
Submitted:
19 December 2025
Posted:
22 December 2025
You are already at the latest version
Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is a complex disorder characterized by the progressive development of renal cysts and systemic complications, including cardiovascular and metabolic comorbidities. Disease progression is linked to ADPKD-driven metabolic and mitochondrial reprogramming that exacerbates ADPKD pathophysiology by impairing central energetic pathways, including mitochondrial respiration and glucose, lipid, and amino acid metabolism. Recent studies have revealed that caloric intake interventions can attenuate disease progression; however, they are difficult to achieve and likely applicable only to those who are overweight. Therefore, a better understanding of the mechanisms that drive metabolic and mitochondrial reprogramming may foster the identification of novel ADPKD severity-modifying drug targets and/or drugs. Such drugs could synergistically complement the benefits of tolvaptan (the only US Food and Drug Administration (FDA)-approved ADPKD therapeutic), or serve as alternatives to it, as tolvaptan is costly and associated with idiosyncratic hepatotoxicity. Repurposing FDA-approved drugs could accelerate clinical testing and subsequent translation to practice by building on prior work to improve the traditional drug development pipeline. To facilitate identification of repurposed drugs for ADPKD, we review ADPKD-driven metabolic and mitochondrial reprogramming from the perspective of drug target identification for repurposing ADPKD therapeutics.
Keywords:
autosomal dominant polycystic kidney disease
; ADPKD
; polycystic kidney disease
; PKD
; metabolic reprogramming
; mitochondrial dysfunction
; cystogenesis
; Warburg effect
; oxidative phosphorylation
; drug repurposing
; drug development
; tolvaptan
; pharmacokinetics
; drug metabolism
; in silico
; drug targets
; precision medicine
Background
Autosomal dominant polycystic kidney disease (ADPKD), the fourth leading cause of renal replacement therapy, is projected to affect around 12 million individuals worldwide.[1] Its manifestations are characterized by the formation of renal cysts and a subsequent progressive decline in renal function. Cysts seem to originate most often in the distal nephron. As abnormal cystogenesis-promoting epithelial cells proliferate, they separate from the parent nephron, secrete chloride-rich fluid, and alter the extracellular matrix.[2] The underlying cause of this process in most ADPKD cases involves a genetic defect in the PKD1 (~80% patients with ADPKD) or PKD2 (~15% patients with ADPKD) genes. These genes encode for polycystins 1 and 2 (PC1/PC2), respectively.[1] Many of their downstream pathways have been identified; however, so far, no single pathway has been broadly accepted as the principal driver of cystogenesis.
Among pathways involved in renal cystogenesis, the most extensively studied are those associated with metabolic and mitochondrial reprogramming. These pathways can directly modify ADPKD pathophysiology by impairing crucial processes central to normal healthy kidney function.[3] In particular, ADPKD pathogenesis and progression revolve around energetic pathway alterations that impact glucose, amino acid, and lipid metabolism, along with mitochondrial respiration.[3] Genetic defects appear to play a role in these alterations. PC1, for example, directly and indirectly affects mitochondrial structure and function.[4] However, the mechanistic explanation is still not fully understood. Along with changes in other key cellular functions, these alterations can explain why ADPKD patients develop comorbidities associated with metabolic dysfunction, such as central obesity and insulin resistance[5], and the success of dietary interventions for symptomatic management.[3] Understanding metabolic reprogramming is therefore crucial for advancing ADPKD patient care and treatment options.
Tolvaptan became the first and only U.S. Food and Drug Administration (FDA)-approved therapeutic for ADPKD in 2018.[6] It is a vasopressin 2 receptor (V2R) antagonist, suggested to reduce the rate of renal function decline in ADPKD by ~30% on average. However, it is expensive (~$27,906 for a 30-day supply)[7], associated with idiosyncratic hepatotoxicity, and has a limited impact on extrarenal manifestations.[8,9] Alternative candidate therapeutics to tolvaptan have entered preclinical and clinical trials, but none have successfully received FDA approval to date. Because de novo drug development is expensive and time-consuming, it may take many years for new ADPKD therapeutics to be approved. High levels of phenotypic, gene locus, and allelic heterogeneity contribute to ADPKD complexity[10], further increasing drug development challenges and creating differences in drug biotransformation between individuals. Both of these challenges can be partially mitigated through drug repurposing. This approach leverages prior studies to reduce development costs and timelines, thereby increasing accessibility in drug research and addressing limitations in current ADPKD treatment options. To foster the identification of ADPKD therapeutics, we review evidence supporting metabolic and mitochondrial reprogramming as targets for ADPKD drug development, and its impact on identifying drug targets and repurposing ADPKD therapeutics.
Common Metabolic Diseases/Disorders Associated with ADPKD
Metabolic syndrome (MetS) describes a cluster of physiological, biochemical, clinical, and metabolic factors that notably increase the risk of cardiovascular diseases and type II diabetes mellitus (DM). Central obesity is the primary risk factor, but MetS diagnostic criteria also include insulin resistance, dyslipidemia, glucose intolerance, hypertension, inflammation, prothrombotic state, and microalbuminuria.[11] ADPKD patients have an increased association with multiple components of MetS, including increased waist circumference, fasting glucose levels, and dyslipidemia.[12,13] Notably, ADPKD patients with DM exhibit worse disease progression than patients with ADPKD alone. These individuals have higher body mass index, greater total kidney volume (TKV), earlier onset of hypertension, increased end-stage renal disease risk, and reduced life expectancy.[14] This increase in MetS components is critical to understand. The risk of developing DM in ADPKD is substantially greater post-transplant, and patients have an approximate threefold increased risk of developing new-onset diabetes after transplantation within the first year compared to non-ADPKD recipients.[15] ADPKD cohorts, however, appear to have a lower incidence of non-transplant-related DM.[16,17] Polycystic lesions may be responsible for the reduced risk by impairing renal gluconeogenesis and insulin degradation, causing hyperinsulinemic blood concentrations that lead to pancreatic beta cell fatigue, suppressing insulin production and release.[16] While studies have reported insulin resistance and hyperinsulinemia[18,19], there remains controversy on whether it is ADPKD-specific or a consequence of general renal disease.[20]
Dietary strategies have established benefits in ADPKD disease management. The prevalence of ADPKD-related metabolic disorders and reprogramming likely explains their efficacy.[3] Daily caloric restriction (10-40%) in ADPKD mouse models (Pkd1RC/RC and PKDcond/cond:NesCre) slowed kidney growth, improved kidney function, and, notably, in the 40% caloric restriction group, reversed disease progression.[21,22] A clinical trial on weight loss using caloric restriction or intermittent fasting in overweight and obese ADPKD patients showed reduced kidney growth correlated with body weight and visceral adiposity loss. Notably, patients with clinically meaningful weight loss achieved kidney growth cessation.[23] Overall, ADPKD is associated with metabolic derangements, and addressing them through dietary strategies, for example, is beneficial to disease progression.
Metabolic Reprogramming in ADPKD
Metabolic reprogramming, or alterations in physiological processes in response to disease and stress, is prevalent in ADPKD (Figure 1). Most notable is the Warburg effect, a preferential shift to glycolysis and lactic acid fermentation in the presence of oxygen that drives cystogenesis by promoting flux into biosynthetic pathways and, thus, uncontrolled cell proliferation.[24] In silico analysis of human PKD1 renal cysts identified pathway alterations favoring glucose uptake and lactate production, with key glycolytic enzymes and regulatory genes upregulated, and mitochondrial energy metabolism and pyruvate oxidation pathways downregulated. Additionally, there was increased expression of anoctamin 1, indicating elevated oxidative damage and impairment of antioxidant pathways, such as glutathione metabolism.[25] A chronic progressive model of PKD using heterozygous Han:SPRD (Cy/+) rats also had glycolytic gene upregulation and gluconeogenesis-related gene downregulation in cystic kidneys. Treatment with 2-deoxyglucose, a glycolytic inhibitor, attenuated disease progression and improved both renal function and cyst index.[26] ADPKD dysregulates oxidative phosphorylation (OXPHOS), contributing to reactive oxygen species (ROS) generation and oxidative stress.[27] Cystic mice (Cys1cpk) and rats (Han:SPRD-Cy) had increased mRNA levels of the renoprotective and oxidative stress marker heme oxygenase (HO)-1, along with reduced antioxidant enzyme mRNAs, proteins, and activity levels.[28] Another study found HO induction mitigated renal cystogenesis in Cys1cpk mice, while inhibition accelerated it, directly connecting oxidative stress to ADPKD pathophysiology.[29] Nitric oxide-related pathways may also increase levels of ROS and oxidative stress in ADPKD. Cystic kidneys of PCK rats exhibited elevated expression of NADPH-oxidase complex-4, likely leading to the observed reduction in endothelial nitric oxide synthase activity, along with upregulated ROS production, DNA damage, protein oxidation, and dysfunction in the mitochondria.[30] Renal oxidative stress increases cell proliferation, extracellular matrix synthesis, inflammatory cell infiltration, and apoptosis.[31] As such, oxidative stress is a likely measure of disease severity. This further highlights the pathogenic role of the Warburg effect in ADPKD cystogenesis and progression by impairing the pentose phosphate pathway (PPP) and mitochondrial respiration.
