
Submitted:
21 November 2025
Posted:
24 November 2025
You are already at the latest version
Abstract
Background: Infections caused by the multidrug-resistant pathogen Mycobacterium abscessus (Mab) are notoriously difficult to treat. The novel β-lactamase inhibitor durlobactam, in combination with β-lactams, shows potent bactericidal activity against Mab, but the potential for acquired resistance remains a clinical concern. Objectives: To identify and characterize mechanisms of acquired resistance to durlobactam in Mab. Methods: In vitro single-step resistance selection was performed by plating wild-type Mab ATCC 19977 on agar containing durlobactam. Resistant mutants were isolated, and their genomes were sequenced. The resistance phenotype was confirmed by constructing a targeted gene deletion mutant and by transcriptional silencing using a CRISPR interference (CRISPRi) system. Minimum inhibitory concentrations (MICs) were determined by both, an agar-based method and broth microdilution. Results: Whole-genome sequencing of durlobactam-resistant mutants identified loss-of-function mutations in ponA1, a gene encoding a class A penicillin-binding protein involved in cell wall synthesis. Targeted deletion of ponA1 (ΔponA1) and CRISPRi-mediated knockdown of ponA1 expression both recapitulated the resistance phenotype, resulting in a significant increase in the durlobactam MIC on solid agar media. Strikingly, broth microdilution MICs remained largely unaffected. Conclusions: Inactivation of the peptidoglycan synthase PonA1 is a novel mechanism of resistance to durlobactam in Mab that is phenotypically expressed only during growth on solid surfaces. This finding identifies a specific genetic pathway for resistance and highlights that standard broth-based susceptibility testing could miss clinically relevant resistance mechanisms.
Keywords:
Mycobacterium abscessus
; durlobactam
; acquired resistance
1. Introduction
Mycobacterium abscessus (Mab) is an emerging opportunistic pathogen responsible for chronic and debilitating pulmonary infections, particularly in individuals with underlying lung conditions [1]. Treatment of Mab is exceptionally challenging due to its extensive intrinsic and acquired resistance to most available antibiotics, leading to poor clinical outcomes and cure rates often below 50% [2]. This clinical reality underscores the urgent need for novel treatment strategies.
The revitalization of β-lactam antibiotics through combination with potent β-lactamase inhibitors (BLIs) represents a promising therapeutic avenue. Durlobactam (DUR), a next-generation diazabicyclooctane (DBO) inhibitor [3], has demonstrated remarkable potency against Mab. Its efficacy stems from a dual mechanism of action: it potently inactivates the major β-lactamase, BlaMab, while also exhibiting intrinsic antibacterial activity through the direct inhibition of cell wall synthesis enzymes, including penicillin-binding proteins (PBPs) and L,D-transpeptidases [4]. This multi-target engagement results in profound synergy with partner β-lactams like imipenem, achieving potent bactericidal activity at clinically relevant concentrations [5,6].
The long-term clinical success of any new antimicrobial agent is contingent upon understanding and anticipating the pathogen's evolutionary pathways to resistance. While several mechanisms that compromise β-lactam-based therapies in Mab have been described – including upregulation of BlaMab, reduced cell wall permeability via porin loss, and induction of drug tolerance through mutations in the stress-response regulator RshA [7,8,9] – mechanisms conferring direct resistance to advanced DBO inhibitors like DUR are not well understood. Since target-based mutations are considered uncommon for β-lactams in Mab due to the functional redundancy of peptidoglycan synthesis enzymes, it is critical to prospectively identify the most likely pathways to resistance.
In this study, we sought to identify and validate novel mechanisms of acquired resistance to DUR in Mab. Through in vitro single-step selection experiments, we discovered that high level resistance consistently arises through mutations in ponA1, encoding a class A PBP that performs both transglycosylase and D,D-transpeptidase activities. We confirmed this unexpected finding by demonstrating that both targeted deletion and CRISPRi-mediated knockdown of ponA1 are sufficient to confer DUR resistance.
