Submitted:
28 November 2025
Posted:
01 December 2025
You are already at the latest version
Abstract
Caveolin-1 (Cav-1) is a protein found in various forms and locations within cells and tissues throughout the body. Studying its structure and function provides valuable in-sights into key cellular processes such as growth, death, and cell signalling. Expanding our understanding of Cav-1 and its roles across different organs and disease conditions may reveal its potential as a target for developing novel treatments and therapeutic strategies. Cav-1 is a master regulator of endothelial nitric oxide synthase (eNOS), tonically inhibiting its activity to ensure balanced nitric oxide (NO) production. While this mechanistic relationship is well defined in endothelial cells, its role in the pathobi-ology of atherosclerosis remains complex, particularly within perivascular adipose tissue (PVAT), a layer of fat surrounding blood vessels. PVAT is not merely a structural support; it is a metabolically active tissue capable of producing NO, adipokines, and inflammatory mediators, and it also expresses Cav-1. These characteristics allow PVAT to directly influence vascular function and may contribute to the paradoxical roles of Cav-1 observed in different stages and sites of atherosclerosis.
The Problem: Human studies present a challenge: loss of function Cav-1 mutations are linked to pulmonary arterial hypertension (PAH), suggesting a protective vascular role. Conversely, in atherosclerosis, Cav-1 is frequently upregulated in diseased vessels but whether this reflects increased expression, altered function, or both remains un-clear. In addition, does this upregulation reflect a compensatory mechanism to regu-late uncoupled eNOS, or a pathological process that aggravates disease progression by further reducing bioactive NO availability?
The vast majority of studies establishing the Cav-1/eNOS interaction (where Cav-1 tonically inhibits eNOS, preventing its hyperactivation) have been conducted in endo-thelial cells from mice, rats, or cultured cell lines. The direct extrapolation of this mechanism to perivascular adipose tissue (PVAT), a distinct tissue with adipocytes, immune cells, and stromal cells, is not fully validated except in a few studies.
Keywords:
caveolin-1
; low-density lipoprotein (LDL)
; transcytosis
; atherosclerosis
; caveolae
; endothelial cells
1. Introduction
1.1. Atherosclerosis: An Overview:
Atherosclerosis is a chronic and multifactorial vascular disease characterized by the progressive accumulation of lipids, inflammatory cells, and fibrous material within the subendothelial space of large and medium-sized arteries. The process begins with endothelial dysfunction, leading to enhanced vessel permeability, leukocyte adhesion, and lipid infiltration. Low-density lipoprotein (LDL) particles become oxidized in the intima, triggering an inflammatory response that recruits monocytes, which differentiate into macrophages and form lipid-laden foam cells. Over time, smooth muscle cells migrate from the media to the intima, contributing to extracellular matrix deposition and fibrous cap formation. These changes result in the development of atherosclerotic plaques, which can gradually narrow the arterial lumen, impair blood flow, and destabilize to cause acute cardiovascular events such as myocardial infarction or stroke. Thus, atherosclerosis represents not only a disorder of lipid metabolism but also a chronic inflammatory condition that integrates vascular, immune, and metabolic pathways [1,2].
1.2. Caveolae and Caveolins in Vascular Biology
The caveolae The caveolae was first described by Eichi Yamada in 1955 in gall bladder epithelial cells using early electron microscopy, as characteristic flask-shaped invaginations of the plasma membrane. He coined the term “caveola intracellularis” (Latin for “little cave of the cell”) to describe these invaginations he observed, thereby giving them the name that remains in use today [3]. Decades later, the discovery of caveolin proteins (Cav-1, Cav-2 and Cav-3) as the principal scaffolding protein of caveolae provided mechanistic insights into their role in vascular biology and disease.
For decades following their discovery, caveolae remained a morphological curiosity. Their function was subject to speculation, with early hypotheses focusing on their role in endocytosis and transcytosis (the transport of materials across cells). It was not until the late 1980s and 1990s, with the advent of molecular biology and the identification of Caveolin-1 as the principal caveolar protein, that a clearer understanding of their function began to emerge.
A fundamental breakthrough came in 1992 with the identification and cloning of the first structural component of caveolae, a 22-kDa protein named Caveolin (now known as Caveolin-1 and abbreviated to Cav-1) by Richard Anderson and colleagues [4]. This discovery provided the impetus for studying these previously elusive structures in more detail. The subsequent generation of Cav-1 knockout mice in the late 1990s was another landmark discovery; these mice completely lacked caveolae, confirming that Cav-1 was crucial for their formation and allowing researchers to directly investigate the functional consequences of their absence [5,6].
Caveolae range in size between 50-100 nm and serve as crucial organizing centres for cellular signalling complexes. To date, three caveolin proteins have been identified Cav-1, Cav-2 and Cav-3, comprising six distinct subtypes, each has two main isoforms Cav-1α/β, Cav-2α/β, and Cav-3α/β, that share high sequence homology but differ in tissue distribution, cellular localization, and likely physiological roles in signal transduction. Notably, caveolin composition varies by cell type: Cav-1 and Cav-2 predominate in endothelial and adipose cells, whereas Cav-3 is largely restricted to skeletal and cardiac muscle. These specialized membrane microdomains are enriched in cholesterol and sphingolipids and are found in most terminally differentiated cells, particularly endothelial cells, adipocytes, and muscle cells [7,8].
Caveolae are abundant in endothelial cells and other vascular cell types and serve as organizing platforms for receptors, ion channels, and signalling complexes that control vascular homeostasis. The structural and functional integrity of caveolae depends primarily on the caveolin family of proteins, which comprise three members with distinct tissue distributions. Caveolin-1 (Cav-1 α/β) and caveolin-2 (Cav-2 α/β/γ) are co-expressed and co-localized in many tissues, whereas caveolin-3 (Cav-3) is predominantly expressed in muscle, including cardiomyocytes and skeletal muscle. Notably, Cav-3 shares less sequence similarity with Cav-2 than with Cav-1 [9,10,11,12,13].
Full-length Cav-1 is a 178-amino-acid protein (Figure 1) and is the major structural component of caveolae in the plasma membrane. Beyond its structural role, Cav-1 functions as a scaffolding protein, binding to signaling molecules and participating in processes such as cell signaling and membrane trafficking [14]. The protein adopts a unique hairpin-like configuration with both N- and C-termini facing the cytoplasm. The protein’s structure includes several critical domains: an N-terminal domain (residues 1-81), a scaffolding domain (residues 82-101), a transmembrane domain (residues 102-134), and a C-terminal domain (residues 135-178) [8].
The scaffolding domain is particularly important as it facilitates interactions with numerous signaling molecules including endothelial nitric oxide synthase (eNOS), heterotrimeric G proteins, Src family kinases, Ras, protein kinase C (PKC), and transforming growth factor-β (TGF-β) receptors, through recognition of specific caveolin-binding motifs, this allows direct protein–protein interactions with molecules that contain caveolin-binding motifs (CBMs), generally defined by aromatic-rich consensus sequences such as ΦXΦXXXXΦ ΦXXXXΦXXΦ (where Φ represents an aromatic residue like tryptophan, phenylalanine, or tyrosine). Through this molecular recognition, the scaffolding domain acts as a regulatory hub, modulating the activity of kinases, G-proteins, eNOS, and other key effectors involved in vascular homeostasis, lipid metabolism, and cellular proliferation. By sequestering these signaling molecules within caveolae, Cav-1 can negatively regulate their activation states, maintaining a controlled balance between stimulation and inhibition under physiological conditions [13]. Dysregulation or mutation of this domain may therefore disrupt critical signaling cascades, leading to altered nitric oxide production, endothelial dysfunction, and the pathogenesis of cardiovascular and metabolic diseases.
