Submitted:
24 December 2025
Posted:
25 December 2025
You are already at the latest version
Abstract
Parkinson’s disease (PD) is a biologically heterogeneous neurodegenerative disorder characterized by progressive motor and non-motor symptoms. Despite decades of research, no pharmacologic intervention has conclusively demonstrated disease-modifying efficacy in late-phase randomized controlled trials. Current strategies primarily target pathogenic mechanisms such as α-synuclein aggregation, mitochondrial dysfunction, impaired autophagy, and neuroinflammation, alongside genetic contributors including LRRK2 and GBA variants. Therapeutic approaches under investigation encompass monoclonal antibodies, small molecules, gene therapy, and metabolic modulators; however, pivotal trials of agents such as isradipine, cinpanemab, prasinezumab, exenatide, and inosine have failed to alter long-term progression, underscoring limitations in trial design, biomarker validation, and patient stratification. Repurposing of approved drugs—representing approximately one-third of disease-modifying therapy trials—offers a cost-efficient strategy, with candidates including GLP-1 receptor agonists, α1-adrenergic antagonists, and lysosomal enhancers such as ambroxol. Emerging paradigms emphasize biomarker-driven and genotype-enriched designs to improve internal validity and enable precision medicine, particularly for GBA- and LRRK2-associated PD. Despite these advances, major unmet needs persist, including validated progression biomarkers, robust adaptive trial frameworks, and strategies for early or prodromal intervention. This review synthesizes biological mechanisms, pharmacological strategies, and clinical pipeline trends, highlighting lessons from past failures and opportunities for translational innovation. Accelerating progress will require global platform trials, integration of molecular staging, and harmonization of regulatory pathways to bridge mechanistic insights with clinically meaningful outcomes.
Keywords:
Parkinson’s disease
; disease-modifying therapy
; α-synuclein aggregation
; neuroinflammation
; biomarker-driven trials
; drug repurposing
; precision medicine
; genotype-enriched design
; platform trials
; gene therapy
1. Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by motor and non-motor symptoms arising from selective dopaminergic neuronal loss in the substantia nigra and widespread synaptic dysfunction. Increasing evidence positions PD as a biologically heterogeneous syndrome rather than a single disease entity, encompassing diverse clinical phenotypes such as tremor-predominant and gait-dominant forms, REM sleep behavior disorder, and variable cognitive trajectories [1]. This heterogeneity is further amplified by genetic variability, with pathogenic variants in LRRK2, GBA, and other loci influencing disease onset, progression, and therapeutic response [2]. Neuropathological diversity adds complexity. While Lewy body deposition remains a hallmark of idiopathic PD, some LRRK2 mutation carriers exhibit minimal or absent α-synuclein (αSyn) pathology, challenging traditional staging models [3].
The molecular pathogenesis of PD reflects a convergence of interrelated mechanisms, including oxidative stress, mitochondrial dysfunction, impaired proteostasis, defective autophagy, excitotoxicity, and neuroinflammation, alongside misfolding and aggregation of αSyn [4,5]. These processes interact with genetic and environmental risk factors to accelerate neuronal vulnerability and clinical decline [6]. Such complexity underscores the need for therapeutic strategies that extend beyond symptomatic dopamine replacement toward interventions capable of modifying disease trajectory.
Disease-modifying therapies (DMT) are defined by their ability to slow clinical progression independent of symptomatic benefit, distinguishing them from neuroprotective interventions that primarily target molecular cascades to prevent neuronal death [7,8]. Despite decades of translational research, no pharmacologic agent has conclusively demonstrated DME in late-phase randomized controlled trials. Current efforts largely emphasize tertiary prevention—initiated after diagnosis—while strategies for prodromal or preclinical stages remain underdeveloped [9,10]. Emerging paradigms prioritize biologically targeted approaches, including gene therapy, molecular chaperones, kinase inhibitors, and immunotherapies, integrated with biomarker-driven designs to enhance precision and interpretability [11]. These advances aim to bridge mechanistic insights with clinically meaningful outcomes, addressing a critical unmet need in PD therapeutics.
2. Unsuccessful Disease-Modifying Therapies in PD
Randomized controlled trials of candidate disease-modifying therapies in PD have consistently yielded negative or non-confirmatory results despite strong mechanistic rationales (Table 1) [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28]. This section provides a structured analysis of these interventions, organized by pharmacologic class, summarizing pivotal phase 2 and 3 trials, dosing strategies, endpoints, and reasons for lack of efficacy [29].
2.1. MAO-B Inhibitors and Dopaminergic Modulators
Evidence from randomized trials and systematic reviews demonstrates that MAO-B inhibitors provide symptomatic benefit without altering the underlying progression of PD. The Cochrane review by Macleod et al. concluded that selegiline and rasagiline improve UPDRS scores and delay the need for levodopa initiation, but long-term follow-up failed to confirm disease modification [30]. Early optimism from the DATATOP trial [31], which reported that selegiline delayed time to levodopa by approximately nine months, was later attributed to symptomatic effects rather than neuroprotection, given the rapid initial improvement and sensitivity of endpoints to treatment [32].
The ADAGIO trial remains the most rigorous test of rasagiline in a delayed-start design [33]. In this 72-week study, rasagiline 1 mg/day met all three hierarchical criteria (Superiority in early slope, superiority in total change, and non-inferiority of later slope) for potential disease modification, whereas 2 mg/day did not. However, the magnitude of benefit—approximately 1.6 to 1.8 UPDRS points at weeks 48–72—was considered insufficient to establish neuroprotection once symptomatic confounding was accounted for. Subsequent analyses and expert commentary judged these findings as consistent with sustained symptomatic benefit rather than true disease modification [34].
Similar conclusions apply to dopaminergic modulators. The LEAP trial tested early versus delayed levodopa initiation over 80 weeks and found no significant difference in UPDRS progression between groups, a result confirmed at three- and five-year follow-up [35]. Dopamine agonists have also failed to demonstrate DME. The PROUD study [36] showed no sustained advantage for early pramipexole initiation, and long-term data from CALM-PD [37] revealed fewer motor complications with initial pramipexole compared to levodopa but no difference in overall progression. Meta-analyses corroborate these findings, indicating that while these agents improve motor symptoms, they do not alter the natural course of the disease [38].
2.2. L-Type Calcium Channel Block
Calcium influx through Cav1.3 L-type channels contributes to dopaminergic neuron vulnerability by promoting mitochondrial calcium overload and oxidative stress. Epidemiological studies have suggested a protective association. A Danish cohort reported a 29% lower PD risk among dihydropyridine calcium channel blocker (CCB) users (IRR 0.71, 95% CI 0.60–0.82) and reduced mortality after PD diagnosis (IRR 0.66, 95% CI 0.47–0.91) [39]. A meta-analysis of nearly 3 million participants confirmed a 22% reduction in PD incidence with CCB use (RR 0.78, 95% CI 0.62–0.99) [18].
Clinical trials have tested this hypothesis. The Phase II STEADY-PD trial evaluated controlled-release isradipine at 5, 10, and 20 mg/day in early, drug-naïve PD patients over 12 months. While post-hoc analysis suggested smaller UPDRS increases at higher doses, significance disappeared after adjusting for symptomatic therapy [40]. The definitive Phase III STEADY-PD III trial randomized 336 early PD patients to 10 mg/day of immediate-release isradipine or placebo for 36 months. Results showed no difference in ON-state MDS-UPDRS I–III progression (LSMD −0.27 points; p = 0.85) and no benefit on secondary endpoints, indicating no DME [41].
Other dihydropyridines have shown preclinical promise. Felodipine induced autophagy and improved mitochondrial clearance in αSyn mouse models and GBA1-mutant cell lines [42]. Nimodipine demonstrated neuro-restorative effects in MPTP zebrafish models and in silico binding to targets such as MAO A/B, CASP3, and GSK3B, suggesting antioxidative and anti-apoptotic mechanisms [43].
2.3. Antioxidants and Bioenergetic Supplements
Coenzyme Q10 showed early promise in phase-2 studies, but the phase-3 QE3 trial (1,200–2,400 mg/day) was terminated for futility after interim analysis revealed no slowing of clinical decline compared to placebo [44]. Similarly, creatine, hypothesized to enhance cellular energy buffering and reduce oxidative damage, was evaluated in the long-duration NET-PD LS-1 trial. This study enrolled over 1,700 participants and was stopped for futility, demonstrating no effect on disease progression [45].
Vitamin E, despite strong preclinical rationale as a lipid-soluble antioxidant, failed to demonstrate benefit in the DATATOP trial, which showed no significant impact on motor progression in early PD [31]. Similar results were found with others vitamins, such as vitamin D [46]. Other mitochondrial-targeted antioxidants such as MitoQ and nicotinamide riboside have been explored in small pilot studies, but none have advanced to phase-3 trials due to lack of robust efficacy signals [47,48].
Metabolic and inflammatory modulation strategies have consistently failed to demonstrate DME in PD. In the phase 3 SURE-PD3 trial, oral inosine titrated to elevate serum urate from ≤5.7 mg/dL to 7.1–8.0 mg/dL over two years in 298 early PD patients achieved robust biochemical target engagement but did not slow progression on the primary MDS-UPDRS I–III outcome [49]. Similarly, randomized caffeine trials have not shown durable benefit despite epidemiological associations with reduced PD risk. The Café-PD study (200 mg twice daily for up to 18 months) reported no improvement in MDS-UPDRS-III scores at six months, with only transient reductions in somnolence and a trend toward increased dyskinesia [50]. A smaller six-week trial suggested borderline symptomatic improvement, but lacked long-term efficacy [50].
2.4. αSyn Immunotherapies
Proteinopathy-targeted and kinase-inhibitor approaches have failed in pivotal settings. Cinpanemab (BIIB054), a human-derived anti-αSyn antibody, was evaluated in the phase 2 SPARK trial involving early PD patients. The study was terminated for lack of efficacy, showing no improvement in MDS-UPDRS progression or DaT-SPECT imaging compared to placebo over 52 weeks [14]. Similarly, prasinezumab (RO7046015), another anti-αSyn antibody, was tested in the PASADENA trial, a randomized, double-blind phase 2 study. Despite achieving target engagement and exploratory signals suggesting slower motor decline in some subgroups, the trial did not meet its primary endpoint of reducing MDS-UPDRS Part I–III progression at 52 weeks, indicating no proven DME [15].
Additional immunotherapy programs have faced similar challenges. ABBV-0805, an αSyn antibody developed by AbbVie, was discontinued after phase 1 due to strategic reprioritization and lack of compelling efficacy signals (https://www.bioarctic.com/en/abbvie-terminates-collaboration-with-bioarctic-on-alpha-synuclein-portfolio/, accessed on 23 December 2025). Other approaches, including vaccines such as AFFITOPE PD01A and PD03A, have demonstrated immunogenicity and safety in early-phase trials but have not advanced to pivotal studies due to insufficient evidence of clinical benefit [51].
2.5. c-Abl Kinase Inhibition
The multi-site phase 2 NILO-PD trial randomized participants with moderate PD to nilotinib 150 mg or 300 mg daily versus placebo for six months. Results showed no clinically meaningful benefit on motor outcomes (MDS-UPDRS Part III) or secondary biomarkers, including dopamine metabolites and αSyn species, despite adequate systemic exposure. Furthermore, exploratory imaging endpoints and CSF pharmacodynamics failed to demonstrate significant changes, constraining further development of nilotinib as a DMT [52].
Other candidates include INNO-406 (NS-187), which crosses the BBB and reduced dopaminergic neuron loss by 40–45% in MPTP mouse models [53], and IkT-148009, a novel CNS-penetrant c-Abl inhibitor that preserved ≥85% of dopaminergic neurons and improved motor function in transgenic and sporadic PD models [54]. IkT-148009 has advanced into early phase II clinical trials, representing a next-generation approach to overcome nilotinib’s pharmacokinetic limitations.
2.6. Iron Chelation
Iron chelation has also failed to demonstrate disease modification. Deferiprone was evaluated in the phase 2 FAIRPARK-II trial, which enrolled 372 patients with early PD. The study demonstrated significant reductions in nigrostriatal iron content on MRI, confirming target engagement; however, clinical outcomes were unfavorable. Patients receiving deferiprone exhibited greater worsening in MDS-UPDRS scores and required earlier initiation of dopaminergic therapy compared to placebo, contradicting the hypothesis of neuroprotection [55]. Earlier pilot studies suggested benefit, with small trials showing improved motor scores and reduced iron deposition [56], but these findings were not replicated in larger controlled settings.