2. Results
To identify genes involved in acquired DUR resistance, we selected spontaneous resistant mutants on 7H10 agar containing 4× the agar MIC of 8 mg/L. We isolated fourteen mutants in two independent rounds of selection, seven of which showed high level resistance, with an agar MIC >128 mg/L. The remainder displayed moderate resistance with agar MICs ranging from 32 to 64 mg/L (Table 1). The frequency of resistance was ~10-7/CFU. Interestingly, all fourteen mutants showed weak (twofold) increase in broth MIC compared to the wild-type strain.
Whole-genome sequencing revealed that high-level resistant mutants all carried mutations in MAB_4901c encoding the bifunctional transglycosylase / transpeptidase PonA1 involved in peptidoglycan synthesis [10]: two frameshift, three nonsense, one in-frame deletion and one missense mutation (Table 1). The nature of the mutations in ponA1 suggested that loss-of-function causes the resistance phenotype. To confirm this hypothesis, we generated a complete deletion of MAB_4901c via allelic exchange, verified by whole genome sequencing (Figure S1). The MIC pattern of the ponA1 knockout strain was identical to the original spontaneous frameshift mutant strain Mab DUR_res2 with high-level (>128 mg/L) resistance on solid medium (Table 2).
Mutations conferring moderate-level resistance were found in either MAB_0505c or MAB_0205c, whose functions are currently unknown (Table 1). From there on, we focused on characterizing the PonA1-mediated mechanism of high-level resistance to DUR.
Since genomic analysis suggests that ponA1 (MAB_4901c) and the downstream gene MAB_4900c, encoding a hypothetical protein, form a putative operon, we hypothesized that the mutations in ponA1 might exert a polar effect on the expression of MAB_4900c. Using the CRISPRi system for gene expression knockdown, we silenced either ponA1 or MAB_4900c in two separate engineered strains (Figure S2) and quantified their respective mRNA levels by qRT-PCR. As expected, induction of ponA1 silencing with 0.5 mg/L anhydro-tetracycline led to > 85% reduction in both ponA1 and MAB_4900c mRNA, while targeting MAB_4900c only suppressed the expression of MAB_4900c, confirming the operon structure (Figure 1A). The polar effect of ponA1 on the transcription of MAB_4900c was recapitulated in the ∆ponA1 knockout strain (Figure 1B). Next, we evaluated the effect of targeted gene silencing on DUR resistance. Silencing of ponA1 resulted in a 4-fold increase in DUR agar MIC, phenocopying the resistance observed in the spontaneous mutants and the ∆ponA1 deletion strain. However, specific silencing of MAB_4900c alone had no effect on DUR susceptibility, indicating that loss of ponA1 function is the primary driver of resistance. The broth MICs of both knockdown strains remained unchanged.
3. Discussion
In this study, we identify a novel mechanism of acquired resistance to DUR in Mab. Through single-step in vitro selection, we consistently isolated mutants with alterations in ponA1, encoding the class A PBP PonA1. This finding is notable because resistance to β-lactam-based therapies in Mab has predominantly been associated with mechanisms that protect the β-lactam or induce a general state of drug tolerance [8,9]. In M. tuberculosis, biochemical studies showed that PonA1 is one of the targets of DUR [11]. Thus, inactivation of PonA1 represents a direct, target-based resistance mechanism, a pathway previously thought to be uncommon for β-lactams in this pathogen.
The observation that inactivating a drug's target confers resistance is counterintuitive. Here, this could be explained by DUR’s multi-target mechanism of action. It is plausible that while DUR inhibits several PBPs and L,D-transpeptidases, binding to PonA1 is a primary driver of its antimicrobial activity. Therefore, complete removal of this key target via a loss-of-function mutation allows the bacterium to evade the drug's most potent effect and may overcome a lethal imbalance in cell wall synthesis. The viability of ponA1 loss-of-function mutants strongly suggests that other PBPs can compensate for its loss. This functional redundancy is a known feature of the mycobacterial cell wall synthesis machinery [12]. Under the selective pressure of DUR, the bacterium can sacrifice the PonA1-mediated pathway – most vulnerable to the drug – and rely on alternative, less-susceptible pathways for survival.
