
Submitted:
01 December 2025
Posted:
02 December 2025
You are already at the latest version
Abstract
p53 tumor suppressor evolved as the critical player in navigating the response to environmental stresses such as DNA, or oxidative damage and drives the cell fate by governing the live and death decision. The p53 protein is encoded by the most commonly mutated gene in human cancers. TP53 gene mutations are associate with worse prognosis and refractory and relapse disease. The most prevalent mutations are of the missense type and often lead to disruption of DNA binding capacity. In healthy cells, p53 protein is tightly regulated by its E3 ubiquitin ligase, MDM2 (HDM2), it’s own transcription target. Mutant p53 therefore escapes the regulation by the negative feedback loop and is often found upregulated in cancer cells. The efforts to exploit mutant p53 for precision medicine has been ongoing in the last decade, yet not successful. One way to target TP53-mutant cancers would be by proximity, where abundant mutp53 protein serves as a molecular glue for a toxin inhibiting essential gene. The strategy might still require the adjustments but emerges as promising strategy for precision oncology.
Keywords:
MDM2
; mutant p53
; proximity
; PLK1
; PROTAC
; RIPTAC
Targeting Cancer Vulnerabilities by Rationally Designed Proximity
p53 is an intrinsically disordered protein with unresolved tertiary structure which makes the structure-based drug discovery a daunting task [1]. Missense mutations in the p53 are prevalent in human cancers, disrupt p53 DNA binding and underly poor prognosis. To date, no specific mutant p53 targeting molecule was approved by the Food and Drug Administration [2].
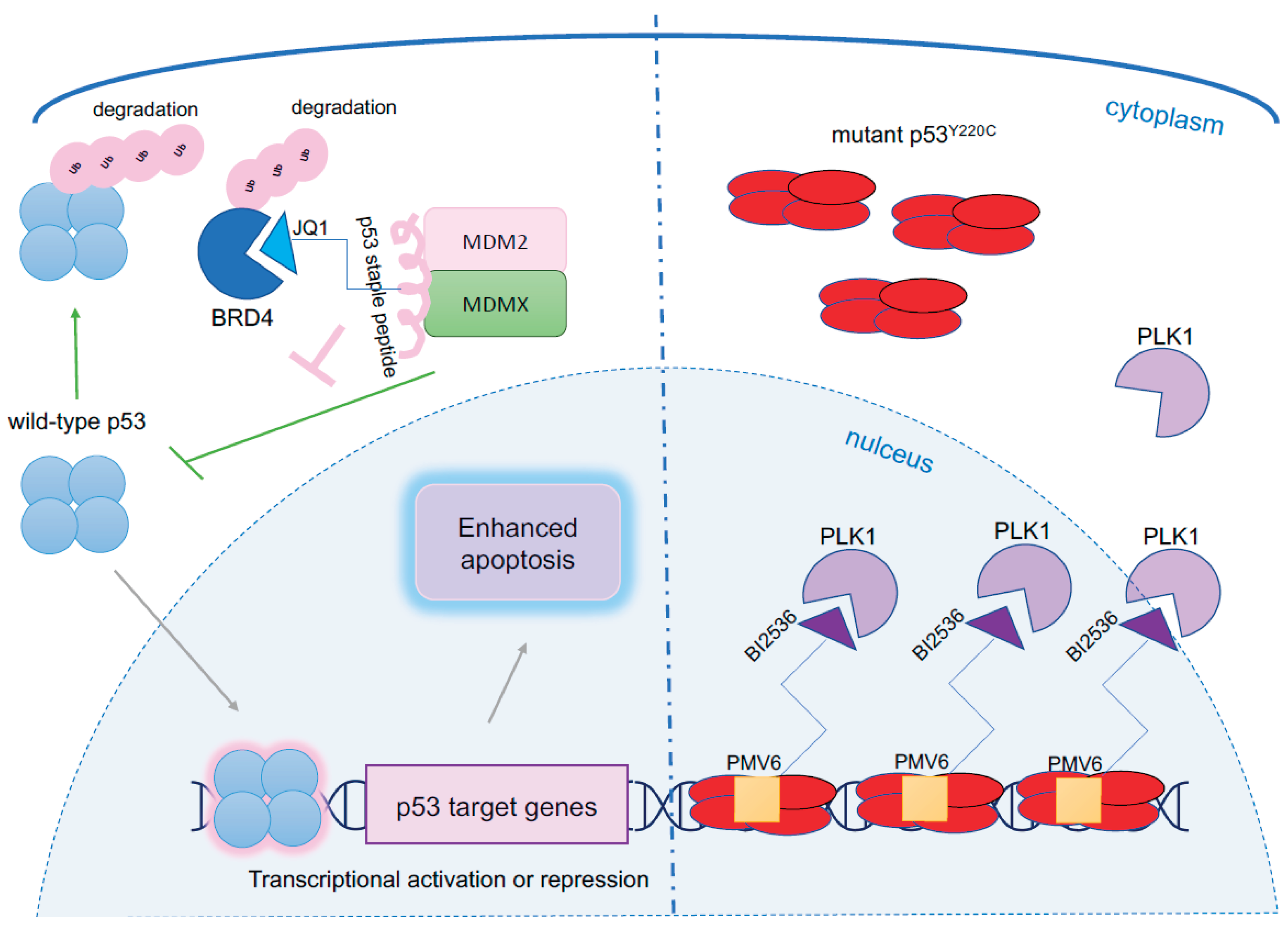
One of the promising approaches identified for targeting oncogenes including MDM2, is through employing targeted degradation using rationally designed proteolysis-targeting chimeras (PROTACs) [3] or harnessing regulated induced proximity-targeting chimeras (RIPTACs) [4]. Recently, a tri-functional PROTAC has been developed to induce apoptosis in wild-type p53 cancer cells. Bird et al. established a triple-action proteolysis targeting chimera (TAPTAC) utilizing stapled peptide technology that efficiently reactivates the biologically active alpha-helical structure to investigated peptide while enabling proteolytic resistance [5]. The authors employed a cell-permeable p53 stapled peptide which targets both, the HDM2 and HDM4(X) oncogenes, making it particularly efficient in restoring p53 activity in cancer cells (Figure 1). Molecules found to have a dual action and inhibit p53/HDM2/HDM4 interactions were recently reported [6] and are in clinical trial studies like sulanemadlin (ALRN-6924) [7]. The second component of the TAPTAC molecule includes JQ1, BET inhibitor which interacts with the BD1 domain of BRD4 (BRD4BD1) and induces its degradation. The resulting 3-in-1 approach delivers a highly precise killing degrader which simultaneously restores wild-type p53 and blocks the critical regulator of oncogenic pathways (Figure 1).