Other iron-modulating strategies have also failed. The iron chelator deferasirox, tested in small open-label studies, showed poor tolerability and no meaningful motor improvement, leading to discontinuation [57]. Similarly, VK-28 demonstrated neuroprotection in preclinical PD models but never advanced beyond early-phase trials due to safety concerns [58].
2.7. GLP-1 Receptor Agonists and Incretin-Based Strategies
The phase 3 Exenatide-PD3 trial (96 weeks, 2 mg weekly) demonstrated no advantage over placebo on OFF-state motor progression, patient-reported outcomes, or DaT-SPECT imaging, effectively closing the door on exenatide as a DMT [59]. These findings underscore the importance of biomarker-anchored designs and stratified cohorts to match mechanisms to PD subtypes and verify target engagement prospectively [60].
Other incretin-based strategies have also failed to translate into clinical benefit. Liraglutide (NCT02953665) and semaglutide (NCT03659682), despite strong preclinical evidence of neuroprotection and anti-inflammatory effects, have not demonstrated significant motor or progression-modifying outcomes in small pilot studies. Collectively, these negative results parallel the futility of pioglitazone, a PPAR-γ agonist evaluated in the FS-ZONE phase 2 trial, where both 15 mg and 45 mg doses over 44 weeks produced UPDRS changes similar to placebo (4.42–5.13 vs. 6.25 points), confirming lack of DME [61].
3. Repurposing Existing Drugs
For central nervous system (CNS) drugs specifically, Wouters et al. estimated an average capitalized pre-launch R&D cost of $1.10 billion, reflecting longer development timelines (approximately 8.6 years) and moderate clinical success rates (15%) compared with other therapeutic areas [62]. Considering that over 130 million chemical structures (https://www.chemspider.com/, accessed on 23 December 2025) are known and more than 19,000 FDA-approved drugs exist (https://www.fda.gov/media/115824/download, accessed on 23 December 2025), it is plausible that at least one compound possesses neuroprotective properties. Notably, more than one-third of DMT PD trials involve repurposed drugs. Repurposing enables trials to begin at Phase 2 (since safety data already exist), thereby reducing both time and cost.
A classical example of drug repurposing relevant to PD include amantadine, originally developed as an antiviral for influenza and now widely used to alleviate motor symptoms and reduce levodopa-induced dyskinesias [63]. Other notable repurposed candidates for PD include selegiline, vitamin D, inosine, creatine, isradipine, pioglitazone, and incretin-based therapies such as exenatide, liraglutide, and lixisenatide. Among these, agents like rasagiline and exenatide have shown promising clinical benefits in trials, suggesting potential disease-modifying properties (Table 2).
Drug repurposing for PD is guided by three fundamental pillars: a plausible mechanistic link to neurodegeneration, measurable biomarkers to confirm target engagement or drug exposure, and a robust epidemiological basis derived from observational or cohort studies [64]. For instance, inosine satisfies all three criteria, supported by its antioxidant mechanism, serum urate as a biomarker, and strong epidemiological association with reduced PD risk [49]. Isradipine, a CCB, offers a well-defined mechanistic rationale and epidemiological support but lacks validated biomarkers for CNS penetration [41]. Conversely, GLP-1 receptor agonists exhibit promising epidemiological signals yet remain limited by incomplete mechanistic validation and absence of reliable biomarkers [65], underscoring the need for integrated translational frameworks before advancing to large-scale trials.
Developing DMTs for PD remains a lengthy and resource-intensive process, characterized by sequential phase 2 and phase 3 randomized controlled trials with a single objective, population, and compound [66]. Between 1999 and 2021, 84 compounds entered early-phase trials, but only 28 progressed to phase 3 and 15 achieved regulatory approval, yielding an overall success rate of approximately 14.9%, with just 13% of trials targeting disease modification [67]. A recurring pattern is that positive phase 2 results often fail to translate into phase 3 efficacy, underscoring the challenges of trial design, patient stratification, and endpoint selection in neurodegenerative disorders.
3.1. Glycolysis-Enhancing Drugs
Dopaminergic neurons have exceptionally high energy demands, making them uniquely vulnerable to genetic, environmental, and aging-related stressors that disrupt energy homeostasis. Impaired cellular energetics promotes protein misfolding and aggregation, accelerating neurodegeneration. Neuronal ATP production relies heavily on glycolysis, with phosphoglycerate kinase 1 (PGK1) catalyzing the critical step of converting ADP to ATP [68]. The physiological significance of PGK1 is highlighted by reports of an X-linked PGK1 mutation associated with a levodopa-responsive parkinsonism epilepsy syndrome [69].
Terazosin activates PGK1, likely through its quinazoline moiety, enhancing glycolytic flux and cellular stress resistance [70]. Experimental models demonstrate that terazosin increases brain ATP levels in iPSC-derived dopaminergic neurons from LRRK2-PD patients, as well as in rotenone-, PINK1-, and PGK1-knockout Drosophila, MPTP and αSyn mouse models, and the 6-OHDA rat model, indicating broad neuroprotective potential [71]. Cognitive benefits have also been reported; in PD models of dementia, interval timing tasks revealed improved coefficient of variation, suggesting modulation of temporal processing [72]. Mechanistically, PGK1 is emerging as a central node in synaptic transmission and neuronal energy homeostasis, reinforcing its relevance as a therapeutic target in PD [73].
Schultz et al. demonstrated that terazosin significantly increased whole-blood ATP levels in healthy adults using a luciferase-based assay [74]. Furthermore, a pilot trial in PD patients showed elevated brain ATP following terazosin administration [75]. Observational analyses using the Parkinson’s Progression Markers Initiative (PPMI) database revealed that PD patients on terazosin exhibited slower clinical progression compared to non-users [71]. Complementing these findings, a meta-analysis of observational studies reported that α1-adrenergic antagonist use was associated with a reduced risk of developing PD (HR 0.82; 95% CI, 0.71–0.94) [76].
3.2. Challenges in Repurposing
Repurposing existing drugs for PD faces several critical challenges. Dosing is a major issue, as the optimal therapeutic dose for neuroprotection may differ substantially from that used for the original indication, requiring new pharmacokinetic and safety evaluations [74]. Brain penetration represents another hurdle; many compounds exhibit poor BBB permeability and may need reformulation or alternative delivery strategies to achieve sufficient CNS exposure [77]. Intellectual property constraints further complicate late-stage development, as repurposed drugs often lack patent protection, reducing commercial incentives for large-scale trials [62]. Trial design poses unique difficulties because PD progresses slowly, making short-duration studies underpowered to detect DME; adaptive or delayed-start designs have been proposed but remain challenging to implement. To improve interpretability, future trials should incorporate strategies for wash-in/ wash-out bias mitigation, robust handling of symptomatic therapy confounding, and composite endpoints that integrate clinical scales (e.g., MDS-UPDRS), imaging biomarkers, and digital metrics [60,78]. Finally, measuring disease modification is limited by the absence of validated biomarkers and imaging endpoints, which hampers the ability to confirm target engagement and interpret clinical outcomes [78].
4. Ongoing Trials
Current DMT trials in PD target diverse pathogenic mechanisms. The most represented category involves αSyn-directed strategies, including monoclonal antibodies, active immunization, and anti-aggregation compounds (Figure 1)(https://drive.google.com/file/d/1NeSyFA37b9IbUzryRRP-EqrgScjCRL-3/view, accessed on 23 December 2025)(Appendix A)[79]. Genetically targeted approaches focus on mutations in GBA and LRRK2, aiming to restore lysosomal function or inhibit kinase activity. Additional interventions address neuroinflammation, mitochondrial dysfunction, oxidative stress, and metabolic pathways through GLP-1/GIP receptor agonists and tyrosine kinase inhibitors [8].
In 2025, approximately 51% of the active clinical pipeline for PD targets DME [80]. Small molecules remain the predominant therapeutic modality, accounting for nearly 66% of all ongoing studies, followed by cell and tissue-based therapies (12%) and peptide-based approaches (6%) [80].
4.1. αSyn Targeted
αSyn aggregation is a central pathogenic hallmark of PD, driving neurodegeneration through misfolding, oligomerization, and propagation across neural circuits [81]. Therapeutic strategies targeting αSyn aim to reduce its aggregation, enhance clearance, or block cell-to-cell transmission, with approaches including small molecules, monoclonal antibodies, and active immunization currently in clinical development [82].
The ORCHESTRA trial (NCT04658186) evaluated minzasolmin, an oral small molecule designed to inhibit αSyn misfolding, in early PD. This Phase 2A study enrolled 496 participants with idiopathic PD diagnosed within two years, H&Y stage ≤2.5, and no prior dopaminergic therapy. Subjects were randomized to two active doses or placebo for 18 months, with the primary endpoint being change in MDS-UPDRS parts I–III. Despite its targeted mechanism, the trial did not meet primary or secondary endpoints, leading to study termination (https://www.ucb.com/newsroom/press-releases/article/findings-from-minzasolmin-proof-of-concept-orchestra-study-shape-next-steps-in-ucb-parkinson-s-research-program, accessed on 23 December 2025).
The PADOVA trial (NCT04777331) investigated prasinezumab in 586 patients with early-stage PD (diagnosis 3 months–3 years, H&Y stage 1–2) receiving stable dopaminergic therapy. The study employed a time-to-event design, defining progression as ≥5-point worsening in MDS-UPDRS part III OFF state over a minimum of 76 weeks. Although the Phase 2B trial missed its primary endpoint, exploratory analyses suggested a potential benefit in slowing motor progression, prompting advancement to Phase III development (https://www.roche.com/media/releases/med-cor-2024-12-19, accessed on 23 December 2025).
Beyond minzasolmin and prasinezumab, multiple αSyn-targeted agents are in the pipeline. These include antibodies (e.g., ABL Bio/Sanofi, Bioarctic, AstraZeneca/Takeda), vaccines (AC Immune, Vaxxinity), and small molecules (Allyx Therapeutics, Alterity Therapeutics, MODAG, WaveBreak, Janssen). Clinical stages range from Phase 1 to Phase 3, reflecting diverse strategies to interfere with αSyn aggregation and propagation [83].
Monoclonal antibodies aim to neutralize extracellular αSyn species and prevent their uptake by neighboring neurons, while vaccines induce adaptive immune responses to enhance clearance. Small molecules, such as minzasolmin, target intracellular misfolding and oligomerization processes. Despite promising preclinical data, translation to clinical efficacy remains challenging, with recent trials highlighting the need for earlier intervention and improved biomarkers for patient stratification [8].
4.2. Inflammation/Immune Pathways
Neuroinflammation mediated by the NLRP3 inflammasome is increasingly recognized as a key contributor to PD pathogenesis, linking αSyn aggregation to microglial activation and dopaminergic neurodegeneration [84]. NLRP3 activation promotes caspase-1–dependent maturation of IL-1β and IL-18, amplifying neuroinflammatory cascades and oxidative stress. Preclinical studies demonstrate that pharmacologic inhibition of NLRP3 reduces αSyn pathology and preserves nigrostriatal integrity, supporting its candidacy as a DMT [85].
Several oral, brain-penetrant NLRP3 inhibitors are now in clinical development for PD. Dapansutrile (OLT-1177), developed by Olatec Therapeutics, is under evaluation in a randomized, placebo-controlled trial assessing safety, tolerability, and biomarker effects over 6–12 months in early PD (NCT07157735). NT-0796 (NodThera) has shown Phase Ib/IIa evidence of neuroinflammation reduction and robust target engagement, marking the first clinical demonstration of NLRP3 pathway modulation in PD (https://www.nodthera.com/news/nodtheras-nlrp3-inhibitor-nt-0796-reverses-neuroinflammation-in-parkinsons-disease-phase-ib-iia-trial/, accessed on 23 December 2025). Selnoflast (RO7568282), Roche’s candidate, is in Phase 1b studies with biomarker-driven endpoints to confirm CNS penetration and pharmacodynamic activity (https://medically.roche.com/global/en/neuroscience/adpd-2025/medical-material/ADPD-2025-presentation-pagano-a-phase-1B-study-pdf.html, accessed on 23 December 2025). VENT-02 (Ventus Therapeutics) completed Phase 1 with full ex vivo IL-1β inhibition and initiated a Phase 2a PD trial incorporating digital motor assessments and CSF biomarkers, with topline results expected in late 2025 (https://www.ventustx.com/ventus-therapeutics-announces-results-from-phase-1-clinical-trial-of-vent-02-a-novel-orally-administered-brain-penetrant-nlrp3-inhibitor/, accessed on 23 December 2025).
