Submitted:
17 November 2025
Posted:
18 November 2025
You are already at the latest version
Abstract
Intervertebral disc (IVD), the largest avascular structure in the human body, contains nucleus pulposus (NP) cells that generate an abundant quantity of lactate from anaerobic glycolysis as an adaptation to hypoxia. Historically, IVD lactate was viewed as a metabolic toxic byproduct necessitating clearance to maintain IVD health. This is because accumulation of lactic acid, the protonated form of lactate, acidifies the IVD microenvironment, impairs cell viability, disrupts extracellular matrix integrity, and promotes degeneration. However, recent studies discovered that lactate serves as an important IVD biofuel in a process known as lactate-dependent metabolic symbiosis in which lactate produced by NP is shuttled into cells of the neighboring annulus fibrosus (AF), and cartilage endplate (CEP) to be metabolized via the tricarboxylic acid cycle and oxidative phosphorylation to generate ATP and amino acids to maintain IVD matrix homeostasis. Additionally, lactate is found to function as a signaling molecule and epigenetic regulator in IVD: it regulates transcription via histone lactylation that modulates ferroptosis and other cell fate decisions. Lactate also modulates senescence, apoptosis, and inflammatory responses through pathways such as Phosphatidylinositol 3-kinase/Protein kinase B (PI3K/Akt) in IVD and other organs. This review synthesizes current knowledge on lactate production, transport, and clearance in the IVD along with the emerging roles of lactate in IVD health and pathophysiology. The review also provides research perspectives and directions aimed at advancing our understanding of lactate biology and evaluating its potential as a therapeutic target for treating intervertebral disc degeneration.
Keywords:
intervertebral disc degeneration
; lactate metabolism
; histone lactylation
; epigenetics regulation
; metabolic symbiosis
1. Introduction
Low back pain (LBP) ranks among the leading causes of global disability [1], with intervertebral disc degeneration (IDD) recognized as a principal pathological driver [2]. Cells in the intervertebral disc (IVD), the largest avascular structure in the human body [3,4], exist in a microenvironment characterized by hypoxia, restricted nutrient supply, acidity, and mechanical strain. To adapt to hypoxia, NP cells rely primarily on anaerobic glycolysis, converting glucose to lactic acid that readily deprotonates into lactate and a proton that contributes to acidification of IVD tissue. Under physiological conditions, IVD lactate is exported via monocarboxylate transporters (MCTs) and cleared across the CEP into the vertebral circulation, a process essential for maintaining pH balance and metabolic homeostasis [5,6]. However, during degeneration, CEP permeability declines about 40% compared with healthy discs, and calcification further impairs clearance, leading to lactate accumulation and progressive acidification of the intradiscal milieu [7]. Excess acidity suppresses anabolic activity, activates catabolic cascades, induces senescence and apoptosis, disrupts extracellular matrix (ECM) integrity, and ultimately exacerbates disc degeneration [8]. Consequently, lactate has traditionally been considered a toxic metabolic byproduct necessitating clearance.
Beyond imparting cytotoxic effects, recent evidence indicates that lactate, the deprotonated form of lactic acid, also serves as an important biofuel and other regulatory functions in disc homeostasis [9,10]. Disc cells from distinct anatomical regions can establish lactate-dependent metabolic symbiosis, in which NP-derived lactate is imported into adjacent AF cells and oxidized through the TCA cycle. This intercellular recycling redefines lactate not as a waste product but as a reusable metabolic substrate [11], a concept validated in multiple experimental models [12,13,14]. Moreover, lactate promotes trans-differentiation of CEP chondrocytes into subchondral bone–like cells via histone H3 lysine 18 (H3K18) lactylation, implicating lactate in CEP ossification and remodeling through epigenetic regulation [15]. In addition, lactate functions as a signaling molecule, activating pathways such as PI3K/Akt [16] , NF-κB [17] , and innate immune responses [18] , thereby modulating inflammatory activity, apoptosis, and cell fate decisions. Importantly, lactate serves as a substrate for lysine lactylation for post-translational modifications of histones and nonhistone proteins that result in modulation of epigenetic regulation as well as enzymatic and signaling activities [19]. In recent years, histone lactylation has emerged as a novel epigenetic mechanism, drawing increasing attention across diverse pathological contexts [20,21,22]. Collectively, these findings suggest that lactate is not merely a metabolic waste byproduct but a metabolic regulator of important IVD biologic processes.
In light of recent advances, this review examines lactate metabolism and functional roles of lactate in disc physiology and pathology to provide a conceptual framework for lactate-targeted therapeutic strategies in IDD. The review summarizes reported literature on lactate-driven metabolic symbiosis among distinct disc cell populations, lactate involvement in signaling pathways and epigenetic regulation, lactate overall effects on disc homeostasis and degeneration. The review also outlines key unresolved questions and prospectives for IVD lactate research.
2. IVD Lactate Production
Disc Structure and Nutrient Supply Mechanisms. IVD, situated between adjacent vertebral bodies, is composed of a central NP, an outer AF, and CEP on both superior and inferior sides [23,24]. As the largest avascular structure in the human body, nutrient transport to disc cells occurs through passive diffusion, driven by concentration gradients established by cellular metabolic activity. In addition, the daily fluctuations in spinal mechanical loading promote fluid exchange in and out of the disc, thereby facilitating nutrient delivery [25]. Cells in the outer AF can exchange metabolites and nutrients with capillaries in the surrounding soft tissues, whereas most IVD cells in the deeper regions rely on trans-osseous blood vessels within the vertebral body. These vessels penetrate the subchondral bone and extend into the marrow cavity, forming capillary loops terminating beneath the CEP. Although bulk fluid flow can facilitate the transport of larger solutes, extensive experimental and theoretical evidence indicates that small molecules such as glucose and oxygen enter the disc primarily by diffusion [26,27,28].This nutrient supply system is essential for maintaining cell viability and metabolic activity, particularly within the central NP region [29,30,31,32,33,34,35,36,37,38].
Production of IVD lactate by anaerobic glycolysis. The IVD is hypoxic due to its avasculature. In healthy IVD, cells located farther from the CEP encounter reduced oxygen and glucose availability, with the central NP exhibiting the lowest oxygen tensions of 5–15 mmHg and highest lactate concentrations of 2–6 mmol/L, thereby establishing a distinct spatial metabolic gradient [30,39]. Under these hypoxic conditions, disc cells rely almost exclusively on anaerobic glycolysis, in which glucose is converted to pyruvate and reduced to lactate by lactate dehydrogenase A (LDH-A, also known as LDH5) [11,40,41]. Each molecule of glucose yields two molecules of lactate, maintaining a consistent 2:1 molar ratio across disc regions [23,42,43]. This reaction, coupled with the oxidation of reduced nicotinamide adenine dinucleotide to oxidized nicotinamide adenine dinucleotide (NAD⁺), is essential for maintaining glycolytic flux under anaerobic conditions [44].
Lactate concentrations within different IVD compartments. Lactate levels in the IVD display a distinct spatial gradient, with the highest concentrations consistently found in the NP and decreasing radially toward the AF [24]. Under resting conditions, lactate levels in human blood are approximately 1 mM, while the physiological range in serum typically lies between 1 and 2 mM [45]. In surgical specimens from patients with scoliosis or low back pain, NP lactate concentrations typically range from 2–6 mM, while the outer AF lactate is approximately 1mM [30]. In more degenerated discs, lactate concentrations as high as 12–16 mM have been reported [30]. Similar trends are observed in canine discs, where lactate concentrations of 4-7 mM in NP and 1-2mM in peripheral AF have been reported [23]. Moreover, localized measurements in human discs indicate a lactate concentration of about 2 mM at the CEP-NP interface and approximately 0.9 mM at the outer AF boundary [46].
Integrated regulation of IVD lactate production. To adapt to the chronically hypoxic and nutrient-limited microenvironment of the IVD, resident cells undergo metabolic reprogramming governed by the hypoxia-inducible factor-1α (HIF-1α) [47]. Stabilized under low oxygen, HIF-1α promotes glycolysis by upregulating glucose transporters (GLUT1/3) and rate-limiting enzymes within the glycolytic pathway such as hexokinase, phosphofructokinase, and pyruvate kinase, thereby enhancing glycolytic flux [48,49]. HIF-1α simultaneously induces lactate dehydrogenase A (LDHA) to convert pyruvate into lactate [50] and pyruvate dehydrogenase kinase 1 (PDK1) to inhibit pyruvate dehydrogenase complex from converting pyruvate to acetyl-CoA for oxidative phosphorylation, collectively reinforcing dependence on anaerobic glycolysis [51].
Lactate production in the IVD depends not only on glucose supply but also on the coordinated influence of oxygen and extracellular pH, with complex nonlinear interactions among these factors [52]. Computational models indicate that lactic acid accumulation acidifies the microenvironment, thereby inhibiting further lactate production, while elevated oxygen tension reduces lactate yield, suggesting a negative feedback loop between oxygen availability and tissue acidification on lactate synthesis [52]. For example, glycolysis and lactate generation in NP cells are markedly suppressed by hypoxia, glucose deprivation, or acidosis. Notably, at pH 6.2, oxygen consumption falls to ~32% of that used by NP cells cultured at pH 7.4, demonstrating that acidity impairs both glycolytic and oxidative phosphorylation pathways [43]. Thus, the interplay among oxygen, glucose, and pH constitutes a central regulatory axis of lactate metabolism and energy homeostasis in NP cells.
