
Submitted:
15 December 2025
Posted:
17 December 2025
You are already at the latest version
Abstract
Background: Lhermitte-Duclos disease (LDD), known as dysplastic cerebellar gangliocytoma, is a hamartomatous lesion that causes progressive mass effect in the posterior fossa. Cowden syndrome (CS) is a rare autosomal dominant disorder characterized by an increased risk of developing various systemic malignancies. Both conditions result from mutations in the PTEN gene, which disrupts normal cell growth and proliferation. Some children with PTEN mutations may present with developmental delay or autism spectrum disorder (ASD), potentially associated with CS. Methods: Herein, we present a rare case involving a mother with LDD/CS and her child with ASD. We discuss recommendations for screening and management of LDD/CS patients and PTEN-related ASD and provide a brief literature review. Comprehensive clinical evaluation, diagnostic imaging, and genetic analysis were performed on a female patient with LDD/CS, who underwent surgical treatment and was followed up for 10 years. Genetic testing was also conducted on her 10-year-old child, diagnosed with ASD. Results: The LDD/CS patient underwent successful partial surgical resection of the dysplastic cerebellar gangliocytoma and achieved full neurological recovery. Ten-year follow-up showed no evidence of tumor recurrence. Genetic testing in both the mother and the child confirmed PTEN gene mutations. Conclusions: This case supports a probable association between an LDD/CS-affected parent and an ASD-affected child, linked to PTEN mutations inherited in an autosomal dominant manner. Given the significant risk of malignancy and neurodevelopmental disorders associated with PTEN mutations, patients suspected of having LDD/CS and children with ASD should undergo regular screening for PTEN-related neoplasms and receive appropriate genetic counseling.
Keywords:
Lhermitte-Duclos disease
; Cowden syndrome
; autism spectrum disorder
; PTEN mutations
; genetic testing/counseling
1. Introduction
Lhermitte-Duclos disease (LDD), or dysplastic cerebellar gangliocytoma, is a rare hamartomatous lesion that causes progressive mass effect in the posterior cranial fossa [1]. Cowden syndrome (CS), also known as multiple hamartoma-neoplasia syndrome, is a rare and often underdiagnosed autosomal dominant disorder characterized by a high incidence of systemic malignancies [1,2]. It is a genetic condition caused by mutations in the PTEN gene, which disrupts normal cell growth and proliferation [3,4] and is closely associated with LDD [5].
In recent years, several reports have described the association between LDD and CS [3,6]. Some recent studies even suggest that LDD may represent a component of CS [2,7]. However, both conditions might represent a distinct form of phakomatosis [5].
Dysplastic gangliocytoma of the cerebellum is a benign lesion that occurs sporadically, with approximately 300 cases reported worldwide [3]. It should be suspected in adults presenting symptoms of progressive posterior fossa compression [8]. The estimated prevalence of CS ranges between 1 in 200,000 and 1 in 250,000 individuals and appears to show a female predominance [3]. The under-recognized coexistence of LDD and CS warrants careful clinical evaluation, early diagnosis, genetic testing, and long-term follow-up to improve awareness and identify possible PTEN-related malignancies [8,9].
Simultaneously, PTEN mutations have been identified as the primary cause of both LDD and CS [10] and have also been observed in some individuals with autism spectrum disorder (ASD) [11], suggesting that PTEN may be a genetic risk factor for ASD [12]. Many children with PTEN mutations experience developmental delay [13], and ASD has been reported in children with CS [10,11,14]. However, because many clinical features of CS manifest only in adulthood, diagnosing the condition in children remains challenging [15]. Therefore, screening for PTEN mutations is strongly recommended in children with a family history of PTEN-related malignancies [16].
Here, we present a rare case of a mother with LDD/CS and her child with ASD, discussing recommendations for screening, diagnosis, and management of LDD/CS and PTEN-related ASD, and reviewing the relevant literature.
2. Case Presentation
We performed a thorough clinical examination and additional diagnostic and genetic testing in a female patient who had undergone surgery in 2015 for dysplastic gangliocytoma of the cerebellum when she was 35 years old to identify possible signs of Cowden syndrome (CS). The evaluation included skin and mucosal inspection for hamartomas, revealing coffee-colored spots on her skin (Figure 1) and melanosis of the sclera (Figure 2). She also performed blood tests, a thyroid hormone assessment, a thyroid ultrasound, a gastrointestinal evaluation (including gastroscopy and colonoscopy), and a breast examination. She had also been diagnosed with a pituitary corticotropin-secreting microadenoma and a breast fibroadenoma, both surgically removed. Over the following years, she developed multiple cutaneous hamartomas and a recurrent benign mesenchymoma in the region of the first metacarpal bone of the left hand, which had been surgically treated in childhood. She also presented with recurrent fibroadenomas of the right breast, which were successfully resected. In addition, she was diagnosed with polycystic ovarian syndrome, a hormonal disorder characterized by androgen imbalance.