A striking and critical feature of this resistance mechanism is its dependence on cultureconditions. While ponA1 mutants exhibit a pronounced increase in MIC on agar, they appeared almost fully susceptible when tested by standard broth microdilution. This disconnect between agar- and broth-based susceptibility suggests that the resistance phenotype is linked to a physiological state specific to surface-based growth. Two non-mutually exclusive hypotheses could explain this phenomenon. First, the loss of PonA1 may alter cell wall architecture and colony properties in a way that limits drug diffusion through the nascent “biofilm” on a solid agar surface [13], a barrier that would not exist in planktonic cells in a well-mixed broth. Second, growth on a solid surface may induce a specific metabolic or stress state in which the loss of PonA1 is less detrimental, or in which compensatory pathways are activated, allowing the resistance phenotype to manifest.
These findings have potential clinical implications. Standard antimicrobial susceptibility testing is typically performed using broth-based methods [14]. Our results demonstrate that such methods would fail to detect this resistance mechanism, potentially leading to the misclassification of a resistant isolate as susceptible and subsequent therapeutic failure. The possible clinical relevance of this agar-dependent resistance is underscored by the fact that mycobacterial growth in the lungs often occurs in biofilm-like aggregates or microcolonies [15], a state that is more closely modeled by growth on a solid surface than in liquid culture [16].
In conclusion, we have identified loss-of-function mutations in ponA1 as a novel and unexpected mechanism of resistance to DUR in Mab. The conditional expression of this resistance highlights the complex interplay between bacterial genetics and physiology and raises important questions about the predictive value of standard susceptibility testing methods. This discovery expands our understanding of the resistance landscape of DUR and emphasizes the need for methodologies that better reflect the in vivo state of the pathogen to ensure the durable success of new therapies.
4. Materials and Methods
Mab subsp. abscessus ATCC 19977 was used as the wild-type strain in all experiments. Spontaneous resistant mutants were selected by plating approximately 10⁹ colony-forming units (CFU) of wild-type ATCC 19977 onto 7H10 solid medium containing 4× the agar minimum inhibitory concentration (MIC) of DUR (Cat# HY-117974, MedChemExpress LLC, USA). Plates were incubated at 37°C for 5 days. Resistant colonies were re-streaked on selective plates to confirm the resistance phenotype. Two independent selections were performed to ensure the reproducibility of the results. Broth and agar minimum inhibitory concentrations (MICs) were determined as previously described [17].
To knockout MAB_4901c, an allelic exchange substrate (AES) was employed as described in Figure S1. The AES contained a 500 bp region upstream of ponA1 (USH, upstream homology region) and a region 500 bp downstream of ponA1 (DSH, downstream homology region), flanking a cassette comprised of an apramycin resistance gene (Apra R), and an mScarlet reporter gene under control of the PLeft* promoter to enable selection and identification of recombinants [18,19]. The synthesized AES was PCR amplified using primers 5’-CGGACCGCCGGTGTGCCGTCGTACTG-3’ and 5’-CTGGTTAGCGTGCGATTGCAGAGAC-3’, electroporated into Mab ATCC 19977 and plated on 7H10 agar containing 50 mg/L apramycin. After 7 days of incubation at 37°C, colonies were screened visually for red color (mScarlet expression). MAB_4901c knockout was confirmed by whole-genome sequencing.
A CRISPR interference (CRISPRi)-dCas9 system provided on the pLJR962 plasmid [20] was utilized to knock down gene expression, as previously described [21]. Single-guide RNAs (sgRNAs) targeting the N-terminal coding region of each gene were designed based on predicted strength using the sgRNA Design Tool (https://pebble.rockefeller.edu/tools/sgrna-design). The sgRNA target sequences and protospacer adjacent motif (PAM) sequences are provided in Figure S2. Complementary oligos for each sgRNA were synthesized by Azenta Life Sciences, South Plainfield, NJ, USA. To construct the sgRNA expression plasmids, the recipient vector pLJR962 was digested with BsmBI (Thermo Fisher Scientific, Cat. No. ER0451). The complementary top and bottom oligos for each target were annealed in a thermocycler by incubating at 95°C for 2 minutes, followed by a gradual ramp-down to 25°C at a rate of -0.1°C per second. The resulting annealed duplexes were ligated into the BsmBI-digested pLJR962 vector using T4 DNA Ligase overnight at 16°C. Correct insertion of the sgRNA cassette was verified by Sanger sequencing. Gene expression knockdown was induced on solid and in liquid medium with anhydrotetracycline (ATc) as described [21].
