Submitted:
03 December 2025
Posted:
04 December 2025
You are already at the latest version
Abstract
Cellular prion protein (PrPC) is a host protein anchored to the outer surface of neurons and, to a lesser extent, to lymphocytes. The transmissible agent (PrPSc), the causative agent for scrapie, is believed to be a modified form of PrPC. However, the role of PrPC in normal, uninfected animals remains unknown. Here, we describe a novel mouse model for dilated cardiomyopathy (DCM), which was observed during the development of pri-on-agent-susceptible transgenic mice (Tg mice). Tg mice were shown to selectively express scimitar-horned oryx (Oryx dammah) PrP (OrPrP) in their tissues, as demonstrated through RT-PCR for mRNA, Western blotting, and immunohistochemistry. High levels of OrPrP expression were observed in the heart, medium levels in the skeletal muscle, low levels in the brain, and barely detectable levels in the lung, liver, spleen, kidney, and thymus. Elec-trocardiogram (ECG) findings showed significantly prolonged cardiac muscle contraction, with abnormalities in the QRS interval. Additionally, histological analysis showed multi-ple intracytoplasmic vacuolation in the heart muscle. Furthermore, an abnormal ECG waveform of was observed in Tg mice following atropine injection. These mice may pro-vide a new model for analyzing experimental DCM. Besides, the Tg mice showed higher expression levels of OrPrP in the heart muscle following increased Doppel (Dpl) gene ex-pression. Overexpression of Dpl gene may occur in the myocardium, controlled by OrPrP. These findings suggest that higher expression of OrPrP and Dpl gene in the heart may cause cardiac abnormalities. In the histopathological studies of myocardium, the cyto-plasmic vacuolation of muscle fibers were seen both in Tg(OrPrP)Prnp+/+ and Tg(OrPrP)Prnp0/0 mice, but not in C57BL/6J mice.
Keywords:
cardiomyopathy
; Doppel-gene
; prion protein
; prionoid
; scimitar-horned oryx (Oryx dammah)
; transgenic mouse
1. Introduction
In transmissible spongiform encephalopathies (TSE), such as scrapie or bovine spongiform encephalopathy (BSE) in animals and Creutzfeldt-Jakob disease (CJD) in humans, the prions appear to be devoid of nucleic acid and are identical to the scrapie isoform of the prion protein (PrPSc), a modified form of the normal cellular isoform PrP (PrPC) [1,2]. PrPC may play physiological roles, as its gene expression has been observed in the brain and other tissues of all mammals. Additionally, PrPC is believed to convert to PrPSc during the course of prion infection through a posttranslational mechanism. However, the role of PrPC in uninfected animals is poorly understood. Notably, a previous study reported that PrPC overexpression results in the formation of PrPSc-like abnormal lesions [3]. The study found that uninoculated older mice expressing high copy numbers of murine PrP developed truncal ataxia and tremors. This suggests that elevated PrP expression is associated with neurologic disease. Therefore, higher PrP expression levels may lead to tissue abnormalities and vacuolation.
Cardiomyopathy manifests as dyspnea, cardiac failure, or sudden death, causing serious morbidity and mortality. Furthermore, although cardiomyopathy has several possible triggers, such as viral infections or alcohol abuse, multiple causes are expected. Clinical features and molecular genetic studies on cardiomyopathy indicate various possible genetic causes of this disease; however, the causative genes and pathogenesis are poorly understood [4]. The lack of a suitable animal model has severely limited physicians’ ability to understand the causes and mechanisms of this disease and to develop novel, more effective treatments. Therefore, the animal cardiomyopathy model is highly valuable for understanding the pathological process and developing novel therapies as well as pharmacological interventions [5]. Additionally, the association between prion disease and cardiomyopathy has not been reported in the literature and remains unclear. There is a report that shows PrPSc growth in skeletal muscle of hamsters orally infected with scrapie [6]. However, cardiomyopathy and neuromuscular abnormalities may coexist [7]. Furthermore, neurodegenerative diseases, such as Friedreich’s ataxia (FA) and Parkinson’s disease (PD), are frequently associated with cardiomyopathy [8]. Here we describe a novel mouse model for cardiomyopathy, which was observed during the development of a prion agent sensitive transgenic mouse. Particularly, it was expected that the Tg mice express scimitar-horned oryx PrP (OrPrP) ubiquitously, including in nerve cells, immune cells, and skeletal muscles, using the β-actin promoter.
2. Materials and Methods
2.1. Mice
Rikn Prnp0/0 mice were provided from RIKEN and crossed with Tg(OrPrP)Prnp+/+ mice to obtain Tg(OrPrP), devoid of the mouse (Mo) PrP gene. Briefly, the entire PrP open reading frame (ORF) and 3’–end of the second intron were replaced with the pgk-neo gene cassette. Prnp0/0 mice showed no obvious behavioral changes at a young age, although they showed neurophysiological disorder and ataxia when they aged [9]. C57BL/6J (B6) mice were used to have the results of untreated control mice.
2.2. Total DNA Isolation and Non-Radioactive Southern Blot Screening of Transgenes
A tail snip from a living mouse was used for genomic DNA extraction. After cutting off 1–2 cm tail clips from the mouse, the tail was incubated in tail lysis buffer containing 50 mM Tris-HCl (pH 8.0), 1 % sodium dodecyl sulfate (SDS), 100 mM ethylenediaminetetraacetic acid (EDTA) (pH8.0), 100 mM NaCl and 2 mg/ml proteinase K for about 5 hours (hr) at 55 °C to digest it. After phenol chloroform extraction, the aqueous phase was transferred into a new tube and mixed with an equal volume of isopropanol by inverting several times. DNA was resolved with Tris-EDTA (TE) and determined DNA concentration and purity by measurement of absorbance at 260 nm and 280 nm.
DNA (20 μg) for Southern blots was first cut with restriction enzymes, BamHI and EcoRI and electrophoresed using 1.0 % agarose gel. After electrophoresis, the gel was depurinated by soaking it for 10 minutes (min) in 50 mM HCl. DNA was transferred to the nitro-cellulose filter (Hybond N+) by traditional capillary blotting. The blot was prehybridized for 30 min and hybridized overnight at 60 °C in hybridization buffer, 5×saline-sodium citrate (SSC), 0.1 % SDS, 5 % dextran sulphate, 0.5 % blocking reagent (Amersham Corp., U.K.) and 15 ng/ml of Fluorescein-dUTP (FI-dUTP) (Amersham Corp., U.K.) labeled probe by using a rolling bottle incubator. After hybridization, the blot was washed with wash solution, 0.5×SSC and 0.1 % SDS three times for 30 min at 60 °C. For detecting the probe-target hybrids, the horseradish peroxidase (HRP) conjugated anti-fluorescein antibody was used. Detective reaction was performed using an enhanced chemiluminescence (ECL) detection system according to the manufacture’s instruction (Amersham Corp., U.K.).
2.3. Total RNA Isolation and Reverse Transcriptase–Polymerase Chain Reaction (RT-PCR) of OrPrP mRNAs
Frozen tissues were homogenized in TRIzol Reagent (Life Technologies, Rockville, MD). The total RNA was extracted according to the manufacturer’s specifications, precipitated, and dissolved in RNase-free water. First-strand cDNA was synthesized from 1 µg of total RNA using avian myeloblastosis virus (AMV) Reverse Transcriptase (Promega, Madison, WI). Taq DNA polymerase was used for second-strand cDNA synthesis and DNA amplification [10]. Primer sets were as follows: The PCR product with primer specific to OrPrP cDNAs were forward 5'–AGAATTCATGGTGAAAAGCCACATAGGCA–3' and reverse 5'–TCGCTCCATGATCTTGATGTCAGT–3' consisting of 0.6kb. Briefly, typical RT-PCR conditions were as follows: 37 °C for 60 minutes (min) (reverse transcription), 95 °C for 3 min (AMV inactivation and RNA / cDNA / hybrid denaturation), 32 cycles at 94 °C for 30 seconds (sec) (denaturation), 60 °C for 30 sec (annealing), 72 °C for 1 min (extension), 1 cycle at 72 °C for 20 min (final extension), and a final soak at 4 °C.
2.4. RT-PCR of Doppel (Dpl) mRNAs
Total RNA was isolated from brain and heart of Prnp+/+, Prnp0/0 and Tg(OrPrP)Prnp0/0 mice. Total RNA isolation and condition of reagents was carried out as described previously [11]. Primer sets were as follows: The PCR product with primer specific to Dpl cDNAs were forward 5'–GCGTCGACCGCCACCATGAAGAACCGGCTGGGTACATGG–3' and reverse 5'–CTCGTCGACCTCTGTGGCTGCCAGCTTCATTGA–3' consisting of 591 base pair (bp). RT-PCR conditions were as follows: 37 °C for 60 min (reverse transcription), 95 °C for 10 min (AMV inactivation and RNA / cDNA / hybrid denaturation), 35 cycles at 94 °C for 30 sec (denaturation), 62 °C for 30 sec (annealing), 72 °C for 2 min (extension), 1 cycle at 72 °C for 7 min (final extension), and a final soak at 4 °C.
2.5. Immunoprecipitation
The brains were homogenized and centrifuged at 1200 rpm for 5 min in sterile phosphate-buffered saline (PBS), and the resultant supernatant was precleared by centrifugation at 20,000×g for 60 min at 4 °C. For detection of PrP, monoclonal 6H4 (10 μg) mouse antibody (Prionics AG, Switzerland) was added to the samples and incubated with the samples at 4 °C for 1 hr. The antigen-antibody complexes were collected on Protein G beads (Amersham Pharmacia Biotech). After the supernatant was discarded, and the beads were washed three times with immunoprecipitation wash buffer (1 % Triton X-100, 0.5 % bovine serum albumin (BSA), 1 mM phenylmethylsulfonyl fluoride (PMSF) in PBS), the beads were mixed with SDS-sample buffer and boiled for 5 min. By Western blotting, the precipitated PrP were analyzed by 12 % polyacrylamide gels, followed by transfer to Polyvinylidene Difluoride (PVDF) membranes (Amersham Pharmacia Biotech) before detection by polyclonal P8 rabbit antibody directed against 92–109 mouse PrP sequence (1:2000 dilution) and anti-rabbit IgG horseradish peroxidase. The blot was developed using an ECL detection system (Amersham Corp., U.K.).
2.6. Electrocardiographic (ECG) Patterns in Tg Mice
ECG recording was performed as described previously [12]. ECG parameters (PR, QRS, and QT time) were obtained using an ML846 PowerLab System (AD Instrument, Dunedin, New Zealand). For the atropine treatment, 30 weeks old Tg(OrPrP)Prnp0/0 (n = 3) and B6 (n = 3) mice were injected with atropine (4 mg/kg intraperitoneal injection [i.p.]) and evaluated by ECG 5 minutes later.
2.7. Hematoxylin and Eosin (H&E) Staining and Immunohistochemistry
Tissue was fixed embedded in paraffin, and 7 µm thick sections were cut. Serial sections were subjected to H&E staining and immunohistological staining for polyclonal α-PrP (P8) rabbit antibody diluted at 1:200.
