
Submitted:
18 December 2025
Posted:
19 December 2025
You are already at the latest version
Abstract
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) is a debilitating multi-system illness with heterogeneity that complicates identifying the pathophysiology, biomarkers, and therapeutic targets. Evidence indicates the importance of immune dysregulation, including the complement system, in ME/CFS. This study investigates the contribution of genetic drivers to potential dysregulation of the complement pathway in ME/CFS. We used protein quantitative trait loci (pQTL) analyses, adjusted for covariates using linear and logistic regression, to identify genetic variants significantly associated with plasma complement protein levels in a study sample identified from the general population (50 ME/CFS and 121 non-fatigued). ME/CFS patients carrying certain pQTLs exhibited dysregulation of the alternative complement pathway, which defined an inflammatory subgroup with a high C3/low Bb profile and established a genetic link to dysregulation of the alternative complement pathway. Six of the significant pQTLs were also found associated with fatigue-related phenotypes in the UK biobank, 4 of which were complement-associated, providing some validation in an independent population. Our findings highlight a mechanism by which risk alleles contribute to ME/CFS heterogeneity, providing evidence of a genetic basis for complement dysregulation in a subgroup. This approach could identify pathway-focused subgroups in ME/CFS and similar illnesses to inform personalized approaches to diagnosis and treatments.
Keywords:
ME/CFS
; complement system
; pQTLs
; myalgic encephalomyelitis/chronic fatigue syndrome
; heterogeneity
1. Introduction
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) is a debilitating multi-system illness characterized by substantial functional impairment accompanied by profound fatigue, post-exertional malaise, unrefreshing sleep, cognitive dysfunction, orthostatic intolerance, and a wide litany of additional symptoms that significantly reduce quality of life [1]. Its etiology remains elusive, with substantial heterogeneity complicating efforts to identify reliable biomarkers and therapeutic targets [2]; however, understanding the molecular basis for genotype/phenotype associations can greatly enhance our ability to tailor diagnostic and treatment strategies. While this approach is considered useful, it has yet to be investigated for many complex and heterogeneous diseases, including ME/CFS. Recently, we and others have tested whole genome or pathway focused genetic markers for their association with ME/CFS [3,4,5,6,7,8,9,10,11,12,13,14]. Many results from these earlier genetic association studies have not been replicated, possibly due to small sample size, lack of consistency in case ascertainment, heterogeneity at the level of genetics, disease time-course, co-morbid illnesses, and their combinatorial interaction with environment, diet, medications, and microbial environments.
There has been growing interest in exploring the relationship between disease-associated genetic variants and the levels of RNA and proteins, known as expression/protein quantitative trait loci (eQTLs/pQTLs) respectively. These intermediate phenotypes bridge the gap between genotype and clinical presentation, offering a powerful framework for understanding complex diseases and informing biomarker and therapeutic target discovery. While pQTL studies have been applied to cardiovascular disease, a variety of autoimmune disorders, and proteins in the human liver [15,16,17], their application to ME/CFS remains limited.
Our group previously identified several genetic variants within the complement pathway that associated with ME/CFS using a focused analysis of immune and inflammation-related genes [3]. The complement system is a critical component of innate immunity, consisting of plasma proteins that defend against infection and mediate inflammation through three activation pathways – classical, alternative, and lectin – all converging at C3 cleavage and tightly regulated to prevent chronic inflammation and immune-mediated damage [18,19]. Notably, our prior study identified two non-synonymous single nucleotide polymorphisms (SNPs) in complement-related genes associated with ME/CFS: rs4151667 in Complement Factor B (CFB) and rs1061170 in Complement Factor H (CFH), both of which regulate the alternative pathway of complement activation. The SNP rs4151667 in CFB results in a leucine-to-histidine substitution at position 9 (L9H) and is in strong linkage disequilibrium (LD) with rs9332739 in C2. Similarly, rs1061170 in CFH causes a tyrosine-to-histidine substitution at position 404 (Y402H) and is in high LD with three other SNPs in CFH (rs1061147, rs7529589, and rs1080155). Both rs9332739 and rs4151667, located in the paralogous C2 and CFB genes, were significantly associated with ME/CFS in allele and haplotype analyses. Likewise, the major alleles of all four SNPs in CFH were associated with ME/CFS, further implicating the complement pathway in disease susceptibility. Interestingly, previous studies have reported complement dysregulation in ME/CFS – including elevated C4a responses, unique correlations between C3 levels and other circulating inflammatory proteins, and altered lectin and classical pathway components in plasma, cerebrospinal fluid, and extracellular vesicles [20,21,22,23,24,25,26]. Integrating genetic and proteomic data has the potential to better understand disease susceptibility, heterogeneity, and progression.
It should be noted that CFH and CFB have opposing roles in the alternative complement pathway, with CFB promoting and CFH inhibiting the decay of C3 convertase [27]. Based on this observation, we hypothesize that alternative pathway components, consisting of precursor proteins (C3 and Factor B), their breakdown products (C3a, C5a, Bb), negative regulator (Factor H), and marker of terminal pathway of complement activation (SC5b-9), would be associated with ME/CFS, and that this association may be driven by the observed genetic variation in the complement proteins. Interestingly, ME/CFS-associated missense variants in these genes are also linked to age-related macular degeneration (AMD) but with inverted risk alleles [28,29]. As these alleles associate with reduced complement activation and protection in AMD [30,31,32], we hypothesize that reduced complement activity contributes to disease risk in ME/CFS. These risk alleles could enable identification of a subgroup of ME/CFS with complement dysregulation.
To extend our prior work, we investigate the association of plasma concentrations of complement proteins and related activation products with ME/CFS by identifying pQTLs among the 9146 SNPs that passed Affymetrix Human Immune and Inflammation Chip quality control [3]. We also identify correlations of complement proteins with measures of function, fatigue, and symptoms. Finally, we integrate these findings with the previously reported genetic variants associated with ME/CFS illness to identify a genetic subgroup of ME/CFS with complement dysregulation. In addition, we sought to validate our identified complement and disease associated pQTLs with the UK biobank’s publicly available datasets for post-viral and fatigue-related phenotypes. This integrative approach has the potential to identify biologically distinct subgroups within the heterogeneous ME/CFS population and should be helpful in studies of other chronic complex illnesses.
2. Results
2.1. Complement System Dynamics and Associations with Demographics and Disease Status
This study included 50 ME/CFS and 121 non-fatigued control Caucasian participants. While the two groups were comparable in age, significant differences were noted in sex distribution and body mass index (BMI) (Figure 1A). Among participants with ME/CFS, the median time since onset of fatigue was 8.97 years (range: 0.39 – 40.2 years), with 82.2% reporting a gradual illness onset [3]. Compared to the non-fatigued subjects, ME/CFS subjects had worse fatigue (higher scores) in all 5 domains of Multidimensional Fatigue Inventory (MFI-20, Figure 1B), greater symptom burdens in CDC symptom inventory (CDC-SI), and worse functional impairments (lower scores) in all 8 subscales of Short Form Health Survey (SF-36v2, Figure 1C).
Associati ons of the plasma concentrations of eight complement-related analytes spanning both core proteins and activation products of the alternative pathway – CRP, C3, Bb, C3a, C5a, Factor B, Factor D, Factor H, and SC5b-9 – with demographic covariates in all subjects, including age, sex, and BMI, are shown in Table 1 and Figure S1A-K. BMI was the most consistent covariate, showing moderate yet significant positive associations with 7 of the 9 measured proteins: CRP, C3, C3a, C5a, Factor B, Factor D, and Factor H (all p < 0.0001), strongest (R2) with C3. In contrast Bb and SC5b-9 were not significantly associated with BMI, though the ratio of Bb to C3 was inversely associated. Sex was significantly associated with CRP and Factor B (Table 1, Figure S1I and J), with lower levels observed in males. Only Factor D levels were associated with age (Figure S1K). The demographic factors listed in Table 1 were considered as covariates in downstream analyses of the associated proteins.
We calculated correlations among all measured complement proteins across all subjects to evaluate protein interactions. Those with Factor H (Figure 1D) demonstrated the broadest pattern of correlation, including moderate relationships with CRP, C3, C3a, Factor B, and Factor D. The correlation of Factor H was much stronger with C3 and Factor B, and weaker with Bb (r = 0.11, p = 0.12). Given that the alternative complement cascade consists of multiple interdependent and tightly regulated components (Figure 1E), the majority of the pairwise comparisons showed significant positive associations.
To evaluate illness-associated differences in complement protein levels, we performed both linear and logistic regression analyses comparing protein concentrations between illness groups while adjusting for covariates. Linear regression assessed differences in protein concentrations as continuous outcomes, while logistic regression modeled the odds of ME/CFS as a function of protein levels. Box plots of log2 transformed values stratified by disease status are shown in Figure 1F-L and Figure S1L-N. Despite several proteins having higher median values by at least 5% in ME/CFS compared to NF, only C3 and Bb/C3 ratio were significantly associated with ME/CFS in both models. The linear model indicates that C3 levels differ significantly between the two groups and the logistic model shows that higher C3 levels are associated with increased odds of ME/CFS.
2.2. Associations Between Complement System Components and Functional Health Scores
Results of the covariate-adjusted linear regression modeling between each analyte and the measures of function (SF-36), fatigue (MFI-20), and symptoms (CDC-SI) are shown as a heatmap Figure 2A and in correlation plots, Figure S2A-D. C3 shows the most consistent pattern among the tested complement proteins, with higher plasma levels associated with greater symptom burden, increased fatigue, and lower functioning scores (Figure S2B). CRP exhibited the largest beta coefficients across several MFI-20 and SF-36 domains, indicating that small increases in severity or functional impairment were associated with relatively large shifts in CRP levels; however, the corresponding R2 values remained low (Figure S2C; R2 ~ 0.32), suggesting that despite strong directional effects, the symptom scores explained only a modest proportion of the overall variance in participants’ CRP levels. Factor D and the Bb-to-C3 ratio also showed significant associations with some MFI and SF-36 metrics. In contrast, Bb, C3a, C5a, Factor B, Factor H, and SC5b-9 showed no significant associations with any individual scores.
2.3. Identification of Genetic Variants Impacting Plasma Levels of Complement Proteins
To investigate if genetic variants influence circulating complement protein levels, we performed a quantitative trait locus (pQTL) analysis using all 9,146 SNPs previously analyzed in this population [3], identifying 3,192 SNPs significantly associated with at least one complement protein (p<0.05). Data on the SNPs associated with each protein is presented in the supplement (Table S1-10). To distill meaningful biologic themes from this large data set, the 776 SNPs with p < 0.01, representing 359 genes, were categorized into one of seven curated functional groups [(1) direct association with the complement system (20 genes), (2) cytokines, chemokines, and their receptors (69 genes), (3) immune-associated transcription factors and intracellular immunomodulators (57 genes), (4) immune cell surface markers and activators (82 genes), (5) metabolic markers (50 genes), (6) apoptosis and other intracellular signaling molecules involved in non-canonical immune pathways (70 genes), and (7) cell structure and adhesion markers (11 genes); Table S11]. The distribution of the 776 significant SNP and 359 genes by functional group is shown graphically in Figure 3A.
This framework enabled analysis of functional group enrichment scores for each complement protein association (C3 in Figure 3B, and all other proteins in Figure S3A-I). For C3, we observed over-representation in the functional groups immune cell surface markers and activators, apoptosis, and cell structure and adhesion. Statistical parameters for each protein’s most significant associations are found in Supplemental Tables S1-S10. Two SNPs, rs800292 in CFH and rs17759529 in DPP4, linked to Bb and C3 levels are among the most significant in this pQTL screen. Genotypes of rs800292/CFH, previously implicated in complement regulation, showed a stepwise decrease in Bb levels with the T allele (Figure 3C). Interestingly, rs800292 shows a similar decrease in Bb/C3 ratios (Figure S4A), with an inverse trend seen in genotype-stratified C3 levels (Figure 3D). The G allele of rs17759529 in DPP4, a top-ranked pQTL for CRP and C3 in the over-represented immune cell surface markers functional group, had higher C3 levels in homozygous carriers (Figure 3E). Notably, DPP4 is a multifunctional protease expressed on immune cells and endothelium that has been implicated in SARS-CoV-2 [33]. Additionally, the rs17611 SNP in C5 was among our top-ranked pQTLs for C5a in this population (Figure S4B) in agreement with previous reports in independent cohorts [34,35]. The major allele T in rs1061170 (CFH) was associated with lower Bb levels (Figure 3F). Bb levels were similarly significantly lower in individuals carrying minor C allele rs9332739 (C2) and T allele rs4151667 (CFB), two SNPs in linkage disequilibrium (LD) (Figure 3G). The box plots in Figure 3C-3G also show the skewed distribution of ME/CFS cases by genotype of these complement pQTLs, suggesting an interaction between complement pQTLs and ME/CFS risk.
2.4. Identification of Genetic Variants Impacting Both Plasma Levels of Complement Proteins and Disease Status
We identified 16 SNPs found in both the 3,192 protein-associated SNPs and the 48 previously reported ME/CFS-associated SNPs [3], 11 of which act in the complement cascade (Table 2). The additional immune and inflammatory pathways represented indicate genetic risk for ME/CFS beyond the complement system.
Among the 16 SNP associations, four were predicted to act in cis (defined as variants located within 1Mb of the transcription start site of the impacted protein’s associated gene), such as variants located in C2, CFB, or CFH impacting levels of Bb, Factor B, or Factor H, respectively. The remaining 12 variants were predicted to be trans-pQTLs, acting elsewhere in the genome (e.g. SERPINA5 impacting C3, GRK4 impacting C3, and PDE4D impacting C3a and C5a). Of note, rs3020729 (CD8A, miRNA binding) was significantly associated with higher levels of CRP and C3 levels, and rs2277680 (CXCL16, missense) impacted Factor H and Factor B levels (Table 2). By contrast, rs9550987 (TNFRSF19, missense) was associated with decreased levels of C3 and Factor H (Table 2).