Additional agents include VTX-3232 (Ventyx Biosciences) and ZYIL1 (Usnoflast) (Zydus), both in Phase 2 for systemic inflammatory conditions, with PD-specific trials anticipated. Collectively, these programs operationalize mechanistic evidence linking αSyn–driven microglial activation to NLRP3 signaling and aim to achieve disease modification by attenuating neuroinflammation. Success will depend on validated progression endpoints and biomarker confirmation of target engagement [85].
4.3. GLP1 Agonists
Exenatide, a glucagon-like peptide-1 (GLP-1) receptor agonist, is being investigated as a potential DMT for PD due to its neuroprotective effects mediated through PI3K/Akt signaling and mitochondrial stabilization [86]. The ongoing Phase 3 trial (NCT04232969) is a randomized, double-blind, placebo-controlled study enrolling 194 patients with idiopathic PD on stable dopaminergic therapy. Participants receive weekly subcutaneous injections of exenatide 2 mg or placebo for 96 weeks, with the primary endpoint being change in MDS-UPDRS part III in the OFF state [59].
4.4. Tyrosine Kinase Inhibitors
IkT-148009 (risvodetinib), an oral c-Abl tyrosine kinase inhibitor, is being evaluated as a DMT for PD due to its ability to reduce αSyn aggregation and mitochondrial stress [87]. The ongoing Phase 2 trial (NCT05424276) randomized 120 patients with idiopathic PD (H&Y stage <3, MoCA ≥24, no prior dopaminergic therapy) to three active doses or placebo for 12 weeks. Primary endpoints include safety, tolerability, and change in MDS-UPDRS parts II and III, with topline results presented at ADPD 2025 (https://www.ablitherapeutics.com/news/detail/9613/abli-therapeutics-reports-final-results-from-the-phase-2-201-trial-evaluating-risvodetinib-for-the-treatment-of-parkinsons-disease, accessed on 23 December 2025).
4.5. Cell-Based Therapies
Cell-replacement strategies aim to restore dopaminergic circuitry by engrafting midbrain dopaminergic progenitors into the putamen. Early fetal-tissue programs (e.g., TRANSEURO, NCT01898390) established procedural feasibility but showed no overall motor benefit at 3 years and highlighted variability driven by tissue source and implantation devices—strengthening the case for standardized stem-cell–derived sources (hfVM scarcity; PET ^18F-fluorodopa signals improved in some but not most participants) [88]. Current iPSC programs from Kyoto University/CiRA report Phase I/II safety with bilateral transplantation of 5–10 million dopaminergic progenitors, short-term dopamine increases, and absence of severe adverse events under time-limited tacrolimus immunosuppression, with peer-reviewed clinical and translational descriptions now available [89,90]. These trials refine surgical navigation, cell manufacturing (floor-plate CORIN+ selection), and imaging endpoints to track graft viability and function, while remaining vigilant about tumorigenicity and immunogenicity—issues that mandate GMP standardization and multi-year follow-up.
4.6. RNA-Based Therapies
Antisense oligonucleotides (ASOs) enable genotype-informed suppression of pathogenic transcripts. LRRK2-targeted ASOs (BIIB094/ION859, REASON; NCT03976349) achieved intrathecal target engagement in Phase I, reducing CSF LRRK2 by 59% and pRab10 by 50%, and modulating lysosomal pathway proteins; the trial completed in 2024 with safety/tolerability readouts, although Biogen subsequently discontinued BIIB094 amid portfolio reprioritization. Next-generation LRRK2 ASOs (e.g., SNP614) show nonhuman primate knockdown and favorable preclinical safety profiles, supporting continued development in PD. SNCA-directed ASOs have demonstrated broad CNS distribution and reductions of αSyn and CSF aSyn in rodent PFF and nonhuman primate models, reversing established pathology, providing a translational rationale for clinical evaluation in synucleinopathies. Collectively, ASO programs leverage intrathecal delivery to bypass the BBB; near-term priorities include durability of knockdown, off-target risk management, and pharmacodynamic biomarker panels (e.g., CSF aSyn species, LRRK2 kinase activity/pRab10) to anchor dose-finding and progression-sensitive endpoints [91].
5. Gene Therapy
The global gene therapy pipeline has expanded substantially, reaching 2,129 active programs by Q3 2025, with the majority in preclinical development (n = 1,346) and smaller proportions in Phase I (n = 377), Phase II (n = 347), and Phase III (n = 47) (https://www.citeline.com/en/resources/q3-2025-gene-cell-and-rna-therapy-report, accessed on 23 December 2025). Viral vectors remain the dominant delivery platform, utilized in 89% of gene therapy programs. Among these, AAV is the most prevalent vector (n = 405), followed by lentivirus (n = 288) and adenovirus (n = 123), while retrovirus, herpes simplex virus, and poxvirus account for fewer studies. Notably, herpes simplex virus-based approaches exhibited the largest quarterly growth, with a 33% increase in ongoing studies (https://www.asgct.org/news-publications/asgct-news/gene-cell-rna-therapy-landscape-q2-2021, accessed on 23 December 2025).
In vivo gene therapy is delivered through three main routes: stereotaxic intracerebral injection, intrathecal or intra-CSF infusion, and intravenous administration, with stereotaxic surgery being the most widely used due to its ability to achieve targeted delivery to basal ganglia structures [92,93]. Recombinant AAV vectors, derived from non-pathogenic parvoviruses, are the preferred platform because they efficiently transduce non-dividing neurons, provide durable transgene expression exceeding 15 years, and exhibit minimal neurotoxicity in primate and human studies; however, their packaging capacity is limited to approximately 4.7 kb, constraining the size of therapeutic genes [94].
Gene therapy strategies for PD target multiple pathogenic mechanisms (Table 3). Approaches to restore dopamine synthesis include delivery of enzymes such as TH, GCH, and AADC [92,93]. Neuroprotective interventions employ trophic factors like glial cell line-derived neurotrophic factor (GDNF) and neurturin (CERE-120), as well as genes implicated in mitochondrial and lysosomal function, including Parkin and glucocerebrosidase (GBA) [95,96]. Additional strategies aim to reduce STN hyperactivity via glutamic acid decarboxylase isoforms (GAD-65, GAD-67) [97] or mitigate αSyn and amyloid-β aggregation through agents such as irisin and neprilysin [98].
5.1. GBA
GBA1-targeted gene therapy for PD aims to restore glucocerebrosidase (GCase) activity, thereby reducing lysosomal dysfunction and αSyn accumulation. Mutations in GBA1 impair GCase, leading to substrate buildup (glucosylceramide, glucosylsphingosine), endoplasmic reticulum stress, and autophagy deficits [99]. Recombinant AAV9 vectors delivering GBA1 via intra-cisterna magna infusion have shown promise in early-phase trials, such as PROPEL (LY3884961), improving lysosomal function and reducing neuroinflammation (Prevail Therapeutics/Eli Lilly) [100]. Alternative strategies include substrate reduction therapy (e.g., venglustat), molecular chaperones, and enzyme replacement; however, venglustat trials were terminated due to motor worsening [101].
5.2. GAD-65 and GAD-67
AAV2-GAD gene therapy for PD targets the STN to reduce pathological hyperactivity by enhancing local GABA synthesis through GAD expression. Delivered via stereotaxic infusion, this approach modulates basal ganglia circuitry, counteracting excessive excitatory output from the STN to the globus pallidus internus [102]. Randomized controlled trials demonstrated significant improvements in UPDRS motor scores and reduced levodopa-induced dyskinesia over 12 months compared to sham surgery [103]. Long-term follow-up and neuroimaging analyses revealed sustained clinical benefit and reorganization of functional connectivity, particularly in motor and associative cortical regions [104,105].
The AAV2-GAD gene therapy trials for PD were not continued primarily due to limited efficacy and strategic considerations rather than major safety concerns. Early-phase studies demonstrated improvements in motor symptoms and reduced dyskinesia, but the magnitude of benefit was modest compared to existing therapies like deep brain stimulation and optimized pharmacological treatment. Additionally, the therapy required invasive stereotaxic delivery to the STN, which posed logistical challenges for widespread adoption. Combined with the emergence of alternative approaches—such as GBA1-targeted therapies and cell-based interventions—companies shifted resources away from AAV2-GAD programs toward strategies with broader applicability and stronger DME potential.
5.3. GDNF and Neurturin (CERE-120)
AAV2-neurturin gene therapy was developed to enhance dopaminergic neuron survival by delivering neurturin to the putamen and substantia nigra in PD. Despite successful vector expression, two randomized, double-blind trials demonstrated no significant improvement in motor function compared to sham surgery at 12 or 24 months, as measured by UPDRS scores. The first trial focused in the putamen [106], and due to concerns of failure due to significant loss in the substantia nigra, and first results being negative, the second trial injected in the putamen and also substantia nigra, but no positive results were observed [107]. Post hoc analyses suggested that advanced disease stage and persistent αSyn pathology may limit neurotrophic efficacy, highlighting the need for earlier intervention guided by genetic diagnosis, biomarkers, and imaging [108].
5.4. TH, GCH, and AADC
In vivo gene therapy targeting dopamine biosynthesis in PD employs AAV vectors encoding TH, GCH, and AADC to restore striatal dopamine production. Preclinical studies in MPTP-treated nonhuman primates demonstrated robust transgene expression persisting for over 15 years, confirmed by TH immunostaining, and sustained behavioral recovery up to six years post-injection [109,110]. Microdialysis experiments revealed significant increases in extracellular dopamine following L-DOPA administration in AAV-treated animals compared to controls, supporting functional enzyme activity [110].
Eladocagene exuparvovec (AAV2-hAADC) is an AAV vector delivering the human AADC gene to the putamen, enabling continuous dopamine synthesis from levodopa. This therapy is approved by the European Medicines Agency (EMA) and Taiwan FDA for children ≥18 months with severe AADC deficiency, and the U.S. FDA has accepted its biologics license application with priority review [111,112]. Clinical trials in AADC deficiency demonstrated sustained improvements in motor and cognitive function, with AIMS scores increasing over 24 months and PET imaging confirming enhanced dopaminergic activity [111].
For PD, AAV-based gene therapy strategies include delivery of TH, GCH, and AADC to restore dopamine biosynthesis. Phase I trials using MRI-guided stereotaxic infusion of AAV2-AADC into the putamen (doses 1.6–2.0 × 1011 vg) showed significant reductions in UPDRS motor scores in both ON and OFF states, with benefits persisting for up to three years [113]. Advantages of this approach include one-time administration and durable transgene expression (>15 years in preclinical models), but challenges remain, such as the need for invasive surgery and limited efficacy for non-motor symptoms [109,113].
6. Future Studies
Progress toward disease modification in PD will hinge on trial architectures that better accommodate biological heterogeneity and slow progression rates. Master protocols, and specifically platform trials that share a common control arm and permit adaptive addition or discontinuation of investigational regimens, offer operational efficiency and statistical advantages while confronting PD-specific complexities (e.g., confounding by symptomatic therapies and absence of validated surrogate endpoints) [114]. Recent methodological work details how these designs can be tailored to PD and illustrates active program development in the field [115]. The Path-to-Prevention (P2P) platform—nested within PPMI—operationalizes biomarker-defined neuronal αSyn disease with dual primary endpoints (DaT SBR slope; MDS-UPDRS III), representing a pragmatic template for biologically staged, perpetual testing in prodromal or early PD [116].
A second pillar is biomarker-anchored enrichment, led by the rapid maturation of the αSyn seed amplification assay (SAA) [117]. In 2024, the FDA issued a letter of support for αSyn-SAA use in PD trials—based on multi-center data—enabling selection of participants with confirmed αSyn pathology and reducing biological misclassification that undermines trial power. Concurrent advances in serum/CSF assay performance and interpretability—including enhanced serum-based SAA chemistries—support broader adoption for stratification and progression monitoring across synucleinopathies [118]. These biomarker strategies should be combined with digital health technologies (e.g., passive mobility, speech, and sleep metrics) already embedded in innovative protocols to generate sensitive, longitudinal endpoints that de-risk phase-2 decisions.
Neurofilament light chain (NfL) has emerged as a robust prognostic marker for accelerated progression, offering utility for stratification and enrichment [119]. Parallel measurement of CSF and plasma inflammatory mediators—such as IL-6 and TNF-α—can capture neuroimmune dynamics and serve as pharmacodynamic endpoints for anti-inflammatory interventions [84,120].
