Submitted:
15 December 2025
Posted:
18 December 2025
You are already at the latest version
Abstract
NKG2D is an activating immunoreceptor expressed by natural killer (NK) cells and subsets of T cells that recognizes multiple stress-induced ligands (e.g., MICA/MICB and ULBP1–6) frequently upregulated in malignant transformation. NKG2D signaling in humans is mediated by the DAP10 adaptor and associated downstream nodes (e.g., PI3K and Grb2–Vav1), which collectively control cytotoxic synapse formation and effector function. NKG2D-based CAR-T cells leverage this multi-ligand recognition to broaden tumor coverage and mitigate single-antigen escape, positioning the approach as a potentially “pan-tumor” CAR strategy. Early clinical experience with autologous NKG2D-CAR products has established feasibility and a manageable safety profile in selected settings; however, objective responses have been inconsistent and durability remains limited. A major set of barriers is intrinsic to the NKG2D axis itself: tumors can downregulate ligand expression, shed MICA/MICB via metalloproteases, and accumulate soluble/vesicular ligands that blunt NKG2D function and promote receptor internalization. These mechanisms synergize with tumor microenvironment (TME) suppression, particularly TGF-β–mediated inhibition of NKG2D pathways. This review synthesizes the mechanistic rationale for NKG2D-CAR designs, summarizes representative clinical programs, and focuses on current challenges and practical mitigation strategies: (i) selectivity and safety when stress ligands are inducible in inflamed normal tissues; (ii) ligand shedding and soluble ligand decoy effects; (iii) TGF-β dominance; (iv) persistence/exhaustion constraints; and (v) manufacturing and development considerations.
Keywords:
NKG2D
; CAR-T
; DAP10
; MICA
; MICB
; ULBP
; ligand shedding
; soluble NKG2D ligands
; TGF-β
; solid tumors
1. Introduction