2.8. Footprint
The hind feet of 15 months old PrP+/+, Prnp0/0 and Tg(OrPrP)Prnp0/0 mice were dipped in ink and mice prompted to walk on the filter paper.
3. Results
Tg mice were used in this study. In a study by Prusiner et al., each pathogen from BSE and scrapie, variant CJD (vCJD) was introduced into Tg mice using the bovine PrP gene, with incubation periods of approximately 200 days [13]. The study found that the three types of pathogens were identical. Furthermore, Collinge et al. used a human PrP gene-transfected mouse model. In these human prion gene TG mice, vCJD appeared at approximately 220 days [14]. Using various animal prions, the incubation period of Tg mice is often shortened or prolonged. This species-barrier phenomenon of the PrP gene is not well studied.
It is well established that wild ruminants such as greater kudu (Tragelaphus strepsiceros) and oryx (Oryx dammah) have shorter incubation periods compared to livestock ruminants such as sheep and cattle; and disease progression following incubation is rapid [15,16]. Studies have reported notable species-level differences in the incubation time of prion diseases. To study disease progression in farm cattle, animals are neonatally infected. Sheep scrapie shows an incubation period of 36–48 month following experimental infection. Cattle experimentally infected with BSE agent show an incubation period of 36–72 months. Disease incidence occurred at 30–48 months of age for sheep scrapie, and 60–80 months for BSE. However, in oryx, disease incidence occurred at 30 months. The incubation period is assumed to be 21 months [15,16]. The incidence age of oryx is very early compared to cattle and sheep. Transmissibility of spongiform encephalopathy and incubation period are controlled by differences in the amino acid sequence of PrP. The gene encoding prion protein has been detected in many vertebrates, such as humans, sheep, cattle, mice, hamsters, oryx, and fishes. In mammals, over 90% amino acid sequence homology has been observed among species [9,10,11,17,18,19]. This suggests the presence of critical amino acid sequences controlling incubation periods and the age of incidence in OrPrP.
Transgenic technology is useful for analyzing the normal function of PrP and genetically contrasting susceptibility to agents. Although the chicken β-actin promoter and CMV enhancer are known to direct widespread gene expression, the transgene expression is not always ubiquitous [20].
3.1. Effect of PrP Genes in Tg Mice
A restricted pattern of OrPrP expression in the heart was observed at higher levels, medium levels in skeletal muscle, and lower levels in the brain. These expression patterns suggest that the localization of transgene expression may lead to sudden death of these mice (Figure 1 and Figure 2).
To detect weak pathological alterations, ECGs from 20- to 70-week-old Tg(OrPrP)Prnp0/0 mice were analyzed. Notably, abnormal electrocardiographic patterns were observed in Tg(OrPrP)Prnp0/0 mice; cardiac muscle contraction was significantly prolonged with an abnormal QRS interval (data not shown). The mice showed multiple intracytoplasmic vacuolations and elongation of muscle fibers in the heart muscle, and scar formation of muscle fibers was also observed. Abnormalities in the QRS waveform suggest that the excitability transfer into the ventricle from the atrium was blocked by injury caused by intracytoplasmic vacuolation. The intracytoplasmic vacuolation of the heart muscle was observed in both Tg(OrPrP)Prnp+/+ and Tg(OrPrP)Prnp0/0 mice (data not shown). Overexpression of OrPrP may cause intracytoplasmic vacuolation of the heart muscle. Although abnormalities in the heart muscle were detected in 30-week-old Tg(OrPrP)Prnp0/0 mice, other organs such as the brain, lung, liver, spleen, skeletal muscle, thymus, and kidney did not show any abnormalities (Figure 3 and Figure 4).
These results show that age-related intracytoplasmic vacuolation of muscle fibers in Tg(OrPrP)Prnp0/0 mice affected the amplitude of the QRS wave in ECGs, resulting from a loss of the electromotive force of the heart muscle. Although abnormal QRS waves can appear in ECGs, none of the Tg(OrPrP)Prnp0/0 mice exhibited the corresponding cardiac symptoms, such as heart failure or death. Previous studies provide an alternate perspective on cardiomyopathy for these findings, suggesting a genetic disorder in genes encoding proteins. Furthermore, the studies found that the hearts of young mice (6 weeks old) did not show any gross histological abnormalities, whereas those of older mice (over 20 weeks old) showed abnormalities such as fiber disarray and hyperdynamic contraction. Comparable results were obtained in the present study using ECGs and histopathology. All the older mice with histologic abnormalities showed delated cardiomyopathy (DCM), macroscopically. Atropine sulfate inhibits actions of acetylcholine at postganglionic parasympathetic neuroeffector sites. Atropine is primarily used to increase the heart rate in bradycardias. For another application, atropine is known as an indicative method of subtle change in heart muscle. In Tg(OrPrP)Prnp0/0 mice, an abnormal waveform of ECG was observed after atropine injection. These results suggest that Tg(OrPrP)Prnp0/0 mice are a useful model for understanding the causes and mechanisms of weaker forms of cardiomyopathy. Abnormal tissue in the heart of mice with PrP overexpression was not reported before. ECG waves were not confirmed in these PrP overexpressed mice (Figure 5).
3.2. Phenotypic Rescue of Dpl-Induced Ataxia by Co-Expression of OrPrP
Dpl mRNA levels were elevated in the brains of Prnp0/0 mice, which developed ataxia and Purkinje cell loss [9]. Using RT-PCR, Dpl mRNA was observed in the brain and heart of Prnp0/0 and Tg(OrPrP)Prnp0/0 mice; however, its expression was not detectable in wild-type B6 (Prnp+/+) mice (Figure 6a). Notably, Dpl mRNA levels of Tg(OrPrP)Prnp0/0 mice were enhanced in the heart muscle compared to Prnp0/0 mice. A weak signal of Dpl mRNA was detected in the heart muscle of Rikn Prnp0/0 mice (Figure 6a).
Prnp0/0 mice exhibited no symptoms until approximately 10 months of age, when they began to develop pronounced behavioral disorders. Prnp0/0 mice showed a coarse tremor and a staggering gait followed by paresis of the hind legs. Furthermore, Prnp0/0 mice were unable to walk straight and showed a generally reduced step length compared with Tg(OrPrP)Prnp0/0 and Prnp+/+ mice (Figure 6b).
Histological examination of 15-month-old mice showed that Prnp0/0, but not Tg(OrPrP)Prnp0/0 and Prnp+/+ mice, showed considerable pathological changes in the cerebellum (Figure 7a–c). No significant pathological abnormalities were detected in the brains of elderly Tg(OrPrP)Prnp0/0 mice. However, compared with Prnp+/+ mice, limited neurodegeneration was observed in the cerebellar Purkinje cells of Tg(OrPrP)Prnp0/0 mice (Figure 7a,c). The intensity of pathological change was weaker compared with Prnp0/0 mice (Figure 7b,c). These results suggest that the protective effect of OrPrP may be present in the cerebellum of 15-month-old Tg(OrPrP)Prnp0/0 mice. In all experiments, comparable results were obtained in littermates from two surviving founders.
In the histopathological studies of myocardium, the cytoplasmic vacuolation of muscle fibers were seen both in Tg(OrPrP)Prnp+/+ and Tg(OrPrP)Prnp0/0 mice, but not in B6 mice. In all experiments, the same results were obtained in littermates from two survived founders. Although the abnormalities in heart muscle were seen in 30 weeks old Tg(OrPrP)Prnp0/0 mice, other organs such as brain, lung, liver, spleen, skeletal muscle, thymus and kidney did not show any abnormality
4. Discussion
These results show that age-related intracytoplasmic vacuolation of muscle fibers in Tg(OrPrP)Prnp0/0 mice affected the amplitude of the QRS wave in ECGs, resulting from a loss of the electromotive force of the heart muscle. Although abnormal QRS waves can appear in ECGs, none of the Tg(OrPrP)Prnp0/0 mice exhibited the corresponding cardiac symptoms, such as heart failure or death. Previous studies provide an alternate perspective on cardiomyopathy for these findings, suggesting a genetic disorder in genes encoding proteins [5,21]. Furthermore, the studies found that the hearts of young mice (6 weeks old) did not show any gross histological abnormalities, whereas those of older mice (over 20 weeks old) showed abnormalities such as fiber disarray and hyperdynamic contraction. Comparable results were obtained in the present study using ECGs and histopathology. The transgenes (OrPrP) expressed at high levels in the heart and skeletal muscle, perhaps because the chicken β-actin promoter was efficient in mouse myoblast as reported previously [22]. Several previous studies suggest that higher level of transgene expression in the heart was seen without detectable ill effects even with minor cardiac pathology [23]. Ma et al. showed that wild-type PrPC showed neurotoxicity upon accumulation in the cytosol [24]. Misfolded PrP molecules were retrogradely transported to the cytosol for degradation by the proteasome [25]. Small amounts of OrPrP may be toxic to heart muscle. Proteosomal degradation prevents the toxic effects of OrPrP following its accumulation. However, when the ability of the proteasome to degrade PrP is compromised due to aging, the increase in cytosolic PrP can damage heart muscle. Aging effects in transgenic mice is also demonstrated in this report [9,26]. A weak signal of Dpl mRNA was detected in the heart muscle of Rikn Prnp0/0 mice. Dpl mRNA levels of Tg(OrPrP)Prnp0/0 mice were enhanced in the heart muscle compared to Prnp0/0 mice. We still do not know the precise mechanisms of Doppel function in the heart muscle, whether this molecule is cytotoxic or not.
Previous studies have demonstrated that Rikn Prnp0/0 mice develop widespread loss of cerebellar Purkinje cells at 60 weeks of age, accompanied by progressive ataxia [27]. However, mating Prnp+/+ mice with Rcm0 and Ngsk lines of PrP knockout mice resulted in a phenotypic rescue of the ataxic syndrome, although Dpl expression levels remained unchanged [28]. Notably, similar results were obtained in this study using OrPrP. Prnp0/0 mice expressing high Dpl mRNA levels were mated with Tg(OrPrP)Prnp+/+ mice to obtain Tg(OrPrP) mice devoid of the mouse (Mo) PrP gene. No significant ataxia was observed. Weak pathological abnormalities were detected in the cerebellar Purkinje cells of Tg(OrPrP)Prnp0/0 mice. In all cases, expression of OrPrP prevented clinical symptoms in Tg(OrPrP)Prnp0/0 mice. These findings demonstrate that ataxic Prnp0/0 mice were rescued by OrPrP expression, implying that OrPrP was also expressed in nerve cells. These results suggest that Tg(OrPrP)Prnp0/0 mice may serve as a novel model to elucidate pathogenesis caused by expression of normal PrP in nerve cells and heart muscles.
myf5 expression promotes the conversion of embryonal stem cells to myogenic cells [29]. Ectopic expression of bovine MyoD in the heart muscle of Tg mice activated certain muscle-specific skeletal sarcomeric genes, resulting in embryonic lethality with severe cardiac abnormalities. These fetal cardiac muscles were heterogeneous for the transgene. Similar phenomenon may develop with oryx Prnp expression in the heart muscle of transgenic mice [9]. Myogenic stem cells develop into cardiac or skeletal muscles. To understand this process, Tg mice with bovine myf5 (bmyf5) transfection in the stem cells were used [29]. Results showed that ectopic expression of bmyf5 activated a skeletal genome program compatible with normal cardiac function. Cardiac and skeletal muscle cells developed from the same myogenic stem cells, sharing common structural features. Several isoforms of proteins were co-expressed from the same sarcomeric component. These myogenic stem cells eventually differentiate into skeletal or cardiac muscle. These expression patterns are restricted and distinctive.