RIPTACs are another example of the molecules that allow for harnessing the proximity for the therapeutic purpose. They are designed so that the heterobifunctional compound facilitates a stable ternary complex between a target which is selectively expressed in tumor cells and a ubiquitously expressed protein essential for cell survival. The rationale behind RPITACs has been recently applied by Sadagopan et al [8] to target mutant p53-loaded cancer cells.
p53 mutants are highly abundant in mutTP53 mono- and biallelic cancer cells as they escape the p53-HDM2 negative regulatory feedback loop. Now, Sadagopan et al., showed and confirmed in a genome-wide analysis of CRISPR dependencies that this abundance appears to be the only genetic or proteomic distinction between TP53-mutant and TP53-wild-type cancer cells and thus, provides a unique therapeutic opportunity. To take advantage of the mutp53 abundance, first the team, generated HEK293-derived and Calu-1-derived cell lines overexpressing Halo-tag mutant p53 (Halo–p53-R273H (FL)–mCherry) or wild-type p53 (Halo–p53 WT (FL)–mCherry–2A–mTagBFP2-V5) and treated them with a novel, bifunctional molecule liganding with the tagged mutp53s and containing PLK1 inhibitor, Halo-PEG2-BI2536. PLK1 has high essentiality and low abundance in cells as identified by comparing the CRISPR gene essentiality scores across Dependency Map (DepMap) and absolute protein abundance profiled in OpenCell. PLK1 is recognized as an oncogene, critical for mitotic progression and BI2536 inhibitor, was shown to induce G2/M arrest. Bifunctional molecule selectively inhibited proliferation of TP53R273H and TP53Y220C-mutant cells with a good therapeutic window which was more prominent in Calu-1 cells.
The Y220C mutation account for around 1% of all missense mutations and is exploited for improved cancer therapy. Thus, next, the authors tested the bifunctional binder containing PMV6 molecule, own by PMV Pharma, which induces thermostabilisation of p53Y220C core domain by 8°C and binds to the unique pocket created by the Y220C substitution. The activity of the bifunctional molecule containing PMV6 and BI2536 (annotated as p53-01) was assessed to be at EC50 = 1.4 μM, and as reflected by the induction of mitotic arrest and apoptosis. Upon p53-01 treatment, PLK1 colocalized with p53Y220C on chromatin. Authors speculated that such mislocalization could significantly contribute to the disruption of the function of PLK1 as a mitotic kinase and thus, beyond simple steric blockade of its active site. The response in cells was mutp53Y220C dependent, yet without reactivation of mutant p53 protein to wild-type protein and induction of p53 response program. Finally, the team performed linker optimization to generate PMV6-C3-BI2536 molecule which was more potent than p53-01 by >1.5 order of magnitude.
Thus, the bifunctionals targeting mutant p53 abundance and essential genes with the previously identified toxins, might open up the new avenues for the development of more selective and specific treatments for mutTP53 cancers.
Acknowledgements
The work was supported by the Cancercentrum Karolinska Stiftelse.
