1. Introduction
The evolution of incretin-based pharmacotherapy reflects a two-decade trajectory of innovation in both molecular engineering and dosing convenience. The first-generation glucagon-like peptide-1 receptor agonists (GLP-1 RAs), exemplified by twice-daily exenatide, established proof-of-principle that pharmacologic incretin enhancement could achieve clinically meaningful improvements in glycemic control with modest weight reduction [
1]. Long-acting analogues such as liraglutide (once-daily) and, subsequently, once-weekly formulations including dulaglutide and semaglutide shifted the therapeutic paradigm, offering sustained receptor engagement with simplified regimens [
2,
3,
4]. Beyond glycemic control, cardiovascular outcome trials such as LEADER, SUSTAIN-6 and REWIND demonstrated cardioprotection, consolidating GLP-1–based therapy as a cornerstone of type 2 diabetes (T2D) care and, more recently, as a central strategy in obesity management [
5,
6,
7].
While the glycemic benefits of GLP-1 RAs are driven largely by incretin-mediated enhancement of glucose-dependent insulin secretion, their effects on body weight arise primarily from central modulation of appetite, satiety and energy intake [
8,
9]. This duality in mechanism, together with robust cardiometabolic outcome data, has contributed to unprecedented global uptake of once-weekly GLP-1–based therapies for both T2D and obesity [
10]. However, the rapid expansion in clinical use has also exposed practical limitations, including recent supply constraints, treatment costs and variability in patient expectations regarding the need for long-term pharmacotherapy in obesity [
11,
12,
13]. These challenges have intensified interest in ultra–long-acting formulations capable of maintaining therapeutic potency while reducing injection burden, setting the stage for the development of once-monthly injectable agents targeting GLP-1, glucose-dependent insulinotropic polypeptide (GIP) and related metabolic pathways [
14,
15,
16].
Against this background, a natural next step in metabolic peptide pharmacotherapy has been to extend dosing intervals beyond the once-weekly standard while preserving the metabolic benefits that made these agents transformative. Pharmacokinetic and pharmacodynamic modeling, together with exploratory clinical data, suggest that substantial weight-loss efficacy can be maintained even when dosing is less frequent, if receptor engagement remains above a critical threshold over time [
17,
18]. Earlier attempts such as efpeglenatide, an exendin-based GLP-1 analogue evaluated in once-monthly regimens, demonstrated that monthly administration is technically feasible, although efficacy and tolerability appeared constrained by trough exposures at the end of the dosing interval [
19]. Maridebart cafraglutide (MariTide, Amgen) is a long-acting peptide–antibody conjugate that combines GLP-1 RA with glucose-dependent insulinotropic polypeptide (GIP) receptor antagonism and is being developed for the treatment of obesity and T2D [
14,
20]. In phase 2 trials, once-monthly subcutaneous administration of maridebart cafraglutide has produced clinically meaningful weight loss and improvements in glycemic control in adults with obesity, with and without T2D, with a safety profile broadly consistent with GLP-1–based therapies [
14,
20].
Alongside maridebart, an emerging pipeline of ultra–long-acting therapies—including albumin-binding GLP-1 analogues, dual GLP-1/GIP agonists, long-acting GIP receptor antagonists, and amylin receptor agonists—is now explicitly testing once-monthly regimens as alternatives or complements to weekly standards [
21,
22,
23,
24,
25]. This shift raises practical questions that are not simply incremental: what PK/PD thresholds are required to avoid end-of-interval loss of effect, how titration should be engineered when “turning off” exposure is slow, and which patients or care settings stand to benefit most from extended-interval strategies. In this review, we summarize the pharmacologic design principles, available clinical efficacy and safety signals, and the development landscape of once-monthly injectable therapies for T2D and obesity, highlighting the uncertainties that will determine their role in routine metabolic care.
2. Pharmacologic Principles of Once-Monthly Injectable Therapies
2.1. Rationale for Extending Dosing Intervals
The rapid uptake of once-weekly GLP-1 receptor agonists and dual GLP-1/GIP agonists in T2D and obesity has highlighted a persistent gap between trial efficacy and real-world treatment persistence. Despite robust effects on HbA1c and body weight, observational cohorts consistently report high discontinuation rates within the first year, driven by gastrointestinal adverse events, treatment fatigue, perceived regimen complexity, evolving expectations regarding the duration of pharmacotherapy, and—in many health systems—intermittent drug shortages that disrupt otherwise stable treatment courses [
26,
27,
28]. Recurrent supply constraints and high out-of-pocket costs further challenge long-term use, particularly in obesity care where treatment is often perceived as discretionary rather than essential [
29,
30]. In this context, extended-interval formulations should be considered not only through the lens of individual adherence but also within supply-chain frameworks capable of reliably delivering each scheduled dose.
Reducing injection frequency from weekly to monthly is an intuitive strategy to lower treatment burden while preserving metabolic control. Fewer injections may reduce psychological load, simplify routines, and better align therapy with common chronic-care rhythms (e.g., monthly refills and periodic monitoring) [
31,
32]. From a logistical perspective, extended intervals may also reduce the number of injection devices and needles used per patient-year; the environmental implications of this potential reduction, however, remain insufficiently characterized. Convenience alone is insufficient: less frequent dosing narrows the margin for missed or delayed injections, reduces flexibility for rapid dose adjustment, and amplifies the consequences of any disruption in drug availability. Extending dosing intervals therefore requires pharmacokinetics (PK) and pharmacodynamics (PD) designs that maintain stable receptor engagement across a 4-week cycle, with predictable safety despite prolonged drug persistence [
18,
31,
32,
33,
34,
35] and must be embedded within health systems that can ensure timely and continuous access to therapy.
2.2. Molecular Design Strategies for Ultra–Long-Acting Injectables
Several structural approaches are being used to achieve pharmacokinetic profiles compatible with once-monthly dosing. Fc-fusion and peptide–antibody conjugates, exemplified by maridebart cafraglutide, prolong half-life by engaging neonatal Fc receptor (FcRn) recycling and reducing renal clearance, enabling sustained GLP-1 receptor activation with infrequent injections [
14,
20,
36,
37,
38]. Similar concepts underpin long-acting strategies targeting the GIP receptor (GIPR), including programs designed to sustain GIPR antagonism, an approach supported by human genetic and physiological data linking reduced GIP signaling to lower adiposity [
20,
39,
40,
41,
42,
43].
A second strategy is albumin binding and lipidation. Fatty acid–modified GLP-1 analogues or albumin-binding constructs extend circulation time through high-affinity interaction with plasma proteins and altered tissue distribution, as has been characterized for currently approved long-acting GLP-1 RAs [
38,
44,
45,
46]. Platforms such as Metsera’s MET-097i apply these principles to generate ultra–long-acting GLP-1 RA, while complementary amylin receptor agonists such as MET-233i are being developed with half-lives compatible with less frequent dosing, either as monotherapy or in fixed-ratio combinations (for example MET-097i plus MET-233i) [
22,
23,
47,
48,
49]. Collectively, Fc-fusion, albumin binding and receptor-selective antibodies provide a modular toolkit for constructing ultra–long-acting metabolic agents tailored to specific mechanistic targets and dosing intervals.
2.3. PK/PD Requirements for Once-Monthly Regimens
Effective once-monthly therapy depends on tight alignment between PK and PD. Agents must achieve a half-life and minimum concentration (C_
min) sufficient to sustain receptor engagement throughout a 4-week interval, avoiding troughs that erode effects on appetite, body weight and glycemic control. Data from existing GLP-1 RA and dual GLP-1/GIP agonists indicate that cumulative exposure and stable signaling, rather than high transient peaks, best predict HbA1c and weight responses [
50,
51,
52]. Ultra–long-acting constructs therefore aim to minimize peak–trough fluctuation while maintaining pharmacodynamic activity across the entire dosing cycle [
18,
38].
Transitioning from weekly to monthly regimens raises practical issues of dose selection, titration and accumulation. Longer half-lives slow the approach to steady state and increase the risk of overexposure if doses are escalated too aggressively, yet underdosing may lead to a perceptible loss of benefit before the next injection [
34,
53,
54]. Thus, PK/PD requirements for monthly dosing extend beyond simply lengthening half-life; they demand dosing schemes that balance durability of action, tolerability and operational flexibility in routine care.
2.4. Safety Considerations for Monthly Dosing
Ultra–long-acting injectables introduce safety nuances that are less pronounced with shorter-acting formulations. Gastrointestinal adverse events remain the dominant side effects of GLP-1–based therapies and are closely related to dose and speed of titration [
33,
55,
56]. With once-monthly dosing, an excessive initial dose or rapid uptitration may result in more prolonged or severe symptoms because drug clearance is slow and the ability to rapidly “switch off” effective exposure is limited. Careful titration and proactive counseling are therefore critical [
34,
57].
Prolonged systemic exposure also raises theoretical concerns about accumulation-related toxicity, off-target effects and immunogenicity, particularly at doses used for obesity [
58,
59,
60,
61]. Managing adverse events is more challenging when pharmacologic effects persist for weeks, especially in older adults or those with significant comorbidities. Finally, once-monthly regimens will often be combined with other antidiabetic or anti-obesity drugs, potentially modifying hypoglycemia risk (for example when used with insulin) and compounding gastrointestinal or gallbladder events [
5,
62]. Robust pharmacovigilance and real-world data will be essential to define the safety profile of these agents and to guide their integration into complex treatment regimens.
Table 1.
Pharmacologic and molecular design features of established weekly incretin therapies and emerging ultra–long-acting injectables enabling once-monthly dosing in T2D and obesity.
Table 1.
Pharmacologic and molecular design features of established weekly incretin therapies and emerging ultra–long-acting injectables enabling once-monthly dosing in T2D and obesity.
