Submitted:
21 November 2025
Posted:
21 November 2025
You are already at the latest version
Abstract
Synthesizing natural substances has always been a significant challenge for organic chemists. The key to a successful total synthesis lies in utilizing reactions that generate molecular complexity with high stereocontrol. Photochemical reactions offer immense potential in this regard, though their complex mechanisms require careful mastery. This review explores recent examples from the literature where light-mediated reactions are crucial, often irreplaceable by thermal alternatives. The manuscript is organized by dif-ferent photochemical processes, each introduced with relevant background. This review does not offer a complete analysis of all recent light-assisted syntheses; rather, it offers a glimpse into the growing trend of using photo-driven transformations to address signifi-cant synthetic challenges.

Keywords:
total synthesis
; photocycloadditions
; photooxygenation
; photorearrangements
; photoredox catalysis
1. Introduction
The total synthesis of natural products stands as a perpetual challenge in organic chemistry, demanding the strategic use of reactions capable of building molecular complexity with high degrees of stereocontrol and efficiency. Photochemical reactions, leveraging light energy to drive transformations often inaccessible by thermal methods, offer exceptional potential in this arena. This review provides a focused look at recent, impactful examples from the literature where light-mediated processes are not just utilised, but prove to be crucial and sometimes irreplaceable for achieving specific synthetic goals. The discussion is systematically organized by distinct photochemical mechanisms—including photocycloadditions, photooxygenation, photorearrangements, and photoredox catalysis—each introduced with fundamental principles. This systematic approach culminates with a detailed examination of a single, powerful total synthesis that strategically integrates multiple advanced photochemical methodologies, such as HAT catalysis and metallophotoredox transformations, illustrating the current pinnacle of strategic photo-driven design.
Ultimately, this work aims to highlight the growing trend and critical value of photo-driven transformations in solving significant synthetic puzzles within the realm of natural product construction.
2. Building Rings with Light: Cycloaddition Strategies
Most photochemists are familiar with the work of Giacomo Ciamician and Paul Silber, who, as early as 1908, described the cycloaddition of carvone upon a year's exposure to sunlight [1]. From the very beginning, this pioneering discovery highlighted the link between this class of reactions and their potential within the natural world. Cycloaddition reactions stand out as essential methods in synthetic organic chemistry, providing convergent and highly selective access to complex molecular architectures. Their broad applicability is particularly evident in natural product synthesis, where challenging ring systems and densely functionalized frameworks are common. As a subclass of pericyclic reactions, cycloadditions enable the efficient construction of cyclic structures with the possibility of achieving remarkable control over regio- and stereoselectivity. Their popularity stems largely from their efficiency and atom economy, both critical for constructing elaborate molecular frameworks characteristic of many natural products.
Among them, the [2+2]-cycloaddition stands out as a powerful method for forming cyclobutane rings—structural motifs that are widespread in natural molecules. In most cases, the use of light has been the key element enabling chemists to achieve this assembly during synthesis. A noteworthy subclass is represented by Paternò–Büchi reactions, whose relevance in this topic has been well recognized. Some recent studies have expanded on their scope and application; therefore, we have devoted a specific subchapter to this topic. Similarly, the [4+2]-cycloaddition has proven to be an invaluable tool in total synthesis, particularly in reactions leading to endoperoxides. The use of singlet oxygen in Diels–Alder reactions involving dienes and furan moieties has emerged as an interesting strategy for achieving high levels of oxygenation along carbon chains, a typical feature of these products. Finally, several other photochemical methods for ring construction have attracted attention, such as the Norrish–Yang cyclization and the Nazarov cyclization. Given their growing relevance and the recent publications on the topic, these transformations are discussed together in a miscellaneous subchapter.
2.1. [2+2]-Photocycloadditions: Accessing Strained Cyclobutane Rings
As mentioned in the introduction, [2+2]-cycloadditions are of vital importance, as they enable the incorporation of four-membered rings into complex molecular frameworks. Despite the inherent strain of the cyclobutane ring, this structural motif is widely distributed and plays a crucial role in a broad range of natural products.
Two reaction pathways are commonly used to photochemically assemble cyclobutane motifs: the first involves the direct excitation of one of the two substrates—typically an α,β-unsaturated ketone, whose excitation leads, via a fast ISC, to a relatively stable triplet state. Accordingly, the reaction pathway is similar to that of the Paternò–Büchi cycloaddition, proceeding stepwise via radical addition to the olefin, intersystem crossing of the resulting biradical to a singlet state, and final recombination to yield the desired cyclobutane product. The regioselectivity for the two possible products, commonly referred to as head-to-head or head-to-tail isomers, depends on the nature of the substituent on the second olefin, preferentially yielding the former with electron-withdrawing and the latter with electron-donating groups.
The second strategy involves photosensitization via energy transfer, using a photocatalyst with an easily accessible triplet state at a higher energy level than that of the target molecule. This approach allows even olefins with relatively low triplet energies to be effectively engaged in cycloadditions. Typical sensitizers include diarylketones (e.g. benzophenone) or common photoredox catalysts (e.g. ruthenium or iridium catalysts). In general, photocycloadditions perform best when the two reactive double bonds are installed within the same precursor, ideally with geometrical constraints to induce high degrees of regio- and stereoselectivity, which necessitates installing them onto complex molecular scaffolds. The utility of this class of reactions, however, is by no means limited to the introduction of the cyclobutane ring into the structure; on the contrary, they are particularly valuable also for enabling further modifications.
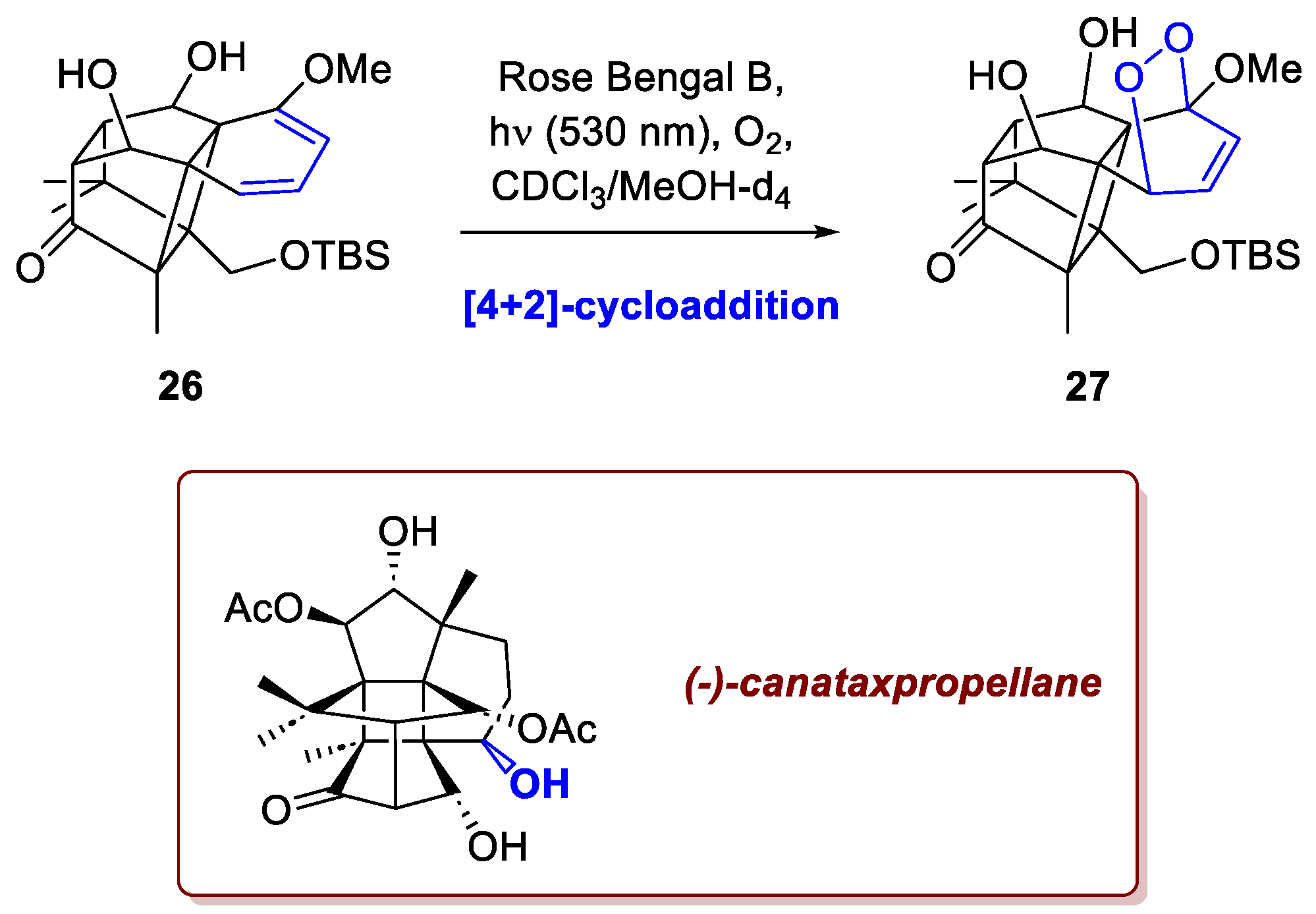
A recent example is the work of Jiao et al. [2], who successfully completed the total synthesis of the potent neurotoxin and NaV channel blocker, (+)-saxitoxin (Scheme 1). Saxitoxin is a formidable synthetic target, characterized by a dense, tricyclic core and the presence of multiple guanidinium groups. The researchers’ success pivoted on a key, novel transformation: an asymmetric intramolecular [2+2]-photocycloaddition involving an alkenylboronate ester as chiral auxiliary. Recognizing that the conventional approach—namely, an intramolecular Michael addition to form the C4–C12 bond—would face significant thermodynamic and kinetic challenges, the researchers devised an unconventional retrosynthetic strategy. Their innovative alternative relied on the aforementioned intramolecular [2+2]-photocycloaddition between the alkenylboronate ester incorporated in an 8-oxoxanthine derivative 1. The success of this crucial step is rooted in the unique properties of the 8-oxoxanthine: its photoexcitation causes it to lose planarity and undergo intersystem crossing to the triplet state. This creative approach offered major advantages beyond simply bypassing the problematic Michael addition. Crucially, it eliminated the need for difficult chemo- and regioselective enol formation and provided excellent stereocontrol over the [2+2]-photocycloaddition. As depicted in Scheme 1, the resulting intermediate 2, upon mild oxidation, was converted into a tertiary alcohol, which subsequently underwent a retro-aldol reaction to 3.
Similarly, in their work toward the total synthesis of epicolactone, Kravina and Carreira strategically exploited the intrinsic ring strain of the cyclobutane unit for a ring expansion methodology executed through a retro-aldol/aldol cascade sequence, which effectively constructed the central five-membered ring of the natural product's core [3]. For the synthesis of this bioactive and structurally intricate secondary metabolite produced by endophytic fungi of the genus Epicoccum associated with cash crops, the [2+2]-cycloaddition served as the key step for introducing the required structural complexity. This reaction between a quinone and an electron-deficient diene, both embedded in intermediate 4, is distinguished by its operational simplicity, proceeding under exceptionally mild conditions (just blue LED irradiation). The desired intermediate 5 is obtained with complete diastereoselectivity, remarkably establishing three quaternary stereocenters in situ from a conformationally flat starting material (Scheme 2). This high level of selectivity is fundamentally attributed to two factors: the favorable polarity matching between the reaction components and the critical role of the covalent tether, which ensures optimal spatial proximity and conformational control through its carefully engineered length and flexibility.
In the recent total synthesis of (–)-canataxpropellane, a structurally intricate diterpenoid natural product from Taxus spp. reported by Schneider et al., the ability to achieve exceptional regio- and stereoselectivity through a [2+2]-photocycloaddition once again proved to be essential [4]. Closely related to the anticancer agent Taxol (paclitaxel), this molecule exhibits high complexity, combining two propellane motifs and twelve contiguous stereocenters—five quaternary and four located on a cyclobutane ring—within a densely oxygenated framework. The authors proposed an alkene–arene ortho-photocycloaddition that delivered the key intermediate in excellent yield (Scheme 3). The efficiency of this transformation relied on both the polarity complementarity between the α,β-unsaturated ketone and the electron-rich arene in 6, and the spatial proximity of these moieties, achieved through the high diastereoselectivity of the preceding Diels–Alder step. As shown in the section dedicated to the [4+2]-cycloadditions (section 2.3 and Scheme 10), the oxidative functionalization of 7 was also performed photochemically. In a total of 26 steps, the total synthesis of canataxpropellane was completed with an overall yield of 0.5%.
Another elegant example in which a [2+2]-cycloaddition has proven valuable for constructing densely functionalized propellane cores, is illustrated by Grünenfelder et al. in the enantioselective total synthesis of (–)-10-hydroxyacutuminine [5]. Although this strategy does not directly afford acutumine or acutuminine, it provides an elegant and efficient route to functionalized propellane cores. The target compounds differ from acutuminine by a hydroxyl at C10 and from acutumine by the absence of one at C1. As shown in Scheme 4, the intramolecular [2+2]-cicloaddition between enone and the electron-rich methyl-substituted furan in 8, leads to the selective formation of a single regioisomer 9. In this synthesis, as in many other examples, the cyclobutane ring is not retained in the final product; nonetheless, its formation is crucial, as it enables the installation of two vicinal quaternary stereocenters in a single step. In the end, the enantioselective synthesis of (−)-10-hydroxyacutuminine was accomplished in 24 steps.
The functionalization of the reaction partners is essential, not only for achieving high regio- and stereocontrol but also for enabling the reaction itself. When less functionalized olefins are used in the construction of cyclobutane rings, direct irradiation of the starting material is often insufficient to promote the reaction. As mentioned in the introduction, another approach relies on the use of a photocatalyst, which can overcome this limitation. Using a suitable photocatalyst also allows the reaction to be carried out at different wavelengths, avoiding UV light, which can be problematic in complex molecules with many functional groups that may absorb UV and interfere with the [2+2]-cycloaddition. While the reaction generally proceeds via energy transfer, it can, in certain cases, also occur through SET.
A remarkable example is provided by the total synthesis of sceptrin by Ma et al. [6]. Sceptrin is a dimeric alkaloid containing pyrrole–imidazole units, produced by a marine sponge that may use an enzyme-promoted single-electron oxidation as a central mechanism to promote the cycloaddition. Its high nitrogen content renders it highly polar, redox-labile, and pH-sensitive, posing significant synthetic challenges. The fundamental concept in their total synthesis involved the efficient construction of the central cyclobutane ring through a SET-mediated photocycloaddition, closely replicating the proposed biosynthetic route. Following a procedure reported by Lu and Yoon [7], they performed the reaction of 10 with 3 mol% of Ir(ppy)3 and a CFL, hence the photoinduced SET generated a radical cation intermediate 11, which smoothly underwent the desired [2+2]-cycloaddition to afford tricyclic adduct 12 in good yield and with an acceptable diastereomeric ratio (Scheme 5). It should be emphasized that this approach also provided insight into the biosynthetic mechanism. When 9-fluorenone was used as a photocatalyst in place of Ir(ppy)₃, the reaction did not proceed, even though the two catalysts have comparable triplet energies. This outcome indicates that the dimerization operates through an oxidation–radical dimerization pathway rather than via energy transfer. Further elaboration led to ent-sceptrin in 16 additional steps, with an overall yield of 0.4%.
Despite the synthetic breakthroughs achieved in batch mode, scaling up these demanding photochemical transformations remains a significant hurdle. It is in this context that the work of Latrache et al. becomes particularly relevant [8]. Their report details a unified, bio-inspired total synthesis for a family of eight dimeric natural products, namely nigramide R, chabamide, dipiperamides F and G, and the notable marine alkaloids sceptrin, dibromosceptrin, ageliferin, and dibromoageliferin. The core of this approach lies in a photocatalytic dimerization of either piperine or fully elaborated hymenidin- and oroidin-type precursors, executed via catalyst-controlled [2+2] or [4+2]-cycloadditions. To overcome the limitations posed by prolonged photochemical exposure and the resulting photodegradation, they proposed a strategic shift from traditional batch processing to low-cost, 3D-printed photoflow reactors, enabling improved efficiency and significantly enhanced scalability compared to batch-based methods.
While most [2+2]-photocycloadditions typically necessitate an intramolecular setup to achieve high regio- and stereocontrol, Liu et al. have demonstrated that this is not strictly required. In the total synthesis of (–)-artochamin J [9], careful substrate design successfully by-passed the need for this covalent tethering. Exploiting boron's inherent oxophilicity, allylic alcohol 13 can undergo highly controlled photocycloadditions with alkenylboronate 14 via a temporary coordination (Scheme 6), thereby ensuring excellent regio- and stereocontrol without a permanent intramolecular link. The reaction employs a straightforward protocol, typically using 1.0 mol % fac-Ir(dFppy)3 blue LED (450 nm) for the photosensitization. The transient association of the allylic alcohol with the Bpin moiety is the mechanistic linchpin that effectively guides the two reacting partners into the correct geometry, enabling the efficient cycloaddition despite their intermolecular nature. Following this key step, the resulting boronate ester intermediate 15 provides a robust handle for subsequent manipulation. Specifically, this intermediate allows for the direct functionalization of the cyclobutane adduct via a Suzuki coupling. This cycloaddition–Suzuki sequence consistently yielded the target product 16 in excellent yield and diastereoselectivity, requiring only a few additional steps to complete the total synthesis of the natural product. The same approach was also used for the synthesis of (–)-piperaborenine B.
A recent application of this class of reaction has been reported by Schoch et al. in their successful semisynthesis of the complex taxane diterpenoid, 1-hydroxytaxuspine C [10], starting from readily available 10-deacetylbaccatin III. Taxanoid diterpenes, commonly known as taxanes, are secondary metabolites produced by slow-growing Taxus species, known for their strong cytotoxic activity, making them highly valuable lead structures for drug discovery and pharmaceutical development. Among them, nonclassical taxanes featuring up to four transannular bonds (C3–C11, C14–C20, C12–C4, C12–C20) within the typical 6/8/6 core are classified as cyclotaxanes. The most common subgroup, the (3,11)-cyclotaxanes, characterized by a C3–C11 transannular bond, includes 32 naturally occurring members identified so far. In their work the authors developed a scale-up-friendly and reproducible route, providing the first gram-scale synthetic access to C1-hydroxylated cyclotaxanes. The final natural product is achieved in 17 steps and for the installation of the pivotal C3-C11 bond, inspired by literature, they adopted a photochemical transannular bond formation from enone 17 (Scheme 7). Upon irradiation of 17 with 254 nm light under an inert atmosphere, 1.04 g of the desired (3,11)-cyclotaxane 18 was obtained, with no detectable formation of byproducts. It is important to emphasize that the mechanism of this transformation remains under investigation. A concerted σ2s + π2s pathway has been proposed as a plausible one, which justifies its inclusion in this subsection. Alternatively, excitation of the enone 17 to its first singlet excited state, followed by intersystem crossing to the triplet state, would generate a biradical intermediate. Facilitated by the close spatial proximity of C3 and C12, this species could undergo a 1,6-hydrogen atom transfer (1,6-HAT), forming a tertiary allylic radical. Subsequent recombination of the resulting C3,C11-diradical intermediate would then lead to the formation of the transannular C3–C11 bond.
2.2. The Paternò-Büchi Reaction: Assembling the Oxetane Ring
As previously mentioned, it is worth to include in this review a brief subsection dedicated to the Paternò–Büchi reaction. Setting aside the well-known controversy between Paternò and Ciamician regarding the origin of this reaction, it is known that in 1909 Paternò investigated the photochemical reaction of benzaldehyde with amylene and reported the formation of the corresponding [2+2]-cycloadduct. However, he was unable to distinguish between the two possible constitutional isomers of the oxetane, and the stereochemistry of the products could not be fully determined. Despite its potential, the Paternò–Büchi reaction was largely overlooked for several decades. It was not until 1954 that Büchi successfully repeated Paternò’s experiment and definitively identified the oxetane product. Traditionally, the Paternò–Büchi reaction is defined as a [2+2]-photocycloaddition between an alkene and the excited state of a carbonyl compound, leading to the formation of a four-membered oxetane ring. Although the reaction typically involves an excited carbonyl species reacting with a ground-state alkene, the reverse situation can also occur [11]. The remarkable potential of this reaction lies in its ability to provide an efficient synthetic route to small heterocyclic structures such as oxetanes, which are commonly found in natural products and biologically active molecules. There can be found different naturally occurring oxetane-containing compounds, such as thromboxane A2, mitrephorone A, and maoecrystal I, that also display a wide range of biological activities, including anticancer and cytotoxic effects. Regarding its role in total synthesis, the Paternò–Büchi reaction continues to hold considerable potential for further innovation and advancement. Although this transformation has been employed less frequently in natural product synthesis than the [2+2]-cyclobutane photocycloaddition, it has nonetheless been successfully applied to the syntheses of (+)-preussin [12], (±)-oxetanocin [13], and (±)-1,13-herbertendiol [14], although the oxetane ring is not always preserved in the final product. Additional noteworthy examples, albeit from earlier studies, are discussed in the comprehensive review by Kärkas et al. [15].
While classical Paternò–Büchi reactions are quite widespread throughout the synthetic records, their aza-variant delivering azetidines is remarkably underdeveloped, mainly due to the fast relaxation via isomerization of the imine excited state that hampers the development of cycloaddition strategies via direct irradiation. In this regard, the recent work of Wearing et al. represents a huge leap forward in accessing 4-membered nitrogen-containing rings in a straightforward fashion [16]. To extend the scope of the aza-Paternò–Büchi cycloaddition to acyclic imine equivalents in an intermolecular fashion, the researcher tailored the reactivity profile of the substrates, using conjugated oximes as imine components and conjugated alkenes as cycloaddition partners. The lowering of the frontier orbital energy gap through conjugation allows for a [2+2]-cycloaddition to occur upon excitation of the alkene via EnT using an iridium photocatalyst under blue light irradiation. This new strategy for accessing 2,3,4-substituted azetidine scaffolds avoids harsh reaction conditions (e.g. reduction of β-lactams), steric constraints (e.g. in nucleophilic cyclizations) for densely substituted products, or long reaction routes that were otherwise limiting the access to installing azetidine rings. Remarkably, this strategy was implemented in the synthetic pathway towards penaresidin A and B, sphingosine alkaloids isolated from the marine sponges Penares spp. known for their biological activity and their cytotoxic effects against tumour cells (Scheme 8). After the assembly of diene 19, the desired azetidine core was assembled with ease using conjugated oxime 20 and standard reaction conditions, and followed by reduction and deprotection, 2,3-epi- and 4-epi-penaresidin B were obtained. Noteworthy, related penaresidin stereoisomers have recently been demonstrated to exhibit similar or improved cytotoxicity compared with the natural diastereomer.
A thia-Paternò–Büchi reaction was used by Wright et al. in the synthetic approaches to calyciphylline A-type daphniphyllum alkaloids, himalensine A and daphenylline [17]. Since the discovery of calyciphylline A, over 50 calyciphylline A-type daphniphyllum alkaloids have been reported, making them the largest subclass of this alkaloid family (Figure 1). Many of these molecules show some structural differences from the original type: himalensine A, isolated in 2016 from Daphniphyllum himalense by Yue and colleagues, lacks the F-ring and a quaternary carbon at C8, while daphenylline, isolated from the fruits of Daphniphyllum longeracemosum in 2009, has a unique benzene-fused ring within the calyciphylline A-type framework (another recent report on the total synthesis of daphenylline is discussed in section 3.3).
The synthesis of daphenylline faced a major difficulty in introducing a crucial quaternary methyl group. Initial attempts to use a standard [2+2]-photocycloaddition/bond cleavage sequence failed because the reaction exclusively yielded unwanted head-to-tail products, which prevented the necessary stereospecific C–C bond cleavage. To overcome this, the team made a key strategic change, successfully exploiting an intermolecular thia-Paternò–Büchi reaction. Inspired by Padwa's work [18], they recognized that thioamides could form C–C bonds via thietane intermediates. This approach fundamentally reversed the course of the synthesis by drastically improving the selectivity. Unlike previous attempts, alkyllithium reagents preferentially react at the sulfur atom of thiones, leading to superior control and enabling the desired transformation; the thietane intermediates can subsequently be desulfurized with Raney nickel. Guided by these insights, irradiation of compound 21 with 400 nm blue LEDs in the presence of trithiocarbonate 22 and [Ir(dF(CF₃)ppy)₂(dtbbpy)]PF₆, as per the previous report by He et al. [19], resulted in a 55% yield for spirocyclic thietane 23 (77% based on recovered starting material). The thietane intermediate 23 was first reduced to 24 with LiAlH₄, followed by quenching with a Raney Ni/H₂O slurry to complete desulfurization to 25. After a few additional steps, this concise 11-step total synthesis was completed (Scheme 9).
2.3. [4+2]-Cycloadditions with Singlet Oxygen: Controlled Oxygenation and Endoperoxide Formation
The [4+2]-cycloaddition is one of the most powerful and versatile synthetic tools derived from singlet oxygen chemistry. This Diels-Alder type reaction is widely exploited for introducing multiple oxygen atoms and forming endoperoxide moieties. These endoperoxides are privileged, strained-ring structures that can be readily transformed into other valuable functionalities, making this method crucial for complex synthesis. In this regard, the widespread use of photosensitizers granted easier access to the very peculiar and highly reactive reagent, singlet oxygen. Singlet oxygen can also be generated through thermal pathways, but photosensitization offers the most appealing conditions. It only requires an oxygen source, a catalyst (methylene blue, rhodamine B, and tetraphenylporphyrin being the most common), and a narrow-band source of irradiation, thereby granting extremely selective and mild conditions. In general, starting from the 1980s, the use of singlet oxygen in total syntheses has become increasingly frequent, as its unique reactivity allows for the engineering of biomimetic approaches toward natural targets. Cycloaddition reactions involving singlet oxygen can target conjugated systems, proceeding either through a [4+2]-pathway to yield endoperoxide products— the focus of this section— or through a [2+2]-pathway to form dioxetanes.
As anticipated in section 2.1, oxidative functionalization was used to elaborate the [2+2]-cycloadduct toward the synthesis of canataxpropellane [4]. The diene moiety is expediently employed to incorporate two additional oxygen atoms with complete stereoselectivity (through an exceptionally mild and selective photoreaction utilizing Rose Bengal B and green light. Due to the rigid conformation of intermediate 26, imposed by the four-membered ring, only the diene's exo-face is accessible. This steric control forces the exclusive formation of the desired endoperoxide product 27, that is further elaborated to afford the natural product.
Scheme 10.
[4+2]-cycloadditions in the synthesis of canataxapropellane.
The recent total synthesis of (+)-tetrodotoxin by Murakami et al. provides another compelling example of this transformation's applicability [20]. Tetrodotoxin, initially isolated from puffer fish (order Tetraodontiformes), possesses an intriguing, highly oxidized structure featuring a dioxaadamantane core fused with a cyclic guanidine moiety. Due to its role as a potent voltage-gated sodium channel blocker, the compound holds significant medicinal importance as a potential scaffold for developing a new class of analgesics. The synthetic route developed by the authors commences with α-methyl-D-mannoside as the primary chiral source. Thanks to a [4+2]-cycloaddition with singlet oxygen on the intermediate 28 (Scheme 11) using Rose Bengal, that proceeded with complete stereoselectivity, the intermediate 29 was obtained as a single isomer following the reductive cleavage of the endoperoxide photoadduct 30. This approach was essential to obtain the resultant allylic diol which permitted further enantioselective oxidation of the bicyclic core via a Sharpless epoxidation, followed by additional modifications, to afford the target compound.
Photooxygenation was also pivotal for Mao et al. in the 15-step, enantioselective total synthesis of fortalpinoid Q [21], a specific member of the Cephalotaxus diterpenoids family. This family is a captivating class of natural products holding significant interest due to both their complex structures and their promising biological activity. Fortalpinoids share a 7-6-5-6-6 pentacyclic ring skeleton as their common structural features, and a key strategic challenge in their synthesis was the construction of the D/E bicyclic ring system. Initially, the authors had envisioned generating this bicyclic system via an aldol-lactonization cascade directly from intermediate 31, following the hydroboration of the cyclopentadiene double bond as described by Frey et al. [22] (Scheme 12). However, all attempts to implement this crucial cascade failed due to a detrimental side-reaction: a vinylogous aldol condensation. This competing, undesired reaction derailed the cyclization sequence. In response to this significant hurdle, a successful, revised strategy was developed. To set the stage for the cascade, the authors introduced an essential modification to the pentacyclic core. This involved a singlet oxygen hetero-Diels-Alder reaction with the cyclopentadiene moiety of intermediate 31. This reaction, followed by the ring-opening of the resulting endoperoxide with thiourea, stereoselectively furnished diol 32. The stereochemical outcome of the Diels-Alder reaction results from the approach of the singlet oxygen to the less sterically hindered face of the cyclopentadiene moiety. This ingenious step served a dual purpose: it introduced the necessary C20 oxidation state and, crucially, it generated an additional C10 hydroxyl group. It was the presence of this C10 hydroxyl group that successfully prevented the parasitic vinylogous aldol reaction that had plagued the previous unsuccessful attempts, allowing the desired cyclization to proceed. The efficient aldol-lactonization cascade ultimately provided the desired fortalpinoid Q in 57% yield from 33.
The research by Zhu et al. on the gram-scale total synthesis of (±)-illisimonin A, a sesquiterpenoid isolated from the Illicium genus, provides a significant example of a planned key step that failed to deliver the desired intermediate [23]. This molecule features a highly congested cage-like 5/5/5/5/5 pentacyclic scaffold, with 7 contiguous stereocenters, over a total of only 15 carbon atoms. Specifically, in their attempt to form the polycyclic framework and introduce the oxygenation pattern, the proposed oxa-[4+2]-cycloaddition between a cyclopentadiene intermediate 34 and singlet oxygen was expected to yield a peroxide bridged intermediate 35 (Scheme 13). However, instead of the desired product, the reaction exclusively furnished a rearranged, ring-expanded product 36. The instability of 35 leads to a consecutive sequence involving O-O bond cleavage, C-C bond cleavage, and hydrogen migration, ultimately resulting in the rearrangement to the conjugated enedione 36. To circumvent this synthetic roadblock, the authors strategically shifted their approach, utilizing a nitroso-Diels–Alder reaction as a crucial alternative. This method successfully enabled the precise installation of the correct oxidation state and the construction of the final molecular architecture, ultimately leading to the completion of the gram-scale total synthesis.
In addition to dienes, there are also furans that are highly effective substrates for singlet oxygen [4+2]-cycloadditions, and the resulting endoperoxides are widely recognized and utilized in chemical synthesis. A striking example of this methodology is demonstrated in the asymmetric total synthesis of (±)-spirochensilide A by Liang et al. [24]. Isolated from Abies chensiensis (an endemic Chinese coniferous tree), spirochensilide A presents a formidable structural challenge, totalling six rings and nine centres of chirality. The molecule features an uncommon spirocyclic core, two pairs of vicinal quaternary carbon stereocenters, and an anomeric spiroketal. The strategic approach designed to address these peculiarities relied on a singlet oxygen [4+2]-cycloaddition followed by cyclization to install the spiroketal ring (Scheme 14). The photocycloaddition took place smoothly from advanced intermediate 37 using methylene blue and a tungsten lamp. This furnished the endoperoxide 38, which was immediately opened under acidic conditions to give compound 39. Final deprotections and manipulations ultimately afforded the target molecule in a total of 22 steps with a 2.2% overall yield.
A similar strategy can be observed in Tuccinardi and Wood's total synthesis of the polycyclic nor-furanocembranoids (+)-ineleganolide and (-)-sinulochmodin C [25], challenging structures that feature seven contiguous stereocenters on a pentacyclic core. The synthesis built the key macrocyclic precursor 40, with the correct stereochemistry, from simple starting materials, subsequently the furan moiety was smoothly oxidized using Rose Bengal (Scheme 15). The resulting endoperoxide was reduced in situ to the ene-dione 41, which then rearranged under acidic conditions to the keto-tetrahydrofuran 42. The synthesis was completed by a final transannular Michael reaction, which delivered a mixture of the two natural products. The entire 20-step sequence concluded with an approximate 1% yield.