In addition, Cav-1 plays a complex role in cellular physiology, contributing to cholesterol distribution, extracellular matrix organization, endocytosis, exocytosis, cell migration and compartmentalization of signal transduction, This is particularly prominent in adipocytes due to their high lipid content and their central role in metabolic storage and endocrine signaling [15,16]. While caveolae are present in most cell types, they are especially abundant in adipocytes, constituting up to 30% of the plasma membrane surface area [17]. In these cells, Cav-1 is the predominant caveolin isoform and is essential for caveolae formation [18]. Furthermore, Cav-1 expression is significantly upregulated during adipocyte differentiation through DNA demethylation, and it has been identified as a key mediator of the insulin signaling pathway., In the adipocyte the Cav-1 interacts directly with the insulin receptor and facilitates its localization within caveolae, promoting efficient signal transduction through the PI3K–Akt pathway. This interaction enhances glucose uptake and lipid accumulation, processes central to adipocyte metabolism and systemic energy homeostasis [19]. Beyond these roles, Cav-1 also critically regulates eNOS activity, highlighting its importance in both metabolic and vascular contexts [7].
Many studies have confirmed that Cav-1 undergoes several post-translational modifications that regulate its function, stability, and role in cellular signaling. Phosphorylation occurs primarily at tyrosine 14 (Tyr14) and serine 80 (Ser80) within the N-terminal domain, influencing both caveolae assembly and signal transduction pathways [14]. Additionally, palmitoylation at three cysteine residues (C133, C143, and C156) near the C-terminal domain anchors Cav-1 to the membrane and facilitates its role in organizing lipid raft microdomains [20]. Furthermore, Cav-1 is subject to ubiquitination, particularly at N-terminal lysine residues, which targets the protein for proteasomal degradation and serves as a key mechanism controlling its turnover and homeostasis (Figure 1), as demonstrated in studies using HEK293 cells [21]. Together, these post-translational enable Cav-1 to function as a dynamic sensor and regulator of cellular homeostasis, rapidly adapting its localization and signaling interactions in response to environmental and metabolic factors [8,22].
Cav-2 is typically co-expressed and co-localized with Cav-1, forming heterooligomeric complexes that are essential for the stabilization and structural integrity of caveolae. These complexes facilitate proper trafficking of caveolar proteins, maintain membrane curvature, and coordinate interactions with multiple signaling molecules. While Cav-2 is often viewed as a structural partner of Cav-1, emerging evidence indicates that Cav-1 and Cav-2 may exhibit distinct or even antagonistic functions depending on the cellular context, particularly in processes such as inflammation, cell proliferation, and vascular remodeling. In practical terms, this means that Cav-2 can sometimes counteract Cav-1-mediated signaling or alter caveolae dynamics, potentially affecting their stability or function. For example, in some cell types, Cav-2 overexpression can inhibit Cav-1-dependent pathways involved in cell proliferation or inflammation, suggesting that the balance between the two isoforms is critical for maintaining normal caveolar activity and vascular homeostasis [23,24].
Cav-3 shares approximately 65% amino acid identity and 85% overall similarity with Cav-1, reflecting a high degree of evolutionary conservation, particularly within the scaffolding and membrane-spanning domains that are essential for caveolae formation [15,25]. Functionally, Cav-3 is the muscle-specific isoform required for the generation of caveolae in myocytes, contributing to mechanoprotection, calcium homeostasis, and regulation of nitric oxide (NO) signaling. Although Cav-3 is restricted to muscle tissue, it can also influence eNOS activity indirectly, for example through paracrine or signaling interactions at the muscle endothelium interface. [26,27]. Mutations or loss of Cav-3 expression have been linked to a range of muscle disorders, including limb-girdle muscular dystrophy type 1C (LGMD-1C), rippling muscle disease, and familial hypertrophic cardiomyopathy, emphasizing its essential structural and signaling roles in muscle integrity [28,29].
Cav-1 is a 178–amino acid membrane protein composed of several distinct regions: an N-terminal domain (red), a scaffolding domain spanning residues 82–101 (black), a hydrophobic hairpin transmembrane region from 102–134 (orange), and a C-terminal domain extending from 135–178. It undergoes several posttranslational modifications, including phosphorylation at Tyr14 and Ser80, ubiquitination of N-terminal residues, and palmitoylation at sites within the C-terminus all of which are critical for its regulation and function.
2. Role of Caveolae Microdomains in eNOS Localization and Activity: A Central Regulatory Mechanism
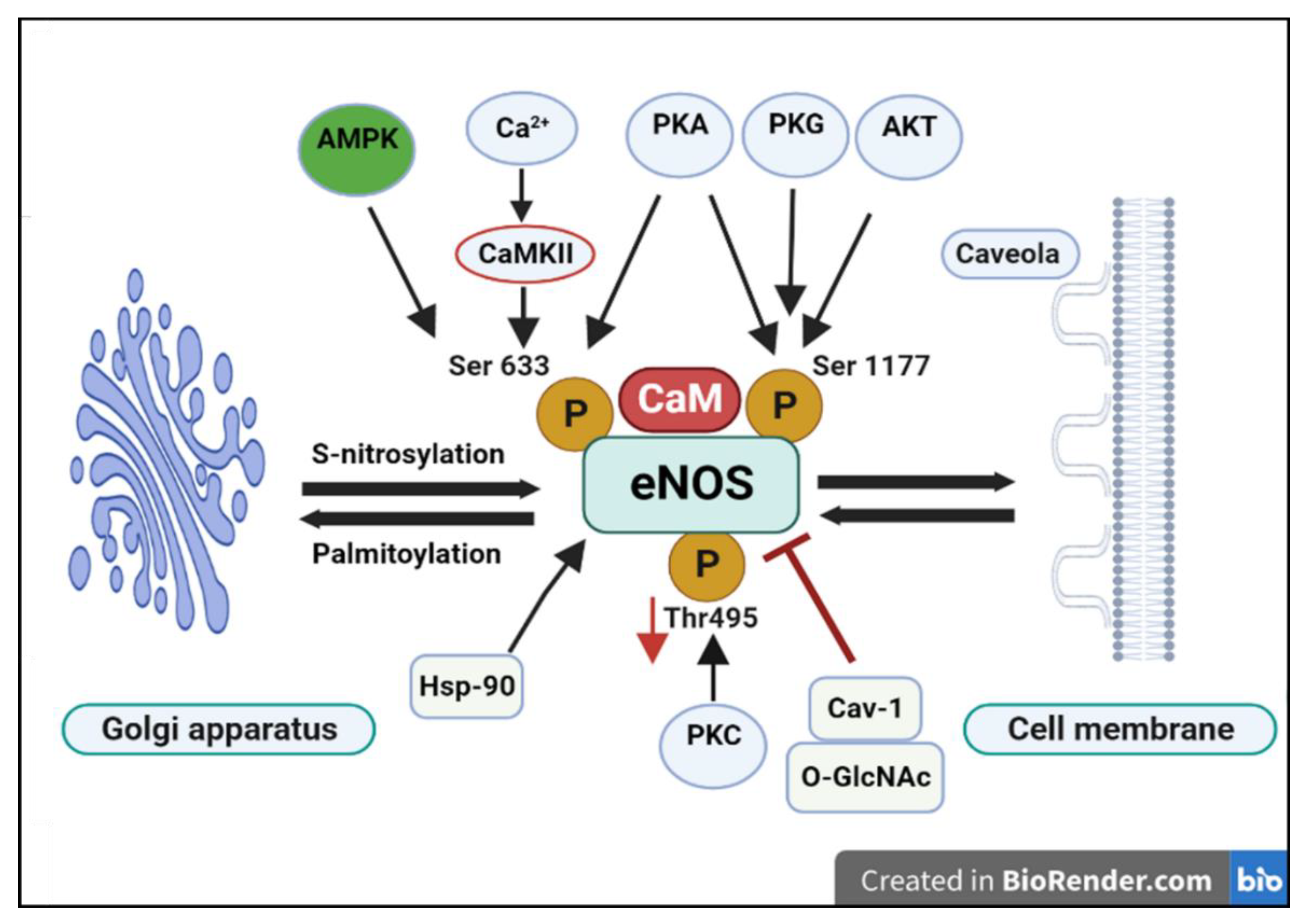
In endothelial cells the subcellular localization of eNOS is a critical determinant of its activity and NO production [30]. Upon translocation to the plasma membrane, eNOS interacts with Cav-1, leading to suppressed enzymatic activity and reduced NO synthesis [7,31]. The interaction between Cav-1 and eNOS represents one of the most extensively studied aspects of caveolar biology in terms of vascular regulation. In endothelial cells, Cav-1 serves as a tonic inhibitor of eNOS by binding to the enzyme via its scaffolding domain and sequestering it within caveolae [32]. This interaction involves specific residues within the scaffolding domain (particularly F92) that disrupt eNOS’s association with Ca²⁺/calmodulin (CaM), thereby maintaining the enzyme in an inactive state under basal conditions. Physiological stimuli (shear stress, agonists) promote eNOS release from caveolae, calmodulin binding, and a consequent increase in NO production. This spatial control of eNOS is critical for healthy blood vessel physiology and inappropriate Cav-1 sequestration reduces NO bioavailability (favouring vasoconstriction and pro-atherogenic signaling), whereas loss of Cav-1 can lead to eNOS hyperactivation and uncoupling, producing superoxide rather than NO and promoting oxidative stress [26,34,35].