3. IVD Lactate Transport, Accumulation, and Clearance
MCT–Mediated Lactate Transmembrane Transport. NP lactic acid must be exported to avoid intracellular accumulation and over acidification. Lactate transport across the cell membrane occurs through three routes: minimal passive diffusion of undissociated lactate, anion exchange systems, and—most importantly—proton-coupled transport mediated by MCTs [17]. MCTs constitute a family of proton-linked carriers that mediate bidirectional lactate flux according to lactate/H⁺ gradients. This process is ATP-independent and relies on passive co-diffusion [53,54]. MCT transport of lactate is coupled with protons (H⁺ ions), i.e., lactate enters or exits the cell with a proton with it, which helps maintain the overall charge balance across the cell membrane. Under hypoxia, MCT activity is essential for lactate transport, maintaining intracellular acid–base balance, and sustaining metabolic stability [55]. Within the hypoxic IVD, MCT1 and MCT4 are the predominant isoforms [56,57,58,59]. Their distribution is region-specific and complementary: MCT4 mediates lactate efflux, whereas MCT1 facilitates lactate influx into cells, thereby sustaining lactate gradients and supporting metabolic equilibrium within disc cells. CD147 (basigin/EMMPRIN) acts as a molecular chaperone required for proper localization and stability of MCT [60,61,62]. However, most insights into MCT–CD147 cooperation are derived from cancer biology [60,63,64,65], and whether a similar interaction exists in IVD cells remains to be elucidated.
MCT4-mediated lactate export in IVD. MCT4, encoded by the solute carrier family 16-member 3 (SLC16A3) gene, is a low-affinity, high-capacity lactate transporter highly expressed in glycolytic tissues [66,67]. Given the glycolytic dependence of NP cells, large amounts of lactate must be exported via MCT4 through a proton-coupled mechanism [68]. Under hypoxia, HIF-1α enhances MCT4 transcription by binding and activating an intronic enhancer within SLC16A3, while simultaneously suppressing mitochondrial respiration, promoting glycolytic flux, and facilitating lactate–proton co-export to maintain intracellular acid–base homeostasis [6,69,70]. In healthy IVDs, lactate released by NP cells via MCT4 diffuses across the cartilage endplate and is cleared by vertebral capillaries [26,29].
The essential role of MCT4 in disc homeostasis has been demonstrated in vivo and in vitro. Silagi et al. [5] showed that MCT4-deficient mice developed age-dependent IDD characterized by disrupted NP structure, reduced glycosaminoglycan (GAG) content, elevated matrix metalloproteinase-13 (MMP13), deterioration of vertebral trabeculae, and reduced bone quality. Metabolomic profiling and [¹³C]-glucose tracing further revealed that short-term inhibition of MCT4 under hypoxia caused intracellular lactate and proton accumulation, reduced extracellular acidification rate (ECAR), and increased pyruvate and tricarboxylic acid (TCA) intermediates, indicating a metabolic shift from glycolysis toward oxidative metabolism primarily via pyruvate dehydrogenase (PDH) activity [5]. Consistent with the glycolytic profile of NP cells, Wang et al. confirmed that MCT4 expression is markedly higher in NP than AF tissue [11], supporting the role of MCT4 in exporting lactate out of NP cells to maintain physiological pH.
MCT1-mediated lactate import in IVDs. MCT1 is a bidirectional, proton-coupled lactate transporter whose expression is upregulated by the transcription factor cellular myelocytomatosis oncogene (c-Myc) [54,71]. It is widely expressed in diverse cell types, with the direction of lactate flux determined by transmembrane gradients of lactate and H⁺. When extracellular lactate concentrations exceed intracellular levels, MCT1 facilitates cellular lactate influx. In IVDs, MCT1 is expressed highly in AF but not in NP tissue [11]. Upon exposure to exogenous lactate, AF cells further upregulate MCT1 expression, accompanied by increased lactate uptake and enhanced metabolic activity. 14C-lactate tracing experiments confirmed that AF cells take up lactate in a concentration-dependent manner, reinforcing the key role of MCT1 in AF cell lactate uptake [11]. Wang et al. [72] developed a five-layer three-dimensional (3D) NP degeneration model using gelatin sponges to mimic the progressively hypoxic and high-lactate microenvironment of the degenerating disc. In this model, inhibition of MCT1-mediated lactate influx by the specific inhibitor AZD3965 reversed the deleterious effects of lactate by restoring GAG accumulation, downregulating matrix metalloproteinase-3 (MMP3) expression, and alleviating NP cell degeneration. These findings suggest that blocking MCT1-dependent lactate uptake by NP cells can mitigate disc degeneration by reducing NP intracellular acid stress and catabolic activity [72]. Hence, lactate export out of NP cell is beneficial while lactate import into NP cells is harmful for IVD health.
Beyond IVD tissue, recent studies have shown that lactate enters dendritic cells via MCT1, stabilizes HIF-1α, and enhances its transcriptional activity⁶⁶. This process is independent of the lactate receptor G protein–coupled receptor 81 (GPR81). These findings provide new insights into metabolic adaptation that could be studied for IVD metabolism, particularly for NP cells residing in the hypoxic, lactate-rich environments. Given the central role of HIF-1α in maintaining NP homeostasis, a similar mechanism—whereby MCT1-mediated lactate uptake stabilizes HIF-1α—has been hypothesized to support NP cell adaptation to chronic hypoxia [73].
Mechanisms of lactate accumulation and its regulation. Excessive lactate accumulation in the IVD reflects an imbalance in production, transport, and or clearance of lactate [5,74,75,76]. Structurally, the CEP is the principal egress route: age or degeneration-related calcification and sclerosis of the CEP that reduces permeability, impairs lactate clearance, and promotes IVD acidification [34,77,78]. CEP Modic changes and Schmorl’s nodes further compromise solute exchange [37,79]. Experiments have demonstrated that lactate diffuses more readily across the CEP than glucose, yet less efficiently than within AF or NP tissues [80]. These findings suggest that the CEP functions as a partial diffusion barrier [80]. Moreover, lactate diffusivity across the CEP correlates strongly with its porosity, underscoring the critical role of CEP microstructural integrity in regulating metabolite transport [80]. Finite element modeling further suggests that impaired CEP permeability disrupts disc nutrient homeostasis, exacerbating the imbalance between lactate accumulation and oxygen/glucose supply [52].
External mechanical loading can modulate the metabolic microenvironment of the IVD, thereby partially alleviating lactate accumulation. Dynamic compression enhances solute convection and diffusion within the tissue, improving oxygen delivery and promoting lactate clearance [83]. Specifically, cyclic loading induces alternating tissue deformation, which enhances solute transport and elevates local oxygen tension. As oxygen concentration increases, the extracellular microenvironment becomes less acidic. This shift, in turn, exerts feedback effects on cellular metabolism, downregulating glycolytic activity, reducing lactate production, and facilitating the establishment of a new metabolic steady state. Additionally, enhanced solute flow promotes lactate clearance across the tissue, thereby minimizing its accumulation within the disc matrix [83]. Computational modeling further supports this mechanism [75]. Simulations revealed that maintaining a state of moderate mechanical–transport coupling—where tissue deformation and porosity changes occur under sustained compressive loading—significantly improves oxygen transport within the IVD and reduces lactate accumulation. Even under continuous compressive loading, lactate levels were lower when mechanical loading was coupled with solute transport, thereby optimizing the disc’s metabolic and nutritional microenvironment [75]. Collectively, these findings suggest that refinement of mechanical loading strategies could represent a promising strategy to mitigate lactate buildup in degenerative disc conditions.
MCT greatly contributes to lactate accumulation and clearance. Beyond CEP permeability, steady-state lactate in the disc depends on MCT4-driven efflux from NP and MCT1-mediated uptake by AF/EP. Insufficient MCT4 elevates intracellular lactate/H⁺ and limits tissue-level clearance; inadequate or spatially mismatched MCT1 reduces downstream “sinks,” curbing redistribution to oxidative/signaling compartments. Thus, the balance of lactate export by MCT4 (NP) and import by MCT1 (AF/EP)—with proper membrane localization (e.g., CD147)—sets net lactate fluxes. In degenerative setting (e.g., CEP sclerosis, chronic hypoxia), even modest shifts favor lactate retention and microenvironmental acidification. Impaired lactate export and glycolytic dysregulation can promote lactate buildup and metabolic stress in NP cells. NP cells are adapted to the hypoxic disc environment, relying predominantly on glycolysis regulated by HIF-1α and its downstream targets such as PDK1 and LDHA [81,82]. When lactate efflux is inhibited, intracellular lactate and intermediates like glucose-6-phosphate (G6P) accumulate, consistent with feedback inhibition of glycolytic enzymes and subsequent acidosis [5].
Acidic stress and cellular consequences of lactate accumulation. Healthy NP has a pH of about 6.8. Thus, given the pKa of 3.9 of lactic acid, any lactic acid molecules produced in the hypoxic NP are readily dissociated into lactate and H+ which contribute to the acidity of the disc microenvironment [11]. Indeed, our study demonstrated that AF cells can tolerate relatively high levels of lactate, showing no adverse effects on cell viability or morphology up to 10mM lactate, with only a modest reduction in viability observed at 20mM [11]. But its lactic acid counterpart is harmful to IVD cells due to its ability to acidify the disc microenvironment. In the IVD, sustained accumulation of lactic acid creates an acidic microenvironment that activates acid-sensing ion channels (ASICs) [84]. This acidic milieu induces inflammatory responses and enhances catabolic activity in disc cells [84].
Evidence indicates that acidic stress not only impairs the physiological functions of NP cells but also upregulates pro-inflammatory cytokines and matrix-degrading enzymes, thereby accelerating ECM degradation and promoting the onset and progression of IDD [85]. Exposure of NP cell cultures to acidic stress (pH 6.5) for seven days upregulates mRNA levels of IL-1β and IL-6 by approximately 81-fold and 7.8-fold, respectively. Expression of neurotrophic factors such as nerve growth factor and brain-derived neurotrophic factor also increase significantly (3.0-fold and 4.6-fold, respectively). These changes are accompanied by activation of the NF-κB signaling pathway and elevated expression of matrix metalloproteinase-2 (MMP2) and matrix metalloproteinase-9 (MMP9), indicating that an acidic environment simultaneously drives inflammatory and catabolic cascades [86,87]. Additional in vitro studies have confirmed that acidic conditions suppress proteoglycan synthesis, upregulate IL-1β expression, and induce metabolic dysfunction and programmed cell injury in NP cells [88,89]. Collectively, these findings demonstrate that low extracellular pH—resulting directly from lactate accumulation—serves as a critical trigger for inflammatory activation and catabolic imbalance within the disc microenvironment.