| Asset |
Primary target |
Secondary target |
Reported half-life* |
Dominant half-life extension strategy |
References |
| Native GLP-1 |
GLP-1R |
— |
~1.5–2 min |
Rapid DPP-4 degradation and renal clearance (no protection) |
[8] |
| Native GIP |
GIPR |
— |
~5–7 min |
Rapid DPP-4 degradation and renal clearance (no protection) |
[9] |
| Liraglutide |
GLP-1R |
— |
~13 h (~0.5 d) |
Fatty-acylation → high albumin binding, reduced clearance |
[45] |
| AT-7687 |
GIPR
antagonism |
— |
~27.4 h (~1.1 d) |
Long-acting peptide engineering (program-specific) |
[25,97] |
| Tirzepatide |
GIPR |
GLP-1R |
~120 h (~5 d) |
Albumin binding via fatty-acid moiety |
[52,56,61] |
| Semaglutide |
GLP-1R |
— |
~168 h (~7 d) |
Albumin binding via fatty-acid moiety |
[50,55,60] |
| VK-2735 |
GLP-1R |
GIPR |
~170–250 h (~7–10 d) |
Long-acting peptide scaffold (program-specific) |
[21,80,81,82,83,84,85,86] |
| Zovaglutide |
GLP-1R |
— |
~260–273 h (~11 d) |
Extended half-life GLP-1 analogue (likely acylation/albumin-binding based) |
[76,77,78] |
| MariTide (maridebart cafraglutide) |
GLP-1R |
GIPR
antagonism |
~343–396 h (~14–16 d) |
Peptide–antibody conjugate with FcRn recycling; markedly reduced clearance |
[14,20] |
| MET-097i |
GLP-1R |
— |
~15–16 d |
NuSH™ HALO proprietary ultra–long-acting engineering (controlled release + reduced clearance) |
[47,48,49,69,70,71,72] |
| ASC36 |
Amylin receptor |
— |
~15 d |
ULAP depot exposure + high intrinsic potency (AISBDD/POTENT) |
[92,93] |
| MET-233i |
Amylin receptor |
— |
~19 d |
NuSH™ HALO proprietary ultra–long-acting engineering |
[22,73,74,75] |
| ASC35 |
GLP-1R |
GIPR |
t½ ≥30 d |
ULAP ultra–long-acting peptide engineering (with AISBDD-guided potency optimization; POTENT) |
[91,92] |
| ASC47-103 |
THRβ |
— |
~26–40 d |
ULAP long-acting depot exposure (with AISBDD-driven adipose-selective THRβ agonism; POTENT) |
[94,95,96] |
| ASC30 |
GLP-1R |
— |
~36 d |
AISBDD-derived potent small-molecule GLP-1RA formulated via ULAP subcutaneous depot |
[87,88,89] |
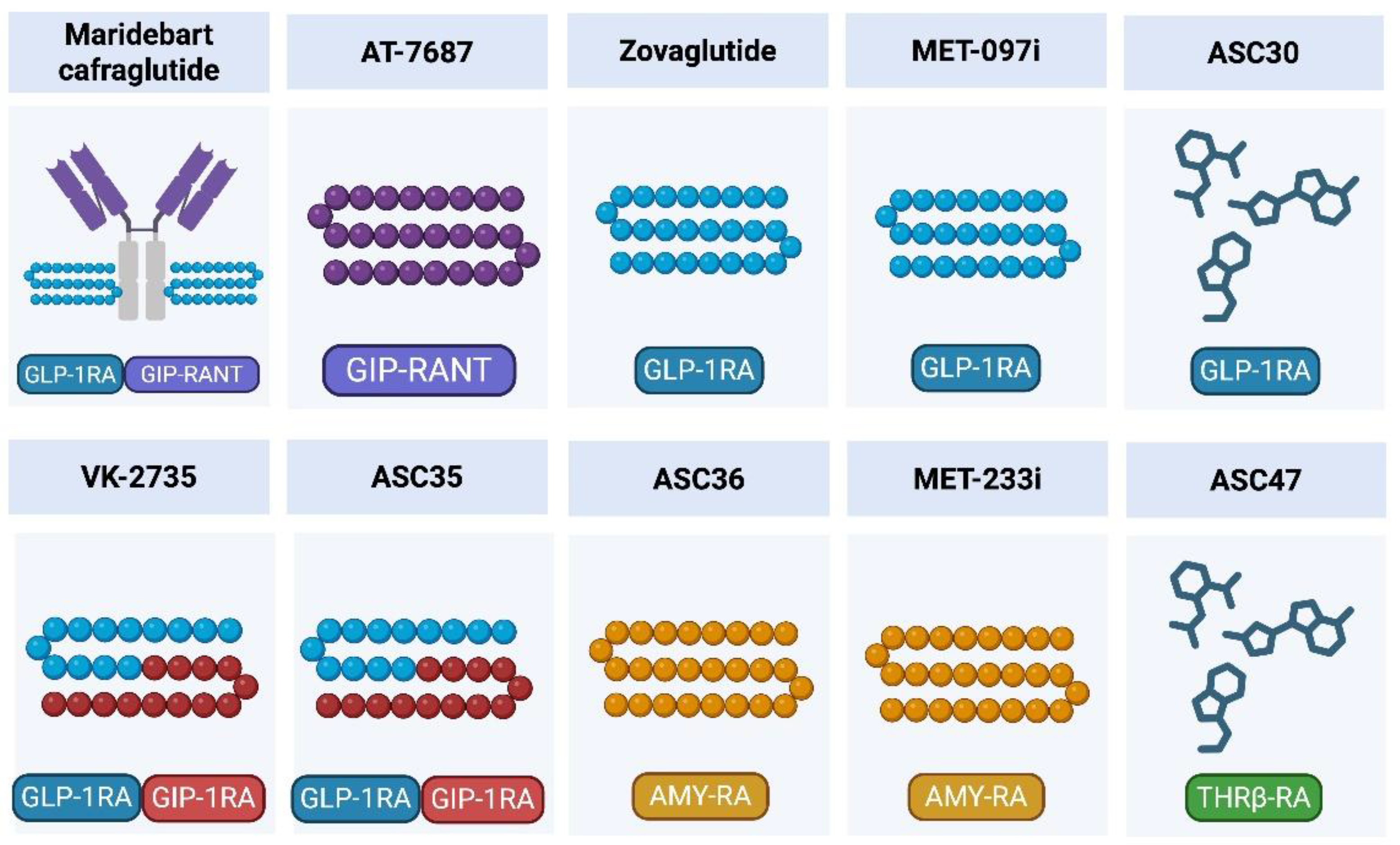
3. Maridebart Cafraglutide (MariTide): Lead Once-Monthly GLP-1 RA Plus GIPR Antagonist
3.1. Mechanistic Rationale: GLP-1 RA Plus GIPR Antagonism
Maridebart cafraglutide (MariTide or AMG133; Amgen) is a long-acting conjugate that combines GLP-1 RA activity with GIPR antagonism in a single molecule. The construct consists of a fully human IgG1 monoclonal antibody directed against GIPR, covalently linked to two GLP-1 analog peptides [
20]. This design leverages high-affinity GLP-1R agonism to drive weight loss and glycemic improvement, while sustained GIPR blockade is expected to counteract GIP-mediated adipogenesis and postprandial lipid storage [
63]. The rationale for targeting GIPR antagonism is supported by human genetic and experimental data [
43]. Loss-of-function variants in GIPR have been associated with lower BMI and improved adiposity profiles, and preclinical studies with GIPR antagonists show reductions in body weight and fat mass [
42]. In this context, MariTide provides a “dual-lever” approach distinct from GLP-1/GIP co-agonists: rather than potentiating both incretin pathways, it amplifies GLP-1 signaling while chronically suppressing GIP signaling. Whether this strategy yields advantages over dual agonism remains an open question, but phase 1–2 data demonstrate that GLP-1 agonism plus GIPR antagonism is sufficient to produce large, sustained reductions in body weight and improvements in glycemic control [
14,
20].
3.2. Preclinical and Early Clinical Development
In preclinical models, maridebart cafraglutide showed robust anti-obesity and metabolic effects. In diet-induced obese mice and cynomolgus monkeys, repeated administration produced marked and sustained reductions in body weight, accompanied by improvements in glycemia and lipid parameters without evidence of off-target toxicity [
14,
20,
64].
In vitro, the molecule behaved as intended, demonstrating potent GLP-1R agonism and GIPR antagonism in cell-based functional assays [
20]. A first-in-human, randomized, double-blind, placebo-controlled phase 1 study in adults with obesity evaluated single and multiple ascending subcutaneous doses of maridebart cafraglutide, with the multiple-dose cohorts receiving injections every 4 weeks [
20]. In the once-monthly multiple-ascending-dose cohorts, maridebart cafraglutide produced dose-dependent weight loss, with mean reductions of about 7% at 140 mg and up to approximately 14–15% at the highest 420-mg regimen after 12 weeks of treatment [
20].
3.3. Phase 2 Trials in Obesity Without T2D
At week 52, mean percentage body-weight reductions in the non-diabetes cohort were approximately 12–16% across the monthly regimens, compared with ~2–3% with placebo [
14]. A high proportion of participants achieved ≥10% and ≥15% weight loss, and weight trajectories generally continued downward through week 52, suggesting that a clear plateau had not yet been reached [
14]. In contrast, the every-8-weeks (q8w) arm showed attenuated efficacy, with smaller absolute and relative weight reductions than the q4w regimens [
14]. This pattern supports the concept that once monthly represents the lower practical limit for dosing frequency if near-maximal efficacy is to be maintained in this pharmacologic class. Beyond weight, maridebart cafraglutide improved multiple cardiometabolic risk markers in participants without diabetes, including modest reductions in blood pressure, triglycerides and high-sensitivity C-reactive protein, consistent with the magnitude of weight loss [
14].
3.4. Phase 2 Trials in T2D with Obesity
A parallel cohort of adults with T2D and obesity or overweight was included in the same phase 2 programme [
14]. Participants (baseline BMI ≈ 36 kg/m², established T2D on stable background therapy) were randomized to once-monthly maridebart cafraglutide at 140, 280 or 420 mg, or placebo, for 52 weeks [
14]. As typically observed with incretin-based therapies, absolute weight loss was somewhat smaller in the diabetes cohort but remained clinically significant. At week 52, mean weight reductions with maridebart cafraglutide ranged from approximately 8–12%, compared with ~2% in the placebo group [
14]. Importantly, these weight effects were accompanied by robust improvements in glycemic control. Across doses, HbA1c decreased by roughly 1.2–1.6 percentage points versus minimal change or slight increases with placebo [
14]. A substantial proportion of participants achieved guideline HbA1c targets despite background therapy [
14]. Together, these data indicate that once-monthly maridebart cafraglutide can deliver dual efficacy in weight and glycemia in people living with T2D and obesity, with effect sizes comparable to those seen with potent weekly GLP-1–based regimens in similar populations [
2,
3,
4,
8,
9,
10,
14].
3.5. Safety and Tolerability Profile
Across phase 1–2, the safety profile of maridebart cafraglutide has been broadly consistent with that of GLP-1 RAs. The most frequent adverse events were GI symptoms—principally nausea, vomiting and constipation—which were generally mild to moderate, occurred predominantly during the early treatment period and tended to resolve spontaneously [
14]. Incidence and severity of GI events were higher with rapid initiation of high doses and mitigated by gradual dose escalation, supporting the use of titration schemes in later-phase trials [
14]. Discontinuation due to GI adverse events occurred in a minority of patients, particularly in titration arms [
14]. No new safety signals specific to GIPR antagonism or the antibody–peptide format have been identified to date. In phase 2, there was no excess of serious cardiovascular events, and maridebart cafraglutide was associated with favourable changes in blood pressure and inflammatory markers [
14]. No adjudicated cases of acute pancreatitis were observed in the phase 2 programme, and no imbalance in pancreatic events was detected relative to placebo. Likewise, no consistent signal for gallbladder disease has emerged to date; however, gallstone formation remains a theoretical consideration with any therapy that induces rapid and sustained weight loss [
14]. Hypoglycaemia rates were low and comparable to placebo when maridebart cafraglutide was used without insulin or sulfonylureas, reflecting the glucose-dependent nature of GLP-1–mediated insulin secretion [
14]. Immunogenicity appears limited so far: development of anti-drug antibodies has been infrequent and without clear impact on PK or efficacy in the timeframe studied [
14,
20]. Overall, maridebart cafraglutide currently appears to have a manageable tolerability profile, dominated by class-typical GI effects, with no overt safety penalty relative to weekly GLP-1 RAs [
14,
20].
3.6. Key Lessons from Maridebart Cafraglutide for Monthly Therapies
Finally, maridebart cafraglutide illustrates practical considerations that will likely apply to other monthly agents: the need for careful titration to balance efficacy with GI tolerability; reduced flexibility to rapidly down-titrate in the event of adverse effects due to prolonged drug persistence; and the importance of understanding how monthly regimens will interface with existing weekly incretin therapies in terms of switching, sequencing or combination use [
14,
15,
16,
20]. Ongoing phase 3 outcome trials will be crucial to clarify durability of weight loss, effects on cardiovascular and heart-failure endpoints, and the real-world role of once-monthly incretin-based therapies within the broader landscape of obesity and T2D management. Building on the phase 2 dataset, Amgen initiated the MARITIME phase 3 program across obesity and T2D, including MARITIME-1 (overweight/obesity), MARITIME-2 (T2D), MARITIME-CV (cardiovascular disease), and MARITIME-HF (heart failure), with additional planned studies in kidney disease and obstructive sleep apnea.
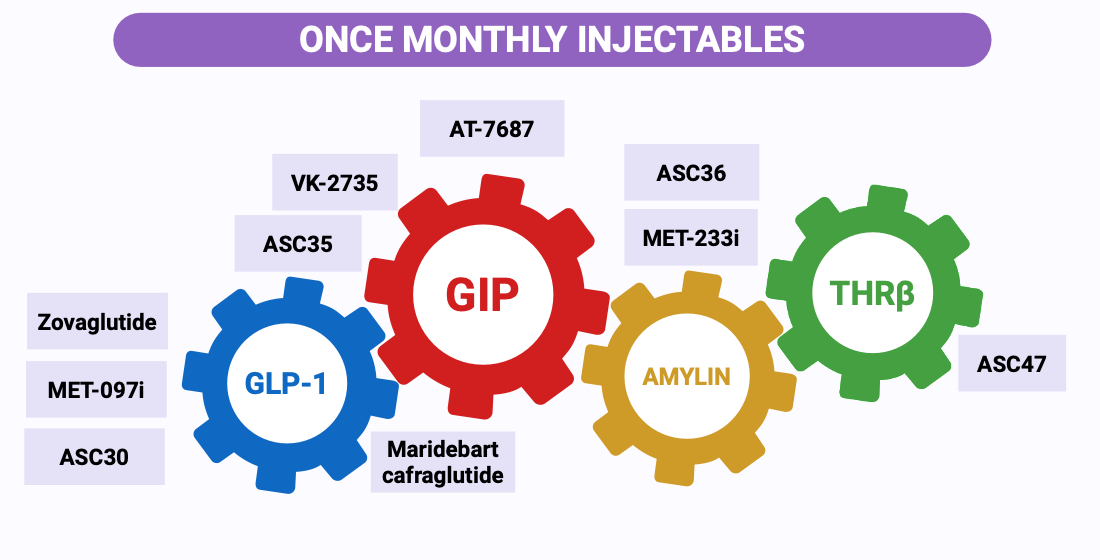
4. Beyond Maridebart: Emerging Once-Monthly Pipelines
The development of maridebart cafraglutide has redefined the therapeutic boundaries of long-acting metabolic pharmacotherapy and catalyzed a new generation of extended-interval agents. Although maridebart remains the only once-monthly incretin-based therapy supported by peer-reviewed phase 2 data, several emerging programs seek to replicate or surpass its efficacy, PK stability, and durability of effect [
14]. As summarized in
Figure 1, the once-monthly pipeline now spans ultra–long-acting GLP-1 RAs, dual GLP-1/GIP agonists, amylin receptor agonists, long-acting GIPR antagonists, and selected non-incretin metabolic targets, including thyroid hormone receptor beta agonist strategies (THRβ-RAs). However, the evidentiary base is uneven: most available data for these agents come from sponsor-released summaries, conference presentations, or early-phase trials, and formal peer-reviewed publications remain scarce. The following narrative synthesizes the mechanistic rationale, developmental progress, and translational potential of these programs, while emphasizing the provisional nature of the available evidence.