At the time of hospital admission in February 2015, she complained of occipital headaches, but her neurological status was intact. Brain MRI revealed a striated, laminar, tiger-striped expansive mass located in the left cerebellar hemisphere, hypointense on T1- and hyperintense on T2-weighted images, consistent with dysplastic cerebellar gangliocytoma (Figure 3 and Figure 4). The patient underwent a left-sided suboccipital craniectomy with partial tumor resection. Her postoperative course was uneventful, and she achieved complete recovery, with no evidence of tumor recurrence during a 10-year follow-up period. Histopathological examination of the tumor confirmed dysplastic ganglion cells in the cerebellum’s molecular and inner granular layers, showing disrupted normal architecture. Immunohistochemical analysis demonstrated synaptophysin- and transmembrane glycoprotein (CD34)-positive ganglion cells with no proliferative activity (Ki-67).
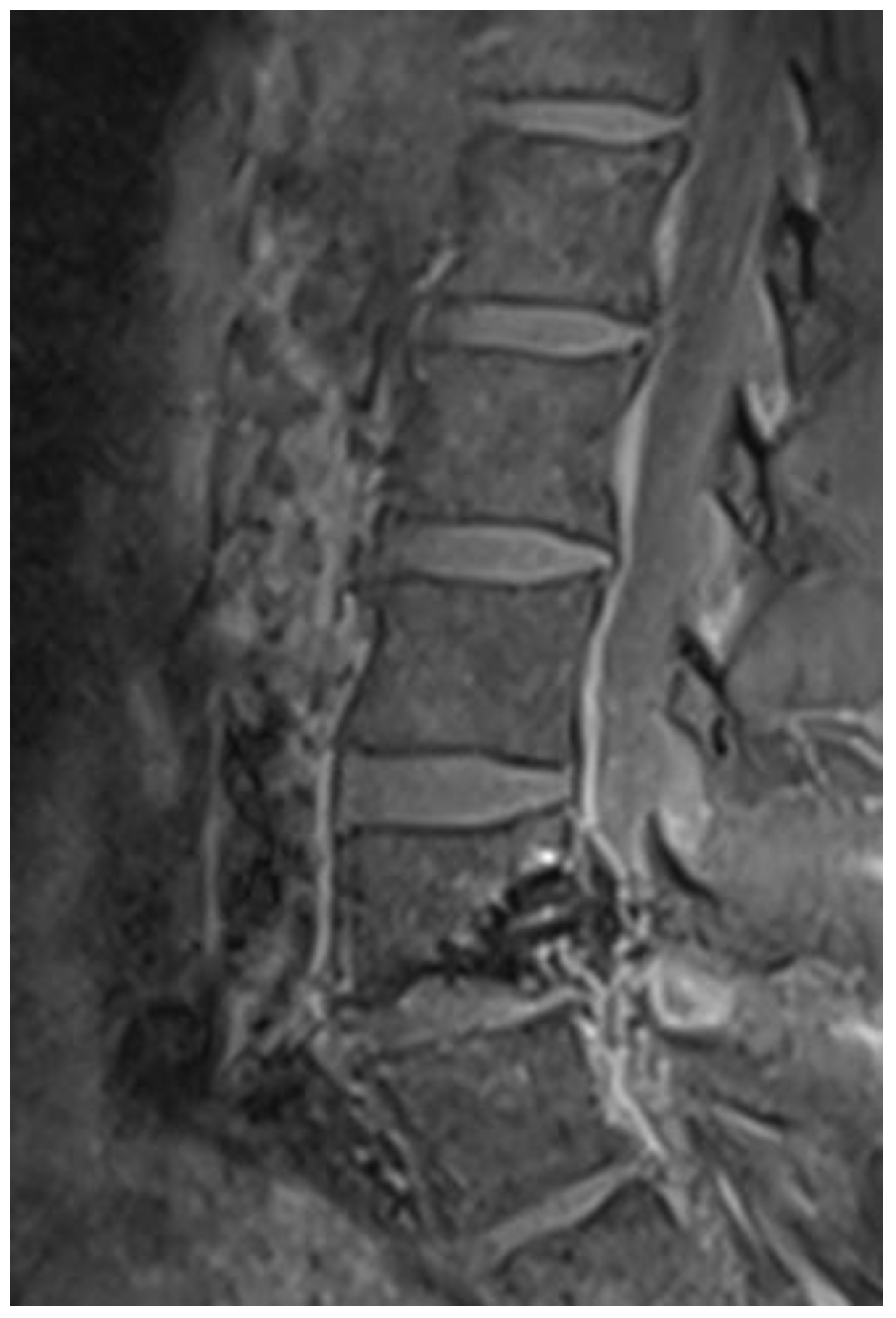
Several years later, a spinal dural venous fistula at the L4/L5 level was detected using spinal angio-MR and angiography and was successfully occluded by endovascular glue embolization (Figure 5).
From family history, it was noted that the patient is the mother of a 14-year-old girl diagnosed with a severe form of autism spectrum disorder (ASD), who was 4 years old when her mother was operated on due to dysplastic cerebellar gangliocytoma.
