
Submitted:
19 December 2025
Posted:
22 December 2025
You are already at the latest version
Abstract
Variants in MYPN encoding a sarcomeric protein, myopalladin, are associated with different types of cardiomyopathies and myopathies. However, molecular mechanisms of MYPN-associated pathologies are still poorly understood. In this study, we generated induced pluripotent stem cells (iPSCs) from a hypertrophic cardiomyopathy patient carried a novel p.N989I (c.2966A>T) variant in MYPN and used iPSC-derived cardiomyocytes to examine the impact of the variant on biophysical characteristics and transcriptomic profile. No significant changes in parameters of calcium transient, sodium current and action potential were found in iPSC-derived cardiomyocytes with p.N989I (c.2966A>T) variant in MYPN compared to control cells from a healthy donor. At the transcriptomic level, MYPN-N989I cardiomyocytes demonstrated an upregulation of genes linked to cell cycle, division springle, microtubule cytoskeleton organization and myogenic program genes. A downregulation of sarcomeric, Z-disc- and cell junction-associated genes and genes involved in ATP synthesis, oxidative phosphorylation and SRF-signaling pathway was also revealed. Our data suggest that the p.N989I (c.2966A>T) variant in MYPN plays a dual role in hypertrophic cardiomyopathy pathogenesis, disrupting not only sarcomeric and cytoskeletal organization but also muscle gene program regulation.
Keywords:
myopalladin
; hypertrophic cardiomyopathy
; genetic variants
; induced pluripotent stem cells
; cardiomyocytes
1. Introduction
Myopalladin encoded by MYPN gene was identified over twenty years ago as one of the key α-actinin binding proteins, which takes part in actin cytoskeleton organization, polymerization and stability [1]. Based on its homology with earlier described palladin and myotillin, myopalladin was demonstrated to have an important role in actin filament assembly of both sarcomeric and cytoskeletal pools of F-actin in muscle cells. Being a striated muscle-specific protein, it is localised in I-band and Z-line of sarcomeric structure and interacts with several structural components of Z-disc, such as nebulette and titin. Myopalladin is also capable to interact with nucleus-translocating proteins of Z-disc area, CARP and ANKRD2. The latter fact arguments for mechanostransduction function of myopalladin, making it one of the important regulators of contractility strength and relaxation through initiating several gene expression programs. The ability of myopalladin to provide a structural integrity of Z-disc [2] and to translocate to the nucleus together with CARP makes it a possible regulator of myogenic gene program through interaction with SRF and regulation of cell cycle-related genes. In addition, MYPN-knockout mice demonstrate the alteration in several signalling pathways and kinase expression such as mitogen-activated protein kinases Erk1/2, JNK, PAK1 as well as Akt-phosphorylation [3]. All together this confirms the important role of MYPN in Z-line architecture and stress-detecting signalling of striated muscle cells.
Being a Z-line associated protein of sarcomere, MYPN attracts attention as a possible cardiomyopathy and myopathy causative gene. Similar to other Z-line proteins, myopalladin has been described in connection to several types of cardiomyopathies – hypertrophic (HCM) [4,5], dilated (DCM) [6,7,8], restrictive (RCM) and left-ventricular non-compaction [9]. It was also detected as a causative gene for slow progressive congenital cap myopathy – a subtype of nemaline myopathy – with autosomal recessive inheritance [10].
Earlier we described a HCM patient carried a novel p.N989I (c.2966A>T) variant in MYPN [11]. The substitution is localized to the alpha-actinin-interacting region of the protein [1] and posesses the pathogenic properties according to in silico analysis data. However, clinical significance of the variant and possible mechanisms underlying its pathogenicity remain unknown. Generating induced pluripotent stem cells (iPSCs) from HCM patients and their subsequent differentiation into cardiomyocytes have been successfully used to create cell models for unraveling pathogenetic mechanisms of the disease [12,13,14,15,16,17,18,19,20,21,22]. In this study, we applied the iPSC-based approach to generate the first cell model for HCM caused by a MYPN variant. To decipher the role of MYPN and the p.N989I (c.2966A>T) variant in HCM pathogenesis, we analysed functional properties (characteristics of calcium transients, sodium currents and action potentials) and transcriptome profile of iPSC-derived cardiomyocytes.
2. Results
2.1. Structural Analysis of MYPN-N989I Variant
A new p.N989I (c.2966A>T) variant in MYPN was earlier described in a patient with HCM and no familial history of inherited cariac disorders [11]. Myopalladin contains five immunoglobulin-like domains. Structural modelling identified this variant to be located in the third Ig domain of myopalladin (Figure 1a). This domain is known as actinin-interacting domain and contributes for direct F-actin binding [3]. The variant resides in a highly conserved position among different species (Figure 1b). Alignment of the myopalladin Ig3 domain with homologous Ig domains shows low similarity of this position (Figure 1c). Myopalladin Ig3 is highly similar with the palladin actin-binding domain Ig3, which consists of seven antiparallel β-strands forming a barrel-like shape (Figure 1c,d) [23]. Within Ig structure, the variant fells in the loop between C and D β-strands (Figure 1d; PDB: 2LQR, chain A). Moreover, ClinPred and AlphaMissense tools predict p.N989I variant as pathogenic.
2.2. Generation of MYPN-N989I iPSC Lines and Directed Differentiation into Cardiomyocytes
Mononuclear cells (MNCs) of the patient carrying a heterozygous p.N989I (c.2966A>T) variant in MYPN were reprogrammed to the pluripotent state using transfection with episomal vectors expressed OCT4, SOX2, KLF4, L-MYC and LIN28 [26]. The cell lines generated demonstrated human pluripotent stem cell-like morphology (Figure 2a) and were negative for mycoplasma presence (Figure 2b). Based on analysis of episome elimination (Figure 2c), two cell lines, FAMRCi015-A and FAMRCi015-B, were selected for further characterization. Pluripotent state of the cell lines was confirmed by expression of pluripotency markers (the OCT4, NANOG, SOX2 genes and SSEA4, TRA-1-60 surface antigens) and capacity to give rise to derivatives of three germ layers during spontaneous differentiation in embryoid bodies (Figure 2d-f). The iPSC lines retained a normal karyotype, 46,XX, (Figure 2g) and p.N989I (c.2966A>T) variant in MYPN (Figure 2h). STR analysis showed that the iPSC lines were identical to the MNCs used for their generation at 26 polimorphic loci (data not shown).
The patient-specific iPSC lines, FAMRCi015-A and FAMRCi015-B, together with a commercially available iPSC line from a healthy donor, WTSIi046-A (Sigma-Aldrich, Darmstadt, Germany, Cat # 66540120), were differentiated into cardiomyocytes. The protocol based on Wnt activation with the GSK3β protein kinase inhibitor (CHIR99021) followed by Wnt inhibition with IWR1 was used first [27]. Zones of spontaneous contractions formed on days 8-9 of differentiation. To isolate pure population of cardiomyocytes, differentiated cells were subjected from day 9 to day 15 of differentiation to metabolic selection in glucose-free medium supplemented with sodium DL-lactate [27]. Purified cardiomyocytes were cultivated for two weeks in the presence of 2µM CHIR99021 for their proliferation and for another two weeks in the medium contained low glucose and high oxidative substrate concentrations [28] to promote maturation of iPSC-derived cardiomyocytes. The fact that the cells obtained were predominantely cardiomyocytes was verified by expression of cardiac troponin I on day 48 of differentiation (Figure S1). iPSC-derived cardiomyocytes from the patient with p.N989I (c.2966A>T) variant in MYPN (MYPN-N989I-CMs) and the healthy donor (Donor-CMs) were used for functional studies and transcriptome analysis to study the impact of the p.N989I (c.2966A>T) variant in MYPN on cardiomyocyte structure and function.
2.3. Calcium Dynamics Is Not Altered in iPSC-Derived Cardiomyocytes with p.N989I Variant in MYPN
MyoPacer field stimulation and IonOptix setup were used to measure parameters of calcium transient in the iPSC-derived cardiomyocytes. Transient duration (time to 90% baseline), pre-peak parameters (time to peak and calcium release velocity), transient Ca2+ amplitude and post-peak parameters (time to baseline and calcium return velocity) did not differ significantly between MYPN-N989I-CMs and Donor-CMs (Figure 3). To sum up, intracellular cytosolic calcium oscillations in MYPN-N989I-CMs were not affected.
2.4. MYPN-N989I-Cardiomyocytes Do Not Demonstrate Any Significant Changes in Characteristics of Sodium Current and Action Ponential
To explore biophysical characteristics of sodium current in MYPN-N989I-CMs, we used patch-clamp technique. Both Donor-CMs and MYPN-N989I-CMs demonstrated typical sodium current traces (Figure 4a). No significant changes were observed in peak sodium current density in MYPN-N989I-CMs versus Donor-CMs (Figure 4b,c, Table 1). Next, we analyzed steady-state activation and steady-state inactivation properties (Figure 4d,e, Table 1). We found a statistically significant depolarizing shift of 4 mV in the half-voltage of steady-state activation (Table 1). Thus, MYPN-N989I-CMs required more depolarization stimuli to action potential initiation and demonstrated features of sodium channel loss-of-function phenotype. In contrast, no statistically significant alteration of the steady-state inactivation was observed (Figure 4e, Table 1).
We also studied the action potentials in MYPN-N989I-CMs and Donor-CMs to clarify the impact of the observed activation shift (Figure 4f). No changes in action potential duration and amplitude were detected (Table 2). Thus, despite the slightly impaired activation in MYPN-N989I-CMs, their overall electrical activity demonstrates no alterations.
2.5. iPSC-Derived Cardiomyocytes with p.N989I Variant in MYPN Revealed a Dysregulation of Several Signaling Pathways
To further study the impact of the p.N989I (c.2966A>T) variant in MYPN on cardiocyomyte properties, we performed transcriptome analysis of MYPN-N989I-CMs and Donor-CMs using RNA sequencing technology. MYPN-N989I-CMs and Donor-CMs could be clearly divided by the principal component analysis (Figure 5a). Differential expression analysis revealed 424 significantly upregulated genes (DEGs) and 226 downregulated DEGs in MYPN-N989I-CMs compared to Donor-CMs (Figure 5b). To investigate the functional relevance of the detected DEGs, GO analysis using the Gene Ontology database (Biological Process, Cellular Components, Molecular Function) was performed. Significantly upregulated DEGs included genes involved in regulation of muscle system process, cell cycle and microtubule cytoskeleton organization (Figure 5c, Table 3). Among genes involved in muscle system process pathway the following genes were detected: CASQ1, MYOM2, P2RX1, MYLK, SMTN, KCNJ8, CHRNA3, GATA5, TNNT3, TNNI3K, OXTR, KBTBD13, ATP8A2, PIK3CG, TNFRSF1B, STRIT1, IGF1, EDNRB, TRIM72, ACTN3 (Figure 4d).
Enrichment analysis revealed upregulation of cell cycle signaling pathways such as nuclear division, regulation of mitotic nuclear division and cell cycle checkpoint. Cyclin-dependent kinase 1 (CDK1) was among the genes involved in these pathways – its expression was increased in MYPN-N989I-CMs. Interestingly, in MYPN-N989I-CMs we detected the concominant decrease in the expression of CDKN1A (p21) gene - an inhibitor of CDK1. Among other genes linked to cell cycle signaling pathway there were genes responsible for formation of Ndc80 complex and structuring the division spindle, such as NDC80, NUF2 and SPC24. In addition, MYPN-N989I-CMs demonstrated activation of genes involved in microtubule-kinetochore conjugation, microtubule assemble and microtubule associated complex such as KIF4A, NDC80, KIFC1, NUF2, CENPE, CDCA8, ESPL1, KIF15, PRC1, SAPCD2, DNAH11, KIF20A, KIF4A, KIFC1, KIF2C, KIF18A, CDCA8, MAP7, KIF15 (Figure 5d, Table 3).