Tg mice transfected with the bmyf5 gene were studied in the myocardium. Heterozygous mice transfected with bmyf5 survived to adulthood. However, with aging, the heterozygous mice showed severe pathology and abnormalities on ECG readings. Ectopic expression of bmyf5 activates a skeletal muscle program compatible with normal murine cardiac function in younger ages.
Tg overexpression of CaMKIIδc in the heart causes dilated cardiomyopathy with reduced contractile function, implying that aberrant signaling kinase expression can induce cardiac pathology [30]. Furthermore, overexpression of desmocollin-2 (DSC2) in cardiomyocytes induces severe cardiac dysfunction, including fibrosis, necrosis, and calcification, suggesting structural protein alterations as a cause of cardiomyopathy [31].
Tg overexpression of Gαq in the heart increases susceptibility to contractile dysfunction and cardiomyopathy in mice, possibly due to the sensitivity of G-protein signaling to transgene dosage [32]. Furthermore, a truncated troponin T transgene model shows that altered sarcomeric protein function can cause familial hypertrophic cardiomyopathy (FHC) characteristics [33]. Moreover, cardiomyocyte-specific overexpression of lysyl oxidase-like 1 (LOXL1) induces cardiac hypertrophy and interstitial fibrosis, indicating another mechanism by which extracellular matrix modifier genes lead to cardiomyopathy [34].
Tg and knock-in mouse models targeting desmosomal proteins such as PKP2 and DSC2 mimic arrhythmogenic cardiomyopathy. These studies suggest that cell to cell adhesion systems are important in maintaining cardiac integrity [35,36].
FHC is a genetic disorder resulting from mutations in the genes encoding sarcomeric proteins [37,38]. A mouse model of FHC was generated by introducing an Arg403 to Gln replacement in the alpha cardiac myosin heavy chain (MHC) gene. This mouse model may help define the cause of FHC. Histological observations of myocardiopathy were more apparent in males than in females and appeared by ages. Ten female alpha MHC sup403/+ mice (two 15 weeks old, one 31 weeks old) showed no cardiomyopathy, whereas all male alpha MHC sup403/+ mice showed cardiomyopathy with myocyte disarray. Myocyte hypertrophy, injury, and fibrosis were observed more frequently in 30-week-old alpha MHC sup403/+ mice than in 15-week-old alpha MHC sup403/+ mice.
Background genotypes and physiological activity of the phenotype were assessed using a Tg mouse model for FHC. Geister-Lawrance et al. showed that both factors affected the clinical progression of FHC [40]. Male mutant mice showed histological alterations earlier than female mice. Phenotypic differences between male and female inbred mice are genetically influenced. Therefore, they propose that other genes could influence the phenotype of the R403Q myosin mutation. These mice may help define therapeutic targets for FHC and other cardiomyopathies [38].
Using the same R403Q myosin mutation mouse model, physiological aspects of the myocardium were further analyzed [39]. An alteration in the excitation–contraction system in the myocardium was observed. Associated changes in intracellular Ca2+ were observed. Defective myofilament structure may cause the differences in intracellular Ca2+ distribution.
There is a correlation between human immunodeficiency virus (HIV) infection and the development of myocardial dysfunction in human patients [40]. Potential mechanism includes direct effects of HIV proteins (gp120 and Tat), and indirect effects of cytokines, autoimmunity, or antiretroviral cytotoxicity [40]. Tg mice expressing the HIV Tat protein showed cardiomyopathy. Under 3 months of age, heart rate was significantly lower in Tg mice (591 ± 47 bpm) than in control mice (FVB) (716 ± 45 bpm). Weak bradycardia and depression of systolic and diastolic function were observed in mice after the expression of HIV Tat [40]. These results show that HIV proteins may directly contribute to myocardial dysfunction. However, this theory does not exclude other possibilities such as the effects of cytokines, nutrition, or co-infection with other pathogens.
The World Health Organization (WHO) surveys viral infections related to cardiovascular disease globally [41]. During 1975–1985, Coxsackie B viruses (CVB) were identified as the biggest contributors to cardiovascular diseases (34.6 per 1,000), followed by influenza B (17.4 per 1,000), influenza A (11.7 per 1,000), Coxsackie A (9.1 per 1,000), and cytomegalovirus (8.0 per 1,000) [42]. Acute CVB infection in humans and mice causes rapid and severe myocarditis. Humans may experience chronic infection and may recover fully. After recovery, they may develop DCM [41].
Typically, infectious virus progeny and viral proteins cannot be detected in patients with DCM through virus isolation and immunohistochemistry. In contrast, Tg mice expressing Coxsackievirus B3 (CVB3) genomes show abnormal excitation–contraction coupling in pressure-overload models of cardiomyopathy [43,44]. According to Wesley, cardiomyopathy is induced in Tg mice through transfection of CVB3 genomes without the production of infectious virions. This disease is similar to human DCM [43]. Further experiments are necessary to clarify the specific viral gene regions that affect myocyte function. Viral proteinases may play a major role in both acute and chronic disease.
Epstein–Barr virus nuclear antigen–leader protein (EBNA-LP) is required for highly efficient B lymphocyte transformation by the viral genome. Here, Tg mice carrying an EBNA-LP cDNA construct were produced. In the construct, the widely expressed metallothionein promoter was used [45]. EBNA-LP was expressed in several organs, such as liver, kidney, heart, lung, and spleen. From 4 months to over a year of age, Tg mice developed symptoms such as congenic heart failure. Fibrillation was not apparent on electrocardiographs. However, a reduction in T-wave amplitude was observed, suggesting abnormal ventricular repolarization. Subsequently, a high frequency of dilated heart failure was unexpectedly observed [45]. The disease produced as a result of EBNA-LP expression in Tg mice shows similarities to human congenic heart failure or DCM. This report suggests that EBNA-LP of Epstein–Barr virus could induce heart failure in mice [45].
Tg models of cardiomyopathy using viral genomes are relevant for understanding the pathogenesis of human cases. The results obtained are valuable for developing novel therapeutic strategies for the treatment and prevention of human cardiomyopathy.
Tg mice developed necrotizing myopathy involving skeletal muscles through the overexpression of the PrP gene derived from hamster or sheep [46]. Tg mice using Prnp showed muscle-specific expression with extremely limited regulation by doxycycline (DOX) [47]. This DOX-induced expression was strictly limited to skeletal muscles. The Tg mice showed a myopathy characterized by an increased variation in the size of myofibers [44]. These typical lesions of myofibers showed nuclei located inside the cytoplasm. Endomysial fibrosis was observed without intracytoplasmic inclusions or rimmed nuclei. No evidence of neurogenic disorder was observed.
Suardi et al. reported another pattern of prion induced myopathy in experimental and natural bovine amyloid spongiform encephalopathy (BASE). Several different skeletal muscles (M. longissimus dorsi, M. intercostalis, and M. gluteus) from BASE cases carried infectivity; however, the spleen, cervical lymph nodes, and kidneys from BASE cases did not show infectivity [48].
Variety of prion diseases showed PrPres deposition in skeletal muscles. These include natural cases of atypical scrapie in sheep, chronic wasting disease in deer, and their experimental infections in mice and hamsters [49,50,51]. These reports agreed that PrPres in murine muscle tissue is associated with terminal nerve endings. In Suardi’s report, however, cattle naturally affected with BASE showed PrPres deposition in the cytoplasm of skeletal muscle [50], not related to terminal nerve endings. The authors conducted bioassays in Tg mice overexpressing bovine PrP (Tgbov XV) and found infectivity in a variety of muscle samples from cattle with natural and experimental BASE. Although their report did not show any involvement of cardiomyocytes, more studies are needed, especially in the final stages of experimental BASE in transgenic mice (Tgbov XV). Apparently, prion agents replicate in skeletal muscle myocytes. Prion gene mRNA levels increased in cardiac myocytes in Tg mice.
In the terms of its role in neurotoxicity and male fertility, Dpl has recently emerged as a potential regulator of vascular biology. Chen et al. showed that endothelial Prnd/Dpl is selectively expressed in angiogenic central nervous system (CNS) endothelial cells and is required for proper brain vessel morphogenesis and blood-brain barrier (BBB) formation [52], suggesting that Dpl plays a role in endothelial signaling pathways to regulate angiogenesis with barrier maturation. In other study, Dpl in tumor endothelial cells has been shown to promote pathological angiogenesis, and pharmacological targeting of the Dpl/VEGFR2 axis selectively suppresses tumor vascularization without affecting normal endothelial cells [53].
Future studies would be required for fully addressing Dpl in terms of prion-related cardiomyopathy. Comparative analyses of Prnd polymorphisms and Dpl expression in human cardiomyopathy cohorts, together with experimental modulation of Prnd in our Tg(OrPrP)Prnp0/0 mice, could contribute to understand whether Dpl play as role of modulator for cardiac disease. Such studies would compensate the recent information from the studies on Prnd variants in companion animals and livestock, which have already revealed species-specific associations between Prnd polymorphisms, and prion disease susceptibility [54,55]. Insights of comparative genetic knowledge on Prnd together with endothelial and myocardial Dpl function might establish a framework link of prion protein family proteins in the terms of vascular homeostasis and cardiomyopathy.
5. Conclusions
Neurodegenerative diseases, such as FA and PD, are frequently associated with cardiomyopathy. We describe a novel mouse model for cardiomyopathy, which was observed during the development of prion agent sensitive Tg mice. Specifically, it was expected that these mice express OrPrP ubiquitously, including in nerve cells, immune cell, and other cells using the β-actin promoter. Tg mouse models are an important resource for the development of cardiac pharmaceuticals for humans and animals. These mice are useful in safety testing before identifying effective measures for humans with underlying heart diseases. To identify new drugs, we require Tg mice, which show abnormal ECG waves. PMDs have been the focus of traditional studies on prion diseases. Understanding aggregation-induced toxicity, abnormal topology or altered trafficking are likely to be a major focus in future prion studies.
Author Contributions
Conceptualization, S.S., H.E., H.T., S.I. and T.O.; writing—original draft preparation, S.S.; writing—review and editing, S.S., A.S., H.E., H.T., S.I. and T.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The animal study protocol was submitted to the Institutional Review Board of the University of Tokyo (notification code: 1818T0061; May 15, 2006).
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding authors.
Acknowledgments
Major thanks are due to Director Prof. Toru Miyazaki (The Institute for AIM Medicine, Shinjuku 162-0054, Tokyo, Japan) for his intensive discussion of Tg mice developments. We would like to express our gratitude to Drs. Yoshitsugu Matsumoto and Keiichi Saeki (Department of Molecular Immunology, School of Agriculture and Life Sciences, the University of Tokyo, Tokyo, Japan) for many helpful discussions and advice throughout this work.