Given that the same chromosomal aberrations (10q23.31 PTEN–related mutations) are known to underlie LDD/CS in adults and ASD in children [17], we hypothesized a possible coexistence of LDD/CS in the mother and ASD in the child, speculating that the same genetic aberration might be responsible for both conditions. Consequently, we proceeded with clinical investigations and genetic PTEN testing for the daughter. Additional investigations included blood tests and clinical assessments such as EEG, psychological testing, and examination of the skin, eyes, and mucous membranes. The girl was born from a normal pregnancy with an uneventful course, delivered by cesarean section. However, her speech development was delayed. At a year and a half, she began to coo (producing a soft murmuring sound) and remained at that speech level, communicating non-verbally and only using stereotyped sounds. There were no behavioral issues until about a year ago (the start of puberty). Otherwise, her personality is calm and withdrawn, never showing hetero-aggressive behavior. She experienced her first epileptic seizure at age 9, which was a generalized tonic-clonic attack. Since then, she has been on anticonvulsants (Trileptal) and was seizure-free until two years ago, when she had her last generalized seizure. A brain MRI was performed when she was 5 years old, and it was normal, but it has not been repeated since. She is now in the care of an educational rehabilitator, no longer sees a speech therapist or psychologist, and has not attended school for the past four years.
During an examination by a pediatrician, logopedist, and geneticist, she appeared emotionally warm and morphologically exhibited pronounced facial dysmorphia, including prominent eyebrows and frontal bossing. She also showed spindle-shaped fingers on both hands and annular discoloration of the conjunctiva in both eyes. Her fine motor skills were clumsy, and her left hand was positioned in a dystonic posture without fixed deviations or contractures. Her posture was accompanied by spinal kyphosis, hypotrophic, hypotonic muscles. and hyperextensibility, while her gait was wider-based with legs in abduction. During EEG recording, while awake with eyes open, alpha and beta activities with isolated bilateral frontal foci were observed.
After obtaining informed consent, genetic analysis of both mother and child confirmed PTEN gene mutations.
3. Discussion
The phosphatase and tensin homolog (PTEN) gene is a tumor suppressor whose mutations result in PTEN Hamartoma Tumor Syndrome (PHTS), an autosomal dominant disorder predisposing individuals to the development of hamartomas [4]. This condition exhibits a broad spectrum of clinical manifestations [18] (Figure 1 and Figure 2), although its natural history during childhood remains poorly understood [19].
The PHTS spectrum includes Lhermitte-Duclos disease (LDD), a hamartomatous tumor of the cerebellum; Cowden syndrome (CS), a PTEN-related familial cancer syndrome characterized by multiple hamartomas and neoplasms; and possibly autism spectrum disorder (ASD), a neurodevelopmental syndrome marked by social and communication deficits, as well as restricted and repetitive behaviors and interests, typically with onset before the age of three years [15]. An ASD also has a strong genetic component, with an inherited susceptibility [20].
Cerebellar involvement in PHTS has long been recognized; however, the fundamental role of the cerebellum as a phenotypic expression in a subset of PHTS patients and its link to ASD pathogenesis has been more recently elucidated [21]. Although environmental factors may contribute to ASD etiology, substantial genetic evidence indicates that cerebellar dysfunction is consistently associated with ASD [22]. Furthermore, PTEN aberrations have been associated with higher rates of developmental delay and ASD among PHTS patients [23]. Therefore, MR proton spectroscopy is recommended as part of the neuroimaging assessment for children of PHTS patients over two years of age who present with developmental delay [24]. However, we did not have an opportunity for an MR spectroscopy in the ASD girl, since she last underwent a brain MRI when she was 5 years old, and her parents did not consent to allow another MRI scan.
A detailed understanding of the radiological and histopathological features of LDD is essential for timely diagnosis [9]. MRI brain imaging typically reveals a distinctive unilateral, striated, “tiger-striped” cerebellar hyperintensity on T2-weighted sequences [25,26]. Our LDD/CS patient demonstrated this characteristic appearance on both T1- and T2-weighted MRI scans (Figure 3 and Figure 4). Histopathological findings of LDD include thickening, vacuolation, and widening of the molecular layer containing abnormal ganglion cells, absence of the Purkinje cell layer, and hypertrophy of the granular layer populated by large dysplastic ganglion cells [8,27]. Immunohistochemically, LDD typically demonstrates loss of PTEN expression, increased neuronal biomarkers such as synaptophysin within dysplastic ganglion cells, and diffuse CD34 staining—findings that confirm the neuronal origin of LDD and its association with PTEN mutations. Our patient’s histopathological and immunohistochemical results were consistent with this profile.
The phenotypic spectrum of PHTS also encompasses various vascular anomalies [28], as PTEN mutations can enhance angiogenesis [29]. This was confirmed in our patient, whose radiological follow-up revealed a spinal dural arteriovenous fistula that was successfully treated with endovascular embolization (Figure 5). Because PTEN mutations significantly increase the risk of tumor development [30], early identification of LDD/CS patients through genetic screening and consistent long-term monitoring is crucial [31].
Although PTEN was originally identified as a tumor suppressor, more recent studies have emphasized its pivotal role in embryonic brain development, regulating cell proliferation, differentiation, migration, neurite outgrowth, synaptogenesis, and myelination [15,18]. In our study, PTEN mutations were confirmed in both the mother and her daughter. Consequently, a PTEN mutation present in a mother with LDD/CS could be transmitted to her child, leading to neurobehavioral manifestations such as ASD and intellectual disability [17]—a lifelong condition characterized by limitations in intellectual functioning and adaptive behavior that begin in childhood, which was confirmed in our ASD girl patient.
Early genetic testing, tailored counseling, and vigilant surveillance of at-risk PHTS patients and their relatives remain challenging yet essential for anticipating transgenerational outcomes [32].