Beyond traditional clinical scales, digital biomarkers are increasingly recognized as sensitive endpoints for DMT in PD. Wearable sensors—such as inertial measurement units embedded in wristbands or shoe insoles—enable continuous quantification of gait parameters (stride length, variability, freezing episodes) and tremor amplitude with millisecond resolution, outperforming episodic in-clinic assessments [121]. Smartphone-based platforms leverage accelerometers and gyroscopes to capture bradykinesia and dyskinesia during daily activities, while touchscreen tapping tasks provide high-frequency motor performance metrics [122]. Speech analysis algorithms extract prosodic and articulatory features (e.g., pitch variability, vowel space area) from passive voice recordings, correlating with motor and cognitive decline [123]. These technologies generate ecologically valid, longitudinal datasets that can serve as progression-sensitive endpoints and pharmacodynamic markers, particularly when integrated with machine-learning models for predictive analytics. Regulatory agencies have begun exploratory qualification of such measures as secondary endpoints, underscoring their potential to complement imaging and fluid biomarkers in adaptive trial designs.
Advancing disease-modifying therapies for Parkinson’s disease requires strategic alignment with evolving regulatory frameworks. The FDA’s 2024 Letter of Support for the αSyn SAA and earlier support for dopamine transporter imaging underscore growing agency acceptance of biomarker-based trial enrichment and progression monitoring (https://www.fda.gov/media/181368/download, accessed on 23 December 2025). Moreover, both the FDA and EMA offer accelerated approval/conditional authorization pathways for serious conditions based on robust surrogate endpoints—provided these biomarkers demonstrate predictive validity and are supported by confirmatory post-approval studies [124]. To fully harness these pathways in PD, rigorous biomarker qualification, mechanistic rationale, and transparent post-market commitments are essential. Ethical and logistical considerations also arise with global, adaptive, multi-arm platform trials. Initiatives such as the Global Alliance for Parkinson’s Platforms (GAPP) aim to harmonize protocols, data standards, and regulatory oversight across jurisdictions (https://cureparkinsons.org.uk/2025/11/launch-of-a-global-alliance-for-parkinsons-platforms-gapp-bringing-together-international-leaders-of-parkinsons-platform-trials/, accessed on 23 December 2025). Engagement of global frameworks and cross-country ethics collaboration will be critical to streamline governance, ensure equitable access, and reduce duplication—thereby accelerating the availability of innovative therapies to patients worldwide. From an economic perspective, platform trials substantially reduce per-arm cost by sharing infrastructure and control groups, with estimates suggesting up to 30–40% savings compared to sequential, single-compound trials [115,125]. However, intellectual property challenges remain a major barrier for repurposed drugs, as lack of patent exclusivity diminishes commercial incentives for late-stage development, often necessitating public–private partnerships or non-profit consortia to sustain funding [126].
Mechanistically, αSyn–directed immunotherapy is poised for decisive testing. After PADOVA Phase 2B mixed results but consistent trends across multiple endpoints and open-label extensions, prasinezumab has been advanced to Phase III in early PD, marking the first large-scale attempt to validate αSyn immunotherapy as a DMT with biomarker-informed design (https://www.roche.com/media/releases/med-cor-2024-12-19, accessed on 23 December 2025). Future studies should ensure longer exposure, progression-sensitive endpoints, and explicit co-enrichment by neuronal αSyn pathology and dopaminergic deficit to maximize the likelihood of detecting clinically meaningful slowing.
Converging evidence also prioritizes neuroinflammation as a modifiable axis. Brain-penetrant NLRP3 inhibitors have demonstrated rapid CSF biomarker normalization—reductions in IL-1β, IL-6, and chemokines—over 28 days in Phase Ib/IIa PD, motivating Phase II planning. In parallel, VENT-02 entered a randomized Phase 2a PD trial in 2025 that integrates plasma/CSF biomarker tiers and digital motor assessments with primary safety endpoints; topline data are expected by late 2025/early 2026 and will inform whether NLRP3 inhibition can yield symptomatic and disease-modifying effects (https://www.nodthera.com/news/nodthera-announces-first-patients-dosed-in-resolve-2-clinical-trial-evaluating-oral-nt-0796-in-combination-with-a-glp-1-receptor-agonist/, accessed on 23 December 2025). Collectively, these programs directly test the mechanistic link between αSyn fibrils, microglial inflammasome activation, and propagation of neurodegeneration.
Finally, genetic and metabolic strategies remain central. On the genetic side, AAV9-GBA1 gene therapy (LY3884961; PROPEL) continues recruitment with intracisternal delivery and long-term biomarker follow-up, offering a durable, mechanism-restorative approach for GBA-associated PD (NCT05669331). On the metabolic side, PGK1 activation via terazosin has human dose-finding data indicating brain penetration and bioenergetic target engagement (whole-blood ATP, FDG-PET, ^31P-MRS), nominating 5 mg/day as a promising dose for PD engagement studies such as TAME-PD (NCT02879136). Incretin-based approaches warrant nuanced reassessment. Lixisenatide produced statistically significant motor slowing at 12 months in early PD but with substantial gastrointestinal adverse effects, highlighting the need for better CNS penetrance, longer exposure, and biomarker-guided subgroup analyses in subsequent trials (NCT04269642). Beyond mechanistic innovation, future progress depends on addressing systemic barriers: insurance coverage for genetic testing remains inconsistent, limiting genotype-based trial enrichment, while equity in trial recruitment—particularly inclusion of underserved populations—must be prioritized to ensure generalizability and fair access to emerging therapies.
7. Conclusion
Advances in trial design for PD increasingly incorporate deep phenotyping, biomarker integration, and adaptive methodologies to address biological heterogeneity and improve interpretability. Master protocols and platform trials offer operational efficiencies by enabling multi-arm evaluations within a unified framework, but require solutions for governance, intellectual property, and regulatory harmonization through global collaboration.
Therapeutically, αSyn–targeted strategies remain central, complemented by gene-based approaches for LRRK2 and GBA variants and emerging interventions targeting neuroinflammation and metabolic dysfunction. Despite these innovations, no agent has demonstrated definitive disease-modifying efficacy in late-phase trials, underscoring the need for translational research that links mechanistic insights to clinically meaningful outcomes.
Genotype-based enrichment can enhance trial efficiency and enable precision medicine, particularly for GBA1- and LRRK2-associated PD, and may facilitate prodromal intervention. However, barriers such as limited genetic testing access, privacy concerns, and insurance gaps constrain implementation. Furthermore, while enrichment improves internal validity, broader inclusion and stratified analyses are essential to ensure generalizability to idiopathic PD and equitable therapeutic benefit.
Future progress will depend on biomarker-driven designs, molecular staging, and harmonized global platform trials to accelerate development and deliver truly DMTs.
Author Contributions
Conceptualization, J.P.R. and A.L.F.C.; methodology, A.L.F.C.; software, A.L.F.C.; validation, A.L.F.C. and J.P.R.; formal analysis, A.L.F.C.; investigation, A.L.F.C.; resources, A.L.F.C.; data cu-ration, J.P.R.; writing—original draft preparation, J.P.R.; writing—review and editing, J.P.R.; visualization, A.L.F.C.; supervision, A.L.F.C.; project administration, A.L.F.C.; funding acquisition, J.P.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data are presented in the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AADC | Aromatic L-amino acid decarboxylase deficiency |
| AAV | Adeno-associated virus |
| ASO | Antisense oligonucleotide |
| BBB | Blood-brain barrier |
| CCB | Calcium channel blocker |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| DME | Disease-modifying effect |
| DMT | Disease-modifying therapy |
| GABA | γ-aminobutyric acid |
| GAD | Glutamic acid decarboxylase |
| GBA | Glucocerebrosidase |
| GCH | GTP cyclohydrolase I |
| H&Y | Hoehn and Yahr |
| IRR | Incidence rate ratio |
| LSMD | Least squares mean difference |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAO | Monoamine oxidase |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| PD | Parkinson’s disease |
| PPAR-γ | Peroxisome Proliferator-Activated Receptor gamma |
| STN | Subthalamic nucleus |
| TH | Tyrosine hydroxylase |
| UPDRS | Unified Parkinson’s Disease Rating Scale |
| αSyn | α-Synuclein |
Appendix A

Table A1.
Distribution of Parkinson’s Disease Clinical Trials by Therapeutic Type and Development Phase (2025).
Table A1.
Distribution of Parkinson’s Disease Clinical Trials by Therapeutic Type and Development Phase (2025).
| Type | Phase 1 | Phase 2 | Phase 3 |
| Cell | 2 | 0 | 0 |
| Gene | 2 | 2 | 0 |
| NCE | 27 | 19 | 2 |
| Reformulation | 4 | 1 | 0 |
| Repurposed | 4 | 15 | 4 |
| Other | 0 | 1 | 0 |
| Total | 39 | 38 | 6 |
References
