; Hou, H.; Khan, N.; Swartz, H.M.; Kuppusamy, P. Real-time, in vivo determination of dynamic changes in lung and heart tissue oxygenation using EPR oximetry. Adv Exp Med Biol 2014, 812, 81-86. [CrossRef]
- Marshall, C.; Marshall, B. Site and sensitivity for stimulation of hypoxic pulmonary vasoconstriction. J Appl Physiol Respir Environ Exerc Physiol 1983, 55, 711-716. [CrossRef]
- Grayson, C.; Mailloux, R.J. Coenzyme Q(10) and nicotinamide nucleotide transhydrogenase: Sentinels for mitochondrial hydrogen peroxide signaling. Free Radic Biol Med 2023, 208, 260-271. [CrossRef]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111-5120. [CrossRef]
- Knock, G.A. NADPH oxidase in the vasculature: Expression, regulation and signalling pathways; role in normal cardiovascular physiology and its dysregulation in hypertension. Free Radic Biol Med 2019, 145, 385-427. [CrossRef]
- Rogers, Z.J.; Flood, D.; Bencherif, S.A.; Taylor, C.T. Oxygen control in cell culture - Your cells may not be experiencing what you think! Free Radic Biol Med 2025, 226, 279-287. [CrossRef]
- Peniche Silva, C.J.; Liebsch, G.; Meier, R.J.; Gutbrod, M.S.; Balmayor, E.R.; van Griensven, M. A New Non-invasive Technique for Measuring 3D-Oxygen Gradients in Wells During Mammalian Cell Culture. Front Bioeng Biotechnol 2020, 8, 595. [CrossRef]
- Rogers, Z.J.; Colombani, T.; Khan, S.; Bhatt, K.; Nukovic, A.; Zhou, G.; Woolston, B.M.; Taylor, C.T.; Gilkes, D.M.; Slavov, N.; et al. Controlling Pericellular Oxygen Tension in Cell Culture Reveals Distinct Breast Cancer Responses to Low Oxygen Tensions. Adv Sci (Weinh) 2024, 11, e2402557. [CrossRef]
- Malconian, M.K.; Rock, P.B.; Reeves, J.T.; Cymerman, A.; Houston, C.S. Operation Everest II: gas tensions in expired air and arterial blood at extreme altitude. Aviat Space Environ Med 1993, 64, 37-42.
- Sutton, J.R.; Reeves, J.T.; Wagner, P.D.; Groves, B.M.; Cymerman, A.; Malconian, M.K.; Rock, P.B.; Young, P.M.; Walter, S.D.; Houston, C.S. Operation Everest II: oxygen transport during exercise at extreme simulated altitude. J Appl Physiol (1985) 1988, 64, 1309-1321. [CrossRef]
- West, J.B.; Hackett, P.H.; Maret, K.H.; Milledge, J.S.; Peters, R.M., Jr.; Pizzo, C.J.; Winslow, R.M. Pulmonary gas exchange on the summit of Mount Everest. J Appl Physiol Respir Environ Exerc Physiol 1983, 55, 678-687. [CrossRef]
- Grocott, M.P.; Martin, D.S.; Levett, D.Z.; McMorrow, R.; Windsor, J.; Montgomery, H.E.; Caudwell Xtreme Everest Research, G. Arterial blood gases and oxygen content in climbers on Mount Everest. N Engl J Med 2009, 360, 140-149. [CrossRef]
- Kemp, P.J.; Telezhkin, V. Oxygen sensing by the carotid body: is it all just rotten eggs? Antioxid Redox Signal 2014, 20, 794-804. [CrossRef]
- Hawkins, C.L.; Davies, M.J. Detection, identification, and quantification of oxidative protein modifications. J Biol Chem 2019, 294, 19683-19708. [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu Rev Biochem 2017, 86, 715-748. [CrossRef]
- Lipinski, B. Hydroxyl radical and its scavengers in health and disease. Oxid Med Cell Longev 2011, 2011, 809696. [CrossRef]
- Bienert, G.P.; Moller, A.L.; Kristiansen, K.A.; Schulz, A.; Moller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem 2007, 282, 1183-1192. [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 2007, 87, 315-424. [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol 2017, 11, 613-619. [CrossRef]
- Rios, E.J.; Fallon, M.; Wang, J.; Shimoda, L.A. Chronic hypoxia elevates intracellular pH and activates Na+/H+ exchange in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2005, 289, L867-874. [CrossRef]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968-983 e924. [CrossRef]
- Ulrich, K.; Jakob, U. The role of thiols in antioxidant systems. Free Radic Biol Med 2019, 140, 14-27. [CrossRef]
- Salsbury, F.R., Jr.; Knutson, S.T.; Poole, L.B.; Fetrow, J.S. Functional site profiling and electrostatic analysis of cysteines modifiable to cysteine sulfenic acid. Protein Sci 2008, 17, 299-312. [CrossRef]
- Mailloux, R.J.; Grayson, C.; Koufos, O. Regulation of Mitochondrial Hydrogen Peroxide Availability by Protein S-glutathionylation. Cells 2022, 12. [CrossRef]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ Res 2012, 111, 1091-1106. [CrossRef]
- Brennan, J.P.; Bardswell, S.C.; Burgoyne, J.R.; Fuller, W.; Schroder, E.; Wait, R.; Begum, S.; Kentish, J.C.; Eaton, P. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem 2006, 281, 21827-21836. [CrossRef]
- Cuello, F.; Eaton, P. Cysteine-Based Redox Sensing and Its Role in Signaling by Cyclic Nucleotide-Dependent Kinases in the Cardiovascular System. Annu Rev Physiol 2019, 81, 63-87. [CrossRef]
- Kettenhofen, N.J.; Wood, M.J. Formation, reactivity, and detection of protein sulfenic acids. Chem Res Toxicol 2010, 23, 1633-1646. [CrossRef]
- Truong, T.H.; Carroll, K.S. Redox regulation of protein kinases. Crit Rev Biochem Mol Biol 2013, 48, 332-356. [CrossRef]
- Ahsan, M.K.; Lekli, I.; Ray, D.; Yodoi, J.; Das, D.K. Redox regulation of cell survival by the thioredoxin superfamily: an implication of redox gene therapy in the heart. Antioxid Redox Signal 2009, 11, 2741-2758. [CrossRef]
- Pillay, C.S.; Rohwer, J.M. Computational models as catalysts for investigating redoxin systems. Essays Biochem 2024, 68, 27-39. [CrossRef]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid Redox Signal 2018, 28, 251-272. [CrossRef]
- Winterbourn, C.C. Biological Production, Detection, and Fate of Hydrogen Peroxide. Antioxid Redox Signal 2018, 29, 541-551. [CrossRef]
- Scherschel, M.; Niemeier, J.O.; Jacobs, L.; Hoffmann, M.D.A.; Diederich, A.; Bell, C.; Hohne, P.; Raetz, S.; Kroll, J.B.; Steinbeck, J.; et al. A family of NADPH/NADP(+) biosensors reveals in vivo dynamics of central redox metabolism across eukaryotes. Nat Commun 2024, 15, 10704. [CrossRef]
- Gebicka, L.; Krych-Madej, J. The role of catalases in the prevention/promotion of oxidative stress. J Inorg Biochem 2019, 197, 110699. [CrossRef]
- Netto, L.E.; Antunes, F. The Roles of Peroxiredoxin and Thioredoxin in Hydrogen Peroxide Sensing and in Signal Transduction. Mol Cells 2016, 39, 65-71. [CrossRef]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Varnai, P.; Hajnoczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol Cell 2016, 63, 240-248. [CrossRef]
- Stocker, S.; Van Laer, K.; Mijuskovic, A.; Dick, T.P. The Conundrum of Hydrogen Peroxide Signaling and the Emerging Role of Peroxiredoxins as Redox Relay Hubs. Antioxid Redox Signal 2018, 28, 558-573. [CrossRef]
- Sobotta, M.C.; Liou, W.; Stocker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol 2015, 11, 64-70. [CrossRef]
- Portillo-Ledesma, S.; Randall, L.M.; Parsonage, D.; Dalla Rizza, J.; Karplus, P.A.; Poole, L.B.; Denicola, A.; Ferrer-Sueta, G. Differential Kinetics of Two-Cysteine Peroxiredoxin Disulfide Formation Reveal a Novel Model for Peroxide Sensing. Biochemistry 2018, 57, 3416-3424. [CrossRef]
- Trujillo, M.; Ferrer-Sueta, G.; Radi, R. Kinetic studies on peroxynitrite reduction by peroxiredoxins. Methods Enzymol 2008, 441, 173-196. [CrossRef]
- Rhee, S.G.; Woo, H.A. Multiple functions of 2-Cys peroxiredoxins, I and II, and their regulations via post-translational modifications. Free Radic Biol Med 2020, 152, 107-115. [CrossRef]
- van Dam, L.; Pages-Gallego, M.; Polderman, P.E.; van Es, R.M.; Burgering, B.M.T.; Vos, H.R.; Dansen, T.B. The Human 2-Cys Peroxiredoxins form Widespread, Cysteine-Dependent- and Isoform-Specific Protein-Protein Interactions. Antioxidants (Basel) 2021, 10. [CrossRef]
- Mailloux, R.J.; Treberg, J.R. Protein S-glutathionlyation links energy metabolism to redox signaling in mitochondria. Redox Biol 2016, 8, 110-118. [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431-435. [CrossRef]
- Pelletier, M.; Lepow, T.S.; Billingham, L.K.; Murphy, M.P.; Siegel, R.M. New tricks from an old dog: mitochondrial redox signaling in cellular inflammation. Semin Immunol 2012, 24, 384-392. [CrossRef]
- Starkov, A.A.; Andreyev, A.Y.; Zhang, S.F.; Starkova, N.N.; Korneeva, M.; Syromyatnikov, M.; Popov, V.N. Scavenging of H2O2 by mouse brain mitochondria. J Bioenerg Biomembr 2014, 46, 471-477. [CrossRef]
- Treberg, J.R.; Munro, D.; Banh, S.; Zacharias, P.; Sotiri, E. Differentiating between apparent and actual rates of H2O2 metabolism by isolated rat muscle mitochondria to test a simple model of mitochondria as regulators of H2O2 concentration. Redox Biol 2015, 5, 216-224. [CrossRef]
- Dey, S.; Sidor, A.; O'Rourke, B. Compartment-specific Control of Reactive Oxygen Species Scavenging by Antioxidant Pathway Enzymes. J Biol Chem 2016, 291, 11185-11197. [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005, 70, 200-214. [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem J 2009, 417, 1-13. [CrossRef]
- Drose, S.; Brandt, U.; Wittig, I. Mitochondrial respiratory chain complexes as sources and targets of thiol-based redox-regulation. Biochim Biophys Acta 2014, 1844, 1344-1354. [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Starkov, A.A. Mitochondrial ROS Metabolism: 10 Years Later. Biochemistry (Mosc) 2015, 80, 517-531. [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 2016, 100, 14-31. [CrossRef]
- Hernansanz-Agustin, P.; Morales-Vidal, C.; Calvo, E.; Natale, P.; Marti-Mateos, Y.; Jaroszewicz, S.N.; Cabrera-Alarcon, J.L.; Acin-Perez, R.; Lopez-Montero, I.; Vazquez, J.; et al. A transmitochondrial sodium gradient controls membrane potential in mammalian mitochondria. Cell 2024, 187, 6599-6613 e6521. [CrossRef]
- Nicholls, D.G.; Ferguson, S.J. Bioenergetics 3, 3rd ed.; Academic Press: San Diego, Calif., 2002; pp. xviii, 297 p.
- Gauthier, L.D.; Greenstein, J.L.; Cortassa, S.; O'Rourke, B.; Winslow, R.L. A computational model of reactive oxygen species and redox balance in cardiac mitochondria. Biophys J 2013, 105, 1045-1056. [CrossRef]
- Castro, L.; Tortora, V.; Mansilla, S.; Radi, R. Aconitases: Non-redox Iron-Sulfur Proteins Sensitive to Reactive Species. Acc Chem Res 2019, 52, 2609-2619. [CrossRef]
- Tahara, E.B.; Navarete, F.D.; Kowaltowski, A.J. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med 2009, 46, 1283-1297. [CrossRef]
- Hoffman, D.L.; Brookes, P.S. Oxygen sensitivity of mitochondrial reactive oxygen species generation depends on metabolic conditions. J Biol Chem 2009, 284, 16236-16245. [CrossRef]
- Balmaceda, V.; Komlodi, T.; Szibor, M.; Gnaiger, E.; Moore, A.L.; Fernandez-Vizarra, E.; Viscomi, C. The striking differences in the bioenergetics of brain and liver mitochondria are enhanced in mitochondrial disease. Biochim Biophys Acta Mol Basis Dis 2024, 1870, 167033. [CrossRef]
- Slade, L.; Chalker, J.; Kuksal, N.; Young, A.; Gardiner, D.; Mailloux, R.J. Examination of the superoxide/hydrogen peroxide forming and quenching potential of mouse liver mitochondria. Biochim Biophys Acta Gen Subj 2017, 1861, 1960-1969. [CrossRef]
- Wong, H.S.; Benoit, B.; Brand, M.D. Mitochondrial and cytosolic sources of hydrogen peroxide in resting C2C12 myoblasts. Free Radic Biol Med 2019, 130, 140-150. [CrossRef]
- Chandel, N.S. Mitochondria. Cold Spring Harb Perspect Biol 2021, 13. [CrossRef]
- Han, D.; Canali, R.; Rettori, D.; Kaplowitz, N. Effect of glutathione depletion on sites and topology of superoxide and hydrogen peroxide production in mitochondria. Mol Pharmacol 2003, 64, 1136-1144. [CrossRef]
- Hoehne, M.N.; Jacobs, L.; Lapacz, K.J.; Calabrese, G.; Murschall, L.M.; Marker, T.; Kaul, H.; Trifunovic, A.; Morgan, B.; Fricker, M.; et al. Spatial and temporal control of mitochondrial H(2) O(2) release in intact human cells. EMBO J 2022, 41, e109169. [CrossRef]
- Goncalves, R.L.S.; Watson, M.A.; Wong, H.S.; Orr, A.L.; Brand, M.D. The use of site-specific suppressors to measure the relative contributions of different mitochondrial sites to skeletal muscle superoxide and hydrogen peroxide production. Redox Biol 2020, 28, 101341. [CrossRef]
- Bleier, L.; Wittig, I.; Heide, H.; Steger, M.; Brandt, U.; Drose, S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic Biol Med 2015, 78, 1-10. [CrossRef]
- Gao, L.; Gonzalez-Rodriguez, P.; Ortega-Saenz, P.; Lopez-Barneo, J. Redox signaling in acute oxygen sensing. Redox Biol 2017, 12, 908-915. [CrossRef]
- Dunham-Snary, K.J.; Wu, D.; Potus, F.; Sykes, E.A.; Mewburn, J.D.; Charles, R.L.; Eaton, P.; Sultanian, R.A.; Archer, S.L. Ndufs2, a Core Subunit of Mitochondrial Complex I, Is Essential for Acute Oxygen-Sensing and Hypoxic Pulmonary Vasoconstriction. Circ Res 2019, 124, 1727-1746. [CrossRef]
- Paddenberg, R.; Ishaq, B.; Goldenberg, A.; Faulhammer, P.; Rose, F.; Weissmann, N.; Braun-Dullaeus, R.C.; Kummer, W. Essential role of complex II of the respiratory chain in hypoxia-induced ROS generation in the pulmonary vasculature. Am J Physiol Lung Cell Mol Physiol 2003, 284, L710-719. [CrossRef]
- Korde, A.S.; Yadav, V.R.; Zheng, Y.M.; Wang, Y.X. Primary role of mitochondrial Rieske iron-sulfur protein in hypoxic ROS production in pulmonary artery myocytes. Free Radic Biol Med 2011, 50, 945-952. [CrossRef]
- Waypa, G.B.; Marks, J.D.; Guzy, R.D.; Mungai, P.T.; Schriewer, J.M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am J Respir Crit Care Med 2013, 187, 424-432. [CrossRef]
- Sommer, N.; Alebrahimdehkordi, N.; Pak, O.; Knoepp, F.; Strielkov, I.; Scheibe, S.; Dufour, E.; Andjelkovic, A.; Sydykov, A.; Saraji, A.; et al. Bypassing mitochondrial complex III using alternative oxidase inhibits acute pulmonary oxygen sensing. Sci Adv 2020, 6, eaba0694. [CrossRef]
- Sommer, N.; Huttemann, M.; Pak, O.; Scheibe, S.; Knoepp, F.; Sinkler, C.; Malczyk, M.; Gierhardt, M.; Esfandiary, A.; Kraut, S.; et al. Mitochondrial Complex IV Subunit 4 Isoform 2 Is Essential for Acute Pulmonary Oxygen Sensing. Circ Res 2017, 121, 424-438. [CrossRef]
- Gibbs, E.T.; Lerner, C.A.; Watson, M.A.; Wong, H.S.; Gerencser, A.A.; Brand, M.D. Site IQ in mitochondrial complex I generates S1QEL-sensitive superoxide/hydrogen peroxide in both the reverse and forward reactions. Biochem J 2023, 480, 363-384. [CrossRef]
- Bleier, L.; Drose, S. Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochim Biophys Acta 2013, 1827, 1320-1331. [CrossRef]
- Guillaud, F.; Drose, S.; Kowald, A.; Brandt, U.; Klipp, E. Superoxide production by cytochrome bc1 complex: a mathematical model. Biochim Biophys Acta 2014, 1837, 1643-1652. [CrossRef]
- Sarewicz, M.; Osyczka, A. Electronic connection between the quinone and cytochrome C redox pools and its role in regulation of mitochondrial electron transport and redox signaling. Physiol Rev 2015, 95, 219-243. [CrossRef]
- Rottenberg, H.; Covian, R.; Trumpower, B.L. Membrane potential greatly enhances superoxide generation by the cytochrome bc1 complex reconstituted into phospholipid vesicles. J Biol Chem 2009, 284, 19203-19210. [CrossRef]
- Markevich, N.I.; Hoek, J.B. Computational modeling analysis of mitochondrial superoxide production under varying substrate conditions and upon inhibition of different segments of the electron transport chain. Biochim Biophys Acta 2015, 1847, 656-679. [CrossRef]
- Quinlan, C.L.; Gerencser, A.A.; Treberg, J.R.; Brand, M.D. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J Biol Chem 2011, 286, 31361-31372. [CrossRef]
- Tsubaki, M. Fourier-transform infrared study of cyanide binding to the Fea3-CuB binuclear site of bovine heart cytochrome c oxidase: implication of the redox-linked conformational change at the binuclear site. Biochemistry 1993, 32, 164-173. [CrossRef]
- Biscoe, T.J.; Duchen, M.R. Cellular basis of transduction in carotid chemoreceptors. Am J Physiol 1990, 258, L271-278. [CrossRef]
- Wilson, D.F. Oxidative phosphorylation: regulation and role in cellular and tissue metabolism. J Physiol 2017, 595, 7023-7038. [CrossRef]
- Ward, J.P. Oxygen sensors in context. Biochim Biophys Acta 2008, 1777, 1-14. [CrossRef]
- Buckler, K.J.; Turner, P.J. Functional Properties of Mitochondria in the Type-1 Cell and Their Role in Oxygen Sensing. Adv Exp Med Biol 2015, 860, 69-80. [CrossRef]
- Wilson, D.F.; Rumsey, W.L.; Green, T.J.; Vanderkooi, J.M. The oxygen dependence of mitochondrial oxidative phosphorylation measured by a new optical method for measuring oxygen concentration. J Biol Chem 1988, 263, 2712-2718.