Total RNA extraction and qRT-PCR were carried out as described [22]. Gene expression levels were normalized to the housekeeping gene sigA (Mab_3009), and relative expression differences were calculated using the 2-ΔΔCt method.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Deletion of ponA1 (MAB_4901c) strategy; Figure S2: Transcriptional silencing of MAB_4901c (ponA1) and MAB_4900c.
Author Contributions
Conceptualization, DAN, MX.; methodology, DAN, WWA, MX; formal analysis, DAN, MX, VD; writing—original draft preparation, DAN, VD; writing—review and editing, all; visualization, DAN, WWA, VD.; supervision, VD, TD; funding acquisition, V.D. and T.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NIH-NIAID awards R01-AI132374 and R01-AI184502 to V.D. and T.D.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data supporting the findings of this study are available within the article and its supplementary materials.
Acknowledgments
We thank our lab colleagues Chui Fann Wong, Aseem Palande and Virgina Saionz for collective brainstorming sessions. During the preparation of this manuscript, the authors used Gemini Pro2.5 for the purpose of refining the language of the first draft. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| DUR | Durlobactam |
| Mab | Mycobacterium abscessus |
References
- Kumar, K.; Loebinger, M.R. Nontuberculous Mycobacterial Pulmonary Disease: Clinical Epidemiologic Features, Risk Factors, and Diagnosis: The Nontuberculous Mycobacterial Series. Chest 2022, 161, 637-646. [CrossRef]
- Dartois, V.; Dick, T. Therapeutic developments for tuberculosis and nontuberculous mycobacterial lung disease. Nat Rev Drug Discov 2024, 23, 381-403. [CrossRef]
- McLeod, S.M.; O'Donnell, J.P.; Narayanan, N.; Mills, J.P.; Kaye, K.S. Sulbactam-durlobactam: a beta-lactam/beta-lactamase inhibitor combination targeting Acinetobacter baumannii. Future Microbiol 2024, 19, 563-576. [CrossRef]
- Dousa, K.M.; Shin, E.; Kurz, S.G.; Rubin, E.J.; Holland, S.M.; Olivier, K.N.; Daley, C.L.; Kreiswirth, B.N.; Pottinger, P.S.; Bonomo, R.A. The Role of beta-Lactam Antibiotics in Treating Mycobacterium abscessus: From Laboratory Insights to Clinical Applications and the Case for Clinical Trials. Clin Infect Dis 2025, online ahead of print. [CrossRef]
- Shin, E.; Dousa, K.M.; Taracila, M.A.; Bethel, C.R.; Nantongo, M.; Nguyen, D.C.; Akusobi, C.; Kurz, S.G.; Plummer, M.S.; Daley, C.L.; et al. Durlobactam in combination with beta-lactams to combat Mycobacterium abscessus. Antimicrob Agents Chemother 2025, 69, e0117424. [CrossRef]
- Shrivastava, A.; Boorgula, G.D.; Singh, S.; Stiles, D.; McShane, P.J.; Devine, M.; Gumbo, T.; Srivastava, S. Sulbactam-durlobactam improves cephalosporin and carbapenem susceptibility and time-kill effect against Mycobacterium abscessus. Microbiol Spectr 2025, e0149225. [CrossRef]
- Negatu, D.A.; Aragaw, W.W.; Gebresilase, T.T.; Paruchuri, S.; Kaya, F.; Shin, S.J.; Sander, P.; Dartois, V.; Dick, T. Durlobactam to boost the clinical utility of standard of care beta-lactams against Mycobacterium abscessus lung disease. Antimicrob Agents Chemother 2025, 69, e0104624. [CrossRef]
- Aragaw, W.W.; Gebresilase, T.T.; Negatu, D.A.; Dartois, V.; Dick, T. Multidrug tolerance conferred by loss-of-function mutations in anti-sigma factor RshA of Mycobacterium abscessus. Antimicrob Agents Chemother 2024, 68, e0105124. [CrossRef]
