
Submitted:
15 December 2025
Posted:
16 December 2025
You are already at the latest version
Abstract
The creation of a specific culture medium for colorectal organoids in 2011 opened a new era in human primary cultures by enabling the indefinite expansion of normal and pathological epithelial organoids. The original formula has been used ever since, with only minor, lab-specific modifications. The goal of culturing organoids from dif-ferent tissues has relied on saving and propagating the pluripotent stem cell. The "magic bullet" and all its subsequent derivatives have pursued this goal. Consequently, agonist and antagonist signals are chronically activated in the organoid medium, forcing organoid cells (as well as any other co-cultured cellular model) into con-strained signaling pathways. This extremely artificial condition is often overlooked in experimental approaches and may bias the results. Furthermore, some molecules in the organoid medium have unpredictable off-target effects that significantly impact the behavior and maturation of certain cell populations. In this short review, we will ex-amine the components of the colorectal organoid medium, describing their activity and necessity for organoid culture. We will also discuss the expected biases in specific experimental settings. While the original organoid medium formula is the gold stand-ard for propagating organoids in vitro, more focused, reliable conditions are necessary for specific organoid-based tests.
Keywords:
colon
; organoid
; culture medium
; A83-01
; SB202190
; nicotinamide
; n-acetylcysteine
; prostaglandin E2
; Primocin
1. The First, Long-Term, Human Colorectal Organoid Culture
In 2011, Sato and Clevers described the first in-vitro method for the long-term expansion of normal and neoplastic human colon organoids [1]. This method allowed for the cultivation of pure epithelial cells embedded in 3D matrix domes, maintaining the stem cell niche indefinitely. The basal medium included advanced DMEM-F12 (a medium designed for culturing cells with low FCS supplementation) with B27 and N2 supplements (both originally developed for neural cells), and N-AcetylCysteine (NAC) (Table 1). The key, added growth factors were Wnt3a and R-spondin, agonists of the β-catenin pathway for stem cell propagation; Noggin, replacing colon Gremlin1 and 2, and neutralizing BMP-mediated differentiation; and EGF empowering Erk1-2 signaling for proliferation and survival. Notably, R-spondin was included in the cocktail following the discovery of its binding to LGR5, the marker of colon stem cells, a few months earlier [2].
Although these growth factors could theoretically suffice for culturing colon organoids, they only permitted short-term propagation. A broad screening of compounds was conducted to refine the formula. The addition of survival modulators (nicotinamide, gastrin, PGE2 and Y27632) increased the plating efficiency of primary cultures and secondary splits. Two essential signaling inhibitors were also identified: the Alk5 inhibitor A83-01, which blocks TGFβ receptor signaling, and the p38 inhibitor SB202190. This final mix enabled the long-term, indefinite propagation of colon crypts.
One limitation of this formula is the requirement of high concentrations of growth factors (Wnt3a 100ng/ml, R-Spondin 500-1000ng/ml, Noggin 100ng/ml, EGF 50ng/ml). Additionally, the poor solubility of recombinant Wnt3a in aqueous solutions can result in reduced or absent biological activity [18]. Currently, most labs have abandoned the use of recombinant proteins in favor of the conditioned medium of cells that have been transfected to overexpress Wnt3a, R-Spondin, or Noggin (Table 1). This modification introduces in the organoid medium an undefined cocktail of additional factors, spontaneously released by the transduced cells. According to the published protocols, up to 80% of the organoid medium volume comes from 4-7-day-old cell culture supernatants made with FCS-containing media. This condition differs greatly from the initial formula, resulting in a loss of standardization and prompting modifications that will be discussed in the following chapters. Since the colon medium recipe formed the basis for developing most organoid media used today, these considerations could be extended to primary cultures of many epithelia.
2. Signal Transduction Pathways Involved in Colorectal Organoids Propagation
Before discussing the numerous effects of the components of the colorectal organoid medium, it is important to understand the signaling pathways that affect organoid physiology and viability (see Figure 1 for a simplified scheme).
The most difficult endpoint for the colorectal organoid medium is maintaining the LGR5 (leucine-rich repeat-containing G-protein-coupled receptor 5) -positive stem cell population. This requires both agonism of positive signals and inhibition of negative signals. The main driver of stem cell persistence and proliferation is β-catenin signaling, which is primarily (though not exclusively) mediated by the canonical Wnt (wingless and int-1) family members (Wnt 1, 2, 3, 8, and 10). Heavy palmitoylation and glycosylation make Wnts insoluble [19], this characteristic confers two apparently contradictory effects on these ligands: it confines them to the site of production (allowing the formation of precise spatial gradients along colorectal crypt axis), but also enables them to bind to serum alpha-albumin and lipoproteins, giving them the ability to exert systemic, endocrine-like activity [20,21]. The main effect of canonical Wnt signaling is the destabilization of the β-catenin destruction complex, allowing β-catenin accumulation and translocation into the nucleus. When CK1 (casein kinase 1), GSK3β (glycogen synthase kinase-3 beta), Axin and APC (adenomatous polyposis coli) assemble the β-catenin destruction complex, CK1 phosphorylates β-catenin at S45 priming successive phosphorylations by GSK3β, directing β-catenin to proteasomal degradation [22]. Wnt, binds to and couples FZD (frizzled) and Lrp5/6 (low-density lipoprotein receptor-related protein 5/6) receptors, causing the sequestration and inactivation of the β-catenin destruction complex. β-catenin then accumulates in the cytoplasm and migrates into the nucleus, activating the TCF/LEF (T-cell factor/lymphoid enhancer factor) transcription factor. This enhances the expression of c-Myc (cellular myelocytomatosis oncogene) and Cyclin D, among other genes. Wnt signaling activation is self-limiting. Once β-catenin activates the TCF/LEF transcription factor, E3 ligases (RNF43/ZNRF3) are transcribed early, driving FZD-LRP complex degradation [23]. What makes the difference in colorectal stem cells, is the binding and activation of LGR5 by R-spondins, causing the sequestration of RNF43/ZNRF3 [24]. R-spondins selectively amplify Wnt signaling in stem cells, activating the ASCL2 (achaete-scute homolog 2) transcription factor, which guarantees the expression of LGR5 and other stem markers [25]. ASCL2 expression is also responsible for regenerating LGR5+ cells in depleted intestinal crypts, in mouse models [26], a phenomenon never observed in vitro.
The activation of the TCF-LEF transcription factor by β-catenin does not only rely on the Wnt pathway. The specific phosphorylation of β-catenin at serine residues 552 and 675 by PKA (protein kinase A) promotes its nuclear translocation and TCF-LEF transactivation [27]. Therefore, any increase in cyclic AMP levels could theoretically activate PKA and enhance β-catenin signaling. Akt (Ak strain transforming) is also able to phosphorylate β-catenin on ser552 [28], creating a direct connection between growth factor receptors and Wnt signaling.
β-catenin signaling is necessary, but not sufficient, for mainteining the LGR5+ stem cell. Along the intestinal crypt, an increasing gradient of BMPs (bone morphogenetic proteins) and TGFβ (transforming growth factor beta) drives cell differentiation by SMAD (mothers against decapentaplegic homolog) intracellular signaling. These factors are absent at the bottom of the crypt, where LGR5+ cells are located. BMPs are sequestered by fibroblast-derived Gremlin-1/2 proteins, which act like Noggin as direct BMP trappers [29,30]. Accordingly, SMAD signaling must be strongly limited to prevent the extinction of stem cells in vitro [4].
P38 is a MAPK (mitogen-activated protein kinase) involved in stress and inflammatory responses [31]. Its role in colorectal organoid culture is controversial, but is generally considered negative, as evidenced by the need for SB202190 (a P38 inhibitor) in the culture medium [1]. A direct consequence of P38 inhibition is the disappearance of Goblet and enteroendocrine cells in cultured organoids [1,32]. Though, the presence of these secretory populations did not affect organoids viability and replacing SB202190 with FGF2 and IGF1 was found to be even more effective in colorectal organoid culture [11]. It appears that the primary beneficial effect of SB202190 is off-target and involves stabilizing EGFR signaling and reducing its degradation [33].
EGFR triggering is the most reliable way to promote organoid proliferation, and EGF is added to almost all organoid media. EGFR directly activates RAS-RAF-MEK-Erk signaling and indirectly activates PI3K-Akt-mTOR signaling [34]. Constitutive Erk1-2 (extracellular signal-regulated kinase 1-2) signaling is necessary for propagating colorectal organoids in vitro by enforcing β-catenin-driven c-Myc and Cyclin D transcription. However, the role of Erk1-2 in the stem cell niche is controversial. Several reports indicate that the absence of Erk1-2 signaling could promote expansion of intestinal stem cells [35,36], perhaps through compensatory activation of Erk5 [37]. Akt plays a central role in the control of cell metabolism and survival, sensing systemic and local signals to balance cellular anabolism/catabolism [38]. Insulin is always overdosed in organoid medium, pushing cells towards a chronic anabolic phase and triggering Akt activation. Therefore, EGF is not the primary activator of this pathway in vitro. Akt reduces GSK3β activity through direct phosphorylation, which blocks GSK3β-triggered inhibition of CyclinD1 and cMyc. This allows the cell to cycle [39]. Conversely, a primary downstream effector of Akt, mTOR (mechanistic target of rapamycin), can exert adverse effects by suppressing Wnt/β-catenin signaling and driving LGR5+ stem cells toward exhaustion [40,41].
The NOTCH (neurogenic locus notch homolog protein 1) pathway is another essential actor of intestinal stem cell persistence and absorptive differentiation [42]. Fortunately, organoids can self-sustain this pathway in vitro through cell-to-cell contact, without the need for any external modulators in the culture medium. Jagged peptide, a NOTCH agonist, has been proposed to improve the survival of organoid-derived single LGR5+ cell suspensions [43], though its true relevance has not been definitively proven. Conversely, the NOTCH pathway inhibitor DAPT is used to promote the differentiation of colon organoids towards a secretory phenotype [44].
A frequently neglected signaling pathway is mediated by integrins. Normal colorectal organoids require proper matrix embedding (3D) or coating (2D) to survive and grow [45]. Integrin-mediated signaling is essential for organoid survival and organization, and laminins are critical components of the LGR5+ stem cell niche [46]. Integrins strongly influence growth factor receptor signaling, and vice versa, activating both Erk1-2 and Akt effectors and forming a complementary activation pathway finely modulated by matrix stiffness [47]. Until now, the Engelbreth-Holm-Swarm (EHS) mouse sarcoma-derived matrix (Matrigel, Geltrex, Cultrex-BME, etc.) is widely used for organoid cultures. However, some complex polymer-peptide hybrid synthetic matrices show promising results for fully standardized, GMP-friendly applications [48,49].
3. Advanced DMEM-F12, B27 and N2
Advanced DMEM-F12, B27, and N2 share several components (see Table 2). An overdose of these components may not necessarily have a positive effect. For example, insulin, transferrin, and sodium selenite could be tripled, resulting in high concentrations. Indeed, N2 has gradually disappeared from the formula of the basal medium in many studies, indirectly suggesting that it has an irrelevant or negative influence on organoid growth.
Insulin can have both positive and negative effects on colorectal organoids. Its ability to induce anabolism and protein synthesis through the PI3K-Akt signaling pathway favors cell proliferation [50]. Akt can also reduce GSK3β activity through direct Ser9 phosphorylation. This inhibition does not affect the GSK3β fraction recruited in the β-catenin destruction complex. However, the contemporary activation of Wnt signaling could reinforce overall GSK3β inhibition, increasing β-catenin accumulation [51]. Conversely, a high concentration of insulin can increase mTORC1 activity inhibiting the autophagic turnover of proteins and mitochondria, causing senescence [52,53]. Thus, the benefits and risks of increased insulin concentrations should be carefully evaluated, to avoid detrimental effects. Any study involving cell metabolism modulation should also consider this constant excess of insulin.
