Submitted:
15 December 2025
Posted:
16 December 2025
You are already at the latest version
Abstract
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are devastating and currently untreatable neurodegenerative diseases, whose genetic and molecular etiologies remain largely unclear. The histopathological hallmark of both diseases is the cytoplasmic deposition of TDP-43 in neurons, which is attributed to both intrinsic (e.g., mutations, aberrant cleavage) and extrinsic factors (e.g., prolonged oxidative stress, impaired clearance pathways). Mutations and certain PTMs (e.g., cysteine oxidation) destabilize RNA binding, promoting monomer misfolding and increasing its half-life. Disruptions to core ubiquitin-proteasome system (UPS) subunits impede efficient processing, contributing to the clearance failure of misfolded TDP-43 monomers. The accumulation of monomers drives phase separation within stress granules, creating nucleation hotspots that eventually bypass the thermodynamic barrier, resulting in exponential growth. This rapid growth then culminates in the failure of the autophagy-lysosome pathway (ALP) to contain the aggregation, resulting in a self-sustaining feed-forward loop. Here, we synthesize these factors into a unified kinetic cascade model. Therapeutic strategies must therefore move beyond simple clearance and focus on targeting these kinetic inflection points (e.g., oligomer seeding, PTM modulation).
Keywords:
TDP-43 proteinopathy
; proteostasis collapse
; ubiquitin-proteasome system (UPS)
; autophagy-lysosome pathway (ALP)
; phase separation
; post-translational modifications (PTMs)
; amyotrophic lateral sclerosis (ALS)
; frontotemporal dementia (FTD)
; neurodegeneration
1. Introduction
Trans-active response DNA-binding protein (TDP-43) is a versatile RNA-binding protein that plays roles in RNA splicing, RNA stabilization, mRNA transport, gene expression control, and the regulation of translation for other proteins [1,2,3]. It is also critical for embryonic and neural development; for instance, TDP-43 knockout models of mice and zebrafish exhibited embryonic lethality and motor deficits [4]. Alternative splicing, maintaining RNA stability, and processing non-coding RNAs in transcripts are all critical for cell differentiation and organogenesis. The loss of TDP-43 disrupts these processes, resulting in failed development [4] and preventing the expression of synaptic proteins necessary for neurodevelopment, which leads to motor deficits [4].
TDP-43 is primarily localized in the nucleus, and a fraction of its total cellular concentration can be found in the cytoplasm, where it performs its other functions. Its subcellular distribution was thought to be regulated by two elements: a nuclear localization signal (NLS) and a nuclear export signal (NES). The NLS (residues 82–98) tags TDP-43 for importation into the nucleus, ensuring that the protein stays there in a soluble state [5,6]. Conversely, the NES (residues 239–250), initially thought to be required for protein egress, was later shown to be non-functional [5,7]. Nevertheless, TDP-43 has been shown to have additional mechanisms, such as the self-regulation of its own mRNA, which allows it to adjust its expression when it becomes overabundant in other cellular regions [8].
Despite this, mutations, prolonged cellular stress, and aberrant post-translational modifications (PTMs) can cause the protein to misfold and form irreversible aggregates that accumulate in the cytoplasm [9,10]. These cytoplasmic aggregates are a hallmark observation in TDP-43 proteinopathies, namely amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) [11,12,13]. Recent neuropathological findings of TDP-43 inclusions concentrated in the hippocampus and limbic structures have also been designated as limbic-predominant age-related TDP-43 encephalopathy (LATE), a condition that becomes more common in older populations (ages beyond 80) and appears to mimic AD but is primarily driven by TDP-43 proteinopathy [14]. In these diseases, the formed TDP-43 oligomers further accelerate the pathological process, acting as seeds and sequestering free and functional TDP-43, eventually leading to nuclear depletion [15,16]. This leads to devastating neurodegenerative consequences, as the protein can no longer perform its regulatory functions required for gene expression and alternative splicing, which are essential for neuronal maintenance and survival.
Nevertheless, several compensatory pathways within the cell help identify and process disordered biomolecules for degradation and clearance. For disordered proteins (e.g., pathologic TDP-43), these checkpoints are in the form of molecular chaperones, addition of PTMs, SUMO-Ubiquitin networks, packaging in lysosomes, aggresomes, and exosomes [9,17,18,19,20]. However, these aggregates possess intrinsic properties and external factors within their immediate cellular environment that enable them to evade these checkpoints and remain in the cytoplasm. In this review, the functional attributes of pathological TDP-43 and their disruptive roles in TDP-43 proteostasis, sequestration, and catabolic clearance will be discussed. Elucidating the mechanistic details of how pathological TDP-43 circumvents these pathways will aid in understanding the pathogenesis of similar proteinopathies and inform the development of actionable targets for therapeutic applications and biomarker discovery.
2. TDP-43 is Intrinsically Aggregation-Prone Due to Its C-Terminal Domain
2.1. Structural Domains and Aggregation-Prone Regions
TDP-43 is a highly conserved protein made up of 414 amino acids. Its full length is organized into four major domains: (1) an N-terminal domain (NTD), that spans approximately 1–80 residues, (2) two RNA Recognition Motifs (RRMs, at residues 105–169 and 194-257 respectively), which are a characteristic feature of heterogeneous nuclear ribonucleoprotein (hnRNP) proteins responsible for RNA recognition and protein interactions, and (3) a highly disordered glycine-rich C-terminal domain (CTD) or prion-like domain (residues 277–414) (Figure 1) [21,22]. Other studies refer to the CTD as the glycine-rich region (GRR) or the low-complexity domain (LCD).
The CTD is unstructured and contains most of the documented ALS-associated mutations, thus it is considered a significant driving force of protein assembly [23,24]. Studies have revealed that the liquid droplet environment, electrostatic repulsion, the presence of CTD segments that act as steric zippers, and low-complexity aromatic-rich kinked segments (LARKS) that can be converted to irreversible interactions by fALS variants are all located in the CTD and play a common role in increasing the rate of TDP-43 aggregation [25,26]. As for the other TDP-43 domains, molecular modeling revealed different segments from the NTD (24GTVLLSTV31), RRM1 (128GEVLMVQV135), and RRM2 (247DLIIKGIS254) as aggregation-prone and were confirmed to have amyloid-fibril propensities through various biophysical techniques and molecular dynamics simulations [27]. The simulations revealed that the RRM2 peptides transitioned from random coil to ß-sheet rich structures, forming parallel and anti-parallel ß-sheets, even in the absence of aromatic residues, underscoring RRM2 as a critical structural region for TDP-43 aggregation [27].
Complementing these earlier findings on aggregation-prone regions, Kumar et al. applied proteolytic cleavage to recombinant TDP-43 filaments, revealing a hidden core sequence within the CTD (residues 279–360), which was found to be necessary for the propagation of TDP-43 proteinopathy [22]. Notably, this core sequence resembled that described by Arseni et al. when they isolated TDP-43 pathological filaments from the brains of ALS/FTLD patients [28]. This core sequence (residues 282–360) had a double-spiral shape fold, whereas the former (in vitro) was buried and flanked by the NTD and RRMs (Figure 1) [22,28]. Despite the striking similarity in the primary sequence, the significant difference in the fold structure underscores the importance of assessing full-length proteins in their native contexts.
2.2. Intrinsic Self-Regulatory Mechanisms
Although its CTD is intrinsically disordered and predisposed to aggregation, Afroz et al. (2017) showed that the NTD also interacts to form physiological oligomers under stress. The formed TDP-43 oligomers result in the spatial separation of the CTD ends, minimizing the chances of contact between these regions and thereby reducing aberrant aggregation [29]. This suggests that TDP-43 has evolved an intrinsic self-regulatory mechanism to counteract its own tendency to aggregate. These findings were supported in a subsequent paper by Jiang et al. (2017), which showed that the NTD is primarily responsible for the formation of homodimers in solution, and further tetramerization occurs through intermolecular disulfide bonds [30]. More importantly, they showed that mutations destabilizing the NTD dimer significantly enhanced the formation of cytoplasmic inclusions, indicating that NTD dimerization is essential in protecting against aggregation [30].
Another study by Wang et al. (2018) further demonstrated the critical role of NTD in enhancing TDP-43 polymeric assembly and liquid-liquid phase separation (LLPS), where proteins and RNA condense into dynamic, droplet-like compartments. Since the interaction of NTD is much weaker and less spontaneous than CTD, the authors hypothesized that TDP-43 phase separation is favored through the synergistic cooperation of NTD and CTD interactions [31]. Solubility shifts of TDP-43 constructs made of NTD alone, CTD alone, and full-length TDP-43 were compared using turbidity assays and microscopic observation of droplet formations. The LLPS of full-length TDP-43 was found to be more pronounced than that of CTD alone, suggesting the contributory role of NTD interaction sites [31]. As for NTD alone, the construct does not readily undergo LLPS but instead forms weak, reversible head-to-tail polymers in a linear arrangement. Most importantly, introducing a phosphomimetic substitution (S48E) in the NTD disrupted head-to-tail assembly, thereby weakening and raising the concentration threshold for LLPS, despite the CTD’s inherent ability to phase separate, further validating the synergistic involvement of the NTD in phase separation [31].
While the cooperation between the NTD and the CTD underscores how TDP-43 regulates the functional assembly of oligomers with pathological aggregation, neuropathological findings reveal that this balance is not maintained equally across disease contexts, as evidenced by the absence of TDP-43 inclusions in certain forms of ALS. Specifically, Tan et al. found TDP-43 inclusions in the brains and spinal cords of sporadic ALS (sALS) and non-mutant superoxide dismutase 1 (SOD1) familial ALS (fALS), but not in two mutant SOD1 cases [32]. Their findings were corroborated by Mackenzie et al., who found TDP-43 inclusions in sALS and non-SOD1 fALS neurons, but not in SOD1 fALS tissue [33,34]. A question was then put forward as to what was making TDP-43 inclusions common in sALS and not as easily observable in fALS tissues [34]. This prompted another group to explore if TDP-43, on its own, was prone to aggregation or sequestered by other aggregated components, thus simply making it a “marker of disease” [35].
In their study, Johnson et al. demonstrated that recombinant TDP-43, without interacting cofactors, rapidly formed aggregates after a lag phase of approximately 5-10 minutes, as indicated by increased solution turbidity and a corresponding increase in pellet size following centrifugation [35]. Identical conditions were applied to control proteins (e.g., BSA, soybean trypsin inhibitor, creatine kinase, and GFP), but no aggregation was observed, indicating that TDP-43 is inherently prone to aggregation [35]. Using a yeast mutant model, the same group showed that reported ALS mutations (e.g., Q331K, M337V, Q343R) accelerated aggregation in vitro and increased the formation of aggregates in vivo.
In summary, these findings highlight that TDP-43 is inherently prone to aggregation, with its sequence and domains already predisposing it to self-assembly. Consequently, even in its native state, the protein exhibits oligomerization hotspots that could ultimately manifest as pathological inclusions. This underscores why the protein is heavily dependent on stringent and continuous quality control. Two distinct degradatory and clearance pathways help clear aberrant, misfolded proteins and the aggregates they form. However, several factors can disrupt their principal components, which will be discussed in the succeeding sections.