Zhan et al. [90] further demonstrated that inflammatory mediators and lactate released from degenerative CEP cells can promote NP cell inflammation via a paracrine mechanism, establishing an acid–inflammation signaling axis between the CEP and NP. To counteract this interaction, they developed a functional hydrogel system (CAP-sEXOs@Gel) composed of endplate-targeting engineered exosomes and a calcium carbonate/chitosan composite. This hydrogel alleviated acidic stress and inflammatory responses in both CEP and NP cells, and effectively delayed disc degeneration in animal models [90].
4. New Roles of Lactate in IVD
Traditionally, IVD lactate has been viewed merely as a toxic metabolic byproduct to be eliminated to maintain IVD health. As discussed below, this idea is challenged by emerging evidence suggesting that lactate serves multiple functions important for maintaining IVD physiology.
Metabolic and Structural Basis for Lactate Exchange within the IVD. Within the IVD, the central NP is concentrically surrounded by the AF, which can be subdivided into an inner AF (IAF) and outer AF (OAF) [91,92]. The IAF, adjacent to the NP, exhibits transitional features in cell composition and extracellular matrix organization, thereby bridging NP and OAF [92,93]. Recent studies further describe a transition zone (TZ) between NP and AF, characterized by gradual changes in collagen orientation and mechanical properties, and integrated with adjacent tissues through anchoring, fiber penetration, or interweaving [94]. This continuous yet regionally specialized architecture provides the structural basis for functional and metabolic interactions between NP and AF.
In their microenvironment, NP cells are chronically exposed to high hydrostatic pressure, hypoxia, and osmolarity, relying on glycolysis and HIF signaling for energy metabolism. By contrast, AF cells—particularly in the OAF—are closer to vasculature, benefiting from greater oxygen and glucose availability that favors oxidative metabolism⁷⁹. This metabolic distinction, together with anatomic proximity, underlies a lactate-mediated communication network that coordinates homeostasis and preserves disc integrity. Physiologically, the IVD is subjected to axial compression while exhibiting intrinsic swelling driven by proteoglycan-associated fixed charges [95,96,97,98]. Although overall hydraulic permeability is low, loading-induced deformation generates interstitial fluid flow, facilitating pressure modulation and metabolite transport, including lactate, across tissue compartments [99,100,101]. Recent computational models have further characterized solute dynamics—including oxygen and lactate in coupled diffusion, as well as nutrients such as glucose—within the IVD [102,103]. Incorporating disc geometry and heterogeneous material properties, these models demonstrate that solute distribution is governed by diffusivity, metabolic activity, concentration gradients, and boundary conditions, and may involve metabolite-mediated coupling between disc regions [102,103].
Lactate as a Biofuel: Metabolic Symbiosis Between NP and AF. Recent studies have revealed a lactate-mediated metabolic symbiosis within the IVD, challenging the traditional notion of lactate as a mere metabolic byproduct. Wang et al. were the first to employ heavy isotope 13C-labeled lactate to trace its metabolic fate in AF cells, demonstrating that lactate is converted into TCA cycle intermediates—including α-ketoglutarate, succinate, and malate—and incorporated into the biosynthesis of amino acids such as alanine and glutamate.
Functionally, lactate exposure increased the mitochondrial oxygen consumption rate of AF cells by approximately 50% and upregulated type I collagen synthesis, indicating efficient utilization of lactate for both energy production and matrix synthesis [11]. At the molecular level, NP and AF cells display complementary expression patterns of lactate transport and metabolizing genes. NP cells predominantly express LDH-A (LDH5) and MCT4, facilitating lactate production and efflux, respectively. In contrast, AF cells are enriched in MCT1, LDH-B, and PDH, enabling them to take up and oxidize lactate efficiently¹¹. This supports a model in which lactate produced by NP cells is exported via MCT4 and subsequently imported into AF cells through MCT1, where it is converted into pyruvate and enters the TCA cycle to fuel oxidative phosphorylation [11]. The schematic of lactate metabolism pathways is shown in Figure 1.
Under hypoxic conditions, HIF-1α is stably expressed and serves as a key transcription factor regulating glycolytic pathways [51]. It upregulates GLUT1 and GLUT3 and rate-limiting glycolytic enzymes such as hexokinase, phosphofructokinase, and pyruvate kinase, thereby enhancing glycolytic flux [49]. Simultaneously, it promotes the expression of LDHA, facilitating lactate production [50]. In addition, HIF-1α induces the expression of PDK1, which inhibits the entry of pyruvate into mitochondrial oxidative pathways, further favoring anaerobic metabolism [51]. MCT1 and MCT4, two major monocarboxylate transporters, interact with the molecular chaperone CD147, which regulates their membrane localization and stability [60,61]. Under hypoxia, HIF-1α also upregulates MCT4 expression by activating an intronic enhancer within the SLC16A3 gene [69,70]. EPs rely on MCT1 to import lactate derived from NP cells, using it both for energy metabolism and histone H3K18 lactylation. This epigenetic modification contributes to the trans-differentiation of EP cells from cartilage toward subchondral bone [15].
This inter-regional metabolic collaboration resembles the “lactate symbiosis” described in solid tumor microenvironments [104,105,106,107]and skeletal muscle [108], highlighting an adaptive strategy that enables the IVD to cope with nutrient limitations. AF cells can tolerate lactate concentrations up to 10 mM—far above normal plasma levels (0.5–1 mM)—allowing them to use lactate as an alternative energy substrate under conditions of glucose deprivation or ischemia, thereby sustaining viability and matrix production [11]. In contrast, the hypoxic NP cells lack the enzymatic machinery required for lactate import and oxidative phosphorylation, with exogenous lactate inhibiting their matrix synthesis. This “unidirectional lactate flow” from NP to AF thus constitutes a critical mechanism by which the disc preserves homeostasis under metabolic stress.
Lactate-mediated metabolic coupling between NP–EP. The NP and CEP are anatomically and functionally integrated: the CEP provides mechanical support and governs solute transport, acting as the primary route for nutrient delivery and waste removal that defines the NP’s metabolic microenvironment and viability [109,110]. CEP stem cells promote NP regeneration and disc homeostasis, while NP-generated loads impose adaptive demands on CEP integrity—together establishing a bidirectional structural–functional coupling essential for disc health and for the disc’s response to degeneration [111,112,113].
Recent studies indicate that lactate functions as a “metabolic currency” among the NP, EP, and AF, sustaining motion-segment homeostasis through an MCT1-dependent, inter-tissue coupling. Specifically, during skeletal growth, NP cells exhibit high glycolytic activity and secrete lactate; this lactate is taken up and reutilized by the adjacent EP (and, to a lesser extent, the AF) via MCT1, thereby supporting EP cartilage ossification and maturation. When MCT1-mediated lactate influx is impaired, a spectrum of phenotypes emerges—including reduced NP cellularity, exacerbated disc degeneration, and persistent, immature EP cartilage—underscoring the essential role of lactate coupling in disc development and health [15].
At the baseline metabolic level, both AF and EP display glycolytic features, yet their substrate preferences diverge in the presence of moderate glucose level (5mM), EP cells efficiently utilize exogenous lactate, whereas AF cells, under the tested conditions, rely predominantly on glucose. 13C-lactate heavy isotope tracing experiment demonstrates that carbon from labeled lactate enters EP pyruvate and tricarboxylic-acid (TCA) intermediates and is incorporated into metabolites such as aspartate and malate, providing direct evidence for a “lactate → pyruvate → TCA” fuel pathway. By contrast, AF cells show markedly weaker lactate uptake and oxidation and instead favor glucose as the primary energy source—a pattern corroborated by Seahorse bioenergetics assays [15]. However, in low glucose (1mM), AF cells readily utilize lactate as carbon source for energy production [11].
Lactate as metabolic epigenetic regulator: histone lactylation in IVD. Post-translational modifications of histones, e.g., acetylation, methylation, ubiquitination, and phosphorylation, represent key forms of epigenetic regulation, as they modulate gene expression by altering chromatin structure and influencing transcription factor accessibility [114]. Modifications such as acetylation, and phosphorylation are generally associated with transcriptional activation, whereas deacetylation or certain types of methylation can lead to gene silencing [114]. These dynamic and reversible modifications constitute the so-called “epigenetic code,” playing critical roles in cell fate determination and disease pathogenesis. Lysine lactylation (Kla) is a relatively recently identified epigenetic modification, first reported by Zhang et al. in macrophages in 2019 [20]. This modification typically occurs at K14 and K18 on histone H3 and is strongly induced under high-lactate conditions such as hypoxia and infection. Functionally, histone lactylation is considered an activating mark that facilitates chromatin accessibility and transcription activation [20]. Subsequent studies have confirmed that Kla is broadly distributed across diverse cell types and is critically involved in regulating tumor proliferation, inflammatory responses, and tissue repair [115,116,117].
In NP cells, elevated lactate is associated with increased histone Kla. This modification contributes to both disc homeostasis and IDD by modulating chromatin structure and gene expression [10,118]. Western blot and immunofluorescence analyses have shown that global Kla (pan-Kla) levels are significantly higher in degenerated NP tissue than in healthy tissue [119]. Moreover, the proportion of Kla-positive cells increases with degeneration severity, suggesting a positive correlation between lactylation and IDD progression [119].