Conflicts of Interest
The authors declare no competing interests.
References
- Condello, C.; Westaway, D; Prusiner, S.B. Expanding the prion paradigm to include Alzheimer and Parkinson Diseases. JAMA Neurol 2024, 81, 1023–1024. [CrossRef]
- Prusiner, S.B. Transgenetic investigations of prion diseases of humans and animals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1993, 339, 239–254. [CrossRef]
- Westaway, D.; DeArmond, S.J.; Cayetano-Canlas, J.; Groth, D.; Foster, D.; Yang, S.L.; Torchia, M.; Carlson, G.A.; Prusiner, S.B. Degeneration of skeletal muscle, peripheral nerves, and the central nervous system in transgenic mice overexpressing wild-type prion proteins. Cell 1994, 76, 117–129. [CrossRef]
- Kelly, D.P.; Strauss, A.W. Inherited cardiomyopathies. N. Engl. J. Med. 1994, 330, 913–919.
- Toyo-oka, T.; Nagayama, K.; Suzuki, J; Sugimoto, T. Noninvasive assessment of cardiomyopathy development with simultaneous measurement of topical1H- and 31P-magnetic resonance spectroscopy. Circulation 1992, 86, 295–301. [CrossRef]
- Thomzig, A.; Kratzel, C.; Lenz, G.; Kruger, D.; Beekes, M. Widespread PrPSc accumulation in muscle of hamsters orally infected with scrapie. EMBO Rep. 2003, 4, 530–533. [CrossRef]
- Marin-Garcia, J.; Goldenthal, M.J.; Filiano, J.J. Cardiomyopathy associated with neurologic disorders and mitochondrial phenotype. J. Child Neurol. 2002, 17, 759–765.
- Bunse, M.; Bit-Avragim, N.; Riefflin, A.; Perrot, A.; Schmidt, O.; Kreuz, F.R.; Dietz, R.; Jung, W.I.; Osterziel, K.J. Cardiac energetics correlates to myocardial hypertrophy in Friedreich's ataxia. Ann. Neurol. 2003, 53, 121–123. [CrossRef]
- Itohara, S.; Onodera, T.; Tsubone, H. Prion gene modified mouse which exhibits heart anomalies. US Patent 6657105, 2003.
- Mastrangelo, P.; Westaway, D. Biology of the prion gene complex. Biochem. Cell Biol. 2001, 79, 613–628.
- Moore, R.C.; Mastrangelo, P.; Bouzamondo, E.; Heinrich, C.; Legname, G.; Prusiner, S.B.; Hood, L.; Westaway, D.; DeArmond, S.J.; Tremblay, P. Doppel-induced cerebellar degeneration in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 15288–15293. [CrossRef]
- Shintaku, T.; Ohba, T.; Niwa, H.; Kushikata, T.; Hirota, K.; Ono, K.; Imaizumi, T.; Kuwasako, K.; Sawamura, D.; Murakami, M. Effects of propofol on electrocardiogram in mice. J. Pharmacol. Sci. 2014, 126, 351–358.
- Scott, M.R.; Will, R.; Nguyen, H.O.; Tremblay, P.; DeArmond, S.J.; Prusiner, S.B. Compelling Transgenic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 15137–15142. [CrossRef]
- Telling, G.C.; Scott, M.; Hsiao, K.K.; Foster, D.; Yang, S.L.; Torchia, M.; Sidle, M.; Collinge, G.; DeArmond, S.J.; Prusiner, S.B. Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 9936–9940.
- Kirkwood, J.K.; Cunningham, A.A. Epidemiological observations on spongiform encephalopathies in captive wild animals in the British islets. Vet. Rec. 1994, 135, 296–303.
- Cunningham, A.A.; Kirkwood, J.K.; Dawson, M.; Spencer, Y.I.; Green, R.B.; Wells, G.A.H. Bovine spongiform encephalopathy infectivity in greater kudu (Tragelaphus strepsiceros). Emerg. Infect. Dis. 2004, 10, 1044–1049. [CrossRef]
- Oesch, B.; Westaway, D.; Prusiner, S.B. Prion protein genes: evolutionary and functional aspects. Curr. Top. Microbiol. Immunol. 1991, 172, 109–124.
- Seo, S.W.; Hara, K.; Kubosaki, A.; Nasu, Y.; Nishimura, T.; Saeki, K.; Matsumoto, Y.; Endo, H.; Onodera, T. Comparative analysis of the prion protein open reading frame nucleotide sequences of two wild ruminants, the moufflon and golden takin. Intervirology 2001, 44, 359–363. [CrossRef]
- Shimada, M.; Shimano, H.; Gotoda, T.; Yamamoto, K.; Kawamura, M.; Inaba, T.; Yazaki, Y.; Yamada, N. Overexpression of human lipoprotein lipase in transgenic mice. Resistance to diet-induced hypertriglyceridemia and hypercholesterolemia. J. Biol. Chem. 1993, 268, 17924–17929. [CrossRef]
- Honda, H.; Mano, H.; Katsuki, M.; Yazaki, Y.; Hirai, H. Increased tyrosine-phosphorylation of 55 KDa proteins in beta-actin / Tec transgenic mice. Biochem. Biophys. Res. Commun. 1995, 206, 287–293.
- Yokoyama, T.; Kimura, K.M.; Ushiki, Y.; Yamada, S.; Morooka, A.; Nakashiba, T.; Sassa, T.; Itohara, S. In vitro conversion of cellular prion protein to pathogenic isoforms, as monitored by conformation-specific antibodies. J. Biol. Chem. 2001, 276, 11265–11271.
- Ishii, S.; Nagase, T.; Tashiro, F.; Ikuta, K.; Sato, S.; Waga, I.; Kume, K.; Miyazaki, J.; Shimizu, T. Bronchial hyperreactivity, increased endotoxin lethality and melanocytic tumorigenesis in transgenic mice overexpressing platelet-activating factor receptor. EMBO J. 1997, 16, 133–142.
- James, J.; Osinska, H.; Hewett, T.E.; Kimball, T.; Klevitsky, R.; Witt, S.; Hall, D. G.; Gulick, J.; Robbins, J. Transgenic over-expression of a motor protein at high levels results in severe cardiac pathology. Transgenic Res. 1999, 8, 9–22.
- Ma, J.; Wollmann, R.; Lindquist, S. Neurotoxicity and neurodegeneration when PrP accumulates in cytosol. Science 2002, 298, 1781–1785.
- Ma, J.; Lindquist, S. Wild-type PrP and a mutant associated with prion diseases are subject to retrograde transport and proteasome degradation. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 14955–14960. [CrossRef]
- Onodera, T. Dual role of cellular prion protein in normal host and Alzheimer’s disease. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 2017, 93, 155–173. [CrossRef]
- Sakudo, A.; Onodera, T. Prion protein (PrP) gene-knockout cell lines: insight into functions of the PrP. Front. Cell Dev. Biol. 2015, 2, 75. [CrossRef]
- Moore, R.C.; Lee, I.Y.; Silverman, G.L.; Harrison, P.M.; Strome, R.; Heinrich, C.; Karunaratne, A.; Pasternak, S.H.; Chishti, M.A.; Liang, Y.; Mastrangelo, P.; Wang, K.; Smit, A.F.; Katamine, S.; Carlson, G.A.; Cohen, F.E.; Prusiner, S.B.; Melton, D.W.; Tremblay, P.; Hood, L.E.; Westaway, D. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J. Mol. Biol. 1999, 292, 797–817. [CrossRef]
- Edwards, J.G.; Lyons, G.E.; Micales, B.K.; Malhorra, A.; Factor, S.; Leinwand, L.A. Cardiomyopathy in transgenic myf5 mice. Circ. Res. 1996, 78, 379–387. [CrossRef]
- Molkentin, J.D.; Robbins, J. With great power comes great responsibility: using mouse genetics to study cardiac hypertrophy and failure. J. Mol. Cell Cardiol. 2009, 46, 130–136. [CrossRef]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.X.; Gordon, PM.; Nygren, A.; Gerull, B. Transgenic mice overexpressing desmocollin-2 (DSC2) develop cardiomyopathy associated with myocardial inflammation and fibrotic remodeling. PLoS One 2017, 12, e0174019.
- D'Angelo, D.D.; Sakata, Y.; Lorenz, J.N.; Boivin, G.P.; Walsh, R.A.; Liggett, S.B.; Dorn, G.W. Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 8121–8126.
- Tardiff, J.C.; Factor, S.M.; Tompkins, B.D.; Hewett, T.E.; Palmer, B.M.; Moore, R.L.; Schwartz, S.; Robbins, J.; Leinwand LA. A truncated cardiac troponin T molecule in transgenic mice suggests multiple cellular mechanisms for familial hypertrophic cardiomyopathy. J. Clin. Invest. 1998, 101, 2800–2811.
- Ohmura, H.; Yasukawa, H.; Minami, T.; Sugi, Y.; Oba, T.; Nagata, T.; Kyogoku, S.; Ohshima, H.; Aoki, H.; Imaizumi, T. Cardiomyocyte-specific transgenic expression of lysyl oxidase-like protein-1 induces cardiac hypertrophy in mice. Hypertens. Res. 2012, 35, 1063–1068. [CrossRef]
- Moncayo-Arlandi, J.; Guasch, E.; Sanz-de la Garza, M.; Casado, M.; Garcia, N.A.; Mont, L.; Sitges, M.; Knöll, R.; Buyandelger, B.; Campuzano, O.; Diez-Juan, A.; Brugada, R. Molecular disturbance underlies to arrhythmogenic cardiomyopathy induced by transgene content, age and exercise in atruncated PKP2 mouse model. Hum. Mol. Genet. 2016, 25, 3676–3688.
- Gerull, B.; Brodehl, A. Genetic Animal Models for Arrhythmogenic Cardiomyopathy. Front. Physiol. 2020, 11:624. [CrossRef]
- Georgakopoulos, D.; Christie, M.E.; Giewat, C.M.; Seidman, J.G.; Kass, D.A. The pathogenesis of familial hypertrophic cardiomyopathy: early and evolving effects from an alpha-cardiac myosin heavy chain missense mutation. Nat. Med. 1999, 5, 327–330. [CrossRef]
- Geisterfer-Lowrance, A.A.; Christe, M.; Conner, D.A.; Ingwall, J.S.; Seidman, C.E.; Seidman, J.G. A mouse model of familial hypertrophic cardiomyopathy. Science 1996, 272, 731–734.
- Gao, W.D.; Perez, N.G.; Seidman, C.E.; Seidman, J.G.; Marban, E. Altered cardiac excitation-contraction coupling in mutant mice with familial hypertrophic cardiomyopathy. J. Clin. Invest. 1999, 103, 661–666.
- Fang, Q.; Kan, H.; Lewis, W.; Chen, F.; Sharmann P.; Finkel, M.S. Dilated cardiomyopathy in transgenic mice expressing HIV Tat. Cardiovasc. Toxicol. 2009, 9, 39–45.