- Buchmann, C.; Bouchard, M. New Perspectives on Parkinson’s Disease Subtyping: A Narrative Review. Can J Neurol Sci 2025, 1–10. [CrossRef]
- Trevisan, L.; Gaudio, A.; Monfrini, E.; Avanzino, L.; Di Fonzo, A.; Mandich, P. Genetics in Parkinson’s Disease, State-of-the-Art and Future Perspectives. Br Med Bull 2024, 149, 60–71. [CrossRef]
- Tanaka, M. Parkinson’s Disease: Bridging Gaps, Building Biomarkers, and Reimagining Clinical Translation. Cells 2025, 14. [CrossRef]
- Wüllner, U.; Borghammer, P.; Choe, C.-U.; Csoti, I.; Falkenburger, B.; Gasser, T.; Lingor, P.; Riederer, P. The Heterogeneity of Parkinson’s Disease. J Neural Transm (Vienna) 2023, 130, 827–838. [CrossRef]
- Pitton Rissardo, J.; McGarry, A.; Shi, Y.; Fornari Caprara, A.L.; Kannarkat, G.T. Alpha-Synuclein Neurobiology in Parkinson’s Disease: A Comprehensive Review of Its Role, Mechanisms, and Therapeutic Perspectives. Brain Sciences 2025, 15, 1260. [CrossRef]
- Smith, L.J.; Lee, C.-Y.; Menozzi, E.; Schapira, A.H.V. Genetic Variations in GBA1 and LRRK2 Genes: Biochemical and Clinical Consequences in Parkinson Disease. Front Neurol 2022, 13, 971252. [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [CrossRef]
- Espay, A.J.; Kalia, L.V.; Gan-Or, Z.; Williams-Gray, C.H.; Bedard, P.L.; Rowe, S.M.; Morgante, F.; Fasano, A.; Stecher, B.; Kauffman, M.A.; et al. Disease Modification and Biomarker Development in Parkinson Disease: Revision or Reconstruction? Neurology 2020, 94, 481–494. [CrossRef]
- Kaitwad, R.; Gulwe, A.; Dipke, C.; Suryawanshi, H.; Murge, N.; Nalage, D. Current and Emerging Therapies for Parkinson’s Disease: Advances toward Disease Modification. Life Res 2025, 8, 22. [CrossRef]
- Rissardo, J.P.; Caprara, A.L. A Narrative Review on Biochemical Markers and Emerging Treatments in Prodromal Synucleinopathies. Clinics and Practice 2025, 15. [CrossRef]
- Stocchi, F.; Bravi, D.; Emmi, A.; Antonini, A. Parkinson Disease Therapy: Current Strategies and Future Research Priorities. Nat Rev Neurol 2024, 20, 695–707. [CrossRef]
- Costa, J.; Lunet, N.; Santos, C.; Santos, J.; Vaz-Carneiro, A. Caffeine Exposure and the Risk of Parkinson’s Disease: A Systematic Review and Meta-Analysis of Observational Studies. J Alzheimers Dis 2010, 20 Suppl 1, S221-238. [CrossRef]
- Torti, M.; Vacca, L.; Stocchi, F. Istradefylline for the Treatment of Parkinson’s Disease: Is It a Promising Strategy? Expert Opin Pharmacother 2018, 19, 1821–1828. [CrossRef]
- Lang Anthony E.; Siderowf Andrew D.; Macklin Eric A.; Poewe Werner; Brooks David J.; Fernandez Hubert H.; Rascol Olivier; Giladi Nir; Stocchi Fabrizio; Tanner Caroline M.; et al. Trial of Cinpanemab in Early Parkinson’s Disease. New England Journal of Medicine 2022, 387, 408–420. [CrossRef]
- Pagano, G.; Taylor, K.I.; Anzures-Cabrera, J.; Marchesi, M.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Azulay, J.-P.; et al. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N Engl J Med 2022, 387, 421–432. [CrossRef]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Agúndez, J.A.G. Coenzyme Q10 and Parkinsonian Syndromes: A Systematic Review. J Pers Med 2022, 12. [CrossRef]
- Attia; Ahmed, H.; Gadelkarim, M.; Morsi, M.; Awad, K.; Elnenny, M.; Ghanem, E.; El-Jaafary, S.; Negida, A. Meta-Analysis of Creatine for Neuroprotection Against Parkinson’s Disease. CNS Neurol Disord Drug Targets 2017, 16, 169–175. [CrossRef]
- Lin, J.; Pang, D.; Li, C.; Ou, R.; Yu, Y.; Cui, Y.; Huang, J.; Shang, H. Calcium Channel Blockers and Parkinson’s Disease: A Systematic Review and Meta-Analysis. Ther Adv Neurol Disord 2024, 17, 17562864241252713. [CrossRef]
- Dooley, M.; Markham, A. Pramipexole. A Review of Its Use in the Management of Early and Advanced Parkinson’s Disease. Drugs Aging 1998, 12, 495–514. [CrossRef]
- Zhu, J.; Chen, M. The Effect and Safety of Ropinirole in the Treatment of Parkinson Disease: A Systematic Review and Meta-Analysis. Medicine (Baltimore) 2021, 100, e27653. [CrossRef]
- Rajendran, A.; Reddy, A.J.; Bisaga, K.; Sommer, D.A.; Prakash, N.; Pokala, V.T.; Yu, Z.; Bachir, M.; Nawathey, N.; Brahmbhatt, T.; et al. A Systematic Review of the Usage of Rotigotine During Early and Advanced Stage Parkinson’s. Cureus 2023, 15, e36211. [CrossRef]
- Hickey, P.; Stacy, M. AAV2-Neurturin (CERE-120) for Parkinson’s Disease. Expert Opin Biol Ther 2013, 13, 137–145. [CrossRef]
- Jankovic, J.; Hunter, C. A Double-Blind, Placebo-Controlled and Longitudinal Study of Riluzole in Early Parkinson’s Disease. Parkinsonism Relat Disord 2002, 8, 271–276. [CrossRef]
- Schapira, A.H.V. Monoamine Oxidase B Inhibitors for the Treatment of Parkinson’s Disease: A Review of Symptomatic and Potential Disease-Modifying Effects. CNS Drugs 2011, 25, 1061–1071. [CrossRef]
- Chen, L.; Tao, Y.; Li, J.; Kang, M. Pioglitazone Use Is Associated with Reduced Risk of Parkinson’s Disease in Patients with Diabetes: A Systematic Review and Meta-Analysis. J Clin Neurosci 2022, 106, 154–158. [CrossRef]
- Yan, J.; Qiao, L.; Tian, J.; Liu, A.; Wu, J.; Huang, J.; Shen, M.; Lai, X. Effect of Statins on Parkinson’s Disease: A Systematic Review and Meta-Analysis. Medicine (Baltimore) 2019, 98, e14852. [CrossRef]
- Xie, X.; Yuan, P.; Kou, L.; Chen, X.; Li, J.; Li, Y. Nilotinib in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front Aging Neurosci 2022, 14, 996217. [CrossRef]
- Basile, M.S.; Bramanti, P.; Mazzon, E. Inosine in Neurodegenerative Diseases: From the Bench to the Bedside. Molecules 2022, 27. [CrossRef]
- McGhee, D.J.M.; Ritchie, C.W.; Zajicek, J.P.; Counsell, C.E. A Review of Clinical Trial Designs Used to Detect a Disease-Modifying Effect of Drug Therapy in Alzheimer’s Disease and Parkinson’s Disease. BMC Neurol 2016, 16, 92. [CrossRef]
- Macleod, A.D.; Counsell, C.E.; Ives, N.; Stowe, R. Monoamine Oxidase B Inhibitors for Early Parkinson’s Disease. Cochrane Database Syst Rev 2005, 2005, CD004898. [CrossRef]
- DATATOP: A Multicenter Controlled Clinical Trial in Early Parkinson’s Disease. Parkinson Study Group. Arch Neurol 1989, 46, 1052–1060. [CrossRef]
- Ward, C.D. Does Selegiline Delay Progression of Parkinson’s Disease? A Critical Re-Evaluation of the DATATOP Study. J Neurol Neurosurg Psychiatry 1994, 57, 217–220. [CrossRef]
- Olanow, C.W.; Rascol, O.; Hauser, R.; Feigin, P.D.; Jankovic, J.; Lang, A.; Langston, W.; Melamed, E.; Poewe, W.; Stocchi, F.; et al. A Double-Blind, Delayed-Start Trial of Rasagiline in Parkinson’s Disease. N Engl J Med 2009, 361, 1268–1278. [CrossRef]
- Jenner, P.; Langston, J.W. Explaining ADAGIO: A Critical Review of the Biological Basis for the Clinical Effects of Rasagiline. Mov Disord 2011, 26, 2316–2323. [CrossRef]
- Verschuur, C.V.M.; Suwijn, S.R.; Boel, J.A.; Post, B.; Bloem, B.R.; van Hilten, J.J.; van Laar, T.; Tissingh, G.; Munts, A.G.; Deuschl, G.; et al. Randomized Delayed-Start Trial of Levodopa in Parkinson’s Disease. N Engl J Med 2019, 380, 315–324. [CrossRef]
- Schapira, A.H.V.; McDermott, M.P.; Barone, P.; Comella, C.L.; Albrecht, S.; Hsu, H.H.; Massey, D.H.; Mizuno, Y.; Poewe, W.; Rascol, O.; et al. Pramipexole in Patients with Early Parkinson’s Disease (PROUD): A Randomised Delayed-Start Trial. Lancet Neurol 2013, 12, 747–755. [CrossRef]
- Parkinson Study Group CALM Cohort Investigators Long-Term Effect of Initiating Pramipexole vs Levodopa in Early Parkinson Disease. Arch Neurol 2009, 66, 563–570. [CrossRef]
- Stowe, R.L.; Ives, N.J.; Clarke, C.; van Hilten, J.; Ferreira, J.; Hawker, R.J.; Shah, L.; Wheatley, K.; Gray, R. Dopamine Agonist Therapy in Early Parkinson’s Disease. Cochrane Database Syst Rev 2008, CD006564. [CrossRef]
- Becker, C.; Jick, S.S.; Meier, C.R. Use of Antihypertensives and the Risk of Parkinson Disease. Neurology 2008, 70, 1438–1444. [CrossRef]
- Parkinson Study Group Phase II Safety, Tolerability, and Dose Selection Study of Isradipine as a Potential Disease-Modifying Intervention in Early Parkinson’s Disease (STEADY-PD). Mov Disord 2013, 28, 1823–1831. [CrossRef]
- Simuni, T. A Phase 3 Study of Isradipine as a Disease Modifying Agent in Patients with Early Parkinson’s Disease (STEADY-PD III): Final Study Results: 206. Mov Disord 2019, 34, S87.
- Siddiqi, F.H.; Menzies, F.M.; Lopez, A.; Stamatakou, E.; Karabiyik, C.; Ureshino, R.; Ricketts, T.; Jimenez-Sanchez, M.; Esteban, M.A.; Lai, L.; et al. Felodipine Induces Autophagy in Mouse Brains with Pharmacokinetics Amenable to Repurposing. Nat Commun 2019, 10, 1817. [CrossRef]
- Zheng, S.; Wang, Y.; Tang, S.; Guo, Y.; Ma, D.; Jiang, X. Mechanism of Nimodipine in Treating Neurodegenerative Diseases: In Silico Target Identification and Molecular Dynamic Simulation. Front Pharmacol 2025, 16, 1549953. [CrossRef]
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; Ravina, B.; et al. A Randomized Clinical Trial of High-Dosage Coenzyme Q10 in Early Parkinson Disease: No Evidence of Benefit. JAMA Neurol 2014, 71, 543–552. [CrossRef]
- Kieburtz, K.; Tilley, B.C.; Elm, J.J.; Babcock, D.; Hauser, R.; Ross, G.W.; Augustine, A.H.; Augustine, E.U.; Aminoff, M.J.; Bodis-Wollner, I.G.; et al. Effect of Creatine Monohydrate on Clinical Progression in Patients with Parkinson Disease: A Randomized Clinical Trial. JAMA 2015, 313, 584–593. [CrossRef]
- Rissardo, J.P.; Caprara, A.L.F. The Role of Vitamin D in Parkinson’s Disease: Evidence from Serum Concentrations, Supplementation, and VDR Gene Polymorphisms. NeuroSci 2025, 6, 130. [CrossRef]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.J.; Murphy, M.P.; Taylor, K.M. A Double-Blind, Placebo-Controlled Study to Assess the Mitochondria-Targeted Antioxidant MitoQ as a Disease-Modifying Therapy in Parkinson’s Disease. Mov Disord 2010, 25, 1670–1674. [CrossRef]
