
Submitted:
13 December 2025
Posted:
15 December 2025
You are already at the latest version
Abstract
Duchenne muscular dystrophy (DMD), Limb-girdle muscular dystrophies (LGMD), and GNE myopathy (GNEM) are progressive neuromuscular rare genetic diseases with differing etiologies either due to defects in structural muscle proteins or impaired metabolism and manifests through progressive weakening and deterioration of skeletal muscle tissues. This review summarizes current and recent clinical trials worldwide, highlighting curative approaches for neuromuscular diseases such as DMD, LGMD, and GNEM. We compiled and summarized latest and most promising outcomes from ongoing global clinical trials, using information from official websites, news sources, trial registries, databases and literature across four key therapeutic modalities: 1) palliative therapies: steroidal and non-steroidal 2) gene therapy and gene-targeted interventions, 3) cell-based regenerative strategies and 4) universal treatments (therapies not limited to the mutation type). We also highlighted the risks associated with gene therapy and discussed key regulatory considerations.Several current clinical trials show promise for muscular dystrophy treatments. RGX-202 gene therapy demonstrates early muscle strength improvement in DMD children. MyoPAXon, a stem cell therapy for DMD muscle regeneration, has FDA clearance to begin trials. Sevasemten (EDG-5506) trials report slowed DMD/BMD progression. SRP-9003 gene therapy targets protein restoration in LGMD. ACENOBEL, approved in Japan for GNEM, may delay disease progression if started early. These trials offer high potential to become widely available treatments improving quality of life and provide life-saving therapeutic options. Additionally, Indian specific founder mutations for the mentioned muscular dystrophies are highlighted to understand the disease etiology in Indian populations for precise diagnostics and therapeutics.
Keywords:
gene therapy
; drug therapy
; cell based therapy
; AAV
; CRISPR/Cas
; muscular dystrophy
; founder mutations
; rare genetic disease
; orphan drugs
Key points:
Comprehensive therapeutic evaluation: This review provides a critical assessment on the promising emerging therapies for the muscular dystrophies “DMD, LGMD and GNEM’ that included curative interventions, with particular emphasis on their clinical implications. Given the similarities in etiology and pathogenesis, we note the similarities in their therapeutic landscape.
Global clinical trial catalogue: We synthesize global clinical trial data from databases and literature across four key therapeutic modalities: 1) palliative therapies: steroidal and non-steroidal 2) gene therapy and gene-targeted interventions, 3) cell-based regenerative strategies and 4) universal treatments (therapies not limited to the mutation type)
India-Specific Genetic Compilation: This review presents, for the first time, a comprehensive census of all reported founder mutations for DMD, LGMD, and GNEM across Indian populations, synthesizing fragmented data from peer-reviewed literature and regional databases.
1. Introduction
Neuromuscular disorders (NMDs) represent a diverse collection of predominantly genetic conditions marked by deteriorating muscle function, progressive physical disability, and reduced life expectancy (Iolascon et al., 2019; Kumar et al., 2025). These disorders encompass conditions that affect the nerves controlling voluntary muscles, the muscles themselves, or the communication between nerves and muscles (e.g., at the neuromuscular junction) (Liang,2012; Mary et al., 2018). Neuromuscular disorders are subclassified into distinct categories based on their etiology (cause), pathogenesis (disease mechanism), and clinical phenotype (presentation). Muscular dystrophies, progressive disorders of muscle degeneration, caused by structural protein defects (e.g., dystrophin in DMD, Calpain3 in LGMD2A) (Murphy and Straub (2015); Falzarno et al 2018; Duan et al., 2021; Bouchard et al., 2023), while myopathies are primary muscle dysfunctions due to metabolic or contractile impairments (e.g., GNE enzyme in GNE Myopathy) (Malicdan et al., 2009) in absence of significant structural damage.
DMD and LGMD, as muscular dystrophies, and GNE myopathy, a metabolic myopathy, represent distinct yet interconnected challenges in neuromuscular medicine. All the three diseases are heritable (Tidball et al., 2018; Salih and Kang, 2020) due to mutations leading defective structural protein or enzyme rather than acquired muscular dystrophies which may be acquired in lifetime due to various factors such as auto-immune response (Preito and Grau, 2010). DMD demonstrates the highest disease burden of the three, with early symptom emergence and aggressive progression (Crisafulli et al., 2020; Duan et al., 2021). LGMD is a genetically heterogeneous group of disorders with variable onset and progression, representing a significant subset of muscular dystrophies worldwide (Murphy and Straub (2015); Nallamilli et al., 2018). GNE myopathy is an adult-onset, progressive distal myopathy with founder population clusters (Malicdan et al., 2009; Carrillo et al., 2018). This review will focus exclusively on three neuromuscular disorders that belong to the rare genetic diseases (RGDs): Duchenne (DMD), limb-girdle (LGMD), and GNE myopathy, due to their clinical and genetic significance in neuromuscular medicine. Recent reviews have extensively characterized the clinical and molecular features of DMD (Verhaart et al., 2012; Yiu et al., 2016; Duan et al., 2021), LGMD (Murphy and Straub (2015); Bouchard et al., 2023), and GNE myopathy (Nishino et al., 2015; Carrillo et al., 2018). While these disorders share overlapping pathogenic mechanisms and therapeutic approaches, this review specifically evaluates emerging treatment strategies through a critical analysis of ongoing and recently completed clinical trials worldwide. We systematically examine both palliative and curative interventions, with particular emphasis on their clinical implications. Furthermore, this work provides the first comprehensive synthesis of founder mutations identified in Indian populations for these three neuromuscular disorders, a critical yet understudied aspect, that has significant implications for developing population-specific therapies and understanding the mutational landscape of these diseases in the Indian subcontinent.
2. Duchenne Muscular Dystrophy (DMD)
As one of the most common rare muscular dystrophies, DMD demonstrates progressive muscle weakness with an estimated global prevalence of 7.1/100,000 males and 2.8/100,000 overall population (Crisafulli et al., 2020). Symptoms start with the weakness of muscles by 4 years of age with rapid progression leading to ambulatory loss (Bushby et al., 2010). Mutations in dystrophin gene result in dystrophinopathies which include DMD, Becker muscular dystrophy (BMD) and Dilated cardiomyopathy (DCM) [19, 20]. The dystrophin protein stabilizes muscle fibers by linking the cytoskeleton to the extracellular matrix, enabling durability to contractile mechanical stress (Verhaart et al., 2012; Del Rio-Pertuz et al., 2022) (Figure 1). BMD is the milder dystrophinopathy with symptoms appearing in the thirties, and much slower loss of mobility. DCM is a frequent age dependent complication in both DMD and BMD with prevalence ranging from 59% to 90% (DMD) and up to 61% in BMD (Nigro et al., 1990, Bello et al., 2016).
While DMD predominantly affects males due to X linkage, female carriers can show some of the symptoms, such as increased risk of developing dilated cardiomyopathy (Suthar et al., 2021).
Earlier approaches to DMD primarily focused on symptom management using steroids, anticonvulsants, and immunosuppressants (Beytia et al., 2012), whereas recent advances in molecular biology facilitate the development of therapies that extend beyond palliative care, as illustrated in Figure 2. These novel approaches, like read-through therapy and exon skipping, aim to restore dystrophin production or function in muscle cells.
The mutation spectrum of DMD mutations in Indian populations are very diverse with cohorts and carriers showing region specific mutational spectra (Basak et al., 2009; Goyal et al., 2021; Nagabhushana et al., 2021). However, there are no reported founder mutations for DMD in Indian populations indicating a need for more pedigree-based studies and wider genome -wide sequencing efforts. The difficulty to identify the founder mutations may also be attributed to the nature of progression of the disease which is characterized by early onset and demise of individuals before reaching adulthood (Bushby et al., 2010; Falzarano et al., 2015) and the genetic heterogeneity of the Indian populations (Papiha, (1996); Indian Genomic variation consortium (2005)). It is also speculated that DMD may be due to more de-novo mutations rather than inherited mutations (Polavarapu et al., 2019).
Searching for clinical trials on NIH site (https://clinicaltrials.gov/) after setting criterion for clinical trials that have not been withdrawn or terminated, we found 332 trials for DMD, 22 trials for LGMD and 14 trials for GNE Myopathy (Figure 2). Of these 332 trials, 220 are completed and 112 are ongoing. 233 of these trials were interventional in nature (i.e. they were examining some therapy or its dosage) while 99 were observational. 37 of these were in advanced stages of clinical trials at Phase 3 (31) and at Phase 4(6). As Figure 1 shows that DMD is most well researched among the neuromuscular diseases with candidate therapies to enter market, if final phases ae cleared. The complete list of clinical trials for the three neuromuscular disorders – DMD, LGMD and GNE Myopathy are given in the Supplementary Table (ST) 1, 2 and 3 respectively. As we note in ST1, ST2 and ST3, for all the three disorders, most of the clinical trials are coming from USA, China or Europe with other developing countries like India lagging behind. We shall elaborate on some of the promising therapies in the following sections.
2.1. Therapies Focused on Symptomatic Management (Palliative Therapies)
Palliative/symptomatic therapies primarily focus on relieving the patient's symptoms rather than addressing the underlying causes of the disease. For neuromuscular disorders there are steroidal and non-steroidal options for symptomatic management.
2.1.1. Steroid-Based Palliative Therapies
Earlier therapies for DMD included corticosteroids such as prednisone, prednisolone, and deflazacort, which interact with glucocorticoid receptors suppressing the NF-κB pathway to control inflammation and muscle fiber necrosis (Matthews et al., 2016, Srinivasan et al., 2022). Patients on corticosteroids maintain muscle strength for longer periods aiding their locomotion. Use of corticosteroids in DMD patients has reduced severity of scoliosis, a curvature of the spine, minimizing the need for surgery (Duan et al., 2021). However, some side effects (Srinivasan et al., 2022) are associated with long-term corticosteroid use including hypertension, weight gain, cataracts, osteoporosis and stunted bone growth (Bianchi et al., 2005, Viviano et al., 2022). Corticosteroid therapy may elevate compression fracture risk, worsening clinical outcomes (Duan et al., 2021. To address the side effects of corticosteroids, new treatment options have emerged. Agamree (vamorolone) (Keam, 2024) and Deflazacort (Emflaza) (McDonald et al., 2020) are FDA-approved corticosteroids specifically designed for DMD. Agamree, a dissociative corticosteroid, aims to reduce the side effects while maintaining the anti-inflammatory properties of corticosteroids. Deflazacort and vamorolone (VBP15) are two more oral corticosteroids with similar objectivesapproved for DMD patients aged 2 and older.
2.1.2. Non-Steroidal Palliative Therapies
In addition to corticosteroid-based treatments, non-steroidal options have also emerged for managing DMD. Duvyzat (givinostat) represents a significant advancement as the first non-steroidal drug approved for treating all genetic variants of DMD in patients aged six and older (Lamb, 2024). It functions as a histone deacetylase (HDAC) inhibitor, dampening chronic inflammation, fat infiltration and muscle fibrosis. Duvyzat was tested in a large-scale, 18-month study with muscle function evaluated using the North Star Ambulatory Assessment (NSAA) scale (Himba et al., 2023).
DMD impairs respiratory function, necessitating monitoring/ventilator support. Other symptoms include spasms, seizures, and contractures managed with anticonvulsants and physiotherapy (Duan et al., 2021). DMD patients have high risk of heart complications for which medications such as ACE (angiotensin converting enzyme) inhibitors, beta-blockers, and ARBs (angiotensin receptor blockers) are often prescribed (Birnkrant et al., 2018, Lee et al., 2022). The emerging technologies of virtual reality have found their applications in biomedical sciences as well, and they have been used to aid rehabilitate and improvise locomotor and co-ordination capabilities of the patients suffering from DMD and other muscular dystrophies (Baeza- Baragan et al., 2020; Kiper et al., 2024).
2.2. Therapies Focused on Restoring Dystrophin Expression and Function: Gene Therapy and Gene-Targeted Therapy
Newer therapeutic approaches such as read-through therapy, exon reframing, gene transfer and CRISPR gene editing aim to directly restore dystrophin expression and function (Shimizu-Motohashi et al., 2016). By targeting the root cause of the disease, these therapies hold the potential in finding a more durable cure for DMDs rather than palliative therapies which may offer only temporary relief.
Genetic studies of DMD cohorts showed that large deletions or duplications affecting multiple exons within the dystrophin gene leading to truncated and non-functional dystrophin proteins (Bladen et al., 2015, Zhang et al., 2019). Nearly 79% of the cases involve large-scale mutations, with large deletions (68%) and duplications (11%), with regions spanning exons 2-20 and 45-55 are particularly susceptible [46]. Rest are small-scale mutations - Nonsense mutations (11%), deletions (5%), insertions (2%), and splice site alterations (3%) (Flanigan et al., 2009, Sun et al., 2020, Zhang et al., 2021).
2.2.1. Read-Through Therapy
Read-through therapy targets nonsense mutations in DMD by bypassing premature stop codons thereby preventing mRNA degradation downstream. Studies have demonstrated that aminoglycosides, such as gentamicin, and compounds like ataluren can bypass these premature stop codons, generating complete dystrophin protein (Shimizu-Motohashi et al., 2016), with limited success in clinical trials (Landfeldt et al., 2020). Not much improvement was seen in timed function tests, though ambulatory loss was delayed (Mercuri et al., 2020).
2.2.2. Exon Skipping Therapy
Synthetic antisense oligonucleotide (ASO) drugs, correct dystrophin reading frames in DMD via exon skipping/reframing, offering targeted treatment for frameshift mutations (Mizobe et al., 2018; Ousterout et al., 2015; Chemello et al., 2021). ASOs target a specific exon, excluding them from the final dystrophin protein with function restored though shorter. It is worth noting that ASO therapy is not a complete cure for DMD, as it just slows the disease progression. AMONDYS 45 (casimersen), EXONDYS 51 (eteplirsen), Vyondys 53 (golodirsen) and Viltolarsen (VILTEPSO) are the few ASO drugs designed for DMD patients (Aartsma-Rus et al., 2020, Assefa et al., 2024;).
FDA-approved ASO therapies for DMD include AMONDYS 45 (casimersen) for exon 45 skipping (Vasterling et al., 2023). and EXONDYS 51 (eteplirsen) for exon 51 skipping, generating truncated dystrophin (Syed, 2016; Lim et al., 2017). XONDYS 51 gained FDA accelerated approval for boosting dystrophin levels, though its clinical efficacy remains under investigation, with reported adverse effects including contusions, rash, and respiratory symptoms [60,61]. Vyondys 53 (golodirsen) and Viltolarsen (VILTEPSO) are other FDA approved ASO therapies for patients amenable to treatment by skipping exon 53 (Heo et al., 2020). All listed ASO drugs were FDA-approved for orphan indications.
2.2.3. Adeno-Associated Virus (AAV) Vector-Based Gene Therapy
ELEVIDYS (delandistrogenemoxeparvovec-rokl) is an AAV vector-based gene therapy delivering microdystrophin (μDys), a truncated (138 kDa instead of 427 kDa) dystrophin variant, for DMD treatment (Hoy, 2023). Developed by Sarepta Therapeutics, ELEVIDYS is based on the results from 3 clinical trial studies: NCT03375164, NCT03769116, NCT04626674, and is administered as a single intravenous infusion and has orphan drug designation.
However, μDys lacks two-thirds of the dystrophin coding sequence, including critical rod and hinge domains required for proper protein interactions, resulting in incomplete protection of muscle integrity (Bostick et al., 2012; Wasala et al., 2023). Delivering full-length dystrophin (14 kb) was hindered by AAV's limited packaging capacity (~4.7 kb) (Srivastava et al., 1983). This was addressed using split intein-mediated protein trans-splicing (Tasfaout et al., 2024), where inteins (self-excising polypeptides) and their flanking exteins reassemble separate protein fragments (Derbyshire et al., 1998; Fernandes et al., 2016).