Pathway analysis on downregulated DEGs revealed disturbances in sarcomere organization, Z-disс and I-band organization (MYH6, DES, CSRP1, ACTN4, MYOZ1, BMP10), cell junction-associated genes (EPHB3, SEMA4A, NRXN1, GABRG2, PKP1, CBLN1, CDH12, SLITRK4, SLITRK2, UGT8, CLDN9) as well as actin cytoskeleton (ACTB, MYH6, ACTN4, MYO1D, LAD1, NTN1, MYOZ1, APC2, ARC, RINL, FHDC1, MISP) (Figure 5 e, f, Table 3).
To deeper uncover transcriptomic signatures of MYPN-N989I-CMs, we also performed a fast gene set enrichment analysis (FGSEA) of non-DEGs using the Gene Ontology database. In addition to the previously described genes, this analysis revealed a suppression of signaling pathways associated with ATP synthesis, oxidative phosphorylation and cellular respiration (Figure 5g). FGSEA analysis using Hallmarks database also revealed downregulation of Oxidative Phosphorylation (Figure 5h).
To compare the changes observed at the transcriptomic level with earlier published data on other MYPN-defective cellular and animal models, we analyzed the expression of several Z-disc associated genes, genes related to Ca2+ homeostasis and genes involved in SRF-signaling pathway [2,3,5]. A decreased expression of DES, DSP, VCL, PALLD and SORBS2 and an increased ANKRD1 expression were detected (Figure 6a). We also observed the increased level of CASQ1 and CASQ2 in MYPN-N989I-CMs compared to Donor-CMs (Figure 6a). In addition, a marked downregulation of genes related to endoplasmic reticulum and calcium homeostasis and upregulation of genes related to sarcoplasmatic reticulum ion transport were detected in line with CASQ1 and CASQ2 upregulation (Figure 6b). Thus, we were able to confirm changes in the expression level for many earlier reported genes including those involved in SRF-signaling pathway (Figure 6c).
To sum up, MYPN-N989I-CMs demonstrate significant upregulation of myogenic program, activation of cell cycle- and division spindle-linked genes, which may contribute to the development of cardiac hypertrophy, along with downregulation of sarcomeric, Z-disc- and cell junction-associated genes as well as ATP synthesis, oxidative phosphorylation-related genes and genes involved in SRF-signaling pathway.
3. Discussion
Mutations in MYPN gene have been demonstrated in connection to various human pathologies since 2008 first being described in patients with dilated cardiomyopathy and later in other types of cardiomyopathies - hypertrophic and restrictive [6,8,29,30]. Along with cardiac disorders, MYPN loss of function or homozygous mutations were reported in congenital and slow-progressive myopathies – similar to other genes commonly expressed in cardiac and skeletal muscle. However, there are still some difficulties in the interpretation of pathogenic role of MYPN variants, especially since they are often detected not solely, but in combination with other pathogenic variants and not always clearly segregate with the disease [4,31]. Apart from the causative role of MYPN in monogenic cardiac and neuromuscular disorders, this gene was described as one of the genetic loci associated with atrial fibrillation and cardiac remodelling [32,33]. All above rise the need for more detailed knowledge on the fine molecular mechanisms of MYPN-associated pathologies and the bases for MYPN-linked cardiomyocyte dysfunction. The described substitution p.N989I (c.2966A>T) is located in the third Ig domain of MYPN. In myopalladin homologue, palladin (PLLD), this region is important for direct binding interaction with F-actin [23]. It is possible that substitutions in a similar region of myopalladin can also lead to changes in actinin binding or interfere with protein-protein interactions, leading to functional and adaptive transcriptional changes. To address this question, we created the first iPSC-derived cardiomyocyte cellular model to search for ion channel dysfunction, calcium homeostasis abnormalities and alterations in gene expression profile due to a novel HCM-associated MYPN-N989I variant.
MYPN function is mainly demonstrated in connection to mechanotransduction process within sarcomeric Z-line and I-band. In a tight connection with ANKRD1 (CARP), ANKRD2, TTN, actinin 2, nebulette and some other Z-line-associated proteins, MYPN facilitates the transformation of mechanical stimuli from contractile apparatus to biochemical signals and adapts the muscle cells to intrinsic and extracellular stress by activating various stress-response pathways and programs [1,3,34]. The potential interaction of MYPN with PDLIM5, LDB3, OBSCN and MYOM1 has also been proposed by a computational approach [35]. This tight incorporation into Z-disc structure makes the protein relevant to the development of protein-surplus skeletal myopathies such as slowly progressive nemaline myopathies. The mechanism underlying these pathologies is mainly based on the interaction disruption of MYPN with multiple Z-line associated binding partners and a “domino effect”. In addition to the structural role, the shuttling function between Z-line and the nucleus in tight interaction with ANKRD2 was demonstrated for MYPN, further supporting its role in mechanotransduction [29,36]. A possible involvement of MYPN in SRF-signaling pathways and ERK cascade regulation was also demonstrated by several research groups [3,29]. However, up and downregulated genes and the signaling pathways activated in response to MYPN mutations are often controversial and depend on the cell line and animal models used and, importantly, type of mutation and the length of the studied protein [2,3,29]. This may be attributed to the different interaction partners of MYPN within the nucleus and Z-line structure as well as different effects of loss-of function and dominant-negative mutations [5]. In spite of several published studies no clear functional consequences apart from the effect on sarcomeric structure and nuclear translocation of MYPN and its partners have been demonstrated for MYPN mutations [2,29].
In our study, we also failed to detect gross functional abnormalities in iPSC-derived cardiomyocytes neither in intracellular calcium dynamics and transients nor in voltage-dependent Nav1.5 sodium channel currents. The slight alteration in Nav1.5 kinetics in a form of depolarizing shift of steady-state activation and features of sodium channel loss-of-function phenotype, however, did not result in significant changes in action potential duration and amplitude, arguing for no remarkable effect of the HCM-associated MYPN-N989I variant on overall electrical activity of cardiomyocytes. However, in an earlier study by Filomena et al., authors demonstrated altered Ca2+ handling, namely a delayed Ca2+ release and reuptake as well as reduced Ca2+ spark amplitude and the velocity of Ca2+ release in MYPN-knockout (KO) mice. This difference needs some explanation in terms of MYPN function and effect on myocyte Ca2+ handling. First, it can be attributed to the different systems used in the studies: while Filomena and co-authors used knockout mouse model and performed Ca2+ release studies in the isolated cardiomyocytes with complete absence of MYPN, we used iPSC-derived cardiomyocytes carrying the heterozygous MYPN-N989I substitution. Knowing complex MYPN interactions with structural and regulating proteins, it is reasonable to suggest that one amino acid substitution is not enough to trigger the cascade of reactions leading to alterations in Ca2+ homeostasis while the total absence of MYPN can significantly influence Ca2+ handling via abnormal differentiation properties of cardiomyocytes and reduced protein-protein interactions. This mechanism was ealier reported for FLNC-knockout cells and, potentially, can be attributed to many actinin-binding structural proteins [37,38]. In addition, the difference in species-dependent properties – mouse adult cardiomyocytes and iPSC-derived human cardiomyocytes can also explain the observed differences.
In spite of scare functional changes detected in MYPN-N989I-CMs we were able to detect significant differences in gene expression profile compared to Donor-CMs. Significant alterations in pathways and processes associated with regulation of muscle system process, differentiation and cell nuclear division along with microtubule cytoskeleton organization correspond to earlier reported data obtained on muscle tissue of MYPN-KO mice [3]. This goes along with a downregulation of Z-disc and sarcomere organisation and actin cytoskeleton associated proteins reported by the same authors, further supporting the role of MYPN in driving myogenic program (Figure 10 from [3]). Of note, in MYPN-N989I-CMs, we detected an upregulation of ANKRD1 similar to that reported earlier by Filomena and co-authors for MYPN-KO skeletal muscle cells and cardiomyocytes and detected by RNA sequencing and real-time PCR [2,3]. The similar observation is valid for PALLD which downregulation was registered both in our and previous studies [2,3]. We suggest that the pathogenic effect of MYPN-N989I variant in terms of ANKRD1 and PALLD regulation is similar to MYPN ablation and, possibly, can be linked to the altered protein-protein interactions. Defective interaction of MYPN and ANKRD1 can cause the decreased shuttling and translocation of ANKRD1 to the nucleus, thus, consequently leading to its upregulation [3,5].
Using RNA sequencing, we also confirmed a downregulation of DES, DSP and VCL in MYPN-N989I-CMs – similar to that detected on a protein level in transgenic MYPN-Y20C mice and, in addition, demonstrated SORBS2 downregulation, further underscorring the role of MYPN in Z-disc, desmosome and cell-contact organisation [5]. We managed to confirm the alterations in mitochondrial respiration and ATP-production processes in MYPN-N989I-CMs – an observation reported earlier at the ultrastructural level in human MYPN-Q529X cardiomyocytes [5]. These findings independently verified in different experimental systems, cell types and for various MYPN mutations provide a strong evidence for MYPN involvement in both Z-disc and cytoskeletal organisation, structure and regulation of cellular contacts and mitochondrial function. In addition, in spite of no functional changes in Ca2+ transients we detected a marked derangements in genes related to calcium homeostasis at the transcriptional level. Thus, MYPN-N989I interferes with calcium-homeostasis related genes – an observation alining with MYPN-KO-related alterations in calcium oscillation in cardiomyocytes [2]. Importantly, we also were able to confirm the downregulation of the SRF-pathway in MYPN-N989I-CMs – phenomenon described for MYPN-KO skeletal muscle cells [3], further supporting the role of MYPN in transcriptional regulation, possibly, through interaction with actin cytoskeleton and ANKRD1. Thus, we deliniated a dual role of MYPN both in sarcomeric and cytoskeletal organisation as well as in muscle program regulation.
4. Materials and Methods
4.1. Sequence and Structural Analysis of Human Myopalladin and Variant Classification
Sequence for the human myopalladin was obtained from the UniProtKB database under the entry Q86TC9. Domain organization of the protein was performed using InterPro database [24] and lollipops tool [25]. Related protein sequences from other organisms for myopalladin were identified by BLASTP [39] search against the Uniref90 database. Similar Ig regions in other human proteins were identified using human proteome (UniProt ID: UP000005640). Multiple sequence alignment was calculated by Clustal Omega [40] and visualized in Jalview 2.11.5 [41]. 3D structure of the Ig-like domain of myopalladin (PDB: 2LQR, chain A) [23] was extracted from the Protein Data Bank [42,43]. Variant p.N989I was mapped on the structure according to the sequence alignment. In silico visualization was conducted using the PyMol 2.5 software. Pathogenicity of variant p.N989I was assessed based on the ClinPred and AlphaMissense prediction scores obtained from the dbNSFP database version 4.5 [44].
4.2. iPSC Generation
8.1 × 105 of mononuclear cells (MNCs) of the HCM patient carrying p.N989I (c.2966A>T) variant in MYPN were electroporated with episomal vectors (0.5 μg each, Addgene IDs #41855-41858, #41813-41814) using Neon Transfection System (Thermo Fisher Scientific, Waltham, MA, USA), program: 1650 V, 10 ms, 3 times. All reprogramming steps were performed according to the Epi5™ Episomal iPSC Reprogramming Kit user guide (Thermo Fisher Scientific, Waltham, MA, USA). Primary iPSC colonies were picked manually, transferred on a feeder layer, and maintained in KnockOut DMEM supplemented with 15% KnockOut Serum Replacement (KoSR), 0.1 mM Non-Essential Amino Acid (NEAA), 1× penicillin-streptomycin, 1 mM GlutaMAX (all Thermo Fisher Scientific, Waltham, MA, USA), 0.05 mM 2-mercaptoethanol (Amresco, Solon, OH, USA), and 10 ng/ml bFGF (SCI-store, Moscow, Russia). Cells were cultured at 37°C in 5% CO2 and passaged at a ratio of 1:10 every 3-5 days with TrypLE™ Express Enzyme (Thermo Fisher Scientific, Waltham, MA, USA).