3. TDP-43 Evasion of Cellular Clearance Systems
3.1. Dual-Pathway Control: The Proteasome and Autophagy Governing TDP-43
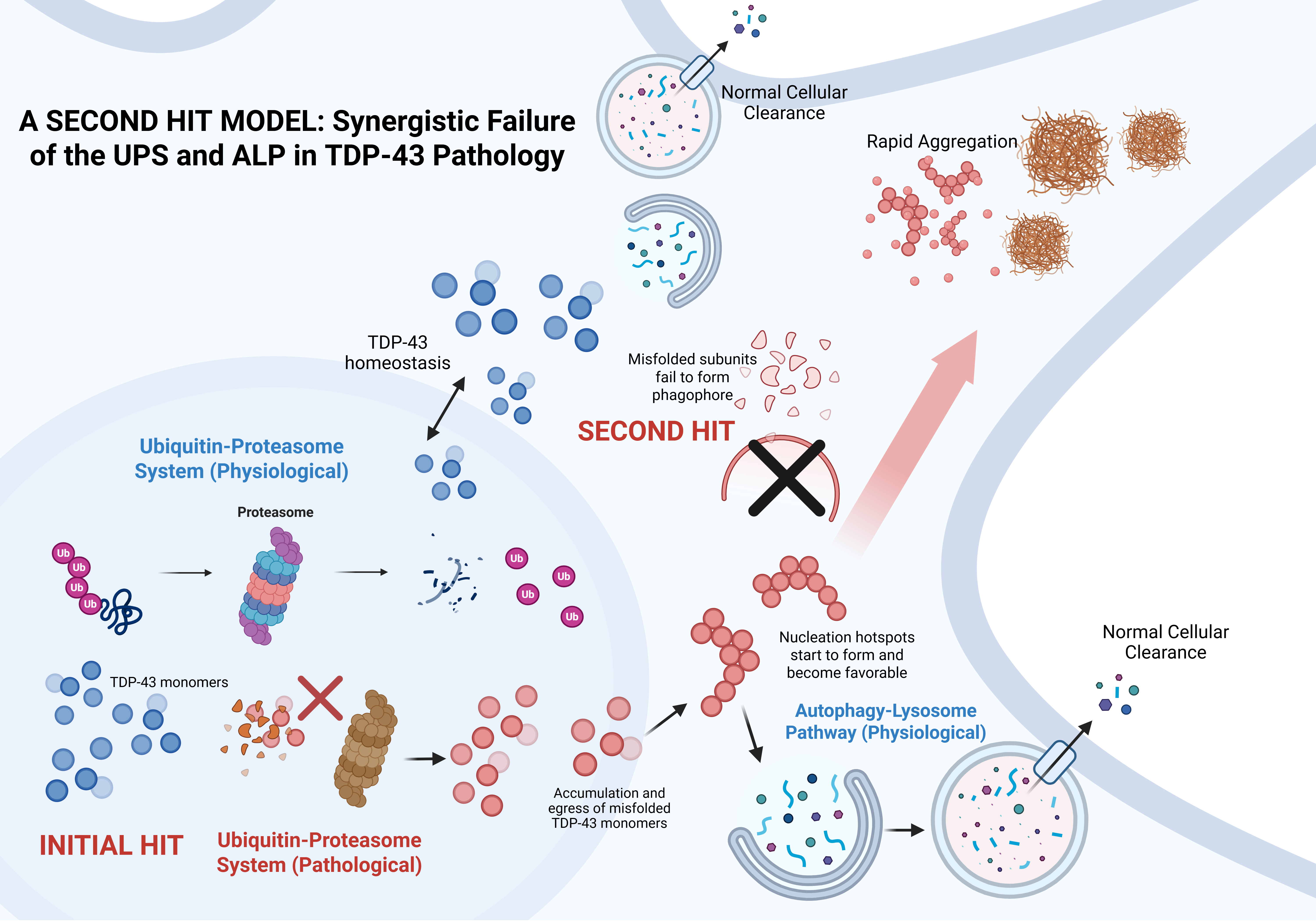
The degradation of aberrant TDP-43 depends on two central quality control systems: (1) the Ubiquitin-Proteasome System (UPS), which degrades soluble, ubiquitinated monomers, and (2) the Autophagy-Lysosome Pathway (ALP), which sequesters larger aggregates in double-membrane vesicles for lysosomal fusion and protease-mediated degradation [36]. For larger, aggregated proteins, the ALP takes over, assisted by adaptor proteins such as sequestosome 1 (p62) and NBR1, which bind to polyubiquitin chains and simultaneously interact with LC3 [36]. Polyubiquitin tags are not limited to UPS, but are also recognized by the ALP via p62 and NBR1 [37,38,39].
While ubiquitin tagging can guide proteins toward either proteasomal or autophagic degradation, the exact functional partitioning between the two systems remained unclear for TDP-43. Scotter et al. (2014) explored this gap, ultimately demonstrating that the UPS processes soluble TDP-43, whereas the ALP handles aggregates to facilitate their clearance [40]. More importantly, they found that cellular macroaggregates of TDP-43 are reversible when both systems are functional, and further postulated that a “second hit” in either of these two systems drives the pathological cascade of TDP-43 aggregation in ALS and FTD [40]. In their study, stable cell lines were generated in which expression of different HA-tagged TDP-43 variants could be induced with doxycycline. The expressed constructs were: (1) wild-type (HA-TDP-43-WT), (2) ΔNLS-truncated mutant (HA-TDP-43-ΔNLS), and (3) C-terminal fragment (aa 181–414) (HA-TDP-43-CTF). Both the localization and expression of the constructs were then observed following treatment with autophagy inhibitors (e.g., 3-methyladenine (3MA), bafilomycin (Baf)) or UPS inhibitors (e.g., MG132 and epomoxin) [40].
The results revealed a distinct separation of TDP-43 processing between the two degradation systems. Autophagy was shown to be the leading player in clearing TDP-43 CTFs, whereas the UPS primarily targeted soluble WT/ΔNLS TDP-43 [40]. Inhibiting UPS resulted in the pronounced accumulation and aggregation of soluble TDP-43 species, while inhibiting autophagy resulted in attenuated effects [40]. Conversely, the activation of autophagy with trehalose or lithium chloride (LiCl) enhanced CTF degradation, validating its principal role in clearing TDP-43 aggregates [40]. When these findings are viewed holistically, they build the following picture: (1) degradation of soluble TDP-43 species is mainly via the UPS, (2) once protein oligomerization is initiated, these species will have become too large to enter the proteasome pore, requiring the intervention of another pathway (i.e., autophagy) [40]. Notably, the study demonstrates that only when both systems are functional can the cells completely reverse TDP-43 macroaggregate formation, further substantiating the cooperative interplay between the UPS and autophagy pathways.
These findings are consistent with previously published work, which shows that different degradative pathways process distinct TDP-43 species. Specifically, Wang et al. (2010) showed that the truncated form of TDP-43 (TDP-25) was more reliant on the autophagy pathway [41]. Furthermore, Urushitani et al. (2010) revealed the synergistic effect of the proteasome and autophagosome in clearing polyubiquitinated TDP-43, corroborating the dual-pathway requirement [42]. Additionally, proteasome inhibition has been shown to drive the cytoplasmic accumulation and aggregation of neuronal TDP-43, supporting the central role of the UPS in degrading soluble TDP-43 species [43]. Conversely, the role of autophagy in clearing truncated or aggregation-prone species was demonstrated through the rapamycin-mediated activation of the pathway, resulting in the rescue of mislocalized TDP-43 and reduction of CTF accumulation in the cytosol [44]. The tight, cooperative interplay between the UPS and ALP systems is critical for TDP-43 proteostasis, providing the mechanistic rationale for how a ‘second hit’ to either system triggers the pathological cascade observed in ALS and FTLD.
3.2. CTD Mutations Increase Structural Stability and Resistance
The domains of TDP-43 are indeed critical in influencing its folding behavior, particularly the highly disordered CTD. Other domains, such as the NTD, were demonstrated to have the capability of counterbalancing this intrinsic disorder through specific structural arrangements that provide spatial separation of the CTD, thereby attenuating uncontrolled folding and aggregation. However, certain conditions, such as the presence of mutations, can enhance the stability of its folding, potentially shifting the balance toward more aggregation-prone conformations.
Particularly, mutations (G298S, Q331K, M337V) in the CTD introduce structural modifications in its primary sequence, which potentially lead to impairment in lysosomal or proteasomal degradation, as well as favoring irreversible aggregation over normal LLPS, thereby decreasing TDP-43’s susceptibility to degradation [45,46,47,48,49]. An early study by Ling et al. (2010) explored the effect of these CTD mutations on the half-life of TDP-43. Here, tetracycline-inducible wild-type and mutant genes were expressed using a cytomegalovirus (CMV) promoter in isogenic cell lines. By randomly cycling the cells through brief incubation with [35S]methionine/cysteine, newly synthesized proteins were radiolabeled and subsequently monitored over time to measure their half-lives [50]. The results showed that mutant proteins were degraded two to four times more slowly than wt-TDP-43 [50]. Moreover, immunoprecipitation results indicated stronger interaction between endogenous wt-FUS/TLS and TDP-43 mutants, suggesting that mutation-induced CTD stabilization not only affects folding but also enhances cross-interaction with FUS/TLS [50].
The structural disruptions resulting from these mutations (e.g., A321G, A321V, Q331K, M337V) were analyzed in detail using NMR spectroscopy, simulation, and microscopy, revealing a CTD subregion that transiently folds into a helix, thereby mediating TDP-43 phase separation [46]. The mutations disrupt this phase separation by inhibiting the interaction and destabilizing the α-helical structure. Notably, A321G and Q331K eliminated the phase separation of the CTD, while M337V decreased solution turbidity by approximately 50% [46]. Removing the region containing the α-helical structure and the mutations (Δ321-343), and the helix-disrupting mutation (A326P) resulted in the loss of phase separation, suggesting the participation of this helical region is essential in LLPS [46]. At low concentrations, the truncated CTD as well as the mutant CTD remained soluble but easily formed insoluble aggregates when their concentrations were increased, demonstrating that this helical region is necessary for stabilizing the liquid-like state of TDP-43 [46]. Zeng et al. (2023) similarly showed the disruptive effect of M337V in the same region with their own simulations, corroborating the above results [48]. Taken together, these findings suggest that residues 321–343 form a critical region that promotes and maintains the reversibility of TDP-43 LLPS through its intermolecular helix-helix interactions with the same region of other TDP-43 molecules. However, mutations within this segment disrupt these interactions, destabilizing LLPS and thereby shifting the equilibrium toward irreversible aggregation.
3.3. Mutations Can Also Increase Vulnerability to Enzymatic Cleavage
So far, we have established that mutations in the CTD are a disruptive force that favors the formation of irreversible aggregates. However, some mutations paradoxically render TDP-43 more vulnerable to protease-mediated cleavage. Investigations into the role of calpains in promoting the cleavage of mutant TDP-43 proteins were prompted by the hypothesis that the downregulation of adenosine deaminase acting on RNA 2 (ADAR2) enzymes enhances the generation of TDP-43 pathology [51]. ADAR2 enzymes convert the adenosine found in the Q/R site of GluA2 (the protein subunit of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid or AMPA receptors) pre-mRNA to inosine, facilitating the impermeability of AMPA receptors to calcium ions (Ca2+) [52,53]. However, the motor neurons of patients with sporadic ALS have reduced levels of ADAR2, resulting in leaky AMPA receptors due to the decreased processing of GluA2 pre-mRNA [54,55]. The reduced ADAR2 leads to increased uptake of Ca2+, which can be neurotoxic. The increased Ca2+ uptake likewise prompted Yamashita et al. to investigate the proportional effect of Ca2+-dependent proteases on TDP-43 pathology, and they found an increased generation of C-terminal fragments (CTFs), with A315T and M337V TDP-43 mutants being cleaved faster than wild-type TDP-43 (Figure 1) [51]. The group further demonstrated that knocking out AR2 in mice led to TDP-43 mislocalization and cleavage, resulting from calpain activation triggered by Ca2+ AMPA receptors. In contrast, AR2res (AR2-restored) mice, which expressed Ca2+-impermeable AMPA receptors even in the absence of AR2, showed no TDP-43 pathology or calpain activation, confirming that Ca2+ influx is the trigger of TDP-43 pathology [51].
A separate study by Chiang et al. (2016) demonstrated that the D169G mutation exhibits higher thermal stability compared to wild-type TDP-43 and is cleaved by caspase-3 more efficiently, resulting in the production of more CTFs (TDP-35) in neuroblastoma cells. The D169G mutation is located in the RRM1 domain and induces a local conformational change in a ß-turn, resulting in increased hydrophobic interactions within the RRM1 core (Figure 1) [56]. The resulting more rigid conformation possibly exposes more caspase-3 cleavage sites to the solvent, thereby enhancing cleavage efficiency. Based on these findings, the effects of TDP-43 cleavage appear paradoxical at first glance; cleavage is generally expected to be a biologically favorable process that facilitates the clearance of degradation of TDP-43 deposits from cells. However, this is not always the case; the cleavage site and the properties of the resulting fragments are critically important, as specific cleavage fragments can, in fact, exacerbate aggregation. Specifically, the generation of CTFs has been shown to promote aggregation due to their increased tendency to self-associate and nucleate TDP-43 oligomers [57,58,59,60,61,62]. In this case, cleavage is not inherently beneficial; however, the type and properties of the resulting fragments dictate the likelihood of their clearance or persistence within the cytoplasm.
3.4. Proteasomal Evasion: Failure to Recognize and Process Soluble Species
As established in the section above, the UPS is the primary pathway for processing soluble TDP-43 monomers. The UPS and the autophagy-lysosome pathways have distinct roles, and the latter cannot compensate for the failure of the former to process TDP-43 soluble species. This functional specialization means that the persistence of pathological TDP-43 in the cytoplasm of affected neurons is a critical indication that the UPS is somehow overwhelmed or circumvented in disease states. Proteasomal evasion, therefore, is not just a consequence of aggregate size exceeding the 20S core aperture but involves specific molecular factors that impede efficient substrate recognition and digestion. The failure of the UPS is linked primarily to two factors that direct the toxic shift towards TDP-43 oligomerization and aggregation: (1) intrinsic substrate characteristics that disrupt ubiquitination, and (2) extrinsic factors that compromise proteasome function.
3.4.1. Intrinsic Substrate Factors That Influence UPS Effectivity
The contribution of each lysine residue to the ubiquitination of TDP-43 was explored by Hans et al. (2018) using single and multiple-site lysine mutants. Their study revealed that the four lysine residues in the CTF, spanning residues 193–414, do not contribute to ubiquitination; however, all four lysines needed to be removed for complete suppression of ubiquitination [63]. Additionally, the substitution of Lys-408 enhanced the pathological phosphorylation of Ser-409/410, a marker commonly observed in TDP-43 pathology [21,63,64,65]. However, these effects were not observed when extended to full-length TDP-43. Mutagenesis of Lys-84 and Lys-95 in the NLS reinforced that these residues had a more significant role in ubiquitination compared to any of the CTD lysines [63]. Overall, these findings suggest that TDP-43 harbors multiple, functionally redundant ubiquitination sites distributed across its domains. Thus, inefficient TDP-43 ubiquitination likely stems not from a lack of modification sites but from other intrinsic properties of the protein.