Tasfaout et al. developed a split intein-mediated protein trans-splicing approach to express large dystrophins, using highly efficient intein pairs to assemble midi- or full-length dystrophin from two or three AAV-delivered fragments. Delivered systemically via the myotropic AAVMYO vector (Weinmann et al., 2020) at low doses (2×10¹³ vg/kg), this method achieved body-wide expression in striated muscles, restoring function in young and old mdx mice more effectively than micro-dystrophin (Tasfaout et al., 2024). Similarly, Zhou et al. designed a triple-AAV system (MyoAAV4A) using orthogonal split inteins to deliver full-length dystrophin, rescuing expression in skeletal and cardiac muscles of mdxAcv mice (Zhou et al., 2024). To overcome neutralizing antibodies and transgene control challenges, emerging strategies include exosome-delivered CRISPR, immune-stealth capsids, and muscle-specific promoters (Saad et al., 2022).
AAV-NoSTOP Gene Therapy
Wang et al. (2022) used rAAV-delivered suppressor tRNAs (rAAV.sup-tRNA) to rescue nonsense mutations in mice by combining stop codon readthrough with NMD inhibition. In HEK293 cells, sup-tRNATyr, -Ser, and -Gln achieved up to 70% readthrough—outperforming G418 (Keeling et al., 2014). Notably, sup-tRNATyr restored full-length IDUA protein in W401X-mutant mIdua mRNA, demonstrating precise amino acid incorporation at PTCs. Optimized AAV capsids and delivery routes enabled therapeutic efficacy in liver, heart, muscle, and brain, with effects lasting >6 months—demonstrating promise as a durable genetic therapy.
2.2.4. CRISPR/CAS9
CRISPR/Cas9 employs sgRNA (guide RNA)-guided DNA cleavage for precise gene editing, enabling mutation correction or gene silencing (Keeling et al., 2014; Tycko et al., 2016; Hsu et al., 2019). A key challenge is off-target editing caused by sgRNA-DNA mismatches, which may disrupt genomic integrity and even trigger cancer (Tycko et al., 2016). This underscores the need for high-fidelity Cas9 variants with strict target specificity for safe clinical application. To address these limitations, engineered Cas9 variants have been developed. Acharya et al. (2024) created enhanced Francisella novicida Cas9 (FnCas9) derivatives with improved on-target efficiency, knock-in rates, and specificity. Their therapeutic potential was demonstrated by correcting RPE65 mutations in LCA2 patient-derived iPSCs using an optimized FnCas9 adenine base editor (Panda & Ray, 2022; Acharya et al., 2024).
CRISPR-Cas9 gene-editing technology has made significant strides in clinical applications, particularly in treating various genetic disorders such as sickle cell disease (Davies et al., 2024), cancer (Wang et al., 2021), cystic fibrosis (Wei et al., 2023) and muscular dystrophy (Laurent et al., 2024). Safe delivery of CRISPR tools to stem cells/iPSCs requires alternatives to DNA-based methods, with mRNA/nanoparticle approaches showing promise (De Masi et al., 2020).
Stadelmann et al. 2022 demonstrated that mRNA-delivered SpCas9 and adenine base editors achieved >90% editing efficiency in human muscle stem cells across donors, while maintaining myogenic function without cell enrichment (Stadelmann et al., 2022). Further, various nanoparticles, including lipid-based, polymer-based, inorganic, and extracellular vesicle-based systems have been explored as efficient delivery systems in advancing CRISPR/Cas9 technology for clinical applications and gene therapy (Emami et al., 2019; Agrawal et al., 2023). Biomineralized and Polyrotaxane nanoparticles have been used to efficiently deliver cas9 systems without muscle damage (Agrawal et al., 2023)
Gustafsson et al. developed hPep cell-penetrating peptides for efficient RNP delivery of CRISPR tools (Gustaffson et al., 2024). These nanoparticles achieved near-complete editing in HEK293T (96%), iPSCs (74%), and muscle stem cells (80%), while maintaining cell functionality. The hPep system offers rapid bench-top preparation, lyophilized storage, and compatibility with standard lab techniques, representing a versatile synthetic delivery platform.
2.2.5. Vector Aided Gene Therapy
RGX-202, a vector-based gene therapy (NCT05693142), delivers a novel microdystrophin transgene containing functional C-terminal domains, showing promising clinical results [89]. A single IV dose of RGX-202 in 4–11-year-old DMD patients produced detectable microdystrophin in muscle by 12 weeks and reduced creatine kinase by 10 weeks, with improved muscle strength—particularly at higher doses. A long-term follow-up study (NCT06491927) is now underway (Braun, 2025).
While promising for ambulatory or younger patients with DMD, AAV-mediated CRISPR therapies carry severe risks from high systemic doses, which can overwhelm an inflamed system and trigger fatal innate immune responses, including acute respiratory distress syndrome (Lek et al., 2023). This unique risk profile emphasises the critical need for optimized dosing and proactive immunosuppression (Horn & Fehse, 2024), a strategy now being pursued to address similar toxicities. Following the exclusion of non-ambulatory patients from Elevidys’ label due to liver toxicity, the FDA has approved a trial of an enhanced sirolimus-based immunosuppressive regimen to mitigate this risk and potentially restore access for this advanced patient population (Chwalenia et al., 2025; Swiderski et al., 2025). A young, ambulatory patient in Pfizer's DMD gene therapy trial suffered a fatal cardiac arrest, showing that severe risks may extend beyond advanced stage patients (https://www.biopharmadive.com/news/pfizer-duchenne-patient-death-gene-therapy-trial/715450/). The fatalities observed in both Pfizer's and Sarepta's gene therapy trials necessitate that regulatory agencies enforce more rigorous safety testing and adopt a more precautionary stance in their approval processes for high-dose AAV vectors.
2.3. Cell Based Therapy
Cell transplantation (e.g., satellite cells, myoblasts) offers potential for DMD by restoring dystrophin production (Siemionow et al., 2018; Tominari et al., 2023). However, challenges such as lack of suitable animal models for human cell studies (Sato et al., 2023) and impaired proliferative capacity of DMD patient-derived satellite cells (Blau et al., 1983) complicate its therapeutic use.
SAT-3247 (Satellos) is an oral AAK1 inhibitor that restores muscle stem cell polarity and regeneration in DMD models, demonstrating improved muscle strength. A completed Phase I study (NCT06565208) in 72 healthy volunteers established safety and translational pharmacokinetics, while an ongoing trial is the assessing the same in 10 DMD patients over 28 days. (https://ir.satellos.com/news/news-details/2024/Satellos-Announces-First-Participant-with-Duchenne-Muscular-Dystrophy-Dosed-in-the-Phase-1b-Clinical-Trial-of-SAT-3247/default.aspx).
2.3.1. Re-Engineering Cellular Medicine:
Vita Therapeutics' VTA-110, an FDA-designated orphan drug, is a first-in-class allogeneic iPSC therapy for DMD. Derived from somatic cells via Yamanaka factor reprogramming, it has demonstrated muscle repair/regeneration in preclinical studies. (https://www.vitatx.com/all-news/vita-therapeutics-receives-orphan-drug-designation-from-fda-for-new-novel-treatment).
2.3.2. Chimeric Cell Therapy:
Dystrogen Therapeutics developed Dystrophic Expressing Chimeric (DEC) cell therapy for DMD—an allogeneic-autologous myoblast fusion approach that restores dystrophin expression independent of mutation type (Heydemann et al., 2023; Siemionow et al., 2024). DEC therapy expands functional myoblasts, reduces inflammation/fibrosis, and minimizes immune rejection. It offers broad applicability across all DMD patients without requiring gene editing or immunosuppression, and can be combined with other therapies (Siemionow et al., 2023, 2024).
2.3.3. One for All Stem Cell-Based Therapy
Myogenica has received FDA approval to initiate a clinical trial for MyoPAXon, a stem cell-based therapy derived from induced pluripotent stem cells (iPSCs) (https://prnmedia.prnewswire.com/news-releases/fda-clears-investigational-new-drug-application-for-muscular-dystrophy-treatment-from-university-of-minnesota-startup-myogenica-302199393.html). MyoPAXon therapy targets skeletal muscle regeneration in DMD patients, with initial trials assessing safety, tolerability, and engraftment. Successful engraftment will be indicated by dystrophin-positive myofibers. The durable persistence of MyoPAXon-derived cells suggests sustained therapeutic potential, demonstrating the utility of iPSC technology in regenerative medicine. Patient-specific iPSCs enable disease modeling and accelerate therapeutic development (Speciale et al., 2020).
2.3.4. Muscle Stem Cells (muSCs) and Piezo1 Ion Channels as Potential Therapeutic for Muscular Dystrophies Including DMD
Adult muscle stem cells (MuSCs) are essential for muscle repair, differentiating into myogenic cells to regenerate fibers. Ma et al. (2022) showed MuSCs transition between 'responsive' and 'sensory' states regulated by the mechanosensitive Piezo1 channel to mediate cell communication and environmental sensing. In DMD mice, the Piezo1 activator Yoda1 restored MuSC state distribution, increasing responsive cells and enhancing regeneration (Ma et al., 2022).
Piezo1 represents a promising therapeutic target for muscular disorders with impaired regeneration (Bernareggi et al., 2022), though further validation in larger DMD animal models is needed (Ma et al., 2022).
2.4. Therapies Not Limited to Dystrophin Mutation Type
Sevasemten (EDG-5506) is an oral myosin inhibitor that protects dystrophin-deficient muscle fibers from contraction-induced damage in BMD/DMD patients (Russel et al., 2023; Donovan et al., 2025). Currently, four clinical trials are evaluating its efficacy across different age groups and treatment backgrounds (NCT05160415, NCT05291091, NCT05540860, NCT06100887).
EDG-5506 significantly slowed DMD/BMD progression (improved NSAA scores, reduced CK/troponin I) with excellent tolerability in NCT05160415. Current pediatric trials include LYNX (ages 4-9; NCT05540860) [103] and FOX (ages 6-14 post-gene therapy; NCT06100887), demonstrating potential therapeutic use. This therapy is designed primarily as monotherapy but may exhibit complementary efficacy when combined with existing treatments.
2.4.1. Utrophin Modulators
Utrophin (a paralog of dystrophin) modulation offers a mutation-independent therapeutic strategy for DMD, as utrophin can functionally compensate for dystrophin deficiency (Blake et al., 1996). Ghosh et al. (2024) developed oral quinazoline/quinoline-based utrophin modulators (SG-02) that increase utrophin expression 2.7-fold in C2C12 cells and 2.3-fold in DMD patient cells enhancing myogenesis, with favorable bioavailability, positioning them as promising universal DMD treatments (Ghosh et al., 2024).
2.4.2. SERCA as a Therapeutic Target for DMD Cardiomyopathy
DMD cardiomyopathy is attributed to the dysregulated cytosolic calcium due to reduced sarcoendoplasmic reticulum calcium ATPase (SERCA) activity (Schneider et al., 2013). Morales et al. (2023) demonstrated a potential treatment for DMD cardiomyopathy by improving the SERCA activity using DWORF gene therapy (Morales et al., 2023). Dworf open reading frame (DWORF) is a regulator for SERCA with significantly reduced expression in mdx mice (Nelson et al., 2016). AAV serotype 9-DWORF vector was delivered to 6-week-old mdx mice (6×1012 vector genome particles/mouse) via the tail vein which was shown to significantly enhance SERCA activity, thereby reducing myocardial fibrosis, improved treadmill running, electrocardiography, and heart hemodynamics
2.5. In Vitro Disease Model Systems
A robust tissue model system is required to model the pathogenesis and response to therapeutic interventions for muscle dystrophies. To address this, Bou Akar et al. (2024) optimized a species-specific protocol to efficiently differentiate myoblasts from human induced pluripotent stem cells (hiPSCs) and murine embryonic stem cells (mESCs) with the help of specific maturation medium (Bou Akar et al., 2024).
3. Limb Girdle Muscular Dystrophies (LGMD)
LGMD are a group of rare progressive genetic disorders with heterogeneous etiology affecting skeletal tissues, mostly proximate muscles of hips and shoulders with different autosomal mutations (dominant or recessive; indicated by D or R respectively) responsible for each subtype (Straub et al., 2018; Muni-Lofra et al., 2023; Bouchard and Tremblay, 2023)]. LGMD is the 4th most common cause of muscular weakness and has a prevalence of around 2 per 100,000 worldwide (Narayanaswami et al., 2014; Wicklund, 2014; Parsipenny, 2018)] with notable variations in the frequencies across the populations (Nallamili et al., 2018; Liu et al., 2019). LGMD molecular markers, pathogenesis, and classifications have been extensively discussed by Bouchard and Tremblay, 2023, Samarasiri, 2023, and Aguti et al., 2024..
With >39 causative genes, LGMD is a highly heterogeneous neuromuscular disorder with subtype-specific etiologies, pathogenesis, and molecular markers (Murphy and Straub, 2015; Liu et al., 2019; Barton et al., 2020; Bouchard and Tremblay, 2023). LGMD treatment begins with genetic diagnosis (Polavarapu et al, 2021; Zidkova et al., 2023), as patients often harbor mutations in multiple interconnected genes (Blandin et al., 2013; Nallamilli et al., 2018) overlapping with other disorders (Uritzberea et al., 2007) alongside biopsy and imaging to characterize disease pathology (Samarasiri, 2023).
3.1. Molecular Spectrum of LGMD Mutations in India
Even amongst the rare genetic disorders the frequency of LGMD mutations have so far been low in Indian populations (Sheth et al., 2024). Khadilkar et al. 2017 provides a comprehensive review of previously identified LGMD mutations in the Indian population (Khadilkar et al. 2017) . Additional mutations, some of them founder mutations (Francis et al., 2014; Khadilkar et al., 2016; Polavarapu et al., 2021; Ganaraja et al., 2022) have been reported in Indian populations since then. Given India's vast ethnic diversity and high rates of endogamy (The Indian Genome Variation Consortium, 2005; Khadilkar et al., 2017; Macha et al., 2025), it is essential to investigate the mutational landscape and distribution of LGMD mutations across Indian subpopulations, (Ankala et al., 2013; Khadilkar, 2015) which is still under sampled in many regions (Sheth et al., 2024).
In the Indian patient cohorts, the predominant phenotypic presentations included LGMDR1 (CAPN3, Calpainopathy), LGMDR4 (SGCG, Sarcoglycanopathy), LGMDR2 (DYSF, Dysferlinopathy), and MMD1 (DMPK, Myotonic Dystrophy Type 1), with LGMDR1 and LGMDR4 being the most frequently identified subtypes (Manjunath et al., 2023). While many mutations observed in Indian populations have also been observed elsewhere in the world, a few novel ones attributed to founder effectsare listed in Table 1. Some of the notable founder mutations in India are - Agarwal (Ankala et al., 2013; Khadilkar et al., 2016), South Indian ethnic communities (Polavarapu et al., 2021), Satwara community (Patel et al., 2024) affecting Calpain, Dysferlin, Lamin genes (Table 1). Agarwal mutations in Calpainopathy are the most extensively studied and well-documented founder mutations in India (Ankala et al., 2013; Khadilkar et al., 2016; Khadilkar et al., 2017).
3.2. LGMD Supportive and Symptomatic Treatments
Despite greater etiological heterogeneity than DMD, LGMD has fewer clinical therapies (Figure 1). However, research interest is growing, with 8 completed and 13 ongoing trials (21 total: 11 interventional, 10 observational). Currently, only 1 LGMD therapy is in late-stage trials.
LGMD patients experience chronic muscular pain and get fatigued easily (Vrist et al., 2022; Reelfs et al., 2023). Palliative \ remains a major domain of research (D’Este et al., 2024). LGMD patients receive tailored exercise regimens—combining strength and aerobic training—adjusted based on the affected protein (e.g., calpain, dysferrin) to mitigate fiber damage (Turken et al., 2022; D’Este et al., 2024). Personalized physiotherapy, including clinically-tested exercise regimens (Angelini et al., 2012; Turken et al., 2022), improves muscle strength, oxidative capacity, and daily function in LGMD patients, particularly subtypes 2I, 2L, and 2A (Siciliano et al., 2015). These supervised programs reduce fatigue and pain, even during pregnancy, while unsupervised eccentric exercise accelerates disease progression (D’Este et al., 2024).