As shown in Figure 3F, the ME/CFS risk-associated major allele T in rs1061170 (CFH) was associated with lower Bb levels. Bb levels were similarly lower in individuals carrying the ME/CFS risk alleles at cis-acting rs9332739 (C2) and rs4151667 (CFB), two SNPs in linkage disequilibrium (LD) (Figure 3G). The inverse trends in genotype-stratified C3 levels and Bb/C3 ratio values for rs800292 (CFH) shown in Figure 3C, are also seen with the rs9332739 (C2) risk allele (Figure S4C and D). The five genotype combinations for these C2 and CFH variants (Figure 3H) show decreasing Bb and increasing C3 levels with increasing risk alleles, with the double risk genotype (GC-TT) exhibiting the lowest Bb and highest C3 levels as well as the highest percentage of ME/CFS in the genotype combination. The correlation analysis of disease-associated SNP beta values and disease odds ratios (Figure S4E and F) agrees with this pattern; variants increasing C3 expression aligned with increased ME/CFS risk (Figure S4E, R2 = 0.78), while those associated with higher Bb showed inverse trends (Figure S4F, R2 = -0.26).
2.5. Use Of Genotype-Defined Complement pQTLs to Identify ME/CFS Subgroups
As the levels of complement proteins are associated with ME/CFS, we assessed their performance as predictive markers using an ROC analysis of all 171 subjects (50 ME/CFS, 121 NF controls, Figure S5A). CRP, Factor B, and C3 had the highest predictive power, with AUCs of 0.75, 0.75, and 0.69, respectively (Figure S5A); only C3 was statistically significant. To incorporate the impact of complement-associated pQTLs, we conducted ROC analyses for complement proteins with ME/CFS as the outcome, stratified by genotype subgroups for single SNPs, selected based on significant associations with complement proteins or disease status (Table 2 and Table S1-10) and restricted to those within the complement system functional group. Results were filtered for AUC >0.75, p<0.05, and sufficient group size (n = 15) and are summarized in Table 3. Heterozygotes for two SNPs, rs9332739 in C2 and rs800292 in CFH, previously examined in Figure 3, showed a high AUC (Table 3). This SNP genotype combination was selected for subgroup analysis.
Subjects were stratified into four groups based on heterozygosity at rs9332739 in C2 and rs800292 in CFH. ME/CFS participants with heterozygous genotypes at one or both SNPs were assigned to CFShet (n = 26, Group A), while the remaining ME/CFS cases were designated as CFSrem (n = 24, Group B). Non-fatigued controls were similarly divided into NFhets (n = 45, Group C) and NFrem (n = 76, Group D), with the distribution and subgroup demographics shown in Figure 4A. Because subgrouping was defined by heterozygosity, two individuals with the double risk genotype for rs800292 but no risk allele at rs9332739 were assigned to the CFSrem and NFrem groups. The CFShet group contained no male participants, and BMI was significantly different between the CFShet group and the other three groups. CRP and C3 exhibited improved AUCs in the heterozygous-defined CFShets vs. NFhets comparison, with AUC values of 0.85 and 0.84, respectively (Table 3, Figure 4B), while Factor H and C3a also had enhanced predictive power (0.78 and 0,74 AUC, respectively) over the non-stratified model (Figure S5A).
To assess complement protein differences across genotype-stratified illness groups, we applied three statistical approaches: a likelihood ratio test to evaluate overall model fit, a multinomial logistic regression to identify proteins associated with subgroup assignment (using NFhet as the reference), and pairwise covariate adjusted linear regressions to examine direct groupwise differences. The likelihood ratio test identified seven proteins – Bb, Bb/C3, C3, C3a, CRP, Factor B, and Factor H – that significantly improved the model, indicating that these proteins contribute meaningfully to group classification (Figure 4C). Multinomial regression revealed that C3, C3a, CRP, and Factor H levels were significantly associated with increased odds of belonging to the CFShet group relative to NFhet (Figure 4C), suggesting potential utility as subgroup classifiers. Pairwise linear regression comparisons confirmed several between-group differences (Figure 4D-I), including lower Bb and Bb/C3 in NFhet compared to NFrem, and lower Bb/C3 in CFShet compared to NFrem. The CFShet group also demonstrated higher C3 and Factor H levels relative to NFhet, with CFShet C3 levels significantly higher than NFrem as well. Collectively, these complementary analyses support a robust association between circulating complement proteins and genotype-defined subgroups – even after adjust adjusting for covariates – highlighting their potential role in distinguishing biologically meaningful ME/CFS subgroups.
We examined the covariate-adjusted association between complement protein levels and scores of symptoms and function for the heterozygous subgroups (CFShet and NFhet, Figure 5A, Figure S5B-D). The covariate-adjusted regression models in this reduced population (n = 71) revealed multiple significant associations between complement protein levels and scores in CDC-SI, MFI-20, and SF-36. In the total study sample population (Figure 2), only four complement-related measurements (Bb/C3, C3, CRP, and Factor D) had significant associations compared to the seven complement proteins with significant associations across at least one of the three survey domains in the genotype-restricted groups (Figure 5A). These included newly significant relationships for C3a, Factor H, and SC5b-9 (Figure 5A, Figure S5B-D). For the total and genotype-restricted samples, the correlations followed the same direction. While exploratory, this expanded set of associations suggest that complement dysregulation may be more tightly linked to reported functional impairments in genetically defined subgroups, reinforcing the relevance of host genotypes in shaping disease heterogeneity in ME/CFS.
2.6. Comparison of Significant SNPs Between Our Population and Fatigue-Related Phenotypes in the UK Biobank
To evaluate the consistency and broader applicability of a pQTL-based approach for identifying genetic contributors to ME/CFS, we compared SNPs associated with each circulating complement protein (Table S1-10, top 50 SNPs from each table with the lowest p-values) identified in our population to SNPs associated with fatigue traits in the publicly available UK biobank GWAS data for chronic fatigue syndrome, post viral fatigue syndrome, a broader fatigue and malaise population, and Ever CFS (individuals reporting recovery from ME/CFS). Six SNPs across 5 genes overlapped between our pQTL dataset and the UK biobank fatigue-related traits (Table 4), mapping to complement-related genes CFB, CFH, and C2, as well as MMP10 (involved in the breakdown of extracellular matrix) and PIK3R5 (which plays roles in cell growth, proliferation, differentiation, motility, survival, and intracellular trafficking). Four of the 6 SNPS (3 genes) were among those we identified as both pQTLs and ME/CFS-associated (rs641153, rs800292, rs9332739 and rs4151667). Interestingly, rs394811 in PIK3R5 was the only SNP to overlap with the UK biobank’s designated CFS group. In contrast, SNPs in CFB, CFH, and C2 appeared in the broader fatigue and malaise and post viral fatigue phenotypes. Moreover, the direction of effect for each overlapping SNP – including risk allele identity and odds ratio – was consistent between the UK biobank phenotypes and our population, providing some replication of the biological relevance of these shared associations.
3. Discussion
This study devised a novel pQTL-based approach to address clinical heterogeneity in ME/CFS using genotype and pathway-focused stratification. Our analysis focused on complement pQTLs and yielded several noteworthy findings. First, we observed significantly higher levels of C3 in ME/CFS compared to NF controls, alongside a decrease in the ratio of Bb to C3, suggesting altered alternative pathway activation in this disease. Second, we identified multiple complement pQTLs that were also associated with disease status. These pQTLs included polymorphisms in C2/CFB (rs9332739 and rs4151667 in LD) and CFH (rs800292). The ability of complement protein levels to distinguish between ME/CFS and NF controls was improved when limiting the analysis to the genotype-restricted subgroup. Cases and controls in this subgroup not only differed in C3 and Factor H levels but also showed distinct patterns across multiple analyses that improved the model’s ability to distinguish between the four subgroups and improved the odds of subgroup assignment, with proteins in the genotype-restricted group also exhibiting more extensive associations with functional impairment and patient health scores. Additionally, several pQTLs that we identified in our population with functional consequences (non-synonymous SNPs: rs4151667, rs9332739, rs641153, rs800292, rs394811 and rs17293607) were found to overlap with SNPs that were significantly associated with fatigue-related phenotypes listed in the UK biobank, demonstrating how integrating approaches may help resolve inconsistencies across genetic studies of this heterogeneous disease. These findings stress a link between complement system genetics, complement protein dysregulation, and ME/CFS pathophysiology, while also providing an approach for the much-needed subgrouping of heterogenous diseases such as ME/CFS.
Our association results reinforce the need to adjust data for covariates, such as age, sex, or BMI when evaluating differences in complement components between ME/CFS and healthy controls; adjustments likely important for other protein associations as well. Our findings of the influence of age, sex, and BMI on the level of these proteins are largely in agreement with recently published results [36,37,38], requiring them to be used as covariates in the analysis of complement proteins using genetic or non-genetic models. The complement dysregulation identified in our study of ME/CFS persisted after covariate adjustment, highlighting its disease-specific nature rather than demographic artifact. While age is known to influence certain classical complement components, such as declining C1q levels in older adults [39,40], we only included age as a covariate in models where it was empirically significant for our specific population, such as for Factor D. In contrast, BMI emerged as a consistent covariate across multiple complement proteins – most notably C3 – likely reflecting adipose-driven complement protein production [41,42,43,44]. This is particularly relevant for ME/CFS, where symptom severity and metabolic shifts may influence body composition. Of note, earlier studies that did not control for BMI reported no differences in C3 between ME/CFS and controls [20,45]. Sex was also considered when statistically warranted (Factor B and CRP) and adjustment ensured complement differences were not driven by the unbalanced sex ratio observed in ME/CFS [46]. Sex hormones and x-linked immune genes shape distinct immunologic profiles [47], and women generally exhibit lower baseline complement levels [48,49]. Notably, the CFShet subgroup contained no male participants, suggesting potential sex-dependent genotype effects that warrant future stratified analyses, in line with other recent suggestions of ME/CFS immune subtyping by sex [50,51].
C3 is the central protein in the complement cascade regulated by three major pathways (classical, lectin and alternate pathways) leading to a pathway-dependent C3 convertase converting C3 to C3b and C3a. The classical/lectin pathways generate C3 convertase by combining C4bC2b, whereas the alternative pathway contributes to C3 convertase (C3bBb) by binding of C3b to Bb (Bb results from cleavage of Factor B by Factor D). An increase in plasma Bb is considered as a marker of complement activation, whereas a decrease in Bb indicates complement inhibition resulting from the inhibitory functions of Factor H [18]. High C3 and C3a, along with high CRP (previously reported in other ME/CFS populations [52,53]) hint at ongoing innate immune activation; however, concurrent high Factor H (an alternative pathway inhibitor) and low Bb suggest increased complement inhibition in our ME/CFS subjects. This could reflect a compensatory response or genetically driven dampening of the alternative pathway in ME/CFS, especially in variant-carrying subgroups. Of note, a recent plasma proteomic analysis in ME/CFS suggests a similar hypotheses of attenuated complement activity, reflecting a compensatory mechanism and subsequent exhausted immune state in ME/CFS [54]. Integrating genetic variant data, we found subjects carrying the risk allele in C2/CFB and CFH (rs9332739 and rs1061170) had lower levels of Bb in plasma, and that Bb levels could be impacted by the pQTL rs800292 in CFH, although this latter trans-pQTL was not directly associated with illness (Armitage P=0.09). Notwithstanding, heterozygotes for rs9332739 in C2 and rs800292 in CFH resulted in the highest AUC for prediction of illness status. Both rs9332739 (E318D) in C2 and rs800292 (V62I) in CFH are missense pQTLs for Bb and both are reported to be protective for AMD [27,28,29,30,31,32,55].
There is limited information on the functional role of these pQTLs in the alternate pathway. Other studies have documented their association with diseases that have seemingly unrelated clinical presentations and pathologies. For example, studies of AMD and atypical hemolytic uremic syndrome (aHUS), clinically different illnesses but both having overactivity of the alternative pathway, identified defective regulation of the alternative pathway [56,57]. This differs from our finding that at least a subset of ME/CFS subjects show inhibition of the alternative pathway through what appears to be a genetically determined low level of Bb. Moreover, when C2 and CFH variants were considered together, Bb and C3 levels showed stepwise opposing trends across the five genotype combinations, most stark in the genotype group with the highest ME/CFS proportion. These reciprocal trends point to a genetically encoded imbalance between complement precursor (C3) and activation product (Bb), potentially reflecting dysregulated convertase activity in ME/CFS. Correlation analyses between C3 or Bb protein levels and disease odds ratios revealed consistent directional concordance across diverse genetic associations, reinforcing a model in which ME/CFS genetic risk is linked to elevated upstream complement activity skewed toward C3 and away from Bb.
Higher complement inhibition and thus reduced Bb, as we see in this study, is also in agreement with reported inhibition of the alternate pathway by the minor allele of rs800292 in CFH, due to the strong binding of the variant to C3b [58,59]. Factor H and C3 protein were highly correlated, perhaps resulting from increased inhibition of alternate pathway by Factor H [60], and both were present in high levels in ME/CFS. Understanding these biological relationships is essential for interpreting circulating levels of complement proteins in ME/CFS, as disease-associated changes may reflect both direct dysregulation and broader shifts in pathway dynamics. For example, rs800292 in CFH is reported as a potent regulator of matrix metalloproteinase-8 (MMP-8), a proinflammatory enzyme conferring risk for cardiovascular diseases, and carriers of the rs800292 minor allele were found to have decreased levels of serum MMP-8 [61]. CFH belongs to a family of 7 genes, denoted as Regulators of Complement Activation (RCA), spanning a region of 360 kbp in chromosome 1 [59]. Mutations seen in RCA can act as both activators and inhibitors, offering the opportunity to regulate their role in different diseases [62]. Notably, we identified one cis-acting pQTL for Factor H, negatively regulating rs1065489 in CFH. This study also identified four disease-associated pQTLs for C3 (rs6108 in SERPINA5, rs3020729 in CD8A, rs1801058 in GRK4, rs9550987 in TNFRSF19). Although C3 levels were significantly different between illness groups, we did not identify any co-localized cis-QTL impacting both illness status and C3 level; however, circulating C3 levels were decreased by the minor alleles of three cis acting intronic pQTLs (rs7257062, rs2241393, rs2241392), all of which performed as strong predictors of illness status, with AUC > 0.78 and p-values below 0.006. Subjects homozygous for major alleles of these three variants had higher levels of C3, but for unclear reasons were not associated with ME/CFS. Of particular interest among pQTLs not associated with ME/CFS are several pQTLs (rs17611, rs7037673, rs1468673, rs992670, rs7040319, rs4837805, rs17220750, rs2416810, rs2300939, rs7026551) in the TRAF1/C5 region in chromosome 9 that impact circulating C5a levels. Interestingly, QTLbase home database (www.mulinlab.org) lists rs7037673 as a pQTL for C5 expression in the prefrontal cortex, supporting further investigation into complement-mediated neuroinflammation in ME/CFS pathogenesis [63]. This variant also lies near TRAF1, a key component of the TNF signaling cascade that promotes inflammatory cytokine expression, suggesting a potential link between complement activity and neuroimmune signaling in ME/CFS. Overall, this approach highlights the potential of complement-associated pQTLs to delineate biologically meaningful markers and targets within ME/CFS.