4.3. Mycoplasma and Episome Detection
Mycoplasma contamination and episome elimination in iPSC lines were detected by PCR on a T100 Thermal Cycler (Bio-Rad, Hercules, CA, USA) with BioMaster HS-Taq PCR-Color (2×) (Biolabmix, Novosibirsk, Russia). Programs: 95°C – 3 min; 35 cycles: 95°C – 15 s, 67°C – 15 s, 72°C – 20 s; 72°C – 5 min (mycoplasma) and 95°C – 5 min; 35 cycles: 95°C – 15 s, 62°C – 15 s, 72°C – 15 s; 72°C – 5 min (episomes). The primers used are given in Table S1.
4.4. Spontaneous In Vitro Differentiation
iPSCs were dissociated with 0.15% Collagenase Type IV (Thermo Fisher Scientific, Waltham, MA, USA), transferred onto dishes coated with 1% agarose, and cultivated for 14 days in DMEM/F12 (1:1) medium supplemented with 15% KoSR, 0.1 mM NEAA, 1× penicillin-streptomycin, 1 mM GlutaMAX (all Thermo Fisher Scientific, Waltham, MA, USA). The embryoid bodies formed were plated onto Matrigel (Corning, New York, NY, USA)-coated 8-well Chambered Coverglass (Thermo Fisher Scientific, Waltham, MA, USA) for additional seven days.
4.5. Immunofluorescent Staining
Cells were fixed in 4% paraformaldehyde (Sigma-Aldrich, Darmstadt, Germany) for 10 min, permeabilized in 0.4% Triton-Х100 (Sigma-Aldrich, Darmstadt, Germany) for 10 min, incubated with 1% BSA (VWR, Solon, OH, USA) for 30 min. Cells were incubated overnight at 4°С with primary antibodies and 1h at room temperature with secondary antibodies. The antibodies used are listed in Table S1. After each incubation with antibodies, the cells were washed twice for 15 min with PBS. Nuclei were counterstained with DAPI (Sigma-Aldrich, Darmstadt, Germany). Staining was analyzed using a Nikon Eclipse Ti-E microscope and NIS Elements software (Nikon, Tokyo, Japan) or Observer.Z1 and ZEISS Efficient Navigation (ZEN) (Zeiss, Oberkochen, Germany).
4.6. RT-qPCR
RNA was isolated with TRIzol Reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Reverse transcription of 1-2 µg of RNA was performed with the M-MuLV reverse transcriptase (Biolabmix, Novosibirsk, Russia). RT-qPCR was carried out on a LightCycler 480 System (Roche, Basel, Switzerland) with BioMaster HS-qPCR SYBR Blue 2× (Biolabmix, Novosibirsk, Russia). Program: 95°C – 5 min; 40 cycles: 95°С – 10 s, 60°С – 1 min. CT values were normalized to ACTB using ΔΔCT method. The primers used are given in Table S1.
4.7. Karyotyping
Colcemid treatment, cell hypotony and fixation were carried out as described previously [45]. Karyotype was analyzed at the Tomsk National Research Medical Center of the Russian Academy of Sciences using a Zeiss Axio Scope.A1 microscope (Zeiss, Oberkochen, Germany) and Ikaros KARyOtyping Software (MetaSystems, Altlussheim, Germany).
4.8. Analysis of the Patient-Specific Variant
Exon 14 of MYPN was amplified by PCR on a T100 Thermal Cycler (Bio-Rad, Hercules, CA, USA) with BioMaster HS-Taq PCR-Color (2×) (Biolabmix, Novosibirsk, Russia). Program: 95°С – 5 min; 35 cycles: 95°С – 30 s, 60°С – 30 s, 72°С – 30 s; 72°С – 5 min. The primers used are given in Table S1. Sanger sequencing was performed using Big Dye Terminator V. 3.1. Cycle Sequencing Kit (Applied Biosystems, Austin, TX, USA) and analyzed at the SB RAS Genomics Core Facility.
4.9. STR Analysis
STR analysis of the patient-specific iPSC lines and MNCs was performed by Genoanalytica (https://www.genoanalytica.ru) using COrDIS EXPERT 26 Kit (Gordiz, Moscow, Russia) on a 3130 Genetic Analyzer (HITACHI, Applied Biosystems, Japan).
4.10. Directed iPSC Differentiation into Cardiomyocytes
iPSCs were cultivated for 2-3 passages in feeder-free conditions - in Essential 8 Medium (Thermo Fisher Scientific, Waltham, MA, USA) on wells coated with Geltrex LDEV-Free hESC-Qualified Reduced Growth Factor Basement Membrane Matrix (Thermo Fisher Scientific, Waltham, MA, USA). iPSCs were dissociated with ReLeSR (STEMCELL Technologies, Vancouver, BC, Canada) and plated at a ratio of 1:5-1:10 on 12-well plates. After cells reached 80-90% confluency, differentiation was induced by adding for 48h RPMI 1640 medium supplemented with 1× penicillin-streptomycin, 1× B27 Supplement minus insulin (all Thermo Fisher Scientific, Waltham, MA, USA) and 6 µM CHIR99021 (Selleckchem, Houston, TX, USA). 72 h after differentiation onset, RPMI 1640 medium supplemented with 1× penicillin-streptomycin, 1× B27 Supplement minus insulin and 5 µM IWR1 (STEMCELL Technologies, Vancouver, BC, Canada) was added for another 48h. Starting from day 7 of differentiation, cells were cultivated in RPMI 1640 medium supplemented with 1× penicillin-streptomycin and 1× B27 Supplement (Thermo Fisher Scientific, Waltham, MA, USA).
On day 9 of differentiation, cells were subjected to metabolic selection for 6 days in RPMI 1640 medium without D-glucose (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 1× penicillin-streptomycin, 213 µg/ml L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate, 500 µg/ml bovine serum albumin and 5 mM sodium DL-lactate (all Sigma-Aldrich, Darmstadt, Germany). Cardiomyocytes were dissociated with 2.5× TrypLE™ Select Enzyme (Thermo Fisher Scientific, Waltham, MA, USA), plated on Geltrex-coated 6-well plates and cultivated for two weeks in RPMI 1640 medium supplemented with 1× penicillin-streptomycin, 1× B27 Supplement and 2 µM CHIR99021. On day 30 of differentiation, cardiomyocytes were plated on Geltrex-coated 12-well plates at a ratio of 9×105 - 1×106 cells per well. Cells were cultivated for two weeks in the medium for cardiomyocyte maturation [28] - DMEM without D-glucose and L-glutamine (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 3 mM glucose, 10 mM L-lactate, 5 µg/ml Vitamin B12, 0.82 µM Biotin, 5 mM Creatine monohydrate, 2 mM Taurine, 2 mM L-carnitine, 0.5 mM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate (all Sigma-Aldrich, Darmstadt, Germany), 0.1 mM NEAA, 1× B27 Supplement, 1% KoSR, 1× penicillin-streptomycin, 2 mM L-glutamine and 0.5% (w/v) Albumax (all Thermo Fisher Scientific, Waltham, MA, USA).
4.11. Calcium Dynamic with STIMULATION
Registration of calcium transients was performed with electrical stimulation using IonOptix setup (IonOptix, Westwood, MA, USA). Calcium measurements were registered using calcium-sensitive fluorescent dye Fura-2AM (Thermo Fisher Scientific, Waltham, MA, USA) and analyzed values constituted the fluorescence ratio between Fura-2AM channels (binding calcium or not). Cells were seeded on Geltrex-treated 170 µm thick coverglasses (Thermo Fisher Scientific, Waltham, MA, USA), pre-washed with PBS to remove the medium and incubated with 2 μM Fura-2AM in calcium-containing buffer (145 mM NaCl, 4.5 mM KCl, 2 mM MgCl2, 10 mM HEPES; 2 mM CaCl2) for 40 minutes prior to imaging. Cells were paced at 0.2 Hz with a 10V square pulse using a MyoPacer field stimulator (IonOptix, Westwood, MA, USA). The images for each cell were recorded during 2–3 minutes and the transients for one cell were pooled and averaged. Data was recorded from each dish for no more than 30 minutes to avoid cell death.
4.12. Electrophysiology
Sodium currents and action potentials were recorded using whole-cell patch-clamp configuration at room temperature. For patch-clamp measurements, single cardiomyocytes were seeded on Geltrex-coated coverslips. We used the solutions described earlier [46] with a slight modification in the extracellular solution (50 mM NaCl and 90 mM CsCl instead of 140 mM NaCl).
Microelectrodes were manufactured using a puller (P-1000, Sutter Instrument, Novato, CA, USA). The electrode resistance ranged from 2.0 to 3.5 MΩ. Data acquisition and junction potential correction were done using Axopatch 200B amplifier and Clampfit software version 10.3 (Molecular Devices, San Jose, CA, USA). Currents were acquired at 20–50 kHz and low-pass filtered at 5 kHz using an analog-to-digital interface (Digidata 1440A acquisition system, Molecular Devices, San Jose, CA, USA). The series resistance was compensated at 75–80%. All pulse protocols were applied more than 5 min after membrane rupture. APs were elicited at 1 Hz by 3 ms, 1.2× threshold current pulses through the patch pipette.
4.13. Patch-Clamp Data Analyses
Pulse protocols were applied with a holding potential of −100 mV. Current-voltage (I-V) curves were assessed by depolarizing voltage steps from −80 to 60 mV during 40 ms in 5 mV increments at 1 Hz frequency. Current densities at each test potential were obtained by dividing the INa by cell capacitance. Cell capacitance values did not differ significantly between experimental groups. The AP amplitude (APA) and AP duration (APD) at 30, 70 and 90% repolarization (APD30, APD70 and APD90, respectively) were analyzed using IgorPro8 software (WaveMetrics, Portland, OR, USA).
4.14. RNA Sequencing and Bioinformatics Analysis
RNA sequencing libraries were prepared using the TruSeq Stranded mRNA kit (Illumina, San Diego, CA, USA) following the manufacturer's guidelines. The libraries' quantity was assessed using the 4150 TapeStation system (Agilent, Santa Clara, CA, USA) with High Sensitivity DNA ScreenTape Analysis. Subsequently, sequencing was carried out on the Illumina NextSeq 2000 (100 cycles). The quality assessment of the raw reads was evaluated using the FastQC tool (v0.11.9), fastp program (v0.12.4) was used to remove overrepresented polyG sequences. Following this, the reads were aligned to the human genome using the STAR aligner (v2.7.9) with the GRCh38.p13 reference genome and the GENCODE annotation (gencode.v41.primary assembly). Mapped reads were count with the featureCounts program (v2.0.3). The identification of differentially expressed genes (DEGs) was carried out using the RStudio package DESeq2 (v1.40.1). The p-values of the genes were adjusted with the Benjamini-Hochberg procedure and filtered with p.adj < 0.05; only genes with a |log2 fold change| ≥ 1.5 were considered as differentially expressed. Subsequently, Gene Set Enrichment Analysis and GO Enrichment Analysis of gene sets were performed to obtain signaling pathways using Gene Ontology database.
4.15. Statistics
At least three independent experiments (independent cardiac differentiations) were performed for each measurement. Statistical analysis was performed using GraphPad Prism version 5.00 for Windows. For comparison of two groups, the Mann–Whitney test was used. An alpha level of 0.05 was used for all statistical analyses. Data are presented as mean ± SD or mean ± SEM.