- Friman, G.; Wesslen, L.; Fohlman, J.; Karjalainen, J.; Rolf, C. The epidemiology of infectious myocarditis, lymphocytic myocarditis and dilated cardiomyopathy. Eur. Heart J. 1996, 16(Supp. O), 36–41. [CrossRef]
- Grist, N.R.; Reid, D. Epidemiology of viral infections of the heart. In Banatvala, J.E. ed. Viral Infections of the Heart; Hodder and Stoughton: London, 1993; pp. 23–31.
- Wessely, R.; Klingel, K.; Santana, L.F.; Dalton, N.; Hongo, M.; Lederer, W.J.; Kandolf, R.; Knowlton, K.U. Transgenic expression of replication-restricted enteroviral genomes in heart muscle induce defective excitation-contraction coupling and dilated cardiomyopathy. J. Clin. Invest. 1998, 102, 1444–1453. [CrossRef]
- Gomez, A.M.; Valdivia, H.H.; Cheng, H.; Lederer, M.R.; Santana, L.F.; Cannell, M.B.; McCune, S.A.; Altschuld, R.A.; Lederer, W.J. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science 1997, 276, 800–806. [CrossRef]
- Huen, D.S.; Fox, A.; Kumar, P.; Searle, P.F. Dilated heart failure in transgenic mice expressing the Epstein-Barr virus nuclear antigen-leader protein. J. Gen. Virol. 1993, 74, 1381–1391. [CrossRef]
- Westaway, D.; DeArmond, S.J.; Canlas-Cayetano, J.; Groth, D.; Foster, D.; Yang, S.L.; Torchia, M.; Calson, G.A.; Prusiner, S.B. Degeneration of skeletal muscle, peripheral nerves, and central nervous system in transgenic mice overexpressing wild-type prion proteins. Cell 1994, 76, 117–129. [CrossRef]
- Huang, S.; Liang, J.; Zheng, M.; Li, X.; Wang, M.; Vanegas, D.; Wu, D.; Chakraborty, B.; Hays, A.P.; Chen, K.; Che, S.G.; Booth, S.; Cohen, M.; Gambetti, P.; Kong, Q-Z. Inducible overexpression of wild-type prion protein in the muscles leads to a primary myopathy in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 6800–6805.
- Suardi, S.; Vimercati, C.; Casalone, C.; Gelmetti, D.; Corona, C.; Lulini, B.; Mazza, M.; Lombardi, G.; Moda, F.; Ruggerone, M.; Campaganani, I.; Piccoli, E.; Catania, M.; Groschup, M.H.; Balkema-Buschmann, A.; Caramelli, M.; Monaco, S.; Zannuso, G.; Tagliavini, F. Infectivity in skeletal muscle of cattle with atypical bovine spongiform encephalopathy. PLoS One 2012, 7, e31449. [CrossRef]
- Andréoletti, O.; Orge, L.; Benestad, S.L.; Beringue, V.; Litaise, G.; Simon, S.; Le Dur, A.; Laude, H.; Simmons, H.; Lugan, S.; Corbière, F.; Costes, P.; Morel, N.; Schelcher, F.; Lacroux, C. Atypical/Nos98 scrapie infectivity in sheep peripheral tissues. PLoS Pathog. 2011, 7, e1001285. [CrossRef]
- Angers, R.C.; Browning, S.R.; Seward, T.S.; Sigurdson, C.J.; Miller, M.W.; Hoover E.A.; Telling G.C. Prions in skeletal muscle of deer with chronic wasting disease. Science 2006, 311, 1117. [CrossRef]
- Bosque, P.J.; Ryou, C.; Telling, G.; Peretz, D.; Legname, G.; DeArmond, S.J.; Prusiner, S.B. Prions in skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 3812–3817.
- Chen, Z.; Morales, J. E.; Avci, N.; Guerrero, P. A.; Rao, G.; Seo, J. H.; McCarty, J. H., The vascular endothelial cell-expressed prion protein doppel promotes angiogenesis and blood-brain barrier development. Development 2020, 147, dev193094.
- Al-Hilal, T. A.; Chung, S. W.; Choi, J. U.; Alam, F.; Park, J.; Kim, S. W.; Kim, S. Y.; Ahsan, F.; Kim, I. S.; Byun, Y., Targeting prion-like protein doppel selectively suppresses tumor angiogenesis. J. Clin. Invest. 2016, 126, 1251–66. [CrossRef]
- Kim, Y. C.; Jeong, B. H., Bovine spongiform encephalopathy (BSE) associated polymorphisms of the prion-like protein gene (PRND) in Korean dairy cattle and Hanwoo. J. Dairy Res. 2018, 85, 7–11. [CrossRef]
- Balbus, N.; Humeny, A.; Kashkevich, K.; Henz, I.; Fischer, C.; Becker, C. M.; Schiebel, K., DNA polymorphisms of the prion doppel gene region in four different German cattle breeds and cows tested positive for bovine spongiform encephalopathy. Mamm. Genome 2005, 16, 884–92. [CrossRef]
- Hogan, B.; Constantini, F.; Lacy, E. Manipulating the Mouse Embryo. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1986.
- Niwa, H.; Yamamura, K.; Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 1991, 108, 193–199.
- Miyazaki, J.; Takai, S.; Araki, K.; Tashiro, F.; Tominaga, A.; Takatsu, K.; Yamamura, K. Expression vector system based on the chicken beta-actin promoter directs efficient production of interleukin-5. Gene 1989, 79, 269–277.
- McKinley, M.P.; Hay, B.; Lingappa, V.R.; Lieberburg, I.; Prusiner, S.B. Developmental expression of prion protein gene. Dev. Biol. 1987, 121, 105–110.
- Bell, J.E.; Gentleman, S.M.; Ironside, J.W.; McCardle, L.; Lantos, P.L.; Doey, L.; Lowe, J.; Fergusson, J.; Luthert, P.; McQuaid, S.; Allen, I.V. Prion protein immunocytochemistry—UK five center consensus report. Neuropathol. Appl. Neurobiol. 1997, 23, 26–35.
- van Keulen, L.J.; Schreuder, B.E.; Meloen, R.H.; Poelen-van den Berg, M.; Mooij-Harkes, G.; Vromans, M.E.; Langeveld, J.P. Immunochemical detection and localization of prion protein in brain tissue of sheep with natural scrapie. Vet. Pathol. 1995, 32, 299–308. [CrossRef]
- Hayashi, H.; Tanaka, M.; Iwamaru, Y.; Ushiki, Y.; Kumura, K.M.; Tagawa, Y.; Shinagawa, M.; Yokoyama, T. Effect of tissue deterioration on postmortem BSE diagnosis by immunobiochemical detection of abnormal isoform of prion protein. J. Vet. Med. Sci. 2004, 66, 515–520. [CrossRef]
Figure 1.
Scheme of transgene construct of OrPrP gene. (a) An EcoRI fragment of OrPrP cDNA indicates the closed bar was inserted into the EcoRI site of the pCAGGS expression vector [9,56,57,58,59,60]. The ATG start codon and TAG stop codon are indicated. A fragment containing human cytomegalovirus immediate early (CMV-IE) enhancer, chicken β-actin promoter, rabbit β-globin splicer, OrPrP cDNA, rabbit β-globin 3’ untranslated region, and polyadenylation [poly(A)] signal was isolated from the vector at a HindIII–SalI site. The HindIII and SalI sites are shown by arrow. The hybridization probe (a 640 bp cDNA) used for Southern blot analysis is also shown. Based upon the DNA sequence [9,10], four lines were generated by microinjection of the construct containing the hybrid promoter plus a 1.0 kb cDNA including the OrPrP open reading frame (ORF). These mice expressed OrPrP containing a nerve cell, an immunity cell, and skeletal muscles by using β-actin promoter [56,58]. (b) In screening of transgene by Southern blot analysis, we have identified four Tg strains carrying oryx PrP gene. An 834 bp band (Lane 1,2,3,4) and 920 bp band (Lane 5,6,7,8) were identified by digestion of restriction enzyme BamHI and EcoRI, respectively. Lane 1 and 5, #34Tg(OrPrP)Prnp+/+ mouse genomic DNA. Loading is 20 μg. Lane 2 and 6, #36Tg(OrPrP)Prnp+/+, 20 μg. Lane 3 and 7, #42Tg(OrPrP)Prnp+/+, 20 μg. Lane 4 and 8, #50Tg(OrPrP)Prnp+/+, 20 μg.
Figure 1.
Scheme of transgene construct of OrPrP gene. (a) An EcoRI fragment of OrPrP cDNA indicates the closed bar was inserted into the EcoRI site of the pCAGGS expression vector [9,56,57,58,59,60]. The ATG start codon and TAG stop codon are indicated. A fragment containing human cytomegalovirus immediate early (CMV-IE) enhancer, chicken β-actin promoter, rabbit β-globin splicer, OrPrP cDNA, rabbit β-globin 3’ untranslated region, and polyadenylation [poly(A)] signal was isolated from the vector at a HindIII–SalI site. The HindIII and SalI sites are shown by arrow. The hybridization probe (a 640 bp cDNA) used for Southern blot analysis is also shown. Based upon the DNA sequence [9,10], four lines were generated by microinjection of the construct containing the hybrid promoter plus a 1.0 kb cDNA including the OrPrP open reading frame (ORF). These mice expressed OrPrP containing a nerve cell, an immunity cell, and skeletal muscles by using β-actin promoter [56,58]. (b) In screening of transgene by Southern blot analysis, we have identified four Tg strains carrying oryx PrP gene. An 834 bp band (Lane 1,2,3,4) and 920 bp band (Lane 5,6,7,8) were identified by digestion of restriction enzyme BamHI and EcoRI, respectively. Lane 1 and 5, #34Tg(OrPrP)Prnp+/+ mouse genomic DNA. Loading is 20 μg. Lane 2 and 6, #36Tg(OrPrP)Prnp+/+, 20 μg. Lane 3 and 7, #42Tg(OrPrP)Prnp+/+, 20 μg. Lane 4 and 8, #50Tg(OrPrP)Prnp+/+, 20 μg.