Ferroptosis is an iron-dependent pathway inducing cell death, by mediating non-enzymatic lipid peroxidation [54]. Transferrin receptor 1 is a key mediator of ferroptosis [55] and excess transferrin likely mediates increased intracellular iron levels. However, unless intracellular iron is complexed with ferritin and glutathione (GSH) is abundant, ferroptosis cannot occur. Both advanced DMEM-F12 and B27 contain Holo-Transferrin (the iron-saturated form) and GSH. However, GSH is a short-lived, spontaneously oxidizing molecule in solution [56], needing a continuous regeneration. The need for constantly high GSH levels to prevent ferroptosis could explain why organoid medium is always supplemented with a high dose of N-acetylcysteine (NAC), a precursor of reduced glutathione and a direct reactive oxygen species scavenger. Conversely, ferritin is involved in a specific degradation pathway by autophagy (ferritinophagy) mediated by nuclear receptor coactivator 4 (NCOA4) [57,58]. NCOA4 overexpression depletes cellular ferritin content, causing the release of free ferrous ions that can trigger a self-propagating lipid peroxidation. Several factors can induce NCOA4 transcription, including steroid and thyroid hormones, glucocorticoids, cAMP/cGMP, and the JNK-JUN pathway [59,60]. These modulators have specific triggers among the organoid medium components (triodo-L-thyronine, progesterone, corticosterone, PGE2, EGF), so an increase in NCOA4 is expected. These observations suggest that transferrin should not be increased in the organoid medium because it could cause negative effects without providing any benefits. Moreover, an equilibrated transferrin content could allow for the reduction or elimination of NAC supplementation, contributing to a simplified organoid medium formula.
Selenium plays an essential role in maintaining redox balance as a coenzyme of glutathione peroxidase and thioredoxin reductase [61]. Cell culture media without FCS are usually deficient in selenium and require a specific supplementation. Despite the fact that seleno-L-methionine has been demonstrated to be less toxic, sodium selenite is used as the source of selenium [62]. The three -fold increase of sodium selenite in organoid culture medium supplemented with DMEM-F12, B27 and N2 (about 216nM) is not expected to be toxic, as the toxic threshold for most cellular models is 2.5-10uM [63,64]. Thus, limited overdosage of this salt should not be detrimental to organoid cultures, even if it is not useful or necessary. Moreover, NAC supplementation could again exert a protective effect [65].
Doubled compounds in the organoid medium are ethanolamine, glutathione, bovine serum albumin, putrescine and progesterone. At this dosage, none of these molecules can have a detrimental effect on organoids, though the presence of doubled progesterone levels should be kept in mind when testing gender-specific effects.
The presence of over-supplemented molecules in the organoid medium is not the only anomaly to consider in biological tests. Some constituents of advanced DMEM-F12 or B27 can strongly modulate cellular responses and affect results in biochemical studies or mixed cultures, but we are not aware of this. In particular, ammonium metavanadate and corticosterone are unknown actors in many organoid studies. Vanadate has been identified as a molecule with specific inhibitory activity toward tyrosine phosphatases [66]; its salts are used to preserve phosphorylated proteins in cell lysates for signal transduction analysis. Several studies have shown that vanadate can increase insulin activity, and many vanadate derivatives have been tested as potential anti-diabetic, anti-parasitic, and anti-cancer drugs [67]. While these drugs showed disappointing activity in clinical trials due to their limited tissue distribution, they remain potent in vitro. Vanadate specifically binds to the Fe++ binding pocket of transferrin and forms nonspecific complexes with the His or Asp side chains of several proteins [66,68]. Vanadate inhibits tyrosine phosphatases by binding to the Cys residues in the enzyme active pocket [69]. Consequently, vanadate causes stochastic amplification of signaling pathways involving tyrosine phosphorylation, affecting any experimental system [70]. This can potentially act as a powerful mechanism of drug resistance against small inhibitors of cell signaling in high-throughput drug screening assays. Although present tests using advanced DMEM-F12 as the basal medium do not consider this bias, a 2010 study showed that treating HeLa cells with EGF or pervanadate could amplify the signaling of 52 and 99 phosphoproteins respectively, on multiple tyrosine residues [71]. The perturbation of cell signaling mediated by vanadate should be carefully reevaluated in organoid-based assays.
Corticosterone is a component of the B27 supplement, used at a final concentration of 20 ng/ml in the medium, corresponding to 57.8 nM. This dose mirrors corticosterone levels in human plasma (58.4 ± 9.2 nM) [72]. Although corticosterone is a well-known immunosuppressant, exposure to chronic high doses increases the innate immune response, causing cytokine secretion and chronic inflammation [73]. Glucocorticoid neo-synthetic enzymes can be directly induced in the intestinal epithelium by LRH-1 (liver receptor homolog-1), expressed by proliferating cells in the intestinal crypts [74,75]. The activation of this pathway is triggered by immune activation and leads to the final production of corticosterone (in the mouse) or cortisol (in humans; see [76] for review). Glucocorticoids are also released by CRC cells, participating in the suppression of T-cell activation in this cancer [77]. Exogenous corticosterone in the organoid medium could mask or influence the synthesis of endogenous glucocorticoids in normal and cancerous colorectal organoids. The influence of autocrine glucocorticoid synthesis in human colorectal organoids is a neglected field in immunology.
4. Primocin
Primocin, is a polyantibiotic mix not included in the original 2011 organoid formula. Although it should be used to prevent contaminations during the initial establishment of organoid primary cultures, Primocin is constitutively added to all organoid media. Therefore, the main activity of Primocin could not be preventing contamination, but rather having a direct, as yet uncharacterized positive effect on organoid cultures. In fact, Primocin has been described as a critical additive for culturing and reprogramming both mouse embryonic and human induced pluripotent stem cells [78,79]. Although the composition of Primocin is not fully disclosed, its formula comprises three cell-permeable antibiotics (targeting bacterial DNA gyrase, 30S and 50S ribosomal subunits) and an antifungal targeting ergosterol. Accordingly, we can hypothesize a combination of one fluoroquinolone, one aminoglycoside or tetracycline, one macrolide, and either amphotericin or an azole compound. Aminoglycosides, amphenicols, lincosamides, tetracyclines, macrolides, oxazolidinones and streptogramins are known inhibitors of bacterial and human mitochondrial polypeptide synthesis. Thus, the constant presence of these compounds in the culture medium could lower the number of mitochondria and/or their efficiency [80]. Interestingly, high-dose nicotinamide, another organoid medium supplement, also decreases the number of mitochondria, causing their fragmentation and autophagy [81]. This suggests that a reduced number or activity of mitochondria could have positive effects on organoids by lowering reactive oxygen species levels. Any metabolic or metabolomic study should be aware of this constant brake on mitochondria.
Fluoroquinolones are metal cation chelators showing a Fe3+ binding ability comparable to deferoxamine [82]. These antibiotics bind to multivalent metal ions by a 1:1 stoichiometry, showing increasing avidity for Mg2+ < Mn2+ < Zn2+ << Fe3+ < Al3+ [83]. Accordingly, fluoroquinolones could lower the availability of metal coenzymes in vitro and reduce organoid metabolism through direct enzymatic inhibition. This has previously been demonstrated in other cell models for histone and DNA demethylases and collagen prolyl 4-hydroxylases [82]. These biological effects do not produce any plausible advantages in organoid cultures and could cause unpredictable effects during tests. Thus, the long-term exposure of organoids to fluoroquinolones should be avoided.
The undisclosed antifungal compound of Primocin targeting ergosterol could act by direct binding (Amphotericin B) or by inhibiting its synthesis (triazoles). Amphotericin B is a powerful molecule directly binding to ergosterol and creating pores in the fungal cell membrane. Unfortunately, it is also highly toxic as its working concentration in vitro (2.5ug/ml) is very close to the cell toxicity threshold (5-10ug/ml) [84]. Amphotericin B also has unpredictable effects on the immune system, activating macrophages and enhancing B, T, NK and Tγδ cell activation in different animal models [85,86,87]. Human colorectal cells (both normal and neoplastic) react to Amphotericin B exposure with an active defense mechanism involving the release of amphotericin B-rich exosomes [88]. This effect is also elicited by the standard 2.5ug/ml dose used in cell cultures; thus, Primocin might interfere with any study on exosomes in colorectal organoids. Alternative ergosterol-targeting compounds in Primocin could be triazoles, the inhibitors of fungal cytochrome P450 (C-14α-dimethylase). Triazoles bind tightly to the iron ion of this enzyme [89]. Triazoles have been reported to cause 600 severe drug-drug interactions and over 1100 moderate interactions with other drugs, requiring dose modifications in clinical practice [90]. Accordingly, the presence of Primocin could strongly bias any high throughput assay in drug screening. These observations suggest that Primocin should be eliminated from the organoid medium, especially during experimental assays.
5. Wnt3a, R-Spondin and Noggin
Recombinant Wnt3a is frequently inactive, due to its low solubility in aqueous solutions [18]. Thus, it is typically supplied to organoids as transfected L-cells (mouse fibroblasts)-derived supernatants. These are prepared by incubating L-cells in FCS-containing medium for 4-7 days. This additive modifies the original formula by adding 50% fully metabolized conditioned medium containing FCS, as well as a variety of fibroblast-secreted factors along with Wnt3a. The situation is even more critical when R-spondin and Noggin are also supplied as HEK cell-derived conditioned media. In this condition, up to the 80% volume of the original formula is substituted by exhausted cells supernatants. Unfortunately (or not?), this broth is effective and has been adopted by the leading lab of H. Clevers and many other organoid researchers [6]. As no one has taken care to characterize the real microenvironment created by this dirty formula, it should be noted that unknown factors can contribute to any experimental outcome. To reduce this bias, R-spondin and Noggin could be supplied as recombinant proteins, though this would increase costs. An interesting alternative to Wnt3a is the WNT surrogate [91]. This water-soluble recombinant protein contains the LRP-binding domain of the extracellular Wnt modulator DKK, which is linked to an ankyrin-repeat protein that binds to FZD receptors. Thus, it acts as a bridge between FZD and LRP5/6, just as Wnt proteins do. This clean reagent could be considered a valuable substitute for Wnt3a.
6. Nicotinamide
Nicotinamide (Nic) was added at a high dose of 10 mM to the original Sato & Clevers organoid medium formula. Nic was described as a necessary constituent to increase plating efficiency [1]. However, T. Sato eliminated Nic from the formula in successive studies conducted in his lab [7]. Although eliminating nicotinamide did not cause any problems for the organoids, studies inspired by Clevers have continued to use this additive, despite it apparently being unnecessary after plating. On the other hand, Nic exerts several biological activities, modulating SIRT1, PARP and mitochondrial turnover [92,93,94]. The initial inclusion of Nic in the organoid medium formula could be linked to its ability to act as a kinase inhibitor targeting ROCK (Rho-associated coiled-coil kinase) and CK1 [95]. Indeed, Nic mimics the ROCK inhibitor Y27632 (well known by all organoid and stem cell enthusiasts) [96], inhibiting the phosphorylation of myosin light chain, thus avoiding programmed cell death by anoikis. Nic also acts as a CK1 inhibitor, preventing beta-catenin degradation by the CK1-APC-AXIN-GSK3B destruction complex. In fact, CK1-mediated phosphorylation of the beta-catenin Ser45 residue is mandatory for GSK3B activity and proteasomal degradation [22]. Conversely, the persistence of Nic in culture media has demonstrated growth-inhibiting and pro-differentiating effects on hESC [95].