Dysregulated complement activity in ME/CFS mirrors persistent complement activation, impaired metabolic and immune responses, and altered inflammatory cytokine profiles, consistent with chronic inflammation and incomplete post-infection resolution illnesses [64,65,66,67,68,69,70,71,72,73] reported in other infection-associated chronic conditions and illnesses (IACCIs). Understanding the nuanced differences in complement profiles across IACCIs may help explain why some individuals recover while others develop chronic sequelae. Related but mechanistically distinct complement system patterns are observed in other chronic immune or neurological conditions: in lupus, low CRP with C3/C4 consumption impairs nuclear antigen clearance, whereas in ME/CFS, elevated CRP alongside high Factor H and reduced Bb suggests suppressed clearance and unresolved inflammation driven by host genetics [74,75,76]. In multiple sclerosis (MS), complement activation contributes to demyelination, with high Factor H observed in some patients, despite a clear complement locus yet to be identified– consistent with predominantly environmental/secondary involvement [79,80]. In Alzheimer’s disease, complement-associated variants and insufficient control of complement-mediated synaptic pruning implicate complement in neurodegeneration [81], pertinent given the cognitive impairments seen in many ME/CFS patients. Notably, our identification of a trans-pQTL for Factor H in TNFRSF19– a gene expressed in the central nervous system – suggests a link between peripheral complement regulation and neuroimmune signaling in ME/CFS. Ultimately, these findings position complement dysregulation not as an isolated feature but as a potential hallmark of chronic immunoinflammatory disease, offering a framework for therapeutic targeting and for refining biological subgroups across heterogeneous conditions.
In summary, our study gives an example of a pQTL-based approach integrating genotype with protein biomarker data that can uncover immune subtypes within a heterogenous disease, offering a path forward for more personalized treatment strategies and mechanistic investigation. In addition, our study identifies inflammation driven by high C3 – likely resulting from increased inhibition of the alternative pathway – as a central feature in a genetically defined subgroup of ME/CFS. This subgroup, identifiable by complement pQTLs, may benefit from targeted therapies such as C3-lowering agents [82]. The complement abnormalities we observe mirror those in other IACCIs, where imbalanced complement activity – whether genetically inherited or acquired – contributes to chronic inflammation and tissue damage. Our findings add to this landscape by highlighting the genetic variants in complement as a heritable contributor to ME/CFS susceptibility, where low basal alternative pathway activity may predispose individuals to persistent inflammation following an infectious or environmental insult. Complement also intersects with metabolic, endothelial, and coagulation pathways, linking dysregulation to hallmark ME/CFS symptoms like fatigue and bioenergetic stress [83].
The strengths of this study include use of participants from the population classified using a standardized approach and employing a novel pQTL analysis. Limitations include the relatively small number of ME/CFS patients, lack of racial diversity and cross-sectional data. Further study is required to evaluate the generalizability of our findings. Future studies should expand this approach genome- and proteome- wide and leverage resources like DecodeME or SearchMECFS biorepository to explore stratification strategies across diverse populations. In parallel, tracking complement dynamics longitudinally – especially in patients with high-risk genotypes or post-infection – could clarify disease trajectories and inform therapeutic targeting.
4. Materials and Methods
4.1. Subject Recruitment and Characteristics
This study included the 50 ME/CFS and 121 non-fatigued (NF) controls subjects from the previously published genetic analysis focusing on inflammatory and immune-related pathways [3]. Briefly, participants were identified in the follow-up phase of the longitudinal study of ME/CFS in Georgia. ME/CFS cases were identified with the operationalized 1994 international research case definition [84]. Sample characteristics such as age, sex, body mass index (BMI), illness duration and onset, and symptom and functioning scores across fatigue (multidimensional fatigue inventory, MFI-20), functional status (SF-36), CDC symptom inventory (CDC-SI), and routine clinical tests results (including hs-CRP) were recorded for every participant [3]. The source study was approved by the Institutional Review Board of the Centers for Disease Control and Prevention and Abt Associates (now Abt Global). All subjects gave written informed consent for participating in the study and for anonymous testing and storage of biologic samples.
4.2. Plasma Protein and Complement Component Assays
Whole blood from each subject was collected in ethylenediaminetetraacetic acid (EDTA) tubes and processed for plasma within 30 minutes to minimize ex-vivo activation of complements. Plasma was separated, aliquoted, and stored at -70°C. A frozen plasma aliquot for each subject was shipped to the CLIA-certified Complement Laboratory at the National Jewish Health, Denver, Colorado for analysis of complement proteins/activation products using their validated clinical assays. Basic assay methods included measuring levels of C3a, C5a, Bb, SC5b-9 and Factor D by ELISA, C3 by nephelometry, and Factor B and Factor H by radial immunodiffusion. Results of hs-CRP testing were retrieved from the study database.
4.3. Statistical Analyses
4.3.1. Data Transformation
Statistical and genetic analyses were performed on log2-transformed values of the measured proteins involved in the complement system (C3a, C5a, C3, Bb, SC5b-9, Factor D, Factor B, Factor H, and CRP). This data transformation was based on the observed variance and distribution of protein levels across all participants, performed using RStudio.
4.3.2. Data analyses and visualizations for group phenotypes and complement protein levels
A Wilcoxon rank-sum test was used to test for significant differences in age and BMI between ME/CFS and NF control subjects. Radar plots made in Microsoft Excel were used to display the mean values for MFI-20 and SF-36 T-scores for both groups. A Pearson correlation test was performed to assess the association of complement factor levels with each other.
4.3.3. Identification of Confounding Factors
A linear regression analysis was used to determine the bivariate association of circulating complement protein levels with demographic features (or variables) recognized to be associated with ME/CFS, such as age, sex, and BMI. Covariate adjustments specific to each circulating complement protein based on these associations and those supported by the literature were included in downstream analyses.
4.3.4. Analysis of Circulating Complement Proteins Between ME/CFS and NF Subjects
Either binary or multinomial logistic regression analyses were performed to test whether individual complement proteins were significantly associated with group classification, maintaining covariate considerations. In the 2-group analysis, ME/CFS cases status (Y = 1) vs. NF controls (Y=0) was modeled as the binary outcome:
In the 4-group multinomial model, subgroup assignment (Y = one of the four groups) was used as the categorical outcome, with NFhet set as the reference category:
For
Likelihood ratio tests (LRTs) were performed to assess the overall contribution of each complement protein to the model. For each protein, we compared the full model (including the protein of interest as a predictor) against a reduced model (excluding the protein) using the following:
Where the full model is:
And the null model is:
Pairwise linear regression analyses were performed to directly compare log2-transfored protein concentrations between groups while adjusting for covariates. These were applied to both the 2-group and 4-group comparisons:
Group was treated as a binary or categorical variable, and pairwise p-values were extracted using estimated marginal means and post hoc contrasts to highlight specific group differences. Boxplots and dot graphs were created using the R package ggPlot2 to visualize protein distributions across groups.
4.3.5. Analysis of Participant Survey Data with Circulating Complement Proteins
Linear regression modeling with previously noted covariate adjustments was performed to assess complement protein levels with participant survey data. Specifically, log2-transformed plasma protein levels were modeled as a function of continuous survey metrics (CDC-SI, MFI-20, and SF-36), using the formula:
RStudio was used to create a heatmap to display the regression β1 coefficient and the p-values for each association. To illustrate specific trends, representative correlation dot plots with corresponding R2 values were created for analytes with significant associations, capturing the distribution of case- and control-level survey responses.
4.3.6. Analysis of SNP Associations with Circulating Complement Proteins
A genotypic logistic regression using an additive model was previously applied to assess 9,146 SNPs that passed Affymetrix Human Immune and Inflammation Chip quality control and their associations with ME/CFS [3]. To investigate genetic variants for associations with complement components, we performed covariate-adjusted linear regression analyses in RStudio comparing full and reduced models for each SNP-protein pair. This analysis was conducted across all subjects, without stratification by illness group, to assess genetic regulation of complement protein expression. For each SNP-protein pair, a full linear model including the SNP genotype and relevant covariates was compared to a reduced model excluding the genotype.
` Full model:
Reduced model:
An F-statistic was calculated to compare the full and reduced models for each SNP-protein pair, testing whether the inclusion of genotype significantly improved model fit. The p-value from the full-versus-reduced (FvR) comparison was used to define significance. Additionally, Bonferroni correction was applied to account for multiple testing across all models, and these results are reported in Supplementary Tables 1-10. Allele frequencies, including the identification of major and minor alleles for the genetic analyses, were determined using data from this study population.
4.3.7. Pathway Enrichment Analysis to Annotate SNP Associations with Circulating Complement Protein Levels
The linear regression analysis identified 3,192 SNPs significantly associated with circulating complement proteins (Full vs. Reduced Linear regression model, p < 0.05). The 3,192 SNPs associated with circulating complement protein levels were further filtered to identify top associations (Full vs. Reduced Linear regression model, p < 0.01) for each measured complement protein. This analysis resulted in 776 significant SNPs linked to complement protein plasma levels, representing a total of 359 unique genes. To investigate the distribution of significant SNPs across functional groups for each protein, data was extracted from Metascape, Golden Helix SVS software, and the literature to create a list of all genes with SNPs significantly associated with each complement protein and their function, allowing assignment of to one of seven functional groups (Table S11). For each complement protein, the number of significant genes and SNPs associated with each functional group was calculated. Enrichment scores were computed by dividing the observed proportion of genes/SNPs in a functional group within each complement factor’s “top hits” list by the expected proportion of genes/SNPs in each functional group based on the overall distribution of significant genes/SNPs across all functional groups and complement factor “top hits” lists. The overall distribution of significant genes/SNPs across all functional groups regardless of complement factor association was visualized by creating a donut plot using Microsoft Excel. Lollipop plots were generated using R to visualize these enrichment scores for each circulating complement protein measured, with the y-axis containing functional groups and overrepresentation in a group indicated by an enrichment score > 1.
4.3.8. Analysis of SNP Associations with Circulating Complement Protein Levels and Disease Status
The 3,192 SNPs significantly associated with circulating complement proteins were compared against the previously published 32 functional SNPs, 10 proxy SNPs, and 6 additional pyro sequenced complement-linked SNPs significantly associated with ME/CFS (with sex and BMI covariate adjustments) [3], identifying 16 SNPs in common with both analyses and therefore associated with both disease status and circulating complement protein levels. These pQTLs were also assessed for whether they were cis or trans-acting using the QTLbase home database (www.mulinlab.org).
4.3.9. Analysis of the Directionality of Genetic Variant Traits with Disease Risk
To investigate the broader pattern of disease-associated SNPs with complement protein levels, we assessed the relationship between the odds ratios (ORs) of significant genetic variants for ME/CFS risk and beta coefficients for circulating complement protein levels. ORs were derived from previously published SNP-disease associations (32 SNPs assessed against C3 beta values, and 27 SNPs assessed against Bb beta values) [3], and beta coefficients were generated from our circulating complement protein-level QTL analyses using the log2-transformed plasma concentrations of each complement protein. All SNPs previously reported as significantly associated with ME/CFS in the prior study [3] were included in this analysis, regardless of functional consequence. Specifically, we plotted ORs on the x-axis and corresponding beta values for C3 or Bb on the y-axis, with each point representing a single SNP significantly associated with ME/CFS status.
4.3.10. Genotype-Stratified Analysis to Identify ME/CFS Subgroup Related to Complement System Dysregulation
The pQTLs identified in this study were evaluated for their ability to discriminate the illness status and subgrouping of the subjects in the study. This was done by first prioritizing pQTLs with high power for prediction of illness status by conducting a series of ROC analyses that split the subjects based on genotypes of QTLs, with log2-transformed plasma complement protein levels as the test variable. Specifically, participants were grouped by genotype at each SNP of interest, and logistic regression models were used to calculate predicted probabilities for ME/CFS status. These probabilities were then used to compute ROC curves and AUC values. Genotypes that resulted in better discrimination of subjects’ illness status as indicated by high Area Under the Curve (AUC ≥0.75) were selected for combined analysis of markers to further improve the AUC for predicting illness status. An example of grouping subjects based on two markers rs9332739 and rs800292 was visualized to evaluate the discriminatory capacity of complement-associated pQTLs for potential subgroup identification within the study population. This combined marker analysis conducted using rs9332739 and rs800292 considered ME/CFS participants with heterozygous genotypes at either SNP as Subgroup A (CFShet), and the remainder (those without heterozygosity at either loci) were assigned to Subgroup B (CFSrem). Similarly, NF controls with heterozygous genotypes at either marker were assigned to Subgroup C (NFhet), and all remaining controls were assigned to Subgroup D (NFrem). Of note, no individuals carried the homozygous risk genotype for rs9332739 (C/C), while five individuals carried the homozygous risk genotype for rs800292 (T//T). Because subgrouping was based on genotypes with the strongest predictive value and sufficiently robust sample sizes, two individuals (one ME/CFS case and one NF control) who were homozygous for the rs800292 risk allele were included in the broader CFSrem or NFrem groups, rather than in the heterozygous subgroup.
This four-group classification was applied to assess refined stratification of disease status and to characterize potential genotype-informed subgroups. Logistic models were rerun using these stratified groups to evaluate differences in complement protein levels, as described in 4.3.4., and ROC performance across identified subgroups.
4.3.11. Comparison of Significant SNPs Between Our Population and the UK Biobank
To assess overlap between our top genetic variant associations and previously reported ME/CFS-related SNPs, we compared the top 50 SNP associations for each of the nine measured complement proteins with publicly available genome-wide association data from the UK biobank [85,86]. Specifically, we extracted SNPs significantly associated with ME/CFS-related phenotypes – including the groups Chronic Fatigue Syndrome, Post Viral Fatigue Syndrome, the broader R53 Fatigue and Malaise category, and Ever CFS (recovered ME/CFS subjects) – from the UK Biobank’s large population-based prospective study. We then evaluated whether any of the top complement protein-associated SNPs in our dataset overlapped with those reported for these fatigue-related phenotypes in the UK biobank, and compared variant details, p-values, and odds ratios for common SNPs.