- Sommer, N.; Pak, O.; Schorner, S.; Derfuss, T.; Krug, A.; Gnaiger, E.; Ghofrani, H.A.; Schermuly, R.T.; Huckstorf, C.; Seeger, W.; et al. Mitochondrial cytochrome redox states and respiration in acute pulmonary oxygen sensing. Eur Respir J 2010, 36, 1056-1066. [CrossRef]
- Michelakis, E.D.; Hampl, V.; Nsair, A.; Wu, X.; Harry, G.; Haromy, A.; Gurtu, R.; Archer, S.L. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ Res 2002, 90, 1307-1315. [CrossRef]
- Roy, A.; Li, J.; Al-Mehdi, A.B.; Mokashi, A.; Lahiri, S. Effect of acute hypoxia on glomus cell Em and psi m as measured by fluorescence imaging. J Appl Physiol (1985) 2002, 93, 1987-1998. [CrossRef]
- Kurokawa, H.; Ito, H.; Inoue, M.; Tabata, K.; Sato, Y.; Yamagata, K.; Kizaka-Kondoh, S.; Kadonosono, T.; Yano, S.; Inoue, M.; et al. High resolution imaging of intracellular oxygen concentration by phosphorescence lifetime. Sci Rep 2015, 5, 10657. [CrossRef]
- Gnaiger, E.; Steinlechner-Maran, R.; Mendez, G.; Eberl, T.; Margreiter, R. Control of mitochondrial and cellular respiration by oxygen. J Bioenerg Biomembr 1995, 27, 583-596. [CrossRef]
- Gnaiger, E.; Lassnig, B.; Kuznetsov, A.; Rieger, G.; Margreiter, R. Mitochondrial oxygen affinity, respiratory flux control and excess capacity of cytochrome c oxidase. J Exp Biol 1998, 201, 1129-1139. [CrossRef]
- Brown, G.C.; Cooper, C.E. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett 1994, 356, 295-298. [CrossRef]
- Palacios-Callender, M.; Quintero, M.; Hollis, V.S.; Springett, R.J.; Moncada, S. Endogenous NO regulates superoxide production at low oxygen concentrations by modifying the redox state of cytochrome c oxidase. Proc Natl Acad Sci U S A 2004, 101, 7630-7635. [CrossRef]
- Edmunds, N.J.; Moncada, S.; Marshall, J.M. Does nitric oxide allow endothelial cells to sense hypoxia and mediate hypoxic vasodilatation? In vivo and in vitro studies. J Physiol 2003, 546, 521-527. [CrossRef]
- Quintero, M.; Colombo, S.L.; Godfrey, A.; Moncada, S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci U S A 2006, 103, 5379-5384. [CrossRef]
- Galkin, A.; Higgs, A.; Moncada, S. Nitric oxide and hypoxia. Essays Biochem 2007, 43, 29-42. [CrossRef]
- Fagan, K.A.; Tyler, R.C.; Sato, K.; Fouty, B.W.; Morris, K.G., Jr.; Huang, P.L.; McMurtry, I.F.; Rodman, D.M. Relative contributions of endothelial, inducible, and neuronal NOS to tone in the murine pulmonary circulation. Am J Physiol 1999, 277, L472-478. [CrossRef]
- Leeman, M.; de Beyl, V.Z.; Delcroix, M.; Naeije, R. Effects of endogenous nitric oxide on pulmonary vascular tone in intact dogs. Am J Physiol 1994, 266, H2343-2347. [CrossRef]
- Persson, M.G.; Gustafsson, L.E.; Wiklund, N.P.; Moncada, S.; Hedqvist, P. Endogenous nitric oxide as a probable modulator of pulmonary circulation and hypoxic pressor response in vivo. Acta Physiol Scand 1990, 140, 449-457. [CrossRef]
- Vaughan, D.J.; Brogan, T.V.; Kerr, M.E.; Deem, S.; Luchtel, D.L.; Swenson, E.R. Contributions of nitric oxide synthase isozymes to exhaled nitric oxide and hypoxic pulmonary vasoconstriction in rabbit lungs. Am J Physiol Lung Cell Mol Physiol 2003, 284, L834-843. [CrossRef]
- Blitzer, M.L.; Loh, E.; Roddy, M.A.; Stamler, J.S.; Creager, M.A. Endothelium-derived nitric oxide regulates systemic and pulmonary vascular resistance during acute hypoxia in humans. J Am Coll Cardiol 1996, 28, 591-596. [CrossRef]
- Leach, R.M.; Robertson, T.P.; Twort, C.H.; Ward, J.P. Hypoxic vasoconstriction in rat pulmonary and mesenteric arteries. Am J Physiol 1994, 266, L223-231. [CrossRef]
- Castello, P.R.; David, P.S.; McClure, T.; Crook, Z.; Poyton, R.O. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab 2006, 3, 277-287. [CrossRef]
- Palacios-Callender, M.; Hollis, V.; Mitchison, M.; Frakich, N.; Unitt, D.; Moncada, S. Cytochrome c oxidase regulates endogenous nitric oxide availability in respiring cells: a possible explanation for hypoxic vasodilation. Proc Natl Acad Sci U S A 2007, 104, 18508-18513. [CrossRef]
- Amdahl, M.B.; DeMartino, A.W.; Gladwin, M.T. Inorganic nitrite bioactivation and role in physiological signaling and therapeutics. Biol Chem 2019, 401, 201-211. [CrossRef]
- Olson, K.R.; Deleon, E.R.; Gao, Y.; Hurley, K.; Sadauskas, V.; Batz, C.; Stoy, G.F. Thiosulfate: a readily accessible source of hydrogen sulfide in oxygen sensing. Am J Physiol Regul Integr Comp Physiol 2013, 305, R592-603. [CrossRef]
- Petersen, L.C. The effect of inhibitors on the oxygen kinetics of cytochrome c oxidase. Biochim Biophys Acta 1977, 460, 299-307. [CrossRef]
- Prieto-Lloret, J.; Snetkov, V.A.; Shaifta, Y.; Docio, I.; Connolly, M.J.; MacKay, C.E.; Knock, G.A.; Ward, J.P.T.; Aaronson, P.I. Role of reactive oxygen species and sulfide-quinone oxoreductase in hydrogen sulfide-induced contraction of rat pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 2018, 314, L670-L685. [CrossRef]
- Szabo, C.; Ransy, C.; Modis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol 2014, 171, 2099-2122. [CrossRef]
- Nicholson, R.A.; Roth, S.H.; Zhang, A.; Zheng, J.; Brookes, J.; Skrajny, B.; Bennington, R. Inhibition of respiratory and bioenergetic mechanisms by hydrogen sulfide in mammalian brain. J Toxicol Environ Health A 1998, 54, 491-507. [CrossRef]
- Levitt, M.D.; Abdel-Rehim, M.S.; Furne, J. Free and acid-labile hydrogen sulfide concentrations in mouse tissues: anomalously high free hydrogen sulfide in aortic tissue. Antioxid Redox Signal 2011, 15, 373-378. [CrossRef]
- Wilson, D.F.; Harrison, D.K.; Vinogradov, A. Mitochondrial cytochrome c oxidase and control of energy metabolism: measurements in suspensions of isolated mitochondria. J Appl Physiol (1985) 2014, 117, 1424-1430. [CrossRef]
- Harrison, D.K.; Fasching, M.; Fontana-Ayoub, M.; Gnaiger, E. Cytochrome redox states and respiratory control in mouse and beef heart mitochondria at steady-state levels of hypoxia. J Appl Physiol (1985) 2015, 119, 1210-1218. [CrossRef]
- Wilson, D.F.; Erecinska, M.; Drown, C.; Silver, I.A. Effect of oxygen tension on cellular energetics. Am J Physiol 1977, 233, C135-140. [CrossRef]
- Wilson, D.F.; Erecinska, M.; Drown, C.; Silver, I.A. The oxygen dependence of cellular energy metabolism. Arch Biochem Biophys 1979, 195, 485-493. [CrossRef]
- Pajuelo Reguera, D.; Cunatova, K.; Vrbacky, M.; Pecinova, A.; Houstek, J.; Mracek, T.; Pecina, P. Cytochrome c Oxidase Subunit 4 Isoform Exchange Results in Modulation of Oxygen Affinity. Cells 2020, 9. [CrossRef]
- Huttemann, M.; Kadenbach, B.; Grossman, L.I. Mammalian subunit IV isoforms of cytochrome c oxidase. Gene 2001, 267, 111-123. [CrossRef]
- Buckler, K.J.; Turner, P.J. Oxygen sensitivity of mitochondrial function in rat arterial chemoreceptor cells. J Physiol 2013, 591, 3549-3563. [CrossRef]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front Physiol 2017, 8, 428. [CrossRef]
- Treberg, J.R.; Quinlan, C.L.; Brand, M.D. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J Biol Chem 2011, 286, 27103-27110. [CrossRef]
- Fernandez-Aguera, M.C.; Gao, L.; Gonzalez-Rodriguez, P.; Pintado, C.O.; Arias-Mayenco, I.; Garcia-Flores, P.; Garcia-Perganeda, A.; Pascual, A.; Ortega-Saenz, P.; Lopez-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab 2015, 22, 825-837. [CrossRef]
- Arias-Mayenco, I.; Gonzalez-Rodriguez, P.; Torres-Torrelo, H.; Gao, L.; Fernandez-Aguera, M.C.; Bonilla-Henao, V.; Ortega-Saenz, P.; Lopez-Barneo, J. Acute O2 Sensing: Role of Coenzyme QH2/Q Ratio and Mitochondrial ROS Compartmentalization. Cell Metab 2018, 28, 145-158 e144. [CrossRef]
- Swiderska, A.; Coney, A.M.; Alzahrani, A.A.; Aldossary, H.S.; Batis, N.; Ray, C.J.; Kumar, P.; Holmes, A.P. Mitochondrial Succinate Metabolism and Reactive Oxygen Species Are Important but Not Essential for Eliciting Carotid Body and Ventilatory Responses to Hypoxia in the Rat. Antioxidants (Basel) 2021, 10. [CrossRef]
- Torres-Lopez, M.; Spiller, P.F.; Gao, L.; Garcia-Flores, P.; Murphy, M.P.; Ortega-Saenz, P.; Lopez-Barneo, J. Acute oxygen sensing by arterial chemoreceptors with a mutant mitochondrial complex I ND6 subunit lacking reverse electron transport. FEBS Lett 2025, 599, 1122-1134. [CrossRef]
- Hernansanz-Agustin, P.; Choya-Foces, C.; Carregal-Romero, S.; Ramos, E.; Oliva, T.; Villa-Pina, T.; Moreno, L.; Izquierdo-Alvarez, A.; Cabrera-Garcia, J.D.; Cortes, A.; et al. Na(+) controls hypoxic signalling by the mitochondrial respiratory chain. Nature 2020, 586, 287-291. [CrossRef]
- Hernansanz-Agustin, P.; Ramos, E.; Navarro, E.; Parada, E.; Sanchez-Lopez, N.; Pelaez-Aguado, L.; Cabrera-Garcia, J.D.; Tello, D.; Buendia, I.; Marina, A.; et al. Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol 2017, 12, 1040-1051. [CrossRef]
- Vinogradov, A.D. Catalytic properties of the mitochondrial NADH-ubiquinone oxidoreductase (complex I) and the pseudo-reversible active/inactive enzyme transition. Biochim Biophys Acta 1998, 1364, 169-185. [CrossRef]
- Roberts, P.G.; Hirst, J. The deactive form of respiratory complex I from mammalian mitochondria is a Na+/H+ antiporter. J Biol Chem 2012, 287, 34743-34751. [CrossRef]
- Babot, M.; Birch, A.; Labarbuta, P.; Galkin, A. Characterisation of the active/de-active transition of mitochondrial complex I. Biochim Biophys Acta 2014, 1837, 1083-1092. [CrossRef]
- Babot, M.; Galkin, A. Molecular mechanism and physiological role of active-deactive transition of mitochondrial complex I. Biochem Soc Trans 2013, 41, 1325-1330. [CrossRef]
- Maklashina, E.; Kotlyar, A.B.; Karliner, J.S.; Cecchini, G. Effect of oxygen on activation state of complex I and lack of oxaloacetate inhibition of complex II in Langendorff perfused rat heart. FEBS Lett 2004, 556, 64-68. [CrossRef]
- Chalmers, S.; Nicholls, D.G. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J Biol Chem 2003, 278, 19062-19070. [CrossRef]
- Gherardi, G.; Monticelli, H.; Rizzuto, R.; Mammucari, C. The Mitochondrial Ca(2+) Uptake and the Fine-Tuning of Aerobic Metabolism. Front Physiol 2020, 11, 554904. [CrossRef]
- Wescott, A.P.; Kao, J.P.Y.; Lederer, W.J.; Boyman, L. Voltage-energized Calcium-sensitive ATP Production by Mitochondria. Nat Metab 2019, 1, 975-984. [CrossRef]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225-236. [CrossRef]
- Lee, I.; Bender, E.; Arnold, S.; Kadenbach, B. New control of mitochondrial membrane potential and ROS formation--a hypothesis. Biol Chem 2001, 382, 1629-1636. [CrossRef]
- Starkov, A.A.; Fiskum, G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem 2003, 86, 1101-1107. [CrossRef]
- Ramzan, R.; Vogt, S.; Kadenbach, B. Stress-mediated generation of deleterious ROS in healthy individuals - role of cytochrome c oxidase. J Mol Med (Berl) 2020, 98, 651-657. [CrossRef]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ Res 2018, 122, 1460-1478. [CrossRef]
- Duong, Q.V.; Hoffman, A.; Zhong, K.; Dessinger, M.J.; Zhang, Y.; Bazil, J.N. Calcium overload decreases net free radical emission in cardiac mitochondria. Mitochondrion 2020, 51, 126-139. [CrossRef]
- Grivennikova, V.G.; Kareyeva, A.V.; Vinogradov, A.D. Oxygen-dependence of mitochondrial ROS production as detected by Amplex Red assay. Redox Biol 2018, 17, 192-199. [CrossRef]
- Stepanova, A.; Konrad, C.; Manfredi, G.; Springett, R.; Ten, V.; Galkin, A. The dependence of brain mitochondria reactive oxygen species production on oxygen level is linear, except when inhibited by antimycin A. J Neurochem 2019, 148, 731-745. [CrossRef]
- Hoffman, D.L.; Salter, J.D.; Brookes, P.S. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: implications for hypoxic cell signaling. Am J Physiol Heart Circ Physiol 2007, 292, H101-108. [CrossRef]
- Archer, S.L.; Dunham-Snary, K.J.; Bentley, R.; Alizadeh, E.; Weir, E.K. Hypoxic Pulmonary Vasoconstriction: An Important Component of the Homeostatic Oxygen Sensing System. Physiol Res 2024, 73, S493-S510. [CrossRef]
- Wu, D.; Dasgupta, A.; Read, A.D.; Bentley, R.E.T.; Motamed, M.; Chen, K.H.; Al-Qazazi, R.; Mewburn, J.D.; Dunham-Snary, K.J.; Alizadeh, E.; et al. Oxygen sensing, mitochondrial biology and experimental therapeutics for pulmonary hypertension and cancer. Free Radic Biol Med 2021, 170, 150-178. [CrossRef]
- Weir, E.K.; Archer, S.L. The role of redox changes in oxygen sensing. Respir Physiol Neurobiol 2010, 174, 182-191. [CrossRef]
- Genova, M.L.; Pich, M.M.; Bernacchia, A.; Bianchi, C.; Biondi, A.; Bovina, C.; Falasca, A.I.; Formiggini, G.; Castelli, G.P.; Lenaz, G. The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann N Y Acad Sci 2004, 1011, 86-100. [CrossRef]
- Read, A.D.; Bentley, R.E.; Archer, S.L.; Dunham-Snary, K.J. Mitochondrial iron-sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox Biol 2021, 47, 102164. [CrossRef]
- Zickermann, V.; Wirth, C.; Nasiri, H.; Siegmund, K.; Schwalbe, H.; Hunte, C.; Brandt, U. Structural biology. Mechanistic insight from the crystal structure of mitochondrial complex I. Science 2015, 347, 44-49. [CrossRef]
- Treberg, J.R.; Braun, K.; Selseleh, P. Mitochondria can act as energy-sensing regulators of hydrogen peroxide availability. Redox Biol 2019, 20, 483-488. [CrossRef]
- Cardoso, A.R.; Chausse, B.; da Cunha, F.M.; Luevano-Martinez, L.A.; Marazzi, T.B.; Pessoa, P.S.; Queliconi, B.B.; Kowaltowski, A.J. Mitochondrial compartmentalization of redox processes. Free Radic Biol Med 2012, 52, 2201-2208. [CrossRef]
- Secilmis, M.; Altun, H.Y.; Pilic, J.; Erdogan, Y.C.; Cokluk, Z.; Ata, B.N.; Sevimli, G.; Zaki, A.G.; Yigit, E.N.; Ozturk, G.; et al. A Co-Culture-Based Multiparametric Imaging Technique to Dissect Local H(2)O(2) Signals with Targeted HyPer7. Biosensors (Basel) 2021, 11. [CrossRef]
- Lim, J.B.; Huang, B.K.; Deen, W.M.; Sikes, H.D. Analysis of the lifetime and spatial localization of hydrogen peroxide generated in the cytosol using a reduced kinetic model. Free Radic Biol Med 2015, 89, 47-53. [CrossRef]
- Archer, S.L.; Reeve, H.L.; Michelakis, E.; Puttagunta, L.; Waite, R.; Nelson, D.P.; Dinauer, M.C.; Weir, E.K. O2 sensing is preserved in mice lacking the gp91 phox subunit of NADPH oxidase. Proc Natl Acad Sci U S A 1999, 96, 7944-7949. [CrossRef]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2004, 24, 677-683. [CrossRef]
- Miller, E.W.; Dickinson, B.C.; Chang, C.J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc Natl Acad Sci U S A 2010, 107, 15681-15686. [CrossRef]
- Oakley, F.D.; Abbott, D.; Li, Q.; Engelhardt, J.F. Signaling components of redox active endosomes: the redoxosomes. Antioxid Redox Signal 2009, 11, 1313-1333. [CrossRef]
- Shaifta, Y.; Snetkov, V.A.; Prieto-Lloret, J.; Knock, G.A.; Smirnov, S.V.; Aaronson, P.I.; Ward, J.P. Sphingosylphosphorylcholine potentiates vasoreactivity and voltage-gated Ca2+ entry via NOX1 and reactive oxygen species. Cardiovasc Res 2015, 106, 121-130. [CrossRef]
- Miyano, K.; Ueno, N.; Takeya, R.; Sumimoto, H. Direct involvement of the small GTPase Rac in activation of the superoxide-producing NADPH oxidase Nox1. J Biol Chem 2006, 281, 21857-21868. [CrossRef]
- Schroder, K.; Weissmann, N.; Brandes, R.P. Organizers and activators: Cytosolic Nox proteins impacting on vascular function. Free Radic Biol Med 2017, 109, 22-32. [CrossRef]
- Streeter, J.; Schickling, B.M.; Jiang, S.; Stanic, B.; Thiel, W.H.; Gakhar, L.; Houtman, J.C.; Miller, F.J., Jr. Phosphorylation of Nox1 regulates association with NoxA1 activation domain. Circ Res 2014, 115, 911-918. [CrossRef]
- Niu, X.L.; Madamanchi, N.R.; Vendrov, A.E.; Tchivilev, I.; Rojas, M.; Madamanchi, C.; Brandes, R.P.; Krause, K.H.; Humphries, J.; Smith, A.; et al. Nox activator 1: a potential target for modulation of vascular reactive oxygen species in atherosclerotic arteries. Circulation 2010, 121, 549-559. [CrossRef]
- Zhang, F.; Jin, S.; Yi, F.; Xia, M.; Dewey, W.L.; Li, P.L. Local production of O2- by NAD(P)H oxidase in the sarcoplasmic reticulum of coronary arterial myocytes: cADPR-mediated Ca2+ regulation. Cell Signal 2008, 20, 637-644. [CrossRef]
- Vendrov, A.E.; Vendrov, K.C.; Smith, A.; Yuan, J.; Sumida, A.; Robidoux, J.; Runge, M.S.; Madamanchi, N.R. NOX4 NADPH Oxidase-Dependent Mitochondrial Oxidative Stress in Aging-Associated Cardiovascular Disease. Antioxid Redox Signal 2015, 23, 1389-1409. [CrossRef]
- Anilkumar, N.; San Jose, G.; Sawyer, I.; Santos, C.X.; Sand, C.; Brewer, A.C.; Warren, D.; Shah, A.M. A 28-kDa splice variant of NADPH oxidase-4 is nuclear-localized and involved in redox signaling in vascular cells. Arterioscler Thromb Vasc Biol 2013, 33, e104-112. [CrossRef]
- Kracun, D.; Lopes, L.R.; Cifuentes-Pagano, E.; Pagano, P.J. NADPH oxidases: redox regulation of cell homeostasis and disease. Physiol Rev 2025, 105, 1291-1428. [CrossRef]
- Sutliff, R.L.; Hilenski, L.L.; Amanso, A.M.; Parastatidis, I.; Dikalova, A.E.; Hansen, L.; Datla, S.R.; Long, J.S.; El-Ali, A.M.; Joseph, G.; et al. Polymerase delta interacting protein 2 sustains vascular structure and function. Arterioscler Thromb Vasc Biol 2013, 33, 2154-2161. [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol 2020, 21, 363-383. [CrossRef]
- Stone, J.R.; Yang, S. Hydrogen peroxide: a signaling messenger. Antioxid Redox Signal 2006, 8, 243-270. [CrossRef]
- Oshino, N.; Chance, B.; Sies, H.; Bucher, T. The role of H 2 O 2 generation in perfused rat liver and the reaction of catalase compound I and hydrogen donors. Arch Biochem Biophys 1973, 154, 117-131. [CrossRef]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol Rev 1979, 59, 527-605. [CrossRef]
- Jones, D.P. Intracellular catalase function: analysis of the catalatic activity by product formation in isolated liver cells. Arch Biochem Biophys 1982, 214, 806-814. [CrossRef]
- Lyublinskaya, O.; Antunes, F. Measuring intracellular concentration of hydrogen peroxide with the use of genetically encoded H(2)O(2) biosensor HyPer. Redox Biol 2019, 24, 101200. [CrossRef]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim Biophys Acta 2014, 1840, 1596-1604. [CrossRef]
- Antunes, F.; Cadenas, E. Estimation of H2O2 gradients across biomembranes. FEBS Lett 2000, 475, 121-126. [CrossRef]
- Makino, N.; Mochizuki, Y.; Bannai, S.; Sugita, Y. Kinetic studies on the removal of extracellular hydrogen peroxide by cultured fibroblasts. J Biol Chem 1994, 269, 1020-1025.