This report contributes to the existing body of literature by reinforcing the probable genetic association between Lhermitte-Duclos disease/Cowden syndrome and autism spectrum disorder in PTEN-related patients—a relationship that has been only rarely documented.
The limitations associated with this paper arise from its single-case report character and its retrospective nature. Therefore, further large-scale, prospective studies are needed to characterize pediatric PTEN-ASD patients better and establish standardized diagnostic and treatment protocols.
4. Conclusions
Our findings suggest a probable association between the LDD/CS-affected parent and the ASD-affected child, as both conditions may be linked to PTEN mutations, which can be inherited in an autosomal dominant manner. Because such mutations confer a significant risk of malignancy, patients suspected of having LDD/CS should undergo regular screening for PTEN-associated neoplasms. In addition, their relatives, including offspring, should receive genetic testing and counseling.
Author Contributions
M.V.: conceptualization, writing—original draft preparation; G.L.: data curation; resources; B.B.: validation; methodology; S.L.: software; formal analysis; S.F.: supervision, validation; B.S.: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki. It did not require ethical approval in accordance with local/national guidelines.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data underlying this study are accessible upon request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
Contains supplementary figures providing illustrations that are essential for a comprehensive understanding of this case report.
References
- Tan, T.C.; Ho, L.C. Lhermitte-Duclos disease associated with Cowden syndrome. J Clin Neurosci 2007, 14, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.; Scholz, M.; Nelen, M.R.; Schwechheimer, K.; Epplen, J.T.; Harders, A.G. Lhermitte-Duclos disease as a component of Cowden’s syndrome. Case report and review of the literature. J Neurosurg 1999, 90, 776–9. [Google Scholar] [CrossRef]
- Alanazi, A.I.; Alanezi, T.; Aljofan, Z.F.; Alarabi, A.; Elwatidy, S. Lhermitte-Duclos disease: A systematic review. Surg Neurol Int 2023, 14, 351. [Google Scholar] [CrossRef]
- Pilarski, R. PTEN hamartoma tumor syndrome: a clinical overview. Cancers 2019, 11, 844. [Google Scholar] [CrossRef]
- Robinson, S.; Cohen, A.R. Cowden disease and Lhermitte-Duclos disease: An update. Case report and review of the literature. Neurosurg Focus 2006, 20, E6. [Google Scholar] [CrossRef] [PubMed]
- Vantomme, N.; Van Calenbergh, F.; Goffin, J.; Sciot, R.; Demaerel, P.; Plets, C. Lhermitte-Duclos disease is a clinical manifestation of Cowden’s syndrome. Surg Neurol 2001, 56, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Núñez, A.; Lagares, A.; Benítez, J.; Urioste, M.; Lobato, R.D.; Ricoy, J.R. Lhermitte-Duclos disease and Cowden disease: Clinical and genetic study in five patients with Lhermitte-Duclos disease and literature review. Acta Neurochir (Wien) 2004, 146, 679–690. [Google Scholar] [CrossRef]
- Nowak, D.A.; Trost, H.A. Lhermitte–Duclos disease (dysplastic cerebellar gangliocytoma): A malformation, hamartoma or neoplasm? Acta Neurol Scand 2002, 105, 137–145. [Google Scholar] [CrossRef]
- Lakicevic, G.; Tinjak-Demic, S.; Lakicevic, S.; Frol, S.; Splavski, B. Two illustrative cases of adult Lhermitte-Duclos disease and a systematic review of literature related to surgical management. Brain Spine 2025, 104258. [Google Scholar] [CrossRef]
- Nelen, M.R.; van Staveren, W.C.; Peeters, E.A.; Hassel, M.B.; Gorlin, R.J.; Hamm, H.; et al. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Hum Mol Genet 1997, 6, 1383–1837. [Google Scholar] [CrossRef]
- Goffin, A.; Hoefsloot, L.H.; Bosgoed, E.; Swillen, A.; Fryns, J.P. PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet 2001, 105, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Skelton, P.D.; Stan, R.V.; Luikart, B.W. The role of PTEN in neurodevelopment. Mol Neuropsychiatry 2020, 5 Suppl 1, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xu, Z.; Qin, J.; Yang, Z. Epilepsy and developmental delay in pediatric patients with PTEN variants and a literature review. Pediatr Neurol 2025, 163, 35–44. [Google Scholar] [CrossRef]