References
- M. Behzadi, J. E. M. Behzadi, J. E. Zawacka, Preprints.org (www.preprints.org) 2025. [CrossRef]
- Tuval, C. Strandgren, A. Heldin, M. Palomar-Siles, K.G. Wiman, Pharmacological reactivation of p53 in the era of precision anticancer medicine., Nat. Rev. Clin. Oncol. 21 (2024) 106–120. [CrossRef]
- C.M. Adams, R. C.M. Adams, R. Mitra, Y. Xiao, P. Michener, J. Palazzo, A. Chao, J. Gour, J. Cassel, J.M. Salvino, C.M. Eischen, Targeted MDM2 Degradation Reveals a New Vulnerability for p53-Inactivated Triple-Negative Breast Cancer., Cancer Discov. 13 (2023) 1210–1229. [CrossRef]
- K. Raina, C.D. K. Raina, C.D. Forbes, R. Stronk, J.P. Rappi, K.J. Eastman, N. Zaware, X. Yu, H. Li, A. Bhardwaj, S.W. Gerritz, M. Forgione, A. Hundt, M.P. King, Z.M. Posner, A.D. Correia, A. McGovern, D.E. Puleo, R. Chenard, J.J. Mousseau, J.I. Vergara, E. Garvin, J. Macaluso, M. Martin, K. Bassoli, K. Jones, M. Garcia, K. Howard, M. Yaggi, L.M. Smith, J.M. Chen, A.B. Mayfield, C.A. De Leon, J. Hines, K.J. Kayser-Bricker, C.M. Crews, Regulated induced proximity targeting chimeras-RIPTACs-A heterobifunctional small molecule strategy for cancer selective therapies., Cell Chem. Biol. 31 (2024) 1490-1502.e42. [CrossRef]
- G.H. Bird, U. G.H. Bird, U. Adhikary, M.J. Schmidt, M. Godes, B. Tesar, C.M. Camara, J.A. Paulo, J.F. Vidlak, T.M. DeAngelo, M. Marquez, P. Gokhale, R. Li, S.J. Ho Sui, M.D. Cameron, S.P. Gygi, L.D. Walensky, A triple-action PROTAC for wild-type p53 cancer therapy., Cell Rep. Med. (2025) 102467. [CrossRef]
- V.V. Grinkevich, A. V.V. Grinkevich, A. Vema, K. Fawkner, N. Issaeva, V. Andreotti, E.R. Dickinson, E. Hedström, C. Spinnler, A. Inga, L.-G. Larsson, A. Karlén, M. Wilhelm, P.E. Barran, A.L. Okorokov, G. Selivanova, J.E. Zawacka-Pankau, Novel Allosteric Mechanism of Dual p53/MDM2 and p53/MDM4 Inhibition by a Small Molecule., Front. Mol. Biosci. 9 (2022) 823195. [CrossRef]
- V. Guerlavais, T.K. V. Guerlavais, T.K. Sawyer, L. Carvajal, Y.S. Chang, B. Graves, J.-G. Ren, D. Sutton, K.A. Olson, K. Packman, K. Darlak, C. Elkin, E. Feyfant, K. Kesavan, P. Gangurde, L.T. Vassilev, H.M. Nash, V. Vukovic, M. Aivado, D.A. Annis, Discovery of Sulanemadlin (ALRN-6924), the First Cell-Permeating, Stabilized α-Helical Peptide in Clinical Development., J. Med. Chem. 66 (2023) 9401–9417. [CrossRef]
- Sadagopan, M. Carson, E.J. Zamurs, N. Garaffo, H.-J. Chang, S.L. Schreiber, M. Meyerson, W.J. Gibson, Mutant p53 protein accumulation is selectively targetable by proximity-inducing drugs., Nat. Chem. Biol. (2025). [CrossRef]
Figure 1.
Harnessing p53 for proximity killing. Left panel: A strategy using tri-functional molecular glue TAPTAC which inhibits MDM2 and MDM4 by hyper-stable, cell-permeable p53-derived staple peptide and uses JQ1 molecule, a potent inhibitor of oncogenic BRD4 for induction of massive apoptosis. Right panel: p53-01, a novel RIPTAC which exploits highly abundant mutant p53Y220C to drive the accumulation of essential gene PLK1 in the nucleus which inhibits proliferation and indues cell death.
Figure 1.
Harnessing p53 for proximity killing. Left panel: A strategy using tri-functional molecular glue TAPTAC which inhibits MDM2 and MDM4 by hyper-stable, cell-permeable p53-derived staple peptide and uses JQ1 molecule, a potent inhibitor of oncogenic BRD4 for induction of massive apoptosis. Right panel: p53-01, a novel RIPTAC which exploits highly abundant mutant p53Y220C to drive the accumulation of essential gene PLK1 in the nucleus which inhibits proliferation and indues cell death.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