- Wagner, B.A.; Witmer, J.R.; van 't Erve, T.J.; Buettner, G.R. An Assay for the Rate of Removal of Extracellular Hydrogen Peroxide by Cells. Redox Biol 2013, 1, 210-217. [CrossRef]
- Huang, B.K.; Sikes, H.D. Quantifying intracellular hydrogen peroxide perturbations in terms of concentration. Redox Biol 2014, 2, 955-962. [CrossRef]
- Adimora, N.J.; Jones, D.P.; Kemp, M.L. A model of redox kinetics implicates the thiol proteome in cellular hydrogen peroxide responses. Antioxid Redox Signal 2010, 13, 731-743. [CrossRef]
- Sobotta, M.C.; Barata, A.G.; Schmidt, U.; Mueller, S.; Millonig, G.; Dick, T.P. Exposing cells to H2O2: a quantitative comparison between continuous low-dose and one-time high-dose treatments. Free Radic Biol Med 2013, 60, 325-335. [CrossRef]
- Marinho, H.S.; Cyrne, L.; Cadenas, E.; Antunes, F. H2O2 delivery to cells: steady-state versus bolus addition. Methods Enzymol 2013, 526, 159-173. [CrossRef]
- Lim, J.B.; Langford, T.F.; Huang, B.K.; Deen, W.M.; Sikes, H.D. A reaction-diffusion model of cytosolic hydrogen peroxide. Free Radic Biol Med 2016, 90, 85-90. [CrossRef]
- Jones, D.P. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol 2008, 295, C849-868. [CrossRef]
- Pardo-Pena, K.; Yanez-Hernandez, A.; Medina-Ceja, L.; Morales-Villagran, A. Ellagic acid and allopurinol decrease H(2)O(2) concentrations, epileptiform activity and astrogliosis after status epilepticus in the hippocampus of adult rats. Exp Brain Res 2022, 240, 1191-1203. [CrossRef]
- Pardo-Pena, K.; Sanchez-Lira, A.; Salazar-Sanchez, J.C.; Morales-Villagran, A. A novel online fluorescence method for in-vivo measurement of hydrogen peroxide during oxidative stress produced in a temporal lobe epilepsy model. Neuroreport 2018, 29, 621-630. [CrossRef]
- Pardo-Pena, K.; Lorea-Hernandez, J.J.; Camacho-Hernandez, N.P.; Ordaz, B.; Villasana-Salazar, B.; Morales-Villagran, A.; Pena-Ortega, F. Hydrogen peroxide extracellular concentration in the ventrolateral medulla and its increase in response to hypoxia in vitro: Possible role of microglia. Brain Res 2018, 1692, 87-99. [CrossRef]
- Puppulin, L.; Hosogi, S.; Sun, H.; Matsuo, K.; Inui, T.; Kumamoto, Y.; Suzaki, T.; Tanaka, H.; Marunaka, Y. Bioconjugation strategy for cell surface labelling with gold nanostructures designed for highly localized pH measurement. Nat Commun 2018, 9, 5278. [CrossRef]
- Hosogi, S.; Marunaka, Y.; Ashihara, E.; Yamada, T.; Sumino, A.; Tanaka, H.; Puppulin, L. Plasma membrane anchored nanosensor for quantifying endogenous production of H(2)O(2) in living cells. Biosens Bioelectron 2021, 179, 113077. [CrossRef]
- Forman, H.J.; Bernardo, A.; Davies, K.J. What is the concentration of hydrogen peroxide in blood and plasma? Arch Biochem Biophys 2016, 603, 48-53. [CrossRef]
- Sousa, T.; Gouveia, M.; Travasso, R.D.M.; Salvador, A. How abundant are superoxide and hydrogen peroxide in the vasculature lumen, how far can they reach? Redox Biol 2022, 58, 102527. [CrossRef]
- Bayer, S.B.; Maghzal, G.; Stocker, R.; Hampton, M.B.; Winterbourn, C.C. Neutrophil-mediated oxidation of erythrocyte peroxiredoxin 2 as a potential marker of oxidative stress in inflammation. FASEB J 2013, 27, 3315-3322. [CrossRef]
- Ezerina, D.; Morgan, B.; Dick, T.P. Imaging dynamic redox processes with genetically encoded probes. J Mol Cell Cardiol 2014, 73, 43-49. [CrossRef]
- Mishra, P.K.; Park, I.; Sharma, N.; Yoo, C.M.; Lee, H.Y.; Rhee, H.W. Enzymatic Recording of Local Hydrogen Peroxide Generation Using Genetically Encodable Enzyme. Anal Chem 2022, 94, 14869-14877. [CrossRef]
- Eid, M.; Barayeu, U.; Sulkova, K.; Aranda-Vallejo, C.; Dick, T.P. Using the heme peroxidase APEX2 to probe intracellular H(2)O(2) flux and diffusion. Nat Commun 2024, 15, 1239. [CrossRef]
- Yamdjeu, O.T.; Begerow, A.; Sommer, N.; Diener, M.; Weissmann, N.; Knoepp, F. H(2)O(2) Sensitivity of K(v) Channels in Hypoxic Pulmonary Vasoconstriction: Experimental Conditions Matter. Int J Mol Sci 2025, 26. [CrossRef]
- Mishina, N.M.; Tyurin-Kuzmin, P.A.; Markvicheva, K.N.; Vorotnikov, A.V.; Tkachuk, V.A.; Laketa, V.; Schultz, C.; Lukyanov, S.; Belousov, V.V. Does cellular hydrogen peroxide diffuse or act locally? Antioxid Redox Signal 2011, 14, 1-7. [CrossRef]
- Mishina, N.M.; Bogdanova, Y.A.; Ermakova, Y.G.; Panova, A.S.; Kotova, D.A.; Bilan, D.S.; Steinhorn, B.; Arner, E.S.J.; Michel, T.; Belousov, V.V. Which Antioxidant System Shapes Intracellular H(2)O(2) Gradients? Antioxid Redox Signal 2019, 31, 664-670. [CrossRef]
- Pak, V.V.; Ezerina, D.; Lyublinskaya, O.G.; Pedre, B.; Tyurin-Kuzmin, P.A.; Mishina, N.M.; Thauvin, M.; Young, D.; Wahni, K.; Martinez Gache, S.A.; et al. Ultrasensitive Genetically Encoded Indicator for Hydrogen Peroxide Identifies Roles for the Oxidant in Cell Migration and Mitochondrial Function. Cell Metab 2020, 31, 642-653 e646. [CrossRef]
- Kritsiligkou, P.; Bosch, K.; Shen, T.K.; Meurer, M.; Knop, M.; Dick, T.P. Proteome-wide tagging with an H(2)O(2) biosensor reveals highly localized and dynamic redox microenvironments. Proc Natl Acad Sci U S A 2023, 120, e2314043120. [CrossRef]
- Potekhina, E.S.; Bass, D.I.; Ezerina, D.; Fleckenstein, D.D.; Chebotarev, A.S.; Sysoeva, V.A.; Maltsev, D.I.; Pak, V.V.; Moshchenko, A.A.; Sokolov, A.I.; et al. A color-tailored fluorogenic sensor for hydrogen peroxide. Nat Chem Biol 2025. [CrossRef]
- Booth, D.M.; Varnai, P.; Joseph, S.K.; Hajnoczky, G. Oxidative bursts of single mitochondria mediate retrograde signaling toward the ER. Mol Cell 2021, 81, 3866-3876 e3862. [CrossRef]
- Joseph, S.K.; Young, M.P.; Alzayady, K.; Yule, D.I.; Ali, M.; Booth, D.M.; Hajnoczky, G. Redox regulation of type-I inositol trisphosphate receptors in intact mammalian cells. J Biol Chem 2018, 293, 17464-17476. [CrossRef]
- King, M.P.; Attardi, G. Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol 1996, 264, 304-313. [CrossRef]
- Firth, A.L.; Gordienko, D.V.; Yuill, K.H.; Smirnov, S.V. Cellular localization of mitochondria contributes to Kv channel-mediated regulation of cellular excitability in pulmonary but not mesenteric circulation. Am J Physiol Lung Cell Mol Physiol 2009, 296, L347-360. [CrossRef]
- Archer, S.L.; Huang, J.; Henry, T.; Peterson, D.; Weir, E.K. A redox-based O2 sensor in rat pulmonary vasculature. Circ Res 1993, 73, 1100-1112. [CrossRef]
- Weir, E.K. Does normoxic pulmonary vasodilatation rather than hypoxic vasoconstriction account for the pulmonary pressor response to hypoxia? Lancet 1978, 1, 476-477. [CrossRef]
- Kilfoil, P.J.; Tipparaju, S.M.; Barski, O.A.; Bhatnagar, A. Regulation of ion channels by pyridine nucleotides. Circ Res 2013, 112, 721-741. [CrossRef]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid Redox Signal 2015, 23, 734-746. [CrossRef]
- Dwenger, M.M.; Raph, S.M.; Reyzer, M.L.; Lisa Manier, M.; Riggs, D.W.; Wohl, Z.B.; Ohanyan, V.; Mack, G.; Pucci, T.; Moore, J.B.t.; et al. Pyridine nucleotide redox potential in coronary smooth muscle couples myocardial blood flow to cardiac metabolism. Nat Commun 2022, 13, 2051. [CrossRef]
- Liu, S.Q.; Jin, H.; Zacarias, A.; Srivastava, S.; Bhatnagar, A. Binding of pyridine coenzymes to the beta-subunit of the voltage sensitive potassium channels. Chem Biol Interact 2001, 130-132, 955-962. [CrossRef]
- Gutscher, M.; Pauleau, A.L.; Marty, L.; Brach, T.; Wabnitz, G.H.; Samstag, Y.; Meyer, A.J.; Dick, T.P. Real-time imaging of the intracellular glutathione redox potential. Nat Methods 2008, 5, 553-559. [CrossRef]
- Swain, L.; Kesemeyer, A.; Meyer-Roxlau, S.; Vettel, C.; Zieseniss, A.; Guntsch, A.; Jatho, A.; Becker, A.; Nanadikar, M.S.; Morgan, B.; et al. Redox Imaging Using Cardiac Myocyte-Specific Transgenic Biosensor Mice. Circ Res 2016, 119, 1004-1016. [CrossRef]
- Weir, E.K.; Lopez-Barneo, J.; Buckler, K.J.; Archer, S.L. Acute oxygen-sensing mechanisms. N Engl J Med 2005, 353, 2042-2055. [CrossRef]
- Weir, E.K.; Will, J.A.; Lundquist, L.J.; Eaton, J.W.; Chesler, E. Diamide inhibits pulmonary vasoconstriction induced by hypoxia or prostaglandin F2 alpha. Proc Soc Exp Biol Med 1983, 173, 96-103. [CrossRef]
- Burghuber, O.; Mathias, M.M.; McMurtry, I.F.; Reeves, J.T.; Voelkel, N.F. Lung edema due to hydrogen peroxide is independent of cyclooxygenase products. J Appl Physiol Respir Environ Exerc Physiol 1984, 56, 900-905. [CrossRef]
- Weir, E.K.; Eaton, J.W.; Chesler, E. Redox status and pulmonary vascular reactivity. Chest 1985, 88, 249S-252S. [CrossRef]
- Archer, S.L.; Will, J.A.; Weir, E.K. Redox status in the control of pulmonary vascular tone. Herz 1986, 11, 127-141.
- Archer, S.L.; Peterson, D.; Nelson, D.P.; DeMaster, E.G.; Kelly, B.; Eaton, J.W.; Weir, E.K. Oxygen radicals and antioxidant enzymes alter pulmonary vascular reactivity in the rat lung. J Appl Physiol (1985) 1989, 66, 102-111. [CrossRef]
- McMurtry, I.F. Angiotensin is not required for hypoxic constriction in salt solution-perfused rat lungs. J Appl Physiol Respir Environ Exerc Physiol 1984, 56, 375-380. [CrossRef]
- Archer, S.L.; Nelson, D.P.; Weir, E.K. Simultaneous measurement of O2 radicals and pulmonary vascular reactivity in rat lung. J Appl Physiol (1985) 1989, 67, 1903-1911. [CrossRef]
- Post, J.M.; Hume, J.R.; Archer, S.L.; Weir, E.K. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am J Physiol 1992, 262, C882-890. [CrossRef]
- Rounds, S.; McMurtry, I.F. Inhibitors of oxidative ATP production cause transient vasoconstriction and block subsequent pressor responses in rat lungs. Circ Res 1981, 48, 393-400. [CrossRef]
- Leach, R.M.; Sheehan, D.W.; Chacko, V.P.; Sylvester, J.T. Effects of hypoxia on energy state and pH in resting pulmonary and femoral arterial smooth muscles. Am J Physiol 1998, 275, L1051-1060. [CrossRef]
- Leach, R.M.; Sheehan, D.W.; Chacko, V.P.; Sylvester, J.T. Energy state, pH, and vasomotor tone during hypoxia in precontracted pulmonary and femoral arteries. Am J Physiol Lung Cell Mol Physiol 2000, 278, L294-304. [CrossRef]
- Buescher, P.C.; Pearse, D.B.; Pillai, R.P.; Litt, M.C.; Mitchell, M.C.; Sylvester, J.T. Energy state and vasomotor tone in hypoxic pig lungs. J Appl Physiol (1985) 1991, 70, 1874-1881. [CrossRef]
- Wu, W.; Platoshyn, O.; Firth, A.L.; Yuan, J.X. Hypoxia divergently regulates production of reactive oxygen species in human pulmonary and coronary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2007, 293, L952-959. [CrossRef]
- Mehta, J.P.; Campian, J.L.; Guardiola, J.; Cabrera, J.A.; Weir, E.K.; Eaton, J.W. Generation of oxidants by hypoxic human pulmonary and coronary smooth-muscle cells. Chest 2008, 133, 1410-1414. [CrossRef]
- Divakaruni, A.S.; Jastroch, M. A practical guide for the analysis, standardization and interpretation of oxygen consumption measurements. Nat Metab 2022, 4, 978-994. [CrossRef]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat Methods 2006, 3, 281-286. [CrossRef]
- Brandt, U. Energy converting NADH:quinone oxidoreductase (complex I). Annu Rev Biochem 2006, 75, 69-92. [CrossRef]
- Hameedi, M.A.; Grba, D.N.; Richardson, K.H.; Jones, A.J.Y.; Song, W.; Roessler, M.M.; Wright, J.J.; Hirst, J. A conserved arginine residue is critical for stabilizing the N2 FeS cluster in mitochondrial complex I. J Biol Chem 2021, 296, 100474. [CrossRef]
- Kashani-Poor, N.; Zwicker, K.; Kerscher, S.; Brandt, U. A central functional role for the 49-kDa subunit within the catalytic core of mitochondrial complex I. J Biol Chem 2001, 276, 24082-24087. [CrossRef]
- Prieur, I.; Lunardi, J.; Dupuis, A. Evidence for a quinone binding site close to the interface between NUOD and NUOB subunits of Complex I. Biochim Biophys Acta 2001, 1504, 173-178. [CrossRef]
- Read, A.D.; Bentley, R.E.T.; Martin, A.Y.; Mewburn, J.D.; Alizadeh, E.; Wu, D.; Lima, P.D.A.; Dunham-Snary, K.J.; Thebaud, B.; Sharp, W.; et al. Electron Leak From the Mitochondrial Electron Transport Chain Complex I at Site I(Q) Is Crucial for Oxygen Sensing in Rabbit and Human Ductus Arteriosus. J Am Heart Assoc 2023, 12, e029131. [CrossRef]
- Huttemann, M.; Sommer, N.; Weissmann, N.; Grossman, L.I. Letter by Huttemann et al Regarding Article, "Ndufs2, a Core Subunit of Mitochondrial Complex I, Is Essential for Acute Oxygen-Sensing and Hypoxic Pulmonary Vasoconstriction". Circ Res 2019, 125, e33-e34. [CrossRef]
- Dunham-Snary, K.J.; Archer, S.L. Response by Dunham-Snary and Archer to Letter Regarding Article, "Ndufs2, a Core Subunit of Mitochondrial Complex I, Is Essential for Acute Oxygen-Sensing and Hypoxic Pulmonary Vasoconstriction". Circ Res 2019, 125, e35-e36. [CrossRef]
- Onukwufor, J.O.; Farooqi, M.A.; Vodickova, A.; Koren, S.A.; Baldzizhar, A.; Berry, B.J.; Beutner, G.; Porter, G.A., Jr.; Belousov, V.; Grossfield, A.; et al. A reversible mitochondrial complex I thiol switch mediates hypoxic avoidance behavior in C. elegans. Nat Commun 2022, 13, 2403. [CrossRef]
- Thomas, G.; Ramwell, P. Induction of vascular relaxation by hydroperoxides. Biochem Biophys Res Commun 1986, 139, 102-108. [CrossRef]
- Morgan, B.; Van Laer, K.; Owusu, T.N.; Ezerina, D.; Pastor-Flores, D.; Amponsah, P.S.; Tursch, A.; Dick, T.P. Real-time monitoring of basal H2O2 levels with peroxiredoxin-based probes. Nat Chem Biol 2016, 12, 437-443. [CrossRef]
- Bandara, A.B.; Drake, J.C.; James, C.C.; Smyth, J.W.; Brown, D.A. Complex I protein NDUFS2 is vital for growth, ROS generation, membrane integrity, apoptosis, and mitochondrial energetics. Mitochondrion 2021, 58, 160-168. [CrossRef]
- Sazanov, L.A. From the 'black box' to 'domino effect' mechanism: what have we learned from the structures of respiratory complex I. Biochem J 2023, 480, 319-333. [CrossRef]
- Pagniez-Mammeri, H.; Loublier, S.; Legrand, A.; Benit, P.; Rustin, P.; Slama, A. Mitochondrial complex I deficiency of nuclear origin I. Structural genes. Mol Genet Metab 2012, 105, 163-172. [CrossRef]
- Burska, D.; Stiburek, L.; Krizova, J.; Vanisova, M.; Martinek, V.; Sladkova, J.; Zamecnik, J.; Honzik, T.; Zeman, J.; Hansikova, H.; et al. Homozygous missense mutation in UQCRC2 associated with severe encephalomyopathy, mitochondrial complex III assembly defect and activation of mitochondrial protein quality control. Biochim Biophys Acta Mol Basis Dis 2021, 1867, 166147. [CrossRef]
- Iuso, A.; Scacco, S.; Piccoli, C.; Bellomo, F.; Petruzzella, V.; Trentadue, R.; Minuto, M.; Ripoli, M.; Capitanio, N.; Zeviani, M.; et al. Dysfunctions of cellular oxidative metabolism in patients with mutations in the NDUFS1 and NDUFS4 genes of complex I. J Biol Chem 2006, 281, 10374-10380. [CrossRef]
- McMurtry, I.F.; Davidson, A.B.; Reeves, J.T.; Grover, R.F. Inhibition of hypoxic pulmonary vasoconstriction by calcium antagonists in isolated rat lungs. Circ Res 1976, 38, 99-104. [CrossRef]
- Harder, D.R.; Madden, J.A.; Dawson, C. A membrane electrical mechanism for hypoxic vasoconstriction of small pulmonary arteries from cat. Chest 1985, 88, 233S-235S. [CrossRef]
- Hasunuma, K.; Rodman, D.M.; McMurtry, I.F. Effects of K+ channel blockers on vascular tone in the perfused rat lung. Am Rev Respir Dis 1991, 144, 884-887. [CrossRef]
- Yuan, X.J. Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ Res 1995, 77, 370-378. [CrossRef]
- Yuan, X.J.; Goldman, W.F.; Tod, M.L.; Rubin, L.J.; Blaustein, M.P. Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am J Physiol 1993, 264, L116-123. [CrossRef]
- Archer, S.L.; Huang, J.M.; Reeve, H.L.; Hampl, V.; Tolarova, S.; Michelakis, E.; Weir, E.K. Differential distribution of electrophysiologically distinct myocytes in conduit and resistance arteries determines their response to nitric oxide and hypoxia. Circ Res 1996, 78, 431-442. [CrossRef]
- Olschewski, A.; Hong, Z.; Nelson, D.P.; Weir, E.K. Graded response of K+ current, membrane potential, and [Ca2+]i to hypoxia in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 2002, 283, L1143-1150. [CrossRef]
- Archer, S.L.; Souil, E.; Dinh-Xuan, A.T.; Schremmer, B.; Mercier, J.C.; El Yaagoubi, A.; Nguyen-Huu, L.; Reeve, H.L.; Hampl, V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 1998, 101, 2319-2330. [CrossRef]
- Archer, S.L.; London, B.; Hampl, V.; Wu, X.; Nsair, A.; Puttagunta, L.; Hashimoto, K.; Waite, R.E.; Michelakis, E.D. Impairment of hypoxic pulmonary vasoconstriction in mice lacking the voltage-gated potassium channel Kv1.5. FASEB J 2001, 15, 1801-1803. [CrossRef]
- Platoshyn, O.; Yu, Y.; Ko, E.A.; Remillard, C.V.; Yuan, J.X. Heterogeneity of hypoxia-mediated decrease in I(K(V)) and increase in [Ca2+](cyt) in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2007, 293, L402-416. [CrossRef]
- Firth, A.L.; Platoshyn, O.; Brevnova, E.E.; Burg, E.D.; Powell, F.; Haddad, G.H.; Yuan, J.X. Hypoxia selectively inhibits KCNA5 channels in pulmonary artery smooth muscle cells. Ann N Y Acad Sci 2009, 1177, 101-111. [CrossRef]
- Olschewski, A.; Li, Y.; Tang, B.; Hanze, J.; Eul, B.; Bohle, R.M.; Wilhelm, J.; Morty, R.E.; Brau, M.E.; Weir, E.K.; et al. Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ Res 2006, 98, 1072-1080. [CrossRef]
- Nagaraj, C.; Tang, B.; Balint, Z.; Wygrecka, M.; Hrzenjak, A.; Kwapiszewska, G.; Stacher, E.; Lindenmann, J.; Weir, E.K.; Olschewski, H.; et al. Src tyrosine kinase is crucial for potassium channel function in human pulmonary arteries. Eur Respir J 2013, 41, 85-95. [CrossRef]
- Fuchs, B.; Dietrich, A.; Gudermann, T.; Kalwa, H.; Grimminger, F.; Weissmann, N. The role of classical transient receptor potential channels in the regulation of hypoxic pulmonary vasoconstriction. Adv Exp Med Biol 2010, 661, 187-200. [CrossRef]
- Archer, S.L.; Michelakis, E.D.; Thebaud, B.; Bonnet, S.; Moudgil, R.; Wu, X.C.; Weir, E.K. A central role for oxygen-sensitive K+ channels and mitochondria in the specialized oxygen-sensing system. Novartis Found Symp 2006, 272, 157-171; discussion 171-155, 214-157.