The disordered nature of the CTD promotes aggregation and phase separation, potentially masking ubiquitination sites and interfering with UPS interaction [10,66]. Specifically, the misfolding of the protein and the formation of aggregates make ubiquitination sites less accessible to E3 ligases and other components of the UPS, thereby decreasing the efficiency of recognizing the tagged proteins and their subsequent degradation. Moreover, the accumulated CTFs in the cytoplasm have been shown to interact with proteasome assembly proteins, thereby disrupting the formation of the proteasome complex and resulting in inefficient degradation [67]. TDP-43 aggregates were also demonstrated to sequester ubiquitin and UPS components into inclusions, depleting the pool of free ubiquitin needed for tagging aberrant proteins [68]. Thus, the impairment of UPS activity creates a vicious cycle, accelerating TDP-43 aggregation and leading to cellular dysfunction and neurotoxicity. While the CTD has been extensively documented for its role in aggregation and contribution to UPS evasion, NTD conformation was also suggested to influence TDP-43 aggregation dynamics, albeit through more subtle and less frequent mechanisms.
A study by Qin et al. (2014), through the use of NMR spectroscopy and CS-Rosetta modeling (supported by 78 unambiguous long-range NOE constraints), revealed that the NTD of TDP-43 adopts a previously unknown ubiquitin-like fold despite not bearing any sequence homology with ubiquitin [69]. NTD conformations were also demonstrated to exist in a concentration-dependent equilibrium, comprising a well-folded state and a highly disordered (unfolded) state. Any disturbance to this equilibrium may favor the unfolded state and possibly promote aggregation. The folded state was also shown to bind single-stranded DNA (ssDNA), and this interaction favors the shift towards the more stable conformation, underscoring a potential safeguard that ensures TDP-43 remains structurally viable. Thus, in an event where DNA/RNA binding is impaired, the unfolded NTD becomes more favorable, leading to increased aggregation propensity and reduced UPS accessibility.
3.4.2. Extrinsic Factors that Modulate UPS Activity
Complementing these intrinsic properties are extrinsic factors, which can modulate or intensify the disruption of UPS activity. The 26S proteasome consists of a 20S core particle, a hollow complex composed of four heteroheptameric rings with an α7-ß7-ß7-α7 orientation [70,71]. A small gate or flap (the N-termini of α-subunits) folds over the 13Å aperture of the core subunit, preventing unregulated access to catalytic sites within [72]. The opening of the gate is controlled by the binding of the HbYX motif from the 19S ATPases to intersubunit pockets on the α-ring, creating a conformational change that permits substrate entry [73]. However, pathologic proteins were shown to bind to these intersubunit pockets, rather than engaging the active site to open the core subunit (as in HbYX), which further stabilizes the closed conformation and impairs gate activation altogether.
This principle of inhibition by extrinsic oligomers has been demonstrated for Aß, α-synuclein, and mutant huntingtin, which share a common structural epitope that interacts with the intersubunit pockets of the 20S core particle to inhibit gate opening allosterically [74]. While TDP-43 oligomers have been detected in patient tissues, their role in inhibiting the proteasome remains unclear. Notably, spherical TDP-43 oligomers have been detected in the hippocampal and frontal cortical tissue of FTD-TDP patients using oligomeric TDP-43-specific antibodies that bind to a different epitope [75]. However, a study by Riemenschneider et al. expressed GFP-TDP-25 (residues 220–414) in primary neurons, showing (via cryo-electron tomography and subtomogram) an eight-fold increase of 26S proteasomes inside TDP-25 inclusions compared to the surrounding cytoplasm. They further demonstrated that these sequestered proteasomes exhibited substrate-processing conformations of the 19S regulatory particle, and ground-state particles were nearly absent, strongly indicating that these proteasomes were stalled mid-cycle due to the interaction [76]. Additionally, an extra density in the 3D average that was not accounted for by known proteasome subunits was observed and regarded as a substrate or adaptor protein bound to the 19S regulatory subunit [76]. Thus, these findings suggest that the CTF of TDP-43 is more likely to interact with the 19S regulatory subunit of the proteasome rather than the 20S core subunit. Other studies pointed to TDP-43 interacting with proteasome assembly factors PSMG2 and PSD13, reducing the pool of fully assembled proteasomes [67]. Nevertheless, by analogy to A11-positive Aß, α-synuclein, and huntingtin oligomers, it is possible that certain TDP-43 oligomeric strains could engage the 20S core and also disturb gate dynamics, but this remains to be tested with purified proteasomes [74].
Taken together, these findings underscore the central role of a common structural epitope found in protein oligomers that arise from common neurodegenerative diseases in inhibiting proteasomal function. This suggests that the specific oligomer type is secondary to the structural conformation causing the interference; thus, targeting this unique shape may restore proteasomal activity. Equally important is considering the significance of the potential impact that comorbid expression of these oligomers will have on the failed clearance of TDP-43 aggregates, as observed in some pathologies [77].
Additionally, other sources of stress can further burden the UPS, particularly the proteasome. Endoplasmic reticulum (ER) stress, for instance, has been shown to compromise proteasomal degradation, as treatment with various ER stressors delayed the turnover of ER-localized reporters and led to the subtle but consistent accumulation of multiple nuclear/cytosolic ones [78]. A similar accumulation was observed in transgenic mice expressing UPS reporter substrates following induction of ER stress. Furthermore, ER-stressed cells failed to clear UBB+1, an aberrant ubiquitin variant associated with conformational diseases, resulting in impaired UPS activity [78].
Oxidative stress represents another primary consideration that undermines UPS efficiency. Some studies have shown that mild oxidative stress can transiently enhance UPS activity; however, chronic or severe oxidative stress can compromise both ubiquitin-conjugating enzymes and the proteasome itself [79,80]. Key residues in the proteasomal core and its regulatory subunits can undergo oxidative structural changes, rendering the complex functionally impaired [81,82]. Oxidation of critical active sites can likewise reduce proteolytic efficiency [83,84]. In addition, cysteine residues in E1, E2, and E3 enzymes, which are responsible for ubiquitin conjugation, are susceptible to oxidation, potentially compromising their activity [79,80]. Nevertheless, in some cases, protein degradation can still proceed independently of ubiquitination [79]. Ultimately, the excessive accumulation of aberrant proteins can obstruct the proteasomal gate, creating a vicious cycle in which progressive impairment of degradation pathways further enhances proteotoxic stress [82,84].
Alongside ER and oxidative stress, other chronic cellular insults such as hypoxia, hyperglycemia associated with diabetes, and persistent neuroinflammation were also reported to contribute to this systemic burden on the UPS [85,86,87,88,89]. Ultimately, the interplay of physical occlusion, resulting from chemical modifications, and chronic cellular stress compromises the biological efficiency of the UPS, allowing soluble TDP-43 to persist and rapidly transition into aggregation-prone species, which subsequently burden the autophagy-lysosomal pathway.
3.5. Autophagic Evasion
Building on what was introduced earlier, protein processing by the UPS can occasionally be inaccurate and exacerbated by the stressors mentioned above. Hence, misfolded TDP-43 oligomers that slip past the UPS could coalesce into larger aggregates that the ALP processes. Autophagy is the cell’s main bulk degradation pathway, which sequesters large, insoluble aggregates, damaged organelles, and aberrant proteins into specialized vesicles called autophagosomes, which then fuse with lysosomes, organelles that contain hydrolytic enzymes for degradation (Figure 2) [90,91,92]. Given its high processing capacity, the ALP should, in theory, be capable of processing and clearing the protein aggregates that escape the UPS. However, the unopposed formation of TDP-43 aggregates is associated with the dysfunction of the ALP at several critical stages, resulting from a failed recognition, impaired influx, and compromised downstream clearance, which will be discussed next.
3.5.1. Impaired Autophagy Initiation and Maturation
Due to the wider variety, larger size, and greater stability of its protein substrates, the ALP requires several more groups of receptors, chaperones, and enzymes to facilitate effective degradation and clearance. The pathway begins with the formation of the initiation (ULK1/ATG1) complex, which is composed of ULK1, ATG13, ATG101, and FIP200, and induces autophagosome formation (Figure 2) [93,94]. Now, C9orf72 (chromosome 9 ORF 72) is a well-documented protein with mutations associated with ALS and FTD [95], and one study has implicated it in Ras-related protein Rab 1a (Rab1a)-dependent trafficking of ULK1 to sites that initiate autophagy [96]. Here, the reduction of C9orf72 expression in HeLa or HEK293 cells via RNA interference directed against different regions of C9orf72, as well as in rat primary cortical neurons using miRNA, resulted in an attenuated increase in autophagosomes in siRNA-treated cells compared to the untreated cells following Torin1 treatment (an autophagy initiator) [96]. Further, the absence of changes in FIP200 puncta from basal levels in C9orf72 knockdown cells and neurons following Torin1 treatment demonstrated the critical role of C9orf72 in the translocation of the ULK1 protein complex to the phagophore [96]. These results suggest that C9orf72 plays a crucial role in regulating autophagy initiation by binding to Rab1a and directing the trafficking of ULK1 during the process. Therefore, failed TDP-43 clearance in ALS and FTD may be associated with the inability to initiate autophagosome formation resulting from C9orf72 haploinsufficiency.
Following the assembly of the initiation (ULK1) complex is the formation of the phosphoinositide 3-kinase (PI3K) complex made up of VPS34, Beclin1, ATG13, and p150/VPS15. The PI3K complex is required to synthesize phosphatidyl inositol 3-phosphate (PI3P), and recruit other effector proteins (e.g., WIPI2, DFCP1) to facilitate membrane expansion and maturation into an autophagosome (Figure 2) [97,98]. This growth is sustained by the Atg12-Atg5/Atg16 complex and the LC3 lipidation system [99]. The impairment of VPS34 was shown to disrupt autophagosome formation and endosomal maturation, which are critical pathways for TDP-43 clearance [100,101]. The loss of VPS34 or Beclin1 (as demonstrated in knockdown experiments) also destabilized other components of the complex, including ATG14, creating a feedback loop for dysfunction [101]. These findings likewise demonstrate that defective TDP-43 clearance may be associated with destabilization of the PI3K complex, resulting in failed autophagosome maturation.
3.5.2. Failed Cargo Recognition
Other components, such as autophagy receptors (e.g., p62/SQSTM1, NBR1, and OPTN), which recognize and bind various protein cargoes to direct them towards the autophagosome, could also be disrupted due to structural mutations (Figure 2). Specifically, mutations linked to ALS/FTD in SQSTM1 interfere with the protein’s biological function by reducing its phosphorylation by ULK1 and TBK1, inhibiting its binding to ubiquitin, and effectively impeding the clearance of TDP-43 aggregates [102]. The accumulation of p62 is also considered a hallmark of disrupted autophagy in TDP-43 proteinopathies. It is correlated with the loss of TDP-43 function, suggesting that TDP-43 plays a regulatory role in the ALP through a feedback loop [103,104]. Optineurin (OPTN) is another selective autophagy receptor involved in the clearance of ubiquitinated proteins and damaged mitochondria [105,106,107]. OPTN mutations, such as the ALS-associated E478G variant, prevent the degradation of TDP-43 by disrupting autophagosome formation [108]. OPTN interacts with the kinase TBK1, and inhibition of this interaction adversely affects ALP downstream processes [105,106,109].
3.5.3. Compromised Downstream Clearance
Another essential component of the ALP is LC-II (lipidated LC3), a microtubule-associated protein involved in sealing the membrane of the mature autophagosome (Figure 2). This protein is also recognized as an indicator of increased autophagosome formation or interrupted autophagosome-lysosome fusion. Separate studies revealed the colocalization of TDP-43 puncta with LC3-positive autophagic compartments, as well as the LC3-II-positive material and autophagic markers associated with extracellular TDP-43 resulting from interrupted lysosomal-autophagic fusion [110,111]. This coaccumulation of LC3-II with TDP-43 aggregates is a clear indication that failed autophagic processes contribute significantly to TDP-43 deposition within cells.
Finally, essential components that facilitate the fusion of the mature autophagosome with the lysosome comprise the SNARE (Soluble NSF Attachment protein REceptor) complex. These proteins complete the fusion of the autophagic and lysosomal membranes, and are made up of STX17 on the autophagosomes, and SNAP29 (cytosolic Qbc-SNARE) and VAMP8 on lysosomes [112]. Despite the limited studies associating TDP-43 with SNARE protein aberration, the disruption of fusion observed in TDP-43 proteinopathies strongly suggests a connection [113]. STX17 recruits SNAP29, which then results in the binding of VAMP8 to create the fusion-capable SNARE complex [112]. When any of the critical components are disrupted, the fusion of the autophagosome-lysosome complex fails and is reflected by the accumulation of unfused autophagosomes [112,114].
Collectively, these findings firmly establish that a robust and cooperative interplay between the UPS and ALP is crucial for maintaining TDP-43 proteostasis. Given the intrinsic propensity of TDP-43 to misfold and form oligomers, along with the myriad of extrinsic factors that could interfere with the UPS and the inherent complexity and number of regulatory steps of the ALP, this dual requirement forms a mechanistic basis to illustrate that just a few destabilizing “second hits” to either system is enough to reduce the clearance of TDP-43 aggregates effectively.
4. Dynamics and Systemic Failure Further The Persistence of Pathologic TDP-43
While intrinsic resistance and mechanistic properties facilitate the evasion of aberrant TDP-43 from protein clearance pathways within the nucleus, these are insufficient to account for the relentless progression of aggregation characteristic of neurodegenerative diseases. The transition from transient evasion to a stable, self-propagating mechanism is a result of a combination of several factors, including a shift in the cellular environment. These intrinsic modifications (e.g., mutations) promote aggregation kinetics and, most importantly, systemic failure of the cell’s quality control infrastructure. This section examines downstream mechanisms following successful evasion, where the addition of PTMs promotes nucleation, and the formed aggregate overwhelms both systems, particularly the ALP, driving the process into a self-sustaining toxic cycle.