Supplementary Materials
The following supporting information can be found in the supplemental data files (Supplementary_Materials_Figures_V4.docx, Suppl_Tables_V2.xlsx)
Author Contributions
Conceptualization: MSR, JM, and ERU; Methodology: MSR and JM; Software: MSR and JM; Validation: MSR and JM; Formal analysis: MSR and JM; Investigation: MSR and JM; Resources: MSR, JM, JSL, and ERU; Data curation: MSR, JSL, and JM; Writing—original draft preparation: MSR and JM; Writing—review and editing: MSR, JM, JSL, and ERU; Visualization: JM and MSR; Supervision: MSR and ERU; Project administration: MSR and ERU; Funding acquisition: MSR and ERU.
Funding
This research received no external funding.
Institutional Review Board Statement
As a requirement of publication, author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the authorship and conflict of interest criteria.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data supporting reported results can be found in supplemental materials. Researchers wanting to access the datasets used in this study should email CDC’s ME/CFS Program (cfs@cdc.gov) and discuss next steps for the data request. The ME/CFS program data review committee will grant access after the review and the data use agreement is finalized.
Acknowledgments
Partial support for JM was provided by the research participation program at the Centers for Disease Control and Prevention (CDC), Division of High-Consequence Pathogens & Pathology, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the CDC. We thank Janna Murray for their invaluable assistance in acquiring and coordinating access to datasets used in this study. We are especially grateful to the patients and non-fatigue healthy controls who generously participated in the research, making this work possible. Their contributions continue to advance our understanding of ME/CFS and related conditions.
Conflicts of Interest
The findings and the conclusions in this report are those of the authors and do not necessarily represent the official position of CDC. Authors declare no conflict of interest.
References
- Medicine, I.o. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness; The National Academies Press: Washington, DC, 2015; p. 304. [Google Scholar]
- Unger, E.R.; Lin, J.-M.S.; Chen, Y.; Cornelius, M.E.; Helton, B.; Issa, A.N.; Bertolli, J.; Klimas, N.G.; Balbin, E.G.; Bateman, L.; et al. Heterogeneity in Measures of Illness among Patients with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Is Not Explained by Clinical Practice: A Study in Seven U.S. Specialty Clinics. J. Clin. Med. 2024, 13, 1369. [Google Scholar] [CrossRef] [PubMed]
- Rajeevan, M.S.; Dimulescu, I.; Murray, J.; Falkenberg, V.R.; Unger, E.R. Pathway-focused genetic evaluation of immune and inflammation related genes with chronic fatigue syndrome. Hum. Immunol. 2015, 76, 553–560. [Google Scholar] [CrossRef]
- Smith, A.K.; Fang, H.; Whistler, T.; Unger, E.R.; Rajeevan, M.S. Convergent Genomic Studies Identify Association of GRIK2 and NPAS2 with Chronic Fatigue Syndrome. Neuropsychobiology 2011, 64, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Jaundoo, R.; Hilton, K.; Del Alamo, A.; Gemayel, K.; Klimas, N.G.; Craddock, T.J.A.; Nathanson, L. Genetic Predisposition for Immune System, Hormone, and Metabolic Dysfunction in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Pilot Study. Front. Pediatr. 2019, 7, 206. [Google Scholar] [CrossRef]
- A Schlauch, K.; Khaiboullina, S.F.; De Meirleir, K.L.; Rawat, S.; Petereit, J.; A Rizvanov, A.; Blatt, N.; Mijatovic, T.; Kulick, D.; Palotás, A.; et al. Genome-wide association analysis identifies genetic variations in subjects with myalgic encephalomyelitis/chronic fatigue syndrome. Psychiatry 2016, 6, e730–e730. [Google Scholar] [CrossRef]
- Marshall-Gradisnik, S.; Huth, T.; Chacko, A.; Johnston, S.; Smith, P.; Staines, D. Natural killer cells and single nucleotide polymorphisms of specific ion channels and receptor genes in myalgic encephalomyelitis/chronic fatigue syndrome. Appl. Clin. Genet. 2016, 9, 39–47. [Google Scholar] [CrossRef]
- Marshall-Gradisnik, S.; Johnston, S.; Chacko, A.; Nguyen, T.; Smith, P.; Staines, D. Single nucleotide polymorphisms and genotypes of transient receptor potential ion channel and acetylcholine receptor genes from isolated B lymphocytes in myalgic encephalomyelitis/chronic fatigue syndrome patients. J. Int. Med Res. 2016, 44, 1381–1394. [Google Scholar] [CrossRef]
- Carlo-Stella, N.; Badulli, C.; De Silvestri, A.; Bazzichi, L.; Martinetti, M.; Lorusso, L.; Bombardieri, S.; Salvaneschi, L.; Cuccia, M. A first study of cytokine genomic polymorphisms in CFS: Positive association of TNF-857 and IFNgamma 874 rare alleles. 2006, 24, 179–82. [Google Scholar]
- Steiner, S.; Becker, S.C.; Hartwig, J.; Sotzny, F.; Lorenz, S.; Bauer, S.; Löbel, M.; Stittrich, A.B.; Grabowski, P.; Scheibenbogen, C. Autoimmunity-Related Risk Variants in PTPN22 and CTLA4 Are Associated With ME/CFS With Infectious Onset. Front. Immunol. 2020, 11, 578. [Google Scholar] [CrossRef]
- Lande, A.; Fluge, Ø.; Strand, E.B.; Flåm, S.T.; Sosa, D.D.; Mella, O.; Egeland, T.; Saugstad, O.D.; Lie, B.A.; Viken, M.K. Human Leukocyte Antigen alleles associated with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rajeevan, M.S.; Smith, A.K.; Dimulescu, I.; Unger, E.R.; Vernon, S.D.; Heim, C.; Reeves, W.C. Glucocorticoid receptor polymorphisms and haplotypes associated with chronic fatigue syndrome. Genes, Brain Behav. 6, 167–176. [CrossRef]
- Tziastoudi, M.; Cholevas, C.; Stefanidis, I.; Theoharides, T.C. Genetics of COVID-19 and myalgic encephalomyelitis/chronic fatigue syndrome: a systematic review. Ann. Clin. Transl. Neurol. 2022, 9, 1838–1857. [Google Scholar] [CrossRef]
- Hajdarevic, R.; Lande, A.; Mehlsen, J.; Rydland, A.; Sosa, D.D.; Strand, E.B.; Mella, O.; Pociot, F.; Fluge, Ø.; Lie, B.A.; et al. Genetic association study in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) identifies several potential risk loci. Brain, Behav. Immun. 2022, 102, 362–369. [Google Scholar] [CrossRef]
- Yao, C.; et al. Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat Commun 2018, 9, 3268. [Google Scholar] [CrossRef]
- Zhao, J.H.; Stacey, D.; Eriksson, N.; Macdonald-Dunlop, E.; Hedman, Å.K.; Kalnapenkis, A.; Enroth, S.; Cozzetto, D.; Digby-Bell, J.; Marten, J.; et al. Genetics of circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. Nat. Immunol. 2023, 24, 1540–1551. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Shi, J.; Wang, X.; Jiang, H.; Zhu, H.-J. Genome-wide pQTL analysis of protein expression regulatory networks in the human liver. BMC Biol. 2020, 18, 1–16. [Google Scholar] [CrossRef]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Mathern, D.R.; Heeger, P.S. Molecules Great and Small: The Complement System. Clin J Am Soc Nephrol 2015, 10, 1636–1650. [Google Scholar] [CrossRef]
- Sorensen, B.; Streib, J.E.; Strand, M.; Make, B.; Giclas, P.C.; Fleshner, M.; Jones, J.F. Complement activation in a model of chronic fatigue syndrome. J. Allergy Clin. Immunol. 2003, 112, 397–403. [Google Scholar] [CrossRef]
- Sorensen, B.; Jones, J.F.; Vernon, S.D.; Rajeevan, M.S. Transcriptional Control of Complement Activation in an Exercise Model of Chronic Fatigue Syndrome. Mol. Med. 2009, 15, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Giloteaux, L.; Glass, K.A.; Germain, A.; Franconi, C.J.; Zhang, S.; Hanson, M.R. Dysregulation of extracellular vesicle protein cargo in female myalgic encephalomyelitis/chronic fatigue syndrome cases and sedentary controls in response to maximal exercise. J. Extracell. Vesicles 2024, 13, e12403. [Google Scholar] [CrossRef]
- Schutzer, S.E.; et al. Distinct cerebrospinal fluid proteomes differentiate post-treatment lyme disease from chronic fatigue syndrome. PLoS One 2011, 6, e17287. [Google Scholar] [CrossRef] [PubMed]
- Germain, A.; Levine, S.M.; Hanson, M.R. In-Depth Analysis of the Plasma Proteome in ME/CFS Exposes Disrupted Ephrin-Eph and Immune System Signaling. Proteomes 2021, 9, 6. [Google Scholar] [CrossRef]
- Guenther, S.; Loebel, M.; Mooslechner, A.A.; Knops, M.; Hanitsch, L.G.; Grabowski, P.; Wittke, K.; Meisel, C.; Unterwalder, N.; Volk, H.-D.; et al. Frequent IgG subclass and mannose binding lectin deficiency in patients with chronic fatigue syndrome. Hum. Immunol. 2015, 76, 729–735. [Google Scholar] [CrossRef]
- Lutz, L.; Rohrhofer, J.; Zehetmayer, S.; Stingl, M.; Untersmayr, E. Evaluation of Immune Dysregulation in an Austrian Patient Cohort Suffering from Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Biomolecules 2021, 11, 1359. [Google Scholar] [CrossRef]
- Ding, X.; Patel, M.; Chan, C.-C. Molecular pathology of age-related macular degeneration. Prog. Retin. Eye Res. 2009, 28, 1–18. [Google Scholar] [CrossRef]
- Despriet, D.D.G.; Klaver, C.C.W.; Witteman, J.C.M.; Bergen, A.A.B.; Kardys, I.; de Maat, M.P.M.; Boekhoorn, S.S.; Vingerling, J.R.; Hofman, A.; Oostra, B.A.; et al. Complement Factor H Polymorphism, Complement Activators, and Risk of Age-Related Macular Degeneration. JAMA 2006, 296, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Gold, B.; E Merriam, J.; Zernant, J.; Hancox, L.S.; Taiber, A.J.; Gehrs, K.; Cramer, K.; Neel, J.; Bergeron, J.; et al.; The AMD Genetics Clinical Study Group Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 2006, 38, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Scholl, H.P.N.; Issa, P.C.; Walier, M.; Janzer, S.; Pollok-Kopp, B.; Börncke, F.; Fritsche, L.G.; Chong, N.V.; Fimmers, R.; Wienker, T.; et al. Systemic Complement Activation in Age-Related Macular Degeneration. PLOS ONE 2008, 3, e2593. [Google Scholar] [CrossRef]
- Hecker, L.A.; Edwards, A.O.; Ryu, E.; Tosakulwong, N.; Baratz, K.H.; Brown, W.L.; Issa, P.C.; Scholl, H.P.; Pollok-Kopp, B.; Schmid-Kubista, K.E.; et al. Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum. Mol. Genet. 2009, 19, 209–215. [Google Scholar] [CrossRef]
- Smailhodzic, D.; Klaver, C.C.; Klevering, B.J.; Boon, C.J.; Groenewoud, J.M.; Kirchhof, B.; Daha, M.R.; Hollander, A.I.D.; Hoyng, C.B. Risk Alleles in CFH and ARMS2 Are Independently Associated with Systemic Complement Activation in Age-related Macular Degeneration. Ophthalmology 2012, 119, 339–346. [Google Scholar] [CrossRef]
- Sebastián-Martín, A.; Sánchez, B.G.; Mora-Rodríguez, J.M.; Bort, A.; Díaz-Laviada, I. Role of Dipeptidyl Peptidase-4 (DPP4) on COVID-19 Physiopathology. Biomedicines 2022, 10, 2026. [Google Scholar] [CrossRef] [PubMed]
- Hoke, M.; Speidl, W.; Schillinger, M.; Minar, E.; Zehetmayer, S.; Schönherr, M.; Wagner, O.; Mannhalter, C. Polymorphism of the complement 5 gene and cardiovascular outcome in patients with atherosclerosis. Eur. J. Clin. Investig. 2012, 42, 921–926. [Google Scholar] [CrossRef]
- Giles, J.L.; Choy, E.; Berg, C.v.D.; Morgan, B.P.; Harris, C.L. Functional Analysis of a Complement Polymorphism (rs17611) Associated with Rheumatoid Arthritis. J. Immunol. 2015, 194, 3029–3034. [Google Scholar] [CrossRef]
- Acar, I.E.; Willems, E.; Kersten, E.; Keizer-Garritsen, J.; Kragt, E.; Bakker, B.; Galesloot, T.E.; Hoyng, C.B.; Fauser, S.; van Gool, A.J.; et al. Semi-Quantitative Multiplex Profiling of the Complement System Identifies Associations of Complement Proteins with Genetic Variants and Metabolites in Age-Related Macular Degeneration. J. Pers. Med. 2021, 11, 1256. [Google Scholar] [CrossRef]
- Copenhaver, M.M.; Yu, C.-Y.; Zhou, D.; Hoffman, R.P. Relationships of complement components C3 and C4 and their genetics to cardiometabolic risk in healthy, non-Hispanic white adolescents. Pediatr. Res. 2020, 87, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Gaya da Costa, M.; et al. Age and Sex-Associated Changes of Complement Activity and Complement Levels in a Healthy Caucasian Population. Front Immunol 2018, 9, 2664. [Google Scholar] [CrossRef] [PubMed]
- LaFon, D.C.; Thiel, S.; Kim, Y.-I.; Dransfield, M.T.; Nahm, M.H. Classical and lectin complement pathways and markers of inflammation for investigation of susceptibility to infections among healthy older adults. Immun. Ageing 2020, 17, 1–9. [Google Scholar] [CrossRef]