Although short-term Nic-induced changes could be beneficial for organoid cultures, Nic has been reported to exert opposite effects on immune cells. Nic inhibits T lymphocytes and CART cells cytotoxicity [12,97], B-lymphocytes activation [98] and reduces dendritic cells super-activation in autoimmune models [99]. However, it can favor the expansion of hematopoietic stem cells [100]. These findings strongly suggest performing Nic-free experiments, particularly in immunological settings.
7. N-Acetyl Cysteine
N-acetyl cysteine (NAC) is added to almost all basal organoid medium formulas, although no study has ever attempted to identify its effects on organoids, or justify the selected working concentration. The 1-1.25 mM dose of NAC that is permanently added to the organoid medium exceeds the maximum dose used in short-term treatment for acute acetaminophen intoxication (150mg/Kg ≈ 0.91mM, 1h). In a mouse model of ulcerative colitis, long-term administration of a 150mg/Kg dose of NAC caused irreversible oxidative and inflammatory damage to the colon, liver, and kidney [101]. This effect was mainly mediated by an increase in malondialdehyde (MDA) and an imbalance in TNFα and IL10. A single dose of the same amount lowered MDA levels in mice treated with organophosphate compounds [102], while a dose of 600 mg twice daily (approximately 0.157 mM) is sufficient to lower MDA in humans [103,104]. In vitro, NAC increases the growth and survival of single circulating tumor cells, though this effect is mediated by an optimized concentration of 0.3 mM [105]. According to these observations, NAC could be overdosed in standard organoid medium. Indeed, NAC elimination showed positive effects on prostate organoids [106]. NAC also contrasts with platinum-based chemotherapy [107]. In fact, when NAC was used in drug screenings that included oxaliplatin, no correlation was found between organoid sensitivity and the matched patient response [108,109]). The opposite was observed in the absence of NAC [110]. Thus, NAC should be excluded from any test involving ROS production or evaluating pro-oxidizing drugs.
8. A83-01
A83-01 is an inhibitor of ALK5/TGFβR1 and ALK4, ALK7 activin receptors [111]. This small molecule is used in human colorectal organoid cultures at a 0.5µM concentration to switch off TGFβ receptor signaling. A83-01 blocks Smad2/3 activation avoiding the terminal differentiation of stem cells. According to Vogt et al., A83-01 is not specific, and targets VEGFR and RIPK2 with the same affinity as ALK5 [112]. A83-01 also inhibits MINK1, p38α MAPK and FGFR1 by more than 50% or 30%, at a 1 or 0.1µM concentration respectively. Because VEGFR inhibition could strongly affect any angiogenesis-related study, A83-01 should be omitted from any multicellular model using endothelial cells. RIPK2 is a master regulator of innate immunity and inflammation, also involved in T helper activation [113], and rectal cancer progression [114]. Thus, its inhibition should be considered in inflammatory bowel disease models and immune studies. MINK1 is a crossroads for several signal transduction pathways, regulating cell cycle, apoptosis, and cell migration [115]. The inhibition of FGFR1 should be considered as an active variable in tests involving organoids alone, or in mixed cultures with fibroblasts and endothelial cells. A83-01 has been also shown to promote the growth of mouse hematopoietic stem and progenitor cells in vitro by blocking mast cell differentiation [116]. Although A83-01 is necessary for long-term organoid culture expansion, it can be omitted from short-term assays to eliminate off-target effects.
9. SB202190
SB202190 is a p38 inhibitor [117], its addition to the organoid medium was based on empirical screening of several compounds [1]. P38 inhibition forces colorectal stem cell differentiation toward enterocytes, while secretory cells, such as Goblet and enteroendocrine cells, are no-longer represented in organoids [1,32]. The absence of secretory populations does not enhance organoids viability; thus, SB202190 compromises the physiologic development of a multi-population organoid inducing an undesirable effect.
Despite being a p38 inhibitor, the fundamental activity of SB202190 on colorectal organoids is Erk1-2 signaling amplification [7]. Initially, SB202190-mediated Erk1-2 signaling agonism was attributed to EGFR stabilization and reduced turnover [7]. Though, SB202190 can also mimic the BRAF inhibitor Dabrafenib, resulting in a well-known paradox: Erk1-2 agonism in BRAF wild-type cells and Erk1-2 inhibition in BRAF-mutated cells [118]. Therefore, SB202190 should not be used with BRAF-mutated CRC organoids. The agonist activity does not rely on EGFR, but rather on increased BRAF/CRAF dimerization induced by the drug [119]. SB202190 also inhibits GSK3β, CK1δ, RIPK2, and GAK off-target, at a concentration of 1 µM, which is far below the 3-10 µM concentration used in organoid cultures [120]. Inhibiting GSK3β and CK1δ can lead to β-catenin accumulation and amplified Wnt signaling. RIPK2, as reported for A83-01, modulates immune responses. GAK (Cyclin G-associated kinase) is involved in clathrin-mediated endocytosis and intracellular trafficking [121], also acting as a positive regulator of mitophagy [122]. Low dose SB202190 also inhibits MLK2/MAP3K10 and MLK3/MAP3K11, direct modulators of the Jun pathway [123]. At a 10µM concentration SB202190 induces autophagy and lysosomal biogenesis by activating PPP3/calcineurin signaling [124]. The multiple, complex activities of SB202190 should be considered in any experimental setting involving organoid biochemistry. This compound should be omitted from short-term tests and studies of secretory cells.
10. EGF, Gastrin, PGE2, Y27632
EGF is a necessary constituent of the organoid medium for culturing cells with a wild-type EGFR pathway. EGF is used at a concentration of 50 ng/ml, which is a high dose that can counteract the efficacy of Cetuximab in vitro [125]. Therefore, when testing or comparing Cetuximab, Panitumumab, or other EGFR-targeting antibodies, the optimal EGF dose in the culture medium should be evaluated in advance to avoid false negative results. Although a high dose of EGF is necessary for long-term expansion of normal mucosa organoids, CRC organoids with a wild-type EGFR pathway perform equally well with a 2 ng/ml EGF concentration (our unpublished results), while organoids carrying RAS/RAF activating mutations do not need EGF supplementation. Accordingly, EGF could be lowered or eliminated when testing CRC organoids.
Gastrin, supplemented as [Leu15]-Gastrin-I heptadecapeptide, was initially reported to marginally increase organoid plating efficiency (tested in the absence of Nic, A83-01 and SB202190). Gastrin activates the G protein-coupled, cholecystokinin B receptor, which increases organoid swelling through the production and release of HCl [126], a mechanism shared with PGE2 and Forskolin. The authors originally included Gastrin in the culture medium as it “did not interfere with intestinal differentiation” [1]. However, its usefulness has never been confirmed in any subsequent study, yet Gastrin continues to be added to most organoid medium formulas. Gastrin is not required in organoid medium; however, its presence is not neutral, as it could alter the response of other target cells. Cholecystokinin B receptor is expressed in macrophages, B and T lymphocytes, and its triggering usually determines inhibitory effects [127]. Accordingly, Gastrin should not be added to organoid medium in immunology tests.
PGE2, is a well-known product of prostaglandin E synthase 2 (PTGS2/COX2), an enzyme strictly associated with CRC progression [128]. Initially, PGE2 10µM was added to the medium as a rescue molecule after the enzymatic disaggregation of organoids, not as a stable component of the medium [1]. Subsequent studies by Clevers' group lowered the PGE2 concentration to 10nM, including it in the standard organoid medium (see Tab.1). PGE2 has several positive effects on the colorectal epithelium: it transactivates EGFR signaling, contrasts apoptosis, and enforces β-catenin pathway activation [129,130,131]. Moreover PGE2 mediates tumor-induced angiogenesis [132] and fibroblast-derived PGE2 acts as a trigger for colorectal cancer initiation [133]. These promoting effects on tumor and stromal cells are counter paired by the immunosuppressive effects on almost all components of the immune system [134]. The pleiotropic activity of PGE2 should be considered in any experimental approach and PGE2 should be eliminated from the organoid medium during testing, unless specific studies on its activity are planned.
Y27632 is an inhibitor of the Rho-associated coiled-coil forming protein serine/threonine kinase (ROCK) family. ROCK1-2 mediate the formation of stress fibers in cultured cells by activating the myosin regulatory light chain [96]. Y27632 exerts anti apoptotic and reprogramming effects on epidermal stem-like cells and pluripotent stem cells cultured without a matrix carrier or stromal support [135,136]. Y27632 increases plating efficiency after the enzymatic disaggregation of organoids and can be stably added to mouse intestinal epithelial cultures to create 2D models [137,138]. Y27632 activity is not limited to ROCK1-2 inhibition, the screening of several kinases revealed that it affects other enzymes involved in cell motility as well (PRK1, PRK2, ULK1). Moreover, also TOPK and PKCε are partially inhibited by a 0.5µM dose [123]. TOPK is a serine/threonine protein kinase involved in cell cycle regulation and mitosis, typically upregulated in many cancers [139]. In CRC TOPK expression is linked to a negative prognosis in patients with KRAS or BRAF mutations [140]. In CRC cells, EGF stimulation induces PKCε-dependent phosphorylation of migration and invasion inhibitory protein (MIIP) facilitating metastasis [141]. Thus, Y27632 is expected to exert an inhibitory effect on CRC organoid proliferation and invasion. At the standard concentration of 10 µM, Y27632 also partially inhibits AMPK, RSK1, MNK1, and MSK1 [142]. Accordingly, the addition of Y27632 should be limited to the minimal time needed to rescue cell cultures after enzymatic split, and should not be added stably during any experimental approach. Of course, Y27632 intrinsic ability to prevent apoptosis/anoikis does not allow it to be associated with any drug testing.
11. Conclusions
According to the reported information, the colorectal organoid medium is still the gold standard for propagating these unique 3D cultures indefinitely. However, it also creates a highly artificial and incompletely defined microenvironment. Purified growth factors should be preferred to the supernatants from super-expressing cell cultures, to prevent the addition of unknown/unwanted paracrine factors. Some components, such as Nicotinamide, Gastrin, N2 supplement and Primocin, could be removed from the standard culture medium without issue, thus simplifying the formula and reducing off-target modulations. Other additives like NAC, PGE2, A83-01, SB202190 can significantly impact cell signaling, metabolism, oxidative stress and immunological responses, creating frequent, yet underrated experimental biases (Figure 2). Unless long-term tests preserving the stem cell component are necessary, excluding most additives is recommended for short-term, high-throughput assays. This is particularly true when CRC organoids are used for drug screening, as some compounds can create an artifactual resistance or sensitization to specific drugs, that do not reflect the patient’s situation.
Funding
This research was funded by Ministero della Salute, Ricerca Corrente 2025-2027.