Corticosteroids like prednisone and dexamethasone provide therapeutic benefits in inflammatory LGMD subtypes, preserving muscle function and slowing disease progression (Aho et al., 2015; Darin et al., 2021). While prednisone shows efficacy similar to DMD treatment (Quattrocelli et al., 2017, 2021; Zelikovich et al., 2022)., dexamethasone enhances muscle contraction (Quatrocelli et al., 2021). However, their pleiotropic effects require careful dosage regulation to balance anti-inflammatory benefits with potential atrophy risks, ideally combined with tailored exercise (Quatrocelli et al., 2021).
LGMD patients—particularly children with early-onset symptoms and pregnant women—receive nutritional counseling as part of comprehensive palliative care. Late-stage sarcoglycanopathies often require ventilator support due to respiratory muscle weakness (Graustein et al., 2023). Ongoing trials are developing assistive devices (e.g., lower-limb exoskeletons) to improve mobility (Supplementary Table S2)
3.3. Therapeutic Approaches and Clinical Trials for LGMD
LGMD therapies fall into two categories: (1) Primary approaches targeting genetic/protein defects (exon skipping, CRISPR, cell therapy) and (2) Secondary approaches managing symptoms (physical therapy, respiratory support, anti-inflammatory drugs). Supplementary Table 2 provides an exhaustive list of clinical therapies targeting LGMD obtained from the data available from https://clinicaltrials.gov. The LGMD subtypes “LGMDR1 and LGMDR4” prevalent in Indian patient cohorts (Manjunath et al., 2023) has been reviewed in detail in next section.

Table 2.
Founder mutations for GNE Myopathy in India.
| Mutation | Mutation Type | Gene | Community | Paper |
|---|---|---|---|---|
| p.Val727Met | Missense | GNE | Gujarat and Rajasthan | Bhattacharya et al., 2018 [187], Khandilkar et al., 2022 [136] |
3.3.1. Treatment of LGMDR1/2A
Pharmacological and Small Molecule Therapies
- Ubiquitin-proteasome as a therapeutic target for LGMDR1
LGMDR1 results from CAPN3 mutations, disrupting calpain-3 protease function (Richard et al.,1995). CAPN3 deficiency in mice triggers UPS-mediated SERCA degradation (Dayanithi et al., 2009). impairing calcium transport (Elgarresta et al., (2022)) and causing cytosolic calcium accumulation. Proteasome inhibition with bortezomib rescues SERCA2 levels and calcium homeostasis in CAPN3-deficient human myotubes (including G567W and R289W/R546L mutants), while also restoring mutant CAPN3 protein in patient cells (Lasa-Elgarresta et al., 2022). However, in CAPN3-KO mice, the same treatment (0.8 mg/kg every 72h for 3 weeks) failed to restore SERCA levels, indicating potential dose-dependent effects that require further investigation. Further studies are needed to validate proteasome inhibition's efficacy across missense mutations, including India-prevalent Agarwal mutations.
- SERCA overexpression for LGMDR1 treatment
Given BTZ's narrow therapeutic index, alternative approaches warrant consideration, such as AAV-mediated SERCA gene therapy or pharmacological SERCA modulation via β-adrenergic agents, hormones, antioxidants, or direct activators (e.g., CDN1163) [(Zsebo et al., 2014; Mázala et al., 2015; Kang et al., 2016). Additionally, targeting ER stress with compounds like tauroursodeoxycholic acid, rapamycin, or DWORF may mitigate muscle atrophy (Kawakami et al, 2019; Luo et al., 2019; Fisher et al., 2021).
- Drug therapies for LGMDR1
The WSiMD pilot trial (NCT04054375) evaluated weekly prednisone (0.75-1 mg/kg) in 20 LGMD/BMD patients over 24 weeks. Results showed trends of increased lean mass, reduced fat mass/CK levels, and improved motor function. Serum proteomics revealed prednisone-responsive markers (decreased muscle proteins, increased immune/matrix proteins), while MRI demonstrated fat fraction correlations with functional status. These findings support further investigation of low-dose weekly steroids (Zelikovich et al., 2019; Willis et al., 2024).
- Small molecule approach
In LGMDR1, calpain 3 deficiency disrupts CaMKIIβ signaling and slow-oxidative muscle programming. The small molecule AMBMP reversed this defect in C3KO mice, restoring oxidative metabolism, increasing slow fiber size, and improving exercise capacity via selective CaMKIIβ activation without affecting other growth pathways (Liu et al., 2020). These findings support CaMKII-targeted therapy development for LGMDR1.
Gene and Genome Editing Therapies
- Gene therapy approach for LGMDR1
While AAV-mediated myostatin inhibition increased muscle mass and power in CAPN3-deficient mice (Bartoli et al., 2007), follistatin-based myostatin blockade in C3KO models worsened exercise tolerance and oxidative capacity despite modest hypertrophy (Kramerova et al., 2020). These contradictory results suggest limited therapeutic efficacy of myostatin inhibitors in LGMDR1, likely due to incomplete understanding of disease mechanisms [163].
- AAV- mediated therapy
Sahenk et al. (2021) demonstrated that systemic AAVrh74.tMCK.hCAPN3 gene therapy (1.17-2.35×10¹⁴ vg/kg) improved muscle function, contractility, and fiber size in CAPN3-KO mice after 20 weeks, with females showing a shift toward oxidative fibers. Safety studies confirmed no toxicity, supporting its therapeutic potential for LGMDR1 (Sahenk et al., 2021).
- ATA-200, a gene therapy for LGMD
Atamyo’s ATA-200, an AAV8-based γ-sarcoglycan gene therapy for LGMD2C/R5, is currently in a Phase 1b pediatric trial (NCT05973630) assessing safety, protein expression, and functional outcomes. Parallel IND-enabling studies explore its application for LGMD2A/R1, building on preclinical evidence of dose-dependent muscle transduction without toxicity. (Atamyo, 2023; trial details: https://atamyo.com/press-releases/ind-for-ata-200-a-gene-therapy-for-the-treatment-of-limb-girdle-muscular-dystrophy-type-2c-r5-lgmd2c-r5-cleared-to-proceed-by-fda/)
- CRISPR/Cas9 approach towards LGMDR1
CRISPR/Cas9 enables direct gene-level editing (vs. mRNA-targeting approaches) by introducing double-strand breaks at specific loci, allowing defective gene excision and wild-type allele insertion for causal correction (Xiao-Jie et al., 2015; Chulanova et al., 2025). While widely used for knock-out/in models, its clinical translation for LGMD was limited by delivery challenges in higher organisms (Kim et al., 2024). Recent advances in non-viral delivery (nanoparticles, lipid particles) now enhance its therapeutic potential (Kim et al., 2024; Chulanova et al., 2025).
CRISPR/Cas9 has enabled generation of KO/KI animal models for studying LGMD pathogenesis and therapy development (Tanihara et al., 2019). Navarro-Serna et al. (2022) optimized electroporation-based CRISPR delivery in porcine oocytes to create LGMDR1 models with high mutation rates (Navarro-Serna et al., 2022). Similarly, a CRISPR-generated LGMDR1 mouse model (Capn3 c.635T>C variant) revealed impaired muscle repair due to mitochondrial dysfunction (Ma et al., 2023).
CRISPR/Cas9 can be integrated with cell replacement strategies. Mavrommatis et al. (2023) demonstrated CRISPR/Cas9-mediated correction of CAPN3 mutations (W130C, 550delA) in LGMDR1 patient-derived iPSCs using two approaches: (1) at the iPSC level, creating a functional chimeric exon 3-4 via CRISPR editing combined with FACS/Tet-based biallelic selection; and (2) in iPSC-derived CD82+/Pax7+ myogenic progenitors, specifically rescuing the prevalent exon 4 mutation. Both restored normal functions enabling autologous therapies (Mavrommatis et al., 2023). CRISPR/Cas9 holds particular promise for LGMD treatment, as correcting even a fraction of nuclei in multinucleated muscle cells can yield therapeutic benefits. (Zhang et al., 2021).
Patient-Specific iPSC-Derived Cellular Models of LGMDR1
Mateos-Aierdi et al. (2021) developed human myogenic models of LGMDR1 with diverse CAPN3 mutations using iPSCs (Mateos-Aierdi et al., 2021). Transient PAX7 overexpression enabled differentiation into myogenic progenitors and myotubes in vitro and in vivo. Despite inter-line variability, all patient-derived cells showed reduced DMD expression—critical for satellite cell regulation—providing a platform to study mutation-specific pathology and test therapies in a human myogenic context also helpful for LGMD.
3.3.2. Treatment of LGMDR4/2E
Gene and Genome Editing Therapies
LGMD2E/R4 or beta-sarcoglycanopathy is attributed to the mutations in the β-sarcoglycan (SGCB) gene, with consequential SGCB deficiency and muscle loss. A gene therapy “bidridistrogenexeboparvovec/SRP-9003” approach was developed based on SCB protein functional replacement. SRP-9003 is currently in Phase 3 evaluation (EMERGENE, NCT0624613), following prior Phase 1 (NCT05876780) and Phase 1/2 (NCT03652259) trials. SRP-9003 utilizes an AAVrh74 vector with MHCK7 promoter for systemic delivery of full-length β-sarcoglycan (SGCB) to skeletal, cardiac, and diaphragm muscles - particularly crucial for LGMD2E/R4 patients who often develop fatal cardiopulmonary complications.
Phase 3 study comprises ambulatory and non-ambulatory patients with LGMD2E/R4, aged 4 and older, monitoring changes in the expression of SGCB at 60 days (https://www.cgtlive.com/view/sarepta-therapeutics-limb-girdle-muscular-dystrophy-gene-therapy-srp-9003-safety-5-year-data) followed by study of secondary outcomes which includes functional outcomes through 60th month (https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-completes-enrollment-emergene-phase-3).
Interim Phase 1/2 data (NCT03652259) demonstrated dose-dependent SGCB expression (Cohort 1 [1.85×10¹³ vg/kg]: 36.2%; Cohort 2 [7.41×10¹³ vg/kg]: 62.1% at 60 days) using a self-complementary AAVrh74 vector with MHCK7 promoter driving codon-optimized full-length SGCB. All patients showed reduced CK levels (Cohort 1: -92.4%; Cohort 2: -94.9%) at 60 days, maintained through 2 years, with preliminary motor function improvements (NSAA) (Mendell et al., 2024).
Therapeutic Approaches Addressing the Secondary Causes of the Disease
- Redox-sensitive HMGB1 as a therapeutic target in sarcoglycanopathies
Recent studies implicate redox-sensitive HMGB1 in sarcoglycanopathies, where its oxidized isoform (dsHMGB1) promotes inflammation over regeneration. In mdx and sgca-null mice, HMGB1 knockout or administration of 3S (a non-oxidizable HMGB1 variant) improved function, enhanced regeneration, and reduced inflammation/fibrosis (Careccia et al., 2021). While 3S-HMGB1 may broadly target sarcoglycanopathy mechanisms, validation in diverse models and humanized systems is needed given subtype variability.
- P2X7 receptor blockade as a therapeutic strategy in sarcoglycanopathies
In sarcoglycanopathies, deficient ecto-ATPase activity elevates extracellular ATP (eATP), excessively activating P2X7 receptors and exacerbating inflammation. In sgca-null mice, blocking this pathway with either broad-spectrum oATP (Gazzero et al., 2019) or selective P2X7 antagonist A438079 (Raffaghelo et al., 2022) attenuated dystrophic progression. This intervention reduced inflammation, increased FOXP3+ regulatory T-cell recruitment, improved muscle strength, and decreased necrosis/fibrosis (Pannicucci et al., 2020; Zablocki et al., 2023)
- Inhibition of FAP-driven fibrosis via nintedanib in sarcoglycanopathy models
Fibro-adipogenic progenitors (FAPs; PDGFRα+) are essential for acute muscle repair but drive fibrosis in chronic dystrophies via persistent PDGF-AA/RhoA signaling (Fernández-Simón et al, 2022). Nintedanib, a PDGFRα/β-FGFR2/3-VEGFR inhibitor, reduced fibrosis and improved muscle function in sgca-null mice by reshaping the inflammatory microenvironment (Alonso- Perez et al., 2022). Similarly, P2X7 antagonists (oATP, A438079) attenuated dystrophic progression in these models. These therapies may extend to other sarcoglycanopathies (LGMDR4/2E, LGMDR1/2A) given shared fibro-inflammatory pathways.
4. GNE Myopathy (GNEM)
GNE myopathy (GNEM) is an ultra-rare (estimated prevalence: 1–9 per million), autosomal recessive disorder marked by progressive muscle wasting and ambulatory loss. While typically presenting with foot drop in the third decade of life (age range: 10–61 years), its hallmark pathological feature is the accumulation of rimmed vacuoles with concentric lamellar bodies, autophagic vacuoles, atrophic fibers, and 14–18 nm filamentous inclusions in skeletal muscle biopsies (Noguchi et al., 2004; Carrillo et al., 2018) Caused by biallelic mutations in the GNE gene—which encodes the bifunctional enzyme UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase—GNEM disrupts the rate-limiting step of sialic acid biosynthesis. This results in systemic hyposialylation of muscle glycoproteins, a key driver of pathology (Noguchi et al., 2004; Sparks et al., 2005; Malicdan et al., 2007; Malicdan et al., 2009).
Detailed mutation analysis of GNE gene showed that GNEM results from predominantly missense mutations (Chaouch et al., 2014, Zhu et al., 2017). Some of these are known to be population specific founder mutation, such as p.Met743Thr in Persian Jews (Eisenberg et al., 2001), p.Asp207Val in the Japanese (Nishino et al., 2002), p.Ile618Thr in Roma Gypsies (Kalaydjieva et al., 2004; Khadilkar et al., 2017) and p.Val727Met in populations of the Indian subcontinent (Nalini et al., 2013). As we can see from Table 2, the p.Val727Met is the only unique founder mutation which was detected in Indian populations and was observed under multiple cohorts (Bhattacharya et al., 2018; Khandilkar et al., 2022).
GNE mutations impair enzymatic function, causing sialic acid deficiency and GNEM pathogenesis (Noguchi et al., 2004). While no cure exists, therapeutic strategies aim to restore sialylation. In GNE mouse models, pre-symptomatic aceneuramic acid administration normalized motor function, muscle contractility, and histopathology (Malicdan et al., 2007, 2009). Similarly, sialic acid metabolites and IVIG (rich in sialylated glycoproteins) showed efficacy in preclinical studies (Sparks et al., 2007), supporting sialic acid supplementation as a viable therapeutic approach.
The candidate biomolecules/drugs evaluated in clinical trials included administration of intravenous immune globulin (IVIG) (NCT00195637, phase 1), Sialic Acid/aceneuramic acid/Neu5Ac (NCT01236898, phase 1), NeuAc extended-release tablet (Ace-ER) NCT01517880 (phase 2), NCT01830972 (phase 2), NCT02731690 (phase 2), NCT02377921 (phase 3),N-Acetylmannosamine (ManNAc) NCT01634750 (phase 1), NCT02346461 (phase 2), NCT04231266 (phase2)) and 6′-Sialyllactose.
GNE Myopathy has 12 completed clinical trials and 2 ongoing. Three of the clinical trials of GNE Myopathy are observational in nature while rest are interventional. Two of these interventional clinical trials are in phase 3.
4.1. Early-Stage Trials (2005–2010)
Early clinical trials (2005–2010) of IVIG and free sialic acid supplements (Neu5Ac/aceneuramic acid) showed limited efficacy due to rapid renal excretion (Sparks et al., 2007; Suzuki et al., 2018). This prompted development of Ace-ER, a slow-release sialic acid formulation designed to improve bioavailability.
4.2. Extended-Form Sialic Acid and Aceneuramic Acid Extended-Release (Ace-ER) Treatment
While a Phase 2 trial of extended-release sialic acid (Ace-ER) showed promise, the Phase 3 study (NCT02377921; n=89) failed to meet primary endpoints. Patients received oral Ace-ER (6 g/day, TID) for 48 weeks, with no significant improvement in upper/lower extremity composite scores versus placebo (Argoy et al., 2016; Lochmuller et al., 2019). However, subsequent research suggests potential efficacy with modified sialic acid formulations.