5. Conclusions
In the current study we aimed to decipher MYPN-associated pathogenetic mechanisms underlying hypertrophic cardiomyopathy. Using the iPSC-technology, we analyzed functional properties and gene expression pattern in cardiomyocytes with a novel p.N989I (c.2966A>T) variant in MYPN. Despite no significant alterations in parameters of calcium transients, sodium current and action potential were found, the p.N989I (c.2966A>T) variant in MYPN had a global impact on the transcriptomic profile of iPSC-derived cardiomyocytes. We observed an upregulation of genes linked to cell cycle, division sprindle, microtubule cytoskeleton organization and myogenic program genes as well as a downregulation of sarcomeric, Z-disc- and cell junction-associated genes. In addition, genes involved in ATP synthesis, oxidative phosphorylation and SRF-signaling pathway were also downregulated. Our results come in a good agreement with the previously published data obtained using different cell and animal models and MYPN variants. Together these shed more light on pathogenesis of MYPN-associated cardiomyopathies and myopathies, supporting MYPN role not only in sarcomeric and cytoskeletal organization but also in regulation of muscle gene program.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Cells obtained in the course of directed cardiac differentiation of the patient-specific iPSC lines, FAMRCi015-A and FAMRCi015-B, express a cardiomyocyte marker, cardiac troponin I, Table S1: Primers and antibodies used in this study.
Author Contributions
Conceptualization, A.A.K. and S.M.Z.; methodology, A.A.K. and S.M.Z.; data analysis and validation, E.V.D., E.S.K., M.Y.S., A.K.Z., M.T.R., E.G.N., A.M.Z., D.A.K., S.I.T., Y.V.V., D.N.S. and A.A.K.; investigation, E.V.D., E.S.K., M.Y.S., A.K.Z., E.G.N., A.M.Z., S.I.T.; resources, A.A.K. and S.M.Z.; writing—original draft preparation, E.V.D., M.Y.S., E.S.K., A.K.Z., M.T.R. and A.A.K.; writing—review and editing, A.A.K., E.V.D. and M.Y.S.; supervision and funding acquisition, A.A.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministry of Science and Higher Education of the Russian Federation (Agreement No. 075-15-2022-301) and the Russian Science Foundation, grant No. 25-15-00552.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of the E.N. Meshalkin National Medical Research Center (Protocol No. 31 dated 28 June 2013).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data on the iPSC line characterization are available in the Human Pluripotent Stem Cell Registry (https://hpscreg.eu/cell-line/FAMRCi015-A, https://hpscreg.eu/cell-line/FAMRCi015-B accessed on 12 August 2025). The raw RNA sequence data and counts table were submitted in the GEO database and are available under accession number GSE305865.
Acknowledgments
The analysis of immunofluorescence staining was performed using resources of the Common Facilities Centre of Microscopic Analysis of Biological Objects supported by the State project of ICG SB RAS (FWNR-2022-0015).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bang, M.L.; Mudry, R.E.; McElhinny, A.S.; Trombitás, K.; Geach, A.J.; Yamasaki, R.; Sorimachi, H.; Granzier, H.; Gregorio, C.C.; Labeit, S. Myopalladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J. Cell Biol. 2001, 153, 413–427. [Google Scholar] [CrossRef]
- Filomena, M.C.; Yamamoto, D.L.; Carullo, P.; Medvedev, R.; Ghisleni, A.; Piroddi, N.; Scellini, B.; Crispino, R.; D'Autilia, F.; Zhang, J.; et al. Myopalladin knockout mice develop cardiac dilation and show a maladaptive response to mechanical pressure overload. Elife 2021, 10, e58313. [Google Scholar] [CrossRef]
- Filomena, M.C.; Yamamoto, D.L.; Caremani, M.; Kadarla, V.K.; Mastrototaro, G.; Serio, S.; Vydyanath, A.; Mutarelli, M.; Garofalo, A.; Pertici, I.; et al. Myopalladin promotes muscle growth through modulation of the serum response factor pathway. J. Cachexia Sarcopenia Muscle 2020, 11, 169–194. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Yeates, L.; Semsarian, C. Analysis of the Z-disc genes PDLIM3 and MYPN in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2010, 145, 601–602. [Google Scholar] [CrossRef]
- Purevjav, E.; Arimura, T.; Augustin, S.; Huby, A.C.; Takagi, K.; Nunoda, S.; Kearney, D.L.; Taylor, M.D.; Terasaki, F.; Bos, J.M.; et al. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum. Mol. Genet. 2012, 21, 2039–2053. [Google Scholar] [CrossRef]
- Meyer, T.; Ruppert, V.; Ackermann, S.; Richter, A.; Perrot, A.; Sperling, S.R.; Posch, M.G.; Maisch, B.; Pankuweit, S. German Competence Network Heart Failure. Novel mutations in the sarcomeric protein myopalladin in patients with dilated cardiomyopathy. Eur. J. Hum. Genet. 2013, 21, 294–300. [Google Scholar] [CrossRef]
- Zhao, Y.; Feng, Y.; Zhang, Y.M.; Ding, X.X.; Song, Y.Z.; Zhang, A.M.; Liu, L.; Zhang, H.; Ding, J.H.; Xia, X.S. Targeted next-generation sequencing of candidate genes reveals novel mutations in patients with dilated cardiomyopathy. Int. J. Mol. Med. 2015, 36, 1479–1486. [Google Scholar] [CrossRef]
- Duboscq-Bidot, L.; Xu, P.; Charron, P.; Neyroud, N.; Dilanian, G.; Millaire, A.; Bors, V.; Komajda, M.; Villard, E. Mutations in the Z-band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovasc. Res. 2008, 77, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Vershinina, T.; Fomicheva, Y.; Muravyev, A.; Jorholt, J.; Kozyreva, A.; Kiselev, A.; Gordeev, M.; Vasichkina, E.; Segrushichev, A.; Pervunina, T.; et al. Genetic Spectrum of Left Ventricular Non-Compaction in Paediatric Patients. Cardiology 2020, 145, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Lornage, X.; Malfatti, E.; Chéraud, C.; Schneider, R.; Biancalana, V.; Cuisset, J.M.; Garibaldi, M.; Eymard, B.; Fardeau, M.; Boland, A.; et al. Recessive MYPN mutations cause cap myopathy with occasional nemaline rods. Ann. Neurol. 2017, 81, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Dementyeva, E.V.; Vyatkin, Y.V.; Kretov, E.I.; Elisaphenko, E.A.; Medvedev, S.P.; Zakian, S.M. Genetic analysis of patients with hypertrophic cardiomyopathy. Genes and Cells 2020, 15, 68–73. [Google Scholar] [CrossRef]
- Bhagwan, J.R.; Mosqueira, D.; Chairez-Cantu, K.; Mannhardt, I.; Bodbin, S.E.; Bakar, M.; Smith, J.G.W.; Denning, C. Isogenic models of hypertrophic cardiomyopathy unveil differential phenotypes and mechanism-driven therapeutics. J. Mol. Cell. Cardiol. 2020, 145, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Shafaattalab, S.; Li, A.Y.; Gunawan, M.G.; Kim, B.; Jayousi, F.; Maaref, Y.; Song, Z.; Weiss, J.N.; Solaro, R.J.; Qu, Z.; Tibbits, G.F. Mechanisms of Arrhythmogenicity of Hypertrophic Cardiomyopathy-Associated Troponin T (TNNT2) Variant I79N. Front. Cell Dev. Biol. 2021, 9, 787581. [Google Scholar] [CrossRef] [PubMed]
- Vander Roest, A.S.; Liu, C.; Morck, M.M.; Kooiker, K.B.; Jung, G.; Song, D.; Dawood, A.; Jhingran, A.; Pardon, G.; Ranjbarvaziri, S.; et al. Hypertrophic cardiomyopathy β-cardiac myosin mutation (P710R) leads to hypercontractility by disrupting super relaxed state. Proc. Natl. Acad. Sci. U S A 2021, 118, e2025030118. [Google Scholar] [CrossRef]
- Escribá, R.; Larrañaga-Moreira, J.M.; Richaud-Patin, Y.; Pourchet, L.; Lazis, I.; Jiménez-Delgado, S.; Morillas-García, A.; Ortiz-Genga, M.; Ochoa, J.P.; Carreras, D.; et al. iPSC-Based Modeling of Variable Clinical Presentation in Hypertrophic Cardiomyopathy. Circ. Res. 2023, 133, 108–119. [Google Scholar] [CrossRef]
- Loiben, A.M.; Chien, W.M.; Friedman, C.E.; Chao, L.S.; Weber, G.; Goldstein, A.; Sniadecki, N.J.; Murry, C.E.; Yang, K.C. Cardiomyocyte Apoptosis Is Associated with Contractile Dysfunction in Stem Cell Model of MYH7 E848G Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2023, 24, 4909. [Google Scholar] [CrossRef]
- Desai, D.; Song, T.; Singh, R.R.; Baby, A.; McNamara, J.; Green, L.C.; Nabavizadeh, P.; Ericksen, M.; Bazrafshan, S.; Natesan, S.; Sadayappan, S. MYBPC3 D389V Variant Induces Hypercontractility in Cardiac Organoids. Cells 2024, 13, 1913. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Wang, L.; Li, X.; Fu, W.; Cao, J.; Zhang, J.; Liu, Y.; Liu, M.; Wang, M.; Zhao, G.; et al. Enhanced myofilament calcium sensitivity aggravates abnormal calcium handling and diastolic dysfunction in patient-specific induced pluripotent stem cell-derived cardiomyocytes with MYH7 mutation. Cell Calcium 2024, 117, 102822. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Mangala, M.M.; Holliday, M.; Cserne Szappanos, H.; Barratt-Ross, S.; Li, S.; Thorpe, J.; Liang, W.; Ranpura, G.N.; Vandenberg, J.I.; et al. Reduced connexin-43 expression, slow conduction and repolarisation dispersion in a model of hypertrophic cardiomyopathy. Dis. Model Mech. 2024, 17, dmm050407. [Google Scholar] [CrossRef]
