Submitted:
21 December 2025
Posted:
22 December 2025
You are already at the latest version
Abstract
Tumor evolution under immune pressure follows the logic of cancer immunoediting—elimination, equilibrium, and escape—in which immune surveillance removes highly immunogenic clones while selecting for variants capable of persistence and progression. Clinically, this evolutionary outcome is often operationalized as an immune-phenotype continuum, frequently simplified into “hot” (immune-inflamed) and “cold” (immune-excluded or immune-desert) tumors. This mini-review uses the provided schematic of elimination–equilibrium–escape (Figure 1) to organize tumor defense strategies into two archetypes: (i) immune hiding (“cold” tumors)—minimizing immune recognition through impaired priming, trafficking, and antigen presentation; and (ii) active immune resistance (“hot” tumors)—withstanding immune attack through adaptive checkpoint induction (e.g., PD-L1), immunosuppressive stromal/myeloid circuits, and genetic/epigenetic escape from interferon and antigen presentation pathways. We highlight mechanistic nodes that repeatedly govern the cold↔hot state and propose a practical framework to match intervention strategy to dominant failure mode: initiate immunity (convert cold to inflamed) versus release brakes (overcome adaptive resistance in hot tumors). We also briefly situate innate-like stress surveillance (e.g., NKG2D-mediated recognition) within immunoediting, emphasizing how tumors can evade both T cell– and NK cell–mediated control by altering ligand availability and microenvironmental context.
Keywords:
cancer immunoediting
; hot tumors
; cold tumors
; immune exclusion
; immune desert
; PD-1/PD-L1
; antigen presentation
; IFN-γ–JAK–STAT
; cGAS–STING
; WNT/β-catenin
; TGF-β
; innate immunity
; NKG2D
1. Conceptual Framework: Immunoediting and Tumor “Defense Doctrines”
Cancer immunoediting describes how the immune system can both protect the host and shape tumor evolution via elimination, equilibrium, and escape phases. The clinical “hot vs cold” dichotomy overlays this framework by describing the immune landscape at a given timepoint—whether immune effectors are present and engaged (hot) or absent/excluded (cold). Contemporary consensus work recommends moving beyond a binary label toward spatially explicit phenotypes such as immune-inflamed, immune-excluded, and immune-desert states.
A useful way to interpret tumor immune evasion is that tumors tend to adopt one (or a mixture) of two defense doctrines:
- Hide (“cold”): avoid detection or prevent immune entry/priming so that elimination never becomes a sustained effector response.
- Fight (“hot”): tolerate immune infiltration but deploy suppressive/adaptive countermeasures that neutralize killing and select for resistant clones.
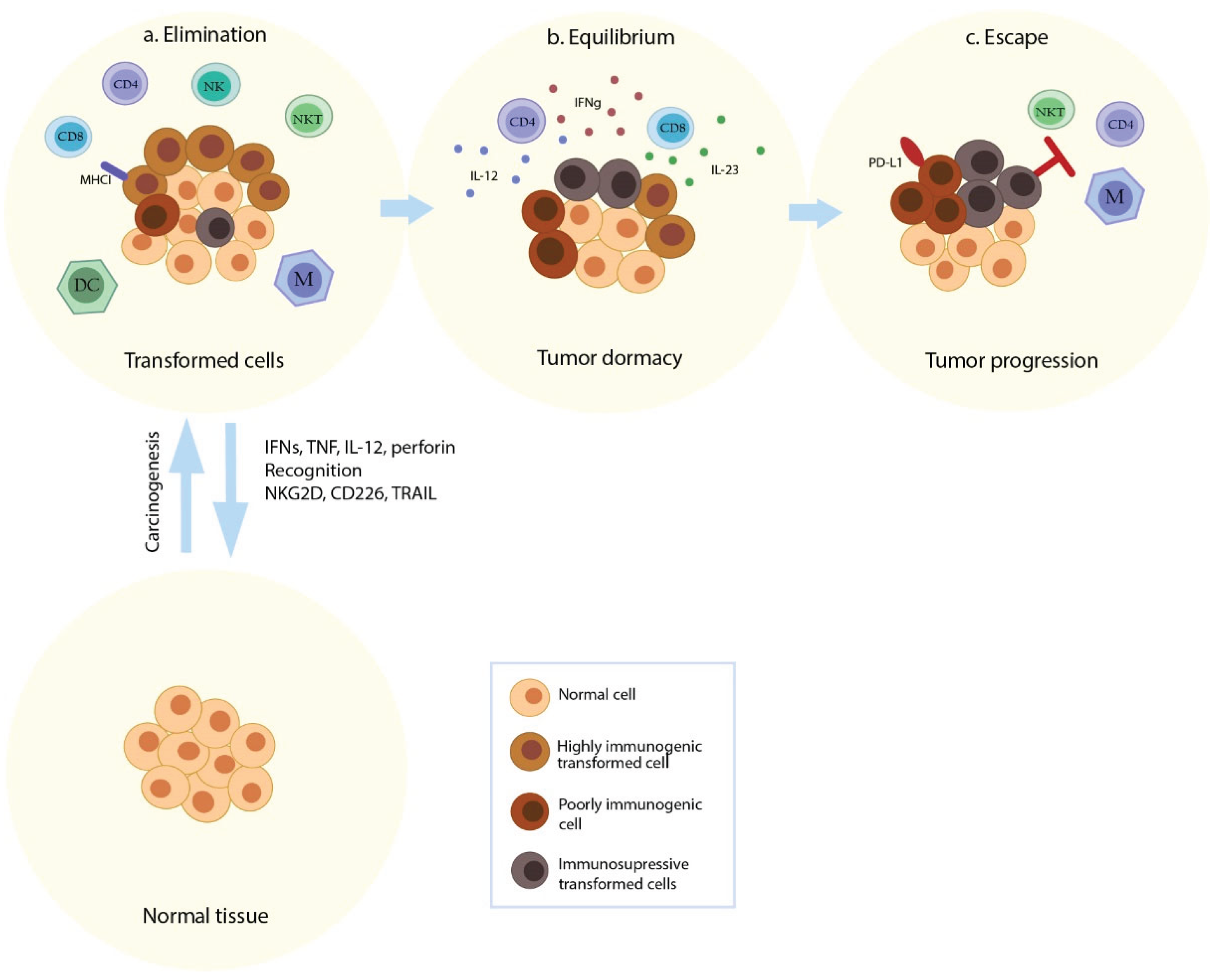
Figure 1 provides an intuitive map of this progression: immune mediators and recognition pathways dominate early (elimination), a constrained steady state can occur (equilibrium), and immune escape mechanisms (e.g., PD-L1; reduced antigen presentation) accompany progression.
2. What “Cold” Means Mechanistically: Tumors That Minimize Immune Engagement
“Cold tumors” typically correspond to immune-desert (few immune cells) or immune-excluded (immune cells present but restricted to stroma/periphery) phenotypes. While diverse, cold states often share failures in one or more of three requirements: (i) antigenicity/visibility, (ii) priming, and (iii) trafficking/entry.