As noted above, amino acid and lipid metabolism are dysregulated in ADPKD patients. Similar to humans with ADPKD, mouse models of ADPKD demonstrated upregulation in glutaminase 1 (GLS1) GAC, a GLS1 isoform associated with glutaminolysis reliance in cancer, suggesting a similar phenomenon that supports hyperproliferation in ADPKD.[32] Glutamine and glutaminase are important for shuttling ammonia in the urea cycle[33], suggesting GLS1 upregulation in ADPKD may affect the urea cycle. Other key players in the urea cycle include argininosuccinate synthase 1 and arginine. Loss of argininosuccinate synthase 1 was found in both murine and human ADPKD tissue, along with an inverse relationship between arginine levels and cystogenesis[34], further highlighting urea cycle dysregulation in ADPKD. Given that the urea-to-plasma ratio in ADPKD patients is significantly associated with the rate of estimated glomerular filtration rate decline (P=0.02)[35], impairment in the urea cycle may have a pathogenic role in ADPKD progression. Lipid metabolism alterations are common in kidney disease and often present as increased fatty acid (FA) uptake, lipid accumulation, and the formation of lipid droplets.[27] For example, metabolomics identified acetylcarnitine, an important metabolite in mitochondrial β-oxidation, as a urine marker in Pkd1cko/cko mice specific to disease progression.[36,37] Another study also demonstrated an intrinsic FA oxidation (FAO) defect in Pkd1cko/cko murine cells lacking glycolysis alterations[38], suggesting ADPKD mitochondrial reprogramming may be related to mitochondrial dysfunction, rather than a preferential shift to aerobic glycolysis. Reduced FAO often occurs alongside increased de novo FA synthesis (FAS) and is associated with rapidly proliferating cells that require membrane production.[37] Pkd1 mouse embryonic fibroblasts and transcriptomic profiling of human PKD1 kidneys exhibited a similar phenomenon with increased FAS and preferential glutamine use.[39] These findings suggest FAO impairment in ADPKD may not only highlight mitochondrial dysfunction, but additionally drive hyperproliferation in cystogenesis.
ADPKD mitochondrial reprogramming is likely influenced heavily by PC1. In HEK293 and LLC-PK1 cell models with modulated PC1 and PC2 expression, oxygen levels controlled the subcellular location and channel activity of polycystin complexes by interacting with Egl-9 Family Hypoxia Inducible Factor 3, the oxygen-sensing prolyl hydroxylase domain containing protein that hydroxylates PC1. Cells lacking PC1 exhibited reduced oxygen consumption and mitochondrial calcium uptake, indicating that PC1 can directly regulate mitochondrial function in response to changes in cellular oxygen levels.[40] Given that ADPKD-associated cystogenesis aggravates regional hypoxia, pathogenic PKD1 variants impairing PC1 function may exacerbate hypoxia-related inflammation and ADPKD progression. One study found that tissue oxygenation is inversely correlated with height-adjusted total kidney volume[41], potentially supporting a connection between PC1 and hypoxia. In Pkd1ko/ko renal epithelial cells, a PC1 C-terminal fragment containing the cytoplasmic tail (CTT) translocated to the mitochondrial matrix. It restored some mitochondrial phenotypes, further confirming PC1 CTT interaction with both mitochondrial function and cellular metabolism. PC1 also modulates mitochondrial morphology, as demonstrated by studies in Pkd1 mouse models and PKD1 ADPKD patients, which found mitochondria had reduced elongation, mass, matrix electron density, and fusion proteins, along with increased mitochondrial network fragmentation, swelling with membrane whorls, and upregulated fission proteins.[4,42] PC2 potentially contributes to the changes in mitochondrial morphology as well, and one study found reduced PC2 expression affected mitochondrial calcium buffering, increasing mitochondrial network fragmentation. The authors proposed that PC2 regulates mitochondrial metabolism at mitochondrial-ER contacts.[43] In another study, truncating PKD2 mutations altered mitochondrial morphology, resulting in a rounded phenotype and dilated cristae, and resembling the effects of a PC1 deficiency.[44] Overall, it is clear that both PC1 and PC2 have a role in mitochondrial function and may exacerbate ADPKD pathophysiology and progression, as the alterations described are indicators of mitochondrial stress in ADPKD[45], which directly enhances oxidative stress.
Established ADPKD Drug Targets
V2R, the main target for tolvaptan, is upregulated in ADPKD and contributes to cell proliferation, fluid secretion, and cystogenesis. Its activation increases cyclic adenosine monophosphate (cAMP) and protein kinase A, which promote aquaporin 2 membrane localization, increasing water reabsorption and fluid secretion.[46] Tolvaptan antagonizes V2R, reducing cAMP levels and thereby inhibiting cyst epithelial cell proliferation and fluid secretion. However, benefits are largely restricted to patients with rapidly progressing ADPKD (Mayo Class ≥ 1C) or to chronic kidney disease (CKD) patients who begin treatment in an early stage (CKD Stage 3A).[9] As such, alternative safe and effective therapeutics are desperately needed. Encouragingly, advances in understanding the metabolic underpinnings of ADPKD have created opportunities to explore potential drug targets and alleviate some of the challenges in ADPKD drug development. Here, we review both targets currently under development and those that could be studied in future analyses.
The mammalian target of rapamycin (mTOR) signaling pathway and its interactors are promising targets for addressing the consequences of the Warburg effect in ADPKD. The mTOR signaling pathway is hyperactivated in cystic lesions and promotes a metabolic shift from OXPHOS to glycolysis.[47] Increased mTOR activation is also suggested to increase insulin resistance and fat accumulation[48], potentially contributing to the prevalence of MetS in ADPKD. Together, the effects of mTOR upregulation may explain why rapamycin inhibition of mTOR complex 1 (mTORC1) attenuated cystic progression in an ADPKD mouse model.[49] The phosphatidylinositol 3-kinase (PI3K)/protein kinase B pathway activates mTOR, initiating cellular processes, including metabolic reprogramming.[47] As such, dual PI3K/mTOR inhibition may have potential in ADPKD over only rapalog inhibition of mTOR.[50] Asparagine (Asn) synthetase (ASNS) is upregulated in ADPKD and has been implicated in mTOR activation and the Warburg effect through Asn, which can stimulate mTORC1 to boost glycolysis.[51] ASNS inhibition in a slowly progressing ADPKD murine model drastically reduced TKV, proliferation, and cyst expansion, and rescued renal function.[52] Activating adenosine monophosphate-activated protein kinase, which in turn inhibits mTOR signaling and cystic fibrosis transmembrane conductance regulator activity, is another strategy that may be able to reverse the Warburg-like metabolic shift in ADPKD cells and reduce cyst growth.[53,54,55,56] Wingless-related integration site (Wnt)/β-catenin signaling is proposed to coordinate metabolic flux consistent with the Warburg effect and activate mTORC1 through interactions with metabolic transcription factors such as cellular myelocytomatosis oncogene, peroxisome proliferator–activated receptor (PPAR) δ, and PPARγ.[57] Transcriptomic profiling of PKD1 ADPKD renal cortical patient tissue samples demonstrated substantial Wnt pathway upregulation specific to cystic cells, and that the free CTT of PC1 physically interacts with β-catenin to inhibit Wnt signaling. The authors suggested that ADPKD-related loss of PC1-CTT β-catenin inhibition causes epithelial cells to revert to a less differentiated phenotype associated with proliferation and cyst formation[58], directly connecting aberrant Wnt/β-catenin signaling to cystogenesis.