- Mori, H.; Xu, D.; Shimoda, Y.; Yuan, Z.; Murakata, Y.; Xi, B.; Sato, K.; Yamamoto, M.; Tajiri, K.; Ishizu, T.; et al. Metabolic remodeling and calcium handling abnormality in induced pluripotent stem cell-derived cardiomyocytes in dilated phase of hypertrophic cardiomyopathy with MYBPC3 frameshift mutation. Sci. Rep. 2024, 14, 15422. [Google Scholar] [CrossRef]
- Pavlova, S.V.; Shulgina, A.E.; Zakian, S.M.; Dementyeva, E.V. Studying Pathogenetic Contribution of a Variant of Unknown Significance, p.M659I (c.1977G > A) in MYH7, to the Development of Hypertrophic Cardiomyopathy Using CRISPR/Cas9-Engineered Isogenic Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2024, 25, 8695. [Google Scholar] [CrossRef]
- Steczina, S.; Mohran, S.; Bailey, L.R.J.; McMillen, T.S.; Kooiker, K.B.; Wood, N.B.; Davis, J.; Previs, M.J.; Olivotto, I.; Pioner, J.M.; et al. MYBPC3-c.772G>A mutation results in haploinsufficiency and altered myosin cycling kinetics in a patient induced stem cell derived cardiomyocyte model of hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2024, 191, 27–39. [Google Scholar] [CrossRef]
- Beck, M.R.; Dixon, R.D.; Goicoechea, S.M.; Murphy, G.S.; Brungardt, J.G.; Beam, M.T.; Srinath, P.; Patel, J.; Mohiuddin, J.; Otey, C.A.; Campbell, S.L. Structure and function of palladin's actin binding domain. J. Mol. Biol. 2013, 425, 3325–3337. [Google Scholar] [CrossRef]
- Blum, M.; Andreeva, A.; Florentino, L.C.; Chuguransky, S.R.; Grego, T.; Hobbs, E.; Pinto, B.L.; Orr, A.; Paysan-Lafosse, T.; Ponamareva, I.; et al. InterPro: the protein sequence classification resource in 2025. Nucleic Acids Res. 2025, 53, D444–D456. [Google Scholar] [CrossRef] [PubMed]
- Jay, J.J.; Brouwer, C. Lollipops in the Clinic: Information Dense Mutation Plots for Precision Medicine. PLoS One 2016, 11, e0160519. [Google Scholar] [CrossRef]
- Okita, K.; Yamakawa, T.; Matsumura, Y.; Sato, Y.; Amano, N.; Watanabe, A.; Goshima, N.; Yamanaka, S. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells 2013, 31, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Feyen, D.A.M.; McKeithan, W.L.; Bruyneel, A.A.N.; Spiering, S.; Hörmann, L.; Ulmer, B.; Zhang, H.; Briganti, F.; Schweizer, M.; Hegyi, B.; et al. Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell Rep. 2020, 32, 107925. [Google Scholar] [CrossRef]
- Huby, A.C.; Mendsaikhan, U.; Takagi, K.; Martherus, R.; Wansapura, J.; Gong, N.; Osinska, H.; James, J.F.; Kramer, K.; Saito, K.; et al. Disturbance in Z-disk mechanosensitive proteins induced by a persistent mutant myopalladin causes familial restrictive cardiomyopathy. J. Am. Coll. Cardiol. 2014, 64, 2765–2776. [Google Scholar] [CrossRef]
- Refaat, M.M.; Hassanieh, S.; Ballout, J.A.; Zakka, P.; Hotait, M.; Khalil, A.; Bitar, F.; Arabi, M.; Arnaout, S.; Skouri, H.; et al. Non-familial cardiomyopathies in Lebanon: exome sequencing results for five idiopathic cases. BMC Med. Genomics 2019, 12, 33. [Google Scholar] [CrossRef]
- Chen, Y.; Barajas-Martinez, H.; Zhu, D.; Wang, X.; Chen, C.; Zhuang, R.; Shi, J.; Wu, X.; Tao, Y.; Jin, W.; et al. Novel trigenic CACNA1C/DES/MYPN mutations in a family of hypertrophic cardiomyopathy with early repolarization and short QT syndrome. J. Transl. Med. 2017, 15, 78. [Google Scholar] [CrossRef]
- Hong, M.; Ebana, Y.; Shim, J.; Choi, E.K.; Lim, H.E.; Hwang, I.; Yu, H.T.; Kim, T.H.; Uhm, J.S.; Joung, B.; et al. Ethnic similarities in genetic polymorphisms associated with atrial fibrillation: Far East Asian vs European populations. Eur. J. Clin. Invest. 2021, 51, e13584. [Google Scholar] [CrossRef]
- Meng, X.; Nie, Y.; Wang, K.; Fan, C.; Zhao, J.; Yuan, Y. Identification of Atrial Fibrillation-Associated Genes ERBB2 and MYPN Using Genome-Wide Association and Transcriptome Expression Profile Data on Left-Right Atrial Appendages. Front. Genet. 2021, 12, 696591. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Wang, K. Interaction of nebulin SH3 domain with titin PEVK and myopalladin: implications for the signaling and assembly role of titin and nebulin. FEBS Lett. 2002, 532, 273–278. [Google Scholar] [CrossRef]
- Gu, Q.; Mendsaikhan, U.; Khuchua, Z.; Jones, B.C.; Lu, L.; Towbin, J.A.; Xu, B.; Purevjav, E. Dissection of Z-disc myopalladin gene network involved in the development of restrictive cardiomyopathy using system genetics approach. World J. Cardiol. 2017, 9, 320–331. [Google Scholar] [CrossRef]
- Kojic, S.; Medeot, E.; Guccione, E.; Krmac, H.; Zara, I.; Martinelli, V.; Valle, G.; Faulkner, G. The Ankrd2 protein, a link between the sarcomere and the nucleus in skeletal muscle. J. Mol. Biol. 2004, 339, 313–325. [Google Scholar] [CrossRef]
- Klimenko, E.S.; Sukhareva, K.S.; Vlasova, Y.; Smolina, N.A.; Fomicheva, Y.; Knyazeva, A.; Muravyev, A.S.; Sorokina, M.Y.; Gavrilova, L.S.; Boldyreva, L.V.; et al. Flnc expression impacts mitochondrial function, autophagy, and calcium handling in C2C12 cells. Exp. Cell Res. 2024, 442, 114174. [Google Scholar] [CrossRef]
- Klimenko, E.S.; Zaytseva, A.K.; Sorokina, M.Yu.; Perepelina, K.I.; Rodina, N.L.; Nikitina, E.G.; Sukhareva, K.S.; Khudiakov, A.A.; Vershinina, T.L.; Muravyev, A.S.; et al. Distinct molecular features of FLNC mutations, associated with different clinical phenotypes. Cytoskeleton 2025, 82, 158–174. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Chen, Z.; Wang, C.; Yan, R.X.; Zhang, Z.; Song, J. Predicting residue-residue contacts and helix-helix interactions in transmembrane proteins using an integrative feature-based random forest approach. PLoS One 2011, 6, e26767. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, C.; Mou, C.; Dong, Y.; Tu, Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020, 12, 103. [Google Scholar] [CrossRef]
- Sorogina, D.A.; Grigor’eva, E.V.; Malakhova, A.A.; Pavlova, S.V.; Medvedev, S.P.; Vyatkin, Y.V.; Khabarova, E.A.; Rzaev, J.A.; Zakian, S.M. Creation of Induced Pluripotent Stem Cells ICGi044-B and ICGi044-C Using Reprogramming of Peripheral Blood Mononuclear Cells of a Patient with Parkinson’s Disease Associated with с.1492T>G Mutation in the GLUD2 Gene. Russ. J. Dev. Biol. 2023, 54, 104–111. [Google Scholar] [CrossRef]
- Khudiakov, A.; Zaytseva, A.; Perepelina, K.; Smolina, N.; Pervunina, T.; Vasichkina, E.; Karpushev, A.; Tomilin, A.; Malashicheva, A.; Kostareva, A. Sodium current abnormalities and deregulation of Wnt/β-catenin signaling in iPSC-derived cardiomyocytes generated from patient with arrhythmogenic cardiomyopathy harboring compound genetic variants in plakophilin 2 gene. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165915. [Google Scholar] [CrossRef]
Figure 1.
Localization of p.N989I variant in immunoglobulin-like domain 3 of myopalladin. (a) Domain organization of myopalladin (UniprotKB: Q86TC9) and localization of the p.N989I variant performed using InterPro database [24] and lollipops tool [25]. Immunoglobulin-like domains are marked as ‘Ig’. p.N989I fell in Ig3 domain of myopalladin; (b) A multiple sequence alignment of human myopalladin with orthologues; (c) A multiple sequence alignment of Ig3 domain of human myopalladin with Ig-domains of homologous human proteins. p.N989I is located at a variable position within the alignment of the domains. Each residue in the alignment is colored using Clustal X Colour Scheme: blue - hydrophobic; red – positive charged; magenta – negative charged; green – polar; pink – cysteines; orange – glycines; yellow – prolines; cyan – aromatic; white – unconserved; (d) 3D structure of palladin Ig3 domain (PDB:2LQR) [23]. p.N989I is mapped on the structure according to the sequence alignment. Substitution fell in the linker (green) between C and D strands (yellow).
Figure 1.
Localization of p.N989I variant in immunoglobulin-like domain 3 of myopalladin. (a) Domain organization of myopalladin (UniprotKB: Q86TC9) and localization of the p.N989I variant performed using InterPro database [24] and lollipops tool [25]. Immunoglobulin-like domains are marked as ‘Ig’. p.N989I fell in Ig3 domain of myopalladin; (b) A multiple sequence alignment of human myopalladin with orthologues; (c) A multiple sequence alignment of Ig3 domain of human myopalladin with Ig-domains of homologous human proteins. p.N989I is located at a variable position within the alignment of the domains. Each residue in the alignment is colored using Clustal X Colour Scheme: blue - hydrophobic; red – positive charged; magenta – negative charged; green – polar; pink – cysteines; orange – glycines; yellow – prolines; cyan – aromatic; white – unconserved; (d) 3D structure of palladin Ig3 domain (PDB:2LQR) [23]. p.N989I is mapped on the structure according to the sequence alignment. Substitution fell in the linker (green) between C and D strands (yellow).
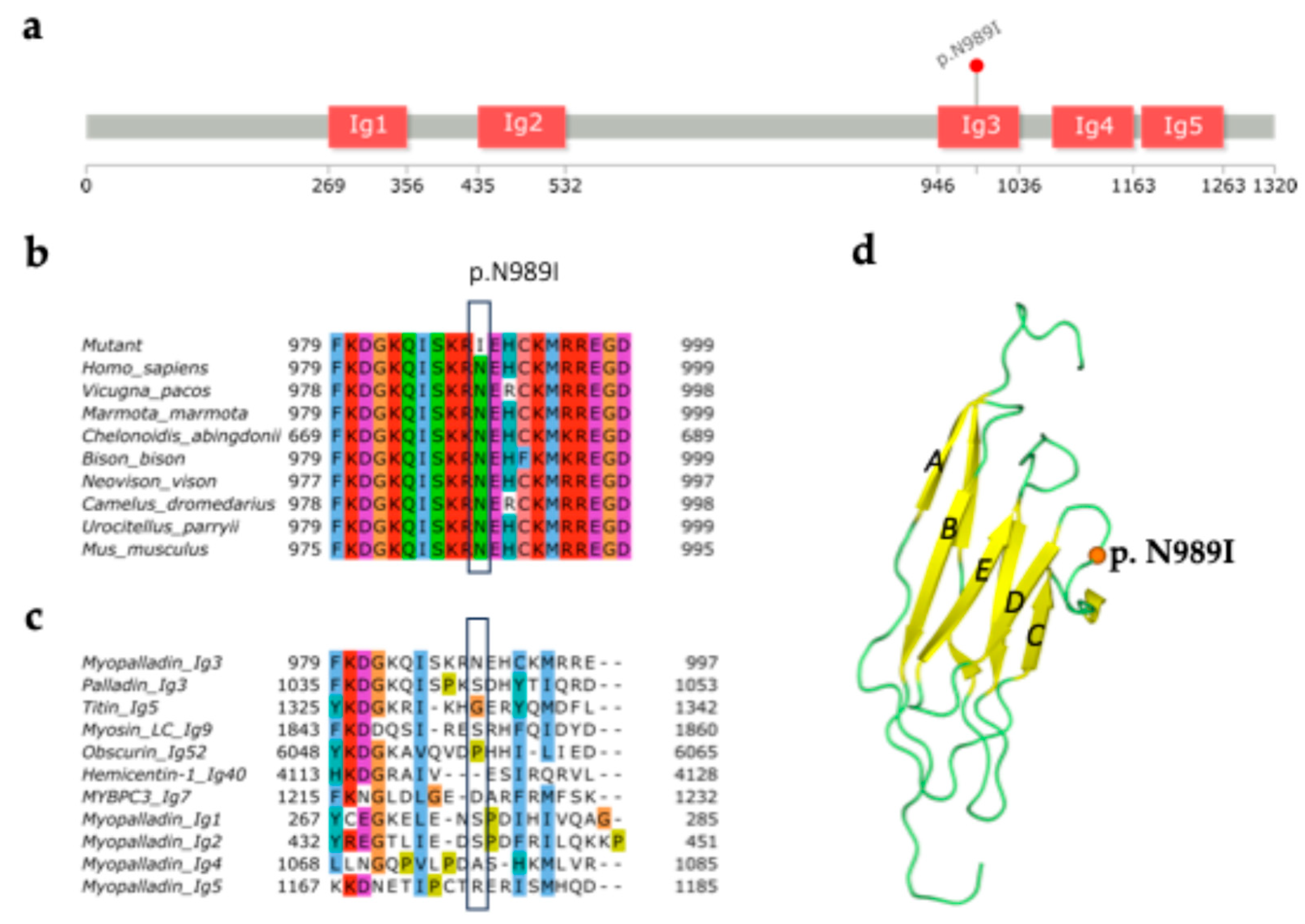
Figure 2.