2.4. Miscellaneous Reactions
As emphasized in the introduction, the use of light to build rings within complex molecular structures often represents a key step in the total synthesis of natural products. Beyond the cycloadditions discussed above, several other cyclization strategies have been ingeniously employed for this purpose. In general, photocatalyzed HAT- and XAT-based methodologies frequently involve radical cyclization processes and the most notable recent examples are discussed in detail in the corresponding section.
Other processes involving the direct excitation of a reagent, such as the photoenolization/Diels–Alder (PEDA) sequence, also represent fundamental methods that proceed through a cyclization step. The Diels–Alder reaction is a cornerstone of organic synthesis, enabling the stereospecific and regioselective construction of six-membered carbocycles through a concerted [4+2]-cycloaddition under thermal condition. If it is combined with light activation, its scope can be elegantly extended. In the PEDA sequence, photoexcitation of an aromatic carbonyl generates a ketyl diradical I, this intermediate can undergo a 1,6-hydrogen atom transfer (HAT) from an ortho-aliphatic C–H bond delivering II and, after subsequent electronic rearrangement, a corresponding enol III is produced. So, this photoenolization leads to the generation of an intermediate that can then participate in different cyclizations, such as a DA reaction (Scheme 16).
Several examples of its use in the total synthesis of natural products have been reported in the literature, with a recent notable case described in the work of Lu et al. on the asymmetric total syntheses of (+)-xestoquinone and (+)-adociaquinones A and B complex natural products from Xestospongia sponges [26]. These compounds feature a pentacyclic fused core with an additional F ring for adociaquinones. Following a desymmetric intramolecular Michael addition catalyzed by an organocatalyst, the [6,6]-bicyclic decalin B–C scaffold 43 bearing an all-carbon quaternary centre at C6 was successfully constructed. Subsequently, formation of the naphthalene diol D–E framework was achieved via a Ti(O-i-Pr)₄-promoted PEDA sequence, which enabled furan ring formation through a cyclization–oxidation cascade (Scheme 17). The titanium catalyst was essential, likely stabilizing a chelated transition state that favoured an endo-selective [4+2]-cycloaddition and promoted dehydration to yield enone 44 with excellent diastereoselectivity. Subsequent transformations furnished (+)-xestoquinone and, through a late-stage cyclization, (+)-adociaquinones A and B in seven steps from the corresponding aldehyde.
The same research group also reported the total syntheses of several hasubanan alkaloids, employing a related PEDA sequence that proceeded intramolecularly [27]. The hasubanan alkaloids constitute a family of biogenetically related natural products characterized by a tricyclic aza[4.4.3]propellane core, primarily isolated from Stephania species used in traditional Chinese medicine. Over forty members of this family have been identified, displaying diverse biological activities, including antiviral, antimicrobial, and cytotoxic effects, making them promising targets for medicinal research. In this work, the authors proposed a synthetic route to periglaucines A–C, N,O-dimethyloxostephine, and oxostephabenine. These molecules represent a subgroup of hasubanan alkaloids, characterized by a higher degree of oxidation and a tetrahydrofuran ring formed via acetalization, along with a densely functionalized and highly oxidised C ring bearing three contiguous quaternary centres (C8, C13, and C14). The PEDA sequence proved to be fundamental for constructing the highly functionalized tricyclic core skeleton bearing a quaternary centre. As described above, the aldehyde underwent rapid photolysis to generate a tetrasubstituted hydroxy-o-quinodimethane intermediate. Upon chelation with the titanium catalyst, this intermediate undergoes the desired intramolecular [4+2]-cycloaddition with the unsaturated ester moiety, effectively constructing the B and C rings of the hasubanan core (Scheme 18). The use of (S)-TADDOL as a chiral ligand for the metal centre established a stereochemically defined environment, enabling smooth reaction progress and affording exclusively the endo-cycloadduct.
Another fundamental reaction in this field is the Norrish–Yang cyclization, which was masterfully exploited by Zheng et al. [28]. The Norrish–Yang cyclization is a photochemical ring closure process wherein a light-activated carbonyl functional group abstracts a hydrogen atom from within the same molecule, creating a 1,4-diradical intermediate. This unstable specie cyclizes to form cycloalkanols. Due to its capacity to introduce functionality into typically unreactive C-H bonds, this transformation has garnered substantial interest within the field of synthetic chemistry, finding wide applicability, notably in the construction of complex natural products. Nevertheless, it should be noted that achieving stereochemical control in the Norrish–Yang reaction often remains challenging, as the coupling of the diradical intermediates typically affords mixtures of both syn- and anti-products. Consequently, the attainment of high diastereoselectivity represents a major hurdle to the broader synthetic application of this transformation. Significant improvements have been realized by controlling the conformation of the diradical intermediate through strategies such as intramolecular hydrogen bonding, steric modulation by adjacent substituents, encapsulation within supramolecular hosts, and exploitation of solid-state crystal-packing effects. Zheng and colleagues studied immunosuppressive plant sesterterpenoids and discovered the gracilisoids, an extremely rare new family of natural products. They isolated these compounds from the ethnomedicinal Lamiaceae plant Eurysolen gracilis. The family includes the known gracilisoids A–E and four new compounds, gracilisoids F–I. Gracilisoid A is a unique monocarbocyclic sesterterpenoid, while the others feature two new types of bicyclo[3.2.0]heptane carbon skeletons, each with six neighboring stereogenic centers. The team used (-)-citronellal as a starting material and a bioinspired approach for the synthesis. The authors proposed gracilisoid A (47) as the biosynthetic precursor to gracilisoid F and gracilisoid H (Scheme 19). The crucial step involved an α-diketone-based Norrish–Yang photocyclization followed by an α-hydroxy ketone rearrangement. They readily performed this transformation by irradiating gracilisoid A with a compact fluorescent lamp under oxygen-free conditions. Treating the resulting crude products with silica gel yielded the desired products. This result suggests a radical Norrish–Yang photocyclization: light excites the α-diketone in 47, creating a 1,2-biradical intermediate that abstracts respectively the C10 and C7 hydrogen atoms (respectively) to form the corresponding 1,4-biradicals. These biradicals then form C–C bonds, stereoselectively producing the bicyclic intermediates 48 and 49 while minimizing steric clash. Intermediates 48 and 49 then undergo an α-hydroxy ketone rearrangement on silica gel (mildly acidic), which relieves ring strain and yields the target bicyclo[3.2.0]heptane skeletons. Further photochemical oxidation steps completed the synthesis of the entire gracilisoid family (A-I).
Another remarkable reaction is the Nazarov cyclization, a 4π-conrotatory electrocyclization, which stereospecifically forms functionalized cyclopentanones, structural motifs commonly found in natural products. It must be reminded that to design an efficient cyclization, it is useful to consider the reaction as a two-stage process: the first stage involves the 4π-electrocyclization forming an allylic cation, and the second stage concerns the fate of this cationic intermediate. As highlighted by Frontier and Hernandez [29], fully understanding the individual steps of the process is crucial for enhancing reactivity and precisely controlling selectivity—an aspect of central importance in total synthesis. A modern emblematic example of its exploitation in natural product synthesis is reported by Rao et al. [30]. In this specific case they exploited a torquoselective interrupted Nazarov reaction to obtain the divergent total synthesis of two phragmalin (moluccensins G and H) and two khayanolide-type (krishnolide F and khayseneganin F) limonoids. After inconclusive attempts with Lewis acid–mediated Nazarov cyclization, a milder photochemical activation proved to be the most suitable approach. As shown in Scheme 20, starting from the dienone 50 and irradiating at 254 nm in the presence of i-PrCO2H, the Nazarov cyclization was triggered, providing a 4.8:1 mixture of the two isomers: 51 (the desired one) and 52 in 75% combined yield. The tertiary alcohol at C14 proved to be essential to avoid photoenolization followed by a 6π-electrocyclization and diene isomerization, which would lead to an undesired by-product (not shown). It can therefore be stated that robust methodology has been establish, by integrating a torque-selective interrupted Nazarov cyclization with a Liebeskind-Srogl coupling, a benzoin condensation, and bidirectional acyloin rearrangements. This strategic combination significantly simplifies the synthetic design. Furthermore, this developed approach offers valuable additional perspectives on the biosynthetic connections between these two separate molecular skeletons.
3. Beyond Cycloadditions: Diverse Photochemical Rearrangements and Oxidations
Following the exploration of photochemical cycloadditions in the previous chapter, Chapter 3 delves into two other powerful classes of light-mediated transformations: rearrangements and oxidations. These reactions are indispensable tools in the total synthesis of natural products, often enabling the construction of complex or strained molecular architectures with exceptional selectivity and efficiency.
3.1. Wolff Rearrangements
Dating back to 1902, the Wolff rearrangement of α-diazoketones – righteously named after Ludwig Wolff who first reported it [31] – has found vast applications in total syntheses. Beside homologations such as the Arndt–Eistert synthesis [32], it proved its versatility in particular as a ring contraction methodology, affording strained rings that would otherwise be hard to synthetize while leaving an exocyclic carboxylic group that can be easily removed or used as further synthetic handle. The process can be induced via metal catalysis (e.g. Ag salts) or even thermally, but selective irradiation of the diazoketone group proved over time to be more orthogonal with respect to other moieties present on the substrate, making it very convenient in the context of a total synthesis.
In the total synthesis of (+)-[5]-ladderanoic acid, Hancock et al. used a Wolff ring contraction to replace the envisioned [2+2]-photocycloaddition that led to unexpected results [33]. Ladderanes are a family of natural products first isolated in 2002 from anammox bacteria, presenting a peculiar skeleton with fused cyclobutane rings. Taking inspiration from the synthesis of similar structures reported by Mascitti and Corey [34,35], the core cyclobutane rings were assembled via successive [2+2]-photocycloaddition/Wolff rearrangements (Scheme 21). Cyclopentenone 53 first underwent [2+2]-photocycloaddition with 1,2-dichloroethylene, affording olefin 54 after a protection/elimination sequence. This intermediate underwent another [2+2]-cycloaddition with 53 affording 55, to which followed diazotization to 56 and Wolff rearrangement, delivering the ring-contracted product 57. After a 3-step sequence to achieve intermediate 58, this latter underwent the same ring contraction on the opposite side delivering the target compound after further elaboration.
Similarly, Sun et al. exploited a Wolff ring contraction in their recent total synthesis of (-)-retigeranic acid A [36]. Found in the lichen Lobaria retigera from Western Himalayas, this compound features a pentacyclic carbon core with eight stereocenters, three of which are bridged quaternary carbons. The group of Ding elaborated a strategy that exploits a late-stage ring contraction delivering the desired CDE ring fusion. The ketone precursor was assembled in 12 steps from commercial starting materials; then, upon in situ diazotization, 60 underwent Wolff ring contraction, delivering ester 61 in good yield, and the researchers could reach the natural target in two more steps (Scheme 22).
3.2. Skeletal Rearrangments and Isomerizations
The interaction of light with matter, in spite of its fundamental role in the evolution of life on our planet, had not been clearly established for a long time, in particular the interaction of light with chemical substances. Among the oldest known interactions are surely those photoinduced processes leading to isomerization of susceptible moieties or molecular rearrangements. We examine here the synthetic evolution and impact of such processes in contemporary syntheses.
To highlight the historical and synthetic importance of this class of phototransformations, it is surely worth mentioning first the approach of Zhang et al. to (+)-berkeleyacetal D and (+)-peniciacetal I [37]. Berkeleyacetals are a class of meroterpenoids featuring a densely functionalized and highly oxidized pentacyclic core, with a 6/7/6/5/6 fused ring system. Berkeleyacetal, in particular, presents 17 of the 18 carbons of its pentacyclic skeleton in an oxidised state, or serving as stereocenters (for a total of eight, two of which quaternary). These characteristics pose a significant challenge to the synthesis of it and its congeners. Remarkably, Li’s group envisioned the synthesis of the 5/7 A/B fused rings of intermediate B by means of a photosantonin rearrangement – that same process that in 1834 puzzled Trommsdorff, who first observed it within the crystals of α-santonin (an antihelminthic drug extracted from plants of the Artemisia family) turning yellow upon sunlight exposure before bursting [38]. Precursor 62 was synthetized as single diastereoisomer in 16 steps from available starting materials. At this point, irradiation with a Hg lamp in AcOH/H2O delivered enone 63 in high yield, which was eventually converted in peniacetal I (7 steps) and berkeleyacetal D (8 steps) (Scheme 23).
The isomerization of C=C bonds is surely one of the oldest photochemical processes known to chemists, yet some limitations are usually inherent to the process itself. First, upon UV irradiation, other functional groups (e.g. carbonyls or other C=C bonds) might undergo undesired processes in parallel. Moreover, depending on the structures of the products, the final ratio of E/Z products might strongly change from scaffold to scaffold. The use of photocatalysis partially overcomes these issues, and a veritable example can be found in Schoch and Gaich’s pathway towards (−)-sodagnitin E [39]. Sodagnitins are chromogenic molecules found in a variety of rainforest fungi (Cortinarius sodagnitus, Cortinarius fulvoincarnatus, and Cortinarius arcuatorum), featuring a strong colour reaction upon exposure to basic media and showing promising biological activity. Sodagnitin E presents a clustered 6/6/5-tricyclic core and a highly oxidized sidechain bearing a six-membered lactol and a 3(2H)-furanone moiety. In the last steps of the synthesis, the authors isolated enal 64 in good yields, but the cyclization to the desired lactol was hampered by the E configuration of the double bond. Photocatalysis allowed them to circumvent the need for direct UV irradiation for the isomerization (detrimental to the sensitive molecule). Upon exposure to riboflavin at 400 nm, isomerization to the Z enal 65 and subsequent cyclization to lactol promptly occurred, leading to the desired final product after a last deprotection step (Scheme 24).
3.3. Oxidations
Photochemistry and photoredox catalysis found vast applications in performing trickier or unprecedented oxidations of benzylic, styrenic, or dienic systems.
Cui et al. heavily relied on photochemistry in their semisynthetic approach to (+)-clionastatins A and B [40]. Clionastatins are polychlorinated steroids, found in the burrowing sponge Cliona nigricans in very low amounts (1.0–1.3 mg/1.7 kg of dry sponge), featuring a highly oxidized tetracyclic androstane core that is further decorated by three or four chlorine atoms. The semi-synthesis presented by the authors features two stages – a former chlorination one and a latter oxidation one – from testosterone, sharing the same core and configuration of the substituents. During the oxidation stage, the authors resorted to photoredox catalysis to perform an elegant anti-Markovnikov oxidation-elimination. The electron-rich C6–C7 double bond of intermediate 66 (also photochemically introduced) was selectively engaged over the electron-deficient C4–C5 one, affording dienone 67 in a single step, all the while leaving C15–OH unoxidized (Scheme 25). Cui and colleagues conveniently adapted a procedure reported by Zhang et al. [41], where a photoredox manifold is effectively coupled with a cobalt catalyst to give the same Wacker-like oxidation on styrenic systems.
Benzylic positions can be selectively engaged in oxidation by means of photoredox catalysis, as portrayed by Yang et al. in their total synthesis of (+)-stephadiamine [42]. Belonging to the family of hasubanan alkaloids, isolated from the plants of genus Stephania and structurally homologous to the morphinan alkaloids, these compounds feature a tetracyclclic aza[4.4.3]propellane core at different degrees of oxidation, in some cases adorned with an oxygen heterocycle (as for (+)-stephadiamine) and four stereocentres, of which two tertiary amines and one quaternary carbon. To address the failure of all thermally-driven protocols, the C10 benzylic position of intermediate 68 was oxidised by means of photoredox catalysis adapting a procedure from Lee et al. [43] (Scheme 26). Overall, the process proceeds by sequential oxidative SETs at the electron-rich arene, delivering ultimately a benzyl cation that, upon addition of water, delivers an alcohol intermediate and ultimately product 69. This carbonyl handle was then elaborated into an alcohol upon diastereoselective reduction, and incorporated in the E-ring of stephadiamine by transesterification.
One more example of remarkable use in terms of photochemical strategies is showcased in the synthesis of (-)-daphenylline by Wu et al. [44]. This hexacyclic Daphniphyllum alkaloid features a central tetrasubstituted arene ring, with six stereocentres and a tertiary bridgehead amine. Most interestingly, intermediate 70 was promptly hydroformylated using a protocol reported by Alektiar et al. [45] (Scheme 27). Upon irradiation, a thiol catalyst delivers a formyl radical via HAT from the corresponding formate salt. After radical addition to the isolated olefin and HAT from the thiol to the alkyl radical, the desired product 71 was achieved in high yield, with this photochemical alternative performing much better than the more common palladium-catalyzed hydroformylation. The photochemical process is not only more tolerant of the carbamate protecting group (R= NHBoc), but also easier to scale up due to the simplicity of the operational conditions. Later in the synthesis, the same hydroformylation was exploited with analogous efficiency on intermediate 72, affording 73, which was eventually elaborated into the final product.
Lastly, a beautiful example of the possibility to tackle significant synthetic challenges is portrayed within the synthesis of dixiamycin A and B by Nandi et al. [46]. The innovation is represented by the late-stage N-N oxidative dimerization of the carbazole-derived core scaffold, which allowed for a smooth synthesis of dimeric indolosesquiterpene alkaloids dixiamycins A/B, isolated from marine Streptomyces sp. and featuring antimicrobial, antiviral, antitumor, immunomodulatory (including anti-HIV), and enzyme inhibitory properties. Most interestingly, these dimers feature a pentacyclic core bearing four contiguous stereocenters on a trans-decalin scaffold annulated to a carbazole moiety. The rare and congested N-N bond between the two monomeric units leads to the emergence of axial chirality and thus to the differentiation of dixiamycins A/B as two different diastereo-atropoisomers. After synthetising a protected version 74 of xyamicin A, this monomer was subjected to photoredox-catalyzed conditions using a suitable Ir-catalyst and a base in the presence of air to exploit oxygen as stoichiometric oxidant (Scheme 28). Upon SET oxidation, the carbazole anion delivers a N-centered radical that rapidly undergoes radical pairing, delivering the dimer (and its atropoisomer) in very high yields, separated easily upon column chromatography. Upon TMS deprotection and ester hydrolysis, the target compound was eventually achieved.
3.4. Ene-Reactions
Photochemical oxidation reactions represent a fundamental class of light-induced transformations in organic chemistry, and the use of singlet oxygen as a reagent plays a critical role at this level. This topic has previously been introduced in the chapter dedicated to cycloadditions, specifically due to the fundamental importance of [4+2]-cycloadditions that yield endoperoxides. More generally, since the 1980s, the application of singlet oxygen in total syntheses has become increasingly frequent, as its unique reactivity allows for the engineering of more biomimetic approaches toward natural targets. Reactions involving singlet oxygen also include ene-reactions (delivering hydroperoxides) and oxidations of heteroatoms. At a time when the demand for sustainability in chemistry has encouraged the use of cheap, abundant, and environmentally friendly resources, the idea of utilizing atmospheric oxygen and visible light for chemical transformations is compelling. More specifically, the Schenck-ene reaction proves particularly useful, as it involves reacting simple, unbiased alkenes reacting with atmospheric oxygen under visible light irradiation in the presence of a catalytic photosensitizer, resulting in the formation of valuable allyl hydroperoxides.
The strategic use of ene reactions with singlet oxygen clearly demonstrates the power of the biomimetic approach in chemical synthesis, notably in the recent production of important natural products like Artemisinin. Artemisinin, an essential antimalarial drug discovered by Nobel laureate Youyou Tu in 1972, showcases this technique. To allow for the synthesis of both enantiomers of Artemisinin and study the dependence of its antimalarial activity on its configuration, Krieger et al. developed a strategy in which the most crucial step involved a highly biomimetic Schenck-ene/Hock cleavage sequence utilizing singlet oxygen (Scheme 29) [47]. In this critical final stage, singlet oxygen performs a Schenck-ene reaction on 75, which generates the hydroperoxide intermediate 76. This intermediate, upon protonation, then undergoes Hock cleavage, triggering a ring expansion to form intermediate 77. This is followed by a cascade of reactions: hydrolysis to intermediate 78, subsequent oxidation using triplet oxygen to form 79 and a final ring closure cascade, and methylation, which ultimately completes the formation of the target molecule. This sequence is considered one of the most biomimetic methods available for synthesizing Artemisinin and its derivatives.
The total synthesis of taxol offers another compelling example of challenging organic chemistry. This famous anticancer drug, originally isolated from the bark of the Taxus brevifolia tree, is renowned among organic chemists not only for its significant biological importance but also for its immense and formidable structural complexity. Its discovery spurred one of the most extensive synthetic efforts in the chemical community. Structurally, taxol features a strained, highly oxidised tetracyclic [6-8-6-4] core, including a bridgehead alkene and eleven stereocentres. While most synthetic strategies historically focused on forming the central eight-membered ring via different bond disconnections (C9-C10 or C10-C11), the recent total synthesis by Hu et al. introduced an unprecedented approach by disconnecting the C1-C2 bond [48]. A key strategic step in the Li synthesis involved using a Schenck-ene reaction to oxidise the C5 position of intermediate 80 (Scheme 30). This reaction successfully installed a hydroxyl group and shifted the double bond, which streamlined the subsequent assembly of the challenging oxetane D ring. The conversion of intermediate 80 in 81 was performed efficiently using TPP as a sensitizer, achieving a high yield. The mild nature of these synthetic conditions led the research team to speculate that an analogous Schenck-ene oxidation could well be the step employed in the natural biosynthetic pathway of Taxol within the Taxus plant itself.
Another interesting example that includes a Schenck-ene reaction is found in the work by Xu et al. during their total synthesis of (-)-illisimonin A. This sesquiterpene is a compound that has been previously described in this review (Scheme 13), as it is a frequent target of interest in the total synthesis of natural products [49]. Remarkably, during their synthesis, intermediate 82 was found to slowly convert into an allylic alcohol when exposed to air. This spontaneous transformation was attributed to a singlet oxygen Schenck-ene reaction. To make this process more efficient and controlled, the reaction was performed using irradiation at 427 nm in the presence of oxygen and a catalytic amount of TPP (Scheme 31). This accelerated photooxygenation was a great success. The resulting peroxide intermediate was then immediately reduced with PMe3, followed by acetylation, ultimately yielding the desired allylic acetate product 83 in an excellent 89% yield.
Another illustrative example is provided by the work of Xiu et al. [50]. In this study, the main focus is on a nickel-catalyzed asymmetric intramolecular [4+1] cycloaddition between a vinylidene species and a 1,3-diene, affording a [4.3.0] azabicyclic framework. The resulting cycloadduct, which contains stereochemically distinct tri- and tetrasubstituted alkenes, serves as a versatile intermediate for the divergent synthesis of various natural alkaloids, namely 7-epi-incarvilline, isoincarvilline, incarvilline, tecomanine, 5-hydroxyskytanthine, tecostanine and hydroxyincarvilline. In the synthesis of the latter, an additional oxygen functionality was required at the ring junction. To achieve this modification, acetate 84 was subjected to photooxidation with singlet oxygen (using Rose Bengal as photosensitizer under blue LED irradiation), followed by in situ reduction of the resulting peroxide intermediate with NaBH₄, which afforded allylic alcohol 85 in 44% yield (Scheme 32).
Rose Bengal, played a key role also in the work of Zhang et al. [51]. They reported the first total synthesis of the marine sesquiterpene (hydro)quinone meroterpenoids dysideanones A and E–G (Scheme 33) in an enantioselective and divergent manner. Sesquiterpene (hydro)quinones are structurally diverse marine meroterpenoids, biosynthetically derived from two distinct origins. Dysideanone A, isolated in 2014 from the sponge Dysidea avara, features a unique 6/6/6/6-fused tetracyclic dysideanane ring system, representing the first meroterpenoid of this type. Dysideanone B shares the same ring system but differs in oxidation state and contains an additional ethoxy group. Dysideanone D, reported in 2015, differs from A mainly by the position of a double bond and the presence of a tertiary alcohol. Dysideanone E, isolated in 2016, differs from A only in the position of a double bond in the decalin motif. More recently, dysideanones F and G were reported; they share the same tetracyclic skeleton as A and E but exist in the phenol form. In their work, the sesquiterpene fragment was coupled to the aromatic moiety via a site- and diastereoselective intermolecular alkylation between a Wieland–Miescher ketone derivative and benzyl bromide, the formation of the 6/6/6/6-fused core relied on an intramolecular radical cyclization. Crucially, the oxidative functionalization of dysideanone G, promoted by visible-light irradiation in the presence of Rose Bengal and molecular oxygen, efficiently delivered dysideanone E in 85% yield, highlighting the central role of photochemically induced oxidation in accessing the target meroterpenoids (Scheme 3He). Interestingly, conversion of dysideanone F (obtained from dysiadeanone G via acid-mediated double bond migration) into dysideanone A using the same procedure afforded only traces of the desired product. This demonstrates how small structural changes, specifically the presence of an endo- or exo-cyclic double bond which can result in a competing pathway, can strongly influence the reaction outcome. Fortunately, dysideanone A was obtained using alternative oxidizing conditions which involved the use of PIFA.
3.5. Carboxyl Group Manipulations
A plethora of processes are now available thanks to the advent of photocatalysis, and the modern synthetic chemists’ toolbox is wider than ever. Herein are reported those processes that do not belong to any of the previous classes, but that represent nonetheless remarkable accomplishments from a synthetic point of view.
Manipulations of carboxyl groups are very common by means of photochemical reactions, and in this example Li et al. operated a convenient iodination from a carboxylic intermediate en route towards the family of diepoxy-ent-kaurane diterpenoids, isolated from plant of the Isodon genus [52]. Namely, the researchers reported the first enantioselective synthesis of (−)-macrocalin B, (−)-acetyl-macrocalin B, (−)-isoadenolin A, and (−)-phyllostacin I, taking advantage of an intramolecular Diels-Alder reaction on anti-Bredt intermediate 89, obtained from 88 via HI elimination. The introduction of an iodine atom on an unactivated quaternary carbon would prove challenging without resorting to a photoactivated metal-free variant of the Hunsdiecker-Borodin reaction. Using NIS as halogen donor, the free carboxylic acid 86 forms an acyl hypoiodite that upon absorption of light decarboxylates and recombines with the iodine radical, delivering the desired intermediate 87 with excellent yield (Scheme 34).
The possibility to manipulate carboxyl groups easily is extremely practical since many natural sources such as amino acids feature this moiety already installed. Moon et al. devised a brillant photoredox-mediated atroposelective Betti reaction to access ancistobrevoline A and B [53]. The key to achieve such transformation relies in the activation of the carboxylic acid of an aminoacid as N-hydroxyphtalimido ester (intermediate 90, achieved from N-acetyl alanine), that upon SET reduction from a suitable photocatalyst and ensuing decarboxylation would deliver an -aminoradical, then promptly oxidised to imine 91 by the oxidised-state photocatalyst, closing its cycle. In presence of a chiral phosphoric acid catalyst, the in situ generated imine 91 reacts atroposelectively with an electron-rich aromatic, delivering enantioselectively the desired Betti product 92 (Scheme 35). Further elaborations afford the desired products.
4. Photoredox Catalysis: A Radical Approach to Molecular Complexity
This section will explore how photoredox catalysis facilitates radical processes, providing new avenues for constructing intricate molecular architectures.
4.1. C-C Bond Formation via Radical Conjugate (Giese) Additions
1,4-radical conjugated additions, also known as Giese additions after the seminal report that first explored them in 1981 [54], are an excellent alternative to their polar counterparts, Michael additions. Most typically, nucleophilic radicals achieved from non--acidic species react with electron-poor olefins. The main advantage relies in the generation method of the carbon-centred radical, that can be obtained via SET reduction or XAT from halides [55,56,57,58,59], carboxylic acids [60,61,62,63], oxalates [64,65,66] and other substrates, most often in an umpolung fashion, and allowing for an easier handling of the precursor when the corresponding organometallic coupling counterpart would either be difficult to prepare, or would not lead to the right selectivity. Moreover, photoredox catalysis circumvents the need for the common AIBN/Bu3SnH initiation system used in the generation of radicals from halides, avoiding the moderately high temperatures (50–70°C) and the use of toxic tin reagents, thus minimizing the environmental impact and the occurrence of side reactions. [cite articles from book chapter]
The recent literature offers several examples of the synthetic utility of this approach. A remarkable example is portrayed in Tharra et al. synthesis of (+)-actinobolin and its analogues [67]. Belonging to the family of bactobolins, densenly functionalized metabolites featuring a broad antibacterial activity, they require a long synthesis of 16 steps, hampering the investigation of SARs properties. A photoredox catalyzed Giese addition allowed the authors to simplify the synthesis, disconnecting the C4-C10 bond, coupling the enone intermediate 93 (available from (-)-quinic acid in 4 steps) with the radical precursor 94 (1 step from L-threonine) (Scheme 36). Optimization of the diol protecting group allowed for the isolation of the desired product 95 with satisfactory yield and diastereoselectivity, the radical directing the facial control which occurs selectively on the opposite side of its methyl group. Overall, the synthesis was shortened to just 9 steps with an optimal 18% overall yield, paving the way for accessing 7 different analogues some of which showed an increased bioactivity.
Li et al. exploited two different photochemical transformations in its pathway towards (25S)-Δ7-dafachronic acid, a steroid hormone of nematode Caenorhabditis elegans [68]. While the main novelty of this approach relies in the photocatalytic stereochemical editing of some stereocenters (vide infra), their synthesis also featured a relevant Giese-type decarboxylative allylation at the C23-C24 bond. Namely, the advanced intermediate 96, achieved from chenodeoxycholic acid, was subjected to blue LEDs irradiation in presence of a base, an Hantzsch ester, and the allylating agent 97 (Scheme 37). Decarboxylation of the radical precursor, Giese addition on the acceptor and ensuing elimination afforded the desired intermediate 98 in 71% yield. It is remarkable to note that this transformation does not rely on a photocatalyst but on an EDA complex between the Hantzsch ester and precursor 96 formed in situ and capable of absorbing light in the visible region.
Capitalizing on a Giese-type reactivity and HAT chemistry Lee et al. developed the novel convergent synthesis of (-)-elodeoidins A and B [69]. Isolated from Hypericum elodeoides, a traditional medical herb used in chinese pharmacopoeia to treat stomatitis, infantile pneumonia, and mastitis. These molecules belong to the class of acylphloroglucinol meroterpenoids, from which they slightly differ due to the presence of a five-membered β-diketone moiety with a gem-dimethyl group. This feature was cleverly introduced through two successive photochemical couplings at the C2-C3 and C1-C7 bonds. Firstly, enedione 99 was converted to the enol intermediate 101 via a TBADT-catalyzed Giese addition of 100, exploiting a direct HAT from the aldehyde substrate 100 to the photocatalyst itself, as al-ready described by Fagnoni [70]. After further elaborations, enetrione 102 was subjected to the cross dehydrogenative coupling conditions altogether with aldehyde 103 (nine steps from geraniol). In this process, the photoexcited copper catalyst generates an O-centered radical from TBPB, to which a HAT on the aldehyde partner follows (Scheme 38). After Giese addition of the acyl radical on the electron-poor olefin, the resulting intermediate 104 undergoes oxidation-elimination, delivering the desired final product.
4.2. Strategic Hydrogen Atom Transfer (HAT) and Halogen Atom Transfer (XAT) Reactions
These processes involve the abstraction of a hydrogen or halogen atom from one of the substrates. While also exploitable through thermal pathways with AIBN and tin reagents, photocatalysis introduced a greater degree of orthogonality to these processes. The catalyst can either directly perform the HAT (direct HAT, dHAT), or it can activate an abstractor (indirect HAT, iHAT). Common dHAT catalysts are tetrabutylammonium decatungstate (TBADT) and aromatic ketones (e.g. benzophenone), while common HAT catalysts are tertiary amines such as quinuclidine derivatives, or thiols. In this section, HAT and XAT-based processes that do not involve addition to electron-poor olefins (Giese additions, vide supra) are examined.