Upon appropriate stimulation (e.g., by shear stress, acetylcholine, or bradykinin), intracellular Ca²⁺ levels rise, promoting CaM binding to eNOS and its dissociation from Cav-1. This activation process is further enhanced by phosphorylation at Ser1177 via various kinases including Akt, PKA, and AMPK. Cav-1 directly binds eNOS via its scaffolding domain (amino acids 82–101; DGIWKASFTTFTVTKYWFY), disrupting the enzyme’s association with Ca²⁺/calmodulin (CaM). Within this region, residues 89–95 (FTTFTVT), particularly F92, are essential for mediating this inhibition [6,26,36,37]. Details on eNOS regulation are shown schematically in Figure 2.
The importance of this interaction has been shown using a cell-permeable mutant peptide corresponding to the Cav-1 scaffolding domain which disrupts Cav-1’s inhibitory effect on eNOS, thereby restoring NO production [7,38]. Additionally, in rat vaginal endothelial cells, estrogen and VEGF have been reported to enhance eNOS phosphorylation at Ser1177 via Akt activation, which reduces the interaction between eNOS and Cav-1 [42]. Beyond direct regulation of eNOS, Cav-1 also negatively modulates the small GTPase Rac1, which influences cell motility, growth, cytoskeletal organization, transcription, and proliferation [40,41,42]. Rac1, in turn, affects the PI3K/Akt/eNOS signaling pathway in endothelial cells [43,44]. Moreover, siRNA-mediated Cav-1 knockdown in bovine aortic endothelial cells (BAECs) has been shown to enhance AMPK phosphorylation, resulting in increased eNOS activity [45].
Despite these well characterized roles in endothelial cells, the role of Cav-1 in perivascular adipose tissue (PVAT), a key paracrine regulator of vascular function has received growing attention and adds complexity to the traditional understanding of Cav-1 regulation of eNOS and vascular function. PVAT surrounds most conduit and resistance vessels and secretes adipokines, cytokines, and reactive oxygen species (ROS) that can either promote anticontractile (vasodilatory) effects or, under metabolic stress, shift to a pro-contractile, pro-inflammatory phenotype. The PVAT microenvironment contains adipocytes, immune cells, and stromal cells whose signaling can influence endothelial eNOS activity indirectly (via adipokines) and possibly directly through changes in Cav-1 expression or post-translational modification. Recent reviews emphasize PVAT’s central role in redox balance and inflammatory crosstalk with the vessel wall, making PVAT a strategic site to re-examine Cav-1 biology in vascular disease [32,46,47].
Recent mechanistic studies have refined this model by demonstrating bidirectional regulation between Cav-1 and eNOS and by mapping phosphorylation and trafficking events that control eNOS between caveolar and non-caveolar pools [48].
eNOS activity is enhanced through phosphorylation at Ser1177 and Ser633, while phosphorylation at Thr495 exerts an inhibitory effect. Ser1177 can be phosphorylated by multiple upstream kinases, including Akt, protein kinase G (PKG), and protein kinase A (PKA). Similarly, Ser633 phosphorylation is mediated by Ca2+/calmodulin-dependent kinase II (CaMKII), PKA, and AMP-activated protein kinase (AMPK). In contrast, protein kinase C (PKC) phosphorylates the inhibitory Thr495 residue. Furthermore, eNOS activity is suppressed by its interaction with Cav-1 and by O-GlcNAc modification, both of which reduce nitric oxide (NO) production. Conversely, binding to heat shock protein 90 (Hsp90) promotes eNOS activation. Post-translational modifications such as S-nitrosylation and palmitoylation facilitate eNOS trafficking from the Golgi apparatus to the plasma membrane. Created with BioRender.com.
3. The Paradox of Cav-1 in Atherosclerosis: Protective vs. Proatherogenic Roles:
The role of Cav-1 in atherosclerosis represents a compelling and cell-type-specific paradox that has become a central focus of recent cardiovascular research. Genetic ablation studies in mice demonstrate that complete loss of Cav-1 confers significant protection against atherosclerotic lesion formation, primarily due to the absence of Cav-1-mediated inhibition of eNOS, resulting in enhanced NO bioavailability and improved endothelial function. Conversely, accumulating evidence indicates that Cav-1 also plays crucial homeostatic roles in vascular integrity, lipid regulation, and mechanotransduction. Under physiological conditions, Cav-1 supports endothelial barrier function, facilitates reverse cholesterol transport, and modulates redox balance, all of which are protective against early vascular injury. However, when Cav-1 is excessively expressed, post-translationally modified, or dysregulated within a pro-inflammatory or insulin-resistant milieu, it can suppress eNOS activity, promote oxidative stress, and enhance vascular inflammation. Thus, Cav-1 functions as a double-edged sword its optimal expression is vital for endothelial stability and vascular health, but its deficiency or overexpression may each promote distinct pathogenic processes that contribute to atherosclerosis. This intricate balance underscores the paradoxical nature of Cav-1 in vascular biology and the need to understand its cell-type–specific and context-dependent effects in atherogenesis. [31,32,49,51,52].
4. PVAT Inflammation and Vascular Dysfunction: Early Event or Late Response?
Perivascular adipose tissue (PVAT) is not merely a passive fat depot; it actively communicates with the vascular wall. In response to metabolic stressors such as obesity, hypertension, or hyperlipidemia, PVAT exhibits early inflammatory changes, including macrophage infiltration, increased cytokine release, and oxidative stress. Evidence suggests that PVAT inflammation can lead to atherosclerotic lesion formation, acting as an initiating factor that primes endothelial dysfunction. Alternatively, in some diseases, PVAT inflammation may be amplified by local vascular injury and lipid accumulation, creating a feed-forward loop that exacerbates lesion progression. These dynamics highlight the importance of temporal and spatial context when studying PVAT’s influence on vascular homeostasis and the Cav-1/eNOS axis. [53,54,55].
5. Anti-Atherogenic / Protective Effects of Cav-1
The relationship between Cav-1 and endothelial nitric oxide synthase (eNOS) in atherosclerosis is best understood as a context-dependent paradox rather than a simple binary effect. On one hand, Cav-1 helps maintain endothelial barrier integrity, regulates mechanosensing, and contributes to cholesterol trafficking; functions that can be atheroprotective by limiting vascular leakage and preserving vascular homeostasis. In several experimental models, loss of Cav-1 integrity has been shown to increase endothelial permeability and inflammation, supporting a protective role for Cav-1 in early vascular health [56].