- Olschewski, A.; Weir, E.K. Redox regulation of ion channels in the pulmonary circulation. Antioxid Redox Signal 2015, 22, 465-485. [CrossRef]
- Rocher, A.; Aaronson, P.I. The Thirty-Fifth Anniversary of K+ Channels in O2 sensing: What We Know and What We Don't Know. Oxygen 2024, 4, 53-89. [CrossRef]
- Firth, A.L.; Yuill, K.H.; Smirnov, S.V. Mitochondria-dependent regulation of Kv currents in rat pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2008, 295, L61-70. [CrossRef]
- Searle, G.J.; Hartness, M.E.; Hoareau, R.; Peers, C.; Kemp, P.J. Lack of contribution of mitochondrial electron transport to acute O(2) sensing in model airway chemoreceptors. Biochem Biophys Res Commun 2002, 291, 332-337. [CrossRef]
- Cogolludo, A.; Frazziano, G.; Cobeno, L.; Moreno, L.; Lodi, F.; Villamor, E.; Tamargo, J.; Perez-Vizcaino, F. Role of reactive oxygen species in Kv channel inhibition and vasoconstriction induced by TP receptor activation in rat pulmonary arteries. Ann N Y Acad Sci 2006, 1091, 41-51. [CrossRef]
- Frazziano, G.; Moreno, L.; Moral-Sanz, J.; Menendez, C.; Escolano, L.; Gonzalez, C.; Villamor, E.; Alvarez-Sala, J.L.; Cogolludo, A.L.; Perez-Vizcaino, F. Neutral sphingomyelinase, NADPH oxidase and reactive oxygen species. Role in acute hypoxic pulmonary vasoconstriction. J Cell Physiol 2011, 226, 2633-2640. [CrossRef]
- Post, J.M.; Gelband, C.H.; Hume, J.R. [Ca2+]i inhibition of K+ channels in canine pulmonary artery. Novel mechanism for hypoxia-induced membrane depolarization. Circ Res 1995, 77, 131-139. [CrossRef]
- Vandier, C.; Delpech, M.; Bonnet, P. Spontaneous transient outward currents and delayed rectifier K+ current: effects of hypoxia. Am J Physiol 1998, 275, L145-154. [CrossRef]
- Snetkov, V.A.; Smirnov, S.V.; Kua, J.; Aaronson, P.I.; Ward, J.P.; Knock, G.A. Superoxide differentially controls pulmonary and systemic vascular tone through multiple signalling pathways. Cardiovasc Res 2011, 89, 214-224. [CrossRef]
- Svoboda, L.K.; Reddie, K.G.; Zhang, L.; Vesely, E.D.; Williams, E.S.; Schumacher, S.M.; O'Connell, R.P.; Shaw, R.; Day, S.M.; Anumonwo, J.M.; et al. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5. Circ Res 2012, 111, 842-853. [CrossRef]
- Raph, S.M.; Dwenger, M.M.; Hu, X.; Nystoriak, M.A. Basal NAD(H) redox state permits hydrogen peroxide-induced mesenteric artery dilatation. J Physiol 2023, 601, 2621-2634. [CrossRef]
- Moreno, L.; Frazziano, G.; Cogolludo, A.; Cobeno, L.; Tamargo, J.; Perez-Vizcaino, F. Role of protein kinase Czeta and its adaptor protein p62 in voltage-gated potassium channel modulation in pulmonary arteries. Mol Pharmacol 2007, 72, 1301-1309. [CrossRef]
- Schumacher, S.M.; McEwen, D.P.; Zhang, L.; Arendt, K.L.; Van Genderen, K.M.; Martens, J.R. Antiarrhythmic drug-induced internalization of the atrial-specific k+ channel kv1.5. Circ Res 2009, 104, 1390-1398. [CrossRef]
- Lee, S.; Park, M.; So, I.; Earm, Y.E. NADH and NAD modulates Ca(2+)-activated K+ channels in small pulmonary arterial smooth muscle cells of the rabbit. Pflugers Arch 1994, 427, 378-380. [CrossRef]
- Reeve, H.L.; Weir, E.K.; Nelson, D.P.; Peterson, D.A.; Archer, S.L. Opposing effects of oxidants and antioxidants on K+ channel activity and tone in rat vascular tissue. Exp Physiol 1995, 80, 825-834. [CrossRef]
- Park, M.K.; Lee, S.H.; Ho, W.K.; Earm, Y.E. Redox agents as a link between hypoxia and the responses of ionic channels in rabbit pulmonary vascular smooth muscle. Exp Physiol 1995, 80, 835-842. [CrossRef]
- Park, M.K.; Bae, Y.M.; Lee, S.H.; Ho, W.K.; Earm, Y.E. Modulation of voltage-dependent K+ channel by redox potential in pulmonary and ear arterial smooth muscle cells of the rabbit. Pflugers Arch 1997, 434, 764-771. [CrossRef]
- Olschewski, A.; Hong, Z.; Peterson, D.A.; Nelson, D.P.; Porter, V.A.; Weir, E.K. Opposite effects of redox status on membrane potential, cytosolic calcium, and tone in pulmonary arteries and ductus arteriosus. Am J Physiol Lung Cell Mol Physiol 2004, 286, L15-22. [CrossRef]
- Schach, C.; Xu, M.; Platoshyn, O.; Keller, S.H.; Yuan, J.X. Thiol oxidation causes pulmonary vasodilation by activating K+ channels and inhibiting store-operated Ca2+ channels. Am J Physiol Lung Cell Mol Physiol 2007, 292, L685-698. [CrossRef]
- Yuan, X.J.; Tod, M.L.; Rubin, L.J.; Blaustein, M.P. Deoxyglucose and reduced glutathione mimic effects of hypoxia on K+ and Ca2+ conductances in pulmonary artery cells. Am J Physiol 1994, 267, L52-63. [CrossRef]
- Weir, E.K.; Hong, Z.; Porter, V.A.; Reeve, H.L. Redox signaling in oxygen sensing by vessels. Respir Physiol Neurobiol 2002, 132, 121-130. [CrossRef]
- Marshall, C.; Mamary, A.J.; Verhoeven, A.J.; Marshall, B.E. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol 1996, 15, 633-644. [CrossRef]
- Killilea, D.W.; Hester, R.; Balczon, R.; Babal, P.; Gillespie, M.N. Free radical production in hypoxic pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2000, 279, L408-412. [CrossRef]
- Abdalla, S.; Will, J.A. Potentiation of the hypoxic contraction of guinea-pig isolated pulmonary arteries by two inhibitors of superoxide dismutase. Gen Pharmacol 1995, 26, 785-792. [CrossRef]
- Duranteau, J.; Chandel, N.S.; Kulisz, A.; Shao, Z.; Schumacker, P.T. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J Biol Chem 1998, 273, 11619-11624. [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A 1998, 95, 11715-11720. [CrossRef]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 1985, 237, 408-414. [CrossRef]
- Heumuller, S.; Wind, S.; Barbosa-Sicard, E.; Schmidt, H.H.; Busse, R.; Schroder, K.; Brandes, R.P. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 2008, 51, 211-217. [CrossRef]
- Leach, R.M.; Hill, H.M.; Snetkov, V.A.; Robertson, T.P.; Ward, J.P. Divergent roles of glycolysis and the mitochondrial electron transport chain in hypoxic pulmonary vasoconstriction of the rat: identity of the hypoxic sensor. J Physiol 2001, 536, 211-224. [CrossRef]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J Biol Chem 2018, 293, 9869-9879. [CrossRef]
- Waypa, G.B.; Marks, J.D.; Mack, M.M.; Boriboun, C.; Mungai, P.T.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ Res 2002, 91, 719-726. [CrossRef]
- Weissmann, N.; Ebert, N.; Ahrens, M.; Ghofrani, H.A.; Schermuly, R.T.; Hanze, J.; Fink, L.; Rose, F.; Conzen, J.; Seeger, W.; et al. Effects of mitochondrial inhibitors and uncouplers on hypoxic vasoconstriction in rabbit lungs. Am J Respir Cell Mol Biol 2003, 29, 721-732. [CrossRef]
- Weissmann, N.; Tadic, A.; Hanze, J.; Rose, F.; Winterhalder, S.; Nollen, M.; Schermuly, R.T.; Ghofrani, H.A.; Seeger, W.; Grimminger, F. Hypoxic vasoconstriction in intact lungs: a role for NADPH oxidase-derived H(2)O(2)? Am J Physiol Lung Cell Mol Physiol 2000, 279, L683-690. [CrossRef]
- Weissmann, N.; Winterhalder, S.; Nollen, M.; Voswinckel, R.; Quanz, K.; Ghofrani, H.A.; Schermuly, R.T.; Seeger, W.; Grimminger, F. NO and reactive oxygen species are involved in biphasic hypoxic vasoconstriction of isolated rabbit lungs. Am J Physiol Lung Cell Mol Physiol 2001, 280, L638-645. [CrossRef]
- Weissmann, N.; Zeller, S.; Schafer, R.U.; Turowski, C.; Ay, M.; Quanz, K.; Ghofrani, H.A.; Schermuly, R.T.; Fink, L.; Seeger, W.; et al. Impact of mitochondria and NADPH oxidases on acute and sustained hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol 2006, 34, 505-513. [CrossRef]
- Liu, J.Q.; Sham, J.S.; Shimoda, L.A.; Kuppusamy, P.; Sylvester, J.T. Hypoxic constriction and reactive oxygen species in porcine distal pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 2003, 285, L322-333. [CrossRef]
- Dikalov, S.; Griendling, K.K.; Harrison, D.G. Measurement of reactive oxygen species in cardiovascular studies. Hypertension 2007, 49, 717-727. [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab 2005, 1, 401-408. [CrossRef]
- Waypa, G.B.; Guzy, R.; Mungai, P.T.; Mack, M.M.; Marks, J.D.; Roe, M.W.; Schumacker, P.T. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ Res 2006, 99, 970-978. [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 2001, 30, 1191-1212. [CrossRef]
- Sabharwal, S.S.; Waypa, G.B.; Marks, J.D.; Schumacker, P.T. Peroxiredoxin-5 targeted to the mitochondrial intermembrane space attenuates hypoxia-induced reactive oxygen species signalling. Biochem J 2013, 456, 337-346. [CrossRef]
- Wang, Q.S.; Zheng, Y.M.; Dong, L.; Ho, Y.S.; Guo, Z.; Wang, Y.X. Role of mitochondrial reactive oxygen species in hypoxia-dependent increase in intracellular calcium in pulmonary artery myocytes. Free Radic Biol Med 2007, 42, 642-653. [CrossRef]
- Yang, Z.; Zhuan, B.; Yan, Y.; Jiang, S.; Wang, T. Roles of different mitochondrial electron transport chain complexes in hypoxia-induced pulmonary vasoconstriction. Cell Biol Int 2016, 40, 188-195. [CrossRef]
- Waypa, G.B.; Marks, J.D.; Guzy, R.; Mungai, P.T.; Schriewer, J.; Dokic, D.; Schumacker, P.T. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res 2010, 106, 526-535. [CrossRef]
- Meyer, A.J.; Dick, T.P. Fluorescent protein-based redox probes. Antioxid Redox Signal 2010, 13, 621-650. [CrossRef]
- Desireddi, J.R.; Farrow, K.N.; Marks, J.D.; Waypa, G.B.; Schumacker, P.T. Hypoxia increases ROS signaling and cytosolic Ca(2+) in pulmonary artery smooth muscle cells of mouse lungs slices. Antioxid Redox Signal 2010, 12, 595-602. [CrossRef]
- Doctrow, S.R.; Huffman, K.; Marcus, C.B.; Musleh, W.; Bruce, A.; Baudry, M.; Malfroy, B. Salen-manganese complexes: combined superoxide dismutase/catalase mimics with broad pharmacological efficacy. Adv Pharmacol 1997, 38, 247-269. [CrossRef]
- Casteels, R.; Raeymaekers, L.; Suzuki, H.; Van Eldere, J. Tension response and 45Ca release in vascular smooth muscle incubated in Ca-free solution. Pflugers Arch 1981, 392, 139-145. [CrossRef]
- Paddenberg, R.; Konig, P.; Faulhammer, P.; Goldenberg, A.; Pfeil, U.; Kummer, W. Hypoxic vasoconstriction of partial muscular intra-acinar pulmonary arteries in murine precision cut lung slices. Respir Res 2006, 7, 93. [CrossRef]
- Sanderson, M.J.; Bai, Y.; Perez-Zoghbi, J. Ca(2+) oscillations regulate contraction of intrapulmonary smooth muscle cells. Adv Exp Med Biol 2010, 661, 77-96. [CrossRef]
- Truong, L.; Zheng, Y.M.; Wang, Y.X. Mitochondrial Rieske iron-sulfur protein in pulmonary artery smooth muscle: A key primary signaling molecule in pulmonary hypertension. Arch Biochem Biophys 2020, 683, 108234. [CrossRef]
- Song, T.; Zheng, Y.M.; Wang, Y.X. Cross Talk Between Mitochondrial Reactive Oxygen Species and Sarcoplasmic Reticulum Calcium in Pulmonary Arterial Smooth Muscle Cells. Adv Exp Med Biol 2017, 967, 289-298. [CrossRef]
- Yang, Z.; Song, T.; Truong, L.; Reyes-Garcia, J.; Wang, L.; Zheng, Y.M.; Wang, Y.X. Important Role of Sarcoplasmic Reticulum Ca(2+) Release via Ryanodine Receptor-2 Channel in Hypoxia-Induced Rieske Iron-Sulfur Protein-Mediated Mitochondrial Reactive Oxygen Species Generation in Pulmonary Artery Smooth Muscle Cells. Antioxid Redox Signal 2020, 32, 447-462. [CrossRef]
- Anso, E.; Weinberg, S.E.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Z.; Steadman, M.; et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat Cell Biol 2017, 19, 614-625. [CrossRef]
- Hughes, B.G.; Hekimi, S. A mild impairment of mitochondrial electron transport has sex-specific effects on lifespan and aging in mice. PLoS One 2011, 6, e26116. [CrossRef]
- Waypa, G.B.; Smith, K.A.; Mungai, P.T.; Dudley, V.J.; Helmin, K.A.; Singer, B.D.; Peek, C.B.; Bass, J.; Nelson, L.; Shah, S.J.; et al. Mitochondria regulate proliferation in adult cardiac myocytes. J Clin Invest 2024, 134. [CrossRef]
- Horvath, A.; Horakova, E.; Dunajcikova, P.; Verner, Z.; Pravdova, E.; Slapetova, I.; Cuninkova, L.; Lukes, J. Downregulation of the nuclear-encoded subunits of the complexes III and IV disrupts their respective complexes but not complex I in procyclic Trypanosoma brucei. Mol Microbiol 2005, 58, 116-130. [CrossRef]
- Owens, K.M.; Kulawiec, M.; Desouki, M.M.; Vanniarajan, A.; Singh, K.K. Impaired OXPHOS complex III in breast cancer. PLoS One 2011, 6, e23846. [CrossRef]
- Kadenbach, B.; Huttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64-76. [CrossRef]
- Zhou, T.; Chien, M.S.; Kaleem, S.; Matsunami, H. Single cell transcriptome analysis of mouse carotid body glomus cells. J Physiol 2016, 594, 4225-4251. [CrossRef]
- Gao, L.; Bonilla-Henao, V.; Garcia-Flores, P.; Arias-Mayenco, I.; Ortega-Saenz, P.; Lopez-Barneo, J. Gene expression analyses reveal metabolic specifications in acute O(2) -sensing chemoreceptor cells. J Physiol 2017, 595, 6091-6120. [CrossRef]
- Pak, O.; Scheibe, S.; Esfandiary, A.; Gierhardt, M.; Sydykov, A.; Logan, A.; Fysikopoulos, A.; Veit, F.; Hecker, M.; Kroschel, F.; et al. Impact of the mitochondria-targeted antioxidant MitoQ on hypoxia-induced pulmonary hypertension. Eur Respir J 2018. [CrossRef]
- Weissmann, N.; Kuzkaya, N.; Fuchs, B.; Tiyerili, V.; Schafer, R.U.; Schutte, H.; Ghofrani, H.A.; Schermuly, R.T.; Schudt, C.; Sydykov, A.; et al. Detection of reactive oxygen species in isolated, perfused lungs by electron spin resonance spectroscopy. Respir Res 2005, 6, 86. [CrossRef]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat Metab 2022, 4, 651-662. [CrossRef]
- Dry, I.B.; Moore, A.L.; Day, D.A.; Wiskich, J.T. Regulation of alternative pathway activity in plant mitochondria: nonlinear relationship between electron flux and the redox poise of the quinone pool. Arch Biochem Biophys 1989, 273, 148-157. [CrossRef]
- El-Khoury, R.; Dufour, E.; Rak, M.; Ramanantsoa, N.; Grandchamp, N.; Csaba, Z.; Duvillie, B.; Benit, P.; Gallego, J.; Gressens, P.; et al. Alternative oxidase expression in the mouse enables bypassing cytochrome c oxidase blockade and limits mitochondrial ROS overproduction. PLoS Genet 2013, 9, e1003182. [CrossRef]
- Maxwell, D.P.; Wang, Y.; McIntosh, L. The alternative oxidase lowers mitochondrial reactive oxygen production in plant cells. Proc Natl Acad Sci U S A 1999, 96, 8271-8276. [CrossRef]
- Moreno-Dominguez, A.; Ortega-Saenz, P.; Gao, L.; Colinas, O.; Garcia-Flores, P.; Bonilla-Henao, V.; Aragones, J.; Huttemann, M.; Grossman, L.I.; Weissmann, N.; et al. Acute O2 sensing through HIF2alpha-dependent expression of atypical cytochrome oxidase subunits in arterial chemoreceptors. Sci Signal 2020, 13. [CrossRef]
- Tanaka, H.; Nishimaru, K.; Aikawa, T.; Hirayama, W.; Tanaka, Y.; Shigenobu, K. Effect of SEA0400, a novel inhibitor of sodium-calcium exchanger, on myocardial ionic currents. Br J Pharmacol 2002, 135, 1096-1100. [CrossRef]
- Ruiz, A.; Alberdi, E.; Matute, C. CGP37157, an inhibitor of the mitochondrial Na+/Ca2+ exchanger, protects neurons from excitotoxicity by blocking voltage-gated Ca2+ channels. Cell Death Dis 2014, 5, e1156. [CrossRef]
- Friedman, S.M. The effects of external sodium substitution on cell sodium and potassium in vascular smooth muscle. J Physiol 1977, 270, 195-208. [CrossRef]
- Nita, II; Hershfinkel, M.; Lewis, E.C.; Sekler, I. A crosstalk between Na(+) channels, Na(+)/K(+) pump and mitochondrial Na(+) transporters controls glucose-dependent cytosolic and mitochondrial Na(+) signals. Cell Calcium 2015, 57, 69-75. [CrossRef]
- Majander, A.; Finel, M.; Wikstrom, M. Diphenyleneiodonium inhibits reduction of iron-sulfur clusters in the mitochondrial NADH-ubiquinone oxidoreductase (Complex I). J Biol Chem 1994, 269, 21037-21042.
- Mohazzab, K.M.; Fayngersh, R.P.; Kaminski, P.M.; Wolin, M.S. Potential role of NADH oxidoreductase-derived reactive O2 species in calf pulmonary arterial PO2-elicited responses. Am J Physiol 1995, 269, L637-644. [CrossRef]
- Grimminger, F.; Weissmann, N.; Spriestersbach, R.; Becker, E.; Rosseau, S.; Seeger, W. Effects of NADPH oxidase inhibitors on hypoxic vasoconstriction in buffer-perfused rabbit lungs. Am J Physiol 1995, 268, L747-752. [CrossRef]
- Diatchuk, V.; Lotan, O.; Koshkin, V.; Wikstroem, P.; Pick, E. Inhibition of NADPH oxidase activation by 4-(2-aminoethyl)-benzenesulfonyl fluoride and related compounds. J Biol Chem 1997, 272, 13292-13301. [CrossRef]
- Lahiri, S.; Ehleben, W.; Acker, H. Chemoreceptor discharges and cytochrome redox changes of the rat carotid body: role of heme ligands. Proc Natl Acad Sci U S A 1999, 96, 9427-9432. [CrossRef]
- Rathore, R.; Zheng, Y.M.; Li, X.Q.; Wang, Q.S.; Liu, Q.H.; Ginnan, R.; Singer, H.A.; Ho, Y.S.; Wang, Y.X. Mitochondrial ROS-PKCepsilon signaling axis is uniquely involved in hypoxic increase in [Ca2+]i in pulmonary artery smooth muscle cells. Biochem Biophys Res Commun 2006, 351, 784-790. [CrossRef]
- Cogolludo, A.; Moreno, L.; Frazziano, G.; Moral-Sanz, J.; Menendez, C.; Castaneda, J.; Gonzalez, C.; Villamor, E.; Perez-Vizcaino, F. Activation of neutral sphingomyelinase is involved in acute hypoxic pulmonary vasoconstriction. Cardiovasc Res 2009, 82, 296-302. [CrossRef]
- Moral-Sanz, J.; Gonzalez, T.; Menendez, C.; David, M.; Moreno, L.; Macias, A.; Cortijo, J.; Valenzuela, C.; Perez-Vizcaino, F.; Cogolludo, A. Ceramide inhibits Kv currents and contributes to TP-receptor-induced vasoconstriction in rat and human pulmonary arteries. Am J Physiol Cell Physiol 2011, 301, C186-194. [CrossRef]
- Wu-Zhang, A.X.; Newton, A.C. Protein kinase C pharmacology: refining the toolbox. Biochem J 2013, 452, 195-209. [CrossRef]
- Veith, C.; Kraut, S.; Wilhelm, J.; Sommer, N.; Quanz, K.; Seeger, W.; Brandes, R.P.; Weissmann, N.; Schroder, K. NADPH oxidase 4 is not involved in hypoxia-induced pulmonary hypertension. Pulm Circ 2016, 6, 397-400. [CrossRef]
- Murtaza, G.; Paddenberg, R.; Pfeil, U.; Goldenberg, A.; Mermer, P.; Kummer, W. Hypoxia-induced pulmonary vasoconstriction of intra-acinar arteries is impaired in NADPH oxidase 4 gene-deficient mice. Pulm Circ 2018, 8, 2045894018808240. [CrossRef]
- Hales, C.A. The site and mechanism of oxygen sensing for the pulmonary vessels. Chest 1985, 88, 235S-240S. [CrossRef]
- Nagaraj, C.; Tabeling, C.; Nagy, B.M.; Jain, P.P.; Marsh, L.M.; Papp, R.; Pienn, M.; Witzenrath, M.; Ghanim, B.; Klepetko, W.; et al. Hypoxic vascular response and ventilation/perfusion matching in end-stage COPD may depend on p22phox. Eur Respir J 2017, 50. [CrossRef]
- Littler, C.M.; Morris, K.G., Jr.; Fagan, K.A.; McMurtry, I.F.; Messing, R.O.; Dempsey, E.C. Protein kinase C-epsilon-null mice have decreased hypoxic pulmonary vasoconstriction. Am J Physiol Heart Circ Physiol 2003, 284, H1321-1331. [CrossRef]
- Robertson, T.P.; Aaronson, P.I.; Ward, J.P. Hypoxic vasoconstriction and intracellular Ca2+ in pulmonary arteries: evidence for PKC-independent Ca2+ sensitization. Am J Physiol 1995, 268, H301-307. [CrossRef]
- Stuart, J.A.; Fonseca, J.; Moradi, F.; Cunningham, C.; Seliman, B.; Worsfold, C.R.; Dolan, S.; Abando, J.; Maddalena, L.A. How Supraphysiological Oxygen Levels in Standard Cell Culture Affect Oxygen-Consuming Reactions. Oxid Med Cell Longev 2018, 2018, 8238459. [CrossRef]
- Wang, X.T.; McCullough, K.D.; Wang, X.J.; Carpenter, G.; Holbrook, N.J. Oxidative stress-induced phospholipase C-gamma 1 activation enhances cell survival. J Biol Chem 2001, 276, 28364-28371. [CrossRef]
- Strielkov, I.V.; Kizub, I.V.; Khromov, A.S.; Soloviev, A.I. Evidence for the role of phosphatidylcholine-specific phospholipase C in sustained hypoxic pulmonary vasoconstriction. Vascul Pharmacol 2013, 58, 292-298. [CrossRef]
- Liao, B.; Zheng, Y.M.; Yadav, V.R.; Korde, A.S.; Wang, Y.X. Hypoxia induces intracellular Ca2+ release by causing reactive oxygen species-mediated dissociation of FK506-binding protein 12.6 from ryanodine receptor 2 in pulmonary artery myocytes. Antioxid Redox Signal 2011, 14, 37-47. [CrossRef]
- Chen, T.X.; Xu, X.Y.; Zhao, Z.; Zhao, F.Y.; Gao, Y.M.; Yan, X.H.; Wan, Y. Hydrogen peroxide is a critical regulator of the hypoxia-induced alterations of store-operated Ca(2+) entry into rat pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2017, 312, L477-L487. [CrossRef]
- Lin, M.J.; Yang, X.R.; Cao, Y.N.; Sham, J.S. Hydrogen peroxide-induced Ca2+ mobilization in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2007, 292, L1598-1608. [CrossRef]
- Park, J.M.; Do, V.Q.; Seo, Y.S.; Kim, H.J.; Nam, J.H.; Yin, M.Z.; Kim, H.J.; Kim, S.J.; Griendling, K.K.; Lee, M.Y. NADPH Oxidase 1 Mediates Acute Blood Pressure Response to Angiotensin II by Contributing to Calcium Influx in Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol 2022, 42, e117-e130. [CrossRef]