- Conti, S.; Condò, M.; Posar, A.; Mari, F.; Resta, N.; Renieri, A.; et al. Phosphatase and tensin homolog (PTEN) gene mutations and autism: literature review and a case report of a patient with Cowden syndrome, autistic disorder, and epilepsy. J Child Neurol 2012, 27, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.D.; Cai, G.; Chaste, P.; Nygren, G.; Goldsmith, J.; Reichert, J.; et al. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet 2007, 144B(4), 484–491. [Google Scholar] [CrossRef]
- Ciaccio, C.; Saletti, V.; D’Arrigo, S.; Esposito, S.; Alfei, E.; Moroni, I.; et al. Clinical spectrum of PTEN mutation in pediatric patients. A bicenter experience. Eur J. Med Genet 2019, 62, 103596. [Google Scholar] [CrossRef]
- Frazier, T.W. Autism spectrum disorder associated with germline heterozygous PTEN mutations. Cold Spring Harb Perspect Med 2019, 9, a037002. [Google Scholar] [CrossRef]
- Dhawan, A.; Liu, D.; Baitamouni, S.; Anthony, K.; Srivastava, S.; Hardan, A.Y.; et al. Neurodevelopmental and neurologic manifestations of PTEN hamartoma tumor syndrome: management recommendations. Neurol Genet 2025, 11, e200299. [Google Scholar] [CrossRef]
- Macken, W.L.; Tischkowitz, M.; Lachlan, K.L. PTEN Hamartoma tumor syndrome in childhood: A review of the clinical literature. Am J Med Genet C Semin Med Genet 2019, 181, 591–610. [Google Scholar] [CrossRef]
- Veenstra-Vanderweele, J; Christian, SL; Cook, EH, Jr. Autism as a paradigmatic complex genetic disorder. Annu Rev Genomics Hum Genet 2004, 5, 379–405. [Google Scholar] [CrossRef]
- Gambini, D.; Ferrero, S.; Bulfamante, G.; Pisani, L.; Corbo, M.; Kuhn, E. Cerebellar phenotypes in germline PTEN mutation carriers. Neuropathol Appl Neurobiol 2024, 50, e12970. [Google Scholar] [CrossRef]
- D’Mello, S.R. Autism spectrum disorder: The cerebellum, genes, and pathways. Neurol Int 2025, 17, 173. [Google Scholar] [CrossRef] [PubMed]
- Cummings, K.; Watkins, A.; Jones, C.; Dias, R.; Welham, A. Behavioural and psychological features of PTEN mutations: a systematic review of the literature and meta-analysis of the prevalence of autism spectrum disorder characteristics. J Neurodev Disord 2022, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Filippi, C.G.; Uluğ, A.M.; Deck, M.D.; Zimmerman, R.D.; Heier, L.A. Developmental delay in children: assessment with proton MR spectroscopy. AJNR Am J Neuroradiol 2002, 23, 882–888. [Google Scholar]
- Joo, G.; Doumanian, J. Radiographic findings of dysplastic cerebellar gangliocytoma (Lhermitte-Duclos Disease) in a woman with Cowden Syndrome: A case study and literature review. J Radiol Case Rep 2020, 14, 1–6. [Google Scholar] [CrossRef]
- Zhang, H.W.; Zhang, Y.Q.; Liu, X.L.; Mo, Y.Q.; Lei, Y.; Lin, F. MR imaging features of Lhermitte-Duclos disease. Medicine (Baltimore) 2022, 101, e28667. [Google Scholar] [CrossRef] [PubMed]
- Kolhe, A.A.; Shenoy, A.; Tayal, S.; Goel, N.A. Lhermitte-Duclos disease: A series of six cases. J Neurosci Rural Pract 2023, 14, 127–131. [Google Scholar] [CrossRef]
- Jong-A-Liem, G.S.; Sarti, T.H.M.; Dos Santos, M.G.; Giacon, L.M.T.; Wuo-Silva, R.; Baeta, A.M.; et al. Case report: Association between PTEN-gene variant and an aggressive case of multiple dAVFs. Front Neurol 2024, 15, 1347289. [Google Scholar] [CrossRef]
- Takei, J.; Tochigi, S.; Arai, M.; Tanaka, T.; Kajiwara, I.; Hatano, K.; et al. Spinal extradural arteriovenous fistula with Cowden syndrome: a case report and literature review regarding pathogenesis and therapeutic strategy. NMC Case Rep J 2018, 5, 83–85. [Google Scholar] [CrossRef]
- Miguelote, S.; Silva, R.; Fougo, J.L.; Barbosa, L.E.; Araújo Teixeira, J.P. Cowden syndrome is a risk factor for multiple neoplasms: a case report. World J Surg Oncol 2020, 18, 211. [Google Scholar] [CrossRef]
- Monjaras-Romo, G; Zavala-Romero, L; Tejada-Pineda, MF; Meraz-Soto, JM; Ballesteros-Herrera, D; Cienfuegos-Meza, J. Lhermitte-Duclos Disease: a case series. Cureus 2023, 15, e44326. [Google Scholar] [CrossRef] [PubMed]
- Ilic, N.; Mitrovic, N.; Radeta, R.; Krasić, S.; Vukomanović, V.; Samardzija, G.; et al. Phenotypic variability of Cowden Syndrome within a single family: impact on diagnosis, management and genetic counseling. Balkan J Med Genet 2025, 27, 95–100. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A photo of skin inspection for hamartomas, revealing coffee-colored spots on the back.