- Kelkar, N.S.; Goldberg, B.S.; Dufloo, J.; Bruel, T.; Schwartz, O.; Hessell, A.J.; Ackerman, M.E. Sex- and species-associated differences in complement-mediated immunity in humans and rhesus macaques. mBio 2024, 15, e0028224. [Google Scholar] [CrossRef]
- Butler, A.E.; Moin, A.S.M.; Sathyapalan, T.; Atkin, S.L. Complement Dysregulation in Obese Versus Nonobese Polycystic Ovary Syndrome Patients. Cells 2023, 12. [Google Scholar] [CrossRef]
- Qiao, Q.; Bouwman, F.G.; van Baak, M.A.; Renes, J.; Mariman, E.C. Glucose Restriction Plus Refeeding in Vitro Induce Changes of the Human Adipocyte Secretome with an Impact on Complement Factors and Cathepsins. Int. J. Mol. Sci. 2019, 20, 4055. [Google Scholar] [CrossRef]
- Fruh, S.M. Obesity: Risk factors, complications, and strategies for sustainable long-term weight management. J Am Assoc Nurse Pract 2017, 29, S3–S14. [Google Scholar] [CrossRef]
- Moreno-Navarrete, J.M.; Fernández-Real, J.M. The complement system is dysfunctional in metabolic disease: Evidences in plasma and adipose tissue from obese and insulin resistant subjects. Semin. Cell Dev. Biol. 2019, 85, 164–172. [Google Scholar] [CrossRef]
- Mawle, A.C.; Nisenbaum, R.; Dobbins, J.G.; Gary, H.E.; Stewart, J.A.; Reyes, M.; Steele, L.; Schmid, D.S.; Reeves, W.C. Immune Responses Associated with Chronic Fatigue Syndrome: A Case-Control Study. J. Infect. Dis. 1997, 175, 136–141. [Google Scholar] [CrossRef]
- Valdez, A.R.; Hancock, E.E.; Adebayo, S.; Kiernicki, D.J.; Proskauer, D.; Attewell, J.R.; Bateman, L.; DeMaria, A., Jr.; Lapp, C.W.; Rowe, P.C.; et al. Estimating Prevalence, Demographics, and Costs of ME/CFS Using Large Scale Medical Claims Data and Machine Learning. Front. Pediatr. 2018, 6, 412. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Marchegiani, F.; Matacchione, G.; Giuliani, A.; Ramini, D.; Fazioli, F.; Sabbatinelli, J.; Bonafè, M. Sex/gender-related differences in inflammaging. Mech. Ageing Dev. 2023, 211, 111792. [Google Scholar] [CrossRef] [PubMed]
- Kamitaki, N.; Sekar, A.; Handsaker, R.E.; de Rivera, H.; Tooley, K.; Morris, D.L.; Taylor, K.E.; Whelan, C.W.; Tombleson, P.; et al.; Schizophrenia Working Group of the Psychiatric Genomics Consortium Complement genes contribute sex-biased vulnerability in diverse disorders. Nature 2020, 582, 577–581. [Google Scholar] [CrossRef]
- Sodhi, E.U.; Philpott, H.T.; Carter, M.M.; Birmingham, T.B.; Appleton, C.T. Sex-Differences and Associations Between Complement Activation and Synovial Vascularization in Patients with Late-Stage Knee Osteoarthritis. Front. Immunol. 2022, 13, 890094. [Google Scholar] [CrossRef]
- Rohrhofer, J.; Hauser, L.; Lettenmaier, L.; Lutz, L.; Koidl, L.; Gentile, S.A.; Ret, D.; Stingl, M.; Untersmayr, E. Immunological Patient Stratification in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Clin. Med. 2024, 13, 275. [Google Scholar] [CrossRef]
- Castro-Marrero, J.; Zacares, M.; Almenar-Pérez, E.; Alegre-Martín, J.; Oltra, E. Complement Component C1q as a Potential Diagnostic Tool for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Subtyping. J. Clin. Med. 2021, 10, 4171. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, D.; Wener, M.H.; Pearlman, T.; Kith, P. Markers of inflammation and immune activation in chronic fatigue and chronic fatigue syndrome. 1997, 24, 372–6. [Google Scholar] [PubMed]
- Nacul, L.; de Barros, B.; Kingdon, C.C.; Cliff, J.M.; Clark, T.G.; Mudie, K.; Dockrell, H.M.; Lacerda, E.M. Evidence of Clinical Pathology Abnormalities in People with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) from an Analytic Cross-Sectional Study. Diagnostics 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.; Vlok, M.; Proal, A.; Kell, D.B.; Pretorius, E. Data-independent LC-MS/MS analysis of ME/CFS plasma reveals a dysregulated coagulation system, endothelial dysfunction, downregulation of complement machinery. Cardiovasc. Diabetol. 2024, 23, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Paun, C.C.; Lechanteur, Y.T.E.; Groenewoud, J.M.M.; Altay, L.; Schick, T.; Daha, M.R.; Fauser, S.; Hoyng, C.B.; Hollander, A.I.D.; de Jong, E.K. A Novel Complotype Combination Associates with Age-Related Macular Degeneration and High Complement Activation Levels in vivo. Sci. Rep. 2016, 6, 26568. [Google Scholar] [CrossRef]
- Loirat, C.; Frémeaux-Bacchi, V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis 2011, 6, 60. [Google Scholar]
- Bogdan, R.-G.; Anderco, P.; Ichim, C.; Cimpean, A.-M.; Todor, S.B.; Glaja-Iliescu, M.; Crainiceanu, Z.P.; Popa, M.L. Atypical Hemolytic Uremic Syndrome: A Review of Complement Dysregulation, Genetic Susceptibility and Multiorgan Involvement. J. Clin. Med. 2025, 14, 2527. [Google Scholar] [CrossRef]
- Tortajada, A.; Montes, T.; Martı́nez-Barricarte, R.; Morgan, B.P.; Harris, C.L.; de Córdoba, S.R. The disease-protective complement factor H allotypic variant Ile62 shows increased binding affinity for C3b and enhanced cofactor activity. Hum. Mol. Genet. 2009, 18, 3452–3461. [Google Scholar] [CrossRef]
- Lucientes-Continente, L.; Márquez-Tirado, B.; de Jorge, E.G. The Factor H protein family: The switchers of the complement alternative pathway. Immunol. Rev. 2022, 313, 25–45. [Google Scholar] [CrossRef]
- Chen, S.-F.; Wang, F.-M.; Li, Z.-Y.; Yu, F.; Zhao, M.-H.; Chen, M. Plasma complement factor H is associated with disease activity of patients with ANCA-associated vasculitis. Arthritis Res. Ther. 2015, 17, 1–9. [Google Scholar] [CrossRef]
- Salminen, A.; et al. Genetic Variants Contributing to Circulating Matrix Metalloproteinase 8 Levels and Their Association With Cardiovascular Diseases: A Genome-Wide Analysis. Circ Cardiovasc Genet 2017, 10. [Google Scholar] [CrossRef]
- Cheng, J.; Clayton, J.S.; Acemel, R.D.; Zheng, Y.; Taylor, R.L.; Keleş, S.; Franke, M.; Boackle, S.A.; Harley, J.B.; Quail, E.; et al. Regulatory Architecture of the RCA Gene Cluster Captures an Intragenic TAD Boundary, CTCF-Mediated Chromatin Looping and a Long-Range Intergenic Enhancer. Front. Immunol. 2022, 13, 901747. [Google Scholar] [CrossRef]
- Tate, W.; Walker, M.; Sweetman, E.; Helliwell, A.; Peppercorn, K.; Edgar, C.; Blair, A.; Chatterjee, A. Molecular Mechanisms of Neuroinflammation in ME/CFS and Long COVID to Sustain Disease and Promote Relapses. Front. Neurol. 2022, 13, 877772. [Google Scholar] [CrossRef]
- Komaroff, A.L.; Lipkin, W.I. ME/CFS and Long COVID share similar symptoms and biological abnormalities: road map to the literature. Front. Med. 2023, 10, 1187163. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Hoag, G.E.; Salerno, J.P.; Hornig, M.; Klimas, N.; Selin, L.K. Identification of CD8 T-cell dysfunction associated with symptoms in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and Long COVID and treatment with a nebulized antioxidant/anti-pathogen agent in a retrospective case series. Brain, Behav. Immun. - Heal. 2024, 36, 100720. [Google Scholar] [CrossRef] [PubMed]
- Iu, D.S.; Maya, J.; Vu, L.T.; Fogarty, E.A.; McNairn, A.J.; Ahmed, F.; Franconi, C.J.; Munn, P.R.; Grenier, J.K.; Hanson, M.R.; et al. Transcriptional reprogramming primes CD8+ T cells toward exhaustion in Myalgic encephalomyelitis/chronic fatigue syndrome. Proc. Natl. Acad. Sci. 2024, 121. [Google Scholar] [CrossRef]
- Lage, S.L.; et al. Persistent immune dysregulation and metabolic alterations following SARS-CoV-2 infection. medRxiv 2025. [Google Scholar]
- Mandarano, A.H.; Maya, J.; Giloteaux, L.; Peterson, D.L.; Maynard, M.; Gottschalk, C.G.; Hanson, M.R. Myalgic encephalomyelitis/chronic fatigue syndrome patients exhibit altered T cell metabolism and cytokine associations. J. Clin. Investig. 2020, 130, 1491–1505. [Google Scholar] [CrossRef]
- Cervia-Hasler, C.; Brüningk, S.C.; Hoch, T.; Fan, B.; Muzio, G.; Thompson, R.C.; Ceglarek, L.; Meledin, R.; Westermann, P.; Emmenegger, M.; et al. Persistent complement dysregulation with signs of thromboinflammation in active Long Covid. Science 2024, 383, eadg7942. [Google Scholar] [CrossRef] [PubMed]
- Baillie, K.; Davies, H.E.; Keat, S.B.; Ladell, K.; Miners, K.L.; Jones, S.A.; Mellou, E.; Toonen, E.J.; Price, D.A.; Morgan, B.P.; et al. Complement dysregulation is a prevalent and therapeutically amenable feature of long COVID. Med 2024, 5, 239–253.e5. [Google Scholar] [CrossRef]
- Ahearn-Ford, S.; Lunjani, N.; McSharry, B.; MacSharry, J.; Fanning, L.; Murphy, G.; Everard, C.; Barry, A.; McGreal, A.; al Lawati, S.M.; et al. Long-term disruption of cytokine signalling networks is evident in patients who required hospitalization for SARS-CoV-2 infection. Allergy 2021, 76, 2910–2913. [Google Scholar] [CrossRef]
- Lai, Y.-J.; Liu, S.-H.; Manachevakul, S.; Lee, T.-A.; Kuo, C.-T.; Bello, D. Biomarkers in long COVID-19: A systematic review. Front. Med. 2023, 10, 1085988. [Google Scholar] [CrossRef] [PubMed]
- Hagiya, H.; Tokumasu, K.; Otsuka, Y.; Sunada, N.; Nakano, Y.; Honda, H.; Furukawa, M.; Otsuka, F. Relevance of complement immunity with brain fog in patients with long COVID. J. Infect. Chemother. 2024, 30, 236–241. [Google Scholar] [CrossRef]
- Shih, P.B.; Manzi, S.; Shaw, P.; Kenney, M.; Kao, A.H.; Bontempo, F.; Barmada, M.M.; Kammerer, C.; Kamboh, M.I. Genetic Variation in C-Reactive Protein (CRP) Gene May Be Associated with Risk of Systemic Lupus Erythematosus and CRP Concentrations. J. Rheumatol. 2008, 35, 2171–2178. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wu, H.; Khosravi, M.; Cui, H.; Qian, X.; Kelly, J.A.; Kaufman, K.M.; Langefeld, C.D.; Williams, A.H.; Comeau, M.E.; et al. Association of Genetic Variants in Complement Factor H and Factor H-Related Genes with Systemic Lupus Erythematosus Susceptibility. PLOS Genet. 2011, 7, e1002079. [Google Scholar] [CrossRef]
- Kelley, J.M.; Edberg, J.C.; Kimberly, R.P. Pathways: Strategies for susceptibility genes in SLE. Autoimmun. Rev. 2010, 9, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Berndtson, K. Review of evidence for immune evasion and persistent infection in Lyme disease. Int. J. Gen. Med. 2013, 6, 291–306. [Google Scholar] [CrossRef]
- Hovis, K.M.; Tran, E.; Sundy, C.M.; Buckles, E.; McDowell, J.V.; Marconi, R.T. Selective Binding of Borrelia burgdorferi OspE Paralogs to Factor H and Serum Proteins from Diverse Animals: Possible Expansion of the Role of OspE in Lyme Disease Pathogenesis. Infect. Immun. 2006, 74, 1967–1972. [Google Scholar] [CrossRef]
- Parnell, G.P.; Booth, D.R. The Multiple Sclerosis (MS) Genetic Risk Factors Indicate both Acquired and Innate Immune Cell Subsets Contribute to MS Pathogenesis and Identify Novel Therapeutic Opportunities. Front. Immunol. 2017, 8, 425. [Google Scholar] [CrossRef]
- Ingram, G.; Hakobyan, S.; Robertson, N.P.; Morgan, B.P. Complement in multiple sclerosis: its role in disease and potential as a biomarker. Clin. Exp. Immunol. 2009, 155, 128–139. [Google Scholar] [CrossRef]
- Negro-Demontel, L.; Maleki, A.F.; Reich, D.S.; Kemper, C. The complement system in neurodegenerative and inflammatory diseases of the central nervous system. Front. Neurol. 2024, 15, 1396520. [Google Scholar] [CrossRef]
- Lamers, C.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Compstatins: the dawn of clinical C3-targeted complement inhibition. Trends Pharmacol. Sci. 2022, 43, 629–640. [Google Scholar] [CrossRef]
- Nunes, J.M.; Kell, D.B.; Pretorius, E. Cardiovascular and haematological pathology in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): A role for viruses. Blood Rev. 2023, 60, 101075. [Google Scholar] [CrossRef] [PubMed]
- Reeves, W.C.; Jones, J.F.; Maloney, E.; Heim, C.; Hoaglin, D.C.; Boneva, R.S.; Morrissey, M.; Devlin, R. Prevalence of chronic fatigue syndrome in metropolitan, urban, and rural Georgia. Popul. Heal. Metrics 2007, 5, 5–5. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M.; et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Dhindsa, R.S.; Carss, K.; Harper, A.R.; Nag, A.; Tachmazidou, I.; Vitsios, D.; Deevi, S.V.V.; Mackay, A.; Muthas, D.; et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature 2021, 597, 527–532. [Google Scholar] [CrossRef]
Figure 1.