- Maier, T.; Follmann, M.; Hessler, G.; Kleemann, H.W.; Hachtel, S.; Fuchs, B.; Weissmann, N.; Linz, W.; Schmidt, T.; Lohn, M.; et al. Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol-sensitive TRPC cation channels. Br J Pharmacol 2015, 172, 3650-3660. [CrossRef]
- Knock, G.A.; Ward, J.P. Redox regulation of protein kinases as a modulator of vascular function. Antioxid Redox Signal 2011, 15, 1531-1547. [CrossRef]
- Knock, G.A.; Snetkov, V.A.; Shaifta, Y.; Drndarski, S.; Ward, J.P.; Aaronson, P.I. Role of src-family kinases in hypoxic vasoconstriction of rat pulmonary artery. Cardiovasc Res 2008, 80, 453-462. [CrossRef]
- Knock, G.A.; Snetkov, V.A.; Shaifta, Y.; Connolly, M.; Drndarski, S.; Noah, A.; Pourmahram, G.E.; Becker, S.; Aaronson, P.I.; Ward, J.P. Superoxide constricts rat pulmonary arteries via Rho-kinase-mediated Ca(2+) sensitization. Free Radic Biol Med 2009, 46, 633-642. [CrossRef]
- MacKay, C.E.; Shaifta, Y.; Snetkov, V.V.; Francois, A.A.; Ward, J.P.T.; Knock, G.A. ROS-dependent activation of RhoA/Rho-kinase in pulmonary artery: Role of Src-family kinases and ARHGEF1. Free Radic Biol Med 2017, 110, 316-331. [CrossRef]
- Forman, H.J.; Ursini, F.; Maiorino, M. An overview of mechanisms of redox signaling. J Mol Cell Cardiol 2014, 73, 2-9. [CrossRef]
- Jernigan, N.L.; Resta, T.C.; Gonzalez Bosc, L.V. Altered Redox Balance in the Development of Chronic Hypoxia-induced Pulmonary Hypertension. Adv Exp Med Biol 2017, 967, 83-103. [CrossRef]
- Pourmahram, G.E.; Snetkov, V.A.; Shaifta, Y.; Drndarski, S.; Knock, G.A.; Aaronson, P.I.; Ward, J.P. Constriction of pulmonary artery by peroxide: role of Ca2+ release and PKC. Free Radic Biol Med 2008, 45, 1468-1476. [CrossRef]
- Genet, N.; Billaud, M.; Rossignol, R.; Dubois, M.; Gillibert-Duplantier, J.; Isakson, B.E.; Marthan, R.; Savineau, J.P.; Guibert, C. Signaling Pathways Linked to Serotonin-Induced Superoxide Anion Production: A Physiological Role for Mitochondria in Pulmonary Arteries. Front Physiol 2017, 8, 76. [CrossRef]
- Poyton, R.O.; Castello, P.R.; Ball, K.A.; Woo, D.K.; Pan, N. Mitochondria and hypoxic signaling: a new view. Ann N Y Acad Sci 2009, 1177, 48-56. [CrossRef]
- Viner, R.I.; Williams, T.D.; Schoneich, C. Peroxynitrite modification of protein thiols: oxidation, nitrosylation, and S-glutathiolation of functionally important cysteine residue(s) in the sarcoplasmic reticulum Ca-ATPase. Biochemistry 1999, 38, 12408-12415. [CrossRef]
- Martinez-Ruiz, A.; Cadenas, S.; Lamas, S. Nitric oxide signaling: classical, less classical, and nonclassical mechanisms. Free Radic Biol Med 2011, 51, 17-29. [CrossRef]
- Andre, F.R.; dos Santos, P.F.; Rando, D.G. Theoretical studies of the role of C-terminal cysteines in the process of S-nitrosylation of human Src kinases. J Mol Model 2016, 22, 23. [CrossRef]
- Minetti, M.; Mallozzi, C.; Di Stasi, A.M. Peroxynitrite activates kinases of the src family and upregulates tyrosine phosphorylation signaling. Free Radic Biol Med 2002, 33, 744-754. [CrossRef]
- Lock, J.T.; Sinkins, W.G.; Schilling, W.P. Effect of protein S-glutathionylation on Ca2+ homeostasis in cultured aortic endothelial cells. Am J Physiol Heart Circ Physiol 2011, 300, H493-506. [CrossRef]
- Sun, J.; Yamaguchi, N.; Xu, L.; Eu, J.P.; Stamler, J.S.; Meissner, G. Regulation of the cardiac muscle ryanodine receptor by O(2) tension and S-nitrosoglutathione. Biochemistry 2008, 47, 13985-13990. [CrossRef]
- Flores-Tamez, V.; Escalante, B.; Rios, A. Peroxynitrite-Induced Intracellular Ca2+ Depression in Cardiac Myocytes: Role of Sarco/Endoplasmic Reticulum Ca2+ Pump. Folia Biol (Praha) 2019, 65, 237-245. [CrossRef]
- Sadowska-Bartosz, I.; Bartosz, G. Peroxiredoxin 2: An Important Element of the Antioxidant Defense of the Erythrocyte. Antioxidants (Basel) 2023, 12. [CrossRef]
- Emerling, B.M.; Weinberg, F.; Snyder, C.; Burgess, Z.; Mutlu, G.M.; Viollet, B.; Budinger, G.R.; Chandel, N.S. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic Biol Med 2009, 46, 1386-1391. [CrossRef]
- Gusarova, G.A.; Trejo, H.E.; Dada, L.A.; Briva, A.; Welch, L.C.; Hamanaka, R.B.; Mutlu, G.M.; Chandel, N.S.; Prakriya, M.; Sznajder, J.I. Hypoxia leads to Na,K-ATPase downregulation via Ca(2+) release-activated Ca(2+) channels and AMPK activation. Mol Cell Biol 2011, 31, 3546-3556. [CrossRef]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab 2010, 11, 554-565. [CrossRef]
- Choi, S.L.; Kim, S.J.; Lee, K.T.; Kim, J.; Mu, J.; Birnbaum, M.J.; Soo Kim, S.; Ha, J. The regulation of AMP-activated protein kinase by H(2)O(2). Biochem Biophys Res Commun 2001, 287, 92-97. [CrossRef]
- Prieto-Lloret, J.; Aaronson, P.I. Hydrogen Sulfide as an O2 Sensor: A Critical Analysis. Adv Exp Med Biol 2017, 967, 261-276. [CrossRef]
- Prieto-Lloret, J.; Shaifta, Y.; Ward, J.P.; Aaronson, P.I. Hypoxic pulmonary vasoconstriction in isolated rat pulmonary arteries is not inhibited by antagonists of H2 S-synthesizing pathways. J Physiol 2015, 593, 385-401. [CrossRef]
- Iciek, M.; Bilska-Wilkosz, A.; Gorny, M. Sulfane sulfur - new findings on an old topic. Acta Biochim Pol 2019, 66, 533-544. [CrossRef]
- Olson, K.R.; Gao, Y.; DeLeon, E.R.; Markel, T.A.; Drucker, N.; Boone, D.; Whiteman, M.; Steiger, A.K.; Pluth, M.D.; Tessier, C.R.; et al. Extended hypoxia-mediated H(2) S production provides for long-term oxygen sensing. Acta Physiol (Oxf) 2020, 228, e13368. [CrossRef]
- Goubern, M.; Andriamihaja, M.; Nubel, T.; Blachier, F.; Bouillaud, F. Sulfide, the first inorganic substrate for human cells. FASEB J 2007, 21, 1699-1706. [CrossRef]
- Skovgaard, N.; Olson, K.R. Hydrogen sulfide mediates hypoxic vasoconstriction through a production of mitochondrial ROS in trout gills. Am J Physiol Regul Integr Comp Physiol 2012, 303, R487-494. [CrossRef]
- Gupte, R.S.; Rawat, D.K.; Chettimada, S.; Cioffi, D.L.; Wolin, M.S.; Gerthoffer, W.T.; McMurtry, I.F.; Gupte, S.A. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J Biol Chem 2010, 285, 19561-19571. [CrossRef]
- Gupte, S.A.; Okada, T.; McMurtry, I.F.; Oka, M. Role of pentose phosphate pathway-derived NADPH in hypoxic pulmonary vasoconstriction. Pulm Pharmacol Ther 2006, 19, 303-309. [CrossRef]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 2010, 31, 194-223. [CrossRef]
- Wilson, H.L.; Dipp, M.; Thomas, J.M.; Lad, C.; Galione, A.; Evans, A.M. Adp-ribosyl cyclase and cyclic ADP-ribose hydrolase act as a redox sensor. a primary role for cyclic ADP-ribose in hypoxic pulmonary vasoconstriction. J Biol Chem 2001, 276, 11180-11188. [CrossRef]
- Moreno-Dominguez, A.; Colinas, O.; Arias-Mayenco, I.; Cabeza, J.M.; Lopez-Ogayar, J.L.; Chandel, N.S.; Weissmann, N.; Sommer, N.; Pascual, A.; Lopez-Barneo, J. Hif1alpha-dependent mitochondrial acute O(2) sensing and signaling to myocyte Ca(2+) channels mediate arterial hypoxic vasodilation. Nat Commun 2024, 15, 6649. [CrossRef]
- Arias-Mayenco, I.; Gonzalez-Rodriguez, P.; Torres-Torrelo, H.; Gao, L.; Fernandez-Aguera, M.C.; Bonilla-Henao, V.; Ortega-Saenz, P.; Lopez-Barneo, J. Acute O(2) Sensing: Role of Coenzyme QH(2)/Q Ratio and Mitochondrial ROS Compartmentalization. Cell Metab 2018, 28, 145-158 e144. [CrossRef]
- Dwenger, M.M.; Ohanyan, V.; Navedo, M.F.; Nystoriak, M.A. Coronary microvascular Kv1 channels as regulatory sensors of intracellular pyridine nucleotide redox potential. Microcirculation 2018, 25. [CrossRef]
- Torres-Torrelo, H.; Ortega-Saenz, P.; Gao, L.; Lopez-Barneo, J. Lactate sensing mechanisms in arterial chemoreceptor cells. Nat Commun 2021, 12, 4166. [CrossRef]
- Michelakis, E.D.; Rebeyka, I.; Wu, X.; Nsair, A.; Thebaud, B.; Hashimoto, K.; Dyck, J.R.; Haromy, A.; Harry, G.; Barr, A.; et al. O2 sensing in the human ductus arteriosus: regulation of voltage-gated K+ channels in smooth muscle cells by a mitochondrial redox sensor. Circ Res 2002, 91, 478-486. [CrossRef]
- Hirst, J.; King, M.S.; Pryde, K.R. The production of reactive oxygen species by complex I. Biochem Soc Trans 2008, 36, 976-980. [CrossRef]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol 2013, 1, 304-312. [CrossRef]
- Reeve, H.L.; Tolarova, S.; Nelson, D.P.; Archer, S.; Weir, E.K. Redox control of oxygen sensing in the rabbit ductus arteriosus. J Physiol 2001, 533, 253-261. [CrossRef]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thebaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630-2641. [CrossRef]
- Ahmad, M.; Kelly, M.R.; Zhao, X.; Kandhi, S.; Wolin, M.S. Roles for Nox4 in the contractile response of bovine pulmonary arteries to hypoxia. Am J Physiol Heart Circ Physiol 2010, 298, H1879-1888. [CrossRef]
- Paky, A.; Michael, J.R.; Burke-Wolin, T.M.; Wolin, M.S.; Gurtner, G.H. Endogenous production of superoxide by rabbit lungs: effects of hypoxia or metabolic inhibitors. J Appl Physiol (1985) 1993, 74, 2868-2874. [CrossRef]
- Wang, Y.X.; Zheng, Y.M.; Abdullaev, I.; Kotlikoff, M.I. Metabolic inhibition with cyanide induces calcium release in pulmonary artery myocytes and Xenopus oocytes. Am J Physiol Cell Physiol 2003, 284, C378-388. [CrossRef]
- Young, T.A.; Cunningham, C.C.; Bailey, S.M. Reactive oxygen species production by the mitochondrial respiratory chain in isolated rat hepatocytes and liver mitochondria: studies using myxothiazol. Arch Biochem Biophys 2002, 405, 65-72. [CrossRef]
- Starkov, A.A.; Fiskum, G. Myxothiazol induces H(2)O(2) production from mitochondrial respiratory chain. Biochem Biophys Res Commun 2001, 281, 645-650. [CrossRef]
- Watson, M.A.; Wong, H.S.; Brand, M.D. Use of S1QELs and S3QELs to link mitochondrial sites of superoxide and hydrogen peroxide generation to physiological and pathological outcomes. Biochem Soc Trans 2019, 47, 1461-1469. [CrossRef]
- Kilbride, S.M.; Telford, J.E.; Davey, G.P. Complex I Controls Mitochondrial and Plasma Membrane Potentials in Nerve Terminals. Neurochem Res 2021, 46, 100-107. [CrossRef]
- Cheung, J.Y.; Wang, J.; Zhang, X.Q.; Song, J.; Davidyock, J.M.; Prado, F.J.; Shanmughapriya, S.; Worth, A.M.; Madesh, M.; Judenherc-Haouzi, A.; et al. Methylene Blue Counteracts H(2)S-Induced Cardiac Ion Channel Dysfunction and ATP Reduction. Cardiovasc Toxicol 2018, 18, 407-419. [CrossRef]
- Cheranov, S.Y.; Jaggar, J.H. Mitochondrial modulation of Ca2+ sparks and transient KCa currents in smooth muscle cells of rat cerebral arteries. J Physiol 2004, 556, 755-771. [CrossRef]
- McCarron, J.G.; Olson, M.L.; Wilson, C.; Sandison, M.E.; Chalmers, S. Examining the role of mitochondria in Ca(2)(+) signaling in native vascular smooth muscle. Microcirculation 2013, 20, 317-329. [CrossRef]
- Sward, K.; Dreja, K.; Lindqvist, A.; Persson, E.; Hellstrand, P. Influence of mitochondrial inhibition on global and local [Ca(2+)](I) in rat tail artery. Circ Res 2002, 90, 792-799. [CrossRef]
- Doege, K.; Heine, S.; Jensen, I.; Jelkmann, W.; Metzen, E. Inhibition of mitochondrial respiration elevates oxygen concentration but leaves regulation of hypoxia-inducible factor (HIF) intact. Blood 2005, 106, 2311-2317. [CrossRef]
- Potter, M.; Badder, L.; Hoade, Y.; Johnston, I.G.; Morten, K.J. Monitoring Intracellular Oxygen Concentration: Implications for Hypoxia Studies and Real-Time Oxygen Monitoring. Adv Exp Med Biol 2016, 876, 257-263. [CrossRef]
- Hagen, T.; Taylor, C.T.; Lam, F.; Moncada, S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science 2003, 302, 1975-1978. [CrossRef]
- Wilcox, C.S.; Pearlman, A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol Rev 2008, 60, 418-469. [CrossRef]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J 2015, 29, 4766-4771. [CrossRef]
- Pedre, B.; Barayeu, U.; Ezerina, D.; Dick, T.P. The mechanism of action of N-acetylcysteine (NAC): The emerging role of H(2)S and sulfane sulfur species. Pharmacol Ther 2021, 228, 107916. [CrossRef]
- Barbosa, N.V.; Nogueira, C.W.; Nogara, P.A.; de Bem, A.F.; Aschner, M.; Rocha, J.B.T. Organoselenium compounds as mimics of selenoproteins and thiol modifier agents. Metallomics 2017, 9, 1703-1734. [CrossRef]
- Olson, K.R.; Gao, Y.; Steiger, A.K.; Pluth, M.D.; Tessier, C.R.; Markel, T.A.; Boone, D.; Stahelin, R.V.; Batinic-Haberle, I.; Straubg, K.D. Effects of Manganese Porphyrins on Cellular Sulfur Metabolism. Molecules 2020, 25. [CrossRef]
- Batinic-Haberle, I.; Tovmasyan, A.; Spasojevic, I. An educational overview of the chemistry, biochemistry and therapeutic aspects of Mn porphyrins--From superoxide dismutation to H2O2-driven pathways. Redox Biol 2015, 5, 43-65. [CrossRef]
- Silveira, L.R.; Pereira-Da-Silva, L.; Juel, C.; Hellsten, Y. Formation of hydrogen peroxide and nitric oxide in rat skeletal muscle cells during contractions. Free Radic Biol Med 2003, 35, 455-464. [CrossRef]
- Messent, M.; Griffiths, M.J.; Quinlan, G.J.; Gutteridge, J.M.; Evans, T.W. Ischaemia--reperfusion injury in the rat is modulated by superoxide generation and leads to an augmentation of the hypoxic pulmonary vascular response. Clin Sci (Lond) 1996, 90, 47-54. [CrossRef]
- Shen, C.Y.; Lee, J.F.; Su, C.L.; Wang, D.; Chen, C.F. Hypoxia and reoxygenation of the lung tissues induced mRNA expressions of superoxide dismutase and catalase and interventions from different antioxidants. Transplant Proc 2008, 40, 2182-2184. [CrossRef]
- Francis, B.N.; Wilkins, M.R.; Zhao, L. Tetrahydrobiopterin and the regulation of hypoxic pulmonary vasoconstriction. Eur Respir J 2010, 36, 323-330. [CrossRef]
- Ahmad, M.; Zhao, X.; Kelly, M.R.; Kandhi, S.; Perez, O.; Abraham, N.G.; Wolin, M.S. Heme oxygenase-1 induction modulates hypoxic pulmonary vasoconstriction through upregulation of ecSOD. Am J Physiol Heart Circ Physiol 2009, 297, H1453-1461. [CrossRef]
- Patel, D.; Alhawaj, R.; Wolin, M.S. Exposure of mice to chronic hypoxia attenuates pulmonary arterial contractile responses to acute hypoxia by increases in extracellular hydrogen peroxide. Am J Physiol Regul Integr Comp Physiol 2014, 307, R426-433. [CrossRef]
- Bopp, C.; Auger, C.; Mebazaa, A.; Joshi, G.P.; Schini-Kerth, V.B.; Diemunsch, P. Urapidil, but not dihydropyridine calcium channel inhibitors, preserves the hypoxic pulmonary vasoconstriction: an experimental study in pig arteries. Fundam Clin Pharmacol 2019, 33, 527-534. [CrossRef]
- Demiryurek, A.T.; Wadsworth, R.M.; Kane, K.A. Pharmacological evidence for the role of mediators in hypoxia-induced vasoconstriction in sheep isolated intrapulmonary artery rings. Eur J Pharmacol 1991, 203, 1-8. [CrossRef]
- Weissmann, N.; Grimminger, F.; Voswinckel, R.; Conzen, J.; Seeger, W. Nitro blue tetrazolium inhibits but does not mimic hypoxic vasoconstriction in isolated rabbit lungs. Am J Physiol 1998, 274, L721-727. [CrossRef]
- Mohazzab, K.M.; Wolin, M.S. Properties of a superoxide anion-generating microsomal NADH oxidoreductase, a potential pulmonary artery PO2 sensor. Am J Physiol 1994, 267, L823-831. [CrossRef]
- Du, W.; Frazier, M.; McMahon, T.J.; Eu, J.P. Redox activation of intracellular calcium release channels (ryanodine receptors) in the sustained phase of hypoxia-induced pulmonary vasoconstriction. Chest 2005, 128, 556S-558S. [CrossRef]
- Mansoori, S.; Moosavi, S.M.S.; Ketabchi, F. The Interaction between Trolox and 4,4'-diisothiocyanatostilbene-2,2'-disulfonic Acid on Hypoxic Pulmonary Vasoconstriction in the Isolated Rabbit Lung. Iran J Med Sci 2017, 42, 284-291.
- Hodyc, D.; Snorek, M.; Brtnicky, T.; Herget, J. Superoxide dismutase mimetic tempol inhibits hypoxic pulmonary vasoconstriction in rats independently of nitric oxide production. Exp Physiol 2007, 92, 945-951. [CrossRef]
- Pak, O.; Scheibe, S.; Esfandiary, A.; Gierhardt, M.; Sydykov, A.; Logan, A.; Fysikopoulos, A.; Veit, F.; Hecker, M.; Kroschel, F.; et al. Impact of the mitochondria-targeted antioxidant MitoQ on hypoxia-induced pulmonary hypertension. Eur Respir J 2018, 51. [CrossRef]
- Go, Y.M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic Biol Med 2015, 84, 227-245. [CrossRef]
- Meyrick, B.; Reid, L. Hypoxia-induced structural changes in the media and adventitia of the rat hilar pulmonary artery and their regression. Am J Pathol 1980, 100, 151-178.
- Burke, T.M.; Wolin, M.S. Hydrogen peroxide elicits pulmonary arterial relaxation and guanylate cyclase activation. Am J Physiol 1987, 252, H721-732. [CrossRef]
- Sheehan, D.W.; Giese, E.C.; Gugino, S.F.; Russell, J.A. Characterization and mechanisms of H2O2-induced contractions of pulmonary arteries. Am J Physiol 1993, 264, H1542-1547. [CrossRef]
- Jin, N.; Rhoades, R.A. Activation of tyrosine kinases in H2O2-induced contraction in pulmonary artery. Am J Physiol 1997, 272, H2686-2692. [CrossRef]
- Jones, R.D.; Thompson, J.S.; Morice, A.H. The effect of hydrogen peroxide on hypoxia, prostaglandin F2 alpha and potassium chloride induced contractions in isolated rat pulmonary arteries. Pulm Pharmacol Ther 1997, 10, 37-42. [CrossRef]
- Pelaez, N.J.; Braun, T.R.; Paul, R.J.; Meiss, R.A.; Packer, C.S. H(2)O(2) mediates Ca(2+)- and MLC(20) phosphorylation-independent contraction in intact and permeabilized vascular muscle. Am J Physiol Heart Circ Physiol 2000, 279, H1185-1193. [CrossRef]
- Kerkar, S.; Speyer, C.; Tyburski, J.; Steffes, C. Reactive oxygen metabolites induce a biphasic contractile response in microvascular lung pericytes. J Trauma 2001, 51, 440-445. [CrossRef]
- Nagi, M.N.; Mansour, M.A.; Al-Shabanah, O.A.; El-Kashef, H.A. Melatonin inhibits the contractile effect of vanadate in the isolated pulmonary arterial rings of rats: possible role of hydrogen peroxide. J Biochem Mol Toxicol 2002, 16, 273-278. [CrossRef]
- Oeckler, R.A.; Arcuino, E.; Ahmad, M.; Olson, S.C.; Wolin, M.S. Cytosolic NADH redox and thiol oxidation regulate pulmonary arterial force through ERK MAP kinase. Am J Physiol Lung Cell Mol Physiol 2005, 288, L1017-1025. [CrossRef]
- Maki, J.; Hirano, M.; Hoka, S.; Kanaide, H.; Hirano, K. Involvement of reactive oxygen species in thrombin-induced pulmonary vasoconstriction. Am J Respir Crit Care Med 2010, 182, 1435-1444. [CrossRef]
- Neo, B.H.; Kandhi, S.; Wolin, M.S. Roles for soluble guanylate cyclase and a thiol oxidation-elicited subunit dimerization of protein kinase G in pulmonary artery relaxation to hydrogen peroxide. Am J Physiol Heart Circ Physiol 2010, 299, H1235-1241. [CrossRef]
- Zaloudikova, M.; Herget, J.; Vizek, M. The contractile response of isolated small pulmonary arteries induced by activated macrophages. Physiol Res 2014, 63, 267-270. [CrossRef]
- Lucchesi, P.A.; Belmadani, S.; Matrougui, K. Hydrogen peroxide acts as both vasodilator and vasoconstrictor in the control of perfused mouse mesenteric resistance arteries. J Hypertens 2005, 23, 571-579. [CrossRef]
- Thakali, K.; Davenport, L.; Fink, G.D.; Watts, S.W. Pleiotropic effects of hydrogen peroxide in arteries and veins from normotensive and hypertensive rats. Hypertension 2006, 47, 482-487. [CrossRef]
- Neo, B.H.; Kandhi, S.; Wolin, M.S. Roles for redox mechanisms controlling protein kinase G in pulmonary and coronary artery responses to hypoxia. Am J Physiol Heart Circ Physiol 2011, 301, H2295-2304. [CrossRef]
- Zhang, D.X.; Borbouse, L.; Gebremedhin, D.; Mendoza, S.A.; Zinkevich, N.S.; Li, R.; Gutterman, D.D. H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circ Res 2012, 110, 471-480. [CrossRef]
- Nishijima, Y.; Cao, S.; Chabowski, D.S.; Korishettar, A.; Ge, A.; Zheng, X.; Sparapani, R.; Gutterman, D.D.; Zhang, D.X. Contribution of K(V)1.5 Channel to Hydrogen Peroxide-Induced Human Arteriolar Dilation and Its Modulation by Coronary Artery Disease. Circ Res 2017, 120, 658-669. [CrossRef]
- Gutterman, D.D.; Miura, H.; Liu, Y. Redox modulation of vascular tone: focus of potassium channel mechanisms of dilation. Arterioscler Thromb Vasc Biol 2005, 25, 671-678. [CrossRef]
- Matoba, T.; Shimokawa, H.; Kubota, H.; Morikawa, K.; Fujiki, T.; Kunihiro, I.; Mukai, Y.; Hirakawa, Y.; Takeshita, A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in human mesenteric arteries. Biochem Biophys Res Commun 2002, 290, 909-913. [CrossRef]
- Yang, W.; Block, E.R. Effect of hypoxia and reoxygenation on the formation and release of reactive oxygen species by porcine pulmonary artery endothelial cells. J Cell Physiol 1995, 164, 414-423. [CrossRef]
- Wojciak-Stothard, B.; Tsang, L.Y.; Haworth, S.G. Rac and Rho play opposing roles in the regulation of hypoxia/reoxygenation-induced permeability changes in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 2005, 288, L749-760. [CrossRef]