The already-examined synthesis of (25S)-Δ7-dafachronic acid by Li et al. showcases the possibility to perform stereochemical editing on the skeletal core by means of a photocatalyzed HAT process [68]. Within their synthesis, Wu and co-workers started from chenodeoxycholic ester 105, whose skeleton features an opposite stereocenter at C5. To tackle this problem, the group submitted 15 to irradiation at 395 nm in the presence of NaDT, a disulfide co-catalyst, and a biphosphate salt. The electron-withdrawing nature of the protecting group on the C7-OH induces a faster HAT on C5, affording the epimerized product 106 with an excellent 96:4 diastereoselective ratio (Scheme 39). In the same work, the group exploited this stereochemical editing also to synthetize demissidine and smilagenin.
The possibility of inducing radical skeletal changes by means of HATs is surely one of the most remarkable tools offered by this family of processes. In analogy to the stereochemical editing exploited by the Li et al. in the aforementioned case, Deng et al. devised a HAT-based skeletal rearrangement en route to (+)-kobusine, (+)spirasine IX, and (+)-orgetine [71]. These C20 hetisine-type diterpenoid alkaloids, found in Aconitum species (with kobusine being first isolated from Aconitum sachalinense), feature a complex, cage-like heptacyclic skeleton, and showed promising antiarrhythmic, analgesic, and anti-inflammatory properties, which explains the efforts of the synthetic community towards streamlining their synthesis. In this regard, Luo group showed how to access the complex core of these molecules exploiting a HAT-initiated 1,2-radical rearrangement on intermediate 107, achieved on gram-scale in 14 steps from commercial starting materials. The four tertiary C-H of 107 are unfavorable to abstraction from an O-centered radical due to electronic and steric constraints. Upon 13C-NMR studies, only C2, C12 and C13 of the remaining eight methylene groups seemed most prone to undergo the desired process. Under TBADT catalysis at 390 nm in presence of a thiol co-catalyst, C12 underwent abstraction smoothly delivering the desired product 108 as a mixture of diastereoisomers in 75% yield (Scheme 40). Starting from 108, kobusine, spirasine IX and orgetine were synthetised in 10, 7, and 8 additional steps, respectively.
HAT catalysis also found application in olefin transpositions, as portrayed in Xu et al. synthesis of (−)-illisimonin A, a sesquiterpenoid already described in Section 2.3 [49]. Pivotal to the synthesis developed by Dai’s group are a total of 5 olefin rearrangements, two of which come from photochemical processes (vide infra for an analysis of the Schenck-ene reaction involved). Particularly challenging proved to be the migration of the C=C double bond from C2-C3 to C3-C4 of intermediate 109. In spite of the greater thermodynamic stability of product 110, the remarkable steric hindrance around the moiety hampered any successful metal-catalyzed isomerization. Dai and co-workers resorted then to a novel protocol proposed by the group of Wendlandt [72,73], employing NaDT as direct photocatalytic abstractor at 370 nm and, replacing the original cobaloxime co-catalyst in favour, yet again, of a thiol. Delightfully, abstraction from C4 proceeded smoothly due to its allylic nature despite the relative hindrance of the surroundings, affording the desired product 110 in a moderate 45% yield, after three cycles of isomerization (Scheme 41).
One last example is portrayed by the asymmetric synthesis of a prostaglandin D2 metabolite (PGDM) methyl ester by Xiao et al. [74]. Prostaglandins are hormone-like lipid compounds featuring a five-membered cyclopentanol core decorated with two aliphatic chains, and they play a crucial role in many physiological processes. To streamline the synthesis of the metabolite methyl ester, the synthesis of the cyclopentanol core was achieved via a novel diastereoselective radical cyclization. The linear precursor A, upon deprotonation, is oxidised to an -carbonyl radical which rapidly undergoes 5-exo-trig cyclization upon addition to the neighbouring C=C double bond. Of pivotal importance is the HAT from the alkyl radical to the thiol co-catalyst, which following SET reduction and protonation, closes the double catalytic cycle. Interestingly, the reaction is extremely diastereoselective, since the radical cyclization seems to proceed through a syn-anti transition state. Using chiral alcohol 111 (2 steps from Chan’s diene) as starting material, intermediate 112 could be diastereoselectively achieved upon photochemical cyclization and ketalization (Scheme 42). Further elaboration gave the desired target molecule in a total of 9 steps with an overall yield of 7%.
5. Total Synthesis of Stemoamide Alkaloids: A Virtuous Example of Incorporating Photochemistry in Retrosynthetic Logic
As we showed throughout the previous sections of this review, the latest years have seen an increase in the use of photochemical steps to advance the current landscape of chemical synthesis. It is worth mentioning, however, that further efforts are being made to elevate photochemistry to a pivotal element of retrosynthetic design. In this regard, the latest approach towards stemoamide alkaloids reported by Akkawi and Nicewicz represents an excellent example of how such an endeavour can be turned into a reality [75]. The Stemona alkaloids, found in the stemonaceae plants, manifest interesting antitussive effects and insecticidal activity; among them, featuring a unique 5/7/5 tricyclic scaffold, an embedded pyrrole ring, and an oxaspirocyclic butanolide, bisdehydrostemoninine, bisdehydrostemoninine A, and bisdehydroneoostemoninine (all from Stemona tuberosa) pose a significant challenge from a synthetic point of view (Scheme 43). The pathway proposed by Nicewicz present seven key steps, five of which are photochemically driven. The synthesis starts with a photochemical hydroarylation, delivering the needed vinylpyridine intermediate 115 in a single step from alkyne 113 and bromopyridine 114. Upon SET reduction, a pyridyl radical is formed, adding to the terminal alkyne and accomplishing the hydroarylation upon HAT on a thiol co-catalyst, using an Ir catalyst under blue LEDs irradiation. Remarkably, the synthesis of 115 was accomplished columnless on a 5 grams scale, with immense advantage over the previously reported 4-step sequence to access the same product. To this step followed one of the pivotal ones of the whole synthesis, a polar radical crossover cycloaddition followed by reduction with β-chlorocrotonic acid 116, combining two different photochemical processes in one-pot and with a single catalyst. Using Mes-tBuAcr upon 450 nm irradiation, the olefin would undergo SET oxidation to radical cation, to which nucleophilic attack of the acid would ensue. The resulting radical would engage in a 5-exo-trig cyclization to a radical lactone, which, upon HAT from a thiol co-catalyst and following HCl elimination, would deliver an intermediate olefin (not shown). This latter intermediate, upon addition of DIPEA and irradiation at 390 nm, would then be reduced to radical anion and, eventually, to saturated lactone 117. The choice of 116 rather than unsubstituted chrotonic acid allowed to isolate the desired Markownikov product over the anti-Markownikov one furnished by a competitive 6-endo-trig cyclization, the chloroolefin being more polarized and prone to undergo elimination and subsequent reduction. It is worth noting that the procedure yielded 117 with excellent (>20:1) diastereoselectivity, owing to the steric effects borne by the alkene substituents in the first process, and the enolizable α-position of the lactone that, with excess base, leads to the formation of the thermodynamically favoured product in the second step, installing three of the contiguous stereocenters of the central B-ring. The carbonyl handle could then be elaborated into an enolether by means of a Petasis olefination, and the resulting olefin 118 was engaged in the second pivotal photochemical step of the pathway, a radical polar crossover cycloaddition, as previously reported by Guo et al. [76]. Reduction of the α-bromocarboxylic acid 119 would result in a radical that would add to the olefin, and following oxidation to a cation and nucleophilic attack by the acidic moiety, would deliver the final product as a mixture of 4 diastereoisomers. Conveniently, treatment of the crude with TFA first and DBU then would lead to epimerization to the thermodynamically stable cyclic lactone, deprotection of the TBS, and elimination of MeOH delivering the desired product 120 as single isomer in just 5 steps. The olefin was then isomerized to its endocyclic isomer 121 by means of HAT catalysis, using TBADT at 390 nm, as similarly described in this review. Thermal N-Alkylation of the pyridine via Mitsunobu reaction and Chen skeletal rearrangement gave the linchpin intermediate 122, from which the final steps ensued. A metallophotoredox deformylation with TBADT at 390 nm with a cobalt co-catalyst delivered in a single step bisdehydroneoostemoninine in satisfactory yield. By means of a HAT-driven Giese addition on a methylacrylate partner and subsequent hydrolysis of the product, bisdehydrostemoninine A was achieved. Notably, the TBADT photocatalyst can generate the desired acyl radical via dHAT on the substrate, whereas the chiral phosphoric acid co-catalyst imparts enantioselectivity to the addition, generating a chiral environment during the protonation step. Lastly, bisdehydrostemoninine was achieved via a deoxygenative 5-endo-trig cyclization from bisdehydrostemoninine A using a methodology previously reported by Stache et al. [77].
6. Conclusions
This review has highlighted the synthetic power and versatility of modern photochemistry in constructing complex natural product scaffolds. The demonstrated applications across a variety of mechanisms—from building strained cyclobutane rings via photocycloadditions (such as [2+2]) to achieving precise oxygenation patterns using singlet oxygen for endoperoxide formation, and executing challenging bond formations through photoredox catalysis—underscore the strategic importance of light-driven reactions. These methodologies frequently offer crucial advantages over their thermal counterparts, including enhanced efficiency, superior regio- and stereocontrol, and the ability to access unique, high-energy intermediates. As demonstrated by recent breakthroughs and the adoption of techniques like flow chemistry to overcome scalability hurdles, the field of synthetic photochemistry continues to advance rapidly. The strategic mastery of these light-mediated tools is becoming increasingly central to the efficient and elegant total synthesis of intricate biologically active molecules, confirming photochemistry's vital and growing role in modern synthetic design.
Author Contributions
Conceptualization, P.C. and A.B.; methodology, P.C. and A.B.; software, C.M.; resources, C.M.; writing—original draft preparation, P.C and C.M.; writing—review and editing, P.C., C.M. and A.B.; supervision, A.B. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
During the preparation of this review, the authors used Gemini (free version) for the purposes of grammatical correction and stylistic improvements, and used ChatGPT (free version) for the purpose of graphical abstract creation. The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 4CzTPN | 2,3,5,6-tetrakis(carbazol-9-yl)-1,4-dicyanobenzene |
| AcCN | acetonitrile |
| AIBN | azobisisobutyronitrile |
| Bpin | boron pinacolate |
| CFL | compact fluorescent lamp |
| DCM | dichloromethane |
| dCF3bpy | 5,5'-bis(trifluoromethyl)-2,2'-bipyridine |
| dF(CF3)ppy | 2-(4,6-difluorophenyl)-3-trifluoromethylpyridine |
| dFppy | 2-(4,6-difluorophenyl)pyridine |
| DIPEA | diisopropylethylamine |
| dtbbpy | 4,4'-di-tert-butyl-2,2'-bipyridine |
| EnT | energy transfer |
| HAT | hydrogen atom transfer |
| ISC | intersystem crossing |
| LED | light-emitting diode |
| NaDT | sodium decatungstate |
| NaV | natrium voltage |
| NIS | N-iodosuccinimide |
| PIFA | bis(trifluoroacethoxy)iodobenzene |
| ppy | 2-phenylpyridine |
| rt | room temperature |
| SET | single electron transfer |
| TBADT | tetrabutylammonium decatungstate |
| TMS | trimethylsilyl |
| TPP | tetraphenylporphyrin |
| TRIPSH | 2,4,6-triisopropylbenzenethiol |
| UV | ultraviolet |
| XAT | halogen atom transfer |
References
- Ciamician, G.; Silber, P. Chemische Lichtwirkungen. Berichte Dtsch. Chem. Ges. 1908, 41, 1928–1935. [Google Scholar] [CrossRef]
- Jiao, Y.; Liu, J.; Mao, W.; Fang, R.; Xia, T.; Lang, Q.; Luo, T. Synthesis of (+)-Saxitoxin Facilitated by a Chiral Auxiliary for Photocycloadditions Involving Alkenylboronate Esters. J. Am. Chem. Soc. 2025, 147, 9091–9097. [Google Scholar] [CrossRef]
- Kravina, A.G.; Carreira, E.M. Total Synthesis of Epicolactone. Angew. Chem. Int. Ed. 2018, 57, 13159–13162. [Google Scholar] [CrossRef]
- Schneider, F.; Samarin, K.; Zanella, S.; Gaich, T. Total Synthesis of the Complex Taxane Diterpene Canataxpropellane. Science 2020, 367, 676–681. [Google Scholar] [CrossRef]
- Grünenfelder, D.C.; Navarro, R.; Wang, H.; Fastuca, N.J.; Butler, J.R.; Reisman, S.E. Enantioselective Synthesis of (−)-10-Hydroxyacutuminine. Angew. Chem. Int. Ed. 2022, 61, e202117480. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, X.; Wang, X.; Rodriguez, R.A.; Moore, C.E.; Gao, S.; Tan, X.; Ma, Y.; Rheingold, A.L.; Baran, P.S.; Chen, C. Asymmetric Syntheses of Sceptrin and Massadine and Evidence for Biosynthetic Enantiodivergence. Science 2014, 346, 219–224. [Google Scholar] [CrossRef]
- Lu, Z.; Yoon, T.P. Visible Light Photocatalysis of [2+2] Styrene Cycloadditions by Energy Transfer. Angew. Chem. Int. Ed. 2012, 51, 10329–10332. [Google Scholar] [CrossRef]
- Latrache, M.; Sesay, A.; Oger, S.; Montaner, M.B.; Zhang, J.; Mellah, M.; Poupon, E.; Hilton, S.T.; Arseniyadis, S.; Evanno, L. Bioinspired Synthesis of Sceptrin, Ageliferin and Six Piperine Dimers by Photo(Flow)catalysis: [2+2] vs [4+2] Cycloaddition. ChemRxiv 2025. preprint. [Google Scholar] [CrossRef]
- Liu, Y.; Ni, D.; Brown, M.K. Boronic Ester Enabled [2 + 2]-Cycloadditions by Temporary Coordination: Synthesis of Artochamin J and Piperarborenine B. J. Am. Chem. Soc. 2022, 144, 18790–18796. [Google Scholar] [CrossRef] [PubMed]
- Schoch, P.; Krivolapova, Y.; Schneider, F.; Pan, L.; Gaich, T. Gram-Scale Access to (3,11)-Cyclotaxanes — Synthesis of 1-Hydroxytaxuspine C. Angew. Chem. Int. Ed. 2025, 64, e202506245. [Google Scholar] [CrossRef] [PubMed]
- Kumarasamy, E.; Raghunathan, R.; Kandappa, S.K.; Sreenithya, A.; Jockusch, S.; Sunoj, R.B.; Sivaguru, J. Transposed Paternò–Büchi Reaction. J. Am. Chem. Soc. 2017, 139, 655–662. [Google Scholar] [CrossRef]
- Bach, T.; Brummerhop, H.; Harms, K. The Synthesis of (+)-Preussin and Related Pyrrolidinols by Diastereoselective Paternò–Büchi Reactions of Chiral 2-Substituted 2,3-Dihydropyrroles. Chem. Eur. J. 2000, 6, 3838–3848. [Google Scholar] [CrossRef]
- Hambalek, R.; Just, G. A Short Synthesis of (±)-Oxetanocin. Tetrahedron Lett. 1990, 31, 5445–5448. [Google Scholar] [CrossRef]
- Boxall, R.J.; Ferris, L.; Grainger, R.S. Synthesis of C-13 Oxidised Cuparene and Herbertane Sesquiterpenes via a Paternò–Büchi Photocyclisation-Oxetane Fragmentation Strategy: Total Synthesis of 1,13- Herbertenediol. Synlett 2004, 2379–2381. [Google Scholar] [CrossRef]
- Kärkas, M.D.; Porco, J.A., Jr.; Stephenson, C.R.J. Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis. Chem. Rev. 2016, 116, 9683–9747. [Google Scholar] [CrossRef]
- Wearing, E.R.; Yeh, Y.-C.; Terrones, G.G.; Parikh, S.G.; Kevlishvili, I.; Kulik, H.J.; Schindler, C.S. Visible Light–Mediated Aza Paternò–Büchi Reaction of Acyclic Oximes and Alkenes to Azetidines. Science 2024, 384, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Wright, B.A.; Okada, T.; Regni, A.; Luchini, G.; Sowndarya, S. V, S.; Chaisan, N.; Kölbl, S.; Kim, S.F.; Paton, R.S.; Sarpong, R. Molecular Complexity-Inspired Synthetic Strategies toward the Calyciphylline A-Type Daphniphyllum Alkaloids Himalensine A and Daphenylline. J. Am. Chem. Soc. 2024, 146, 33130–33148. [Google Scholar] [CrossRef] [PubMed]
- Padwa, A.; Jacquez, M.N.; Schmidt, A. An Approach toward Azacycles Using Photochemical and Radical Cyclizations of N -Alkenyl Substituted 5-Thioxopyrrolidin-2-Ones. J. Org. Chem. 2004, 69, 33–45. [Google Scholar] [CrossRef]
- He, J.; Bai, Z.-Q.; Yuan, P.-F.; Wu, L.-Z.; Liu, Q. Highly Efficient Iridium-Based Photosensitizers for Thia-Paternò–Büchi Reaction and Aza-Photocyclization. ACS Catal. 2021, 11, 446–455. [Google Scholar] [CrossRef]
- Murakami, K.; Toma, T.; Fukuyama, T.; Yokoshima, S. Total Synthesis of Tetrodotoxin. Angew. Chem. Int. Ed. 2020, 59, 6253–6257. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.-K.; Wang, Q.; Xu, J. Enantioselective Total Synthesis of Fortalpinoid Q via a TEMPO+ BF4– -Mediated Dehydrative Nazarov Cyclization. J. Am. Chem. Soc. 2025, 147, 9079–9084. [Google Scholar] [CrossRef]
- Frey, B.; Wells, A.P.; Rogers, D.H.; Mander, L.N. Synthesis of the Unusual Diterpenoid Tropones Hainanolidol and Harringtonolide. J. Am. Chem. Soc. 1998, 120, 1914–1915. [Google Scholar] [CrossRef]
- Zhu, L.; Li, J.; Lu, Z. Gram-Scale Total Synthesis of Illisimonin A. J. Am. Chem. Soc. 2025, 147, 23417–23421. [Google Scholar] [CrossRef]
- Liang, X.-T.; Chen, J.-H.; Yang, Z. Asymmetric Total Synthesis of (−)-Spirochensilide A. J. Am. Chem. Soc. 2020, 142, 8116–8121. [Google Scholar] [CrossRef]
- Tuccinardi, J.P.; Wood, J.L. Total Syntheses of (+)-Ineleganolide and (−)-Sinulochmodin C. J. Am. Chem. Soc. 2022, 144, 20539–20547. [Google Scholar] [CrossRef]
- Lu, X.-L.; Qiu, Y.; Yang, B.; He, H.; Gao, S. Asymmetric Total Synthesis of (+)-Xestoquinone and (+)-Adociaquinones A and B. Chem. Sci. 2021, 12, 4747–4752. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Shi, Y.; Yang, B.; Hou, M.; He, H.; Gao, S. Asymmetric Total Synthesis of Hasubanan Alkaloids: Periglaucines A–C, N,O -Dimethyloxostephine and Oxostephabenine. Angew. Chem. Int. Ed. 2023, 62, e202214873. [Google Scholar] [CrossRef]
- Zheng, Y.; Teng, L.; Zhou, T.; Liu, Z.; Guo, K.; Li, H.; Li, T.; Wang, L.; Liu, Y.; Li, S. Discovery and Total Synthesis of a New Class of Minor Immunosuppressive Plant Sesterterpenoids. Angew. Chem. Int. Ed. 2025, 64, e202421497. [Google Scholar] [CrossRef] [PubMed]
- Frontier, A.J.; Hernandez, J.J. New Twists in Nazarov Cyclization Chemistry. Acc. Chem. Res. 2020, 53, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.; Tang, D.; Xia, Q.; Hu, J.; Lin, X.; Xuan, J.; Ding, H. Divergent Total Syntheses of Phragmalin and Khayanolide-Type Limonoids: A Torquoselective Interrupted Nazarov Approach. J. Am. Chem. Soc. 2025, 147, 3003–3009. [Google Scholar] [CrossRef]
- Wolff, L. Ueber Diazoanhydride. Justus Liebigs Ann. Chem. 1902, 325, 129–195. [Google Scholar] [CrossRef]
- Arndt, F.; Eistert, B. Ein Verfahren zur Überführung von Carbonsäuren in ihre höheren Homologen bzw. deren Derivate. Berichte Dtsch. Chem. Ges. B Ser. 1935, 68, 200–208. [Google Scholar] [CrossRef]
- Hancock, E. N.; Kuker, E. L.; Tantillo, D. J.; Brown, M. K. Lessons in Strain and Stability: Enantioselective Synthesis of (+)-[5]-Ladderanoic Acid. Angew. Chem. Int. Ed. 2020, 59, 436–441. [Google Scholar] [CrossRef]
- Mascitti, V.; Corey, E.J. Total Synthesis of (±)-Pentacycloanammoxic Acid. J. Am. Chem. Soc. 2004, 126, 15664–15665. [Google Scholar] [CrossRef]
- Mascitti, V.; Corey, E.J. Enantioselective Synthesis of Pentacycloanammoxic Acid. J. Am. Chem. Soc. 2006, 128, 3118–3119. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Chen, R.; Tang, D.; Xia, Q.; Zhao, Y.; Liu, C.-H.; Ding, H. Total Synthesis of (−)-Retigeranic Acid A: A Reductive Skeletal Rearrangement Strategy. J. Am. Chem. Soc. 2023, 145, 11927–11932. [Google Scholar] [CrossRef]
- Zhang, J.; Luo, X.; Zhang, J.; Li, C. Total Synthesis of DMOA-Derived Meroterpenoids: Achieving Selectivity in the Synthesis of (+)-Berkeleyacetal D and (+)-Peniciacetal I. J. Am. Chem. Soc. 2025, 147, 5933–5942. [Google Scholar] [CrossRef]
- Trommsdorff, H. Ueber Santonin. Ann. Pharm. 1834, 11, 190–207. [Google Scholar] [CrossRef]
- Schoch, P.; Gaich, T. Total Synthesis and Structural Revision of (−)-Sodagnitin E. Angew. Chem. Int. Ed. 2025, 64, e202506247. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Shen, Y.; Chen, Y.; Wang, R.; Wei, H.; Fu, P.; Lei, X.; Wang, H.; Bi, R.; Zhang, Y. Two-Stage Syntheses of Clionastatins A and B. J. Am. Chem. Soc. 2022, 144, 8938–8944. [Google Scholar] [CrossRef]
- Zhang, G.; Hu, X.; Chiang, C.-W.; Yi, H.; Pengkun, P.; Singh, A. K.; Lei, A. Anti-Markovnikov Oxidation of β-Alkyl Styrenes with H₂O as the Terminal Oxidant. J. Am. Chem. Soc. 2016, 138, 12037–12040. [Google Scholar] [CrossRef]
- Yang, B.; Li, G.; Wang, Q.; Zhu, J. Enantioselective Total Synthesis of (+)-Stephadiamine. J. Am. Chem. Soc. 2023, 145, 5001–5006. [Google Scholar] [CrossRef]
- Lee, B.J.; DeGlopper, K.S.; Yoon, T.P. Site-Selective Alkoxylation of Benzylic C–H Bonds by Photoredox Catalysis. Angew. Chem. Int. Ed. 2020, 59, 197–202. [Google Scholar] [CrossRef]
- Wu, B.-L.; Yao, J.-N.; Long, X.-X.; Tan, Z.-Q.; Liang, X.; Feng, L.; Wei, K.; Yang, Y.-R. Enantioselective Total Synthesis of (−)-Daphenylline. J. Am. Chem. Soc. 2024, 146, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Alektiar, S.N.; Han, J.; Dang, Y.; Rubel, C.Z.; Wickens, Z.K. Radical Hydrocarboxylation of Unactivated Alkenes via Photocatalytic Formate Activation. J. Am. Chem. Soc. 2023, 145, 10991–10997. [Google Scholar] [CrossRef] [PubMed]
- Nandi, R.; Murmu, R.; Sadhukhan, S.; Pal, D.; Biswas, S.; Das, B.; Bisai, A. Total Synthesis of Dixiamycins A and B via a Late-Stage N–N Bond Formation under Visible Light Photoredox Catalysis. Org. Lett. 2025, 27, 1531–1536. [Google Scholar] [CrossRef]
- Krieger, J.; Smeilus, T.; Kaiser, M.; Seo, E.-J.; Efferth, T.; Giannis, A. Total Synthesis and Biological Investigation of (-)-Artemisinin: The Antimalarial Activity of Artemisinin Is Not Stereospecific. Angew. Chem. Int. Ed. 2018, 57, 8293–8296. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-J.; Gu, C.-C.; Wang, X.-F.; Min, L.; Li, C.-C. Asymmetric Total Synthesis of Taxol. J. Am. Chem. Soc. 2021, 143, 17862–17870. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, Z.; Dai, M. Total Synthesis of (−)-Illisimonin A Enabled by Pattern Recognition and Olefin Transposition. J. Am. Chem. Soc. 2025, 147, 17592–17597. [Google Scholar] [CrossRef]
- Xiu, W.; Huffman, C.D.; Swann, W.A.; Li, C.W.; Uyeda, C. A Catalytic Asymmetric Intramolecular [4 + 1]-Cycloaddition for the Total Synthesis of Terpene Alkaloid Natural Products. J. Am. Chem. Soc. 2025, 147, 17510–17516. [Google Scholar] [CrossRef]
- Zhang, Q.; Kang, J.; Tan, T.; Dong, G.; Chen, J.; Lu, Z. Total synthesis of 1’-epi-septosones B and C and the originally assigned structures of spiroetherones A and B. ChemRxiv 2025, preprint. [Google Scholar] [CrossRef]
- Li, Y.; Xue, Q.; Zhao, X.; Ma, D. Total Syntheses of Diepoxy- Ent- Kaurane Diterpenoids Enabled by a Bridgehead-Enone-Initiated Intramolecular Cycloaddition. J. Am. Chem. Soc. 2025, 147, 1197–1206. [Google Scholar] [CrossRef]
- Moon, J.; Shin, E.; Kwon, Y. Enantioselective Desymmetrization of Biaryls via Cooperative Photoredox/Brønsted Acid Catalysis and Its Application to the Total Synthesis of Ancistrobrevolines. J. Am. Chem. Soc. 2025, 147, 12800–12810. [Google Scholar] [CrossRef]
- Giese, B.; Lachhein, S. Steric Effects in the Addition of Alkyl Radicals to Alkenes. Angew. Chem. Int. Ed. Engl. 1981, 20, 967–967. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Z.; Tang, J.-C.; Che, J.-T.; Zhang, H.-Y.; Chen, J.-H.; Yang, Z. Total Synthesis of (+)-Haperforin G. J. Am. Chem. Soc. 2020, 142, 19487–19492. [Google Scholar] [CrossRef]
- Park, K.H. (Kenny); Chen, D.Y.-K. A Desymmetrization-Based Approach to Morphinans: Application in the Total Synthesis of Oxycodone. Chem. Commun. 2018, 54, 13018–13021. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, R.; Zhang, W.; Li, A. Total Synthesis of Indotertine A and Drimentines A, F, and G. Angew. Chem. Int. Ed. 2013, 52, 9201–9204. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Guo, Z.; Lu, J.-T.; Fang, R.; Chen, S.-C.; Luo, T. Total Synthesis of (−)-Batrachotoxinin A: A Local-Desymmetrization Approach. J. Am. Chem. Soc. 2020, 142, 3675–3679. [Google Scholar] [CrossRef]
- Lu, T.; Jiang, Y.-T.; Ma, F.-P.; Tang, Z.-J.; Kuang, L.; Wang, Y.-X.; Wang, B. Bromide-Mediated C–H Bond Functionalization: Intermolecular Annulation of Phenylethanone Derivatives with Alkynes for the Synthesis of 1-Naphthols. Org. Lett. 2017, 19, 6344–6347. [Google Scholar] [CrossRef]
- Samame, R.A.; Owens, C.M.; Rychnovsky, S.D. Concise Synthesis of (+)-Fastigiatine. Chem. Sci. 2016, 7, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Burtea, A.; DeForest, J.; Li, X.; Rychnovsky, S.D. Total Synthesis of (−)-Himeradine A. Angew. Chem. Int. Ed. 2019, 58, 16193–16197. [Google Scholar] [CrossRef]
- Schnermann, M. J.; Overman, L. E. A concise synthesis of (−)-aplyviolene facilitated by a strategic tertiary radical conjugate addition. Angew. Chem. Int. Ed. 2012, 51, 9576–9580. [Google Scholar] [CrossRef]
- Tao, D.J.; Slutskyy, Y.; Overman, L.E. Total Synthesis of (−)-Chromodorolide B. J. Am. Chem. Soc. 2016, 138, 2186–2189. [Google Scholar] [CrossRef] [PubMed]
- Slutskyy, Y.; Jamison, C.R.; Zhao, P.; Lee, J.; Rhee, Y.H.; Overman, L.E. Versatile Construction of 6-Substituted Cis -2,8-Dioxabicyclo[3.3.0]Octan-3-Ones: Short Enantioselective Total Syntheses of Cheloviolenes A and B and Dendrillolide C. J. Am. Chem. Soc. 2017, 139, 7192–7195. [Google Scholar] [CrossRef]
- Müller, D.S.; Untiedt, N.L.; Dieskau, A.P.; Lackner, G.L.; Overman, L.E. Constructing Quaternary Stereogenic Centers Using Tertiary Organocuprates and Tertiary Radicals. Total Synthesis of Trans -Clerodane Natural Products. J. Am. Chem. Soc. 2015, 137, 660–663. [Google Scholar] [CrossRef]
- Allred, T.K.; Dieskau, A.P.; Zhao, P.; Lackner, G.L.; Overman, L.E. Enantioselective Total Synthesis of Macfarlandin C, a Spongian Diterpenoid Harboring a Concave-Substituted Cis -Dioxabicyclo[3.3.0]Octanone Fragment. Angew. Chem. Int. Ed. 2020, 59, 6268–6272. [Google Scholar] [CrossRef] [PubMed]
- Tharra, P.R.; Mikhaylov, A.A.; Švejkar, J.; Gysin, M.; Hobbie, S.N.; Švenda, J. Short Synthesis of (+)-Actinobolin: Simple Entry to Complex Small-Molecule Inhibitors of Protein Synthesis. Angew. Chem. Int. Ed. 2022, 61, e202116520. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Z.; Wu, J. Photocatalytic Stereochemical Editing for the Concise Syntheses of (25S)-Δ⁷-Dafachronic Acid, Demissidine, and Smilagenin. Angew. Chem. Int. Ed. 2025, 64, e202500341. [Google Scholar] [CrossRef]
- Lee, C.; Kang, G.; You, J.; Kim, T.; Lee, H.-S.; Park, Y.; Han, S. Total Synthesis of (−)-Elodeoidins A and B. JACS Au 2025, 5, 1096–1103. [Google Scholar] [CrossRef]
- Esposti, S.; Dondi, D.; Fagnoni, M.; Albini, A. Acylation of Electrophilic Olefins Through Decatungstate-Photocatalyzed Activation of Aldehydes. Angew. Chem. Int. Ed. 2007, 46(14), 2531–2534. [Google Scholar] [CrossRef]
- Deng, M.; Wu, F.; Liu, T.; Jiang, Z.; Luo, T. Enantioselective Total Syntheses of (+)-Kobusine, (+)-Spirasine IX and the Purported Structure of (+)-Orgetine: Strategic Use of C–H Bonds. J. Am. Chem. Soc. 2025, 147, 8132–8137. [Google Scholar] [CrossRef]
- Occhialini, G.; Palani, V.; Wendlandt, A.E. Catalytic, Contra -Thermodynamic Positional Alkene Isomerization. J. Am. Chem. Soc. 2022, 144, 145–152. [Google Scholar] [CrossRef]
- Palani, V.; Wendlandt, A.E. Strain-Inducing Positional Alkene Isomerization. J. Am. Chem. Soc. 2023, 145, 20053–20061. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Shang, Q.; Pu, L.; Wang, Z.; Zhu, L.; Yang, Z.; Huang, J. Photoredox-Catalyzed Radical Cyclization of Unactivated Alkene-Substituted β-Ketoesters Enabled Asymmetric Total Synthesis of Tricyclic Prostaglandin D₂ Metabolite Methyl Ester. JACS Au 2025, 5, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Akkawi, N.R.; Nicewicz, D.A. Photochemically Enabled Total Syntheses of Stemoamide Alkaloids. J. Am. Chem. Soc. 2025, 147, 15482–15489. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Bao, R.; Li, Y.; Li, Y.; Zhang, J.; Tang, Y. Tailored Synthesis of Skeletally Diverse Stemona Alkaloids through Chemoselective Dyotropic Rearrangements of β-Lactones. Angew. Chem. Int. Ed. 2021, 60, 14545–14553. [Google Scholar] [CrossRef]
- Stache, E.E.; Ertel, A.B.; Rovis, T.; Doyle, A.G. Generation of Phosphoranyl Radicals via Photoredox Catalysis Enables Voltage–Independent Activation of Strong C–O Bonds. ACS Catal. 2018, 8, 11134–11139. [Google Scholar] [CrossRef]
Scheme 1.