As misfolded TDP-43 accumulates outside the nucleus, a significant shift in its cellular distribution occurs. The aggregation dynamics involved in this cytoplasmic relocation further favor its sequestration into irreversible aggregates; the factors that drive this process forward will be discussed next.
4.1. Shifting the Environment: Nuclear Depletion and Aggregation Kinetics
TDP-43 normally resides and performs its regulatory functions within the nucleus. Still, a small fraction of its concentration is regularly shuttled to the cytoplasm to perform functions in RNA metabolism and regulation of translation [11]. In the nucleus, the RRM domains of TDP-43 bind with high affinity, stabilizing the protein and maintaining it in a soluble state [115,116,117]. One study demonstrated that the removal of the RRM domains using site-directed mutagenesis significantly reduced TDP-43 levels within the nucleoplasm, underscoring the valuable role of RRM domains in TDP-43 nuclear localization [117]. This cooperative binding produces a higher-order assembly on mRNA, facilitating the reduction of intermolecular interactions between the aggregate-prone NTD and CTD, which could result in aggregation [115].
However, once TDP-43 exits the nucleus, multiple factors further reduce its RNA-binding capacity. PTMs, such as the acetylation of lysines (e.g., K136 and K145), disrupt RNA binding and enhance phase separation through the CTD, leading to the formation of insoluble aggregates [10,118]. Acetylation mimicking mutants (K145Q) exhibited insoluble, hyperphosphorylated, and ubiquitinated aggregates [21]. Another mutation (K181E) found in the RRM-linking region also reduced RNA-binding affinity and enhanced aggregate formation [119]. These findings confirm that the solubility and conformational integrity of TDP-43 are significantly dependent on sustained RNA engagement.
Once RNA engagement is weakened, the resulting consequence significantly contributes to the increased formation of TDP-43 aggregates [66,120]. Additional studies have described the formation of anisotropic intranuclear liquid droplets exhibiting HSP70 chaperones, which translates to a state of pathological assembly [121]. Furthermore, RNA-binding-deficient TDP-43 was shown to undergo demixing in stress granules (SGs), where the TDP-43-rich phase has significantly reduced RNA levels compared to the G3BP1-rich phase [122]. G3BP1 (Ras GTPase-activating protein-binding protein 1) functions as a scaffolding protein that initiates stress granule assembly. Together with other RBPs and RNAs, it contributes to the formation of a dynamic, liquid-like phase that constitutes the structural matrix of normal SGs. TDP-43 is also included in this phase, but prolonged oxidative stress and the buildup of misfolded TDP-43 result in its “demixing” from the G3BP1 matrix, creating its own subphase within the granule [122]. Thus, the observed loss of RNA in the TDP-43 phase corroborates that maintaining TDP-43 solubility highly depends on its RNA-binding capability.
Additionally, Yan et al. (2024) concluded that although the formation of SGs increased TDP-43 concentration above a critical threshold, this alone was insufficient for aggregation. Conversely, when prolonged oxidative stress resulting in cysteine oxidation and structural destabilization within the RRM domain (and increased hydrophobic interactions in the CTD) was the only variable treated, but TDP-43 levels failed to reach the critical threshold in SGs, no aggregation was observed [122]. Therefore, both variables (the crucial concentration of TDP-43 in SGs and prolonged oxidation) are essential for the phase separation of TDP-43 to occur within the granules and form irreversible aggregates with pathological hallmarks.
Mechanistically, RNA binding increases the stability of the RRM domains and inhibits the conformational plasticity of the protein. Molecular dynamics simulations demonstrated that the absence of RNA promotes the tandem RRMs to exhibit enhanced conformational plasticity, particularly RRM1, which displays an increased likelihood of adopting partially unfolded conformations [123]. The interaction with RNA introduces constraints to TDP-43, increasing intradomain stability and inhibiting the structural transitions that lead to uncontrolled aggregation [123].
Moreover, single-molecule tracking revealed a significant reduction in TDP-43 mobility with increasing stress duration, where the effective diffusion coefficients dropped from a baseline of 3 μm2/s to 0.75 μm2/s after two hours of arsenite-induced stress [124]. This was observed for both free cytoplasmic TDP-43 and TDP-43 sequestered into SGs, suggesting that oligomeric hotspots, which could potentially lead to irreversible aggregates, may result from cytoplasmic TDP-43, not just those already sequestered into SGs.
Finally, the cytoplasm fundamentally lacks the quality control mechanisms that maintain TDP-43 homeostasis and solubility in the nucleus, fostering an environment that predisposes TDP-43 to misfolding and aggregation. Nuclear TDP-43 is delicately retained by forming large RNA-mediated macromolecular complexes that block its cytoplasmic diffusion in a size-dependent manner [116]. These nuclear compartments are also stabilized by RNA binding, oligomerization, and phase separation, where TDP-43 is organized into compartments that maintain its solubility and functionality [125].
Studies have also demonstrated that passive diffusion is the primary force driving TDP-43 cytoplasmic egress, where the protein continuously leaks into the cytoplasm but is actively shuttled back to the nucleus through nucleoporins and import factors [5]. Investigation of the interactome of TDP-43 insoluble aggregates revealed their enrichment in the components of the nuclear pore complex and nucleocytoplasmic machinery, indicating the direct sequestration of these essential karyopherins, leading to the impairment of the nuclear pore complex [126,127]. The result is a feed-forward mechanism in which the increased levels of aberrant cytoplasmic TDP-43 progressively sequester more components, disrupting nuclear import and leading to further accumulation of cytoplasmic TDP-43.
Another study revealed that the monomerization of TDP-43 is key in driving the nuclear export of the protein. Monomeric TDP-43 was demonstrated to preferentially interact with nuclear RNA export factor 1 (Nxf1), compared to its dimeric form [128]. Additionally, the monomerization was shown to be triggered by cellular stresses or dysfunctions (i.e., spliceosomal impairment), compared to physiological conditions, where TDP-43 dimerizes or forms higher-order oligomers to facilitate its nuclear retention and solubility [128].
Taken together, reduced affinity to RNA due to structural mutations or PTMs, reaching critical concentration thresholds within SGs (demixing), prolonged oxidative stress, the absence of quality control mechanisms in the cytoplasm, and the resulting vicious cycle induced by the sequestration of critical importins by the increased egress of aberrant TDP-43 are all significant drivers that produce a self-reinforcing cascade that drive irreversible aggregation.
4.2. The Aggregation Cascade: Acceleration and Propagation Dynamics
Now that we have established the key molecular players involved in the mislocalization of TDP-43, we will discuss more factors that further accelerate the formation of TDP-43 aggregates once the initial stages have been fulfilled. These factors are critical to understand, as they substantially increase the rate of TDP-43 entanglement. Several steps within this process may also serve as therapeutic targets to slow or interrupt the pathological cascade.
4.2.1. PTMs as Structural Destabilizers and Pathological Markers
The key mechanism driving the acceleration of aggregation is the reduction of the energy threshold required for the formation of oligomeric hotspots, as well as the stabilization of the insoluble core of the fibril. One of the mechanisms exploited by the addition of PTMs involves reducing the RNA-binding affinity within the RRM domains.
Acetylation is one PTM recognized for reducing the binding affinity of the RRMs, as discussed in the previous sections. The loss of the interacting RNA further exposes the CTD, significantly lowering the nucleation barrier or phase separation. The acetylation of crucial lysine residues (e.g., K145, K192, and K136) was demonstrated to enhance the tendency for TDP-43 to form solid-like or oligomeric assemblies [16]. This phase separation was described as anisomes, which are nuclear membraneless organelles that act as seeds for aggregation, and are pathologically observed in ALS/FTD tissue [16]. Physiologically, the nucleus fosters an environment that maintains the solubility and homeostasis of TDP-43. Still, the introduction of structural mutations and charge-neutralizing PTMs reduces RNA affinity of nuclear TDP-43, leading to the creation of these membraneless compartments.
Oxidative stress is another mechanism that can structurally modify cysteine residues in the RRMs (e.g., C173, C175, C198, and C244), inducing local unfolding or domain swapping, ultimately resulting in rapid and stable ß-sheet-rich oligomers through the formation of intermolecular disulfide bridges [129]. Another PTM is citrullination, which functions similarly by neutralizing the positive charge on arginines, weakening RNA affinity, and favoring the shift to phase separation. These modifications drive TDP-43 past the solubility threshold, increasing the rate of oligomer growth and aggregation [130].
As discussed previously, ubiquitination is a critical PPTM that tags misfolded TDP-43 for degradation via the ALP. However, due to the impaired cascade reminiscent of ALS and FTD pathologies, ubiquitinated TDP-43 substantially accumulates in the cytoplasm, with the ubiquitin tags acting as markers of failed autophagy [13]. While phosphorylation is consistently observed in ALS/FTD tissues [131,132,133,134], its precise role remains debated, with some evidence suggesting it may initially function as a protective clearance signal before contributing to aggregate insolubility [9,104,132].
4.2.2. Lowering the Nucleation Barrier: Accumulation of Misfolded Monomers Eventually Leads to Uncontrolled Aggregation and Systemic Collapse
The net balance between TDP-43 aggregate formation and clearance modulates whether its deposition or removal from the cell is favored. For instance, as discussed earlier, the half-life of the soluble phase of TDP-43 significantly determines its clearance rate, owing to the increased structural stability of the misfolded form. Wild-type TDP-43 has been shown to have a half-life ranging from 4 to 12 hours in immortalized cells; however, it has also been reported to last as long as 18 hours in primary neurons [135]. Depending on various experimental conditions, the reported values can range from 4 to 34 hours [40,50,136,137]. However, in the presence of mutations, the half-lives of monomeric TDP-43 generally increase due to the conferred resistance to temperature-induced unfolding, aggregation, and degradation, resulting in elevated steady-state TDP-43 concentrations within the cell [138]. PTMs, such as citrullination, acetylation, and oxidation, also contribute to pathogenesis by destabilizing the interaction of TDP-43 monomers with RNA, promoting the misfolded state, and rendering the protein more resistant to proteolytic processing by the UPS [16,129,130].
This elevation of steady-state concentrations is critical because it leads to the initial accumulation of misfolded aberrant monomers that escape UPS processing, subsequently serving as potential substrates for the next onslaught. While the rate-limiting step in TDP-43 pathogenesis is the elevated nucleation barrier that monomers must overcome to form stable and insoluble fibrils, this persistent monomer accumulation increases the likelihood of intermolecular interactions, leading to the formation of small soluble oligomers [138,139]. The resulting oligomers act as potent seeds that facilitate surmounting the thermodynamic barrier, leading to the recruitment and conversion of the remaining soluble monomers into insoluble aggregates at a significantly accelerated rate [25,140,141,142]. This abrupt shift from spontaneous nucleation (lag phase) to seeded growth (exponential phase) corresponds to the kinetic inflection point that leads to uncontrollable self-propagation. Finally, aggregation kinetics can also be promoted extrinsically through cross-seeding. Oligomers of co-pathologies, such as α-synuclein, can further reduce the lag time by acting as heterologous molecular scaffolds to support the growing TDP-43 oligomers [75,143].
Taken together, the complex interplay between these variables indicates a debilitating kinetic cascade, summarized visually in Figure 3. Pathological mutations or PTMs (e.g., citrullination, oxidation, acetylation) weaken RNA binding, resulting in a pool of proteolysis-resistant monomers that the UPS could not process. The accumulated monomers eventually bypass the nucleation barrier, further facilitated by the formation of catalytic sites, such as stress granules, that foster LLPS irreversibility. The resulting oligomers (including C-terminal fragments and co-pathologies) function as seeds that lower the thermodynamic barrier, driving the pathology from gradual accumulation to exponential fibril growth. Following the full maturation of the aggregates, the rate at which the fibrils form overwhelms the sequestration capacity of the ALP, leading to a feed-forward loop where the impairment of nuclear import mechanisms further saturates dysfunctional TDP-43 in the cytoplasm, sealing the cell’s fate.
5. Conclusions
The pathogenicity of TDP-43 originates not only from its intrinsic tendency to aggregate but is compounded by external factors that allow it to evade the cell’s quality control mechanisms and progress towards systemic collapse. The clearance of TDP-43 aggregates requires the participation of various chaperones, ligases, and enzymes to retain TDP-43 homeostasis as it is continuously shuttled between the nucleus and the cytoplasm. However, these can be disturbed by mutations, structural modifications by PTMs, and chronic cellular stress, potentially initiating the pathologic cascade.
Crucially, distinct TDP-43 species are processed by separate pathways: the UPS handles misfolded soluble monomers, and the ALP sequesters and clears the larger insoluble aggregates. This functional partitioning underscores that these two systems are not fully redundant. Consequently, the ALP cannot effectively compensate for UPS failure and vice versa. Synergistic function is required to effectively clear the proteins, meaning a ‘second hit’ compromising either system is sufficient to trigger the pathological cascade.