PPARα, a transcription factor vital for OXPHOS, is a potent target for addressing ADPKD-related mitochondrial dysfunction.[59,60] Studies have noted reduced PPARɑ levels in CKD, aging kidneys, and ADPKD, which impair FAO, leading to lipid accumulation in the kidney and worsened renal fibrosis.[61,62,63] Researchers are also exploring inducing PPARγ signalling to target lipid metabolism, glucose homeostasis, and oxidative stress.[64] PPARγ coactivator 1-ɑ (PGC-1ɑ), a key regulator of mitochondrial biogenesis and function, is downregulated in ADPKD and linked to several PKD and cilia-related genes.[65] One study found that reduced PGC-1ɑ was associated with increased mitochondrial superoxide and reduced antioxidative effect, suggesting that inducing PGC-1ɑ activity may reduce oxidative stress in ADPKD.[66] Another avenue for rescuing mitochondrial function is the microRNA (miR)-17 family, which is upregulated in both human and murine ADPKD models.[67] ADPKD mouse models have shown anti-miR-17 oligonucleotides can rescue OXPHOS defects and attenuate disease progression regardless of variant location (Pkd1 or Pkd2) or type (null or hypomorphic).[59,68] Finally, the hormone somatostatin inhibits intracellular cAMP production and, in part, could regulate cellular energy turnover, which is heavily altered in ADPKD.[69] Targeting somatostatin may additionally improve mitochondrial function, as cAMP signaling is involved in mitochondrial dynamics like fusion, fission, motility, and mitophagy.[70] These drug targets may offer the opportunity to mitigate ADPKD metabolic and mitochondrial reprogramming, and advance options for patients.
ADPKD Impact on Drug Development and Metabolism
Traditional drug development is an extensive process that can take up to 17 years and $2-3 billion, with approval rates around 11%.[71] These associated costs and low approval rates create a barrier to entrance for designing novel therapeutics, further confounded in ADPKD by heterogeneity and limited research support (Figure 2). As such, drug repurposing has emerged as a strategic alternative. It leverages computational tools and prior studies to provide new indications for therapeutics, increasing approval rates to ~30% and reducing time and costs as low as 3 years and $300 million, respectively (Figure 3).[71] Tolvaptan is an example of the potential of drug repurposing in ADPKD, as it was originally indicated for hypervolemic or euvolemic hyponatremia from the syndrome of inappropriate antidiuretic hormone, chronic heart failure, or cirrhosis.[72] In one drug repurposing study from our lab, we prioritized 16 drug repurposing candidates and 29 drug targets, including many related to metabolic and mitochondrial pathways. We also demonstrated that protein interactions within oxidoreductase activity were involved in modulating drug metabolism activity, specifically within the cytochrome P450 (CYP450) family.[73] Many repurposed candidates proposed by the field target cellular metabolism, a key driver of cystogenesis in ADPKD (Table 1). Given the high level of metabolic and overall dysregulation in ADPKD, precision medicine approaches that account for its metabolic heterogeneity are essential as the field advances.
Hepatic and renal dysfunction can have significant consequences on drug metabolism and excretion, leading to the toxic accumulation of the drug and its metabolites.[74] This is particularly relevant in ADPKD, as 81.7% of adult patients develop hepatic cysts[75] that can cause hepatomegaly, loss of liver parenchyma, and lower quality of life in advanced stages.[76] Patients with renal disease commonly exhibit altered pharmacokinetics due to decreased renal excretion. They are particularly susceptible to volume of distribution and clearance changes, potentially requiring adjustments in dosing regimens to avoid adverse events.[77] Chronic renal failure has been shown to affect hepatic, renal, and intestinal mechanisms of drug metabolism and transport with a 40-85% downregulation of hepatic and intestinal CYP450 metabolism, along with interference in phase II reactions and drug transporters like P-glycoprotein and organic anion transporting polypeptide.[78] ADPKD-specific effects on hepatic and renal drug metabolism and transporter-mediated drug expression are not well characterized, but are likely, as CKD has been shown to change the protein expression and activity of kidney and liver drug metabolism enzymes and transporters.[79] One study on early- and late-stage ADPKD human kidney samples found significant reductions in several drug metabolism enzymes. In the end-stage group, organic anion transporter (OAT) 1, OAT3, and organic cation transporter 2 were below the limit of quantification.[80] These changes could contribute to tolvaptan hepatotoxicity, as demonstrated by an ADPKD ex-situ perfused rat liver study that found decreased hepatobiliary clearance and greater hepatocellular concentrations of tolvaptan within their prediction model.[81] Impaired renal function has additionally been associated with changes in absorption, plasma protein binding, and drug distribution, along with altered liver and gut drug metabolism and transport pathways. As such, the FDA recommends that renal function effects are characterized early in drug development to avoid later-stage issues in dosing, efficacy, and safety.[82] While currently understudied, ADPKD alterations in drug metabolism are likely and could explain some of the challenges in finding safe and effective therapeutic options.
Conclusions, Limitations, and Future Directions
Here, we outlined the metabolic and mitochondrial alterations associated with ADPKD, in relation to potential drug targets and the challenges in drug development. Further exploration is critical for understanding ADPKD pathophysiology and uncovering promising mechanisms for pharmacological evaluation. While there are numerous ADPKD models that offer many benefits, they do not fully capture the complexity of ADPKD pathophysiology, translatability to humans, and resources for model generation and upkeep.[83] Moving forward, in silico drug repurposing methods leveraging genomic, structural, and phenotypic data, combined with improved drug screening efficiency through options like high-throughput organoid drug screening, may help identify context-specific therapies and improve translational success.[84] Techniques such as network/graph theory, molecular docking, and large language models offer promising avenues to refine target selection and predict off-target effects, potentially avoiding pitfalls seen with mTOR inhibitors and others. This, coupled with better characterization of metabolic and mitochondrial alterations, could improve outcomes for ADPKD patients.
Disclosures
MM reports additional research support from Otsuka, Sanofi, Vertex, and AbbVie, and has been a member of advisory boards for Sanofi, Santa Barbara Nutrients, and PKD Foundation. MM has provided consultancy for Otsuka, Sanofi, Vertex, AbbVie, and Regulus.
Author Contributions
JMF, MM, and BNL conceived the project, and ST, EJW, and TCH contributed to its conceptualization; JMF and ST prepared the figures; JMF and EJW drafted the manuscript; JMF, ST, EJW, TCH, PC, MM, and BNL edited and revised the manuscript; JMF, ST, EJW, TCH, PC, MM, and BNL approved the final version of the manuscript.
Acknowledgements
This work was supported in part by National Institute of General Medical Sciences (NIGMS) of the National Institute of Health (NIH) (T32GM146611; JMF), the UAB Pilot Center for Precision Animal Modeling (C-PAM) (1U54OD030167; BNL), NIH-funded PKD Research Resource Consortium (U54DK126087; BNL and MM), PKD Foundation Award (BNL), NIDDK NIH-funded grant (R01DK134310; BNL and MM), the UAB Dean’s 1R01 Award (BNL), from the Office of Research and Development, Medical Research Service, Department of Veterans Affairs (1-I01-BX006266-01; MM), and the Detraz Endowed Research Fund in Polycystic Kidney Disease (MM). We would also like to thank other members of the Lasseigne Lab, especially Dr. Tabea Soelter, and external collaborators, especially Dr. Robert E. Van Sciver, for their thoughtful feedback. The funders had no role in the design and writing of the manuscript.
References
- Chapman, AB; Devuyst, O; Eckardt, KU; et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015, 88(1), 17–27. [Google Scholar] [CrossRef] [PubMed]
- Mahboob, M; Rout, P; Leslie, SW; Bokhari, SRA. Autosomal dominant polycystic kidney disease. In StatPearls; StatPearls Publishing, 2025; Available online: https://www.ncbi.nlm.nih.gov/books/NBK532934/.
- Nowak, KL; Hopp, K. Metabolic reprogramming in autosomal dominant polycystic kidney disease: Evidence and therapeutic potential. Clin J Am Soc Nephrol. 2020, 15(4), 577–584. [Google Scholar] [CrossRef] [PubMed]
- Lin, CC; Kurashige, M; Liu, Y; et al. A cleavage product of Polycystin-1 is a mitochondrial matrix protein that affects mitochondria morphology and function when heterologously expressed. Sci Rep. 2018, 8(1), 2743. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z; Xie, G; Ong, A. Metabolic abnormalities in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2015, 30(2), 197–203. [Google Scholar] [CrossRef]
- Available Treatment. Polycystic kidney disease | PKD treatment research | PKD Foundation. 11 April 2020. Available online: https://pkdcure.org/about-the-disease/living-with-pkd/treatments/ (accessed on 22 August 2025).