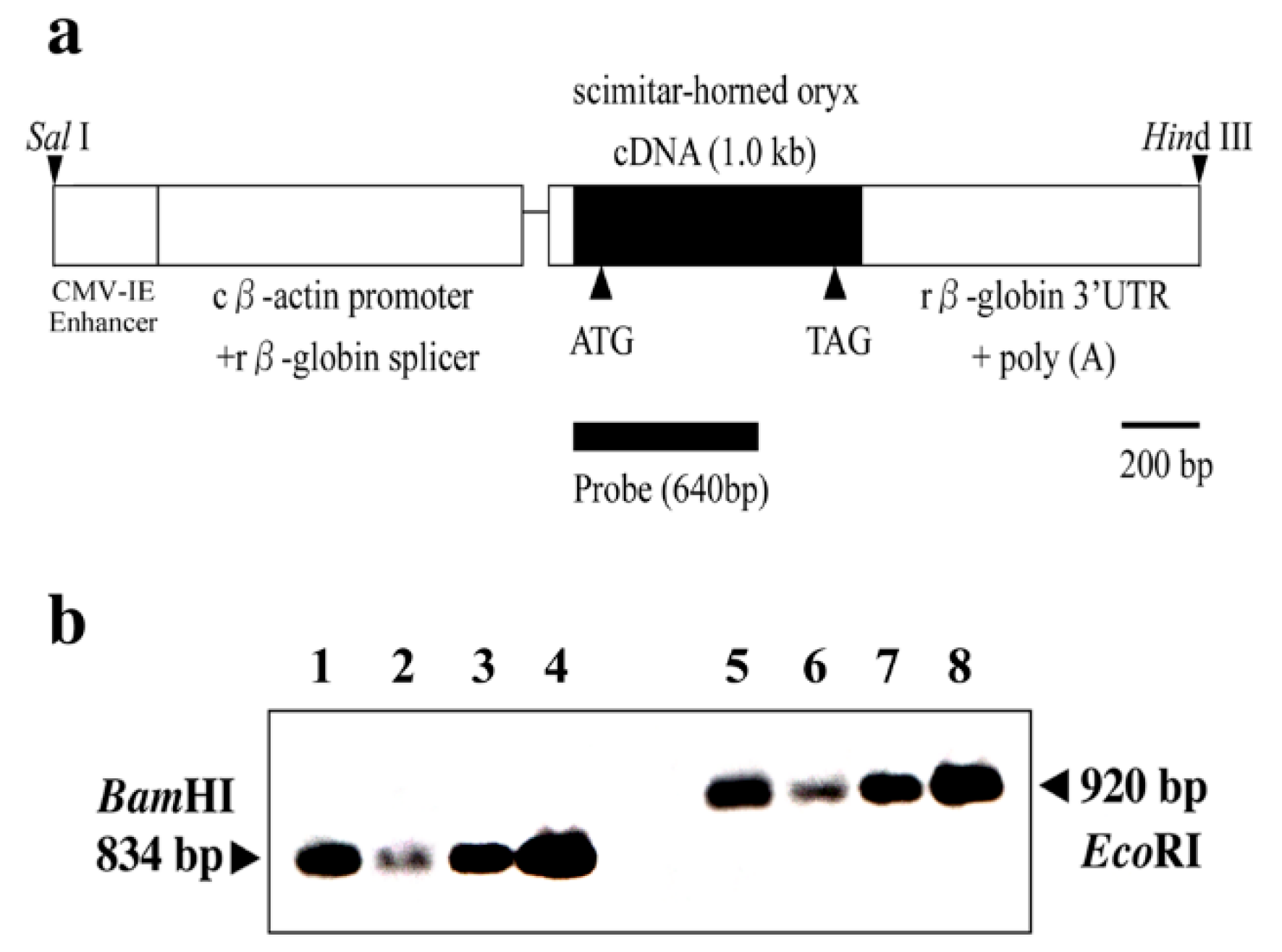
Figure 2.
Characterization of transgenic mice expressing OrPrP. In our studies, Tg(OrPrP)Prnp+/+ mice were mated to Prnp0/0 mice to obtain animals carrying the OrPrP on a murine Prnp knockout genotype. (a) Expression of the OrPrP mRNA by RT-PCR analysis. OrPrP mRNA was detected in the heart, skeletal muscle, and brain, but not detectable in the lung, liver, spleen, kidney, and thymus. No band was observed without reverse transcriptase in each tissue. The size marker (in kb) of OrPrP mRNA observed in brain, heart and skeletal muscle was 0.6-kb. (b) Immunoprecipitation of OrPrP in the brain, heart, and skeletal muscle from the Tg(OrPrP)Prnp0/0 mice [60,61,62]. OrPrP levels in the heart (Lane 4) were compared with the brain (Lane 1). Precipitation of total brain and heart homogenates from the Tg(OrPrP)Prnp0/0 mice showed higher levels of OrPrP expression in the brain and was detectable in the heart. Distinct bands corresponding to OrPrP showed equivalent size of the 31-kilodalton protein in the brain and heart. No bands are seen in the brain and heart from the Prnp0/0 mouse brain (Lane 3) and heart (Lane 6). Wild type B6 mice showed distinct band in the brain (Lane 2) and heart (lane 5). Numbered arrows indicate molecular size markers (kilodaltons).
Figure 2.
Characterization of transgenic mice expressing OrPrP. In our studies, Tg(OrPrP)Prnp+/+ mice were mated to Prnp0/0 mice to obtain animals carrying the OrPrP on a murine Prnp knockout genotype. (a) Expression of the OrPrP mRNA by RT-PCR analysis. OrPrP mRNA was detected in the heart, skeletal muscle, and brain, but not detectable in the lung, liver, spleen, kidney, and thymus. No band was observed without reverse transcriptase in each tissue. The size marker (in kb) of OrPrP mRNA observed in brain, heart and skeletal muscle was 0.6-kb. (b) Immunoprecipitation of OrPrP in the brain, heart, and skeletal muscle from the Tg(OrPrP)Prnp0/0 mice [60,61,62]. OrPrP levels in the heart (Lane 4) were compared with the brain (Lane 1). Precipitation of total brain and heart homogenates from the Tg(OrPrP)Prnp0/0 mice showed higher levels of OrPrP expression in the brain and was detectable in the heart. Distinct bands corresponding to OrPrP showed equivalent size of the 31-kilodalton protein in the brain and heart. No bands are seen in the brain and heart from the Prnp0/0 mouse brain (Lane 3) and heart (Lane 6). Wild type B6 mice showed distinct band in the brain (Lane 2) and heart (lane 5). Numbered arrows indicate molecular size markers (kilodaltons).
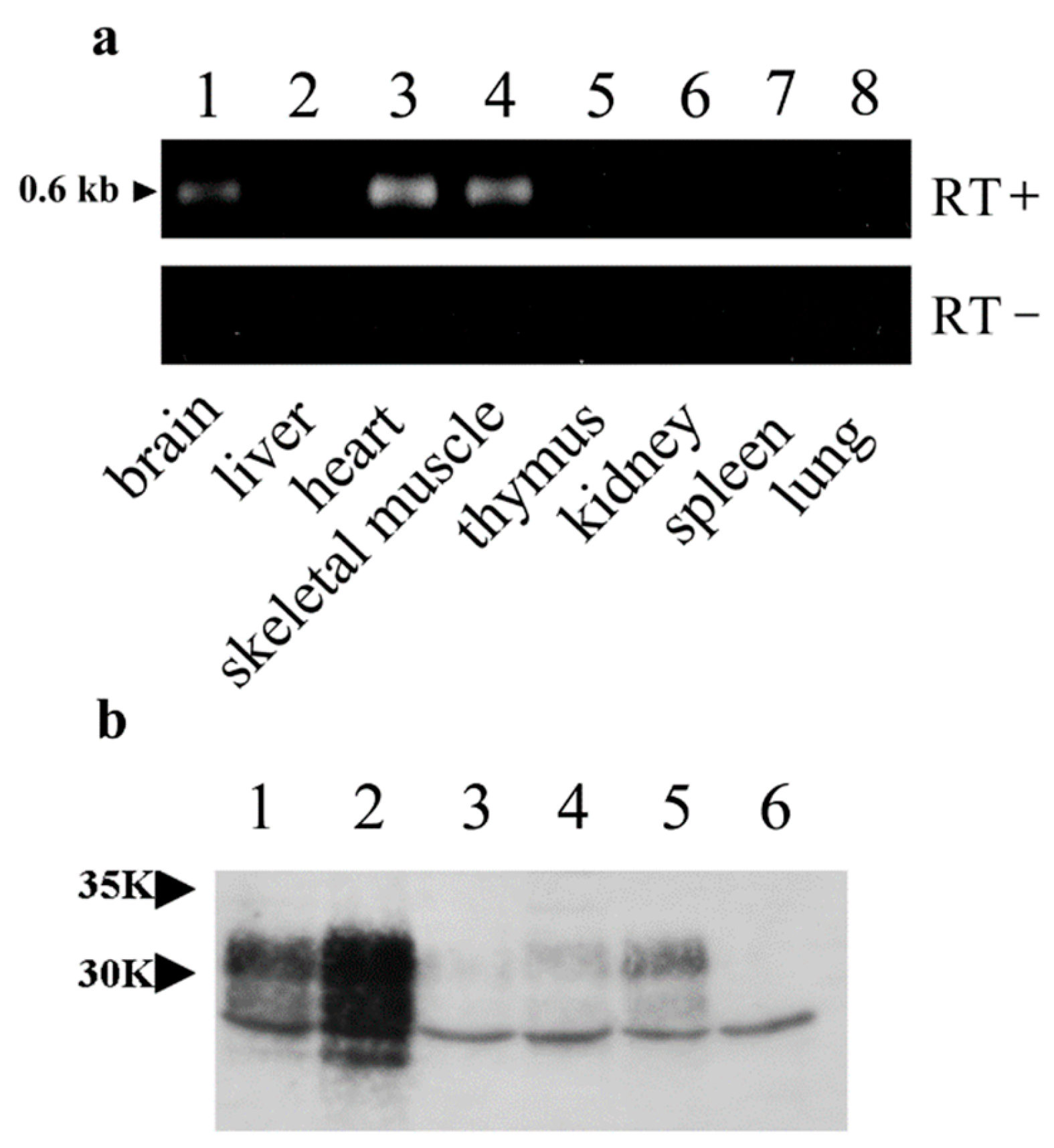
Figure 3.
Alterations in electrocardiographic patterns in the 20 to 70 weeks old Tg(OrPrP)Prnp0/0 mice [12]. ECGs in lead II recorded from the Tg(OrPrP)Prnp0/0 mice at 20, 30, 40, 50, 60, and 70 weeks old. ECG and pulse rate traces from 30 weeks old (n = 5) and 40 weeks old (n = 5) Tg(OrPrP)Prnp0/0 mice were distinguishable from the control B6 mice. The QRS complex showed that the durations of the R wave were significantly abnormal in 30 weeks old (n = 5) and 40 weeks old (n = 5) Tg(OrPrP)Prnp0/0 mice, but the duration of the PQ interval was not. However, normal waveforms were seen with 20 weeks old (n = 10) Tg(OrPrP)Prnp0/0 mice. The notch in the R waves in the QRS complex was observed after 50 weeks old (n = 5), 60 weeks old (n = 5), and 70 weeks old (n = 5) Tg(OrPrP)Prnp0/0 mice. The P waveform of the PR interval reversed in 70 weeks old Tg(OrPrP)Prnp0/0 mice (first arrow). However, this change was not observed in younger Tg(OrPrP)Prnp0/0 mice. All arrows indicate the abnormal waveform (R and P waveform) of ECG in Tg(OrPrP)Prnp0/0 mice.
Figure 3.