Characterization of iPSC lines generated by reprogramming of the mononuclear cells from the patient with p.N989I (c.2966A>T) variant in MYPN. (a) FAMRCi015-A and FAMRCi015-B iPSC lines have a morphology characteristic to human pluripotent stem cells. Scale bar – 200 µm; (b) Absence of mycoplasma contamination in the patient-specific iPSC lines. Contr+, positive control for mycoplasma contamination. H2O, negative control; (c) Episome elimination in the FAMRCi015-A and FAMRCi015-B iPSC lines. Contr+, positive control for episome presence, an iPSC line at an early passage. H2O, negative control; (d) Patient-specific iPSC lines express a number of pluripotent state markers – the OCT4 and NANOG transcription factors and SSEA4 and TRA-1-60 surface antigens. Scale bar – 100 µm; (e) Positive expression of pluripotency genes, OCT4, NANOG and SOX2, in the FAMRCi015-A and FAMRCi015-B iPSC lines. MNCs, patient’s mononuclear cells. ESM04, a line of human embryonic stem cells used as a positive control of OCT4, NANOG and SOX2 expression. Data are presented as mean ± SD; (f) Patient-specific iPSC lines are able to be differentiated into derivatives of three germ layers – ectoderm (TUBB3, βIII-tubulin), mesoderm (αSMA, smooth muscle alpha-actinin) and endoderm (HNF3β, hepatocyte nuclear factor 3 beta). Scale bar – 50 µm; (g) FAMRCi015-A and FAMRCi015-B iPSC lines have a normal karyotype – 46,XX; (h) iPSC lines retain the patient-specific heterozygous p.N989I (c.2966A>T) substitution. The nucleotide sequence of a part of MYPN exon 14 in a healthy donor and the patient’s mononuclear cells (MNCs) are given for comparison. The position where the c.2966A>T substitution occurs in the HCM patient is indicated by red arrows.
Figure 2.
Characterization of iPSC lines generated by reprogramming of the mononuclear cells from the patient with p.N989I (c.2966A>T) variant in MYPN. (a) FAMRCi015-A and FAMRCi015-B iPSC lines have a morphology characteristic to human pluripotent stem cells. Scale bar – 200 µm; (b) Absence of mycoplasma contamination in the patient-specific iPSC lines. Contr+, positive control for mycoplasma contamination. H2O, negative control; (c) Episome elimination in the FAMRCi015-A and FAMRCi015-B iPSC lines. Contr+, positive control for episome presence, an iPSC line at an early passage. H2O, negative control; (d) Patient-specific iPSC lines express a number of pluripotent state markers – the OCT4 and NANOG transcription factors and SSEA4 and TRA-1-60 surface antigens. Scale bar – 100 µm; (e) Positive expression of pluripotency genes, OCT4, NANOG and SOX2, in the FAMRCi015-A and FAMRCi015-B iPSC lines. MNCs, patient’s mononuclear cells. ESM04, a line of human embryonic stem cells used as a positive control of OCT4, NANOG and SOX2 expression. Data are presented as mean ± SD; (f) Patient-specific iPSC lines are able to be differentiated into derivatives of three germ layers – ectoderm (TUBB3, βIII-tubulin), mesoderm (αSMA, smooth muscle alpha-actinin) and endoderm (HNF3β, hepatocyte nuclear factor 3 beta). Scale bar – 50 µm; (g) FAMRCi015-A and FAMRCi015-B iPSC lines have a normal karyotype – 46,XX; (h) iPSC lines retain the patient-specific heterozygous p.N989I (c.2966A>T) substitution. The nucleotide sequence of a part of MYPN exon 14 in a healthy donor and the patient’s mononuclear cells (MNCs) are given for comparison. The position where the c.2966A>T substitution occurs in the HCM patient is indicated by red arrows.
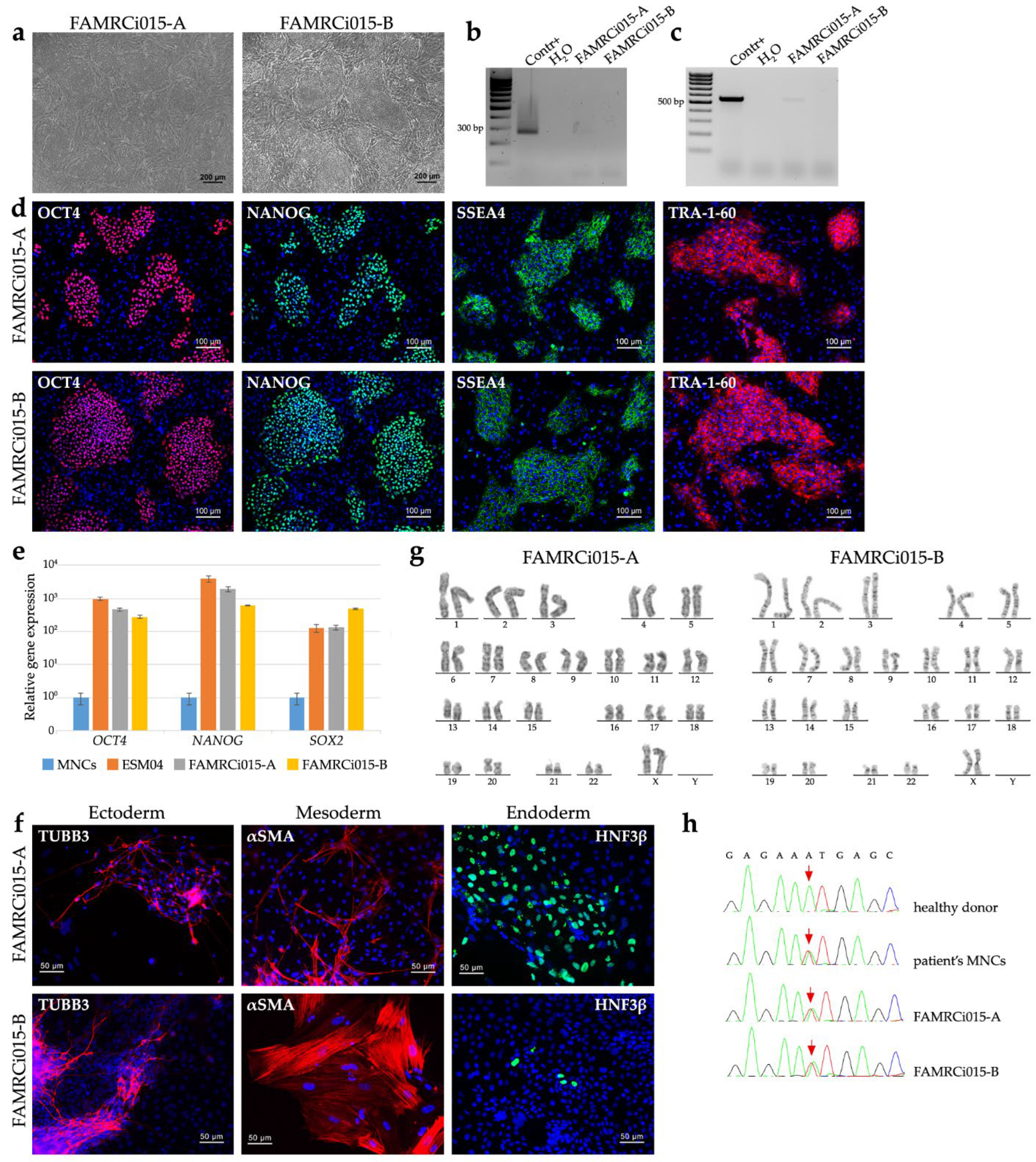
Figure 3.
Parameters of calcium transients in iPSC-derived cardiomyocytes of the HCM patient and healthy donor. Black columns — healthy donor CMs (n>60); red columns — MYPN-N989I-CMs (n>90). (a) Duration of calcium transient; (b) Time to transient’s peak; (c) Calcium rise velocity; (d) Amplitude of calcium transient; (e) Time from peak to baseline; (f) Calcium decay velocity. p-value: ns − not significant. The data are presented as mean ± SD.
Figure 3.
Parameters of calcium transients in iPSC-derived cardiomyocytes of the HCM patient and healthy donor. Black columns — healthy donor CMs (n>60); red columns — MYPN-N989I-CMs (n>90). (a) Duration of calcium transient; (b) Time to transient’s peak; (c) Calcium rise velocity; (d) Amplitude of calcium transient; (e) Time from peak to baseline; (f) Calcium decay velocity. p-value: ns − not significant. The data are presented as mean ± SD.
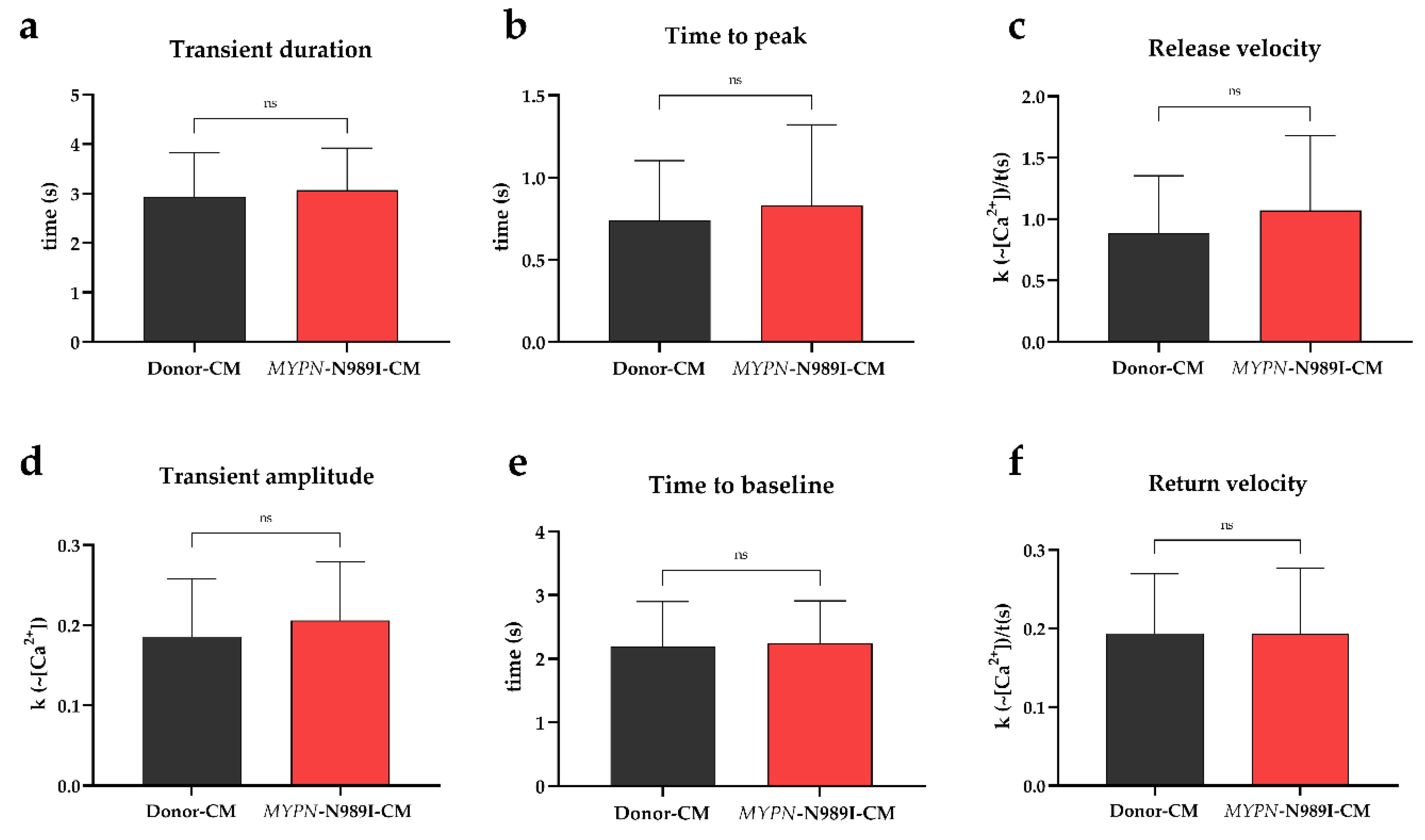
Figure 4.
