
Submitted:
19 November 2025
Posted:
21 November 2025
You are already at the latest version
Abstract
Recent advances in large language models (LLMs) have unlocked new possibilities for scientific discovery, yet most remain limited to text summarization or hallucination-prone dialogue. Here, we present LongevityLLM—a function-driven AI agent engineered to execute real, reproducible analyses in structural bioinformatics, comparative genomics, and aging biology. Unlike conventional chatbots, LongevityLLM maps natural language queries to deterministic bioinformatics pipelines, producing structured outputs (FASTA, PDB, XLSX, phylogenetic trees, aging clock reports) while grounding all responses in empirical data. The system retrieves and summarizes scientific information from peer-reviewed literature (via Europe PMC) and biological databases (e.g., UniProt). It integrates five major epigenetic clocks—Horvath, Hannum, PhenoAge, Brunet, and Wyss-Coray—as well as AlphaFold2-based structural mutation impact prediction, cross-species ortholog retrieval with phylogenetic analysis, and curated mammalian life-history traits from the AnAge database and incorporates a time-calibrated mammalian phylogeny and the AROCM (Average Rate of Change in Methylation) metric—a cross-species epigenetic biomarker of aging derived from conserved CpG sites. Built on open-source tools and designed for full auditability, LongevityLLM enables researchers to explore questions such as “How is IFI27 implicated across different aging clock models?” or “What is the structural effect of the IL17A-E100K mutation?” through a single natural language query, without compromising scientific rigor. We release LongevityLLM as an open framework to accelerate hypothesis generation, education, and collaborative geroscience.
Keywords:
AI agent
; literature retrieval
; biological databases
; bioinformatics
; structural biology
; aging research
; mammalian lifespan evolution
; aging clocks
Introduction
The application of artificial intelligence (AI) in biomedical research has grown rapidly, yet its practical utility remains constrained by two persistent challenges.
First, while large language models (LLMs) can summarize literature or suggest hypotheses, they are prone to factual hallucinations—especially when dealing with nuanced, data-intensive domains like aging biology or protein structure-function relationships. In scientific contexts, where reproducibility and precision are paramount, such unreliability limits their use to ideation at best, and introduces risk at worst.
Second, and perhaps more fundamentally, modern computational biology is operationally inefficient. Even for technically proficient researchers, executing standard analyses—be it bulk or single-cell RNA-seq data analysis, molecular docking, or structural variant impact prediction—requires navigating a fragmented landscape of command-line tools, poorly documented databases, and brittle pipelines that frequently fail due to version mismatches, missing dependencies, or opaque error messages. Each step demands time, debugging, and deep familiarity with domain-specific conventions. This overhead consumes valuable researcher hours that could otherwise be spent on interpretation, experimental design, or hypothesis generation.
Platforms like PandaOmics (Insilico Medicine) have demonstrated a promising alternative: integrating multi-omics evidence, literature mining, and AI-driven prioritization into a unified, user-accessible interface. By grounding LLM-like reasoning in structured biological knowledge and precomputed analyses, PandaOmics reduces the barrier to target discovery—particularly for non-bioinformaticians—while maintaining scientific traceability [1].
Inspired by this vision, we present LongevityLLM: a prototype AI agent designed to bridge natural language queries with executable bioinformatics workflows in the domain of aging and protein biology. Unlike conventional chatbots, LongevityLLM does not generate answers from internal parameters alone. Instead, it calls deterministic functions—for protein annotation, cross-species alignment, epigenetic clock interrogation, or AlphaFold-based mutation modeling—and synthesizes responses from the actual outputs. All intermediate files (FASTA, PDB, XLSX, phylogenetic trees) are saved and made available, ensuring full auditability.
We emphasize that this is an early-stage prototype. Its current scope is narrow: it supports a limited set of aging clocks, a basic structural prediction pipeline, and curated life-history data from public sources. It does not yet handle single-cell data, Hi-C, or complex multi-omics integration—though these are planned extensions. Nevertheless, even in its current form, LongevityLLM illustrates a viable path toward delegating routine computational labor to machines, freeing researchers to focus on scientific insight rather than pipeline maintenance.
We release this work not as a finished product, but as a proof of concept: that function-driven, transparent AI agents can begin to address the inefficiencies of modern bioinformatics—making rigorous, reproducible analysis more accessible and ultimately, more human-centered.