Intramolecular [2+2]-cycloaddition between an alkenyl boronate and an 8-oxoxantine moiety in the synthesis of saxitoxin.
Scheme 1.
Intramolecular [2+2]-cycloaddition between an alkenyl boronate and an 8-oxoxantine moiety in the synthesis of saxitoxin.
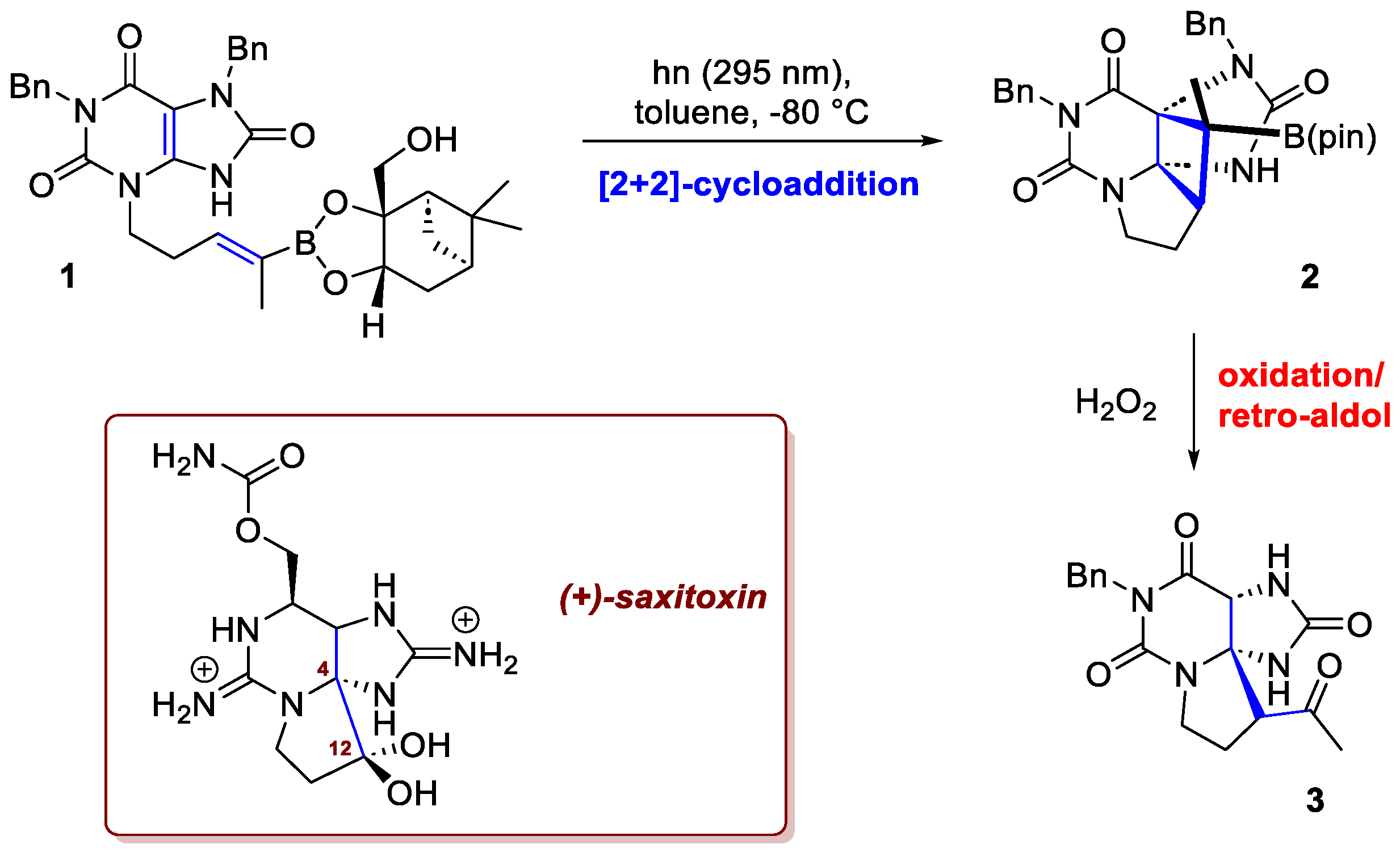
Scheme 2.
Intramolecular [2+2] cycloaddition to the synthesis of epicolactone.
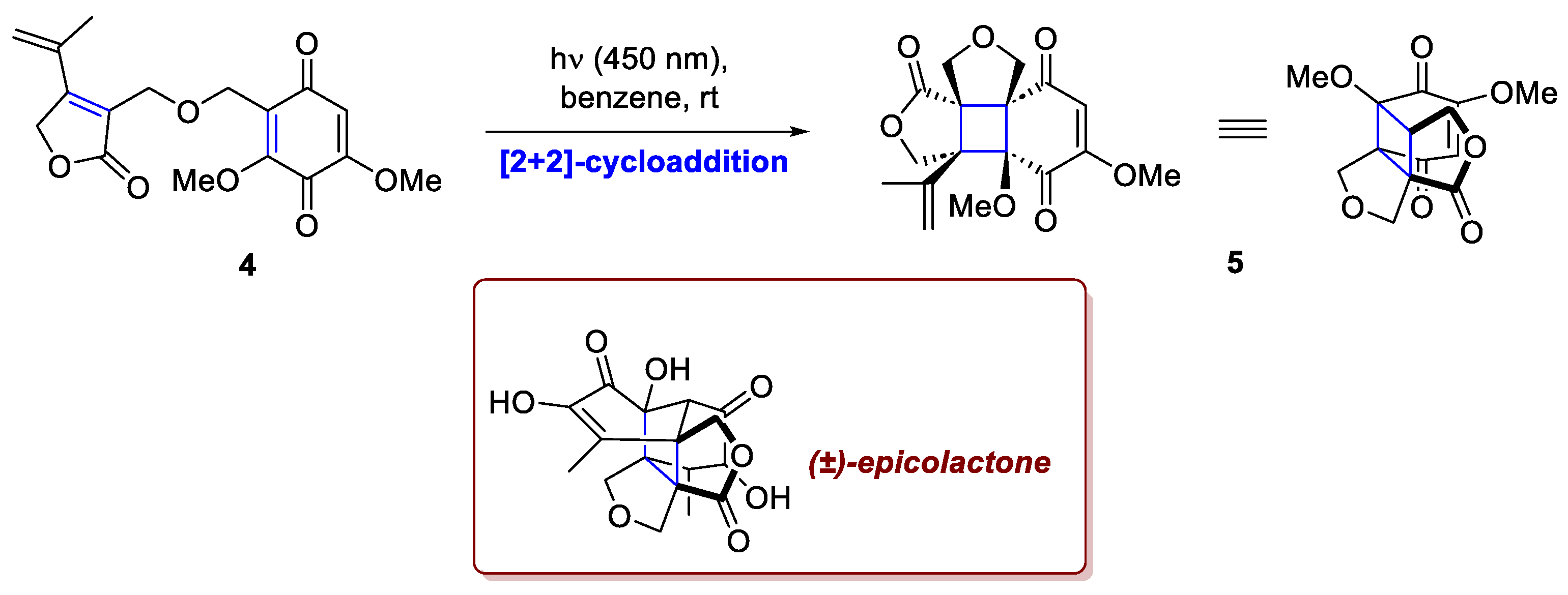
Scheme 3.
Intramolecular [2+2] cycloaddition in the synthesis of canataxapropellane.
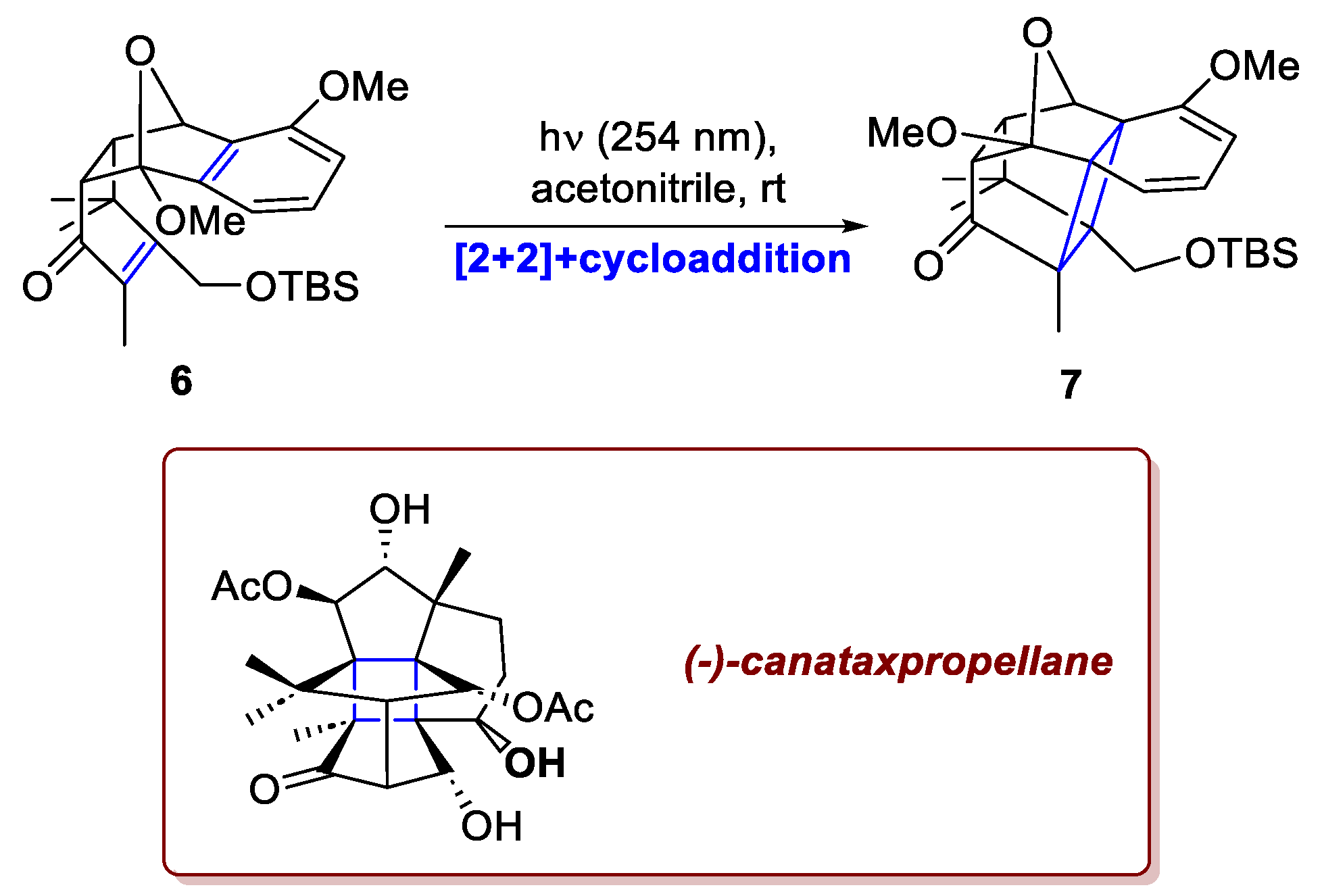
Scheme 4.
An intramolecular [2+2]-cycloaddition between an enone and an electron-rich furan is exploited in the synthesis of 10-hydroxyacutuminine.
Scheme 4.
An intramolecular [2+2]-cycloaddition between an enone and an electron-rich furan is exploited in the synthesis of 10-hydroxyacutuminine.
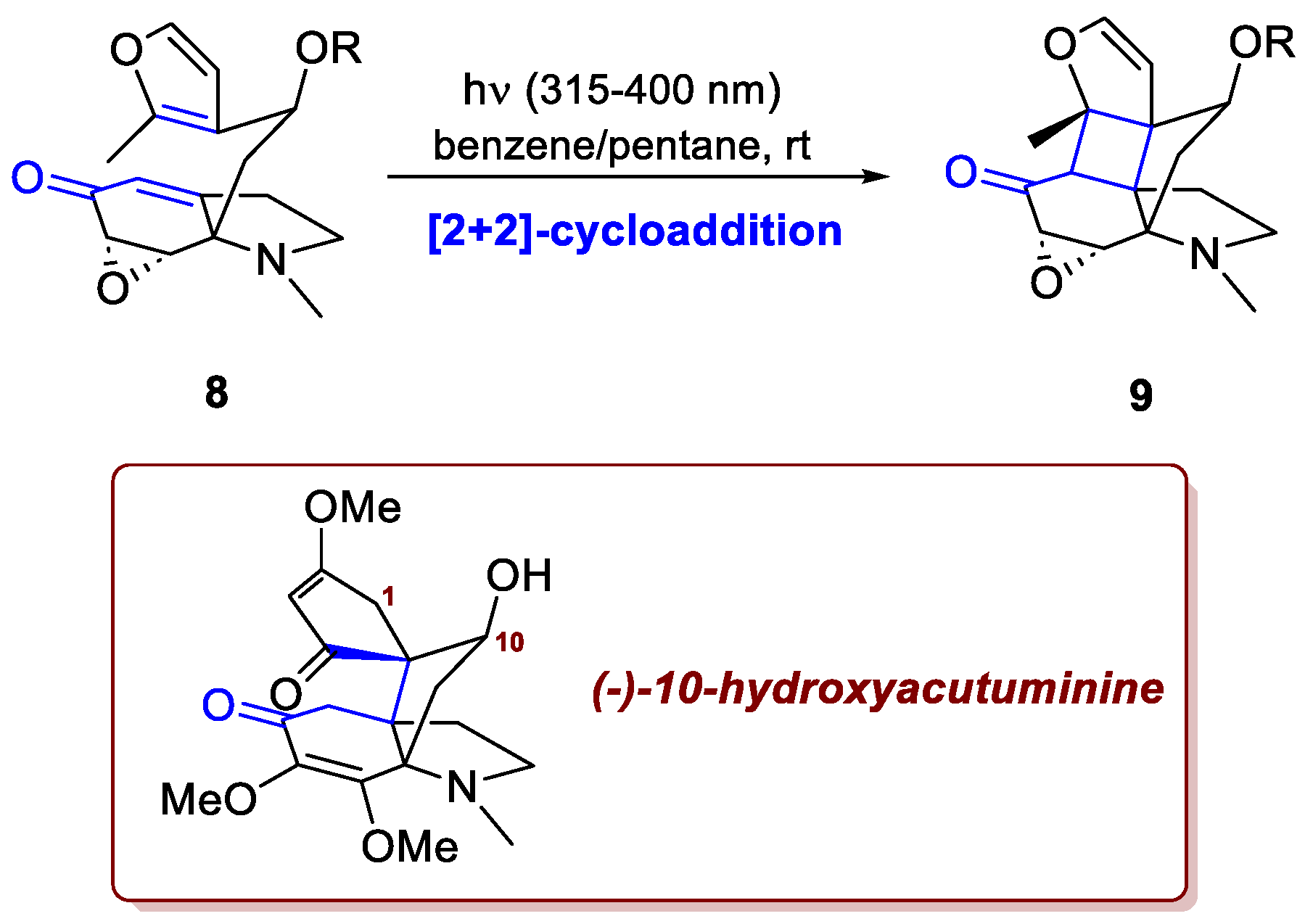
Scheme 5.
A SET-mediated [2+2] photocycloaddition of an imidazole precursor in the total synthesis of sceptrin.
Scheme 5.
A SET-mediated [2+2] photocycloaddition of an imidazole precursor in the total synthesis of sceptrin.
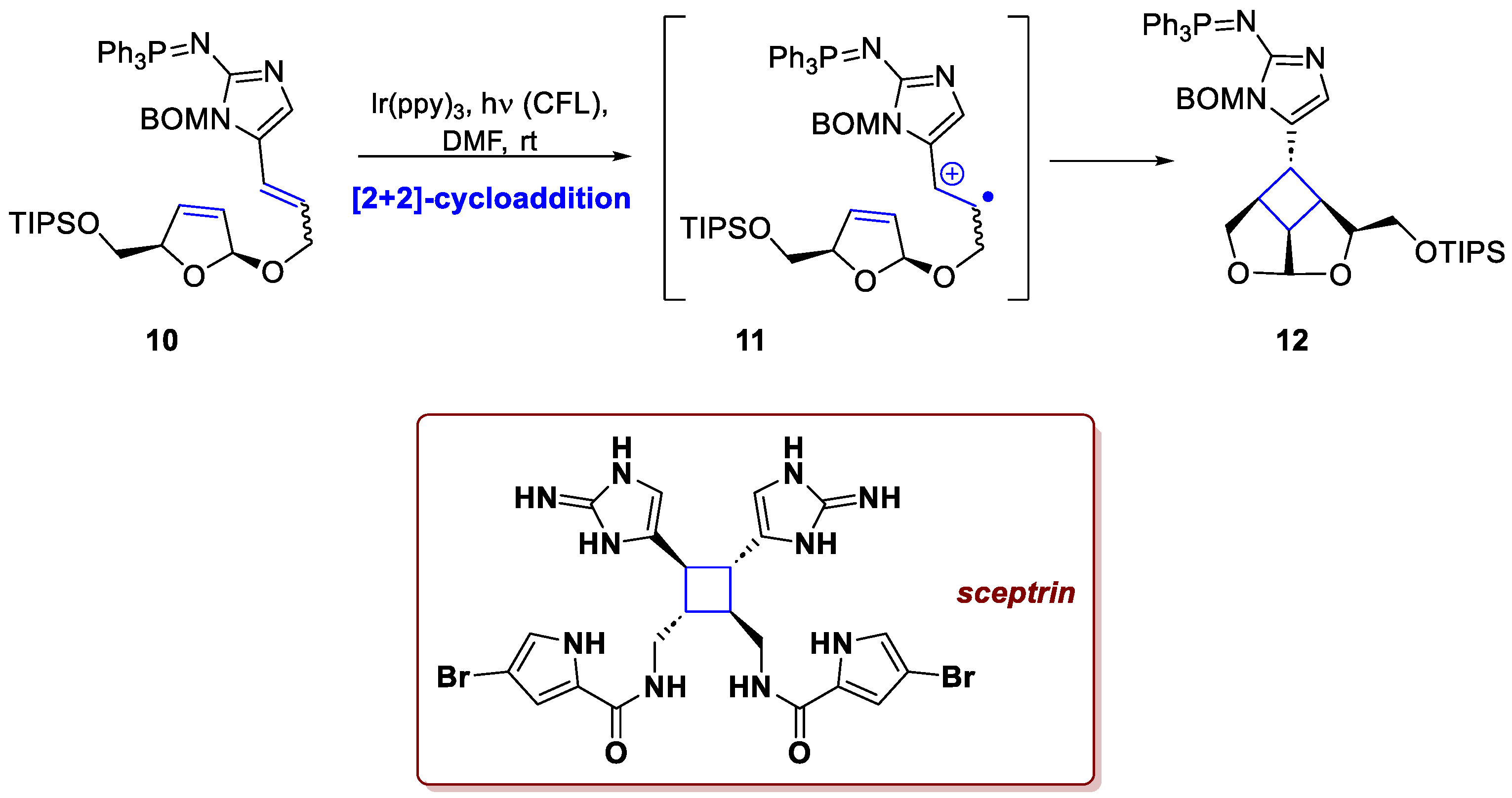
Scheme 6.
Intermolecular [2+2] cycloaddition exploiting a temporary B-O tether to regio- and stereocontrol, in the synthesis of (-)-artochamin J.
Scheme 6.
Intermolecular [2+2] cycloaddition exploiting a temporary B-O tether to regio- and stereocontrol, in the synthesis of (-)-artochamin J.
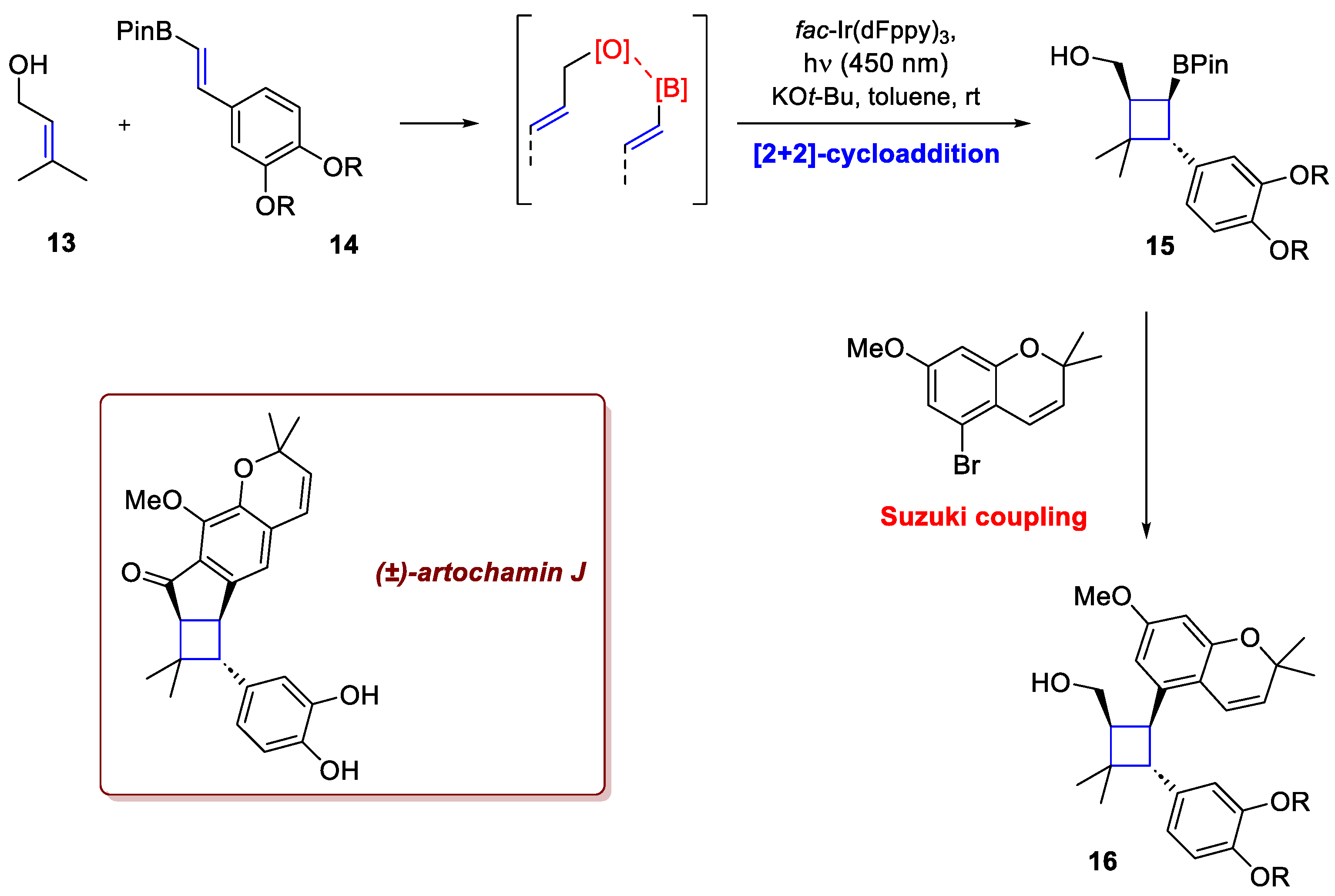
Scheme 7.
[2σ+2π] cycloaddition in the synthesis of cyclotaxanes.
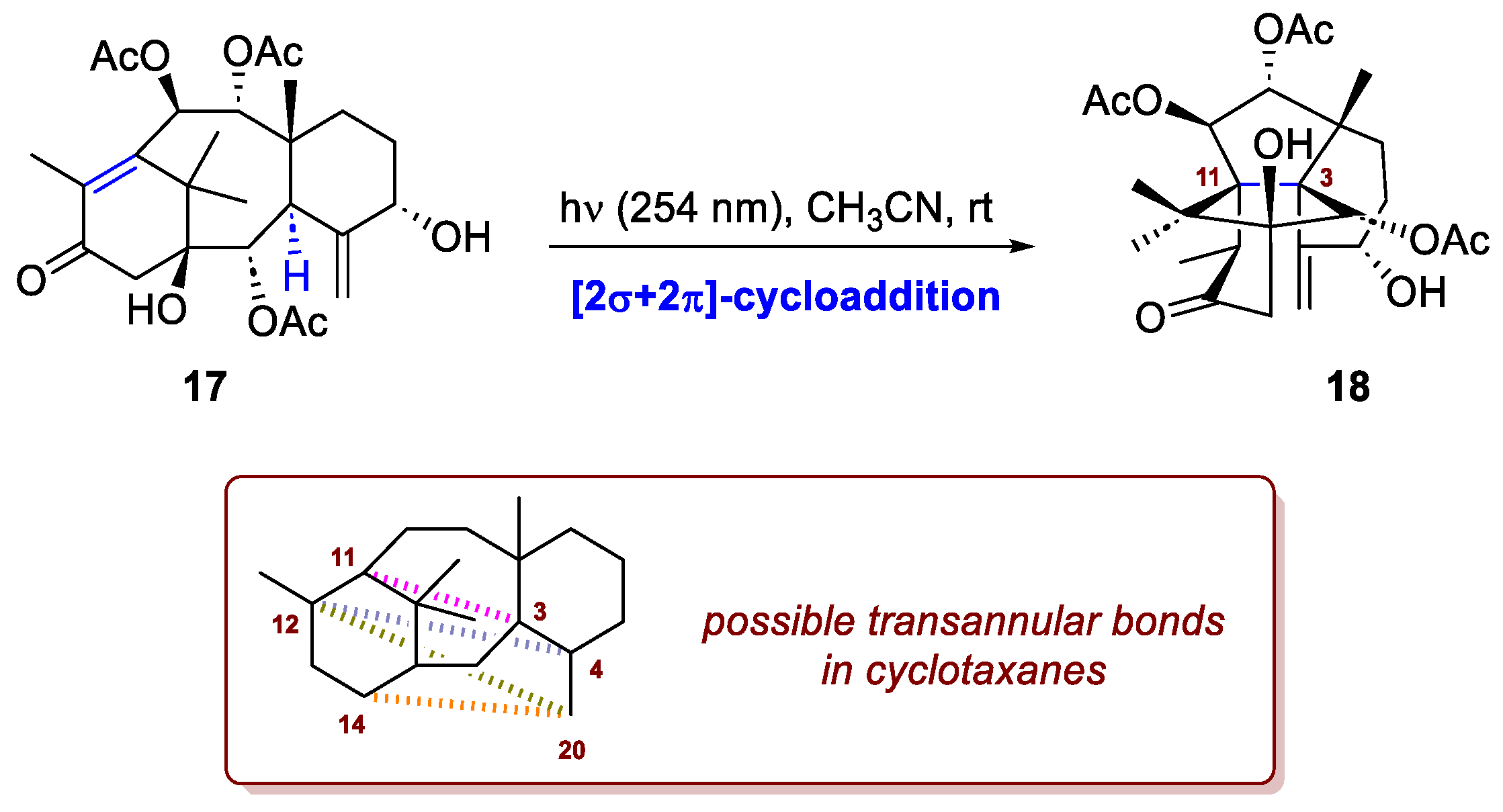
Scheme 8.
Aza-Paternò–Büchi cycloaddition in the synthesis of stereoisomers of penaresidin B.
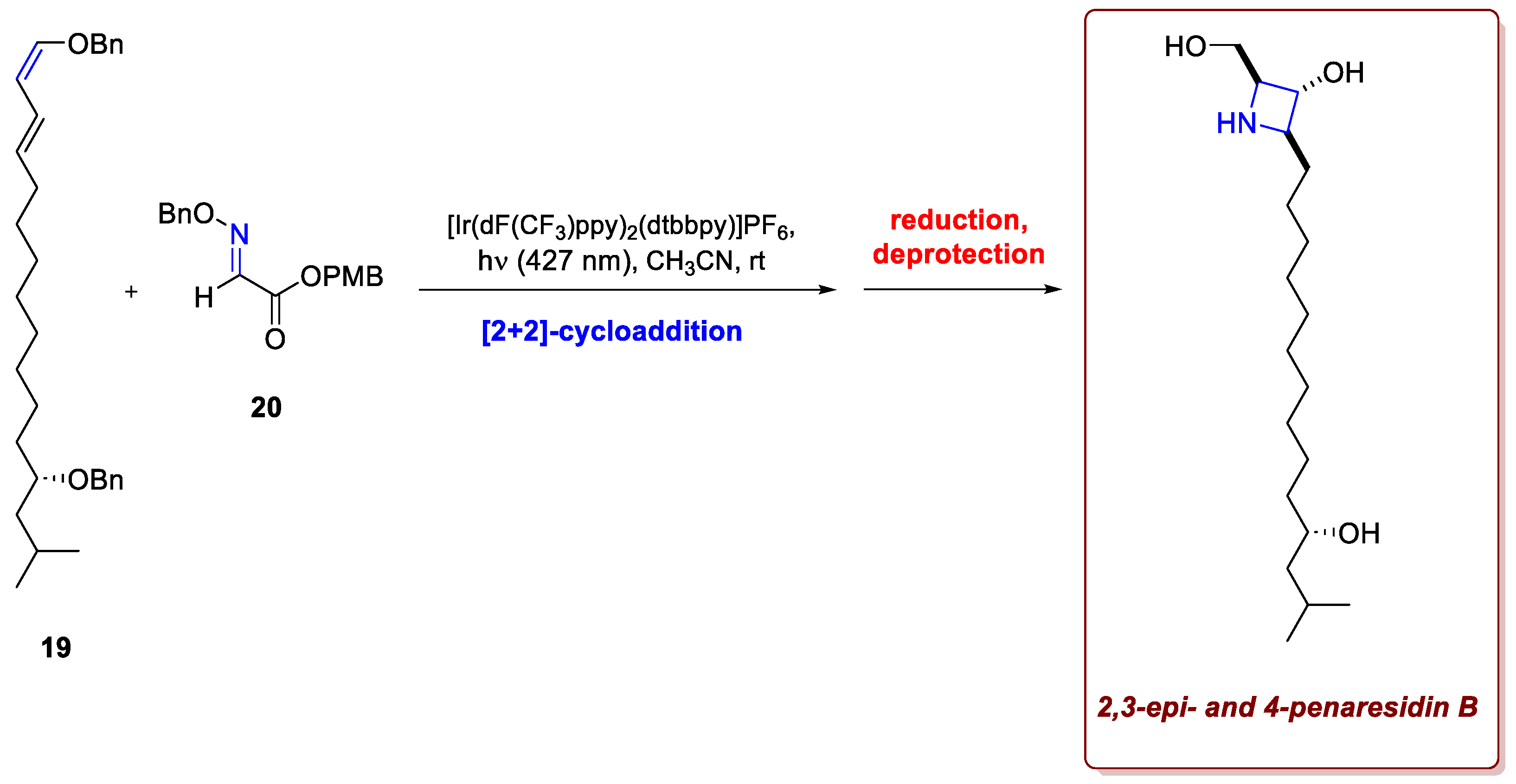
Figure 1.
