Submitted:
19 November 2025
Posted:
20 November 2025
You are already at the latest version
Abstract
The rapid adaptation of microorganisms to antibiotics, including those previously re-garded as last-resort choices, led to intractable superbugs among both Gram-positive and Gram-negative species, causing increasing antibiotic resistance (AR). AR triggers the worldwide propagation of tenacious to lethal infections, which need urgent development of novel antibacterial agents, active also on worrying superbugs. To this end, totally inac-tive betulin (BET) and betulinic acid (BA), as well as ursolic acid (UA) already active on bacteria Gram-positive, have been chemically modified achieving derivatives 1-7. Com-pounds 1 and 4-7 contained the triphenyl phosphonium (TPP) group, promoting antibac-terial effects, while 2 and 3 did not. 1-7 and all synthetic intermediates were characterized by, chemometric-assisted FTIR and NMR spectroscopy, as well as by other analytical techniques, which confirmed their structure and high purity. Determinations of the min-imum inhibitory concentration values (MICs) of 1-7, BET, BA and UA using a selection of Gram-positive and Gram-negative clinically isolated superbugs, evidenced that com-pounds 4-7 had potent antibacterial effects against Gram-positive strains, higher than those reported so far for other BET, BA and UA derivatives, mainly considering the com-plex pattern of resistance of isolates used here and their clinical source. For the first time, due to the use of TPP, a real activity (MICs 2-16 µg/mL) was conferred to inactive BET and BA (original MICs > 1024 and 256 µg/mL). Moreover, the antibacterial effects of UA were 16 and 32-fold improved against MRSE and MRSA (MICs = 2 vs. 23 and 64 μg/mL). These early, but very promising microbiologic results, pave the way for further experiments with the best performant compounds 5 and 7 (MICs = 2 μg/mL) on an enlarged number of Gram-positive isolates, to assess their time-killing curves, to evaluate their cytotoxicity on eukaryotic cells and to assess their possible antibiofilm activity.
Keywords:
multidrug resistant bacteria (superbugs)
; natural triterpenoids
; triphenyl phosphonium (TPP) group
; minimum inhibitory concentration (MIC)
; strong antibacterial effects
1. Introduction
A superbug is usually defined as a multidrug resistant microorganism, which have become resistant to multiple antibiotics [1]. The number of different antibiotics, to which it can be resistant, establishes the degree of its being a superbug [1]. The superbug of all superbugs is a bacterium which has developed resistance to all available antibiotics, causing increased morbidity, mortality rate and economic loss [1]. Major superbugs develop in hospital settings and include methicillin-resistant Staphylococcus aureus (MRSA), multidrug-resistant Gram-negative bacilli including Pseudomonas aeruginosa, carbapenem-resistant Enterobacteriaceae, ESBL-producing Enterobacteriaceae, vancomycin-resistant enterococci (VRE), E. coli Hx30 and others [1].
Major risk factors for the development of antibiotic-resistant superbugs consists in over prescription and misuse of antibiotics, their use without medical indication, poor drug quality, genetic mutation among micro-organisms, patient not completing the entire antibiotic dosage or not strictly following the correct antibiotic regimen, long term hospitalization, prophylactic antibiotic therapy and poor hygiene and sanitation [2].
According to a report by Mancuso et al. on Pathogens in 2021, antibiotic-resistant bacteria caused 700,000 deaths worldwide each year [3]. The World Health Organization (WHO) predicted that without new and better treatments, this number could rise to 10 million by 2050, highlighting a health concern of paramount importance [3].
Recently, the increasing development of antibiotic resistance has led some Gram-negative bacteria to become tolerant also to last-resort antibiotics, used in the treatment of non-fermentative species in critically ill patients [4]. In this regard, drug resistance has been reported versus colistin and the recently approved cefiderocol (FDC) [4]. Particularly, colistin is an older polycationic antibiotic, traditionally employed in the management of infections sustained by Enterobacteriaceae that have developed resistance practically to all other antibiotics [5]. Since 2016, many Gram-negative bacteria have been shown to possess genes that also confer resistance to colistin, thus further reducing the available weapons to treat the infections they cause [5] Furthermore, FDC is a strategically catechol-substituted siderophore cephalosporin, capable to kill Gram-negative bacteria [6]. Nevertheless, cases of in vivo emerging FDC resistance are increasingly being reported [7]. Moreover, some Gram-negative superbugs show a complex pattern of resistance, including cross resistances to carbapenems, colistin, the combination ceftazidime-avibactam (Zavicefta) and FDC [8].
Anyway, infections sustained by Gram-positive superbugs are of serious concern, too. Although there are many more drugs available to combat them, much more drug-resistant infections from Gram-positive bacteria exist compared with those by Gram-negative strains [9].
Dr Mark Blaskovich, a senior research chemist at the Institute for Molecular Bioscience (IMB) at the University of Queensland in Australia, described MRSA as the “poster child” of Gram-positive superbugs [10].
Blaskovich points to a report from the Centers for Disease Control (CDC) in the US from 2013, which counted the number of infections and the number of deaths from different types of drug-resistant bacteria [10].
The number of infections from drug-resistant Gram-positive MRSA or strep pneumonia were over a million, versus about 30,000 from Gram negative bacterial infections.
Also, drug-resistant Gram-positive bacteria were by far the biggest killers in the report, too. The number of deaths from resistant bacteria were about five-fold higher for Gram-positive compared to Gram-negative infections [10]. Particularly, VRE enterococcal species are MDR bacteria that, in addition to resistance to vancomycin, have already developed a variety of mechanisms of resistance to several other antibiotics, like aminoglycosides, β-lactams, tetracyclines, and quinolones. Additionally, they can produce β-lactamases and have decreased cellular permeability, thus being the cause of severe hospital-acquired infections [11]. VREs are reported as the main responsible for central line-associated bloodstream infections (CLABSI), catheter-associated urinary tract infection (CAUTI), ventilator-associated pneumonia (VAP), surgical site infections (SSI). VREs are categorized as a “serious threat” by Centers for Disease Control (CDC) and Prevention [12]. Moreover, staphylococci, especially MRSA, are the first cause of nosocomial infections, antibiotic-resistant diseases, central line-associated bacteraemia, and hospital-associated endocarditis in the USA [13,14]. Notably, both MRSA and VRE are reported as the first most common cause of community-acquired endocarditis in North America [15]. Very common in hospitals, prisons, and nursing homes, where immunocompromised patients and people with open wounds and/or invasive devices such as catheters are at greater risk of hospital-acquired infections, MRSA represents a global health threat and a clear ‘One Health’ problem. Moreover, MRSA can spread between and impact the environment, animals, and several human sectors [16]. Concerning the current available armamentarium to counteract these superbugs, vancomycin is successful only in about 49% of cases of MRSA infection. [8]. Its use is complicated by its inconvenient route of administration [17] and since the late 1990s, the emergence of several vancomycin and teicoplanin resistant strains of MRSA has been reported [18]. Oxazolidinones such as linezolid (LNZ), available from the 1990s, were initially beneficial in limiting the widespread infections by Gram-positive superbugs, representing one of the last-line therapeutic options for serious infections caused by VRE, MRSE and MDR Enterococcus and Staphylococcus species, but cases of bacteria tolerant to LNZ have been reported since 2001 [19]. According to data from recent large-scale studies, isolates of MDR S. epidermidis genus have demonstrated resistance to LNZ [8]. Additionally, epidemiologic data have shown a 2.5-fold increase in the prevalence of clinical LNZ-resistant enterococci (LRE) over the past decade with a global detection rate of 1.1% for LNZ-resistant E. faecium (LREfm) and 2.2% for LNZ-resistant E. faecalis (LREfs) [20]. Most reported cases have originated from China, followed by South Korea and the United States [20].
For surgical site infections (SSIs) by MRSA [21] and for MRSA colonization in nonsurgical wounds, such as traumatic wounds, burns, and chronic ulcers (i.e., diabetic ulcer, pressure ulcer, arterial insufficiency ulcer, and venous ulcer), no conclusive evidence has been found regarding the best antibiotic regimen to be used[22]. Although infection control and antimicrobial stewardship are important tools for combating the development and spreading of lethal infections, their uncontrolled development will lead to a point where conducting surgeries such as c-sections and transplants will be too dangerous, due to the risk of superbug infection, which would have huge implications for the health of people and economical safety around the world. Collectively, we are living in an era of missing epidemiologic evidence and of a plethora of uncertainties, due to the interindividual responses of patients to existing antibiotics, where the decreasing efficacy of available drugs requires urgently the development of new curative options against difficult-to-treat superbugs.
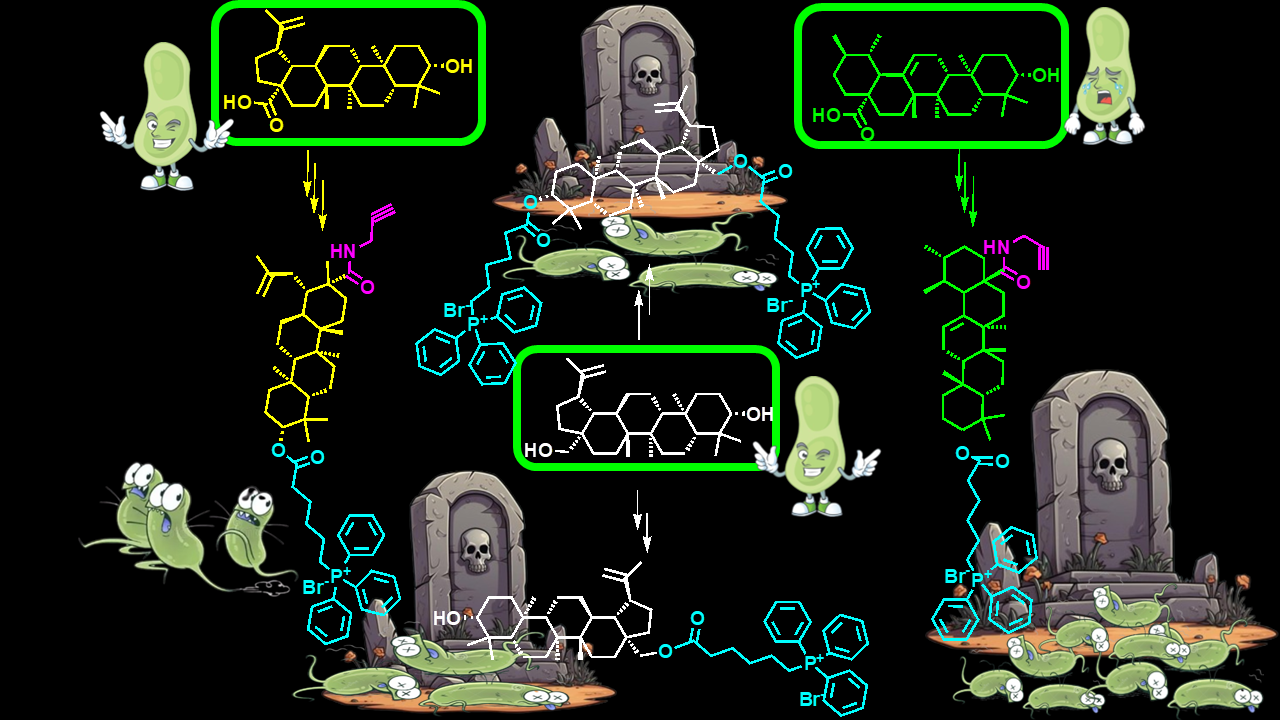
To meet these needs, in this study, seven BET, BA and UA derivatives (1-7) have been synthesized and fully characterized. Compounds 1, 4, 5 and 7 contained 1 triphenyl phosphonium (TPP) group. In the case of compound 5, the TPP-group was inserted as ester in C-28 of pristine BET, while in the other cases it was inserted as ester in C-3 of BET, BA and UA, since position C-28 was functionalized with propargyl amine carbamate (1), and propargyl amide (4, 7) residues. Compound 6 contained 2 TPP group as esters in both C-3 and C-28. Compounds 2 and 3 did not bear the TPP groups, but only a propiolic ester (2) or a propargyl amide (3) residue inserted in C-28 of BET or BA, respectively. Compounds 5 and 6 did not contain the propine derivatives, but only the TPP group (Figure 1).
Compounds 1-7 and all synthetic intermediates were characterized by chemometric-assisted FTIR and NMR spectroscopy, as well as by other analytical techniques. The minimum inhibitory concentration values (MICs) of 1-7, BET, BA and UA were assessed by the agar dilution method according to EUCAST [23], using a selection of Gram-positive and Gram-negative clinically isolated superbugs. Potent antibacterial effects against Gram-positive strains were observed for compounds 4-7 (MICs = 2–16 μg/mL) bearing the TPP group.
2. Materials and Methods
2.1. Chemicals and Instruments
All solvents (Acros Organics, Geele, Belgium) were dried and purified according to standard procedures prior to use. When required, reactions were performed under inert atmosphere (Ar) in pre-flamed glassware. Anhydrous Na2SO4 was used for drying solutions, and the solvents were then routinely removed at ca. 40 °C under reduced pressure (ca 10–20 mmHg), using a rotary evaporator. All reagents employed in the present work were commercially available and used without further purification. Flash column chromatography (FCC) was performed on silica gel (70–230 and 230–400 mesh, Merck, Darmstadt, Germany) and analytical thin layer chromatography (TLC) on silica gel 60-F254 precoated aluminium foils (0.2 mm film, Merck, Germany). Spots on the TLC plates were visualized with UV light at 254 nm and using ninhydrin, p-anisaldehyde or charring solution. Attenuated total reflectance (ATR) Fourier transform infrared (FTIR) analyses were carried out using a Spectrum Two FT-IR Spectrometer (PerkinElmer, Inc., Waltham, MA, USA) as previously reported [24]. 1H NMR spectra were recorded in CDCl3 at 600 MHz and 13C spectra at 151 MHz. on a Bruker AVANCEIII HD spectrometer. Additionally, a Jeol 400 MHz spectrometer (JEOL USA, Inc., Peabody, MA, USA) at 400, 100 and 162 MHz respectively, was used to acquire some 1H, 13C and 31P NMR spectra. Fully Chemical shifts (δ) are indicated in parts per million downfield from TMS and coupling constants (J) are reported in Hz. Gas chromatography-mass spectrometry (GC-MS) spectra were performed on a Varian Saturn 2000 ion trap GC-MS instrument (Artisan Technology Group ®, Champaign, Illinois, USA) equipped with a DB-5MS column (30 m, i.d. 0.25 mm) (Agilent, Santa Clara, CA, USA). ESI mass spectra were recorded at 30 V on amaZon SL ion trap mass spectrometer Bruker Daltonics using MeOH as solvent. Elemental analyses were carried out using an Elemental Analyzer (Fison Instruments Ltd., Farnborough, Hampshire, UK).
2.2. Synthesis of Triterpenoids Derivatives
With the aim at finding novel compounds effectively active against difficult-to-treat clinically isolated superbugs, seven triterpenoid derivatives (1-7) were synthesized via chemical modifications of BET, BA and UA.
2.2.1. Synthesis of Betulin (BET) Derivatives
BET derivatives were synthesized according to Scheme 1a via intermediates 8, 9, 11, 12. Reagents used have been described in the legend available in Scheme 1a caption.
(3β)-Lup-20(29)-ene-3,28-diol, 28-(4-nitrophenyl carbonate) 8. To a stirring solution of BET (200 mg, 0.45 mmol) in dry THF (10 mL), under Ar atmosphere, pyridine (36 μL, 0.45 mmol) was added, and the mixture was cooled to 0 °C. p-nitro-phenyl-chloroformate (94.3 mg, 0.47 mmol) was inserted in 3 portions and the reaction mixture was stirred for 24 h at room temperature (r. t.). Following, the mixture was concentrated under reduced pressure to dryness. The obtained solid residue was diluted in DCM and washed sequentially with 5% aqueous ice-cold citric acid, water, and brine, dried over anhydrous Na2SO4, filtered and concentrated to dryness. The desired product was afforded as a white foam (227 mg, 0.37 mmol, 83% yield) after flash column chromatography (FCC) purification using PhMe:AcOEt 95:5 as eluent. Rf (PhMe:AcOEt 95:5) = 0.14.
1H NMR (600 MHz, CDCl3) δ 8.29 (s, 1 H), 8.28 (s, 1 H), 7.41 (s, 1 H), 7.39 (s, 1 H), 4.71 (br s, 1 H), 4.61 (t, J = 1.7 Hz, 1 H), 4.51 (dd, J = 10.8 Hz, 2.0 Hz, 1 H), 4.08 (d, J = 10.7 Hz, 1 H), 3.19 (dd, J = 11.4, 4.8 Hz, 1 H), 2.45 (td, J = 10.8, 5.4 Hz, 1 H), 2.03 (dtd, J = 14.2, 10.5, 4.9 Hz, 1 H), 1.95–1.84 (m, 2 H), 1.75 (td, J = 13.9, 4.7 Hz, 1 H), 1.70 (s, 3 H), 1.68–1.08 (m, 19 H), 1.05 (s, 3 H), 1.00 (s, 3 H), 0.97 (s, 3 H), 0.91 (td, J = 12.5, 3.7 Hz, 1 H), 0.83 (s, 3 H), 0.76 (s, 3 H), 0.69 (d, J = 11.2 Hz, 1 H);. 13C NMR (151 MHz, CDCl3) δ 155.6, 153.0, 149.7, 145.3, 125.3, 121.8, 110.1, 78.9, 68.3, 55.3, 50.3, 48.8, 47.7, 46.7, 42.7, 40.9, 38.9, 38.7, 37.7, 37.1, 34.4, 34.2, 29.6, 29.5, 28.0, 27.4, 27.0, 25.2, 20.7, 18.3, 16.1, 16.0, 15.3, 14.8.
(3β)-Lup-20(29)-ene-3,28-diol, 28-(N-propargyl carbamate) 9. To an ice-cooled solution of propargylamine (10 μL, 8.6 mg, 0.156 mmol, 1.9 equiv.) in THF (0.42 mL), 8 (50 mg, 0.0823 mmol) and Et3N (0.030 mL, 0.22 mmol, 2.6 equiv.) were portion-wise added and the resulting mixture was left under stirring at 0 °C to r.t. Upon consumption of the starting material, the reaction mixture was concentrated to dryness. Then, the residue was diluted in EtOAc and washed twice with NaHCO3 (aq) 5%, water, K2CO3 (aq) 10%, water and NaCl (sat.). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated to dryness and the pure product was afforded as colourless oil (28 mg, 65% yield) after FCC purification using PhMe:EtOAc 97:3 as eluent. Rf (PhMe:EtOAc 97:3) = 0.12.
1H NMR (CDCl3, 600 MHz) δ 4.95 (br s, 1 H), 4.67 (d, J=2.3 Hz, 1 H), 4.57 (d, J=1.9 Hz, 1 H), 4.27 (d, J=10.8 Hz, 1 H), 3.98 (br s, 2 H), 3.86 (d, J=10.8 Hz, 1 H), 3.18 (dd, J=11.5, 4.7 Hz, 1 H), 2.43 (td, J=11.2, 5.7 Hz, 1 H), 2.24 (t, J=2.4 Hz, 1 H), 1.97 (dq, J=14.0, 10.2 Hz, 1 H), 1.81 (d, J=13.6 Hz, 1 H), 1.76 (m, 1 H), 1.73 (m, 1 H), 1.67 (s, 3 H), 1.64-1.49 (m, 8 H), 1.38 (m, 5 H), 1.25 (dd, J=12.6, 2.3 Hz, 2 H), 1.19 (qd, J=12.5, 4.1 Hz, 2 H), 1.04 (m, 1 H), 1.02 (s, 3 H), 0.96 (s, 6 H), 0.88 (td, J=13.4, 4.4 Hz, 2 H), 0.81 (s, 3 H), 0.75 (s, 3 H), 0.66 (d, J=9.5 Hz, 1 H); 13C NMR (CDCl3, 151 MHz) δ 157.0, 150.5, 129.4, 128.6, 126.5, 116.0, 110.2, 80.1, 79.3, 71.9, 64.0, 55.6, 50.7, 49.1, 48.0, 46.9, 43.0, 41.2, 39.2, 37.9, 37.5, 34.5, 31.2, 29.9, 28.3, 27.7, 27.4, 25.5, 21.1, 19.5, 18.6, 16.4, 15.7, 15.1.
(3β)-Lup-20(29)-Ene-3,28-Diol-3-(6-Triphenyl Phosphonium Hexanoate)-28-(N-propargyl carbamate) 1. To a solution of 9 (28 mg, 0.053 mmol) in DCM (0.180 mL), 6-triphenyl phosphonium hexanoic acid 10 (6-TPPHA), synthesized as described in Scheme 1a (24 mg, 0.052 mmol, 0.99 equiv.) and DMAP (0.64 mg, 0.0053 mmol, 0.1 equiv.) were added and the mixture was cooled to 0 °C. Then, DCC (12 mg, 0.058 mmol, 1.1 equiv.) was portion-wise added and the resulting mixture was stirred at 0 °C to r. t. Upon completion of the reaction, the mixture was filtered under vacuum, and the solid residue was washed twice with EtOAc. The filtrate was concentrated to dryness, and the oily residue was subjected to FCC purification using EtOAc:methanol (MeOH):acetic acid (AcOH) 9:1:0.1 as eluent, to afford the pure product as colourless oil (28 mg, 54% yield). Rf (AcOEt:MeOH:AcOH 9:1:0.1) = 0.13.
ATR-FTIR (ṽ, cm-1): 3060 (=C-H), 2941, 2869 (Csp3-H), 1715 (NHC=OO), 1438 (C-H), 1246, 1112 (C-O), 723, 689 (C-P). 1H NMR (CDCl3, 600 MHz) δ 7.84 (ddd, J=12.6, 8.3, 1.3 Hz, 6 H), 7.77 (td, J=7.4, 1.7 Hz, 3 H), 7.68 (td, J=7.8, 3.4 Hz, 6 H), 4.88 (br s, 1 H), 4.66 (d, J=2.3 Hz, 1 H), 4.56 (t, J=2.0 Hz, 1 H), 4.39 (m, 1 H), 4.27 (d, J=10.8 Hz, 1 H), 3.97 (m, 2 H), 3.84 (ddt, J=21.0, 15.6, 8.4 Hz, 3 H), 2.42 (td, J=10.8, 5.4 Hz, 1 H), 2.24 (m, 3 H), 1.96 (dq, J=14.0, 10.4 Hz, 1 H), 1.80 (m, 1 H), 1.71 (ddd, J=13.7, 9.3, 6.2 Hz, 4 H), 1.66 (s, 3 H), 1.65-1.51 (m, 9 H), 1.46 (m, 1 H), 1.37 (m, 5 H), 1.25(m, 2 H), 1.18 (qd, J=12.6, 4.2 Hz, 2 H), 1.01 (s, 6 H), 0.94 (s, 3 H), 0.88 (m, 1 H), 0.81 (s, 3 H), 0.77 (s, 3 H), 0.76 (s, 3 H), 0.73 (d, J=9.6 Hz, 1 H); 13C NMR (CDCl3, 151 MHz) δ 173.6, 150.0, 135.2, 135.2, 134.0, 133.9, 130.7, 130.7, 118.9, 118.3, 110.0, 81.0, 80.0, 71.8, 55.6, 50.5, 49.0, 47.9, 46.8, 42.9, 41.1, 38.6, 38.0, 37.7, 37.3, 34.7, 34.3, 34.3, 30.0, 29.9, 28.2, 27.3, 25.4, 24.7, 23.9, 23.0, 22.7, 22.7, 22.7, 21.0, 19.3, 18.4, 16.8, 16.4, 16.2, 14.9; 31P NMR (CDCl3, 243 MHz) δ 24.38. GC-MS: m/e: 963.48 (100.0%), 961.48 (84.1%), 962.48 (55.4%), 964.48 (54.4%), 965.48 (18.3%), 964.49 (3.8%), 966.49 (3.8%). ESI-MS (30 eV): m/z 882.75 [(M-Br-)+], 962.54 [M+H+]; Anal. Calcd. for C58H77BrO4P (Mol. Wt.: 963.11): C, 72.33; H, 8.06; Br, 8.30; N, 1.45; O, 6.64; P, 3.22. Found: C, 72.13; H, 8.38; Br, 8.38; O, 6.52; P, 3.43.
(3β)-Lup-20(29)-Ene-3,28-Diol, 28-(2-Propynoate) 2. To an ice-cooled (-10 ° C) suspension of BET (40 mg, 0.09 mmol) and propiolic acid (10 μL, 0.161 mmol, 1.79 equiv.) in DCM (0.45 mL), a solution of DCC (21 mg, 0.102 mmol, 1.13 eq.) and DMAP (1 mg, 0.008 mmol, 0.09 eq.) in DCM (0.2 mL) was portion-wise added and the resulting brown mixture was stirred at -10 °C to r. t. Upon completion of the reaction, the mixture was diluted with cold EtOAc and filtered under vacuum. The filtrate was concentrated to dryness, and the oily residue was subjected to FCC purification using PhMe:EtOAc 97:3 as eluent, to afford the pure product as colourless oil (17 mg, 38% yield). Rf (PhMe:EtOAc 97:3) = 0.18.
ATR-FTIR (ṽ, cm-1): 3284 (OH), 3074 (=C-H), 2941, 2867 (Csp3-H), 2120 (Csp-H ), 1716 (OC=O), 1455 (C-H), 1221 (C-O). 1H NMR (CDCl3, 600 MHz) δ 4.68 (d, J=2.2 Hz, 1 H), 4.58 (t, J=1.8 Hz, 1 H), 4.37 (dd, J=11.1, 2.0 Hz, 1 H), 3.98 (d, J=11.0 Hz, 1 H), 3.17 (dd, J=11.5, 4.7 Hz, 1 H), 2.89 (s, 1 H), 2.42 (td, J=10.7, 5.8 Hz, 1 H), 1.97 (dtd, J=14.1, 10.6, 8.4 Hz, 1 H), 1.88 (ddd, J=13.6, 4.6, 2.6 Hz, 1 H), 1.81 (dd, J=12.7, 8.3 Hz, 1 H), 1.68 (s, 4 H), 1.66-1.58 (m, 5 H), 1.57-1.48 (m, 3 H), 1.44-1.37 (m, 6 H), 1.32-1.24 (m, 2 H), 1.20 (qd, J=12.8, 4.3 Hz, 1 H), 1.13 (ddd, J=12.6, 10.4, 2.1 Hz, 1 H), 1.07 (ddd, J=14.1, 4.4, 2.7 Hz, 1 H), 1.03 (s, 3 H), 0.97 (s, 3 H), 0.96 (s, 3 H), 0.90 (td, J=12.9, 4.2 Hz, 1 H), 0.81 (s, 3 H), 0.75 (s, 3 H), 0.67 (m, 1 H); 13C NMR (CDCl3, 151 MHz) δ 153.5, 150.1, 110.2, 79.2, 75.0, 74.9, 65.1, 55.5, 50.6, 49.0, 47.9, 46.6, 42.9, 41.1, 39.1, 38.9, 37.9, 37.4, 34.7, 34.4, 29.8, 29.7, 28.2, 27.6, 27.2, 25.4, 21.0, 19.3, 18.5, 16.3, 16.2, 15.6, 15.0. GC-MS: m/e: 494.38 (100.0%), 495.38 (37.6%), 496.38 (7.2%). ESI-MS (30 eV): m/z 493.43 [M-H-]; Anal. Calcd. for C33H50O3 (Mol. Wt.: 494.75): C, 80.11; H, 10.19; O, 9.70. Found: C, 79.93; H, 10.02; O, 9.99.