- Cloutier, M; Manceur, AM; Guerin, A; Aigbogun, MS; Oberdhan, D; Gauthier-Loiselle, M. The societal economic burden of autosomal dominant polycystic kidney disease in the United States. BMC Health Serv Res. 2020, 20(1), 126. [Google Scholar] [CrossRef]
- Bellos, I. Safety profile of tolvaptan in the treatment of autosomal dominant polycystic kidney disease. Ther Clin Risk Manag. 2021, 17, 649–656. [Google Scholar] [CrossRef]
- Chebib, FT; Perrone, RD; Chapman, AB; et al. A practical guide for treatment of rapidly progressive ADPKD with tolvaptan. J Am Soc Nephrol. 2018, 29(10), 2458–2470. [Google Scholar] [CrossRef]
- Claverie-Martin, F; Gonzalez-Paredes, FJ; Ramos-Trujillo, E. Splicing defects caused by exonic mutations in PKD1 as a new mechanism of pathogenesis in autosomal dominant polycystic kidney disease. RNA Biol. 2015, 12(4), 369–374. [Google Scholar] [CrossRef]
- Kaur, J. A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014, 2014, 943162. [Google Scholar] [CrossRef]
- Pietrzak-Nowacka, M; Safranow, K; Byra, E; Bińczak-Kuleta, A; Ciechanowicz, A; Ciechanowski, K. Metabolic syndrome components in patients with autosomal-dominant polycystic kidney disease. Kidney Blood Press Res. 2009, 32(6), 405–410. [Google Scholar] [CrossRef]
- Veeramuthumari, P; Isabel, W. Clinical study on autosomal dominant polycystic kidney disease among south Indians. Int J Clin Med. 2013, 04(04), 200–204. [Google Scholar] [CrossRef]
- Chen, LC; Chu, YC; Lu, T; Lin, HYH; Chan, TC. Cardiometabolic comorbidities in autosomal dominant polycystic kidney disease: a 16-year retrospective cohort study. BMC Nephrol. 2023, 24(1), 333. [Google Scholar] [CrossRef] [PubMed]
- de Mattos, AM; Olyaei, AJ; Prather, JC; Golconda, MS; Barry, JM; Norman, DJ. Autosomal-dominant polycystic kidney disease as a risk factor for diabetes mellitus following renal transplantation. Kidney Int. 2005, 67(2), 714–720. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak-Nowacka, M; Rózanski, J; Safranow, K; Kedzierska, K; Dutkiewicz, G; Ciechanowski, K. Autosomal dominant polycystic kidney disease reduces the risk of diabetes mellitus. Arch Med Res. 2006, 37(3), 360–364. [Google Scholar] [CrossRef]
- Reed, B; Helal, I; McFann, K; Wang, W; Yan, XD; Schrier, RW. The impact of type II diabetes mellitus in patients with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2012, 27(7), 2862–2865. [Google Scholar] [CrossRef]
- Vareesangthip, K; Tong, P; Wilkinson, R; Thomas, TH. Insulin resistance in adult polycystic kidney disease. Kidney Int. 1997, 52(2), 503–508. [Google Scholar] [CrossRef]
- Pietrzak-Nowacka, M; Safranow, K; Byra, E; Nowosiad, M; Marchelek-Myśliwiec, M; Ciechanowski, K. Glucose metabolism parameters during an oral glucose tolerance test in patients with autosomal dominant polycystic kidney disease. Scand J Clin Lab Invest. 2010, 70(8), 561–567. [Google Scholar] [CrossRef]
- Fliser, D; Pacini, G; Engelleiter, R; et al. Insulin resistance and hyperinsulinemia are already present in patients with incipient renal disease. Kidney Int. 1998, 53(5), 1343–1347. [Google Scholar] [CrossRef]
- Kipp, KR; Rezaei, M; Lin, L; Dewey, EC; Weimbs, T. A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease. Am J Physiol Renal Physiol. 2016, 310(8), F726–F731. [Google Scholar] [CrossRef]
- Warner, G; Hein, KZ; Nin, V; et al. Food restriction ameliorates the development of polycystic kidney disease. J Am Soc Nephrol. 2016, 27(5), 1437–1447. [Google Scholar] [CrossRef]
- Hopp, K; Catenacci, VA; Dwivedi, N; et al. Weight loss and cystic disease progression in autosomal dominant polycystic kidney disease. iScience 2022, 25(1), 103697. [Google Scholar] [CrossRef] [PubMed]
- Liberti, MV; Locasale, JW. The Warburg Effect: How does it benefit cancer cells? Trends Biochem Sci. 2016, 41(3), 211–218. [Google Scholar] [CrossRef] [PubMed]
- Song, X; Pickel, L; Sung, HK; Scholey, J; Pei, Y. Reprogramming of energy metabolism in human PKD1 polycystic kidney disease: A systems biology analysis. Int J Mol Sci. 2024, 25(13), 7173. [Google Scholar] [CrossRef] [PubMed]
- Riwanto, M; Kapoor, S; Rodriguez, D; Edenhofer, I; Segerer, S; Wüthrich, RP. Inhibition of aerobic glycolysis attenuates disease progression in polycystic kidney disease. PLoS One 2016, 11(1), e0146654. [Google Scholar] [CrossRef]
- Saxena, S; Dagar, N; Shelke, V; Lech, M; Khare, P; Gaikwad, AB. Metabolic reprogramming: Unveiling the therapeutic potential of targeted therapies against kidney disease. Drug Discov Today 2023, 28(11), 103765. [Google Scholar] [CrossRef]
- Maser, RL; Vassmer, D; Magenheimer, BS; Calvet, JP. Oxidant stress and reduced antioxidant enzyme protection in polycystic kidney disease. J Am Soc Nephrol. 2002, 13(4), 991–999. [Google Scholar] [CrossRef]
- Zhou, J; Ouyang, X; Schoeb, TR; et al. Kidney injury accelerates cystogenesis via pathways modulated by heme oxygenase and complement. J Am Soc Nephrol. 2012, 23(7), 1161–1171. [Google Scholar] [CrossRef]
- Kahveci, AS; Barnatan, TT; Kahveci, A; et al. Oxidative stress and mitochondrial abnormalities contribute to decreased endothelial nitric oxide synthase expression and renal disease progression in early experimental Polycystic Kidney Disease. Int J Mol Sci. 2020, 21(6), 1994. [Google Scholar] [CrossRef]
- Nath, KA; Grande, J; Croatt, A; Haugen, J; Kim, Y; Rosenberg, ME. Redox regulation of renal DNA synthesis, transforming growth factor-beta1 and collagen gene expression. Kidney Int. 1998, 53(2), 367–381. [Google Scholar] [CrossRef]
- Soomro, I; Sun, Y; Li, Z; et al. Glutamine metabolism via glutaminase 1 in autosomal-dominant polycystic kidney disease. Nephrol Dial Transplant. 2018, 33(8), 1343–1353. [Google Scholar] [CrossRef]
- Barmore, W; Azad, F; Stone, WL. Physiology, urea cycle. In StatPearls; StatPearls Publishing, 2025; Available online: https://www.ncbi.nlm.nih.gov/books/NBK513323/ (accessed on 2 September 2025).