- Berven, H.; Kverneng, S.; Sheard, E.; Søgnen, M.; Af Geijerstam, S.A.; Haugarvoll, K.; Skeie, G.-O.; Dölle, C.; Tzoulis, C. NR-SAFE: A Randomized, Double-Blind Safety Trial of High Dose Nicotinamide Riboside in Parkinson’s Disease. Nat Commun 2023, 14, 7793. [CrossRef]
- Schwarzschild, M.A.; Ascherio, A.; Casaceli, C.; Curhan, G.C.; Fitzgerald, R.; Kamp, C.; Lungu, C.; Macklin, E.A.; Marek, K.; Mozaffarian, D.; et al. Effect of Urate-Elevating Inosine on Early Parkinson Disease Progression: The SURE-PD3 Randomized Clinical Trial. JAMA 2021, 326, 926–939. [CrossRef]
- Postuma, R.B.; Lang, A.E.; Munhoz, R.P.; Charland, K.; Pelletier, A.; Moscovich, M.; Filla, L.; Zanatta, D.; Rios Romenets, S.; Altman, R.; et al. Caffeine for Treatment of Parkinson Disease: A Randomized Controlled Trial. Neurology 2012, 79, 651–658. [CrossRef]
- Volc, D.; Poewe, W.; Kutzelnigg, A.; Lührs, P.; Thun-Hohenstein, C.; Schneeberger, A.; Galabova, G.; Majbour, N.; Vaikath, N.; El-Agnaf, O.; et al. Safety and Immunogenicity of the α-Synuclein Active Immunotherapeutic PD01A in Patients with Parkinson’s Disease: A Randomised, Single-Blinded, Phase 1 Trial. Lancet Neurol 2020, 19, 591–600. [CrossRef]
- Simuni, T.; Fiske, B.; Merchant, K.; Coffey, C.S.; Klingner, E.; Caspell-Garcia, C.; Lafontant, D.-E.; Matthews, H.; Wyse, R.K.; Brundin, P.; et al. Efficacy of Nilotinib in Patients With Moderately Advanced Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol 2021, 78, 312–320. [CrossRef]
- Imam, S.Z.; Trickler, W.; Kimura, S.; Binienda, Z.K.; Paule, M.G.; Slikker, W.J.; Li, S.; Clark, R.A.; Ali, S.F. Neuroprotective Efficacy of a New Brain-Penetrating C-Abl Inhibitor in a Murine Parkinson’s Disease Model. PLoS One 2013, 8, e65129. [CrossRef]
- Karuppagounder, S.S.; Wang, H.; Kelly, T.; Rush, R.; Nguyen, R.; Bisen, S.; Yamashita, Y.; Sloan, N.; Dang, B.; Sigmon, A.; et al. The C-Abl Inhibitor IkT-148009 Suppresses Neurodegeneration in Mouse Models of Heritable and Sporadic Parkinson’s Disease. Sci Transl Med 2023, 15, eabp9352. [CrossRef]
- Devos, D.; Labreuche, J.; Rascol, O.; Corvol, J.-C.; Duhamel, A.; Guyon Delannoy, P.; Poewe, W.; Compta, Y.; Pavese, N.; Růžička, E.; et al. Trial of Deferiprone in Parkinson’s Disease. N Engl J Med 2022, 387, 2045–2055. [CrossRef]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; et al. Targeting Chelatable Iron as a Therapeutic Modality in Parkinson’s Disease. Antioxid Redox Signal 2014, 21, 195–210. [CrossRef]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain Iron Chelation by Deferiprone in a Phase 2 Randomised Double-Blinded Placebo Controlled Clinical Trial in Parkinson’s Disease. Sci Rep 2017, 7, 1398. [CrossRef]
- Shachar, D.B.; Kahana, N.; Kampel, V.; Warshawsky, A.; Youdim, M.B.H. Neuroprotection by a Novel Brain Permeable Iron Chelator, VK-28, against 6-Hydroxydopamine Lession in Rats. Neuropharmacology 2004, 46, 254–263. [CrossRef]
- Vijiaratnam, N.; Girges, C.; Auld, G.; McComish, R.; King, A.; Skene, S.S.; Hibbert, S.; Wong, A.; Melander, S.; Gibson, R.; et al. Exenatide Once a Week versus Placebo as a Potential Disease-Modifying Treatment for People with Parkinson’s Disease in the UK: A Phase 3, Multicentre, Double-Blind, Parallel-Group, Randomised, Placebo-Controlled Trial. Lancet 2025, 405, 627–636. [CrossRef]
- Zeissler, M.-L.; Boey, T.; Chapman, D.; Rafaloff, G.; Dominey, T.; Raphael, K.G.; Buff, S.; Pai, H.V.; King, E.; Sharpe, P.; et al. Investigating Trial Design Variability in Trials of Disease-Modifying Therapies in Parkinson’s Disease: A Scoping Review Protocol. BMJ Open 2023, 13, e071641. [CrossRef]
- NINDS Exploratory Trials in Parkinson Disease (NET-PD) FS-ZONE Investigators Pioglitazone in Early Parkinson’s Disease: A Phase 2, Multicentre, Double-Blind, Randomised Trial. Lancet Neurol 2015, 14, 795–803. [CrossRef]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. JAMA 2020, 323, 844–853. [CrossRef]
- Mahlknecht, P.; Poewe, W. Pharmacotherapy for Disease Modification in Early Parkinson’s Disease: How Early Should We Be? J Parkinsons Dis 2024, 14, S407–S421. [CrossRef]
- Stocchi, F.; Olanow, C.W. Neuroprotection in Parkinson’s Disease: Clinical Trials. Ann Neurol 2003, 53 Suppl 3, S87-97; discussion S97-99. [CrossRef]
- Kalinderi, K.; Papaliagkas, V.; Fidani, L. GLP-1 Receptor Agonists: A New Treatment in Parkinson’s Disease. Int J Mol Sci 2024, 25. [CrossRef]
- Dhruv, N.T.; Robinson Schwartz, S.; Swanson-Fischer, C.; Cho, H.J.; Corlew, R.; Jakeman, L.; Laboissonniere, L.A.; Price, R.; Sarraf, S.; Sieber, B.-A.; et al. Mapping the Developmental Path for Parkinson’s Disease Therapeutics. NPJ Parkinsons Dis 2025, 11, 313. [CrossRef]
- Boucherie, D.M.; Duarte, G.S.; Machado, T.; Faustino, P.R.; Sampaio, C.; Rascol, O.; Ferreira, J.J. Parkinson’s Disease Drug Development Since 1999: A Story of Repurposing and Relative Success. J Parkinsons Dis 2021, 11, 421–429. [CrossRef]
- Kulkarni, S.R.; Thokchom, B.; Abbigeri, M.B.; Bhavi, S.M.; Singh, S.R.; Metri, N.; Yarajarla, R.B. The Role of L-DOPA in Neurological and Neurodegenerative Complications: A Review. Mol Cell Biochem 2025, 480, 5221–5242. [CrossRef]
- Guimarães, T.G.; Parmera, J.B.; Castro, M.A.A.; Cury, R.G.; Barbosa, E.R.; Kok, F. X-Linked Levodopa-Responsive Parkinsonism-Epilepsy Syndrome: A Novel PGK1 Mutation and Literature Review. Mov Disord Clin Pract 2024, 11, 556–566. [CrossRef]
- Chen, X.; Zhao, C.; Li, X.; Wang, T.; Li, Y.; Cao, C.; Ding, Y.; Dong, M.; Finci, L.; Wang, J.-H.; et al. Terazosin Activates Pgk1 and Hsp90 to Promote Stress Resistance. Nat Chem Biol 2015, 11, 19–25. [CrossRef]
- Cai, R.; Zhang, Y.; Simmering, J.E.; Schultz, J.L.; Li, Y.; Fernandez-Carasa, I.; Consiglio, A.; Raya, A.; Polgreen, P.M.; Narayanan, N.S.; et al. Enhancing Glycolysis Attenuates Parkinson’s Disease Progression in Models and Clinical Databases. J Clin Invest 2019, 129, 4539–4549. [CrossRef]
- Weber, M.A.; Sivakumar, K.; Tabakovic, E.E.; Oya, M.; Aldridge, G.M.; Zhang, Q.; Simmering, J.E.; Narayanan, N.S. Glycolysis-Enhancing α(1)-Adrenergic Antagonists Modify Cognitive Symptoms Related to Parkinson’s Disease. NPJ Parkinsons Dis 2023, 9, 32. [CrossRef]
- Kokotos, A.C.; Antoniazzi, A.M.; Unda, S.R.; Ko, M.S.; Park, D.; Eliezer, D.; Kaplitt, M.G.; De Camilli, P.; Ryan, T.A. Phosphoglycerate Kinase Is a Central Leverage Point in Parkinson’s Disease-Driven Neuronal Metabolic Deficits. Sci Adv 2024, 10, eadn6016. [CrossRef]
- Schultz, J.L.; Gander, P.E.; Workman, C.D.; Boles Ponto, L.L.; Cross, S.; Nance, C.S.; Groth, C.L.; Taylor, E.B.; Ernst, S.E.; Xu, J.; et al. A Dose-Finding Study Shows Terazosin Enhanced Energy Metabolism in Neurologically Healthy Adults. J Parkinsons Dis 2025, 15, 1253–1263. [CrossRef]
- Schultz, J.L.; Brinker, A.N.; Xu, J.; Ernst, S.E.; Tayyari, F.; Rauckhorst, A.J.; Liu, L.; Uc, E.Y.; Taylor, E.B.; Simmering, J.E.; et al. A Pilot to Assess Target Engagement of Terazosin in Parkinson’s Disease. Parkinsonism Relat Disord 2022, 94, 79–83. [CrossRef]
- Lamichhane, P.; Tariq, A.; Akhtar, A.N.; Raza, M.; Lamsal, A.B.; Agrawal, A. Risk of Parkinson’s Disease among Users of Alpha-Adrenergic Receptor Antagonists: A Systematic Review and Meta-Analysis. Ann Med Surg (Lond) 2024, 86, 3409–3415. [CrossRef]
- Lang, A.E.; Espay, A.J. Disease Modification in Parkinson’s Disease: Current Approaches, Challenges, and Future Considerations. Mov Disord 2018, 33, 660–677. [CrossRef]
- Vijiaratnam, N.; Foltynie, T. How Should We Be Using Biomarkers in Trials of Disease Modification in Parkinson’s Disease? Brain 2023, 146, 4845–4869. [CrossRef]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Pitzer, K.; Fiske, B.; Navangul, A.; Beissert, K.; Pilcicka, A.; Fuest, R.; Wyse, R.K.; et al. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2024 Update. J Parkinsons Dis 2024, 14, 899–912. [CrossRef]
- Fiske, B.; Klapper, K.; Schorken, Z.; Sweet-Byrnes, M. Parkinson’s Priority Therapeutic Clinical Pipeline Report 2025.
- Pitton Rissardo, J.; Fornari Caprara, A.L. Protein Aggregation and Proteostasis Failure in Neurodegenerative Diseases: Mechanisms, Structural Insights, and Therapeutic Strategies. Preprints 2025. [CrossRef]
- Wong, Y.C.; Krainc, D. α-Synuclein Toxicity in Neurodegeneration: Mechanism and Therapeutic Strategies. Nat Med 2017, 23, 1–13. [CrossRef]
- Fabbri, M.; Corvol, J.C.; Rascol, O. Disease-Modifying Therapies in Parkinson’s Disease. Neurol Clin 2025, 43, 319–340. [CrossRef]
- Han, Q.-Q.; Le, W. NLRP3 Inflammasome-Mediated Neuroinflammation and Related Mitochondrial Impairment in Parkinson’s Disease. Neurosci Bull 2023, 39, 832–844. [CrossRef]
- Yao, J.; Wang, Z.; Song, W.; Zhang, Y. Targeting NLRP3 Inflammasome for Neurodegenerative Disorders. Mol Psychiatry 2023, 28, 4512–4527. [CrossRef]
- Athauda, D.; Foltynie, T. Insulin Resistance and Parkinson’s Disease: A New Target for Disease Modification? Prog Neurobiol 2016, 145–146, 98–120. [CrossRef]
- Werner, M.H.; Olanow, C.W. Parkinson’s Disease Modification Through Abl Kinase Inhibition: An Opportunity. Mov Disord 2022, 37, 6–15. [CrossRef]
- Barker, R.A.; Lao-Kaim, N.P.; Guzman, N.V.; Athauda, D.; Bjartmarz, H.; Björklund, A.; Church, A.; Cutting, E.; Daft, D.; Dayal, V.; et al. The TransEuro Open-Label Trial of Human Fetal Ventral Mesencephalic Transplantation in Patients with Moderate Parkinson’s Disease. Nat Biotechnol 2025. [CrossRef]
- Barker, R.A.; Parmar, M.; Studer, L.; Takahashi, J. Human Trials of Stem Cell-Derived Dopamine Neurons for Parkinson’s Disease: Dawn of a New Era. Cell Stem Cell 2017, 21, 569–573. [CrossRef]
- Takahashi, J. Stem Cell Therapy for Parkinson’s Disease. Expert Rev Neurother 2007, 7, 667–675. [CrossRef]