Mixture (3β)-Lup-20(29)-Ene-3,28-Diol, 28-(6-Bromo-Hexanoate) 11 + (3β)-Lup-20(29)-Ene-3,28-Diol, 3,28-di-(6-Bromo-Hexanoate) 12. To an ice-cooled suspension of BET (100 mg, 0.226 mmol) and 6-bromohexanoic acid (133 mg, 0.68 mmol, 3.0 equiv.) in a mixture of DCM (1.15 mL) and THF (0.5 mL), DMAP (3 mg, 0.02 mmol, 0.1 equiv.) and DCC (119 mg, 0.57 mmol, 2.55 eq.) were added and the resulting mixture was stirred at 0 °C to r. t. Upon completion of the reaction, the mixture was diluted with cold EtOAc and filtered under vacuum. The filtrate was concentrated to dryness, and the oily residue was subjected to FCC purification using gradient PhMe:EtOAc 97:3 to 95:5 as eluent, to afford the pure product 12 as colourless oil (55 mg, 31% yield), Rf (PhMe:EtOAc 97:3) = 0.70, and the pure product 11 as colourless oil (80 mg, 57% yield), Rf (PhMe:EtOAc 97:3) = 0.15.
(3β)-Lup-20(29)-Ene-3,28-Diol, 28-(6-Bromo-Hexanoate) 11. 1H NMR (CDCl3, 600 MHz) δ 4.68 (d, J=2.2 Hz, 1 H), 4.58 (m, 1 H), 4.26 (dd, J=11.1, 2.0 Hz, 1 H), 3.84 (dd, J=11.1, 1.4 Hz, 1 H), 3.40 (t, J=6.8 Hz, 2 H), 3.18 (d, J=11.3 Hz, 1 H), 2.44 (td, J=11.2, 5.8 Hz, 1 H), 2.34 (m, 2 H), 1.96 (m, 1 H), 1.88 (m, 2 H), 1.82 (ddd, J=13.4, 4.5, 2.5 Hz, 1 H), 1.75 (ddd, J=12.4, 8.5, 1.3 Hz, 1 H), 1.67 (s, 6 H), 1.65-1.57 (m, 5 H), 1.56-1.43 (m, 5 H), 1.42-1.30 (m, 6 H), 1.26 (qt, J=7.3, 4.0 Hz, 2 H), 1.20 (td, J=12.9, 4.4 Hz, 1 H), 1.13-1.04 (m, 2 H), 1.02 (s, 3 H), 0.97 (s, 3 H), 0.96 (s, 3 H), 0.89 (td, J=13.4, 4.5 Hz, 1 H), 0.81 (s, 3 H), 0.75 (s, 3 H), 0.67 (dd, J=9.1, 2.2 Hz, 1 H); 13C NMR (CDCl3, 151 MHz) δ 174.1, 150.4, 110.1, 79.2, 62.9, 55.5, 50.6, 49.0, 47.9, 46.6, 42.9, 41.1, 39.1, 38.9, 37.8, 37.4, 34.8, 34.4, 34.4, 33.7, 32.6, 30.0, 28.2, 27.9, 27.6, 27.8, 27.3, 24.4, 24.2, 21.6, 21.0, 18.5, 16.3, 16.3, 15.6, 15.0.
(3β)-Lup-20(29)-Ene-3,28-Diol, 3,28-di-(6-Bromo-Hexanoate) 12. 1H NMR (CDCl3, 600 MHz) δ 4.68 (d, J=2.2 Hz, 1 H), 4.59 (t, J=1.9 Hz, 1 H), 4.47 (dd, J=10.6, 5.8 Hz, 1 H), 4.27 (dd, J=11.1, 1.9 Hz, 1 H), 3.84 (d, J=11.0 Hz, 1 H), 3.40 (td, J=6.8, 2.3 Hz, 4 H), 2.44 (td, J=11.2, 5.8 Hz, 1 H), 2.33 (m, 4 H), 1.96 (dtd, J=14.1, 10.6, 8.5 Hz, 1 H), 1.88 (pd, J=6.9, 2.2 Hz, 4 H), 1.82 (ddd, J=13.3, 4.5, 2.5 Hz, 1 H), 1.76 (m, 1 H), 1.68 (s, 5 H), 1.65 (m, 6 H), 1.59 (m, 4 H), 1.49 (tdd, J=9.9, 6.6, 2.2 Hz, 5 H), 1.39 (m, 5 H), 1.28 (m, 2 H), 1.21 (m, 1 H), 1.07 (m, 3 H), 1.03 (s, 3 H), 0.97 (s, 3 H), 0.84 (s, 3 H), 0.83 (s, 6 H), 0.79 (m, 1 H); 13C NMR (CDCl3, 151 MHz) δ 174.1, 173.5, 150.3, 110.1, 81.0, 62.9, 55.6, 50.5, 49.0, 47.9, 46.6, 42.9, 41.1, 38.6, 38.1, 37.8, 37.3, 34.8, 34.4, 34.3, 33.7, 32.6, 32.6, 30.0, 29.8, 28.2, 28.1, 27.9, 27.3, 25.4, 24.5, 24.4, 24.0, 21.0, 19.3, 18.4, 16.8, 16.4, 16.3, 15.0.
(3β)-Lup-20(29)-Ene-3,28-Diol, 28-(6-Triphenyl Phosphonium-Hexanoate) 5. A solution of 11 (30 mg, 0.048 mmol) and Ph3P (15 mg, 0.057 mmol, 1.2 equiv.) in PhMe (100 μL) was heated under reflux. Upon consumption of the starting material the mixture was subjected to FCC purification using chloroform (CHCl3):MeOH 92:8 as eluent, to afford the pure product as colourless oil (15 mg, 36% yield). Rf (CHCl3:MeOH 92:8) = 0.08.
ATR-FTIR (ṽ, cm-1): 3362 (OH), 3057 (=C-H), 2958, 2867 (Csp3-H), 1726 (C=OO), 1438 (C-H), 1112 (C-O), 723, 690 (C-P). 1H NMR (CDCl3, 600 MHz) δ 7.84 (dd, J=12.7, 7.8, Hz, 6 H), 7.78 (td, J=7.5, 1.8 Hz, 3 H), 7.69 (td, J=7.8, 3.4 Hz, 6 H), 4.66 (d, J=2.4 Hz, 1 H), 4.56 (s, 1 H), 4.21 (dd, J=11.1, 1.9 Hz, 1 H), 3.87 (td, J=12.5, 11.9, 6.1 Hz, 2 H), 3.78 (d, J=11.0 Hz, 1 H), 3.17 (dd, J=11.6, 4.7 Hz, 1 H), 2.40 (td, J=11.2, 6.0 Hz, 1 H), 2.29 (t, J=7.4 Hz, 2 H), 1.90 (dtd, J=14.1, 10.7, 8.6 Hz, 2 H), 1.77 (ddd, J=13.9, 4.6, 2.6 Hz, 1 H), 1.70 (m, 4 H), 1.66 (s, 3 H), 1.62 (m, 6 H), 1.54 (m, 5 H), 1.38 (m, 5 H), 1.21 (m, 4 H), 1.03 (m, 2 H), 1.00 (s, 3 H), 0.95 (s, 6 H), 0.88 (tt, J=14.0, 4.8 Hz, 1 H), 0.80 (s, 3 H), 0.74 (s, 3 H), 0.66 (d, J=9.7 Hz, 1 H); 13C NMR (CDCl3, 151 MHz) δ 174.0, 150.2, 135.0, 135.0, 133.8, 133.7, 133.7, 133.6, 130.5, 130.5, 130.4, 129.0, 128.2, 125.3, 118.7, 118.1, 110.1, 78.9, 62.5, 55.3, 50.3, 48.8, 47.7, 46.4, 42.7, 40.9, 38.8, 38.7, 37.6, 37.1, 34.5, 34.2, 33.7, 29.8, 29.7, 29.6, 28.0, 27.4, 27.0, 25.2, 24.5, 22.5, 22.5, 21.4, 20.8, 19.1, 18.3, 16.1, 16.0, 15.4, 14.7; 31P NMR (CDCl3, 243 MHz) δ 24.41. GC-MS: m/e: 880.46 (100.0%), 882.45 (97.3%), 881.46 (61.3%), 883.46 (60.0%), 884.46 (18.5%), 882.46 (18.4%), 885.46 (3.7%), 883.47 (3.6%). ESI-MS (30 eV): m/z 801.67 [(M-Br-)+]; Anal. Calcd. for C54H74BrO3P (Mol. Wt.: 882.04): C, 73.53; H, 8.46; Br, 9.06; O, 5.44; P, 3.51. Found: C, 73.13; H, 8.50; Br, 9.38; O, 5.42; P, 3.73.
(3β)-Lup-20(29)-Ene-3,28-Diol, 3-28-di-(6-Triphenyl Phosphonium-Hexanoate) 6. A solution of 12 (37 mg, 0.046 mmol) and Ph3P (30 mg, 0.112 mmol, 2.4 equiv.) in PhMe (100 μL) was heated under reflux. Upon consumption of the starting material the mixture was subjected to FCC purification using CHCl3:MeOH 92:8 as eluent, to afford the pure product as colourless oil (18 mg, 30% yield). Rf (CHCl3:MeOH) 92:8 = 0.08.
ATR-FTIR (ṽ, cm-1): 3054 (=C-H), 2941, 2867 (Csp3-H), 1721 (C=OO), 1436 (C-H), 1112 (C-O), 745, 722, 690 (C-P). 1H NMR (CDCl3, 600 MHz) δ 7.83 (m, 12 H), 7.78 (t, J=7.6 Hz, 6 H), 7.69 (m, 12 H), 4.65 (d, J=2.4 Hz, 1 H), 4.55 (s, 1 H), 4.38 (dd, J=9.0, 7.4 Hz, 1 H), 4.20 (dd, J=11.1, 1.9 Hz, 1 H), 3.83 (tt, J=12.3, 7.6 Hz, 4 H), 3.77 (d, J=11.0 Hz, 1 H), 2.39 (td, J=11.1, 5.7 Hz, 1 H), 2.28 (t, J=7.4 Hz, 2 H), 2.24 (td, J=7.4, 1.9 Hz, 2 H), 1.89 (m, 4 H), 1.72 (m, 6 H), 1.67 (m, 1 H), 1.65 (s, 3 H), 1.61 (m, 10 H), 1.53 (m, 3 H), 1.45 (m, 1 H), 1.35 (m, 5 H), 1.25 (dd, J=12.6, 2.6 Hz, 1 H), 1.19 (td, J=12.9, 6.3 Hz, 2 H), 1.02 (m, 2 H), 0.99 (s, 3 H), 0.93 (s, 3 H), 0.80 (s, 3 H), 0.77 (s, 3 H), 0.76 (s, 3 H), 0.73 (d, J=9.7 Hz, 1 H); 13C NMR (CDCl3, 151 MHz) δ 176.4, 174.2, 173.6, 150.1, 135.2, 135.2, 133.9, 133.9, 130.7, 130.7, 118.9, 118.4, 118.4, 110.1, 81.0, 64.3, 62.8, 55.6, 50.5, 49.0, 47.9, 46.6, 42.9, 41.1, 38.6, 38.0, 37.8, 37.3, 34.3, 34.0, 30.0, 29.9, 29.9, 29.8, 28.2, 27.2, 25.4, 24.7, 24.6, 23.9, 23.6, 22.7, 22.7, 22.6, 22.3, 21.0, 18.4, 16.8, 16.3, 16.2, 14.9; 31P NMR (CDCl3, 243 MHz) δ 24.36. GC-MS: m/e: 1320.53 (100.0%), 1321.53 (88.0%), 1318.53 (51.2%), 1322.53 (49.2%), 1319.53 (44.5%), 1323.53 (42.8%), 1322.54 (39.7%), 1320.54 (19.7%), 1324.53 (18.5%), 1323.54 (11.7%), 1321.54 (6.0%), 1325.54 (5.5%), 1324.54 (3.3%), 1326.54 (1.3%). ESI-MS (30 eV): m/z 580.82 [(M-2Br-)/2]+; Anal. Calcd. for C78H98Br2O4P2 (Mol. Wt.: 1321.37): C, 70.90; H, 7.48; Br, 12.09; O, 4.84; P, 4.69. Found: C, 70.13; H, 7.10; Br, 12.38; O, 4.51; P, 4.73.
Synthesis of 6-TPPHA (10)
Scheme 1b.
Synthesis of 6-TPPHA (10). Legend to scheme: (i) Ph3P, PhMe, 85 °C overnight, 53%.
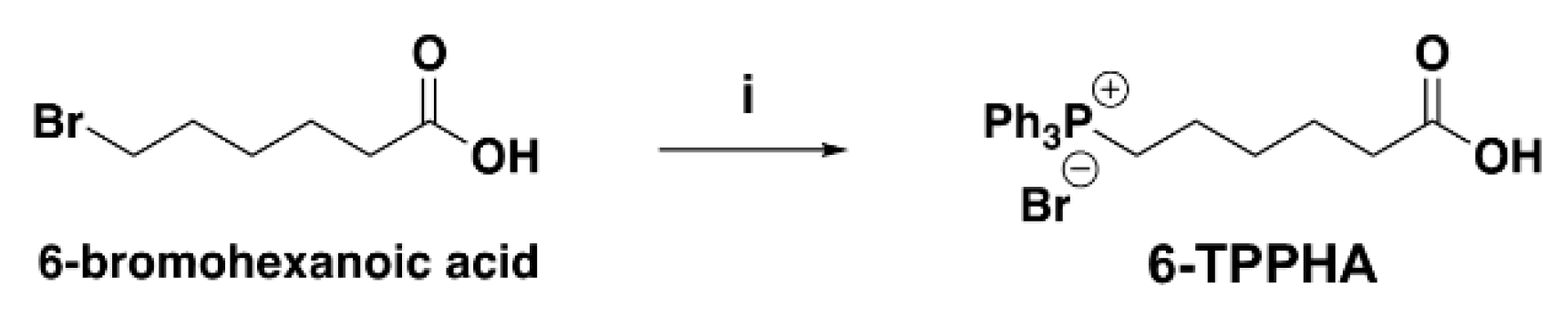
6-Tri Phenyl Phosphonium Hexanoic Acid 10. To a solution of Ph3P (806 mg, 3.07 mmol, 1.2 equiv.) in PhMe (4.27 mL, 0.6M), 6-bromohexanoic acid (500 mg, 2.56 mmol) was added and the resulting mixture was heated, under stirring, at 85 °C overnight. Following, the resulting suspension was left to cool down at r. t., then cooled to 0 °C and filtered under vacuum. The solid residue was washed twice with diethyl ether (Et2O) and dried. The pure product was afforded without further purification as white solid (621 mg, 53% yield).
2.2.2. Synthesis of Betulinic Acid (BA) Derivatives
BA derivatives 3 and 4 were synthesized according to Scheme 2 using reagents described in the Scheme 2 caption.
(3β)-3-Hydroxy-N-2-Propyn-1-Yllup-20(29)-En-28-Amide 3. To an ice-cooled solution of BA (40 mg, 0.088 mmol) and propargylamine (10 μL, 0.157 mmol, 1.79 equiv.) in THF (0.3 mL), 2-(7-azobenzotriazole)-N, N, N’, N’-tetramethyluronium hexafluorophosphate (HATU, 37 mg, 0.097 mmol, 1.11 equiv.) and Et3N (50 μL, 0.36 mmol, 4.08 equiv.) were added and the resulting mixture was stirred at 0 °C to r. t. Upon completion of the reaction the mixture was concentrated to dryness and the oily residue was subjected to FCC purification using gradient PhMe:EtOAc 97:3 to 95:5 as eluent, to afford the pure product as colourless oil (30 mg, 69% yield). Rf (PhMe:EtOAc 97:3) = 0.09.
ATR-FTIR (ṽ, cm-1): 3309 (OH), 3073 (=C-H), 2940, 2867 (Csp3-H), 1639 (C=ONH), 1505, 1447 (C-H). 1H NMR (CDCl3, 600 MHz) δ 5.71 (t, J=5.3 Hz, 1 H), 4.73 (d, J=2.3 Hz, 1 H), 4.59 (dd, J=2.4, 1.4 Hz, 1 H), 4.08 (ddd, J=17.5, 5.4, 2.5 Hz, 1 H), 3.98 (ddd, J=17.5, 5.1, 2.5 Hz, 1 H), 3.18 (dd, J=11.5, 4.8 Hz, 1 H), 3.13 (td, J=11.2, 4.6 Hz, 1 H), 2.42 (ddd, J=13.1, 11.5, 3.5 Hz, 1 H), 2.20 (t, J=2.5 Hz, 1 H), 1.94 (m, 2 H), 1.75 (dd, J=12.2, 7.8 Hz, 1 H), 1.71 (m, 1 H), 1.68 (s, 3 H), 1.65 (t, J=3.6 Hz, 1 H), 1.59 (m, 3 H), 1.54 (m, 1 H), 1.50 (m, 2 H), 1.45 (m, 1 H), 1.42 (m, 2 H), 1.38 (m, 1 H), 1.35 (m, 2 H), 1.25 (m, 2 H), 1.16 (dt, J=13.4, 3.2 Hz, 1 H), 1.00 (m, 1 H), 0.97 (s, 3 H), 0.96 (s, 3 H), 0.94 (s, 3 H), 0.90 (dd, J=13.2, 4.1 Hz, 1 H), 0.86 (d, J=7.1 Hz, 1 H), 0.81 (s, 3 H), 0.75 (s, 3 H), 0.67 (m, 1 H); 13C NMR (CDCl3, 151 MHz) δ 176.1, 151.1, 129.3, 128.4, 109.6, 80.4, 79.2, 71.4, 55.9, 55.6, 50.9, 50.4, 47.0, 42.7, 41.0, 39.1, 38.0, 37.4, 34.6, 33.9, 31.0, 29.6, 29.2, 28.2, 27.7, 25.8, 21.1, 19.7, 18.5, 16.4, 15.6, 14.9. GC-MS: m/e: 493.39 (100.0%), 494.40 (37.5%), 495.40 (7.2%). ESI-MS (30 eV): m/z 494.45 [M+H+]; Anal. Calcd. for C33H51NO2 (Mol. Wt.: 493.76): C, 80.27; H, 10.41; N, 2.84; O, 6.48. Found: C, 80.13; H, 10.50; N, 3.02; O, 6.42.
(3β)-3-Hydroxy-3-(6-Triphenyl Phosphonium-Hexanoate)-N-2-Propyn-1-Ylurs-20(29)-En-28-Amide 4. To an ice-cooled (-10 °C) suspension of 3 (23 mg, 0.047 mmol) and 10 (23 mg, 0.050 mmol, 1.07 equiv.) in DCM (0.45 mL), a solution of DCC (10.3 mg, 0.050 mmol, 1.06 equiv.) and DMAP (2 mg, 0.016 mmol, 0.35 equiv.) in DCM (0.2 mL) was portion-wise added and the resulting mixture was stirred at -10 °C to r. t. Upon completion of the reaction, the mixture was diluted with EtOAc and washed with HCl (aq) 1M, water and brine. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated to dryness and the oily residue was subjected to FCC purification using CHCl3:MeOH 92:8 as eluent, to afford the pure product as colourless oil (15 mg, 34% yield). Rf (CHCl3:MeOH 92:8) = 0.08.
ATR-FTIR (ṽ, cm-1): 3061 (=C-H), 2940, 2868 (Csp3-H), 1721 (C=OO), 1639 (C=ONH), 1439 (C-H), 1112 (C-O), 723, 690 (C-P). 1H NMR (CDCl3, 600 MHz) δ 7.85 (m, 6H), 7.77 (td, J=7.4, 1.7 Hz, 3 H), 7.69 (td, J=7.8, 3.3 Hz, 6 H), 5.79 (br s, 1 H), 4.72 (d, J=2.3 Hz, 1 H), 4.58 (m, 1 H), 4.39 (m, 1 H), 4.06 (ddd, J=17.4, 5.4, 2.5 Hz, 1 H), 3.96 (ddd, J=17.5, 5.1, 2.5 Hz, 1 H), 3.90 (br s, 2 H), 3.12 (td, J=11.1, 4.5 Hz, 1 H), 2.52 (t, J=2.5 Hz, 1 H), 2.41 (td, J=12.0, 11.5, 3.6 Hz, 1 H), 2.25 (td, J=7.3, 2.1 Hz, 2 H), 1.94 (dtd, J=13.4, 11.0, 7.9 Hz, 3 H), 1.72 (m, 4 H), 1.67 (s, 3 H), 1.64-1.51 (m, 9 H), 1.45 (m, 3 H), 1.35 (ttd, J=15.3, 11.5, 9.3, 3.8 Hz, 5 H), 1.25 (m, 3 H), 1.14 (dt, J=13.4, 3.2 Hz, 1 H), 0.94 (s, 3 H), 0.92 (s, 3 H), 0.81 (s, 3 H), 0.77 (s, 6 H), 0.73 (m, 1 H); 13C NMR (CDCl3, 151 MHz) δ 176.2, 173.7, 151.2, 135.2, 134.1, 134.0, 130.8, 130.7, 119.2, 118.6, 109.7, 81.2, 80.6, 80.5, 71.4, 56.0, 55.8, 50.9, 50.6, 47.1, 42.8, 41.2, 38.7, 38.4, 38.2, 38.1, 37.5, 34.7, 34.4, 33.9, 31.2, 30.1, 30.0, 29.7, 29.3, 28.3, 25.9, 24.8, 24.1, 22.9, 22.8, 22.8, 22.6, 21.3, 19.8, 18.5, 16.9, 16.5, 15.0; 31P NMR (CDCl3, 243 MHz) δ 24.39.GC-MS: m/e: 931.47 (100.0%), 933.46 (97.3%), 932.47 (64.6%), 934.47 (63.3%), 935.47 (20.8%), 933.47 (20.7%), 936.47 (4.4%), 934.48 (4.3%). ESI-MS (30 eV): m/z 852.72 [(M-Br-)+]; Anal. Calcd. for C57H75BrNO3P (Mol. Wt.: 933.09): C, 73.37; H, 8.10; Br, 8.56; N, 1.50; O, 5.14; P, 3.32. Found: C, 73.23; H, 8.47; Br, 8.38; N, 1.74; O, 5.32; P, 3.70.
2.2.3. Synthesis of Ursolic Acid (UA) Derivatives
UA derivative 7 was synthesized according to Scheme 3 via intermediate 13. Reagents used have been described in the legend in Scheme 3 caption.
(3β)-3-Hydroxy-N-2-Propyn-1-Ylurs-12-En-28-Amide 13. To an ice-cooled suspension of UA (50 mg, 0.109 mmol) and propargylamine (10 μL, 0.157 mmol, 1.44 equiv.) in THF (0.55 mL), HBTU (46 mg, 0.121 mmol, 1.11 equiv.) and Et3N (50 μL, 0.36 mmol, 3.3 equiv.) were added and the resulting mixture was stirred at -10 °C to r. t. for 72 hours. The reaction the mixture was concentrated to dryness and the oily residue was subjected to FCC purification using PhMe:EtOAc 97:3 as eluent, to afford both the unreacted UA-active ester and the pure product 13 as colourless oil (12 mg, 22% yield – 51% based on recovered unreacted material). Rf (PhMe:EtOAc 97:3) = 0.06.
1H NMR (CDCl3, 600 MHz) δ 6.07 (t, J=4.8 Hz, 1 H), 5.36 (t, J=3.6 Hz, 1 H), 4.03 (ddd, J=17.6, 5.3, 2.6 Hz, 1 H), 3.89 (ddd, J=17.6, 4.2, 2.6 Hz, 1 H), 3.22 (dd, J=11.4, 4.6 Hz, 1 H), 2.20 (t, J=2.6 Hz, 1 H), 1.98 (m, 3 H), 1.88 (m, 2 H), 1.73 (ddt, J=13.6, 4.7, 2.3 Hz, 1 H), 1.63 (ddt, J=13.9, 10.1, 4.2 Hz, 3 H), 1.58 (m, 2 H), 1.54 (m, 2 H), 1.46 (m, 4 H), 1.37 (dd, J=12.4, 3.3 Hz, 1 H), 1.30 (m, 3 H), 1.10 (s, 3 H), 1.06 (ddd, J=14.0, 4.2, 2.6 Hz, 1 H), 1.01 (m, 1 H), 0.99 (s, 3 H), 0.95 (s, 3 H), 0.93 (s, 3 H), 0.87 (d, J=6.5 Hz, 3 H), 0.80 (s, 3 H), 0.78 (s, 3 H), 0.72 (dd, J=11.7, 1.9 Hz, 1 H); 13C NMR (CDCl3, 151 MHz) δ 178.2, 140.0, 126.3, 80.0, 79.2, 71.8, 55.4, 54.0, 48.0, 47.8, 42.7, 40.0, 39.8, 39.3, 39.0, 38.9, 37.2, 37.1, 32.9, 31.1, 29.6, 28.4, 28.1, 27.4, 25.2, 23.6, 23.5, 21.4, 18.5, 17.4, 17.1, 15.8, 15.7.
(3β)-3-Hydroxy-(6-Triphenyl Phosphonium-Hexanoate)-N-2-Propyn-1-Ylurs-12-En-28-Amide 7. To an ice-cooled suspension of 13 (12 mg, 0.024 mmol) and 10 (12 mg, 0.026 mmol, 1.09 equiv.) in DCM (0.1 mL), DCC (6.0 mg, 0.029 mmol, 1.2 equiv.) and DMAP (2 mg, 0.016 mmol, 0.68 equiv.) were added and the resulting mixture was stirred at 0 °C to r. t. Upon completion of the reaction, the mixture was diluted with EtOAc and washed with HCl (aq) 1M, water and brine. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated to dryness and the oily residue was subjected to FCC purification using CHCl3:MeOH 92:8 as eluent, to afford the pure product as colourless oil (10 mg, 45% yield). Rf (CHCl3:MeOH 92:8) = 0.08.