4.1. MET-097i: Ultra–Long-Acting GLP-1 RA (NuSH Platform)
Metsera, a clinical-stage biotech that Pfizer has agreed to acquire as part of its obesity strategy [
65], is developing an ultra–long-acting “nutrient-stimulated hormone” (NuSH) platform centered on two injectable peptides engineered for extended dosing intervals: the fully biased GLP-1 RA MET-097i and the ultra–long-acting amylin analogue MET-233i [
66]. Both candidates are designed as next-generation NuSH analogues with closely aligned PK profiles intended to support once-monthly dosing and, ultimately, fixed-ratio co-formulation. MET-097i uses Metsera’s HALO™ peptide lipidation technology to enhance albumin binding and prolong systemic exposure, whereas MET-233i incorporates dual fatty-acid modifications to extend its half-life; together, they are being developed to provide complementary control of appetite, gastric emptying, and postprandial metabolism in people with overweight or obesity, with or without T2D [
22,
23,
66,
67,
68].
MET-097i is a fully biased, subcutaneously administered GLP-1 RA with a reported human half-life of approximately 380 hours (~16 days), providing a strong PK rationale for exploring once-monthly maintenance regimens [
66,
68]. To date, however, all efficacy data have been generated under once-weekly schedules. In an early phase 1/2 study in adults with overweight or obesity without diabetes, five once-weekly injections of MET-097i (up to 1.2 mg) given without dose titration produced dose-dependent weight loss, with mean reductions of approximately 7–8% by day 36 and about 8.1% by day 57—four weeks after the last injection—indicating sustained pharmacodynamic activity beyond the formal dosing period [
66,
68,
69]. This continued weight loss after treatment cessation is a key argument supporting the feasibility of extended-interval dosing. Building on these findings, the randomized, placebo-controlled VESPER-1 phase 2b trial evaluated 28 weeks of once-weekly MET-097i (0.4–1.2 mg) in adults with overweight or obesity without diabetes, reporting placebo-subtracted mean weight loss of up to 14.1% at the highest dose, individual reductions as high as 26.5%, a total study discontinuation rate of 2.9%, and a gastrointestinal tolerability profile that appears manageable in the context of GLP-1–based therapies [
47,
66,
68,
70]. In parallel, the VESPER-3 program and a dedicated phase 2b study (NCT06973720) are evaluating multiple once-monthly MET-097i regimens, generally following an initial weekly induction phase, with the aim of characterizing the durability, safety, and tolerability of true monthly maintenance dosing [
47,
71].
At present, these phase 2 results are available mainly through sponsor materials and conference disclosures, and detailed peer-reviewed reports on glycemic outcomes, body-composition changes, and long-term durability remain limited. Consequently, positioning MET-097i as a once-monthly therapy is currently supported primarily by its PK profile, the observation of continued weight loss after weekly dosing is stopped, and the design of ongoing trials, rather than by fully adjudicated clinical data.
4.2. MET-233i: Ultra–Long-Acting Amylin Analogue
MET-233i is Metsera’s ultra–long-acting, subcutaneously administered amylin analogue, developed within its next-generation nutrient-stimulated hormone (NuSH) portfolio and specifically engineered for potential once-monthly dosing [
22,
48,
66]. Amylin receptor agonism complements GLP-1 signalling by reducing appetite, slowing gastric emptying and enhancing postprandial satiety, providing a mechanistically distinct but synergistic axis within NuSH-based weight-loss strategies [
22,
67]. MET-233i is the lead next-generation NuSH peptide designed for combination and co-formulation with the fully biased GLP-1 RA MET-097i; both molecules were engineered using Metsera’s HALO™ peptide stabilisation and lipidation platform, yielding matched solubility profiles and extended half-lives compatible with monthly regimens [
48,
66]. In a randomized, double-blind, placebo-controlled phase 1 trial in 80 adults with overweight or obesity without T2D, MET-233i was evaluated as single ascending doses (0.15–2.4 mg) and multiple ascending doses (0.15–1.2 mg) administered once weekly over five weeks, without titration [
48,
66,
72].
MET-233i showed dose-linear PK with an observed half-life of approximately 19 days—the most durable profile reported to date for an amylin analogue—which, together with an exposure pattern that closely matched MET-097i after multiple doses, supports the feasibility of once-monthly dosing and fixed-ratio co-formulation. In the multiple-dose cohorts, five weekly injections of 1.2 mg produced placebo-subtracted mean weight loss of up to 8.4% at day 36, with individual reductions approaching 10% [
48,
66,
72]. In the single-dose cohorts, a single injection produced clinically meaningful weight loss that was maintained for more than four weeks, consistent with the ultra-long PK profile [
48,
66,
72].
Tolerability in phase 1 was favourable: GI adverse events in the multiple-dose portion were mild, dose-dependent and largely confined to the first week of treatment, despite roughly three-fold accumulation of exposure over five weeks, and no severe or serious adverse events or concerning safety signals were reported [
48,
66,
72]. Anticipated starting doses of 0.15–0.3 mg showed placebo-like tolerability. This profile, aligned with the historically lower GI burden reported with amylin analogues relative to GLP-1 RAs, supports the use of MET-233i as a tolerable backbone for chronic, high-exposure regimens and combination therapy. MET-233i is now being investigated in a 12-week monotherapy study employing weekly titration followed by an exposure-matched once-monthly dose at week 13, designed to characterize the maintenance potential, safety and tolerability of true monthly administration, and to inform dose selection for later-phase development [
48,
66,
72].
4.3. MET-233i + MET-097i: First-in-Class Monthly Multi-NuSH Combination
A central element of Metsera’s development strategy is the co-formulation of the ultra–long-acting amylin analogue MET-233i with the fully biased GLP-1 RA MET-097i as a potential once-monthly multi–nutrient-stimulated hormone (NuSH) injectable therapy for chronic weight management. Both peptides were deliberately engineered using Metsera’s proprietary HALO™ lipidation platform to achieve compatible solubility characteristics and closely aligned PK profiles, with observed terminal half-lives of approximately 19 days for MET-233i and ~380 hours (~16 days) for MET-097i [
48,
66]. This alignment is intended to enable coordinated exposure over a multi-week dosing interval and supports their development as a co-formulated injectable combination [
48,
66].
The mechanistic rationale for this dual-NuSH approach lies in the complementary metabolic actions of GLP-1 and amylin signaling. GLP-1 RA activity provides potent incretin-mediated effects on glycemic control and central appetite suppression, while amylin agonism reinforces satiety, slows gastric emptying and modulates postprandial nutrient handling. In combination, these mechanisms may offer additive or synergistic effects on body-weight reduction and metabolic regulation, with the potential for dose-sparing and possibly improved GI tolerability relative to escalation of a single pathway [
48,
66]. Evidence supporting the combination remains early but has been substantiated by multiple sponsor-reported sources [
72]. Phase 1 data indicate that MET-233i achieves placebo-adjusted mean weight loss of up to 8.4% at day 36 following five weekly doses, with individual responses approaching 10%, and demonstrates an observed half-life compatible with extended-interval dosing [
72]. Importantly, sponsor-reported PK analyses suggest that exposure profiles of MET-233i after multiple doses closely match those of MET-097i, supporting practical co-formulation [
72]. GI adverse events observed to date have been predominantly mild, dose-dependent and largely confined to the initial dosing period, with no severe or serious adverse events reported in early studies [
72].
The co-administration strategy is currently being evaluated in a randomized, double-blind, placebo-controlled phase 1 trial (NCT06924320) enrolling adults with overweight or obesity, including a cohort with T2D [
73]. This study incorporates single- and multiple-ascending dose cohorts with extended safety follow-up and is designed to characterize safety, tolerability, PK, PD and preliminary weight-loss effects of MET-233i administered together with MET-097i [
73]. In parallel, Metsera has disclosed plans to explore regimens that transition from an induction phase to exposure-matched monthly maintenance dosing, aiming to balance rapid onset of weight loss with long-term convenience and scalability. From a regulatory perspective, the sponsor has indicated an intent to advance the MET-233i/MET-097i combination through the U.S. FDA biologics pathway, targeting eventual submission under a biologics license application framework. While the pharmacological rationale and early clinical signals are encouraging, positioning this combination as a leading monthly therapy remains provisional [
48,
66]. Robust phase 2 and phase 3 trials will be required to define incremental efficacy over single-mechanism NuSH agents, long-term safety, and effects on body-composition endpoints, metabolic risk factors and treatment persistence relative to established weekly GLP-1–based regimens.
4.4. Zovaglutide (ZT002): A Once-Monthly Long-Acting GLP-1 RA
Zovaglutide (ZT002; QL Biopharmaceutical) is an ultra–long-acting GLP-1 RA being developed for true once-monthly subcutaneous administration. The molecule incorporates a dual fatty-acid chain modification strategy that markedly enhances albumin binding, thereby extending systemic exposure and enabling prolonged PK durability. This structural design differentiates zovaglutide from earlier acylated GLP-1 analogues and supports a terminal half-life in the range of approximately 260–273 h, consistent with its intended four-week dosing interval [
74]. The clinical development of zovaglutide is led by Beijing QL Biopharmaceutical Co., Ltd., reportedly founded by former Novo Nordisk scientists, with integrated capabilities spanning peptide discovery, recombinant E. coli–based manufacturing, and GMP-scale production [
74]. Zovaglutide is described as a selective GLP-1 RA with no reported activity on GIP or glucagon receptors, exerting its metabolic effects through established central and peripheral GLP-1 pathways that promote satiety, reduce caloric intake, and enhance glucose-dependent insulin secretion [
74].
Phase 2 efficacy data were presented at the European Association for the Study of Diabetes (EASD) Annual Meeting in 2025 from a randomized, double-blind, placebo-controlled, multicenter trial enrolling 303 adults with overweight or obesity [
75]. Participants received zovaglutide at doses of 80 mg or 160 mg administered either once monthly (Q4W) or every two weeks (Q2W) for 24 weeks. Mean placebo-subtracted body-weight reductions ranged from 10.6% to 14.4% at week 24, with the 160 mg Q4W regimen achieving a mean reduction of 13.8%, closely comparable to the 160 mg Q2W regimen (14.4%) [
75]. No plateau in weight loss was observed at the end of the treatment period, suggesting ongoing efficacy beyond six months [
75]. A high proportion of participants achieved clinically meaningful weight loss, with approximately 90–97% reaching ≥5% weight reduction and 77–85% achieving ≥10% weight loss across active treatment arms, compared with 13% and ~5%, respectively, in the placebo group [
75]. These magnitudes of effect approach those reported for high-dose weekly semaglutide over longer treatment durations, highlighting the potential of zovaglutide to deliver substantial efficacy with reduced injection frequency.
Zovaglutide demonstrated a safety and tolerability profile consistent with the GLP-1 RA class. GI adverse events, primarily nausea, vomiting, and diarrhea, were the most commonly reported events and occurred predominantly during the dose-escalation phase. Treatment discontinuation due to GI adverse events was low (approximately 1.3%) in this trial [
75]. The once-monthly 160 mg regimen was associated with fewer GI events during maintenance compared with bi-weekly dosing, suggesting that extended dosing intervals may mitigate peak-related tolerability in some settings [
75]. Based on these findings, zovaglutide has advanced into a pivotal phase 3 program. The ongoing HORIZON-1 trial (NCT07230119) is a randomized, double-blind, placebo-controlled phase 3 study enrolling approximately 620 adults with overweight or obesity, evaluating the efficacy and safety of once-monthly zovaglutide over 52 weeks, with primary endpoints including percent change in body weight and the proportion of participants achieving ≥5% weight loss [
76]. The results of this study will be critical in determining long-term durability, safety, and regulatory viability of zovaglutide as a monthly GLP-1 RA.
4.5. VK-2735: Dual GLP-1/GIP Receptor Agonist with a Maintenance-Oriented Path toward Monthly Dosing
VK-2735 (Viking Therapeutics, San Diego, California, USA) is a peptide-based dual agonist of the GLP-1 and GIP receptors developed for metabolic disease, conceptually aligned with the central premise of this review: that pharmacologic engineering can shift incretin therapy from weekly standards toward longer dosing intervals without surrendering clinically meaningful efficacy [
61]. By co-activating GLP-1R and GIPR, VK-2735 is positioned to couple established GLP-1–mediated satiety and glucose-dependent insulin secretion with a complementary incretin axis that may further modulate nutrient handling and adiposity-related biology, consistent with mechanistic work across the dual-agonist class [
77]. A key feature relevant to extended-interval development is VK-2735’s predictable and prolonged exposure profile in humans. In early clinical development, Viking reported a terminal half-life of ~170–250 hours (≈7–10 days) and a t_
max of ~75–90 hours, consistent with a gradual rise in systemic exposure and a PK profile described as amenable to extended dosing strategies [
78,
79].