Recent research elucidates a critical pathway linking Sirtuin1 (Sirt1), microRNA-204 (miR-204), Cav-1, and endothelial function. This study by Kassan et al. (2017) demonstrates that endothelial deletion of Sirt1 leads to increased miR-204 expression, resulting in reduced Cav-1 levels and impaired endothelium-dependent vasorelaxation. These effects were reversed by administering a miR-204 inhibitor or by overexpressing Cav-1, indicating that Sirt1 preserves endothelial function by suppressing miR-204-mediated Cav-1 downregulation, reduced Cav-1 expression impairs endothelium dependent vasorelaxation in both large (conductance) and small (resistance) arteries [67].
Caveolae and Cav-1 play a central role in reverse cholesterol transport (RCT) via interaction with transporters such as ATP-binding cassette subfamily G member 1 (ABCG1), facilitating cholesterol efflux and protecting against lipid accumulation in the vessel wall, while simultaneously modulating inflammatory signaling through pathways such as PPARγ and TLR-mediated responses [47,58]. Inflammation can impair the RCT pathway, promoting foam cell formation and plaque development, whereas elevated circulating lipids particularly low-density lipoprotein (LDL) cholesterol can activate pro-inflammatory signaling, underscoring a bidirectional interplay between lipid metabolism and vascular inflammation. Thus, although Cav-1 has emerged as a key integrator linking RCT and inflammatory signaling, its net effect in atherosclerosis depends on the balance between lipid handling, inflammatory status, and endothelial function.
Overexpression of Cav-1 in hepatic cells enhances cholesterol efflux, which may help prevent cholesterol accumulation in the arterial wall and slow the development of atherosclerosis. While the effect appears cell type-specific and may be most relevant to liver cells, overexpression of Cav-1 in the liver has also been shown to increase plasma HDL levels, potentially enhancing RCT and offering further anti-atherogenic protection. The protective effects of Cav-1 may be additive with ABCA1-mediated cholesterol efflux, and proper intracellular localization and interaction with other caveolins (e.g., Cav-2) may be critical for its function. Additionally, Cav-1 overexpression inhibits vascular smooth muscle cell (VSMC) proliferation, which constitutes a pivotal step in neointimal hyperplasia and plaque growth. By limiting VSMC proliferation and migration, Cav-1 may therefore exert a stabilizing influence on the vessel wall and slow atherosclerotic progression. These findings suggest that targeted modulation of Cav-1 expression could be a promising strategy for the prevention and treatment of atherosclerosis [59,60].
Evidence from studies on diabetic human coronary arterioles demonstrates that oxidative stress-induced peroxynitrite (ONOO⁻) formation disrupts Cav-1 rich caveolae, leading to eNOS uncoupling and impaired flow-mediated dilation (FMD) [61]. These alterations are characterized by reduced membrane localized Cav-1, loss of endothelial caveolae, and displacement of eNOS from its functional microdomain. Importantly, endothelial function could be restored either by ONOO⁻ sequestration, reconstruction of caveolae, or BH₄ supplementation, underscoring the essential role of Cav-1 and caveolae integrity in maintaining NO bioavailability and vascular homeostasis. Collectively, these findings indicate that loss or nitration of Cav-1 contributes to endothelial dysfunction, highlighting its protective, anti-atherogenic role under physiological conditions and emphasizing the bidirectional interplay between oxidative stress and Cav-1 dependent NO signaling. Further studies are needed to elucidate the molecular cross-talk pathways among Cav-1, RCT, and inflammation to better understand its paradoxical role in atherosclerosis [36].
6. Pro-Atherogenic and Pathological Roles of Caveolin-1 (Cav-1)
While Cav-1 can exert several protective effects in lipid metabolism and vascular smooth muscle homeostasis, accumulating evidence demonstrates that excessive or dysregulated Cav-1 expression particularly in endothelial cells can promote atherosclerosis.
Cav-1 can directly inhibit eNOS through its scaffolding domain, and chronic or compartmentalized upregulation of Cav-1 particularly within a pro-inflammatory environment may suppress NO bioavailability or promote eNOS uncoupling. These processes contribute to oxidative stress and endothelial dysfunction, thereby supporting an atherogenic role for Cav-1 [62]. An interesting study by Lobysheva et al. provided important mechanistic insight into how Cav-1 contributes to both endothelial dysfunction and vascular protection, dependent on its expression level. Their findings revealed that Cav-1 abundance critically modulates the assembly of NADPH oxidase and its interaction with eNOS within caveolae. Under angiotensin II stimulation conditions that promote oxidative stress and vascular injury, excess Cav-1 facilitates the colocalization of NADPH oxidase and eNOS, leading to increased reactive oxygen species (ROS) formation and eNOS uncoupling. Also, they found that moderate downregulation of Cav-1 disrupted this harmful interaction, preserved eNOS coupling, and enhanced NO bioavailability without triggering uncontrolled NO release. This partial reduction of Cav-1 therefore offers a dual therapeutic advantage: attenuating oxidative stress while maintaining physiological eNOS regulation. The study suggested that controlled modulation of Cav-1 expression, such as through statins, could help counteract the adverse vascular effects of the renin angiotensin system and provide a potential anti-atherogenic strategy [63].
Under conditions of insulin resistance (IR), Cav-1 appears to have a pro-atherogenic role. Cav-1 interacts with PKCζ to antagonize the IRS-1/PI3K/Akt signaling pathway, leading to impaired eNOS activity, reduced NO bioavailability, oxidative stress, and endothelial dysfunction; all events which promote atherogenesis. In a recent study, Tan et al showed that persistent hyperglycemia and lipid abnormalities further exacerbated endothelial damage through inflammation and ROS generation and that modulation of Cav-1/PKCζ interactions in endothelial cells restored NO signaling. Thus, Cav-1 could represent a novel target to counteract atherosclerosis development in metabolic disease contexts [64].
Another study demonstrated that endothelial-specific overexpression of Cav-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Mechanistically, Cav-1 overexpression reduces endothelial cell proliferation and migration, suppresses NO production, and enhances inflammatory signaling, including upregulation of VCAM-1, which facilitates leukocyte adhesion and infiltration into the arterial wall. These findings, together with previous studies using global Cav-1 knockout models, suggest that Cav-1 promotes atherogenesis in vessels with intact caveolae, likely via its scaffolding function and regulation of endothelial barrier properties. So, while Cav-1 may exert anti-atherogenic effects in other vascular cells, such as smooth muscle cells, its overexpression in endothelial cells highlights its context dependent pro-atherogenic role, emphasizing the complex and paradoxical functions of Cav-1 in atherosclerosis [75].
Recent studies have added further complexity by implicating Cav-1 dependent caveolae in the transcytosis of low-density lipoprotein (LDL) across endothelial cells and in the subsequent subendothelial lipid accumulation. Using cultured endothelial cells, the study demonstrated that Cav-1 knock-down or pharmacological disruption of caveolae markedly reduces LDL flux from the luminal to the abluminal side, thereby limiting subendothelial lipid deposition. This mechanism provides a direct link between Cav-1 and the initiation of atherosclerotic plaque formation that is independent of nitric oxide (NO) signaling [66].
7. Caveolin-1, PVAT, and Endothelial Dysfunction
PVAT and its secretome, comprising adipokines, cytokines, and reactive oxygen species (ROS), can modulate the endothelial Cav-1/eNOS signaling axis, thereby influencing local nitric oxide (NO) bioavailability and vascular tone in a depot and disease stage-dependent manner. In physiological conditions, PVAT-derived factors such as adiponectin help maintain endothelial homeostasis by supporting eNOS activation and NO production. However, under pathological conditions such as obesity or hypertension, the altered PVAT secretome promotes oxidative stress and inflammation, leading to Cav-1 overexpression and eNOS uncoupling. These changes impair endothelial NO signaling and contribute to vascular dysfunction and atherogenesis [67].