ATR-FTIR (ṽ, cm-1): 3058 (=C-H), 2962, 2924, 2869 (Csp3-H), 1723 (C=OO), 1644 (C=ONH), 1439 (C-H), 1112 (C-O), 746, 729, 690 (C-P). 1H NMR (CDCl3, 600 MHz) δ 7.84 (ddd, J=12.6, 8.4, 1.3 Hz, 6 H), 7.78 (td, J=7.4, 1.7 Hz, 3 H), 7.69 (td, J=7.8, 3.3 Hz, 6 H), 6.07 (t, J=4.8 Hz, 1 H), 5.34 (t, J=3.7 Hz, 1 H), 4.49 (d, J=8.1 Hz, 2 H), 4.42 (m, 1 H), 4.01 (dd, J=17.6, 5.3 Hz, 1 H), 3.88 (m, 1 H), 3.82 (m, 1 H), 3.47 (tdt, J=11.0, 7.9, 3.9 Hz, 2 H), 2.26 (td, J=7.5, 1.5 Hz, 1 H), 2.20 (t, J=2.6 Hz, 1 H), 1.97 (m, 2 H), 1.89 (dd, J=14.6, 5.5 Hz, 3 H), 1.84 (m, 1 H), 1.79 (s, 1 H), 1.72 (m, 2 H), 1.68 (t, J=4.0 Hz, 1 H), 1.65 (m, 1 H), 1.62 (d, J=7.0 Hz, 1 H), 1.59 (m, 1 H), 1.56 (m, 1 H), 1.55 (d, J=3.9 Hz, 1 H), 1.50 (m, 1 H), 1.48 (m, 1 H), 1.45 (m, 1 H), 1.42 (m, 1 H), 1.35 (m, 1 H), 1.31 (m, 2 H),M 1.28 (m, 1 H), 1.14 (m, 1 H), 1.11 (m, 1 H), 1.10 (m, 1 H), 1.08 (s, 3 H), 1.04 (m, 1 H), 0.94 (s, 3 H), 0.92 (s, 3 H), 0.86 (d, J=6.4 Hz, 3 H), 0.80 (s, 3 H), 0.80 (s, 3 H), 0.79 (s, 3 H); 13C NMR (CDCl3, 151 MHz) δ 178.1, 173.6, 157.3, 140.0, 135.2, 135.2, 134.0, 133.9, 130.7, 130.7, 126.2, 118.9, 118.3, 81.0, 79.9, 71.8, 55.4, 53.9, 49.2, 48.0, 47.7, 42.7, 39.9, 39.8, 39.3, 38.5, 37.9, 37.1, 37.0, 34.3, 34.1, 32.8, 31.0, 30.0, 29.9, 29.6, 28.3, 28.0, 25.9, 25.2, 25.2, 24.8, 23.8, 23.6, 23.5, 22.7, 21.4, 18.3, 17.5, 17.1, 17.0, 15.8; 31P NMR (CDCl3, 242.9 MHz) δ 24.38. GC-MS: m/e: 931.47 (100.0%), 933.46 (97.3%), 932.47 (64.6%), 934.47 (63.3%), 935.47 (20.8%), 933.47 (20.7%), 936.47 (4.4%), 934.48 (4.3%). ESI-MS (30 eV): m/z 852.71 [(M-Br-)+]; Anal. Calcd. for C57H75BrNO3P (Mol. Wt.: 933.09): C, 73.37; H, 8.10; Br, 8.56; N, 1.50; O, 5.14; P, 3.32. Found: C, 73.15; H, 8.43; Br, 8.37; N, 1.78; O, 5.43; P, 3.63.
2.3. ATR-FTIR Spectroscopy of BA, BET, UA and Compounds 1-7
FTIR spectra of BA, BET, UA and compounds 1-7 were recorded in triplicate directly on the solid samples in attenuated total reflection (ATR) mode. Acquisitions were made from 4000 to 600 cm-1, with 1 cm-1 spectral resolution, co-adding 32 interferograms, with a measurement accuracy in the frequency data at each measured point of 0.01 cm-1, due to the laser internal reference of the instrument. The frequency of each band was obtained automatically by using the “find peaks” command of the instrument software.
2.4. Multivariate Analysis of ATR-FTIR and 13C NMR Spectral Data
2.4.1. ATR-FTIR Spectral Data
ATR-FTIR data (transmittance, %) of all acquired spectra were arranged in a matrix of 3401 (wavenumbers cm-1) × 10 (compounds) = 34,010 measurable variables. Then, spectral data of previously reported triphenyl phosphonium salt (BPPB) were added to this matrix [8], obtaining a second dataset of 3401 (wavenumbers cm-1) × 11 (compounds) = 37,411 measurable variables. For each sample, the variables consisted of the values of transmittance (%) associated with the wavenumbers (3401) in the range 4000–600 cm-1. The systems were simplified by exploiting the multivariate analysis, named principal component analysis (PCA), processing each matrix of spectral data using CAT (Chemometric Agile Tool freely available online at https://www.gruppochemiometria.it/index.php/software/19-download-the-r-based-chemometric-software), accessed on 11 November 2025). Before PCA, ATR-FTIR spectral data were scaled and centred. The results were reported as score plot of PC1 vs. PC2 and discussed in Section 3.
2.4.2.13. C NMR Spectral Data
13C NMR values of δ (ppm) of all peaks present in the NMR spectra of analysed samples were first arranged in a matrix of 52 (δ, ppm) × 15 (compounds) = 780 measurable variables. Then, since the number of 13C NMR signals were very different for analysed compounds, the resulting matrix contained a too high number of missing data, which did not allow to calculate the system variance and carry out PCA. To address this issue, the number of missing data was reduced, by reducing the range of possible δ (ppm). A second matrix was then constructed made of 34 (δ, ppm) × 15 (compounds) = 510 measurable variables. For each sample, the variables consisted of the values of δ (ppm) of all peaks present in its 13C NMR spectrum. The system was simplified by exploiting PCA, processing the matrix of spectral data using CAT (Chemometric Agile Tool freely available online at https://www.gruppochemiometria.it/index.php/software/19-download-the-r-based-chemometric-software), accessed on 10 November 2025). Before PCA, 13C NMR spectral data were scaled and centred. The results were reported as score plot of PC1 vs. PC3 and discussed in Section 3.
2.5. Potentiometric Titrations of Compound 1 and 4-7
The potentiometric titration of TPP-containing compounds was carried out in non-aqueous medium (mixture of anhydrous acetic acid (AcOH) and acetic anhydride (Ac2O) 30:70 (v:v)) with HClO4, performing a slightly modified procedure previously described by us for the volumetric titration of ammonium salts [25,26,27]. A similar protocol was in fact described by Pifer and Wollish, who applied this method for salts of weaky organic bases [28]. Briefly, exacted weighted samples of compounds 1 and 4-7 were dissolved in AcOH:Ac2O 30:70, treated with a solution of mercury acetate (1.5 g) in AcOH (25 mL), and titrated with a standardized 0.1 N solution of HClO4 in AcOH:Ac2O, prepared as described in the following section, using potentiometric endpoint detection. The titrations were performed under efficient stirring with a magnetic stirrer, at room temperature (25 ± 2 ◦C). Millivolts were measured at fixed points up to the addition of 6 mL of 0.1 N HClO4. Titrations were made in triplicate, and the measurements were reported as mean ± SD.
2.5.1. Preparation of a 0.1 M Perchloric Acid Volumetric Solution
The 0.1 M perchloric acid volumetric solution was prepared by diluting 8.5 mL of 70–73 wt% perchloric acid with 900 mL of anhydrous acetic acid and 30 mL of acetic anhydride and then diluting to 1000 mL with anhydrous acetic acid. Perchloric acid was standardized by titration against potassium hydrogen phthalate [29].
2.6. Optical Microscopy Analyses
m-Q Water solutions of compounds 1 and 4-7, possessing the triphenyl phosphonium (TPP) salt group, were investigated via optical microscopy (OM) analysis, to assess their possible capability to give spherical vesicles. In the performed experiments, 0.6-1.5 mg of solid compounds were dissolved in m-Q water (44-110 µL) by gentle heating, obtaining clear solutions with concentration 13.6 mg/mL. Upon cooling, the obtained solutions were observed using a Leica DM750 optical microscope (Leica Italy, Milan, Italy) equipped with 40× and 100× objectives. The camera used for image capture was a Leica ICC50W (Leica Italy, Milan, Italy). All images were processed using LAS EZ 3.4.0. software (Leica Italy, Milan, Italy).
2.7. Dynamic Light Scattering (DLS) Analysis
The particle size intended as hydrodynamic diameter distribution, polydispersity index (PDI), and zeta potential (ζ-p) (mV) of compound 6 which demonstrated spherical vesicles at OM were measured at 25 ◦C, at a scattering angle of 90◦ in m-Q water using a Malvern Nano ZS90 light scattering apparatus (Malvern Instruments Ltd., Worcestershire, UK). The solution of 6 used for OM (9.8 mM) was diluted to a final concentration of 5 mM (8.4 kcps) and analysed. The ζ-p value of BPPB was recorded using the same apparatus at a count rate of 20–59 kcps. The results of the experiments are presented as the mean of 3 independent determinations, made of 10 runs (particle size) or 12 runs (ζ-p), each one ± SD. Intensity-based results have been reported to express particle size distribution.
2.8. Microbiology
With the aim of developing novel compounds active against MDR clinical isolates, preliminary experiments were conducted to evaluate the inhibitory effects of the synthesized compounds 1-7, as well as those of pure BET, BA and UA by determining their minimum inhibitory concentrations (MICs) on a selection of Gram-positive and Gram-negative species. All bacteria used in this study were clinical isolates that had developed resistance to at least one or two antibiotics, including also ESCAPE species. The main objectives were understanding if chemical modifications would have improved the activity of pristine compounds and then select the best performing compounds to be used for further experimentation.
2.8.1. Microorganisms
A total of 7 isolates belonging to a collection of MDR Gram-positive and Gram-negative species of the University of Genova were used in this study. All were clinical strains isolated from human specimens and identified using VITEKR 2 (Biomerieux, Firenze, Italy) or matrix-assisted laser desorption-ionization time-of-flight (MALDI-TOF) mass spectrometric technique (Biomerieux, Firenze, Italy). The 7 MDR isolates included 4 Gram-positive and 3 Gram-negative bacteria of different genera. Among bacteria of Gram-positive species, 2 were enterococci (1 E. faecalis and 1 E. faecium), while 2 were staphylococci (1 S. aureus and 1 S. epidermidis). All enterococci were MDR isolates with resistance to vancomycin (VRE) and teicoplanin, while all staphylococci were MDR strains with resistance to methicillin (MRSA and MRSE). Gram-negative species included 1 non-fermenting isolate of P. aeruginosa isolated from cystic fibrosis patients with resistance to carbapenems. 2 strains were Enterobacteriaceae, including 1 E. coli and 1 K. pneumoniae which were resistant to carbapenems and K. pneumoniae carbapenemase (KPC)-producing bacteria.
2.8.2. Determination of the MICs
To investigate the antibacterial activity of BPPB on the described pathogens, their Minimal Inhibitory Concentrations (MICs) were determined by following the microdilution procedures detailed by the European Committee on Antimicrobial Susceptibility Testing EUCAST [23] and reported in our previous works [30].
3. Results and Discussion
3.1. Synthesis of Triterpenoids Derivatives
With the aim at finding novel compounds effectively active against difficult-to-treat clinically isolated superbugs, seven triterpenoid derivatives (1-7) were synthesized by chemical modifications of BET, BA and UA. On literature indication, which evidenced that several UA, BET and BA derivatives with enhanced potency, bioavailability and water solubility (including esters, amides, oxadiazole quinolone, etc.), mostly derived from modifications at positions C-3 (hydroxyl), C-12-C-13 (double bonds) and C-28 (carboxylic acid) [31], we carried out chemical modification of parent compounds at position C-28 or C-28 and C-3 as reported in a recent study for preparing BET [32] and BA derivatives [33]. Particularly, it has been reported that structural modifications which transformed the C-28 carboxyl group in ester or amide groups, as well as ester modifications or oxidations of C-3 hydroxyl could enhance the cytotoxicity of BA [33]. Organic fragments such as propargyl amine (PAM), propiolic acid (PA, and 6-triphenyl phosphonium hexanoic acid (6-TPPHA), bearing the triphenyl phosphonium group (TPP), were selected as modifying groups. PA was selected and introduced on hydroxyl in C-28 of BET obtaining the ester derivatives 2. This choice was promoted by papers by Chrobak et al. [32] and Csuk et al. [34]. Authors reported that the introduction of PA in such position or a carbonyl group at C-28 and a short substituent with a terminal triple bond, improved significantly BET activity against most of the tested cancer lines at concentration in the range 0.35–18.7 µM [32]. PAM was selected to modify BA and UA on hydroxyl in C-28 thus preparing the C-28 propargyl amide BA derivatives 3 and the C-28 propargyl UA derivative 13, which were also the intermediates to obtain the TPP-BA derivative 4 and the final UA derivative 7. These projects were based on a paper, that reported compound 3 to have high anticancer activity against human T47D (breast cancer), SW707 (colorectal adenocarcinoma) and mouse P388 (leukaemia) cell lines [35]. Compound 3 was four times more cytotoxic than BA against the human SW707 cell line, also establishing that BA derivatives having shorter alkynyl chain are the more cytotoxic [35]. The prop-2-ynyl-carbamate group was chosen to insert on BET scaffold a carbamate residue in a two-step reaction and achieve BET-carbamate derivative 9. In this regard, Wiemann et al. have reported that BET derived carbamates are interesting scaffolds for the synthesis of novel cytotoxic compounds [36]. Specifically, authors evidenced that anticancer activity of some BET derived mono and bis-carbamates against different cancer cell lines was higher than that of pristine BET, while toxicity on normal fibroblasts was lower [36]. 6-TPPHA was selected as the carboxylic acid moiety for esterification of the BET derivative 9, BA derivative 3 and UA derivative 13 achieving the TPP-containing triterpenoids 1, 4, and 7. This further modification on C-3 hydroxyl was carried out because it has been recently reported that compounds bearing the TPP group possess potent antibacterial effects against several clinical superbugs, despite their complex pattern of resistance [8,30]. On this evidence, the TPP-hexanoate group was inserted only in C-3, as well as in both C-3 and C-28 positions of BET, via a two-step reaction, achieving BET derivatives without triple bonds 5 and 6. In fact, it also observed that a compound having 2 TPP groups, as in the case of 6, linked by a C12 chain and capable of giving nanovesicles in water (BPPB), possessed antibacterial effects [8], remarkably higher than those of another compound (namely 1 in the paper) possessing only a 1 TPP moiety [30]. Finally, it was observed that, differently from the previously reported compound 1, active only against Gram-positive species, BPPB bearing 2 TPP groups was very potent also against superbugs of Gram-negative species [8]. Additionally, the cytotoxicity of all TPP-bearing compounds (1 and BPPB), assessed using several mammalian cell lines, evidenced from >1 to high values of therapeutic index, thus making appear the TPP group as an appealing candidate to chemically modify BET, BA and UA [8,30]. Concerning the selected triterpenoids nuclei, BET, BA and UA were chosen because naturally occurring and deprived of evident intrinsic cytotoxicity. These characteristics could have possibly softened the sharp cationic characteristic of the TPP group, rationally responsible for the residual cytotoxicity of previously reported compounds containing it, thus reducing or nullifying it, while maintaining the antibacterial properties. This approach, in sight of a future clinical development of the most performant antibacterial compound possibly found in this early study. We selected BA, since it was found deprived of toxicity in normal cells [37,38,39]. When it was used in the 24h treatment of human immortalized keratinocytes (HaCaT), the percentage of viable cells calculated at the highest concentration tested (50 μM) was > 81% [40]. BET was selected, since it has been reported to be selectively toxic towards neoplastic cells, but only weakly toxic towards normal cells [41,42,43]. UA was finally selected, because it is reported to have only weak cytotoxic effects on normal cells [44]. There are examples of single dose subcutaneous injections of 300 mg/kg of UA, which did not cause any deviations to the clinical haematology parameters and tissue morphology of animal models. Additionally, 5-day short-term toxicity studies about the combined UA and oleanolic acid administration at a dose of 1.0 mg/kg, did not lead to any morbidity or mortality[45]. Moreover, when UA, in a recent long-term (90 days) oral toxicity in vivo study, was given to Han-Wistar rats at a repeated dose of 0, 100, 300, or 1000 mg/kg/day to assess its safety and toxicity, no mortality, uncharacteristic body weight changes, or tissue architecture variations at all the analysed test doses were observed, as well as no changes in behaviour, or haematological and clinical parameters was evidenced, suggesting safe and non-toxic nature of UA [45]. Moreover, BET and BA, while excellent multi-target natural compounds for several pharmacological applications, are totally inactive and not usable against both Gram-positive and Gram-negative species, as effective antibacterial agents. Therefore, they were selected to evaluate if upon our chemical modifications, it could be possible reevaluate them also as possible potent new antibacterials. Conversely, UA already known as remarkable antibacterial for treating Gram-positive bacteria, was selected to assess if by inserting the TPP group, its intrinsic antibacterial effects could be enhanced and directed also versus Gram-negative superbugs. In the following Section 3.1.1, Section 3.1.2 and Section 3.1.3 the synthetic procedures and NMR characterization of BET, BA and UA intermediates and final derivatives tested as antibacterial agents have been discussed. Copies of NMR spectra of compounds discussed in the following Section 3.1.1, Section 3.1.2 and Section 3.1.3 are available in Section S1.2 (Supplementary Materials) as Figures S1.2.1-S1.2.27.
3.1.1. Synthesis of BET Derivatives
(3β)-Lup-20(29)-Ene-3,28-Diol, 28-(4-Nitrophenyl Carbonate) 8. BET derivative intermediate 8 is known and was prepared in 83% yield, following the procedure described by Laiolo et al.[46]. The white foam obtained after FCC purification as described in the experimental Section was subjected to 1H and 13C NMR analysis which provided spectral data in agreement with those reported by Laiolo et al., thus confirming the structure and high level of purity of 8 [46]. In this reaction stoichiometry of reagents and temperature are of paramount importance to obtain the desired C-28 mono-substituted derivative. In fact, a procedure like that of Laiolo et al. but carried out working at room temperature instead of 0 °C and with 2.2 equivalents excess of 4-nitrophenyl chloroformate, instead of 1.04 equivalents, allowed Liu et al. to obtain the C-3, C-28 di-(4-nitrophenyl carbonate) BET derivative (3β)-lup-20(29)-ene-3,28-diol, 3,28-di-(4-nitrophenyl carbonate) in good yield of 74% [47]. NMR characterization of 8 evidenced that, respect to the 1H NMR spectrum of BET, that of 8 presented new peaks at 8.29, 8.28, 7.41, 7.39 ppm, due to the AA’-BB’ aromatic p-di-substituted system given by the introduction of the nitrophenyl carbamate group on C-28. The double doublet signal of the diastereotopic protons of the CH2OH group, which were at 3.34 and 3.81 ppm in the spectrum of BET with a J = 10.8 Hz [48], shifted to higher value of δ in the spectrum of 8 (4.51 and 4.08 ppm), maintaining the same multiplicity with a J of about 10.8 Hz. Similarly, in the 13C NMR spectrum of 8, signals for the aromatic carbon atoms were detected at 155.6, 145.3 ppm (quaternary C) and at 125.3, 121.8 (CH=CH), while the signal of C=O of carbamate group was found at 153.0 ppm. The C-28 methylene signal, respect to the position occupied in the spectrum of BET (60.47 ppm) [48], in the spectrum of 8 shifted to higher value of δ (68.30 ppm).
(3β)-Lup-20(29)-Ene-3,28-Diol, 28-(N-Propargyl Carbamate) 9. BET derivative intermediate 9 was unknown. Anyway, it was prepared via aminolysis of the BET derivative 8 following the modified procedure described by Kemmer et al. [49]. By this approach, the authors prepared a novel dextran norbornene methylcarbamate, completely converting the dextran 4-nitrophrnyl carbonate (DNPC), used as starting material, by its reaction with 5-norbornene-2-methylamine in ratio 1:2, at 20°C for 5h in DMF [49]. Here, 8 and propargylamine were reacted in similar ratio 1:1.9 in presence of Et3N, but DMF was replaced by THF, since is easier to be removed under vacuum after reaction. The reaction mixture was stirred at room temperature to completion. According to what reported by different authors [36,50], the reaction proceeds according to the following two-step process (Figure 2).
Practically, the carbamates formation involves the acyl nucleophilic substitution of 4-nitro-phenoxyl group, by the amine reagent, with expulsion of 4-nitro-phenoxide, thus obtaining a protonated carbamate, which transforms in the final carbamate by deprotonation operated by 4-nitro-phenoxide providing 4-nitro-phenol [50]. The reactions are proposed to proceed through a stepwise mechanism with a change in the rate-determining step (RDS), based on the curved Brønsted-type plots [50]. Concerning NMR analyses of 9, in both 1H and 13C NMR spectra, except for small traces, the aromatic signals disappeared. On the contrary, the signal of NH appeared in the form of broad singlet at 4.95 ppm, the double doublet signal of diastereotopic proton atoms of C-28 methylene (J=10.8 Hz) was now detected at 4.27 and 3.86 ppm, the signal of methylene of propargyl group was detected at 3.98 ppm in form of broad singlet due to the coupling with the NH proton, while the signal of ≡C−H was found at 2.24 ppm. In the 13C NMR, the signal of C=O of carbonate of 8 at 153 ppm disappeared and was substituted by the new signal of carbamate of 9 at 157.0 ppm. The signal of methylene in C-28 was found at 64.0 ppm, that of C≡C−H was found at 80.1 ppm, while that of ≡C−H at 71.9 ppm. Copies of spectra of 9 are available in Section 1.2 (Supplementary Materials) as Figure S1.2.1 and S1.2.2.
(3β)-Lup-20(29)-Ene-3,28-Diol-(6-Triphenyl Phosphonium Hexanoate)-28-(N-propargyl carbamate) 1. BET derivative 1 was unknown and it was synthesized starting from 9 by a Steglich esterification reaction, carried out as in other synthesis of this study. In this case, 6-triphenyl phosphonium hexanoic acid 10 was the acid component, while DCC and DMAP were the condensation promoter and the catalyst, respectively. Compound 1 was achieved in > 50% yield and with high level of purity. The structure of 1 was confirmed by ATR-FTIR, 1H, 13C and 31P NMR, was well as by GC-MS, ESI-MS, elemental analysis and potentiometric titrations, which confirmed its molecular weight. In the 1H NMR spectrum of compound 1, aromatic signals integrable for 15 proton atoms appeared at 7.84, 7.77, 7.68 ppm, due to the introduction of triphenyl phosphonium (TPP) group present on C-6 of the new hexanoate inserted on C-3. The signal of C-3 CH shifted from the δ value of 3.18 ppm, found in the spectrum of compound 9, to 4.39 ppm in the spectrum of 1. Five additional methylene signals appeared in the range 3.84-1.25 ppm, due to the alkyl chain of hexanoate. They were dispersed among the signals of CH and CH2 groups of pentacyclic structure of BET and of CH2 groups already present in 9, thus being of critical attribution. Anyway, the signals at 3.84 and 2.24 ppm, accounting for 3 proton atoms each one, unequivocally contained the signals of the two proton atoms of CH2P+ and CH2C=O groups, respectively. Similarly, in the 13C NMR of 1, despite the C=O of carbamate was no longer visible, for the low intensity of the spectrum, the new signal of the C=OO of hexanoate was clearly observable at 173.6 ppm. New aromatic signals, some of which in form of doublet, due to the coupling with phosphorous atom, appeared at 135.2 (3 equivalent CH=), 134.0, 133.9 (6 equivalent CH=), 130.7 (6 equivalent CH=), 118.9, 118.3 (3 equivalent quaternary C) ppm. The signal of the C-3 was found at 81.0 ppm, the signals of CH2C=O and of the CH2P+ groups were found at 34.3 and at 22.7 ppm respectively, while in the 31P NMR, a singlet signal was observed at 24.38 ppm. Copies of spectra of 1 are available in Section 1.2 (Supplementary Materials) as Figure S1.2.3-S1.2.5.
(3β)-Lup-20(29)-Ene-3,28-Diol, 28-(2-Propynoate) 2. BET derivative 2 was prepared according to the procedure described by Boryczka et al. in the year 2013 [51] and reproposed later by Bȩbenek et al. [52], achieving the desired compound in 38% yield vs. the 60% claimed by Boryczka et al. [51]. Another discrepancy with compounds previously prepared consisted in physical state of 2, which was obtained as colourless oil after FCC by us, instead as white solid with melting points of 132-134 °C [52] and 133-135°C [51], reported by other authors. Anyway, the structure and purity of 2 was confirmed by more reliable analyses, including ATR-FTIR, GC-MS, ESI-MS, elemental analyses, as well as 1H and 13C NMR. Specifically, respect to the 1H NMR spectrum of BET, where the double doublet signal of diastereotopic proton atoms of methylene C-28 (CH2OH) is usually present at 3.81 and 3.34 ppm, with a J of about 10.80 Hz [48], in the spectrum of 2, where the propyne ester has been inserted in C-28, such signal shifted to higher values of δ (4.37 and 3.98 ppm) with J = 11.1 Hz, maintaining the same multiplicity. Moreover, a new signal at 2.89 ppm was found, due to the new ≡C−H group of propine. In the 13C NMR spectrum of 2, new signals respect to those observed in the spectrum of BET, were found at 153.5 (C=OO), 75.0 (-C≡C−H) and 74.9 (-C≡C−H), ppm, while the signal of methylene C-28, usually at 60.47 ppm in BET [48], in the spectrum of 2 was found at 65.1 ppm. Copies of spectra of 2 are available in Section 1.2 (Supplementary Materials) as Figure S1.2.6 and S1.2.7.