- Zhao, H.T.; John, N.; Delic, V.; Ikeda-Lee, K.; Kim, A.; Weihofen, A.; Swayze, E.E.; Kordasiewicz, H.B.; West, A.B.; Volpicelli-Daley, L.A. LRRK2 Antisense Oligonucleotides Ameliorate α-Synuclein Inclusion Formation in a Parkinson’s Disease Mouse Model. Mol Ther Nucleic Acids 2017, 8, 508–519. [CrossRef]
- Christine, C.W.; Starr, P.A.; Larson, P.S.; Eberling, J.L.; Jagust, W.J.; Hawkins, R.A.; VanBrocklin, H.F.; Wright, J.F.; Bankiewicz, K.S.; Aminoff, M.J. Safety and Tolerability of Putaminal AADC Gene Therapy for Parkinson Disease. Neurology 2009, 73, 1662–1669. [CrossRef]
- Bartus, R.T. Gene Therapy for Parkinson’s Disease: A Decade of Progress Supported by Posthumous Contributions from Volunteer Subjects. Neural Regen Res 2015, 10, 1586–1588. [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol Ther 2021, 29, 464–488. [CrossRef]
- Kordower, J.H.; Bjorklund, A. Trophic Factor Gene Therapy for Parkinson’s Disease. Mov Disord 2013, 28, 96–109. [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Glucocerebrosidase and Parkinson Disease: Molecular, Clinical, and Therapeutic Implications. Neuroscientist 2018, 24, 540–559. [CrossRef]
- LeWitt, P.A.; Rezai, A.R.; Leehey, M.A.; Ojemann, S.G.; Flaherty, A.W.; Eskandar, E.N.; Kostyk, S.K.; Thomas, K.; Sarkar, A.; Siddiqui, M.S.; et al. AAV2-GAD Gene Therapy for Advanced Parkinson’s Disease: A Double-Blind, Sham-Surgery Controlled, Randomised Trial. Lancet Neurol 2011, 10, 309–319. [CrossRef]
- Kim, E.; Kim, H.; Jedrychowski, M.P.; Bakiasi, G.; Park, J.; Kruskop, J.; Choi, Y.; Kwak, S.S.; Quinti, L.; Kim, D.Y.; et al. Irisin Reduces Amyloid-β by Inducing the Release of Neprilysin from Astrocytes Following Downregulation of ERK-STAT3 Signaling. Neuron 2023, 111, 3619-3633.e8. [CrossRef]
- Mullin, S.; Stokholm, M.G.; Hughes, D.; Mehta, A.; Parbo, P.; Hinz, R.; Pavese, N.; Brooks, D.J.; Schapira, A.H.V. Brain Microglial Activation Increased in Glucocerebrosidase (GBA) Mutation Carriers without Parkinson’s Disease. Mov Disord 2021, 36, 774–779. [CrossRef]
- Menozzi, E.; Schapira, A.H.V. Prospects for Disease Slowing in Parkinson Disease. Annu Rev Pharmacol Toxicol 2025, 65, 237–258. [CrossRef]
- Schiffmann, R.; Cox, T.M.; Dedieu, J.-F.; Gaemers, S.J.M.; Hennermann, J.B.; Ida, H.; Mengel, E.; Minini, P.; Mistry, P.; Musholt, P.B.; et al. Venglustat Combined with Imiglucerase for Neurological Disease in Adults with Gaucher Disease Type 3: The LEAP Trial. Brain 2023, 146, 461–474. [CrossRef]
- Kaplitt, M.G.; Feigin, A.; Tang, C.; Fitzsimons, H.L.; Mattis, P.; Lawlor, P.A.; Bland, R.J.; Young, D.; Strybing, K.; Eidelberg, D.; et al. Safety and Tolerability of Gene Therapy with an Adeno-Associated Virus (AAV) Borne GAD Gene for Parkinson’s Disease: An Open Label, Phase I Trial. Lancet 2007, 369, 2097–2105. [CrossRef]
- Niethammer, M.; Tang, C.C.; LeWitt, P.A.; Rezai, A.R.; Leehey, M.A.; Ojemann, S.G.; Flaherty, A.W.; Eskandar, E.N.; Kostyk, S.K.; Sarkar, A.; et al. Long-Term Follow-up of a Randomized AAV2-GAD Gene Therapy Trial for Parkinson’s Disease. JCI Insight 2017, 2, e90133. [CrossRef]
- Niethammer, M.; Tang, C.C.; Vo, A.; Nguyen, N.; Spetsieris, P.; Dhawan, V.; Ma, Y.; Small, M.; Feigin, A.; During, M.J.; et al. Gene Therapy Reduces Parkinson’s Disease Symptoms by Reorganizing Functional Brain Connectivity. Sci Transl Med 2018, 10, eaau0713. [CrossRef]
- Khan, S.; Aldawood, Y.O.; Kazi, N.; Sideeque, S.; Ansari, N.; Mohammed, H.; Byroju, V.V.; Caprara, A.L.F.; Rissardo, J.P. Brain Structural and Functional Alteration in Movement Disorders: A Narrative Review of Markers and Their Dynamic Changes. NeuroMarkers 2025, 100130. [CrossRef]
- Marks, W.J.J.; Bartus, R.T.; Siffert, J.; Davis, C.S.; Lozano, A.; Boulis, N.; Vitek, J.; Stacy, M.; Turner, D.; Verhagen, L.; et al. Gene Delivery of AAV2-Neurturin for Parkinson’s Disease: A Double-Blind, Randomised, Controlled Trial. Lancet Neurol 2010, 9, 1164–1172. [CrossRef]
- Warren Olanow, C.; Bartus, R.T.; Baumann, T.L.; Factor, S.; Boulis, N.; Stacy, M.; Turner, D.A.; Marks, W.; Larson, P.; Starr, P.A.; et al. Gene Delivery of Neurturin to Putamen and Substantia Nigra in Parkinson Disease: A Double-Blind, Randomized, Controlled Trial. Ann Neurol 2015, 78, 248–257. [CrossRef]
- Witt, J.; Marks, W.J.J. An Update on Gene Therapy in Parkinson’s Disease. Curr Neurol Neurosci Rep 2011, 11, 362–370. [CrossRef]
- Sehara, Y.; Fujimoto, K.-I.; Ikeguchi, K.; Katakai, Y.; Ono, F.; Takino, N.; Ito, M.; Ozawa, K.; Muramatsu, S.-I. Persistent Expression of Dopamine-Synthesizing Enzymes 15 Years After Gene Transfer in a Primate Model of Parkinson’s Disease. Hum Gene Ther Clin Dev 2017, 28, 74–79. [CrossRef]
- Bankiewicz, K.S.; Forsayeth, J.; Eberling, J.L.; Sanchez-Pernaute, R.; Pivirotto, P.; Bringas, J.; Herscovitch, P.; Carson, R.E.; Eckelman, W.; Reutter, B.; et al. Long-Term Clinical Improvement in MPTP-Lesioned Primates after Gene Therapy with AAV-hAADC. Mol Ther 2006, 14, 564–570. [CrossRef]
- Kojima, K.; Nakajima, T.; Taga, N.; Miyauchi, A.; Kato, M.; Matsumoto, A.; Ikeda, T.; Nakamura, K.; Kubota, T.; Mizukami, H.; et al. Gene Therapy Improves Motor and Mental Function of Aromatic L-Amino Acid Decarboxylase Deficiency. Brain 2019, 142, 322–333. [CrossRef]
- Hwu, W.-L.; Muramatsu, S.; Tseng, S.-H.; Tzen, K.-Y.; Lee, N.-C.; Chien, Y.-H.; Snyder, R.O.; Byrne, B.J.; Tai, C.-H.; Wu, R.-M. Gene Therapy for Aromatic L-Amino Acid Decarboxylase Deficiency. Sci Transl Med 2012, 4, 134ra61. [CrossRef]
- Christine, C.W.; Bankiewicz, K.S.; Van Laar, A.D.; Richardson, R.M.; Ravina, B.; Kells, A.P.; Boot, B.; Martin, A.J.; Nutt, J.; Thompson, M.E.; et al. Magnetic Resonance Imaging-Guided Phase 1 Trial of Putaminal AADC Gene Therapy for Parkinson’s Disease. Ann Neurol 2019, 85, 704–714. [CrossRef]
- Pitton Rissardo, J.; Fornari Caprara, A.L. Disease-Modifying Trials in Parkinson’s Disease: Challenges, Lessons, and Future Directions. Preprints 2025. [CrossRef]
- Fabbri, M.; Rascol, O.; Foltynie, T.; Carroll, C.; Postuma, R.B.; Porcher, R.; Corvol, J.C. Advantages and Challenges of Platform Trials for Disease Modifying Therapies in Parkinson’s Disease. Mov Disord 2024, 39, 1468–1477. [CrossRef]
- Simuni, T.; Coffey, C.; Siderowf, A.; Tanner, C.; Marek, K.; Brumm, M.; Investigators, P.; Chowdhury, S.; Sherer, T.; Kopil, C. Path To Prevention (P2P) Therapeutics Platform Trial in Biomarker Defined Prodromal Parkinson’s Disease: Study Design. Mov Disord 2023, 38, S50–S51.
- Rissardo, J.P.; Fornari Caprara, A.L. Alpha-Synuclein Seed Amplification Assays in Parkinson’s Disease: A Systematic Review and Network Meta-Analysis. Clin Pract 2025, 15. [CrossRef]
- Parnetti, L.; Bellomo, G.; Gaetani, L. Unlocking the Full Clinical Potential of the α-Synuclein Seed Amplification Assay. Lancet Neurol 2025, 24, 559–561. [CrossRef]
- Pedersen, C.C.; Ushakova, A.; Alves, G.; Tysnes, O.-B.; Blennow, K.; Zetterberg, H.; Maple-Grødem, J.; Lange, J. Serum Neurofilament Light at Diagnosis: A Prognostic Indicator for Accelerated Disease Progression in Parkinson’s Disease. NPJ Parkinsons Dis 2024, 10, 162. [CrossRef]
- Williams-Gray, C.H.; Wijeyekoon, R.; Yarnall, A.J.; Lawson, R.A.; Breen, D.P.; Evans, J.R.; Cummins, G.A.; Duncan, G.W.; Khoo, T.K.; Burn, D.J.; et al. Serum Immune Markers and Disease Progression in an Incident Parkinson’s Disease Cohort (ICICLE-PD). Mov Disord 2016, 31, 995–1003. [CrossRef]
- Pardoel, S.; Kofman, J.; Nantel, J.; Lemaire, E.D. Wearable-Sensor-Based Detection and Prediction of Freezing of Gait in Parkinson’s Disease: A Review. Sensors (Basel) 2019, 19. [CrossRef]
- Lipsmeier, F.; Taylor, K.I.; Postuma, R.B.; Volkova-Volkmar, E.; Kilchenmann, T.; Mollenhauer, B.; Bamdadian, A.; Popp, W.L.; Cheng, W.-Y.; Zhang, Y.-P.; et al. Reliability and Validity of the Roche PD Mobile Application for Remote Monitoring of Early Parkinson’s Disease. Sci Rep 2022, 12, 12081. [CrossRef]
- Wayland, R.; Meyer, R.; Tang, K. Speech Markers of Parkinson’s Disease: Phonological Features and Acoustic Measures. Brain Sci 2025, 15. [CrossRef]
- Sharma, R.; Gulati, A.; Chopra, K. Era of Surrogate Endpoints and Accelerated Approvals: A Comprehensive Review on Applicability, Uncertainties, and Challenges from Regulatory, Payer, and Patient Perspectives. Eur J Clin Pharmacol 2025, 81, 605–623. [CrossRef]
- Getz, K.A.; Campo, R.A. New Benchmarks Characterizing Growth in Protocol Design Complexity. Ther Innov Regul Sci 2018, 52, 22–28. [CrossRef]
- Wyse, R.K.; Isaacs, T.; Barker, R.A.; Cookson, M.R.; Dawson, T.M.; Devos, D.; Dexter, D.T.; Duffen, J.; Federoff, H.; Fiske, B.; et al. Twelve Years of Drug Prioritization to Help Accelerate Disease Modification Trials in Parkinson’s Disease: The International Linked Clinical Trials Initiative. J Parkinsons Dis 2024, 14, 657–666. [CrossRef]
Figure 1.
Clinical Pipeline of Disease-Modifying Therapies in Parkinson’s Disease. Nested donut chart illustrating the distribution of investigational programs by therapeutic category (outer ring) and clinical phase (inner ring). Data derived from The Parkinson’s Hope List (last updated November 2025 by McFarthing et al. [79]. Abbreviations: NCE, new chemical entity; Ref, reformulation; Rep, repurposed.
Figure 1.
Clinical Pipeline of Disease-Modifying Therapies in Parkinson’s Disease. Nested donut chart illustrating the distribution of investigational programs by therapeutic category (outer ring) and clinical phase (inner ring). Data derived from The Parkinson’s Hope List (last updated November 2025 by McFarthing et al. [79]. Abbreviations: NCE, new chemical entity; Ref, reformulation; Rep, repurposed.
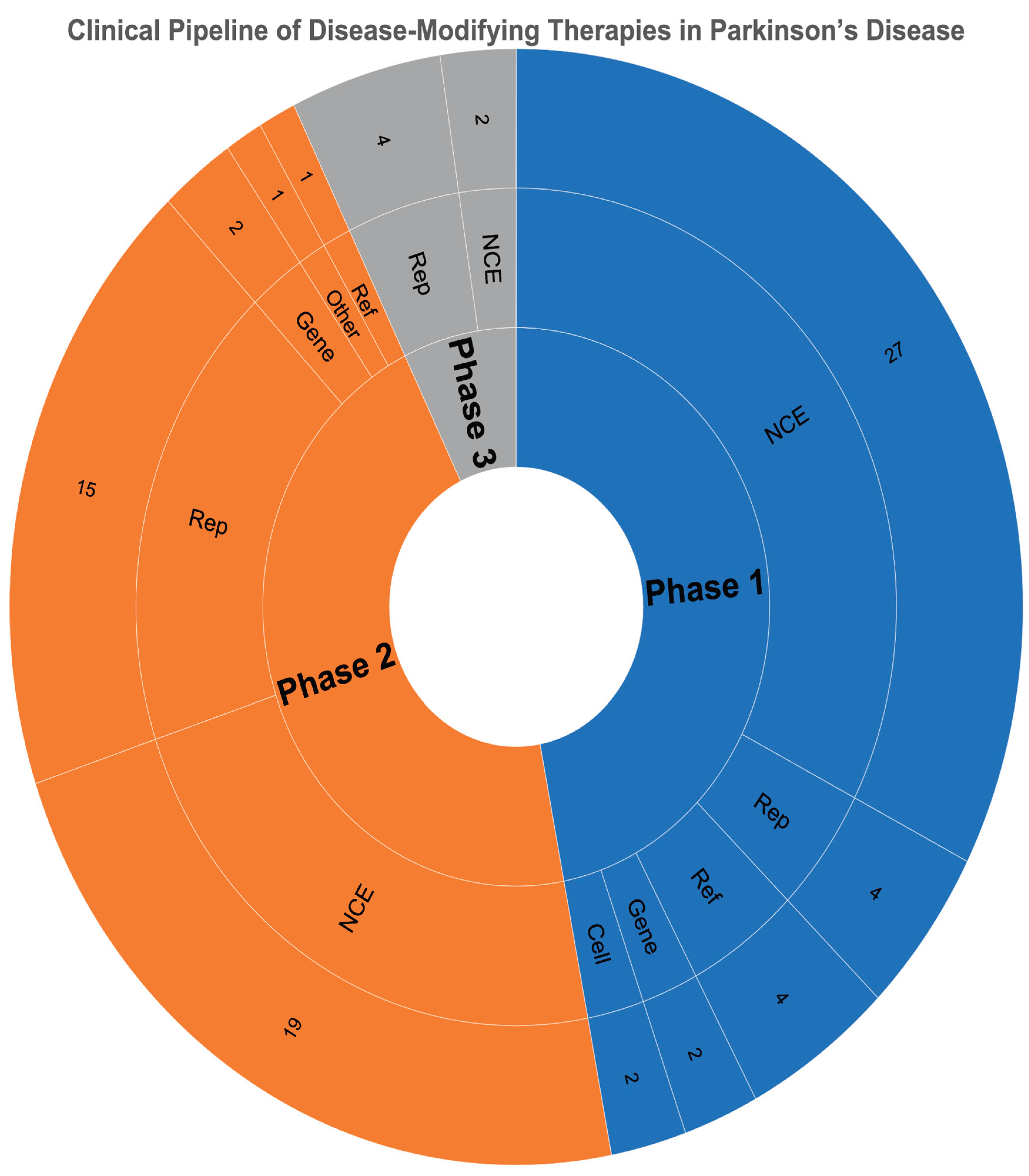
Table 1.