Alterations in electrocardiographic patterns in the 20 to 70 weeks old Tg(OrPrP)Prnp0/0 mice [12]. ECGs in lead II recorded from the Tg(OrPrP)Prnp0/0 mice at 20, 30, 40, 50, 60, and 70 weeks old. ECG and pulse rate traces from 30 weeks old (n = 5) and 40 weeks old (n = 5) Tg(OrPrP)Prnp0/0 mice were distinguishable from the control B6 mice. The QRS complex showed that the durations of the R wave were significantly abnormal in 30 weeks old (n = 5) and 40 weeks old (n = 5) Tg(OrPrP)Prnp0/0 mice, but the duration of the PQ interval was not. However, normal waveforms were seen with 20 weeks old (n = 10) Tg(OrPrP)Prnp0/0 mice. The notch in the R waves in the QRS complex was observed after 50 weeks old (n = 5), 60 weeks old (n = 5), and 70 weeks old (n = 5) Tg(OrPrP)Prnp0/0 mice. The P waveform of the PR interval reversed in 70 weeks old Tg(OrPrP)Prnp0/0 mice (first arrow). However, this change was not observed in younger Tg(OrPrP)Prnp0/0 mice. All arrows indicate the abnormal waveform (R and P waveform) of ECG in Tg(OrPrP)Prnp0/0 mice.
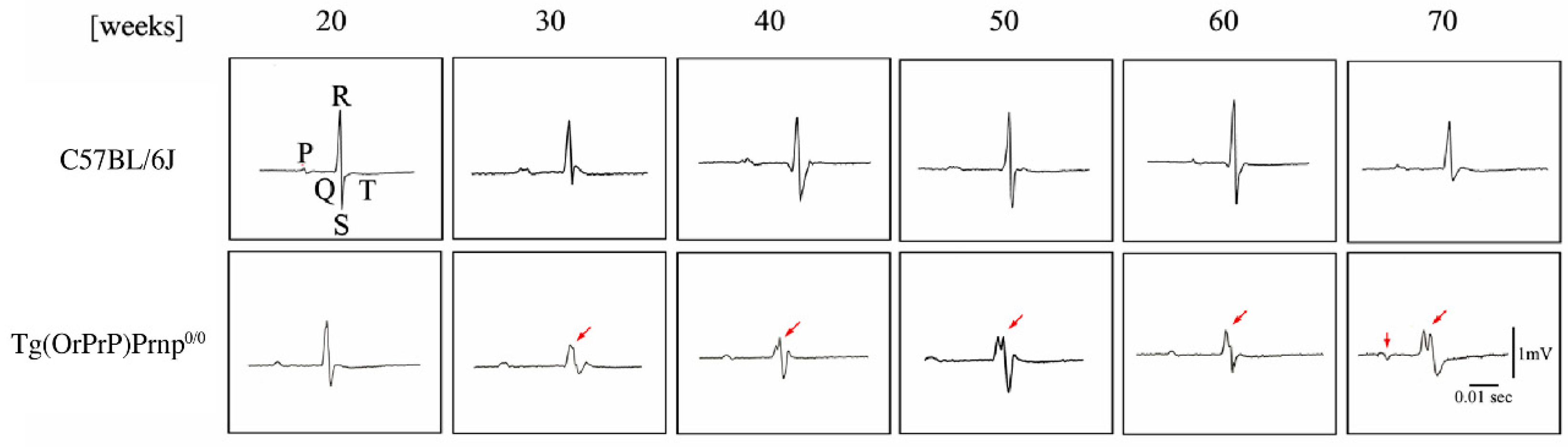
Figure 4.
Spongiform cardiomyopathy in 30 to 60 weeks aged Tg(OrPrP)Prnp0/0 mice. Mild vacuolation was seen in the heart muscle of 30 (a) and 40 (b) weeks old Tg(OrPrP)Prnp0/0 mice. (c) Vacuolation in the atrium area of the heart muscle of 60 weeks old Tg(OrPrP)Prnp0/0 mice. No inflammatory lesion was observed. H&E staining, Magnifications: (a) x400; (b) x400; (c) x200.
Figure 4.
Spongiform cardiomyopathy in 30 to 60 weeks aged Tg(OrPrP)Prnp0/0 mice. Mild vacuolation was seen in the heart muscle of 30 (a) and 40 (b) weeks old Tg(OrPrP)Prnp0/0 mice. (c) Vacuolation in the atrium area of the heart muscle of 60 weeks old Tg(OrPrP)Prnp0/0 mice. No inflammatory lesion was observed. H&E staining, Magnifications: (a) x400; (b) x400; (c) x200.
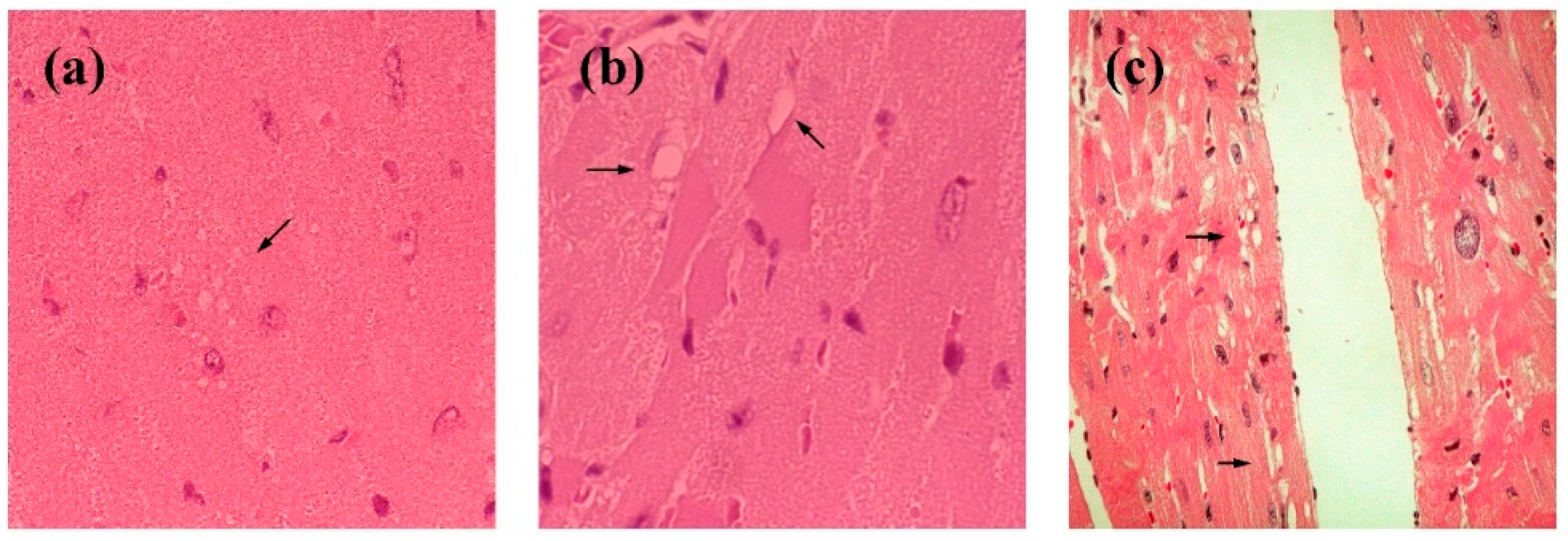
Figure 5.
Abnormal waveform of ECG in Tg(OrPrP)Prnp0/0 mice after atropine injection [12]. (a) Thirty-week aged Tg(OrPrP)Prnp0/0 (n = 3) and B6 (n = 3) mice were injected with atropine (4 mg/kg i.p.) and evaluated by ECG 5 minutes later. Arrows indicate the abnormal waveform, which is located on the QRS complex duration and P wave duration. (b) B6 control mice showed normal waveform in the same condition.
Figure 5.
Abnormal waveform of ECG in Tg(OrPrP)Prnp0/0 mice after atropine injection [12]. (a) Thirty-week aged Tg(OrPrP)Prnp0/0 (n = 3) and B6 (n = 3) mice were injected with atropine (4 mg/kg i.p.) and evaluated by ECG 5 minutes later. Arrows indicate the abnormal waveform, which is located on the QRS complex duration and P wave duration. (b) B6 control mice showed normal waveform in the same condition.
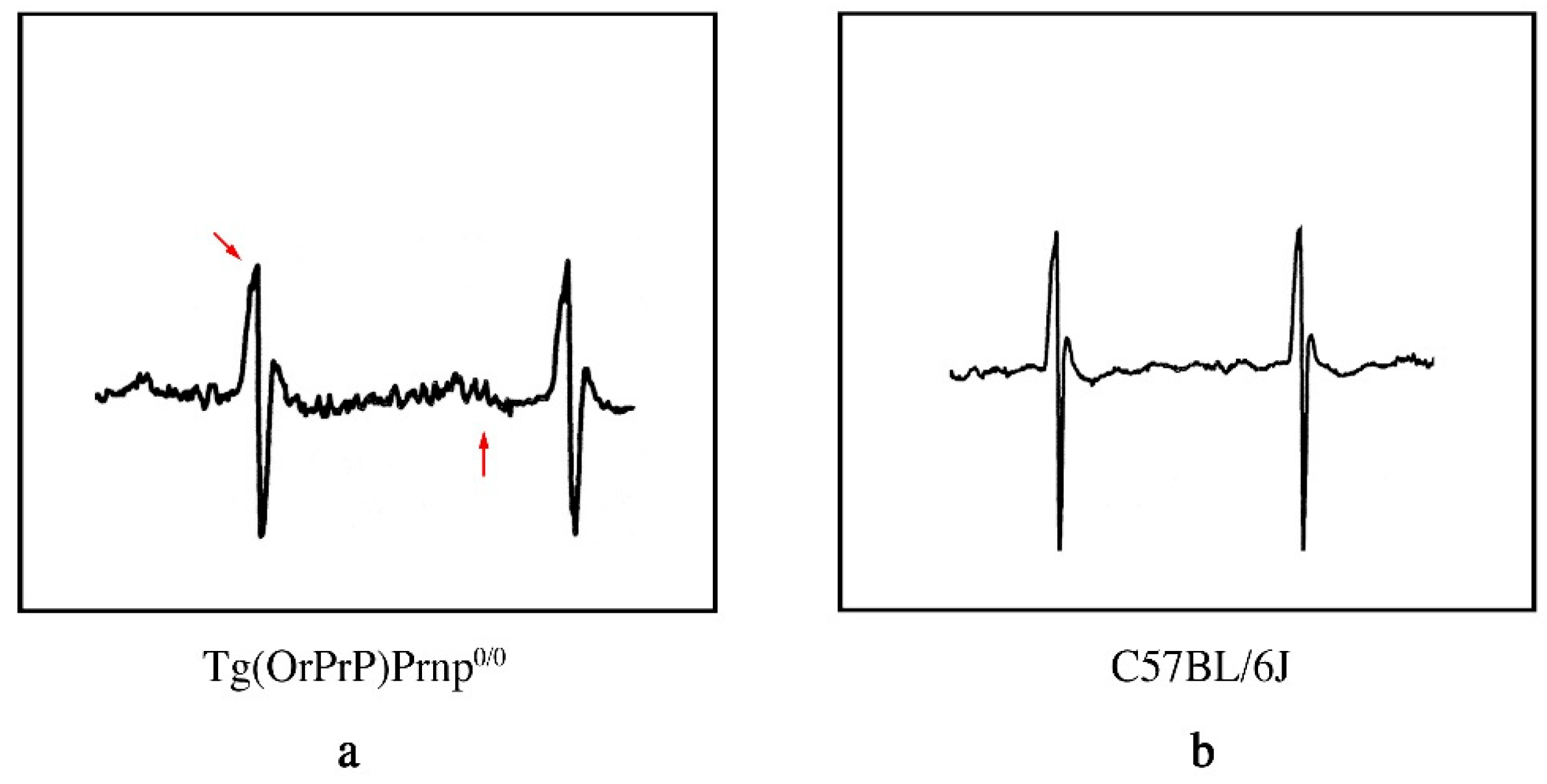
Figure 6.