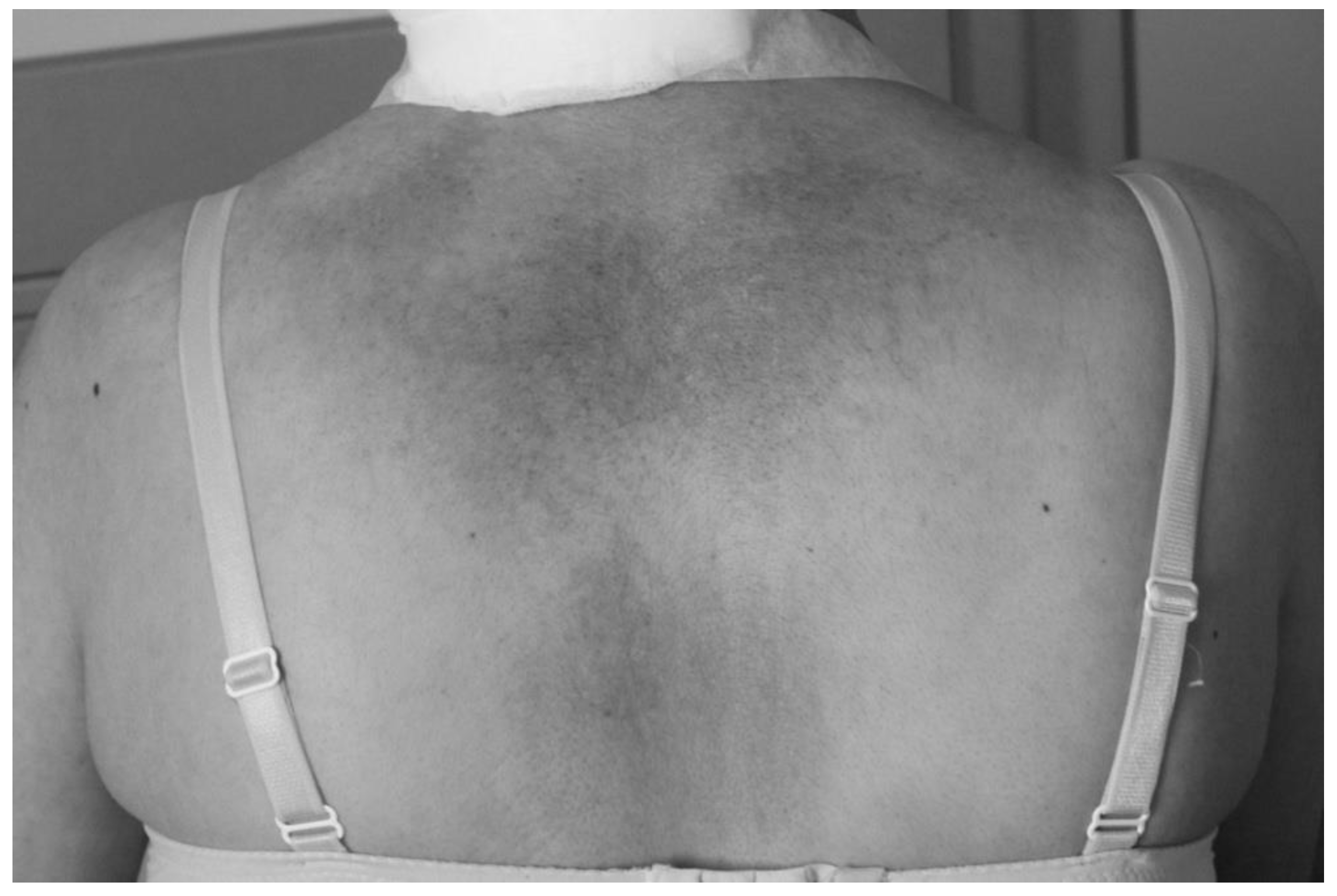
Figure 2.
A photo of mucosal inspection, revealing melanosis of the sclera.
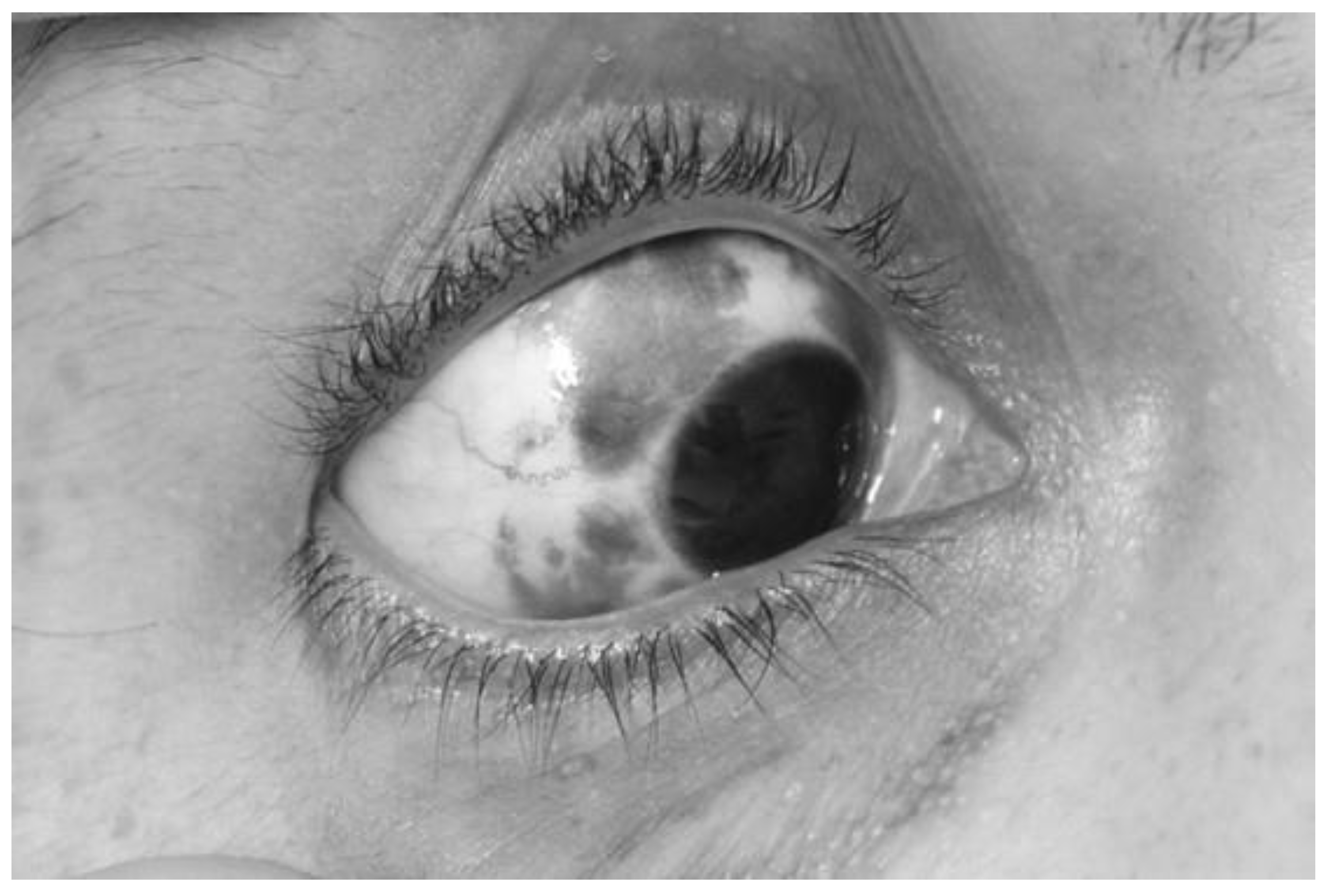
Figure 3.
Axial T1-weighted native MRI brain scan showing a characteristic hypointense expanding mass of the