(3β)-Lup-20(29)-Ene-3,28-Diol, 28-(6-Bromo Hexanoate) 11. BET derivative 11 was unknown, but analogous compounds with shorter alkyl chains, such as (3β)-lup-20(29)-ene-3,28-diol, 28-(5-bromopentanoate) [53,54,55], (3β)-lup-20(29)-ene-3,28-diol, 28-(4-bromobutanoate) [54,55], (3β)-lup-20(29)-ene-3,28-diol, 28-(3-bromopropanoate) [55], (3β)-lup-20(29)-ene-3,28-diol, 28-(2-bromoethanoate) [54] and (3β)-lup-20(29)-ene-3,28-diol, 28-chloroacetate) [56] have been reported. Except for the case of (3β)-lup-20(29)-ene-3,28-diol, 28-chloroacetate), which was prepared by Kommera et al. by reacting BET with chloroacetic anhydride in DCM and stirring with full microwave power (250 W) at 90 °C under a maximum pressure of 10 bar for 20 min [56], all other BET derivatives were prepared starting by the proper brome alkyl acid in DCM, by Steglich type esterification, using a condensing agent, which was EDC for Ye et al. [53] and DCC for Tsepaeva et al. [54,55] and DMAP as catalyst. The monoesters were achieved in very variable yields, from 40% for Ye et al. [53], to yield in the range of 45-95% as reported by Tsepaeva et al. [54,55]. Specifically, we followed the procedure described by Tsepaeva et al. using immediately 3 equivalents excess of bromoacid to promote in a single reaction the formation of both mono- and diester derivatives, thus achieving the never reported monoester 11 (57% isolated yield) in mixture with the unknown diester 12 (31% isolated yield) with an overall yield of 88%, which were separated by FCC. Note that, while the formation of diesters were reported by Tsepaeva et al.[54,55], when an excess of brome acids 1:2 was used, the BET di-brome-pentanoate was not reported by Ye et al., despite their excess 4:1 of 5-bromo-pentanoic acid [53]. Isolated compound 11 was characterized by 1H and 13C NMR spectroscopy analysis, which confirmed its structure and high level of purity. Particularly, while in 1H NMR spectrum of BET, the diastereotopic proton atoms of methylene C-28 provided a signal at 3.81 and 3.34 ppm (J = 10.80 Hz) [48], in the spectrum of 11, such signal shifted at 4.26 and 3.84 ppm (J=11.1 Hz), as reported by Tsepaeva et al. [54]. Five additional methylene signals appeared, due to the alkyl chain of 6-bromo-hexanoate group on C-28, which were dispersed among the signals of CH and CH2 groups of BET, thus being of critical attribution. Anyway, the attribution was possible for the CH2Br and CH2C=O groups, whose signals were found at 3.40 and 2.34 ppm, respectively. Similarly, in the 13C NMR of 11, the signal of C-28 appeared at 62.9 ppm (60.47 ppm in BET), in agreement with findings of Tsepaeva et al. for their BET pentanoate derivative [54]. Moreover, the signals of C=OO, CH2C=O and CH2Br groups were found at 174.1, 33.7 and 32.6 ppm, respectively. Copies of spectra of 11 are available in Section 1.2 (Supplementary Materials) as Figure S1.2.13 and S1.2.14.
(3β)-Lup-20(29)-Ene-3,28-Diol, 28-(6-Triphenyl Phosphonium-Hexanoate) 5. BET-TPP-derivative 5 was unknown, but analogous compounds with shorter alkyl chains, such as (3β)-lup-20(29)-ene-3,28-diol, 28-(5-triphenyl phosphonium pentanoate) [53,54], (3β)-lup-20(29)-ene-3,28-diol, 28-(4-triphenyl phosphonium butanoate) [52] and (3β)-lup-20(29)-ene-3,28-diol, 28-(2-bromoethanoate) [54] have been reported. They were prepared following the same procedure, based on a SN2 nucleophilic substitution of bromo atoms, using Ph3P as nucleophile, in CH3CN at reflux. Such procedure was that followed by us but replacing acetonitrile with toluene. Compound 5 was obtained in 36% yield after FCC isolation, which was a yield significantly higher than that reported by Ye et. al. (15%) [53] but lower than those reported by Tsepaeva et al. (95%) [54]. The structure of 5 and its high level of purity was confirmed by ATR-FTIR, 1H, 13C and 31P NMR, was well as by GC-MS, ESI-MS, elemental analysis and potentiometric titrations, which confirmed its molecular weight. Concerning the NMR spectra, the 1H and 13C NMR signals of CH2 Br group were substituted by those of CH2P+ groups, which were found at 3.87 ppm instead of 3.40 ppm in the proton spectrum and at 22.5 instead of 32.6 ppm in the carbon analysis. More important, new aromatic CH signals were found in the 1H NMR spectrum of 5 at 7.84, 7.78 and 7.69 ppm, due to the 15 proton atoms of the new TPP group. Similarly, new signals sometimes in form of doublet, for the coupling with phosphorous atom, were found in the 13C NMR spectrum of 5 at 135.0 (3 equivalent CH=), 133.8, 133.7, 133.7, 133.6 (6 equivalent CH=), 130.5, 130.5, 130.4 (6 equivalent CH=), 118.7, 118.1 (3 equivalent quaternary C) ppm. The 31P NMR spectrum of 5 evidenced a single signal at δ = 24.41. Copies of spectra of 5 are available in Section 1.2 (Supplementary Materials) as Figure S1.2.17-S1.2.19.
(3β)-Lup-20(29)-Ene-3,28-Diol, 3,28-di-(6-Bromo Hexanoate) (12). BET derivative 12 was herein prepared for the first time. Anyway, analogous compounds with shorter alkyl chains, such as (3β)-lup-20(29)-ene-3,28-diol, 28-di-(5-bromopentanoate), (3β)-lup-20(29)-ene-3,28-diol, 28-di-(4-bromobutanoate) and (3β)-lup-20(29)-ene-3,28-diol, 28-(2-bromoethanoate) have been reported by Tsepaeva et al. [54,55]. Authors achieved the desired diesters starting from BET and an excess 2:1 of the proper brome alkyl acid dissolved in DCM, using DCC as condensing agent and DMAP as catalyst. The di esters were achieved in 85, 60 and 70% yields [54,55]. BET di-(6-bromo hexanoate) prepared by us using an excess 3:1 of bromo acid in the same condition of Tsepaeva et al. was not prepared by previously, and the attempt of Tsepaeva et al. to prepare the BET-di-3-bromo propionate failed [55]. Compound 12 was characterized by 1H and 13C NMR spectroscopy analysis which confirmed its structure and high level of purity. In the 1H NMR spectrum of 12, the value of δ of the signal of methylene C-28 did not change significantly (4.27 and 3.84 ppm, J=11 Hz). The signals for the two CH2C=O groups were found in form of multiplet integrable for 4 protons at 2.33 ppm, while those of CH2Br groups appeared as triple doublet, integrable for 4 protons at 3.40 ppm. The signal of CH-3 shifted from δ value of 2.19 ppm (BET) [48] to δ = 2.44 ppm. In the 13C NMR spectrum of 12, distinct signals at 174.1 and 173.5 ppm were found for the 2 C=OO groups, at 34.4 and 34.3 ppm for the CH2C=O groups and as repeated signal at 32.6 ppm for the CH2Br groups, while the signal of C-3 shifted from the δ value typical of BET (78.96 ppm)[48] to an higher value (δ = 81.0 ppm). Copies of spectra of 12 are available in Section 1.2 (Supplementary Materials) as Figure S1.2.15 and S1.2.16.
(3β)-Lup-20(29)-Ene-3,28-Diol, 3-28-di-(6-Triphenyl Phosphonium-Hexanoate) 6. As in the case of BET-TPP-derivative 5, BET TPP-derivative 6 was unknown. Three compounds like 6 were prepared by Tsepaeva et al. starting from the (3β)-lup-20(29)-ene-3,28-diol, 28-di-(5-bromopentanoate), (3β)-lup-20(29)-ene-3,28-diol, 28-di-(4-bromobutanoate) and (3β)-lup-20(29)-ene-3,28-diol, 28-(2-bromoethanoate), via their rection with Ph3P in acetonitrile at refluxing temperature, achieving the desired products in 90%, 83% and 80% yield [54]. Using the same procedure, but replacing acetonitrile with toluene, we prepared compound 5 in 30% yield after FCC isolation. The structure of 6 and its high level of purity was confirmed by ATR-FTIR, 1H, 13C and 31P NMR, was well as by GC-MS, ESI-MS, elemental analysis and potentiometric titrations, which confirmed its molecular weight. Concerning the NMR spectra of 6, the 1H and 13C NMR signals of two CH2Br groups were substituted by those of two CH2P+ groups, which were found at 3.83 ppm in form of triple doublet (4H) instead of at 3.40 ppm (proton spectrum) and at 22.6 ppm instead of 32.6 ppm in the carbon analysis. More important, new aromatic CH signals were found in the 1H NMR spectrum of 6 at 7.83, 7.78 and 7.69 ppm, due to the 30 proton atoms of the new TPP groups. Similarly, new signals sometimes in form of doublet, for the coupling with phosphorous atom, were found in the 13C NMR spectrum of 6 at 135.2 (6 equivalent CH), 133.9 (12 equivalent CH=), 130.7 (12 equivalent CH), 118.9, 118.4, 118.4 (6 equivalent quaternary C), ppm. The 31P NMR spectrum of 6 evidenced a single signal at δ = 24.36 ppm. Copies of spectra of 6 are available in Section 1.2 (Supplementary Materials) as Figure S1.2.20-S1.2.22.
6-(Tri-Phenyl Phosphonium)-Hexanoic Acid (10). Compound 10 is known and commercially available. It was previously prepared by different authors following similar procedures, with slight differences in terms of stoichiometry, temperature, times of reaction and purification work-up [57,58,59,60]. Additionally, the synthesis of 10 was described in paragraph 000561-000563 of patent by Han, Okamoto and Olson (Current Patent Assignee: REGENERON PHARMACEUTICALS – WO2022/56494, 2022, A1), which was followed by us. The method consisted of reacting 6-bromohexanoic acid with a slight excess of PPh3 in toluene under stirring at 120 °C for 12 h. It is curious that according to what reported in the literature articles, compound 10 was always obtained in very high yield (> 90%), regardless the different conditions [57,58,59,60], while in the patent it was obtained in 56.4% yield as in our case. Compound 10 was isolated as white solid and used as such in subsequent reactions.
3.1.2. Synthesis of BA Derivatives
(3β)-3-Hydroxy-N-2-Propyn-1-Yllup-20(29)-En-28-Amide 3. BA derivative 3 was known and it, as well as its betulonic analogous were prepare by Deng et al. and Bȩbenek et al. [35,61]. Without transforming BA in its chloride derivative to be reacted with propargyl amine as reported by Bȩbenek et al., to prepare the amide derivative of betulonic acid, compound 3 was directly obtained from BA following a procedure like that used by Thi et al., with substantial differences. While Thi et al. used DCC, 1-hydroxybenzotriazole and N,N-diisopropylethylamine (DIPEA) in DMF at room temperature for 12 h, HATU and Et3N in THF at 0 °C to r. t. were employed by us [61]. Compound 3, which was isolated as white solid with melting point value of 236-238 °C, was obtained as colourless oil after FCC with high level of purity by us. The structure of 3 and its high level of purity was confirmed by ATR-FTIR, 1H and 13C NMR spectroscopy, GC-MS, ESI-MS, elemental analysis and potentiometric titrations, which confirmed its molecular weight. Observing the 1H NMR spectrum of 3, new signals were found, which are missing in the spectrum of its precursor UA, due to the introduction of the propargyl amine on C-28 carboxyl to form the propargyl amide function. Such signals included a triplet at 5.71 ppm (NH), a double doublet signal at 4.08 and 3.98 ppm with a J = 17.5 Hz (diastereotopic proton atoms of the methylene of propargyl group) and a triplet at 2.20 ppm was detected due the ≡C−H group, whose multiplicity indicated couplings with the methylene (J3 = 2.5 Hz). Similarly, in the 13C NMR of 3, new signals, due to the introduction of the propargyl amine were detected at 80.4 ppm (C≡C−H), 71.4 p (≡C−H) and 28.2 ppm (-CH2-C≡C−H), while the signal of C=O, which in the spectrum of UA was at 177.30 ppm [62], shifted at lower value of δ (176.1 ppm). Copies of spectra of 3 are available in Section 1.2 (Supplementary Materials) as Figure S1.2.8 and S1.2.9.
(3β)-3-Hydroxy-3-(6-Triphenyl Phosphonium Hexanoate)-N-2-Propyn-1-Ylurs-20(29)-En-28-Amide 4. Compound 4 was unknown and was prepared for the first time by us, following the same procedure carried out for the esterification reactions of BET, using derivative 3 having only one free hydroxyl to be esterified as starting material, in place of BET and 10 in place of 6-bromo hexanoic acid, as the acidic component. After isolation of pure 4 by FCC, it was obtained as colourless oil in 34% yield. The structure of 4 and its high level of purity was confirmed by ATR-FTIR, 1H, 13C and 31P NMR, was well as by GC-MS, ESI-MS, elemental analysis and potentiometric titrations, which confirmed its molecular weight. In the 1H NMR spectrum of 4, the signal of CH-3 shifted from the typical δ value reported for BA (3.19 ppm) [62] to 4.72 ppm. Five new methylene signals, due to the TPP-hexanoate alkyl chain, were dispersed in the region under 3.00 ppm. About these signals, those of CH2C=O (2.25 ppm) and of CH2P+ (3.90 ppm) groups, were unequivocally assigned, while the signals of the 15 aromatic proton atoms of the TPP group were clearly visible at 7.85, 7.77 and 7.69 ppm. As in the proton spectrum, in the 13C NMR of 4 the signal of C-3, which in the spectrum of BA is typically detectable at 76.80 ppm [62], was found at higher value of δ (81.2 ppm), due to the esterification of hydroxyl in C-3. The signal of C=ONH and C=OO groups were observed at 176.2 and 173.7 ppm, respectively, while those of three aromatic rings, sometimes in form of doublets, due to the coupling with P atom, were detected at 135.2 (3 equivalent CH=), 134.1, 134.0 (6 equivalent CH=), 130.8, 130.7 (6 equivalent CH=), 119.2, 118.6 (3 equivalent quaternary C), ppm. More signals were observed for the CH2P+ group at 22.9, 22.8, 228 and 22.6 ppm, due to the coupling with P atom, while the CH2C=O group of hexanoate gave a signal at 34.4 ppm, as observed also in the spectrum of 6. In the 31P NMR a single signal was observed at δ = 24.39 ppm. Copies of spectra of 4 are available in Section 1.2 (Supplementary Materials) as Figure S1.2.10-S1.2.12.
3.1.3. Synthesis of UA Derivatives
(3β)-3-Hydroxy-N-2-Propyn-1-Ylurs-12-En-28-Amide 13. The UA derivative intermediate 13 was known and was previously prepared and characterized by different research groups [63,64,65]. Here 13 was prepared avoiding the two-step procedure proposed by Xiao et al. in the years 2014 and 2016 [64,65]. In the papers, authors prepared and isolated first the 1-benzotriazolyl triterpene ester treating UA in THF with 1.5 equivalents excess of TBTU in the presence of DIPEA at room temperature, for 5 h under nitrogen, achieving the desired product after purification by FCC. Secondly, they transformed the 1-benzotriazolyl triterpene ester previously prepared, via its reaction with K2CO3 and 2-propargylamine in DMF at room temperature, under stirring for 1 h under nitrogen atmosphere [64,65]. Our procedure was like the one-step synthesis proposed by Thi et al. with significant modifications. While authors reacted UA with DCC, HOBt and DIPEA in DMF, for 30 min. at room temperature, followed by the addition of propargyl bromide and stirring for 12 h at room temperature [61], we cooled a suspension of UA and treated it with propargylamine in THF, HBTU and Et3N and stirred the mixture at -10 °C to r. t., upon completion of the reaction. As in other reported cases, while Thi et al. isolated 13 as white solid with melting point of 202-204°C in 96% yield, we obtain a colourless oil in 22% yield (56% yield based on unreacted starting material). The structure of 13 and its high level of purity was confirmed by 1H and 13C NM spectroscopy. Particularly, in the 1H NMR spectrum of 13, new signals appeared respect to the spectrum of UA, such as a triplet for the proton atom linked to the nitrogen of amide group (6.07 ppm), two complex signals of the diastereotopic protons of methylene of the propargyl group (4.03 and 3.89 ppm, J = 17.6 Hz) and the triplet of the proton atom of alkyne group, coupled with protons of methylene (2.20 ppm). In the 13C NMR spectrum of 13, the signal of C=ONH group shifted to a slightly lower value of chemical shift (178.2 ppm), respect to that of C=OOH of UA, typically found at 178.7 ppm [66]. New signals appeared at 80.0 ppm (C≡C−H), 71.8 (≡C−H) and 28.4 (CH2-C≡C−H) ppm, due to the introduction of new propargyl group. Copies of spectra of 13 are available in Section 1.2 (Supplementary Materials) as Figure S1.2.23 and S1.2.24.
(3β)-3-Hydroxy-(6-Triphenyl Phosphonium-Hexanoate)-N-2-Propyn-1-Ylurs-12-En-28-Amide 7. Compound 7 was unknown and was prepared by reacting an ice-cooled suspension of intermediate 13 in DCM previously prepared, with a slight excess of 10, in the presence of DCC and DMAP, following the usual esterification procedure carried out for preparing compounds 1, 4, 11 and 12. After 24h stirring at r. t., the reaction was completed and the pure compound 7 was isolated as colourless oil after FCC. The structure of 7 and its high level of purity was confirmed by ATR-FTIR, 1H, 13C and 31P NMR, was well as by GC-MS, ESI-MS, elemental analysis and potentiometric titrations, which confirmed its molecular weight. Particularly, in the 1H NMR spectrum of 7, the signal of CH-3, which was reported at 3.01 ppm for UA [66] and was detected at 3.22 ppm in the spectrum of compound 13, shifted to higher value of δ (4.49 ppm). New signals appeared respect to the spectrum of 13, due to the introduction of the 6-TPP-exanoate in C-3. Specifically, signals for the 15 aromatic proton atoms of the triphenyl group were detected at 7.84, 7.78 and 7.69 ppm, while the signals of the protons of the five methylene groups of the alkyl chain were dispersed in the region were the also several other signals of the UA scaffold can be found. Anyway, the attribution of the signals of CH2C=O and CH2P+ groups was possible, with a high degree of certainty. In this regard, the methylene group linked to the P atom provided two multiplets at 3.88 and 3.82 ppm, while that linked to the C=OO group gave two signals at 2.26 (td) and 2.20 (t) ppm. In the 13C NMR spectrum of 7, the signal of C-3, reported at 77.3 ppm for the UA [66] and found at 79.2 ppm in the spectrum of 13, was detected at 79.9 ppm. New signals were observed for the C=OO group at 173.6 ppm, for the aromatic carbons of the TPP group at 135.2 (3 equivalent CH=), 134.0, 133.9 (6 equivalent CH=), 130.7 (6 equivalent CH=), 118.9, 118.3 (3 equivalent quaternary C), some of which in form of doublets for the coupling with the phosphorous atom, and new signals were observed for the new methylene groups of the alkyl chain of the hexanoate. Of these signals, the signals of CH2C=O (34.3 and 34.2 ppm) and of CH2P+ (22.7 ppm) groups could be attributed with certainty. As for other TPP-containing compounds the 31P NMR of 7 showed a single signal (δ = 24.38). Copies of spectra of 7 are available in Section 1.2 (Supplementary Materials) as Figure S1.2.25-S1.2.27.
3.2. ATR-FTIR Spectra of BA, BET, UA and Compounds 1-7
ATR-FTIR spectra of all samples were acquired as detailed in the experimental parts and the related images are available in the Supplementary Materials in Figure S1-S10. Despite known, the ATR-FTIR spectra of BA, BET and UA were anyway run for a more reliable comparison with those of their derivatives by multivariate analysis as detailed in the experimental Section and later discussed in this section. Bands assignment to functional groups of the chemical structures of analysed samples, reported in the experimental part, was made according to Infrared Spectroscopy Absorption Table available online at https://chem.libretexts.org/Ancillary_Materials/Reference/Reference_Tables/Spectroscopic_Reference_Tables/Infrared_Spectroscopy_Absorption_Table (accessed on 01 November 2025). Bands of stretching aliphatic C-P were assigned according to what reported by Daasch and Smith [67]. Specifically, in BA spectrum weak bands at 3445 and 3079 cm-1 due to the O-H and =C-H stretching respectively, were observable. Bands related to the C-H stretching of methyl and methylene groups were present at 2939, 2965 and 2869 cm-1, while the C=O stretching of carboxylic acid group in C-28 was detectable at 1684 cm-1. Bands observed at 1451, 1043 and 885 cm-1 were assigned to the C-H banding, C-O stretching and -C=C- banding, respectively. The spectra of BA derivative 3 showed bands similar to those of BA for the O-H and =C-H stretching (3309 and 3073 cm-1), for the C-H stretching of methyl and methylene groups (2940 and 2867 cm-1), for the C-H banding (1505, 1447 cm-1) and for the -C=C- bond (881 cm-1). In addition, a new band at 1639 cm-1 was observable for the C=O stretching of the amide group, which substituted that of C=O of carboxylic acid previously observable at 1684 cm-1. The stretching band of C≡C−H bond of the propyne group was not detectable, as in other compounds containing the propargyl amide group (4 and 7). This fact was due to the weak acidity of proton atoms of the methylene propargyl groups (CH2-C≡C−H), which can tautomerize from form I to provide allenyl derivatives (II), which no longer contain the triple bond [68] (Scheme 3).
Scheme 3.
Propargyl–allenyl tautomerization process [68]. The Scheme is a reproduction from an open access article published on Molecules, and no special permission is required to reuse all or part of article, including figures and tables, since published under an open access Creative Common CC BY license.
Scheme 3.
Propargyl–allenyl tautomerization process [68]. The Scheme is a reproduction from an open access article published on Molecules, and no special permission is required to reuse all or part of article, including figures and tables, since published under an open access Creative Common CC BY license.
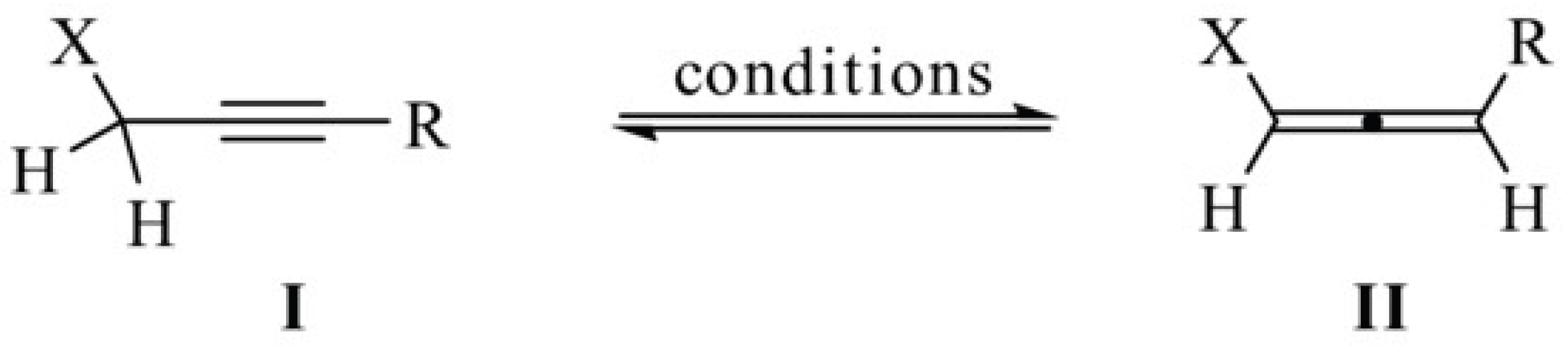
In our specific case, tautomerism was further promoted by the presence of the amide function, where the lone pair on the nitrogen atom can delocalize towards the carboxyl group to form new compounds with a negative formal charge on the oxygen and a positive one on the nitrogen, thus augmenting the acidity of the H atoms in CH2, necessary for tautomerism. In the spectrum of the other BA derivative 4, having new groups on both the secondary OH group on C-3 and on the carboxylic acid group in C-28, in addition to bands already detected in the spectrum of BA for the =C-H stretching (3061 cm-1), for the C-H stretching of methyl and methylene groups (2940, 2868 cm-1) and for the C-H banding (1439 cm-1), new bands of stretching C=O of the amide and ester groups were observable at 1639 cm-1 and 1721 cm-1. Moreover, two very strong bands due to the aliphatic C-P stretching were present at 723 and 690 cm-1, the C-O band shifted to 1112 cm-1 and like for 3, the C≡C−H bond of the propyne group was not detectable. Similarly to BA the spectrum of BET showed weak bands at 3486 and 3077 cm-1 due to the O-H and =C-H stretching respectively, bands related to the C-H stretching of methyl and methylene groups at 2970, 2931 and 2868 cm-1, no stretching C=O band was present due to the absence of carboxylic acid group, replaced by a primary alcohol (CH2OH) group in C-28, while at 1452, 1009 and 887 cm-1 were visible the C-H banding, the C-O stretching and the -C=C- banding, respectively. BET derivatives 2 and 5 showed bands like those of BET for the O-H and =C-H stretching (3362, 3339 and 3057, 3054 cm-1), for the C-H stretching of methyl and methylene groups (2938, 2867 and 2941, 2867 cm-1) and for the C-H banding (1438 and 1436 cm-1), while the C-O stretching shifted to 1112 cm-1 as in the case of compound 4. In addition, in the spectrum of 2, containing the propargyl ester in C-28, the C≡C−H stretching of the triple bond was this time clearly visible at 2120 cm-1 as weak band. Conversely, in the spectrum of 5 containing the TPP-hexyl ester, new bands at 1726 and at 723, 690 cm-1 were observed, due to the C=O stretching of ester and to the stretching of aliphatic C-P. BET derivatives 1 and 6 showed bands like those of BET for the =C-H and C-H stretching (3060, 2941, 2869 and 3054, 2941, 2867 cm-1) and for the C-H banding (1438 and 1436 cm-1), while the C-O stretching shifted to 1112 cm-1 as in the case of compound 2, 4 and 5. In addition, in the spectrum of 1, an additional band for the C-O stretching was visible at 1246 cm-1. In the spectrum of 1, containing both the propargyl carbamate group in C-28, and the TPP-hexyl ester in C-3, the strong and large band of C=O stretching of carbamate and ester groups was visible at 1714 cm-1, while the bands of the aliphatic C-P stretching were observable at 723, 689 cm-1. As in the cases of compounds 3 and 4, the C≡C−H stretching of the triple bond was not visible. Conversely, in the spectrum of 6 containing two TPP-hexyl ester groups in C-3 and C-28, the band of C=O stretching of ester groups was visible at 1721 cm-1, while the bands of the aliphatic C-P stretching were observable at 745, 722 and 690 cm-1. Similarly to BA, the spectrum of UA showed a weak band at 3423 cm-1 due to the O-H stretching, bands related to the C-H stretching of methyl and methylene groups at 2976, 2951, 2928 and 2869 cm-1, a C=O stretching band at 1686 cm-1, associated to a new band at 1714 cm-1, due to carboxylic acid group in C-28, while at 1452 and 1030, 1042 cm-1 bands were visible for the C-H banding and the C-O stretching. The band of =C-H stretching and of the -C=C- banding were not visible. UA derivative 7 showed bands like those of UA for the C-H stretching of methyl and methylene groups (2962, 2924, 2869 cm-1) and for the C-H banding (1439 cm-1), while the C-O stretching shifted to 1112 cm-1 as in the case of compounds 1, 2, 4, 5 and 6. In addition, the band of =C-H stretching, not detected in the spectrum of UA, was visible at 3058 cm-1. New bands were then detected at 1722, 1644 and 746, 729, 690 cm-1, related to the C=O stretching of the ester (1722) and amide (1644) groups and to the aliphatic stretching C-P. As in the case of compounds 3 and 4, the CΞCH stretching of the triple bond was not visible.