The Architecture of LongevityLLM
Overall Structure
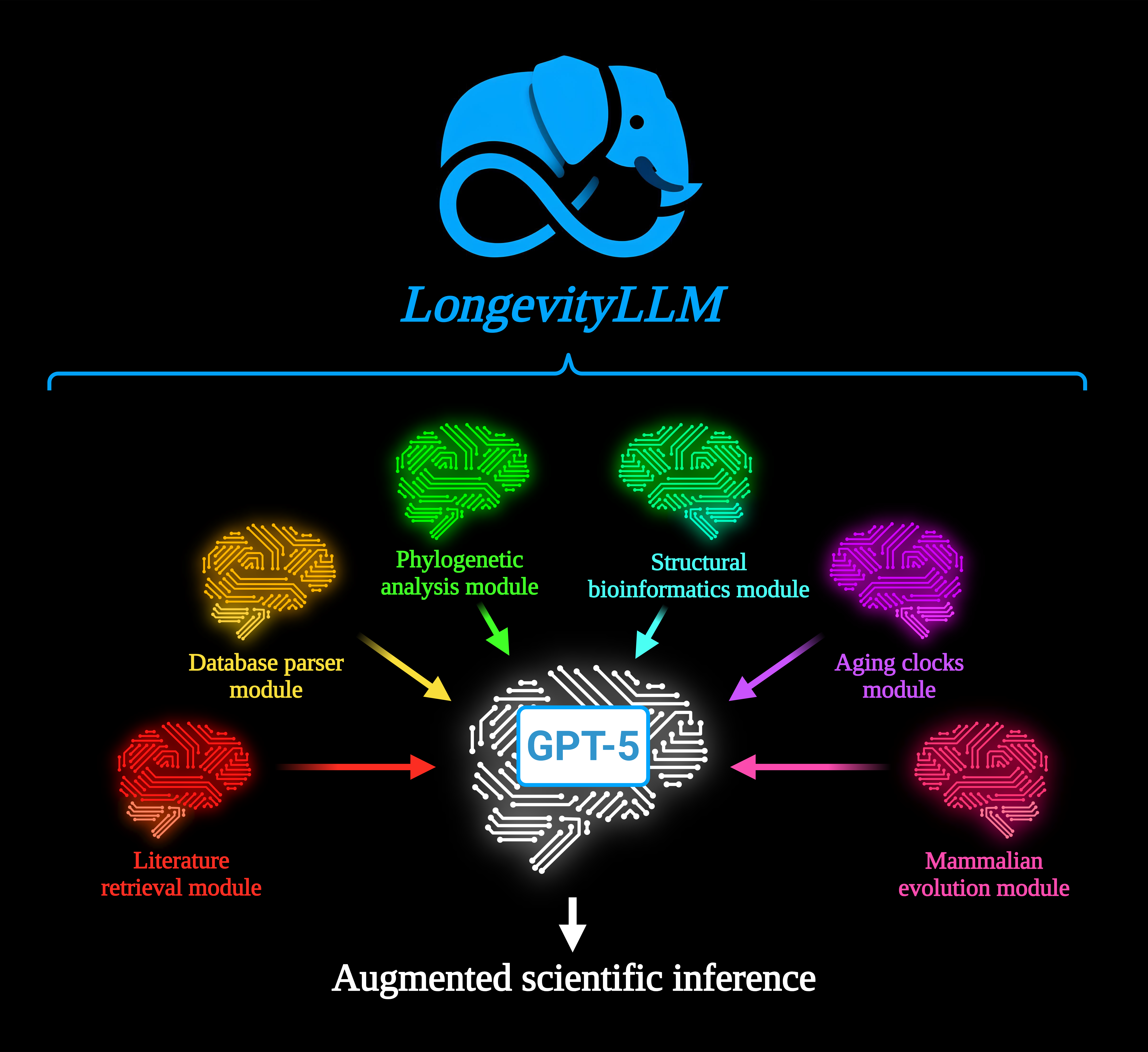
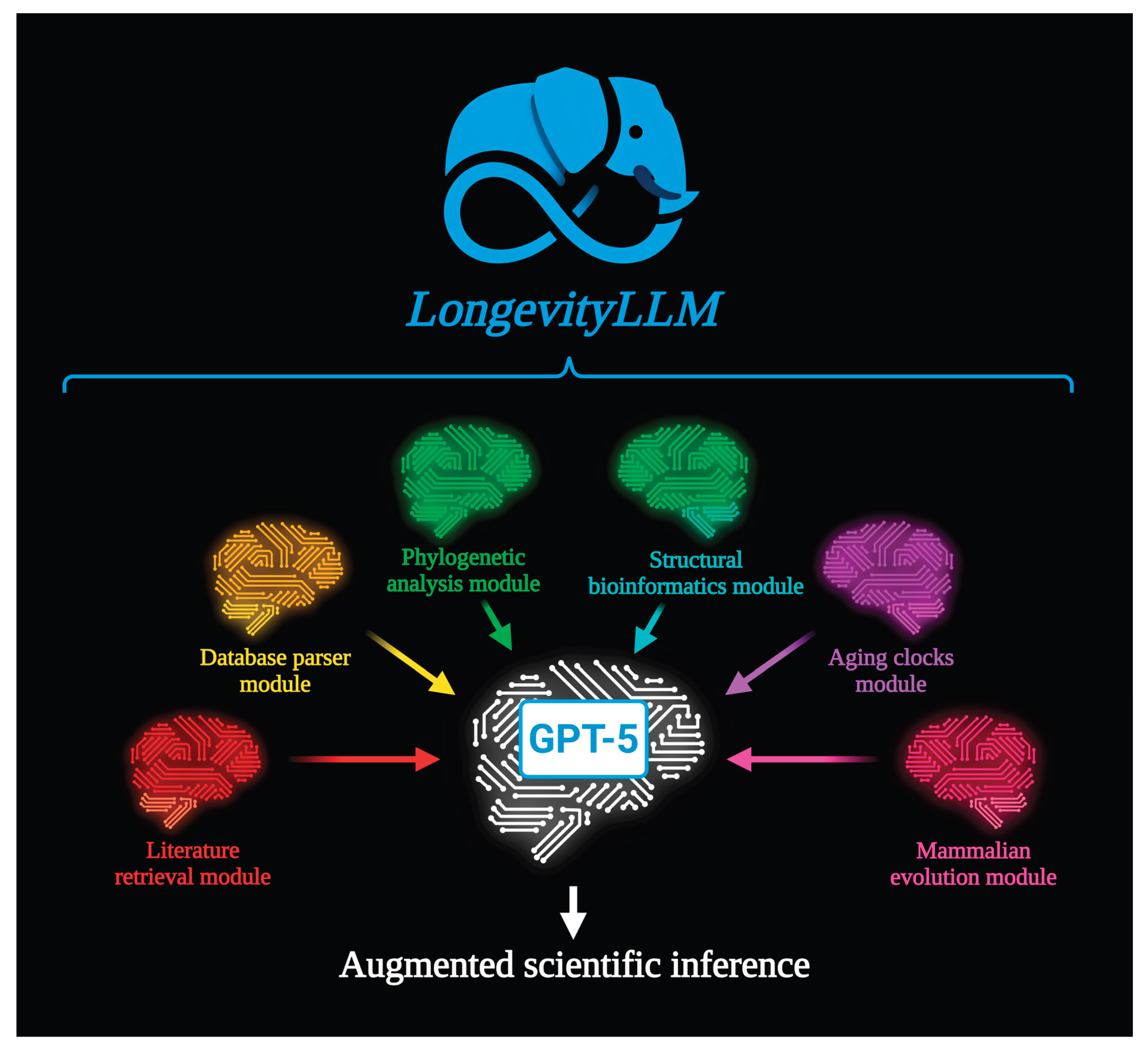
LongevityLLM is built around a large language model (GPT-5), which serves as the user-facing interface and orchestration layer [2]. Rather than generating answers from internal knowledge alone, GPT-5 interprets natural language queries and selectively invokes a set of pre-defined, deterministic functions. These functions are organized into six logical modules: (1) Literature retrieval – fetches and summarizes relevant publications from Europe PMC; (2) Database parser – retrieves protein annotations, pathways, variants, and expression profiles from UniProt, DrugBank, HPA, and other public resources; (3) Phylogenetic analysis – performs cross-species sequence alignment and builds evolutionary trees; (4) Structural bioinformatics – visualizes protein structure (both experimental and predicted), predicts and compares 3D protein structures for wild-type and mutant sequences; (5) Aging clocks – interrogates five major aging clock models (Horvath, Hannum, PhenoAge, Brunet, Wyss-Coray); (6) Mammalian evolution – integrates life-history traits from AnAge, a calibrated species phylogeny, and the AROCM epigenetic metric.
Each module executes its pipeline independently, saves all intermediate and final outputs as structured files (FASTA, PDB, XLSX, PNG, TXT, etc.), and returns a summary to GPT-5. The model then synthesizes a concise, evidence-grounded response based solely on these generated artifacts. Crucially, all output files are preserved and made available to the user, ensuring full transparency, reproducibility, and the ability to inspect or reuse results downstream. This tight coupling of language understanding with executable bioinformatics constitutes the core of LongevityLLM.
Figure 1.
Architecture of LongevityLLM. A GPT-5-based interface interprets user queries and dispatches tasks to six specialized, function-driven modules: literature retrieval, database parser, phylogenetic analysis, structural bioinformatics, aging clocks, and mammalian evolution. Each module produces structured, downloadable outputs that are both interpreted by GPT-5 for response generation and made directly accessible to the user. This design ensures auditability, reproducibility, and seamless integration of multi-domain biological knowledge.