left cerebellar hemisphere resembling a gangliocytoma.
Figure 3.
Axial T1-weighted native MRI brain scan showing a characteristic hypointense expanding mass of the left cerebellar hemisphere resembling a gangliocytoma.
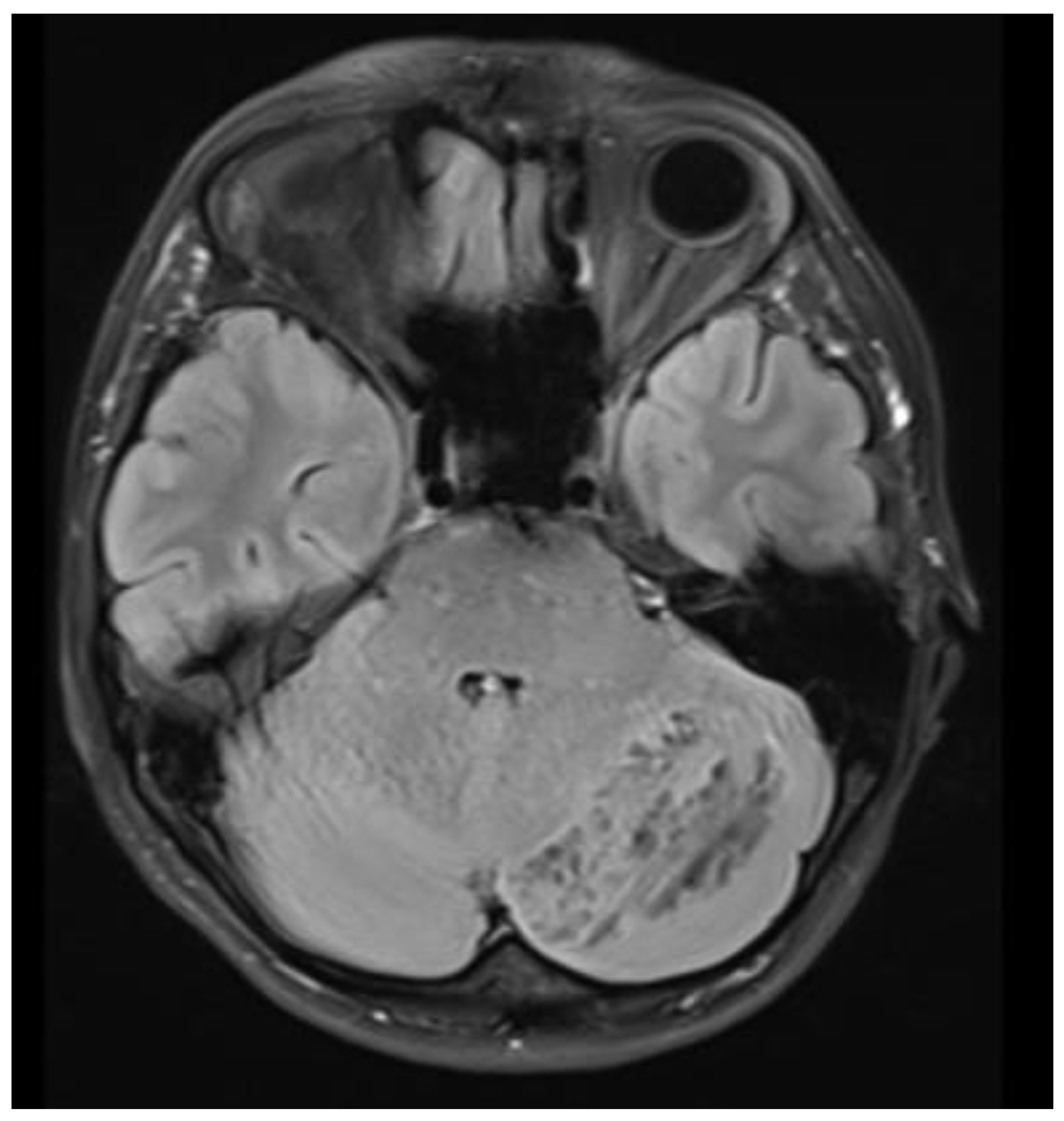
Figure 4.