Demographics and complement protein levels in ME/CFS compared to non-fatigued control subjects (A) Demographic comparisons of sex, age (p=0.96), and BMI (p=0.007) between non-fatigued controls (NF = red) and ME/CFS (blue) subjects (n = 171 total; 50 ME/CFS, 121 NF). Statistical comparisons for age and BMI were performed using a Wilcoxon rank-sum test. (B) Radar plot showing mean scores on Multidimensional Fatigue Inventory (MFI-20) subscales between NF and ME/CFS subjects, where lower MFI-20 scores indicate better health scores.: GF = general fatigue, PF = physical fatigue, RA = reduced activity, RM = reduced motivation, MF = mental fatigue. (C) Radar plot showing mean T-scores on Short Form 36 survey (SF-36) subscales between NF and ME/CFS subjects, where a T-score of 50 refers to the norm of the US general population and lower SF-36 T scores signify a worse functional score: PF = physical functioning, RP = role: physical, BP = bodily pain, V = vitality, GH = general health, RE = role: emotional, SF = social functioning, MH = mental health. (D) Pearson correlation dot plots depicting the extent of correlation of log2-transformed Factor H with (left to right): CRP (p=6.1E-12), C3 (p=9.9E-28), C3a (p=4.3E-9), Factor B (p=1.4E-20), and Factor D (p=6.9E-4) in all subjects (NF = red, ME/CFS = blue). Linear regression lines with 95% confidence intervals are overlaid. (E) Schematic of the complement system and associated components, depicting the three activation pathways (color coded yellow – classical, orange – lectin, brown – alternative, and maroon – C3 and subsequent products). The same color coding is used in other figures to facilitate linkage back to the complement pathway. (F-L) Log2-transformed plasma levels of: (F) CRP (mg/L; plin=0.15, plog=0.14), (G) C3 (mg/ml; plin=0.002, plog=0.002), (H) Bb (mg/ml; plin=0.78, plog=0.77), (I) Bb/C3 (plin=0.038, plog=0.038), (J) Factor B (µg/ml; plin=0.71, plog=0.69), (K) F actor D (µg/ml; plin=0.16, plog=0.14), and (L) Factor H (µg/ml; plin=0.07, plog=0.07) compared between NF and ME/CFS. Boxplots display the five-number summary: minimum, first quartile, median, third quartile, and maximum. The central rectangle spans from the first quartile to the third quartile (the interquartile range (IQR)), a segment inside the rectangle shows the median, the diamond shows the mean, the vertical lines (sometimes referred to as whiskers 1.5xIQR) are extended to the extrema of the distribution in the data set, and the outliers are values outside the whisker range. Statistical comparisons of circulating complement protein levels were performed using a covariate-adjusted (Table 1) linear (plin) and logistic (plog) regression analyses. *p ≤ 0.05, **p ≤ 0.01.
Figure 1.
Demographics and complement protein levels in ME/CFS compared to non-fatigued control subjects (A) Demographic comparisons of sex, age (p=0.96), and BMI (p=0.007) between non-fatigued controls (NF = red) and ME/CFS (blue) subjects (n = 171 total; 50 ME/CFS, 121 NF). Statistical comparisons for age and BMI were performed using a Wilcoxon rank-sum test. (B) Radar plot showing mean scores on Multidimensional Fatigue Inventory (MFI-20) subscales between NF and ME/CFS subjects, where lower MFI-20 scores indicate better health scores.: GF = general fatigue, PF = physical fatigue, RA = reduced activity, RM = reduced motivation, MF = mental fatigue. (C) Radar plot showing mean T-scores on Short Form 36 survey (SF-36) subscales between NF and ME/CFS subjects, where a T-score of 50 refers to the norm of the US general population and lower SF-36 T scores signify a worse functional score: PF = physical functioning, RP = role: physical, BP = bodily pain, V = vitality, GH = general health, RE = role: emotional, SF = social functioning, MH = mental health. (D) Pearson correlation dot plots depicting the extent of correlation of log2-transformed Factor H with (left to right): CRP (p=6.1E-12), C3 (p=9.9E-28), C3a (p=4.3E-9), Factor B (p=1.4E-20), and Factor D (p=6.9E-4) in all subjects (NF = red, ME/CFS = blue). Linear regression lines with 95% confidence intervals are overlaid. (E) Schematic of the complement system and associated components, depicting the three activation pathways (color coded yellow – classical, orange – lectin, brown – alternative, and maroon – C3 and subsequent products). The same color coding is used in other figures to facilitate linkage back to the complement pathway. (F-L) Log2-transformed plasma levels of: (F) CRP (mg/L; plin=0.15, plog=0.14), (G) C3 (mg/ml; plin=0.002, plog=0.002), (H) Bb (mg/ml; plin=0.78, plog=0.77), (I) Bb/C3 (plin=0.038, plog=0.038), (J) Factor B (µg/ml; plin=0.71, plog=0.69), (K) F actor D (µg/ml; plin=0.16, plog=0.14), and (L) Factor H (µg/ml; plin=0.07, plog=0.07) compared between NF and ME/CFS. Boxplots display the five-number summary: minimum, first quartile, median, third quartile, and maximum. The central rectangle spans from the first quartile to the third quartile (the interquartile range (IQR)), a segment inside the rectangle shows the median, the diamond shows the mean, the vertical lines (sometimes referred to as whiskers 1.5xIQR) are extended to the extrema of the distribution in the data set, and the outliers are values outside the whisker range. Statistical comparisons of circulating complement protein levels were performed using a covariate-adjusted (Table 1) linear (plin) and logistic (plog) regression analyses. *p ≤ 0.05, **p ≤ 0.01.
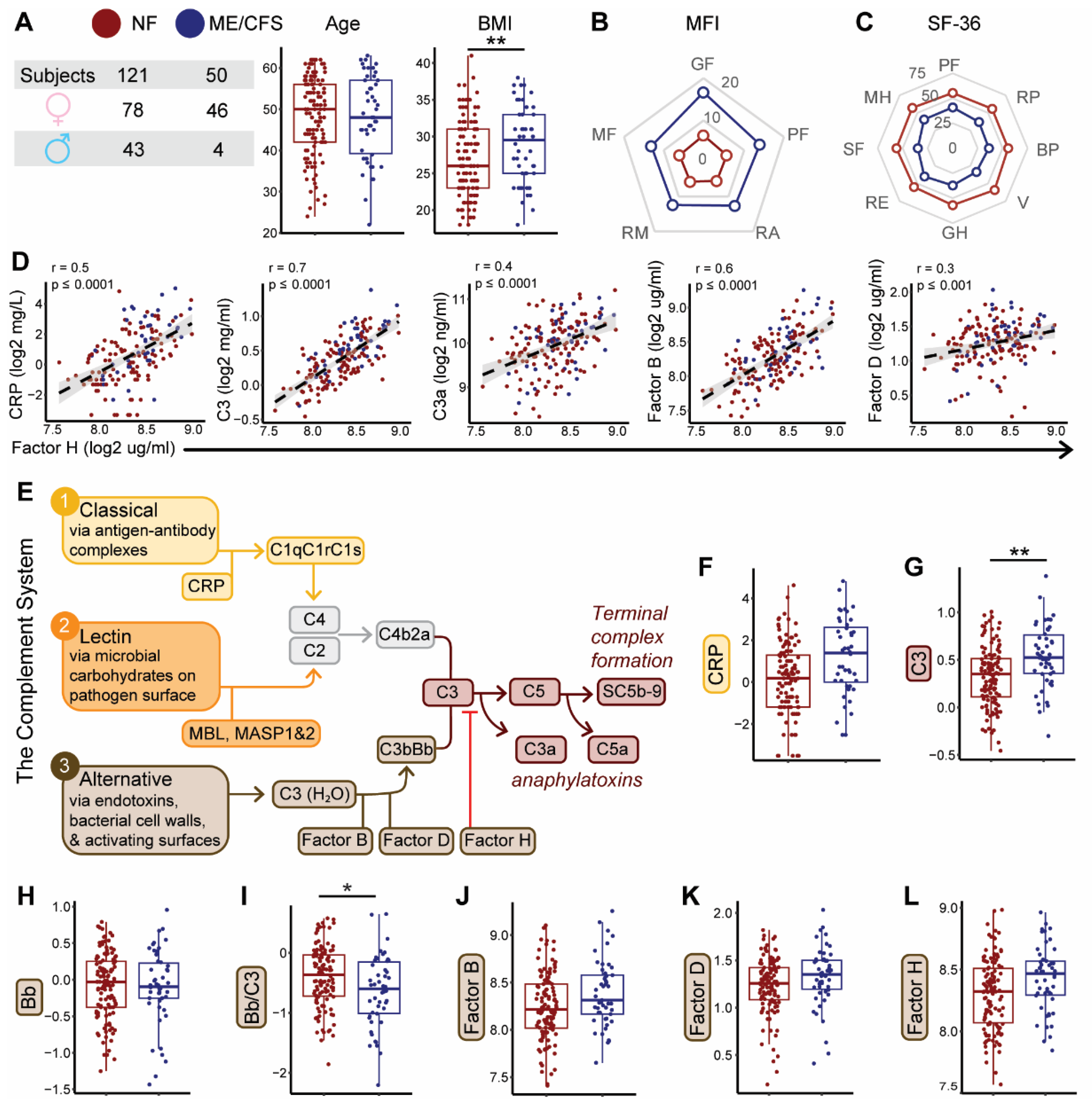
Figure 2.
Association of circulating complement protein levels with participants’ function and symptoms scores. Heatmap depicting covariate-adjusted associations between plasma complement protein levels and scores from three questionnaires: Symptom Inventory (CDC-SI, top), Multidimensional Fatigue Inventory (MFI-20, middle), and Short Form 36 survey (SF-36 T scores, bottom). Lower SF-36 T scores signify a worse functional score, while lower MFI-20 and CDC-SI scores indicate better health scores. Rows represent individual domains or subscales for each instrument, and columns represent complement-related proteins. Color represents the coefficient, or magnitude, of each association, with blue indicating a positive association, while red/maroon indicates a negative association. Statistical comparisons were performed using covariate-adjusted linear regression analysis. *p < 0.05, **p < 0.01, ***p < 0.001. (SI score= CDC Symptom Inventory; MFI: MF = mental fatigue, RM = reduced motivation, RA = reduced activity, PF = physical fatigue, GF = general fatigue; SF-36: MH = mental health, SF = social functioning, RE = role: emotional, GH = general health, V = vitality, BP = bodily pain, RP = role: physical, PF = physical functioning). NOTE: Complement protein labels are color coded to match components of complement system illustrated in Figure 1E.
Figure 2.
Association of circulating complement protein levels with participants’ function and symptoms scores. Heatmap depicting covariate-adjusted associations between plasma complement protein levels and scores from three questionnaires: Symptom Inventory (CDC-SI, top), Multidimensional Fatigue Inventory (MFI-20, middle), and Short Form 36 survey (SF-36 T scores, bottom). Lower SF-36 T scores signify a worse functional score, while lower MFI-20 and CDC-SI scores indicate better health scores. Rows represent individual domains or subscales for each instrument, and columns represent complement-related proteins. Color represents the coefficient, or magnitude, of each association, with blue indicating a positive association, while red/maroon indicates a negative association. Statistical comparisons were performed using covariate-adjusted linear regression analysis. *p < 0.05, **p < 0.01, ***p < 0.001. (SI score= CDC Symptom Inventory; MFI: MF = mental fatigue, RM = reduced motivation, RA = reduced activity, PF = physical fatigue, GF = general fatigue; SF-36: MH = mental health, SF = social functioning, RE = role: emotional, GH = general health, V = vitality, BP = bodily pain, RP = role: physical, PF = physical functioning). NOTE: Complement protein labels are color coded to match components of complement system illustrated in Figure 1E.
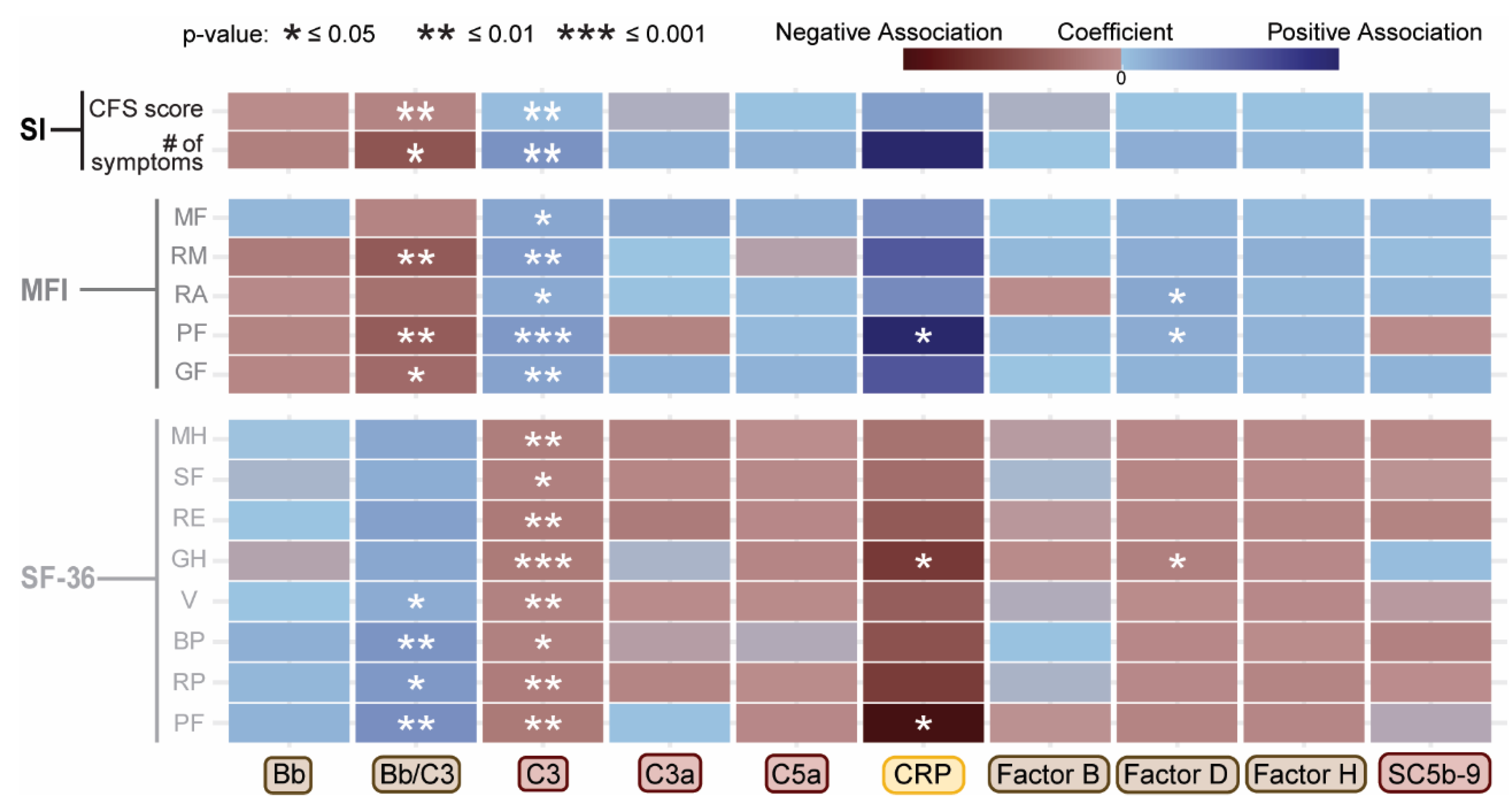
Figure 3.