Figure 1.
Architecture of LongevityLLM. A GPT-5-based interface interprets user queries and dispatches tasks to six specialized, function-driven modules: literature retrieval, database parser, phylogenetic analysis, structural bioinformatics, aging clocks, and mammalian evolution. Each module produces structured, downloadable outputs that are both interpreted by GPT-5 for response generation and made directly accessible to the user. This design ensures auditability, reproducibility, and seamless integration of multi-domain biological knowledge.
Literature Retrieval Module
The Literature Retrieval Module provides a targeted, aging-aware interface to the biomedical literature via the Europe PMC REST API. Given a user query—typically a gene symbol—the module first expands it into a set of context-aware Boolean expressions by combining the input with a curated list of aging-related terms (e.g., aging, longevity, senescence) and, where applicable, gene-specific ortholog aliases (e.g., mapping APOE to E2/E3/E4). This expansion ensures broader recall of biologically relevant publications while maintaining semantic precision.
The system then retrieves abstracts from the past five years (2021–2025 by default), processing each year independently to preserve temporal balance. For each year, it fetches up to a user-defined number of top-relevance results per expanded query, deduplicates across queries, and saves every retrieved paper as a plain-text file containing metadata (title, authors, journal, PMID/DOI, direct link), abstract, and a short list of heuristic key findings.
These summaries are generated automatically by scanning the abstract for sentences that mention aging-related concepts and contain outcome-oriented verbs (e.g., increase, decrease, extend, protect). Sentences are scored (higher weight for keyword + verb co-occurrence), sorted by relevance, and the top 4 are included as a concise, interpretable summary of the paper’s aging-related conclusions.