Disruptions to the UPS originate from two convergent pathways: the intrinsic aggregation propensity of monomeric TDP-43 and external insults, particularly chronic oxidative stress, hypoxia, and neuroinflammation. These insults can compromise critical UPS components, such as ubiquitin-conjugating enzymes and proteasomal core subunits, preventing their assembly into functional degradation complexes. This vulnerability demonstrates that TDP-43 is not the sole player in initiating the pathological process. Nevertheless, a pool of misfolded monomers arises, priming the cellular environment for the next stage: the growth of oligomers, marked by the phase separation of TDP-43 in stress granules. These oligomeric assemblies act as seeds, enabling TDP-43 to circumvent the energy barrier for uncontrolled self-assembly. This relentless feed-forward cycle marks the exponential phase of the cascade, which eventually overwhelms the autophagy-lysosome pathway with the rapid growth of fibrils and aggregates.
Due to the complexity and multiplicity of the pathways involved, the roles of some processes in the aggravation or suppression of TDP-43 clearance remain undefined. Future studies should validate the kinetic contribution of co-pathologies, such as amyloid beta, a-synuclein, and huntingtin, as well as the role of CTFs in sequestering other essential but understudied components of the UPS or ALP. A growing number of studies are also focusing on the role of protein oligomers, which are considered more toxic than fibrils, and how the pathological cascade can be quelled more effectively if targeted in its earlier stages. Finally, a consensus is yet to be reached on whether the accumulation of TDP-43 aggregates is the primary toxic driver or if the loss of protein function is the principal culprit, rendering aggregates a physical, inert manifestation of the dysfunction.
Regardless of the specific toxic species, these findings strongly suggest TDP-43 pathogenesis is a collapse in kinetic regulation. Future interventions must extend beyond simple clearance strategies and explore options for breaking the feed-forward loops that drive the transition from regulatable monomers to self-propagating oligomers.
Author Contributions
Conceptualization, A.J.; Writing – Original Draft Preparation, A.J.; Writing – Review and Editing, A.J., S.A., and J.H.; Supervision, S.A. and J.H.; Funding Acquisition, S.A.
Funding
This research was funded by the Korea Institute of Marine Science & Technology Promotion (KIMST), funded by the Ministry of Oceans and Fisheries (RS-2025-02292973), and by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (RS-2021-NR060117).
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Song, J. Molecular Mechanisms of Phase Separation and Amyloidosis of ALS/FTD-Linked FUS and TDP-43. Aging Dis. 2024, 15, 2084–2112. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA Targets and Position-Dependent Splicing Regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef]
- Bedja-Iacona, L.; Richard, E.; Marouillat, S.; Brulard, C.; Alouane, T.; Beltran, S.; Andres, C.R.; Blasco, H.; Corcia, P.; Veyrat-Durebex, C.; et al. Post-Translational Variants of Major Proteins in Amyotrophic Lateral Sclerosis Provide New Insights into the Pathophysiology of the Disease. Int. J. Mol. Sci. 2024, 25, 8664. [Google Scholar] [CrossRef] [PubMed]
- Versluys, L.; Ervilha Pereira, P.; Schuermans, N.; De Paepe, B.; De Bleecker, J.L.; Bogaert, E.; Dermaut, B. Expanding the TDP-43 Proteinopathy Pathway from Neurons to Muscle: Physiological and Pathophysiological Functions. Front. Neurosci. 2022, 16, 815765. [Google Scholar] [CrossRef] [PubMed]
- Pinarbasi, E.S.; Cağatay, T.; Fung, H.Y.J.; Li, Y.C.; Chook, Y.M.; Thomas, P.J. Active Nuclear Import and Passive Nuclear Export Are the Primary Determinants of TDP-43 Localization. Sci. Rep. 2018, 8, 7083. [Google Scholar] [CrossRef]
- Doll, S.G.; Meshkin, H.; Bryer, A.J.; Li, F.; Ko, Y.-H.; Lokareddy, R.K.; Gillilan, R.E.; Gupta, K.; Perilla, J.R.; Cingolani, G. Recognition of the TDP-43 Nuclear Localization Signal by Importin α1/β. Cell Rep. 2022, 39, 111007. [Google Scholar] [CrossRef]
- Ederle, H.; Funk, C.; Abou-Ajram, C.; Hutten, S.; Funk, E.B.E.; Kehlenbach, R.H.; Bailer, S.M.; Dormann, D. Nuclear Egress of TDP-43 and FUS Occurs Independently of Exportin-1/CRM1. Sci. Rep. 2018, 8, 7084. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 Regulates Its mRNA Levels through a Negative Feedback Loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef] [PubMed]
- da Silva, L.G.; Simonetti, F.; Hutten, S.; Riemenschneider, H.; Sternburg, E.L.; Pietrek, L.M.; Gebel, J.; Dötsch, V.; Edbauer, D.; Hummer, G.; et al. Disease-Linked TDP-43 Hyperphosphorylation Suppresses TDP-43 Condensation and Aggregation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Garcia Morato, J.; Hans, F.; von Zweydorf, F.; Feederle, R.; Elsässer, S.J.; Skodras, A.A.; Gloeckner, C.J.; Buratti, E.; Neumann, M.; Kahle, P.J. Sirtuin-1 Sensitive Lysine-136 Acetylation Drives Phase Separation and Pathological Aggregation of TDP-43. Nat. Commun. 2022, 13, 1223. [Google Scholar] [CrossRef]
- Birsa, N.; Bentham, M.P.; Fratta, P. Cytoplasmic Functions of TDP-43 and FUS and Their Role in ALS. Semin. Cell Dev. Biol. 2020, 99, 193–201. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-Predominant Age-Related TDP-43 Encephalopathy (LATE): Consensus Working Group Report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef]
- Budini, M.; Romano, V.; Quadri, Z.; Buratti, E.; Baralle, F.E. TDP-43 Loss of Cellular Function through Aggregation Requires Additional Structural Determinants beyond Its C-Terminal Q/N Prion-like Domain. Hum. Mol. Genet. 2015, 24, 9–20. [Google Scholar] [CrossRef]
- Keating, S.S.; Bademosi, A.T.; San Gil, R.; Walker, A.K. Aggregation-Prone TDP-43 Sequesters and Drives Pathological Transitions of Free Nuclear TDP-43. Cell. Mol. Life Sci. 2023, 80, 95. [Google Scholar] [CrossRef]
- Suk, T.R.; Part, C.E.; Zhang, J.L.; Nguyen, T.T.; Heer, M.M.; Caballero-Gómez, A.; Grybas, V.S.; McKeever, P.M.; Nguyen, B.; Ali, T.; et al. A Stress-Dependent TDP-43 SUMOylation Program Preserves Neuronal Function. Mol. Neurodegener. 2025, 20, 38. [Google Scholar] [CrossRef]
- Verde, E.M.; Antoniani, F.; Mediani, L.; Secco, V.; Crotti, S.; Ferrara, M.C.; Vinet, J.; Sergeeva, A.; Yan, X.; Hoege, C.; et al. SUMO2/3 Conjugation of TDP-43 Protects against Aggregation. Sci. Adv. 2025, 11, eadq2475. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kwon, Y.; Kim, S.; Jo, M.; Jeon, Y.-M.; Cheon, M.; Lee, S.; Kim, S.R.; Kim, K.; Kim, H.-J. The Role of HDAC6 in TDP-43-Induced Neurotoxicity and UPS Impairment. Front. Cell Dev. Biol. 2020, 8, 581942. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, J.; Spalloni, A.; Cappelli, S.; Ciaiola, F.; Orlando, V.; Buratti, E.; Longone, P. TDP-43 Epigenetic Facets and Their Neurodegenerative Implications. Int. J. Mol. Sci. 2023, 24, 13807. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Hwang, A.W.; Restrepo, C.R.; Yuan, C.-X.; Trojanowski, J.Q.; Lee, V.M.Y. An Acetylation Switch Controls TDP-43 Function and Aggregation Propensity. Nat. Commun. 2015, 6, 5845. [Google Scholar] [CrossRef]
- Kumar, S.T.; Nazarov, S.; Porta, S.; Maharjan, N.; Cendrowska, U.; Kabani, M.; Finamore, F.; Xu, Y.; Lee, V.M.-Y.; Lashuel, H.A. Seeding the Aggregation of TDP-43 Requires Post-Fibrillization Proteolytic Cleavage. Nat. Neurosci. 2023, 26, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Staderini, T.; Bigi, A.; Mongiello, D.; Cecchi, C.; Chiti, F. Biophysical Characterization of Full-Length TAR DNA-Binding Protein (TDP-43) Phase Separation. Protein Sci. 2022, 31, e4509. [Google Scholar] [CrossRef]
- Verde, E.M.; Secco, V.; Ghezzi, A.; Mandrioli, J.; Carra, S. Molecular Mechanisms of Protein Aggregation in ALS-FTD: Focus on TDP-43 and Cellular Protective Responses. Cells 2025, 14, 680. [Google Scholar] [CrossRef]
- Babinchak, W.M.; Haider, R.; Dumm, B.K.; Sarkar, P.; Surewicz, K.; Choi, J.-K.; Surewicz, W.K. The Role of Liquid-Liquid Phase Separation in Aggregation of the TDP-43 Low-Complexity Domain. J. Biol. Chem. 2019, 294, 6306–6317. [Google Scholar] [CrossRef]
- Guenther, E.L.; Cao, Q.; Trinh, H.; Lu, J.; Sawaya, M.R.; Cascio, D.; Boyer, D.R.; Rodriguez, J.A.; Hughes, M.P.; Eisenberg, D.S. Atomic Structures of TDP-43 LCD Segments and Insights into Reversible or Pathogenic Aggregation. Nat. Struct. Mol. Biol. 2018, 25, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Wahiduzzaman; Prakash, A.; Tomar, A.K.; Srivastava, A.; Kundu, B.; Lynn, A.M.; Imtaiyaz Hassan, M. Exploring the Aggregation-Prone Regions from Structural Domains of Human TDP-43. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 286–296. [Google Scholar] [CrossRef]
- Arseni, D.; Chen, R.; Murzin, A.G.; Peak-Chew, S.Y.; Garringer, H.J.; Newell, K.L.; Kametani, F.; Robinson, A.C.; Vidal, R.; Ghetti, B.; et al. TDP-43 Forms Amyloid Filaments with a Distinct Fold in Type A FTLD-TDP. Nature 2023, 620, 898–903. [Google Scholar] [CrossRef]
- Afroz, T.; Hock, E.-M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P.; et al. Functional and Dynamic Polymerization of the ALS-Linked Protein TDP-43 Antagonizes Its Pathologic Aggregation. Nat. Commun. 2017, 8, 45. [Google Scholar] [CrossRef]
- Jiang, L.-L.; Xue, W.; Hong, J.-Y.; Zhang, J.-T.; Li, M.-J.; Yu, S.-N.; He, J.-H.; Hu, H.-Y. The N-Terminal Dimerization Is Required for TDP-43 Splicing Activity. Sci. Rep. 2017, 7, 6196. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Conicella, A.E.; Schmidt, H.B.; Martin, E.W.; Rhoads, S.N.; Reeb, A.N.; Nourse, A.; Ramirez Montero, D.; Ryan, V.H.; Rohatgi, R.; et al. A Single N-Terminal Phosphomimic Disrupts TDP-43 Polymerization, Phase Separation, and RNA Splicing. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Tan, C.-F.; Eguchi, H.; Tagawa, A.; Onodera, O.; Iwasaki, T.; Tsujino, A.; Nishizawa, M.; Kakita, A.; Takahashi, H. TDP-43 Immunoreactivity in Neuronal Inclusions in Familial Amyotrophic Lateral Sclerosis with or without SOD1 Gene Mutation. Acta Neuropathol. 2007, 113, 535–542. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 Distinguishes Sporadic Amyotrophic Lateral Sclerosis from Amyotrophic Lateral Sclerosis with SOD1 Mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef]
- Rothstein, J.D. TDP-43 in Amyotrophic Lateral Sclerosis: Pathophysiology or Patho-Babel? Ann. Neurol. 2007, 61, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 Is Intrinsically Aggregation-Prone, and Amyotrophic Lateral Sclerosis-Linked Mutations Accelerate Aggregation and Increase Toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef]
- Kubota, H. Quality Control against Misfolded Proteins in the Cytosol: A Network for Cell Survival. J. Biochem. 2009, 146, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. P62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Kirkin, V.; Lamark, T.; Sou, Y.-S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.-A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A Role for NBR1 in Autophagosomal Degradation of Ubiquitinated Substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective Autophagy Mediated by Autophagic Adapter Proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.-B.; Chen, H.-J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential Roles of the Ubiquitin Proteasome System and Autophagy in the Clearance of Soluble and Aggregated TDP-43 Species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef]
- Wang, X.; Fan, H.; Ying, Z.; Li, B.; Wang, H.; Wang, G. Degradation of TDP-43 and Its Pathogenic Form by Autophagy and the Ubiquitin-Proteasome System. Neurosci. Lett. 2010, 469, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Urushitani, M.; Sato, T.; Bamba, H.; Hisa, Y.; Tooyama, I. Synergistic Effect between Proteasome and Autophagosome in the Clearance of Polyubiquitinated TDP-43. J. Neurosci. Res. 2010, 88, 784–797. [Google Scholar] [CrossRef] [PubMed]