Functional group distribution of complement protein-associated SNPs/genes and complement protein levels by genotype and illness status. (A) Donut plot summarizing the distribution across 7 functional groups of single-nucleotide polymorphisms (SNPs - dark grey outer ring) and corresponding genes (light grey inner ring) associated with complement proteins (p ≤ 0.01 after covariate-adjustment). Functional group assignments were based on annotated gene functions for gene and associated SNPs (Tables S1-11). (B) Lollipop plot showing functional group enrichment among SNPs and genes significantly associated with C3 plasma levels. Dot size reflects the proportion of significant functional group genes (light grey) and SNPs (dark grey). Enrichment score > 1 (dotted red line) indicates overrepresentation. (C-G) Boxplots of plasma protein levels (log2, mg/ml) by genotype. Dot color indicates disease status (NF = red; ME/CFS = blue); x-axis = percentage of ME/CFS subjects within each genotype group. (C) Bb for rs800292/CFH genotypes (p=0.003); (D) C3 for rs800292/CFH genotypes (p = 0.61); (E) C3 for rs17759529/DPP4 genotypes (p=0.0002); (F) = Bb for rs1061170/CFH genotypes (p=0.025); (G) left: Bb for rs4151667/CFB genotypes (p=0.002), right: Bb for rs9332739/C2 genotypes (p=0.002). (H) Overlay boxplots of Bb (brown, primary y-axis, log2, mg/ml) and C3 (maroon, secondary y-axis, log2, mg/ml) by genotype combinations of rs9332739/C2 and rs1061170/CFH (NF = empty dot, ME/CFS = filled dot). Boxplots represent the median ± 25th and 75th quartiles. Whiskers represent 1.5x the interquartile ranges. Outliers are values outside the whisker range.
Figure 3.
Functional group distribution of complement protein-associated SNPs/genes and complement protein levels by genotype and illness status. (A) Donut plot summarizing the distribution across 7 functional groups of single-nucleotide polymorphisms (SNPs - dark grey outer ring) and corresponding genes (light grey inner ring) associated with complement proteins (p ≤ 0.01 after covariate-adjustment). Functional group assignments were based on annotated gene functions for gene and associated SNPs (Tables S1-11). (B) Lollipop plot showing functional group enrichment among SNPs and genes significantly associated with C3 plasma levels. Dot size reflects the proportion of significant functional group genes (light grey) and SNPs (dark grey). Enrichment score > 1 (dotted red line) indicates overrepresentation. (C-G) Boxplots of plasma protein levels (log2, mg/ml) by genotype. Dot color indicates disease status (NF = red; ME/CFS = blue); x-axis = percentage of ME/CFS subjects within each genotype group. (C) Bb for rs800292/CFH genotypes (p=0.003); (D) C3 for rs800292/CFH genotypes (p = 0.61); (E) C3 for rs17759529/DPP4 genotypes (p=0.0002); (F) = Bb for rs1061170/CFH genotypes (p=0.025); (G) left: Bb for rs4151667/CFB genotypes (p=0.002), right: Bb for rs9332739/C2 genotypes (p=0.002). (H) Overlay boxplots of Bb (brown, primary y-axis, log2, mg/ml) and C3 (maroon, secondary y-axis, log2, mg/ml) by genotype combinations of rs9332739/C2 and rs1061170/CFH (NF = empty dot, ME/CFS = filled dot). Boxplots represent the median ± 25th and 75th quartiles. Whiskers represent 1.5x the interquartile ranges. Outliers are values outside the whisker range.
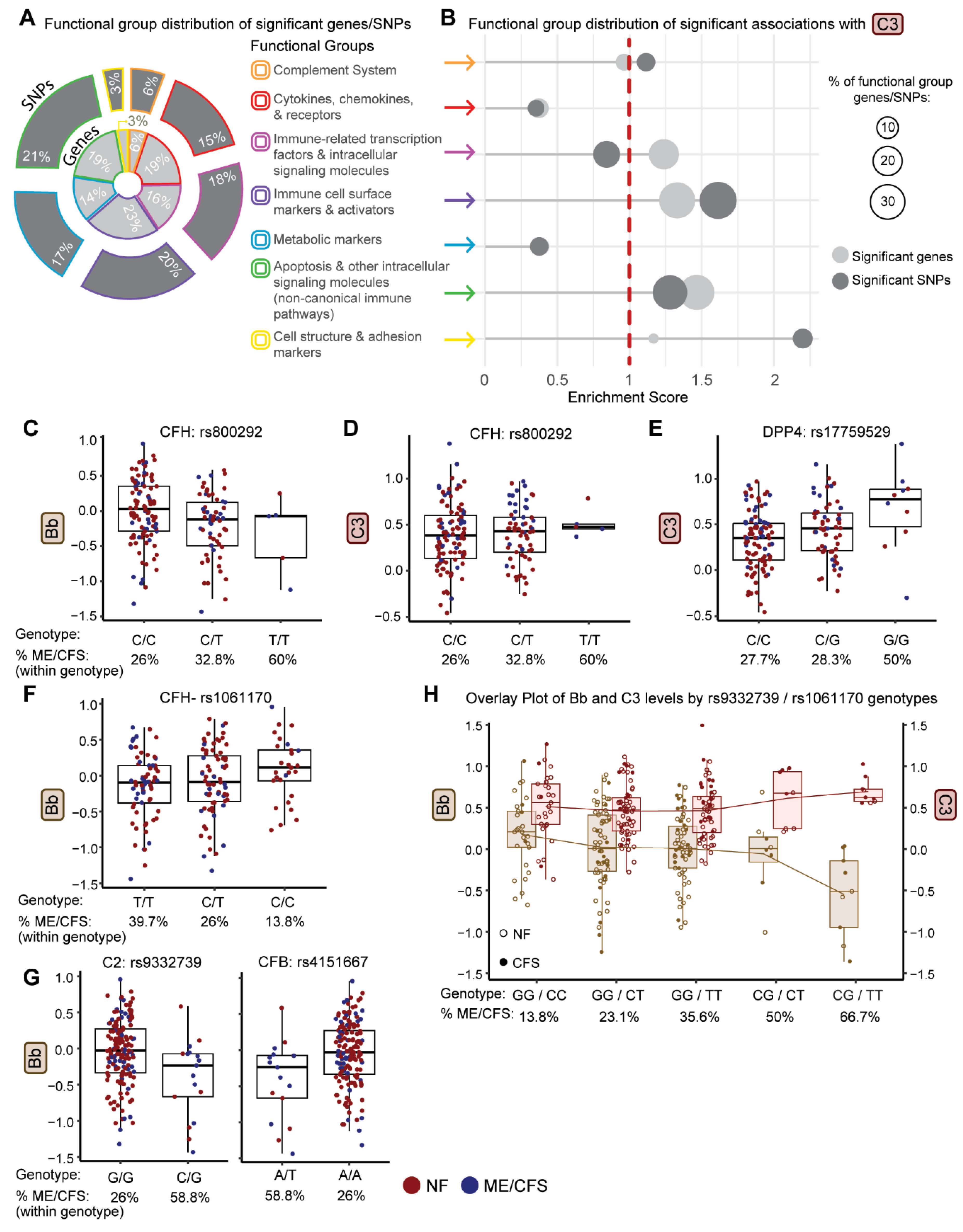
Figure 4.
Stratification of ME/CFS and non-fatigued control subjects using complement protein-associated pQTL genotypes. (A) Subgroup distribution and demographics (age, sex, BMI) based on genotype status of rs9332739 (C2) and rs800292 (CFH). Group A (CFShet, dark blue) = ME/CFS heterozygous at one or both loci; Group B (CFSrem, light blue) = Remainder of ME/CFS subjects; Group C (NFhet, dark red) = NF controls heterozygous at one or both loci; Group D (NFrem, pink) = remainder of NF controls. (B) Complement protein ROC curves for ME/CFS, restricted to heterozygous groups (Group A (CFS het) and Group C (NFhet). (C) Heatmap based on the log2-transformed mean range for each complement protein, low (blue) to high (yellow). Mean values for genotype-defined subgroups (CFShet, CFSrem, NFhet, NFrem) are shown. Asterisks indicate statistical significance, adjusted for covariates (grey = likelihood ratio test with p ≤ 0.05, red = subgroup-specific multinomial logistic regression with reference = NFhet and p ≤ 0.05). (D-I) Boxplots showing distributions for plasma levels of complement protein (log2, mg/ml) by genotype group, CFShet = dark blue, CFSrem = light blue, NFhet = dark red, NFrem = pink. (D) Bb (NFhet vs. NFrem p=0.002), (E) Bb/ C3 (CFShet vs. NFrem p=0.003; NFhet vs. NFrem p=0.03), (F) C3 (CFShet vs. NFhet p=0.005, CFShet vs. NFrem p=0.02), (G) C3a, (H) CRP, and (I) Factor H (CFShet vs. NFhet p=0.05). Boxplots display the five-number summary: minimum, first quartile, median, third quartile, and maximum. The central rectangle spans from the first quartile to the third quartile (the interquartile range (IQR)), a segment inside the rectangle shows the median, the diamond shows the mean, the vertical lines (sometimes referred to as whiskers 1.5xIQR) are extended to the extrema of the distribution in the data set, and the outliers are values outside the whisker range. Statistical comparisons of circulating complement protein levels (black asterisk) were performed using a covariate-adjusted (Table 1) pairwise linear regression analysis. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Figure 4.
Stratification of ME/CFS and non-fatigued control subjects using complement protein-associated pQTL genotypes. (A) Subgroup distribution and demographics (age, sex, BMI) based on genotype status of rs9332739 (C2) and rs800292 (CFH). Group A (CFShet, dark blue) = ME/CFS heterozygous at one or both loci; Group B (CFSrem, light blue) = Remainder of ME/CFS subjects; Group C (NFhet, dark red) = NF controls heterozygous at one or both loci; Group D (NFrem, pink) = remainder of NF controls. (B) Complement protein ROC curves for ME/CFS, restricted to heterozygous groups (Group A (CFS het) and Group C (NFhet). (C) Heatmap based on the log2-transformed mean range for each complement protein, low (blue) to high (yellow). Mean values for genotype-defined subgroups (CFShet, CFSrem, NFhet, NFrem) are shown. Asterisks indicate statistical significance, adjusted for covariates (grey = likelihood ratio test with p ≤ 0.05, red = subgroup-specific multinomial logistic regression with reference = NFhet and p ≤ 0.05). (D-I) Boxplots showing distributions for plasma levels of complement protein (log2, mg/ml) by genotype group, CFShet = dark blue, CFSrem = light blue, NFhet = dark red, NFrem = pink. (D) Bb (NFhet vs. NFrem p=0.002), (E) Bb/ C3 (CFShet vs. NFrem p=0.003; NFhet vs. NFrem p=0.03), (F) C3 (CFShet vs. NFhet p=0.005, CFShet vs. NFrem p=0.02), (G) C3a, (H) CRP, and (I) Factor H (CFShet vs. NFhet p=0.05). Boxplots display the five-number summary: minimum, first quartile, median, third quartile, and maximum. The central rectangle spans from the first quartile to the third quartile (the interquartile range (IQR)), a segment inside the rectangle shows the median, the diamond shows the mean, the vertical lines (sometimes referred to as whiskers 1.5xIQR) are extended to the extrema of the distribution in the data set, and the outliers are values outside the whisker range. Statistical comparisons of circulating complement protein levels (black asterisk) were performed using a covariate-adjusted (Table 1) pairwise linear regression analysis. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.

Figure 5.
Associations between complement protein levels and symptom/function scores for genotype-restricted subgroup (CFShet, NFhet, n = 71). Heatmap of covariate-adjusted associations between complement protein levels and scores: Symptom Inventory (SI, top), Multidimensional Fatigue Inventory (MFI, middle), and Short Form 36 survey (SF-36 T scores, bottom). Rows represent individual domains or subscales for each survey, and columns represent complement-related proteins. Color represents the coefficient, or magnitude, of each association, with blue indicating a positive association, while red indicates a negative association. Statistical comparisons were performed using coviariate-adjusted linear regression analysis. *p < 0.05, **p < 0.01, ***p < 0.001. (SI = CDC Symptom Inventory; MFI: MF = mental fatigue, RM = reduced motivation, RA = reduced activity, PF = physical fatigue, GF = general fatigue; SF-36: MH = mental health, SF = social functioning, RE = role: emotional, GH = general health, V = vitality, BP = bodily pain, RP = role: physical, PF = physical functioning).
Figure 5.
Associations between complement protein levels and symptom/function scores for genotype-restricted subgroup (CFShet, NFhet, n = 71). Heatmap of covariate-adjusted associations between complement protein levels and scores: Symptom Inventory (SI, top), Multidimensional Fatigue Inventory (MFI, middle), and Short Form 36 survey (SF-36 T scores, bottom). Rows represent individual domains or subscales for each survey, and columns represent complement-related proteins. Color represents the coefficient, or magnitude, of each association, with blue indicating a positive association, while red indicates a negative association. Statistical comparisons were performed using coviariate-adjusted linear regression analysis. *p < 0.05, **p < 0.01, ***p < 0.001. (SI = CDC Symptom Inventory; MFI: MF = mental fatigue, RM = reduced motivation, RA = reduced activity, PF = physical fatigue, GF = general fatigue; SF-36: MH = mental health, SF = social functioning, RE = role: emotional, GH = general health, V = vitality, BP = bodily pain, RP = role: physical, PF = physical functioning).