Acknowledgments
The draft of Table 1 was implemented by Dr. Martina Taglieri.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| EGF | epithelial growth factor |
| NAC | n-acetylcysteine |
| Nic | nicotinamide |
| PGE2 | prostaglandin E2 |
References
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-Term Expansion of Epithelial Organoids from Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Carmon, K.S.; Gong, X.; Lin, Q.; Thomas, A.; Liu, Q. R-Spondins Function as Ligands of the Orphan Receptors LGR4 and LGR5 to Regulate Wnt/Beta-Catenin Signaling. Proc Natl Acad Sci U S A 2011, 108, 11452–11457. [Google Scholar] [CrossRef] [PubMed]
- Jung, P.; Sato, T.; Merlos-Suárez, A.; Barriga, F.M.; Iglesias, M.; Rossell, D.; Auer, H.; Gallardo, M.; Blasco, M.A.; Sancho, E.; et al. Isolation and in Vitro Expansion of Human Colonic Stem Cells. Nat Med 2011, 17, 1225–1227. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.; Wharton, N.; Parris, A.; Mitchell, E.; Sobolewski, A.; Kam, C.; Bigwood, L.; El Hadi, A.; Münsterberg, A.; Lewis, M.; et al. Canonical Wnt Signals Combined with Suppressed TGFβ/BMP Pathways Promote Renewal of the Native Human Colonic Epithelium. Gut 2014, 63, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Matano, M.; Nanki, K.; Sato, T. Efficient Genetic Engineering of Human Intestinal Organoids Using Electroporation. Nat Protoc 2015, 10, 1474–1485. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective Derivation of a Living Organoid Biobank of Colorectal Cancer Patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef]
- Boehnke, K.; Iversen, P.W.; Schumacher, D.; Lallena, M.J.; Haro, R.; Amat, J.; Haybaeck, J.; Liebs, S.; Lange, M.; Schäfer, R.; et al. Assay Establishment and Validation of a High-Throughput Screening Platform for Three-Dimensional Patient-Derived Colon Cancer Organoid Cultures. J Biomol Screen 2016, 21, 931–941. [Google Scholar] [CrossRef]
- Verissimo, C.S.; Overmeer, R.M.; Ponsioen, B.; Drost, J.; Mertens, S.; Verlaan-Klink, I.; van Gerwen, B.; van der Ven, M.; de Wetering, M. van; Egan, D.A.; et al. Targeting Mutant RAS in Patient-Derived Colorectal Cancer Organoids by Combinatorial Drug Screening. eLife 2016, 5, e18489. [Google Scholar] [CrossRef]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and Targeting of LGR5+ Human Colon Cancer Stem Cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef]
- Fujii, M.; Matano, M.; Toshimitsu, K.; Takano, A.; Mikami, Y.; Nishikori, S.; Sugimoto, S.; Sato, T. Human Intestinal Organoids Maintain Self-Renewal Capacity and Cellular Diversity in Niche-Inspired Culture Condition. Cell Stem Cell 2018, 23, 787–793.e6. [Google Scholar] [CrossRef]
- Schnalzger, T.E.; de Groot, M.H.; Zhang, C.; Mosa, M.H.; Michels, B.E.; Röder, J.; Darvishi, T.; Wels, W.S.; Farin, H.F. 3D Model for CAR-Mediated Cytotoxicity Using Patient-Derived Colorectal Cancer Organoids. EMBO J 2019, 38, e100928. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.; Yang, L.; Zhu, J.; Wan, J.; Shen, L.; Xia, F.; Fu, G.; Deng, Y.; Pan, M.; et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell 2020, 26, 17–26.e6. [Google Scholar] [CrossRef] [PubMed]
- Toshimitsu, K.; Takano, A.; Fujii, M.; Togasaki, K.; Matano, M.; Takahashi, S.; Kanai, T.; Sato, T. Organoid Screening Reveals Epigenetic Vulnerabilities in Human Colorectal Cancer. Nat Chem Biol 2022, 18, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Teijeira, A.; Migueliz, I.; Garasa, S.; Karanikas, V.; Luri, C.; Cirella, A.; Olivera, I.; Cañamero, M.; Alvarez, M.; Ochoa, M.C.; et al. Three-Dimensional Colon Cancer Organoids Model the Response to CEA-CD3 T-Cell Engagers. Theranostics 2022, 12, 1373–1387. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Fujii, M.; Takahashi, S.; Takano, A.; Nanki, K.; Matano, M.; Hanyu, H.; Saito, M.; Shimokawa, M.; Nishikori, S.; et al. Cell–Matrix Interface Regulates Dormancy in Human Colon Cancer Stem Cells. Nature 2022, 608, 784–794. [Google Scholar] [CrossRef]
- Wang, R.; Mao, Y.; Wang, W.; Zhou, X.; Wang, W.; Gao, S.; Li, J.; Wen, L.; Fu, W.; Tang, F. Systematic Evaluation of Colorectal Cancer Organoid System by Single-Cell RNA-Seq Analysis. Genome Biol 2022, 23, 106. [Google Scholar] [CrossRef]
- Wilson, S.S.; Mayo, M.; Melim, T.; Knight, H.; Patnaude, L.; Wu, X.; Phillips, L.; Westmoreland, S.; Dunstan, R.; Fiebiger, E.; et al. Optimized Culture Conditions for Improved Growth and Functional Differentiation of Mouse and Human Colon Organoids. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef]
- Kurayoshi, M.; Yamamoto, H.; Izumi, S.; Kikuchi, A. Post-Translational Palmitoylation and Glycosylation of Wnt-5a Are Necessary for Its Signalling. Biochem J 2007, 402, 515–523. [Google Scholar] [CrossRef]
- Neumann, S.; Coudreuse, D.Y.M.; Van Der Westhuyzen, D.R.; Eckhardt, E.R.M.; Korswagen, H.C.; Schmitz, G.; Sprong, H. Mammalian Wnt3a Is Released on Lipoprotein Particles. Traffic 2009, 10, 334–343. [Google Scholar] [CrossRef]
- Mihara, E.; Hirai, H.; Yamamoto, H.; Tamura-Kawakami, K.; Matano, M.; Kikuchi, A.; Sato, T.; Takagi, J. Active and Water-Soluble Form of Lipidated Wnt Protein Is Maintained by a Serum Glycoprotein Afamin/α-Albumin. eLife 2016, 5, e11621. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.L.; Weis, W.I. The β-Catenin Destruction Complex. Cold Spring Harb Perspect Biol 2013, 5, a007898. [Google Scholar] [CrossRef] [PubMed]
- Colozza, G.; Koo, B.-K. Wnt/β-Catenin Signaling: Structure, Assembly and Endocytosis of the Signalosome. Development, Growth & Differentiation 2021, 63, 199–218. [Google Scholar] [CrossRef]
- Nagano, K. R-Spondin Signaling as a Pivotal Regulator of Tissue Development and Homeostasis. Japanese Dental Science Review 2019, 55, 80–87. [Google Scholar] [CrossRef]
- Schuijers, J.; Junker, J.P.; Mokry, M.; Hatzis, P.; Koo, B.-K.; Sasselli, V.; van der Flier, L.G.; Cuppen, E.; van Oudenaarden, A.; Clevers, H. Ascl2 Acts as an R-Spondin/Wnt-Responsive Switch to Control Stemness in Intestinal Crypts. Cell Stem Cell 2015, 16, 158–170. [Google Scholar] [CrossRef]
- Murata, K.; Jadhav, U.; Madha, S.; van Es, J.; Dean, J.; Cavazza, A.; Wucherpfennig, K.; Michor, F.; Clevers, H.; Shivdasani, R.A. Ascl2-Dependent Cell Dedifferentiation Drives Regeneration of Ablated Intestinal Stem Cells. Cell Stem Cell 2020, 26, 377–390.e6. [Google Scholar] [CrossRef]
- Taurin, S.; Sandbo, N.; Qin, Y.; Browning, D.; Dulin, N.O. Phosphorylation of β-Catenin by Cyclic AMP-Dependent Protein Kinase*. Journal of Biological Chemistry 2006, 281, 9971–9976. [Google Scholar] [CrossRef]
- Fang, D.; Hawke, D.; Zheng, Y.; Xia, Y.; Meisenhelder, J.; Nika, H.; Mills, G.B.; Kobayashi, R.; Hunter, T.; Lu, Z. Phosphorylation of β-Catenin by AKT Promotes β-Catenin Transcriptional Activity*. Journal of Biological Chemistry 2007, 282, 11221–11229. [Google Scholar] [CrossRef]
- Kosinski, C.; Li, V.S.W.; Chan, A.S.Y.; Zhang, J.; Ho, C.; Tsui, W.Y.; Chan, T.L.; Mifflin, R.C.; Powell, D.W.; Yuen, S.T.; et al. Gene Expression Patterns of Human Colon Tops and Basal Crypts and BMP Antagonists as Intestinal Stem Cell Niche Factors. Proceedings of the National Academy of Sciences 2007, 104, 15418–15423. [Google Scholar] [CrossRef]
- Spit, M.; Koo, B.-K.; Maurice, M.M. Tales from the Crypt: Intestinal Niche Signals in Tissue Renewal, Plasticity and Cancer. Open Biology 2018, 8, 180120. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and Functions of P38 MAPK Signalling. Biochem J 2010, 429, 403–417. [Google Scholar] [CrossRef]
- Otsuka, M.; Kang, Y.J.; Ren, J.; Jiang, H.; Wang, Y.; Omata, M.; Han, J. Distinct Effects of P38α Deletion in Myeloid Lineage and Gut Epithelia in Mouse Models of Inflammatory Bowel Disease. Gastroenterology 2010, 138, 1255–1265.e9. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.R.; Dise, R.S.; Edelblum, K.L.; Polk, D.B. P38 Kinase Regulates Epidermal Growth Factor Receptor Downregulation and Cellular Migration. The EMBO Journal 2006, 25, 5683–5692. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Wei, G.; Gao, N.; Chen, J.; Fan, L.; Zeng, Z.; Gao, G.; Li, L.; Fang, G.; Hu, K.; Pang, X.; et al. Erk and MAPK Signaling Is Essential for Intestinal Development through Wnt Pathway Modulation. Development 2020, 147, dev185678. [Google Scholar] [CrossRef]
- Zhan, T.; Ambrosi, G.; Wandmacher, A.M.; Rauscher, B.; Betge, J.; Rindtorff, N.; Häussler, R.S.; Hinsenkamp, I.; Bamberg, L.; Hessling, B.; et al. MEK Inhibitors Activate Wnt Signalling and Induce Stem Cell Plasticity in Colorectal Cancer. Nat Commun 2019, 10, 2197. [Google Scholar] [CrossRef]