4.3. Sialic Acid Precursors
4.3.1. ManNAc and Sialyllactose
The poor efficacy of sialic acid may be due to its low uptake, therapeutic focus shifted to precursors like N-acetylmannosamine (ManNAc) prodrugs (targeting de novo synthesis) and sialyllactose, which offer improved lipophilicity and bioavailability (Yonekawa et al., 2014; Xu et al., 2017; Carrillo et al., 2021; Pertusati and Morewood, 2022). ManNAc serves as the first committed precursor in sialic acid biosynthesis.
A Phase 1 trial (NCT01634750) demonstrated that oral ManNAc (3-10 g/day) significantly increased plasma sialic acid levels in humans (Xu et al., 2017). An open-label Phase 2 trial (NCT02346461) in 12 GNEM patients demonstrated that oral ManNAc was safe, slowed extremity strength decline, and increased plasma Neu5Ac/sarcolemmal sialylation (Carrillo et al., 2021). The ongoing MAGiNE trial (NCT04231266), a Phase 2 randomized, double-blind, multicenter study by Leadiant Biosciences, is evaluating long-term safety and efficacy of ManNAc (12 g/day) in 51 GNEM patients (2:1 randomization) over 24 months. Assessments include QMA, IBMFRS, PROs, and functional tests, with completion expected October 2025.
4.3.2. ManNAc and Neu5Ac
Neu et al. (2024) demonstrated tissue-specific sialic acid metabolism in GNE-KO models: while HEK293 cells utilized ManNAc for sialic acid synthesis, murine myoblasts (C2C12/Sol8) required Neu5Ac for sialylation rescue (Neu et al., 2024). GNE deficiency also caused irreversible myogenesis defects, suggesting fundamental limitations of precursor supplementation therapies for skeletal muscle.
4.3.3. 6′-Sialyllactose (6SL) Supplements
6 g/day 6′-sialyllactose (6SL) supplements outperformed 3 g/day in maintaining muscle strength and reducing fatty infiltration in GNEM patients over 48 weeks (Park et al., 2023). Their follow-up placebo-controlled study (Park et al., 2025; n=11, 6SL=5/placebo=6) confirmed these findings: MRI revealed significantly increased fat fraction in placebo thigh muscles (P=0.0004), while the 6SL group showed slower progression (24–36 weeks). Biochemical and lectin-binding assays confirmed successful resialylation with 6SL.
4.4. ACENOBEL, a Sustained Release of Aceneuramic Acid Tablet Approved in Japan for GNEM
Japan recently approved ACENOBEL, a sustained-release aceneuramic acid tablet for GNEM, based on a Phase II/III trial (n=20) showing significant improvement in upper limb function versus placebo. (Suzuki et al., 2023). A subsequent 72-week open-label extension study found SA-ER was well-tolerated but showed gradual UEC decline in both SA-ER/SA-ER and placebo/SA-ER groups, suggesting partial disease-modifying effects (Suzuki et al., 2024).
Pre-symptomatic treatment in animal models show normalized muscle function, contraction, and histopathology, highlighting the potential benefits of early intervention (Mori-Yoshimura et al., 2023; Suzuki et al., 2024).
4.5. Gene Delivery Approaches for GNEM
Gene therapy approaches for GNEM include liposomal (Nemunaitis et al., 2011) and AAV-mediated (Mitrani-Rosenbaum et al., 2012; Crowe et al., 2021) delivery of wild-type GNE. While AAV vectors show long-term efficacy in murine/human muscle cells, challenges remain: 50% of patients have pre-existing AAV antibodies (Zygmunt et al., 2017), and risks of toxicity/oncogenicity (Berns et al., 2017). Recent work Zygmunt et al., 2023 demonstrates that rAAVrh74.CMV.GNE can systemically target skeletal muscle at safe doses, using a MyoD-inducible Lec3MyoDI cell line to quantify sialic acid production and vector potency .
Solve GNE (www.solvegne.org) and Genosera are developing a novel bicistronic AAV gene therapy for GNE myopathy, to halt disease progression and potentially reverse muscle damage. (https://www.globenewswire.com/news-release/2023/08/14/2724542/0/en/Solve-GNE-and-Genosera-Sign-Agreement-to-Develop-a-Novel-Bicistronic-AAV-Gene-Therapy-Targeting-HIBM.html).
4.6. Other Emerging Strategies
In addition to sialic acid supplementation, GNE myopathy therapeutic strategies include N-acetylcysteine (NAC) and mutant enzyme activation. Rimmed vacuoles (RVs), resulting from protein misfolding (Raben et al., 2012), impair muscle function by promoting oxidative stress. NAC alleviates this pathology in mouse models by reducing oxidative damage (Cho et al., 2017). Its broader application in mitochondrial disorders (Sahasrabuddhe et al., 2017) further supports its therapeutic potential. These approaches complement efforts to restore sialic acid biosynthesis through targeted enzyme modulation. While GNE p.Met743Thr mutation reduces epimerase activity without causing hyposialylation in cultured muscle cells (Salama et al., 2005; Sharma et al., 2022) identified mutation-specific enhancement of GNE enzymatic activity using plant phenolics and antidiabetic compounds, with metformin showing particular efficacy in restoring epimerase function.
Supplementary Table S3 gives compete list of ongoing and completed clinical trials focused on GNE Myopathy.
5. Conclusions
Muscular dystrophies (DMD, LGMD, GNEM) represent a spectrum of genetic disorders characterized by progressive muscle degeneration. While current therapies primarily aim to slow disease progression, recent advances in gene editing and regenerative medicine have enabled curative strategies targeting underlying genetic defects. These include AAV-mediated gene replacement, CRISPR-Cas9 genome editing, exon skipping, and innovative approaches like split intein-mediated large-gene delivery. Notably, for AAV-mediated gene therapy, stringent caution is required regarding vector dosage and the patient's disease stage, as these factors are critical to mitigate the risk of severe adverse events, including clinical fatalities. Cell-based therapies utilizing iPSCs and small molecule modulators targeting muscle repair pathways (e.g., Piezo1 channels) show particular promise. However, significant challenges remain, including immune responses to viral vectors, off-target effects of gene editing, tissue-specific delivery limitations, and the complexity of personalized therapies. Additionally, translating preclinical success to diverse patient populations requires addressing genetic heterogeneity and founder mutations. Prioritizing therapies for high-prevalence mutations while ensuring scalable manufacturing will maximize population level impact for neuromuscular disorders.
6. Future Directions
The integration of AI/ML with multi-omics profiling holds significant potential to identify novel therapeutic targets and enable personalized treatment strategies for muscular dystrophies. Future innovation will likely focus on three key areas: (1) enhancing the safety and efficiency of gene delivery systems, (2) developing non-viral delivery platforms, and (3) optimizing genome editing precision. Realizing this potential will require sustained interdisciplinary collaboration among clinicians, researchers, and regulatory bodies to translate scientific advances into accessible, durable therapies.
For efficient therapy development, regulatory frameworks must account for national variations in rare genetic disease (RGD) definitions and orphan drug designation criteria. Given disparities in healthcare infrastructure and resources between developed and developing nations, uniform global regulations may prove ineffective. Instead, each country should establish tailored RGD strategies that align with local healthcare priorities, economic realities, and ethical considerations to ensure equitable therapeutic access for patients.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
DK: conceptualization, data curation, writing (original draft, review and editing), supervision, funding acquisition, visualization. AA: data curation, writing (original draft, review and editing), visualization. All authors have read and agreed to the final version of the manuscript.
Funding
This work was supported by DBT BUILDER grant (BT/INF/22/SP45382/2022) and PAIR grant (number to be added) awarded to School of Life Sciences, JNU.
Clinical trial number
Not applicable.
Ethics approval
Not applicable.
Consent (participation and publication)
Not applicable.
Data availability
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study.
Code availability
Not applicable.
Acknowledgments
The authors acknowledge Prof. Sudha Bhattacharya, FASc. FNASc. FNA (INSA Senior Scientist, Ashoka University, Sonipat, India) for her critical suggestions and comments to refine this manuscript. We also acknowledge G. Chandana (Department of Biotechnology, Vignan's Foundation for Science, Technology & Research, Guntur, Andhra Pradesh) for her inputs in Figure 1.:
Conflicts of Interests
The authors (DK and AA) declare that they have no conflicts of interest that are directly relevant to the content of this article.
References
- Aartsma-Rus, Annemieke. "The future of exon skipping for Duchenne muscular dystrophy." Human Gene Therapy 34, no. 9-10 (2023): 372-378. [CrossRef]
- Acharya, S., A.H. Ansari, and P. Kumar Das. “PAM-Flexible Engineered FnCas9 Variants for Robust and Ultra-Precise Genome Editing and Diagnostics.” Nat Commun 15 (2024): 5471. [CrossRef]
- Agrawal, Pooja, Vancha Harish, Sharfuddin Mohd, Sachin Kumar Singh, Devesh Tewari, Ramanjireddy Tatiparthi, Sukriti Vishwas, Srinivas Sutrapu, Kamal Dua, and Monica Gulati. “Role of CRISPR/Cas9 in the Treatment of Duchenne Muscular Dystrophy and Its Delivery Strategies.” Life Sciences 330 (2023): 122003. [CrossRef]
- Aguti, Sara, Gian Nicola Gallus, Silvia Bianchi, Simona Salvatore, Anna Rubegni, Gianna Berti, Patrizia Formichi, Nicola Stefano, Alessandro Malandrini, and Diego Lopergolo. “Novel Biomarkers for Limb Girdle Muscular Dystrophy (LGMD.” Cells 13, no. 4 (2024): 329. [CrossRef]
- Ahmed, H.Shafeeq. “Limb-Girdle Muscular Dystrophy in Pregnancy: A Narrative Review.” Archives of Gynecology and Obstetrics 310, no. 5 (2024): 2373–86. [CrossRef]
- Aho, Anna Carin, Sally Hultsjö, and Katarina Hjelm. "Young adults’ experiences of living with recessive limb-girdle muscular dystrophy from a salutogenic orientation: An interview study." Disability and rehabilitation 37, no. 22 (2015): 2083-2091. [CrossRef]
- Aitken, Murry, E. J. Mercer, and A. I. I. McKemey. "Understanding Neuromuscular Disease Care." IQVIA Institute. Parsippany, NJ (2018).
- Alhamadani, Feryal, Kristy Zhang, Rajvi Parikh, Hangyu Wu, Theodore P. Rasmussen, Raman Bahal, Xiao-bo Zhong, and José E. Manautou. "Adverse drug reactions and toxicity of the food and drug administration–approved antisense oligonucleotide drugs." Drug Metabolism and Disposition 50, no. 6 (2022): 879-887. [CrossRef]
- Alonso-Pérez, Jorge, Ana Carrasco-Rozas, Maria Borrell-Pages, Esther Fernández-Simón, Patricia Piñol-Jurado, Lina Badimon, Lutz Wollin et al. "Nintedanib reduces muscle fibrosis and improves muscle function of the alpha-sarcoglycan-deficient mice." Biomedicines 10, no. 10 (2022): 2629. [CrossRef]
- Angelini, Corrado, and Elisabetta Tasca. “Fatigue in Muscular Dystrophies.” Neuromuscular Disorders 22 (2012): 214–20.
- Ankala, Arunkanth, Jordan N. Kohn, Rashna Dastur, Pradnya Gaitonde, Satish V. Khadilkar, and Madhuri R. Hegde. “Ancestral Founder Mutations in Calpain-3 in the Indian Agarwal Community: Historical, Clinical, and Molecular Perspective.” Muscle & Nerve 47, no. 6 (2013): 931–37. [CrossRef]
- Argov, Zohar, Yoseph Caraco, Heather Lau, Alan Pestronk, Perry B. Shieh, Alison Skrinar, Tony Koutsoukos, Ruhi Ahmed, Julia Martinisi, and Emil Kakkis. "Aceneuramic acid extended release administration maintains upper limb muscle strength in a 48-week study of subjects with GNE myopathy: results from a phase 2, randomized, controlled study." Journal of neuromuscular diseases 3, no. 1 (2016): 49-66. [CrossRef]
- Assefa, Milyard, Addison Gepfert, Meesam Zaheer, Julia M. Hum, and Brian W. Skinner. "Casimersen (AMONDYS 45™): an antisense oligonucleotide for Duchenne muscular dystrophy." Biomedicines 12, no. 4 (2024): 912. [CrossRef]
- Baeza-Barragán, Maria Rosa, Maria Teresa Labajos Manzanares, Carmen Ruiz Vergara, María Jesús Casuso-Holgado, and Rocío Martín-Valero. “The Use of Virtual Reality Technologies in the Treatment of Duchenne Muscular Dystrophy: Systematic Review.” JMIR mHealth and uHealth 8, no. 12 (2020): 21576. [CrossRef]
- Bardhan, Mainak, Ram Murthy Anjanappa, Kiran Polavarapu, Veeramani Preethish-Kumar, Seena Vengalil, Saraswati Nashi, Shamita Sanga et al. "Clinical, genetic profile and disease progression of sarcoglycanopathies in a large cohort from India: high prevalence of SGCB c. 544A> C." neurogenetics 23, no. 3 (2022): 187-202. [CrossRef]
- Barton, Elisabeth R., Christina A. Pacak, Whitney L. Stoppel, and Peter B. Kang. “The Ties That Bind: Functional Clusters in Limb-Girdle Muscular Dystrophy.” Skeletal Muscle 10 (2020): 1–13. [CrossRef]
- Basak, Jayasri, Uma B. Dasgupta, Subhash Chandra Mukherjee, Shyamal Kumar Das, Asit Kumar Senapati, and Tapas Kumar Banerjee. “Deletional Mutations of Dystrophin Gene and Carrier Detection in Eastern India.” The Indian Journal of Pediatrics 76 (2009): 1007–12. [CrossRef]
- Baskar, Dipti, Veeramani Preethish-Kumar, Kiran Polavarapu, Seena Vengalil, Saraswati Nashi, Deepak Menon, Valakunja Harikrishna Ganaraja et al. "Clinical and genetic heterogeneity of nuclear envelopathy related muscular dystrophies in an Indian cohort." Journal of Neuromuscular Diseases 11, no. 5 (2024): 969-979. [CrossRef]
- Bello, L., P. Campadello, A. Barp, M. Fanin, C. Semplicini, G. Sorarù, L. Caumo, C. Calore, C. Angelini, and E. Pegoraro. “Functional Changes in Becker Muscular Dystrophy: Implications for Clinical Trials in Dystrophinopathies.” Sci. Rep 6 (2016): 32439. [CrossRef]
- Bernareggi, Annalisa, Alessandra Bosutti, Gabriele Massaria, Rashid Giniatullin, Tarja Malm, Marina Sciancalepore, and Paola Lorenzon. "The state of the art of Piezo1 channels in skeletal muscle regeneration." International journal of molecular sciences 23, no. 12 (2022): 6616. [CrossRef]
- Berns, K.I., and N. Muzyczka. “AAV: An Overview of Unanswered Questions.” Hum Gene Ther 28 (2017): 308–13. [CrossRef]
- BeytíaMdeL, Vry J., and Kirschner J. “Drug Treatment of Duchenne Muscular Dystrophy: Available Evidence and Perspectives.” Acta Myol 31 (2012): 4–8.
- Bhattacharya, Sudha, Satish V. Khadilkar, Atchayaram Nalini, Aparna Ganapathy, Ashraf U. Mannan, Partha P. Majumder, and Alok Bhattacharya. “Mutation Spectrum of GNE Myopathy in the Indian Sub-Continent.” Journal of Neuromuscular Diseases 5, no. 1 (2018): 85–92. [CrossRef]
- Bianchi, B., D.J. Matthews, G.H. Clayton, and T. Carry. “Corticosteroid Treatment and Functional Improvement in Duchenne Muscular Dystrophy: Long-Term Effect.” Am. J. Phys. Med. Rehabil 84 (2005): 843–50.