- Le Run, E.; Tettelin, H.; Holland, S.M.; Zelazny, A.M. Evolution toward extremely high imipenem resistance in Mycobacterium abscessus outbreak strains. Antimicrob Agents Chemother 2024, e0067324. [CrossRef]
- Rifat, D.; Chen, L.; Kreiswirth, B.N.; Nuermberger, E.L. Genome-Wide Essentiality Analysis of Mycobacterium abscessus by Saturated Transposon Mutagenesis and Deep Sequencing. mBio 2021, 12, e0104921. [CrossRef]
- Nantongo, M.; Nguyen, D.C.; Bethel, C.R.; Taracila, M.A.; Li, Q.; Dousa, K.M.; Shin, E.; Kurz, S.G.; Nguyen, L.; Kreiswirth, B.N.; et al. Durlobactam, a Diazabicyclooctane beta-Lactamase Inhibitor, Inhibits BlaC and Peptidoglycan Transpeptidases of Mycobacterium tuberculosis. ACS Infect Dis 2024, 10, 1767-1779. [CrossRef]
- Nguyen, D.C.; Dousa, K.M.; Kurz, S.G.; Brown, S.T.; Drusano, G.; Holland, S.M.; Kreiswirth, B.N.; Boom, W.H.; Daley, C.L.; Bonomo, R.A. "One-Two Punch": Synergistic ss-Lactam Combinations for Mycobacterium abscessus and Target Redundancy in the Inhibition of Peptidoglycan Synthesis Enzymes. Clin Infect Dis 2021, 73, 1532-1536. [CrossRef]
- Aguilera-Correa, J.J.; Boudehen, Y.M.; Kremer, L. Characterization of Mycobacterium abscessus colony-biofilms based on bi-dimensional images. Antimicrob Agents Chemother 2023, 67, e0040223. [CrossRef]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing. 2020, M100.
- Fennelly, K.P.; Ojano-Dirain, C.; Yang, Q.; Liu, L.; Lu, L.; Progulske-Fox, A.; Wang, G.P.; Antonelli, P.; Schultz, G. Biofilm Formation by Mycobacterium abscessus in a Lung Cavity. Am J Respir Crit Care Med 2016, 193, 692-693. [CrossRef]
- Sriramulu, D.D.; Lunsdorf, H.; Lam, J.S.; Romling, U. Microcolony formation: a novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J Med Microbiol 2005, 54, 667-676. [CrossRef]
- Sarathy, J.; Xie, M.; Wong, C.; Negatu, D.; Rodriguez, S.; Zimmerman, M.; Jimenez, D.; Alshiraihi, I.; Gonzalez-Juarrero, M.; Dartois, V.; et al. Towards a bactericidal oral drug combination for the treatment of Mycobacterium abscessus lung disease. ACS Inf Dis 2025. [CrossRef]
- Ganapathy, U.S.; Lan, T.; Krastel, P.; Lindman, M.; Zimmerman, M.D.; Ho, H.; Sarathy, J.P.; Evans, J.C.; Dartois, V.; Aldrich, C.C.; et al. Blocking Bacterial Naphthohydroquinone Oxidation and ADP-Ribosylation Improves Activity of Rifamycins against Mycobacterium abscessus. Antimicrob Agents Chemother 2021, 65, e0097821. [CrossRef]
- Kolbe, K.; Bell, A.C.; Prosser, G.A.; Assmann, M.; Yang, H.J.; Forbes, H.E.; Gallucci, S.; Mayer-Barber, K.D.; Boshoff, H.I.; Barry Iii, C.E. Development and Optimization of Chromosomally-Integrated Fluorescent Mycobacterium tuberculosis Reporter Constructs. Front Microbiol 2020, 11, 591866. [CrossRef]
- Wong, A.I.; Rock, J.M. CRISPR Interference (CRISPRi) for Targeted Gene Silencing in Mycobacteria. Methods Mol Biol 2021, 2314, 343-364. [CrossRef]
- Kurepina, N.; Chen, L.; Composto, K.; Rifat, D.; Nuermberger, E.L.; Kreiswirth, B.N. CRISPR Inhibition of Essential Peptidoglycan Biosynthesis Genes in Mycobacterium abscessus and Its Impact on beta-Lactam Susceptibility. Antimicrob Agents Chemother 2022, 66, e0009322. [CrossRef]
- Sarathy, J.P.; Xie, M.; Jones, R.M.; Chang, A.; Osiecki, P.; Weiner, D.; Tsao, W.S.; Dougher, M.; Blanc, L.; Fotouhi, N.; et al. A Novel Tool to Identify Bactericidal Compounds against Vulnerable Targets in Drug-Tolerant M. tuberculosis found in Caseum. mBio 2023, 14, e0059823. [CrossRef]
Figure 1.