- Munzel, T.; Afanas'ev, I.B.; Kleschyov, A.L.; Harrison, D.G. Detection of superoxide in vascular tissue. Arterioscler Thromb Vasc Biol 2002, 22, 1761-1768. [CrossRef]
- Wardman, P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic Biol Med 2007, 43, 995-1022. [CrossRef]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J., 2nd; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med 2012, 52, 1-6. [CrossRef]
- Zielonka, J.; Kalyanaraman, B. Small-molecule luminescent probes for the detection of cellular oxidizing and nitrating species. Free Radic Biol Med 2018, 128, 3-22. [CrossRef]
- Li, Y.; Zhu, H.; Trush, M.A. Detection of mitochondria-derived reactive oxygen species production by the chemilumigenic probes lucigenin and luminol. Biochim Biophys Acta 1999, 1428, 1-12. [CrossRef]
- Rembish, S.J.; Trush, M.A. Further evidence that lucigenin-derived chemiluminescence monitors mitochondrial superoxide generation in rat alveolar macrophages. Free Radic Biol Med 1994, 17, 117-126. [CrossRef]
- Janiszewski, M.; Souza, H.P.; Liu, X.; Pedro, M.A.; Zweier, J.L.; Laurindo, F.R. Overestimation of NADH-driven vascular oxidase activity due to lucigenin artifacts. Free Radic Biol Med 2002, 32, 446-453. [CrossRef]
- Budd, S.L.; Castilho, R.F.; Nicholls, D.G. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett 1997, 415, 21-24. [CrossRef]
- Adesina, S.E.; Kang, B.Y.; Bijli, K.M.; Ma, J.; Cheng, J.; Murphy, T.C.; Michael Hart, C.; Sutliff, R.L. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic Biol Med 2015, 87, 36-47. [CrossRef]
- Sen, B.; Benoit, B.; Brand, M.D. Hypoxia decreases mitochondrial ROS production in cells. Free Radic Biol Med 2024, 224, 1-8. [CrossRef]
- Fang, J.; Wong, H.S.; Brand, M.D. Production of superoxide and hydrogen peroxide in the mitochondrial matrix is dominated by site I(Q) of complex I in diverse cell lines. Redox Biol 2020, 37, 101722. [CrossRef]
- Dooley, C.T.; Dore, T.M.; Hanson, G.T.; Jackson, W.C.; Remington, S.J.; Tsien, R.Y. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem 2004, 279, 22284-22293. [CrossRef]
- Lukyanov, K.A.; Belousov, V.V. Genetically encoded fluorescent redox sensors. Biochim Biophys Acta 2014, 1840, 745-756. [CrossRef]
- Pouvreau, S. Genetically encoded reactive oxygen species (ROS) and redox indicators. Biotechnol J 2014, 9, 282-293. [CrossRef]
- Nash, K.M.; Rockenbauer, A.; Villamena, F.A. Reactive nitrogen species reactivities with nitrones: theoretical and experimental studies. Chem Res Toxicol 2012, 25, 1581-1597. [CrossRef]
- Dikalov, S.I.; Polienko, Y.F.; Kirilyuk, I. Electron Paramagnetic Resonance Measurements of Reactive Oxygen Species by Cyclic Hydroxylamine Spin Probes. Antioxid Redox Signal 2018, 28, 1433-1443. [CrossRef]
- Gonzalez, C.; Sanz-Alfayate, G.; Obeso, A.; Agapito, M.T. Role of glutathione redox state in oxygen sensing by carotid body chemoreceptor cells. Methods Enzymol 2004, 381, 40-71. [CrossRef]
- Dikalov, S.I.; Kirilyuk, I.A.; Voinov, M.; Grigor'ev, I.A. EPR detection of cellular and mitochondrial superoxide using cyclic hydroxylamines. Free Radic Res 2011, 45, 417-430. [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457-470 e413. [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 1997, 416, 15-18. [CrossRef]
- Brand, M.D. Riding the tiger - physiological and pathological effects of superoxide and hydrogen peroxide generated in the mitochondrial matrix. Crit Rev Biochem Mol Biol 2020, 55, 592-661. [CrossRef]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 2007, 177, 1029-1036. [CrossRef]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Schumacker, P.T.; Chandel, N.S. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol Cell Biol 2007, 27, 5737-5745. [CrossRef]
- Chandel, N.S.; Schumacker, P.T. Cellular oxygen sensing by mitochondria: old questions, new insight. J Appl Physiol (1985) 2000, 88, 1880-1889. [CrossRef]
- Dada, L.A.; Chandel, N.S.; Ridge, K.M.; Pedemonte, C.; Bertorello, A.M.; Sznajder, J.I. Hypoxia-induced endocytosis of Na,K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-zeta. J Clin Invest 2003, 111, 1057-1064. [CrossRef]
- Hernansanz-Agustin, P.; Izquierdo-Alvarez, A.; Sanchez-Gomez, F.J.; Ramos, E.; Villa-Pina, T.; Lamas, S.; Bogdanova, A.; Martinez-Ruiz, A. Acute hypoxia produces a superoxide burst in cells. Free Radic Biol Med 2014, 71, 146-156. [CrossRef]
- Loor, G.; Kondapalli, J.; Iwase, H.; Chandel, N.S.; Waypa, G.B.; Guzy, R.D.; Vanden Hoek, T.L.; Schumacker, P.T. Mitochondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim Biophys Acta 2011, 1813, 1382-1394. [CrossRef]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab 2005, 1, 393-399. [CrossRef]
- Mironov, S.L.; Langohr, K. Modulation of synaptic and channel activities in the respiratory network of the mice by NO/cGMP signalling pathways. Brain Res 2007, 1130, 73-82. [CrossRef]
- Na, N.; Chandel, N.S.; Litvan, J.; Ridge, K.M. Mitochondrial reactive oxygen species are required for hypoxia-induced degradation of keratin intermediate filaments. FASEB J 2010, 24, 799-809. [CrossRef]
- Schafer, M.; Schafer, C.; Ewald, N.; Piper, H.M.; Noll, T. Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circ Res 2003, 92, 1010-1015. [CrossRef]
- Debski, D.; Smulik, R.; Zielonka, J.; Michalowski, B.; Jakubowska, M.; Debowska, K.; Adamus, J.; Marcinek, A.; Kalyanaraman, B.; Sikora, A. Mechanism of oxidative conversion of Amplex(R) Red to resorufin: Pulse radiolysis and enzymatic studies. Free Radic Biol Med 2016, 95, 323-332. [CrossRef]
- Ferrer-Sueta, G.; Radi, R. Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem Biol 2009, 4, 161-177. [CrossRef]
- Eid, M.; Barayeu, U.; Dick, T.P. Chemogenetic detection and quantitation of H(2)O(2) in living cells. Nat Protoc 2025. [CrossRef]
- Gupte, S.A.; Li, K.X.; Okada, T.; Sato, K.; Oka, M. Inhibitors of pentose phosphate pathway cause vasodilation: involvement of voltage-gated potassium channels. J Pharmacol Exp Ther 2002, 301, 299-305. [CrossRef]
- Burke-Wolin, T.; Wolin, M.S. H2O2 and cGMP may function as an O2 sensor in the pulmonary artery. J Appl Physiol (1985) 1989, 66, 167-170. [CrossRef]
- Burke-Wolin, T.M.; Wolin, M.S. Inhibition of cGMP-associated pulmonary arterial relaxation to H2O2 and O2 by ethanol. Am J Physiol 1990, 258, H1267-1273. [CrossRef]
- Omar, H.A.; Wolin, M.S. Endothelium-dependent and independent cGMP mechanisms appear to mediate O2 responses in calf pulmonary resistance arteries. Am J Physiol 1992, 262, L560-565. [CrossRef]
- Mohazzab, K.M.; Wolin, M.S. Sites of superoxide anion production detected by lucigenin in calf pulmonary artery smooth muscle. Am J Physiol 1994, 267, L815-822. [CrossRef]
- Monaco, J.A.; Burke-Wolin, T. NO and H2O2 mechanisms of guanylate cyclase activation in oxygen-dependent responses of rat pulmonary circulation. Am J Physiol 1995, 268, L546-550. [CrossRef]
- Wolin, M.S.; Burke-Wolin, T.M.; Mohazzab, H.K. Roles for NAD(P)H oxidases and reactive oxygen species in vascular oxygen sensing mechanisms. Respir Physiol 1999, 115, 229-238. [CrossRef]
- Cross, A.R.; Jones, O.T. Enzymic mechanisms of superoxide production. Biochim Biophys Acta 1991, 1057, 281-298. [CrossRef]
- Cross, A.R.; Henderson, L.; Jones, O.T.; Delpiano, M.A.; Hentschel, J.; Acker, H. Involvement of an NAD(P)H oxidase as a pO2 sensor protein in the rat carotid body. Biochem J 1990, 272, 743-747. [CrossRef]
- Griendling, K.K.; Ushio-Fukai, M. NADH/NADPH Oxidase and Vascular Function. Trends Cardiovasc Med 1997, 7, 301-307. [CrossRef]
- Lambeth, J.D.; Cheng, G.; Arnold, R.S.; Edens, W.A. Novel homologs of gp91phox. Trends Biochem Sci 2000, 25, 459-461. [CrossRef]
- Gupte, S.A.; Kaminski, P.M.; Floyd, B.; Agarwal, R.; Ali, N.; Ahmad, M.; Edwards, J.; Wolin, M.S. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol 2005, 288, H13-21. [CrossRef]
- Gupte, S.A.; Rupawalla, T.; Phillibert, D., Jr.; Wolin, M.S. NADPH and heme redox modulate pulmonary artery relaxation and guanylate cyclase activation by NO. Am J Physiol 1999, 277, L1124-1132. [CrossRef]
- Gupte, S.A.; Arshad, M.; Viola, S.; Kaminski, P.M.; Ungvari, Z.; Rabbani, G.; Koller, A.; Wolin, M.S. Pentose phosphate pathway coordinates multiple redox-controlled relaxing mechanisms in bovine coronary arteries. Am J Physiol Heart Circ Physiol 2003, 285, H2316-2326. [CrossRef]
- Mohazzab, H.K.; Kaminski, P.M.; Fayngersh, R.P.; Wolin, M.S. Oxygen-elicited responses in calf coronary arteries: role of H2O2 production via NADH-derived superoxide. Am J Physiol 1996, 270, H1044-1053. [CrossRef]
- Gupte, S.A.; Wolin, M.S. Hypoxia promotes relaxation of bovine coronary arteries through lowering cytosolic NADPH. Am J Physiol Heart Circ Physiol 2006, 290, H2228-2238. [CrossRef]
- Mallet, R.T.; Olivencia-Yurvati, A.H.; Bunger, R. Pyruvate enhancement of cardiac performance: Cellular mechanisms and clinical application. Exp Biol Med (Maywood) 2018, 243, 198-210. [CrossRef]
- Wolin, M.S.; Gupte, S.A.; Mingone, C.J.; Neo, B.H.; Gao, Q.; Ahmad, M. Redox regulation of responses to hypoxia and NO-cGMP signaling in pulmonary vascular pathophysiology. Ann N Y Acad Sci 2010, 1203, 126-132. [CrossRef]
- Mingone, C.J.; Gupte, S.A.; Ali, N.; Oeckler, R.A.; Wolin, M.S. Thiol oxidation inhibits nitric oxide-mediated pulmonary artery relaxation and guanylate cyclase stimulation. Am J Physiol Lung Cell Mol Physiol 2006, 290, L549-557. [CrossRef]
- Wolin, M.S.; Ahmad, M.; Gupte, S.A. Oxidant and redox signaling in vascular oxygen sensing mechanisms: basic concepts, current controversies, and potential importance of cytosolic NADPH. Am J Physiol Lung Cell Mol Physiol 2005, 289, L159-173. [CrossRef]
- Wolin, M.S. Reactive oxygen species and the control of vascular function. Am J Physiol Heart Circ Physiol 2009, 296, H539-549. [CrossRef]
- Pandolfi, P.P.; Sonati, F.; Rivi, R.; Mason, P.; Grosveld, F.; Luzzatto, L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J 1995, 14, 5209-5215. [CrossRef]
- Gupte, R.S.; Ata, H.; Rawat, D.; Abe, M.; Taylor, M.S.; Ochi, R.; Gupte, S.A. Glucose-6-phosphate dehydrogenase is a regulator of vascular smooth muscle contraction. Antioxid Redox Signal 2011, 14, 543-558. [CrossRef]
- Neo, B.H.; Kandhi, S.; Ahmad, M.; Wolin, M.S. Redox regulation of guanylate cyclase and protein kinase G in vascular responses to hypoxia. Respir Physiol Neurobiol 2010, 174, 259-264. [CrossRef]
- Wolin, M.S.; Burke, T.M. Hydrogen peroxide elicits activation of bovine pulmonary arterial soluble guanylate cyclase by a mechanism associated with its metabolism by catalase. Biochem Biophys Res Commun 1987, 143, 20-25. [CrossRef]
- Mingone, C.J.; Gupte, S.A.; Iesaki, T.; Wolin, M.S. Hypoxia enhances a cGMP-independent nitric oxide relaxing mechanism in pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 2003, 285, L296-304. [CrossRef]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schoneich, C.; Cohen, R.A. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med 2004, 10, 1200-1207. [CrossRef]
- Burgoyne, J.R.; Oka, S.; Ale-Agha, N.; Eaton, P. Hydrogen peroxide sensing and signaling by protein kinases in the cardiovascular system. Antioxid Redox Signal 2013, 18, 1042-1052. [CrossRef]
- Meurer, S.; Pioch, S.; Pabst, T.; Opitz, N.; Schmidt, P.M.; Beckhaus, T.; Wagner, K.; Matt, S.; Gegenbauer, K.; Geschka, S.; et al. Nitric oxide-independent vasodilator rescues heme-oxidized soluble guanylate cyclase from proteasomal degradation. Circ Res 2009, 105, 33-41. [CrossRef]
- Neo, B.H.; Patel, D.; Kandhi, S.; Wolin, M.S. Roles for cytosolic NADPH redox in regulating pulmonary artery relaxation by thiol oxidation-elicited subunit dimerization of protein kinase G1alpha. Am J Physiol Heart Circ Physiol 2013, 305, H330-343. [CrossRef]
- Chettimada, S.; Rawat, D.K.; Dey, N.; Kobelja, R.; Simms, Z.; Wolin, M.S.; Lincoln, T.M.; Gupte, S.A. Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery. Am J Physiol Lung Cell Mol Physiol 2012, 303, L64-74. [CrossRef]
- Daiber, A.; Oelze, M.; Steven, S.; Kroller-Schon, S.; Munzel, T. Taking up the cudgels for the traditional reactive oxygen and nitrogen species detection assays and their use in the cardiovascular system. Redox Biol 2017, 12, 35-49. [CrossRef]
- Hyman, A.L.; Lippton, H.L.; Kadowitz, P.J. Methylene blue prevents hypoxic pulmonary vasoconstriction in cats. Am J Physiol 1991, 260, H586-592. [CrossRef]
- Cremona, G.; Wood, A.M.; Hall, L.W.; Bower, E.A.; Higenbottam, T. Effect of inhibitors of nitric oxide release and action on vascular tone in isolated lungs of pig, sheep, dog and man. J Physiol 1994, 481 ( Pt 1), 185-195. [CrossRef]
- Dumas, J.P.; Dumas, M.; Sgro, C.; Advenier, C.; Giudicelli, J.F. Effects of two K+ channel openers, aprikalim and pinacidil, on hypoxic pulmonary vasoconstriction. Eur J Pharmacol 1994, 263, 17-23. [CrossRef]
- Mazmanian, G.M.; Baudet, B.; Brink, C.; Cerrina, J.; Kirkiacharian, S.; Weiss, M. Methylene blue potentiates vascular reactivity in isolated rat lungs. J Appl Physiol (1985) 1989, 66, 1040-1045. [CrossRef]
- Fouty, B.; Komalavilas, P.; Muramatsu, M.; Cohen, A.; McMurtry, I.F.; Lincoln, T.M.; Rodman, D.M. Protein kinase G is not essential to NO-cGMP modulation of basal tone in rat pulmonary circulation. Am J Physiol 1998, 274, H672-678. [CrossRef]
- Rodman, D.M.; Yamaguchi, T.; O'Brien, R.F.; McMurtry, I.F. Methylene blue enhances hypoxic contraction in isolated rat pulmonary arteries. Chest 1988, 93, 93S-94S. [CrossRef]
- Vermeersch, P.; Buys, E.; Pokreisz, P.; Marsboom, G.; Ichinose, F.; Sips, P.; Pellens, M.; Gillijns, H.; Swinnen, M.; Graveline, A.; et al. Soluble guanylate cyclase-alpha1 deficiency selectively inhibits the pulmonary vasodilator response to nitric oxide and increases the pulmonary vascular remodeling response to chronic hypoxia. Circulation 2007, 116, 936-943. [CrossRef]
- Spohr, F.; Busch, C.J.; Teschendorf, P.; Weimann, J. Selective inhibition of guanylate cyclase prevents impairment of hypoxic pulmonary vasoconstriction in endotoxemic mice. J Physiol Pharmacol 2009, 60, 107-112.
- Rodman, D.M.; Yamaguchi, T.; Hasunuma, K.; O'Brien, R.F.; McMurtry, I.F. Effects of hypoxia on endothelium-dependent relaxation of rat pulmonary artery. Am J Physiol 1990, 258, L207-214. [CrossRef]
- Ye, L.; Liu, J.; Liu, H.; Ying, L.; Dou, D.; Chen, Z.; Xu, X.; Raj, J.U.; Gao, Y. Sulfhydryl-dependent dimerization of soluble guanylyl cyclase modulates the relaxation of porcine pulmonary arteries to nitric oxide. Pflugers Arch 2013, 465, 333-341. [CrossRef]
- Harteneck, C.; Koesling, D.; Soling, A.; Schultz, G.; Bohme, E. Expression of soluble guanylyl cyclase. Catalytic activity requires two enzyme subunits. FEBS Lett 1990, 272, 221-223. [CrossRef]
- Le Cras, T.D.; McMurtry, I.F. Nitric oxide production in the hypoxic lung. Am J Physiol Lung Cell Mol Physiol 2001, 280, L575-582. [CrossRef]
- Manoury, B.; Idres, S.; Leblais, V.; Fischmeister, R. Ion channels as effectors of cyclic nucleotide pathways: Functional relevance for arterial tone regulation. Pharmacol Ther 2020, 209, 107499. [CrossRef]
- Rudyk, O.; Prysyazhna, O.; Burgoyne, J.R.; Eaton, P. Nitroglycerin fails to lower blood pressure in redox-dead Cys42Ser PKG1alpha knock-in mouse. Circulation 2012, 126, 287-295. [CrossRef]
- Prysyazhna, O.; Rudyk, O.; Eaton, P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat Med 2012, 18, 286-290. [CrossRef]
- Rudyk, O.; Rowan, A.; Prysyazhna, O.; Krasemann, S.; Hartmann, K.; Zhang, M.; Shah, A.M.; Ruppert, C.; Weiss, A.; Schermuly, R.T.; et al. Oxidation of PKGIalpha mediates an endogenous adaptation to pulmonary hypertension. Proc Natl Acad Sci U S A 2019, 116, 13016-13025. [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Gruning, N.M.; Kruger, A.; Tauqeer Alam, M.; et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc 2015, 90, 927-963. [CrossRef]
- Mullarky, E.; Cantley, L.C. Diverting Glycolysis to Combat Oxidative Stress. In Innovative Medicine: Basic Research and Development, Nakao, K., Minato, N., Uemoto, S., Eds.; Tokyo, 2015; pp. 3-23.
- Gupte, R.; Dhagia, V.; Rocic, P.; Ochi, R.; Gupte, S.A. Glucose-6-phosphate dehydrogenase increases Ca(2+) currents by interacting with Ca(v)1.2 and reducing intrinsic inactivation of the L-type calcium channel. Am J Physiol Heart Circ Physiol 2020, 319, H144-H158. [CrossRef]
- Chang, H.H.; Bennett, A.M.; Cameron, W.D.; Floro, E.; Au, A.; McFaul, C.M.; Yip, C.M.; Rocheleau, J.V. Targeting Apollo-NADP(+) to Image NADPH Generation in Pancreatic Beta-Cell Organelles. ACS Sens 2022, 7, 3308-3317. [CrossRef]
- Han, H.H.; Ge, P.X.; Li, W.J.; Hu, X.L.; He, X.P. Recent Advancement in Fluorescent Probes for Peroxynitrite (ONOO(-)). Sensors (Basel) 2025, 25. [CrossRef]
- Hung, Y.P.; Albeck, J.G.; Tantama, M.; Yellen, G. Imaging cytosolic NADH-NAD(+) redox state with a genetically encoded fluorescent biosensor. Cell Metab 2011, 14, 545-554. [CrossRef]
- Zanetti, C.; Gaspar, R.D.L.; Zhdanov, A.V.; Maguire, N.M.; Joyce, S.A.; Collins, S.G.; Maguire, A.R.; Papkovsky, D.B. Heterosubstituted Derivatives of PtPFPP for O(2) Sensing and Cell Analysis: Structure-Activity Relationships. Bioconjug Chem 2022, 33, 2161-2169. [CrossRef]
Figure 1.
Oxidative modifications of reactive cysteines. Deprotonated cysteine thiols (thiolates) can react with a variety of oxidizing species, leading to the posttranslational modifications which can affect protein function. Reactions of thiolates with glutathione (GSH), reactive sulfur species (RSS) and nitric oxide (NO) leads to their (a) glutathionylation, (b) persulfidation and (c) nitrosylation, which can further lead (d) to a thiol exchange reaction leading to glutathionylation or the formation of an intra-or inter-protein disulfide bond. Thiolate oxidation by H2O2 can also lead to the formation of intra-or inter-protein disulfide bonds (e and f, respectively). The reaction of H2O2 with thiolates can also produce sulfenic acid (g). Although not shown in the diagram, cysteine sulfenic acid can react with GSH, RSS and NO, leading to its glutathionylation, persulfidation and nitrosylation respectively, and can also react with thiols to form disulfide bonds [83] (not shown). Notably, cysteine sulfenic acid can be formed (j) by the oxidation of thiols with peroxynitrite (ONOO-), or by the reaction of peroxynitrous acid (ONOOH) with thiolates. These oxidative modifications can be reversed by cellular antioxidant mechanisms. Under more highly oxidizing conditions, sulfenic acid is further converted to (h) sulfinic acid and then (i) sulfonic acid; these reactions are largely irreversible. The types of oxidative modifications of protein tholates depicted in green have been shown to alter the activities of many proteins (e.g.[84]).
Figure 1.
Oxidative modifications of reactive cysteines. Deprotonated cysteine thiols (thiolates) can react with a variety of oxidizing species, leading to the posttranslational modifications which can affect protein function. Reactions of thiolates with glutathione (GSH), reactive sulfur species (RSS) and nitric oxide (NO) leads to their (a) glutathionylation, (b) persulfidation and (c) nitrosylation, which can further lead (d) to a thiol exchange reaction leading to glutathionylation or the formation of an intra-or inter-protein disulfide bond. Thiolate oxidation by H2O2 can also lead to the formation of intra-or inter-protein disulfide bonds (e and f, respectively). The reaction of H2O2 with thiolates can also produce sulfenic acid (g). Although not shown in the diagram, cysteine sulfenic acid can react with GSH, RSS and NO, leading to its glutathionylation, persulfidation and nitrosylation respectively, and can also react with thiols to form disulfide bonds [83] (not shown). Notably, cysteine sulfenic acid can be formed (j) by the oxidation of thiols with peroxynitrite (ONOO-), or by the reaction of peroxynitrous acid (ONOOH) with thiolates. These oxidative modifications can be reversed by cellular antioxidant mechanisms. Under more highly oxidizing conditions, sulfenic acid is further converted to (h) sulfinic acid and then (i) sulfonic acid; these reactions are largely irreversible. The types of oxidative modifications of protein tholates depicted in green have been shown to alter the activities of many proteins (e.g.[84]).
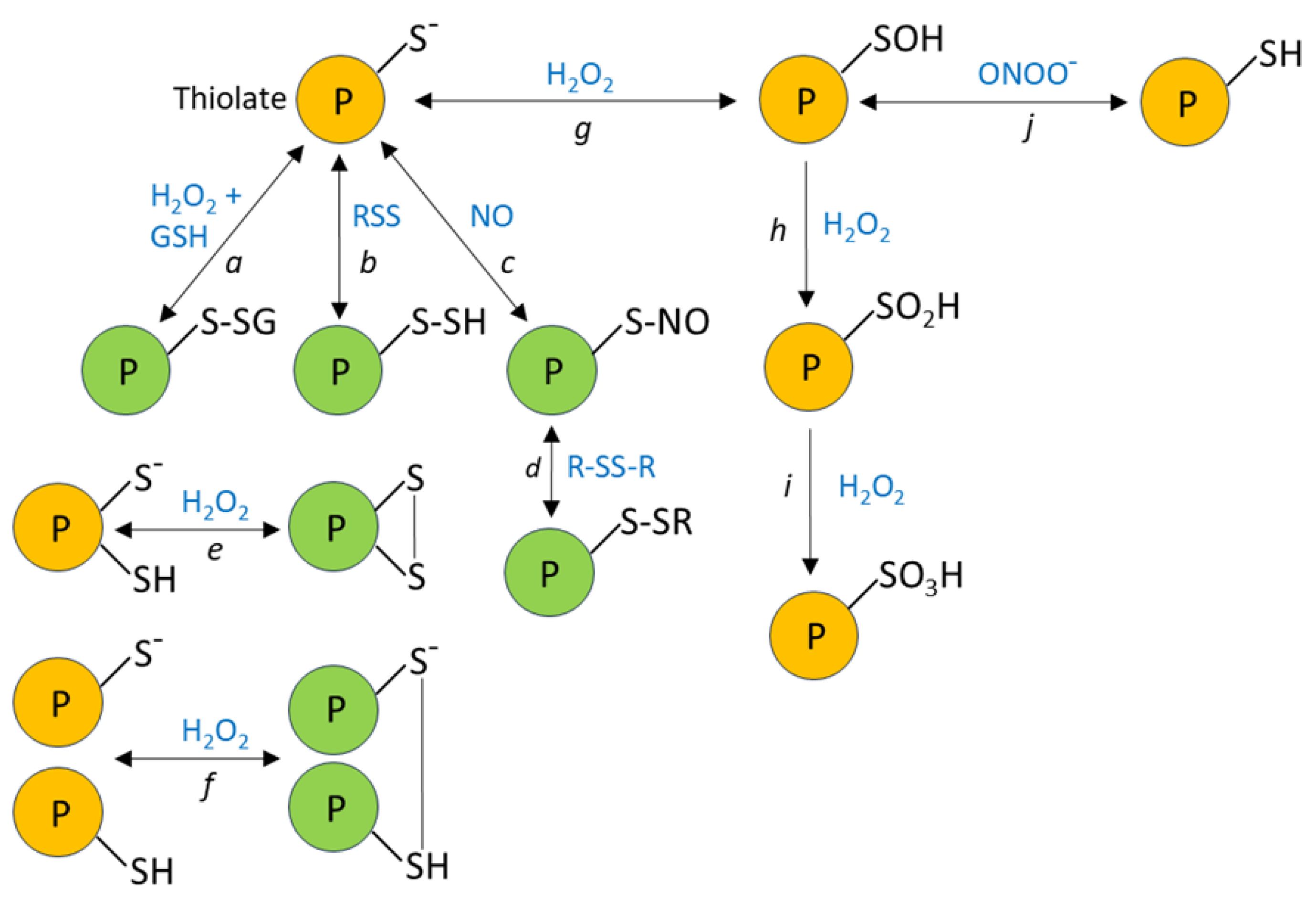
Figure 2.