- Birnkrant, D.J., K. Bushby, C.M. Bann, B.A. Alman, S.D. Apkon, A. Blackwell, L.E. Case, L. Cripe, S. Hadjiyannakis, and A.K. Olson. “Diagnosis and Management of Duchenne Muscular Dystrophy, Part 2: Respiratory, Cardiac, Bone Health, and Orthopaedic Management.” Lancet Neurol 17 (2018): 347–61. [CrossRef]
- Bladen, C.L. “The TREAT-NMD DMD Global Database: Analysis of More than 7000 Duchenne Muscular Dystrophy Mutations.” Hum. Mutat 36, no. 4 (2015): 395–402. [CrossRef]
- Blake, Derek J., Jonathon M. Tinsley, and Kay E. Davies. "Utrophin: a structural and functional comparison to dystrophin." Brain pathology 6, no. 1 (1996): 37-47. [CrossRef]
- Blake, Derek J., Jonathon M. Tinsley, and Kay E. Davies. "Utrophin: a structural and functional comparison to dystrophin." Brain pathology 6, no. 1 (1996): 37-47. [CrossRef]
- Blandin, Gaëlle, Sylvie Marchand, Karine Charton, Nathalie Danièle, Evelyne Gicquel, Jean-Baptiste Boucheteil, and AzéddineBentaib. “A Human Skeletal Muscle Interactome Centered on Proteins Involved in Muscular Dystrophies: LGMD Interactome.” Skeletal Muscle 3 (2013): 1–19. [CrossRef]
- Blau, H.M., C. Webster, and G.K. Pavlath. “Defective myoblasts identified in Duchenne muscular dystrophy.” Proc Natl Acad Sci U S A 80 (1983): 4856–60. [CrossRef]
- Bostick, B. “AAV Micro-Dystrophin Gene Therapy Alleviates Stress-Induced Cardiac Death but Not Myocardial Fibrosis in >21-Mold Mdx Mice, an End-Stage Model of Duchenne Muscular Dystrophy Cardiomyopathy.” J. Mol. Cell Cardiol 53 (2012): 217–22. [CrossRef]
- Bouchard, Camille, and Jacques P. Tremblay. “Limb–Girdle Muscular Dystrophies Classification and Therapies.” Journal of Clinical Medicine 12, no. 14 (2023): 4769. [CrossRef]
- Braun, Serge. "Duchenne muscular dystrophy, one of the most complicated diseases for gene therapy." Journal of Translational Genetics and Genomics 9, no. 1 (2025): 35-47. [CrossRef]
- Bushby, K., R. Finkel, D.J. Birnkrant, L.E. Case, P.R. Clemens, L. Cripe, A. Kaul, K. Kinnett, C. McDonald, and S. Pandya. “Diagnosis and Management of Duchenne Muscular Dystrophy, Part 1: Diagnosis, and Pharmacological and Psychosocial Management.” Lancet Neurol 9 (2010): 77–93. [CrossRef]
- Careccia, G., M. Saclier, and M. Tirone. “Rebalancing Expression of HMGB1 Redox Isoforms to Counteract Muscular Dystrophy.” Sci Transl Med 13, no. 596 (2021). [CrossRef]
- Carrillo, Nuria, May C. Malicdan, and Marjan Huizing. “GNE Myopathy: Etiology, Diagnosis, and Therapeutic Challenges.” Neurotherapeutics 15, no. 4 (2018): 900–914. [CrossRef]
- Carrillo, Nuria, May C. Malicdan, Petcharat Leoyklang, Joseph A. Shrader, Galen Joe, Christina Slota, John Perreault et al. "Safety and efficacy of N-acetylmannosamine (ManNAc) in patients with GNE myopathy: an open-label phase 2 study." Genetics in Medicine 23, no. 11 (2021): 2067-2075. [CrossRef]
- Chaouch, Amina, Kathryn M. Brennan, Judith Hudson, Cheryl Longman, John McConville, Patrick J. Morrison, Maria E. Farrugia et al. "Two recurrent mutations are associated with GNE myopathy in the North of Britain." Journal of Neurology, Neurosurgery & Psychiatry 85, no. 12 (2014): 1359-1365. [CrossRef]
- Chemello, F., A.C. Chai, H. Li, C. Rodriguez-Caycedo, E. Sanchez-Ortiz, A. Atmanli, A.A. Mireault, N. Liu, R. Bassel-Duby, and E.N. Olson. “Precise Correction of Duchenne Muscular Dystrophy Exon Deletion Mutations by Base and Prime Editing.” Sci. Adv 7 (2021): 4910. [CrossRef]
- Cho, Anna, May Christine, V. Malicdan, Miho Miyakawa, Ikuya Nonaka, Ichizo Nishino, and Satoru Noguchi. "Sialic acid deficiency is associated with oxidative stress leading to muscle atrophy and weakness in GNE myopathy." Human molecular genetics 26, no. 16 (2017): 3081-3093. [CrossRef]
- Chwalenia, Katarzyna, Vivi-Yun Feng, Nicole Hemmer, Hans J. Friedrichsen, Ioulia Vorobieva, Matthew JA Wood, and Thomas C. Roberts. "AAV microdystrophin gene replacement therapy for Duchenne muscular dystrophy: progress and prospects." Gene Therapy (2025): 1-15. [CrossRef]
- Chulanova, Yulia, Dor Breier, and Dan Peer. “Delivery of Genetic Medicines for Muscular Dystrophies.” Cell Reports Medicine, 2025. [CrossRef]
- Crisafulli, S., J. Sultana, A. Fontana, F. Salvo, S. Messina, and G. Trifirò. “Global Epidemiology of Duchenne Muscular Dystrophy: An Updated Systematic Review and Meta-Analysis.” Orphanet J Rare Dis 15, no. 141 (2020). [CrossRef]
- Crowe, K.E., D.A. Zygmunt, and K. Heller. “Visualizing Muscle Sialic Acid Expression in the GNED207VTgGne-/- Cmah-/- Model of GNE Myopathy: A Comparison of Dietary and Gene Therapy Approaches.” J Neuromuscul Dis 9 (2021): 53–71. [CrossRef]
- D’Este, Giorgia, Mattia Spagna, Sara Federico, Luisa Cacciante, Błażej Cieślik, Pawel Kiper, and Rita Barresi. Limb-girdle Muscular Dystrophies: A Scoping Review and Overview of Currently Available Rehabilitation Strategies. Muscle & Nerve, n.d. [CrossRef]
- Dahl, Russell, and Ilya Bezprozvanny. "SERCA pump as a novel therapeutic target for treating neurodegenerative disorders." Biochemical and Biophysical Research Communications 734 (2024): 150748. [CrossRef]
- Darin, N., A-K. Kroksmark, A-C. Åhlander, A-R. Moslemi, A. Oldfors, and M. Tulinius. "Inflammation and response to steroid treatment in limb-girdle muscular dystrophy 2I." European Journal of Paediatric Neurology 11, no. 6 (2007): 353-357. [CrossRef]
- Davies, K., A. Philippidis, and R. Barrangou. “Five Years of Progress in CRISPR Clinical Trials (2019-2024.” CRISPR J 7, no. 5 (2024): 227–30. [CrossRef]
- De Masi, Claudia, Paola Spitalieri, Michela Murdocca, Giuseppe Novelli, and Federica Sangiuolo. "Application of CRISPR/Cas9 to human-induced pluripotent stem cells: from gene editing to drug discovery." Human genomics 14 (2020): 1-12. [CrossRef]
- Del Rio-Pertuz, G., C. Morataya, K. Parmar, S. Dubay, and E. Argueta-Sosa. “Dilated Cardiomyopathy as the Initial Presentation of Becker Muscular Dystrophy: A Systematic Review of Published Cases.” Orphanet J. Rare Dis 17 (2022): 194. [CrossRef]
- Donovan, J., H. Phan, A. Russell, B. Barthel, L. Thaler, N. Kilburn, M. Amato, and J. MacDougall. "351P Sevasemten, a fast myosin inhibitor, in adults with Becker muscular dystrophy results in reduced muscle damage biomarkers and functional stabilization." Neuromuscular Disorders 43 (2024): 104441-682. [CrossRef]
- Donovan, Joanne, Jeffrey A. Silverman, Ben Barthel, Michael DuVall, Molly Madden, James MacDougall, Nicole Rempel Kilburn et al. "A Phase 1, Double-Blind, Placebo-Controlled Trial of Sevasemten (EDG-5506), a Selective Modulator of Fast Skeletal Muscle Contraction, in Healthy Volunteers and Adults With Becker Muscular Dystrophy." Muscle & Nerve (2025). [CrossRef]
- Duan, Dongsheng, Nathalie Goemans, Shin’ichi Takeda, Eugenio Mercuri, and Annemieke Aartsma-Rus. “Duchenne Muscular Dystrophy.” Nature Reviews Disease Primers 7, no. 1 (2021): 13. [CrossRef]
- E, Vasterling M., Maitski R. J, and Davis B. A. “AMONDYS 45 (Casimersen), a Novel Antisense Phosphorodiamidate Morpholino Oligomer: Clinical Considerations for Treatment in Duchenne Muscular Dystrophy.” Cureus 15, no. 12 (December 28, 2023): 51237. [CrossRef]
- Emami, Michael R., Courtney S. Young, Ying Ji, Xiangsheng Liu, Ekaterina Mokhonova, April D. Pyle, Huan Meng, and Melissa J. Spencer. “Polyrotaxane Nanocarriers Can Deliver CRISPR/Cas9 Plasmid to Dystrophic Muscle Cells to Successfully Edit the DMD Gene.” Advanced Therapeutics 2, no. 7 (2019): 1900061. [CrossRef]
- Ervasti, James M., and Kevin P. Campbell. “Membrane Organization of the Dystrophin-Glycoprotein Complex.” Cell 66, no. 6 (1991): 1121–31. [CrossRef]
- Falzarano, Maria Sofia, Chiara Scotton, Chiara Passarelli, and Alessandra Ferlini. “Duchenne Muscular Dystrophy: From Diagnosis to Therapy.” Molecules 20, no. 10 (2015): 18168–84. [CrossRef]
- Fernandes, José AL, Tâmara HR Prandini, Maria da Conceicao A. Castro, Thales D. Arantes, Juliana Giacobino, Eduardo Bagagli, and Raquel C. Theodoro. "Evolution and application of inteins in Candida species: a review." Frontiers in Microbiology 7 (2016): 1585. [CrossRef]
- Fernández-Simón, Esther, Xavier Suárez-Calvet, Ana Carrasco-Rozas, Patricia Piñol-Jurado, Susana López-Fernández, Gemma Pons, Joan Josep Bech Serra et al. "RhoA/ROCK2 signalling is enhanced by PDGF-AA in fibro-adipogenic progenitor cells: implications for Duchenne muscular dystrophy." Journal of Cachexia, Sarcopenia and Muscle 13, no. 2 (2022): 1373-1384. [CrossRef]
- Fisher, M.E., E. Bovo, and R. Aguayo-Ortiz. “Dwarf Open Reading Frame (DWORF) Is a Direct Activator of the Sarcoplasmic Reticulum Calcium Pump SERCA.” Elife 10:e65545 (2021). [CrossRef]
- Flanigan, K.M. “Mutational Spectrum of DMD Mutations in Dystrophinopathy Patients: Application of Modern Diagnostic Techniques to a Large Cohort.” Hum. Mutat 30, no. 12 (2009): 1657–66. [CrossRef]
- Francis, Amirtharaj, Balaraju Sunitha, Kandavalli Vinodh, Kiran Polavarapu, Shiva Krishna Katkam, M.M.Srinivas Bharath Sailesh Modi, Narayanappa Gayathri, Atchayaram Nalini, and Kumarasamy Thangaraj. “Novel TCAP mutation c. 32C> A causing limb girdle muscular dystrophy 2G.” PLoS One 9, no. 7 (2014): 102763. [CrossRef]
- G., Dayanithi, Richard I., Viero C., Mazuc E., Mallie S., and Valmier J. “Alteration of Sarcoplasmic ReticulumCa2+Release in Skeletal Muscle from Calpain 3-Deficient Mice.” Int. J. Cel Biol, 2009, 1–12. [CrossRef]
- Ganaraja, Valakunja H., Kiran Polavarapu, Mainak Bardhan, Veeramani Preethish-Kumar, Shingavi Leena, Ram M. Anjanappa, and Seena Vengalil. “Disease Progression and Mutation Pattern in a Large Cohort of LGMD R1/LGMD 2A Patients from India.” Global Medical Genetics 9, no. 01 (2022): 034–041. [CrossRef]
- Gazzerro, Elisabetta. "Role of Extracellular ATP in the Progression of Muscle Damage in Sarcoglycanopathies." PhD diss., 2021.
- Ghosh, S., M.U. Arshi, and S. Ghosh. “Discovery of Quinazoline and Quinoline-Based Small Molecules as Utrophin Upregulators via AhR Antagonism for the Treatment of Duchenne Muscular Dystrophy.” J Med Chem 67, no. 11 (2024): 9260–76. [CrossRef]
- Goyal, Manisha, Ashok Gupta, Kamlesh Agarwal, Seema Kapoor, and Somesh Kumar. “Duchenne Muscular Dystrophy: Genetic and Clinical Profile in the Population of Rajasthan, India.” Annals of Indian Academy of Neurology 24, no. 6 (2021): 873–78.
- Graustein, Andrew, Hugo Carmona, and Joshua O. Benditt. “Noninvasive Respiratory Assistance as Aid for Respiratory Care in Neuromuscular Disorders.” Frontiers in Rehabilitation Sciences 4 (2023): 1152043. [CrossRef]
- Gustafsson, Oskar, Supriya Krishna, Sophia Borate, Marziyeh Ghaeidamini, Xiuming Liang, Osama Saher, Raul Cuellar et al. "Advanced Peptide Nanoparticles Enable Robust and Efficient delivery of gene editors across cell types." bioRxiv (2024): 2024-11. [CrossRef]
- Heydemann, Ahlke, Grzegorz Bieganski, Jacek Wachowiak, Jarosław Czarnota, Adam Niezgoda, Krzysztof Siemionow, Anna Ziemiecka et al. "Dystrophin Expressing Chimeric (DEC) cell therapy for Duchenne muscular dystrophy: A first-in-human study with minimum 6 months follow-up." Stem Cell Reviews and Reports 19, no. 5 (2023): 1340-1359. [CrossRef]
- Hsu, P.D., E.S. Lander, and F. Zhang. “Development and Applications of CRISPR-Cas9 for Genome Engineering.” Cell 157 (2014): 1262–78. [CrossRef]
- I., Richard, Broux O., Allamand V., Fougerousse F., Chiannilkulchai N., and Bourg N. “Mutations in the Proteolytic Enzyme Calpain 3 Cause Limb-Girdle Muscular Dystrophy Type 2A.” Cell 81, no. 1 (1995): 27-40. [CrossRef]
- Indian Genome Variation Consortium+ 91-11-27667806+ 91-11-27667471 skb@ igib. res. in. "The Indian genome variation database (IGVdb): a project overview." Human genetics 118 (2005): 1-11.