3.3. Principal Component Analysis (PCA) of ATR-FTIR and 13C NMR Spectral Data
PCA is a chemometric tool, making part of multivariate analysis (MVA), able to reduce many variables of a certain dataset to a small number of new variables, called principal components (PCs)[69]. PCA allows to reduce a high number of correlated variables (organized in a matrix of n columns x n rows), also including those variables providing non-significant and redundant information, to a limited number of uncorrelated variables, namely Principal Components (PCs), providing the most important information[70]. The number of PCs is always lower than the number of samples analysed. In the following Sections, we have presented and discussed the results obtained by processing both ATR-FTIR and 13C NMR spectroscopic data by PCA, presented as score plots. In both cases, in the score plot the scores were the new coordinates of the processed samples in the new space of the PCs. Note that, PCA was applied to process spectral data intended as transmittance (%) (ART-FTIT) or δ (ppm) values (13C NMR), which are correlated to the functional groups present in the samples’ structures and with their structures themselves, in the first case and to the number and type of carbon atoms present in the samples’ structures and with their structures themselves, in the second case. In this regard, within the score plot, each sample assumed a position depending on its chemical composition and structure[71]. Samples located close to each other shared similar characteristics, while those placed far apart differed[71].
3.3.1. PCA of ATR-FTIR Spectral Data
The matrix of 34,010 variables (A) and that of 37,411 variables (B), where variable were the transmittance (%) values of each sample in the ATR-FTIT spectrum, were constructed as described in the experimental Section. The results from the PCA on both A and B have been reported as score plots explaining a total variance of 82.5% and 90.7%, respectively. In the first case, PC1 explained 69.6% of total variance vs. PC2 which explained 12.9% (Figure 3a), while in the second case, PC1 explained 84.0% of total variance vs. PC2 which explained the 6.7% (Figure 3b).
As observable in Figure 3a, BA and UA, which sheared the carboxylic group, despite in opposite locations on PC1 (negative score for BA and positive for UA), due to the significantly different triterpene structure, assumed similar scores close to 0 on PC2. Conversely, BET not bearing the carboxylic group was located distant from either BA and UA, both on PC1 (close to 0 score) and PC2 (negative score). Anyway, shearing the same triterpene structure with BA, BET was positioned closer to BA than to UA on PC1 and had negative score on PC2 as BA, while UA had positive. The PC unequivocally evidenced that compounds 3 and 7 are BA and UA derivatives respectively, since on both CP1 and CP2, they have score very similar to those of BA and UA. As UA, 7 had negative scores on PC1 and positive on PC2, while as BA, 3 had positive scores on PC1 and close to zero on PC2. On PC1, compounds 1, 2, 5 and 4 were positioned between BET and BA, since they sheared with them the same triterpene structure, while were positioned at opposite scores (positive) respect to UA (negative) due to its significantly different triterpene structure. Anyway, while BA was positioned at score > 50 as 3, compounds 1, 2 and 5 were at scores < 50, closer to BET, thus confirming their BET origin. Additionally, compounds 1 and 5 were very close to score 0 and to each other on PC2 since, differently from 2 they contained the TPP-hexyl ester group. The BET origin of 6 was evident on PC2, since both BET and 6 occupied similar negative scores. The fact that compound 6 positioned at negative scores > -50 on PC1 as compound 7 could depend by their double functionalization in C-3 and C-28 with the same groups, while the presence of a different triterpene nucleus in such compounds was evidenced on PC2 where while 7 had positive score, 6 had negative. Moreover, despite its double functionalization, compound 1 was located significantly distant from both 6 and 7, positive score on PC1 and close to zero on PC2 because of the presence of carbamate functional group absent in both 6 and 7. The initially unexpected location of compound 4 very close to BET, which can make think to a BET origin for 4 despite its parent is BA, can be explained with the same triterpene core for both BET and BA and with its double functionalization presenting the same TPP-hexyl ester group of 6, which rendered it structurally more similar to 6 than to BA and caused its location at a score similar to that of 6 on PC2. Anyway, 4 positioned at positive score on PC1 as BA, while BET at negative ones. Notably, adding to the dataset the spectral data of BPPB, which sheared with compounds 1, 4, 5, 6 and 7 only the presence of TPP group, the PCA score plot of PC1 vs. PC2, well separated BPPB from all other triterpene compound on PC1. While BPPB located at negative score > -100, while all other compounds were at positive scores or in the case of UA, 7 and 6, they were located at negative scores, but very close to zero. Curiously, the relative positions occupied by the triterpene compounds were identical to those occupied by them in the entire score plot in Figure 3a, except that in this new score plot, they grouped on its right side to distance themselves from BPPB. Anyway, a greater similarity between compound 6 and BPPB, due to the presence of two TPP groups was evidenced on PC2 where compounds had practically identical negative scores.
3.3.2. PCA of 13C NMR Spectral Data
To perform PCA on a data set of NMR spectral data, 13C NMR data were chosen in place of 1H NMR ones, because total-decouple carbon NMR provide in the most case a single peak for a values of δ (ppm), thus avoiding multiplicity The matrix of 510 variables, where variables were the values of δ (ppm) of signals in a selected region of the 13C NMR spectrum of each analysed sample, was constructed as described in the experimental Section. The results from PCA have been reported as score plots explaining a total variance of 79%. In the score plot, PC1, explaining the 73.7% of total variance has been reported against PC3, explaining the 5.4 of total variance (Figure 4).
As observable in Figure 4, the three pristine triterpenoids were located very close to each other at the most negative scores of about -5 on PC1, thus evidencing their differences form their derivatives. Anyway, since BA and UA sheared the carboxylic group, while BET no, they were very close to each other on PC3 at positive scores, while BET was positioned at negative score. Moreover, all samples bearing the 6-triphenyl phosphonium hexanoyl group (1, 4, 5, 6 and 7) were well separated form compounds not bearing it (BET, BA, UA, 2, 3, 11, 12, 8 and 9) on PC1. Precisely, they located all at positive scores, while compound without such group positioned all at negative scores. Among the TPP-group at positive scores, apart from compound 1, which located curiously close to 10, PC1 separated well compounds having the triterpene nucleus, from the not triterpene 10. Anyway, the clear separation of 1 from 4, 5, 6 and 7 on both PC1 and PC3, could indicate that 1 is the only TPP-compound containing the carbamate group. Among the not TPP-compounds at negative scores on PC1, PC3 made separated well the most part of compounds containing the triple carbon-carbon bond (2, 3 and 9), which located at negative scores from those not owing it (11 and 12) positioned at positive scores. As exceptions, despite containing the triple bond, 13 located at positive score, because of its structural identity with UA, which was its origin, while 8 located at negative scores, because of its structural identity with BET, which was its origin.
3.4. Optical Microscopy Analyses
It has been reported that compounds bearing two triphenyl phosphonium groups (TPP) linked by a hydrophobic chain of 12 carbon atoms, defined as bola-amphiphiles, when dispersed in aqueous solution at proper concentration, spontaneously self-assembled into spherical vesicles. Depending on the objective used (40× or 100×), vesicles appeared as spherical aggregates (10-30 µm) of smaller spheres (1-3 µm) or well dispersed small vesicles of 1-3 µm [8,72]. In this regard, leaving apart compounds 2 and 3 not possessing the TPP groups, the water solutions prepared according to the procedure detailed in the experimental Section, provided by all compounds bearing TPP, were investigated using optical microscopy. The solutions were examined with a 40× and 100× objective, observing spherical poly dispersed vesicles only for compound 6 carrying two TPP groups, linked to the hydrophobic structure of betulin (BET) via spacers of 7 and 8 atoms. This finding confirmed that the capability to form spontaneously vesicles in water is peculiar of bola-amphiphilic compounds [72]. Figure 4 shows the spherical vesicles provided by 6 as they appear when observed with the 40× (Figure 4a) and 100× (Figure 4b) objectives.
At the used concentration of 6, large spherical vesicles of 24 μm or larger were clearly visible using a 40× objective. Anyway, as previously reported by us for a much simpler bola amphiphilic compound (BPPB), encompassing two TPP groups as 6, but linked by a less complex spacer, made of a chain of 12 carbon atoms [8], these spheres revealed to be formed of smaller vesicles of about 4 μm, which also appeared not clearly visible in the background (Figure 4a). Using the 100× objective, it was possible to bring to the forefront these smaller vesicles of 1.58 ± 0.13 μm, leaving on the background the larger aggregates (Figure 4b). Despite optical microscopy is not a precise method to evaluate the morphological and topographical characteristics of particles, as well as their size, because observations are strictly limited by the focus distance, these findings confirmed that compounds bearing two cationic heads linked by hydrophobic spacers form spherical vesicles of different sizes in water, which can aggregate in larger one depending on the solution concentration and coexist with them [8]. As reported, 6 was able to self-assemble in spherical vesicles, due to its planar cationic TPP headgroups because self-assembly properties are strongly dependent on the complex interplay of non-covalent interactions (ionic, hydrophobic, and π-π) inside the aggregate [8]. In this regard, the π-π stacking between the three aromatic rings on TPP heads of 6 was crucial for the final aggregate morphology.
3.5. Particle Size, ζ-p, and PDI of 6
Using the m-Q water solution of 6 (5 mM), the hydrodynamic size (diameter) (Z-AVE) and polydispersity index (PDI) of 6 vesicles were determined by DLS analysis, to assess the dimensional distribution of its particles and how much their distribution could be uniform. Additionally, zeta potential (ζ-p) measurements were carried out on the same solution, to determine their surface charge. According to results reported in Figure 5a, 6 the analysis evidenced the presence of more than one dimensional family. Precisely, small dimensional families made of nanovesicles were detected with average size of 473 nm, together with larger families made of microparticles up to 2.4 µm. Collectively, the calculated average vesicles dimension was 1.6 µm, while PDI was 0.277, thus confirming the scenario already observed in OM.
Differently from what observed in our recent publication, where it was observed that the microparticles observed for BPPB in OM analysis, as for 6, at the DLS investigation appeared nano dimensioned (49 nm), particles formed by 6 in water resulted micro dimensioned also in DLS analysis. We suppose that the fact that vesicles from 6 were micro- and not nano-dimensioned could depend on the nature of linker which was more complex than the simple carbon chain of BPPB, encompassing also atoms different from carbon and mainly the tri-dimensional structure of BET, which influenced the final dimensions of vesicular spheres. As evidenced in Figure 5b, vesicles self-formed by 6 in aqueous solution demonstrated various ζ-p distributions as in the case of size. Anyway, all distributions had a positive value. Collectively, the average ζ-p of 6 was 19.6±10.2 mV. Size of particles strongly influences their distribution, cytotoxicity, and targeting ability when in vivo administered [24,73,74]. Generally, in biomedical applications, compounds for systemic administration should require sizes lower than 200 nm, with an optimal size of 100–200 nm [24]. In this regard, envisioning a possible future clinical administration of 6 as an antibacterial agent, due to the micrometric size of its particles, it could be suggestable for topical administration to treat skin infections. Concerning ζ-p, 6 showed positive values, which are desirable for effective drugs. In fact, based on the studies published so far, the internalization of positively charged nanoparticles (NPs) is more efficient than that of neutral and anionic ones[75,76,77,78,79,80]. It was in fact discovered that after electrostatic interaction with anionic components of the cell membrane as phospholipids, positively charged materials can be enter the cells by both pore formation, micropinocytosis and clathrin- or dynamin-dependent endocytosis [76].
3.6. Non-Aqueous Potentiometric Titration of Compounds 1 and 4-7
To have further confirmation of the molecular weight (MW) and structure of cationic compounds 1 and 4-7, we titrated their phosphonium groups to experimentally determine the P+ equivalents contained in an exactly weighted sample. Comparing the obtained results with those calculated according to the molecular weight of 1 and 4-7, we would have had confirmation of their mass and structure. In this regard, non-aqueous titrations emerged in the middle of the last century, enabled the possibility of titrating both weak acids and bases, not measurable in aqueous media [81,82,83]. Especially, the titration with perchloric acid in glacial acetic acid medium is widely used to determine salt of weak basic drugs. Titrations in acetic anhydride-acetic acid mixture allow the direct non-aqueous titration of halide salts (mainly hydrochlorides) of organic bases and quaternary ammonium salts. Additionally, by adding mercury (II) acetate reagent to the quaternary ammonium salts solution, stable mercury (II) halide complexes and free acetate ions (equivalent to the base) form, which can be titrated with perchloric acid [28,84]. On these considerations, we carried out the potentiometric titration of quaternary phosphonium salts 1 and 4-7 in a mixture of anhydrous acetic acid (AcOH) and acetic anhydride (Ac2O) 30:70 (v:v) with 0.1 N HClO4, performing the slightly modified procedure previously described by us for the volumetric titration of ammonium salts and described in the experimental Section. Briefly, we adapted the protocol described by Pifer and Wollish, who applied this method for titrating quaternary ammonium salts [28] and used micropipettes and micro-burettes for measuring volumes. By plotting the measured mV values against the volumes of 0.1 N HClO4 solution added, we obtained the titration curves of all compounds and the related first derivative (FD) curves. The maxima of the FD represent the titration end points, which allowed us to find the volumes of titrating solution needed to titrate the phosphonium groups of our samples and then their P+ equivalents. Table 1 reports the experimental details of titrations, the calculated P+ equivalents for all samples according to their molecular weight (MW), the experimentally determined P+ equivalents obtained by titrations, the experimental MW, the residuals, and the percentage error (%).
The experimental MWs were perfectly in agreement with the calculated ones, with a maximum error (%) of 0.78% (<1%), thus further confirming the structure of 1 and 4-7.
3.7. Antibacterial Properties
With screening purposes, the antibacterial properties of the synthesized triterpenoids derivatives 1-7 were assessed by determining their minimum inhibitory concentration (MICs) against selected clinical isolates of both Gram-positive and Gram-negative species. Additionally, the antibacterial effects of the 3 not modified triterpenoids betulin (BET), betulinic acid (BA) and ursolic acid (UA) used to prepare compounds 1-7, were also investigated for comparison purposes. For MICs evaluation, 7 strains, all MDR isolates were exploited. The selected 7 MDR isolates included 4 Gram-positive and 3 Gram-negative bacteria of different genera. Among enterococci, strains of E. faecium and E. faecalis were VRE bacteria, also resistant to teicoplanin, while staphylococci were MDR strains with resistance to methicillin (MRSA and MRSE). The non-fermenting P. aeruginosa strain was isolated from cystic fibrosis patients and possessed resistance to carbapenems, while both Enterobacteriaceae possessed resistance to carbapenems and were K. pneumoniae carbapenemase (KPC)-producers. We selected these superbugs, since currently there is a large variety of mechanisms of resistance, which bacteria could develop, to inactivate available antibiotics and make it difficult to manage infections sustained by mutated bacteria. They include β-lactamases (especially NDM, KPC, and AmpC variants conferring resistance to ceftazidime-avibactam, OXA-427, and PER- and SHV-type ESBLs) production, porin mutations, and mutations affecting siderophore receptors, efflux pumps, and target (PBP-3) modifications [7]. Therefore, to improve this condition, limit the emergence of further resistance and to counteract the spread of worrying infections, we have urgent need for novel compounds functioning also on superbugs, as those tested in this study. Importantly, all bacteria used in this study were clinical isolates that had developed resistance to at least one or two antibiotics. Particularly, multidrug-resistant (MDR) E. faecium as well as S. aureus as those used here are included by WHO among the ESKAPE bacteria. ESKAPE strains are a group of Gram-positive and Gram-negative bacteria capable of evading or ‘escape’ commonly used antibiotics due to their increasing multi-drug resistance (MDR) [85]. As a result, they are the major cause of life-threatening opportunistic and hospital-acquired infections throughout the world [86] Table S1 in Section S2 of Supplementary Materials (SM) reports all observed MICs for compounds 1-7, BET, BA and UA on both Gram-positive and Gram-negative species tested, as well as those of reference antibiotics currently available to counteract the considered clinical isolates. Conversely, the following Table 2 is a refined form of Table S1, where data concerning Gram-negative species, weakly active compound 1 and not active compounds 2 and 3, were not further reported.
It is noteworthy that, despite in literature several articles report as active against bacteria new compounds with MICs over 128 µg/mL, in a limited number of studies, only compounds with MIC = 32 µg/mL are retained endowed with weaky antibacterial effects [87]. In this regard, as a compromise between these two trends, here, only compounds with MICs ≤ 64 µg/mL were considered endowed with a certain antibacterial effect. On these guidelines, all compounds tested in this study were inactive against Gram-negative species (MIC > 64 µg/mL), while 6 out of 10 compounds displayed from weak (MICs = 32-64 µg/mL) to strong antibacterial effects (MICs = 2-16 µg/mL), against here-considered Gram-positive strains. These observations align with the fundamental challenges associated with discovering compounds with activity against Gram-negative bacteria [88]. In fact, the attenuated activity of potential antimicrobial agents against E. coli, K. pneumoniae, P. aeruginosa and other bacteria representative of Gram-negative specie, can be related to the reduced permeability of their outer membrane, often acting as a barrier to the entry of antimicrobial drugs or drug candidates [89]. Conversely, the cell wall of Gram-positive bacteria makes the permeation of these compounds easier, allowing their transporte and enter in the bacterial cell[89]. However, it has also been reported that strongly cationic compounds and macromolecules, including antimicrobial cationic peptides (CAMP) such as colistin[90,91], dendrimers with a high number of peripheral ammonium groups [92], cationic benzyl ammonium-based co-polymers [24,93,94], quaternary phosphonium and ammonium polymers[91], and bola-amphiphilic bis-phosphonium nanovesicles [8], can exhibit antibacterial effects against Gram-negative species, comparable or superior to those observed on Gram-positive isolates. These molecules act not specifically as membrane disruptors on contact, without the need to enter cells. Colistin is a classic example of antibacterial drug which is active only on Gram-negative bacteria and not on Gram-positive ones, since it is less attracted by the surface of latter [95]. In fact, the antibacterial potency of strongly cationic materials is mainly due to their greater affinity for interacting with the membrane of Gram-negative bacteria than with that of Gram-positive ones, where the anionic character is weaker [90,91,92]. Additionally, it has been demonstrated that compounds bearing TPP groups, linked to hydrophobic carbon chains of different length, can deviate from this rule, exhibiting very low MICs = 0.25-8 [87] and 1-32 [8] µg/mL also against E. coli, K. pneumoniae and P. aeruginosa, regardless their complicate pattern of resistance [8], thus being promising as new antibacterial devices to counteract the difficult-to-treat infections sustained by these Gram-negative species. On these results, according to MICs > 1024, = 256-512 and >128 µg/mL, already reported for BET [96], BA [97], and UA [98], against Gram-negative species, it was expected that such pristine triterpenoids could be inactive against E coli, P. aeruginosa and K. pneumoniae tested in this paper. But it was unexpected that, according to literature data [87], [8], their synthetic derivatives containing the TPP group could be ineffective as well. A compound recently reported bearing two TPP groups linked by a C12 alkyl chain (BPPB in the study), despite inferior to those displayed against Gram-positive isolates, demonstrated very low MICs = 1-2 µg/mL on E coli, 16-32 µg/mL on P. aeruginosa and 16 µg/mL on K. pneumoniae [8]. Nunes et al. evidenced that among a series of quaternary ammonium and phosphonium salts bearing different cationic heads and carbon chains, those bearing the TPP group demonstrated the most effective antimicrobial activity and the broadest antimicrobial spectrum, providing in the best cases MICs low to 1 (E. coli) and 8 (P. aeruginosa and K. pneumoniae) µg/mL [87]. Anyway, in another less recent study by us [30], a differently structured TPP-bearing compound (PPB), having a 1-undecanol chain linked to a single TPP group, was active against Gram-positive strains including staphylococci and enterococci with low MICs = 4-16 µg/mL, while it was considered inactive against Gram-negative isolates (>128 µg/mL) [30], like TPP derivatives 1 and 4-7 of this study. To explain this evidence, it must be considered that the antibacterial efficacy of quaternary cationic salts (QCSs) is intricately linked to their physicochemical properties, including hydrophobic-hydrophilic balance, water solubility, adsorption efficiency, and critical micelle concentration which could be very different in different TPP-containing compounds [26,27]. Generically, the possible antibacterial effects of TPP containing QCSs depends on their capacity to establish electrostatic interactions with the negatively charged bacterial cell surfaces, due to the presence of the TPP group and to create hydrophobic interaction with bacterial membranes, due to the π-π system of phenyl rings[8,30]. Upon early interactions, such compounds can therefore readily penetrate the protein-lipid biological membranes of bacteria, thus compromising their structural integrity and function [2,3,28], which lead to bacterial protein denaturation, nucleoprotein complex disruption etc., ultimately resulting in irreversible harm to the pathogenic cells [2,3,28]. It derives that the weight that the TPP group assumes within the whole molecule that contains it, becomes of extreme importance for its antibacterial effects and its spectrum of activity against bacteria. Especially, for efficacious effects against Gram-negative species, having a more negative and difficult to penetrate outer membrane, stronger π-π and electrostatic interactions, promoted by more than one cationic TPP groups, are needed and could translate in higher membrane damage causing cell death. Therefore, among previously reported BPPB and PPB, PPB, containing only one TPP residue and possessing a more hydrophilic OH-terminated shorter carbon chain and probably unable to self-assemble nanovesicles in water was not active against Gram-negatives isolates, while strongly active against Gram-positive strains [30]. Conversely, BPPB, owning two TPP heads, linked by a simple and hydrophobic C12 chain and giving nanovesicles of 49 nm in water was active versus both species [8]. Collectively, the structural differences between BPPB and PPB, led PPB to have lower capability to interact with strongly negative surface of Gram-negative bacteria and kill them on contact, thus resulting not active on these species [30], while enabled BPPB to be significantly more active than PPB against Gram-positive isolates (0.25-0.50 vs 4-16 µg/mL) and to be active also against E coli, P. aeruginosa and K. pneumoniae with MICs = 1-32 µg/mL )[8]. Translating these considerations to compounds 1, 4, 5 and 7 of this study, they contained only a TPP group as PPB, did not provide nanovesicles in solution and were constructed on original triterpenoids (BET and BA) known to be inactive against Gram-negative species (MICs from >128 to > 1024 µg/mL), thus justifying their inactivity against E coli, P. aeruginosa and K. pneumoniae (MICs > 64 µg/mL). The BET derivative 6, despite possessing two TPP groups as BPPB and providing spherical aggregates in water, was constructed on BET skeleton, which is reported as the triterpenoid here considered with the lower antibacterial effects and the highest MICs against both Gram-positive and negative species (MICs > 1024 µg/mL) [96]. Additionally, the aggregates formed in water by 6 were micro dimensioned, thus not possessing the high penetrating capacity of nanoparticles formed by BPPB, thus not reaching its antibacterial potency and failing against Gram-negative isolates (MICs > 64 µg/mL). Pristine BET and BA were inactive also against Gram-positive strains, as well as their derivatives 2 and 3 (MICs > 64 µg/mL) not containing the TPP group, thus confirming literature reports where BET demonstrated MICs > 1024 µg/mL against S. aureus [96] and BA showed MICs = 256 µg/mL against S. aureus and S. epidermidis [97]. Collectively, despite the various pharmacological properties of BET and BA, which make them promising compounds as multitarget agents [102,103], they are not encouraging as novel antibacterial agents, as well as their derivatives 2 and 3, thus not meeting our research needs. In this regard, the overall merit of this study consists of having found the correct approach to recovering these compounds for possible use as future antibiotics conferring them from weak (1) to strong antibacterial effects (4-6) at least against Gram-positive superbugs, including ESCAPE bacteria, via insertion of the TPP moiety. Conversely, according to what reported by us concerning the antibacterial effects of UA on the same strains used in this study, using the EUCAST approved cations-adjusted Muller Hinton broth, it possesses per se week antibacterial activity on both staphylococci isolates (MICs = 32-64 µg/mL) and potent antibacterial effects on enterococci (MICs = 2-4 µg/mL) [98]. Our results were in accordance with those of Farjardo et al. concerning S. aureus (31.3 μg/mL), while MICs observed by us concerning E. faecalis were lower than those of Farajado et al. by about 8 times [104]. On the contrary, Sun et al. found MICs inferior to us by 8 times (MICs = 8 µg/mL), 16 times (MICs = 4 µg/mL) and 4 times (MICs = 16 µg/mL) on S. aureus MRSA, S. aureus ATCC 6538 and S. aureus ATCC 29213 respectively and inferior to us by 16 times on S. epidermidis ATCC 12228 (MICs = 2 µg/mL) [105]. Also, Do Nascimento et al. reported for UA, MICs like lower by 2 times (MICs = 32 µg/mL) when tested on S. aureus [106]. Additional different MICs, anyway lower than those observed by us for MRSA and VRE by 21, 8 and 8 times, were reported in the past by Volska et al. for UA (MICs = 3, 4, 8 µg/mL), when tested on MRSA, VRE and S. aureus ATCC 25923, respectively [107]. This scenario unequivocally evidences that literature data concerning the antibacterial effects on Gram-positive species of UA are strongly contrasting, depending on the UA origin and the broth used. Specifically, compound 1 was constructed on BET core, by inserting a propyne carbamate residue on C-28 and a TPP-bearing group on C-3. It was weakly active against both staphylococci and enterococci, exhibiting MICs in the range 32-64 µg/mL, that is at our threshold of inactivity, despite the presence of the TPP group. A slightly higher antibacterial activity was observed on S. epidermidis and E. faecium (MICs = 32 µg/mL), rather than on S. aureus and E. faecalis (MICs = 64 µg/mL), which demonstrated to be the most potent superbugs, as observed for all other compounds, except for compounds 4, 6 and 7 which against S. aureus and S. epidermidis, were equally strongly active. Anyway, despite in our opinion the antibacterial effects of 1 could be considered in general poor, they were significantly higher than those of 9 out of 12 BET derivatives reported as promising antibacterial devices and synthesized by Den et al. for conferring BET a certain antibacterial effect [96]. Specifically, MICs of 1 against MRSA were lower than those of different BET-derived thioether and S-alkylated sulfonium BET derivatives, by 2-387 times [96]. MICs of 1 on MRSA were 4 times lower than that reported by Serrafi et al., for 6 BET-derivatives when tested on S. aureus ATCC 25923 [108]. Moreover, 1 was better performant of 4 out of 5 BET derivatives synthesized by Haque et al. screened by a wider library of 51 BET derived compounds [109]. Specifically, MICs of 1 against superbugs of S. aureus and E. faecalis genus were lower than those of 4 compounds by Haque et at. by 1.6 times [109]. When the BET-derived core of 1 was replaced with the BA-derived one, to give 4 analogously functionalized with the same TPP-bearing group of 1 on C-3 and having a propyne amide group on C-28, the antibacterial activity resulted drastically enhanced, thus providing a compound with MIC = 4 µg/mL on all strains, except for potent E. faecalis where the observed MIC was 16 µg/mL. Practically, by replacing the BET nucleus with that of BA, which intrinsically has MICs decidedly lower than those of BET, the MICs of 1 were lowered by 2-16 times, thus achieving a compound promising for further experimentation as antimicrobial agent. Compound 4 demonstrated antibacterial activity against MRSA extraordinarily higher than that of two novel BA semisynthetic derivatives ((17 S)-17-(((dimethoxyphosphoryl)methoxy)carbonyl)-3β-hydroxy-28-norlup-20(29)-ene as BPm and sodium (3β-hydroxy-(17R)-17-28-norlup-20(29)-en)-2-oxoethyl-phosphonate as BP), which were synthesized and characterized by Lugiņina et al. [110]. Particularly, 4 was more potent of 2 BA derivatives by 136 times, despite such compounds being tested on S. aureus strains without a defined pattern of resistance [111]. When the unmodified BET nucleus was functionalized by esterification reaction of the hydroxyl on C-28 with the usual TPP-bearing moiety, in place of using the propyne derivative previously inserted and living free the hydroxyl on C-3, compound 5 was achieved. Curiously, despite the BET nucleus, previously considered the most responsible for the weak antibacterial effects of 1, 5 displayed antibacterial effects equal to those of 4 on S. epidermidis (MIC = 4 µg/mL), higher on E. faecium (MIC = 2 µg/mL) and E. faecalis (MIC = 8 µg/mL), and slightly lower than those of 4 on S. aureus (MIC = 8 µg/mL). These findings evidence important structural characteristics which are to be considered to have triterpenoids derivatives based on BET or BA with strong antibacterial effects, at least on Gram-positive species. The presence of propyne containing groups on C-28 could be dangerous rather than advantageous. Such modification has in fact left unchanged the antibacterial inactivity of BET and BA, in absence of the TPPG, has still allowed the TPP-containing group inserted on C-3 in 4, to render BA derivative 3 significantly active, but has strongly limited the antibacterial effects of 1, furtherly modified with the TPP-containing group inserted on C-3, respect to derivative 2. At least in the BET derivatives, propyne derivatives on C-28 are not suggestable, both in the presence and absence of TPP-containing moieties, which should be inserted preferably, by esterification reaction of hydroxyl on C-28 rather than of that on C-3, which should be left free. This was confirmed by compound 6, where the further hydroxyl esterification on C-3, despite with an additional TPP cationic group, has lowered the antibacterial effects of 5 by two times rather than enhancing them. Anyway, both compounds 5, and 6 outperformed the antibacterial efficiency of all BET derivatives synthesized by Deng and Serrafy et al. [96,108] and of 4 out of 5 BET derived compounds proposed by Haque et al. by 14 times against S. aureus (5 and 6) and by 7 times on E. faecalis [109]. The best performant compound was 7, as could be expected, since it was constructed using natural scaffold UA, which was already endowed with antibacterial activity against Gram-positive strains, especially of Enterococcus genus [98]. Although the presence on C-28 of the propyne carbamate derivative, which as above mentioned could be a not advantageous modification, in place of the TPP-containing moiety, which was instead inserted on the hydroxyl on C-3 as in compounds 1 and 4, 7 revealed strongly enhanced antibacterial effects respect to pristine UA against staphylococci, where it displayed the lowest MICs (2 µg/mL), thus reducing those of UA, by 16-32 times. Concerning enterococci, the antibacterial effects did not change. Anyway, it is rational to think that the insertion of the TPP-containing group on the hydroxyl on C-28 in place of the propyne derivative leaving free the hydroxyl on C-3 could provide a better performant compound with lower MICs also on enterococci. Anyway, comparing the results of this study with those obtained by us encapsulating UA in G4K nanoparticles, which demonstrated MICs > 128 µg/mL against both staphylococci species tested here, 64 µg/mL against E. faecalis and 32 µg/mL against E. faecium, 7 demonstrated MICs lower by 16 to much more than 64 times [98]. On the other hand, compared to cyclodextrin β-UA (UA-CDβ) nanocomposites proposed by Farjardo et al. as macromolecules with improved antibacterial effects respect to pristine UA, 7 outperformed their activity by 2 times on E. faecalis and by 4 times on S. aureus [104]. Also, compared to the antibacterial activities of UA derivatives synthesized recently by Sun et al., when tested on S. aureus MRSA, 7 outperformed 33 out of 50 compounds and provided results equal to those reported by Sun et al. for 11 compounds among the prepared ones, as well as 7 was more active than norfloxacin by 2 times [105]. Additionally, when tested on S. epidermidis MRSA, 7 was more performant of 31 out of 50 compounds by Sun et al., equally performant to 13 compounds and norfloxacin, which were instead tested on S. epidermidis ATCC 12228 [105]. Despite these results have demonstrated that none of these new compounds was more potent than BPPB recently reported by us [8], at least compound 4, 5, 6 and 7 outperformed the most part of naturally derived antibacterial agents reported so far, especially considering the complex pattern of resistance of bacteria selected by us and their clinical origin. For the first time, we have succeeded in conferring real (MICs = 2-16 µg/mL) antibacterial activity to BET and BA, thus recovering these natural compounds, already endowed with several pharmacological properties, which make them possible multi-target drugs, also for possible antibacterial uses against worrying Gram-positive ESCAPE bacteria. Moreover, despite BPPB revealed acceptable toxicity on several cell lines [8,112,113,114,115], it is possible that the insertion of similarly structured groups on not cytotoxic natural scaffolds, could have enhanced further the therapeutic index.