Quantification of ponA1 and downstream mRNA transcripts following CRISPRi-mediated gene knockdown and knockout. (A). Relative expression of MAB_4901c (ponA1), MAB_4900c (hypothetical protein, separated by 4 bp from ponA1), and MAB_4899c (rpsF; 30S subunit ribosomal protein S6, separated by ~170 bp from MAB_4900c) in the CRISPRi knockdown strains without and with inducer ATc. (B) Relative expression of MAB_4901c, MAB_4900C and MAB_4899c in ∆ponA1 compared to parental wild-type strain. *: no PCR product detected. Expression levels were normalized to sigA mRNA and are shown relative to the corresponding control strain. Details of strain engineering are provided in Figure S1 and S2. .
Figure 1.
Quantification of ponA1 and downstream mRNA transcripts following CRISPRi-mediated gene knockdown and knockout. (A). Relative expression of MAB_4901c (ponA1), MAB_4900c (hypothetical protein, separated by 4 bp from ponA1), and MAB_4899c (rpsF; 30S subunit ribosomal protein S6, separated by ~170 bp from MAB_4900c) in the CRISPRi knockdown strains without and with inducer ATc. (B) Relative expression of MAB_4901c, MAB_4900C and MAB_4899c in ∆ponA1 compared to parental wild-type strain. *: no PCR product detected. Expression levels were normalized to sigA mRNA and are shown relative to the corresponding control strain. Details of strain engineering are provided in Figure S1 and S2. .
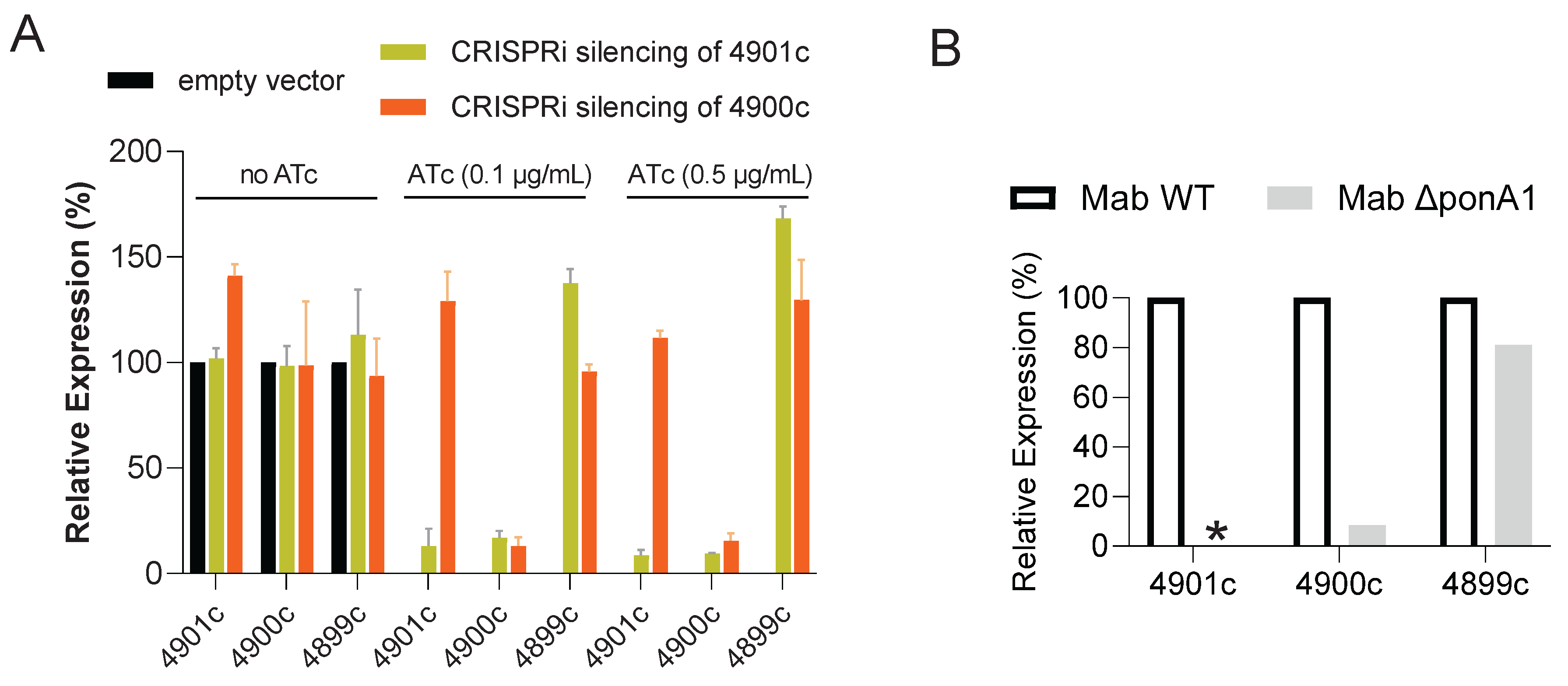

Table 1.
Characterization of DUR-resistant Mycobacterium abscessus mutants.
| Strain | Round of selection |
Agar MIC * (mg/L) |
Broth MIC * (mg/L) |
Mutations | Gene function | ||