Clinically, VK-2735 has demonstrated robust short-term efficacy with once-weekly subcutaneous dosing. In the phase 2 VENTURE trial, weekly VK-2735 administered for 13 weeks resulted in mean weight loss of up to 14.7% from baseline, with placebo-adjusted reductions reaching 13.1%, and with up to 88% of treated participants achieving ≥10% weight loss [
80]. Weight reduction was progressive throughout the treatment period, with no clear plateau at week 13 [
80]. Treatment was generally well tolerated, with GI adverse events predominating and largely mild to moderate in severity, low discontinuation rates, and no unexpected safety signals [
80]. Data presented at ObesityWeek 2024 provided additional insight relevant to maintenance strategies. In a PK subset of VENTURE participants, most of the weight loss was maintained four weeks after the final weekly dose, including 98% maintenance at the lowest evaluated dose, with continued maintenance observed up to seven weeks post-dose in combined analyses [
81]. These findings support the biological plausibility of once-monthly dosing in a maintenance setting, provided that substantial weight loss is first achieved during a weekly induction phase.
This concept has now been operationalized within Viking’s development program. In October 2025, the company initiated an exploratory maintenance-dosing trial in adults with obesity, in which all participants receive weekly subcutaneous VK-2735 for 19 weeks before transitioning to maintenance regimens that include monthly subcutaneous dosing, alongside oral weekly or daily schedules [
82]. Importantly, Viking’s pivotal phase 3 program (VANQUISH-1 and VANQUISH-2) continues to advance VK-2735 on a weekly injectable schedule, underscoring that monthly dosing remains investigational and positioned as a potential persistence-enhancing strategy rather than a primary therapeutic paradigm [
21,
82,
83,
84]. VK-2735 therefore represents a bridge-to-monthly incretin program rather than a true once-monthly agent at present. Its human PK, durable pharmacodynamic effects after dose cessation, and explicit maintenance-focused clinical strategy make it highly relevant to the discussion of extended-interval incretin therapy [
21,
82]. However, whether monthly maintenance can reliably preserve weight loss trajectories and glycemic control comparable to weekly dosing—without compromising tolerability or increasing interindividual variability in response—remains a central unanswered question that ongoing studies are designed to address.
4.6. ASC30: Ultra–Long-Acting Small-Molecule GLP-1 RA with Monthly and Quarterly Injectable Potential
ASC30 is Ascletis’ lead incretin program and represents a distinctive approach within the GLP-1 RA landscape as a fully biased small-molecule GLP-1R agonist being developed for both oral once-daily administration and ultra–long-acting subcutaneous depot formulations designed for once-monthly treatment and once-quarterly maintenance [
85]. ASC30 was discovered and developed in-house using Ascletis’ AI-Assisted Structure-Based Drug Discovery (AISBDD) platform and advanced through proprietary drug-delivery technologies that differentiate it from peptide-based incretin therapies [
85]. Mechanistically, ASC30 is characterized by potent cAMP signaling with minimal β-arrestin recruitment, a signaling bias proposed to preserve metabolic efficacy while potentially improving tolerability and durability of response [
85]. While this property is relevant across both oral and injectable programs, it is particularly consequential for extended-interval regimens, where excessive peak-related adverse events or receptor desensitization could undermine feasibility [
85].
The injectable formulation of ASC30 leverages Ascletis’ Ultra-Long-Acting Platform (ULAP) to generate slow-release subcutaneous depots with tunable release kinetics [
85]. In a randomized, double-blind, placebo-controlled phase Ib study conducted in the United States, ASC30 subcutaneous injection was evaluated in adults with obesity following a single 100-mg dose administered as either a treatment formulation or a maintenance formulation. PK analyses reported exceptionally prolonged apparent half-lives, reaching approximately 46 days (~1,100 hours) for the treatment formulation and 75 days (~1,810 hours) for the maintenance formulation [
85]. These exposure profiles support once-monthly dosing for active treatment and once-quarterly dosing for maintenance, with favorable peak-to-trough ratios (C_
max-to-C_
day29 ratio of 1.5:1 for monthly treatment; C_
max-to-C_
day85 ratio of 2.5:1 for quarterly maintenance) [
85].
Safety and tolerability findings from the phase Ib subcutaneous study were encouraging and relevant to extended-interval dosing. No serious adverse events or grade ≥3 adverse events were reported [
85]. GI events—including nausea, vomiting, diarrhea, and constipation—were mild to moderate in severity, with no treatment discontinuations. Laboratory parameters, vital signs, electrocardiograms (including QTc), and physical examinations revealed no hepatic or cardiovascular safety signals, supporting the translational plausibility of chronic ultra–long-acting exposure [
85]. Parallel clinical development includes an oral formulation enabled by Ascletis’ POTENT platform, which has progressed through phase I and completed a 13-week phase II study in obesity, reporting placebo-adjusted weight loss up to ~7.7%, favorable cardiometabolic effects, and a low discontinuation rate driven primarily by mild GI events [
85,
86]. While the oral program provides important evidence of pharmacologic activity and tolerability, the injectable ASC30 platform is strategically positioned as a long-interval therapy intended to address adherence and persistence challenges that remain unmet even with weekly GLP-1 RAs [
86].
At the time of writing, ASC30 subcutaneous injection is advancing through phase II clinical development for once-monthly administration, while a separate ultra–long-acting quarterly injectable formulation is being evaluated in phase I clinical studies [
87]. If replicated in larger and longer trials, ASC30 would represent a small-molecule GLP-1 RA capable of sustaining meaningful metabolic effects over multi-month dosing intervals. The ASC30 program therefore exemplifies a next-generation incretin strategy in which signaling bias, depot engineering, and ultra-long PK converge, offering a credible path toward reducing injection burden while preserving efficacy and safety in chronic weight management.
4.7. ASC35: Once-Monthly Subcutaneous GLP-1R/GIPR Dual Peptide Agonist
ASC35, previously referred to as ASC31 in early preclinical disclosures, is Ascletis’ once-monthly subcutaneous dual peptide agonist of GLP-1R and GIPR, discovered and developed in-house using the company’s AISBDD platform and ULAP [
88,
89,
90]. ASC35 is engineered to combine high intrinsic potency at both incretin receptors with extended apparent half-life and high bioavailability per milligram of peptide, enabling a low-volume (≤1 mL) once-monthly injection strategy [
8][
90]. The preclinical characterization disclosed by Ascletis is unusually quantitative for a pre-IND incretin asset. In head-to-head non-human primate studies, ASC35 demonstrated an average observed half-life of approximately 14 days, described as roughly six-fold longer than that of subcutaneously administered tirzepatide [
88,
89,
90]. Systemic drug exposure, assessed by area under the curve, was reported to be approximately 80% higher following intravenous administration and 70% higher following subcutaneous administration compared with tirzepatide [
88,
89,
90]. Using cross-species PK scaling anchored to published tirzepatide absorption, distribution, metabolism, and excretion data, Ascletis projects that ASC35 could plausibly achieve a human half-life approaching or exceeding 30 days, supporting a once-monthly dosing paradigm [
88,
89,
90].
In vitro, ASC35 is described as approximately four-fold more potent than tirzepatide at both GLP-1R and GIPR [
88,
89,
90].
These potency and PK properties translated into marked efficacy signals
in vivo. In a 14-day diet-induced obese mouse study using equal molar dosing, ASC35 produced a −33.6% change in body weight compared with −19.6% for tirzepatide, corresponding to a 71% greater relative weight reduction [
88,
89,
90]. While short-duration rodent studies require cautious interpretation given species differences and dosing intensity, the head-to-head design and magnitude of separation support ASC35’s positioning as a high-potency monthly dual incretin agonist candidate at the preclinical stage [
88,
89,
90]. Regulatory intent is explicitly stated, with Ascletis planning to submit an Investigational New Drug application to the U.S. FDA in the second quarter of 2026 for the treatment of obesity [
88,
89,
90]. Beyond monotherapy, the company has articulated a broader strategic vision in which ASC35 serves as a backbone component of once-monthly combination regimens, most notably with ASC36, a long-acting amylin receptor agonist, and potentially with ASC47, an adipose-targeted thyroid hormone receptor beta agonist. This positioning reflects an effort to construct integrated monthly metabolic interventions that align incretin signaling, amylin-mediated satiety, and THRβ biology [
88,
89,
90]. The central translational question for ASC35 remains whether the apparent advantages in potency and PK will translate into clinically meaningful incremental weight loss and improved tolerability compared with established weekly incretin therapies. These hypotheses are mechanistically plausible and testable, but definitive validation awaits first-in-human PK/PD and dose-ranging studies [
88,
89,
90].
4.8. ASC36: Once-Monthly Subcutaneous Amylin Receptor Peptide Agonist
ASC36 is Ascletis’ once-monthly subcutaneous amylin receptor peptide agonist selected for clinical development as a high-potency amylin-pathway candidate and a foundational component for combination regimens in obesity [
90,
91]. Like ASC35, ASC36 was discovered and engineered in-house using Ascletis’ AI-assisted structure-based drug discovery platform and its ultra–long-acting formulation technology, with explicit design objectives that include extended apparent half-life, high subcutaneous bioavailability, low injection volume (≤1 mL), and manufacturing scalability [
90,
91]. The principal differentiators disclosed for ASC36 relate to both PK behavior and formulation stability. In head-to-head non-human primate studies, slow-release subcutaneous depot formulations of ASC36 exhibited an average observed half-life of approximately 15 days, described as roughly threefold longer than petrelintide [
90,
91]. Based on established cross-species PK relationships for peptide hormones, these data support the feasibility of once-monthly subcutaneous dosing in humans.
In a head-to-head diet-induced obese rat study using equal molar dosing, ASC36 reduced body weight by 10.01% after 7 days, compared with 5.25% for petrelintide, corresponding to an approximately 91% greater relative weight reduction [
90,
91]. While short-duration preclinical studies may overestimate clinical separation, the magnitude and head-to-head nature of these data support ASC36’s positioning as an amylin agonist optimized for long-interval administration [
90,
91]. A particularly consequential attribute of ASC36 is its reported chemical and physical stability at near-neutral pH, with sponsor-reported absence of fibrillation or aggregation under the tested conditions [
90,
91]. This feature is positioned to enable co-formulation with other peptide agents, including the once-monthly GLP-1R/GIPR dual agonist ASC35, without reliance on acidic formulations [
90,
91]. Given the historical challenges of amylin aggregation and device compatibility, this stability profile—if confirmed under formal stability programs—could reduce formulation, device, and immunogenicity risks for fixed-dose or co-formulated monthly products [
90,
91].
Ascletis has stated its intention to submit an investigational new drug application to the U.S. FDA in the second quarter of 2026 for ASC36 in obesity [
90,
91]. The company positions ASC36 both as a monotherapy candidate and as a central platform for combination strategies in cardiometabolic disease, with explicit plans to pursue co-development with ASC35 [
90,
91]. Key translational uncertainties remain those inherent to amylin biology: whether extended-interval exposure can preserve or improve tolerability—particularly GI adverse effects—and whether hypothesized benefits of exposure smoothing translate into superior clinical persistence [
90,
91]. Nonetheless, among early-stage once-monthly amylin programs, ASC36 stands out for the granularity of its disclosed PK, efficacy, and formulation data as it advances toward first-in-human evaluation [
90,
91].
4.9. ASC47: Adipose-Targeted, Once-Monthly Subcutaneous THRβ Agonist as a Muscle-Preserving Adjunct to Incretin Therapy
ASC47 is Ascletis’ adipose-targeted, once-monthly subcutaneous small-molecule agonist of thyroid hormone receptor beta (THRβ), positioned as a “muscle-preserving” adjunct to incretin therapy rather than a stand-alone competitor to high-efficacy GLP-1–based regimens [
92]. Conceptually, ASC47 extends the company’s monthly dosing thesis beyond gut–brain hormones toward metabolic remodeling: THRβ engagement is framed as a lever to enhance lipid handling and energy expenditure while incretins primarily suppress intake, creating a rationale for combination regimens that could improve weight-loss trajectory, mitigate rebound after discontinuation, and potentially enable lower incretin exposure with improved tolerability [
92,
93].
The most mature human signal disclosed to date comes from ASC47-103 (NCT06972992), a randomized, double-blind, placebo-controlled U.S. study in adults with obesity evaluating a single ultra–long-acting ASC47 depot dose combined with four weekly doses of semaglutide 0.5 mg, versus placebo plus the same semaglutide schedule, over a 4-week treatment period with 6-week follow-up (n=28) [
94]. Given the short duration, the primary translational value is proof-of-concept for add-on efficacy, tolerability, and pharmacology rather than definitive body-composition outcomes (fat versus lean mass was explicitly not an objective over 28 days) [
94]. At day 29, the semaglutide-only arm achieved ~2.5% weight reduction from baseline, while ASC47 add-on effects were dose-dependent: 30 mg ASC47 plus semaglutide (n=6) was reported to produce a 56.2% greater relative reduction in body weight compared with semaglutide alone (n=7); 60 mg ASC47 plus semaglutide (n=9) produced a 15.1% greater relative reduction; and 10 mg did not add benefit, consistent with sponsor-reported biomarker evidence of insufficient target engagement at that dose [
94]. Mechanistically, Ascletis used sex hormone–binding globulin (SHBG) as a PD readout for THRβ engagement, reporting that SHBG responses at 10 mg were below the threshold required for clinical effect, whereas both 30 mg and 60 mg showed target engagement [
94]. Cardiometabolic directionality was also emphasized: LDL-C reductions were reported as significant in the 30 mg and 60 mg groups versus semaglutide monotherapy, again suggesting that the 10 mg dose was sub-therapeutic [
94].