- Trott, JF; Hwang, VJ; Ishimaru, T; et al. Arginine reprogramming in ADPKD results in arginine-dependent cystogenesis. Am J Physiol Renal Physiol. 2018, 315(6), F1855–F1868. [Google Scholar] [CrossRef] [PubMed]
- Heida, JE; Gansevoort, RT; Messchendorp, AL; et al. Use of the urine-to-plasma urea ratio to predict ADPKD progression. Clin J Am Soc Nephrol. 2021, 16(2), 204–212. [Google Scholar] [CrossRef] [PubMed]
- Menezes, LF; Zhou, F; Patterson, AD; et al. Network analysis of a Pkd1-mouse model of autosomal dominant polycystic kidney disease identifies HNF4α as a disease modifier. PLoS Genet. 2012, 8(11), e1003053. [Google Scholar] [CrossRef] [PubMed]
- Podrini, C; Cassina, L; Boletta, A. Metabolic reprogramming and the role of mitochondria in polycystic kidney disease. Cell Signal. 2020, 67(109495), 109495. [Google Scholar] [CrossRef]
- Menezes, LF; Lin, CC; Zhou, F; Germino, GG. Fatty acid oxidation is impaired in an orthologous mouse model of autosomal dominant polycystic kidney disease. EBioMedicine 2016, 5, 183–192. [Google Scholar] [CrossRef]
- Podrini, C; Rowe, I; Pagliarini, R; et al. Dissection of metabolic reprogramming in polycystic kidney disease reveals coordinated rewiring of bioenergetic pathways. Commun Biol. 2018, 1(1), 194. [Google Scholar] [CrossRef]
- Padovano, V; Kuo, IY; Stavola, LK; et al. The polycystins are modulated by cellular oxygen-sensing pathways and regulate mitochondrial function. Mol Biol Cell. 2017, 28(2), 261–269. [Google Scholar] [CrossRef]
- Buchholz, B; Eckardt, KU. Role of oxygen and the HIF-pathway in polycystic kidney disease. Cell Signal. 2020, 69(109524), 109524. [Google Scholar] [CrossRef]
- Cassina, L; Chiaravalli, M; Boletta, A. Increased mitochondrial fragmentation in polycystic kidney disease acts as a modifier of disease progression. FASEB J 2020, 34(5), 6493–6507. [Google Scholar] [CrossRef]
- Kuo, IY; Brill, AL; Lemos, FO; et al. Polycystin 2 regulates mitochondrial Ca2+ signaling, bioenergetics, and dynamics through mitofusin 2. Sci Signal. 2019, 12(580), eaat7397. [Google Scholar] [CrossRef]
- Chumley, P; Zhou, J; Mrug, S; et al. Truncating PKHD1 and PKD2 mutations alter energy metabolism. Am J Physiol Renal Physiol. 2019, 316(3), F414–F425. [Google Scholar] [CrossRef] [PubMed]
- Picard, M; McEwen, BS. Psychological stress and mitochondria: A systematic review. Psychosom Med. 2018, 80(2), 141–153. [Google Scholar] [CrossRef] [PubMed]
- Devlin, L; Dhondurao Sudhindar, P; Sayer, JA. Renal ciliopathies: promising drug targets and prospects for clinical trials. Expert Opin Ther Targets 2023, 27(4-5), 325–346. [Google Scholar] [CrossRef] [PubMed]
- Margaria, JP; Campa, CC; De Santis, MC; Hirsch, E; Franco, I. The PI3K/Akt/mTOR pathway in polycystic kidney disease: A complex interaction with polycystins and primary cilium. Cell Signal. 2020, 66(109468), 109468. [Google Scholar] [CrossRef]
- Panwar, V; Singh, A; Bhatt, M; et al. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct Target Ther. 2023, 8(1), 375. [Google Scholar] [CrossRef]
- Li, A; Fan, S; Xu, Y; et al. Rapamycin treatment dose-dependently improves the cystic kidney in a new ADPKD mouse model via the mTORC1 and cell-cycle-associated CDK1/cyclin axis. J Cell Mol Med. 2017, 21(8), 1619–1635. [Google Scholar] [CrossRef]
- Liu, Y; Pejchinovski, M; Wang, X; et al. Dual mTOR/PI3K inhibition limits PI3K-dependent pathways activated upon mTOR inhibition in autosomal dominant polycystic kidney disease. Sci Rep. 2018, 8(1), 5584. [Google Scholar] [CrossRef]
- Xu, Y; Shi, T; Cui, X; et al. Asparagine reinforces mTORC1 signaling to boost thermogenesis and glycolysis in adipose tissues. EMBO J 2021, 40(24), e108069. [Google Scholar] [CrossRef]
- Clerici, S; Podrini, C; Stefanoni, D; et al. Inhibition of asparagine synthetase effectively retards polycystic kidney disease progression. EMBO Mol Med. 2024, 16(6), 1379–1403. [Google Scholar] [CrossRef]
- Bais, T; Gansevoort, RT; Meijer, E. Drugs in clinical development to treat autosomal dominant polycystic kidney disease. Drugs 2022, 82(10), 1095–1115. [Google Scholar] [CrossRef]
- King, JD, Jr.; Fitch, AC; Lee, JK; et al. AMP-activated protein kinase phosphorylation of the R domain inhibits PKA stimulation of CFTR. Am J Physiol Cell Physiol. 2009, 297(1), C94–C101. [Google Scholar] [CrossRef] [PubMed]
- Hallows, KR; Raghuram, V; Kemp, BE; Witters, LA; Foskett, JK. Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP-activated protein kinase. J Clin Invest. 2000, 105(12), 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, DM; Shackelford, DB; Egan, DF; et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008, 30(2), 214–226. [Google Scholar] [CrossRef] [PubMed]
- Sethi, JK; Vidal-Puig, A. Wnt signalling and the control of cellular metabolism. Biochem J 2010, 427(1), 1–17. [Google Scholar] [CrossRef]
- Lal, M; Song, X; Pluznick, JL; et al. Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Hum Mol Genet. 2008, 17(20), 3105–3117. [Google Scholar] [CrossRef]
- Pagliarini, R; Podrini, C. Metabolic reprogramming and reconstruction: Integration of experimental and computational studies to set the path forward in ADPKD. Front Med (Lausanne) 2021, 8, 740087. [Google Scholar] [CrossRef]
- Fan, W; Evans, R. PPARs and ERRs: molecular mediators of mitochondrial metabolism. Curr Opin Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef]
- Chung, KW; Lee, EK; Lee, MK; Oh, GT; Yu, BP; Chung, HY. Impairment of PPARα and the fatty acid oxidation pathway aggravates renal fibrosis during aging. J Am Soc Nephrol. 2018, 29(4), 1223–1237. [Google Scholar] [CrossRef]
- Kang, HM; Ahn, SH; Choi, P; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015, 21(1), 37–46. [Google Scholar] [CrossRef]
- Lakhia, R. The role of PPARα in autosomal dominant polycystic kidney disease. Curr Opin Nephrol Hypertens. 2020, 29(4), 432–438. [Google Scholar] [CrossRef]
- Mao, Z; Valluru, MK; Ong, ACM. Drug repurposing in autosomal dominant polycystic kidney disease: back to the future with pioglitazone. Clin Kidney J. 2021, 14(7), 1715–1718. [Google Scholar] [CrossRef] [PubMed]
- Chambers, JM; Wingert, RA. PGC-1α in disease: Recent renal insights into a versatile metabolic regulator. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, Y; Inagi, R; Yoshihara, D; et al. Mitochondrial abnormality facilitates cyst formation in autosomal dominant polycystic kidney disease. Mol Cell Biol. 2017, 37(24). [Google Scholar] [CrossRef] [PubMed]
- Lee, EC; Valencia, T; Allerson, C; et al. Discovery and preclinical evaluation of anti-miR-17 oligonucleotide RGLS4326 for the treatment of polycystic kidney disease. Nat Commun. 2019, 10(1), 4148. [Google Scholar] [CrossRef]
- Hajarnis, S; Lakhia, R; Yheskel, M; et al. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat Commun. 2017, 8(1), 14395. [Google Scholar] [CrossRef]
- Messchendorp, AL; Casteleijn, NF; Meijer, E; Gansevoort, RT. Somatostatin in renal physiology and autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2020, 35(8), 1306–1316. [Google Scholar] [CrossRef]
- Di Benedetto, G; Gerbino, A; Lefkimmiatis, K. Shaping mitochondrial dynamics: The role of cAMP signalling. Biochem Biophys Res Commun. 2018, 500(1), 65–74. [Google Scholar] [CrossRef]
- Pinzi, L; Bisi, N; Rastelli, G. How drug repurposing can advance drug discovery: challenges and opportunities. Front Drug Discov (Lausanne) 2024, 4, 1460100. [Google Scholar] [CrossRef]
- Dubois, EA; Rissmann, R; Cohen, AF. Tolvaptan: New drug mechanisms. Br J Clin Pharmacol. 2012, 73(1), 9–11. [Google Scholar] [CrossRef]
- Wilk, EJ; Howton, TC; Fisher, JL; et al. Prioritized polycystic kidney disease drug targets and repurposing candidates from pre-cystic and cystic mouse Pkd2 model gene expression reversion. Mol Med. 2023, 29(1), 67. [Google Scholar] [CrossRef]
- Garza, AZ; Park, SB; Kocz, R. Drug elimination. In StatPearls; StatPearls Publishing, 2025; Available online: https://www.ncbi.nlm.nih.gov/books/NBK547662/ (accessed on 11 July 2025).
- Arjune, S; Todorova, P; Bartram, MP; Grundmann, F; Müller, RU. Liver manifestations in autosomal dominant polycystic kidney disease (ADPKD) and their impact on quality of life. Clin Kidney J. 2025, 18(1), sfae363. [Google Scholar] [CrossRef]
- Hogan, MC; Abebe, K; Torres, VE; et al. Liver involvement in early autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol. 2015, 13(1), 155–164.e6. [Google Scholar] [CrossRef]
- Lea-Henry, TN; Carland, JE; Stocker, SL; Sevastos, J; Roberts, DM. Clinical pharmacokinetics in kidney disease: Fundamental principles. Clin J Am Soc Nephrol. 2018, 13(7), 1085–1095. [Google Scholar] [CrossRef]
- Dreisbach, AW; Lertora, JJL. The effect of chronic renal failure on drug metabolism and transport. Expert Opin Drug Metab Toxicol. 2008, 4(8), 1065–1074. [Google Scholar] [CrossRef]
- Tillmann, AC; Rostami-Hodjegan, A; Barber, J; Al-Majdoub, ZM. Pharmacotherapy in kidney disease: what it takes to move from general guidance to specific recommendations to stratified subgroups of patients - the tale of autosomal dominant polycystic kidney disease (ADPKD). Expert Opin Drug Metab Toxicol. 2025, 21(6), 677–687. [Google Scholar] [CrossRef]
- Tillmann AC, Peters DJM, Rostami-Hodjegan A, et al. Changes in protein expression of renal drug transporters and drug-metabolizing enzymes in autosomal dominant polycystic kidney disease patients. Clin Pharmacol Ther. Published online May 15, 2025. [CrossRef]
- Beaudoin, JJ; Bezençon, J; Cao, Y; et al. Altered hepatobiliary disposition of tolvaptan and selected tolvaptan metabolites in a rodent model of polycystic kidney disease. Drug Metab Dispos. 2019, 47(2), 155–163. [Google Scholar] [CrossRef]
- Center for Drug Evaluation; Research. Pharmacokinetics in Patients with Impaired Renal Function — Study Design, Data Analysis, and Impact on Dosing. U.S. Food and Drug Administration. 29 July 2024. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/pharmacokinetics-patients-impaired-renal-function-study-design-data-analysis-and-impact-dosing (accessed on 11 July 2025).