| Gene | DNA alteration | Amino acid sequence alteration | |||||
| WT | 8 | 4 | wt | wt | wt | - | |
| DUR_res1 | 1st | >128 | 8 | MAB_4901c (ponA1) |
1312delC | Q438fs | Penicillin-binding protein |
| DUR_res2 | 1st | >128 | 8 | 369_370del | D123fs | ||
| DUR_res3 | 1st | >128 | 8 | C1156T | Q386stop | ||
| DUR_res4 | 2nd | >128 | 8 | G2027T | G676V | ||
| DUR_res5 | 2nd | >128 | 8 | C1333T | Q445stop | ||
| DUR_res6 | 2nd | >128 | 8 | C1333T | Q445stop | ||
| DUR_res7 | 2nd | >128 | 8 | 776_778del | 259_260del | ||
| DUR_res8 | 1st | 32 | 8 | MAB_0505c | 439dupC | R147fs |
Hypothetical protein |
| DUR_res9 | 1st | 64 | 8 | T608C | L203P | ||
| DUR_res10 | 2nd | 32 | 8 | 439dupC | R147fs | ||
| DUR_res11 | 2nd | 32 | 8 | 71_82del | 24_28del | ||
| DUR_res12 | 1st | 64 | 8 | MAB_0205c | G124C | G42R | Hypothetical protein |
| DUR_res13 | 1st | 64 | 8 | G124C | G42R | ||
| DUR_res14 | 2nd | 64 | 8 | G124C | G42R | ||
MIC determinations were carried out twice independently yielding the same results.
Table 2.
DUR susceptibility of spontaneous and engineered M. abscessus mutants.
| Mab strain | Agar MIC (mg/L) | Broth MIC (mg/L) | Strain characteristics |
| Wild-type | 8 | 4 | Wild-type ATCC 19977 |
| DUR_res2 | > 128 | 8 | Spontaneous DUR resistant ponA1 (MAB_4901c) frameshift mutant (Table 1) |
| ∆ ponA1 | > 128 | 4 | Engineered ponA1 deletion mutant (Figure S1) |
| ponA1 KD | 32 | 4 | CRISPRi ponA1 knockdown (Figure S2) |
| MAB_4900c KD | 8 | 4 | CRISPRi MAB_4900c (hypothetical protein) knockdown (Figure S2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