Recent proteomic studies have profiled Kla in NP cells. By comparing Kla expression under normoxic conditions (mimicking degeneration) and hypoxic conditions (reflecting the physiological disc environment), researchers found that hypoxia markedly upregulated global protein lactylation levels [120]. Using proteomics and high-resolution liquid chromatography–tandem mass spectrometry (LC-MS/MS), they identified 3,510 Kla sites across 1,052 proteins in NP cells, among which 27 proteins with 117 Kla sites were specifically regulated by the hypoxic environment [120]. These proteins were mainly enriched in pathways related to the ribosome, spliceosome, and VEGFA–VEGFR2 signaling, implicating their involvement in protein translation, RNA splicing, and angiogenesis relevant to disc homeostasis. These findings highlight the close association between Kla and the hypoxic microenvironment of the disc and suggest that dynamic changes in Kla expression may serve as potential indicators of intervertebral disc functional status.
Extending beyond NP, recent evidence indicates that EP cells are direct recipients of NP-derived lactate via MCT1, linking metabolic flux to epigenetic regulation [15]. In endplate (EP) cells, lactate acts as a metabolic epigenetic cue: lactate exposure increases total protein lactylation (pan-Kla) and histone H3K18 lactylation without altering global acetylation (pan-Kac) [15]. In vivo, nuclear H3K18 lactylation is evident in controls but markedly reduced when MCT1-mediated lactate uptake is ablated, indicating dependence on endogenous lactate flux. RNA-seq after 20 mM lactate for 24 h identifies 131 DEGs (80 down, 51 up), with downregulation of fatty-acid–related genes/pathways and shifts consistent with chondrogenic/osteogenic program adjustment [15]. Collectively, lactate-driven H3K18la links NP-derived lactate, MCT1 transport, and EP transcriptional remodeling within the IVD.
Potential Targeted Interventions of Lactylation in IVD. The functional role of Kla in IDD is only beginning to be elucidated. Shi et al. [119] performed an integrative analysis of bulk RNA-seq data from IDD tissues, combining differential expression and weighted gene co-expression network analysis (WGCNA) to identify lactylation-related genes (LRGs). Six hub LRGs were identified that correlated closely with disease severity across both training and validation cohorts. Among them, chromobox protein homolog 3 (CBX3) was the most significantly upregulated in degenerative tissues and showed a strong positive correlation with global lysine lactylation (pan-Kla) levels in an IL-1β–stimulated NP cell model [119]. Molecular docking predicted atosiban acetate as a potential small-molecule inhibitor targeting CBX3. In vitro, this compound suppressed CBX3 expression as well as lactate metabolism–related enzymes LDHA and PKM2, thereby reducing Kla levels, restoring aggrecan (ACAN) and collagen type II alpha 1 (COL2A1) synthesis, and inhibiting matrix-degrading enzymes MMP3 and ADAMTS5. Histological analysis further confirmed that atosiban acetate significantly alleviated IL-1β–induced IDD. This study provided the first characterization of CBX3-mediated lactylation mechanisms in IDD and proposed CBX3 and its inhibitor as novel diagnostic and therapeutic targets [119].
In addition, Zhang et al. [121] recently elucidated a “glutamine–lactate–AMPKα lactylation axis” as a critical regulatory mechanism in IDD progression. Metabolomic profiling revealed reduced glutamine levels accompanied by lactate accumulation and increased Kla modifications in severely degenerated human NP tissues and senescent rat models [121]. Under this metabolic milieu, AMPKα displayed elevated lysine lactylation and reduced phosphorylation, indicating lactylation-mediated inhibition of its kinase activity [121]. Both in vitro and in vivo experiments demonstrated that glutamine supplementation attenuated glycolytic flux, reduced lactate production and AMPKα lactylation, and restored AMPKα phosphorylation [121]. This reactivated the Nrf2/HO-1 antioxidant pathway, reduced intracellular ROS accumulation, downregulated p21 and p16, and enhanced autophagy (LC3-II/I and Beclin1), thereby delaying NPC senescence [121].
Taken together, lactylation functions as a crucial molecular bridge linking metabolic states with transcriptional and signaling outcomes. Future research is needed to confirm whether disc protein lactylation drives phenotypic reprogramming by upregulating catabolic mediators (e.g., inflammatory cytokines and matrix-degrading enzymes) while repressing anabolic and protective factors (e.g., matrix synthesis enzymes and antioxidant proteins). If so, interventions that modulate lactylation—whether by enhancing lactate clearance or targeting key lactylation regulators—hold promise for altering the trajectory of disc degeneration. Given the inherent plasticity of epigenetic modifications, such strategies may provide reversible and therapeutically exploitable approaches to IDD.
Mechanisms of Lactate–Acetylation Crosstalk in IVD Homeostasis and IDD. Lysine acetylation is an important post-translational modification catalyzed by lysine acetyltransferases (KATs). By modulating stability, activity, and interaction of both histone and non-histone proteins, acetylation broadly regulates biological processes such as cell proliferation, inflammatory responses, and chromatin remodeling [122]. Recent studies have shown that acetylation is closely linked to local inflammation, cellular senescence, and ECM degradation during IDD [123].
Sirtuins, a family of NAD⁺-dependent deacetylases, act as key epigenetic regulators of chromatin structure and transcription [124]. Among them, Sirt1 is the most extensively studied and is significantly downregulated in IDD. Sirt1 exerts both anti-inflammatory and anti-degenerative effects [125,126,127]. Wei et al. [123] reported that Sirt1 expression is markedly reduced in degenerated NP tissue. Overexpression of Sirt1 suppresses NF-κB pathway activation, decreases the secretion of pro-inflammatory cytokines such as IL-6 and TNF-α, and mitigates ECM degradation, thereby preserving NP homeostasis [123]. Mechanistically, Sirt1 deacetylates the NF-κB p65 subunit at lysine 310 (K310), attenuating its transcriptional activity, and simultaneously inhibits phosphorylation at serine 536 (S536), thus broadly suppressing NF-κB–dependent inflammatory transcriptional programs [123]. Furthermore, Sirt1 downregulates the transcriptional target lipocalin-2, which promotes M1 macrophage polarization. This disruption of the NP–immune cell feedback loop alleviates the inflammatory microenvironment [123].
Beyond Sirt1, conditional knockout of Sirt6 in IVD-specific mouse models resulted in elevated H3K9 acetylation across NP, AF, and CEP regions. This was accompanied by increased expression of γH2AX and cell cycle inhibitors (p21/p19), together with enhanced secretion of senescence-associated secretory phenotype (SASP) factors including IL-6, IL-1β, and TGF-β. These pathological alterations were associated with reduced disc height and aggravated histological degeneration [128]. Collectively, these findings highlight the essential role of Sirt6-mediated histone deacetylation in maintaining disc homeostasis and delaying age-related IDD progression.
Within the framework of metabolism–epigenetic crosstalk, a complex and dynamic interplay has been identified between lactylation and acetylation [20,129,130,131,132,133]. Both modifications utilize acyl-CoA intermediates derived from central carbon metabolism—lactyl-CoA and acetyl-CoA—reflecting the balance between glycolytic fermentation and oxidative phosphorylation [134,135]. Emerging evidence further indicates that Kla and lysine acetylation (Kac) can occur on identical histone residues (e.g., H3K18, H3K14), yet elicit distinct transcriptional outcomes through differential recruitment of reader proteins and competition for acyl-group donors [134,136]. In the hypoxic microenvironment of the IVD, the conversion of pyruvate to lactate is markedly enhanced. This metabolic shift reduces acetyl-CoA availability for acetylation while expanding the precursor pool for lactyl-CoA, thereby promoting lactylation [120,137,138]. Excessive lactate can also be converted into lactyl-CoA through the lactate dehydrogenase–acyltransferase system, further intensifying Kla [139].
At the enzymatic level, classical acetyltransferases such as p300/CBP have been shown to possess lactyltransferase activity under specific conditions. Likewise, histone deacetylases (HDACs) and sirtuins such as SIRT3 can catalyze both deacetylation and delactylation [140,141]. Consequently, Kla and Kac may compete or cooperate at the same or adjacent residues. For example, HDAC1/3 not only remove acetyl groups but also partially erase lactyl marks [132,142,143]. Interestingly, lactate has been shown to upregulate HDAC-related gene expression while paradoxically inhibiting HDAC enzymatic activity, ultimately promoting global histone hyperacetylation [138]. Hence, further research is needed to elucidate the interplay between Kla and Kac in IVD metabolic and epigenetic regulation.
Lactate-Mediated Cellular Signaling Pathways in the IVD. In addition to serving as a biofuel and substrate for histone lactylation, lactate through several recent studies also acts as a critical signaling molecule. In IVD cells, lactate—and the acidic microenvironment it generates—can activate multiple signaling pathways that regulate gene expression, cellular phenotype, and fate decisions. The following sections summarize the major signaling pathways through which lactate influences cellular behavior within the IVD.
Role of ASICs in Acid Sensing and Stress Responses in the IVD.ASICs are proton-gated sodium channels highly sensitive to reductions in extracellular pH. They are widely expressed throughout the IVD, including the NP, AF, CEP [144]. Within an acidic microenvironment, ASICs permit Na⁺ influx and, for certain subtypes (e.g., ASIC1a), promote Ca²⁺ entry, thereby activating intracellular signaling pathways that regulate stress responses, programmed cell death, cellular senescence, and inflammation [145]. During IDD, ASIC expression is dysregulated, with ASIC1 and ASIC3 showing the most pronounced upregulation¹¹⁰. Histopathological analyses indicate that ASIC1 and ASIC3 expression in NP and AF cells increases with IDD severity, whereas ASIC2 and ASIC4 exhibit little or no change [146]. This pattern suggests that ASIC1 and ASIC3 play central regulatory roles in acid-induced degenerative responses within the disc.