General structure of calyciphylline A-type daphniphyllum alkaloids and structures of himalensine A and daphenylline.
Figure 1.
General structure of calyciphylline A-type daphniphyllum alkaloids and structures of himalensine A and daphenylline.
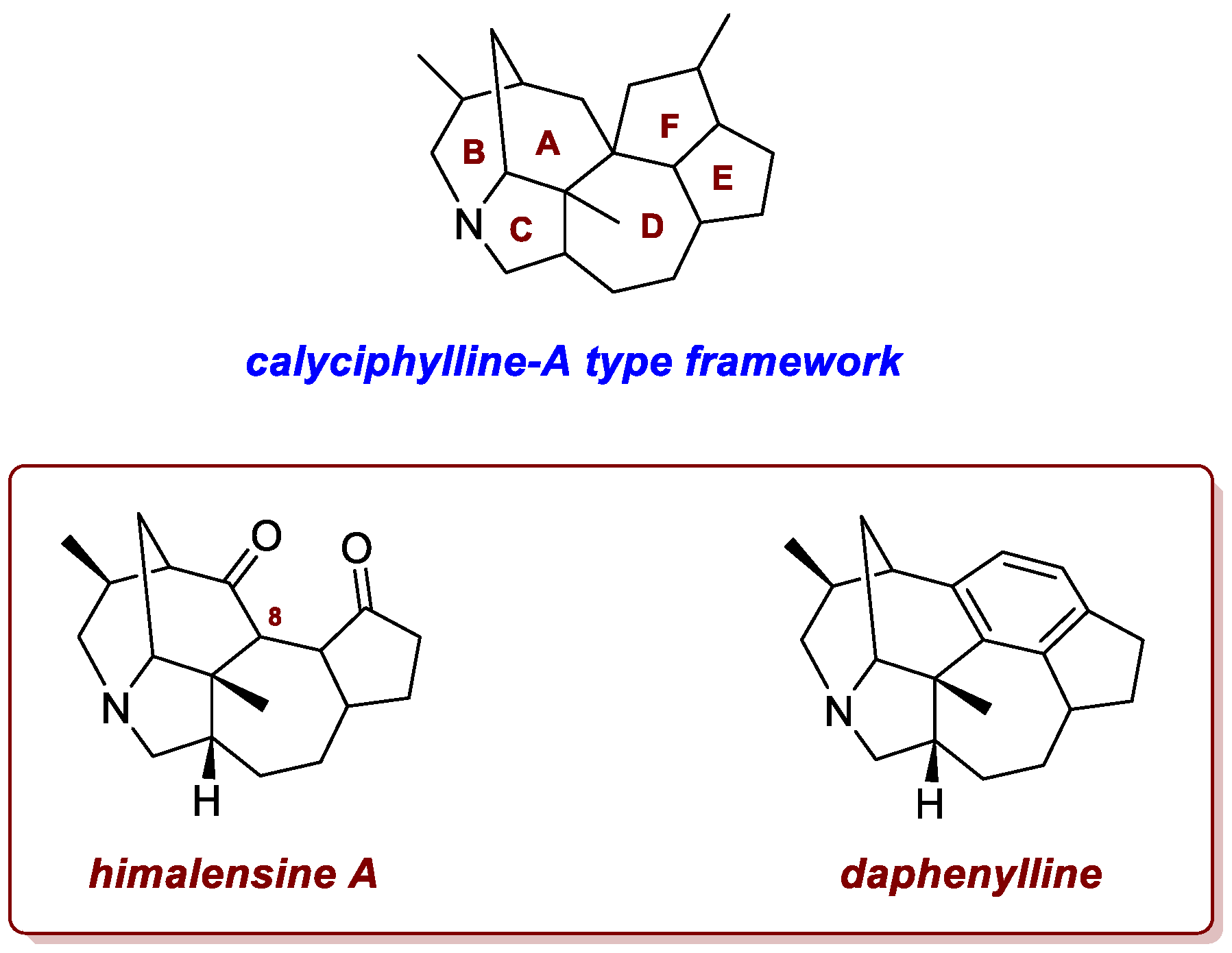
Scheme 9.
Synthesis of daphniphylline exploiting a thia-Paternò–Büchi cycloaddition.
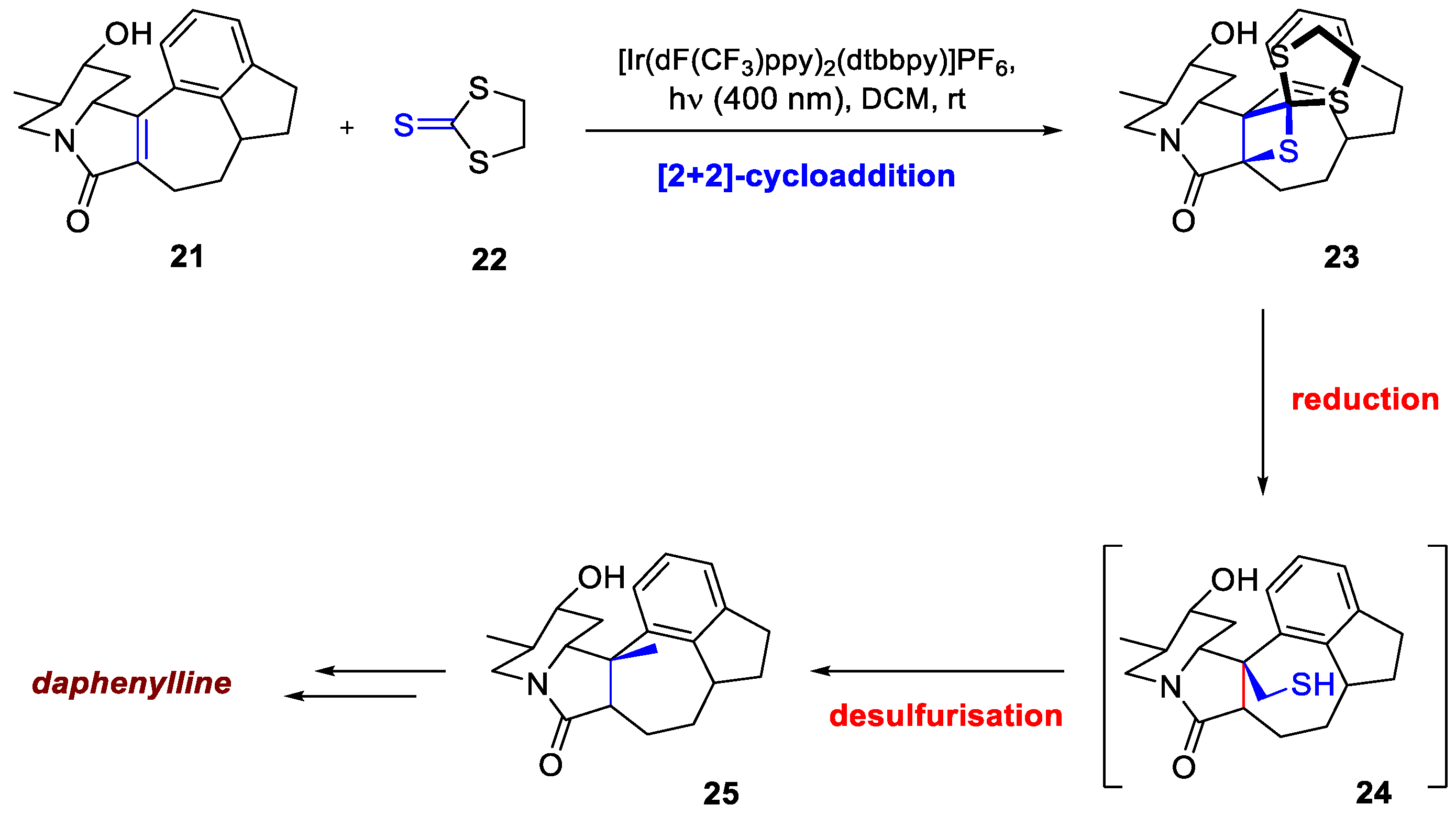
Scheme 11.
Key photocycloaddition/endoperoxide cleavage in the synthesis of tetradotoxin.
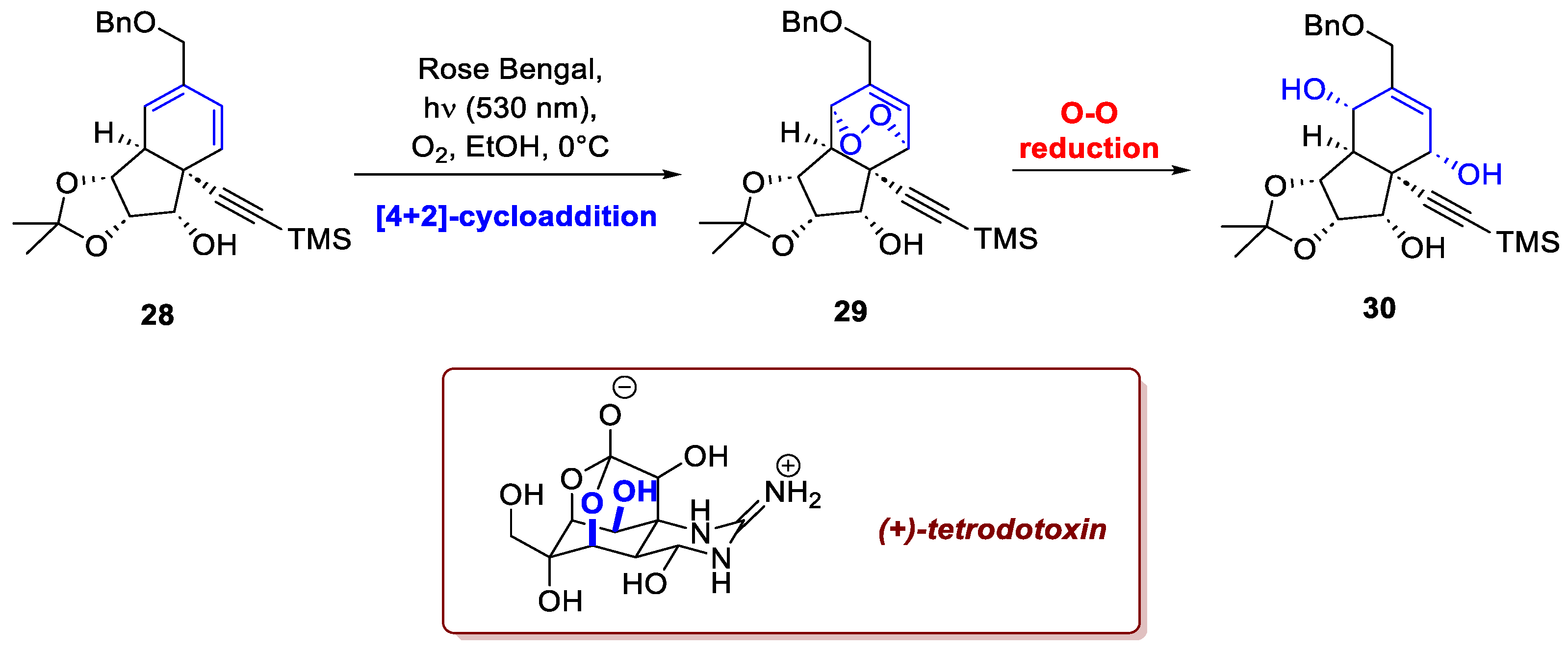
Scheme 12.
Synthesis of fortalpinoid Q exploiting a singlet oxygen Diels-Alder reaction.
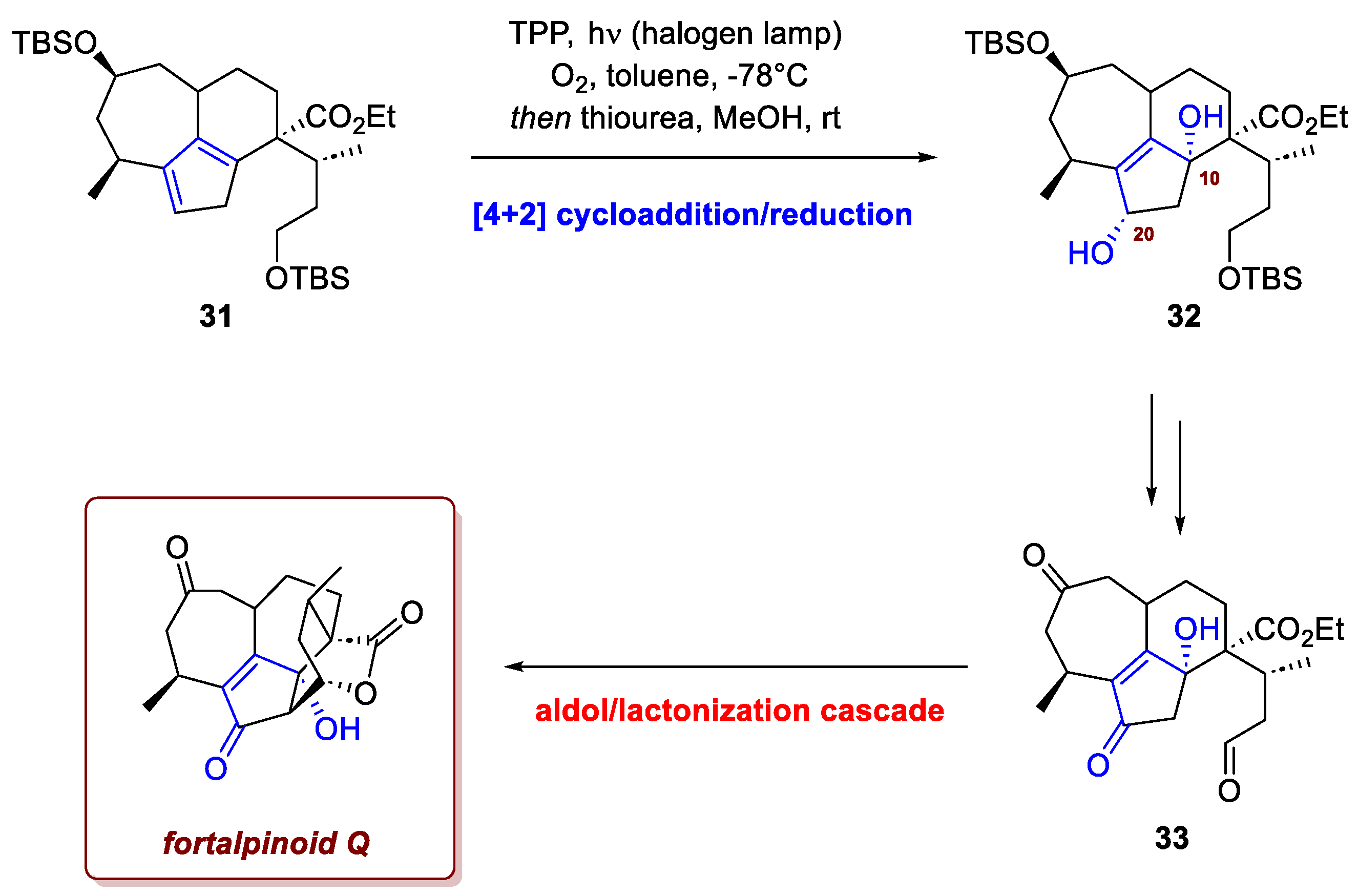
Scheme 13.
Attempted synthesis of illisimonin A via a photooxygenation reaction.
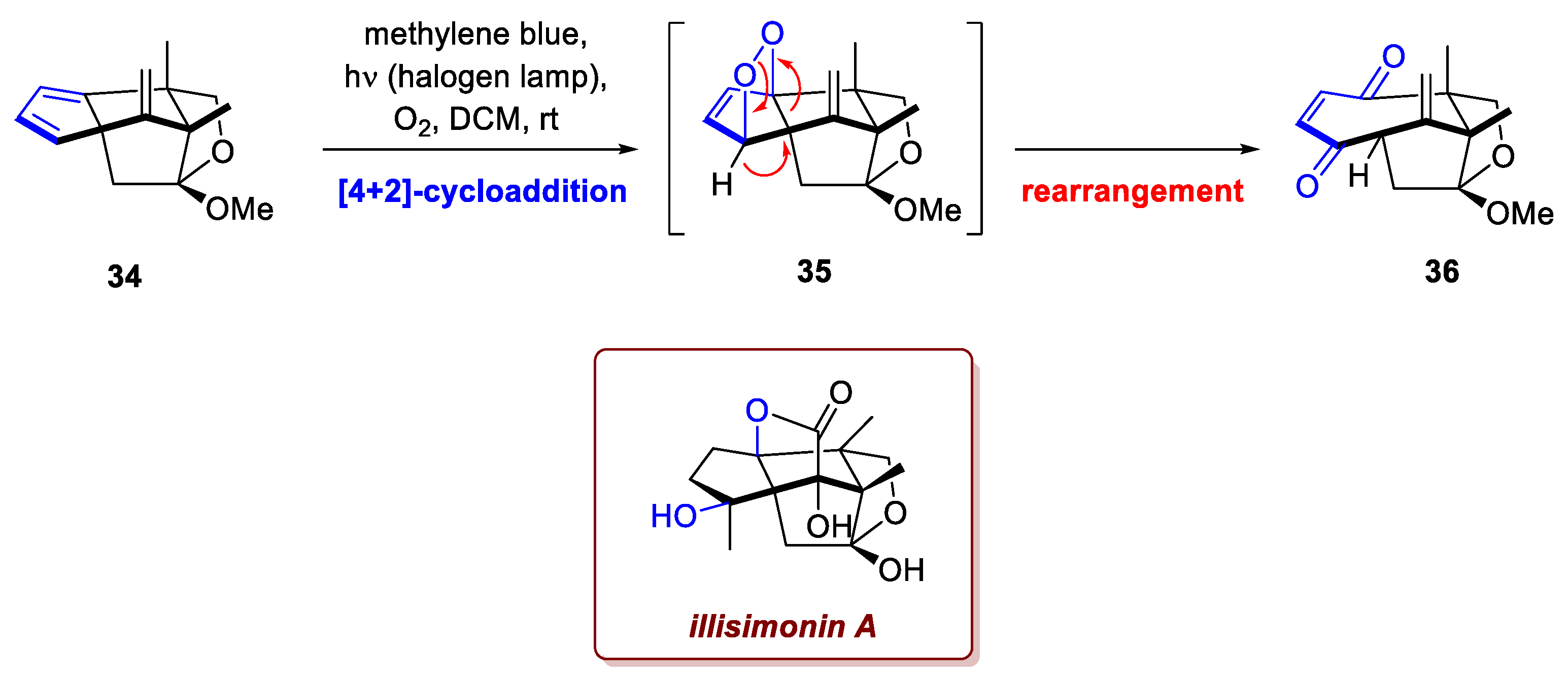
Scheme 14.
Synthesis of spirochensilide A via the photooxygenation of a furan ring.
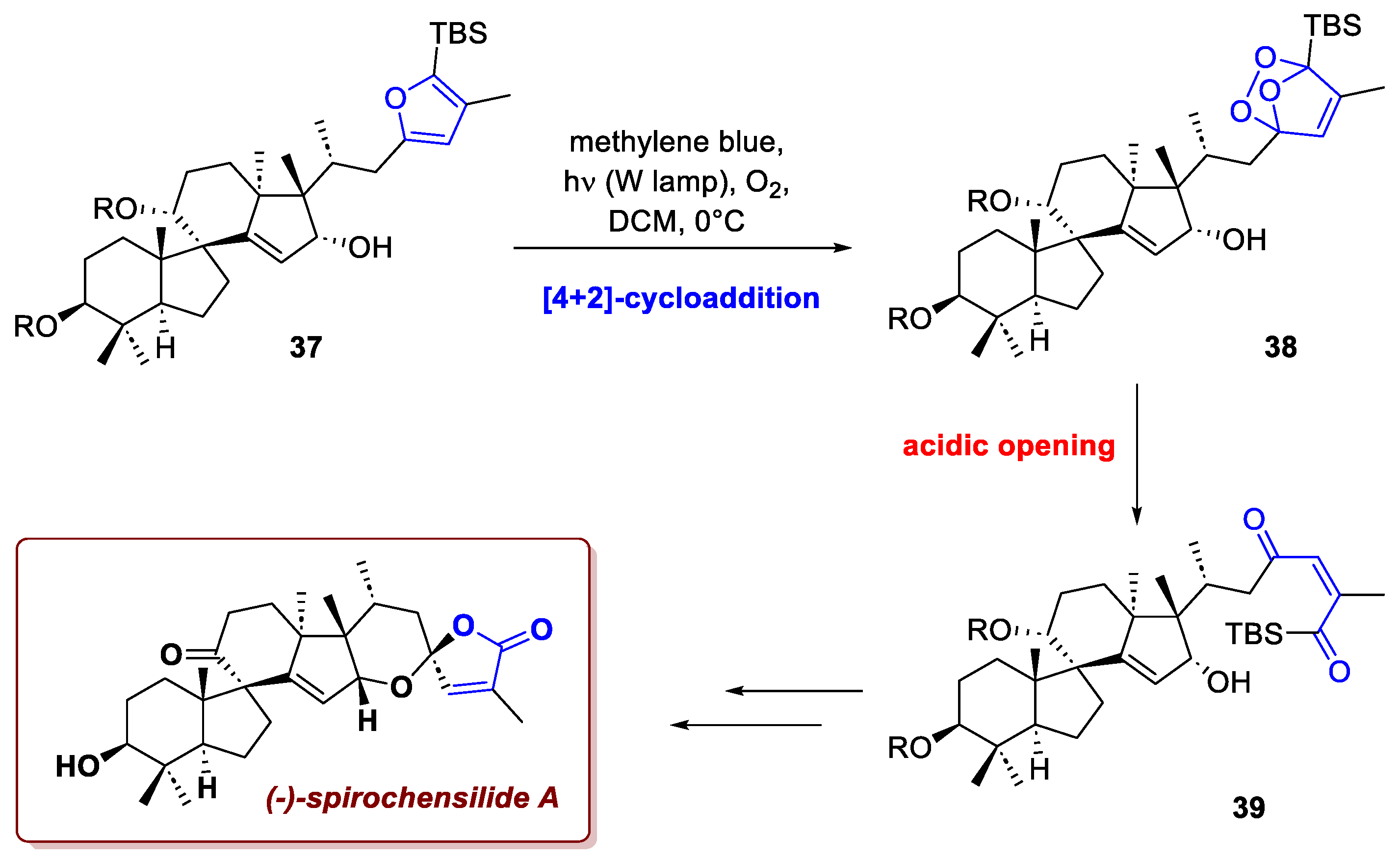
Scheme 15.
Synthesis of spirochensilide A via the photooxygenation of a furan ring.
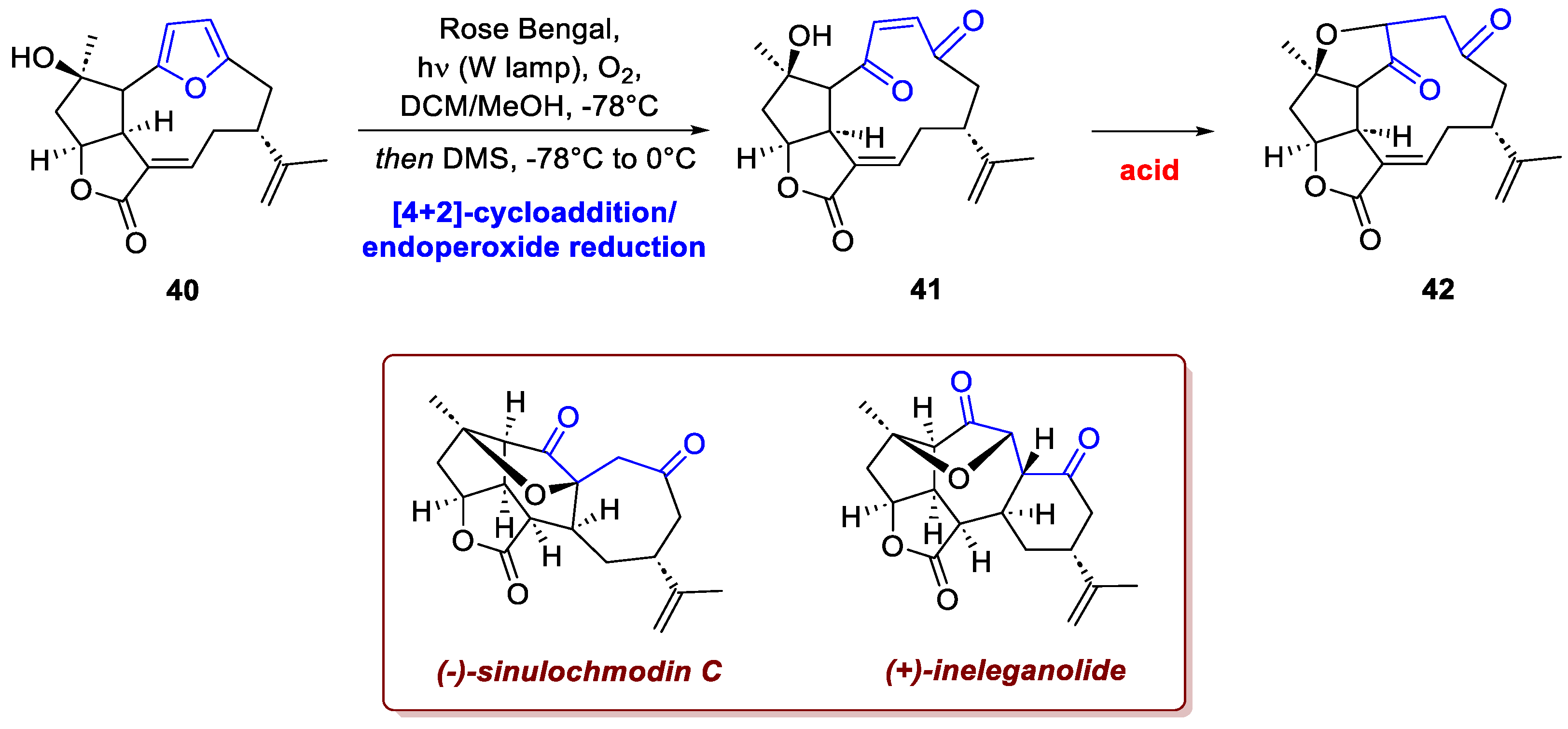
Scheme 16.
Light-mediated generation of o-quino-π-dimethane intermediate.
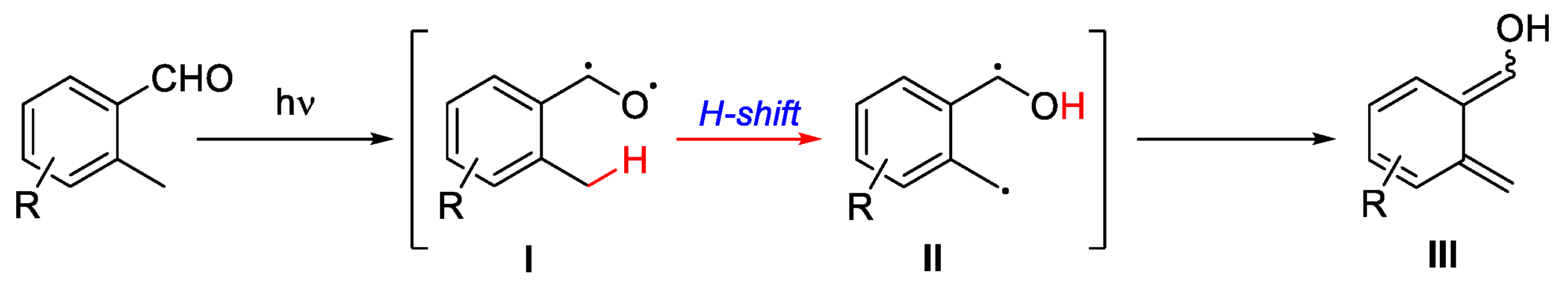
Scheme 17.
Synthesis of (+)-xestoquinone, (+)-adociaquinones A and B through a Ti(O-i-Pr)₄-promoted photoenolization/Diels–Alder (PEDA) sequence.
Scheme 17.
Synthesis of (+)-xestoquinone, (+)-adociaquinones A and B through a Ti(O-i-Pr)₄-promoted photoenolization/Diels–Alder (PEDA) sequence.
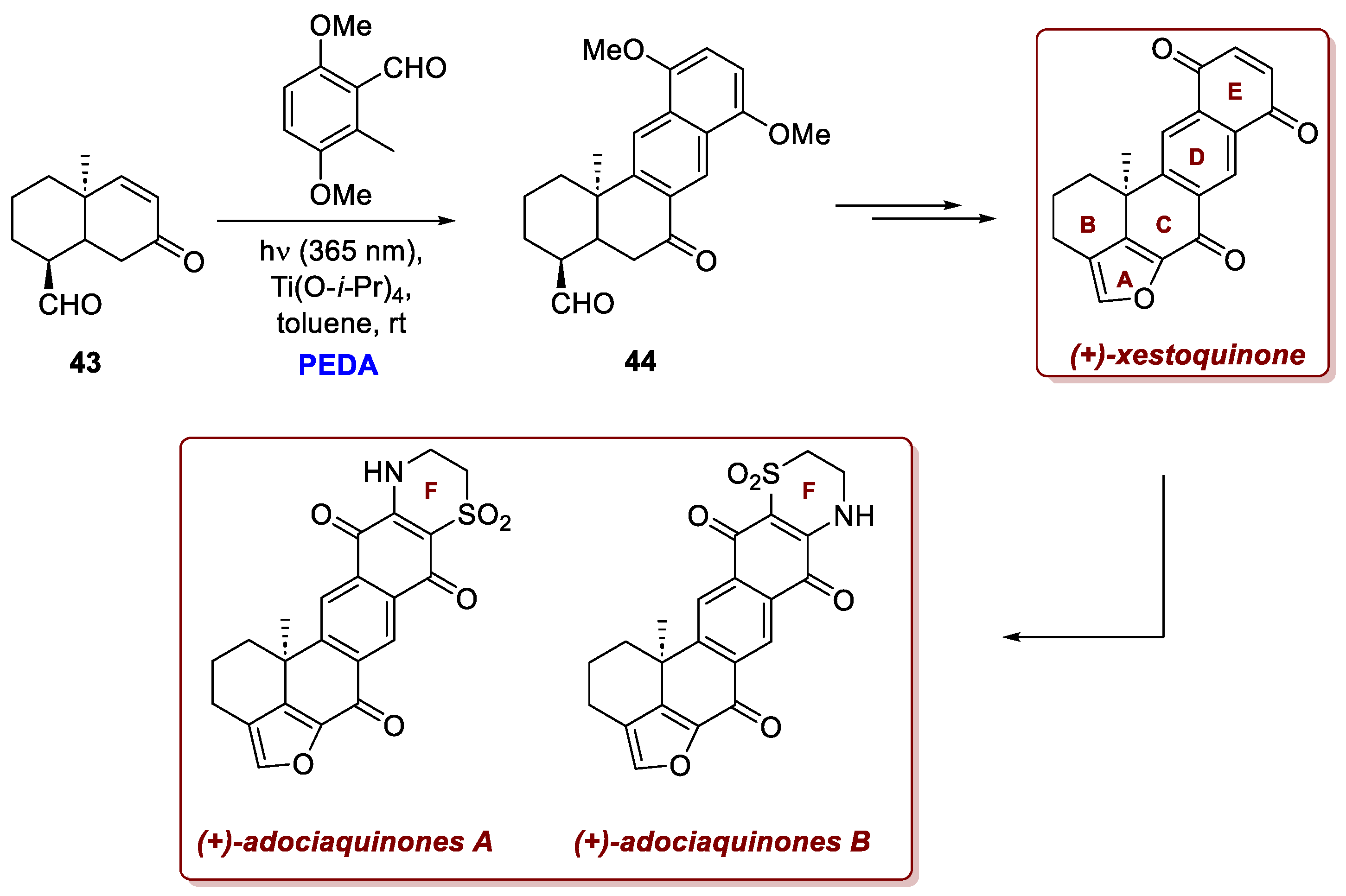
Scheme 18.