4. Conclusions
The scope of this study was to find novel compounds effectively active against worrying superbugs, difficult or impossible to inhibit with available antibiotics and responsible of lethal infections. Such infections develop mainly in nosocomial settings, where immunocompromised patients are highly exposed, causing longer hospitalization times and long-period treatments, with high social and economic impact. To this end, three natural triterpenoids, deprived of cytotoxic effects on eukaryotic cells, including totally inactive BET and BA, as well as already active on Gram-positive isolates UA, have been chemically modified by multipart and laborious synthetic procedures, as well as arduous purification work up, achieving derivatives 1-7, five of which contained the triphenyl phosphonium (TPP) group, recently reported to promote antibacterial effects. All compounds and synthetic intermediates were characterized by chemometric-assisted FTIR and NMR spectroscopy, as well as by other analytical techniques, which all confirmed their structure and high purity. When tested for evaluating their antibacterial effects by MICs determinations, using a selection of Gram-positive and Gram-negative clinically isolated superbugs, compounds bearing the TPP group resulted active on all MDR staphylococci and enterococci tested. Specifically, MICs observed for compounds 4-7 were lower than those reported so far for other BET, BA and UA derivatives. For the first time, due to the use of TPP, a real activity (MICs 2-16 µg/mL) was conferred to inactive BET and BA (original MICs > 1024 and 256 µg/mL). Moreover, the antibacterial effects of UA against MRSA and MRSE was improved by 32 and 16 times, respectively (MICs = 2 vs. 32 and 64 μg/mL). Structure-activity relationships (SAR) studies evidenced that while the introduction of residues containing the carbon-carbon triple bond as ester or amide did not enhance the antibacterial profile of BET and BA (compound 2 and 3), the additional insertion of a TPP-group as hexanoate in C-3 of BA, already containing the propargyl amide group, led to significant antibacterial effects (compound 4). While the combinate presence of TPP-hexanoate in C-3 and propargyl carbamate in C-28 of BET led only to weak antibacterial activity (compound 1), the replacement of propargyl carbamate group with the TPP-hexanoate moiety, leaving free the hydroxyl in C-3 of BET (compound 5) led to excellent antibacterial effects, thus evidencing that the presence of groups containing the triple bond should be avoided. On the other hand, it seems to be essential leaving free the hydroxyl in C-3, as confirmed by BET derivative containing two TPP-hexanoate groups on both C-3 and C-28 (compound 6), which demonstrated good antibacterial activity, but inferior to that of 5. Despite the presence of propargyl amide in C-28 and the TPP-hexanoate groups in C-3, as BET derivative 4, UA derivative 7 showed the highest antibacterial effects, probably due to the intrinsic antibacterial activity of UA core. Our question now is: “Could the antibacterial effects of a new UA derivative functionalized as compound 5 be even more active?” Works are in progress to find the answer. Collectively, these preliminary but very promising microbiologic results pave the way for further experiments with the best performing compounds 5 and 7 (MICs = 2 μg/mL) on a larger number of Gram-positive isolates, to evaluate their mechanism of action by setting up by time-killing curves and to assess their cytotoxicity on eukaryotic cells and their possible antibiofilm activity.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1.1.1. ATR-FTIR spectrum of BA. Figure S1.1.2. ATR-FTIR spectrum of BET. Figure S1.1.3. ATR-FTIR spectrum of UA. Figure S1.1.4. ATR-FTIR spectrum of 1. Figure S1.1.5. ATR-FTIR spectrum of 2. Figure S1.1.6. ATR-FTIR spectrum of 3. Figure S1.1.7. ATR-FTIR spectrum of 4. Figure S1.1.8. ATR-FTIR spectrum of 5. Figure S1.1.9. ATR-FTIR spectrum of 6. Figure S1.1.10. ATR-FTIR spectrum of 7. Figure S1.2.1. 1H NMR spectrum (600 MHz, CHCl3) of compound 9. Figure S1.2.2. 13C NMR spectrum (151 MHz, CHCl3) of compound 9. Figure S1.2.3. 1H NMR spectrum (600 MHz, CHCl3) of compound 1. Figure S1.2.4. 13C NMR spectrum (151 MHz, CHCl3) of compound 1. Figure S1.2.5. 31P NMR spectrum (243 MHz, CHCl3) of compound 1. Figure S1.2.6. 1H NMR spectrum (600 MHz, CHCl3) of compound 2. Figure S1.2.7. 13C NMR spectrum (151 MHz, CHCl3) of compound 2. Figure S1.2.8. 1H NMR spectrum (600 MHz, CHCl3) of compound 3. Figure S1.2.9. 13C NMR spectrum (151 MHz, CHCl3) of compound 3. Figure S1.2.10. 1H NMR spectrum (600 MHz, CHCl3) of compound 4. Figure S1.2.11. 13C NMR spectrum (151 MHz, CHCl3) of compound 4. Figure S1.2.12. 31P NMR spectrum (243 MHz, CHCl3) of compound 4. Figure S1.2.13. 1H NMR spectrum (600 MHz, CHCl3) of compound 11. Figure S1.2.14. 13C NMR spectrum (151 MHz, CHCl3) of compound 11. Figure S1.2.15. 1H NMR spectrum (600 MHz, CHCl3) of compound 12. Figure S1.2.16. 13C NMR spectrum (151 MHz, CHCl3) of compound 12. Figure S1.2.17. 1H NMR spectrum (600 MHz, CHCl3) of compound 5. Figure S1.2.18. 13C NMR spectrum (151 MHz, CHCl3) of compound 5. Figure S1.2.19. 31P NMR spectrum (243 MHz, CHCl3) of compound 5. Figure S1.2.20. 1H NMR spectrum (600 MHz, CHCl3) of compound 6. Figure S1.2.21. 13C NMR spectrum (151 MHz, CHCl3) of compound 6. Figure S1.2.22. 31P NMR spectrum (243 MHz, CHCl3) of compound 6. Figure S1.2.23. 1H NMR spectrum (600 MHz, CHCl3) of compound 13. Figure S1.2.24. 13C NMR spectrum (151 MHz, CHCl3) of compound 13. Figure S1.2.25. 1H NMR spectrum (600 MHz, CHCl3) of compound 7. Figure S1.2.26. 13C NMR spectrum (151 MHz, CHCl3) of compound 7. Figure S1.2.27. 31P NMR spectrum (243 MHz, CHCl3) of compound 7. Table S2.1. MICs of compounds 1-7, BET, BA and UA against MDR clinical isolates of Gram-positive and Gram-negative species obtained from experiments conducted at least in triplicate.
Author Contributions
Conceptualization, methodology, software, validation, formal analysis, investigation, resources, data curation, writing—original draft preparation, writing—review and editing, project administration, S.A., C.M.A. and A.M.S.; D.G. cured the synthesis of all compounds and chemical intermediates reported in this study under the supervision of C.M.A and wrote the synthetic procedures part. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support to D.G. (PhD grant/ call 2022-23) by Andreas Mentzelopoulos Foundation.
Data Availability Statement
All research data related to this study are available in the main text and in the Supplementary Materials file associated to this article available at https://www.mdpi.com/article/doi/s1.
Acknowledgments
Authors are very grateful to Professor Paolo Oliveri of the Analytical Chemistry group of the Pharmacy Department (University of Genoa) for the acquisition of ATR-FTIR. Also, we would like to thank the Instrumental Analysis Laboratory (IAL, School of Natural Sciences, University of Patras) for recording the NMR and ESI-MS spectra.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chandrasekhar, D.; Joseph, C.M.; parambil, J.C.; Murali, S.; Yahiya, M.; K, S. Superbugs: An Invicible Threat in Post Antibiotic Era. Clin Epidemiol Glob Health 2024, 28. [Google Scholar] [CrossRef]
- Rajendran, R. Superbug Infection. J Drug Metab Toxicol 2018, 09. [Google Scholar] [CrossRef]
- Mancuso, G.; Midiri, A.; Gerace, E.; Biondo, C. Bacterial Antibiotic Resistance: The Most Critical Pathogens. Pathogens 2021, 10. [Google Scholar] [CrossRef]
- Wicky, P.-H.; Poiraud, J.; Alves, M.; Patrier, J.; d’Humières, C.; Lê, M.; Kramer, L.; de Montmollin, É.; Massias, L.; Armand-Lefèvre, L.; et al. Cefiderocol Treatment for Severe Infections Due to Difficult-to-Treat-Resistant Non-Fermentative Gram-Negative Bacilli in ICU Patients: A Case Series and Narrative Literature Review. Antibiotics 2023, 12, 991. [Google Scholar] [CrossRef]
- Mondal, A.H.; Khare, K.; Saxena, P.; Debnath, P.; Mukhopadhyay, K.; Yadav, D. A Review on Colistin Resistance: An Antibiotic of Last Resort. Microorganisms 2024, 12, 772. [Google Scholar] [CrossRef]
- Silva, J.T.; López-Medrano, F. Cefiderocol, a New Antibiotic against Multidrug-Resistant Gram-Negative Bacteria. Rev Esp Quimioter 2021, 34 Suppl 1, 41–43. [Google Scholar] [CrossRef]
- Karakonstantis, S.; Rousaki, M.; Kritsotakis, E.I. Cefiderocol: Systematic Review of Mechanisms of Resistance, Heteroresistance and In Vivo Emergence of Resistance. Antibiotics 2022, 11, 723. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Zuccari, G.; Bacchetti, F.; Torazza, C.; Milanese, M.; Siciliano, C.; Athanassopoulos, C.M.; Piatti, G.; Schito, A.M. Synthesized Bis-Triphenyl Phosphonium-Based Nano Vesicles Have Potent and Selective Antibacterial Effects on Several Clinically Relevant Superbugs. Nanomaterials 2024, 14, 1351. [Google Scholar] [CrossRef] [PubMed]
- Gajic, I.; Tomic, N.; Lukovic, B.; Jovicevic, M.; Kekic, D.; Petrovic, M.; Jankovic, M.; Trudic, A.; Mitic Culafic, D.; Milenkovic, M.; et al. A Comprehensive Overview of Antibacterial Agents for Combating Multidrug-Resistant Bacteria: The Current Landscape, Development, Future Opportunities, and Challenges. Antibiotics 2025, 14, 221. [Google Scholar] [CrossRef] [PubMed]
- Institute for Molecular Bioscience Explainer: What Is a Superbug and Why Should We Be Worried?; Australia, 2017.
- García-Solache, M.; Rice, L.B. The Enterococcus: A Model of Adaptability to Its Environment. Clin Microbiol Rev 2019, 32. [Google Scholar] [CrossRef]
- Levitus, M.; Rewane, A.; Perera, T.B. Vancomycin-Resistant Enterococci; 2024.
- Chiang, H.-Y.; Perencevich, E.N.; Nair, R.; Nelson, R.E.; Samore, M.; Khader, K.; Chorazy, M.L.; Herwaldt, L.A.; Blevins, A.; Ward, M.A.; et al. Incidence and Outcomes Associated With Infections Caused by Vancomycin-Resistant Enterococci in the United States: Systematic Literature Review and Meta-Analysis. Infect Control Hosp Epidemiol 2017, 38, 203–215. [Google Scholar] [CrossRef]
- Miller, W.R.; Murray, B.E.; Rice, L.B.; Arias, C.A. Vancomycin-Resistant Enterococci. Infect Dis Clin North Am 2016, 30, 415–439. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Hidalgo, N.; Escolà-Vergé, L. Enterococcus Faecalis Bacteremia. J Am Coll Cardiol 2019, 74, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Rosa, T.F.; Coelho, S.S.; Foletto, V.S.; Bottega, A.; Serafin, M.B.; Machado, C. de S.; Franco, L.N.; Paula, B.R.; Hörner, R. Alternatives for the Treatment of Infections Caused by ESKAPE Pathogens. J Clin Pharm Ther 2020, 45, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Brullo, C.; Caviglia, D.; Piatti, G.; Zorzoli, A.; Marimpietri, D.; Zuccari, G.; Schito, A.M. Pyrazole-Based Water-Soluble Dendrimer Nanoparticles as a Potential New Agent against Staphylococci. Biomedicines 2021, 10, 17. [Google Scholar] [CrossRef]
- Bozdogan, B. Antibacterial Susceptibility of a Vancomycin-Resistant Staphylococcus Aureus Strain Isolated at the Hershey Medical Center. Journal of Antimicrobial Chemotherapy 2003, 52, 864–868. [Google Scholar] [CrossRef]
- Tsiodras, S.; Gold, H.S.; Sakoulas, G.; Eliopoulos, G.M.; Wennersten, C.; Venkataraman, L.; Moellering, R.C.; Ferraro, M.J. Linezolid Resistance in a Clinical Isolate of Staphylococcus Aureus. The Lancet 2001, 358, 207–208. [Google Scholar] [CrossRef]
- Peykov, S.; Kirov, B.; Strateva, T. Linezolid in the Focus of Antimicrobial Resistance of Enterococcus Species: A Global Overview of Genomic Studies. Int J Mol Sci 2025, 26, 8207. [Google Scholar] [CrossRef]
- Liu, C.; Bayer, A.; Cosgrove, S.E.; Daum, R.S.; Fridkin, S.K.; Gorwitz, R.J.; Kaplan, S.L.; Karchmer, A.W.; Levine, D.P.; Murray, B.E.; et al. Clinical Practice Guidelines by the Infectious Diseases Society of America for the Treatment of Methicillin-Resistant Staphylococcus Aureus Infections in Adults and Children. Clinical Infectious Diseases 2011, 52, e18–e55. [Google Scholar] [CrossRef]
- Nour El-Din, H.T.; Yassin, A.S.; Ragab, Y.M.; Hashem, A.M. Phenotype-Genotype Characterization and Antibiotic-Resistance Correlations Among Colonizing and Infectious Methicillin-Resistant Staphylococcus Aureus Recovered from Intensive Care Units. Infect Drug Resist 2021, Volume 14, 1557–1571. [Google Scholar] [CrossRef]
- EUCAST. European Committee on Antimicrobial Susceptibility Testing. Available online: Https://Www.Eucast.Org/Ast_of_bacteria/ (accessed on 20 January 2024).