- Sieben, CJ; Harris, PC. Experimental models of polycystic kidney disease: Applications and therapeutic testing. Kidney360 2023, 4(8), 1155–1173. [Google Scholar] [CrossRef]
- Jones E, Taluri S, Wilk E, Lasseigne B. Streamlining drug repurposing: Optimizing candidate prioritization to facilitate clinical adoption and minimize attrition rates. Preprints. Published online November 21, 2024. [CrossRef]
Figure 1.
Metabolic and mitochondrial reprogramming in autosomal dominant polycystic kidney disease. This figure shows the interactions and consequences of the alterations discussed in this review. Upregulated molecules and processes are colored in red, and downregulated molecules and processes are colored in blue. Black solid lines denote upregulated interactions in ADPKD, and downregulated interactions are marked with gray dashed lines. Abbreviations: Akt = protein kinase B; AMPK = adenosine monophosphate-activated protein kinase; Arg = arginine; ASA = argininosuccinic acid; Asn = asparagine; ASNS = Asparagine synthetase; Asp = aspartate; ASS1 = argininosuccinate synthetase 1; ATP = adenine triphosphate; AVP = arginine vasopressin; AQP2 = aquaporin-2; BCAA = branched chain amino acid; cAMP = cyclic adenosine monophosphate; Cl- = chlorine; CoA = carboxylic acid; Cyt C = cytochrome C; FA = fatty acid; FAO = fatty acid oxidation; FAS = fatty acid synthesis; FFA = free fatty acid; Gln = glutamine; GLS1 GAC = Glutaminase C; Glu = glutamate; H2O = water; HIF-1ɑ = hypoxia-inducible factor 1-alpha; miR-17 = microRNA-17; mTOR = mammalian target of rapamycin; NADPH = nicotinamide adenine dinucleotide phosphate; NH4+ = ammonia; NO = nitric oxide; O2 = oxygen; OAA = oxaloacetic acid; OXPHOS = oxidative phosphorylation; PGC1ɑ = peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PI3K = phosphatidylinositol 3-kinase; PKA = protein kinase A; PPAR = peroxisome proliferator–activated receptor; PPP = pentose phosphate pathway; PTEN = phosphatase and tensin homolog; R5P = ribose-5-phosphate; ROS = reactive oxygen species; SST = somatostatin; TCA = tricarboxylic acid; V2R = vasopressin receptor 2; Wnt= wingless-related integration site; ɑ = alpha; ɑ-KG = alpha-ketoglutarate; β = beta; γ = gamma.
Figure 1.
Metabolic and mitochondrial reprogramming in autosomal dominant polycystic kidney disease. This figure shows the interactions and consequences of the alterations discussed in this review. Upregulated molecules and processes are colored in red, and downregulated molecules and processes are colored in blue. Black solid lines denote upregulated interactions in ADPKD, and downregulated interactions are marked with gray dashed lines. Abbreviations: Akt = protein kinase B; AMPK = adenosine monophosphate-activated protein kinase; Arg = arginine; ASA = argininosuccinic acid; Asn = asparagine; ASNS = Asparagine synthetase; Asp = aspartate; ASS1 = argininosuccinate synthetase 1; ATP = adenine triphosphate; AVP = arginine vasopressin; AQP2 = aquaporin-2; BCAA = branched chain amino acid; cAMP = cyclic adenosine monophosphate; Cl- = chlorine; CoA = carboxylic acid; Cyt C = cytochrome C; FA = fatty acid; FAO = fatty acid oxidation; FAS = fatty acid synthesis; FFA = free fatty acid; Gln = glutamine; GLS1 GAC = Glutaminase C; Glu = glutamate; H2O = water; HIF-1ɑ = hypoxia-inducible factor 1-alpha; miR-17 = microRNA-17; mTOR = mammalian target of rapamycin; NADPH = nicotinamide adenine dinucleotide phosphate; NH4+ = ammonia; NO = nitric oxide; O2 = oxygen; OAA = oxaloacetic acid; OXPHOS = oxidative phosphorylation; PGC1ɑ = peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PI3K = phosphatidylinositol 3-kinase; PKA = protein kinase A; PPAR = peroxisome proliferator–activated receptor; PPP = pentose phosphate pathway; PTEN = phosphatase and tensin homolog; R5P = ribose-5-phosphate; ROS = reactive oxygen species; SST = somatostatin; TCA = tricarboxylic acid; V2R = vasopressin receptor 2; Wnt= wingless-related integration site; ɑ = alpha; ɑ-KG = alpha-ketoglutarate; β = beta; γ = gamma.
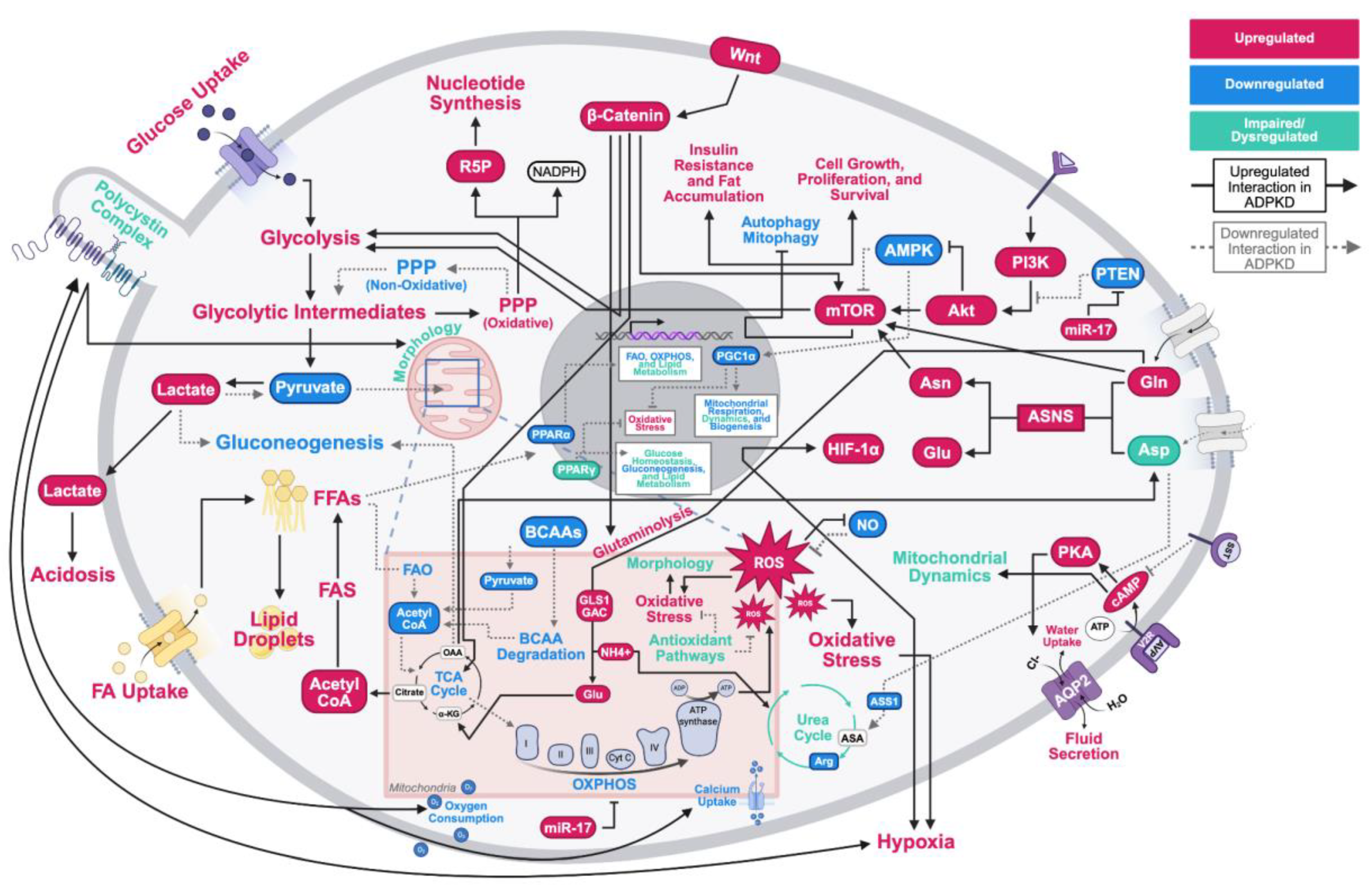
Figure 2.