All results are aggregated into three global artifacts: a machine-readable table (results.csv), a JSONL stream for programmatic consumption, and a human-readable SUMMARY.txt that lists, year by year, the retrieved papers along with their key findings and links. This design ensures both rapid triage for the user and full reproducibility: every claim in the final response is traceable to an actual abstract, and all intermediate data remain accessible for downstream analysis or manual verification.
Database Parser Module
The Database Parser Module provides unified access to key biological databases through a single, gene-centric interface. Given a user query (e.g., a gene symbol, alias, or Entrez ID), the module first normalizes the input to a canonical human gene symbol using a precomputed alias map derived from HGNC, Ensembl, and NCBI Gene. This ensures robust handling of synonyms, historical identifiers, and common naming variations.
Once normalized, the system retrieves structured annotations from multiple authoritative sources: (1) UniProt: canonical protein name, reviewed (Swiss-Prot) FASTA sequence, functional description, Gene Ontology (GO) terms, Reactome pathways, DrugBank interactions, and domain architecture [3,4,5,6]. Sequence is saved in FASTA format, features in XLSX, other reports are saved in the TXT format; (2) Human Protein Atlas (HPA): tissue- and single-cell-type RNA expression profiles, prognostic summaries in cancer, and subcellular localization predictions [7], the output is saved in TXT format; (3) Protein Variants: human missense, nonsense, and splice variants from UniProt’s variation API, including PolyPhen/SIFT pathogenicity scores, disease associations, genomic coordinates, and somatic status [3] (exported as a detailed Excel report).
All outputs are saved as human-readable files (FASTA, TXT, XLSX) with consistent naming, enabling both immediate interpretation by the LLM and direct inspection or reuse by the user. This module eliminates the need to navigate disparate database interfaces or write custom parsers—turning complex, multi-source annotation into a single, reproducible step.
Phylogenetic Analysis Module
The Phylogenetic Analysis Module enables comparative evolutionary studies of protein sequences across species. Given a gene symbol and a list of organisms (specified by common or Latin names), the module first resolves each species to its NCBI taxonomic identifier using the UniProt taxonomy API, including subspecies variants where relevant. It then retrieves the corresponding protein sequences from UniProt, prioritizing reviewed (Swiss-Prot) entries and falling back to unreviewed records only when necessary. Each sequence is saved as a standalone FASTA file with a standardized, filesystem-safe name.
For two sequences, the module performs a global pairwise alignment using the Needleman–Wunsch algorithm with BLOSUM62 scoring and affine gap penalties, producing a human-readable alignment report. For three or more sequences, it constructs a multiple sequence alignment via MAFFT and builds a phylogenetic tree using either neighbor-joining (based on BLOSUM62 distances) or maximum likelihood (via IQ-TREE with automatic model selection and 1000 ultrafast bootstrap replicates) [8,9,10,11,12,13,14]. All trees are rendered as publication-ready PNG images with simplified, readable tip labels (e.g., Homo sapiens instead of full database identifiers) and saved in both Newick and graphical formats.
This module is designed to be used in conjunction with the Mammalian Evolution Module. By overlaying phylogenetic relationships with species-specific life-history traits—such as maximum lifespan, metabolic rate, or the AROCM (Average Rate of Change in Methylation) metric—researchers can explore whether specific protein features (e.g., conserved domains, lineage-specific substitutions, or structural motifs) correlate with longevity or other aging-related phenotypes. Such integrative analyses support hypothesis generation about the molecular evolution of aging and the functional relevance of sequence variation across the mammalian tree of life.
Structural Bioinformatics Module
The Structural Bioinformatics Module enables rapid exploration of protein structure—both experimentally determined and computationally predicted—in the context of aging-related genes and mutations. Given a gene symbol, the module first normalizes the query to a canonical human gene and retrieves its UniProt identifier. It then downloads all available experimental 3D structures from the RCSB Protein Data Bank (PDB) that are cross-referenced in UniProt, saving each as a separate PDB file in a dedicated folder (e.g., SPP1_pdb/).
For cases where no experimental structure exists—or when assessing the impact of non-synonymous variants—the module leverages AlphaFold2 (via ColabFold) to generate high-confidence structural models of both wild-type and mutant sequences [15,16]. Mutations are applied programmatically using a flexible syntax that supports substitutions, insertions, and deletions, and the resulting sequences are folded independently.
In parallel with structural modeling, the module computes physicochemical properties directly from the input FASTA sequences, including the isoelectric point (pI) and GRAVY (Grand Average of Hydropathy) score, which serve as proxies for protein solubility and hydrophobicity. These metrics are compared between wild-type and mutant variants and reported in a plain-text summary, offering a first-pass assessment of potential biophysical consequences.
All structures—experimental or predicted—are visualized through an interactive, publication-ready viewer based on py3Dmol that supports multiple coloring schemes: (1) hydrophobicity (Kyte–Doolittle scale), (2) electrostatic charge (acidic vs. basic residues), (3) confidence scores (lDDT from AlphaFold’s B-factor column), or (4) simple monochrome rendering [17].
Users can toggle side chains, molecular surfaces (van der Waals or solvent-accessible), and chain selection, enabling detailed inspection of mutation-induced changes in surface properties, charge distribution, or local folding confidence.