Functionally, ASIC activation elevates intracellular Ca²⁺—either directly via Ca²⁺-permeable ASIC1a or indirectly through downstream channels—thereby initiating cellular stress programs. For example, ASIC1a activation in CEP cells can induce programmed cell death, whereas ASIC3 activation under combined acidity and hyperosmolarity engages the NGF/p75 neurotrophin receptor /extracellular signal-regulated kinase pathway, promoting NP cell survival and resistance to apoptosis [147]. Moreover, ASIC1 and ASIC3 activate canonical senescence pathways—including the p53–p21/p27 and p16–retinoblastoma protein axes—thereby driving NP-MSCs into senescence, diminishing their regenerative capacity, and accelerating IDD progression [146]. In summary, ASICs—particularly ASIC1 and ASIC3—serve as critical acid sensors in the IVD. They can amplify tissue-damage and inflammatory signaling yet also exert context-dependent protective effects. Defining subtype-specific functions and downstream networks of ASICs may inform therapeutic strategies targeting acid-stress–related disc degeneration.
Dual Role of Lactate in Inflammation Regulation: From Pathological Stress to Immunometabolic Signaling. Lactate not only contributes to tissue acidification and pro-inflammatory responses but also exerts substantial immunomodulatory and protective effects [148]. During inflammation, enhanced glycolysis often produces excess lactate, thereby driving extracellular acidification and activating pro-inflammatory pathways [17]. Nevertheless, accumulating evidence indicates that lactate exerts multifaceted regulation of immune-cell metabolism and gene expression [17,65,148]. Mechanistically, lactate signals through diverse routes, including receptor binding—e.g., GPR81 (also known as hydroxycarboxylic acid receptor 1) [149,150,151,152]-activation of transcription factors such as NF-κB [153,154,155] and HIF-1α [154], and epigenetic modifications, notably protein (lysine) lactylation [133]. Collectively, these pathways modulate the expression of cytokines, chemokines, and metabolic enzymes, underscoring the highly context-dependent nature of lactate signaling across disease states [17].In disc cells, Zhao et al. reported that lactate stimulation upregulates ASIC1a and ASIC3 in NP cells, increasing intracellular Ca²⁺ influx and reactive oxygen species (ROS) generation; this cascade activates NF-κB, promoting NLRP3 inflammasome assembly and release of IL-1β [84]. Furthermore, lactate can induce necroptosis, thereby exacerbate tissue damage and highlight its potential role as a danger-associated signal that facilitates disc degeneration under specific conditions [84].
Collectively, these findings reposition lactate as a critical immunometabolism signaling molecule rather than solely a metabolic byproduct or pro-inflammatory mediator. This conceptual shift provides new mechanistic insight and opens translational avenues toward the development of targeted interventions for autoimmune and degenerative diseases.
Lactate-Related Akt Signaling and Cellular Senescence in the IVD. In IDD, the PI3K/Akt signaling pathway exerts a context-dependent dual role, functioning either as a protective or a pro-degenerative regulator depending on the cellular microenvironment. Attenuation of this pathway can be beneficial: Genkwanin alleviates intervertebral disc degeneration by inhibiting the overactivation of the ITGA2/PI3K/Akt/mTOR signaling pathway, thereby suppressing inflammation, apoptosis, and senescence in nucleus pulposus cells [156]. Similarly, evidence indicates that melatonin mitigates nucleus pulposus cell apoptosis and matrix degradation through downregulation of PI3K/Akt and NF-κB signaling cascades mediated by its receptors [157].
Conversely, appropriate activation of PI3K/Akt under stress conditions is cytoprotective: Bradykinin enhances the antioxidant capacity of nucleus pulposus cells, maintains mitochondrial homeostasis, and reduces apoptosis by activating the B2R–PI3K/Akt signaling axis, thereby delaying the progression of IVDD [158]. Moreover, studies have shown that, in a hypoxic environment, rat nucleus pulposus cells enhance their survival by modulating key gene expression and suppressing apoptosis through activation of the PI3K/Akt and MAPK signaling pathways [159].
High concentrations of lactic acid were shown to trigger oxidative stress and cellular senescence in NP cells [160].Transcriptomic and bioinformatic analyses indicated that these effects are associated with the activation of the PI3K/Akt signaling pathway [160]. Subsequent mechanistic studies confirmed that lactic acid interacts directly with Akt, modulating its downstream cascades—including Akt/p21/p27/cyclin D1 and Akt/Nrf2/HO-1 pathways—to promote oxidative stress and senescence in NP cells [160]. Molecular docking, site-directed mutagenesis, and microscale thermophoresis experiments further demonstrated that lactic acid binds to the Lys39 and Leu52 residues within the PH domain of Akt, thereby regulating its kinase activity [160].
Lactate-Mediated Regulation of Ferroptosis: Signaling Pathways and Molecular Mechanisms in IVD. Ferroptosis is a form of regulated cell death characterized by iron-dependent metabolic dysregulation, lipid peroxidation, and impaired antioxidant defenses. It has been implicated in cancer, metabolic disorders, and autoimmune diseases [161]. Recent evidence suggests that ferroptosis also plays a pivotal role in IDD. In NP cells, elevated oxidative stress and lipid peroxidation can trigger ferroptosis, leading to cellular dysfunction, ECM degradation, and progressive structural deterioration of the IVD [3,162]. Xiang et al. [163] performed a comprehensive transcriptomic analysis of IDD tissues and controls using the Gene Expression Omnibus database and identified 80 ferroptosis-related differentially expressed genes (FRDEGs). Functional enrichment analysis indicated that these genes were mainly involved in responses to chemical stimuli and cellular stress, along with ferroptosis, TNF, HIF-1, NOD-like receptor, and IL-17 signaling pathways. Protein–protein interaction network analysis further identified 10 hub FRDEGs [163]. Receiver operating characteristic curve analysis based on the GSE124272 dataset demonstrated strong diagnostic potential of these core genes for IDD, and their expression trends were validated by RT-qPCR in a TBHP-induced NP cells degeneration model [163]. This study provided the first systematic characterization of ferroptosis-related genes in IDD and suggested potential molecular targets for early diagnosis and therapeutic intervention.
Several groups have further proposed ferroptosis-targeted therapeutic strategies by modulating iron homeostasis. Li et al. [164] reported that 1,25-dihydroxyvitamin D₃ attenuated IDD progression by activating the vitamin D receptor pathway and suppressing ferroptosis in NP cells. Lu et al. [165] demonstrated that under oxidative stress, downregulation of the iron exporter ferroportin leads to intracellular iron accumulation and enhanced lipid peroxidation. Conversely, FPN overexpression upregulated glutathione peroxidase 4 (GPX4)—a key ferroptosis suppressor—reduced ROS levels, improved disc structure and function, and thereby delayed IDD progression [165].
Recent studies demonstrate that lactate functions as a critical signaling molecule regulating ferroptosis through multiple mechanisms—particularly within the degenerative IVD microenvironment. Sun et al. [166] established a puncture-induced rat model of IDD and performed in vitro experiments with human NP cells to investigate the role of glycolysis-derived lactate in ferroptosis during disc degeneration. Multi-omics analyses revealed significantly elevated lactate levels during IDD, together with upregulation of acyl-CoA synthetase long-chain family member 4 (ACSL4), a key pro-ferroptotic enzyme. Mechanistically, lactate enhanced ACSL4 activity via two complementary pathways: (1) by inducing histone H3K18 lactylation, thereby promoting transcriptional activation of ACSL4, and (2) by inhibiting sirtuin 3 which increased ACSL4 protein lactylation and stability. These processes culminated in lipid peroxide accumulation and ferroptosis in NP cells [166]. In vivo, lentivirus-mediated overexpression of lactate oxidase significantly reduced lactate accumulation, suppressed ACSL4 expression and ferroptosis, and improved disc structure, thereby delaying degeneration. This study reveals that lactate contributes significantly to the progression of IVDD by triggering ferroptosis [166]. Lactate as a metabolic signal capable of modulating cell fate in IDD and propose potential targets for metabolic reprogramming–based therapies [166].
Beyond lactate-induced histone lactylation, other epigenetic mechanisms also contribute to ferroptosis regulation in the IVD. For instance, elevated homocysteine has been shown to induce ferroptosis in NP cells by suppressing GPX4 via DNA methylation [167]. Moreover, ferroptosis-associated lipid peroxidation and iron overload during IDD can lead to the release of intracellular contents that act as damage-associated molecular patterns, triggering inflammatory responses in neighboring cells and exacerbating tissue injury and degeneration [3]. Collectively, the lactate–lactylation–ACSL4 axis, together with other stress-induced epigenetic mechanisms, constitutes a complex ferroptosis network in IDD progression and offers promising targets for disease-modifying interventions. The schematic of lactate-mediated cellular signaling pathways in the IVD is shown in Figure 2.
The left panel represents a healthy IVD, while the right panel illustrates a degenerative disc. In the degenerative disc, lactate functions as a key signaling molecule involved in multiple pathological pathways, promoting NP cell senescence, apoptosis, autophagy, and inflammatory responses. Conversely, in the healthy disc, lactate exerts bidirectional regulatory effects and plays a crucial role in maintaining intradiscal homeostasis.
5. Future Perspectives
The metabolism and new functional roles of lactate in the IVD have only recently emerged as a focus of scientific inquiries. This ongoing research of IVD lactate has reshaped our understanding of disc physiology and pathophysiology, while simultaneously opening new avenues for the prevention and treatment of IDD.
Integrating Mechanisms that Regulate IVD Lactate Metabolism. Lactate metabolism plays a pivotal role in maintaining the physiological and metabolic homeostasis of the IVD. Current evidence indicates that the CEP serves as the principal pathway for the influx of nutrients and the efflux of metabolic byproducts such as lactate. Meanwhile, MCTs are crucial mediators of lactate trafficking across disc cells, ensuring the balance between production and clearance. However, the precise interplay between CEP-mediated diffusion and MCT-facilitated transport remains largely unresolved. It is unclear how these mechanisms coordinate under different physiological and pathological conditions—such as aging, degeneration, or altered loading environments—to regulate disc homeostasis. Future studies employing integrative approaches combining advanced imaging, single-cell transcriptomics, and metabolomic profiling are warranted to delineate the spatial–temporal regulation of lactate metabolism and its contribution to the maintenance or disruption of disc health.