Unilaterally altered left cerebellar cortex with a distinctive hyperintense striate, laminar tiger-striped pattern visible on native T2-weighted MRI brain scan.
Figure 4.
Unilaterally altered left cerebellar cortex with a distinctive hyperintense striate, laminar tiger-striped pattern visible on native T2-weighted MRI brain scan.
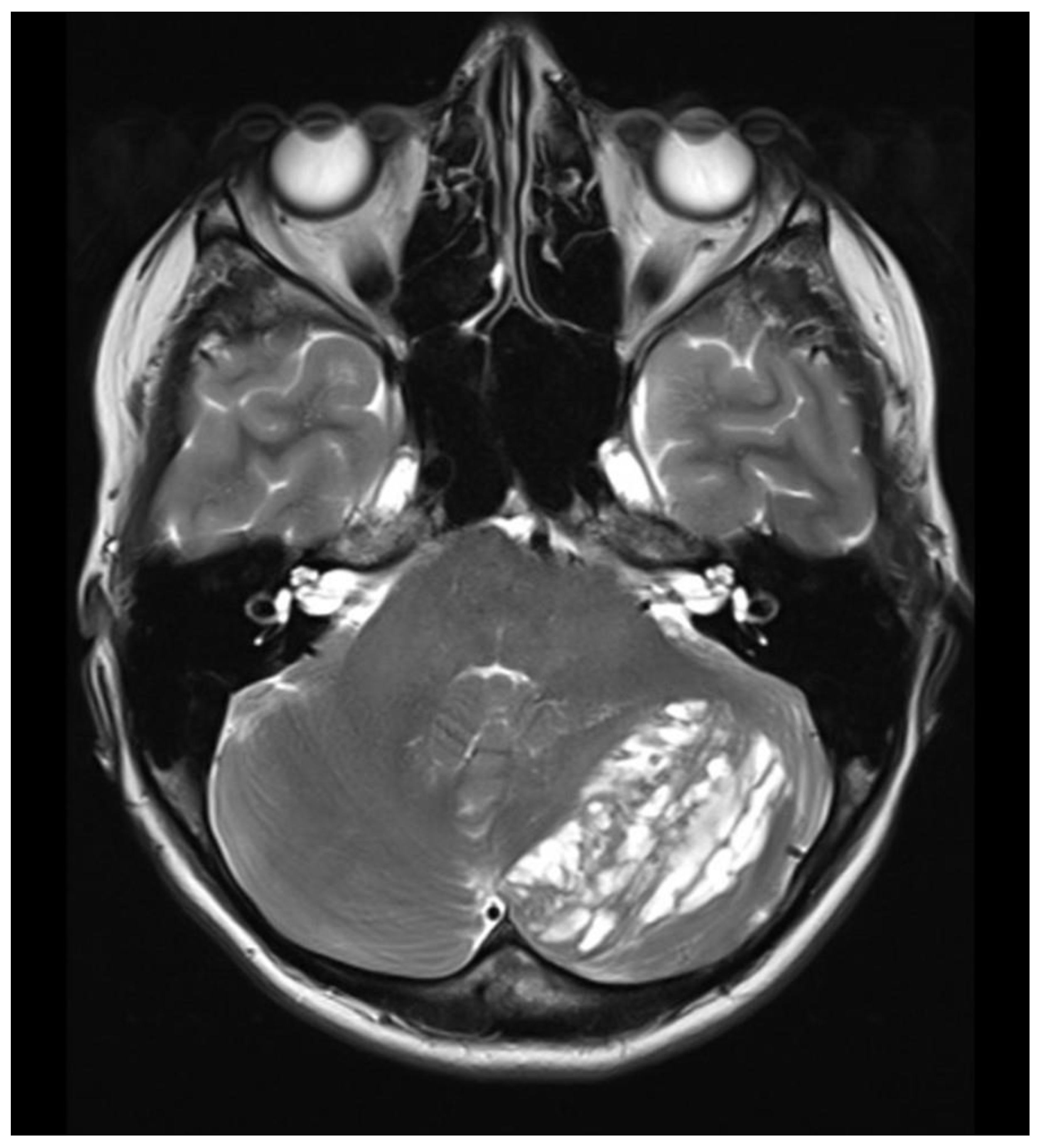
Figure 5.
Figure 5. Postprocedural spinal sagittal T1-weighted (STIR) MRI showing a successfully occluded dural arterio-venous fistula at the L4/5 level.
Figure 5.
Figure 5. Postprocedural spinal sagittal T1-weighted (STIR) MRI showing a successfully occluded dural arterio-venous fistula at the L4/5 level.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