While the module does not perform physics-based stability calculations (e.g., FoldX or Rosetta [18,19]), it includes a lightweight heuristic to compare wild-type and mutant models based on hydrophobic core formation and burial of charged residues. Structural alignment (via Bio.PDB) provides RMSD estimates and superimposed PDB files for direct visual comparison.
It is important to emphasize that all structural predictions and biophysical estimates generated by this module are hypothesis-generating tools, not substitutes for experimental validation. AlphaFold2 models, while highly accurate for many monomeric proteins, may be less reliable for intrinsically disordered regions, multimeric complexes (unless explicitly modeled as such), or conformational states induced by ligands, post-translational modifications, or cellular context. Similarly, GRAVY and pI comparisons offer only coarse-grained insights and do not capture folding kinetics, aggregation propensity, or in vivo behavior.
This module is designed to lower the barrier to structural interpretation: rather than requiring manual PDB searches, model building, or visualization scripting, users can obtain a complete structural assessment—from sequence to 3D insight—through a single natural language request. All intermediate files (FASTA, PDB, alignment reports, stability estimates) are saved for inspection, ensuring full reproducibility and downstream reuse.
Aging Clocks Module
The Aging Clocks Module provides gene-centric interrogation of five major epigenetic and transcriptomic aging models: Horvath, Hannum, PhenoAge, Brunet, and Wyss-Coray. The description for each clock is given below.
Horvath – the first multi-tissue epigenetic clock, developed in 2013, estimates biological age using DNA methylation levels at 353 CpG sites. Trained on ~8,000 samples from 51 healthy human tissues and cell types, it accurately predicts chronological age across most somatic tissues (median absolute error ≈ 3.6 years) without requiring tissue-specific calibration. The model was trained using elastic net regression (a combination of L1 and L2 regularization [20]), which selected 353 CpG sites from a large initial set while balancing sparsity and predictive accuracy. The clock yields near-zero age estimates for embryonic and induced pluripotent stem cells, and it is also applicable to chimpanzee tissues [21]. Horvath age even today is one of the most accurate predictors of chronological age, but is outperformed by second-generation clocks in predicting biological age and clinical outcomes.
The Hannum clock is a first-generation epigenetic aging model based on 71 CpGs from whole blood. Hannum et al. also built tissue-specific methylation clocks for breast (50 CpGs), kidney (62 CpGs), and lung (14 CpGs), plus a blood transcriptomic model using 54 age-associated genes. All Hannum methylation clocks were trained using elastic net regression, enabling robust feature selection from high-dimensional methylation data. Notably, ELOVL2 emerged as a top age-predictive gene shared across multiple tissues [22]. These five models—whole blood, breast, kidney, lung (methylation), and blood transcriptome—are all integrated into the LongevityLLM’s Aging Clocks Module.
PhenoAge – second-generation epigenetic aging model developed in 2018 that estimates biological age using DNA methylation at 513 CpG sites. Unlike first-generation clocks trained on chronological age, PhenoAge was trained to predict “phenotypic age”—a composite of clinical biomarkers (e.g., albumin, creatinine, glucose, CRP) and chronological age—better reflecting morbidity, mortality, and healthspan. Trained on whole blood from the InCHIANTI cohort (n=456), it robustly predicts all-cause mortality, cancer, Alzheimer’s disease, and physical decline across multiple cohorts. DNAm PhenoAge was developed using elastic net regression to select 513 CpG sites predictive of phenotypic age from whole-blood methylation profiles. Although blood-derived, it correlates with age in diverse tissues (brain, liver, lung, etc.) and outperforms Horvath and Hannum clocks in clinical outcome prediction by ~1.5-fold [23]. Among second-generation epigenetic clocks, PhenoAge remains one of the most robust predictors of multimorbidity, functional decline, and mortality, and in recent benchmarking its updated version (PhenoAgeV2 [24]) achieved the highest cumulative performance across diverse aging-accelerating conditions [25].
Brunet: the first single-cell RNA-seq–based transcriptomic aging framework, developed using 21,458 cells from the subventricular zone (SVZ) of 28 mice aged 3.3 to 29 months. It includes two complementary models: Chronological Bootstrap (first-generation), trained to predict chronological age, and Biological Bootstrap (second-generation), trained on SVZ proliferative capacity—a functional proxy for biological age. Both models were built separately for six neural cell types: (1) oligodendrocytes, (2) microglia, (3) endothelial cells, (4) astrocytes & quiescent neural stem cells (qNSCs), (5) activated NSCs & neural progenitor cells (aNSC-NPCs), and (6) neuroblasts. Both Chronological and Biological Bootstrap models were built using LASSO regression (L1 regularization [26]) applied separately to each neural cell type to identify sparse, cell-type-specific gene signatures of aging. Among the top age-predictive genes, IFI27—an interferon-inducible gene—was selected by clocks in five of the six cell types [27].