Taken together, the data argue for a spatiotemporal model: moderate, properly localized Cav-1 supports vascular homeostasis, whereas dysregulated or excessive Cav-1 especially within an inflamed PVAT/endothelial microenvironment contributes to NO deficiency and atherogenesis. Testing this model requires experiments that resolve Cav-1 function by cell type, vascular bed, and disease stage (endothelial vs. PVAT vs. macrophage), and which measure NO coupling/uncoupling and LDL trafficking in parallel.
Interestingly, this atheroprotective effect persists even in Cav-1/LDLR double knockout mice lacking eNOS, suggesting mechanisms beyond NO signaling. The absence of Cav-1 markedly reduces LDL transcytosis across the endothelium, thereby limiting subendothelial lipid deposition and foam cell formation. Concurrently, enhanced autophagic flux in Cav-1 deficient vascular cells promotes clearance of oxidized lipids and damaged organelles, mitigating oxidative stress. Moreover, loss of Cav-1 attenuates proinflammatory signaling through pathways such as TLR4/NF-κB, collectively contributing to reduced plaque burden [8,66]. Within the PVAT: vascular interface, these adaptations disrupt the proinflammatory and lipid exchange normally amplified by PVAT-derived cytokines, suggesting that Cav-1 deficiency confers atheroprotection through diminished lipid entry and inflammation rather than enhanced eNOS activity.
Human and animal studies provide compelling evidence that Cav-1 expression and function vary markedly between vascular cell types and disease contexts, suggesting that its role in atherosclerosis is highly cell-type-specific. For example, loss of function mutations in the CAV1 gene have been identified in patients with Pulmonary Arterial Hypertension (PAH) and cardiomyopathy, with endothelial Cav-1 levels markedly reduced in the pulmonary vasculature of such patients [68].
In contrast, in the setting of atherosclerotic vessels, Cav-1 expression is often found to be upregulated in endothelial cells and smooth muscle cells in early-stage lesions. For example, endothelial-specific overexpression of Cav-1 in apolipoprotein E-deficient mice accelerated lesion development, likely by promoting LDL transcytosis and suppressing NO signalling. [70]. Mechanistically, elevated Cav-1 in endothelial cells may also increase pro-inflammatory signalling (e.g., adhesion molecule expression), thereby promoting atherogenesis [69].

Table 1.
Cell-Type-Specific Effects of Caveolin-1 in Atherosclerosis*.
| Cell Type | Primary Effect | Mechanisms | Overall Impact |
|---|---|---|---|
| Endothelial Cells | Proatherogenic | Increases LDL transcytosis, inhibits eNOS activity, promotes inflammation | Atherosclerosis progression |
| Macrophages | Protective | Regulates cholesterol homeostasis, limits foam cell formation, modulates inflammation | Atherosclerosis suppression |
| Smooth Muscle Cells | Protective | Inhibits proliferation and migration, stabilizes plaques |
Recent evidence suggests that PVAT can directly impair endothelial function through a Cav-1 dependent mechanism. In rat aorta, PVAT was found to enhance Cav-1 expression in endothelial cells, leading to decreased eNOS activity and reduced NO bioavailability. This suppression of NO production resulted in enhanced vasoconstriction and impaired endothelium dependent vasodilation, without changes in eNOS protein levels or phosphorylation at key regulatory sites. Importantly, disruption of caveolae with cholesterol-depleting agents reversed these effects, confirming the central role of Cav-1 in mediating PVAT-induced endothelial dysfunction [47].
In the context of atherosclerosis, PVAT dysfunction represents an early and critical event linking metabolic stress to vascular injury. Under physiological conditions, PVAT exerts anti-atherogenic actions by releasing adiponectin and other vasorelaxant factors that maintain endothelial homeostasis and suppress inflammation. However, in obesity, diabetes, and hypertension, PVAT undergoes a phenotypic shift toward a pro-inflammatory and oxidative state characterized by increased secretion of cytokines (TNF-α, IL-6), reactive oxygen species (ROS), and decreased adiponectin. These alterations amplify endothelial Cav-1 expression and promote eNOS uncoupling, further diminishing NO bioavailability and facilitating endothelial activation and monocyte adhesion [46,70].
In a recent study in non-atherosclerotic mice it was found that Cav-1 plays a crucial modulatory role in regulating eNOS activity and NO bioavailability within PVAT. Thoracic PVAT produces more NO than abdominal PVAT, largely dependent on AMPKα1 activity. In AMPKα1-deficient mice, NO production was reduced despite unchanged eNOS levels, due to increased eNOS Cav-1 interaction, which inhibits eNOS activity. Disrupting this interaction with Thoracic PVAT produces more NO than abdominal PVAT, largely dependent on AMPKα1 activity. In AMPKα1-deficient mice, NO production was reduced despite unchanged eNOS levels, due to increased eNOS Cav-1 interaction, which inhibits eNOS activity. Disrupting this interaction with (CAV-AP) restored NO production and normalized vascular tone, highlighting Cav-1’s inhibitory role under impaired AMPK signaling. CAV-AP restored NO production and normalized vascular tone, highlighting Cav-1’s inhibitory role under impaired AMPK signaling. These results suggest that AMPK maintains PVAT’s vasoprotective effects by limiting Cav-1 mediated eNOS inhibition and that targeting this pathway could help prevent endothelial dysfunction and ultimately the development of atherosclerosis [7].
8. Conclusion and Therapeutic Implications
Cav-1 plays a complex, context-dependent role in vascular biology and atherosclerosis. Its effects are highly cell type specific: in endothelial cells, excessive or dysregulated Cav-1 can promote atherogenesis by inhibiting eNOS, enhancing LDL transcytosis, and amplifying inflammatory signaling; in macrophages and smooth muscle cells, Cav-1 often exerts protective effects by promoting cholesterol efflux, regulating inflammation, and stabilizing plaques. The balance of these opposing actions, combined with the local vascular microenvironment, determines its net impact on disease progression.
Perivascular adipose tissue (PVAT) has emerged as a critical regulator of endothelial function through Cav-1 dependent mechanisms. Under physiological conditions, PVAT maintains vascular homeostasis by supporting eNOS activity and NO production. In contrast, metabolic stress, obesity, diabetes, and hypertension shift PVAT toward a pro-inflammatory and oxidative phenotype, promoting Cav-1 overexpression, eNOS uncoupling, and endothelial dysfunction. Dysregulation of the AMPK/Cav-1/eNOS axis in PVAT further contributes to impaired vasorelaxation and atherogenesis.
Therapeutically, Cav-1 represents a promising target for interventions aimed at restoring endothelial function and preventing vascular disease. Potential strategies include:
Modulating Cav-1 expression or function in a cell type and vascular bed specific manner, e.g., partial downregulation in endothelial cells to preserve eNOS coupling without impairing physiological signaling.
Targeting the AMPK/Cav-1/eNOS pathway in PVAT to restore NO bioavailability and maintain vasoprotective effects.
Enhancing reverse cholesterol transport (RCT) via Cav-1 interaction with cholesterol transporters (ABCG1, ABCA1), particularly in macrophages and hepatic cells, to reduce lipid accumulation and inflammation.
Addressing PVAT inflammation in metabolic disease states (obesity, diabetes) through anti-inflammatory or antioxidant therapies to mitigate Cav-1 mediated endothelial dysfunction.
Overall, understanding the spatiotemporal regulation of Cav-1, the cross-talk between PVAT and endothelial cells, and the interplay with metabolic stressors offers new avenues for preventing and treating atherosclerosis and associated cardiovascular complications. Future studies integrating cell-type specificity, disease stage, and metabolic context will be essential to translate Cav-1 targeted interventions into effective therapies.