- Alfei, S.; Caviglia, D.; Piatti, G.; Zuccari, G.; Schito, A.M. Synthesis, Characterization and Broad-Spectrum Bactericidal Effects of Ammonium Methyl and Ammonium Ethyl Styrene-Based Nanoparticles. Nanomaterials 2022, 12, 2743. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Castellaro, S.; Taptue, G.B. Synthesis and NMR Characterization of Dendrimers Based on 2, 2-Bis-(Hydroxymethyl)-Propanoic Acid (Bis-HMPA) Containing Peripheral Amino Acid Residues for Gene Transfection. Organic Communications 2017, 10, 144–177. [Google Scholar] [CrossRef]
- Alfei, S.; Castellaro, S. Synthesis and Characterization of Polyester-Based Dendrimers Containing Peripheral Arginine or Mixed Amino Acids as Potential Vectors for Gene and Drug Delivery. Macromol Res 2017, 25, 1172–1186. [Google Scholar] [CrossRef]
- Alfei, S.; Taptue, G.B.; Catena, S.; Bisio, A. Synthesis of Water-Soluble, Polyester-Based Dendrimer Prodrugs for Exploiting Therapeutic Properties of Two Triterpenoid Acids. Chinese Journal of Polymer Science 2018, 36, 999–1010. [Google Scholar] [CrossRef]
- Pifer, C.W.; Wollish, E.G. Potentiometric Titration of Salts of Organic Bases in Acetic Acid. Anal Chem 1952, 24, 300–306. [Google Scholar] [CrossRef]
- Alfei, S.; Catena, S. Synthesis and Characterization of Fourth Generation Polyester-based Dendrimers with Cationic Amino Acids-modified Crown as Promising Water Soluble Biomedical Devices. Polym Adv Technol 2018, 29, 2735–2749. [Google Scholar] [CrossRef]
- Bacchetti, F.; Schito, A.M.; Milanese, M.; Castellaro, S.; Alfei, S. Anti Gram-Positive Bacteria Activity of Synthetic Quaternary Ammonium Lipid and Its Precursor Phosphonium Salt. Int J Mol Sci 2024, 25, 2761. [Google Scholar] [CrossRef]
- Mlala, S.; Oyedeji, A.O.; Gondwe, M.; Oyedeji, O.O. Ursolic Acid and Its Derivatives as Bioactive Agents. Molecules 2019, 24, 2751. [Google Scholar] [CrossRef]
- Chrobak, E.; Świtalska, M.; Wietrzyk, J.; Bębenek, E. New Difunctional Derivatives of Betulin: Preparation, Characterization and Antiproliferative Potential. Molecules 2025, 30, 611. [Google Scholar] [CrossRef]
- Xu, Q.; Xie, Y.; Qi, J.; Ren, Z.; Coluccini, C.; Coghi, P. Development of New Amide Derivatives of Betulinic Acid: Synthetic Approaches and Structural Characterization. Molbank 2025, 2025, M2072. [Google Scholar] [CrossRef]
- Csuk, R.; Barthel, A.; Sczepek, R.; Siewert, B.; Schwarz, S. Synthesis, Encapsulation and Antitumor Activity of New Betulin Derivatives. Arch Pharm (Weinheim) 2011, 344, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Bębenek, E.; Chrobak, E.; Wietrzyk, J.; Kadela, M.; Chrobak, A.; Kusz, J.; Książek, M.; Jastrzębska, M.; Boryczka, S. Synthesis, Structure and Cytotoxic Activity of Acetylenic Derivatives of Betulonic and Betulinic Acids. J Mol Struct 2016, 1106, 210–219. [Google Scholar] [CrossRef]
- Wiemann, J.; Heller, L.; Perl, V.; Kluge, R.; Ströhl, D.; Csuk, R. Betulinic Acid Derived Hydroxamates and Betulin Derived Carbamates Are Interesting Scaffolds for the Synthesis of Novel Cytotoxic Compounds. Eur J Med Chem 2015, 106, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Drag Betulinic Acid Exhibits Stronger Cytotoxic Activity on the Normal Melanocyte NHEM-Neo Cell Line than on Drug-Resistant and Drug-Sensitive MeWo Melanoma Cell Lines. Mol Med Rep 2009, 2. [CrossRef]
- Martins, W.K.; Gomide, A.B.; Costa, É.T.; Junqueira, H.C.; Stolf, B.S.; Itri, R.; Baptista, M.S. Membrane Damage by Betulinic Acid Provides Insights into Cellular Aging. Biochimica et Biophysica Acta (BBA) - General Subjects 2017, 1861, 3129–3143. [Google Scholar] [CrossRef]
- Zuco, V.; Supino, R.; Righetti, S.C.; Cleris, L.; Marchesi, E.; Gambacorti-Passerini, C.; Formelli, F. Selective Cytotoxicity of Betulinic Acid on Tumor Cell Lines, but Not on Normal Cells. Cancer Lett 2002, 175, 17–25. [Google Scholar] [CrossRef]
- Coricovac, D.; Dehelean, C.A.; Pinzaru, I.; Mioc, A.; Aburel, O.-M.; Macasoi, I.; Draghici, G.A.; Petean, C.; Soica, C.; Boruga, M.; et al. Assessment of Betulinic Acid Cytotoxicity and Mitochondrial Metabolism Impairment in a Human Melanoma Cell Line. Int J Mol Sci 2021, 22, 4870. [Google Scholar] [CrossRef]
- Król, S.K.; Kiełbus, M.; Rivero-Müller, A.; Stepulak, A. Comprehensive Review on Betulin as a Potent Anticancer Agent. Biomed Res Int 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Hordyjewska, A.; Ostapiuk, A.; Horecka, A.; Kurzepa, J. Betulin and Betulinic Acid: Triterpenoids Derivatives with a Powerful Biological Potential. Phytochemistry Reviews 2019, 18, 929–951. [Google Scholar] [CrossRef]
- Csuk, R. Targeting Cancer by Betulin and Betulinic Acid. In Novel Apoptotic Regulators in Carcinogenesis; Springer Netherlands: Dordrecht, 2012; pp. 267–287. [Google Scholar]
- Bildziukevich, U.; Kvasnicová, M.; Šaman, D.; Rárová, L.; Šlouf, M.; Wimmer, Z. Cytotoxicity and Nanoassembly Characteristics of Aromatic Amides of Oleanolic Acid and Ursolic Acid. ACS Omega 2025, 10, 20938–20948. [Google Scholar] [CrossRef]
- Subramanian, C.; Solairaja, S.; Dunna, N.R.; Venkatabalasubramanian, S. Toxicity, Safety, and Pharmacotherapeutic Properties of Ursolic Acid: Current Status, Challenges, and Future Perspectives against Lung Cancer. Curr Bioact Compd 2023, 19. [Google Scholar] [CrossRef]
- Laiolo, J.; Graikioti, D.G.; Barbieri, C.L.; Joray, M.B.; Antoniou, A.I.; Vera, D.M.A.; Athanassopoulos, C.M.; Carpinella, M.C. Novel Betulin Derivatives as Multidrug Reversal Agents Targeting P-Glycoprotein. Sci Rep 2024, 14, 70. [Google Scholar] [CrossRef]
- Liu, J.; Yin, F.; Hu, J.; Ju, Y. Cu 2+ -Triggered Shrinkage of a Natural Betulin-Derived Supramolecular Gel to Fabricate Moldable Self-Supporting Gel. Mater Chem Front 2021, 5, 4764–4771. [Google Scholar] [CrossRef]
- Boryczka, S.; Michalik, E.; Jastrzebska, M.; Kusz, J.; Zubko, M.; Bębenek, E. X-Ray Crystal Structure of Betulin–DMSO Solvate. J Chem Crystallogr 2012, 42, 345–351. [Google Scholar] [CrossRef]
- Kemmer, A.; Heinze, T. Reactive Norbornene- and Phenyl Carbonate-Modified Dextran Derivatives: A New Approach to Selective Functionalization. React Funct Polym 2025, 208, 106144. [Google Scholar] [CrossRef]
- Um, I.-H.; Kim, E.Y.; Park, H.-R.; Jeon, S.-E. Aminolyses of 4-Nitrophenyl Phenyl Carbonate and Thionocarbonate: Effect of Modification of Electrophilic Center from CO to CS on Reactivity and Mechanism. J Org Chem 2006, 71, 2302–2306. [Google Scholar] [CrossRef] [PubMed]
- Boryczka, S.; Bębenek, E.; Wietrzyk, J.; Kempińska, K.; Jastrzębska, M.; Kusz, J.; Nowak, M. Synthesis, Structure and Cytotoxic Activity of New Acetylenic Derivatives of Betulin. Molecules 2013, 18, 4526–4543. [Google Scholar] [CrossRef] [PubMed]
- Bębenek, E.; Jastrzębska, M.; Kadela-Tomanek, M.; Chrobak, E.; Orzechowska, B.; Zwolińska, K.; Latocha, M.; Mertas, A.; Czuba, Z.; Boryczka, S. Novel Triazole Hybrids of Betulin: Synthesis and Biological Activity Profile. Molecules 2017, 22, 1876. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, T.; Yuan, H.; Li, D.; Lou, H.; Fan, P. Mitochondria-Targeted Lupane Triterpenoid Derivatives and Their Selective Apoptosis-Inducing Anticancer Mechanisms. J Med Chem 2017, 60, 6353–6363. [Google Scholar] [CrossRef]
- Tsepaeva, O.V.; Nemtarev, A.V.; Abdullin, T.I.; Grigor’eva, L.R.; Kuznetsova, E.V.; Akhmadishina, R.A.; Ziganshina, L.E.; Cong, H.H.; Mironov, V.F. Design, Synthesis, and Cancer Cell Growth Inhibitory Activity of Triphenylphosphonium Derivatives of the Triterpenoid Betulin. J Nat Prod 2017, 80, 2232–2239. [Google Scholar] [CrossRef]
- Tsepaeva, O.V.; Nemtarev, A.V.; Grigor’eva, L.R.; Voloshina, A.D.; Mironov, V.F. Esterification of Betulin with ω-Bromoalkanoic Acids. Russian Journal of Organic Chemistry 2015, 51, 1318–1323. [Google Scholar] [CrossRef]
- Kommera, H.; Kaluđerović, G.N.; Kalbitz, J.; Paschke, R. Synthesis and Anticancer Activity of Novel Betulinic Acid and Betulin Derivatives. Arch Pharm (Weinheim) 2010, 343, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Migglautsch, A.K.; Willim, M.; Schweda, B.; Glieder, A.; Breinbauer, R.; Winkler, M. Aliphatic Hydroxylation and Epoxidation of Capsaicin by Cytochrome P450 CYP505X. Tetrahedron 2018, 74, 6199–6204. [Google Scholar] [CrossRef]
- Srinivas, J.; Namito, Y.; Matsubara, R.; Hayashi, M. Synthesis of Enantiomerically Pure (8 S,9 S,10 R,6 Z )-Trihydroxyoctadec-6-Enoic Acid. J Org Chem 2017, 82, 5146–5154. [Google Scholar] [CrossRef]
- Zhang, Q.; Cao, R.; Fei, H.; Zhou, M. Mitochondria-Targeting Phosphorescent Iridium( <scp>iii</Scp> ) Complexes for Living Cell Imaging. Dalton Trans. 2014, 43, 16872–16879. [Google Scholar] [CrossRef]
- Pathak, R.K.; Marrache, S.; Harn, D.A.; Dhar, S. Mito-DCA: A Mitochondria Targeted Molecular Scaffold for Efficacious Delivery of Metabolic Modulator Dichloroacetate. ACS Chem Biol 2014, 9, 1178–1187. [Google Scholar] [CrossRef]
- Dang Thi, T.A.; Kim Tuyet, N.T.; Pham The, C.; Thanh Nguyen, H.; Ba Thi, C.; Thi Phuong, H.; Van Boi, L.; Van Nguyen, T.; D’hooghe, M. Synthesis and Cytotoxic Evaluation of Novel Amide–Triazole-Linked Triterpenoid–AZT Conjugates. Tetrahedron Lett 2015, 56, 218–224. [Google Scholar] [CrossRef]
- Pohjala, L.; Alakurtti, S.; Ahola, T.; Yli-Kauhaluoma, J.; Tammela, P. Betulin-Derived Compounds as Inhibitors of Alphavirus Replication. J Nat Prod 2009, 72, 1917–1926. [Google Scholar] [CrossRef]
- Dang Thi, T.A.; Kim Tuyet, N.T.; Pham The, C.; Thanh Nguyen, H.; Ba Thi, C.; Thi Phuong, H.; Van Boi, L.; Van Nguyen, T.; D’hooghe, M. Synthesis and Cytotoxic Evaluation of Novel Amide–Triazole-Linked Triterpenoid–AZT Conjugates. Tetrahedron Lett 2015, 56, 218–224. [Google Scholar] [CrossRef]
- Xiao, S.; Wang, Q.; Si, L.; Zhou, X.; Zhang, Y.; Zhang, L.; Zhou, D. Synthesis and Biological Evaluation of Novel Pentacyclic Triterpene α -Cyclodextrin Conjugates as HCV Entry Inhibitors. Eur J Med Chem 2016, 124, 1–9. [Google Scholar] [CrossRef]
- Xiao, S.; Wang, Q.; Si, L.; Shi, Y.; Wang, H.; Yu, F.; Zhang, Y.; Li, Y.; Zheng, Y.; Zhang, C.; et al. Synthesis and Anti-HCV Entry Activity Studies of Β-Cyclodextrin–Pentacyclic Triterpene Conjugates. ChemMedChem 2014, 9, 1060–1070. [Google Scholar] [CrossRef]
- Ludeña Huaman, M.A.; Tupa Quispe, A.L.; Huamán Quispe, R.I.; Serrano Flores, C.A.; Robles Caycho, J. A Simple Method to Obtain Ursolic Acid. Results Chem 2021, 3. [Google Scholar] [CrossRef]
- Daasch, L.; Smith, D. Infrared Spectra of Phosphorus Compounds. Anal Chem 1951, 23, 853–868. [Google Scholar] [CrossRef]
- Abonia, R.; Insuasty, D.; Laali, K.K. Recent Advances in the Synthesis of Propargyl Derivatives, and Their Application as Synthetic Intermediates and Building Blocks †. Molecules 2023, 28. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Brullo, C.; Caviglia, D.; Zuccari, G. Preparation and Physicochemical Characterization of Water-Soluble Pyrazole-Based Nanoparticles by Dendrimer Encapsulation of an Insoluble Bioactive Pyrazole Derivative. Nanomaterials 2021, 11, 2662. [Google Scholar] [CrossRef]
- Alfei, S.; Schito, A.M.; Zuccari, G. Considerable Improvement of Ursolic Acid Water Solubility by Its Encapsulation in Dendrimer Nanoparticles: Design, Synthesis and Physicochemical Characterization. Nanomaterials 2021, 11, 2196. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Oliveri, P.; Malegori, C. Assessment of the Efficiency of a Nanospherical Gallic Acid Dendrimer for Long-Term Preservation of Essential Oils: An Integrated Chemometric-Assisted FTIR Study. ChemistrySelect 2019, 4, 8891–8901. [Google Scholar] [CrossRef]
- Ceccacci, F.; Sennato, S.; Rossi, E.; Proroga, R.; Sarti, S.; Diociaiuti, M.; Casciardi, S.; Mussi, V.; Ciogli, A.; Bordi, F.; et al. Aggregation Behaviour of Triphenylphosphonium Bolaamphiphiles. J Colloid Interface Sci 2018, 531, 451–462. [Google Scholar] [CrossRef]
- Alfei, S.; Marengo, B.; Valenti, G.; Domenicotti, C. Synthesis of Polystyrene-Based Cationic Nanomaterials with Pro-Oxidant Cytotoxic Activity on Etoposide-Resistant Neuroblastoma Cells. Nanomaterials 2021, 11, 977. [Google Scholar] [CrossRef]
- Alfei, S.; Zuccari, G.; Caviglia, D.; Brullo, C. Synthesis and Characterization of Pyrazole-Enriched Cationic Nanoparticles as New Promising Antibacterial Agent by Mutual Cooperation. Nanomaterials 2022, 12, 1215. [Google Scholar] [CrossRef]
- Fröhlich, E. The Role of Surface Charge in Cellular Uptake and Cytotoxicity of Medical Nanoparticles. Int J Nanomedicine 2012, 5577. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Spallarossa, A.; Lusardi, M.; Zuccari, G. Successful Dendrimer and Liposome-Based Strategies to Solubilize an Antiproliferative Pyrazole Otherwise Not Clinically Applicable. Nanomaterials 2022, 12, 233. [Google Scholar] [CrossRef] [PubMed]
- Petri-Fink, A.; Chastellain, M.; Juillerat-Jeanneret, L.; Ferrari, A.; Hofmann, H. Development of Functionalized Superparamagnetic Iron Oxide Nanoparticles for Interaction with Human Cancer Cells. Biomaterials 2005, 26, 2685–2694. [Google Scholar] [CrossRef] [PubMed]
- Jambhrunkar, S.; Yu, M.; Yang, J.; Zhang, J.; Shrotri, A.; Endo-Munoz, L.; Moreau, J.; Lu, G.; Yu, C. Stepwise Pore Size Reduction of Ordered Nanoporous Silica Materials at Angstrom Precision. J Am Chem Soc 2013, 135, 8444–8447. [Google Scholar] [CrossRef]
- Chen, L.; Mccrate, J.M.; Lee, J.C.-M.; Li, H. The Role of Surface Charge on the Uptake and Biocompatibility of Hydroxyapatite Nanoparticles with Osteoblast Cells. Nanotechnology 2011, 22, 105708. [Google Scholar] [CrossRef]
- Akinc, A.; Battaglia, G. Exploiting Endocytosis for Nanomedicines. Cold Spring Harb Perspect Biol 2013, 5, a016980–a016980. [Google Scholar] [CrossRef]
- Kratochvil, Byron. Titrations in Nonaqueous Solvents. Anal Chem 1982, 54, 105–121. [Google Scholar] [CrossRef]
- Seher, A. Dr. I. Gyenes, C. Sc. (Chim.), Titrationen in Nichtwäßrigen Medien, 3. Neubearb. u. Erg. Aufl., 701 S., 206 Abb., 108 Tab., Gln., Ferdinand Enke Verlag, Stuttgart 1970, Preis: 84.—DM. Fette, Seifen, Anstrichmittel 1973, 75, 232–232. [Google Scholar] [CrossRef]
- Šafařík, L.; Stránský, Z.; Svehla, G.; Burns, D.T. Titrimetric Analysis in Organic Solvents (Comprehensive Analytical Chemistry, Vol. XXII). Anal Chim Acta 1987, 201, 367. [Google Scholar] [CrossRef]
- Mascellani, Giuseppe. ; Casalini, Claudio. Use of Mercuric Acetate in Potentiometric Titrations in a Nonaqueous Medium. Anal Chem 1975, 47, 2468–2470. [Google Scholar] [CrossRef]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front Microbiol 2019, 10. [Google Scholar] [CrossRef]
- Rice, L.B. Federal Funding for the Study of Antimicrobial Resistance in Nosocomial Pathogens: No ESKAPE. J Infect Dis 2008, 197, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Nunes, B.; Cagide, F.; Borges, F.; Simões, M. Antimicrobial Activity and Cytotoxicity of Novel Quaternary Ammonium and Phosphonium Salts. J Mol Liq 2024, 401, 124616. [Google Scholar] [CrossRef]
- Frei, A.; Zuegg, J.; Elliott, A.G.; Baker, M.; Braese, S.; Brown, C.; Chen, F.; G. Dowson, C.; Dujardin, G.; Jung, N.; et al. Metal Complexes as a Promising Source for New Antibiotics. Chem Sci 2020, 11, 2627–2639. [Google Scholar] [CrossRef] [PubMed]
- Frei, A.; Ramu, S.; Lowe, G.J.; Dinh, H.; Semenec, L.; Elliott, A.G.; Zuegg, J.; Deckers, A.; Jung, N.; Bräse, S.; et al. Platinum Cyclooctadiene Complexes with Activity against Gram-positive Bacteria. ChemMedChem 2021, 16, 3165–3171. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Schito, A.M. From Nanobiotechnology, Positively Charged Biomimetic Dendrimers as Novel Antibacterial Agents: A Review. Nanomaterials 2020, 10, 2022. [Google Scholar] [CrossRef]
- Alfei, S.; Schito, A.M. Positively Charged Polymers as Promising Devices against Multidrug Resistant Gram-Negative Bacteria: A Review. Polymers (Basel) 2020, 12, 1195. [Google Scholar] [CrossRef]
- Schito, A.M.; Alfei, S. Antibacterial Activity of Non-Cytotoxic, Amino Acid-Modified Polycationic Dendrimers against Pseudomonas Aeruginosa and Other Non-Fermenting Gram-Negative Bacteria. Polymers (Basel) 2020, 12, 1818. [Google Scholar] [CrossRef]
- Schito, A.M.; Piatti, G.; Caviglia, D.; Zuccari, G.; Alfei, S. Broad-Spectrum Bactericidal Activity of a Synthetic Random Copolymer Based on 2-Methoxy-6-(4-Vinylbenzyloxy)-Benzylammonium Hydrochloride. Int J Mol Sci 2021, 22, 5021. [Google Scholar] [CrossRef]
- Alfei, S.; Piatti, G.; Caviglia, D.; Schito, A. Synthesis, Characterization, and Bactericidal Activity of a 4-Ammoniumbuthylstyrene-Based Random Copolymer. Polymers (Basel) 2021, 13, 1140. [Google Scholar] [CrossRef]
- Gurjar, M. Colistin for Lung Infection: An Update. J Intensive Care 2015, 3, 3. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, R.; Ma, Z.; Zuo, W.; Zhu, M. S-Alkylated Sulfonium Betulin Derivatives: Synthesis, Antibacterial Activities, and Wound Healing Applications. Bioorg Chem 2025, 154, 108056. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.C.S.; dos Santos Maia, M.; de Souza, T.A.; de Oliveira Lima, E.; dos Santos, L.E.C.G.; Silva, S.L.; da Silva, M.S.; Filho, J.M.B.; da Silva Rodrigues Junior, V.; Scotti, L.; et al. Antimicrobial Potential of Betulinic Acid and Investigation of the Mechanism of Action against Nuclear and Metabolic Enzymes with Molecular Modeling. Pathogens 2023, 12, 449. [Google Scholar] [CrossRef] [PubMed]
- Schito, A.M.; Caviglia, D.; Piatti, G.; Zorzoli, A.; Marimpietri, D.; Zuccari, G.; Schito, G.C.; Alfei, S. Efficacy of Ursolic Acid-Enriched Water-Soluble and Not Cytotoxic Nanoparticles against Enterococci. Pharmaceutics 2021, 13, 1976. [Google Scholar] [CrossRef] [PubMed]
- Seferyan, M.A.; Saverina, E.A.; Frolov, N.A.; Detusheva, E.V.; Kamanina, O.A.; Arlyapov, V.A.; Ostashevskaya, I.I.; Ananikov, V.P.; Vereshchagin, Anatoly.N. Multicationic Quaternary Ammonium Compounds: A Framework for Combating Bacterial Resistance. ACS Infect Dis 2023, 9, 1206–1220. [CrossRef]
- Haykir, N.I.; Nizan Shikh Zahari, S.M.S.; Harirchi, S.; Sar, T.; Awasthi, M.K.; Taherzadeh, M.J. Applications of Ionic Liquids for the Biochemical Transformation of Lignocellulosic Biomass into Biofuels and Biochemicals: A Critical Review. Biochem Eng J 2023, 193, 108850. [Google Scholar] [CrossRef]
- Alfei, S. Shifting from Ammonium to Phosphonium Salts: A Promising Strategy to Develop Next-Generation Weapons against Biofilms. Pharmaceutics 2024, 16, 80. [Google Scholar] [CrossRef]
- Adepoju, F.O.; Duru, K.C.; Li, E.; Kovaleva, E.G.; Tsurkan, M.V. Pharmacological Potential of Betulin as a Multitarget Compound. Biomolecules 2023, 13, 1105. [Google Scholar] [CrossRef]
- Alakurtti, S.; Mäkelä, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological Properties of the Ubiquitous Natural Product Betulin. European Journal of Pharmaceutical Sciences 2006, 29, 1–13. [Google Scholar] [CrossRef]
- Fajardo, J.B.; Vianna, M.H.; Ferreira, T.G.; de, O. Lemos, A.S.; Souza, T. de F.; Campos, L.M.; Paula, P. de L.; Andrade, N.B.; Gamarano, L.R.; Queiroz, L.S.; et al. Enhanced Antitumor and Antibacterial Activities of Ursolic Acid through β-Cyclodextrin Inclusion Complexation. ACS Omega 2025, 10, 12906–12916. [Google Scholar] [CrossRef]
- Sun, Y.; Li, X.; Wang, Y.; Shang, X.; Huang, W.; Ang, S.; Li, D.; Wong, W.-L.; Hong, W.D.; Zhang, K.; et al. In Vitro and in Vivo Evaluation of Novel Ursolic Acid Derivatives as Potential Antibacterial Agents against Methicillin-Resistant Staphylococcus Aureus (MRSA). Bioorg Chem 2025, 154, 107986. [Google Scholar] [CrossRef] [PubMed]
- Do Nascimento, P.G.G.; Lemos, T.L.G.; Bizerra, A.M.C.; Arriaga, A.M.C.; Ferreira, D.A.; Santiago, G.M.P.; Braz-Filho, R.; Costa, J.G.M. Antibacterial and Antioxidant Activities of Ursolic Acid and Derivatives. Molecules 2014, 19. [Google Scholar] [CrossRef] [PubMed]
- Wolska, K.; Grudniak, A.; Fiecek, B.; Kraczkiewicz-Dowjat, A.; Kurek, A. Antibacterial Activity of Oleanolic and Ursolic Acids and Their Derivatives. Open Life Sci 2010, 5, 543–553. [Google Scholar] [CrossRef]
- Serrafi, A.; Wasilewski, A. Synthesis and Antimicrobial Activity of New Betulin Derivatives. Sci Rep 2025, 15, 17719. [Google Scholar] [CrossRef]
- Haque, S.; Nawrot, D.A.; Alakurtti, S.; Ghemtio, L.; Yli-Kauhaluoma, J.; Tammela, P. Screening and Characterisation of Antimicrobial Properties of Semisynthetic Betulin Derivatives. PLoS One 2014, 9. [Google Scholar] [CrossRef]
- Lugiņina, J.; Kroškins, V.; Lācis, R.; Fedorovska, E.; Demir, Ö.; Dubnika, A.; Loca, D.; Turks, M. Synthesis and Preliminary Cytotoxicity Evaluation of Water Soluble Pentacyclic Triterpenoid Phosphonates. Sci Rep 2024, 14, 28031. [Google Scholar] [CrossRef]
- Ungureanu (Similie), D.; Bora, L.; Avram, Ștefana; Turks, M.; Muntean, D.; Danciu, C. Assessment of the Antiproliferative, Cytotoxic, Antimigratory, Antimicrobial, and Irritative Potential of Novel Phosphonate Derivatives of Betulinic Acid. In Proceedings of the INT-DOC-RES.; MDPI: Basel Switzerland, September 16 2025; p. 1.
- Alfei, S.; Torazza, C.; Bacchetti, F.; Milanese, M.; Passalaqua, M.; Khaledizadeh, E.; Vernazza, S.; Domenicotti, C.; Marengo, B. TPP-Based Nanovesicles Kill MDR Neuroblastoma Cells and Induce Moderate ROS Increase, While Exert Low Toxicity To-Wards Primary Cell Cultures: An in Vitro Study. IJMS 2025, 26, 4991. [Google Scholar] [CrossRef]
- Silvana Alfei; Marco Milanese; Carola Torazza; Maria Grazia Signorello; Mario Passalacqua; Cinzia Domenicotti; Barbara Marengo Tri-Phenyl-Phosphonium-Based Nano Vesicles: A New In Vitro Nanomolar-Active Weapon to Eradicate PLX-Resistant Mela-Noma Cells by a Time-Dependent Mechanism. International Journal of Molecular Science 2025.
- Alfei, S.; Giannoni, P.; Signorello, M.G.; Torazza, C.; Zuccari, G.; Athanassopoulos, C.M.; Domenicotti, C.; Marengo, B. The Remarkable and Selective In Vitro Cytotoxicity of Synthesized Bola-Amphiphilic Nanovesicles on Etoposide-Sensitive and -Resistant Neuroblastoma Cells. Nanomaterials 2024, 14, 1505. [Google Scholar] [CrossRef]
- Alfei, S.; Zuccari, G.; Athanassopoulos, C.M.; Domenicotti, C.; Marengo, B. Strongly ROS-Correlated, Time-Dependent, and Selective Antiproliferative Effects of Synthesized Nano Vesicles on BRAF Mutant Melanoma Cells and Their Hyaluronic Acid-Based Hydrogel Formulation. Int J Mol Sci 2024, 25, 10071. [Google Scholar] [CrossRef]
Figure 1.
Compounds encountered in this work.
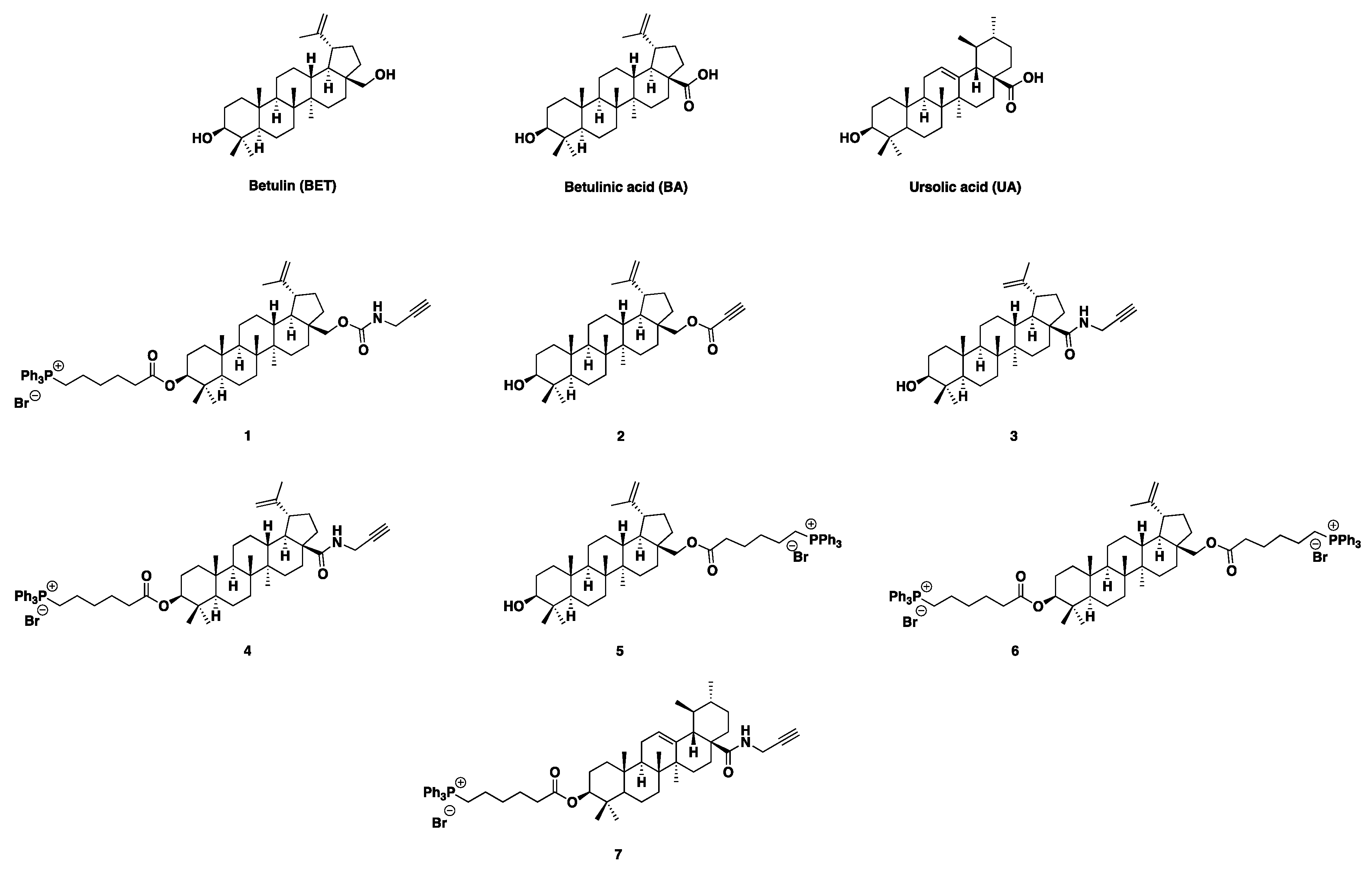
Scheme 1a.
Synthesis of the BET derivatives 1, 2, 5 and 6 and intermediates 8, 9, 11, 12. Legend to scheme: (i) 6-bromohexanoic acid, dichloromethane (DCM)-tetra-hydro-furane (THF), 4-di-methylamino pyridine (DMAP), dicyclohexyl carbodiimide (DCC), 0 °C to r. t., 48 h, 88% (11, 57% and 12, 31%) ; (ii) propiolic acid, DCC, DMAP, DCM, -10 °C to r. t., 24 h, 38%; (iii) p-nitro-phenyl-chloroformate, THF, pyridine, 0°C to r. t., 24 h, 83%; (iv) propargylamine, triethylamine (Et3N); THF, 0°C to r. t., 3 h, 65%; (v) 6-triphenyl phosphonium hexanoic acid 10, DCM, DCC, DMAP, 0 °C, to r. t., 90 min, 54%; (vi), (vii) triphenylphosphine (Ph3P), toluene (PhMe), reflux, 48 h, 30-36%.
Scheme 1a.