A distinctive element relevant to a “monthly maintenance” narrative is the sponsor-reported rebound signal after discontinuation. In ASC47-103, the ultra–long-acting ASC47 depot was reported to have an observed half-life up to ~30 days, accompanied by less post-treatment rebound versus semaglutide alone [
94]. Four weeks after discontinuation (day 57), the semaglutide-only arm’s initial day-29 weight loss was reported to rebound by 68% (to ~0.8% below baseline) [
94]. In contrast, the 30 mg ASC47 arm was reported to show a 157.1% greater relative reduction in body weight versus semaglutide alone at day 57, and the 60 mg arm a 110.4% greater relative reduction; pooled (30+60 mg) analysis was described as a 129.9% greater relative reduction [
94]. These relative metrics require cautious interpretation in small cohorts and short trials; nonetheless, the pattern is directionally consistent with the company’s hypothesis that a long-acting THRβ component could serve as a “maintenance scaffold” when incretin exposure stops or is interrupted [
94].
Tolerability signals—if reproducible—are among the most differentiating sponsor-reported claims. Under the study’s non-titrated semaglutide schedule, vomiting incidence was reported as 6.7% with ASC47 add-on versus 57.1% in the semaglutide-only group [
94]. The sponsor also reported no thyroid-related adverse events; thyroid function tests within normal limits (TSH, FT3/TT3, FT4/TT4); normal telemetry and ECGs; and no increases in heart rate or QTc—points directly relevant to historic regulatory sensitivities around thyroid-axis pharmacology [
94]. If sustained in larger studies, this combination profile would support ASC47’s most plausible clinical role: not as a high-efficacy monotherapy, but as a monthly add-on aimed at improving tolerability and persistence while preserving (or extending) weight-loss effects across dosing gaps [
94].
Finally, ASC47’s positioning is tightly linked to Ascletis’ broader once-monthly metabolic strategy, but combination claims must be interpreted within the limits of current evidence. To date, combinations of ASC47 with a GLP-1R/GIPR dual agonist (ASC35) have been demonstrated only in preclinical diet-induced obese mouse models, where ASC47 plus the dual incretin peptide reportedly outperformed ASC47 combined with tirzepatide [
88]. In contrast, human combination data exist only for ASC47 with semaglutide, as shown in ASC47-103 [
94]. Accordingly, ASC47 should presently be viewed as an early clinically supported adjunct to incretin therapy via semaglutide, while ASC35-based combinations remain preclinical and hypothesis-generating [
88,
94]. ASC47 therefore represents one of the first attempts to integrate monthly metabolic remodeling with satiety-based pharmacology within a single long-interval therapeutic framework.
4.10. AT-7687: GIPR Peptide Antagonist with Extended-Interval Development Intent
AT-7687 is Antag Therapeutics’ peptide-based antagonist of GIPR, developed as a complementary strategy to incretin agonism rather than as a stand-alone weight-loss agent. The program targets suppression of GIP signaling, a pathway genetically and pharmacologically linked to adiposity, insulin resistance, and dyslipidemia, while remaining compatible with GLP-1–based therapies [
24,
25]. Preclinical data provide the primary rationale for its development. In non-human primates, AT-7687 showed high antagonistic potency at GIPR (pK_B ≈ 9.5), an observed half-life of ~27 hours, and favorable metabolic effects without reported GI adverse events [
25]. In these models, AT-7687 prevented weight gain as monotherapy and enhanced weight loss when combined with a GLP-1 RA, alongside improvements in glucose and lipid parameters [
25]. Collectively, these findings position AT-7687 as a potential tolerability- and durability-enabling adjunct rather than a maximal-efficacy monotherapy [
25].
In 2025, Antag initiated a first-in-human, randomized, double-blind, placebo-controlled phase 1a study to assess safety, tolerability, and PK in healthy individuals and participants with obesity [
95]. While efficacy data remain pending, the development strategy explicitly prioritizes future combination studies with GLP-1 RAs, particularly for patients who are inadequate responders or intolerant to incretin-associated GI effects [
95]. AT-7687 is currently being evaluated as a once-weekly injectable, and the sponsor has stated that a once-monthly formulation is under development [
95]. Within the context of extended-interval obesity pharmacotherapy, AT-7687 represents a mechanistically orthogonal approach aimed at improving the durability and tolerability of incretin-based regimens rather than competing directly on maximal weight-loss efficacy.
5. Clinical Positioning and Future Role of Once-Monthly Therapies
The emergence of once-monthly injectable incretin-, amylin-, and THRβ-based agents is not merely a formulation upgrade; it is a deliberate attempt to reframe chronic metabolic pharmacotherapy around persistence. Weekly GLP-1–based regimens demonstrate that sustained receptor engagement can transform weight and glycaemic trajectories, yet real-world discontinuation, intermittent access, and treatment fatigue remain major frictions in long-term care [
11,
12,
29,
30]. Monthly dosing should therefore be viewed as a strategy to reduce regimen burden while preserving durable pharmacodynamic coverage—an objective that is technically feasible but clinically non-trivial, because longer intervals narrow the margin for missed doses, limit titration flexibility, and prolong exposure when adverse effects occur [
18,
53,
54,
55,
56,
57]. In this context, the pivotal question is not whether monthly dosing is possible (it is, as shown by maridebart cafraglutide), but where it adds incremental clinical value relative to established weekly standards, and for whom [
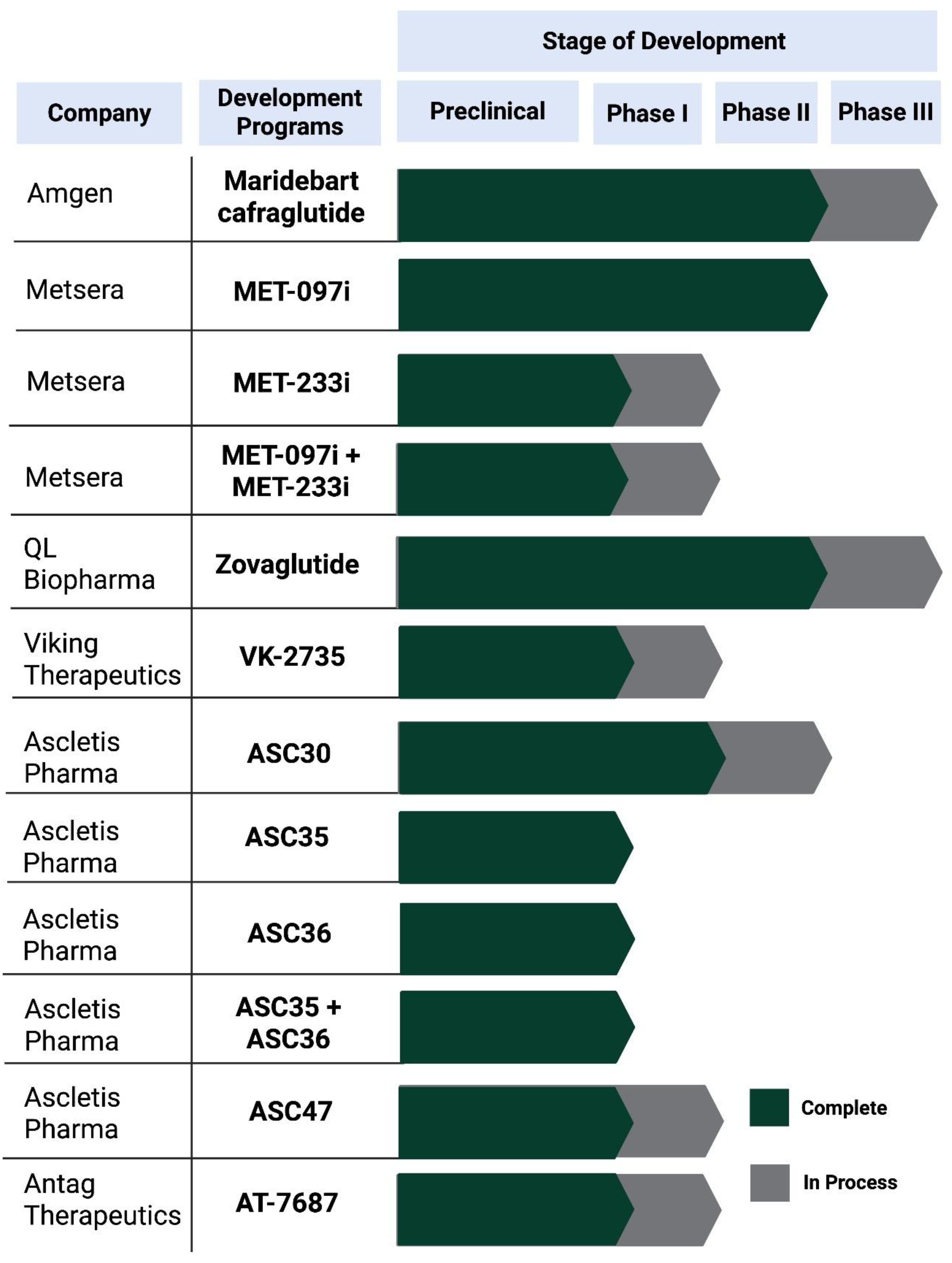
14]. The developmental maturity of these programs is highly heterogeneous, with maridebart cafraglutide currently representing the most advanced clinical example of true once-monthly dosing, while most other candidates remain in early clinical or preclinical stages.
Figure 2.
Clinical development status of extended-interval injectable therapies (monthly or longer) and maintenance-to-monthly programs for obesity and T2D. Dark green bars indicate development stages completed, and grey bars indicate stages ongoing at the time of writing. Program stage is based on publicly available sponsor disclosures and trial registries and may change as studies progress.
Figure 2.
Clinical development status of extended-interval injectable therapies (monthly or longer) and maintenance-to-monthly programs for obesity and T2D. Dark green bars indicate development stages completed, and grey bars indicate stages ongoing at the time of writing. Program stage is based on publicly available sponsor disclosures and trial registries and may change as studies progress.
5.1. Who Benefits Most from Monthly Dosing?
In routine practice, the most compelling candidates for monthly regimens may not be those seeking maximal short-term weight loss, but those in whom persistence is the limiting factor. Patients who have already demonstrated benefit from incretin therapy yet struggle with weekly injection routines, device fatigue, or recurrent disruptions in access may derive disproportionate value from a monthly schedule—if efficacy and tolerability are maintained across the full dosing cycle [
11,
12,
31,
32,
35]. The value proposition is strongest when monthly dosing converts intermittent, stop–start use into stable longitudinal treatment, particularly in obesity care where pharmacotherapy is frequently perceived as optional despite the chronicity of the disease [
29,
30]. At the same time, extended intervals create a paradox: fewer injections may improve acceptability, but any missed or delayed administration can carry larger clinical consequences if trough exposure at the end of the interval permits appetite or glycaemic rebound [
18,
53,
54,
55,
56,
57]. As such, monthly regimens will likely require explicit adherence scaffolding (structured follow-up, reminder systems, proactive refills) to prevent “silent discontinuation” that becomes apparent only after weight regain or glycaemic drift.
5.2. Induction-to-Monthly Maintenance and Switching Strategies
A central practical insight from current pipelines is that “true monthly from day 1” may not be the dominant use case. Several programs implicitly support an induction-to-maintenance paradigm: initial weekly titration to establish early momentum and tolerability, followed by exposure-matched monthly maintenance once a stable response is achieved [
14,
70,
71,
72,
84]. This is aligned with PK/PD realities—ultra–long-acting agents approach steady state slowly, and overly rapid escalation can amplify GI adverse events that may then persist for weeks [
18,
53,
54,
55,
56,
57]. MariTide also provides an important boundary condition: extending intervals beyond monthly (e.g., every 8 weeks) appears to attenuate benefit, consistent with a threshold model in which receptor engagement must remain above a critical minimum across the dosing cycle to preserve metabolic effects [
14]. Switching from weekly incretin therapy to a monthly agent similarly raises under-studied practical questions. Ultra–long-acting exposure limits rapid reversibility; overlap during switching could prolong intolerance, while excessive washout could permit symptomatic rebound. Until formal switching studies exist, transitions should prioritize tolerability, avoid unnecessary exposure stacking, and allow longer observation windows before concluding that a stable monthly “maintenance equilibrium” has been reached [
18,
53,
54,
55,
56,
57].