Advancing Research on Lactate-dependent metabolic symbiosis in IVD. Recent studies suggest a lactate-centered symbiotic/coupled metabolic network among the NP, AF, and EP, identifying MCTs—particularly MCT4 and MCT1—as key executors and challenging the notion of lactate as a mere metabolic waste product [11,15]. However, the existing evidence base remains narrow in scope and limited in depth. Compared with oncology and other fields, where the lactate–MCT–CD147 (EMMPRIN)–HIF-1α–LDH axis has been systematically delineated, biology of intervertebral disc still lacks a comprehensive mapping of mechanistic pathways, hierarchical regulation, and actionable intervention nodes. Accordingly, there is a need for more systematic, quantitative, and cross-scale investigations in the IVD context to broaden and deepen our understanding of lactate symbiotic/coupled metabolism and to define its physiological significance and translational potential.
Elucidating Histone Lactylation Regulatory Network in IVD. As a novel form of epigenetic modification, Kla remains in the early stages of investigation in IVD biology. Key unanswered questions include (1) which histones are lactylated and on which lysine sites under normal and stress conditions, (2) which gene sets are most sensitive to epigenetic regulation by histone lactylation, (3) what are the key histone lactylases, delactylases, and readers of lactylated histones in IVD, (4) which non-histone proteins serve as lactylation targets and how their functions are modulated by such post-translational modification, (5) whether dysregulated protein lactylation drive IDD. Addressing these questions will require more sensitive detection techniques and systematic experimental strategies. For example, integrating quantitative mass spectrometry with chromatin immunoprecipitation sequencing could reveal lactylation profiles in healthy versus degenerated discs to identify lactylation-enriched promoters and regulatory networks.
Lactate-Mediated Signaling Pathways in the IVD: Emerging Insights and Future Directions. Emerging evidence suggests that lactate is a signaling molecule that modulates diverse cellular pathways within the IVD. Recent studies have implicated lactate-mediated signaling in the regulation of ferroptosis, inflammation, oxidative stress, and extracellular matrix remodeling, highlighting its potential role in disc degeneration. Nevertheless, compared with other tissues and disease models—such as cancer, skeletal muscle, and the central nervous system—the mechanistic understanding of lactate-driven signaling in the IVD remains limited. Future research should aim to elucidate how lactate interacts with key signaling pathways, including PI3K/Akt, MAPK, and NF-κB, and whether it functions as a context-dependent regulator of cell survival or death. Exploring these mechanisms will be critical for clarifying the dual role of lactate in maintaining disc homeostasis versus promoting degeneration.
6. Conclusions
In recent years, IVD lactate has gained increasing recognition. The prevailing view has shifted from considering lactate merely as a “metabolic waste product” to acknowledging its multifaceted functions in energy metabolism, signal transduction, and epigenetic regulation in IVD biology. Under pathological conditions, excessive lactic acid contributes to cellular dysfunction and tissue degeneration, demonstrating a detrimental effect. Conversely, lactate under normal physiological condition supports cellular homeostasis and modulates key signaling pathways, thereby exerting protective influences. Further investigation is required to elucidate mechanisms of the dual roles of lactate as a driver of IDD and protector of IVD health.
Looking ahead, the integration of multi-omics technologies, advanced molecular imaging, and functional validation studies will be crucial to constructing a comprehensive map of lactate metabolic and signaling networks in the IVD. Such advances could provide both a framework and translational opportunities for the management of degenerative disc disease. Therapeutic strategies targeting lactate production, transport, and downstream signaling may enable modulation of the intradiscal microenvironment and deceleration of disease progression.
Author Contributions
Ting Zhang and Peng Feng contributed equally to this work and co-wrote the original draft of the manuscript. Peng Feng also prepared all the figures included in the manuscript. Nam Vo conceived and designed the manuscript, and oversaw the overall revision and supervision. Gwendolyn Sowa, Joon Lee, and Peter G. Alexander contributed to the critical revision of the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported by the National Institutes of Health (NIH), National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) under grant number R01AR081234, the Bethel Musculoskeletal Research Center, the Ferguson Laboratory for Orthopaedic and Spine Research, and the Department of Physical Medicine and Rehabilitation (PM&R).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The figures were created using Adobe illustrator and Bio-render.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| Abbreviation | Full Name |
| ACAN | Aggrecan |
| AF | Annulus fibrosus |
| ASICs | Acid-sensing ion channels |
| ACSL4 | Acyl-CoA synthetase long chain family member 4 |
| BDNF | Brain-derived neurotrophic factor |
| B2R | B2 receptor |
| CBX3 | Chromobox protein homolog 3 |
| CEP | Cartilage endplate |
| CEPCs | Cartilage endplate progenitor cells |
| CD147 | Cluster of differentiation 147 |
| CEST | Chemical exchange saturation transfer |
| c-Myc | Cellular myelocytomatosis oncogene |
| COL2A1 | Collagen type II alpha 1 |
| DCs | Dendritic cells |
| EAE | Experimental autoimmune encephalomyelitis |
| ECM | Extracellular matrix |
| ECAR | Extracellular acidification rate |
| EMMPRIN | Matrix metalloproteinase inducer |
| EPs | Endplate chondrocytes |
| ERK | Extracellular signal-regulated kinase |
| FOXO1 | Forkhead Box Protein O1 |
| FRDEGs | Ferroptosis-related differentially expressed genes |
| GLUT1 | Glucose transporter 1 |
| GLUT3 | Glucose transporter 3 |
| GPX4 | Glutathione peroxidase 4 |
| GAG | Glycosaminoglycan |
| GPR81 | G protein–coupled receptor 81 |
| GSK-3β | Glycogen synthase kinase-3β |
| G6P | Glucose-6-phosphate |
| H3K18 | Histone H3 lysine 18 |
| HDACs | Histone deacetylases |
| HIF | Hypoxia-inducible factor |
| HIF-1α | Hypoxia-Inducible Factor-1 alpha |
| IVD | Intervertebral disc |
| IDD | Intervertebral disc degeneration |
| IAF | Inner annulus fibrosus |
| IL-1β | Interleukin-1 beta |
| Kla | Lysine lactylation |
| Kac | Lysine acetylation |
| K310 | Lysine 310 |
| KATs | Lysine acetyltransferases |
| LBP | Low back pain |
| LDHA | Lactate dehydrogenase A |
| LDHB | Lacate dehydrogenase B |
| LDH5 | Lactate dehydrogenase 5 |
| LOx | Lactate oxidase |
| LPS | Lipopolysaccharide |
| LRGs | Lactylation-related genes |
| MCT | Monocarboxylate transporter |
| MMP3 | Matrix metalloproteinase 3 |
| MMP2 | Matrix metalloproteinase 2 |
| MMP9 | Matrix metalloproteinase 9 |
| MMP13 | Matrix metalloproteinase 13 |
| MRS | Magnetic resonance spectroscopy |
| NP | Nucleus pulposus |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| NAD⁺ | Nicotinamide adenine dinucleotide |
| NGF | Nerve Growth Factor |
| OAF | Outer annulus fibrosus |
| OCR | Oxygen consumption rate |
| PI3K/Akt | Phosphatidylinositol 3-kinase / Protein kinase B |
| pan-Kla | Global lysine lactylation |
| PDK1 | Pyruvate dehydrogenase kinase 1 |
| PDH | Pyruvate dehydrogenase |
| PC | Pyruvate carboxylase |
| PH | Pleckstrin homology |
| p75NTR | p75 neurotrophin receptor |
| p53 | Tumor protein p53 |
| p21 | Cyclin-dependent kinase inhibitor 1A |
| p27 | Cyclin-dependent kinase inhibitor 1B |
| p16 | Cyclin-dependent kinase inhibitor 2A |
| RCD | Regulated cell death |
| ROC | Receiver operating characteristic |
| ROS | Reactive oxygen species |
| SLC16A3 | Solute Carrier Family 16 Member 3 |
| SIRT3 | Sirtuin 3 |
| S536 | Serine 536 |
| SASP | Senescence-associated secretory phenotype |
| TBHP | Tert-butyl hydroperoxide |
| TCA | Tricarboxylic acid |
| TZ | Transition zone |
| TGF-β | Transforming Growth Factor-beta |
| VDR | Vitamin D receptor |
References
- Murray, C. J.; et al. The state of US health, 1990-2010: burden of diseases, injuries, and risk factors. JAMA 2013, 310, 591–608. [Google Scholar] [CrossRef]