Synthesis of periglaucines A–C, N,O-dimethyloxostephine, and oxostephabenine.
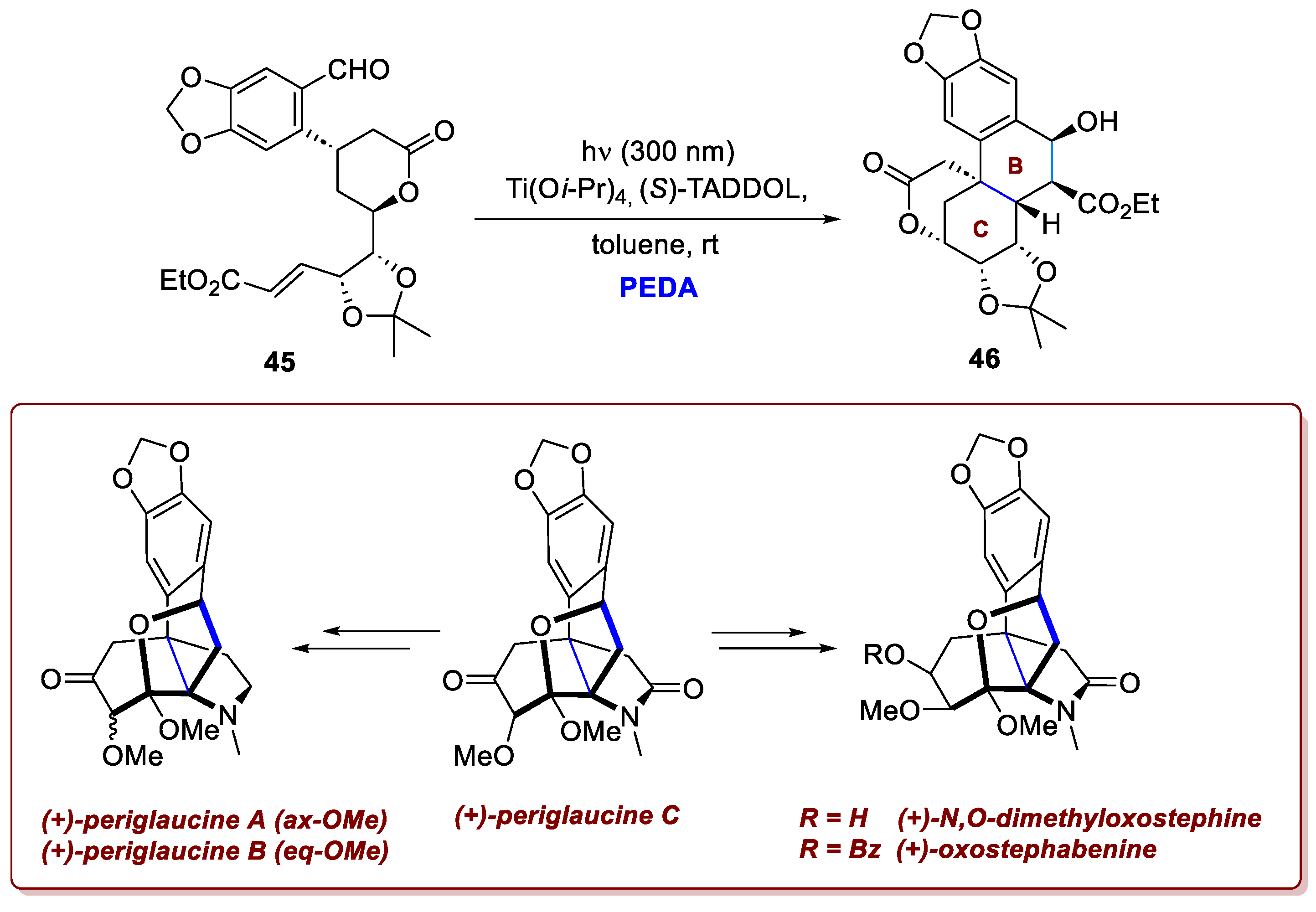
Scheme 19.
Synthesis of gracillisoid F and H through α-diketone–based Norrish–Yang photocyclization followed by an α-hydroxy ketone rearrangement.
Scheme 19.
Synthesis of gracillisoid F and H through α-diketone–based Norrish–Yang photocyclization followed by an α-hydroxy ketone rearrangement.
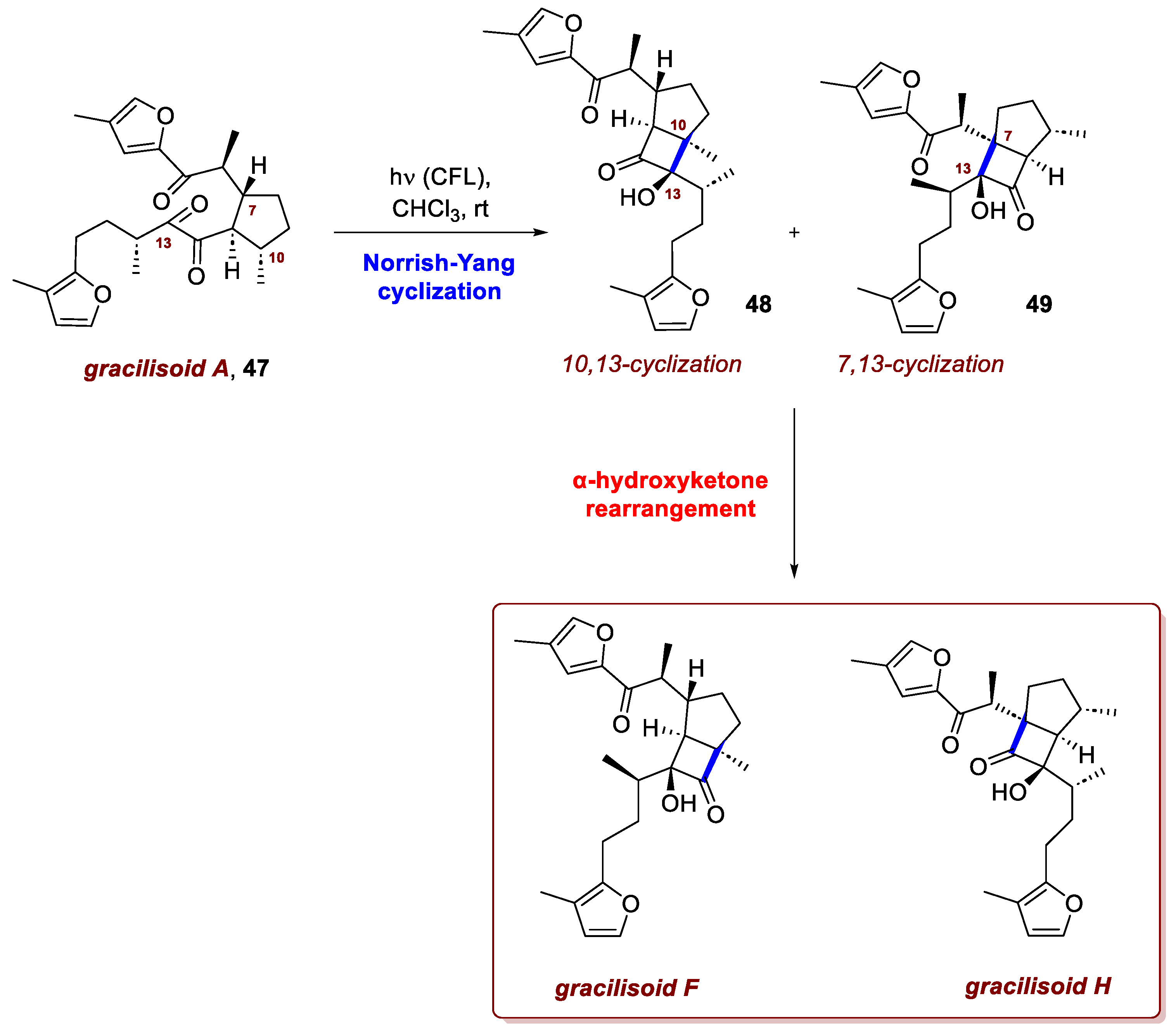
Scheme 20.
Divergent total synthesis of moluccensins G and H, krishnolide F, and khayseneganin F.
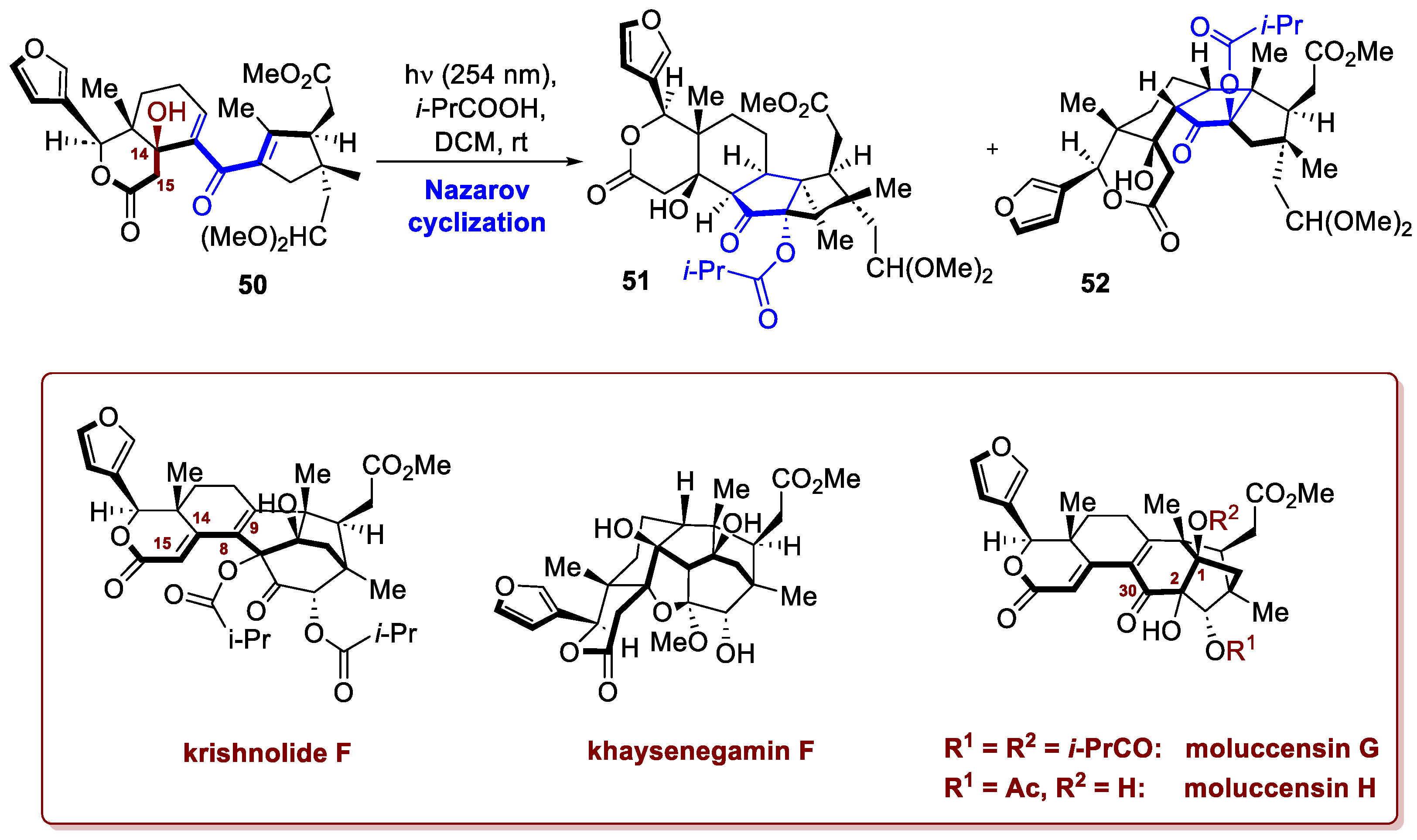
Scheme 21.
Synthesis of ladderanoic acid via the combination of [2+2]-cycloadditions followed by Wolff rearrangements.
Scheme 21.
Synthesis of ladderanoic acid via the combination of [2+2]-cycloadditions followed by Wolff rearrangements.
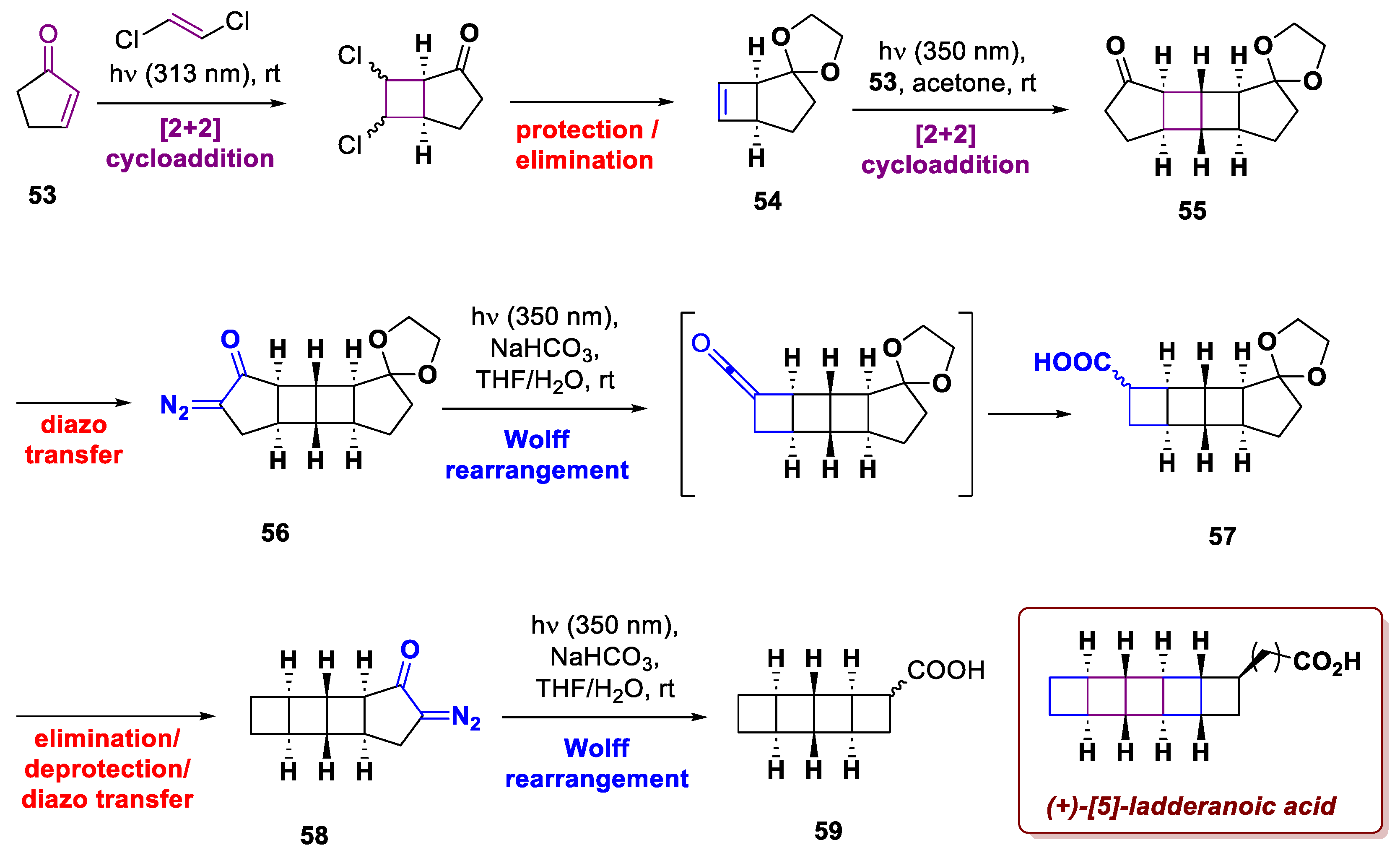
Scheme 22.
Construction of ring C of retigeranic acid A via ring contraction.
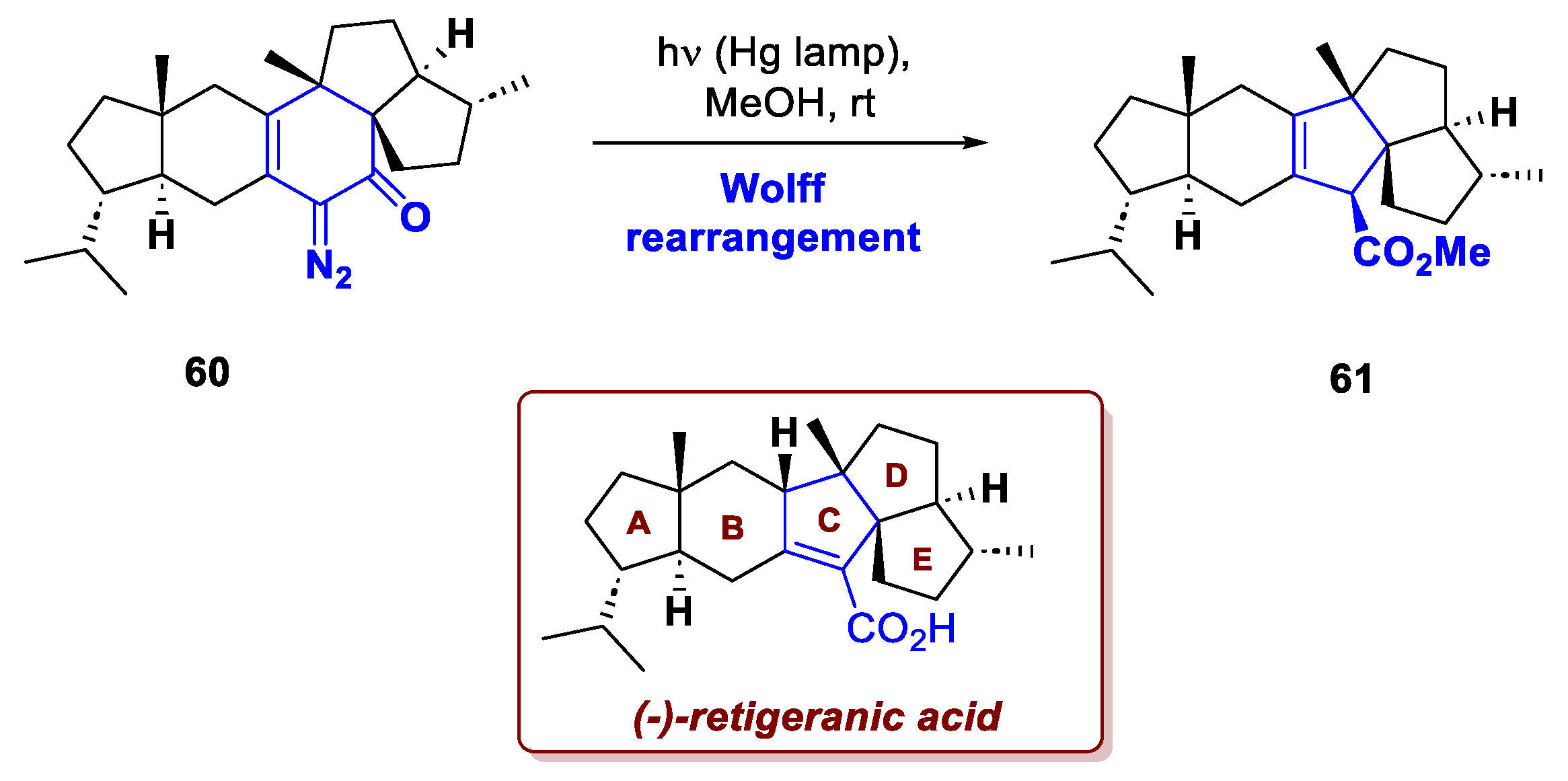
Scheme 23.
Santonin-like photoisomerization in the construction of A/B rings of barkeleyacetal D and peniciacetal I. The mechanism of santonin photoisomerization is also displayed.
Scheme 23.
Santonin-like photoisomerization in the construction of A/B rings of barkeleyacetal D and peniciacetal I. The mechanism of santonin photoisomerization is also displayed.
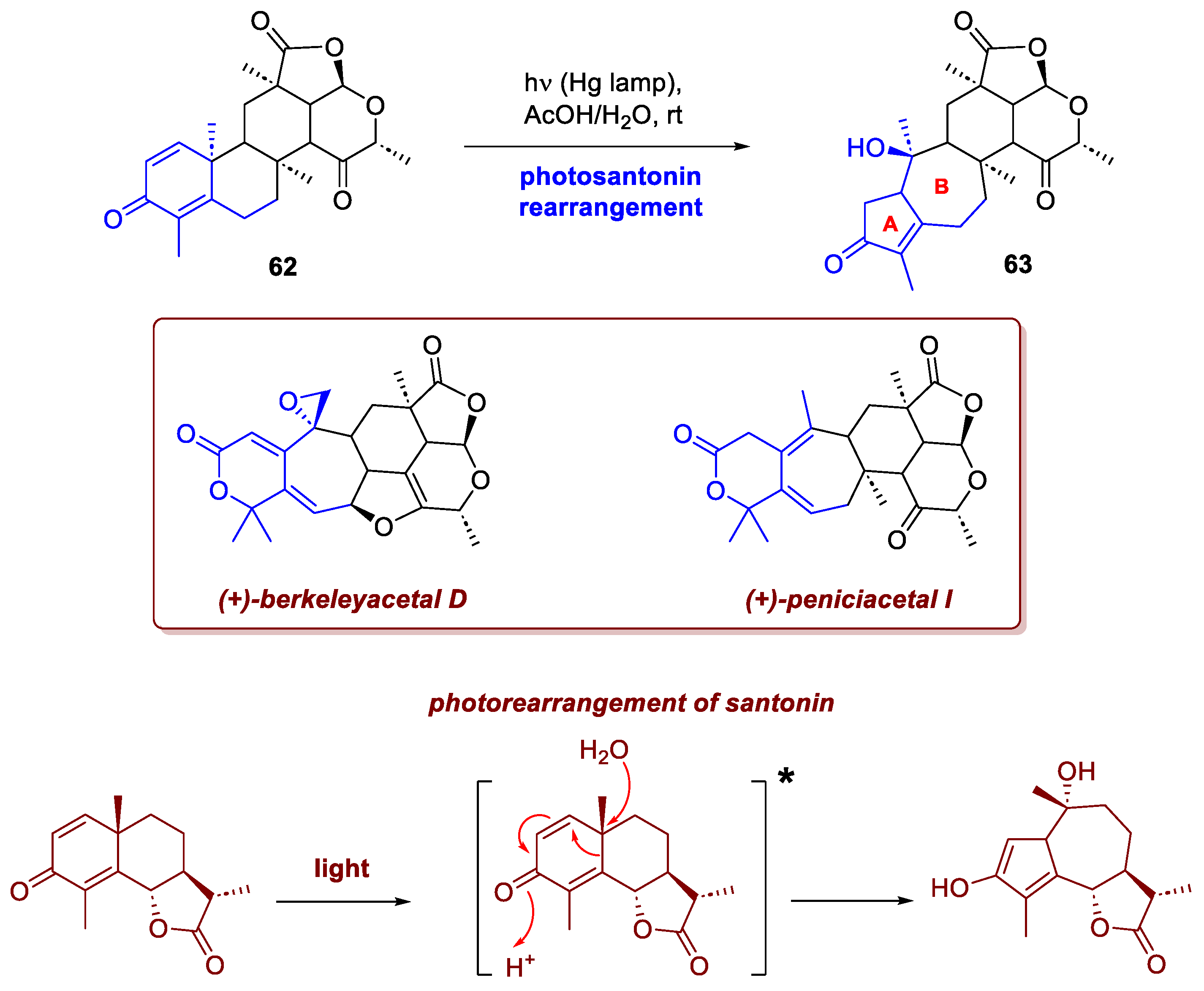
Scheme 24.
Riboflavin-mediated double bond isomerisation in the synthesis of sodagnitin E.
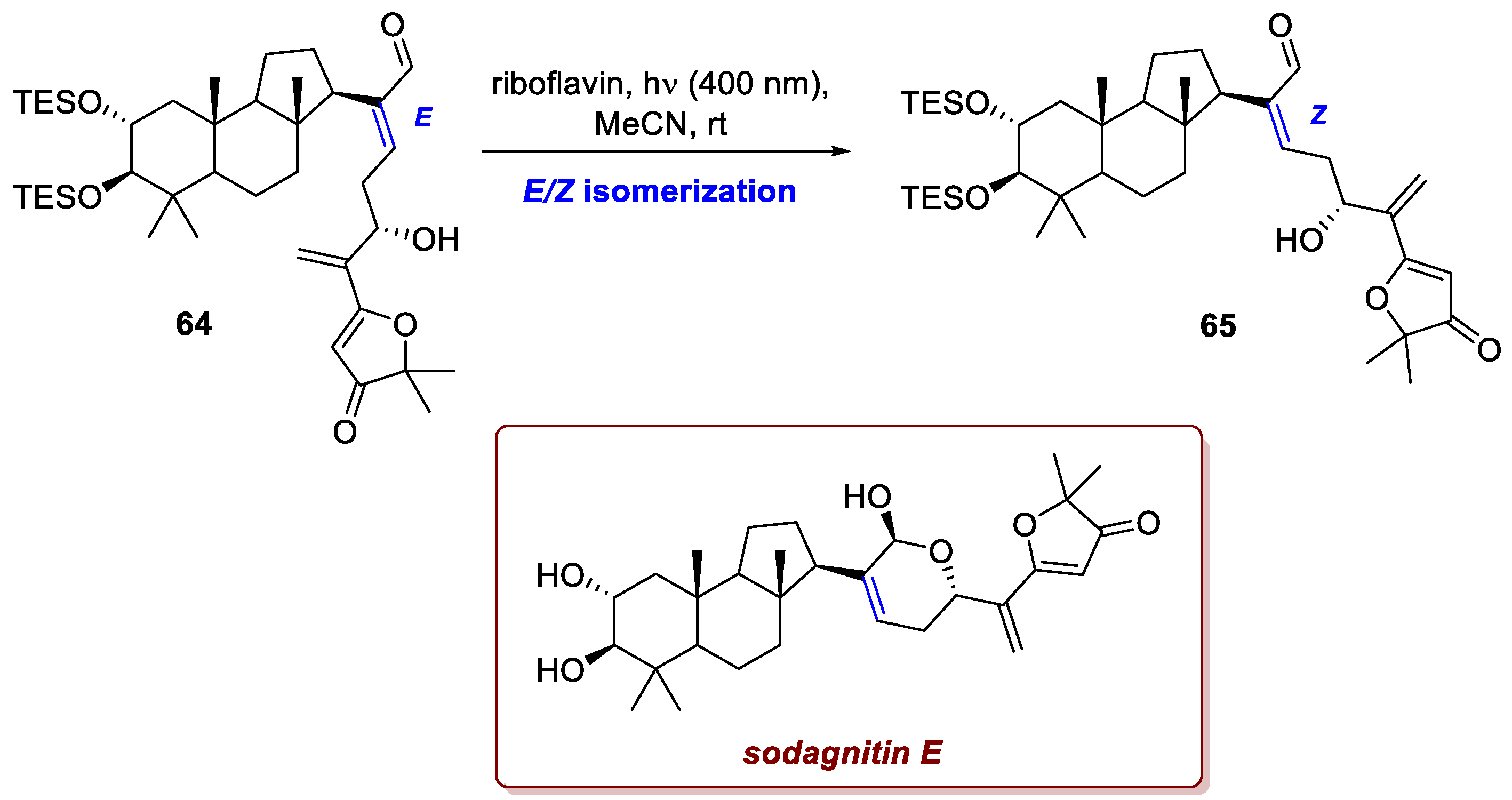
Scheme 25.
Anti-Markovnikov oxidation-elimination towards the synthesis of clionastatins.
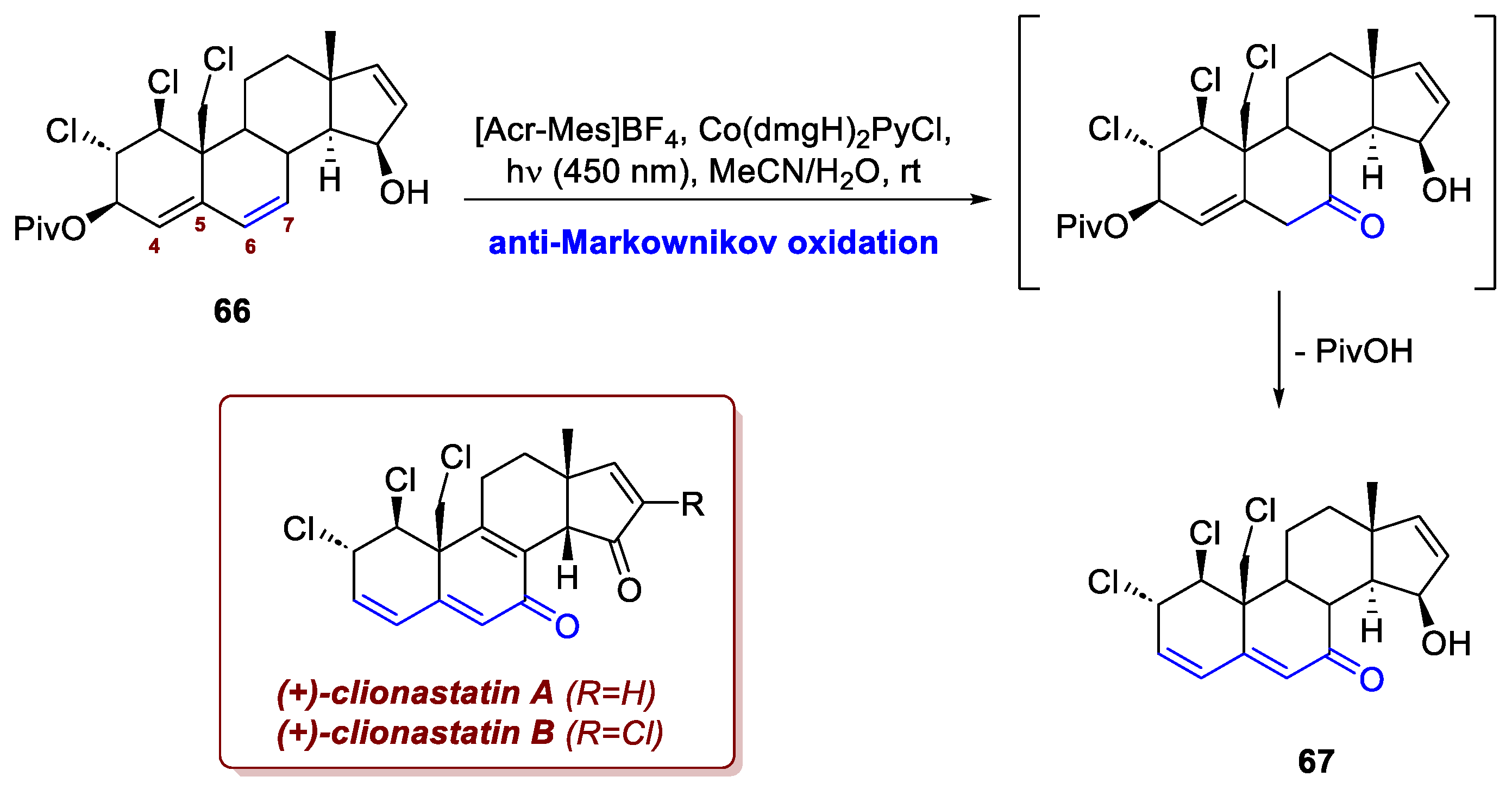
Scheme 26.
Benzylic oxidation using photoredox catalysis.