- de Jong, P.R.; Taniguchi, K.; Harris, A.R.; Bertin, S.; Takahashi, N.; Duong, J.; Campos, A.D.; Powis, G.; Corr, M.; Karin, M.; et al. ERK5 Signalling Rescues Intestinal Epithelial Turnover and Tumour Cell Proliferation upon ERK1/2 Abrogation. Nat Commun 2016, 7, 11551. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Nat Rev Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Maurer, U.; Preiss, F.; Brauns-Schubert, P.; Schlicher, L.; Charvet, C. GSK-3 – at the Crossroads of Cell Death and Survival. J Cell Sci 2014, 127, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Lu, B.; Zamponi, R.; Yang, Z.; Wetzel, K.; Loureiro, J.; Mohammadi, S.; Beibel, M.; Bergling, S.; Reece-Hoyes, J.; et al. MTORC1 Signaling Suppresses Wnt/β-Catenin Signaling through DVL-Dependent Regulation of Wnt Receptor FZD Level. Proceedings of the National Academy of Sciences 2018, 115, E10362–E10369. [Google Scholar] [CrossRef]
- Matozaki, T.; Kotani, T.; Murata, Y.; Saito, Y. Roles of Src Family Kinase, Ras, and MTOR Signaling in Intestinal Epithelial Homeostasis and Tumorigenesis. Cancer Science 2021, 112, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Pizarro, T.; Zhou, L. Organoids as a Model System for Studying Notch Signaling in Intestinal Epithelial Homeostasis and Intestinal Cancer. The American Journal of Pathology 2022, 192, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Farin, H.F.; van Es, J.H.; Clevers, H.; Langer, R.; Karp, J.M. Niche-Independent High-Purity Cultures of Lgr5+ Intestinal Stem Cells and Their Progeny. Nat Methods 2014, 11, 106–112. [Google Scholar] [CrossRef]
- Gu, W.; Colarusso, J.L.; Dame, M.K.; Spence, J.R.; Zhou, Q. Rapid Establishment of Human Colonic Organoid Knockout Lines. STAR Protocols 2022, 3, 101308. [Google Scholar] [CrossRef]
- Rezakhani, S.; Gjorevski, N.; Lutolf, M.P. Extracellular Matrix Requirements for Gastrointestinal Organoid Cultures. Biomaterials 2021, 276, 121020. [Google Scholar] [CrossRef]
- Ramadan, R.; Wouters, V.M.; van Neerven, S.M.; de Groot, N.E.; Garcia, T.M.; Muncan, V.; Franklin, O.D.; Battle, M.; Carlson, K.S.; Leach, J.; et al. The Extracellular Matrix Controls Stem Cell Specification and Crypt Morphology in the Developing and Adult Mouse Gut. Biol Open 2022, 11, bio059544. [Google Scholar] [CrossRef]
- Sarker, F.A.; Prior, V.G.; Bax, S.; O’Neill, G.M. Forcing a Growth Factor Response – Tissue-Stiffness Modulation of Integrin Signaling and Crosstalk with Growth Factor Receptors. J Cell Sci 2020, 133, jcs242461. [Google Scholar] [CrossRef]
- Hernandez-Gordillo, V.; Kassis, T.; Lampejo, A.; Choi, G.; Gamboa, M.E.; Gnecco, J.S.; Brown, A.; Breault, D.T.; Carrier, R.; Griffith, L.G. Fully Synthetic Matrices for in Vitro Culture of Primary Human Intestinal Enteroids and Endometrial Organoids. Biomaterials 2020, 254, 120125. [Google Scholar] [CrossRef]
- Bergenheim, F.; Fregni, G.; Buchanan, C.F.; Riis, L.B.; Heulot, M.; Touati, J.; Seidelin, J.B.; Rizzi, S.C.; Nielsen, O.H. A Fully Defined 3D Matrix for Ex Vivo Expansion of Human Colonic Organoids from Biopsy Tissue. Biomaterials 2020, 262, 120248. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Röhling, M.; Martin, S. Insulin: Too Much of a Good Thing Is Bad. BMC Med 2020, 18, 224. [Google Scholar] [CrossRef] [PubMed]
- Ding, V.W.; Chen, R.-H.; McCormick, F. Differential Regulation of Glycogen Synthase Kinase 3β by Insulin and Wnt Signaling*. Journal of Biological Chemistry 2000, 275, 32475–32481. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Galizzi, G.; Di Carlo, M. Insulin and Its Key Role for Mitochondrial Function/Dysfunction and Quality Control: A Shared Link between Dysmetabolism and Neurodegeneration. Biology 2022, 11, 943. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat Rev Mol Cell Biol 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Reports 2020, 30, 3411–3423.e7. [Google Scholar] [CrossRef]
- Nagana Gowda, G.A.; Pascua, V.; Raftery, D. Extending the Scope of 1H NMR-Based Blood Metabolomics for the Analysis of Labile Antioxidants: Reduced and Oxidized Glutathione. Anal. Chem. 2021, 93, 14844–14850. [Google Scholar] [CrossRef]
- Sun, K.; Li, C.; Liao, S.; Yao, X.; Ouyang, Y.; Liu, Y.; Wang, Z.; Li, Z.; Yao, F. Ferritinophagy, a Form of Autophagic Ferroptosis: New Insights into Cancer Treatment. Front. Pharmacol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Das, N.K.; Jain, C.; Sankar, A.; Schwartz, A.J.; Santana-Codina, N.; Solanki, S.; Zhang, Z.; Ma, X.; Parimi, S.; Rui, L.; et al. Modulation of the HIF2α-NCOA4 Axis in Enterocytes Attenuates Iron Loading in a Mouse Model of Hemochromatosis. Blood 2022, 139, 2547–2552. [Google Scholar] [CrossRef]
- Kollara, A.; Brown, T.J. Expression and Function of Nuclear Receptor Co-Activator 4: Evidence of a Potential Role Independent of Co-Activator Activity. Cell. Mol. Life Sci. 2012, 69, 3895–3909. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Hou, L.; Guo, Z.; Wang, G.; Guo, J.; Xu, J.; Zhang, X.; Guo, F. JNK-JUN-NCOA4 Axis Contributes to Chondrocyte Ferroptosis and Aggravates Osteoarthritis via Ferritinophagy. Free Radical Biology and Medicine 2023, 200, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Lewin, M.H.; Arthur, J.R.; Riemersma, R.A.; Nicol, F.; Walker, S.W.; Millar, E.M.; Howie, A.F.; Beckett, G.J. Selenium Supplementation Acting through the Induction of Thioredoxin Reductase and Glutathione Peroxidase Protects the Human Endothelial Cell Line EAhy926 from Damage by Lipid Hydroperoxides. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2002, 1593, 85–92. [Google Scholar] [CrossRef]
- Stolwijk, J.; Wagner, B.; McCormick, M.; Zakharia, Y.; Spitz, D.; Buettner, G. 290 - Optimization of Selenium in Cell Culture Media to Maximize Selenoenzyme Activity. Free Radical Biology and Medicine 2018, 128, S121. [Google Scholar] [CrossRef]
- Stewart, M.S.; Davis, R.L.; Walsh, L.P.; Pence, B.C. Induction of Differentiation and Apoptosis by Sodium Selenite in Human Colonic Carcinoma Cells (HT29). Cancer Letters 1997, 117, 35–40. [Google Scholar] [CrossRef]
- Selenius, M.; Fernandes, A.P.; Brodin, O.; Björnstedt, M.; Rundlöf, A.-K. Treatment of Lung Cancer Cells with Cytotoxic Levels of Sodium Selenite: Effects on the Thioredoxin System. Biochemical Pharmacology 2008, 75, 2092–2099. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Yu, S.; He, W.; Li, J.; Xu, T.; Xue, J.; Shi, P.; Chen, S.; Li, Y.; Hong, S.; et al. Selenite Induces Cell Cycle Arrest and Apoptosis via Reactive Oxygen Species-Dependent Inhibition of the AKT/MTOR Pathway in Thyroid Cancer. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Hulley, P.; Davison, A. Regulation of Tyrosine Phosphorylation Cascades by Phosphatases: What the Actions of Vanadium Teach Us. The Journal of Trace Elements in Experimental Medicine 2003, 16, 281–290. [Google Scholar] [CrossRef]
- Levina, A.; Lay, P.A. Stabilities and Biological Activities of Vanadium Drugs: What Is the Nature of the Active Species? Chemistry – An Asian Journal 2017, 12, 1692–1699. [Google Scholar] [CrossRef] [PubMed]
- Sanna, D.; Micera, G.; Garribba, E. Interaction of Insulin-Enhancing Vanadium Compounds with Human Serum Holo-Transferrin. Inorg. Chem. 2013, 52, 11975–11985. [Google Scholar] [CrossRef]
- Crans, D.C. Antidiabetic, Chemical, and Physical Properties of Organic Vanadates as Presumed Transition-State Inhibitors for Phosphatases. J. Org. Chem. 2015, 80, 11899–11915. [Google Scholar] [CrossRef]
- Klarlund, J.K. Transformation of Cells by an Inhibitor of Phosphatases Acting on Phosphotyrosine in Proteins. Cell 1985, 41, 707–717. [Google Scholar] [CrossRef]
- Boersema, P.J.; Foong, L.Y.; Ding, V.M.Y.; Lemeer, S.; van Breukelen, B.; Philp, R.; Boekhorst, J.; Snel, B.; den Hertog, J.; Choo, A.B.H.; et al. In-Depth Qualitative and Quantitative Profiling of Tyrosine Phosphorylation Using a Combination of Phosphopeptide Immunoaffinity Purification and Stable Isotope Dimethyl Labeling. Mol Cell Proteomics 2010, 9, 84–99. [Google Scholar] [CrossRef]
- Raubenheimer, P.J.; Young, E.A.; Andrew, R.; Seckl, J.R. The Role of Corticosterone in Human Hypothalamic– Pituitary–Adrenal Axis Feedback. Clinical Endocrinology 2006, 65, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, B.; Ao, H. Corticosterone Effects Induced by Stress and Immunity and Inflammation: Mechanisms of Communication. Front. Endocrinol. 2025, 16. [Google Scholar] [CrossRef]
- Botrugno, O.A.; Fayard, E.; Annicotte, J.-S.; Haby, C.; Brennan, T.; Wendling, O.; Tanaka, T.; Kodama, T.; Thomas, W.; Auwerx, J.; et al. Synergy between LRH-1 and β-Catenin Induces G1 Cyclin-Mediated Cell Proliferation. Molecular Cell 2004, 15, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; Cima, I.; Noti, M.; Fuhrer, A.; Jakob, S.; Dubuquoy, L.; Schoonjans, K.; Brunner, T. The Nuclear Receptor LRH-1 Critically Regulates Extra-Adrenal Glucocorticoid Synthesis in the Intestine. J Exp Med 2006, 203, 2057–2062. [Google Scholar] [CrossRef]
- Bouguen, G.; Dubuquoy, L.; Desreumaux, P.; Brunner, T.; Bertin, B. Intestinal Steroidogenesis. Steroids 2015, 103, 64–71. [Google Scholar] [CrossRef]