Conventional CAR-T therapies have achieved durable remissions in select hematologic malignancies, yet efficacy in solid tumors remains limited by antigen heterogeneity, impaired trafficking, and an immunosuppressive TME. NKG2D-based CAR-T cells offer a distinct targeting paradigm: instead of a single antigen, they target multiple stress-induced ligands shared across tumor types (Figure 1). This feature is conceptually attractive for mitigating antigen escape; however, it introduces unique liabilities because NKG2D ligands can be induced dynamically during inflammation and tissue stress.
2. The NKG2D–Ligand Axis: Biology that Defines Both Opportunity and Risk
2.1. NKG2D in NK Cells Versus T Cells (Updated with Figure 1)
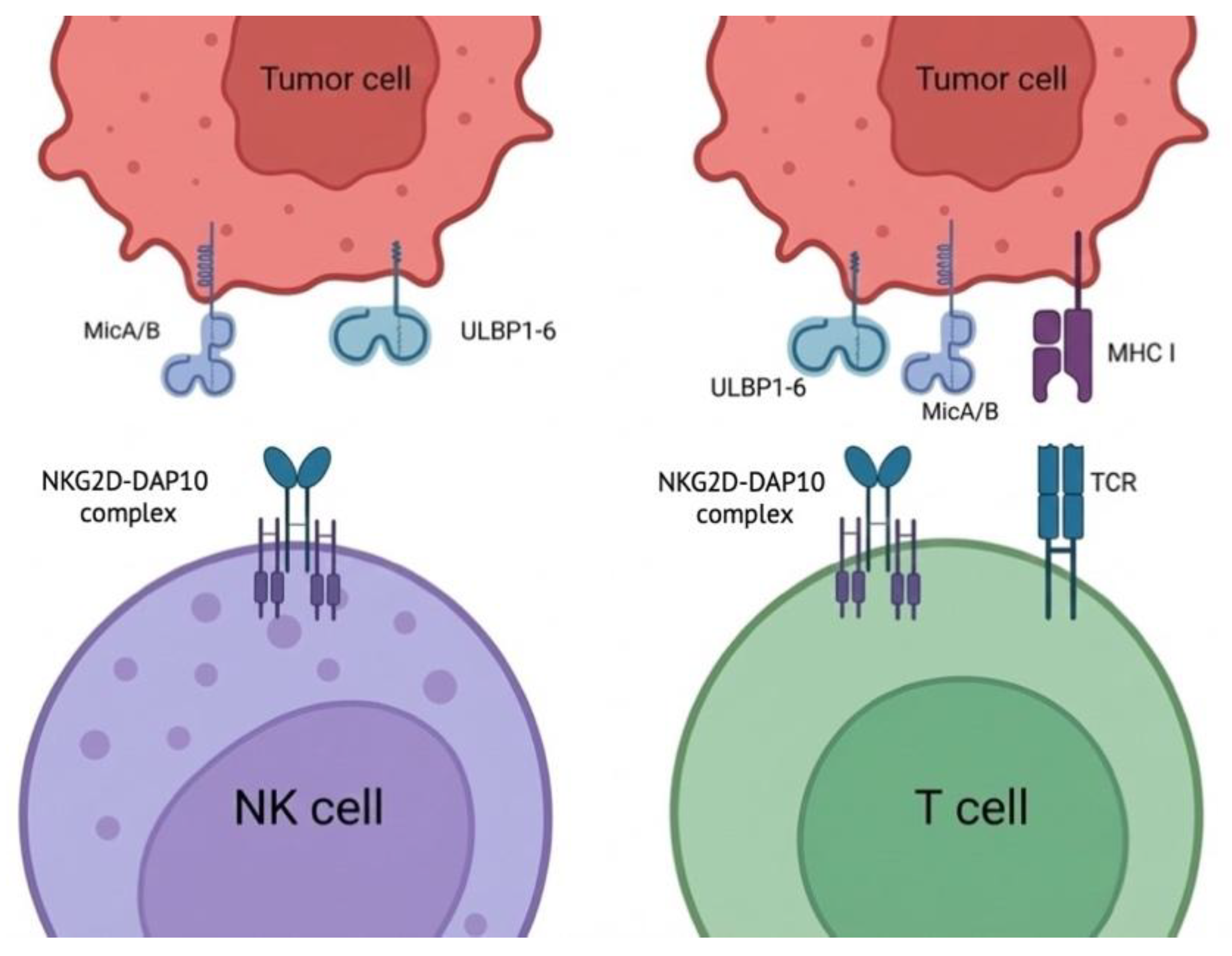
NKG2D is constitutively expressed on NK cells and also on subsets of T cells (notably cytotoxic T cells), where its functional role differs by lineage context. In NK cells, NKG2D acts as a potent activating receptor, while in T cells it typically serves as a co-stimulatory receptor cooperating with TCR signaling (Figure 1). Mechanistically, human NKG2D signals through the DAP10 adaptor, which provides a tyrosine-based motif that recruits PI3K and Grb2-associated signaling nodes (including Vav1), supporting cytoskeletal remodeling and cytotoxic synapse formation.
2.2. Ligand Diversity and Inducible Expression
NKG2D ligands (NKG2DL) include MICA/MICB and the ULBP family (ULBP1–6), which are commonly upregulated by DNA damage responses, oncogenic stress, and inflammatory cues. The multi-ligand nature broadens tumor coverage but complicates target validation because the therapeutic window may narrow when healthy tissues upregulate stress ligands during infection, inflammation, or treatment-related tissue injury.
2.3. Tumor Immune Evasion Through Ligand Shedding and Soluble Decoys
A recurring immune-evasion mechanism is proteolytic shedding of MICA/MICB, which reduces surface ligand density and generates soluble ligands that can impair effector function and promote NKG2D down-modulation. Strategies that inhibit shedding have shown the ability to restore NKG2D-axis antitumor immunity in preclinical models, supporting the view that shedding is not merely correlative but mechanistically consequential.