Antioxidant mechanisms. Superoxide spontaneously dismutates to H2O2 at a high rate, but this process is reinforced by cellular (and extracellular) SODs. Once formed H2O2 is thought to have a half-life of ~10 s. It is reduced to H2O, most importantly by peroxiredoxins (Prxs), with the oxidizing equivalent then being passed ultimately to NADPH via thioredoxin (Trx) and thioredoxin reductase (TrxR). H2O2 is also reduced by glutathione peroxidase, which also passes the oxidizing equivalent to NADPH, in this case via glutathione peroxidase (Gpx), glutaredoxin (Grx), glutathione (GSH) and glutathione reductase (Gr). NADP+ is reduced back to NADPH by several enzymes, including malic enzyme (ME) and isocitrase dehydrogenase (IDH) in the mitochondria and by glucose-6-phosphate-dehydrogenase (G-6-PD) and also 6- phosphogluconate dehydrogenase in the cytoplasm. NADP+ reduction to NADPH in the mitochondria is also coupled to the oxidation of NADH to NAD+ by the NAM nucleotide transhydrogenase in the inner mitochondrial membrane, a reaction which is driven by the protein gradient across this membrane (see Section 3.1.1.1). These processes ensure that the NADP+: NADPH redox couple is very highly reduced in both cytoplasm and mitochondria [87], thereby maintaining the effectiveness of the NADPH requiring antioxidant systems. At higher concentrations, H2O2 is also scavenged by catalase, an enzyme mainly expressed in lysosomes. Moreover, Prx is present in micromolar concentrations in cells while H2O2 concentrations probably remain submicromolar during signaling [74]. Consequently, antioxidant mechanisms are predicted to scavenge H2O2 before it can react with thiolates on target proteins.
Figure 2.
Antioxidant mechanisms. Superoxide spontaneously dismutates to H2O2 at a high rate, but this process is reinforced by cellular (and extracellular) SODs. Once formed H2O2 is thought to have a half-life of ~10 s. It is reduced to H2O, most importantly by peroxiredoxins (Prxs), with the oxidizing equivalent then being passed ultimately to NADPH via thioredoxin (Trx) and thioredoxin reductase (TrxR). H2O2 is also reduced by glutathione peroxidase, which also passes the oxidizing equivalent to NADPH, in this case via glutathione peroxidase (Gpx), glutaredoxin (Grx), glutathione (GSH) and glutathione reductase (Gr). NADP+ is reduced back to NADPH by several enzymes, including malic enzyme (ME) and isocitrase dehydrogenase (IDH) in the mitochondria and by glucose-6-phosphate-dehydrogenase (G-6-PD) and also 6- phosphogluconate dehydrogenase in the cytoplasm. NADP+ reduction to NADPH in the mitochondria is also coupled to the oxidation of NADH to NAD+ by the NAM nucleotide transhydrogenase in the inner mitochondrial membrane, a reaction which is driven by the protein gradient across this membrane (see Section 3.1.1.1). These processes ensure that the NADP+: NADPH redox couple is very highly reduced in both cytoplasm and mitochondria [87], thereby maintaining the effectiveness of the NADPH requiring antioxidant systems. At higher concentrations, H2O2 is also scavenged by catalase, an enzyme mainly expressed in lysosomes. Moreover, Prx is present in micromolar concentrations in cells while H2O2 concentrations probably remain submicromolar during signaling [74]. Consequently, antioxidant mechanisms are predicted to scavenge H2O2 before it can react with thiolates on target proteins.
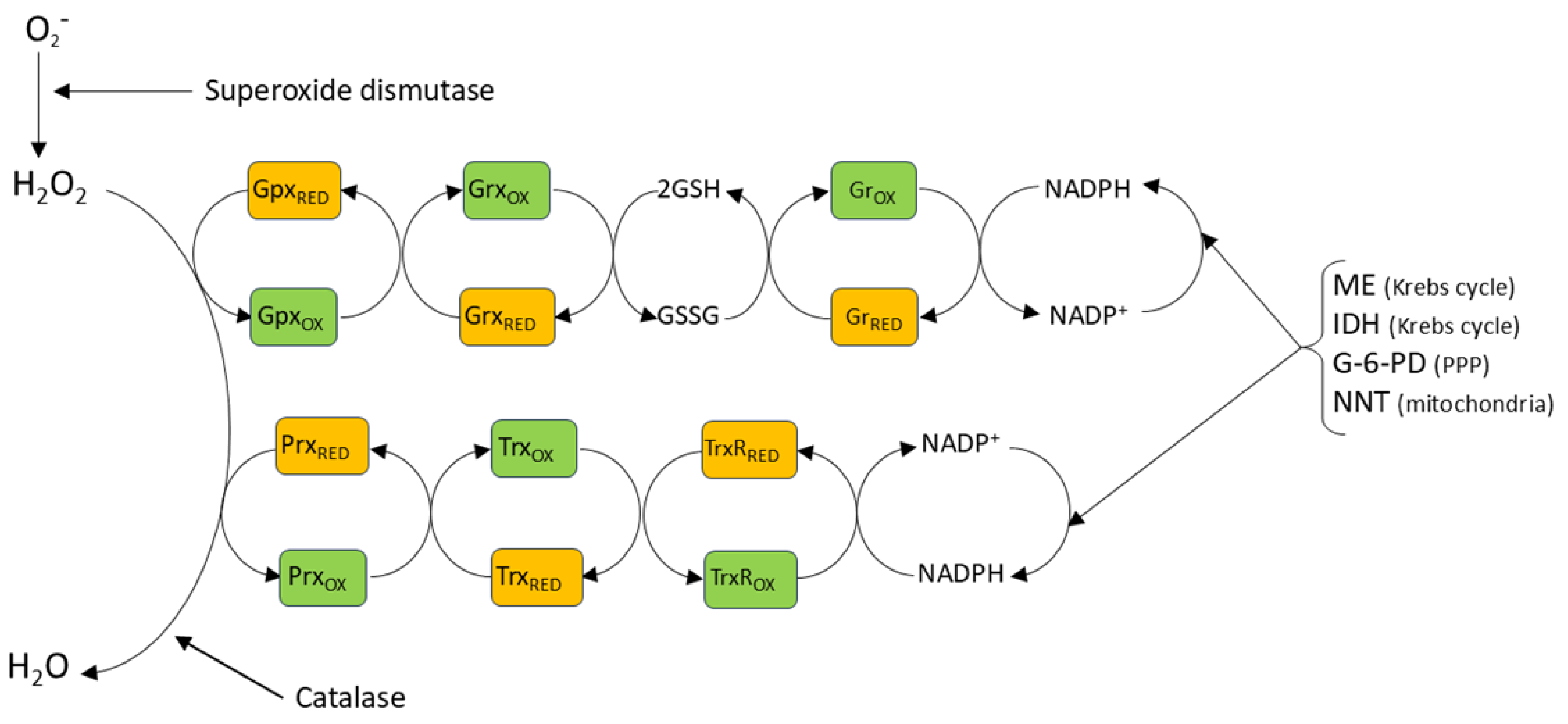
Figure 3.
Redox relays. Rather than oxidizing reactive cysteines directly, H2O2 generally does so indirectly via redox relays which transfer oxidizing equivalents from H2O2 (and also peroxynitrite) to signaling proteins (SP). Prxs can oxidize signaling proteins directly or can do so by passing an oxidizing equivalent to Trx, which is generally responsible for restoring several Prx isoforms to their reduced state following their oxidation. Oxidizing equivalents can also be passed from H2O2 to signaling proteins via glutathione peroxidase[93].
Figure 3.
Redox relays. Rather than oxidizing reactive cysteines directly, H2O2 generally does so indirectly via redox relays which transfer oxidizing equivalents from H2O2 (and also peroxynitrite) to signaling proteins (SP). Prxs can oxidize signaling proteins directly or can do so by passing an oxidizing equivalent to Trx, which is generally responsible for restoring several Prx isoforms to their reduced state following their oxidation. Oxidizing equivalents can also be passed from H2O2 to signaling proteins via glutathione peroxidase[93].
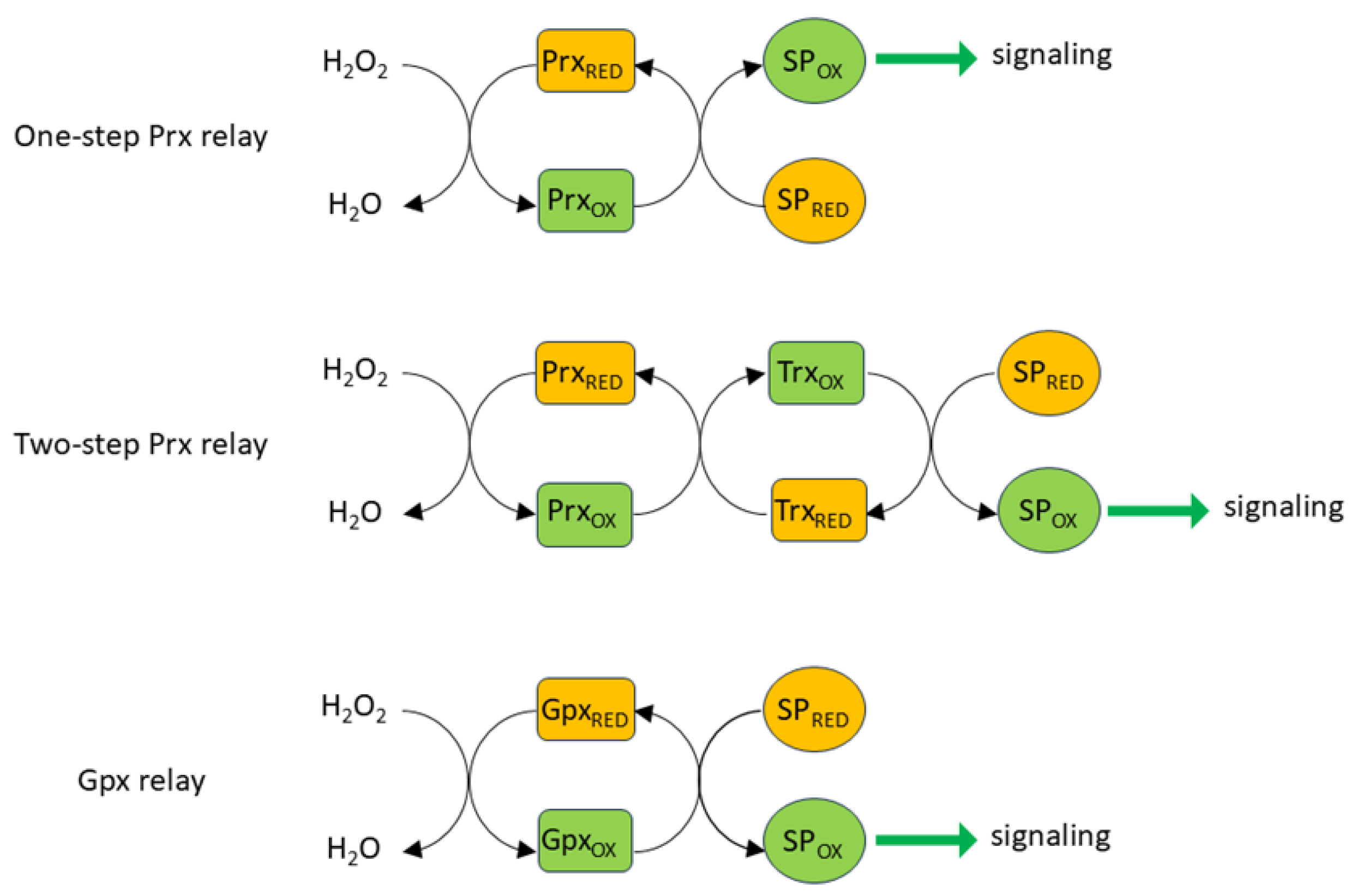
Figure 4.
Oxidative phosphorylation. The electron transport chain consists of four protein complexes (I -V). Complex V, the ATP synthase, is not part of the ETC per se. Complexes I and II accept electrons (e-) from NADH and FADH2, respectively, which have been reduced by reactions occurring during glycolysis, fatty acid metabolism and the Krebs cycle. The electrons from complexes I and II reduce co-enzyme Q (CoQ; ubiquinone) to ubiquinol (CoQH2). Electrons from CoQH2 are then passed to complex III, and then via (cyt c) to complex IV, where they combine with O2 to form H2O2. This process is driven by the successively less negative redox potentials of the complexes, and the energy lost by the electrons as they fall down this energetic redox gradient is used by complexes 1, 3 and 4 to create an electrochemical proton gradient across the inner mitochondrial wall (DP). The energy stored in DP is used by complex V to phosphorylate ADP, thereby producing ATP which is used by cells to fuel energy-requiring reactions.
Figure 4.
Oxidative phosphorylation. The electron transport chain consists of four protein complexes (I -V). Complex V, the ATP synthase, is not part of the ETC per se. Complexes I and II accept electrons (e-) from NADH and FADH2, respectively, which have been reduced by reactions occurring during glycolysis, fatty acid metabolism and the Krebs cycle. The electrons from complexes I and II reduce co-enzyme Q (CoQ; ubiquinone) to ubiquinol (CoQH2). Electrons from CoQH2 are then passed to complex III, and then via (cyt c) to complex IV, where they combine with O2 to form H2O2. This process is driven by the successively less negative redox potentials of the complexes, and the energy lost by the electrons as they fall down this energetic redox gradient is used by complexes 1, 3 and 4 to create an electrochemical proton gradient across the inner mitochondrial wall (DP). The energy stored in DP is used by complex V to phosphorylate ADP, thereby producing ATP which is used by cells to fuel energy-requiring reactions.
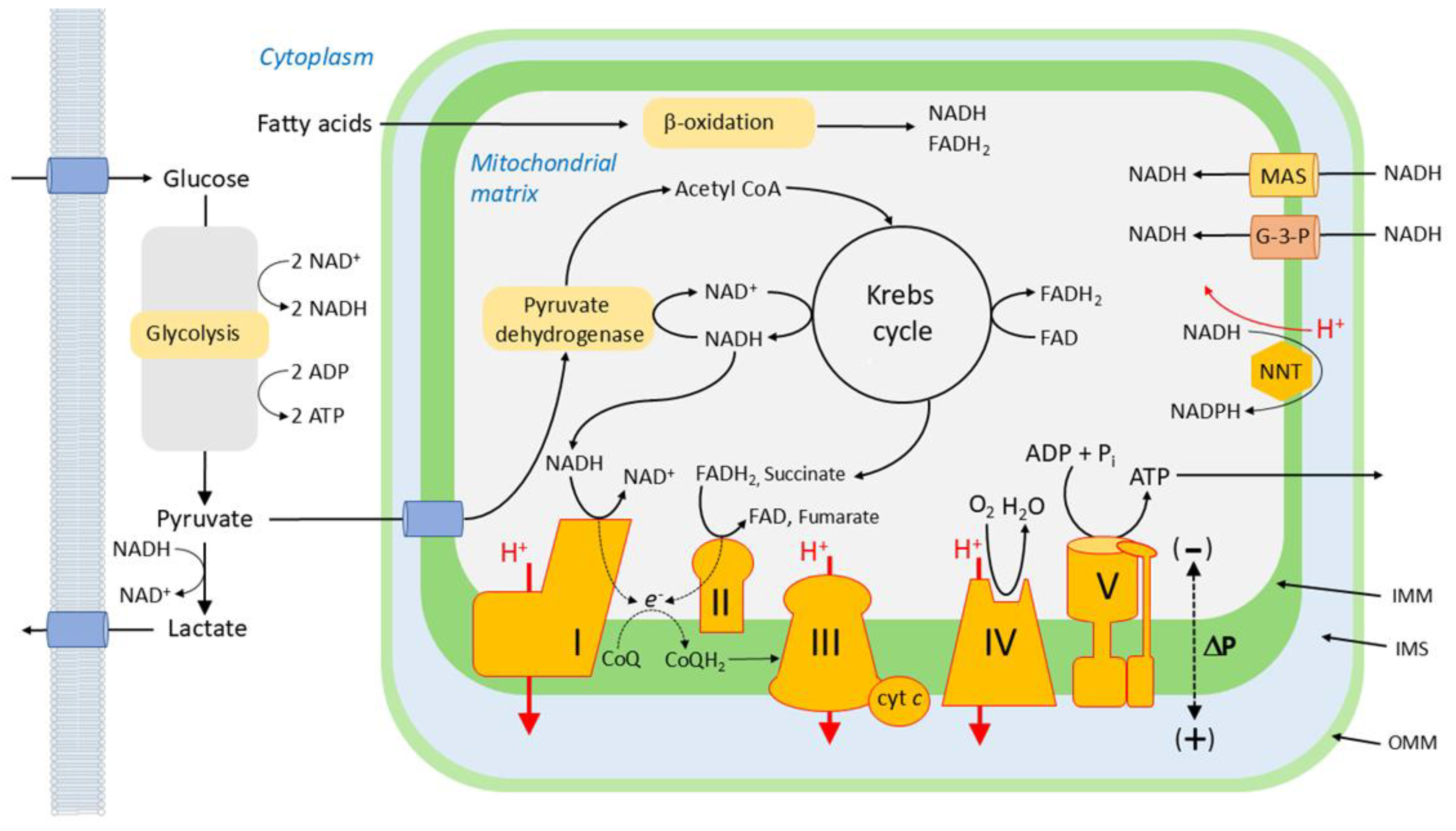
Figure 6.
The Redox theory of HPV. This is based on the concept that under normoxic conditions mitochondria in PASMCs are generating a higher level of ROS than in systemic VSMCs. The resulting higher level of H2O2 and/or oxidation of cytoplasmic redox couples activates KV channels, causing a relatively hyperpolarized Em which limits Ca2+ influx through voltage-gated Ca2+ channels, thereby contributing to a tonic vasodilation. Other as yet unidentified redox-regulated vasodilating pathways might also be similarly recruited by cytoplasmic oxidation. By diminishing mitochondrial ROS production, hypoxia decreases this vasodilating influence, causing an increase in vascular tone. See the text for further details.
Figure 6.
The Redox theory of HPV. This is based on the concept that under normoxic conditions mitochondria in PASMCs are generating a higher level of ROS than in systemic VSMCs. The resulting higher level of H2O2 and/or oxidation of cytoplasmic redox couples activates KV channels, causing a relatively hyperpolarized Em which limits Ca2+ influx through voltage-gated Ca2+ channels, thereby contributing to a tonic vasodilation. Other as yet unidentified redox-regulated vasodilating pathways might also be similarly recruited by cytoplasmic oxidation. By diminishing mitochondrial ROS production, hypoxia decreases this vasodilating influence, causing an increase in vascular tone. See the text for further details.
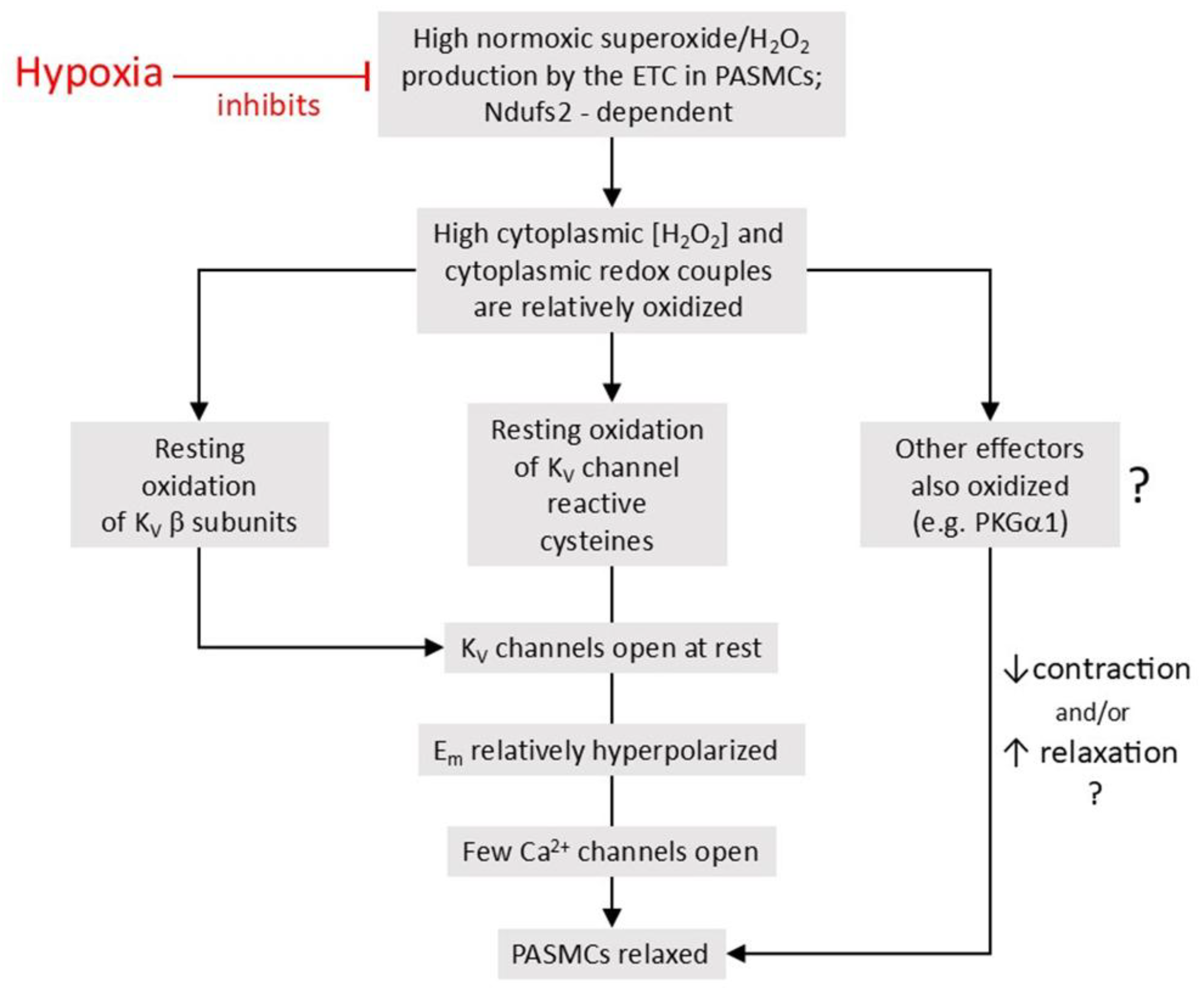
Figure 7.