5.3. Practical Constraints: Tolerability, Safety Monitoring, Access and Cost
Monthly regimens magnify a core feature of long-acting incretin pharmacology: adverse effects can last longer because clearance is slow [
18,
53,
54,
55,
56,
57]. GI symptoms remain class-defining and are strongly influenced by dose and titration speed; therefore, monthly products will require disciplined titration frameworks and proactive counselling to prevent early discontinuation [
18,
53,
54,
55,
56,
57]. In parallel, rapid and sustained weight loss increases the background risk of gallbladder events, and while no consistent unexpected pancreatic or biliary signals have dominated leading programs to date, long-term surveillance and outcomes-grade evidence remain decisive for definitive clinical positioning [
5,
6,
7,
60,
61,
62]. Access and economics may ultimately be as determinative as biology. Monthly dosing could simplify distribution and reduce device count per patient-year, but it also concentrates therapeutic value into fewer administrations—meaning supply interruptions may be more clinically disruptive if a scheduled monthly dose is missed [
11,
12]. Cost-effectiveness will depend on whether monthly regimens measurably improve persistence and long-term outcomes relative to weekly comparators, not merely whether they reduce injection frequency [
29,
30]. Finally, the pipeline’s move beyond incretins toward amylin and THRβ suggests that once-monthly therapy may evolve primarily as combination architecture rather than as a single-mechanism alternative. For monthly dosing to become a new standard rather than a niche convenience, later-phase trials must demonstrate durable maintenance, acceptable long-term safety, and real-world persistence advantages that withstand cost, access, and behavioral constraints [
14,
18,
53,
54,
55,
56,
57].
6. Discussion
The transition from once-weekly to once-monthly injectable therapies in obesity and T2D is being driven by a clinically credible premise: durable metabolic benefit requires sustained pharmacologic coverage, yet sustained coverage in real-world care is frequently constrained by persistence, access, and treatment fatigue. Extended-interval dosing should therefore not be framed as a purely cosmetic convenience upgrade, but rather as a deliberate redesign of chronic metabolic pharmacotherapy around continuity of exposure—an objective that is technically feasible but clinically demanding, as longer dosing intervals inherently reduce flexibility for rapid dose adjustment and prolong exposure when adverse effects occur. Within this evolving landscape, maridebart cafraglutide (MariTide) currently represents the clearest proof-of-concept that clinically meaningful metabolic efficacy can be achieved with a true once-monthly regimen. In a phase 2 trial, once-monthly maridebart produced substantial weight reduction in adults with obesity with or without T2D, with mean weight loss in the obesity cohort in the ~12–16% range and in the obesity–T2D cohort in the ~8–12% range at 52 weeks, accompanied by clinically relevant HbA1c reductions in the T2D cohort. Importantly, exploratory every-8-week regimens demonstrated attenuated efficacy relative to monthly dosing, supporting a threshold model in which receptor engagement must remain above a critical minimum across the entire dosing interval to preserve appetite and glycaemic effects [
1,
2,
3,
4,
14,
18].
However, MariTide establishes feasibility rather than definitive clinical positioning. Several uncertainties remain central. First, durability beyond one year is unresolved, particularly in obesity where long-term maintenance and relapse prevention ultimately determine clinical value. Second, comparative advantage over best-in-class once-weekly GLP-1 RA–based regimens cannot be inferred from separate trials and will require either direct head-to-head studies or robust comparative-effectiveness analyses. Third, ultra–long-acting exposure alters the practical management calculus: titration discipline, anticipatory tolerability counseling, and conservative dose escalation become more consequential, as adverse effects may persist longer and rapid pharmacodynamic de-escalation is inherently limited.
A second defining feature of the once-monthly field is that pipeline expansion is currently outpacing the peer-reviewed evidence base. Beyond MariTide, many monthly-intent programs rely predominantly on sponsor communications, conference disclosures, or early-phase trial descriptions rather than fully adjudicated publications. This evidentiary asymmetry is particularly consequential for ultra–long-acting agents, as short-term tolerability snapshots may fail to capture prolonged symptom burden, accumulation dynamics, delayed-onset events, or the clinical consequences of missed or delayed doses within a long-interval schedule. If monthly dosing requires clinicians and patients to accept fewer administrations with greater pharmacologic commitment per dose, transparency regarding dose selection, titration logic, immunogenicity, and discontinuation management must be at least as rigorous as that established during the once-weekly GLP-1 RA era [
18,
33,
34,
35,
55,
56,
57,
58].
Biological heterogeneity in treatment response represents a third axis that may ultimately determine whether once-monthly therapies become broadly transformative or selectively useful. The marked interindividual variability observed with GLP-1 RAs and GLP-1/GIPR dual agonists reinforces that obesity is not a physiologically homogeneous condition. A plausible explanatory framework positions individuals along a secretion–responsiveness spectrum of incretin effectiveness, ranging from relative hyposecretion to functional forms of incretin resistance, in which circulating incretin levels may be preserved or elevated but downstream insulinotropic and metabolic effects are attenuated—classically reflected in an impaired incretin effect involving both secretion and β-cell responsiveness [
5,
6,
7,
8,
9,
59,
64]. Monthly dosing may amplify this heterogeneity: individuals near the exposure threshold required for stable appetite suppression may be more vulnerable to end-of-interval troughs, whereas others with prolonged pharmacodynamic carryover may maintain benefit with lower-frequency maintenance once a new weight equilibrium is achieved.
This variability reframes why “monthly” may matter most as a platform for mechanistic diversification rather than as a simple alternative to once-weekly incretin therapy. MariTide itself already exemplifies a qualitative shift within the incretin axis—GLP-1 RA biology combined with sustained GIPR antagonism—underscoring that altering pathway biology, not merely dosing frequency, can be clinically meaningful [
1,
2,
3,
20]. In parallel, long-acting amylin analogues, THRβ agonists, and non-peptide or biased small-molecule GLP-1 RAs expand beyond classical incretin biology, raising the possibility that some patients with attenuated incretin responsiveness may preferentially benefit from therapies that engage satiety, energy handling, or adipose-selective pathways through complementary mechanisms [
22,
23,
24,
40,
96,
97]. As monthly regimens mature, the most clinically informative trials may therefore be those that move beyond mean efficacy and incorporate response-dispersion metrics, end-of-interval stability, body-composition outcomes, and biomarker-enabled stratification strategies that support mechanism-matched therapy selection.
Ultimately, several inflection points will determine whether once-monthly therapies redefine routine metabolic care or remain a niche convenience layer. The first is durability and maintenance: sustained efficacy beyond one year, accompanied by clear guidance on missed doses and discontinuation in long half-life contexts. The second is long-term safety and outcomes: extended surveillance and, where appropriate, outcomes programs capable of establishing benefit–risk profiles comparable in credibility to the evidence that legitimized once-weekly GLP-1 RA standards in T2D. The third is real-world value: demonstration that monthly schedules meaningfully improve persistence and effectiveness relative to weekly comparators, sufficiently to justify likely premium pricing and to withstand access and supply constraints that have challenged the current incretin era [
11,
26,
27,
28,
29,
30,
96]. Until these conditions are met, once-monthly therapies should be viewed as a promising, mechanistically expanding platform—anchored by MariTide proof-of-concept—but still in the process of earning their definitive clinical role.
7. Conclusions
Once-monthly injectable therapies are emerging as a credible next step in chronic pharmacologic management of obesity and T2D, aiming to reduce injection burden while preserving sustained metabolic efficacy. Maridebart cafraglutide currently provides the strongest proof-of-concept, demonstrating clinically meaningful improvements in body weight and glycaemic control with monthly dosing, while also suggesting that extending dosing intervals beyond monthly may attenuate efficacy in this format.
Beyond this lead program, the pipeline is rapidly expanding across incretin-, amylin-, and THRβ-based platforms, but definitive clinical positioning will depend on evidence that monthly regimens deliver durable long-term maintenance, acceptable safety under prolonged exposure, and real-world persistence advantages that justify cost and access constraints. As these programs mature, physiology-informed stratification and response-stability metrics—particularly end-of-interval control—may be essential to match extended-interval therapies to the biological heterogeneity that underlies variable treatment response in obesity and T2D.
Author Contributions
Conceptualization, H.I.S.-C.; methodology, H.I.S.-C.; formal analysis, H.I.S.-C.; investigation, H.I.S.-C.; resources, H.I.S.-C.; data curation, H.I.S.-C.; writing—original draft preparation, H.I.S.-C.; writing—review and editing, H.I.S.-C.; visualization, H.I.S.-C.; supervision, H.I.S.-C.; project administration, H.I.S.-C. The author has read and agreed to the published version of the manuscript.
Funding
This research was funded by the Consejo Mexiquense de Ciencia y Tecnología (COME- CYT), grant numbers FICDTEM-2023-131 (awarded to H.I.S.-C.) and by the Universidad Nacional Autónoma de México (UNAM), through grants PAPIIT IA201725 and PAPIME 203825 (H.I.S.-C.).
Abbreviations
The following abbreviations are used in this manuscript:
| AI |
Artificial intelligence |
| AISBDD |
AI-Assisted Structure-Based Drug Discovery |
| BMI |
Body mass index |
| cAMP |
Cyclic adenosine monophosphate |
| EASD |
European Association for the Study of Diabetes |
| ECG |
Electrocardiogram |
| GI |
Gastrointestinal |
| GIP |
Glucose-dependent insulinotropic polypeptide |
| GIPR |
Glucose-dependent insulinotropic polypeptide receptor |
| GLP-1 |
Glucagon-like peptide-1 |
| GLP-1R |
Glucagon-like peptide-1 receptor |
| GLP-1 RA |
Glucagon-like peptide-1 receptor agonist |
| HbA1c |
Glycated hemoglobin |
| IND |
Investigational New Drug |
| LDL-C |
Low-density lipoprotein cholesterol |
| NCT |
ClinicalTrials.gov identifier |
| NuSH |
Nutrient-stimulated hormone |
| PD |
Pharmacodynamics |
| PK |
Pharmacokinetics |
| PK/PD |
Pharmacokinetics/pharmacodynamics |
| Q2W |
Once every 2 weeks |
| Q4W |
Once every 4 weeks |
| QTc |
Corrected QT interval |
| SHBG |
Sex hormone–binding globulin |
| SQ |
Subcutaneous |
| T2D |
Tpe 2 diabetes |
| THRβ |
Thyroid hormone receptor beta |
| TSH |
Thyroid-stimulating hormone |
| ULAP |
Ultra-Long-Acting Platform |
References