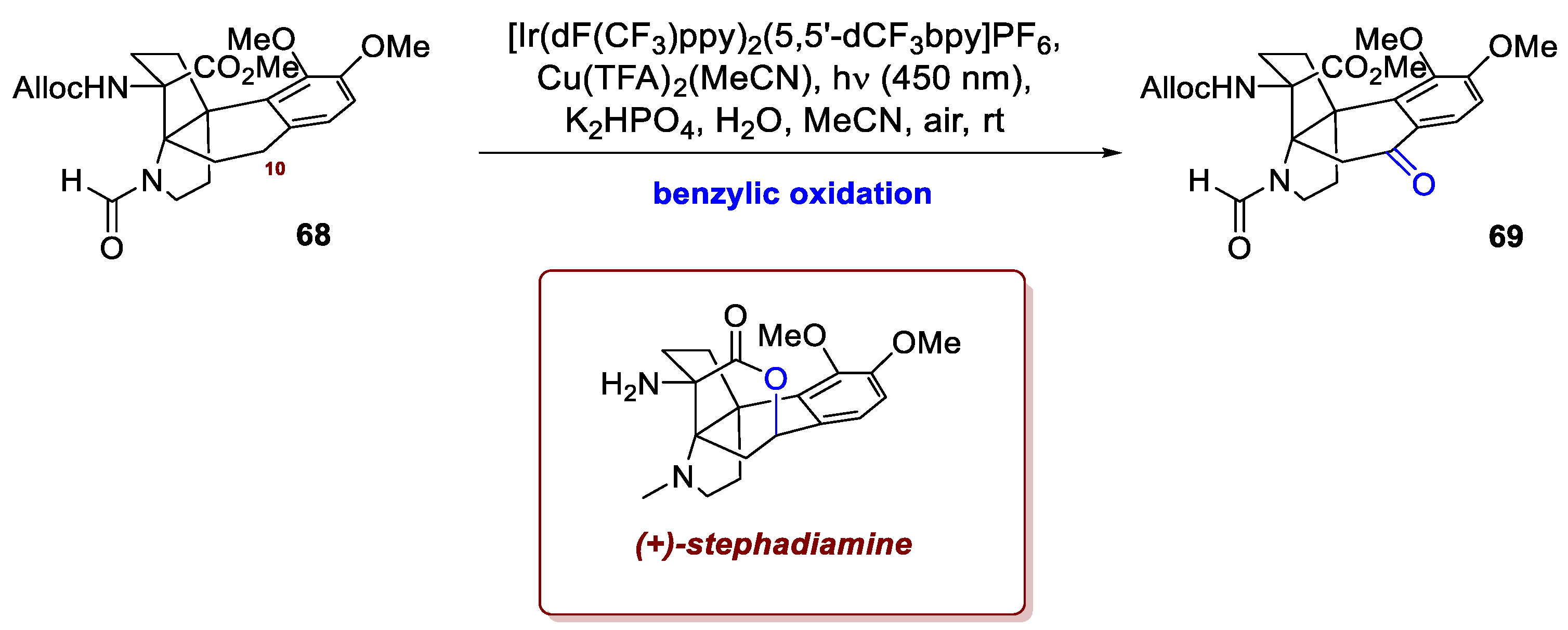
Scheme 27.
Thiol catalyzed hydroformylation of isolated double bonds in the synthesis of daphenylline.
Scheme 27.
Thiol catalyzed hydroformylation of isolated double bonds in the synthesis of daphenylline.
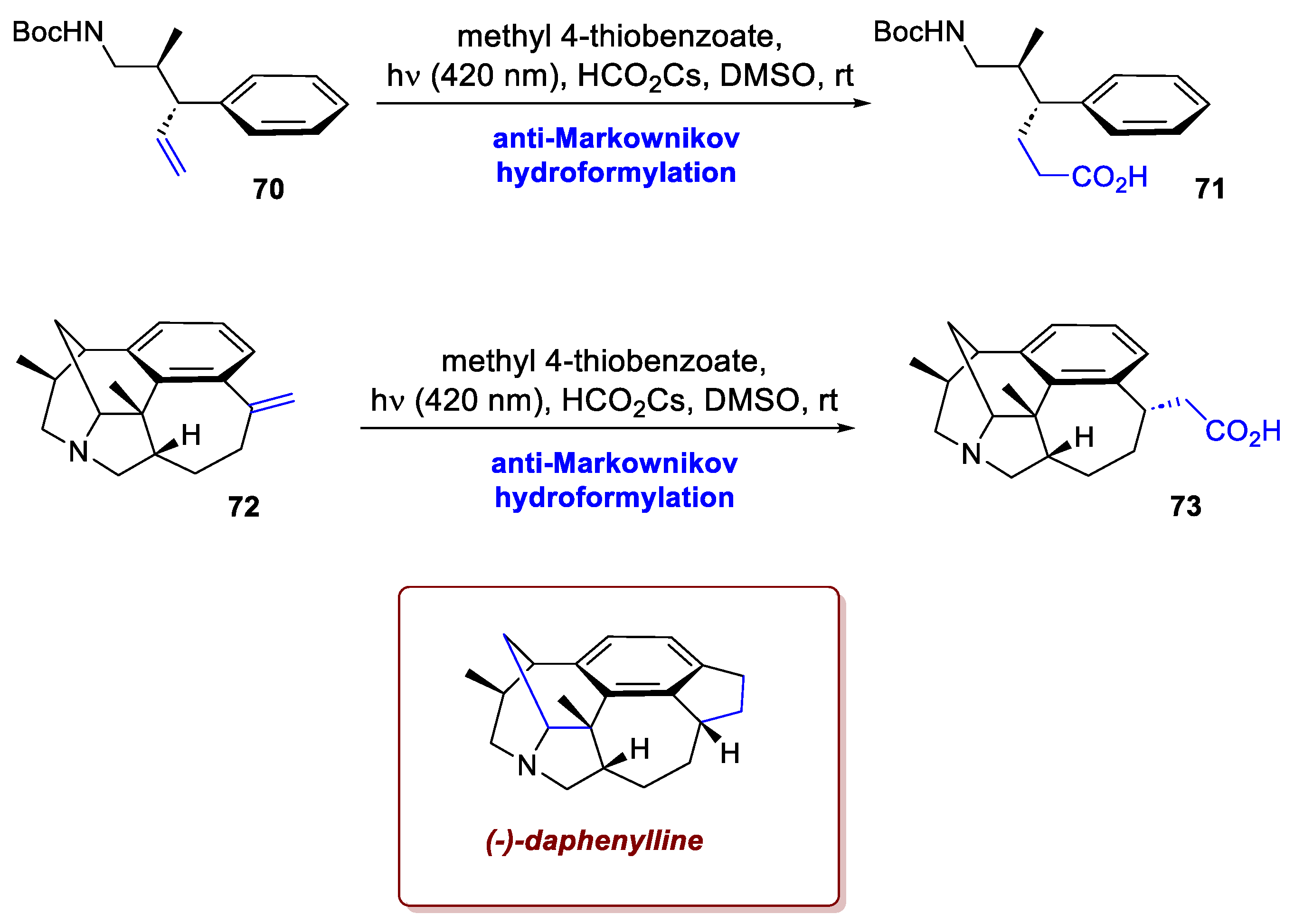
Scheme 28.
Synthesis of dixiamycin A via N-N oxidative dimerization.
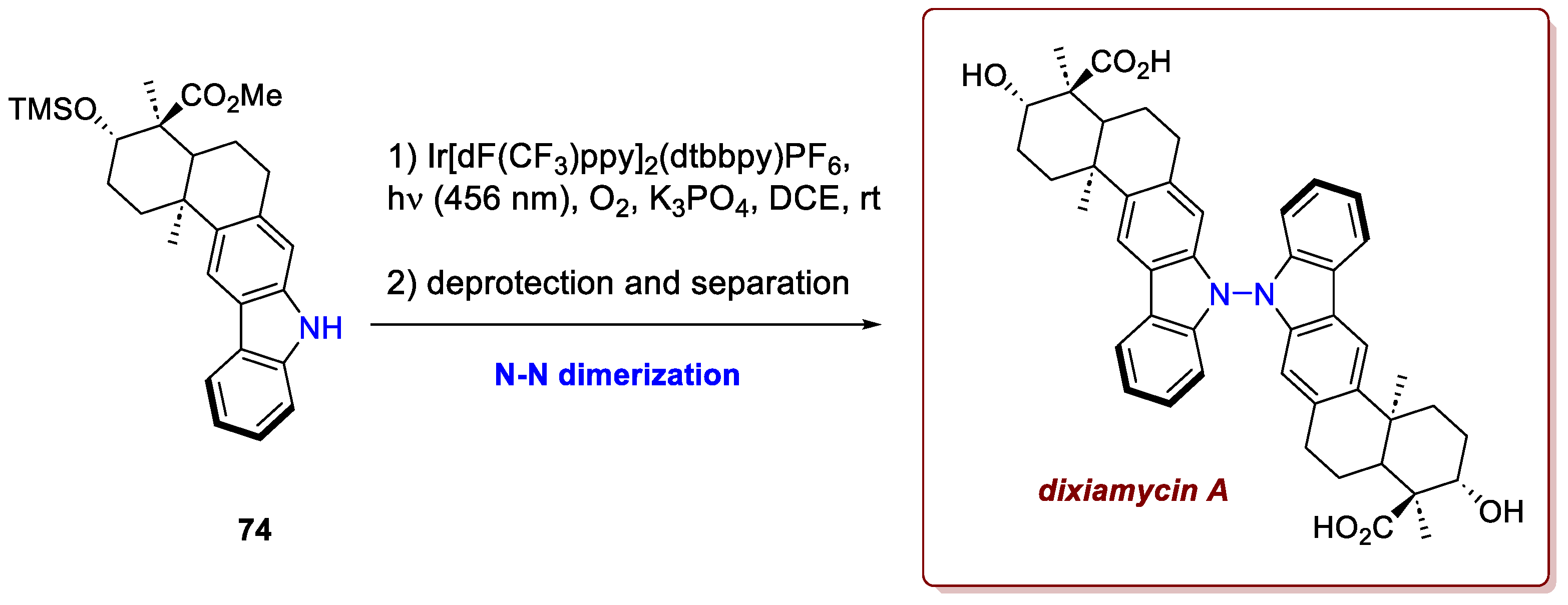
Scheme 29.
Synthesis of artemisinin exploiting a Schenck-ene reaction.
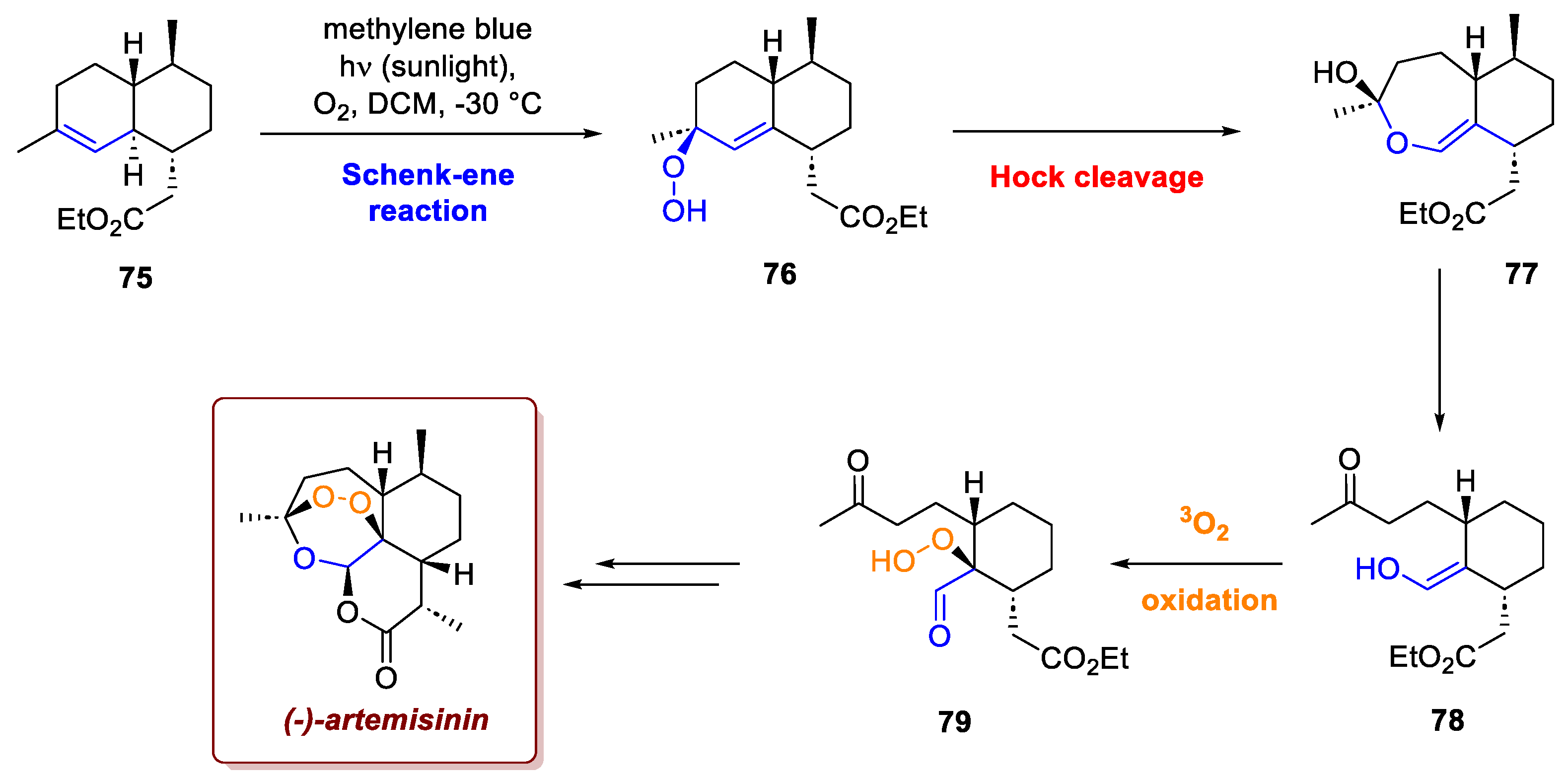
Scheme 30.
Synthesis of taxol exploiting a Schenck-ene reaction to construct the D ring.
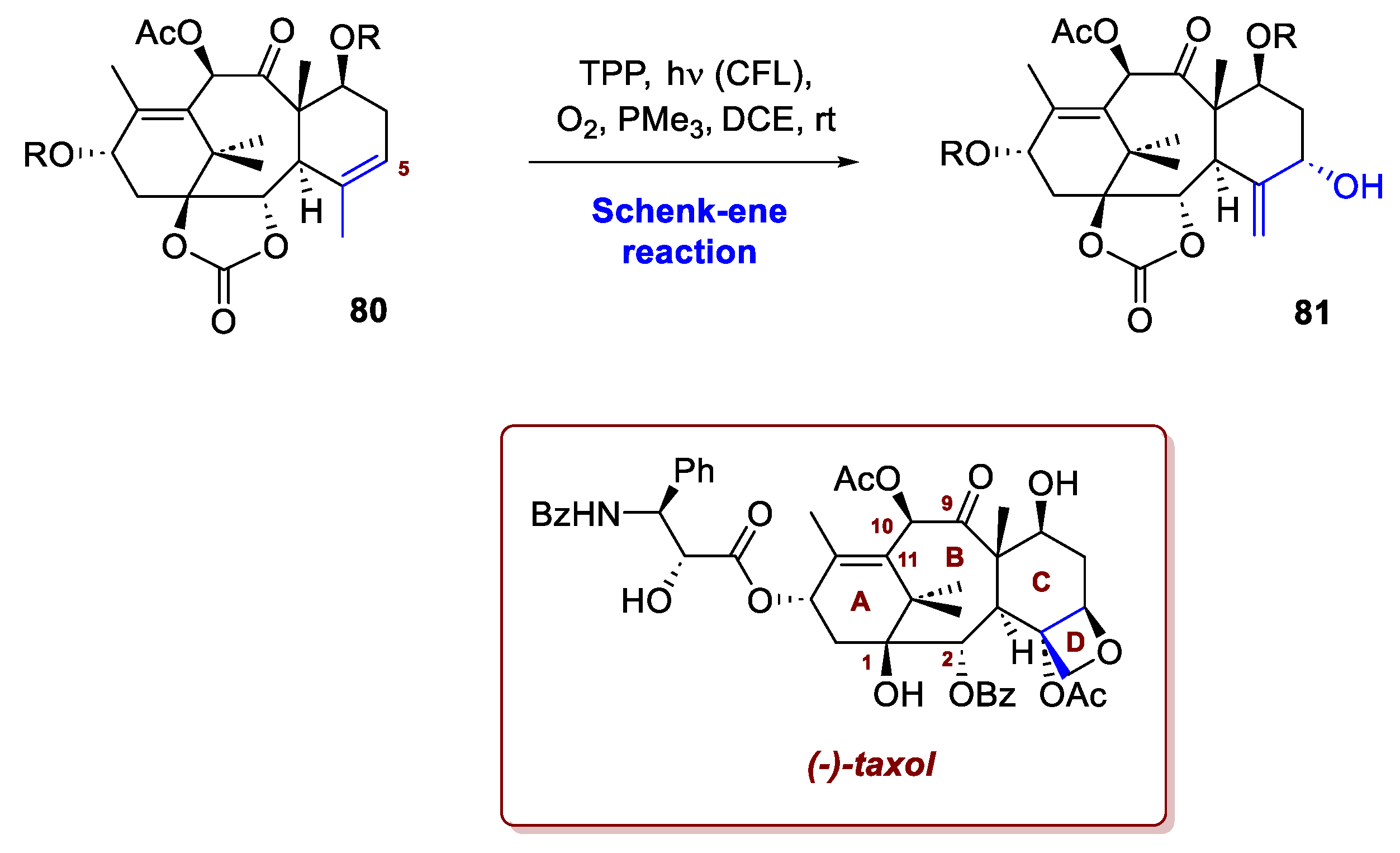
Scheme 31.
Synthesis of hydroxyincarvilline exploiting a Schenck-ene reaction.
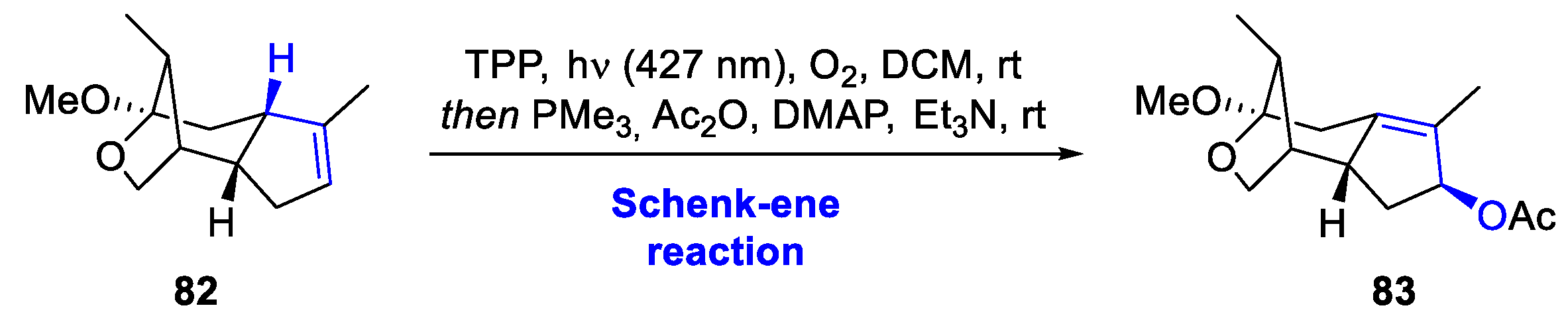
Scheme 32.
Synthesis of hydroxyincarvilline exploiting a Schenck-ene reaction.
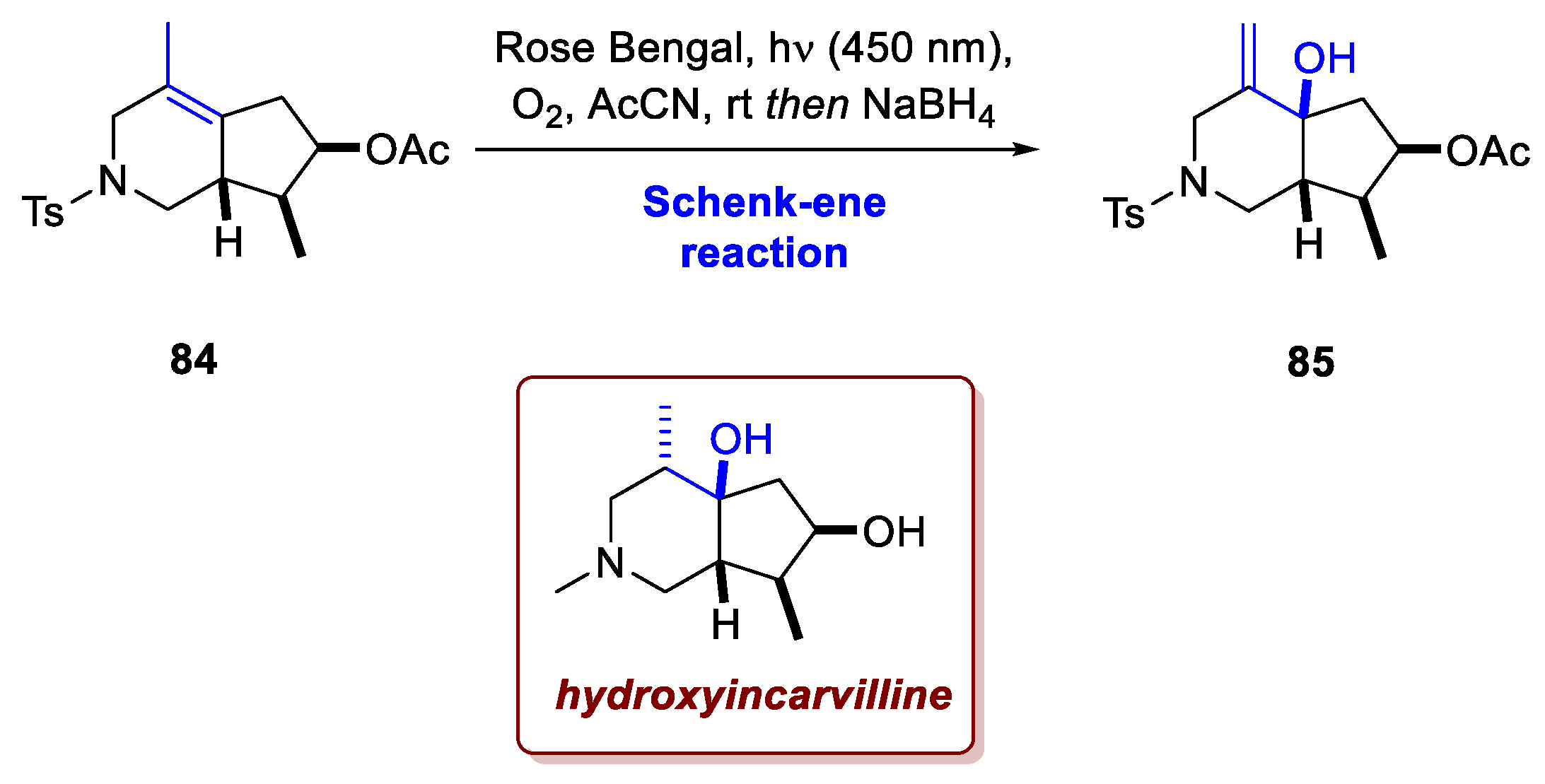
Scheme 33.
Synthesis of dysideanone E exploiting a Schenck-ene reaction. The same approach could not be applied to the synthesis of dysideanone A.
Scheme 33.
Synthesis of dysideanone E exploiting a Schenck-ene reaction. The same approach could not be applied to the synthesis of dysideanone A.
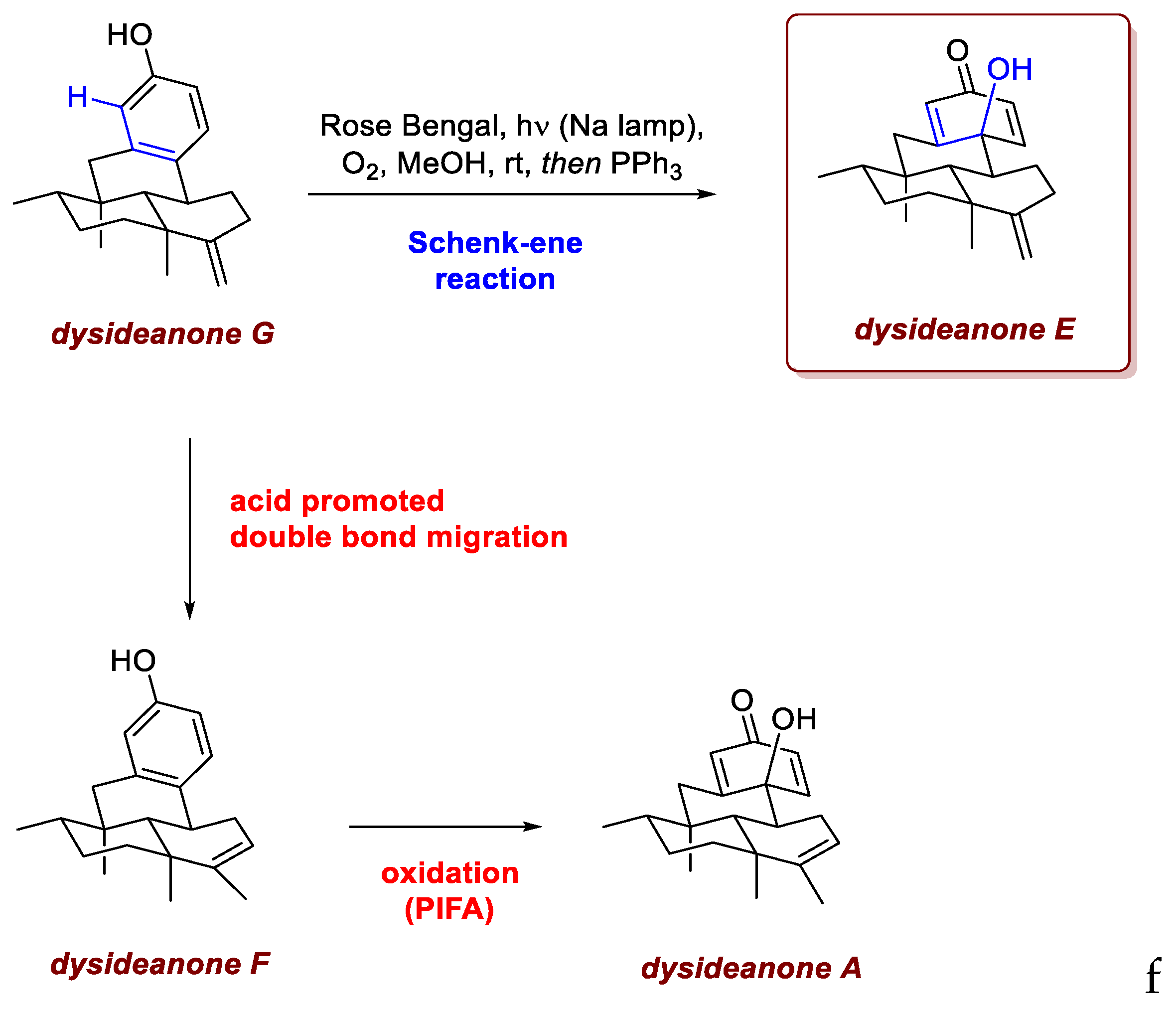
Scheme 34.
Photoinduced iododecarboxylation in the synthesis of macrocalin B.
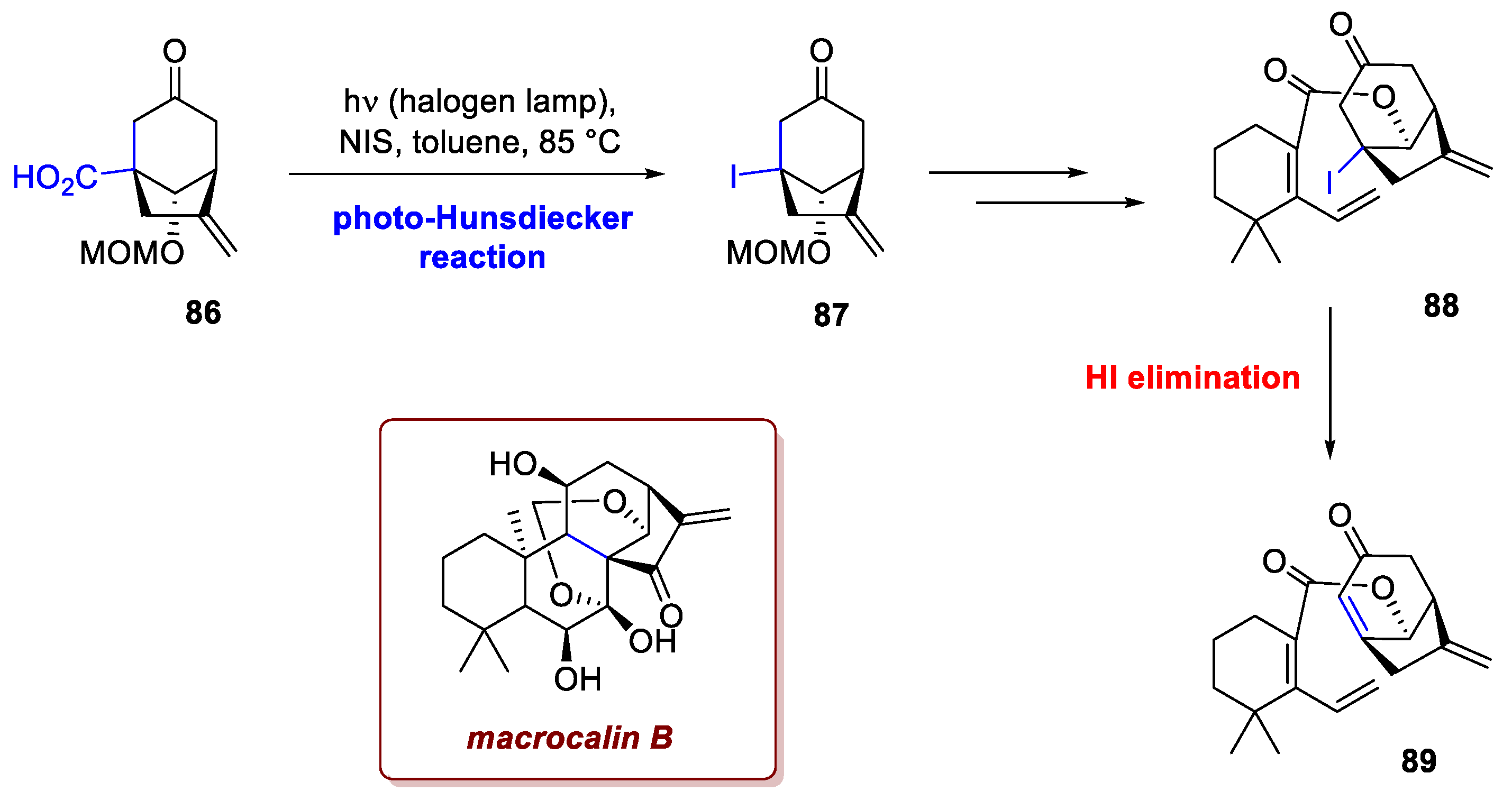
Scheme 35.
Photocatalyzed decarboxylation in the synthesis of ancistrobrevolines.