Overview of complications in autosomal dominant polycystic kidney disease (ADPKD) drug development. Like a puzzle, ADPKD pathophysiology and external influences combine to complicate therapeutic design, efficacy, and toxicity. This creates obstacles in developing efficacious and safe therapeutic options for ADPKD patients.
Figure 2.
Overview of complications in autosomal dominant polycystic kidney disease (ADPKD) drug development. Like a puzzle, ADPKD pathophysiology and external influences combine to complicate therapeutic design, efficacy, and toxicity. This creates obstacles in developing efficacious and safe therapeutic options for ADPKD patients.
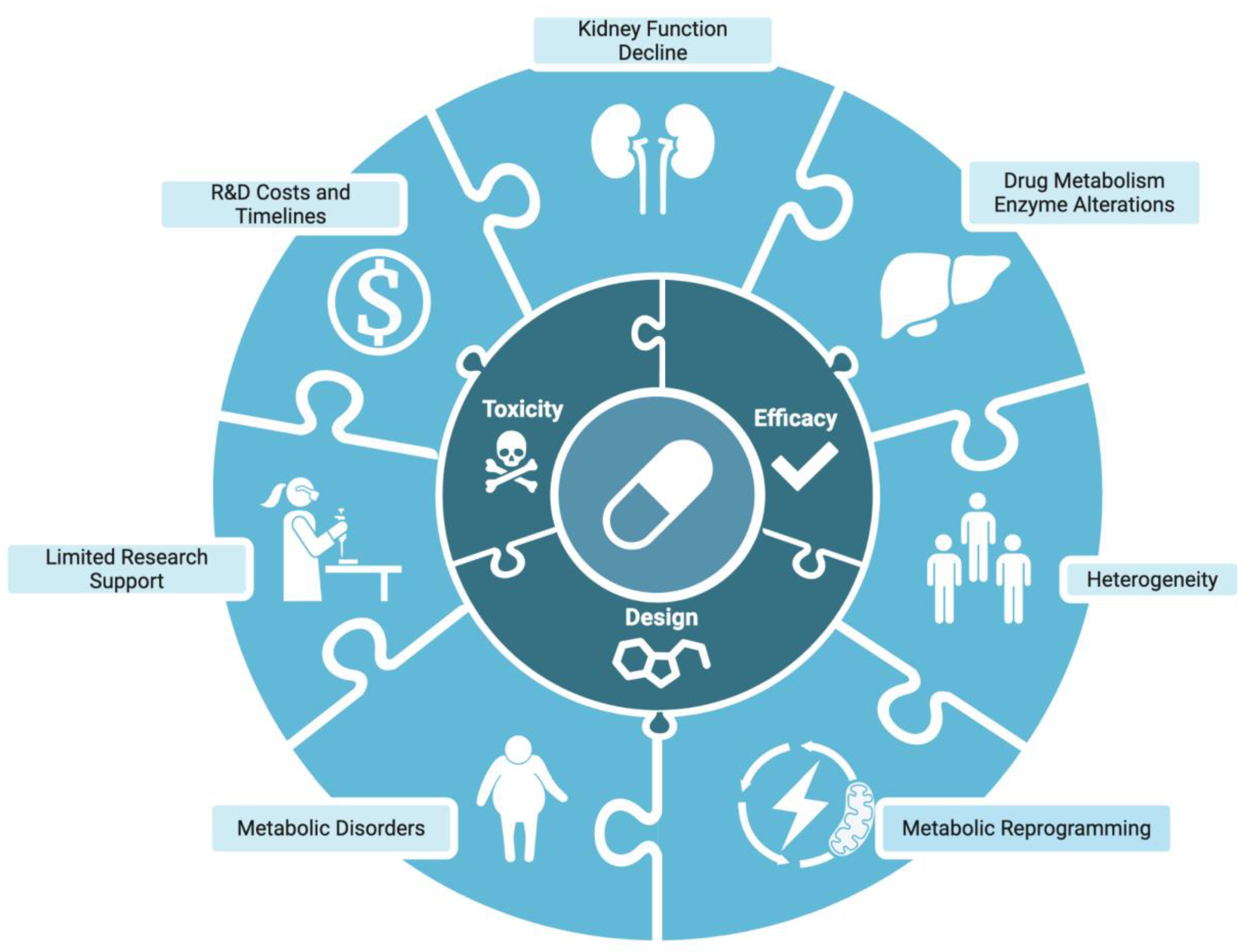
Figure 3.
Traditional drug development compared to drug repurposing. Drug repurposing leverages prior studies and computational tools to reduce the time and costs associated with de novo development. While traditional drug development can take 10-15 years and up to $2 billion, drug repurposing takes 3-12 years and up to $300 million. Drug repurposing also increases approval rates from 12% to over 30%.
Figure 3.
Traditional drug development compared to drug repurposing. Drug repurposing leverages prior studies and computational tools to reduce the time and costs associated with de novo development. While traditional drug development can take 10-15 years and up to $2 billion, drug repurposing takes 3-12 years and up to $300 million. Drug repurposing also increases approval rates from 12% to over 30%.
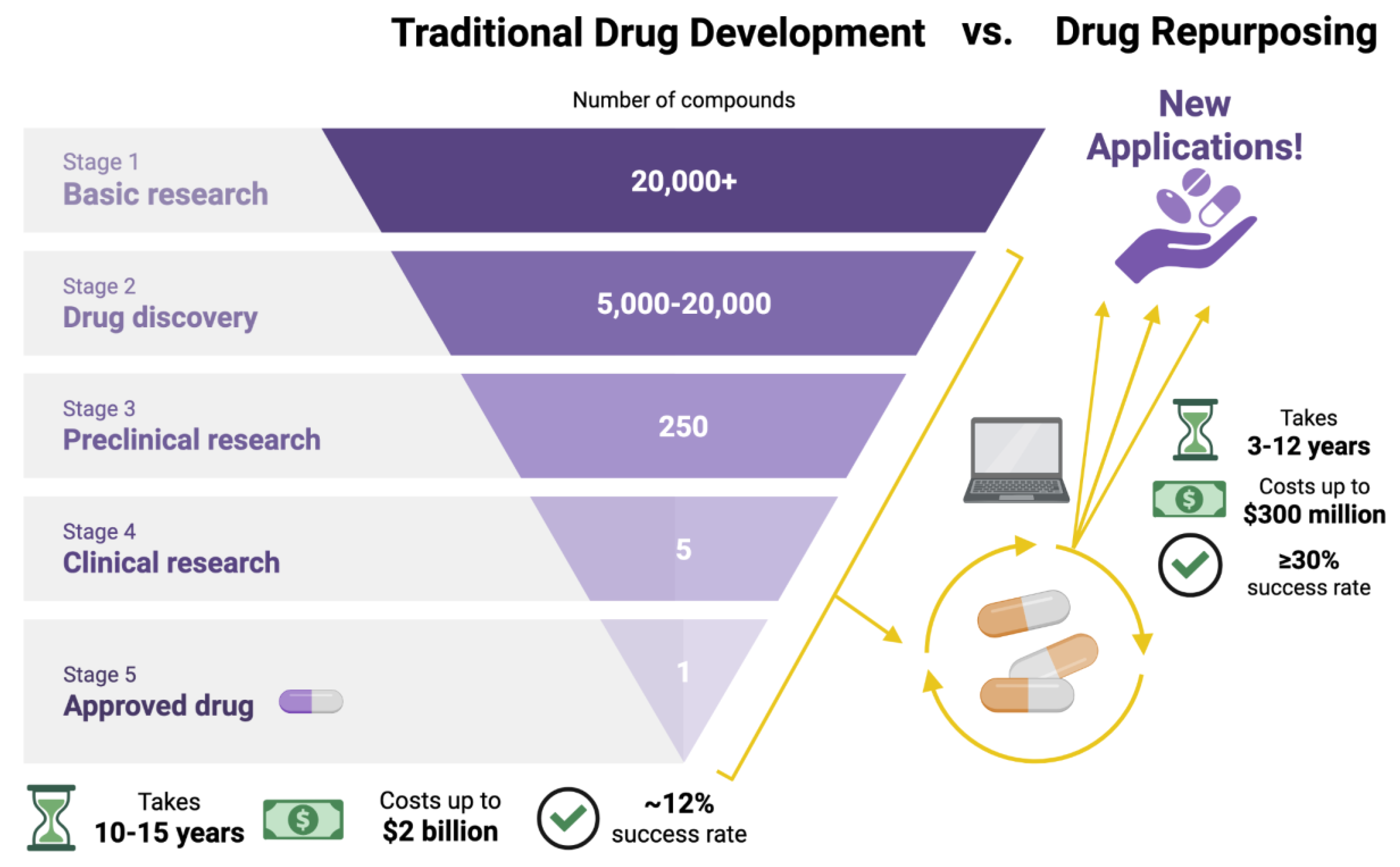

Table 1.