Author Contributions
Abdmajid Hwej: Writing – review & editing, Writing – original draft preparation, Visualization, Validation, Conceptualization. Mohammed Abdullah Alsharif: review & editing. Ali Al-Ferjani: review & editing,. Simon Kennedy: Writing – review & editing, Supervision, Project administration, Conceptualization.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wojtasińska, A., Frąk, W., Lisińska, W., Sapeda, N., Młynarska, E., Rysz, J., & Franczyk, B. (2023). Novel Insights into the Molecular Mechanisms of Atherosclerosis. International Journal of Molecular Sciences, 24(17), 13434. [CrossRef]
- Zubirán, R., Neufeld, E. B., Dasseux, A., Remaley, A. T., & Sorokin, A. V. (2024). Recent Advances in Targeted Management of Inflammation In Atherosclerosis: A Narrative Review. Cardiology and therapy, 13(3), 465–491. [CrossRef]
- YAMADA E. (1955). The fine structure of the gall bladder epithelium of the mouse. The Journal of biophysical and biochemical cytology, 1(5), 445–458. [CrossRef]
- Rothberg, K. G., Heuser, J. E., Donzell, W. C., Ying, Y. S., Glenney, J. R., & Anderson, R. G. (1992). Caveolin, a protein component of caveolae membrane coats. Cell, 68(4), 673–682. [CrossRef]
- Drab, M., Verkade, P., Elger, M., Kasper, M., Lohn, M., Lauterbach, B., Menne, J., Lindschau, C., Mende, F., Luft, F. C., Schedl, A., Haller, H., & Kurzchalia, T. V. (2001). Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science (New York, N.Y.), 293(5539), 2449–2452. [CrossRef]
- Razani, B., Engelman, J. A., Wang, X. B., Schubert, W., Zhang, X. L., Marks, C. B., Macaluso, F., Russell, R. G., Li, M., Pestell, R. G., Di Vizio, D., Hou, H., Jr, Kneitz, B., Lagaud, G., Christ, G. J., Edelmann, W., & Lisanti, M. P. (2001). Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. The Journal of biological chemistry, 276(41), 38121–38138. [CrossRef]
- Hwej, A.; Al-Ferjani, A.; Alshuweishi, Y.; Naji, A.; Kennedy, S.; Salt, I.P. Lack of AMP-activated protein kinase-α1 reduces nitric oxide synthesis in thoracic aorta perivascular adipose tissue. Vasc. Pharmacol. 2024, *157*, 107437. [CrossRef]
- Puddu, A., Montecucco, F., & Maggi, D. (2023). Caveolin-1 and Atherosclerosis: Regulation of LDLs Fate in Endothelial Cells. International Journal of Molecular Sciences, 24(10), 8869. [CrossRef]
- Kawabe, J. I., Grant, B. S., Yamamoto, M., Schwencke, C., Okumura, S., & Ishikawa, Y. (2001). Changes in caveolin subtype protein expression in aging rat organs. Molecular and cellular endocrinology, 176(1-2), 91–95. [CrossRef]
- Williams, T. M., & Lisanti, M. P. (2004). The caveolin proteins. Genome biology, 5(3), 214. [CrossRef]
- Fridolfsson, H. N., Roth, D. M., Insel, P. A., & Patel, H. H. (2014). Regulation of intracellular signaling and function by caveolin. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 28(9), 3823–3831. [CrossRef]
- Zipes, D.P.; Jalife, J.; Stevenson, W.G. Cardiac Electrophysiology: From Cell to Bedside; Elsevier: Philadelphia, PA, USA, 2017.
- de Almeida C. J. G. (2017). Caveolin-1 and Caveolin-2 Can Be Antagonistic Partners in Inflammation and Beyond. Frontiers in immunology, 8, 1530. [CrossRef]
- Campos, A., Burgos-Ravanal, R., González, M. F., Huilcaman, R., Lobos González, L., & Quest, A. F. G. (2019). Cell Intrinsic and Extrinsic Mechanisms of Caveolin-1-Enhanced Metastasis. Biomolecules, 9(8), 314. [CrossRef]
- Parton, R. G., & Simons, K. (2007). The multiple faces of caveolae. Nature reviews. Molecular cell biology, 8(3), 185–194. [CrossRef]
- Nwosu, Z. C., Ebert, M. P., Dooley, S., & Meyer, C. (2016). Caveolin-1 in the regulation of cell metabolism: a cancer perspective. Molecular cancer, 15(1), 71. [CrossRef]
- Fan, J.Y.; Carpentier, J.L.; van Obberghen, E.; Grunfeld, C.; Gorden, P.; Orci, L. Morphological changes of the 3T3-L1 fibroblast plasma membrane upon differentiation to the adipocyte form. J. Cell Sci. 1983, *61*, 219–230.
- Scherer, P. E., Lisanti, M. P., Baldini, G., Sargiacomo, M., Mastick, C. C., & Lodish, H. F. (1994). Induction of caveolin during adipogenesis and association of GLUT4 with caveolin-rich vesicles. The Journal of cell biology, 127(5), 1233–1243. [CrossRef]
- Palacios-Ortega, S., Varela-Guruceaga, M., Algarabel, M., Ignacio Milagro, F., Alfredo Martínez, J., & de Miguel, C. (2015). Effect of TNF-Alpha on Caveolin-1 Expression and Insulin Signaling During Adipocyte Differentiation and in Mature Adipocytes. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology, 36(4), 1499–1516. [CrossRef]
- Krishna, A., & Sengupta, D. (2019). Interplay between Membrane Curvature and Cholesterol: Role of Palmitoylated Caveolin-1. Biophysical journal, 116(1), 69–78. [CrossRef]
- Hayer, A., Stoeber, M., Bissig, C. and Helenius, A., 2010. Biogenesis of caveolae: stepwise assembly of large caveolin and cavin complexes. Traffic, 11(3), pp.361-382. [CrossRef]
- Ashford, F., Kuo, C. W., Dunning, E., Brown, E., Calagan, S., Jayasinghe, I., Henderson, C., Fuller, W., & Wypijewski, K. (2024). Cysteine post-translational modifications regulate protein interactions of caveolin-3. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 38(5), e23535. [CrossRef]
- Gokani, S., & Bhatt, L. K. (2022). Caveolin-1: A Promising Therapeutic Target for Diverse Diseases. Current molecular pharmacology, 15(5), 701–715. [CrossRef]
- Huo, J., Mo, L., Lv, X., Du, Y., & Yang, H. (2025). Ion Channel Regulation in Caveolae and Its Pathological Implications. Cells, 14(9), 631. [CrossRef]
- Yu, L. Nerve Growth Factor Signaling from Membrane Microdomain to Nucleus: Differential Regulation by Caveolins. Doctoral Dissertation, Ecole Normale Supérieure de Lyon / East China Normal University, Lyon, France/Shanghai, China, 2012.