- Buse, J.B.; Henry, R.R.; Han, J.; Kim, D.D.; Fineman, M.S.; Baron, A.D. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care 2004, 27, 2628–2635. [Google Scholar] [CrossRef]
- Buse, J.B.; Rosenstock, J.; Sesti, G.; Schmidt, W.E.; Montanya, E.; Brett, J.H.; Zychma, M.; Blonde, L. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet 2009, 374, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Dungan, K.M.; Povedano, S.T.; Forst, T.; González, J.G.; Atisso, C.; Sealls, W.; Fahrbach, J.L. Once-weekly dulaglutide versus once-daily liraglutide in metformin-treated patients with type 2 diabetes (AWARD-6): a randomised, open-label, phase 3, non-inferiority trial. Lancet 2014, 384, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Pratley, R.E.; Aroda, V.R.; Lingvay, I.; Lüdemann, J.; Andreassen, C.; Navarria, A.; Viljoen, A. Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): a randomised, open-label, phase 3b trial. Lancet Diabetes Endocrinol 2018, 6, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med 2016, 375, 1834–1844. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef]
- Moiz, A.; Filion, K.B.; Tsoukas, M.A.; Yu, O.H.Y.; Peters, T.M.; Eisenberg, M.J. Mechanisms of GLP-1 Receptor Agonist-Induced Weight Loss: A Review of Central and Peripheral Pathways in Appetite and Energy Regulation. Am J Med 2025, 138, 934–940. [Google Scholar] [CrossRef]
- Zheng, Z.; Zong, Y.; Ma, Y.; Tian, Y.; Pang, Y.; Zhang, C.; Gao, J. Glucagon-like peptide-1 receptor: mechanisms and advances in therapy. Signal Transduct Target Ther 2024, 9, 234. [Google Scholar] [CrossRef]
- Moiz, A.; Filion, K.B.; Tsoukas, M.A.; Yu, O.H.Y.; Peters, T.M.; Eisenberg, M.J. The expanding role of GLP-1 receptor agonists: a narrative review of current evidence and future directions. EClinicalMedicine 2025, 86, 103363. [Google Scholar] [CrossRef]
- Rodriguez, P.J.; Zhang, V.; Gratzl, S.; Do, D.; Goodwin Cartwright, B.; Baker, C.; Gluckman, T.J.; Stucky, N.; Emanuel, E.J. Discontinuation and Reinitiation of Dual-Labeled GLP-1 Receptor Agonists Among US Adults With Overweight or Obesity. JAMA Netw Open 2025, 8, e2457349. [Google Scholar] [CrossRef] [PubMed]
- Altabas, V.; Orlović, Z.; Baretić, M. Addressing the Shortage of GLP-1 RA and Dual GIP/GLP-1 RA-Based Therapies—A Systematic Review. Diabetology 2025, 6, 52. [Google Scholar] [CrossRef]
- World Health Organization. WHO guideline on the use of glucagon-like peptide-1 (GLP-1) therapies for the treatment of obesity in adults; World Health Organization: Geneva, Switzerland, 2025. [Google Scholar]
- Jastreboff, A.M.; Ryan, D.H.; Bays, H.E.; Ebeling, P.R.; Mackowski, M.G.; Philipose, N.; Ross, L.; Liu, Y.; Burns, C.E.; Abbasi, S.A.; et al. Once-Monthly Maridebart Cafraglutide for the Treatment of Obesity—A Phase 2 Trial. N Engl J Med 2025, 393, 843–857. [Google Scholar] [CrossRef]
- Wu, C.C.; Cengiz, A.; Lawley, S.D. Less frequent dosing of GLP-1 receptor agonists as a viable weight maintenance strategy. Obesity (Silver Spring) 2025, 33, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, A.; Wu, C.C.; Lawley, S.D. Alternative dosing regimens of GLP-1 receptor agonists may reduce costs and maintain weight loss efficacy. Diabetes Obes Metab 2025, 27, 2251–2258. [Google Scholar] [CrossRef]
- Belančić, A.; Al-Sallami, H.S. Spotlight commentary: Changes in pharmacokinetics following significant weight loss. Br J Clin Pharmacol 2025, 91, 678–680. [Google Scholar] [CrossRef]
- Min, J.S.; Jo, S.J.; Lee, S.; Kim, D.Y.; Kim, D.H.; Lee, C.B.; Bae, S.K. A Comprehensive Review on the Pharmacokinetics and Drug-Drug Interactions of Approved GLP-1 Receptor Agonists and a Dual GLP-1/GIP Receptor Agonist. Drug Des Devel Ther 2025, 19, 3509–3537. [Google Scholar] [CrossRef]
- Del Prato, S.; Kang, J.; Trautmann, M.E.; Stewart, J.; Sorli, C.H.; Derwahl, M.; Soto, A.; Yoon, K.H. Efficacy and safety of once-monthly efpeglenatide in patients with type 2 diabetes: Results of a phase 2 placebo-controlled, 16-week randomized dose-finding study. Diabetes Obes Metab 2020, 22, 1176–1186. [Google Scholar] [CrossRef]
- Véniant, M.M.; Lu, S.C.; Atangan, L.; Komorowski, R.; Stanislaus, S.; Cheng, Y.; Wu, B.; Falsey, J.R.; Hager, T.; Thomas, V.A.; et al. A GIPR antagonist conjugated to GLP-1 analogues promotes weight loss with improved metabolic parameters in preclinical and phase 1 settings. Nat Metab 2024, 6, 290–303. [Google Scholar] [CrossRef]
- Modesto, K.; Chung, K.; Ji, S.; Stubbe, S.; Albers, K.; Lian, B. Abstract 4367152: VANQUISH-2: Phase 3, randomized, double-blind, placebo-controlled trial of weekly subcutaneous administration of VK2735 in obese or overweight adults with type 2 diabetes. Circulation 2025, 152, A4367152–A4367152. [Google Scholar] [CrossRef]
- Minnion, J.S.; Hinds, C.; Reglinska, B. 894-P: MET-233 is an ultra-long-acting amylin receptor agonist. Diabetes 2025, 74 (Suppl. 1), 894–P. [Google Scholar] [CrossRef]
- Hinds, C.; Minnion, J.S.; Zoumpoulidou, G. 794-P: MET-097: Preclinical characterization of a potent and ultra-long-acting GLP-1 receptor agonist. Diabetes 2025, 74 Suppl. 1, 794–P. [Google Scholar] [CrossRef]
- Rosenkilde, M.M.; George, J.T.; Véniant, M.M.; Holst, J.J. GIP Receptor Antagonists in the Pharmacotherapy of Obesity: Physiologic, Genetic, and Clinical Rationale. Diabetes 2025, 74, 1334–1338. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.H.; Sanni, S.J.; Riber, D.; Holst, J.J.; Rosenkilde, M.M.; Sparre-Ulrich, A.H. AT-7687, a novel GIPR peptide antagonist, combined with a GLP-1 agonist, leads to enhanced weight loss and metabolic improvements in cynomolgus monkeys. Mol Metab 2024, 88, 102006. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yu, S.; Jin, X.; Sheng, L.; YanMu, M.R.; Gao, J.; Lu, J.; Lei, T. The Clinical Application of GLP-1RAs and GLP-1/GIP Dual Receptor Agonists Based on Pharmacological Mechanisms: A Review. Drug Des Devel Ther 2025, 19, 10383–10409. [Google Scholar] [CrossRef]
- Xu, Y.; Drucker, D.J.; Traverso, G.; Beloqui, A. Innovative molecules and delivery technologies enabling the future of GLP-1-based therapies. Endocr Rev 2025, Epub ahead of print. [Google Scholar] [CrossRef]
- Li, Z.; Han, Z.; Sun, R.; Xuan, X.; Huang, C. Long-Term Efficacy Trajectories of GLP-1 Receptor Agonists: A Systematic Review and Network Meta-Analysis. Diabetes Metab Syndr Obes 2025, 18, 3611–3624. [Google Scholar] [CrossRef]
- Ferreira, K.; Kont, E.; Abdelkhalik, A.; Jones, D.; Baker-Knight, J. The out-of-pocket cost of living with obesity: Results from a survey in Spain, South Korea, Brazil, India, Italy, and Japan. Obes Sci Pract 2024, 10, e70000. [Google Scholar] [CrossRef]
- Okunogbe, A.; Nugent, R.; Spencer, G.; Ralston, J.; Wilding, J. Economic impacts of overweight and obesity: current and future estimates for eight countries. BMJ Glob Health 2021, 6, e006351. [Google Scholar] [CrossRef]
- Hauber, A.B.; Nguyen, H.; Posner, J.; Kalsekar, I.; Ruggles, J. A discrete-choice experiment to quantify patient preferences for frequency of glucagon-like peptide-1 receptor agonist injections in the treatment of type 2 diabetes. Curr Med Res Opin 2016, 32, 251–262. [Google Scholar] [CrossRef]
- Gelhorn, H.L.; Poon, J.L.; Davies, E.W.; Paczkowski, R.; Curtis, S.E.; Boye, K.S. Evaluating preferences for profiles of GLP-1 receptor agonists among injection-naïve type 2 diabetes patients in the UK. Patient Prefer Adherence 2015, 9, 1611–1622. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.M.; Nuffer, W.; Ellis, S.L. GLP-1 receptor agonists: a review of head-to-head clinical studies. Ther Adv Endocrinol Metab 2015, 6, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Denimal, D. Emerging perspectives on once-weekly insulins in type 1 and type 2 diabetes: a mini-review. Front Endocrinol (Lausanne) 2025, 16, 1656884. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Chen, S.; Flood, E.; Shaunik, A.; Romero, B.; de la Cruz, M.; Alvarez, C.; Grandy, S. Glucagon-like Peptide-1 Receptor Agonist Treatment Attributes Important to Injection-Naïve Patients with Type 2 Diabetes Mellitus: A Multinational Preference Study. Diabetes Ther 2017, 8, 321–334. [Google Scholar] [CrossRef]
- Wan, W.; Qin, Q.; Xie, L.; Zhang, H.; Wu, F.; Stevens, R.C.; Liu, Y. GLP-1R Signaling and Functional Molecules in Incretin Therapy. Molecules 2023, 28, 0751. [Google Scholar] [CrossRef]
- Deng, W.; Zhao, Z.; Zou, T.; Kuang, T.; Wang, J. Research Advances in Fusion Protein-Based Drugs for Diabetes Treatment. Diabetes Metab Syndr Obes 2024, 17, 343–362. [Google Scholar] [CrossRef]
- Peri, R.V.; Anchan, H.; Jonnalagadda, K.; Varghese, R.; Gupta, P. Designing GLP-1 delivery: structural perspectives and formulation approaches for optimized therapy. Nutr Diabetes 2025, 15, 53. [Google Scholar] [CrossRef]
- Lu, S.C.; Chen, M.; Atangan, L.; Killion, E.A.; Komorowski, R.; Cheng, Y.; Netirojjanakul, C.; Falsey, J.R.; Stolina, M.; Dwyer, D.; et al. GIPR antagonist antibodies conjugated to GLP-1 peptide are bispecific molecules that decrease weight in obese mice and monkeys. Cell Rep Med 2021, 2, 100263. [Google Scholar] [CrossRef]
- Anastasiou, I.A.; Argyrakopoulou, G.; Dalamaga, M.; Kokkinos, A. Dual and Triple Gut Peptide Agonists on the Horizon for the Treatment of Type 2 Diabetes and Obesity. An Overview of Preclinical and Clinical Data. Curr Obes Rep 2025, 14, 34. [Google Scholar] [CrossRef]
- Emanuelsson, F.; Nordestgaard, B.G.; Benn, M. Genetic variants of glucose-dependent insulinotropic polypeptide (GIP) signalling as proxy for body weight reduction and cardiovascular risk. Eur Heart J 2025. [Google Scholar] [CrossRef]
- Kizilkaya, H.S.; Sørensen, K.V.; Kibsgaard, C.J.; Gasbjerg, L.S.; Hauser, A.S.; Sparre-Ulrich, A.H.; Grarup, N.; Rosenkilde, M.M. Loss of Function Glucose-Dependent Insulinotropic Polypeptide Receptor Variants Are Associated With Alterations in BMI, Bone Strength and Cardiovascular Outcomes. Front Cell Dev Biol 2021, 9, 749607. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E. Targeting the GIPR for obesity: To agonize or antagonize? Potential mechanisms. Mol Metab 2021, 46, 101139. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Su, W.; Zhang, W.; Zhang, J.; Sattler, M.; Zou, P. Albumin-binding domain extends half-life of glucagon-like peptide-1. Eur J Pharmacol 2021, 890, 173650. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, L.B.; Lau, J. The Discovery and Development of Liraglutide and Semaglutide. Front Endocrinol (Lausanne) 2019, 10, 155. [Google Scholar] [CrossRef]
- Přáda Brichtová, E.; Edu, I.A.; Li, X.; Becher, F.; Gomes Dos Santos, A.L.; Jackson, S.E. Effect of Lipidation on the Structure, Oligomerization, and Aggregation of Glucagon-like Peptide 1. Bioconjug Chem 2025, 36, 401–414. [Google Scholar] [CrossRef]
- Metsera, Inc. Metsera Announces Positive Topline Phase 2a Clinical Data for its Ultra-Long Acting GLP-1 Receptor Agonist MET-097i. Business Wire 2025. Available online: https://www.businesswire.com/news/home/20250106419531/en/Metsera-Announces-Positive-Topline-Phase-2a-Clinical-Data-for-its-Ultra-Long-Acting-GLP-1-Receptor-Agonist-MET-097i (accessed on 17 Dec 2025).
- Metsera, Inc. Metsera Pipeline: Advancing Next-Generation Obesity Therapies. Metsera 2024. Available online: https://metsera.com/pipeline/ (accessed on 17 Dec 2025).
- Metsera, Inc. Metsera Reports Positive Phase 2b Results for First-and-Best-in-Class Ultra-Long Acting GLP-1 RA Candidate MET-097i Enabling Rapid Transition into Phase 3. GlobeNewswire 2025. Available online: https://www.globenewswire.com/news-release/2025/09/29/3158095/0/en/Metsera-Reports-Positive-Phase-2b-Results-for-First-and-Best-in-Class-Ultra-long-Acting-GLP-1-RA-Candidate-MET-097i-Enabling-Rapid-Transition-into-Phase-3.html (accessed on 17 Dec 2025).