Wyss-Coray – a second-generation plasma proteomic aging model developed in 2023 that estimates organ-specific biological age using levels of organ-enriched plasma proteins. Trained on 4,778 proteins quantified by the SomaScan aptamer-based platform, it models aging in 11 major organs: adipose tissue, artery, brain, heart, immune tissue, intestine, kidney, liver, lung, muscle, and pancreas. Each protein is represented by one or more aptamer-detected epitopes, which may be unique to a single protein or shared across isoforms or related proteins. The framework includes both baseline (“organ age”) models and cognition-optimized variants (e.g., CognitionBrain, CognitionArtery), refined via the FIBA algorithm to prioritize proteins whose plasma levels best associate with cognitive decline and Alzheimer’s disease pathology. In parallel, the study also introduced an organismal aging model (based on organ-nonspecific proteins) and a conventional proteomic clock (using all 4,778 proteins), serving as global references for shared and composite aging signals. The organ-specific and cognition-optimized models were trained using a bagged ensemble of LASSO regressors, enhancing stability and reducing overfitting in proteomic feature selection. Although trained on chronological age, these models robustly predict organ-specific morbidity, multimorbidity patterns, and mortality, with the CognitionBrain clock performing comparably to pTau-181 in forecasting dementia progression [28].
For a given gene symbol (or alias), the system first normalizes the input to a canonical human gene symbol using the same alias map as other modules. It then queries preprocessed, model-specific coefficient tables to identify all associated CpG sites (for DNA methylation clocks), gene expression features (for transcription-based clocks), or plasma protein epitopes (for proteomic clocks).
For each association, the module computes: (1) the effect direction (accelerating or decelerating aging), (2) the absolute and signed coefficient value, and two levels of ranking: (1) among all features in the model (by absolute coefficient magnitude), (2) within the subset of features that share the same effect direction.
All results are compiled into human-readable reports. Horvath, PhenoAge, Hannum, and Brunet outputs are saved as plain-text files with structured, interpretable summaries. Wyss-Coray results are rendered as a publication-ready PDF that includes coefficient rankings, shared epitope status, and age- and sex-dependent protein trajectory plots.
The module is designed to support cross-clock comparison: by running all five functions in parallel, users can assess whether a gene consistently accelerates or decelerates biological age across tissues, cell types, and molecular modalities. All intermediate data files are preserved, ensuring full traceability from query to conclusion.
Mammalian Evolution Module
The Mammalian Evolution Module integrates curated life-history data from the AnAge database—a widely used, expert-curated compendium of aging and life-history traits across vertebrates [29] with the the Average Rate of Change in Methylation (AROCM)—a cross-species epigenetic biomarker of aging derived from 552 CpG sites in the BivProm2+ chromatin state—a bivalent promoter region bound by Polycomb Repressive Complex 2 (PRC2); in this context, AROCM exhibits a robust inverse correlation with species maximum lifespan (AROCM ∝ 1/Lifespan), making it a cross-species epigenetic biomarker of biological aging that is conserved across mammals [30]. Given a species query (e.g., Loxodonta africana), the module first resolves the input to a standardized binomial name and maps it onto a time-calibrated phylogeny of 5,881 mammalian species.
This phylogeny is drawn from the “completed” node-dated set of Upham, Esselstyn & Jetz (2019)—specifically, a single representative tree sampled from their credible set of 10,000 Bayesian posterior trees. That study employed a “backbone-and-patch” approach that deliberately avoids enforcing a single consensus topology; instead, it explicitly preserves phylogenetic uncertainty in contentious regions (e.g., the root of Placentalia) and eliminates branch-length artifacts common in older supertree methods [31]. While the full uncertainty is best represented by the entire distribution of 10,000 trees, our implementation uses one fixed tree to enable concrete, reproducible inference and user-friendly interpretation. This single tree remains fully credible—it adheres to the same modeling framework and fossil calibrations as the full set—and provides a robust scaffold for comparative analyses of mammalian life-history and aging traits.
If exact life-history or AROCM data are unavailable for the queried species, the system automatically identifies the closest phylogenetic relative with available measurements and explicitly reports this substitution, along with the evolutionary distance.
The resulting report includes: (1) phylogenetic classification (order, family, common name), (2) lifespan and reproductive traits (maximum longevity, gestation, litter size, age at maturity), (3) physiological metadata (body mass, metabolic rate, growth rate, temperature), and (4) the AROCM value, which quantifies the rate of epigenetic drift and is inversely correlated with species lifespan.
Additionally, the module can visualize custom subtrees (e.g., a user-defined list of species or an entire taxonomic clade such as FELIDAE) while preserving the original tree topology and replacing internal labels with human-readable binomial names.
All outputs are saved as plain-text reports and publication-ready PNG figures, enabling direct comparison of aging-related traits across evolutionary lineages and supporting hypothesis generation about the molecular and ecological determinants of longevity.
Discussion and Future Plans
LongevityLLM is a proof-of-concept prototype, not a production-ready platform. Its current implementation demonstrates the feasibility of coupling a large language model with deterministic, domain-specific bioinformatics pipelines—but it remains limited in scope, depth, and robustness. The system should be viewed as an early step toward a broader vision: an open, reproducible, and user-friendly AI layer for geroscience.
Several key limitations define the current state of the system. First, database coverage is narrow: while core resources like UniProt, HPA, and AnAge are integrated, many other critical repositories (e.g., GTEx, ARCHS4, DisGeNET, ClinVar, or structural variant databases) remain absent [32,33,34,35]. Expanding this coverage would significantly improve annotation completeness and biological context.