- García-Cardeña, G., Martasek, P., Masters, B. S., Skidd, P. M., Couet, J., Li, S., Lisanti, M. P., & Sessa, W. C. (1997). Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. The Journal of biological chemistry, 272(41), 25437–25440. [CrossRef]
- Razani, B., Woodman, S. E., & Lisanti, M. P. (2002). Caveolae: from cell biology to animal physiology. Pharmacological reviews, 54(3), 431–467. [CrossRef]
- Minetti, C., Sotgia, F., Bruno, C., Scartezzini, P., Broda, P., Bado, M., Masetti, E., Mazzocco, M., Egeo, A., Donati, M. A., Volonte, D., Galbiati, F., Cordone, G., Bricarelli, F. D., Lisanti, M. P., & Zara, F. (1998). Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nature genetics, 18(4), 365–368. [CrossRef]
- Galbiati, F., Razani, B., & Lisanti, M. P. (2001). Caveolae and caveolin-3 in muscular dystrophy. Trends in molecular medicine, 7(10), 435–441. [CrossRef]
- Chakraborty, S., & Ain, R. (2017). Nitric-oxide synthase trafficking inducer is a pleiotropic regulator of endothelial cell function and signaling. The Journal of biological chemistry, 292(16), 6600–6620. [CrossRef]
- Wang, X., Lian, Y., Wen, X., Guo, J. and Chen, T. (2014) Caveolin-1: an essential modulator of eNOS function and vascular homeostasis. In: Advances in Experimental Medicine and Biology. [e-book] Vol. 857. Springer, pp. 1-21. [CrossRef]
- Frank P. G. (2010). Endothelial caveolae and caveolin-1 as key regulators of atherosclerosis. The American journal of pathology, 177(2), 544–546. [CrossRef]
- Jia, G., & Sowers, J. R. (2015). Caveolin-1 in Cardiovascular Disease: A Double-Edged Sword. Diabetes, 64(11), 3645–3647. [CrossRef]
- Wang, L., Wang, B., Jia, L., Yu, H., Wang, Z., Wei, F., & Jiang, A. (2023). Shear stress leads to the dysfunction of endothelial cells through the Cav-1-mediated KLF2/eNOS/ERK signaling pathway under physiological conditions. Open life sciences, 18(1), 20220587. [CrossRef]
- Figueroa, X. F., González, D. R., Puebla, M., Acevedo, J. P., Rojas-Libano, D., Durán, W. N., & Boric, M. P. (2013). Coordinated endothelial nitric oxide synthase activation by translocation and phosphorylation determines flow-induced nitric oxide production in resistance vessels. Journal of vascular research, 50(6), 498–511. [CrossRef]
- Li, S., Couet, J., & Lisanti, M. P. (1996). Src tyrosine kinases, Galpha subunits, and H-Ras share a common membrane-anchored scaffolding protein, caveolin. Caveolin binding negatively regulates the auto-activation of Src tyrosine kinases. The Journal of biological chemistry, 271(46), 29182–29190. [CrossRef]
- Cohen, A. W., Razani, B., Schubert, W., Williams, T. M., Wang, X. B., Iyengar, P., Brasaemle, D. L., Scherer, P. E., & Lisanti, M. P. (2004). Role of caveolin-1 in the modulation of lipolysis and lipid droplet formation. Diabetes, 53(5), 1261–1270. [CrossRef]
- Bernatchez, P., Sharma, A., Bauer, P. M., Marin, E., & Sessa, W. C. (2011). A noninhibitory mutant of the caveolin-1 scaffolding domain enhances eNOS-derived NO synthesis and vasodilation in mice. The Journal of clinical investigation, 121(9), 3747–3755. [CrossRef]
- Musicki, B., Liu, T., Lagoda, G. A., Bivalacqua, T. J., Strong, T. D., & Burnett, A. L. (2009). Endothelial nitric oxide synthase regulation in female genital tract structures. The journal of sexual medicine, 6 Suppl 3(S3PROCEEDINGS), 247–253. [CrossRef]
- Kinsella, B. T., Erdman, R. A., & Maltese, W. A. (1991). Carboxyl-terminal isoprenylation of ras-related GTP-binding proteins encoded by rac1, rac2, and ralA. The Journal of biological chemistry, 266(15), 9786–9794.Burridge, K.; Wennerberg, K. Rho and Rac take center stage. *Cell 2004, *116*, 167–179. [CrossRef]
- Burridge, K., & Wennerberg, K. (2004). Rho and Rac take center stage. Cell, 116(2), 167–179. [CrossRef]
- Ramadoss, J., Pastore, M. B., & Magness, R. R. (2013). Endothelial caveolar subcellular domain regulation of endothelial nitric oxide synthase. Clinical and experimental pharmacology & physiology, 40(11), 753–764. [CrossRef]
- Gonzalez, E., Nagiel, A., Lin, A.J., Golan, D.E. and Michel, T., 2004. Small interfering RNA-mediated down-regulation of caveolin-1 differentially modulates signaling pathways in endothelial cells. Journal of Biological Chemistry, 279(39), pp.40659-40669. [CrossRef]
- Gonzalez, E., Kou, R., Lin, A. J., Golan, D. E., & Michel, T. (2002). Subcellular targeting and agonist-induced site-specific phosphorylation of endothelial nitric-oxide synthase. The Journal of biological chemistry, 277(42), 39554–39560. [CrossRef]
- Levine, Y. C., Li, G. K., & Michel, T. (2007). Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells. Evidence for an AMPK -> Rac1 -> Akt -> endothelial nitric-oxide synthase pathway. The Journal of biological chemistry, 282(28), 20351–20364. [CrossRef]
- Chang, L., Garcia-Barrio, M. T., & Chen, Y. E. (2020). Perivascular Adipose Tissue Regulates Vascular Function by Targeting Vascular Smooth Muscle Cells. Arteriosclerosis, thrombosis, and vascular biology, 40(5), 1094–1109. [CrossRef]
- Lee, M. H., Chen, S. J., Tsao, C. M., & Wu, C. C. (2014). Perivascular adipose tissue inhibits endothelial function of rat aortas via caveolin-1. PloS one, 9(6), e99947. [CrossRef]
- Chen, Z., D S Oliveira, S., Zimnicka, A. M., Jiang, Y., Sharma, T., Chen, S., Lazarov, O., Bonini, M. G., Haus, J. M., & Minshall, R. D. (2018). Reciprocal regulation of eNOS and caveolin-1 functions in endothelial cells. Molecular biology of the cell, 29(10), 1190–1202. [CrossRef]
- Fernández-Hernando, C., Yu, J., Suárez, Y., Rahner, C., Dávalos, A., Lasunción, M. A., & Sessa, W. C. (2009). Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell metabolism, 10(1), 48–54. [CrossRef]