- Sidler, D.; Renzulli, P.; Schnoz, C.; Berger, B.; Schneider-Jakob, S.; Flück, C.; Inderbitzin, D.; Corazza, N.; Candinas, D.; Brunner, T. Colon Cancer Cells Produce Immunoregulatory Glucocorticoids. Oncogene 2011, 30, 2411–2419. [Google Scholar] [CrossRef]
- Kogata, N.; Bland, P.; Tsang, M.; Oliemuller, E.; Lowe, A.; Howard, B.A. Sox9 Regulates Cell State and Activity of Embryonic Mouse Mammary Progenitor Cells. Commun Biol 2018, 1, 228. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Mostoslavsky, G. Generation of Human Induced Pluripotent Stem Cells Using a Defined, Feeder-Free Reprogramming System. Current Protocols in Stem Cell Biology 2018, 45, e48. [Google Scholar] [CrossRef]
- Wang, X.; Ryu, D.; Houtkooper, R.H.; Auwerx, J. Antibiotic Use and Abuse: A Threat to Mitochondria and Chloroplasts with Impact on Research, Health, and Environment. BioEssays 2015, 37, 1045–1053. [Google Scholar] [CrossRef]
- Jang, S.; Kang, H.T.; Hwang, E.S. Nicotinamide-Induced Mitophagy: EVENT MEDIATED BY HIGH NAD+/NADH RATIO AND SIRT1 PROTEIN ACTIVATION*. Journal of Biological Chemistry 2012, 287, 19304–19314. [Google Scholar] [CrossRef] [PubMed]
- Badal, S.; Her, Y.F.; Maher, L.J. Nonantibiotic Effects of Fluoroquinolones in Mammalian Cells *. Journal of Biological Chemistry 2015, 290, 22287–22297. [Google Scholar] [CrossRef]
- Ma, H.H.; Chiu, F.C.; Li, R.C. Mechanistic Investigation of the Reduction in Antimicrobial Activity of Ciprofloxacin by Metal Cations. Pharm Res 1997, 14, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, S.; McLaren, A.C.; Pauken, C.; McLemore, R. Amphotericin B Is Cytotoxic at Locally Delivered Concentrations. Clinical Orthopaedics and Related Research® 2011, 469, 3016. [Google Scholar] [CrossRef]
- Wolf, J.E.; Massof, S.E. In Vivo Activation of Macrophage Oxidative Burst Activity by Cytokines and Amphotericin B. Infect Immun 1990, 58, 1296–1300. [Google Scholar] [CrossRef] [PubMed]
- Hammarström, L.; Smith, E. Mitogenic Properties of Polyene Antibiotics for Murine B Cells. Scand J Immunol 1976, 5, 37–43. [Google Scholar] [CrossRef]
- Hedges, J.F.; Mitchell, A.M.; Jones, K.; Kimmel, E.; Ramstead, A.G.; Snyder, D.T.; Jutila, M.A. Amphotericin B Stimulates Γδ T and NK Cells, and Enhances Protection from Salmonella Infection. Innate Immun 2015, 21, 598–608. [Google Scholar] [CrossRef]
- Grela, E.; Piet, M.; Luchowski, R.; Grudzinski, W.; Paduch, R.; Gruszecki, W.I. Imaging of Human Cells Exposed to an Antifungal Antibiotic Amphotericin B Reveals the Mechanisms Associated with the Drug Toxicity and Cell Defence. Sci Rep 2018, 8, 14067. [Google Scholar] [CrossRef]
- Warrilow, A.G.; Parker, J.E.; Kelly, D.E.; Kelly, S.L. Azole Affinity of Sterol 14α-Demethylase (CYP51) Enzymes from Candida Albicans and Homo Sapiens. Antimicrob Agents Chemother 2013, 57, 1352–1360. [Google Scholar] [CrossRef]
- Lewis, R.; Niazi-Ali, S.; McIvor, A.; Kanj, S.S.; Maertens, J.; Bassetti, M.; Levine, D.; Groll, A.H.; Denning, D.W. Triazole Antifungal Drug Interactions—Practical Considerations for Excellent Prescribing. J Antimicrob Chemother 2024, 79, 1203–1217. [Google Scholar] [CrossRef]
- Miao, Y.; Ha, A.; de Lau, W.; Yuki, K.; Santos, A.J.M.; You, C.; Geurts, M.H.; Puschhof, J.; Pleguezuelos-Manzano, C.; Peng, W.C.; et al. Next-Generation Surrogate Wnts Support Organoid Growth and Deconvolute Frizzled Pleiotropy In Vivo. Cell Stem Cell 2020, 27, 840–851.e6. [Google Scholar] [CrossRef] [PubMed]
- Avalos, J.L.; Bever, K.M.; Wolberger, C. Mechanism of Sirtuin Inhibition by Nicotinamide: Altering the NAD+ Cosubstrate Specificity of a Sir2 Enzyme. Molecular Cell 2005, 17, 855–868. [Google Scholar] [CrossRef]
- Salech, F.; Ponce, D.P.; Paula-Lima, A.C.; SanMartin, C.D.; Behrens, M.I. Nicotinamide, a Poly [ADP-Ribose] Polymerase 1 (PARP-1) Inhibitor, as an Adjunctive Therapy for the Treatment of Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12. [Google Scholar] [CrossRef]
- Song, S.B.; Jang, S.-Y.; Kang, H.T.; Wei, B.; Jeoun, U.; Yoon, G.S.; Hwang, E.S. Modulation of Mitochondrial Membrane Potential and ROS Generation by Nicotinamide in a Manner Independent of SIRT1 and Mitophagy. Molecules and Cells 2017, 40, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Ren, Z.; Xu, F.; Zhou, X.; Song, C.; Wang, V.Y.-F.; Liu, W.; Lu, L.; Thomson, J.A.; Chen, G. Nicotinamide Promotes Cell Survival and Differentiation as Kinase Inhibitor in Human Pluripotent Stem Cells. Stem Cell Reports 2018, 11, 1347–1356. [Google Scholar] [CrossRef]
- Ishizaki, T.; Uehata, M.; Tamechika, I.; Keel, J.; Nonomura, K.; Maekawa, M.; Narumiya, S. Pharmacological Properties of Y-27632, a Specific Inhibitor of Rho-Associated Kinases. Molecular Pharmacology 2000, 57, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; de Haar, J. van; Fanchi, L.F.; Slagter, M.; der Velden, D.L. van; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-Culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef]
- Daniel, J.; Marechal, Y.; Van Gool, F.; Andris, F.; Leo, O. Nicotinamide Inhibits B Lymphocyte Activation by Disrupting MAPK Signal Transduction. Biochemical Pharmacology 2007, 73, 831–842. [Google Scholar] [CrossRef]
- Cao, A.-P.; Wang, Y.-Y.; Shen, Y.-Y.; Liu, Y.-H.; Liu, J.-Y.; Wang, Y.; Guo, Y.; Wang, R.-B.; Xie, B.-Y.; Pan, X.; et al. Nicotinamide Suppresses Hyperactivation of Dendritic Cells to Control Autoimmune Disease through PARP Dependent Signaling. Nutrients 2024, 16. [Google Scholar] [CrossRef]
- Ren, Y.; Cui, Y.-N.; Wang, H.-W. Effects of Different Concentrations of Nicotinamide on Hematopoietic Stem Cells Cultured in Vitro. World Journal of Stem Cells 2024, 16, 163–175. [Google Scholar] [CrossRef]
- da Paz Martins, A.S.; de Andrade, K.Q.; de Araújo, O.R.P.; da Conceição, G.C.M.; da Silva Gomes, A.; Goulart, M.O.F.; Moura, F.A. Extraintestinal Manifestations in Induced Colitis: Controversial Effects of N-Acetylcysteine on Colon, Liver, and Kidney. Oxidative Medicine and Cellular Longevity 2023, 2023, 8811463. [Google Scholar] [CrossRef] [PubMed]
- Yurumez, Y.; Cemek, M.; Yavuz, Y.; Birdane, Y.O.; Buyukokuroglu, M.E. Beneficial Effect of N-Acetylcysteine against Organophosphate Toxicity in Mice. Biological and Pharmaceutical Bulletin 2007, 30, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Khalatbari Mohseni, G.; Hosseini, S.A.; Majdinasab, N.; Cheraghian, B. Effects of N-Acetylcysteine on Oxidative Stress Biomarkers, Depression, and Anxiety Symptoms in Patients with Multiple Sclerosis. Neuropsychopharmacology Reports 2023, 43, 382–390. [Google Scholar] [CrossRef]
- Trimarchi, H.; Mongitore, M.R.; Baglioni, P.; Forrester, M.; Freixas, E. a. R.; Schropp, M.; Pereyra, H.; Alonso, M. N-Acetylcysteine Reduces Malondialdehyde Levels in Chronic Hemodialysis Patients--a Pilot Study. Clin Nephrol 2003, 59, 441–446. [Google Scholar] [CrossRef]
- Teng, T.; Kamal, M.; Iriondo, O.; Amzaleg, Y.; Luo, C.; Thomas, A.; Lee, G.; Hsu, C.-J.; Nguyen, J.D.; Kang, I.; et al. N-Acetyl-L-Cysteine Promotes Ex Vivo Growth and Expansion of Single Circulating Tumor Cells by Mitigating Cellular Stress Responses. Mol Cancer Res 2021, 19, 441–450. [Google Scholar] [CrossRef]
- Beshiri, M.L.; Tice, C.M.; Tran, C.; Nguyen, H.M.; Sowalsky, A.G.; Agarwal, S.; Jansson, K.H.; Yang, Q.; McGowen, K.M.; Yin, J.; et al. A PDX/Organoid Biobank of Advanced Prostate Cancers Captures Genomic and Phenotypic Heterogeneity for Disease Modeling and Therapeutic Screening. Clin Cancer Res 2018, 24, 4332–4345. [Google Scholar] [CrossRef]
- Laoukili, J.; Constantinides, A.; Wassenaar, E.C.E.; Elias, S.G.; Raats, D.A.E.; van Schelven, S.J.; van Wettum, J.; Volckmann, R.; Koster, J.; Huitema, A.D.R.; et al. Peritoneal Metastases from Colorectal Cancer Belong to Consensus Molecular Subtype 4 and Are Sensitised to Oxaliplatin by Inhibiting Reducing Capacity. Br J Cancer 2022, 126, 1824–1833. [Google Scholar] [CrossRef]
- Narasimhan, V.; Wright, J.A.; Churchill, M.; Wang, T.; Rosati, R.; Lannagan, T.R.M.; Vrbanac, L.; Richardson, A.B.; Kobayashi, H.; Price, T.; et al. Medium-Throughput Drug Screening of Patient-Derived Organoids from Colorectal Peritoneal Metastases to Direct Personalized Therapy. Clin Cancer Res 2020, 26, 3662–3670. [Google Scholar] [CrossRef]
- Ooft, S.N.; Weeber, F.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van Werkhoven, E.; Schipper, L.; Hoes, L.; Vis, D.J.; van de Haar, J.; et al. Patient-Derived Organoids Can Predict Response to Chemotherapy in Metastatic Colorectal Cancer Patients. Science Translational Medicine 2019, 11, eaay2574. [Google Scholar] [CrossRef]
- Smabers, L.P.; Wensink, E.; Verissimo, C.S.; Koedoot, E.; Pitsa, K.-C.; Huismans, M.A.; Higuera Barón, C.; Doorn, M.; Valkenburg-van Iersel, L.B.; Cirkel, G.A.; et al. Organoids as a Biomarker for Personalized Treatment in Metastatic Colorectal Cancer: Drug Screen Optimization and Correlation with Patient Response. J Exp Clin Cancer Res 2024, 43, 61. [Google Scholar] [CrossRef] [PubMed]