(a) Dpl mRNA expression by RT-PCR. Dpl mRNA was seen in the brain and heart of Prnp0/0 (Lane 1, 2) and Tg(OrPrP)Prnp0/0 (Lane 5, 6) mice, but expression was not detectable in Prnp+/+ (Lane 3, 4) mice. The size marker (in bp) of Dpl mRNAs observed in the brain and heart was approximately 591 bp. Abbreviations: B, Brain; H, Heart. (b) Footprints of 15 months old Tg(OrPrP)Prnp0/0 (Lane 3), Prnp0/0 (Lane 1) and Prnp+/+ (Lane 2) mice. The posterior footpads of these mice were dipped in ink and the mice were allowed to walk on the filter paper. Prnp0/0 mouse is droopy due to low tonus and has bedraggled fur, perhaps because of deficient grooming. However, the Tg(OrPrP)Prnp0/0 mouse, by re-introducing OrPrP as well as Prnp+/+, was able to walk straight and show a normal step length.
Figure 6.
(a) Dpl mRNA expression by RT-PCR. Dpl mRNA was seen in the brain and heart of Prnp0/0 (Lane 1, 2) and Tg(OrPrP)Prnp0/0 (Lane 5, 6) mice, but expression was not detectable in Prnp+/+ (Lane 3, 4) mice. The size marker (in bp) of Dpl mRNAs observed in the brain and heart was approximately 591 bp. Abbreviations: B, Brain; H, Heart. (b) Footprints of 15 months old Tg(OrPrP)Prnp0/0 (Lane 3), Prnp0/0 (Lane 1) and Prnp+/+ (Lane 2) mice. The posterior footpads of these mice were dipped in ink and the mice were allowed to walk on the filter paper. Prnp0/0 mouse is droopy due to low tonus and has bedraggled fur, perhaps because of deficient grooming. However, the Tg(OrPrP)Prnp0/0 mouse, by re-introducing OrPrP as well as Prnp+/+, was able to walk straight and show a normal step length.
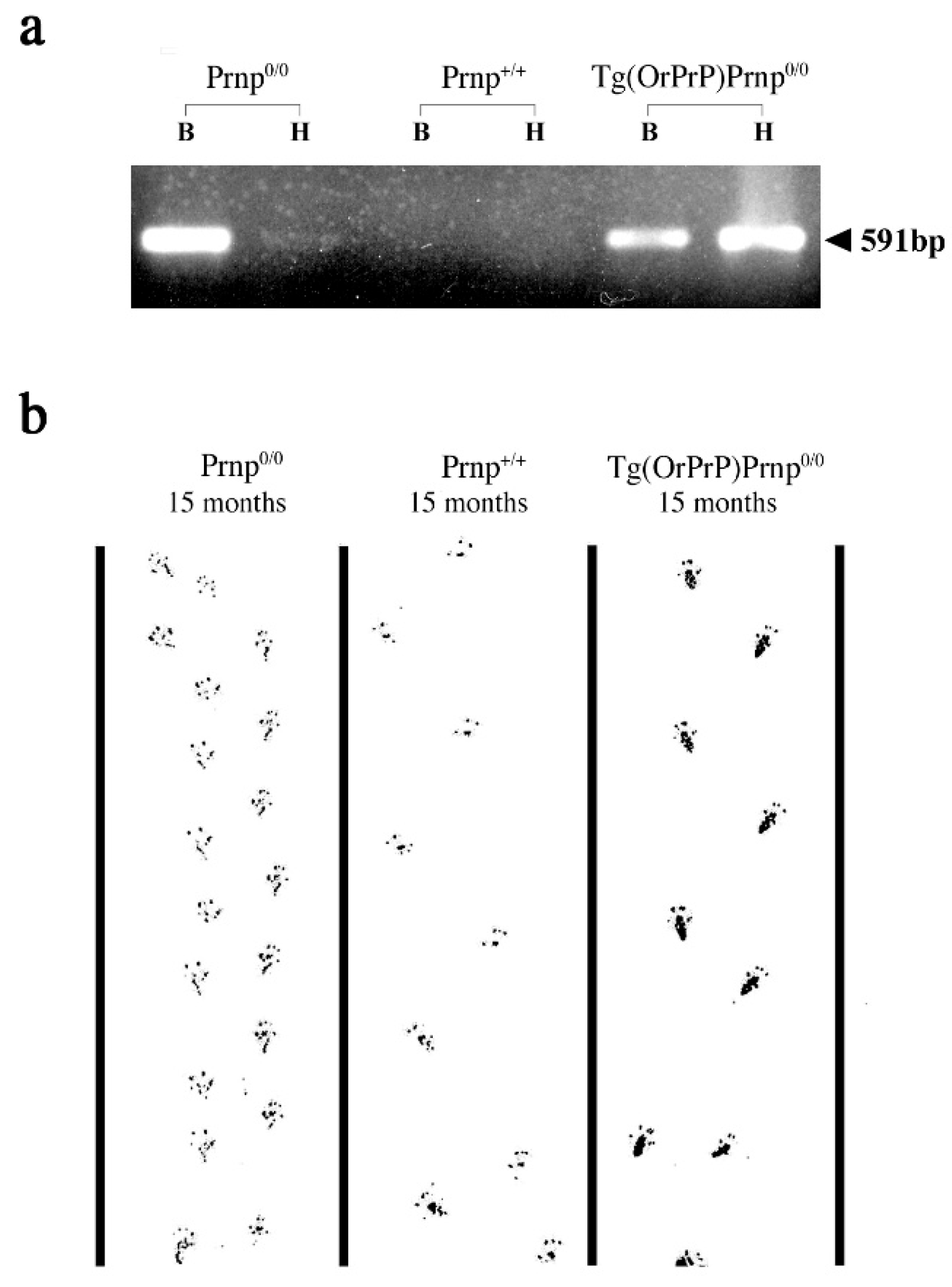
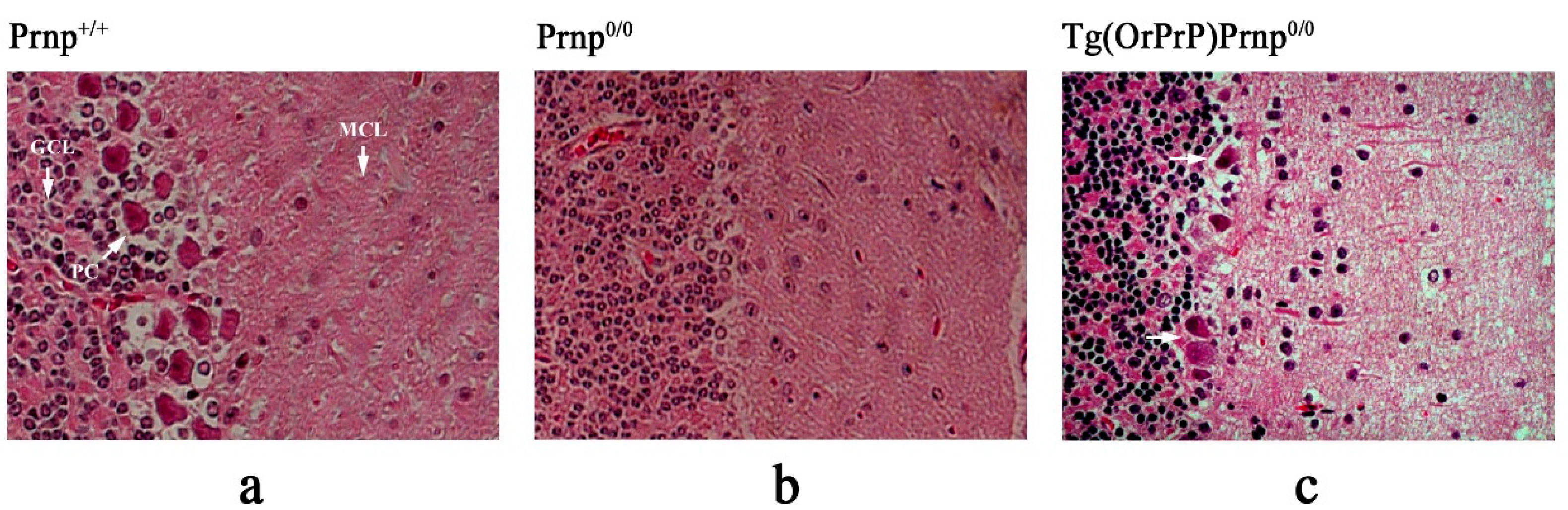
Figure 7.
Histological examination of the brains from 15 months old Tg(OrPrP)Prnp0/0, Prnp0/0 and Prnp+/+ mice. (a) No significant pathological changes were apparent in the cerebellar Purkinje cells from Prnp+/+ mice. Abbreviations: GCL, Granular Cell Layer; PC, Purkinje Cell; MCL, Molecular Cell Layer. (b) Prnp0/0 mice showed a widespread loss of cerebellar Purkinje cell at this age. (c) No significant pathological abnormalities were apparent in the brains from the elderly Tg(OrPrP)Prnp0/0 mice. However, compared with Prnp+/+ mice, limited neurodegeneration was seen in the cerebellar Purkinje cells of the Tg(OrPrP)Prnp0/0 mouse. Since similar lesions were seen in Prnp0/0 mice more severely, the protective effect of OrPrP may be present in this old Tg(OrPrP)Prnp0/0 mice. Arrows indicate the Purkinje cell. H&E staining. Magnifications: (a), x400; (b), x400; (c), x400.
Figure 7.
Histological examination of the brains from 15 months old Tg(OrPrP)Prnp0/0, Prnp0/0 and Prnp+/+ mice. (a) No significant pathological changes were apparent in the cerebellar Purkinje cells from Prnp+/+ mice. Abbreviations: GCL, Granular Cell Layer; PC, Purkinje Cell; MCL, Molecular Cell Layer. (b) Prnp0/0 mice showed a widespread loss of cerebellar Purkinje cell at this age. (c) No significant pathological abnormalities were apparent in the brains from the elderly Tg(OrPrP)Prnp0/0 mice. However, compared with Prnp+/+ mice, limited neurodegeneration was seen in the cerebellar Purkinje cells of the Tg(OrPrP)Prnp0/0 mouse. Since similar lesions were seen in Prnp0/0 mice more severely, the protective effect of OrPrP may be present in this old Tg(OrPrP)Prnp0/0 mice. Arrows indicate the Purkinje cell. H&E staining. Magnifications: (a), x400; (b), x400; (c), x400.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