2.1. Failed Innate Sensing and Deficient Priming
A T cell–inflamed microenvironment is strongly linked to effective antigen presentation and dendritic-cell (DC)–driven priming. Reviews of the cGAS–STING axis describe how cytosolic DNA sensing can promote type I interferon programs, chemokine production, and DC activation that support T-cell inflamed tumors. When these pathways are weak or suppressed, tumors can remain immunologically quiet even if neoantigens exist.
2.2. Tumor-Intrinsic Programs That Enforce Non-Inflamed (“Excluded/desert”) States
Tumor-intrinsic signaling can actively prevent immune infiltration. The WNT/β-catenin pathway is repeatedly linked to non–T cell-inflamed tumors and immune exclusion, including through impaired recruitment or function of key DC subsets and chemokine programs needed for effector entry.
2.3. Stromal and Vascular “Physical Immunology”
A frequent cold-tumor mechanism is not the absence of immunity, but its mislocalization—immune cells trapped in stroma or vasculature-adjacent regions without tumor penetration. A broad literature implicates TGF-β–driven stromal programs and cancer-associated fibroblasts (CAFs) in immune-excluded phenotypes and immunotherapy resistance. The practical implication is that “coldness” can reflect spatial barriers as much as immunologic ignorance.
3. What “Hot” Means Mechanistically: Tumors That Withstand Immune Attack
Hot (immune-inflamed) tumors are characterized by abundant immune infiltration and inflammatory signaling, but they often persist due to adaptive resistance and suppressive network effects. A common pattern is that immune pressure itself induces tumor defenses—particularly checkpoint ligand induction—contributing to the escape phase in Figure 1.
3.1. Adaptive Immune Resistance: PD-L1 As a Feedback Response to Inflammation
PD-L1 expression can be induced by interferon signaling within the tumor microenvironment, linking immune infiltration to checkpoint-mediated suppression. This biology explains why many hot tumors show high PD-L1 and why checkpoint blockade can be effective—yet also why tumors may evolve additional suppressive layers when PD-1/PD-L1 is blocked.
3.2. Escape via Antigen Presentation Loss and Interferon Pathway Disruption
Two recurrent escape routes in hot tumors are:
- Loss of antigen presentation, especially via MHC class I pathway disruption (e.g., B2M alterations), which impairs TCR-mediated recognition.
- Loss of IFN-γ responsiveness, including alterations in the IFN-γ–JAK–STAT axis, which can reduce immune-mediated growth control and alter inducible immune programs.
These mechanisms align with Figure 1’s “escape” panel where tumor progression emerges in the presence of immune elements.
3.3. Myeloid and Stromal Suppression Layered onto Inflammation
Even in infiltrated tumors, suppressive macrophage/DC states, CAF programs, and cytokine milieus can neutralize cytotoxic function. TGF-β-mediated mechanisms are again central here, operating not only as a “cold-making” factor but also as a brake within hot tumors.
4. Integrating Innate Surveillance: How “Hide vs Fight” Applies Beyond T Cells
Figure 1 explicitly includes innate-like recognition pathways (e.g., NKG2D, CD226, TRAIL) in elimination and equilibrium. This matters because tumors can escape T cell control via MHC class I loss, potentially increasing susceptibility to NK cell surveillance, but tumors frequently counter-adapt through microenvironmental suppression and altered ligand presentation.