- Tojo, M.; Hamashima, Y.; Hanyu, A.; Kajimoto, T.; Saitoh, M.; Miyazono, K.; Node, M.; Imamura, T. The ALK-5 Inhibitor A-83-01 Inhibits Smad Signaling and Epithelial-to-Mesenchymal Transition by Transforming Growth Factor-β. Cancer Science 2005, 96, 791–800. [Google Scholar] [CrossRef]
- Vogt, J.; Traynor, R.; Sapkota, G.P. The Specificities of Small Molecule Inhibitors of the TGFß and BMP Pathways. Cellular Signalling 2011, 23, 1831–1842. [Google Scholar] [CrossRef]
- Pham, A.-T.; Ghilardi, A.F.; Sun, L. Recent Advances in the Development of RIPK2 Modulators for the Treatment of Inflammatory Diseases. Front. Pharmacol. 2023, 14. [Google Scholar] [CrossRef]
- Flebbe, H.; Hamdan, F.H.; Kari, V.; Kitz, J.; Gaedcke, J.; Ghadimi, B.M.; Johnsen, S.A.; Grade, M.; Flebbe, H.; Hamdan, F.H.; et al. Epigenome Mapping Identifies Tumor-Specific Gene Expression in Primary Rectal Cancer. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Kot, A.; Koszewska, D.; Ochman, B.; Świętochowska, E.; Kot, A.; Koszewska, D.; Ochman, B.; Świętochowska, E. Clinical Potential of Misshapen/NIKs-Related Kinase (MINK) 1—A Many-Sided Element of Cell Physiology and Pathology. Current Issues in Molecular Biology 2024, 46, 13811–13845. [Google Scholar] [CrossRef]
- Fleischauer, J.; Bastone, A.L.; Selich, A.; John-Neek, P.; Weisskoeppel, L.; Schaudien, D.; Schambach, A.; Rothe, M.; Fleischauer, J.; Bastone, A.L.; et al. TGFβ Inhibitor A83-01 Enhances Murine HSPC Expansion for Gene Therapy. Cells 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.P.; McCaffrey, P.G.; Hsiao, K.; Pazhanisamy, S.; Galullo, V.; Bemis, G.W.; Fitzgibbon, M.J.; Caron, P.R.; Murcko, M.A.; Su, M.S.S. The Structural Basis for the Specificity of Pyridinylimidazole Inhibitors of P38 MAP Kinase. Chemistry & Biology 1997, 4, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.; Venè, R.; Coco, S.; Longo, L.; Tosetti, F.; Scabini, S.; Mastracci, L.; Grillo, F.; Poggi, A.; Benelli, R. SB202190 Predicts BRAF-Activating Mutations in Primary Colorectal Cancer Organoids via Erk1-2 Modulation. Cells 2023, 12, 664. [Google Scholar] [CrossRef]
- Lavoie, H.; Thevakumaran, N.; Gavory, G.; Li, J.J.; Padeganeh, A.; Guiral, S.; Duchaine, J.; Mao, D.Y.L.; Bouvier, M.; Sicheri, F.; et al. Inhibitors That Stabilize a Closed RAF Kinase Domain Conformation Induce Dimerization. Nat Chem Biol 2013, 9, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.C.; Alessi, D.R.; Cohen, P. The Selectivity of Protein Kinase Inhibitors: A Further Update. Biochem J 2007, 408, 297–315. [Google Scholar] [CrossRef]
- He, K.; Song, E.; Upadhyayula, S.; Dang, S.; Gaudin, R.; Skillern, W.; Bu, K.; Capraro, B.R.; Rapoport, I.; Kusters, I.; et al. Dynamics of Auxilin 1 and GAK in Clathrin-Mediated Traffic. J Cell Biol 2020, 219, e201908142. [Google Scholar] [CrossRef]
- Munson, M.J.; Mathai, B.J.; Ng, M.Y.W.; Trachsel-Moncho, L.; de la Ballina, L.R.; Schultz, S.W.; Aman, Y.; Lystad, A.H.; Singh, S.; Singh, S.; et al. GAK and PRKCD Are Positive Regulators of PRKN-Independent Mitophagy. Nat Commun 2021, 12, 6101. [Google Scholar] [CrossRef]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive Assay of Kinase Catalytic Activity Reveals Features of Kinase Inhibitor Selectivity. Nat Biotechnol 2011, 29, 1039–1045. [Google Scholar] [CrossRef]
- Yang, C.; Zhu, Z.; Tong, B.C.-K.; Iyaswamy, A.; Xie, W.-J.; Zhu, Y.; Sreenivasmurthy, S.G.; Senthilkumar, K.; Cheung, K.-H.; Song, J.-X.; et al. A Stress Response P38 MAP Kinase Inhibitor SB202190 Promoted TFEB/TFE3-Dependent Autophagy and Lysosomal Biogenesis Independent of P38. Redox Biology 2020, 32, 101445. [Google Scholar] [CrossRef] [PubMed]
- Garvey, C.M.; Lau, R.; Sanchez, A.; Sun, R.X.; Fong, E.J.; Doche, M.E.; Chen, O.; Jusuf, A.; Lenz, H.-J.; Larson, B.; et al. Anti-EGFR Therapy Induces EGF Secretion by Cancer-Associated Fibroblasts to Confer Colorectal Cancer Chemoresistance. Cancers 2020, 12, 1393. [Google Scholar] [CrossRef] [PubMed]
- Dockray, G.J. Gastrin. Best Practice & Research Clinical Endocrinology & Metabolism 2004, 18, 555–568. [Google Scholar] [CrossRef]
- van Niekerk, G.; Kelchtermans, L.; Broeckhoven, E.; Coelmont, L.; Alpizar, Y.A.; Dallmeier, K. Cholecystokinin and Gastrin as Immune Modulating Hormones: Implications and Applications. Cytokine & Growth Factor Reviews 2024, 80, 37–46. [Google Scholar] [CrossRef]
- Benelli, R.; Venè, R.; Ferrari, N. Prostaglandin-Endoperoxide Synthase 2 (Cyclooxygenase-2), a Complex Target for Colorectal Cancer Prevention and Therapy. Transl Res 2018, 196, 42–61. [Google Scholar] [CrossRef] [PubMed]
- Pai, R.; Soreghan, B.; Szabo, I.L.; Pavelka, M.; Baatar, D.; Tarnawski, A.S. Prostaglandin E2 Transactivates EGF Receptor: A Novel Mechanism for Promoting Colon Cancer Growth and Gastrointestinal Hypertrophy. Nat Med 2002, 8, 289–293. [Google Scholar] [CrossRef]
- Leone, V.; di Palma, A.; Ricchi, P.; Acquaviva, F.; Giannouli, M.; Di Prisco, A.M.; Iuliano, F.; Acquaviva, A.M. PGE2 Inhibits Apoptosis in Human Adenocarcinoma Caco-2 Cell Line through Ras-PI3K Association and CAMP-Dependent Kinase A Activation. American Journal of Physiology-Gastrointestinal and Liver Physiology 2007, 293, G673–G681. [Google Scholar] [CrossRef]
- Shao, J.; Jung, C.; Liu, C.; Sheng, H. Prostaglandin E2 Stimulates the β-Catenin/T Cell Factor-Dependent Transcription in Colon Cancer*. Journal of Biological Chemistry 2005, 280, 26565–26572. [Google Scholar] [CrossRef]
- Xu, L.; Stevens, J.; Hilton, M.B.; Seaman, S.; Conrads, T.P.; Veenstra, T.D.; Logsdon, D.; Morris, H.; Swing, D.A.; Patel, N.L.; et al. COX-2 Inhibition Potentiates Antiangiogenic Cancer Therapy and Prevents Metastasis in Preclinical Models. Science Translational Medicine 2014, 6, 242ra84–242ra84. [Google Scholar] [CrossRef]
- Roulis, M.; Kaklamanos, A.; Schernthanner, M.; Bielecki, P.; Zhao, J.; Kaffe, E.; Frommelt, L.-S.; Qu, R.; Knapp, M.S.; Henriques, A.; et al. Paracrine Orchestration of Intestinal Tumorigenesis by a Mesenchymal Niche. Nature 2020, 580, 524–529. [Google Scholar] [CrossRef]
- Santiso, A.; Heinemann, A.; Kargl, J. Prostaglandin E2 in the Tumor Microenvironment, a Convoluted Affair Mediated by EP Receptors 2 and 4. Pharmacological Reviews 2024, 76, 388–413. [Google Scholar] [CrossRef]
- Witkowski, T.A.; Li, B.; Andersen, J.G.; Kumar, B.; Mroz, E.A.; Rocco, J.W. Y-27632 Acts beyond ROCK Inhibition to Maintain Epidermal Stem-like Cells in Culture. J Cell Sci 2023, 136, jcs260990. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.K.-L.; Chen, X.; Lim, Y.M.; Reuveny, S.; Oh, S.K.W. Inhibition of ROCK-Myosin II Signaling Pathway Enables Culturing of Human Pluripotent Stem Cells on Microcarriers without Extracellular Matrix Coating. Tissue Eng Part C Methods 2014, 20, 227–238. [Google Scholar] [CrossRef]
- Liu, Y.; Qi, Z.; Li, X.; Du, Y.; Chen, Y.-G. Monolayer Culture of Intestinal Epithelium Sustains Lgr5+ Intestinal Stem Cells. Cell Discov 2018, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Kozuka, K.; He, Y.; Koo-McCoy, S.; Kumaraswamy, P.; Nie, B.; Shaw, K.; Chan, P.; Leadbetter, M.; He, L.; Lewis, J.G.; et al. Development and Characterization of a Human and Mouse Intestinal Epithelial Cell Monolayer Platform. Stem Cell Reports 2017, 9, 1976–1990. [Google Scholar] [CrossRef] [PubMed]
- Herbert, K.J.; Ashton, T.M.; Prevo, R.; Pirovano, G.; Higgins, G.S. T-LAK Cell-Originated Protein Kinase (TOPK): An Emerging Target for Cancer-Specific Therapeutics. Cell Death Dis 2018, 9, 1089. [Google Scholar] [CrossRef]
- Zlobec, I.; Molinari, F.; Kovac, M.; Bihl, M.P.; Altermatt, H.J.; Diebold, J.; Frick, H.; Germer, M.; Horcic, M.; Montani, M.; et al. Prognostic and Predictive Value of TOPK Stratified by KRAS and BRAF Gene Alterations in Sporadic, Hereditary and Metastatic Colorectal Cancer Patients. Br J Cancer 2010, 102, 151–161. [Google Scholar] [CrossRef]
- Chen, T.; Li, J.; Xu, M.; Zhao, Q.; Hou, Y.; Yao, L.; Zhong, Y.; Chou, P.-C.; Zhang, W.; Zhou, P.; et al. PKCε Phosphorylates MIIP and Promotes Colorectal Cancer Metastasis through Inhibition of RelA Deacetylation. Nat Commun 2017, 8, 939. [Google Scholar] [CrossRef] [PubMed]
- Barcelo, J.; Samain, R.; Sanz-Moreno, V. Preclinical to Clinical Utility of ROCK Inhibitors in Cancer. Trends in Cancer 2023, 9, 250–263. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
the main signaling pathways involved in long-term organoid culture and their agonist or inhibitors included in the organoid medium formula. R-spo (R-spondin); Nog (Noggin); cAMP (cyclic AMP).
Figure 1.
the main signaling pathways involved in long-term organoid culture and their agonist or inhibitors included in the organoid medium formula. R-spo (R-spondin); Nog (Noggin); cAMP (cyclic AMP).
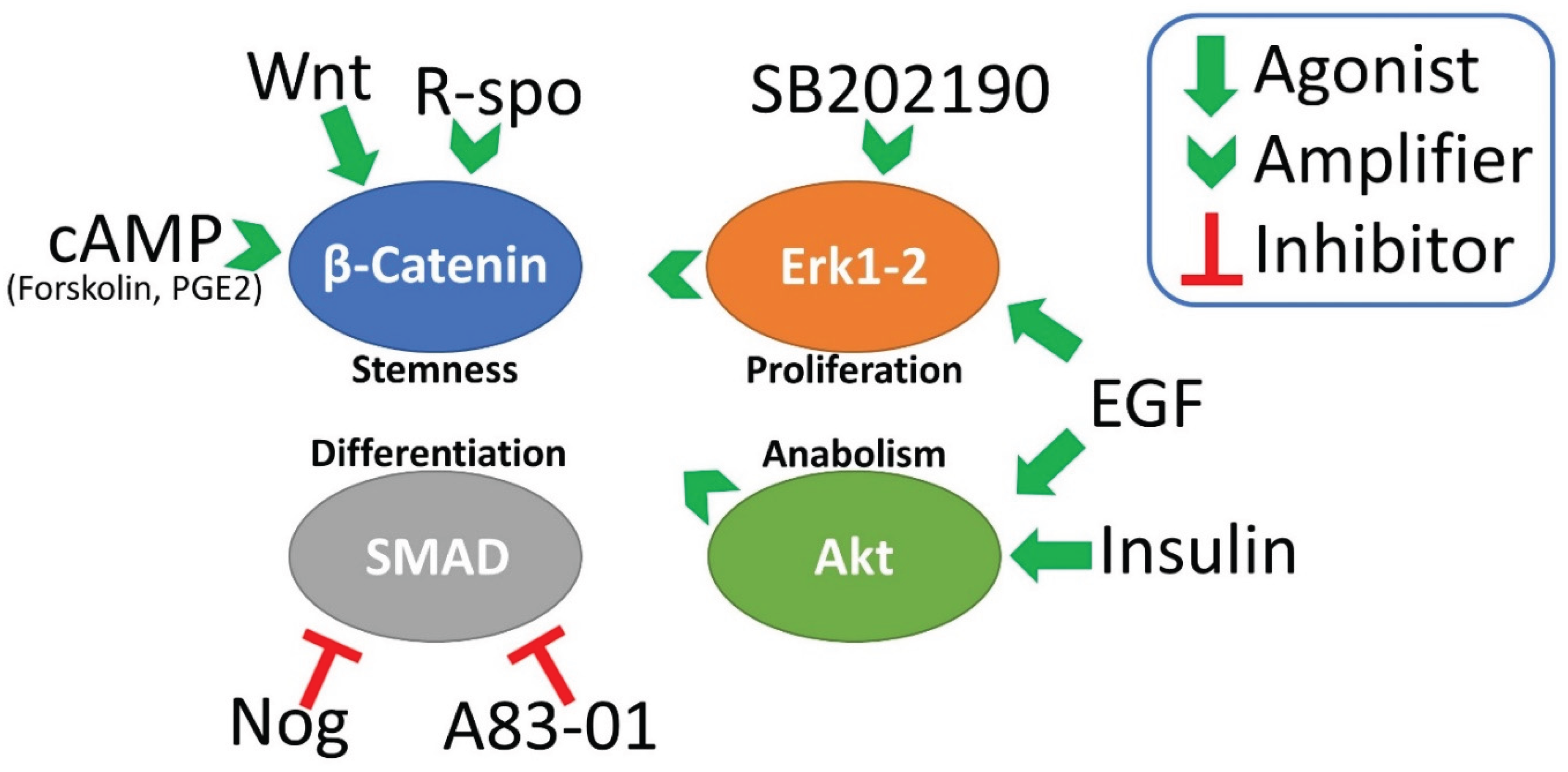
Figure 2.