Second, the phylogenetic module currently supports only basic ortholog retrieval and tree inference. Future work should incorporate synteny-aware orthology detection, branch-length-aware trait mapping, and integration with comparative genomics frameworks (e.g., CAFE for gene family evolution).
Third, structural predictions rely on AlphaFold2 via ColabFold, which—while powerful—lacks the ligand- and complex-aware capabilities of AlphaFold3 (whose open implementation is not yet available) [36]. Moreover, the current stability heuristic is intentionally simplistic; replacing it with physics-informed or deep learning–based predictors (e.g., ThermoMPNN, PROSTATA, or ESMFold-based ΔΔG estimators) would greatly enhance mutation impact assessment [37,38,39].
Regarding aging clocks, LongevityLLM currently supports five well-established models (Horvath, Hannum, PhenoAge, Brunet, Wyss-Coray). However, several important clocks—such as GrimAge [40], Zhang [41], and especially DamAge/AdaptAge—are not yet included. The latter pair is particularly compelling, as it attempts to disentangle detrimental (“damage”) from adaptive epigenetic changes with age [42]. Unfortunately, these models are not yet publicly annotated at the CpG-gene level, which prevents automated gene-level interpretation.
Looking ahead, our long-term vision is to automate the full spectrum of modern omics analysis—from raw single-cell RNA-seq or ATAC-seq data to integrated multi-omics aging signatures—within the same natural-language-driven framework. This includes cell-type deconvolution and trajectory inference, gene regulatory network reconstruction (e.g., via hdWGCNA [43] or LINGER [44]), spatial transcriptomics integration, ChIP-seq, ATAC-seq, Hi-C, lipidomics, proteomics, protein–ligand docking and binding affinity prediction, and cross-species meta-analysis of aging interventions.
Ultimately, we aim to build not just a tool for longevity research, but a general-purpose AI assistant for computational biology—one that lowers the barrier to rigorous, reproducible science for students, wet-lab biologists, and bioinformaticians alike. LongevityLLM is merely the first scaffold of that future system.
Data and Code Availability
All code, documentation, and example notebooks for LongevityLLM are publicly available under an open-source license:
- GitHub repository: https://github.com/davidzheglov/longevity-knowledge-base;
- Interactive Colab notebook (with all functions preloaded): https://colab.research.google.com/drive/1Pxlebzb4bFAEs7FMZur0xvnzAq0Abxw6#scrollTo=7ngdOSklAg7H;
- Live demo website: http://167.99.194.255:8080/chat.
Note: Not all functions work properly in the current version of the website.
The backend relies exclusively on publicly available data sources, such as Europe PMC, UniProt, Human Protein Atlas, RCSB PDB, AnAge and other standard biological databases.
All preprocessed aging clocks model coefficients (Horvath, Hannum, PhenoAge, Brunet, Wyss-Coray), AROCM values, and mammalian reference evolutionary tree are derived from the Supplementary files of the original publications (see References). Users may replace or extend these files to incorporate updated clock models.
Author Contributions
Maxim A. Kovalev conceived the LongevityLLM project, designed the overall architecture, implemented the Database Parser, Phylogenetic Analysis, Structural Bioinformatics, Aging Clocks, and Mammalian Evolution modules, and wrote the majority of the manuscript.
Ekaterina Leskina developed the design and implementation of the LongevityLLM web interface.
David Zheglov implemented the Literature Retrieval module and developed the frontend implementation of the web interface.
Dmitry Galatenko and Timofey Fedoseev developed and fine-tuned the large language model integration layer, including function-calling orchestration and response synthesis.
All authors reviewed and approved the final manuscript.
References
- Kamya, P.; Ozerov, I.V.; Pun, F.W.; Tretina, K.; Fokina, T.; Chen, S.; Naumov, V.; Long, X.; Lin, S.; Korzinkin, M.; et al. PandaOmics: An AI-Driven Platform for Therapeutic Target and Biomarker Discovery. J. Chem. Inf. Model. 2024, 64, 3961–3969. [Google Scholar] [CrossRef]
- OpenAI. GPT-5 System Card. Available online: https://cdn.openai.com/gpt-5-system-card.pdf (accessed on 13 August 2025).