- Iolascon, Giovanni, Marco Paoletta, Sara Liguori, Claudio Curci, and Antimo Moretti. “Neuromuscular Diseases and Bone.” Frontiers in Endocrinology 10 (2019): 794. [CrossRef]
- J, Keam S. “Vamorolone: First Approval.” Drugs 84, no. 1 (2024): 111–17. [CrossRef]
- Kang, Soojeong, Russell Dahl, Wilson Hsieh, Andrew Shin, Krisztina M. Zsebo, Christoph Buettner, Roger J. Hajjar, and Djamel Lebeche. "Small molecular allosteric activator of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) attenuates diabetes and metabolic disorders." Journal of Biological Chemistry 291, no. 10 (2016): 5185-5198. [CrossRef]
- Kawakami, Y., W.S. Hambright, and K. Takayama. “Rapamycin Rescues Age-Related Changes in Muscle-Derived Stem/Progenitor Cells from Progeroid Mice.” Mol Ther Methods Clin Dev 14 (2019): 64–76. [CrossRef]
- Keeling, K.M., X. Xue, G. Gunn, and D.M. Bedwell. “Therapeutics Based on Stop Codon Readthrough.” Annu Rev Genomics Hum Genet 15 (2014): 371–94. [CrossRef]
- Khadilkar, S.V., B.R.R. Nallamilli, and A. Bhutada. “A Report on GNE Myopathy: Individuals of Rajasthan Ancestry Share the Roma Gene.” J Neurol Sci 375 (2017): 239–40. [CrossRef]
- Khadilkar, Satish V. "Limb girdle muscular dystrophies in India." Neurology India 63, no. 4 (2015): 495-496. [CrossRef]
- Khadilkar, Satish V., Chetan R. Chaudhari, Rashna S. Dastur, Pradnya S. Gaitonde, and Jayendra G. Yadav. “Limb-Girdle Muscular Dystrophy in the Agarwals: Utility of Founder Mutations in CAPN3 Gene.” Annals of Indian Academy of Neurology 19, no. 1 (2016): 108–11. [CrossRef]
- Khadilkar, Satish Vasant, Hiral Amrut Halani, Rashna Dastur, Pradnya Satish Gaitonde, Harsh Oza, and Madhuri Hegde. “Genetic Appraisal of Hereditary Muscle Disorders in a Cohort from Mumbai, India.” Journal of Neuromuscular Diseases 9, no. 4 (2022): 571–80. [CrossRef]
- Kim, Minse, Youngwoo Hwang, Seongyu Lim, Hyeon-Ki Jang, and Hyun-Ouk Kim. “Advances in nanoparticles as non-viral vectors for efficient delivery of CRISPR/Cas9.” Pharmaceutics 16, no. 9 (2024): 1197. [CrossRef]
- Kiper, Pawel, Sara Federico, Joanna Szczepańska-Gieracha, Patryk Szary, Adam Wrzeciono, Justyna Mazurek, and Carlos Luque-Moreno. “A Systematic Review on the Application of Virtual Reality for Muscular Dystrophy Rehabilitation: Motor Learning Benefits.” Life 14, no. 7 (2024): 790. [CrossRef]
- Kumar, Prasann, and Padmanabh Dwivedi. “Diagnosis of Neuromuscular Disorder.” In Computational Intelligence for Genomics Data, 225–40. Academic Press, 2025. [CrossRef]
- Laing, Nigel G. “Genetics of Neuromuscular Disorders.” Critical Reviews in Clinical Laboratory Sciences 49, no. 2 (2012): 33–48. [CrossRef]
- Landfeldt, E., C. Lindberg, and T. Sejersen. “Improvements in Health Status and Utility Associated with Ataluren for the Treatment of Nonsense Mutation Duchenne Muscular Dystrophy.” Muscle Nerve 61 (2020): 363–68. [CrossRef]
- Lasa-Elgarresta, Jaione, Laura Mosqueira-Martín, Neia Naldaiz-Gastesi, Amets Sáenz, Adolfo Lopez de Munain, and Ainara Vallejo-Illarramendi. "Calcium mechanisms in limb-girdle muscular dystrophy with CAPN3 mutations." International journal of molecular sciences 20, no. 18 (2019): 4548. [CrossRef]
- Laurent, M., M. Geoffroy, G. Pavani, and S. Guiraud. “CRISPR-Based Gene Therapies: From Preclinical to Clinical Treatments.” Cells 13, no. 10 (2024). [CrossRef]
- Lee, H., J. Song, I.S. Kang, J. Huh, J.A. Yoon, and Y.B. Shin. “Early Prophylaxis of Cardiomyopathy with Beta-Blockers and Angiotensin Receptor Blockers in Patients with Duchenne Muscular Dystrophy.” Clin. Exp. Pediatr 65 (2022): 507–9. [CrossRef]
- Lim, K.R., R. Maruyama, and T. Yokota. “Eteplirsen in the Treatment of Duchenne Muscular Dystrophy.” Drug Design, Development and Therapy 11 (2017): 533–45. [CrossRef]
- Liu, J., J. Campagna, and V. John. “A Small-Molecule Approach to Restore a Slow-Oxidative Phenotype and Defective CaMKIIβ Signaling in Limb Girdle Muscular Dystrophy.” Cell Rep Med 1, no. 7 (2020). [CrossRef]
- Liu, Wei, Sander Pajusalu, Nicole J. Lake, Geyu Zhou, Nilah Ioannidis, Plavi Mittal, and Nicholas E. Johnson. “Estimating Prevalence for Limb-Girdle Muscular Dystrophy Based on Public Sequencing Databases.” Genetics in Medicine 21, no. 11 (2019): 2512–20. [CrossRef]
- Liu, Wei, Sander Pajusalu, Nicole J. Lake, Geyu Zhou, Nilah Ioannidis, Plavi Mittal, Nicholas E. Johnson et al. "Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases." Genetics in Medicine 21, no. 11 (2019): 2512-2520. [CrossRef]
- LochmüllerH, Behin A., and Caraco Y. “A Phase 3 Randomized Study Evaluating Sialic Acid Extended-Release for GNE Myopathy.” Neurology 92, no. 18 (2019). [CrossRef]
- Luo, H., C. Zhou, and J. Chi. “The Role of Tauroursodeoxycholic Acid on Dedifferentiation of Vascular Smooth Muscle Cells by Modulation of Endoplasmic Reticulum Stress and as an Oral Drug Inhibiting In-Stent Restenosis.” Cardiovasc Drugs Ther 33, no. 1 (2019): 25–33. [CrossRef]
- M, Hoy S. “DelandistrogeneMoxeparvovec: First Approval.” Drugs 83, no. 14 (2023): 1323–29. [CrossRef]
- Ma, H.S., X.L. Gong, and W.X. Li. “Missense mutation of c.635 T > C in CAPN3 impairs muscle injury repair in a Limb-Girdel Muscular Dystropy Model.” Clin Genet 103, no. 6 (2023): 663–71. [CrossRef]
- Ma, Nuoying, Delia Chen, Ji-Hyung Lee, Paola Kuri, Edward Blake Hernandez, Jacob Kocan, Hamd Mahmood, Elisia D. Tichy, Panteleimon Rompolas, and Foteini Mourkioti. "Piezo1 regulates the regenerative capacity of skeletal muscles via orchestration of stem cell morphological states." Science advances 8, no. 11 (2022): eabn0485. [CrossRef]
- Machha, Pratheusa, Amirtha Gopalan, Yamini Elangovan, Sarath Chandra Mouli Veeravalli, Divya Tej Sowpati, and Kumarasamy Thangaraj. "Endogamy and high prevalence of deleterious mutations in India: evidence from strong founder events." Journal of Genetics and Genomics 52, no. 4 (2025): 570-582. [CrossRef]
- Malicdan, M.C., S. Noguchi, Y.K. Hayashi, I. Nonaka, and I. Nishino. “Prophylactic Treatment with Sialic Acid Metabolites Precludes the Development of the Myopathic Phenotype in the DMRV-hIBM Mouse Model.” Nat Med 15, no. 6 (2009): 690–95. [CrossRef]
- Malicdan, May Christine V., Satoru Noguchi, Yukiko K. Hayashi, Ikuya Nonaka, and Ichizo Nishino. "Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model." Nature medicine 15, no. 6 (2009): 690-695. [CrossRef]
- Manjunath, V., S.G. Thenral, B.R. Lakshmi, A.Bassi Atchayaram Nalini, K.Priya Karthikeyan, and K. Piyusha. “Large Region of Homozygous (ROH) Identified in Indian Patients with Autosomal Recessive Limb-Girdle Muscular Dystrophy with p. Thr182Pro Variant in SGCB Gene.” Human Mutation, no. 1 (2023): 4362273. [CrossRef]
- Mary, P., Laurent Servais, and Raphaël Vialle. “Neuromuscular Diseases: Diagnosis and Management.” Orthopaedics& Traumatology: Surgery & Research 104, no. 1 (2018): 89–95. [CrossRef]
- Mateos-Aierdi, A.J., M. Dehesa-Etxebeste, and M. Goicoechea. “Patient-Specific iPSC-Derived Cellular Models of LGMDR1.” Stem Cell Res 53, no. 102333 (2021). [CrossRef]
- Matthews, E., R. Brassington, T. Kuntzer, F. Jichi, and A.Y. Manzur. “Corticosteroids for the Treatment of Duchenne Muscular Dystrophy.” Cochrane Database Syst. Rev, 2016, 003725. [CrossRef]
- Mavrommatis, L., A. Zaben, and U. Kindler. “CRISPR/Cas9 Genome Editing in LGMD2A/R1 Patient-Derived Induced Pluripotent Stem and Skeletal Muscle Progenitor Cells.” Stem Cells Int 2023, no. 9246825 (2023). [CrossRef]
- McDonald, C.M., G. Sajeev, Z. Yao, E. McDonnell, G. Elfring, M. Souza, S.W. Peltz, et al. “Deflazacort vs Prednisone Treatment for Duchenne Muscular Dystrophy: A Meta-Analysis of Disease Progression Rates in Recent Multicenter Clinical Trials.” Muscle & Nerve 61, no. 1 (2020): 26–35. [CrossRef]
- Mendell, J.R., L.R. Rodino-Klapac, Z. Sahenk, K. Roush, L. Bird, L.P. Lowes, L. Alfano, et al. “Eteplirsen for the treatment of Duchenne muscular dystrophy.” Annals of neurology 74, no. 5 (2013): 637–47. [CrossRef]
- Mendell, Pozsgai, JR, Lewis ER, and S. “Gene Therapy with Bidridistrogenexeboparvovec for Limb-Girdle Muscular Dystrophy Type 2E/R4: Phase 1/2 Trial Results.” Nat Med 30, no. 1 (2024): 199–206. [CrossRef]
- Mercuri, E., F. Muntoni, and A.N. Osorio. “Safety and Effectiveness of Ataluren: Comparison of Results from the STRIDE Registry and CINRG DMD Natural History Study.” J Comp Eff Res 9 (2020): 341–60. [CrossRef]
- Mitrani-Rosenbaum, S., L. Yakovlev, and M. Becker Cohen. “Sustained Expression and Safety of Human GNE in Normal Mice after Gene Transfer Based on AAV8 Systemic Delivery.” NeuromusculDisord 22 (2012): 1015–24. [CrossRef]
- Mizobe, Y., S. Miyatake, H. Takizawa, Y. Hara, T. Yokota, A. Nakamura, S. Takeda, and Y. Aoki. “In Vivo Evaluation of Single-Exon and Multiexon Skipping in Mdx52 Mice.” Methods Mol. Biol, 2018, 275–92.
- Molaei, Negar, Parnian Alagha, Ali Khanbazi, Maryam Beheshtian, Fatemeh Ahangari, Shima Dehdahsi, Mahsa Fadaee et al. "Genetic spectrum among 2009 Iranian individuals with neuromuscular disorders using next generation sequencing and multiple ligation dependent probe amplification methods." Scientific Reports 15, no. 1 (2025): 42736. [CrossRef]
- Morales, E.D., Y. Yue, and T.B. Watkins. “Dwarf Open Reading Frame (DWORF) Gene Therapy Ameliorated Duchenne Muscular Dystrophy Cardiomyopathy in Aged Mdx Mice.” J Am Heart Assoc 12, no. 3 (2023). [CrossRef]
- Mori-Yoshimura, M., N. Suzuki, and M. Katsuno. “Efficacy Confirmation Study of Aceneuramic Acid Administration for GNE Myopathy in Japan.” Orphanet J Rare Dis 18 (2023): 241. [CrossRef]
- Mullen, Jeffrey, Khalid Alrasheed, and Tahseen Mozaffar. "GNE myopathy: History, etiology, and treatment trials." Frontiers in Neurology 13 (2022): 1002310. [CrossRef]
- Muni-Lofra, Robert, Eduard Juanola-Mayos, Marianela Schiava, Dionne Moat, Maha Elseed, Jassi Michel-Sodhi, and Elizabeth Harris. “Longitudinal Analysis of Respiratory Function of Different Types of Limb Girdle Muscular Dystrophies Reveals Independent Trajectories.” Neurology: Genetics 9, no. 4 (2023): 200084. [CrossRef]
- Murphy, Alexander Peter, and Volker Straub. “The Classification, Natural History and Treatment of the Limb Girdle Muscular Dystrophies.” Journal of Neuromuscular Diseases 2, no. s2 (2015): 7–19. [CrossRef]
- N, Elangkovan, and Dickson G. “Gene Therapy for Duchenne Muscular Dystrophy.” J Neuromuscul Dis 8:S303–S316 (2021).
- N, Lamb Y. “Givinostat: First Approval.” In Drugs, 10.1007/S40265-024-02052-1. Advance Online Publication, 2024. [CrossRef]
- Nagabushana, Divya, Kiran Polavarapu, Mainak Bardhan, Gautham Arunachal, Swetha Gunasekaran, Veeramani Preethish-Kumar, and Ram Murthy Anjanappa. “Comparison of the Carrier Frequency of Pathogenic Variants of DMD Gene in an Indian Cohort.” Journal of Neuromuscular Diseases 8, no. 4 (2021): 525–35. [CrossRef]
- Nallamilli, Babi Ramesh Reddy, Samya Chakravorty, Akanchha Kesari, Alice Tanner, Thomas Schneider ArunkanthAnkala, and Cristina Silva. “Genetic Landscape and Novel Disease Mechanisms from a Large LGMD Cohort of 4656 Patients.” Annals of Clinical and Translational Neurology 5, no. 12 (2018): 1574–87.
- Narayanaswami, P. “Evidence-Based Guideline Summary?: Diagnosis and Treatment of Limb-Girdle and Distal Dystrophies.” Neurology, 2014. [CrossRef]
- Navarro-Serna, S., A. Hachem, and A. Canha-Gouveia. “Generation of Nonmosaic, Two-Pore Channel 2 Biallelic Knockout Pigs in One Generation by CRISPR-Cas9 Microinjection Before Oocyte Insemination.” CRISPR J 4, no. 1 (2021): 132–46. [CrossRef]
- Nelson, B.R., C.A. Makarewich, and D.M. Anderson. “A Peptide Encoded by a Transcript Annotated as Long Noncoding RNA Enhances SERCA Activity in Muscle.” Science 351, no. 6270 (2016): 271–75. [CrossRef]
- Neu, Carolin T., Linus Weilepp, Kaya Bork, Astrid Gesper, and Rüdiger Horstkorte. "GNE deficiency impairs Myogenesis in C2C12 cells and cannot be rescued by ManNAc supplementation." Glycobiology 34, no. 3 (2024): cwae004. [CrossRef]
- Nigro, G., L.I. Comi, L. Politano, and R.J. Bain. “The Incidence and Evolution of Cardiomyopathy in Duchenne Muscular Dystrophy.” Int. J. Cardiol 26 (1990): 271–77. [CrossRef]
- Noguchi, S., Y. Keira, and K. Murayama. “Reduction of UDP-N-Acetylglucosamine 2-Epimerase/N-Acetylmannosamine Kinase Activity and Sialylation in Distal Myopathy with Rimmed Vacuoles.” J Biol Chem 279, no. 12 (2004): 11402–7. [CrossRef]
- Ousterout, D.G., A.M. Kabadi, P.I. Thakore, W.H. Majoros, T.E. Reddy, and C.A. Gersbach. “Multiplex CRISPR/Cas9-Based Genome Editing for Correction of Dystrophin Mutations That Cause Duchenne Muscular Dystrophy.” Nat. Commun 6 (2015): 6244. [CrossRef]
- Panda, G., and A. Ray. “Comparative Structural and Dynamics Study of Free and gRNA-Bound FnCas9 and SpCas9 Proteins.” Comput Struct Biotechnol J 20 (2022): 4172–84. [CrossRef]
- Panicucci, Chiara, Lizzia Raffaghello, Santina Bruzzone, Serena Baratto, Elisa Principi, Carlo Minetti, Elisabetta Gazzerro, and Claudio Bruno. "eATP/P2X7R axis: an orchestrated pathway triggering inflammasome activation in muscle diseases." International Journal of Molecular Sciences 21, no. 17 (2020): 5963. [CrossRef]
- Papiha, S.S. “Genetic Variation in India.” Human Biology, 1996, 607–28.