Sodium current parameters and action potential dynamics in iPSC-derived cardiomyocytes of the healthy donor (black) and HCM patient (red). (a) Representative sodium current traces; (b) Peak current density; (c) Voltampere characteristics of current density; (d) Steady-state activation of sodium current; (e) Steady-state inactivation of sodium current; (f) Action potential dynamics.
Figure 4.
Sodium current parameters and action potential dynamics in iPSC-derived cardiomyocytes of the healthy donor (black) and HCM patient (red). (a) Representative sodium current traces; (b) Peak current density; (c) Voltampere characteristics of current density; (d) Steady-state activation of sodium current; (e) Steady-state inactivation of sodium current; (f) Action potential dynamics.
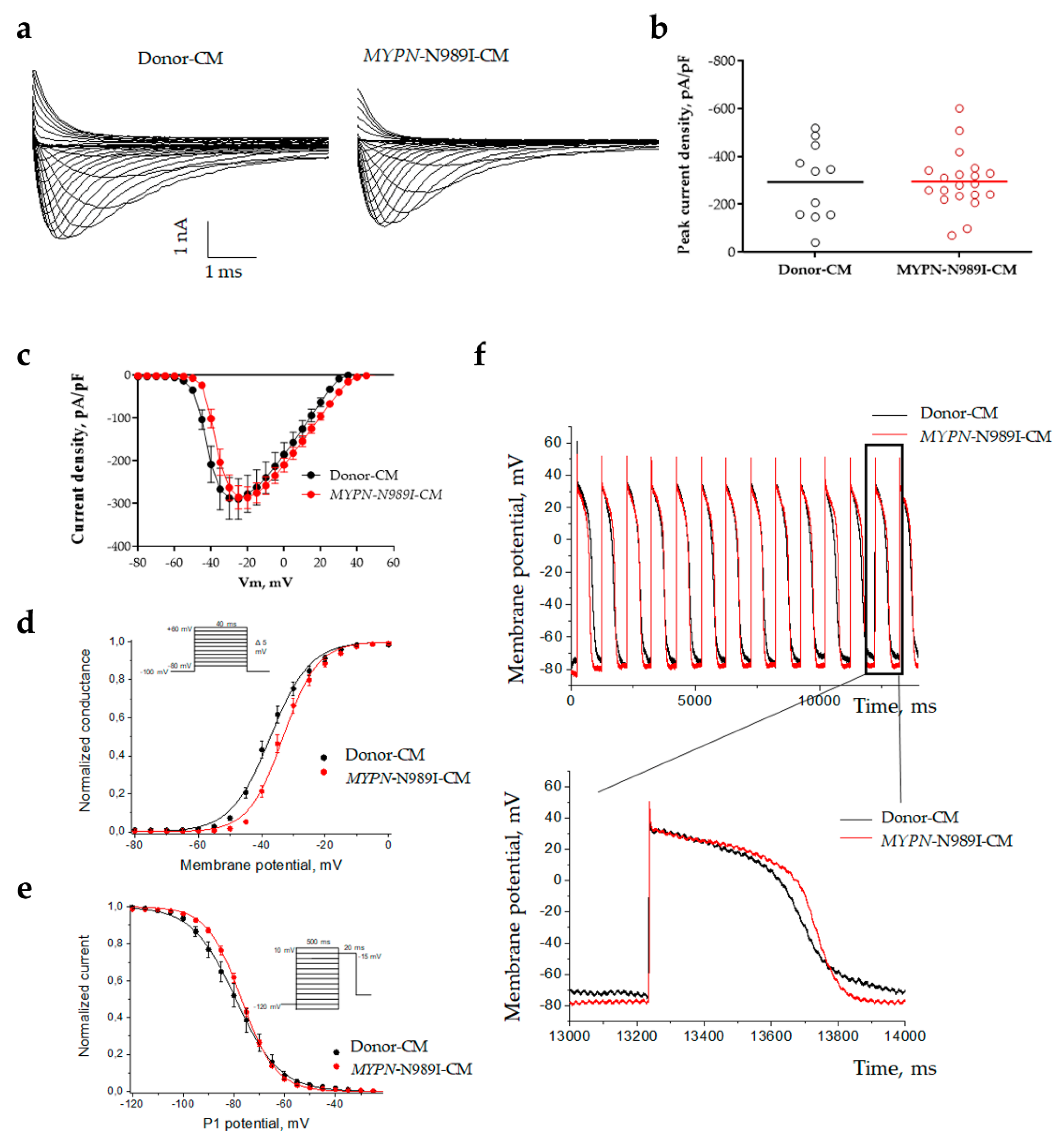
Figure 5.
Transcriptome analysis of MYPN-N989I-CMs compared to Donor-CMs. (a) PCA plot; (b) Volcano-plot of differentially expressed genes (DEGs, |LFC| > 1.5 and p.adj < 0.05); (c) Gene set enrichment analysis of upregulated DEGs (p.adj < 0.05); (d) Heatmap of genes involved in Muscle system process and Microtubule associated complex signaling pathways; (e) Gene set enrichment analysis of downregulated DEGs (p.adj < 0.05); (f) Heatmap of genes involved in Sarcomere, Z-disc and I-band organization, Cell junction-associated genes and Actin cytoskeleton signaling pathways; (g) Results of FGSEA analysis; (h) GSEA plot of the Oxidative phosphorylation signaling pathways from Hallmark Gene Ontology database (NES = -2.05 and p.adj < 0.05).
Figure 5.
Transcriptome analysis of MYPN-N989I-CMs compared to Donor-CMs. (a) PCA plot; (b) Volcano-plot of differentially expressed genes (DEGs, |LFC| > 1.5 and p.adj < 0.05); (c) Gene set enrichment analysis of upregulated DEGs (p.adj < 0.05); (d) Heatmap of genes involved in Muscle system process and Microtubule associated complex signaling pathways; (e) Gene set enrichment analysis of downregulated DEGs (p.adj < 0.05); (f) Heatmap of genes involved in Sarcomere, Z-disc and I-band organization, Cell junction-associated genes and Actin cytoskeleton signaling pathways; (g) Results of FGSEA analysis; (h) GSEA plot of the Oxidative phosphorylation signaling pathways from Hallmark Gene Ontology database (NES = -2.05 and p.adj < 0.05).
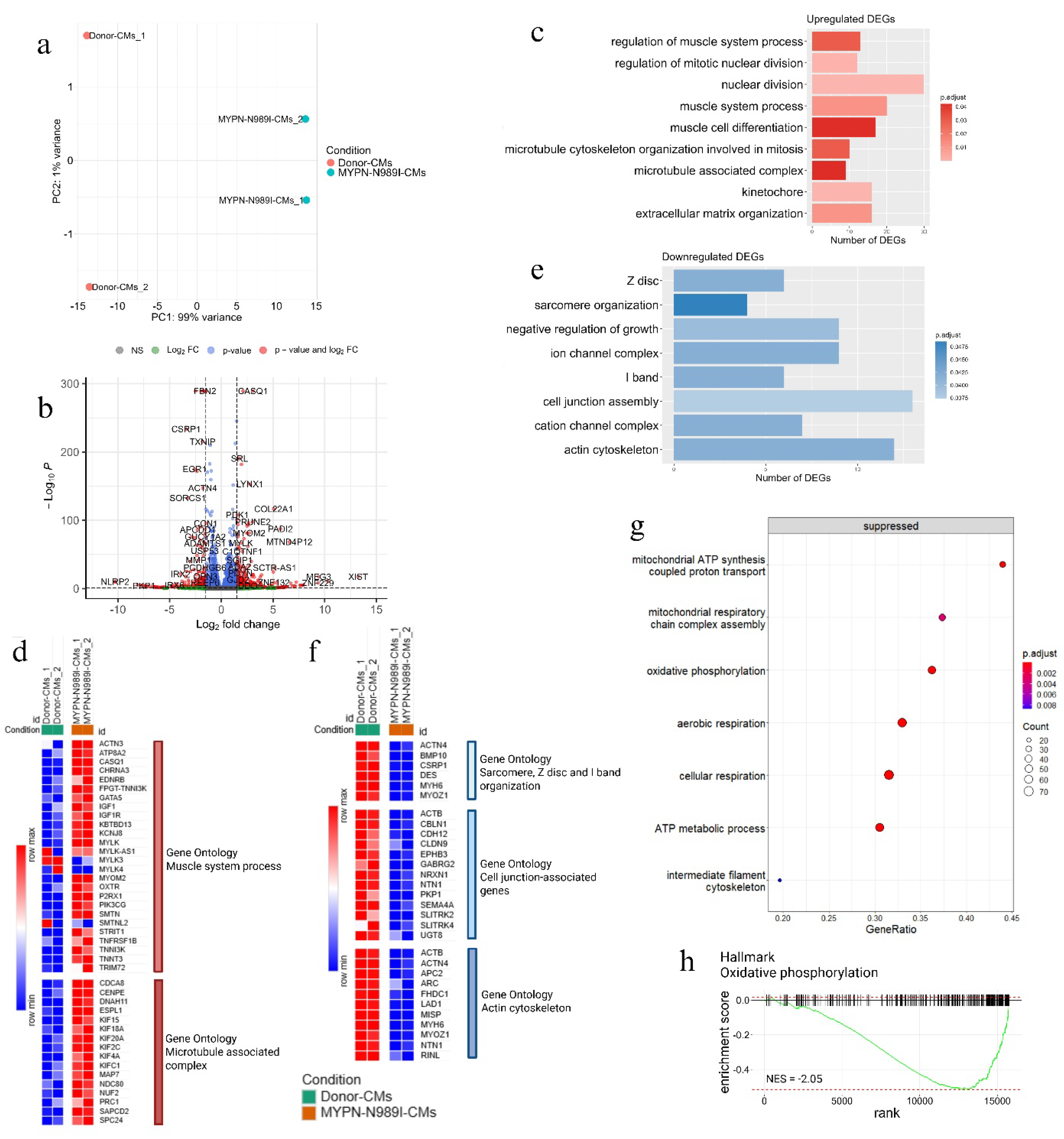
Figure 6.
(a) Heatmap of Z-disc-associated genes and genes related to Ca2+ homeostasis; (b) GSEA plot of the Endoplasmic and Sarcoplasmic reticulum calcium ion homeostasis signaling pathways from Hallmark Gene Ontology database (NES = -2.02, NES = -1.78 and p.adj < 0.05).; (c) Heatmap of genes involved in SRF-signaling pathway.
Figure 6.