- van Eersel, J.; Ke, Y.D.; Gladbach, A.; Bi, M.; Götz, J.; Kril, J.J.; Ittner, L.M. Cytoplasmic Accumulation and Aggregation of TDP-43 upon Proteasome Inhibition in Cultured Neurons. PLoS One 2011, 6, e22850. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Deng, J.J.; Bai, Y.; Thornton, F.B.; Oddo, S. Rapamycin Rescues TDP-43 Mislocalization and the Associated Low Molecular Mass Neurofilament Instability. J. Biol. Chem. 2009, 284, 27416–27424. [Google Scholar] [CrossRef]
- Sampognaro, P.J.; Arya, S.; Knudsen, G.M.; Gunderson, E.L.; Sandoval-Perez, A.; Hodul, M.; Bowles, K.; Craik, C.S.; Jacobson, M.P.; Kao, A.W. Mutations in α-Synuclein, TDP-43 and Tau Prolong Protein Half-Life through Diminished Degradation by Lysosomal Proteases. Mol. Neurodegener. 2023, 18, 29. [Google Scholar] [CrossRef]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef]
- Li, H.-R.; Chen, T.-C.; Hsiao, C.-L.; Shi, L.; Chou, C.-Y.; Huang, J.-R. The Physical Forces Mediating Self-Association and Phase-Separation in the C-Terminal Domain of TDP-43. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 214–223. [Google Scholar] [CrossRef]
- Zeng, J.; Tang, Y.; Dong, X.; Li, F.; Wei, G. Influence of ALS-Linked M337V Mutation on the Conformational Ensembles of TDP-43321-340 Peptide Monomer and Dimer. Proteins 2024, 92, 1059–1069. [Google Scholar] [CrossRef]
- Hallegger, M.; Chakrabarti, A.M.; Lee, F.C.Y.; Lee, B.L.; Amalietti, A.G.; Odeh, H.M.; Copley, K.E.; Rubien, J.D.; Portz, B.; Kuret, K.; et al. TDP-43 Condensation Properties Specify Its RNA-Binding and Regulatory Repertoire. Cell 2021, 184, 4680–4696.e22. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.-C.; Albuquerque, C.P.; Han, J.S.; Lagier-Tourenne, C.; Tokunaga, S.; Zhou, H.; Cleveland, D.W. ALS-Associated Mutations in TDP-43 Increase Its Stability and Promote TDP-43 Complexes with FUS/TLS. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 13318–13323. [Google Scholar] [CrossRef]
- Yamashita, T.; Hideyama, T.; Hachiga, K.; Teramoto, S.; Takano, J.; Iwata, N.; Saido, T.C.; Kwak, S. A Role for Calpain-Dependent Cleavage of TDP-43 in Amyotrophic Lateral Sclerosis Pathology. Nat. Commun. 2012, 3, 1307. [Google Scholar] [CrossRef]
- Kwak, S.; Kawahara, Y. Deficient RNA Editing of GluR2 and Neuronal Death in Amyotropic Lateral Sclerosis. J. Mol. Med. 2005, 83, 110–120. [Google Scholar] [CrossRef]
- Kawahara, Y.; Ito, K.; Sun, H.; Aizawa, H.; Kanazawa, I.; Kwak, S. Glutamate Receptors: RNA Editing and Death of Motor Neurons. Nature 2004, 427, 801. [Google Scholar] [CrossRef]
- Aizawa, H.; Sawada, J.; Hideyama, T.; Yamashita, T.; Katayama, T.; Hasebe, N.; Kimura, T.; Yahara, O.; Kwak, S. TDP-43 Pathology in Sporadic ALS Occurs in Motor Neurons Lacking the RNA Editing Enzyme ADAR2. Acta Neuropathol. 2010, 120, 75–84. [Google Scholar] [CrossRef]
- Hideyama, T.; Yamashita, T.; Aizawa, H.; Tsuji, S.; Kakita, A.; Takahashi, H.; Kwak, S. Profound Downregulation of the RNA Editing Enzyme ADAR2 in ALS Spinal Motor Neurons. Neurobiol. Dis. 2012, 45, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-H.; Grauffel, C.; Wu, L.-S.; Kuo, P.-H.; Doudeva, L.G.; Lim, C.; Shen, C.-K.J.; Yuan, H.S. Structural Analysis of Disease-Related TDP-43 D169G Mutation: Linking Enhanced Stability and Caspase Cleavage Efficiency to Protein Accumulation. Sci. Rep. 2016, 6, 21581. [Google Scholar] [CrossRef]
- Nonaka, T.; Kametani, F.; Arai, T.; Akiyama, H.; Hasegawa, M. Truncation and Pathogenic Mutations Facilitate the Formation of Intracellular Aggregates of TDP-43. Hum. Mol. Genet. 2009, 18, 3353–3364. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Sivalingam, V.; Bharathi, V.; Girdhar, A.; Patel, B.K. The Amyloidogenicity of a C-Terminal Region of TDP-43 Implicated in Amyotrophic Lateral Sclerosis Can Be Affected by Anions, Acetylation and Homodimerization. Biochimie 2018, 150, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Xu, Y.-F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.-L.; Tong, J.; Castanedes-Casey, M.; Ash, P.; et al. Aberrant Cleavage of TDP-43 Enhances Aggregation and Cellular Toxicity. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 7607–7612. [Google Scholar] [CrossRef]
- Shimonaka, S.; Nonaka, T.; Suzuki, G.; Hisanaga, S.-I.; Hasegawa, M. Templated Aggregation of TAR DNA-Binding Protein of 43 kDa (TDP-43) by Seeding with TDP-43 Peptide Fibrils. J. Biol. Chem. 2016, 291, 8896–8907. [Google Scholar] [CrossRef]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.A.; Akiyama, H.; et al. Prion-like Properties of Pathological TDP-43 Aggregates from Diseased Brains. Cell Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef]
- Saini, A.; Chauhan, V.S. Delineation of the Core Aggregation Sequences of TDP-43 C-Terminal Fragment. Chembiochem 2011, 12, 2495–2501. [Google Scholar] [CrossRef]
- Hans, F.; Eckert, M.; von Zweydorf, F.; Gloeckner, C.J.; Kahle, P.J. Identification and Characterization of Ubiquitinylation Sites in TAR DNA-Binding Protein of 43 kDa (TDP-43). J. Biol. Chem. 2018, 293, 16083–16099. [Google Scholar] [CrossRef]
- Tomé, S.O.; Vandenberghe, R.; Ospitalieri, S.; Van Schoor, E.; Tousseyn, T.; Otto, M.; von Arnim, C.A.F.; Thal, D.R. Distinct Molecular Patterns of TDP-43 Pathology in Alzheimer’s Disease: Relationship with Clinical Phenotypes. Acta Neuropathol. Commun. 2020, 8, 61. [Google Scholar] [CrossRef]
- Wang, P.; Wander, C.M.; Yuan, C.-X.; Bereman, M.S.; Cohen, T.J. Acetylation-Induced TDP-43 Pathology Is Suppressed by an HSF1-Dependent Chaperone Program. Nat. Commun. 2017, 8, 82. [Google Scholar] [CrossRef]
- Pérez-Berlanga, M.; Wiersma, V.I.; Zbinden, A.; De Vos, L.; Wagner, U.; Foglieni, C.; Mallona, I.; Betz, K.M.; Cléry, A.; Weber, J.; et al. Loss of TDP-43 Oligomerization or RNA Binding Elicits Distinct Aggregation Patterns. EMBO J. 2023, 42, e111719. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Bai, D.; Zhu, L.; Deng, F.; Guo, X.; Li, B.; Chen, L.; Li, S.; Li, X.-J. Cytoplasmic TDP-43 Impairs the Activity of the Ubiquitin-Proteasome System. Exp. Neurol. 2021, 345, 113833. [Google Scholar] [CrossRef]
- Farrawell, N.E.; McAlary, L.; Lum, J.S.; Chisholm, C.G.; Warraich, S.T.; Blair, I.P.; Vine, K.L.; Saunders, D.N.; Yerbury, J.J. Ubiquitin Homeostasis Is Disrupted in TDP-43 and FUS Cell Models of ALS. iScience 2020, 23, 101700. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Lim, L.-Z.; Wei, Y.; Song, J. TDP-43 N Terminus Encodes a Novel Ubiquitin-like Fold and Its Unfolded Form in Equilibrium That Can Be Shifted by Binding to ssDNA. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 18619–18624. [Google Scholar] [CrossRef] [PubMed]
- Voges, D.; Zwickl, P.; Baumeister, W. The 26S Proteasome: A Molecular Machine Designed for Controlled Proteolysis. Annu. Rev. Biochem. 1999, 68, 1015–1068. [Google Scholar] [CrossRef]
- Groll, M.; Ditzel, L.; Löwe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S Proteasome from Yeast at 2.4Å Resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Bajorek, M.; Köhler, A.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.H.; Finley, D. A Gated Channel into the Proteasome Core Particle. Nat. Struct. Biol. 2000, 7, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.M.; Chang, S.-C.; Park, S.; Finley, D.; Cheng, Y.; Goldberg, A.L. Docking of the Proteasomal ATPases’ Carboxyl Termini in the 20S Proteasome's Alpha Ring Opens the Gate for Substrate Entry. Mol. Cell 2007, 27, 731–744. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A Common Mechanism of Proteasome Impairment by Neurodegenerative Disease-Associated Oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Fang, Y.-S.; Tsai, K.-J.; Chang, Y.-J.; Kao, P.; Woods, R.; Kuo, P.-H.; Wu, C.-C.; Liao, J.-Y.; Chou, S.-C.; Lin, V.; et al. Full-Length TDP-43 Forms Toxic Amyloid Oligomers That Are Present in Frontotemporal Lobar Dementia-TDP Patients. Nat. Commun. 2014, 5, 4824. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, H.; Guo, Q.; Bader, J.; Frottin, F.; Farny, D.; Kleinberger, G.; Haass, C.; Mann, M.; Hartl, F.U.; Baumeister, W.; et al. Gel-like Inclusions of C-terminal Fragments of TDP-43 Sequester Stalled Proteasomes in Neurons. EMBO Reports 2022, 23, e53890. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.-L.; Zhang, X.-L.; Hu, H.-Y. Co-Aggregation of TDP-43 with Other Pathogenic Proteins and Their Co-Pathologies in Neurodegenerative Diseases. Int. J. Mol. Sci. 2024, 25, 12380. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Benito, V.; Verhoef, L.G.G.C.; Masucci, M.G.; Dantuma, N.P. Endoplasmic Reticulum Stress Compromises the Ubiquitin-Proteasome System. Hum. Mol. Genet. 2005, 14, 2787–2799. [Google Scholar] [CrossRef] [PubMed]
- Shringarpure, R.; Grune, T.; Mehlhase, J.; Davies, K.J.A. Ubiquitin Conjugation Is Not Required for the Degradation of Oxidized Proteins by Proteasome. J. Biol. Chem. 2003, 278, 311–318. [Google Scholar] [CrossRef]
- Pickering, A.M.; Davies, K.J.A. Degradation of Damaged Proteins: The Main Function of the 20S Proteasome. Prog. Mol. Biol. Transl. Sci. 2012, 109, 227–248. [Google Scholar]
- Grune, T.; Merker, K.; Sandig, G.; Davies, K.J.A. Selective Degradation of Oxidatively Modified Protein Substrates by the Proteasome. Biochem. Biophys. Res. Commun. 2003, 305, 709–718. [Google Scholar] [CrossRef]
- Stadtman, E.R. Protein Oxidation and Aging. Free Radic. Res. 2006, 40, 1250–1258. [Google Scholar] [CrossRef]
- Davies, K.J. Degradation of Oxidized Proteins by the 20S Proteasome. Biochimie 2001, 83, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Reinheckel, T.; Davies, K.J. Degradation of Oxidized Proteins in Mammalian Cells. FASEB J. 1997, 11, 526–534. [Google Scholar] [CrossRef]
- Svikle, Z.; Peterfelde, B.; Sjakste, N.; Baumane, K.; Verkauskiene, R.; Jeng, C.-J.; Sokolovska, J. Ubiquitin-Proteasome System in Diabetic Retinopathy. PeerJ 2022, 10, e13715. [Google Scholar] [CrossRef]
- Shruthi, K.; Reddy, S.S.; Reddy, G.B. Ubiquitin-Proteasome System and ER Stress in the Retina of Diabetic Rats. Arch. Biochem. Biophys. 2017, 627, 10–20. [Google Scholar] [CrossRef]
- Broca, C.; Varin, E.; Armanet, M.; Tourrel-Cuzin, C.; Bosco, D.; Dalle, S.; Wojtusciszyn, A. Correction: Proteasome Dysfunction Mediates High Glucose-Induced Apoptosis in Rodent Beta Cells and Human Islets. PLoS One 2014, 9, e102652. [Google Scholar] [CrossRef] [PubMed]
- Goetzke, C.C.; Ebstein, F.; Kallinich, T. Role of Proteasomes in Inflammation. J. Clin. Med. 2021, 10, 1783. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, E.; Díaz-García, E.; García-Tovar, S.; Zamarrón, E.; Mangas, A.; Galera, R.; López-Collazo, E.; García-Rio, F.; Cubillos-Zapata, C. Upregulated Proteasome Subunits in COVID-19 Patients: A Link with Hypoxemia, Lymphopenia and Inflammation. Biomolecules 2022, 12, 442. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of Mammalian Autophagy in Physiology and Pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Ryter, S.W.; Cloonan, S.M.; Choi, A.M.K. Autophagy: A Critical Regulator of Cellular Metabolism and Homeostasis. Mol. Cells 2013, 36, 7–16. [Google Scholar] [CrossRef]
- Nixon, R.A.; Rubinsztein, D.C. Mechanisms of Autophagy-Lysosome Dysfunction in Neurodegenerative Diseases. Nat. Rev. Mol. Cell Biol. 2024, 25, 926–946. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.N.; Fujioka, Y. Atg1 Family Kinases in Autophagy Initiation. Cell. Mol. Life Sci. 2015, 72, 3083–3096. [Google Scholar] [CrossRef] [PubMed]
- Zachari, M.; Ganley, I.G. The Mammalian ULK1 Complex and Autophagy Initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef]