- Frank, P. G., & Lisanti, M. P. (2004). Caveolin-1 and caveolae in atherosclerosis: differential roles in fatty streak formation and neointimal hyperplasia. Current opinion in lipidology, 15(5), 523–529. [CrossRef]
- Cohen, A. W., Hnasko, R., Schubert, W., & Lisanti, M. P. (2004). Role of caveolae and caveolins in health and disease. Physiological reviews, 84(4), 1341–1379. [CrossRef]
- Murata, T., Lin, M. I., Huang, Y., Yu, J., Bauer, P. M., Giordano, F. J., & Sessa, W. C. (2007). Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. The Journal of experimental medicine, 204(10), 2373–2382. [CrossRef]
- Keane, J., & Longhi, M. P. (2025). Perivascular Adipose Tissue Niches for Modulating Immune Cell Function. Arteriosclerosis, thrombosis, and vascular biology, 45(6), 857–865. [CrossRef]
- Adachi, Y., Ueda, K., & Takimoto, E. (2023). Perivascular adipose tissue in vascular pathologies-a novel therapeutic target for atherosclerotic disease?. Frontiers in cardiovascular medicine, 10, 1151717. [CrossRef]
- Cheng, C. K., Ding, H., Jiang, M., Yin, H., Gollasch, M., & Huang, Y. (2023). Perivascular adipose tissue: Fine-tuner of vascular redox status and inflammation. Redox biology, 62, 102683. [CrossRef]
- Dalton, C. M., Schlegel, C., & Hunter, C. J. (2023). Caveolin-1: A Review of Intracellular Functions, Tissue-Specific Roles, and Epithelial Tight Junction Regulation. Biology, 12(11), 1402. [CrossRef]
- Kassan, M., Vikram, A., Kim, Y. R., Li, Q., Kassan, A., Patel, H. H., Kumar, S., Gabani, M., Liu, J., Jacobs, J. S., & Irani, K. (2017). Sirtuin1 protects endothelial Caveolin-1 expression and preserves endothelial function via suppressing miR-204 and endoplasmic reticulum stress. Scientific reports, 7, 42265. [CrossRef]
- Qin, L., Zhu, N., Ao, B. X., Liu, C., Shi, Y. N., Du, K., Chen, J. X., Zheng, X. L., & Liao, D. F. (2016). Caveolae and Caveolin-1 Integrate Reverse Cholesterol Transport and Inflammation in Atherosclerosis. International journal of molecular sciences, 17(3), 429. [CrossRef]
- Fu, Y., Hoang, A., Escher, G., Parton, R. G., Krozowski, Z., & Sviridov, D. (2004). Expression of caveolin-1 enhances cholesterol efflux in hepatic cells. The Journal of biological chemistry, 279(14), 14140–14146. [CrossRef]
- Rodriguez-Feo, J. A., Hellings, W. E., Moll, F. L., De Vries, J. P., van Middelaar, B. J., Algra, A., Sluijter, J., Velema, E., van den Broek, T., Sessa, W. C., De Kleijn, D. P., & Pasterkamp, G. (2008). Caveolin-1 influences vascular protease activity and is a potential stabilizing factor in human atherosclerotic disease. PloS one, 3(7), e2612. [CrossRef]
- Cassuto, J., Dou, H., Czikora, I., Szabo, A., Patel, V. S., Kamath, V., Belin de Chantemele, E., Feher, A., Romero, M. J., & Bagi, Z. (2014). Peroxynitrite disrupts endothelial caveolae leading to eNOS uncoupling and diminished flow-mediated dilation in coronary arterioles of diabetic patients. Diabetes, 63(4), 1381–1393. [CrossRef]
- Janaszak-Jasiecka, A., Płoska, A., Wierońska, J. M., Dobrucki, L. W., & Kalinowski, L. (2023). Endothelial dysfunction due to eNOS uncoupling: molecular mechanisms as potential therapeutic targets. Cellular & molecular biology letters, 28(1), 21. [CrossRef]
- Lobysheva, I., Rath, G., Sekkali, B., Bouzin, C., Feron, O., Gallez, B., Dessy, C., & Balligand, J. L. (2011). Moderate caveolin-1 downregulation prevents NADPH oxidase-dependent endothelial nitric oxide synthase uncoupling by angiotensin II in endothelial cells. Arteriosclerosis, thrombosis, and vascular biology, 31(9), 2098–2105. [CrossRef]
- Tan, J., Li, X., & Dou, N. (2024). Insulin Resistance Triggers Atherosclerosis: Caveolin 1 Cooperates with PKCzeta to Block Insulin Signaling in Vascular Endothelial Cells. Cardiovascular drugs and therapy, 38(5), 885–893. [CrossRef]
- Fernández-Hernando, C., Yu, J., Dávalos, A., Prendergast, J., & Sessa, W. C. (2010). Endothelial-specific overexpression of caveolin-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. The American journal of pathology, 177(2), 998–1003. [CrossRef]
- Zhang, Y., Jia, X., Wang, Y., & Zheng, Q. (2025). Caveolin-1-mediated LDL transcytosis across endothelial cells in atherosclerosis. Atherosclerosis, 402, 119113. [CrossRef]
- Valentini, A., Cardillo, C., Della Morte, D., & Tesauro, M. (2023). The Role of Perivascular Adipose Tissue in the Pathogenesis of Endothelial Dysfunction in Cardiovascular Diseases and Type 2 Diabetes Mellitus. Biomedicines, 11(11), 3006. [CrossRef]
- Zaborska, K. E., Wareing, M., Edwards, G., & Austin, C. (2016). Loss of anti-contractile effect of perivascular adipose tissue in offspring of obese rats. International journal of obesity (2005), 40(8), 1205–1214. [CrossRef]
- Ramírez, C. M., Zhang, X., Bandyopadhyay, C., Rotllan, N., Sugiyama, M. G., Aryal, B., Liu, X., He, S., Kraehling, J. R., Ulrich, V., Lin, C. S., Velazquez, H., Lasunción, M. A., Li, G., Suárez, Y., Tellides, G., Swirski, F. K., Lee, W. L., Schwartz, M. A., Sessa, W. C., … Fernández-Hernando, C. (2019). Caveolin-1 Regulates Atherogenesis by Attenuating Low-Density Lipoprotein Transcytosis and Vascular Inflammation Independently of Endothelial Nitric Oxide Synthase Activation. Circulation, 140(3), 225–239. [CrossRef]
- Victorio, J. A., Fontes, M. T., Rossoni, L. V., & Davel, A. P. (2016). Different Anti-Contractile Function and Nitric Oxide Production of Thoracic and Abdominal Perivascular Adipose Tissues. Frontiers in physiology, 7, 295. [CrossRef]
Figure 1.
A schematic diagram of Cav-1 structure.
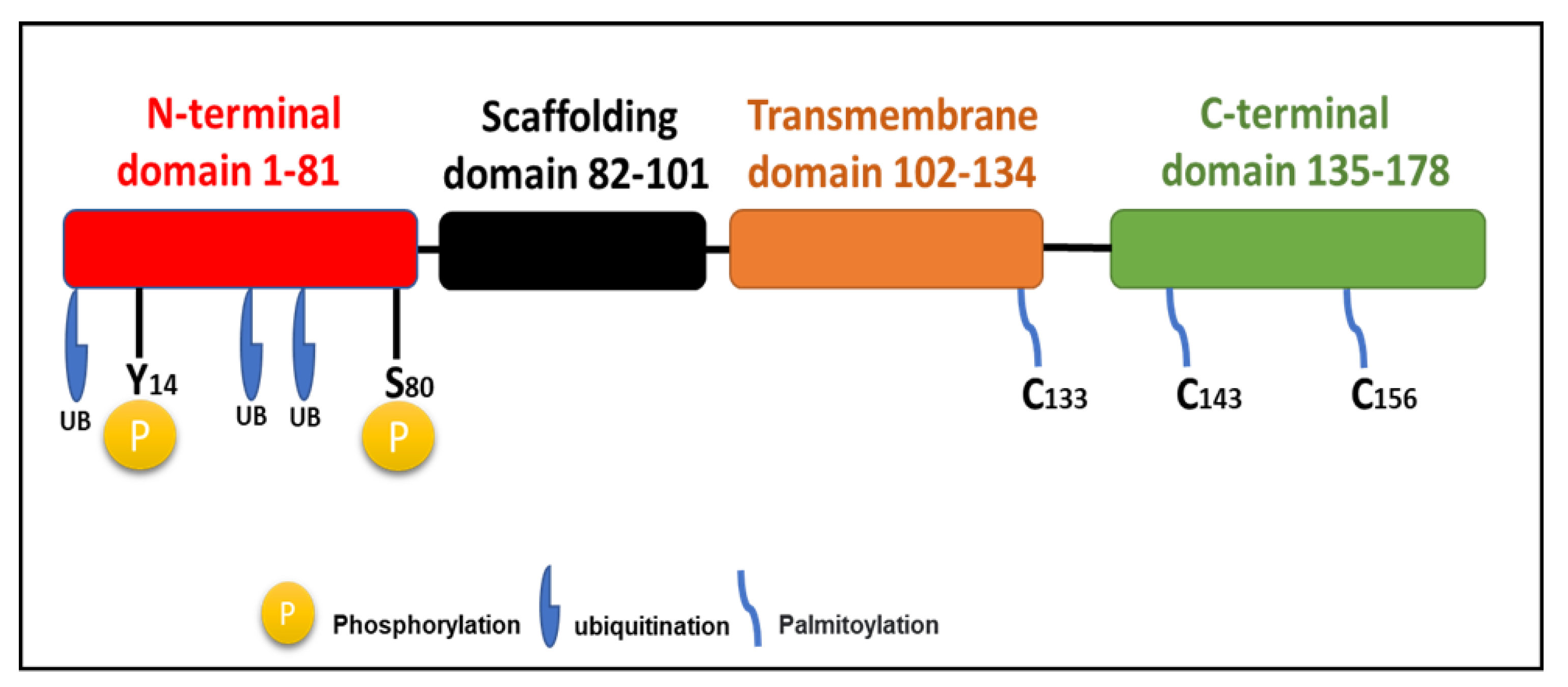
Figure 2.
A schematic diagram showing eNOS regulation and activation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