- Sorli, C.; Harashima, S.I.; Tsoukas, G.M.; Unger, J.; Karsbøl, J.D.; Hansen, T.; Bain, S.C. Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol 2017, 5, 251–260. [Google Scholar] [CrossRef]
- Granhall, C.; Donsmark, M.; Blicher, T.M.; Golor, G.; Søndergaard, F.L.; Thomsen, M.; Bækdal, T.A. Safety and Pharmacokinetics of Single and Multiple Ascending Doses of the Novel Oral Human GLP-1 Analogue, Oral Semaglutide, in Healthy Subjects and Subjects with Type 2 Diabetes. Clin Pharmacokinet 2019, 58, 781–791. [Google Scholar] [CrossRef]
- Nauck, M.A.; D’Alessio, D.A. Tirzepatide, a dual GIP/GLP-1 receptor co-agonist for the treatment of type 2 diabetes with unmatched effectiveness regarding glycaemic control and body weight reduction. Cardiovasc Diabetol 2022, 21, 169. [Google Scholar] [CrossRef]
- Brønden, A.; Knop, F.K.; Christensen, M.B. Clinical Pharmacokinetics and Pharmacodynamics of Albiglutide. Clin Pharmacokinet 2017, 56, 719–731. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Li, Y.; Zhao, X.; Zhou, W.; Loghin, C.; Tham, L.S.; Cui, X.; Cui, Y.; Wang, W. Pharmacokinetics, Pharmacodynamics, and Safety of Dulaglutide After Single or Multiple Doses in Chinese Healthy Subjects and Patients with T2DM: A Randomized, Placebo-Controlled, Phase I Study. Adv Ther 2022, 39, 488–503. [Google Scholar] [CrossRef]
- Ahmann, A.J.; Capehorn, M.; Charpentier, G.; Dotta, F.; Henkel, E.; Lingvay, I.; Holst, A.G.; Annett, M.P.; Aroda, V.R. Efficacy and Safety of Once-Weekly Semaglutide Versus Exenatide ER in Subjects With Type 2 Diabetes (SUSTAIN 3): A 56-Week, Open-Label, Randomized Clinical Trial. Diabetes Care 2018, 41, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Frías, J.P.; Davies, M.J.; Rosenstock, J.; Pérez Manghi, F.C.; Fernández Landó, L.; Bergman, B.K.; Liu, B.; Cui, X.; Brown, K. Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes. N Engl J Med 2021, 385, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Geiser, J.S.; Heathman, M.A.; Cui, X.; Martin, J.; Loghin, C.; Chien, J.Y.; de la Peña, A. Clinical Pharmacokinetics of Dulaglutide in Patients with Type 2 Diabetes: Analyses of Data from Clinical Trials. Clin Pharmacokinet 2016, 55, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Puig, M.; Shubow, S. Immunogenicity of therapeutic peptide products: bridging the gaps regarding the role of product-related risk factors. Front Immunol 2025, 16, 1608401. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. Management of Endocrine Disease: Are all GLP-1 agonists equal in the treatment of type 2 diabetes? Eur J Endocrinol 2019, 181, R211–R234. [Google Scholar] [CrossRef]
- Wilding, J.P.H.; Batterham, R.L.; Calanna, S.; Davies, M.; Van Gaal, L.F.; Lingvay, I.; McGowan, B.M.; Rosenstock, J.; Tran, M.T.D.; Wadden, T.A.; et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N Engl J Med 2021, 384, 989–1002. [Google Scholar] [CrossRef]
- Jastreboff, A.M.; Aronne, L.J.; Ahmad, N.N.; Wharton, S.; Connery, L.; Alves, B.; Kiyosue, A.; Zhang, S.; Liu, B.; Bunck, M.C.; et al. Tirzepatide Once Weekly for the Treatment of Obesity. N Engl J Med 2022, 387, 205–216. [Google Scholar] [CrossRef]
- Davies, M.J.; Aroda, V.R.; Collins, B.S.; Gabbay, R.A.; Green, J.; Maruthur, N.M.; Rosas, S.E.; Del Prato, S.; Mathieu, C.; Mingrone, G.; et al. Management of Hyperglycemia in Type 2 Diabetes, 2022. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2022, 45, 2753–2786. [Google Scholar] [CrossRef]
- Douros, J.D.; Mowery, S.A.; Knerr, P.J. The Premise of the Paradox: Examining the Evidence That Motivated GIPR Agonist and Antagonist Drug Development Programs. J Clin Med 2025, 14, 3812. [Google Scholar] [CrossRef]
- Wolfe, M.M.; Boylan, M.O.; Chin, W.W. Glucose-Dependent Insulinotropic Polypeptide in Incretin Physiology: Role in Health and Disease. Endocr Rev 2025, 46, 479–500. [Google Scholar] [CrossRef]
- Pfizer, Inc. Pfizer Completes Acquisition of Metsera. Pfizer Press Release 2025. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-completes-acquisition-metsera (accessed on 17 Dec 2025).
- Metsera, Inc. Registration Statement on Form S-1. U.S. Securities and Exchange Commission (SEC) 2025. Available online: https://www.sec.gov/Archives/edgar/data/2040807/000119312525004504/d900229ds1.htm (accessed on 17 Dec 2025).
- Hansford, R.; Hinds, C.; Adams, W.J. 765-P: Therapeutic NuSH Cocktails—Coadministration of Ultra-Long-Acting GLP-1, GIP, Glucagon, and Amylin Peptide Analogs Induce Profound Weight Loss in DIO Mice. Diabetes 2025, 74 Suppl. 1, 765–P. [Google Scholar] [CrossRef]
- Metsera, Inc. Metsera Pipeline MET-097i. Metsera 2024. Available online: https://metsera.com/pipeline/#met-097 (accessed on 17 Dec 2025).
- Stoekenbroek, R.; Bisch, J.; Kolluri, S.; Noor, M.A.; Mallory, J.; Cunningham, R.; Hubbard, B.; Marso, S.P. 788-P: A Twelve-Week Trial of MET097—A Potent and Ultra-Long-Acting GLP-1 Receptor Agonist. Diabetes 2025, 74 Suppl. 1, 788–P. [Google Scholar] [CrossRef]
- Metsera, Inc. A Phase 2b Study to Examine the Safety and Efficacy of Four Different Regimens of Once-weekly MET-097 in Adults With Obesity or Overweight. ClinicalTrials.gov 2024. Available online: https://clinicaltrials.gov/study/NCT06712836 (accessed on 17 Dec 2025).
- Metsera, Inc. A Phase 2b Study to Evaluate the Efficacy and Safety of Once-monthly MET-097 in Adults With Obesity or Overweight. ClinicalTrials.gov 2025. Available online: https://www.clinicaltrials.gov/study/NCT06973720 (accessed on 17 Dec 2025).
- Metsera, Inc. Metsera Announces Positive Phase 1 Data of First-in-Class Once-Monthly Amylin Candidate MET-233i. BioSpace 2025. Available online: https://www.biospace.com/press-releases/metsera-announces-positive-phase-1-data-of-first-in-class-once-monthly-amylin-candidate-met-233i (accessed on 17 Dec 2025).
- ClinicalTrials.gov. A Study of MET233 in Combination With MET097 in Individuals With Obesity or Overweight With or Without Diabetes. Available online: https://clinicaltrials.gov/study/NCT06924320 (accessed on 17 Dec 2025).
- QL Biopharm reports Phase 2 results of a monthly dose study evaluating zovaglutide, a novel, extended half-life GLP-1 receptor agonist at EASD 2025. BioSpace (Press Releases) 2025. Available online: https://www.biospace.com/press-releases/ql-biopharm-reports-phase-2-results-of-a-monthly-dose-study-evaluating-zovaglutide-a-novel-extended-half-life-glp-1-receptor-agonist-at-easd-2025 (accessed on 17 Dec 2025).
- 61st EASD Annual Meeting of the European Association for the Study of Diabetes: Vienna, Austria, 15–19 September 2025. Diabetologia 2025, 68 Suppl. 1, 1–754. [CrossRef]
- Beijing QL Biopharmaceutical Co; Ltd. A Study of Zovaglutide in Subjects With Overweight or Obesity (HORIZON-1). Available online: https://clinicaltrials.gov/study/NCT07230119 (accessed on 17 Dec 2025).
- Liu, Q.K. Mechanisms of action and therapeutic applications of GLP-1 and dual GIP/GLP-1 receptor agonists. Front Endocrinol (Lausanne) 2024, 15, 1431292. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Phase 1 Study to Evaluate the Safety and Tolerability of VK2735. Available online: https://www.clinicaltrials.gov/study/NCT05203237 (accessed on 15 Dec 2025).
- Viking Therapeutics; Inc. Viking Therapeutics Announces Results from Phase 1 Clinical Trial of Dual GLP-1/GIP Receptor Agonist VK2735. Available online: https://ir.vikingtherapeutics.com/2023-03-28-Viking-Therapeutics-Announces-Results-from-Phase-1-Clinical-Trial-of-Dual-GLP-1-GIP-Receptor-Agonist-VK2735 (accessed on 17 Dec 2025).
- Viking Therapeutics; Inc. VK2735 (Subcutaneous & Oral Formulations): Dual GLP-1/GIP Receptor Agonist. Available online: https://vikingtherapeutics.com/pipeline/metabolic-disease-program/vk2735/ (accessed on 17 Dec 2025).
- Viking Therapeutics; Inc. Viking Therapeutics Presents Preclinical Data on Novel Dual GLP-1/GIP Agonists at ObesityWeek® 2021. Available online: https://ir.vikingtherapeutics.com/2021-11-01-Viking-Therapeutics-Presents-Preclinical-Data-on-Novel-Dual-GLP-1-GIP-Agonists-at-ObesityWeek-R-2021 (accessed on 17 Dec 2025).
- Viking Therapeutics; Inc. Viking Therapeutics Announces Initiation of VK2735 Maintenance Dosing Clinical Trial in Patients with Obesity. Available online: https://ir.vikingtherapeutics.com/2025-10-21-Viking-Therapeutics-Announces-Initiation-of-VK2735-Maintenance-Dosing-Clinical-Trial-in-Patients-with-Obesity (accessed on 17 Dec 2025).
- ClinicalTrials.gov. VK2735 for Weight Management Phase 3 (VANQUISH 1). Available online: https://clinicaltrials.gov/study/NCT07104500 (accessed on 17 Dec 2025).
- ClinicalTrials.gov. VK2735 for Weight Management Type 2 Diabetes Phase 3 (VANQUISH 2). Available online: https://clinicaltrials.gov/study/NCT07104383 (accessed on 17 Dec 2025).
- Wu, J.J.; Wang, V. ASC30, a Once-Monthly SQ Injected Small Molecule GLP-1RA in Participants with Obesity: A Phase Ib Study. Proceedings of ObesityWeek®, Atlanta, GA, USA, 2–6 November 2025; p. Poster-174. [Google Scholar]
- Wu, J.J. ASC30, an oral GLP-1 receptor biased small-molecule agonist in participants with obesity: Phase I clinical evaluation. In Proceedings of the 85th Scientific Sessions of the American Diabetes Association (ADA 2025), Chicago, IL, USA, 20–23 June 2025. [Google Scholar]
- Ascletis Pharma Inc. Ascletis’ Oral Small Molecule GLP-1, ASC30, Demonstrated Placebo-Adjusted Weight Loss of 7.7% with Better Gastrointestinal Tolerability in Its 13-Week U.S. Phase II Study in Participants with Obesity or Overweight. Available online: https://www.ascletis.com/single/145.html (accessed on 17 December 2025).
- Wu, J.J. GLP-1R/GIPR Peptide Agonist ASC31 plus ASC47 Shows Superior Weight Loss Compared with Tirzepatide in Diet-Induced Obese Mice. Proceedings of ObesityWeek®, Atlanta, GA, USA, 2–6 November 2025; p. Poster-139. [Google Scholar]
- Ascletis Pharma Inc. Ascletis Selects a Best-in-Class Once-Monthly Subcutaneously Administered GLP-1R/GIPR Dual Peptide Agonist, ASC35, for Clinical Development. Available online: https://www1.hkexnews.hk/listedco/listconews/sehk/2025/1013/2025101300079.pdf (accessed on 17 December 2025).
- Ascletis Pharma Inc. Ascletis Announces Co-formulation of ASC36, Once-Monthly Next-Generation Amylin Receptor Agonist and ASC35, Once-Monthly Next-Generation GLP-1R/GIPR Dual Agonist for Clinical Development. Available online: https://markets.ft.com/data/announce/detail?dockey=600-202511121830PR_NEWS_USPRX____CN22524-1 (accessed on 17 December 2025).
- Ascletis Pharma Inc. Ascletis Selects a Best-in-Class Once-Monthly Subcutaneously Administered Amylin Receptor Agonist, ASC36, for Clinical Development. Available online: https://www.prnewswire.com/apac/news-releases/ascletis-selects-a-best-in-class-once-monthly-subcutaneously-administered-amylin-receptor-agonist-asc36-for-clinical-development-302598393.html (accessed on 17 December 2025).
- Wu, J.J. ASC47, an Adipose-Targeted, Muscle-Preserving Weight Loss Drug Candidate for Obesity, Demonstrated Significant Weight Loss and Preserved Muscle in Diet-Induced Obese Mice. Abstract 0254; Proceedings of the 32nd European Congress on Obesity (ECO 2025). Malaga, Spain, 11-14 May 2025. [Google Scholar]
- Wu, J.J.; Wu, C. 847-P: ASC47, a Muscle-Preserving Weight Loss Drug Candidate for Obesity, in Combination with Semaglutide, Demonstrated Superior Weight Loss to Semaglutide Monotherapy in a Preclinical Model. Diabetes 2025, 74 Suppl 1, 847–P. [Google Scholar] [CrossRef]
- Ascletis Pharma Inc. Ascletis Announces ASC47 in Combination with Semaglutide Demonstrated Up to 56.2% Greater Relative Reduction in Body Weight in Participants with Obesity Compared to Semaglutide Monotherapy. Available online: https://www1.hkexnews.hk/listedco/listconews/sehk/2025/0922/2025092200091.pdf (accessed on 17 December 2025).
- Therapeutics, Antag. Antag Therapeutics initiates Phase 1a trial of AT-7687, a First-in-Class GIPR Antagonist Designed to Address Key Gaps in Obesity Treatment. Available online: https://www.globenewswire.com/news-release/2025/04/02/3054121/0/en/Antag-Therapeutics-initiates-Phase-1a-trial-of-AT-7687-a-first-in-class-GIPR-antagonist-designed-to-address-key-gaps-in-obesity-treatment.html (accessed on 17 December 2025).
- Drucker, D.J. GLP-1-based therapies for diabetes, obesity and beyond. Nat. Rev. Drug Discov. 2025, 24, 631–650. [Google Scholar] [CrossRef]
- Saldívar-Cerón, H.I.; Vargas-Camacho, J.A.; León-Cabrera, S.; Briseño-Díaz, P.; Castañeda-Ramírez, A.E.; Muciño-Galicia, A.E.; Díaz-Domínguez, M.R. Oral Small-Molecule GLP-1 Receptor Agonists: Mechanistic Insights and Emerging Therapeutic Strategies. Sci. Pharm. 2025, 93, 26. [Google Scholar] [CrossRef]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).