(a) Heatmap of Z-disc-associated genes and genes related to Ca2+ homeostasis; (b) GSEA plot of the Endoplasmic and Sarcoplasmic reticulum calcium ion homeostasis signaling pathways from Hallmark Gene Ontology database (NES = -2.02, NES = -1.78 and p.adj < 0.05).; (c) Heatmap of genes involved in SRF-signaling pathway.
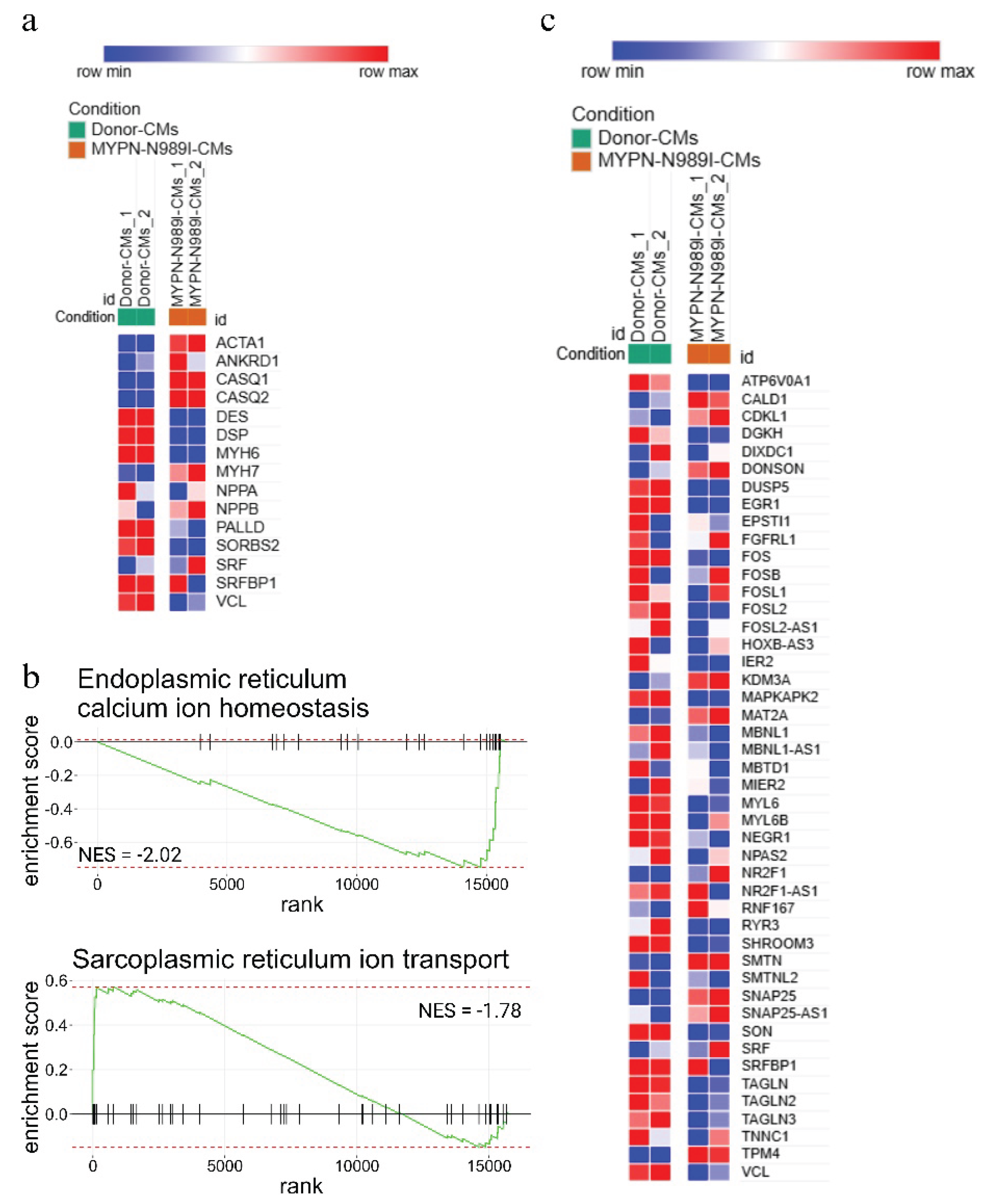

Table 1.
Biophysical characteristics of sodium current. Data are represented as mean ± SEM.
| Donor-CMs | n | MYPN-N989I-CMs | n | p | ||
| Current density at -20mV | pA/pF | -292.1 ± 47.9 | 11 | -294.5 ± 27.0 | 20 | 0.9506 |
| Steady-state activation | V1/2, mV | -37.1 ± 1.1 | 11 | -33.1 ± 0.9 | 20 | 0.0121 |
| k | 5.7 ± 0.3 | 4.6 ± 0.2 | 0.0194 | |||
| Steady-state inactivation | V1/2, mV | -79.2 ± 2.0 | 10 | -77.1 ± 0.7 | 20 | 0.3012 |
| k | 7.7 ± 0.5 | 6.5 ± 0.2 | 0.0114 |
Table 2.
Action potential parameters. Data are represented as mean ± SEM.
|
Donor-CMs, n=12 |
MYPN-N989I-CMs, n=9 | p | |
| APA, mV | 130,2 ± 5,876 | 125,0 ± 3,314 | 0,6187 |
| APD90, ms | 572,6 ± 52,65 | 546,8 ± 54,58 | 0,859 |
| APD70, ms | 472,0 ± 47,57 | 459,5 ± 46,29 | 0,9717 |
| APD30, ms | 338,8 ± 37,13 | 318,8 ± 42,18 | 0,594 |
Table 3.
Differently expressed genes from the detected signalling pathways.
| Signalingpathway | Gene symbol | Gene name | LFC | p.adj |
| Muscle system process | ACTN3 | actinin alpha 3 | 2.24 | 3.91*10-2 |
| ATP8A2 | ATPase phospholipid transporting 8A2 | 1.83 | 6.62*10-4 | |
| CASQ1 | calsequestrin 1 | 3.06 | 0 | |
| CHRNA3 | cholinergic receptor nicotinic alpha 3 subunit | 1.66 | 4.57*10-22 | |
| EDNRB | endothelin receptor type B | 4.29 | 0.01 | |
| GATA5 | GATA binding protein 5 | 1.55 | 1.62*10-8 | |
| IGF1 | insulin like growth factor 1 | 1.92 | 6.24*10-3 | |
| KBTBD13 | kelch repeat and BTB domain containing 13 | 2.38 | 7.47*10-5 | |
| KCNJ8 | potassium inwardly rectifying channel subfamily J member 8 | 1.57 | 6.09*10-27 | |
| MYLK | myosin light chain kinase | 1.90 | 1.66*10-67 | |
| MYOM2 | myomesin 2 | 2.70 | 3.46*10-82 | |
| OXTR | oxytocin receptor | 2.19 | 2.75*10-5 | |
| P2RX1 | purinergic receptor P2X 1 | 1.55 | 9.67*10-81 | |
| PIK3CG | phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit gamma | 6.38 | 1.84*10-4 | |
| SMTN | smoothelin | 1.55 | 3.28*10-40 | |
| STRIT1 | small transmembrane regulator of ion transport 1 | 6.16 | 4.03*10-3 | |
| TNFRSF1B | TNF receptor superfamily member 1B | 1.99 | 3.96*10-3 | |
| TNNI3K | TNNI3 interacting kinase | 1.83 | 1.03*10-6 | |
| TNNT3 | troponin T3, fast skeletal type | 3.13 | 9.81*10-8 | |
| TRIM72 | tripartite motif containing 72 | 1.65 | 0.02 | |
| Microtubule associated complex | CDCA8 | cell division cycle associated 8 | 1.51 | 2.35*10-6 |
| CENPE | centromere protein E | 1.52 | 1.21*10-6 | |
| DNAH11 | dynein axonemal heavy chain 11 | 5.04 | 2.64*10-25 | |
| ESPL1 | extra spindle pole bodies like 1, separase | 1.68 | 3.59*10-5 | |
| KIF15 | kinesin family member 15 | 1.64 | 4.95*10-3 | |
| KIF18A | kinesin family member 18A | 2.00 | 3.19*10-7 | |
| KIF20A | kinesin family member 20A | 1.67 | 1.61*10-15 | |
| KIF2C | kinesin family member 2C | 1.71 | 2.67*10-9 | |
| KIF4A | kinesin family member 4A | 1.86 | 8.12*10-13 | |
| KIFC1 | kinesin family member C1 | 1.80 | 4.95*10-10 | |
| MAP7 | microtubule associated protein 7 | 1.59 | 2.68*10-3 | |
| NDC80 | NDC80 kinetochore complex component | 2.32 | 1.50*10-11 | |
| NUF2 | NUF2 component of NDC80 kinetochore complex | 2.27 | 1.49*10-9 | |
| PRC1 | protein regulator of cytokinesis 1 | 2.18 | 1.43*10-2 | |
| SAPCD2 | suppressor APC domain containing 2 | 1.56 | 0.02 | |
| SPC24 | kinetochore-associated Ndc80 complex subunit SPC24 | 1.59 | 1.06*10-5 | |
| Sarcomere, Z-disc and I-band organization | ACTN4 | actinin alpha 4 | -1.79 | 4.7*10-148 |
| BMP10 | bone morphogenetic protein 10 | -5.37 | 4.91*10-2 | |
| CSRP1 | cysteine and glycine rich protein 1 | -3.40 | 1.86*10-234 | |
| DES | desmin | -1.51 | 4.73*10-289 | |
| MYH6 | myosin heavy chain 6 | -1.95 | 0 | |
| MYOZ1 | myozenin 1 | -1.68 | 1.11*10-24 | |
| Cell junction associated genes | ACTB | actin beta | -1.81 | 0 |
| CBLN1 | cerebellin 1 precursor | -1.70 | 1.83*10-3 | |
| CDH12 | cadherin 12 | -3.26 | 5.18*10-3 | |
| CLDN9 | claudin 9 | -1.66 | 0.04 | |
| EPHB3 | EPH receptor B3 | -1.56 | 1.75*10-81 | |
| GABRG2 | gamma-aminobutyric acid type A receptor subunit gamma2 | -7.74 | 1.81*10-5 | |
| NRXN1 | neurexin 1 | -1.71 | 5.66*10-7 | |
| NTN1 | netrin 1 | -1.97 | 3.61*10-63 | |
| PKP1 | plakophilin 1 | -7.47 | 5.99*10-5 | |
| SEMA4A | semaphorin 4A | -2.48 | 3.31*10-12 | |
| SLITRK2 | SLIT and NTRK like family member 2 | -5.83 | 0.02 | |
| SLITRK4 | SLIT and NTRK like family member 4 | -1.58 | 0.01 | |
| UGT8 | UDP glycosyltransferase 8 | -1.81 | 0.04 | |
| Actin cytoskeleton | ACTB | actin beta | -1.81 | 0 |
| ACTN4 | actinin alpha 4 | -1.79 | 4.67*10-148 | |
| APC2 | APC regulator of Wnt signaling pathway 2 | -3.62 | 4.09*10-5 | |
| ARC | activity regulated cytoskeleton associated protein | -3.93 | 1.94*10-4 | |
| FHDC1 | FH2 domain containing 1 | -2.85 | 0.03 | |
| LAD1 | ladinin 1 | -2.58 | 6.27*10-75 | |
| MISP | mitotic spindle positioning | -5.56 | 0.03 | |
| MYH6 | myosin heavy chain 6 | -1.95 | 0 | |
| MYO1D | myosin ID | -2.04 | 7,09*10-91 | |
| MYOZ1 | myozenin 1 | -1.68 | 1.11*10-24 | |
| NTN1 | netrin 1 | -1.97 | 3.61*10-63 | |
| RINL | Ras and Rab interactor like | -1.84 | 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