Practically, the cold/hot framing extends to innate immunity:
- Cold with respect to innate sensing: weak danger signaling and low activation cues limit NK/DC engagement.
- Hot but NK-resistant: inflammatory niches may still suppress NK/T cell cytotoxic programs via cytokines, metabolic constraints, and ligand modulation.
This helps explain why durable control often requires either (i) restoring priming and entry (warming cold tumors) or (ii) dismantling adaptive suppression and escape routes (disarming hot tumors).
5. Cold-to-Hot Transitions and the Inevitability of Secondary Resistance
Hot and cold states are not fixed categories but dynamic phases of the immunoediting trajectory. Interventions that successfully inflame a cold tumor can generate a predictable next problem: adaptive resistance (e.g., PD-L1 induction) and selection for antigen presentation/IFN-pathway escape variants. Therefore, the practical objective is often not simply “make it hot,” but “make it hot and prevent or rapidly treat escape.”
6. Therapeutic Implications: Matching Strategy to Dominant Failure mode
A clinically actionable interpretation of Figure 1 is that interventions should be matched to the immunoediting bottleneck:
6.1. If the Tumor Is “Cold”: Initiate Immunity and Enable Entry
Mechanistic priorities include restoring innate sensing/priming (e.g., pathways linked to STING and DC function) and overcoming immune exclusion barriers (often stromal/TGF-β-linked) or tumor-intrinsic exclusion programs (e.g., WNT/β-catenin-associated non-inflamed states).
6.2. If the Tumor Is “Hot”: Release Brakes and Prevent Genetic/Epigenetic Escape
In hot tumors, the priority is to counter adaptive resistance (PD-L1/PD-1 axis and related checkpoints) and anticipate escape routes (MHC class I loss, IFN-γ pathway disruption).
7. Conclusions
Tumor immune evasion can be productively summarized as either immune hiding (cold tumors that prevent priming/entry) or active resistance (hot tumors that survive inflammation via checkpoints, suppressive networks, and escape mutations). The elimination–equilibrium–escape framework provides an evolutionary map: initial immune pressure shapes tumor composition and selects for variants capable of either avoiding immune engagement altogether or resisting it once engaged. A key implication for translational strategy is sequential logic: cold tumors often require initiation (turn immunity on and allow entry), while hot tumors require disinhibition and containment (remove adaptive brakes and limit emergence of antigen presentation/IFN-pathway escape).
Funding
This work was funded by the subsidy allocated to Kazan Federal University for the state assignment in the sphere of scientific activities (project number FZSM-2025-0001).
Conflicts of Interest
The author declares no conflicts of interest.
References
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat Immunol 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell 2020, 37, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. New England Journal of Medicine 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Ghahremanifard, P.; Chanda, A.; Bonni, S.; Bose, P. TGF-β Mediated Immune Evasion in Cancer—Spotlight on Cancer-Associated Fibroblasts. Cancers (Basel) 2020, 12, 3650. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e4. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhang, B.; Li, B.; Wu, H.; Jiang, M. Cold and Hot Tumors: From Molecular Mechanisms to Targeted Therapy. Signal Transduct Target Ther 2024, 9, 274. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Oravecz, T.; Dillon, L.A.; Italiano, A.; Audoly, L.; Fridman, W.H.; Clifton, G.T. Towards a Consensus Definition of Immune Exclusion in Cancer. Front Immunol 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New Insights into Cancer Immunoediting and Its Three Component Phases — Elimination, Equilibrium and Escape. Curr Opin Immunol 2014, 27, 16–25. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Cancer immunoediting and immune phenotypes across elimination, equilibrium, and escape. Schematic overview of tumor–immune interaction showing (a) elimination of immunogenic transformed cells by immune effectors, (b) equilibrium (tumor dormancy under immune pressure) shaped by cytokines such as IFN-γ and immune modulators, and (c) escape culminating in tumor progression via immune inhibitory mechanisms (e.g., PD-L1) and selection of poorly immunogenic or immunosuppressive tumor variants. The figure highlights innate and adaptive contributors (CD4, CD8, NK, NKT, DC, macrophages) and recognition/effector axes (e.g., NKG2D, CD226, TRAIL; perforin; inflammatory cytokines) that shape each phase.
Figure 1.
Cancer immunoediting and immune phenotypes across elimination, equilibrium, and escape. Schematic overview of tumor–immune interaction showing (a) elimination of immunogenic transformed cells by immune effectors, (b) equilibrium (tumor dormancy under immune pressure) shaped by cytokines such as IFN-γ and immune modulators, and (c) escape culminating in tumor progression via immune inhibitory mechanisms (e.g., PD-L1) and selection of poorly immunogenic or immunosuppressive tumor variants. The figure highlights innate and adaptive contributors (CD4, CD8, NK, NKT, DC, macrophages) and recognition/effector axes (e.g., NKG2D, CD226, TRAIL; perforin; inflammatory cytokines) that shape each phase.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