- Livshits, G.; Popham, M.; Malkin, I.; Sambrook, P.N.; Macgregor, A.J.; Spector, T.; Williams, F.M. Lumbar disc degeneration and genetic factors are the main risk factors for low back pain in women: the UK Twin Spine Study. Ann Rheum Dis 2011, 70, 1740–1745. [Google Scholar] [CrossRef]
- Fan, C.; Chu, G.; Yu, Z.; Ji, Z.; Kong, F.; Yao, L.; Wang, J.; Geng, D.; Wu, X.; Mao, H. The role of ferroptosis in intervertebral disc degeneration. Front Cell Dev Biol 2023, 11, 1219840. [Google Scholar] [CrossRef]
- Pattappa, G.; Li, Z.; Peroglio, M.; Wismer, N.; Alini, M.; Grad, S. Diversity of intervertebral disc cells: phenotype and function. J Anat 2012, 221, 480–496. [Google Scholar] [CrossRef] [PubMed]
- Silagi, E.S.; Novais, E.J.; Bisetto, S.; Telonis, A.G.; Snuggs, J.; Le Maitre, C.L.; Qiu, Y.; Kurland, I.J.; Shapiro, I.M.; Philp, N.J.; et al. Lactate Efflux From Intervertebral Disc Cells Is Required for Maintenance of Spine Health. J Bone Miner Res 2020, 35, 550–570. [Google Scholar] [CrossRef] [PubMed]
- Silagi, E.S.; Schipani, E.; Shapiro, I.M.; Risbud, M.V. The role of HIF proteins in maintaining the metabolic health of the intervertebral disc. Nat Rev Rheumatol 2021, 17, 426–439. [Google Scholar] [CrossRef]
- Ren, P.; Chen, P.; Reeves, R.A.; Buchweitz, N.; Niu, H.; Gong, H.; Mercuri, J.; Reitman, C.A.; Yao, H.; Wu, Y. Diffusivity of Human Cartilage Endplates in Healthy and Degenerated Intervertebral Disks. J Biomech Eng 2023, 145. [Google Scholar] [CrossRef]
- Ohshima, H.; Urban, J.P. The effect of lactate and pH on proteoglycan and protein synthesis rates in the intervertebral disc. Spine (Phila Pa 1976) 1992, 17, 1079–1082. [Google Scholar] [CrossRef]
- Pushpa, B.T.; Rajasekaran, S.; Easwaran, M.; Murugan, C.; Algeri, R.; Sri Vijay Anand, K.S.; Mugesh Kanna, R.; Shetty, A.P. ISSLS PRIZE in basic science 2023: Lactate in lumbar discs-metabolic waste or energy biofuel? Insights from in vivo MRS and T2r analysis following exercise and nimodipine in healthy volunteers. Eur Spine J 2023, 32, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Liang, H.; Du, Z.; Liu, Q.; Li, G.; Zhang, W.; Wu, D.; Zhou, X.; Song, Y.; Yang, C. Altered Metabolism and Inflammation Driven by Post-translational Modifications in Intervertebral Disc Degeneration. Research 2024, 7, 0350. [Google Scholar] [CrossRef]
- Wang, D.; Hartman, R.; Han, C.; Zhou, C.M.; Couch, B.; Malkamaki, M.; Roginskaya, V.; Van Houten, B.; Mullett, S.J.; Wendell, S.G.; et al. Lactate oxidative phosphorylation by annulus fibrosus cells: evidence for lactate-dependent metabolic symbiosis in intervertebral discs. Arthritis Res Ther 2021, 23, 145. [Google Scholar] [CrossRef]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A.; Curl, C.C.; Leija, R.G.; Osmond, A.D.; Duong, J.J.; Arevalo, J.A. Tracing the lactate shuttle to the mitochondrial reticulum. Exp Mol Med 2022, 54, 1332–1347. [Google Scholar] [CrossRef]
- Tsingas, M.; Tsingas, K.; Zhang, W.; Goldman, A.R.; Risbud, M.V. Lactate metabolic coupling between the endplates and nucleus pulposus via MCT1 is essential for intervertebral disc health. bioRxiv 2025. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.F.; Wang, G.; Ding, L.; Bai, Z.R.; Leng, Y.; Tian, J.W.; Zhang, J.Z.; Li, Y.Q.; Ahmad; Qin, Y.H.; et al. Lactate-upregulated NADPH-dependent NOX4 expression via HCAR1/PI3K pathway contributes to ROS-induced osteoarthritis chondrocyte damage. Redox Biol 2023, 67, 102867. [CrossRef]
- Manosalva, C.; Quiroga, J.; Hidalgo, A.I.; Alarcon, P.; Anseoleaga, N.; Hidalgo, M.A.; Burgos, R.A. Role of Lactate in Inflammatory Processes: Friend or Foe. Front Immunol 2021, 12, 808799. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, G.; Xu, Z.G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189. [Google Scholar] [CrossRef]
- Hu, Y.; He, Z.; Li, Z.; Wang, Y.; Wu, N.; Sun, H.; Zhou, Z.; Hu, Q.; Cong, X. Lactylation: the novel histone modification influence on gene expression, protein function, and disease. Clin Epigenetics 2024, 16, 72. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Wang, N.; Wang, W.; Wang, X.; Mang, G.; Chen, J.; Yan, X.; Tong, Z.; Yang, Q.; Wang, M.; Chen, L.; et al. Histone Lactylation Boosts Reparative Gene Activation Post-Myocardial Infarction. Circ Res 2022, 131, 893–908. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, J.; Zhai, L.; Zhang, T.; Yin, H.; Gao, H.; Zhao, F.; Wang, Z.; Yang, X.; Jin, M.; et al. Metabolic regulation of homologous recombination repair by MRE11 lactylation. Cell 2024, 187, 294–311. [Google Scholar] [CrossRef]
- Holm, S.; Maroudas, A.; Urban, J.P.; Selstam, G.; Nachemson, A. Nutrition of the intervertebral disc: solute transport and metabolism. Connect Tissue Res 1981, 8, 101–119. [Google Scholar] [CrossRef]
- Urban, J.P.; Smith, S.; Fairbank, J.C. Nutrition of the intervertebral disc. Spine (Phila Pa 1976) 2004, 29, 2700–2709. [Google Scholar] [CrossRef] [PubMed]
- Laffosse, J.M.; Accadbled, F.; Molinier, F.; Bonnevialle, N.; de Gauzy, J.S.; Swider, P. Correlations between effective permeability and marrow contact channels surface of vertebral endplates. J Orthop Res 2010, 28, 1229–1234. [Google Scholar] [CrossRef]
- Urban, J.P.; Holm, S.; Maroudas, A.; Nachemson, A. Nutrition of the intervertebral disc: effect of fluid flow on solute transport. Clin Orthop Relat Res 1982, 296–302. [Google Scholar] [CrossRef]
- Katz, M.M.; Hargens, A.R.; Garfin, S.R. Intervertebral disc nutrition. Diffusion versus convection. Clin Orthop Relat Res 1986, 243–245. [Google Scholar] [CrossRef]
- Ferguson, S.J.; Ito, K.; Nolte, L.P. Fluid flow and convective transport of solutes within the intervertebral disc. J Biomech 2004, 37, 213–221. [Google Scholar] [CrossRef]
- Roberts, S.; Menage, J.; Urban, J.P. Biochemical and structural properties of the cartilage end-plate and its relation to the intervertebral disc. Spine (Phila Pa 1976) 1989, 14, 166–174. [Google Scholar] [CrossRef]
- Bartels, E.M.; Fairbank, J.C.; Winlove, C.P.; Urban, J.P. Oxygen and lactate concentrations measured in vivo in the intervertebral discs of patients with scoliosis and back pain. Spine (Phila Pa 1976) 1998, 23, 1–7. [Google Scholar] [CrossRef]
- Nerlich, A.G.; Schaaf, R.; Walchli, B.; Boos, N. Temporo-spatial distribution of blood vessels in human lumbar intervertebral discs. Eur Spine J 2007, 16, 547–555. [Google Scholar] [CrossRef]
- Grunhagen, T.; Shirazi-Adl, A.; Fairbank, J.C.; Urban, J.P. Intervertebral disk nutrition: a review of factors influencing concentrations of nutrients and metabolites. Orthop Clin North Am 2011, 42, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Sakai, D.; Grad, S. Advancing the cellular and molecular therapy for intervertebral disc disease. Adv Drug Deliv Rev 2015, 84, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Nachemson, A.; Lewin, T.; Maroudas, A.; Freeman, M.A. In vitro diffusion of dye through the end-plates and the annulus fibrosus of human lumbar inter-vertebral discs. Acta Orthop Scand 1970, 41, 589–607. [Google Scholar] [CrossRef]
- Maroudas, A.; Stockwell, R.A.; Nachemson, A.; Urban, J. Factors involved in the nutrition of the human lumbar intervertebral disc: cellularity and diffusion of glucose in vitro. J Anat 1975, 120, 113–130. [Google Scholar]
- Urban, J.P.; Holm, S.; Maroudas, A. Diffusion of small solutes into the intervertebral disc: as in vivo study. Biorheology 1978, 15, 203–221. [Google Scholar] [CrossRef]
- Roberts, S.; Urban, J.P.; Evans, H.; Eisenstein, S.M. Transport properties of the human cartilage endplate in relation to its composition and calcification. Spine (Phila Pa 1976) 1996, 21, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Horner, H.A.; Urban, J.P. 2001 Volvo Award Winner in Basic Science Studies: Effect of nutrient supply on the viability of cells from the nucleus pulposus of the intervertebral disc. Spine (Phila Pa 1976) 2001, 26, 2543–2549. [Google Scholar] [CrossRef]
- Shalash, W.; Ahrens, S.R.; Bardonova, L.A.; Byvaltsev, V.A.; Giers, M.B. Patient-specific apparent diffusion maps used to model nutrient availability in degenerated intervertebral discs. JOR Spine 2021, 4, e1179. [Google Scholar] [CrossRef]
- Comandatore, A.; Franczak, M.; Smolenski, R.T.; Morelli, L.; Peters, G.J.; Giovannetti, E. Lactate Dehydrogenase and its clinical significance in pancreatic and thoracic cancers. Semin Cancer Biol 2022, 86, 93–100. [Google Scholar] [CrossRef]