- UniProt Consortium. UniProt: The universal protein knowledgebase in 2025. Nucleic Acids Res. 2025, 53, D609–D617. [Google Scholar] [CrossRef]
- Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; Hill, D.P. The gene ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [CrossRef]
- Milacic, M.; Beavers, D.; Conley, P.; Gong, C.; Gillespie, M.; Griss, J.; Haw, R.; Jassal, B.; Matthews, L.; May, B.; et al. The Reactome Pathway Knowledgebase 2024. Nucleic Acids Res. 2023, 52, D672–D678. [Google Scholar] [CrossRef] [PubMed]
- Knox, C.; Wilson, M.; Klinger, C.M.; Franklin, M.; Oler, E.; Wilson, A.; Pon, A.; Cox, J.; Chin, N.E.; Strawbridge, S.A.; et al. DrugBank 6.0: The DrugBank Knowledgebase for 2024. Nucleic Acids Res. 2023, 52, D1265–D1275. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 1992, 89, 10915–10919. [Google Scholar] [CrossRef]
- Kazutaka, K.; Misakwa, K.; Kei-ichi, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing evolutionary trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Rego, N.; Koes, D. 3Dmol.js: Molecular visualization with WebGL. Bioinformatics 2014, 31, 1322–1324. [Google Scholar] [CrossRef]
- Delgado, J.; Radusky, L.G.; Cianferoni, D.; Serrano, L. FoldX 5.0: Working with RNA, small molecules and a new graphical interface. Bioinformatics 2019, 35, 4168–4169. [Google Scholar] [CrossRef]
- Thieker, D.F.; Maguire, J.B.; Kudlacek, S.T.; Leaver-Fay, A.; Lyskov, S.; Kuhlman, B. Stabilizing proteins, simplified: A Rosetta-based webtool for predicting favorable mutations. Protein Sci. 2022, 31, e4428. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Stat. Methodol. Ser. B 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2015, 14, R115. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Higgins-Chen, A.T.; Thrush, K.L.; Wang, Y.; Minteer, C.J.; Kuo, P.-L.; Wang, M.; Niimi, P.; Sturm, G.; Lin, J.; Moore, A.Z.; et al. A computational solution for bolstering reliability of epigenetic clocks: Implications for clinical trials and longitudinal tracking. Nat. Aging 2022, 2, 644–661. [Google Scholar] [CrossRef]
- Kriukov, D.; Efimov, E.; Kuzmina, E.; Dudkovskaia, A.; Khrameeva, E.E.; Dylov, D.V. ComputAgeBench: Epigenetic Aging Clocks Benchmark. In Proceedings of the 31st ACM SIGKDD Conference on Knowledge Discovery and Data Mining V. 2; ACM: New York, NY, USA, 2025; pp. 5560–5570. [Google Scholar]
- Tibshirani, R. Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. Ser. B Stat. Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Buckley, M.T.; Sun, E.D.; George, B.M.; Liu, L.; Schaum, N.; Xu, L.; Reyes, J.M.; Goodell, M.A.; Weissman, I.L.; Wyss-Coray, T.; et al. Cell-type-specific aging clocks to quantify aging and rejuvenation in neurogenic regions of the brain. Nat. Aging 2022, 3, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.S.-H.; Rutledge, J.; Nachun, D.; Pálovics, R.; Abiose, O.; Moran-Losada, P.; Channappa, D.; Urey, D.Y.; Kim, K.; Sung, Y.J.; et al. Organ aging signatures in the plasma proteome track health and disease. Nature 2023, 624, 164–172. [Google Scholar] [CrossRef] [PubMed]
- De Magalhães, J.P.; Costa, J. A database of vertebrate longevity records and their relation to other life-history traits. J. Evol. Biol. 2009, 22, 1770–1774. [Google Scholar] [CrossRef]
- Horvath, S.; Zhang, J.; Haghani, A.; Lu, A.T.; Fei, Z. Fundamental equations linking methylation dynamics to maximum lifespan in mammals. Nat. Commun. 2024, 15, 1–17. [Google Scholar] [CrossRef]
- Upham, N.S.; Esselstyn, J.A.; Jetz, W. Inferring the mammal tree: Species-level sets of phylogenies for questions in ecology, evolution, and conservation. PLOS Biol. 2019, 17, e3000494. [Google Scholar] [CrossRef]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lachmann, A.; Torre, D.; Keenan, A.B.; Jagodnik, K.M.; Lee, H.J.; Wang, L.; Silverstein, M.C.; Ma’ayan, A. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun. 2018, 9, 1366. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; I Furlong, L. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Dieckhaus, H.; Brocidiacono, M.; Randolph, N.Z.; Kuhlman, B. Transfer learning to leverage larger datasets for improved prediction of protein stability changes. Proc. Natl. Acad. Sci. 2024, 121. [Google Scholar] [CrossRef]
- Umerenkov, D.; Nikolaev, F.; Shashkova, T.I.; Strashnov, P.V.; Sindeeva, M.; Shevtsov, A.; Ivanisenko, N.V.; Kardymon, O.L. PROSTATA: A framework for protein stability assessment using transformers. Bioinformatics 2023, 39. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Zhang, Q.; Vallerga, C.L.; Walker, R.M.; Lin, T.; Henders, A.K.; Montgomery, G.W.; He, J.; Fan, D.; Fowdar, J.; Kennedy, M.; et al. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 2019, 11, 1–11. [Google Scholar] [CrossRef]
- Ying, K.; Liu, H.; Tarkhov, A.E.; Sadler, M.C.; Lu, A.T.; Moqri, M.; Horvath, S.; Kutalik, Z.; Shen, X.; Gladyshev, V.N. Causality-enriched epigenetic age uncouples damage and adaptation. Nat. Aging 2024, 4, 231–246. [Google Scholar] [CrossRef]
- Morabito, S.; Reese, F.; Rahimzadeh, N.; Miyoshi, E.; Swarup, V. hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data. Cell Rep. Methods 2023, 3, 100498. [Google Scholar] [CrossRef]
- Yuan, Q.; Duren, Z. Inferring gene regulatory networks from single-cell multiome data using atlas-scale external data. Nat. Biotechnol. 2024, 43, 247–257. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