- Gao, J.; Douglas, A.G.L.; Chalitsios, C.V.; Scaber, J.; Talbot, K.; Turner, M.R.; Thompson, A.G. Neurodegenerative Disease in C9orf72 Repeat Expansion Carriers: Population Risk and Effect of UNC13A. Brain 2025, 148, 3865–3871. [Google Scholar] [CrossRef]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 Protein Interacts with Rab1a and the ULK1 Complex to Regulate Initiation of Autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Fracchiolla, D.; Chang, C.; Hurley, J.H.; Martens, S. A PI3K-WIPI2 Positive Feedback Loop Allosterically Activates LC3 Lipidation in Autophagy. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Nähse, V.; Raiborg, C.; Tan, K.W.; Mørk, S.; Torgersen, M.L.; Wenzel, E.M.; Nager, M.; Salo, V.T.; Johansen, T.; Ikonen, E.; et al. ATPase Activity of DFCP1 Controls Selective Autophagy. Nat. Commun. 2023, 14, 4051. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, H.; Xu, H. Autophagy-Lysosome Pathway in Insulin & Glucagon Homeostasis. Front. Endocrinol. (Lausanne) 2025, 16, 1541794. [Google Scholar]
- Liu, G.; Coyne, A.N.; Pei, F.; Vaughan, S.; Chaung, M.; Zarnescu, D.C.; Buchan, J.R. Endocytosis Regulates TDP-43 Toxicity and Turnover. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Deng, Z.; Lim, J.; Wang, Q.; Purtell, K.; Wu, S.; Palomo, G.M.; Tan, H.; Manfredi, G.; Zhao, Y.; Peng, J.; et al. ALS-FTLD-Linked Mutations of SQSTM1/p62 Disrupt Selective Autophagy and NFE2L2/NRF2 Anti-Oxidative Stress Pathway. Autophagy 2020, 16, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Bose, J.K.; Huang, C.-C.; Shen, C.-K.J. Regulation of Autophagy by Neuropathological Protein TDP-43. J. Biol. Chem. 2011, 286, 44441–44448. [Google Scholar] [CrossRef] [PubMed]
- Brady, O.A.; Meng, P.; Zheng, Y.; Mao, Y.; Hu, F. Regulation of TDP-43 Aggregation by Phosphorylation and p62/SQSTM1: Regulation of TDP-43 Aggregation by Phosphorylation and p62. J. Neurochem. 2011, 116, 248–259. [Google Scholar] [CrossRef]
- Qiu, Y.; Wang, J.; Li, H.; Yang, B.; Wang, J.; He, Q.; Weng, Q. Emerging Views of OPTN (optineurin) Function in the Autophagic Process Associated with Disease. Autophagy 2022, 18, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Holzbaur, E.L.F. Dynamic Recruitment and Activation of ALS-Associated TBK1 with Its Target Optineurin Are Required for Efficient Mitophagy. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E3349–E3358. [Google Scholar] [CrossRef]
- Shen, W.-C.; Li, H.-Y.; Chen, G.-C.; Chern, Y.; Tu, P.-H. Mutations in the Ubiquitin-Binding Domain of OPTN/optineurin Interfere with Autophagy-Mediated Degradation of Misfolded Proteins by a Dominant-Negative Mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of Optineurin in Amyotrophic Lateral Sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 Enhances Its Binding to Ub Chains and Promotes Selective Autophagy of Damaged Mitochondria. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 4039–4044. [Google Scholar] [CrossRef]
- Erwin, A.L.; Chang, M.L.; Fernandez, M.G.; Attili, D.; Russ, J.E.; Sutanto, R.; Pinarbasi, E.S.; Bekier, M.; Brant, T.S.; Hahn, T.; et al. Molecular Visualization of Neuronal TDP43 Pathology in Situ. bioRxivorg 2024. [Google Scholar]
- Tanaka, Y.; Ito, S.-I.; Honma, Y.; Hasegawa, M.; Kametani, F.; Suzuki, G.; Kozuma, L.; Takeya, K.; Eto, M. Dysregulation of the Progranulin-Driven Autophagy-Lysosomal Pathway Mediates Secretion of the Nuclear Protein TDP-43. J. Biol. Chem. 2023, 299, 105272. [Google Scholar] [CrossRef] [PubMed]
- Hegedűs, K.; Takáts, S.; Kovács, A.L.; Juhász, G. Evolutionarily Conserved Role and Physiological Relevance of a STX17/Syx17 (syntaxin 17)-Containing SNARE Complex in Autophagosome Fusion with Endosomes and Lysosomes. Autophagy 2013, 9, 1642–1646. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Wang, H.; Hao, Z.; Fu, C.; Hu, Q.; Gao, F.; Ren, H.; Chen, D.; Han, J.; Ying, Z.; et al. TDP-43 Loss of Function Increases TFEB Activity and Blocks Autophagosome-Lysosome Fusion. EMBO J. 2016, 35, 121–142. [Google Scholar] [CrossRef] [PubMed]
- Dafsari, H.S.; Schuler, J.; Schober, E.; Möller, B.; Antebi, A.; Fanto, M.; Jungbluth, H. The Space-Time Continuum in Neurological Disorders of the Autophagosome-Lysosome Fusion Machinery. Autophagy Rep. 2025, 4, 2560903. [Google Scholar] [CrossRef]
- Rengifo-Gonzalez, J.C.; El Hage, K.; Clément, M.-J.; Steiner, E.; Joshi, V.; Craveur, P.; Durand, D.; Pastré, D.; Bouhss, A. The Cooperative Binding of TDP-43 to GU-Rich RNA Repeats Antagonizes TDP-43 Aggregation. Elife 2021, 10. [Google Scholar] [CrossRef]
- Dos Passos, P.M.; Hemamali, E.H.; Mamede, L.D.; Hayes, L.R.; Ayala, Y.M. RNA-Mediated Ribonucleoprotein Assembly Controls TDP-43 Nuclear Retention. PLoS Biol. 2024, 22, e3002527. [Google Scholar] [CrossRef]
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.-F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural Determinants of the Cellular Localization and Shuttling of TDP-43. J. Cell Sci. 2008, 121, 3778–3785. [Google Scholar] [CrossRef]
- Necarsulmer, J.C.; Simon, J.M.; Evangelista, B.A.; Chen, Y.; Tian, X.; Nafees, S.; Marquez, A.B.; Jiang, H.; Wang, P.; Ajit, D.; et al. RNA-Binding Deficient TDP-43 Drives Cognitive Decline in a Mouse Model of TDP-43 Proteinopathy. Elife 2023, 12. [Google Scholar] [CrossRef]
- Chen, H.-J.; Topp, S.D.; Hui, H.S.; Zacco, E.; Katarya, M.; McLoughlin, C.; King, A.; Smith, B.N.; Troakes, C.; Pastore, A.; et al. RRM Adjacent TARDBP Mutations Disrupt RNA Binding and Enhance TDP-43 Proteinopathy. Brain 2019, 142, 3753–3770. [Google Scholar] [CrossRef]
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 2019, 102, 321–338.e8. [Google Scholar] [CrossRef]
- Yu, H.; Lu, S.; Gasior, K.; Singh, D.; Vazquez-Sanchez, S.; Tapia, O.; Toprani, D.; Beccari, M.S.; Yates, J.R., 3rd; Da Cruz, S.; et al. HSP70 Chaperones RNA-Free TDP-43 into Anisotropic Intranuclear Liquid Spherical Shells. Science 2021, 371, eabb4309. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Kuster, D.; Mohanty, P.; Nijssen, J.; Pombo-García, K.; Rizuan, A.; Franzmann, T.M.; Sergeeva, A.; Passos, P.M.; George, L.; et al. Intra-Condensate Demixing of TDP-43 inside Stress Granules Generates Pathological Aggregates. bioRxiv 2024. [Google Scholar] [CrossRef] [PubMed]
- Ozguney, B.; Mohanty, P.; Mittal, J. RNA Binding Tunes the Conformational Plasticity and Intradomain Stability of TDP-43 Tandem RNA Recognition Motifs. Biophys. J. 2024, 123, 3844–3855. [Google Scholar] [CrossRef]
- Streit, L.; Kuhn, T.; Vomhof, T.; Bopp, V.; Ludolph, A.C.; Weishaupt, J.H.; Gebhardt, J.C.M.; Michaelis, J.; Danzer, K.M. Stress Induced TDP-43 Mobility Loss Independent of Stress Granules. Nat. Commun. 2022, 13, 5480. [Google Scholar] [CrossRef]
- Scherer, N.M.; Maurel, C.; Graus, M.S.; McAlary, L.; Richter, G.; Radford, R.A.W.; Hogan, A.; Don, E.K.; Lee, A.; Yerbury, J.; et al. RNA-Binding Properties Orchestrate TDP-43 Homeostasis through Condensate Formation in Vivo. Nucleic Acids Res. 2024, 52, 5301–5319. [Google Scholar] [CrossRef]
- Chou, C.-C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 Pathology Disrupts Nuclear Pore Complexes and Nucleocytoplasmic Transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef]
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z. ’ev; Guo, L.; Shorter, J.; Da Cruz, S.; Cleveland, D.W. Cytoplasmic TDP-43 DE-Mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 2019, 102, 339–357.e7. [Google Scholar] [CrossRef] [PubMed]
- Oiwa, K.; Watanabe, S.; Onodera, K.; Iguchi, Y.; Kinoshita, Y.; Komine, O.; Sobue, A.; Okada, Y.; Katsuno, M.; Yamanaka, K. Monomerization of TDP-43 Is a Key Determinant for Inducing TDP-43 Pathology in Amyotrophic Lateral Sclerosis. Sci. Adv. 2023, 9, eadf6895. [Google Scholar] [CrossRef]
- Rabdano, S.O.; Izmailov, S.A.; Luzik, D.A.; Groves, A.; Podkorytov, I.S.; Skrynnikov, N.R. Onset of Disorder and Protein Aggregation due to Oxidation-Induced Intermolecular Disulfide Bonds: Case Study of RRM2 Domain from TDP-43. Sci. Rep. 2017, 7, 11161. [Google Scholar] [CrossRef]
- Saunders, C.; Rocha-Rangel, P.; Desai, R.; Quadri, Z.; Lui, H.; Hunt, J.B., Jr.; Liang, H.; Rogers, C.; Nash, K.; Tsoi, P.S.; et al. Citrullination of TDP-43 Is a Key Post-Translation Modification Associated with Structural and Functional Changes and Progressive Pathology in TDP-43 Mouse Models and Human Proteinopathies. bioRxiv 2025. [Google Scholar] [CrossRef]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M.; et al. Phosphorylated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Ann. Neurol. 2008, 64, 60–70. [Google Scholar] [CrossRef]
- Mosna, S.; Dormann, D. TDP-43 Phosphorylation: Pathological Modification or Protective Factor Antagonizing TDP-43 Aggregation in Neurodegenerative Diseases? Bioessays 2025, e70084. [Google Scholar] [CrossRef]
- Guedes, Á.C.B.; Santin, R.; Costa, A.S.R.; Reiter, K.C.; Hilbig, A.; Fernandez, L.L. Distinct Phospho-TDP-43 Brain Distribution in Two Cases of FTD, One Associated with ALS. Dement. Neuropsychol. 2017, 11, 249–254. [Google Scholar] [CrossRef]
- Forman, M.S.; Trojanowski, J.Q.; Lee, V.M.-Y. TDP-43: A Novel Neurodegenerative Proteinopathy. Curr. Opin. Neurobiol. 2007, 17, 548–555. [Google Scholar] [CrossRef]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy Induction Enhances TDP43 Turnover and Survival in Neuronal ALS Models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef]
- Pesiridis, G.S.; Tripathy, K.; Tanik, S.; Trojanowski, J.Q.; Lee, V.M.-Y. A “Two-Hit” Hypothesis for Inclusion Formation by Carboxyl-Terminal Fragments of TDP-43 Protein Linked to RNA Depletion and Impaired Microtubule-Dependent Transport. J. Biol. Chem. 2011, 286, 18845–18855. [Google Scholar] [CrossRef]
- Watanabe, S.; Kaneko, K.; Yamanaka, K. Accelerated Disease Onset with Stabilized Familial Amyotrophic Lateral Sclerosis (ALS)-Linked Mutant TDP-43 Proteins. J. Biol. Chem. 2013, 288, 3641–3654. [Google Scholar] [CrossRef]
- Austin, J.A.; Wright, G.S.A.; Watanabe, S.; Grossmann, J.G.; Antonyuk, S.V.; Yamanaka, K.; Hasnain, S.S. Disease Causing Mutants of TDP-43 Nucleic Acid Binding Domains Are Resistant to Aggregation and Have Increased Stability and Half-Life. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 4309–4314. [Google Scholar] [CrossRef] [PubMed]
- Ceron-Codorniu, M.; Torres, P.; Fernàndez-Bernal, A.; Rico-Rios, S.; Serrano, J.C.; Miralles, M.P.; Beltran, M.; Garcera, A.; Soler, R.M.; Pamplona, R.; et al. TDP-43 Dysfunction Leads to Bioenergetic Failure and Lipid Metabolic Rewiring in Human Cells. Redox Biol. 2024, 75, 103301. [Google Scholar] [CrossRef] [PubMed]
- French, R.L.; Reeb, A.N.; Aligireddy, H.; Kedia, N.; Dhavale, D.D.; Grese, Z.R.; Kotzbauer, P.T.; Bieschke, J.; Ayala, Y.M. TDP-43 Oligomers Detected as Initial Intermediate Species during Aggregate Formation. bioRxiv 2018. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kaneko, K.; Watanabe, S.; Yamanaka, K.; Nukina, N. A Seeding Reaction Recapitulates Intracellular Formation of Sarkosyl-Insoluble Transactivation Response Element (TAR) DNA-Binding Protein-43 Inclusions. J. Biol. Chem. 2011, 286, 18664–18672. [Google Scholar] [CrossRef] [PubMed]
- Audrain, M.; Egesipe, A.-L.; Tentillier, N.; Font, L.; Ratnam, M.; Mottier, L.; Clavel, M.; Le Roux-Bourdieu, M.; Fenyi, A.; Ollier, R.; et al. Targeting Amyotrophic Lateral Sclerosis by Neutralizing Seeding-Competent TDP-43 in CSF. Brain Commun. 2023, 5, fcad306. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, S.; Wyant, C.E.; George, H.E.; Morgan, S.E.; Rangachari, V. Prion-like C-Terminal Domain of TDP-43 and α-Synuclein Interact Synergistically to Generate Neurotoxic Hybrid Fibrils. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structural overview and pathogenic hotspots of TDP-43 discussed in this review. The full-length protein (414 amino acids) comprises the N-terminal domain (NTD), two RNA recognition motifs (RRM1 and RRM2), and the highly disordered C-terminal domain (CTD). The NLS, which is responsible for maintaining nuclear localization of the protein, is shown near the NTD. The NLS is within the RRM2 and was shown to be nonfunctional. The amyloid core within the CTD is an essential sequence required for aggregate formation. Mutations such as D169G, M337V, and A315T indicate sites that become more vulnerable to cleavage by either caspase-3 or calpains. Cysteine oxidation sites that destabilize the interaction between RNA and the RRM domains are also shown.