2.4. TGF-β as a Dominant Suppressor of NKG2D Pathways
TGF-β can downregulate NKG2D expression and suppress NKG2D-mediated tumor immunity across NK and CD8+ T-cell compartments, and this is widely considered a central resistance mechanism in solid tumors. This has direct implications for NKG2D-CAR-T efficacy, as the therapy relies on sustained effector competence under TME cytokine pressure.
3. Engineering NKG2D-CAR-T Cells: Design Implications from Figure 1 Biology
3.1. Recognition Module: “Multi-Ligand Targeting” and Activation Tuning
Most NKG2D-CAR designs use NKG2D as the ligand-binding domain fused to intracellular activation modules (commonly CD3ζ with additional costimulation). Because Figure 1 highlights the breadth of ligand recognition (MICA/B and ULBP1–6), a key development challenge is activation tuning: increasing sensitivity enough to overcome heterogeneous ligand density without creating unacceptable activity against inflamed normal tissues.
3.2. Addressing Fratricide and Self-Ligand Induction
A specific, practical challenge for NKG2D-ligand CAR approaches is that activated immune cells can transiently express stress ligands, contributing to fratricide and reduced persistence in some settings. Next-generation concepts have explored reducing MICA/MICB expression in the product to support persistence.
3.3. Countering Ligand Shedding and Soluble Ligand Suppression
Given the established role of MICA/MICB shedding and soluble ligand decoys, CAR development and clinical protocols may benefit from incorporating (i) baseline and on-treatment soluble ligand measurements and (ii) combination approaches that reduce shedding or neutralize soluble ligand activity.
3.4. TGF-β Resistance and Armored Designs
Because TGF-β can directly blunt NKG2D-axis function, engineering strategies that confer TGF-β resistance (or combination therapies that reduce TGF-β signaling) are particularly relevant for NKG2D-CAR-T in solid tumors.
3.5. Controllability and Safety Engineering
The inducible nature of stress ligands motivates enhanced control layers such as safety switches and logic-gating—especially for clinical scenarios with high baseline inflammation risk.
4. Translational and Clinical Experience (Representative Programs)
Autologous NKG2D-CAR-T therapy (CYAD-01 / NKR-2) has been evaluated clinically, including in the THINK Phase I study. These data support feasibility and provide a safety/dose framework, while also illustrating field-wide challenges in durability and reproducibility of clinical responses.
Allogeneic NKG2D-based CAR-T programs have also reached clinical testing (e.g., alloSHRINK; CYAD-101), with the trial registration documenting key design and treatment context. Program discontinuations in this space highlight that operational, strategic, and clinical-development constraints can terminate programs even when biology remains compelling.
5. Current Challenges in NKG2D-CAR-T Development (Field-Facing)

Table 1.
Major challenges and representative mitigation strategies (updated to reflect Figure 1 biology).
Table 1.
Major challenges and representative mitigation strategies (updated to reflect Figure 1 biology).
| Challenge | Mechanism (NKG2D-axis specific) | Consequence | Mitigation strategies |
|---|---|---|---|
| Inducible stress ligands on normal tissues | Inflammation/tissue injury induces MICA/B, ULBPs | Safety risk; variable therapeutic window | Conservative escalation; inflammation-aware eligibility; controllable CARs; logic gating |
| Ligand heterogeneity & dynamics | Spatial/temporal variation in surface NKG2DL | Mixed responses; early relapse | Multi-site profiling; longitudinal biomarker monitoring; combinations that increase surface ligands |
| Ligand shedding and soluble/vesicular decoys | ADAM-mediated shedding; soluble MICA/B reduces function and down-modulates NKG2D | Reduced potency; systemic/local suppression | Add soluble ligand biomarkers; shedding inhibition/neutralization strategies |
| TGF-β dominance in TME | Downregulates NKG2D pathways and cytotoxic function | Low activity in solid tumors | TGF-β–resistant designs; TGF-β pathway combinations |
| Persistence/exhaustion constraints | Chronic stimulation + metabolic stress | Limited durability | Memory-biased manufacturing; signaling tuning; cytokine/chemokine armoring |
| Development/manufacturing complexity | Patient variability and cost; allogeneic immunologic constraints | Program viability risk | Biomarker-enriched trials; fit-for-purpose potency assays; early go/no-go endpoints |
6. Future Directions
Development priorities that map directly onto the Figure 1 biology include: (i) standardized quantification of tumor surface NKG2DL and soluble ligands; (ii) rational combinations to stabilize surface ligands and counter shedding; (iii) engineering to resist TGF-β–mediated suppression; and (iv) controllable CAR architectures that protect the therapeutic index in inflammatory clinical contexts.