- Park, Y.E., J. Choi, L. Kim, E. Park, H. Go, and J. Shin. “A Pilot Trial for Efficacy Confirmation of 6’-Sialyllactose Supplementation in GNE Myopathy: Randomized, Placebo-Controlled Trial.” Mol Genet Metab 144, no. 1 (2025). [CrossRef]
- Patel, Alpesh, Shaishav Shah, Shiva Shankaran Chettiar, Devendrasinh Jhala, Siddharth Shah, and Alpesh Patel. “A Novel Founder Mutation in the SGCB Gene (Exon 5) Causes Limb Girdle Muscular Dystrophy (LGMDR4.” In Sathwara Community. Gujarat, India, n.d. [CrossRef]
- Pertusati, F., and J. Morewood. “Synthesis of 2-Acetamido-1,3,4-Tri-o-Acetyl-2-Deoxy-d-Mannopyranose −6-Phosphate Prodrugs as Potential Therapeutic Agents.” Curr Protoc 2022, no. 2 (n.d.). [CrossRef]
- Piccolo, F., Marc Jeanpierre, F. Leturcq, C. Dode, K. Azibi, A. Toutain, L. Merlini et al. "A founder mutation in the γ-sarcoglycan gene of Gypsies possibly predating their migration out of India." Human molecular genetics 5, no. 12 (1996): 2019-2022. [CrossRef]
- Polavarapu, Kiran, Aradhna Mathur, Aditi Joshi, Saraswati Nashi, Veeramani Preethish-Kumar, Mainak Bardhan, and Pooja Sharma. “A Founder Mutation in the GMPPB Gene [c. 1000G> A (p. Asp334Asn)] Causes a Mild Form of Limb-Girdle Muscular Dystrophy/Congenital Myasthenic Syndrome (LGMD/CMS) in South Indian Patients.” Neurogenetics 22 (2021): 271–85. [CrossRef]
- Polavarapu, Kiran, Veeramani Preethish-Kumar, Deepha Sekar, Seena Vengalil, Saraswati Nashi, Niranjan P. Mahajan, and Priya Treesa Thomas. “Mutation Pattern in 606 Duchenne Muscular Dystrophy Children with a Comparison between Familial and Non-Familial Forms: A Study in an Indian Large Single-Center Cohort.” Journal of Neurology 266 (2019): 2177–85. [CrossRef]
- Prieto, Sergio, and Josep M. Grau. “The geoepidemiology of autoimmune muscle disease.” Autoimmunity reviews 9, no. 5 (2010): 330–34. [CrossRef]
- Quattrocelli, Mattia, Aaron S. Zelikovich, Isabella M. Salamone, Julie A. Fischer, and Elizabeth M. McNally. "Mechanisms and clinical applications of glucocorticoid steroids in muscular dystrophy." Journal of neuromuscular diseases 8, no. 1 (2021): 39-52. [CrossRef]
- Quattrocelli, Mattia, Isabella M. Salamone, Patrick G. Page, James L. Warner, Alexis R. Demonbreun, and Elizabeth M. McNally. "Intermittent glucocorticoid dosing improves muscle repair and function in mice with limb-girdle muscular dystrophy." The American Journal of Pathology 187, no. 11 (2017): 2520-2535. [CrossRef]
- R, Bou Akar, Lama C, and Aubin D. “Generation of Highly Pure Pluripotent Stem Cell-Derived Myogenic Progenitor Cells and Myotubes.” Stem Cell Reports 19, no. 1 (2024): 84–99. [CrossRef]
- Raben, N., A. Wong, E. Ralston, and R. Myerowitz. “Autophagy and Mitochondria in Pompe Disease: Nothing Is so New as What Has Long Been Forgotten.” Am J Med Genet Part C, 2012. [CrossRef]
- Raffaghello, Lizzia, Elisa Principi, Serena Baratto, Chiara Panicucci, Sara Pintus, Francesca Antonini, Genny Del Zotto et al. "P2X7 receptor antagonist reduces fibrosis and inflammation in a mouse model of alpha-sarcoglycan muscular dystrophy." Pharmaceuticals 15, no. 1 (2022): 89. [CrossRef]
- Reelfs, Anna M., Carrie M. Stephan, Shelley R.H. Mockler, Katie M. Laubscher, M.Bridget Zimmerman, and Katherine D. Mathews. “Pain Interference and Fatigue in Limb-Girdle Muscular Dystrophy R9.” Neuromuscular Disorders 33, no. 6 (2023): 523–30. [CrossRef]
- Russell, Alan J., Mike DuVall, Ben Barthel, Ying Qian, Angela K. Peter, Breanne L. Newell-Stamper, Kevin Hunt et al. "Modulating fast skeletal muscle contraction protects skeletal muscle in animal models of Duchenne muscular dystrophy." The Journal of clinical investigation 133, no. 10 (2023). [CrossRef]
- Saad, F.A., J.F. Saad, G. Siciliano, L. Merlini, and C. Angelini. “Duchenne Muscular Dystrophy Gene Therapy.” Curr. Gene Ther, 2022. [CrossRef]
- Sahasrabudhe, S.A., M.R. Terluk, and R.V. Kartha. “N-Acetylcysteine Pharmacology and Applications in Rare Diseases-Repurposing an Old Antioxidant.” Antioxidants (Basel Jun 21;12(7):1316 (2023). [CrossRef]
- Sahenk, Z., B. Ozes, and D. Murrey. “Systemic Delivery of AAVrh74.tMCK.hCAPN3 Rescues the Phenotype in a Mouse Model for LGMD2A/R1.” Mol Ther Methods Clin Dev 22 (2021): 401–14. [CrossRef]
- Şahin, İzem Olcay, Yusuf Özkul, and Munis Dündar. "Current and future therapeutic strategies for limb girdle muscular dystrophy type R1: Clinical and experimental approaches." Pathophysiology 28, no. 2 (2021): 238-249. [CrossRef]
- Salama, Ilan, Stephan Hinderlich, Zipora Shlomai, Iris Eisenberg, Sabine Krause, Kevin Yarema, Zohar Argov et al. "No overall hyposialylation in hereditary inclusion body myopathy myoblasts carrying the homozygous M712T GNE mutation." Biochemical and biophysical research communications 328, no. 1 (2005): 221-226. [CrossRef]
- Salih, Mustafa A.M., and Peter B. Kang. “Hereditary and Acquired Myopathies.” Clinical Child Neurology, 2020, 1281–1349.
- Samarasiri, Udari. “Limb-girdle muscular dystrophies: An update,” 2023. [CrossRef]
- Schneider, J.S., M. Shanmugam, and J.P. Gonzalez. “Increased Sarcolipin Expression and Decreased Sarco(Endo)Plasmic Reticulum Ca2+ Uptake in Skeletal Muscles of Mouse Models of Duchenne Muscular Dystrophy.” J Muscle Res Cell Motil 34, no. 5–6 (2013): 349–56. [CrossRef]
- Sharma, S., P. Chanana, R. Bharadwaj, S. Bhattacharya, and R. Arya. “Functional Characterization of GNE Mutations Prevalent in Asian Subjects with GNE Myopathy, an Ultra-Rare Neuromuscular Disorder.” Biochimie 199 (2022): 36–45. [CrossRef]
- Sheth, Jayesh, Aadhira Nair, Frenny Sheth, Manali Ajagekar, Tejasvi Dhondekar, Inusha Panigrahi, and Ashish Bavdekar. “Burden of Rare Genetic Disorders in India: Twenty-Two Years’ Experience of a Tertiary Centre.” Orphanet Journal of Rare Diseases 19, no. 1 (2024): 295. [CrossRef]
- Siciliano, Gabriele, Costanza Simoncini, Stefano Giannotti, Virna Zampa, Corrado Angelini, and Giulia Ricci. "Muscle exercise in limb girdle muscular dystrophies: pitfall and advantages." Acta Myologica 34, no. 1 (2015): 3.
- Siemionow, M., J. Cwykiel, A. Heydemann, J. Garcia-Martinez, K. Siemionow, and E. Szilagyi. “Creation of Dystrophin Expressing Chimeric Cells of Myoblast Origin as a Novel Stem Cell Based Therapy for Duchenne Muscular Dystrophy.” Stem Cell Rev Rep 14 (2018): 189–99. [CrossRef]
- Siemionow, M., K. Budzynska, and K. Zalants. “Amelioration of Morphological Pathology in Cardiac, Respiratory, and Skeletal Muscles Following Intraosseous Administration of Human Dystrophin Expressing Chimeric (DEC) Cells in Duchenne Muscular Dystrophy Model.” Biomedicines 12, no. 3 (2024). [CrossRef]
- Siemionow, M., P. Langa, and S. Brodowska. “Long-Term Protective Effect of Human Dystrophin Expressing Chimeric (DEC.” Cell Therapy on Amelioration of Function of Cardiac, Respiratory and Skeletal Muscles in Duchenne Muscular Dystrophy [Published Correction Appears in Stem Cell Rev Rep Feb;19(2):582-583 (2023). [CrossRef]
- Singh, Reema, and Ranjana Arya. "GNE myopathy and cell apoptosis: a comparative mutation analysis." Molecular neurobiology 53, no. 5 (2016): 3088-3101. [CrossRef]
- Sparks, Susan, Goran Rakocevic, Galen Joe, Irini Manoli, Joseph Shrader, Michael Harris-Love, Barbara Sonies et al. "Intravenous immune globulin in hereditary inclusion body myopathy: a pilot study." BMC neurology 7, no. 1 (2007): 3. [CrossRef]
- Speciale, Alfina A., Ruth Ellerington, Thomas Goedert, and Carlo Rinaldi. "Modelling neuromuscular diseases in the age of precision medicine." Journal of personalized medicine 10, no. 4 (2020): 178. [CrossRef]
- Srinivasan, M., and C.Circadian Clock Walker. “Glucocorticoids and NF-κB Signaling in Neuroinflammation- Implicating Glucocorticoid Induced Leucine Zipper as a Molecular Link.” ASN Neuro 14 (2022): 175909142211201.
- Srivastava, A., E.W. Lusby, and K.I. Berns. “Nucleotide Sequence and Organization of the Adeno-Associated Virus 2 Genome.” J. Virol 45 (1983): 555–64. [CrossRef]
- Stadelmann, C., S. Francescantonio, A. Marg, S. Müthel, S. Spuler, and H. Escobar. “mRNA-Mediated Delivery of Gene Editing Tools to Human Primary Muscle Stem Cells.” Mol Ther Nucleic Acids 28 (2022): 47–57. [CrossRef]
- Sun, C., L. Shen, Z. Zhang, and X. Xie. “Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update.” Genes (Basel 11, no. 8 (2020): 1–25. [CrossRef]
- Suthar, R., S. Kesavan, I.K. Sharawat, M. Malviya, T. Sirari, B.K. Sihag, A.G. Saini, V. Jyothi, and N. Sankhyan. “The Expanding Spectrum of Dystrophinopathies: HyperCKemia to Manifest Female Carriers.” J. Pediatr. Neurosci 16 (2021): 206–11. [CrossRef]
- Suzuki, N., M. Mori-Yoshimura, and M. Katsuno. “Phase II/III Study of Aceneuramic Acid Administration for GNE Myopathy in Japan.” J Neuromuscul Dis 10, no. 4 (2023): 555–66. [CrossRef]
- Suzuki, Naoki, Madoka Mori-Yoshimura, Masahisa Katsuno, Masanori P. Takahashi, Satoshi Yamashita, Yasushi Oya, Atsushi Hashizume, et al. “Safety and Efficacy of Aceneuramic Acid in GNE Myopathy: Open-Label Extension Study.” Journal of Neurology, Neurosurgery, and Psychiatry, 2024. [CrossRef]
- Suzuki, Naoki, Masaaki Kato, Hitoshi Warita, Rumiko Izumi, Maki Tateyama, Hiroshi Kuroda, Ryuta Asada et al. "Phase I clinical trial results of aceneuramic acid for GNE myopathy in Japan." Translational Medicine Communications 3, no. 1 (2018): 7. [CrossRef]
- Sveen, Marie-Louise, Søren P. Andersen, Lina H. Ingelsrud, Sarah Blichter, Niels E. Olsen, Simon Jønck, Thomas O. Krag, and John Vissing. “Resistance Training in Patients with Limb-girdle and Becker Muscular Dystrophies.” Muscle & Nerve 47, no. 2 (2013): 163–69. [CrossRef]
- Swiderski, Kristy, and Gordon S. Lynch. "Translational progress in the development of pharmacotherapies for Duchenne muscular dystrophy." Regenerative Medicine 20, no. 10 (2025): 489-500. [CrossRef]
- Syed, Y.Y. “Eteplirsen: first global approval.” Drugs 76 (2016): 1699–1704. [CrossRef]
- Tanihara, F., M. Hirata, N.T. Nguyen, Q.A. Le, M. Wittayarat, M. Fahrudin, and T. Otoi. “Generation of CD163-edited pig via electroporation of the CRISPR/Cas9 system into porcine in vitro-fertilized zygotes.” Animal Biotechnology 32, no. 2 (2019): 147–54. [CrossRef]
- Tasfaout, H., C.L. Halbert, and T.S. McMillen. “Split Intein-Mediated Protein Trans-Splicing to Express Large Dystrophins.” Nature 632, no. 8023 (2024): 192–200. [CrossRef]
- Tasfaout, H., C.L. Halbert, and T.S. McMillen. “Split Intein-Mediated Protein Trans-Splicing to Express Large Dystrophins.” Nature 632, no. 8023 (2024): 192–200. [CrossRef]
- Tidball, James G., Steven S. Welc, and Michelle Wehling-Henricks. “Immunobiology of Inherited Muscular Dystrophies.” Comprehensive Physiology 8, no. 4 (2018): 1313–56.
- Tominari, T., and Y. Aoki. “Clinical Development of Novel Therapies for Duchenne Muscular Dystrophy—Current and Future Neurology and Clinical Neuroscience,” 2023.
- Turken, Askeri, Maria Luckas, Vatan Kavak, Ömer Satıcı, and Dildan Kavak. “The effects of home exercise program on limb-girdle disease: a cohort study.” Folia Neuropathologica 60, no. 1 (2022): 48–59. [CrossRef]
- Tycko, J., V.E. Myer, and P.D. Hsu. “Methods for Optimizing CRISPR-Cas9 Genome Editing Specificity.” Molecular Cell 63, no. 3 (2016): 355–70. [CrossRef]
- Urtizberea, J. Andoni, and France Leturcq. "Limb girdle muscular dystrophies: The clinicopathological viewpoint." Annals of Indian Academy of Neurology 10, no. 4 (2007): 214-224. [CrossRef]
- V., Derbyshire, and Belfort M. “Lightning Strikes Twice: Intron–Intein Coincidence.” Proc. Natl. Acad. Sci. USA 95 (1998): 1356–57. [CrossRef]
- V., Mariot, Joubert R., Hourdé C., Féasson L., Hanna M., Muntoni F., Maisonobe T., Servais L., Bogni C., and Le Panse R. “Downregulation of Myostatin Pathway in Neuromuscular Diseases May Explain Challenges of Anti-Myostatin Therapeutic Approaches.” Nat. Commun 8 (2017): 1–8. [CrossRef]
- Veerapandiyan, Aravindhan. "Early Insights on 3 Patients With DMD From Trial Assessing Gene Therapy RGX-202: Aravindhan Veerapandiyan, MD." Neurology Live (2023): NA-NA.
- Verhaart, I.E., R.J. Duijn, B. Adel, A.A. Roest, J.J. Verschuuren, A. Aartsma-Rus, and L. Weerd. “Assessment of Cardiac Function in Three mouseDystrophinopathies by Magnetic Resonance Imaging.” Neuromuscul. Disord 22 (2012): 418–26. [CrossRef]
- Verhaart, I.E., R.J. Duijn, B. Adel, A.A. Roest, J.J. Verschuuren, A. Aartsma-Rus, and L. Weerd. “Assessment of Cardiac Function in Three mouseDystrophinopathies by Magnetic Resonance Imaging.” Neuromuscul. Disord 22 (2012): 418–26. [CrossRef]
- Viviano, K.R.Glucocorticoids, and Azathioprine Cyclosporine. “Chlorambucil, and Mycophenolate in Dogs and Cats: Clinical Uses, Pharmacology, and Side Effects.” Vet. Clin. N. Am. Small Anim. Pract 52 (2022): 797–817.