a visual summary of the main experimental biases that could be triggered by different components of the organoid medium. These molecules should be eliminated or limited, at least in short-term drug testing and immunoassays.
Figure 2.
a visual summary of the main experimental biases that could be triggered by different components of the organoid medium. These molecules should be eliminated or limited, at least in short-term drug testing and immunoassays.
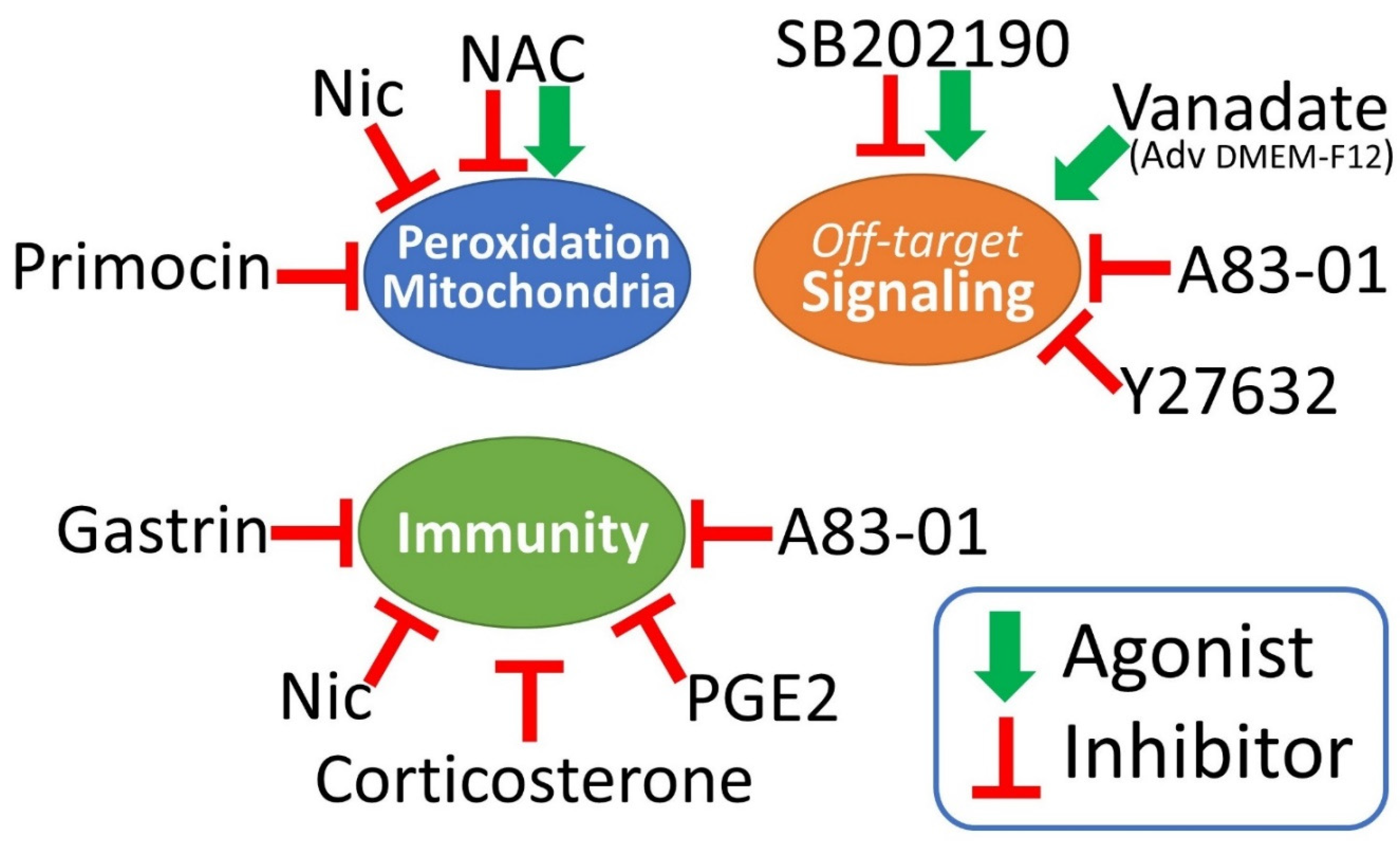

Table 1.
synopsis of human intestinal organoid culture medium modifications used in different studies.
Table 1.
synopsis of human intestinal organoid culture medium modifications used in different studies.
| Medium | Additives | EGF ng/ml | Wnt3a ng/ml; or %CM | R-spondin ng/ml or %CM | Noggin ng/ml or %CM | A83-01 µM | SB202190 µM | Nic mM | NAC mM | Gast nM | PGE2 µM | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Advanced DMEM/F12 |
B27 +N2 | 50 | 100 | 1000 | 100 | 0.5 | 10 | 10 | 1 | 10** | 10* | [1] |
| Advanced DMEM/F12 |
B27noA +N2 | 50 | 50% CM | 1000*** | 100 | LY2157299 0.5uM | 10 | 10 | 1 | 48 1ug/ml | 0.01 | [3] |
| Advanced DMEM/F12 |
B27+N2 | 50 +IGF1 50ng/ml |
100 | 500 | 100 | 0.5 | 1 | [4] | ||||
| Advanced DMEM/F12 |
B27 | 50 | 50% CM | 10% CM | 100 | 0.5 | 10 | 1 | 10 | [5] | ||
| Advanced DMEM/F12 |
B27 | 50 | 50% CM or omitted in CRC |
20% CM | 10% CM | 0.5 | 3 | 10 | 1.25 | 10 | 0.01 | [6] |
| Advanced DMEM/F12 |
B27 | 50 | 50% CM or omitted in most CRC |
10% CM omitted in most CRC |
100 | 0.5 | 10 | 1 | 10 | [7] | ||
| Advanced DMEM/F12 | B27+N2 | 50 +bFGF 20ng/ml |
omitted (CRC) | omitted (CRC) | omitted (CRC) | 1 | [8] | |||||
| Advanced DMEM/F12 | B27 | 50 | 50% CM or omitted (CRC) | 20% CM | 10% CM | 0.5 | 10 | 10 | 1.25 nM | [9] | ||
| Advanced DMEM/F12 | B27 | 50 | omitted (CRC) | omitted (CRC) | 100 | 0.5 | 1 | 10 | [10] | |||
| Advanced DMEM/F12 | B27 | 50 | 50% CM | 1000 | 100 | 0.5 | 10 | 1 | 10 | [11] | ||
| Avanced DMEM/F12 | B27 | 50 | 50% CM or omitted (CRC) |
20% CM | 10% CM | 0.5 | 10 | 10 | 12.5 err? | [12] | ||
| Advanced DMEM/F12 | B27 | 50 | omitted (CRC) | 500 | 100 | 0.5 | 3 | 1 | 10 | 0.01 | [13] | |
| Advanced DMEM/F12 | B27 | 50 | 20% Afamin-Wnt3A serum-free | 10% CM | 100 | 0.5 | 10 | 1 | 10 | [14] | ||
| Avanced DMEM/F12 | B27 | 50 +FGF10 100ng/ml |
omitted (CRC) | 20% CM | 100 | 0.5 | 3 | 10 | 1.25 | 10 | 0.01 | [15] |
| Avanced DMEM/F12 | B27 | 50 | omitted (CRC) | omitted (CRC) | 100 | 0.5 | 1 | 10 | [16] | |||
| Avanced DMEM/F12 | B27 | 50 +bFGF 10ng/ml +FGF10 10ng/ml |
100 | 500 | 100 | 0.5 | 5 | 4 | 10 | 0.1 | [17] |
Legend: EGF (epithelial growth factor); IGF (insulin-like growth factor); FGF (fibroblast growth factor); Wnt3a (Wingless/int 3a); A83-01 (TGF-ß type I receptor/ALK5 kinase inhibitor; also inhibits ALK4, ALK7); LY2157299 (TGF-ß type I receptor/ALK5 kinase inhibitor; also empowers VEGF and bFGF signaling); SB202190 (p38 inhibitor); Nic (nicotinamide); NAC (n-acetylcysteine); Gast ([Leu15]-Gastrin I); PGE2 (prostaglandin E2). CM: transfected cells conditioned medium. CRC: colorectal cancer derived cultures. * used to increase plating efficiency during the first 48h of culture; ** originally described to slightly increase plating efficiency, but stably let in the medium formula as it did not cause differentiation; *** Home-made RSPO1-His6, purified from HEK293T cells. err? (erroneously reported?).
Table 2.
A list of the components of Advanced DMEM-F12, B27 and N2.
| Reagent | Unique components | Shared by all | Shared by two |
|---|---|---|---|
| Advanced DMEM-F12 | DMEM/F12, Ascorbic acid, Ammonium metavanadate, Cupric sulfate, Manganous chloride, (+Hepes) | Insulin, Holo-Transferrin, Sodium selenite | Ethanolamine, Glutathione, Bovine serum albumin (AlbuMAX® II, lipid-rich) |
| B27 w/woVit.A | Catalase, Superoxide dismutase, Triodo-L-thyronine, L-carnitine, D-galactose, Corticosterone, Linoleic acid, Linolenic acid, Retinol acetate (not included in the retinol-free formula), DL-alpha tocopherol, DL-alpha tocopherol acetate, Biotin, Vitamin B12, Zinc sulfate, Selenium, Sodium pyruvate, Lipoic acid, L-Alanine, L-Glutamate, L-Glutamine, L-Proline | Insulin, Holo-Transferrin, Sodium selenite | Ethanolamine, Glutathione-reduced, Bovine serum albumin (Fraction V IgG free, fatty-acid poor), Putrescine, Progesterone |
| N2 | - | Insulin, Transferrin, Sodium selenite | Putrescine, Progesterone |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