7. Conclusions
NKG2D-based CAR-T cells provide an inherently multi-ligand targeting strategy (Figure 1) with the potential to reduce single-antigen escape. However, the same stress-ligand biology that enables breadth also drives key liabilities: inducible expression on stressed tissues, ligand shedding and soluble decoy effects, and potent suppression by TGF-β in the TME. Evidence to date indicates feasibility and manageable safety in early studies, but durable efficacy will likely require biomarker-guided clinical positioning plus next-generation engineering focused on shedding/soluble ligand biology, TGF-β resistance, and controllability.
Funding
This work was funded by the subsidy allocated to Kazan Federal University for the state assignment in the sphere of scientific activities (project number FZSM-2023-0011).
Conflicts of Interest
The author(s) declare no conflicts of interest.
References
- Crane, C. A. et al. TGF-downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro-Oncology 12, 7–13 (2010). [CrossRef]
- Ferrari de Andrade, L. et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell–driven tumor immunity. Science 359, 1537–1542 (2018).
- Xing, S. & Ferrari de Andrade, L. NKG2D and MICA/B shedding: a ‘tag game’ between NK cells and malignant cells. Clinical & Translational Immunology 9, (2020).
- Michaux, A. et al. Clinical Grade Manufacture of CYAD-101, a NKG2D-based, First in Class, Non–Gene-edited Allogeneic CAR T-Cell Therapy. Journal of Immunotherapy 45, 150–161 (2022).
- Sallman, D. A. et al. CYAD-01, an autologous NKG2D-based CAR T-cell therapy, in relapsed or refractory acute myeloid leukaemia and myelodysplastic syndromes or multiple myeloma (THINK): haematological cohorts of the dose escalation segment of a phase 1 trial. The Lancet Haematology 10, e191–e202 (2023). [CrossRef]
- Curio, S., Jonsson, G. & Marinović, S. A summary of current NKG2D-based CAR clinical trials. Immunotherapy Advances 1, (2021). [CrossRef]
- Zingoni, A., Vulpis, E., Loconte, L. & Santoni, A. NKG2D Ligand Shedding in Response to Stress: Role of ADAM10. Frontiers in Immunology 11, (2020). [CrossRef]
- Wensveen, F. M., Jelenčić, V. & Polić, B. NKG2D: A Master Regulator of Immune Cell Responsiveness. Frontiers in Immunology 9, (2018). [CrossRef]
- Upshaw, J. L. et al. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nature immunology 7, 524–32 (2006). [CrossRef]
Figure 1.
NKG2D recognition of stress ligands in NK and T cells (schematic). Tumor cells can express NKG2D ligands, including MICA/B and ULBP1–6. Left: in NK cells, ligand engagement triggers activation through the NKG2D–DAP10 complex. Right: in T cells, NKG2D signaling (via DAP10) cooperates with TCR recognition of peptide–MHC I, consistent with a co-stimulatory role.
Figure 1.
NKG2D recognition of stress ligands in NK and T cells (schematic). Tumor cells can express NKG2D ligands, including MICA/B and ULBP1–6. Left: in NK cells, ligand engagement triggers activation through the NKG2D–DAP10 complex. Right: in T cells, NKG2D signaling (via DAP10) cooperates with TCR recognition of peptide–MHC I, consistent with a co-stimulatory role.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