- Vrist, Louise T.H., Lone F. Knudsen, and Charlotte Handberg. “‘It Becomes the New Everyday Life’–Experiences of Chronic Pain in Everyday Life of People with Limb-Girdle Muscular Dystrophy.” Disability and Rehabilitation 45, no. 23 (2023): 3875–82.
- Wang, J., Y. Zhang, C.A. Mendonca, O. Yukselen, K. Muneeruddin, L. Ren, J. Liang, et al. “AAV-delivered suppressor tRNA overcomes a nonsense mutation in mice.” Nature 604, no. 7905 (2022): 343–48. [CrossRef]
- Wang, X., C. Tokheim, and S.S. Gu. “In Vivo CRISPR Screens Identify the E3 Ligase Cop1 as a Modulator of Macrophage Infiltration and Cancer Immunotherapy Target.” Cell 184, no. 21 (2021): 5357–74. [CrossRef]
- Wang, Zhonglei, Xin Sun, Mingyu Sun, Chao Wang, and Liyan Yang. "Game Changers: Blockbuster Small-Molecule Drugs Approved by the FDA in 2024." Pharmaceuticals 18, no. 5 (2025): 729. [CrossRef]
- Wasala, L.P. “The Implication of Hinge 1 and Hinge 4 in Microdystrophin Gene Therapy for Duchenne Muscular Dystrophy.” Hum. Gene Ther 34 (2023): 459–70. [CrossRef]
- Wei, T., Y. Sun, and Q. Cheng. “Lung SORT LNPs Enable Precise Homology-Directed Repair Mediated CRISPR/Cas Genome Correction in Cystic Fibrosis Models.” Nat Commun 14, no. 1 (2023). [CrossRef]
- Weinmann, J. “Identification of a Myotropic AAV by Massively Parallel in Vivo Evaluation of Barcoded Capsid Variants.” Nat. Commun 11 (2020): 5432. [CrossRef]
- Wicklund, M.P. “Limb-Girdle Muscular Dystrophies.” In Encyclopedia of the Neurological Sciences, 2014. [CrossRef]
- Willis, Alexander B., Aaron S. Zelikovich, Robert Sufit, Senda Ajroud-Driss, Krista Vandenborne, Alexis R. Demonbreun, Abhinandan Batra, Glenn A. Walter, and Elizabeth M. McNally. "Serum protein and imaging biomarkers after intermittent steroid treatment in muscular dystrophy." Scientific reports 14, no. 1 (2024): 28745. [CrossRef]
- Xiao-Jie, Lu, Xue Hui-Ying, Ke Zun-Ping, Chen Jin-Lian, and Ji Li-Juan. “CRISPR-Cas9: A New and Promising Player in Gene Therapy.” Journal of Medical Genetics 52, no. 5 (2015): 289–96. [CrossRef]
- Xu, X., A.Q. Wang, and L.L. Latham. “Safety, Pharmacokinetics and Sialic Acid Production after Oral Administration of N-Acetylmannosamine (ManNAc) to Subjects with GNE Myopathy.” Mol Genet Metab 122, no. 1–2 (2017): 126–34. [CrossRef]
- Yiu, Eppie M., and Andrew J. Kornberg. “Duchenne Muscular Dystrophy.” Journal of Paediatrics and Child Health 51, no. 8 (2015): 759–64.
- Yonekawa, T., M.C. Malicdan, and A. Cho. “Sialyllactose ameliorates myopathic phenotypes in symptomatic GNE myopathy model mice.” Brain 137, no. Pt 10 (2014): 2670–79. [CrossRef]
- Zabłocki, Krzysztof, and Dariusz C. Górecki. "The role of P2X7 purinoceptors in the pathogenesis and treatment of muscular dystrophies." International Journal of Molecular Sciences 24, no. 11 (2023): 9434. [CrossRef]
- Zelikovich, A.S., B.C. Joslin, P. Casey, E.M. McNally, and S. Ajroud-Driss. “An Open Label Exploratory Clinical Trial Evaluating Safety and Tolerability of Once-Weekly Prednisone in Becker and Limb-Girdle Muscular Dystrophy.” J Neuromuscul Dis 9, no. 2 (2022): 275–87. [CrossRef]
- Zelikovich, Aaron S., Benjamin C. Joslin, Patricia Casey, Elizabeth M. McNally, and Senda Ajroud-Driss. "An open label exploratory clinical trial evaluating safety and tolerability of once-weekly prednisone in becker and limb-girdle muscular dystrophy." Journal of neuromuscular diseases 9, no. 2 (2022): 275-287. [CrossRef]
- Zhang, J. “Genetic Analysis of 62 Chinese Families with Duchenne Muscular Dystrophy and Strategies of Prenatal Diagnosis in a Single Center.” BMC Med. Genet, 2019. [CrossRef]
- Zhang, S. “Genotype Characterization and Delayed Loss of Ambulation by Glucocorticoids in a Large Cohort of Patients with Duchenne Muscular Dystrophy.” Orphanet. J. Rare Dis, 2021. [CrossRef]
- Zhang, Yu, Takahiko Nishiyama, Eric N. Olson, and Rhonda Bassel-Duby. “CRISPR/Cas Correction of Muscular Dystrophies.” Experimental Cell Research 408, no. 1 (2021): 112844. [CrossRef]
- Zhou, Y., C. Zhang, and W. Xiao. “Systemic Delivery of Full-Length Dystrophin in Duchenne Muscular Dystrophy Mice.” Nat Commun 15 (2024): 6141. [CrossRef]
- Zídková, Jana, Tereza Kramářová, Johana Kopčilová, Kamila Réblová, Jana Haberlová, Radim Mazanec, Stanislav Voháňka et al. "Genetic findings in Czech patients with limb girdle muscular dystrophy." Clinical Genetics 104, no. 5 (2023): 542-553. [CrossRef]
- Zygmunt, D.A., P. Lam, and A. Ashbrook. “Development of Assays to Measure GNE Gene Potency and Gene Replacement in Skeletal Muscle.” J Neuromuscul Dis 10, no. 5 (2023): 797–812. [CrossRef]
Figure 1.
Schematic representation of effect of normal and mutated dystrophin proteins on muscle integrity and clinical phenotype. (A) Wild-type dystrophin protein maintains sarcolemma stability while (B) mutations trigger progressive muscle dysfunction.
Figure 1.
Schematic representation of effect of normal and mutated dystrophin proteins on muscle integrity and clinical phenotype. (A) Wild-type dystrophin protein maintains sarcolemma stability while (B) mutations trigger progressive muscle dysfunction.
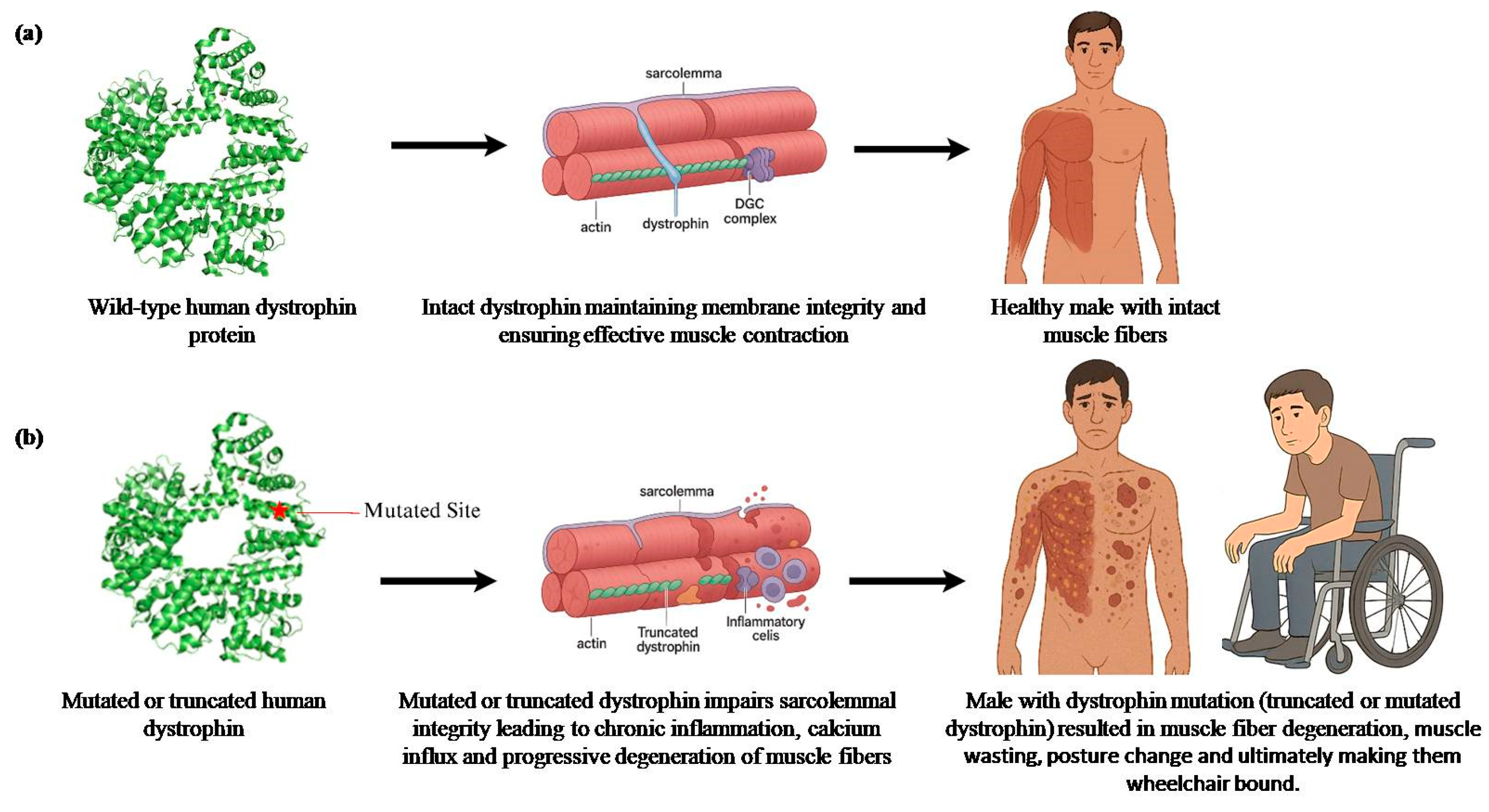
Figure 2.
Statistics of clinical trials of DMD, LGMD and GNEM. (a) Study status of clinical trials (completed/ongoing) (b) The study type of the trial (interventional/observational), (c) The phase of the completed/ongoing trial (d) The enrollment distribution of the trials.
Figure 2.
Statistics of clinical trials of DMD, LGMD and GNEM. (a) Study status of clinical trials (completed/ongoing) (b) The study type of the trial (interventional/observational), (c) The phase of the completed/ongoing trial (d) The enrollment distribution of the trials.
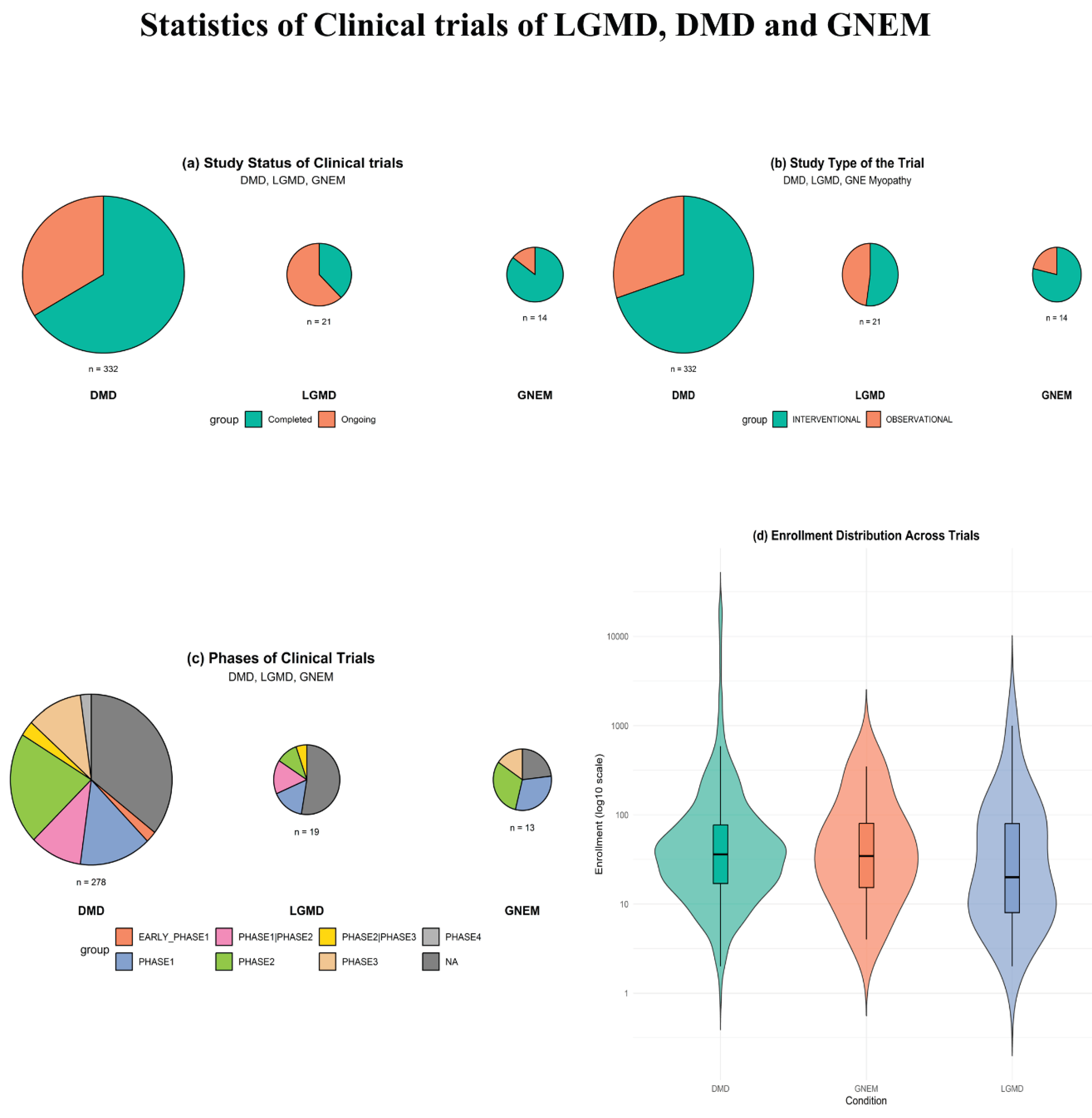
Table 1.
LGMD Founder mutations catalogued so far in Indian populations.
| Mutation | Mutation Type | Gene | LGMD type | Community | Reference |
|---|---|---|---|---|---|
| c.1000G > A | Missense | GMPPB (GDP-mannose pyrophosphorylase B) | LGMDR23 | South Indian | Polavarapu et al., 2021 [32] |
| intron 18/exon 19 c.2051-1G>T | Missense | CAPN3 | LGMD2A/R1 | Agarwal Community | Khadilkar et al., 2016 [125] |
| exon 22 c.2338G>C | Missense | CAPN3 | LGMD2A/R1 | Agarwal Community | Khadilkar et al., 2016 [125] |
| c.2051–1G > T and c.2338G > C in 9.7%, c.1343G > A, c.802–9G > A, and c.1319G > A | Missense | CAPN3 | LGMD2A/R1 | South Indian | Ganaraja et al., 2022 [127] |
| Chr 4:52894204C>T; | Missense | SGCB | LGMD2E/R4 | Sathwara | Patel et al., 2024 [132] |
| EMD c.654_658dup*, | duplication | LMNA | LGMD1B | Not mentioned | Baskar et al., 2024 [133] |
| c.847G>A | Missense | SGCG | LGMD2C/R5 | Gypsies (Indian origin community) | Piccolo et al., 1996 [134] |
| SGCB c.544A>C | Missense | SGCB | LGMD2E/R4 | 11 different states in India and 1 individual from Bangladesh | Bardhan et al., 2022 [135] |
| Not available | NA | CAPN3 | LGMDR1/2A | Mumbai | Khadilkar et al., 2022 [136] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