The Long List of Failed Disease-Modifying Trials in PD.
| Class | Medication | MOA | Possible failure cause | Note | NCT/ References |
| Adenosine A₂A receptors | Caffeine | Non-selective adenosine receptor antagonist | Epidemiological signal not replicated in RCTs | Observational benefit not confirmed | NCT01738178 Costa et al. [12] |
| Istradefylline | Selective A₂A receptor antagonist | Symptomatic benefit only; no DME | Approved for motor fluctuations, not progression | NCT00199433 Torti et al. [13] |
|
| Anti-αSyn monoclonal antibody | Cinpanemab (BIIB054) | Binds the N-terminal region of αSyn | Lack of target engagement or insufficient CNS penetration | SPARK (Phase II) stopped for lack of efficacy | NCT03318523 Lang et al. [14] |
| Prasinezumab (RO7046015/PRX002) | Targets the C-terminal region of αSyn | Possible late intervention; limited clinical effect | PASADENA did not meet primary endpoint (signals on Part III; ongoing OLE); PADOVA primary negative | NCT03100149 Pagano et al. [15] |
|
| Coenzyme Q10 (CoQ10) | Mitochondrial electron transport cofactor; antioxidant | Poor CNS bioavailability; inadequate oxidative stress modulation | QE3 stopped for futility; no benefit | NCT00740714 Jiménez-Jiménez et al. [16] |
|
| Creatine | Cellular energy buffer (phosphocreatine system) | No measurable neuroprotection | NET-PD LS-1 terminated for futility | NCT00449865 Attia et al. [17] |
|
| Calcium channel blocker | Isradipine | Dihydropyridine L-type Ca²⁺ channel blocker | Dose limited by hypotension; insufficient nigral protection | Phase III (STEADY-PD III) negative for slowing progression | NCT02168842 Lin et al. [18] |
| Dopamine agonists | Pramipexole | D2/D3 receptor agonist | Delayed-start design; no biomarker confirmation | Phase III (PROUD) negative for slowing progression | NCT00321854 Dooley et al. [19] |
| Ropinirole | D2/D3 receptor agonist | Symptomatic improvement appears slower; no biomarker confirmation | REAL-PD trial negative; no evidence of neuroprotection | NCT00243855 Zhu et al. [20] |
|
| Rotigotine | Non-ergoline dopamine agonist | Symptomatic improvement appears slower; no biomarker confirmation | No significant difference in progression; exploratory analyses inconclusive | NCT00474058 Rajendran et al. [21] |
|
| Gene therapy | CERE-120 (AAV2-neurturin) | Gene therapy delivering neurturin | Limited axonal transport; poor distribution | Phase 2 trials failed primary endpoint; post-mortem shows limited expression | NCT00985517 Hickey et al. [22] |
| Glutamate antagonists | Riluzole | Glutamate release inhibitor | Insufficient effect on excitotoxicity | Small trials negative; no large confirmatory study | NCT00013624 Jankovic et al. [23] |
| MAO-B inhibitors | Selegiline / Rasagiline | Irreversible MAO-B inhibition (dopamine metabolism) | Symptomatic only; delayed-start designs inconclusive | ADAGIO failed to confirm DME | NCT00256204 Schapira et al. [24] |
| PPAR-γ agonists | Pioglitazone | PPAR-γ agonist (metabolic/inflammatory modulation) | Weak CNS penetration; insufficient anti-inflammatory effect | Phase II futility trial: unlikely to modify progression | NCT01280123 Chen et al. [25] |
| Statins | Simvastatin | HMG-CoA reductase inhibitor; pleiotropic anti-inflammatory effect | Observational signal not replicated; possible off-target toxicity | PD STAT trial negative | NCT02787590 Yan et al. [26] |
| Tyrosine kinase inhibitor | Nilotinib | c-Abl inhibitor (proteostasis/ autophagy) | Biomarker changes without clinical benefit | Multicenter RCT: no efficacy; biomarker shifts without clinical benefit | NCT03205488 Xie et al. [27] |
| Urate precursor | Inosine | Antioxidant hypothesis | No clinical benefit despite biomarker elevation | SURE-PD3: no clinical benefit | NCT02642393 Basile et al. [28] |
Table 2.
Repurposing Drugs for Disease-Modifying Trials in PD.
| Medication | Mechanistic rationale in PD | Preclinical evidence | Epidemiological evidence | NCT |
| Ambroxol | Chaperone for GCase; ↑ lysosomal function; ↓ αSyn aggregation | ↑ GCase activity & ↓ αSyn pathology in GBA-mutant PD mouse models | Observational studies suggest benefit | NCT06193421; NCT05830396; NCT05778617; NCT05287503; NCT02941822; NCT02914366 |
| Allopregnanolone | ↑ GABAergic signaling & neurogenesis; ↓ DA neuronal loss & mitochondrial dysfunction | ↑ DA neuron survival; ↓ inflammation in toxin-induced PD models | Limited population data | NCT06263010 |
| Carvedilol | β-adrenergic blockade can ↓ ROS & inflammation | ↓ DA neuronal loss & oxidative markers in MPTP mouse models | β-blocker use linked to lower PD incidence in some cohorts | NCT03775096 |
| Cilostazol | PDE-3 inhibition ↑ cAMP, ↑ cerebral perfusion, and ↓ apoptosis in DA neurons | ↑ DA neuron survival & motor function in 6-OHDA rat models | Stroke prevention cohorts suggest ↓ PD risk with PDE inhibitors | NCT06612593 |
| Doxycycline | ↓ microglial activation & αSyn oligomerization | ↓ αSyn oligomerization and inflammation in transgenic PD models | Antibiotic exposure studies show mixed associations with PD risk | NCT05492019 |
| DPP-4 inhibitors (sitagliptin, vildagliptin) | ↑ GLP-1 signaling, ↑ insulin sensitivity, ↓ inflammation | ↑ motor function & ↓ DA loss in diabetic PD models | Diabetes cohorts suggest GLP-1 agonists ↓ PD incidence | NCT06263673; NCT06951334 |
| Febuxostat | ↓ ROS & UA imbalance, ↓ oxidative stress in DA neurons | ↓ ROS and preserved DA neurons in rotenone-induced PD models | ↑ UA associated with ↓ PD risk | NCT07170475 |
| Fexofenadine | ↓ inflammation & glial activation | ↓ microglial activation and ROS in PD rodent models | Antihistamine use linked to ↓ PD risk in populational studies | NCT06785298 |
| Gemfibrozil | PPAR-α activation ↑ lipid metabolism & mitochondrial function | ↓ inflammation & ↑ lipid metabolism in PD models | Lipid-lowering drugs show mixed associations with PD risk | NCT05931484 |
| Hydroxychloroquine | Modulates autophagy and lysosomal clearance of misfolded proteins, potentially ↓ αSyn burden | ↑ clearance of misfolded proteins and ↓ αSyn burden in vitro | Limited data; autoimmune cohorts show variable PD risk | NCT06816810 |
| Lithium | GSK-3β inhibition ↑ neurotrophic signaling & autophagy | ↑ autophagy & DA neuron survival in PD models | Bipolar disorder cohorts suggest lithium may ↓ PD risk | NCT06592014; NCT06339034; NCT06099886; NCT04273932 |
| Metformin | AMPK activation ↑ mitochondrial bioenergetics & ↓ ROS | ↓ ROS & ↑ motor function in PD rodent models | Diabetes cohorts show metformin use linked to ↓ PD risk | NCT07229651, NCT07055958 |
| Montelukast | ↓ inflammation & BBB disruption in PD | ↓ inflammation; preserve DA neurons in PD models | Asthma cohorts suggest leukotriene antagonists may reduce PD risk | NCT06113640 |
| Nicotinamide riboside | NAD⁺ precursor ↑ mitochondrial function & sirtuin activity | ↑ neuronal survival & ROS in PD models | Limited epidemiological data | NCT05589766; NCT05546567; NCT05344404; NCT04044131; NCT03816020; NCT03568968 |
| Sargramostim | GM-CSF modulates microglial phenotype toward neuroprotection | ↑ neuroprotective microglia & ↓ DA loss in PD models | No large-scale epidemiological data | NCT05677633; NCT03790670; NCT01882010 |
| Semaglutide | ↓ inflammation & ROS, supporting DA neuron survival | ↑ DA neuron survival & motor function in PD models | Diabetes cohorts show GLP-1 agonists associated with ↓ PD risk | NCT03659682 |
| Telmisartan | Angiotensin II receptor blockade and PPAR-γ activation ↓ inflammation & ROS in PD | ↓ROS and ↑ mitochondrial function in PD models | Hypertension cohorts suggest ARBs may lower PD risk | NCT07207057 |
| Terazosin | Activates PGK1, ↑ glycolysis & ATP production, improving neuronal energy metabolism in PD | ↑ neuronal energy metabolism & survival in PD models | Observational studies show α1-blockers linked to reduced PD risk | NCT07207057; NCT05855577; NCT05109364; NCT04386317; NCT03905811 |
| Tocotrienols | Potent antioxidant properties ↓ lipid peroxidation & ROS in DA neurons | Protected DA neurons from oxidative damage in PD models | Vitamin E intake inversely associated with PD risk | NCT04491383; NCT01923584 |
| Vinpocetine | PDE inhibition ↑ cerebral blood flow and ↓ inflammation, supporting neuronal metabolism | ↓ inflammation & ↑ motor function in PD models | Limited population data | NCT07229664 |
Abbreviations: DA, dopamine; GCase, glucocerebrosidase; ROS, reactive oxygen species; UA, uric acid; ↑, increase/upregulate; ↓, decrease/downregulate.
Table 3.
Gene therapy targets and key features in PD.
| Mechanism | Drug | Key Feature | Key Challenges | Development Stage |
| Local production of dopamine | TH | Rate-limiting enzyme for dopamine production | Invasive surgery; limited non-motor benefit | Phase I/II trials |
| GCH | Cofactor synthesis for TH | Phase I/II trials | ||
| AADC | Approved for AADC deficiency; tested in PD | Phase I/II trials | ||
| Protection of dopaminergic neurons | GDNF | Neurotrophic factor delivery | Advanced disease limits efficacy | Phase II (negative) |
| Neurturin (CERE-120) | Dual-site infusion (putamen + SN) | Poor axonal transport | Phase II (negative) | |
| GBA | Targets glucocerebrosidase activity | Biomarker validation | Early-phase trials | |
| Suppression of STN hyperactivity | GAD-65 and GAD67 | Boosts GABA synthesis for circuit modulation | Modest benefit vs DBS | Phase I/II trials |
| Reduction of αSyn | Irisin | Stimulates neprilysin release | Translational gap | Preclinical |
| Neprilysin | Direct proteolytic degradation of aggregates | Off-target effects | Preclinical |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