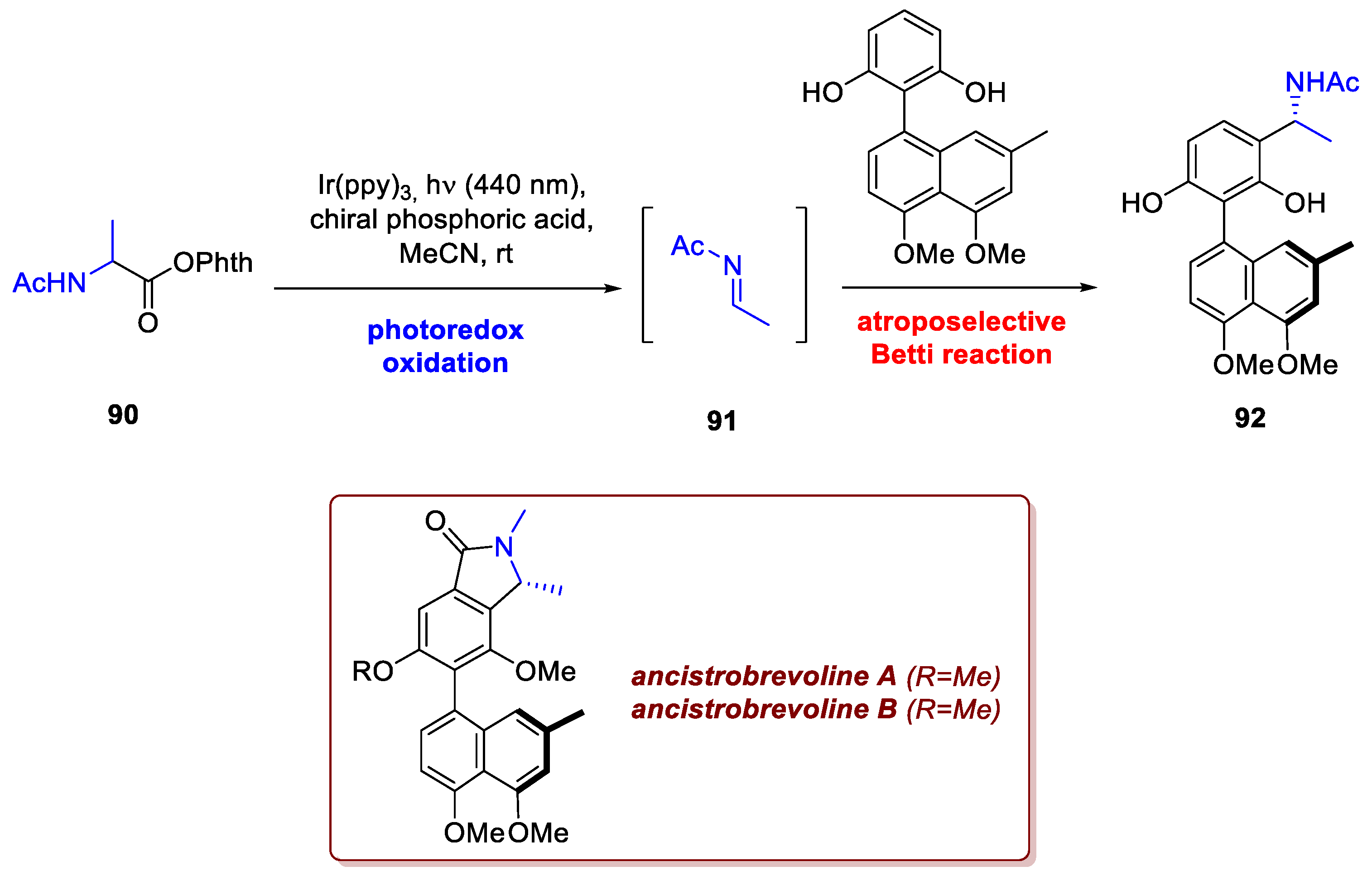
Scheme 36.
Synthesis of (+)-actinobolin exploiting a diastereoselective Giese radical addition.
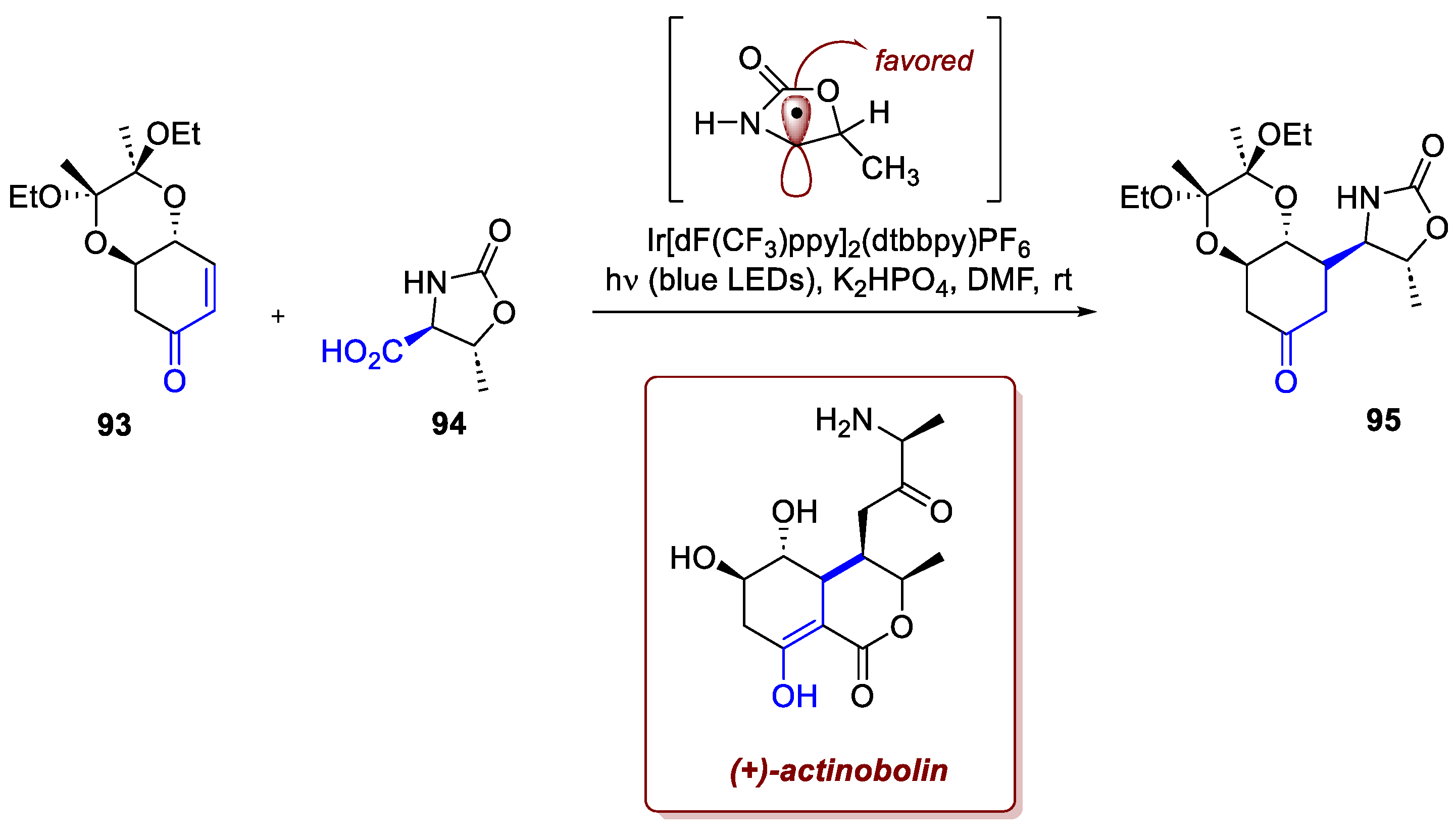
Scheme 37.
Giese-type decarboxylative allylation towards the synthesis of dafachronic acid.
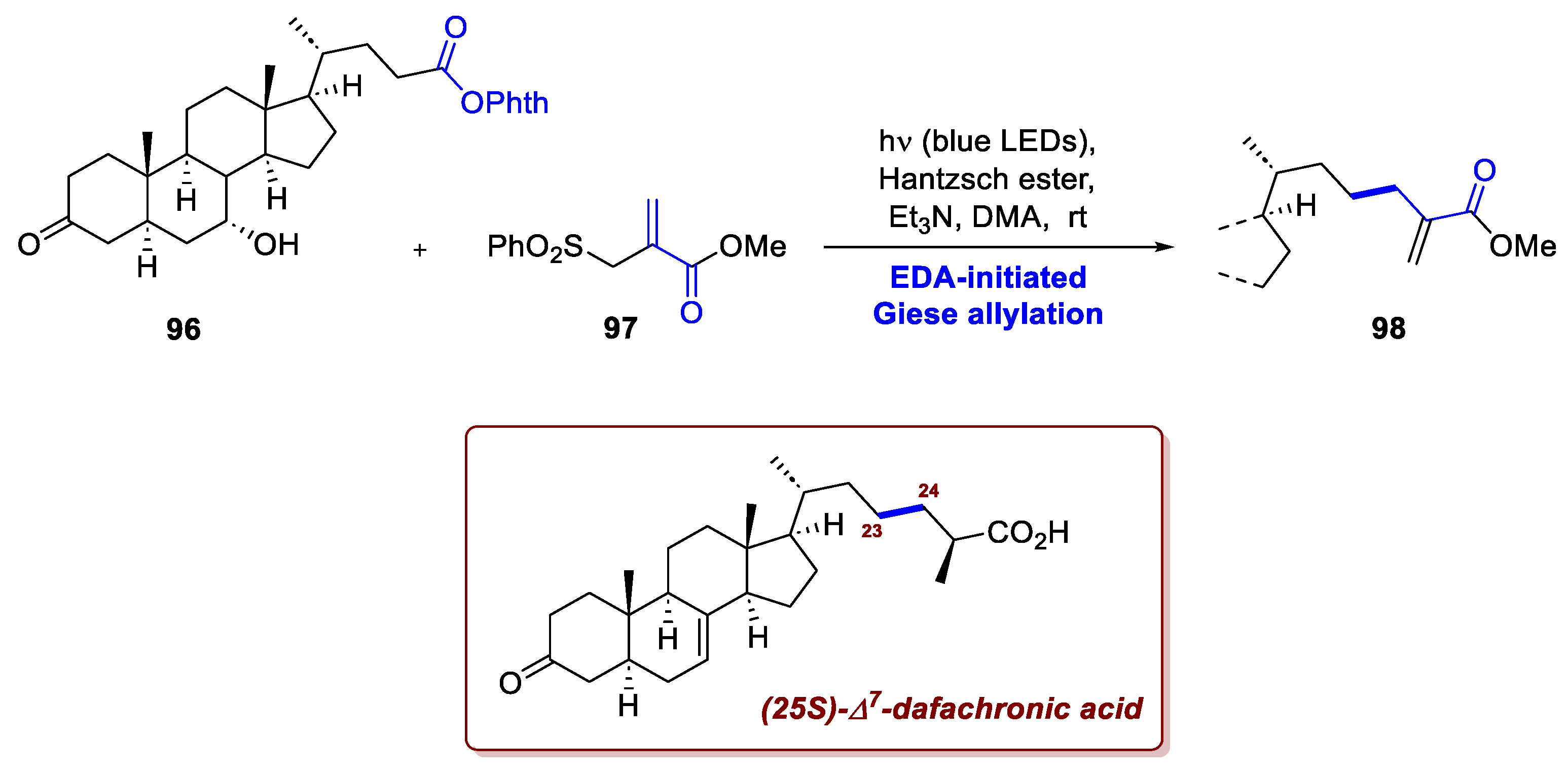
Scheme 38.
Double Giese-acylation of an enedione scaffold for the synthesis of elodeoidins A and B.
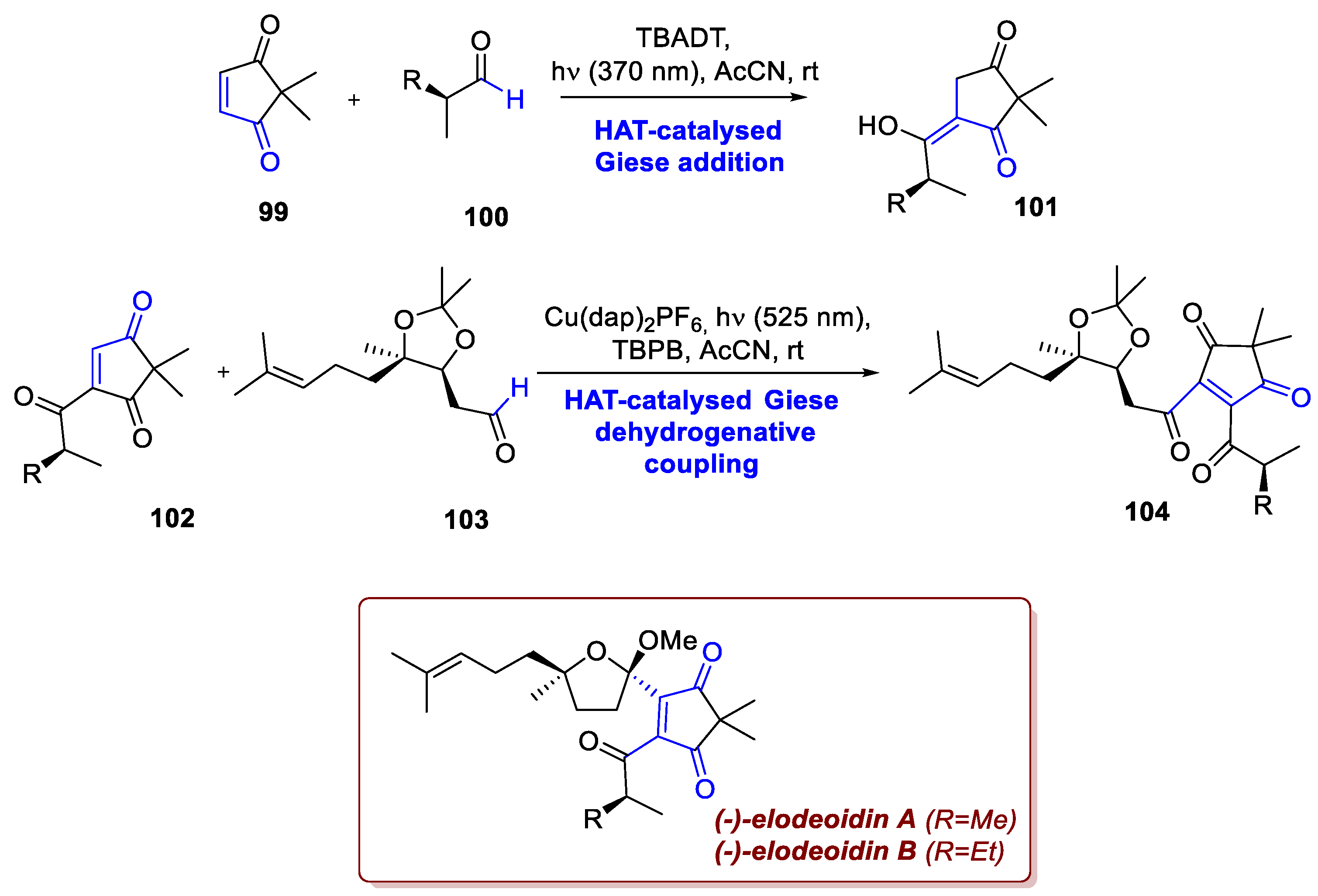
Scheme 39.
Epimerization of chenodeoxycholic ester in the synthesis of (25S)-Δ7-dafachronic acid.
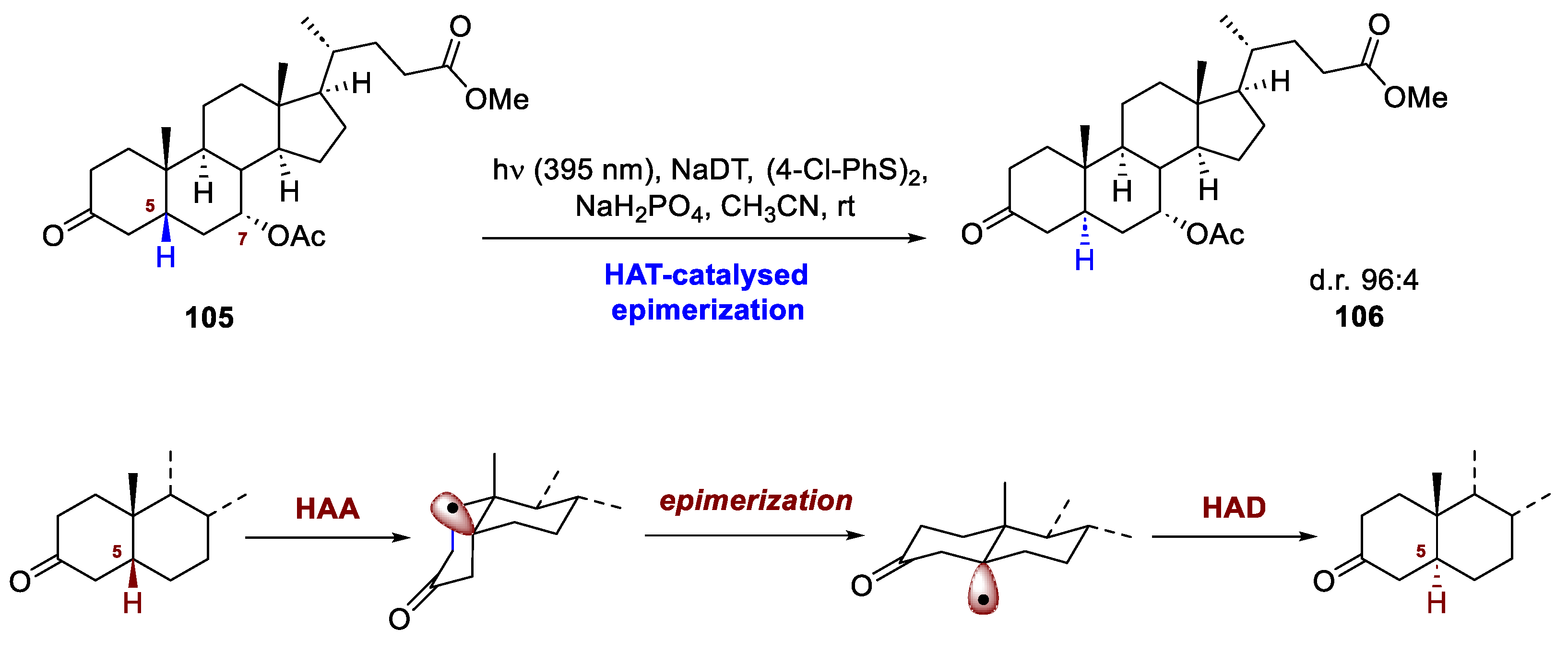
Scheme 40.
HAT-initiated 1,2-radical rearrangement in the synthesis of hetisine-type diterpenoid alkaloids.
Scheme 40.
HAT-initiated 1,2-radical rearrangement in the synthesis of hetisine-type diterpenoid alkaloids.
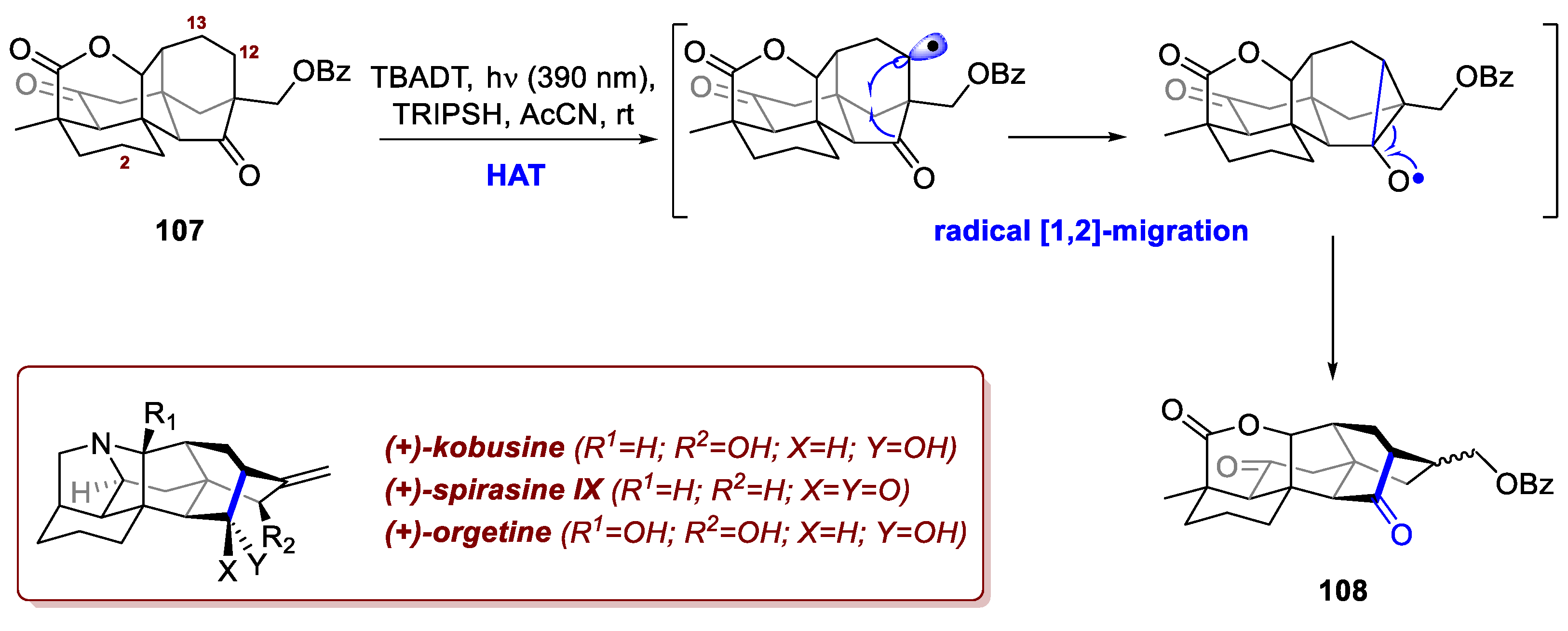
Scheme 41.
NaDT-mediated double bond transposition in the synthesis of illisimonin A.
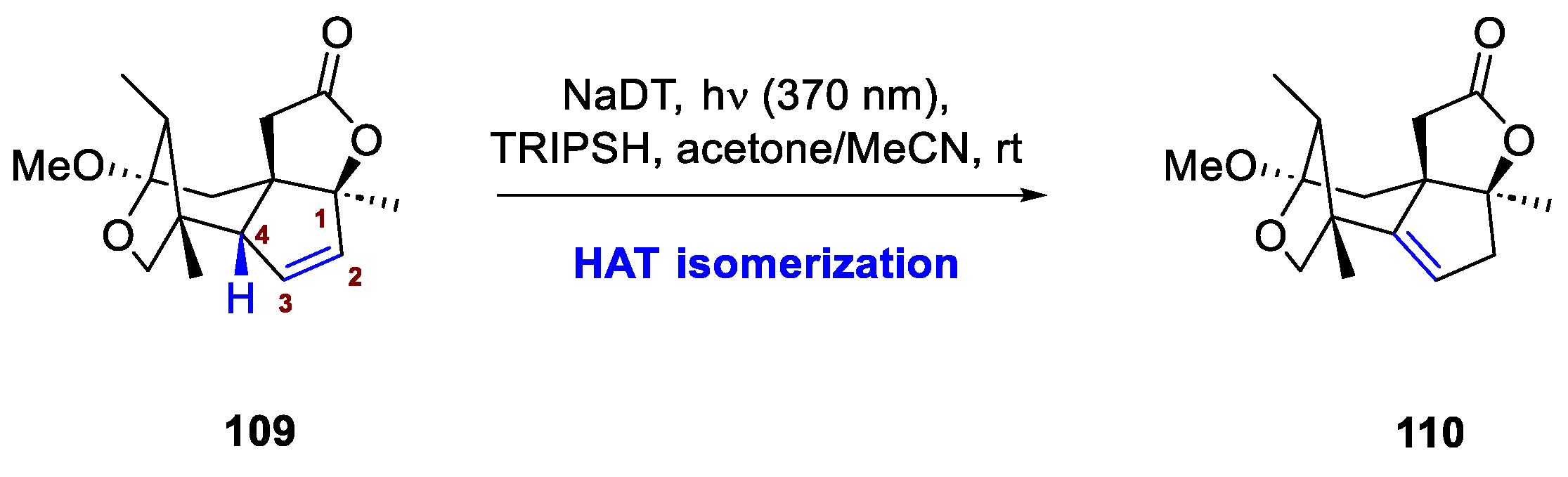
Scheme 42.
HAT-initiated radical cyclization in the synthesis of a prostaglandin D2 metabolite.
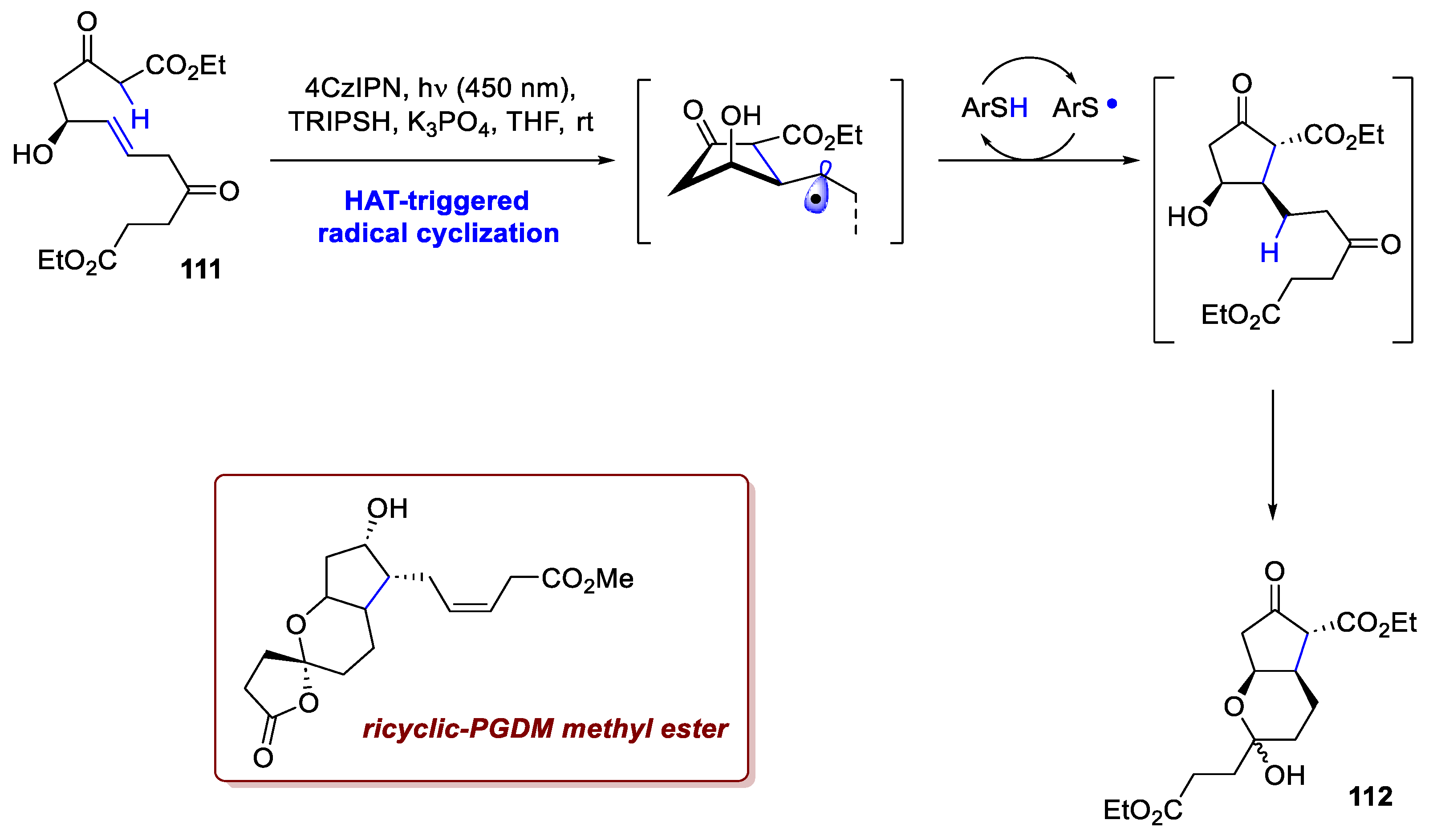
Scheme 43.
Nicewicz photo-driven synthetic route towards stemoamide alkaloids.
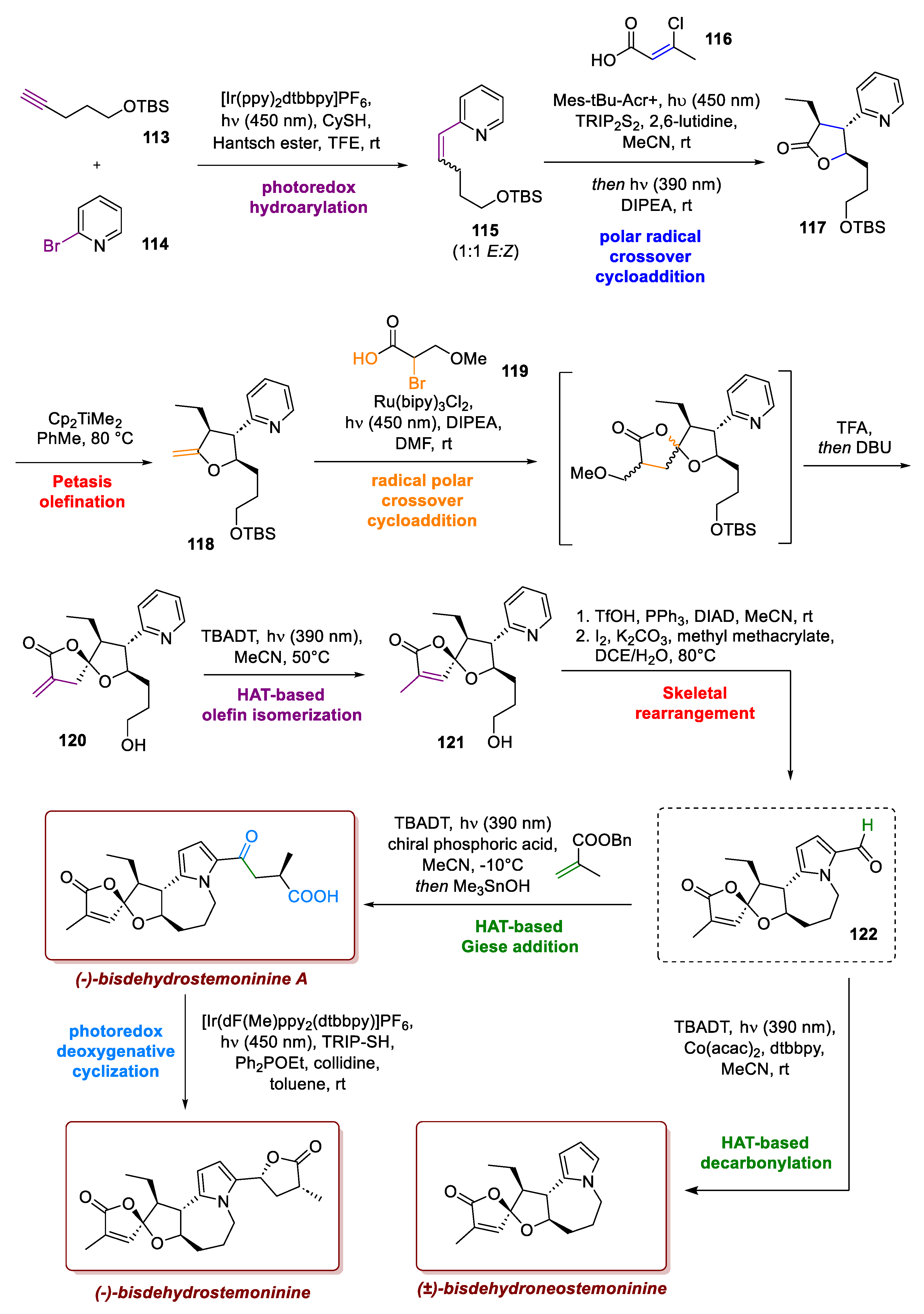
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