The mitochondrial ROS hypothesis of HPV. According to this proposal, hypoxia causes an increased ROS production in the intermembrane space by complex III. This leads to a cytoplasmic ROS signal which causes contraction by by acting on several HPV effectors to increase SR Ca2+ release, Ca2+ influx and Ca2+ sensitization (see Section 5.2.5 for a brief discussion of these effectors and references supporting their ROS-sensitivity. A comprehensive description of the evidence implicating these effectors in HPV can be found in [3].
Figure 7.
The mitochondrial ROS hypothesis of HPV. According to this proposal, hypoxia causes an increased ROS production in the intermembrane space by complex III. This leads to a cytoplasmic ROS signal which causes contraction by by acting on several HPV effectors to increase SR Ca2+ release, Ca2+ influx and Ca2+ sensitization (see Section 5.2.5 for a brief discussion of these effectors and references supporting their ROS-sensitivity. A comprehensive description of the evidence implicating these effectors in HPV can be found in [3].
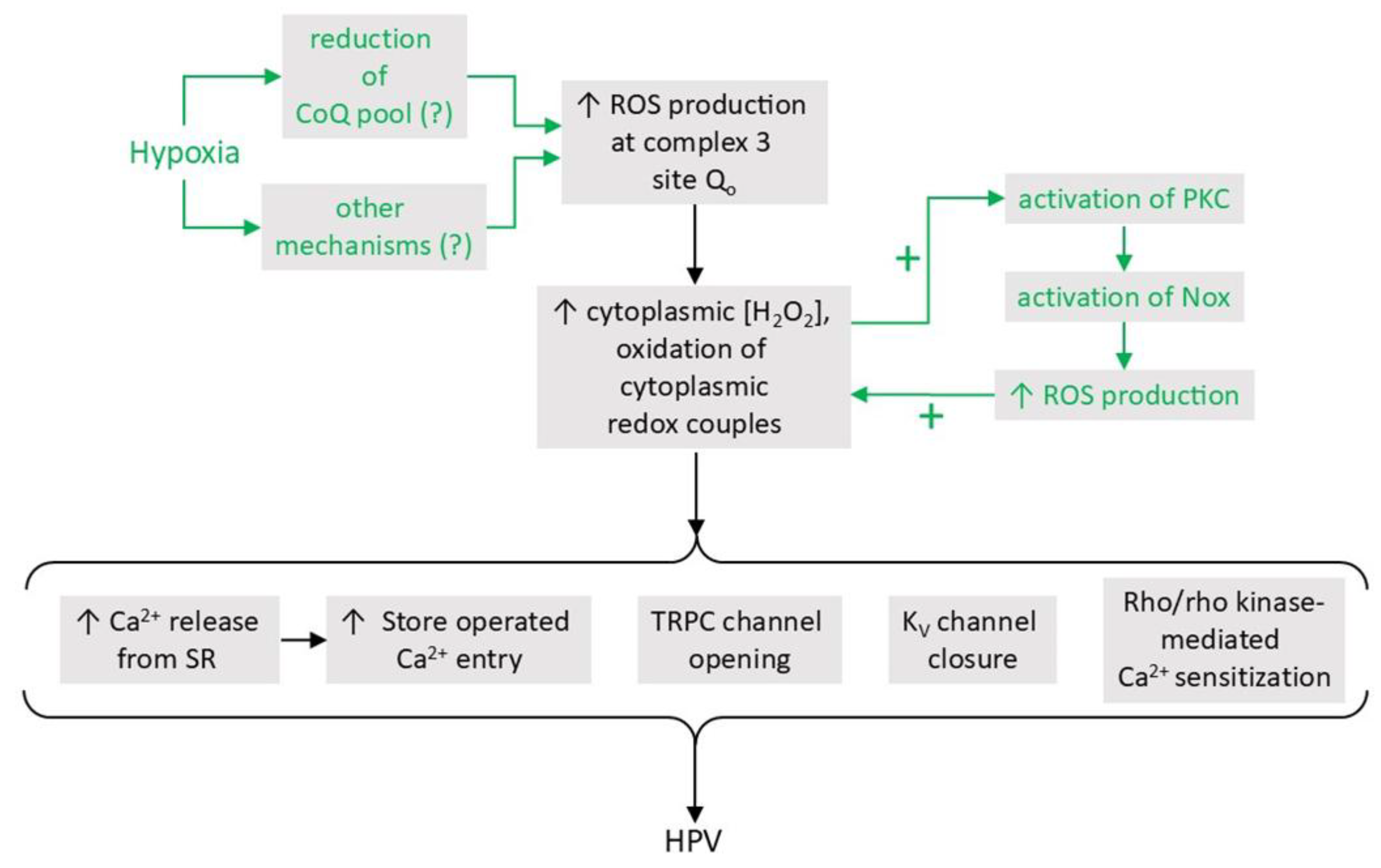
Figure 8.
A summary of the experimental evidence reported in Sommer et al [130]. The flow chart on the left shows Text in black shows results related to the characterization of HPV mechanisms, text in red shows the sequence of events proposed to cause HPV. The evidence supporting each step in the sequence is shown to the right in black font, and the red font shows the results of negative control experiments.
Figure 8.
A summary of the experimental evidence reported in Sommer et al [130]. The flow chart on the left shows Text in black shows results related to the characterization of HPV mechanisms, text in red shows the sequence of events proposed to cause HPV. The evidence supporting each step in the sequence is shown to the right in black font, and the red font shows the results of negative control experiments.
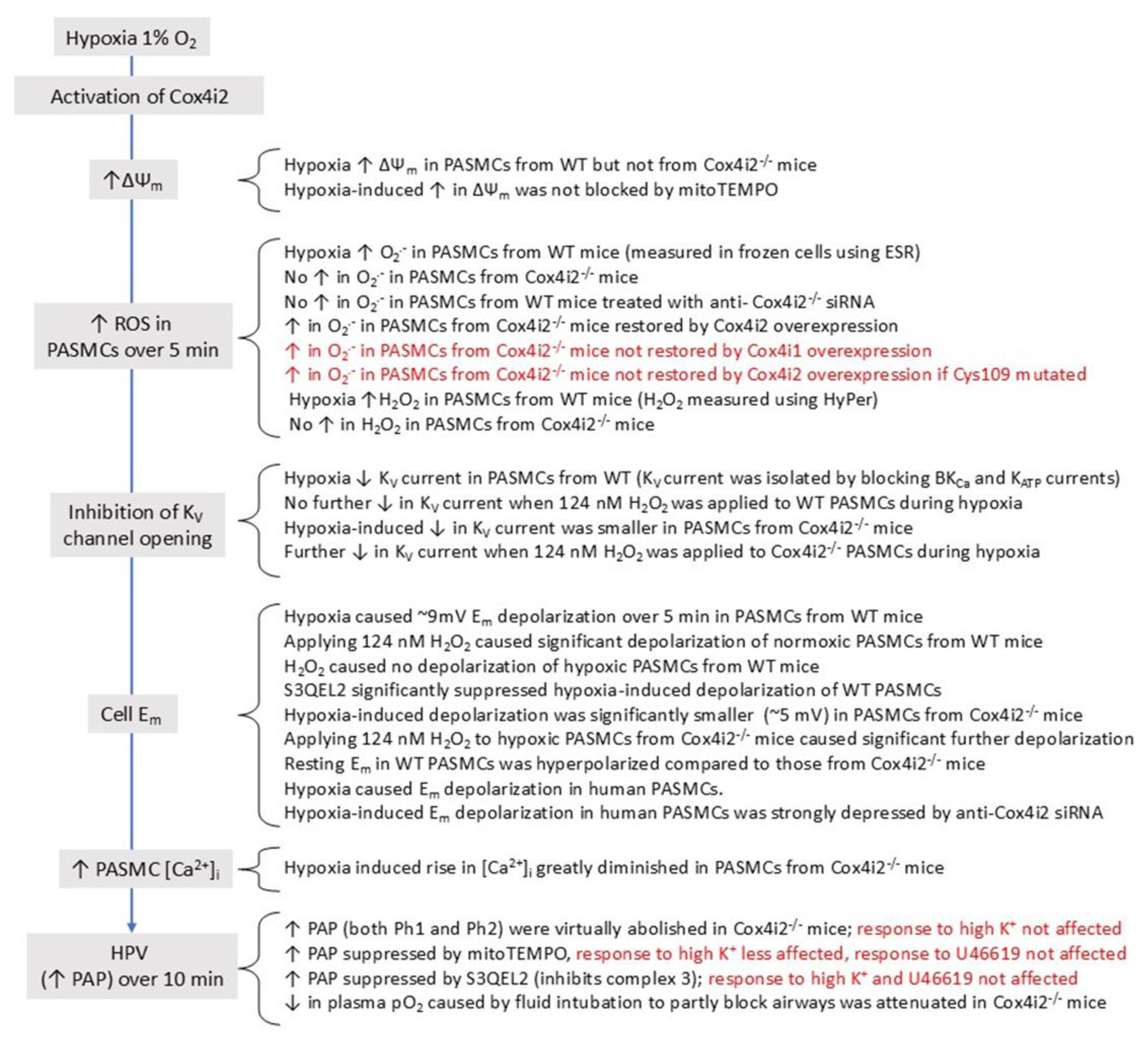
Figure 9.
The role of the PPP in HPV as described by Gupte et al [440]. According to their model, hypoxia induces changes in glucose metabolism and cytoplasmic [ROS]. This causes an increased activity of PPP which, acting through NADPH causes a further rise in [ROS]. This activates rho kinase and other pro-contractile pathways, evoking HPV. The figure is based closely on that presented in [440]. See the text for further details.
Figure 9.
The role of the PPP in HPV as described by Gupte et al [440]. According to their model, hypoxia induces changes in glucose metabolism and cytoplasmic [ROS]. This causes an increased activity of PPP which, acting through NADPH causes a further rise in [ROS]. This activates rho kinase and other pro-contractile pathways, evoking HPV. The figure is based closely on that presented in [440]. See the text for further details.
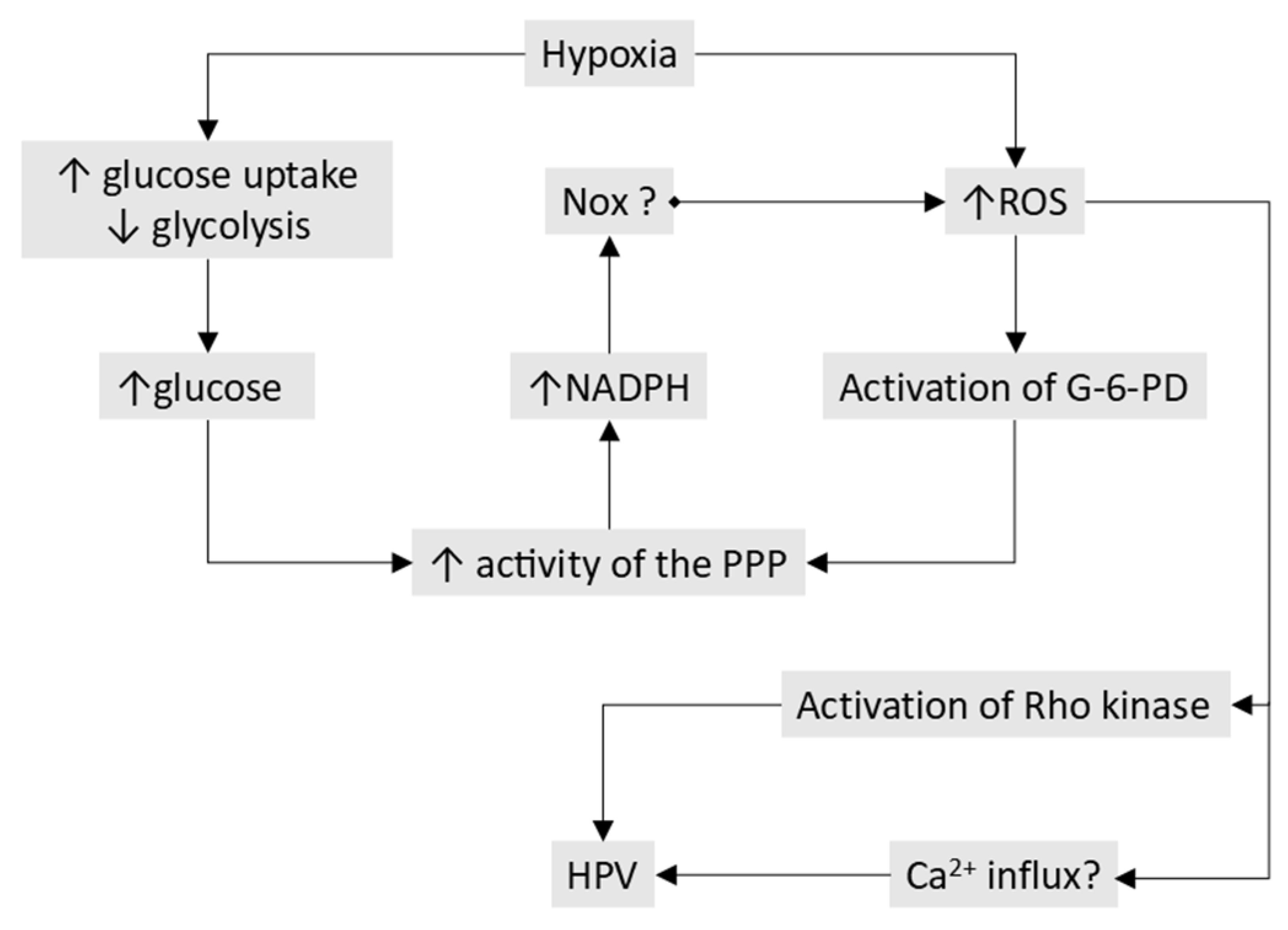
Figure 10.
Proposed role of PKGa1 and sGC in HPV. According to this model, HPV is largely due to the removal of a basal vasodilating mechanism which is caused by an ongoing activation of sGC and PKGa1. Hypoxia removes this influence via two pathways. Firstly, by activating the PPP, it increases the NADPH/NADP+ ratio. The increase in NADPH, causes the reduction of TrxR-1 and Trx-1, thereby reversing the resting oxidation and activation of PKGa1, diminishing its vasorelaxing influence. Secondly, the fall in PO2 decreases the ambient intracellular concentration of H2O2, further decreasing the activation of PKGa1 both directly by promoting its and indirectly by inhibiting sGC. This figure is largely based on information present in [572].
Figure 10.
Proposed role of PKGa1 and sGC in HPV. According to this model, HPV is largely due to the removal of a basal vasodilating mechanism which is caused by an ongoing activation of sGC and PKGa1. Hypoxia removes this influence via two pathways. Firstly, by activating the PPP, it increases the NADPH/NADP+ ratio. The increase in NADPH, causes the reduction of TrxR-1 and Trx-1, thereby reversing the resting oxidation and activation of PKGa1, diminishing its vasorelaxing influence. Secondly, the fall in PO2 decreases the ambient intracellular concentration of H2O2, further decreasing the activation of PKGa1 both directly by promoting its and indirectly by inhibiting sGC. This figure is largely based on information present in [572].
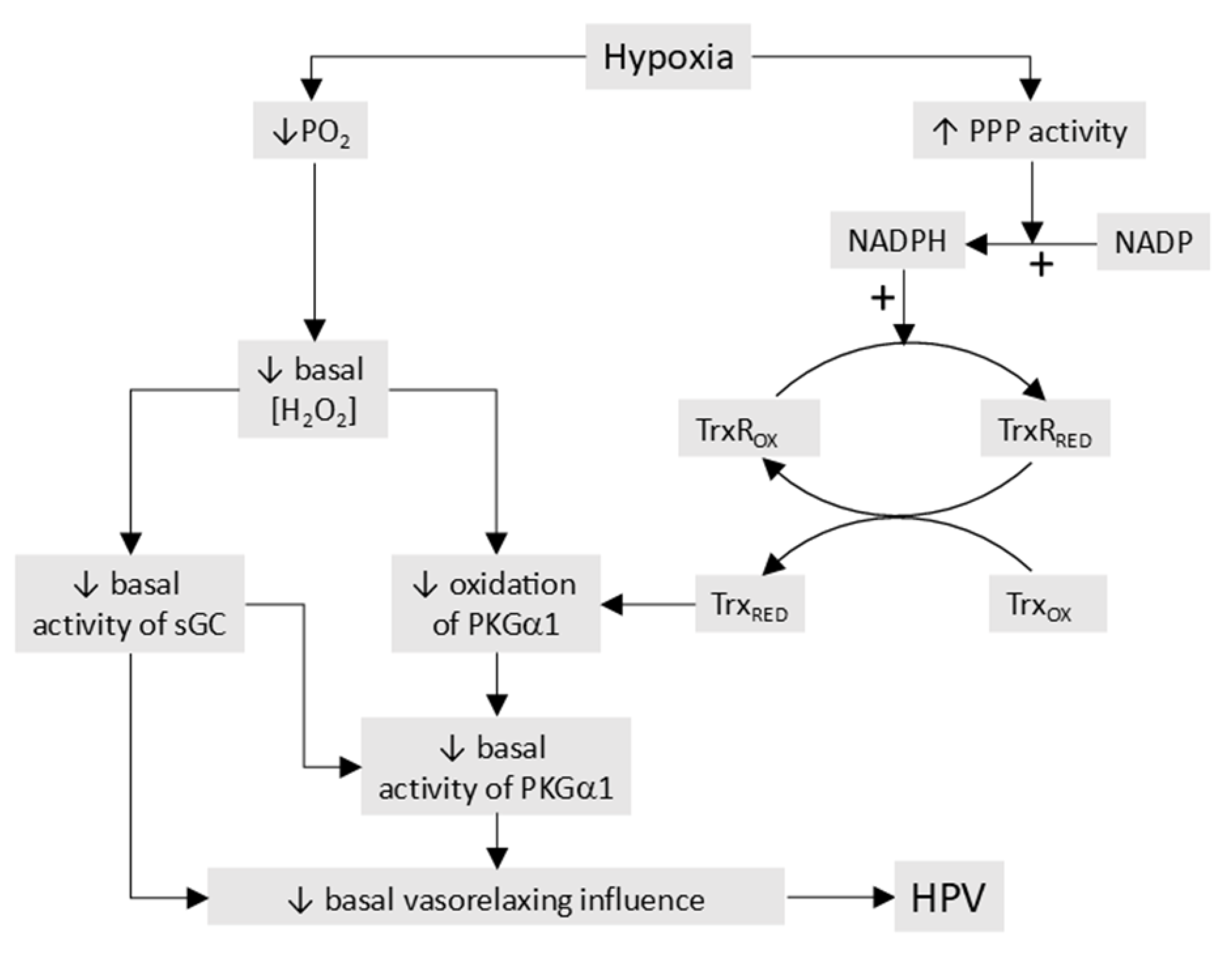
Figure 11.
HPV was evoked by gassing small endothelium-intact PA from rats in a conventional myograph with 95% N/5%CO2/0% O2 as previously described; the PO2 during hypoxia was shown to be 15-20 Torr under these conditions[10]. A low concentration of PGF2α was applied before hypoxia to create a small degree of pre-tone; this increases the amplitude of HPV, particularly the transient initial phase (phase 1). Application of DHEA significantly (p < 0.05 by 2- way ANOVA and also paired T-test) depressed the amplitude of phase 1 and also of phase 2, which is sustained. In the absence of pre-tone (in which case phase 1 is not evident), DHEA also virtually abolished HPV in 7 arteries (not shown) (taken from Prieto-Lloret et al, unpublished results).
Figure 11.
HPV was evoked by gassing small endothelium-intact PA from rats in a conventional myograph with 95% N/5%CO2/0% O2 as previously described; the PO2 during hypoxia was shown to be 15-20 Torr under these conditions[10]. A low concentration of PGF2α was applied before hypoxia to create a small degree of pre-tone; this increases the amplitude of HPV, particularly the transient initial phase (phase 1). Application of DHEA significantly (p < 0.05 by 2- way ANOVA and also paired T-test) depressed the amplitude of phase 1 and also of phase 2, which is sustained. In the absence of pre-tone (in which case phase 1 is not evident), DHEA also virtually abolished HPV in 7 arteries (not shown) (taken from Prieto-Lloret et al, unpublished results).
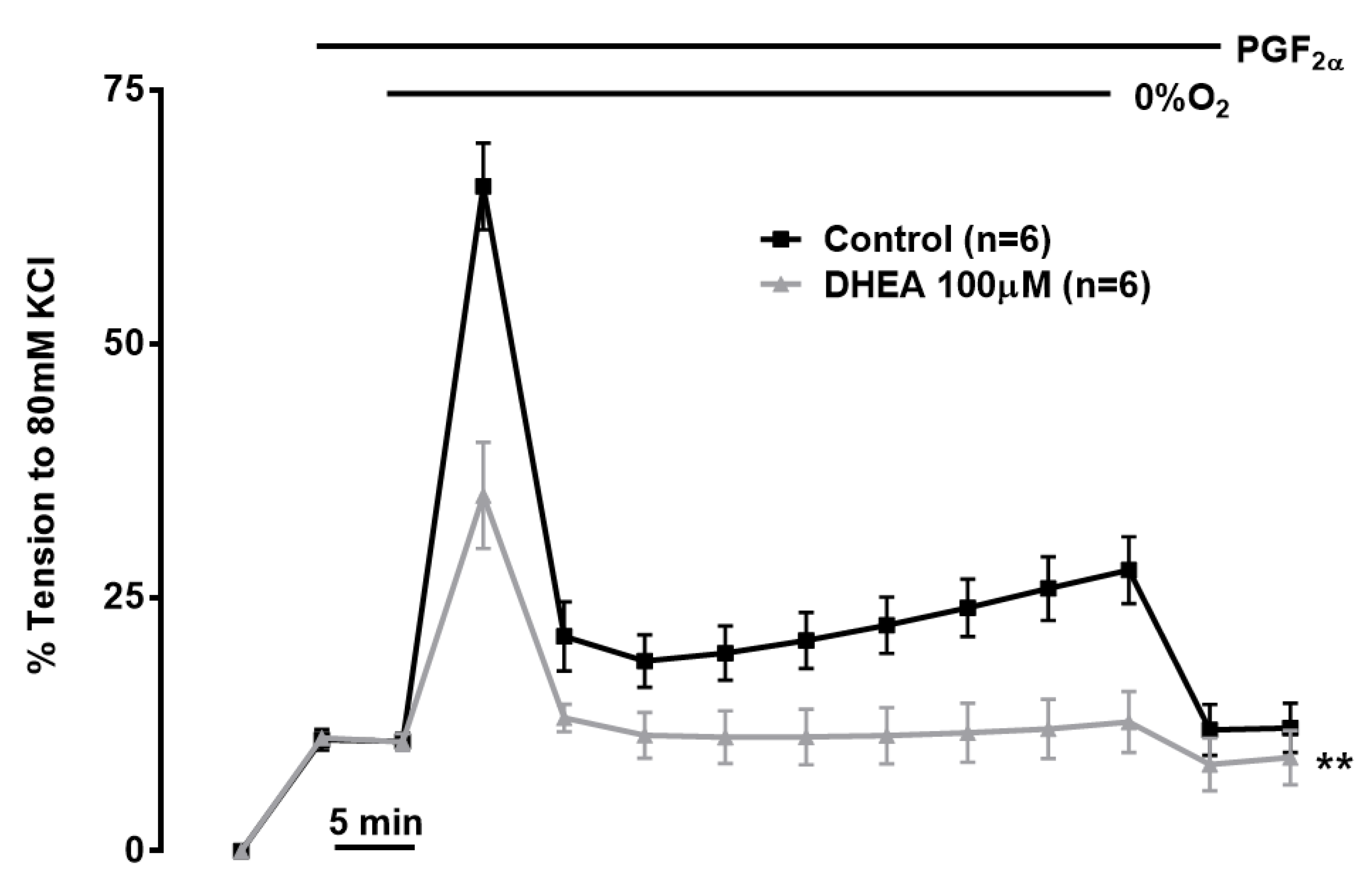
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