Repurposed autosomal dominant polycystic kidney disease (ADPKD) drugs with their targets and clinical trial outcomes. Drugs in this table were repurposed for molecular targets discussed in this review. Original indications were found using the U.S. Food and Drug Administration (FDA) Drugs@FDA search tool. As shown, metabolic reprogramming is being extensively studied as a therapeutic target with promising results. Conclusive evidence of a successful repurposed drug targeting ADPKD metabolic reprogramming is still lacking.
Table 1.
Repurposed autosomal dominant polycystic kidney disease (ADPKD) drugs with their targets and clinical trial outcomes. Drugs in this table were repurposed for molecular targets discussed in this review. Original indications were found using the U.S. Food and Drug Administration (FDA) Drugs@FDA search tool. As shown, metabolic reprogramming is being extensively studied as a therapeutic target with promising results. Conclusive evidence of a successful repurposed drug targeting ADPKD metabolic reprogramming is still lacking.
| Drug Class | Molecular Target / MOA | Drug Compound | Original Indication | Results | Clinical Trials / Relevant DOIs |
| Vaptans | |||||
| V2R / Antagonist | Tolvaptan | Hypervolemic or euvolemic hyponatremia | Tolvaptan significantly reduced the rate of TKV growth and eGFR decline (P<0.001) compared to the placebo. Patients in the tolvaptan group had a higher frequency of aquaresis- and hepatic-related adverse events. | TEMPO (NCT00428948); REPRISE (NCT02160145); doi:10.1056/NEJMoa120551; doi:10.1056/NEJMoa1710030 | |
| Lixivaptan | Euvolemic hyponatremia | Terminated early by sponsor due to concerns about commercial potential and hepatic safety. | ACTION (NCT04064346); ALERT (NCT04152837) | ||
| Rapalogs | |||||
| mTORC1 / Antagonist | Everolimus | Advanced renal cell carcinoma after failure of sunitinib or sorafenib treatment | Everolimus significantly slowed TKV increase (P=0.02) in the first year but was associated with a significantly greater decline in eGFR (P=0.004) compared with placebo. | NCT00414440; doi:10.1056/NEJMoa1003491 | |
| Sirolimus | Prophylaxis of organ rejection in renal transplant patients | Sirolimus treatment increased TKV and TCV and did not significantly affect eGFR. Incidence of gastrointestinal adverse events in treated patients was high (94%). SIRENA-II was terminated early due to increased adverse events and ESRD progression in treated participants. A later study found that low-dose oral sirolimus is associated with ovarian toxicity, menstrual cycle disturbances, and ovarian cysts. | SIRENA-II (NCT01223755); NCT00346918; doi:10.2215/CJN.09900915; doi:10.1056/NEJMoa0907419; doi:10.1093/ndt/gfp280; doi:10.1371/journal.pone.0045868 | ||
| Polyunsaturated Fatty Acids | |||||
| PPAR family / Agonist | Gamolenic acid | Atopic dermatitis/eczema; Asthma; Rheumatoid arthritis | Identified as a potential ADPKD treatment option in a drug repurposing study. Reduced cyst size in renal epithelial cells grown in a 3D-gel matrix. | doi:10.1016/j.ebiom.2019.11.046 | |
| Icosapent ethyl / Eicosapentaenoic acid (EPA) | Hypertriglycidemia | Part of a larger clinical trial investigating the use of calcium channel blockers for the treatment of hypertension in ADPKD patients. EPA administration over 2 years did not have a significant effect on renal function decline or kidney enlargement. | NCT00541853; doi:10.1093/ndt/gfn144 | ||
| Fibrates | |||||
| PPAR-α / Agonist | Fenofibrate | Hypertriglyceridemia; Primary hypercholesterolemia; Mixed dyslipidemia | In a preclinical trial on Pkd1^RC/RC mice, treatment with fenofibrate reduced TKV by 26.9%, TCV by 60.9%, liver fibrosis by 40%, and cyst proliferation by 71.6% compared to the control group. | doi:10.1152/ajprenal.00352.2017 | |
| Thiazolidinediones | |||||
| PPAR-γ / Agonist | Pioglitazone | T2DM | Low-dose pioglitazone was associated with reduced TKV growth rate compared to the placebo; however, the study was underpowered. Due to concerns over side effects with higher doses and preclinical data showing maximal efficacy at a low dose, the study did not examine higher dosages. | NCT02697617; doi:10.1093/ckj/sfaa232 | |
| Rosiglitazone | In human cystic epithelial cell culture models, rosiglitazone reduced the expression of fibrotic markers and cell proliferation. Male heterozygous Han:SPRD rats treated with rosiglitazone had improved kidney-to-body weight ratios and delayed ADPKD progression. | doi:10.1152/ajprenal.00194.2015 | |||
| Biguanides | |||||
| AMPK / Agonist | Metformin | Malaria; T2DM | Metformin treatment was well tolerated in ADPKD patients. Changes in htTKV (p=0.9) and eGFR (p=0.2) were not statistically significant, but eGFR decline was reduced in the metformin group compared to placebo (-0.41±1.81 vs -3.35±1.70 mL/min/1.73 m^2). Starting kidney size, hypertension, and disease stage may have influenced this difference. | TAME-PKD (NCT02656017); IMPEDE-PKD (NCT04939935); NCT02903511; doi:10.34067/KID.0004002020; doi:10.1053/j.ajkd.2021.06.026 | |
| Glucose Analogs | |||||
| Glycolysis / Antagonist | 2-deoxy-d-glucose (2DG) | Cancer | In a preclinical Pkd1^(ΔC/flox)TmCre murine model, 2DG administration restored AMPK activation, reduced kidney-to-body weight ratio, and was associated with improved kidney function preservation. Mice treated with 2DG additionally had reduced cyst expansion in both distal tubules and collecting ducts. | doi:10.1681/ASN.2015030231 | |
| Somatostatin Analogs | |||||
| SSTR1-5 / Agonist | Octreotide | Acromegaly; Thyrotrophinomas | Octreotide-LAR significantly decreased TKV growth in both studies after one year (P=0.002, P=0.04) and at three years in ALADIN (P=0.04). ALADIN found the octreotide-LAR group had reduced eGFR decline, while ALADIN 2 found a higher rate of eGFR decline compared to the placebo (11.3% vs 7.0%). Octreotide treatment was associated with cholelithiasis and cholecystitis adverse events. | ALADIN (NCT00309283); ALADIN 2 (NCT01377246); doi:10.1016/S0140-6736(13)61407-5; doi:10.1371/journal.pmed.1002777 | |
| Lanreotide | Acromegaly; Gastroenteropancreatic neuroendocrine tumors | Lanreotide treatment significantly lowered the percent change in htTKV (P=0.02), with a 24% reduction in htTKV growth rate compared to the placebo. There was a high incidence of adverse events in the treatment group, leading 10% of patients to withdraw from the study. 28% of treated patients experienced serious adverse events. An interim analysis of DIPAK-1 concluded somatostatin analog use is associated with an increased risk of hepatic cyst infection. | DIPAK-1 (NCT01616927); doi:10.1001/jama.2018.15870; doi:10.1007/s40264-016-0486-x | ||
| Pasireotide | Acromegaly; Cushing's disease | In patients with ADPKD/ADPLD with severe liver involvement, pasireotide-LAR treatment was associated with a significant decrease in TLV (P<0.001) and TKV (P=0.02). Adverse events were most commonly related to hyperglycemia, and 19/32 (59%) of treated patients developed diabetes compared to 1/15 (7%) of patients in the placebo group (P<0.001). | NCT01670110; doi:10.2215/CJN.13661119 | ||
Abbreviations: ADPKD = autosomal dominant polycystic kidney disease; ADPLD = autosomal dominant polycystic liver disease; AMPK = adenosine monophosphate-activated protein kinase; eGFR = estimated glomerular filtration rate; ESRD = end-stage renal disease; htTKV = height-adjusted total kidney volume; LAR = long-acting release; mTORC1 = mammalian target of rapamycin complex 1; PPAR = peroxisome proliferator–activated receptor; SSTR = somatostatin receptor; T2DM = type-2 diabetes mellitus; TCV = total cyst volume; TKV = total kidney volume; TLV = total liver volume; V2R = vasopressin 2 receptor.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