Figure 1.
Structural overview and pathogenic hotspots of TDP-43 discussed in this review. The full-length protein (414 amino acids) comprises the N-terminal domain (NTD), two RNA recognition motifs (RRM1 and RRM2), and the highly disordered C-terminal domain (CTD). The NLS, which is responsible for maintaining nuclear localization of the protein, is shown near the NTD. The NLS is within the RRM2 and was shown to be nonfunctional. The amyloid core within the CTD is an essential sequence required for aggregate formation. Mutations such as D169G, M337V, and A315T indicate sites that become more vulnerable to cleavage by either caspase-3 or calpains. Cysteine oxidation sites that destabilize the interaction between RNA and the RRM domains are also shown.
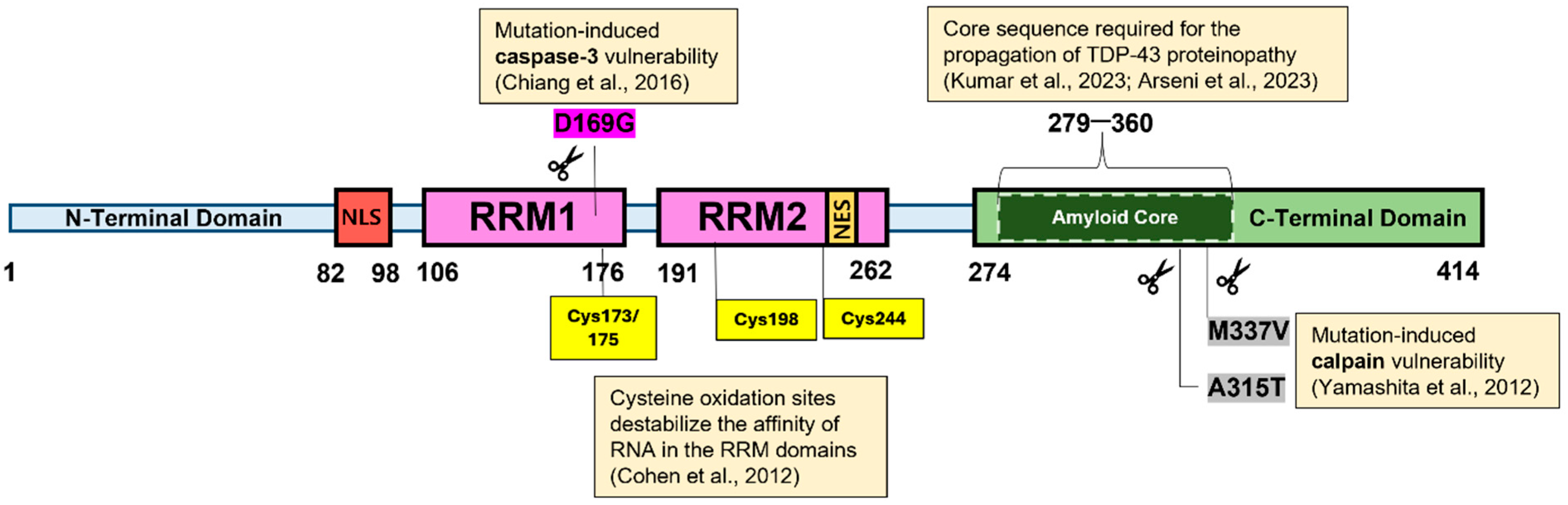
Figure 2.
Overview of the Autophagy-Lysosomal Pathway and Its Complexes.
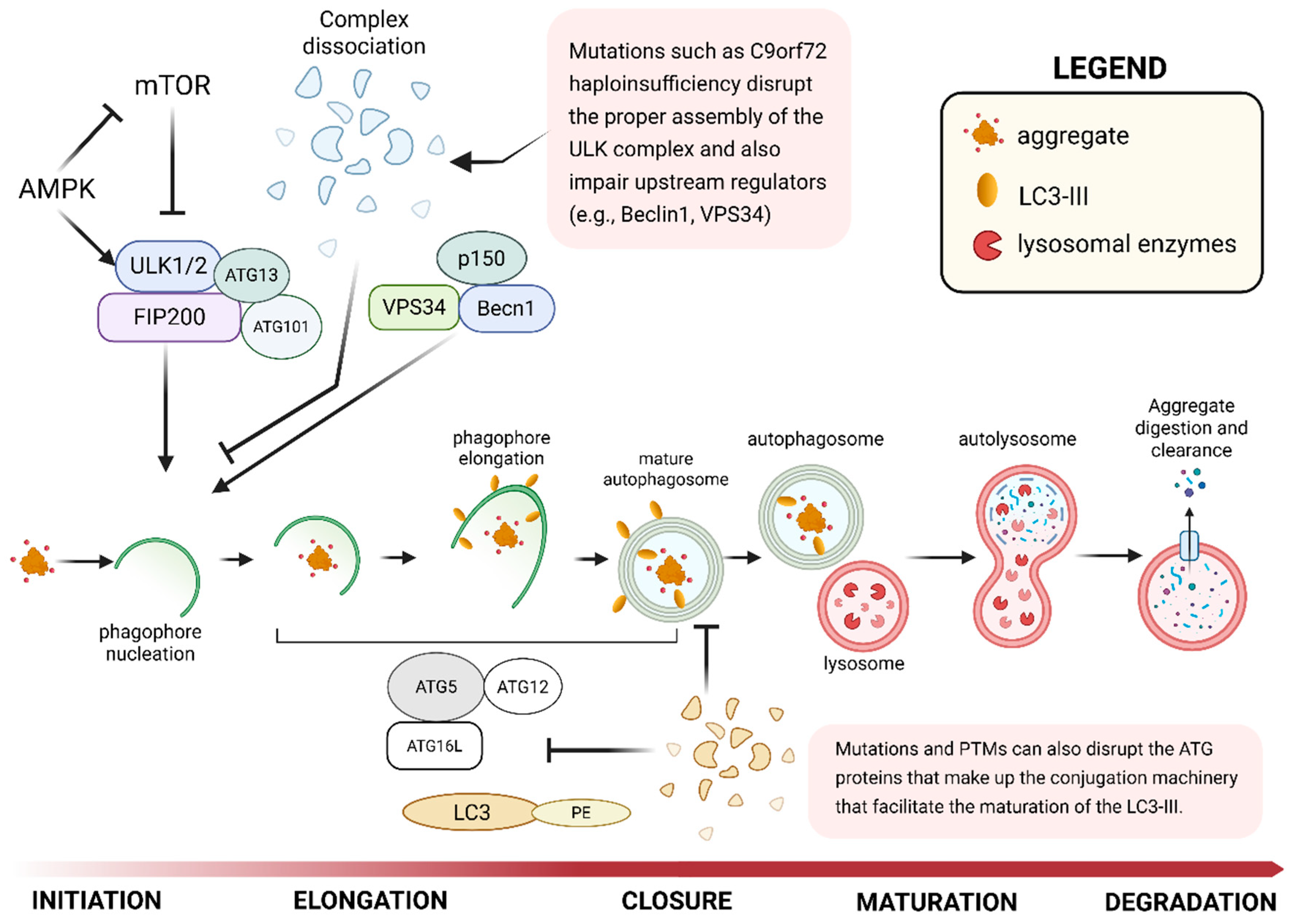
Figure 3.
The Kinetic Cascade of TDP-43.
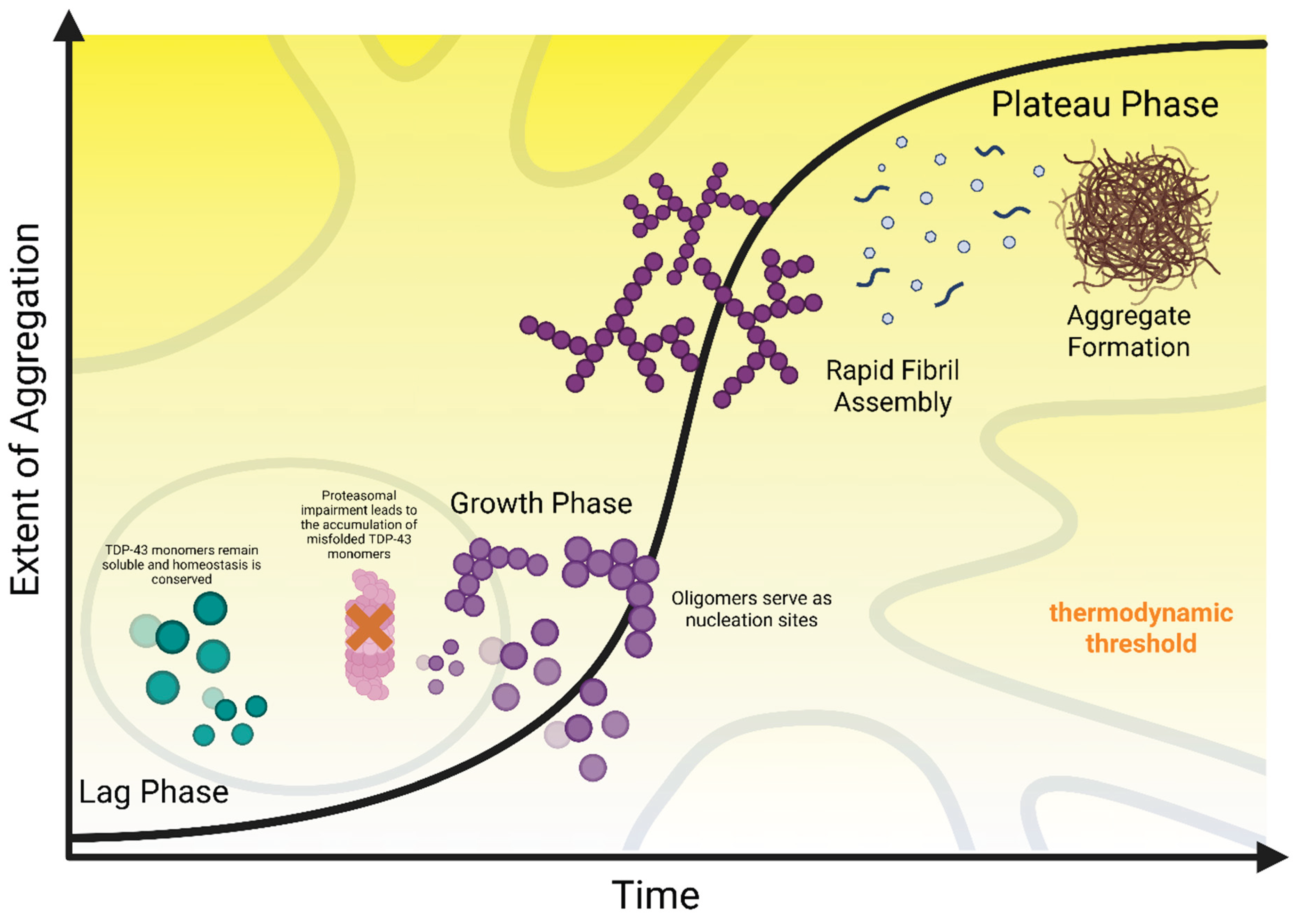
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