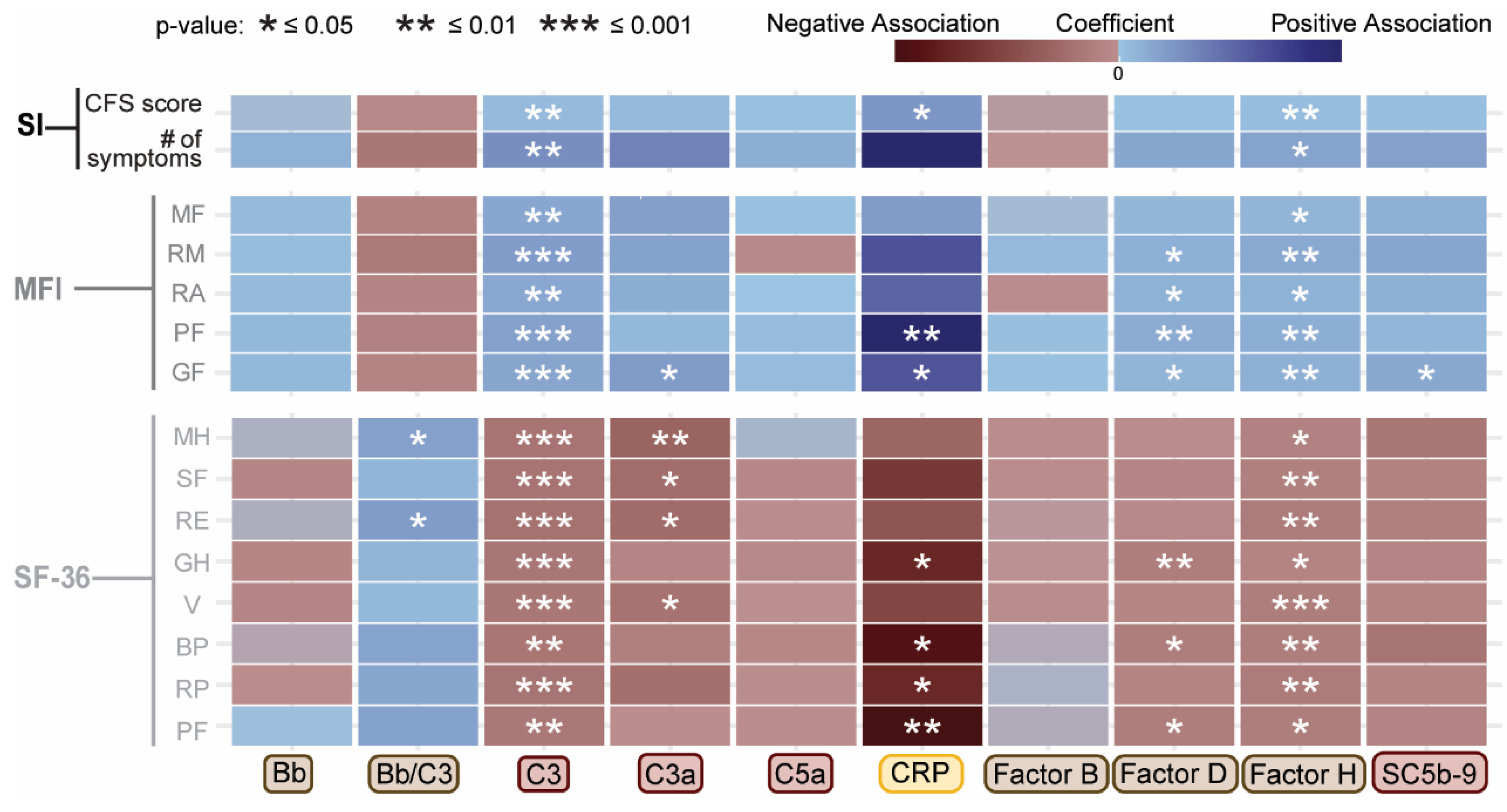

Table 1.
Association of demographic factors with plasma complement proteins and activation products (log2 transformed).
Table 1.
Association of demographic factors with plasma complement proteins and activation products (log2 transformed).
| Plasma component or fragment | Covariate | Coefficient | Std Error | Adjusted R² | F Statistic | p-value |
| CRP | BMI | 0.169 | 0.024 | 0.230 | 47.47 | 1.3E-10 |
| C3 | 0.033 | 0.004 | 0.269 | 63.11 | 2.7E-13 | |
| C3a | 0.036 | 0.008 | 0.102 | 20.28 | 1.2E-05 | |
| C5a | 0.025 | 0.005 | 0.106 | 21.06 | 8.7E-06 | |
| Factor B | 0.026 | 0.005 | 0.142 | 28.32 | 3.3E-07 | |
| Factor D | 0.020 | 0.004 | 0.113 | 22.62 | 4.2E-06 | |
| Factor H | 0.023 | 0.004 | 0.181 | 38.47 | 4.2E-09 | |
| Bb/C3 | -0.027 | 0.008 | 0.066 | 12.92 | 4.3E-04 | |
| CRP | Sex | 0.645 | 0.318 | 0.019 | 4.10 | 4.5E-02 |
| Factor B | 0.159 | 0.061 | 0.034 | 6.84 | 9.8E-03 | |
| Factor D | Age | 0.011 | 0.002 | 0.117 | 23.44 | 2.9E-06 |
*No association of Bb and Sc5b-9 with any of the demographic factors tested in this study.
Table 2.
Genetic variants impacting both disease status and plasma levels of measured complement proteins and activation factors.
Table 2.
Genetic variants impacting both disease status and plasma levels of measured complement proteins and activation factors.
| Pathway | Chromosome #: Gene name | SNP rsID |
ME/CFS risk allele (nt) |
Variant consequence |
Impacted complement protein (cis/trans) | β ± SE | p-value |
| Complement cascade | 6: C2 | rs9332739~ | Minor (C) | Missense (E318D) | Bb/C3 | -0.49 ± 0.13 | 1.9E-04 |
| Bb (cis) | -0.36 ± 0.12 | 0.002 | |||||
| Factor B (cis) | -0.23 ± 0.08 | 0.007 | |||||
| 6: CFB | rs4151667~ | Minor (T) | Missense (L9H) | Bb/C3 | -0.49 ± 0.13 | 1.9E-04 | |
| Bb (cis) | -0.36 ± 0.12 | 0.002 | |||||
| Factor B (cis) | -0.23 ± 0.08 | 0.007 | |||||
| rs641153 | Major (C) | Missense (R32L) | Factor B (cis) | 0.24 ± 0.07 | 0.001 | ||
| Bb/C3 | -0.24 ± 0.10 | 0.021 | |||||
| Factor D (trans) | -0.11 ± 0.05 | 0.046 | |||||
| 1: CFH | rs7529589* | Major (G) | Intronic | Factor H (cis) | 0.06 ± 0.03 | 0.020 | |
| Bb (trans) | 0.10 ± 0.05 | 0.036 | |||||
| rs1061147* | Major (G) | Codon-synonymous (A307A); splicing regulation | Bb (trans) | 0.12 ± 0.05 | 0.015 | ||
| rs800292 | Minor (T) | Missense (V62I) | Bb (trans) | -0.19 ± 0.06 | 0.003 | ||
| Bb/C3 | -0.21 ± 0.07 | 0.003 | |||||
| rs1061170* | Major (T) | Missense (H402Y) | Bb (trans) | 0.11 ± 0.05 | 0.025 | ||
| rs10801555 | Major (G) | Intronic | Bb (trans) | 0.12 ± 0.05 | 0.014 | ||
| 3: MASP1 | rs3774268 | Minor (T) | Missense (S445R) | Bb/C3 | -0.24 ± 0.08 | 0.003 | |
| Bb (trans) | -0.17 ± 0.07 | 0.026 | |||||
| 14: SERPINA5 | rs6115 | Minor (G) | Missense (S64N) | Bb/C3 | -0.11 ± 0.05 | 0.047 | |
| rs6108 | Minor (A) | UTR-3; miRNA binding | C3 (trans) | 0.07 ± 0.03 | 0.026 | ||
| T cell signaling | 2: CD8A | rs3020729 | Major (T) | UTR-3; miRNA binding | CRP (trans) | -0.67 ± 0.25 | 0.008 |
| C3 (trans) | -0.10 ± 0.05 | 0.028 | |||||
| Chemokine | 17: CXCL16 | rs2277680 | Major (G) | Missense (I142T; A200V) | Factor H (trans) | -0.06 ± 0.03 | 0.024 |
| Factor B (trans) | -0.08 ± 0.04 | 0.031 | |||||
| G-protein coupled receptor signaling |
5: PDE4D | rs2014012 | Major (A) | Intronic | C3a (trans) | -0.13 ± 0.06 | 0.036 |
| C5a (trans) | -0.09 ± 0.04 | 0.038 | |||||
| 4: GRK4 | rs1801058 | Major (C) | Missense (V486A); splicing regulation | C3 (trans) | -0.07 ± 0.03 | 0.034 | |
| TNF super family signaling |
13: TNFRSF19 | rs9550987 | Minor (T) | Missense (S31T); splicing regulation | C3 (trans) | 0.09 ± 0.03 | 0.011 |
| Factor H (trans) | 0.07 ± 0.03 | 0.027 |
SNP IDs underlined indicate pQTLs that have not been previously reported (no match in the QTL database: http://www.mulinlab.org/qtlbase).~ and * indicate SNPs that are in LD
Table 3.
Summary of ROC analysis stratified by genotype-defined pQTLs for identifying ME/CFS subgroups with predictive complement protein markers.
Table 3.
Summary of ROC analysis stratified by genotype-defined pQTLs for identifying ME/CFS subgroups with predictive complement protein markers.
| Variant | Gene | Genotype | Test variable |
Covariates | # of CFS | # of NF | # of subjects |
AUC ± SE | 95% CI | p-value |
| Single marker analysis | ||||||||||
| rs9332739 | C2/CFB | CG | Factor H | BMI | 10 | 7 | 17 | 0.89 ± 0.09 | 0.71 - 1 | 0.05 |
| rs800292 | CFH | CT | C3 | BMI | 20 | 41 | 61 | 0.82 ± 0.06 | 0.74 - 0.94 | 0.001 |
| Factor H | 0.78 ± 0.06 | 0.7 - 0.94 | 0.007 | |||||||
| CRP | Sex, BMI | 17 | 40 | 57 | 0.84 ± 0.05 | 0.72 - 1 | 0.04 | |||
| rs1061170 | TT | Factor B | Sex, BMI | 27 | 40 | 67 | 0.81 ± 0.06 | 0.7 - 0.92 | 0.03 | |
| rs10801555 | GG | Factor B | Sex, BMI | 26 | 38 | 64 | 0.81 ± 0.06 | 0.7 - 0.92 | 0.03 | |
| rs395544 | CC | Factor B | Sex, BMI | 20 | 36 | 56 | 0.83 ± 0.05 | 0.73 - 0.94 | 0.03 | |
| rs1065489 | GG | C3 | BMI | 27 | 61 | 88 | 0.75 ± 0.06 | 0.64 - 0.86 | 0.02 | |
| rs7135975 | C1R | AG | C3 | BMI | 16 | 43 | 59 | 0.76 ± 0.08 | 0.6 - 0.92 | 0.01 |
| rs7257062 | C3 | CC | C3 | BMI | 20 | 43 | 63 | 0.78 ± 0.06 | 0.65 - 0.91 | 0.006 |
| rs2241393 | CC | C3 | BMI | 18 | 38 | 56 | 0.81 ± 0.06 | 0.69 - 0.93 | 0.003 | |
| rs2241392 | CC | C3 | BMI | 19 | 43 | 62 | 0.78 ± 0.07 | 0.65 - 0.91 | 0.006 | |
| rs7037673 | C5 | CT | C3 | BMI | 16 | 45 | 61 | 0.82 ± 0.05 | 0.72 - 0.93 | 0.005 |
| Bb/C3 | 0.81 ± 0.06 | 0.69 - 0.93 | 0.006 | |||||||
| rs261753 | C9 | CC | CRP | Sex, BMI | 31 | 76 | 107 | 0.78 ± 0.05 | 0.69 - 0.87 | 0.04 |
| rs1986158 | CR1 | CT | C3 | BMI | 11 | 23 | 34 | 0.83 ± 0.09 | 0.65 - 1 | 0.009 |
| 2-marker analysis | ||||||||||
| rs9332739 & rs800292 | C2/CFH | CG/CT | CRP | Sex, BMI | 23 | 43 | 66 | 0.85 ± 0.05 | 0.76 - 0.94 | 0.046 |
| C3 | BMI | 26 | 45 | 71 | 0.84± 0.05 | 0.74 - 0.94 | 0.0004 | |||
| Factor H | 0.78 ± 0.06 | 0.67 - 0.89 | 0.003 | |||||||
For single marker and 2-marker analyses, test variables with AUC >0.75 and p < 0.05 are listed.
Table 4.
Overlapping SNP associations between circulating complement proteins and UK biobank fatigue-related phenotypes.
Table 4.
Overlapping SNP associations between circulating complement proteins and UK biobank fatigue-related phenotypes.
| UK Biobank fatigue-related phenotype |
Chr #: Gene name |
Variant | Variant consequence (reference nt - variant nt) |
UK biobank: cases / healthy controls |
SNP - UK biobank phenotype odds ratio |
SNP - UK biobank phenotype odds ratio confidence interval |
SNP – UK biobank phenotype p-value |
SNP – Impacted Complement protein p-value |
| Chronic Fatigue Syndrome | 17: PIK3R5 | rs394811 | Synonymous (G-A) | 2047 / 307792 | 0.847 | 0.75 - 0.96 | 0.007 | 0.002 (C3) |
| Post Viral Fatigue Syndrome | 6: CFB | rs641153 | Missense (G-A) | 4363 / 216118 | 0.571 | 0.38 - 0.86 | 0.005 | 0.001 (CFB) |
| 11: MMP10 | rs17293607 | Missense (C-T) | 4360 / 216027 | 0.920 | 0.86 - 0.98 | 0.009 | 0.004 (C5a) | |
| 41202/R53 Fatigue & Malaise |
1: CFH | rs800292 | Missense (G-A) | 2132 / 283735 | 0.754 | 0.61 - 0.94 | 0.010 | 0.003 (Bb) |
| Ever CFS | 6: C2 | rs9332739 | Missense (G-C) | 2547 / 121864 | 1.186 | 1.05 - 1.34 | 0.008 | 0.002 (Bb) |
| 6: CFB | rs4151667 | Missense (T-A) | 2547 / 121863 | 1.185 | 1.05 - 1.34 | 0.009 | 0.002 (Bb) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