Synthesis of the BET derivatives 1, 2, 5 and 6 and intermediates 8, 9, 11, 12. Legend to scheme: (i) 6-bromohexanoic acid, dichloromethane (DCM)-tetra-hydro-furane (THF), 4-di-methylamino pyridine (DMAP), dicyclohexyl carbodiimide (DCC), 0 °C to r. t., 48 h, 88% (11, 57% and 12, 31%) ; (ii) propiolic acid, DCC, DMAP, DCM, -10 °C to r. t., 24 h, 38%; (iii) p-nitro-phenyl-chloroformate, THF, pyridine, 0°C to r. t., 24 h, 83%; (iv) propargylamine, triethylamine (Et3N); THF, 0°C to r. t., 3 h, 65%; (v) 6-triphenyl phosphonium hexanoic acid 10, DCM, DCC, DMAP, 0 °C, to r. t., 90 min, 54%; (vi), (vii) triphenylphosphine (Ph3P), toluene (PhMe), reflux, 48 h, 30-36%.
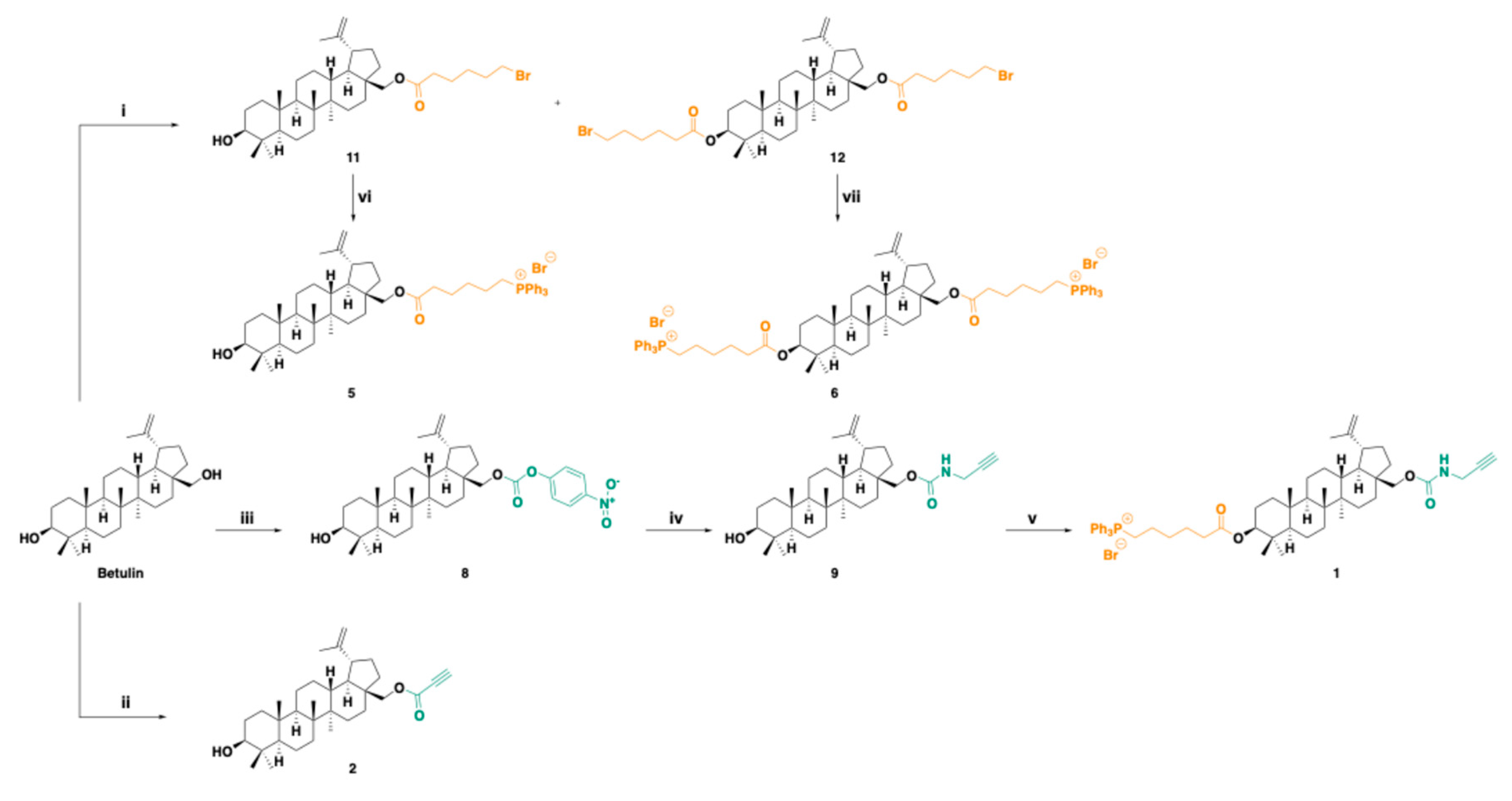
Scheme 2.
Synthesis of the BA derivatives 3 and 4. Legend to scheme (i) propargylamine, THF, 2-(7-azobenzotriazole)-N, N, N’, N’-tetramethyluronium hexafluorophosphate (HATU), Et3N, 0 °C to r. t.; (ii) 6-TPPHA (10), DCM, DCC, DMAP, -10 °C to r. t.
Scheme 2.
Synthesis of the BA derivatives 3 and 4. Legend to scheme (i) propargylamine, THF, 2-(7-azobenzotriazole)-N, N, N’, N’-tetramethyluronium hexafluorophosphate (HATU), Et3N, 0 °C to r. t.; (ii) 6-TPPHA (10), DCM, DCC, DMAP, -10 °C to r. t.
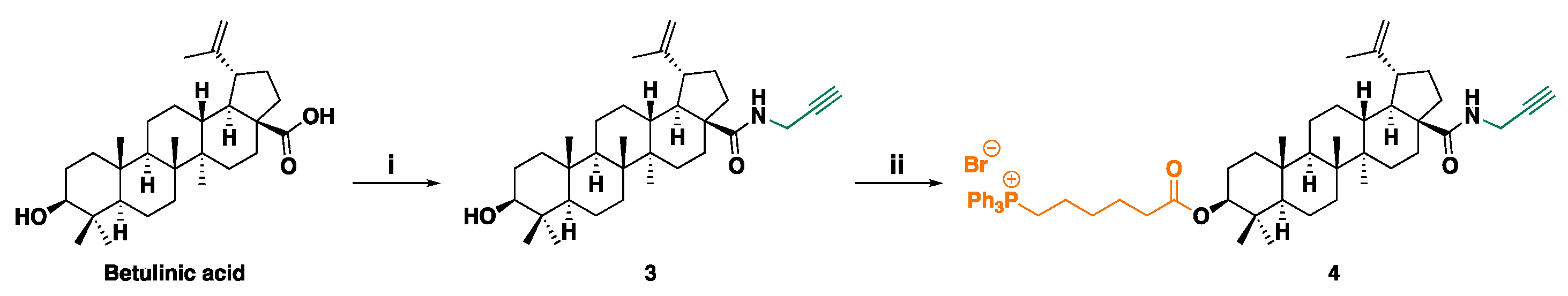
Scheme 3.
Synthesis of the UA derivative 7 and intermediate 13. Legend to scheme: (i) propargylamine, THF, HATU, Et3N, 0 °C to r. t.; (ii) 10, DCM, DCC, DMAP, -10 °C to r. t.
Scheme 3.
Synthesis of the UA derivative 7 and intermediate 13. Legend to scheme: (i) propargylamine, THF, HATU, Et3N, 0 °C to r. t.; (ii) 10, DCM, DCC, DMAP, -10 °C to r. t.
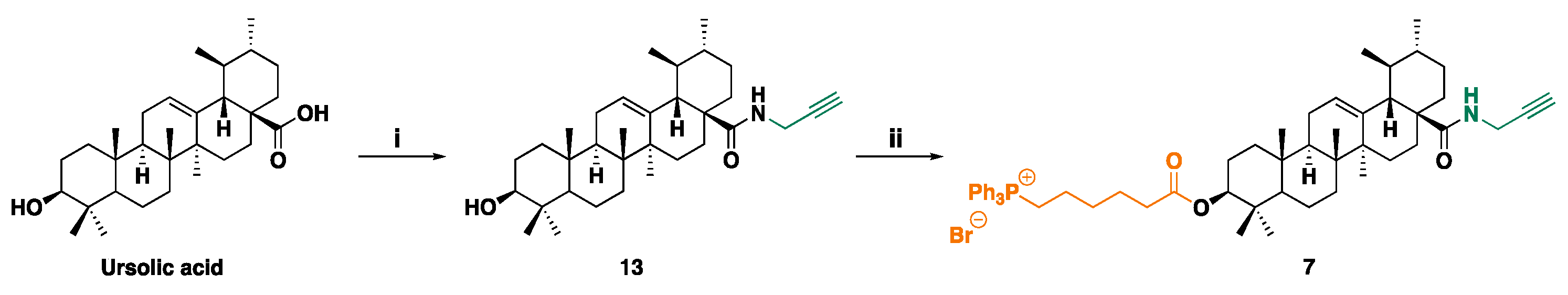
Figure 2.
Mechanism of carbonate aminolysis. R = organic residues.
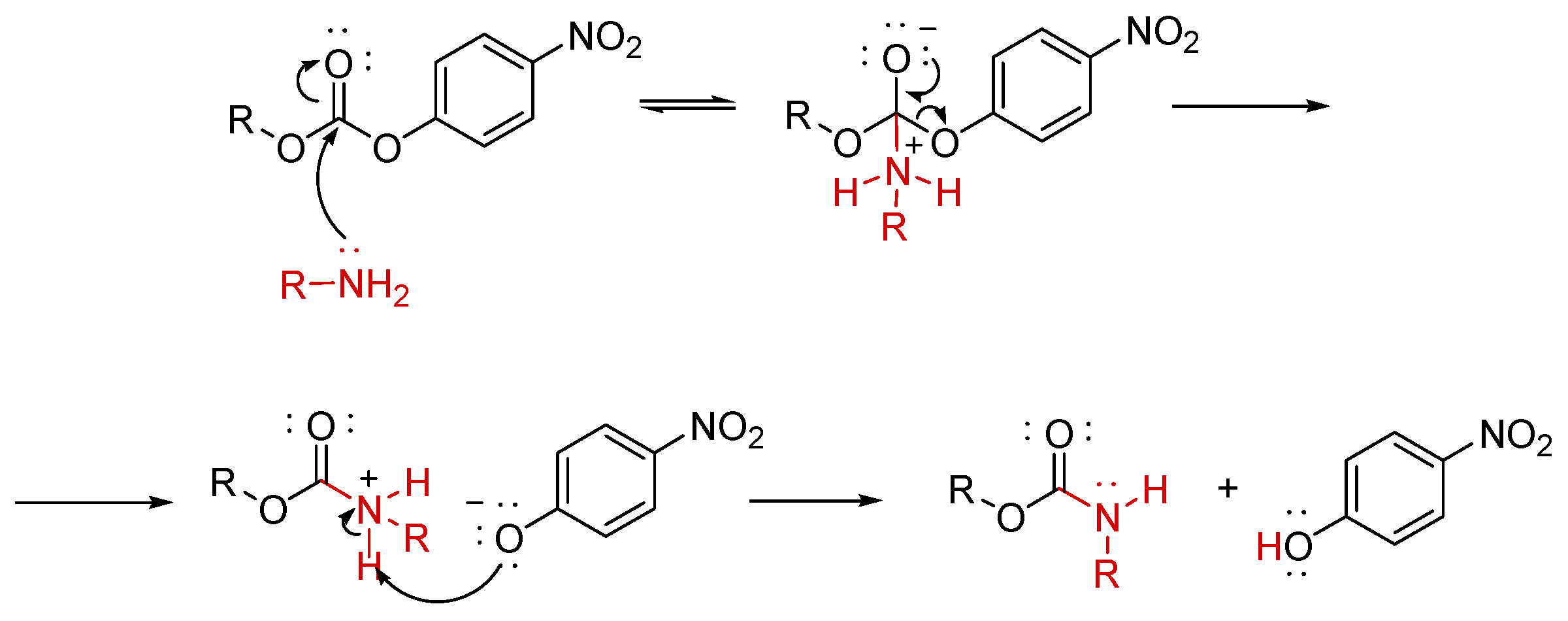
Figure 3.
Score plot of PCA on the matrix of 34,010 variables (spectral data of BA, BET, UA and compound 1-7 (a) and that of PCA on the matrix of 37,411 variables (spectral data of BA, BET, UA, compounds 1-7 and BPPB (b).
Figure 3.
Score plot of PCA on the matrix of 34,010 variables (spectral data of BA, BET, UA and compound 1-7 (a) and that of PCA on the matrix of 37,411 variables (spectral data of BA, BET, UA, compounds 1-7 and BPPB (b).
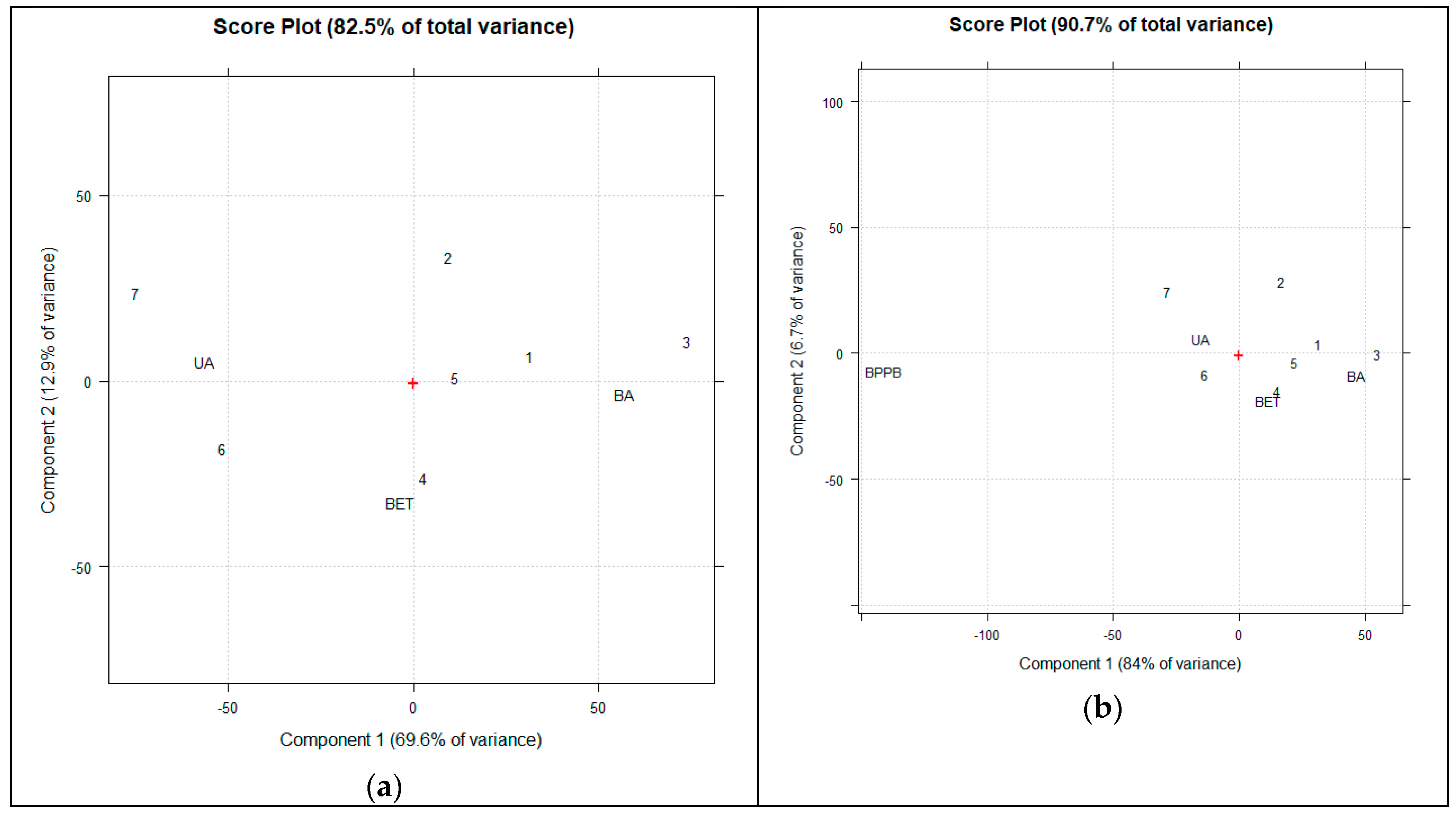
Figure 4.
Score plot of PCA on the matrix of 510 variables consisting in the values of δ (ppm) of the 13C NMR spectral data of BA, BET, UA, compound 1-7 and intermediate 8, 9, 10, 11 and 12.
Figure 4.
Score plot of PCA on the matrix of 510 variables consisting in the values of δ (ppm) of the 13C NMR spectral data of BA, BET, UA, compound 1-7 and intermediate 8, 9, 10, 11 and 12.
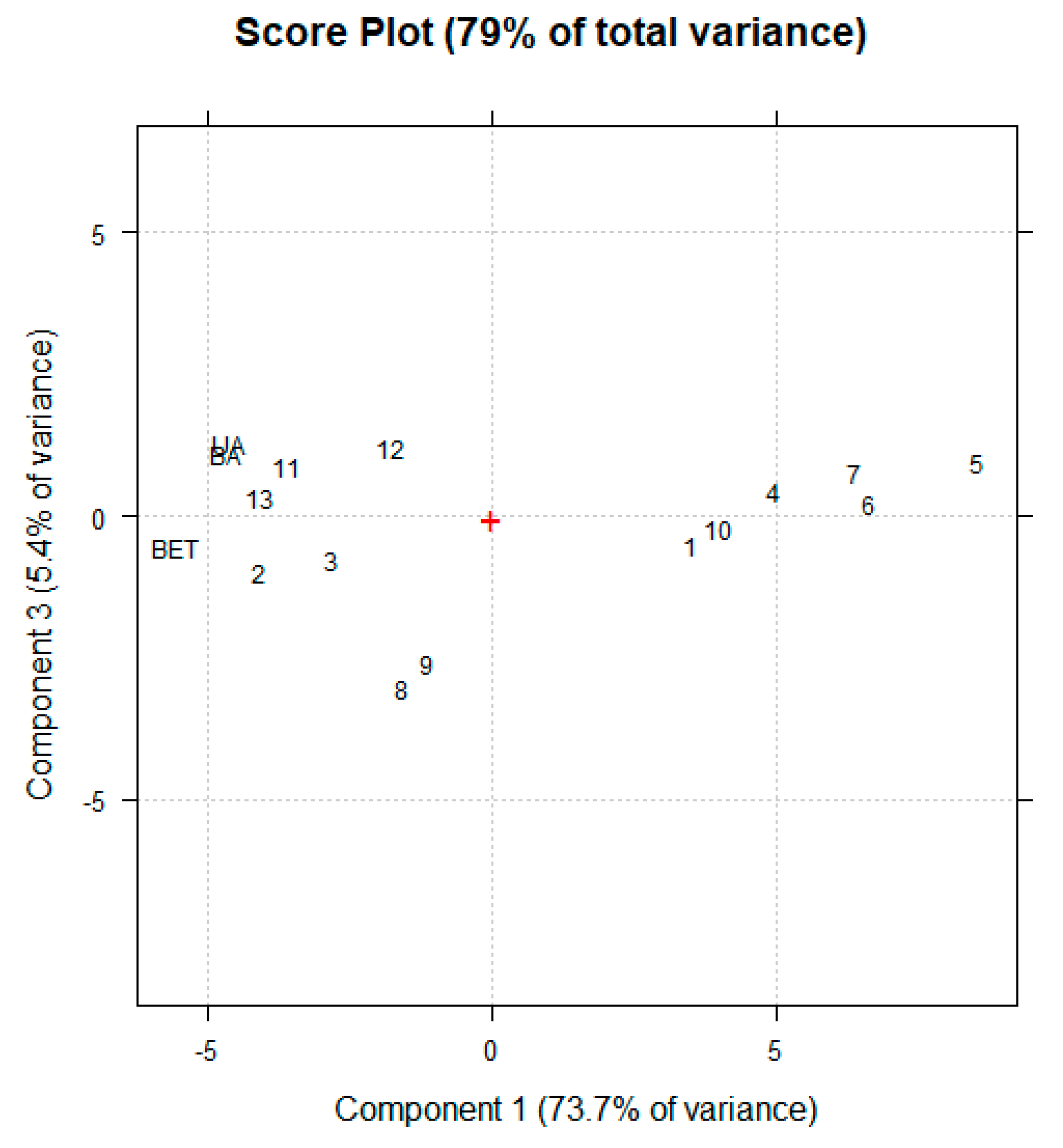
Figure 4.
Vesicular aggregates of 6 in water solution observed with a 40× objective (a). Smaller spherical vesicles of 6 were better evidenced using the 100× objective (b).
Figure 4.
Vesicular aggregates of 6 in water solution observed with a 40× objective (a). Smaller spherical vesicles of 6 were better evidenced using the 100× objective (b).

Figure 5.
Representative images of the particle size distribution (nm) with DPI of 6 (a)and of its ζ-p (b).
Figure 5.
Representative images of the particle size distribution (nm) with DPI of 6 (a)and of its ζ-p (b).
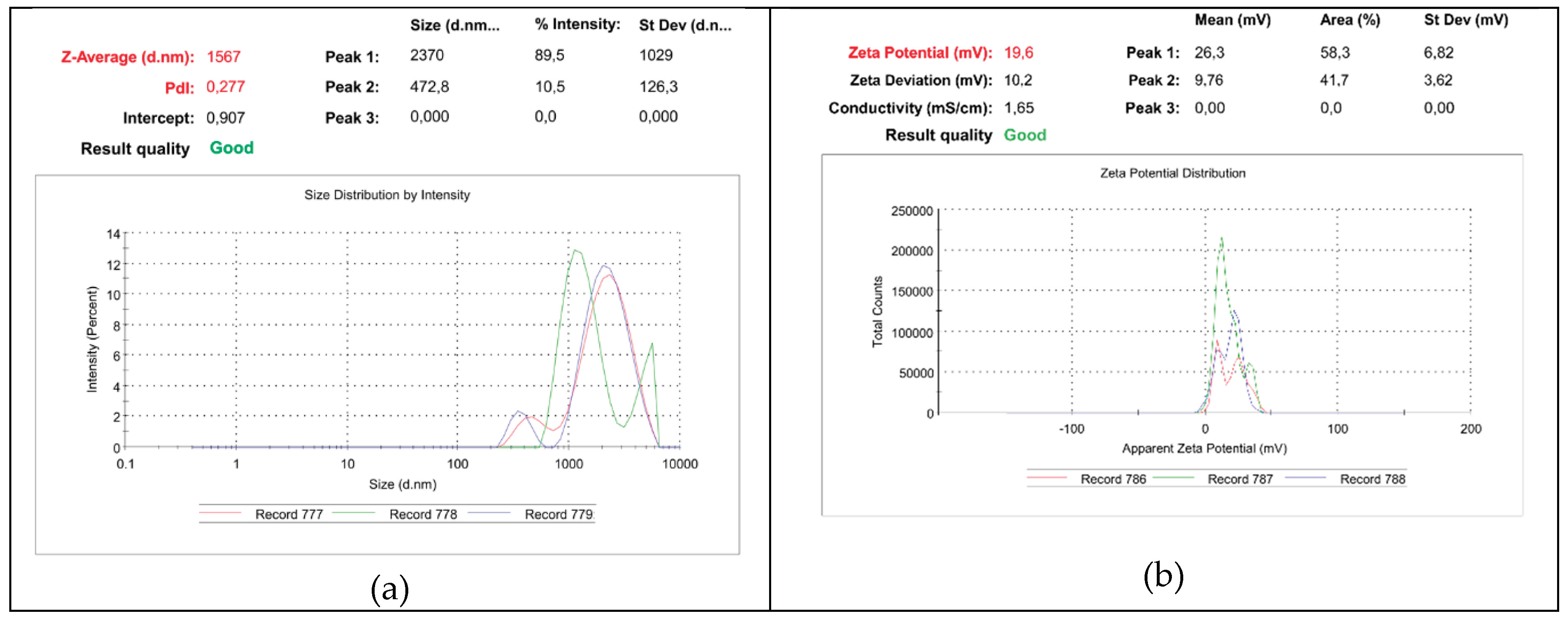

Table 1.
Data of potentiometric titrations of 1 and 6.
| Sample | Weight (mg) | * MW | * P+ (µmol) | 0.1 N HClO4 § | ** P+ (µmol) | ** MW | *** Residuals | Error (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1.0 | 963.1 | 1.0383 | 10.40 | 1.0400 | 961.5 | 1.6 | 0.16 |
| 4 | 0.6 | 933.1 | 0.6430 | 6.44 | 0.6440 | 931.7 | 0.8 | 0.14 |
| 5 | 0.7 | 882.0 | 0.7936 | 7.96 | 0.7960 | 879.4 | 2.6 | 0.29 |
| 6 | 1.1 | 1321.4 | 1.6648 | 16.60 | 1.6600 | 1325.3 | 3.9 | 0.30 |
| 7 | 1.5 | 933.1 | 1.6075 | 16.20 | 1.6202 | 925.8 | 7.3 | 0.78 |
* Calculated; ** experimental; *** refers to MW; § = in µL.
Table 2.
MICs of compounds 4-7, BET, BA and UA against MDR clinical isolates of Gram-positive species obtained from experiments conducted at least in triplicate.
Table 2.
MICs of compounds 4-7, BET, BA and UA against MDR clinical isolates of Gram-positive species obtained from experiments conducted at least in triplicate.
| Gram positive isolates | 4 | 5 | 6 | 7 | BET | BA | UA | R.A. |
|---|---|---|---|---|---|---|---|---|
| MICs (µg/mL) | ||||||||
| S. aureus MRSA 18 | 4 | 8 | 8 | 2 | >64 | >64 | 64 | 256 (O) |
| S. epidermidis MRSE 22 | 4 | 4 | 8 | 2 | >64 | >64 | 32 | 128 (O) |
| E. faecalis VRE 1 * | 16 | 8 | 16 | 4 | >64 | >64 | 4 | 256 (V); 64 (T) |
| E. faecium VRE 152 * | 4 | 2 | 8 | 2 | >64 | >64 | 2 | 128 (V); 64 (T) |
RA = Reference antibiotics; * Resistant to teicoplanin; VRE = vancomycin-resistant enterococci; MRSA = methicillin resistant S. aureus; MRSE = methicillin resistant S. epidermidis; ° From patients with cystic fibrosis; °° resistant to carbapenems; °°° colistin resistant; KPC = K. pneumoniae carbapenemase-producing bacteria; R.A. = reference antibiotic; O = oxacillin; T = teicoplanin; V = vancomycin; BET = betulin; BA = betulinic acid; UA = ursolic acid; M = meropenem.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