- Yao, S.; Xu, M.D.; Wang, Y.; Zhao, S.T.; Wang, J.; Chen, G.F.; Chen, W.B.; Liu, J.; Huang, G.B.; Sun, W.J.; et al. Astrocytic lactate dehydrogenase A regulates neuronal excitability and depressive-like behaviors through lactate homeostasis in mice. Nat Commun 2023, 14, 729. [Google Scholar] [CrossRef]
- Lee, R.B.; Urban, J.P. Evidence for a negative Pasteur effect in articular cartilage. Biochem J 1997, 321 Pt 1, 95–102. [Google Scholar] [CrossRef]
- Bibby, S.R.; Jones, D.A.; Ripley, R.M.; Urban, J.P. Metabolism of the intervertebral disc: effects of low levels of oxygen, glucose, and pH on rates of energy metabolism of bovine nucleus pulposus cells. Spine (Phila Pa 1976) 2005, 30, 487–496. [Google Scholar] [CrossRef]
- Rigoulet, M.; Bouchez, C.L.; Paumard, P.; Ransac, S.; Cuvellier, S.; Duvezin-Caubet, S.; Mazat, J.P.; Devin, A. Cell energy metabolism: An update. Biochim Biophys Acta Bioenerg 2020, 1861, 148276. [Google Scholar] [CrossRef]
- Vavricka, J.; Broz, P.; Follprecht, D.; Novak, J.; Krouzecky, A. Modern Perspective of Lactate Metabolism. Physiol Res 2024, 73, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Gao, X.; Levene, H.B.; Brown, M.D.; Gu, W. Influences of Nutrition Supply and Pathways on the Degenerative Patterns in Human Intervertebral Disc. Spine (Phila Pa 1976) 2016, 41, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Guttapalli, A.; Narayan, S.; Albert, T.J.; Shapiro, I.M.; Risbud, M.V. Normoxic stabilization of HIF-1alpha drives glycolytic metabolism and regulates aggrecan gene expression in nucleus pulposus cells of the rat intervertebral disk. Am J Physiol Cell Physiol 2007, 293, C621–631. [Google Scholar] [CrossRef]
- Xiong, S.; Liu, Z.; Yao, J.; Huang, S.; Ding, X.; Yu, H.; Lin, T.; Zhang, X.; Zhao, F. HIF-1alpha regulated GLUT1-mediated glycolysis enhances Treponema pallidum-induced cytokine responses. Cell Commun Signal 2025, 23, 219. [Google Scholar] [CrossRef] [PubMed]
- Risbud, M.V.; Schipani, E.; Shapiro, I.M. Hypoxic regulation of nucleus pulposus cell survival: from niche to notch. Am J Pathol 2010, 176, 1577–1583. [Google Scholar] [CrossRef]
- Wang, F.; Chen, L.; Kong, D.; Zhang, X.; Xia, S.; Liang, B.; Li, Y.; Zhou, Y.; Zhang, Z.; Shao, J.; et al. Canonical Wnt signaling promotes HSC glycolysis and liver fibrosis through an LDH-A/HIF-1alpha transcriptional complex. Hepatology 2024, 79, 606–623. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Soukane, D.M.; Shirazi-Adl, A.; Urban, J.P. Computation of coupled diffusion of oxygen, glucose and lactic acid in an intervertebral disc. J Biomech 2007, 40, 2645–2654. [Google Scholar] [CrossRef]
- Pereira-Nunes, A.; Simoes-Sousa, S.; Pinheiro, C.; Miranda-Goncalves, V.; Granja, S.; Baltazar, F. Targeting lactate production and efflux in prostate cancer. Biochim Biophys Acta Mol Basis Dis 2020, 1866, 165894. [Google Scholar] [CrossRef]
- Bonglack, E.N.; Messinger, J.E.; Cable, J.M.; Ch'ng, J.; Parnell, K.M.; Reinoso-Vizcaino, N.M.; Barry, A.P.; Russell, V.S.; Dave, S.S.; Christofk, H.R.; et al. Monocarboxylate transporter antagonism reveals metabolic vulnerabilities of viral-driven lymphomas. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef]
- Drozdzik, M.; Szelag-Pieniek, S.; Grzegolkowska, J.; Lapczuk-Romanska, J.; Post, M.; Domagala, P.; Mietkiewski, J.; Oswald, S.; Kurzawski, M. Monocarboxylate Transporter 1 (MCT1) in Liver Pathology. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Narumi, K.; Furugen, A.; Iseki, K. Transport function, regulation, and biology of human monocarboxylate transporter 1 (hMCT1) and 4 (hMCT4). Pharmacol Ther 2021, 226, 107862. [Google Scholar] [CrossRef]
- Wang, N.; Jiang, X.; Zhang, S.; Zhu, A.; Yuan, Y.; Xu, H.; Lei, J.; Yan, C. Structural basis of human monocarboxylate transporter 1 inhibition by anti-cancer drug candidates. Cell 2021, 184, 370–383. [Google Scholar] [CrossRef]
- Singh, M.; Afonso, J.; Sharma, D.; Gupta, R.; Kumar, V.; Rani, R.; Baltazar, F.; Kumar, V. Targeting monocarboxylate transporters (MCTs) in cancer: How close are we to the clinics? Semin Cancer Biol 2023, 90, 1–14. [Google Scholar] [CrossRef] [PubMed]
- D'Aria, S.; Maquet, C.; Li, S.; Dhup, S.; Lepez, A.; Kohler, A.; Van Hee, V.F.; Dadhich, R.K.; Freniere, M.; Andris, F.; et al. Expression of the monocarboxylate transporter MCT1 is required for virus-specific mouse CD8(+) T cell memory development. Proc Natl Acad Sci U S A 2024, 121, e2306763121. [Google Scholar] [CrossRef] [PubMed]
- Kirk, P.; Wilson, M.C.; Heddle, C.; Brown, M.H.; Barclay, A.N.; Halestrap, A.P. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J 2000, 19, 3896–3904. [Google Scholar] [CrossRef]
- Walters, D.K.; Arendt, B.K.; Jelinek, D.F. CD147 regulates the expression of MCT1 and lactate export in multiple myeloma cells. Cell Cycle 2013, 12, 3175–3183. [Google Scholar] [CrossRef]
- Bovenzi, C.D.; Hamilton, J.; Tassone, P.; Johnson, J.; Cognetti, D.M.; Luginbuhl, A.; Keane, W.M.; Zhan, T.; Tuluc, M.; Bar-Ad, V.; et al. Prognostic Indications of Elevated MCT4 and CD147 across Cancer Types: A Meta-Analysis. Biomed Res Int 2015, 2015, 242437. [Google Scholar] [CrossRef]
- Meng, S.; Sorensen, E.E.; Ponniah, M.; Thorlacius-Ussing, J.; Crouigneau, R.; Larsen, T.; Borre, M.T.; Willumsen, N.; Flinck, M.; Pedersen, S.F. MCT4 and CD147 colocalize with MMP14 in invadopodia and support matrix degradation and invasion by breast cancer cells. J Cell Sci 2024, 137. [Google Scholar] [CrossRef]
- Combs, J.E.; Murray, A.B.; Lomelino, C.L.; Mboge, M.Y.; Mietzsch, M.; Horenstein, N.A.; Frost, S.C.; McKenna, R.; Becker, H.M. Disruption of the Physical Interaction Between Carbonic Anhydrase IX and the Monocarboxylate Transporter 4 Impacts Lactate Transport in Breast Cancer Cells. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Fang, Y.; Li, Z.; Yang, L.; Li, W.; Wang, Y.; Kong, Z.; Miao, J.; Chen, Y.; Bian, Y.; Zeng, L. Emerging roles of lactate in acute and chronic inflammation. Cell Commun Signal 2024, 22, 276. [Google Scholar] [CrossRef]
- Halestrap, A.P. Monocarboxylic acid transport. Compr Physiol 2013, 3, 1611–1643. [Google Scholar] [CrossRef]
- Payen, V.L.; Mina, E.; Van Hee, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol Metab 2020, 33, 48–66. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Baeza, Y.; Sandoval, P.Y.; Alarcon, R.; Galaz, A.; Cortes-Molina, F.; Alegria, K.; Baeza-Lehnert, F.; Arce-Molina, R.; Guequen, A.; Flores, C.A.; et al. Monocarboxylate transporter 4 (MCT4) is a high affinity transporter capable of exporting lactate in high-lactate microenvironments. J Biol Chem 2019, 294, 20135–20147. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed]
- Monsorno, K.; Ginggen, K.; Ivanov, A.; Buckinx, A.; Lalive, A.L.; Tchenio, A.; Benson, S.; Vendrell, M.; D'Alessandro, A.; Beule, D.; et al. Loss of microglial MCT4 leads to defective synaptic pruning and anxiety-like behavior in mice. Nat Commun 2023, 14, 5749. [Google Scholar] [CrossRef]
- Wang, C.Y.; Hsieh, M.K.; Hu, Y.J.; Bit, A.; Lai, P.L. Monocarboxylate transporter 1-mediated lactate accumulation promotes nucleus pulposus degeneration under hypoxia in a 3D multilayered nucleus pulposus degeneration model. Eur Cell Mater 2022, 43, 53–65. [Google Scholar] [CrossRef]
- Sanmarco, L.M.; Rone, J.M.; Polonio, C.M.; Fernandez Lahore, G.; Giovannoni, F.; Ferrara, K.; Gutierrez-Vazquez, C.; Li, N.; Sokolovska, A.; Plasencia, A.; et al. Lactate limits CNS autoimmunity by stabilizing HIF-1alpha in dendritic cells. Nature 2023, 620, 881–889. [Google Scholar] [CrossRef]
- Junger, S.; Gantenbein-Ritter, B.; Lezuo, P.; Alini, M.; Ferguson, S.J.; Ito, K. Effect of limited nutrition on in situ intervertebral disc cells under simulated-physiological loading. Spine (Phila Pa 1976) 2009, 34, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Malandrino, A.; Noailly, J.; Lacroix, D. The effect of sustained compression on oxygen metabolic transport in the intervertebral disc decreases with degenerative changes. PLoS Comput Biol 2011, 7, e1002112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, H.; Tan, Q.; Huang, J.; Zhou, S.; Luo, F.; Zhang, D.; Yang, J.; Li, C.; Chen, B.; et al. Inhibition of aberrant Hif1alpha activation delays intervertebral disc degeneration in adult mice. Bone Res 2022, 10, 2. [Google Scholar] [CrossRef]
- Benneker, L.M.; Heini, P.F.; Alini, M.; Anderson, S.E.; Ito, K. 2004 Young Investigator Award Winner: vertebral endplate marrow contact channel occlusions and intervertebral disc degeneration. Spine (Phila Pa 1976) 2005, 30, 167–173. [Google Scholar] [CrossRef]
- Hristova, G.I.; Jarzem, P.; Ouellet, J.A.; Roughley, P.J.; Epure, L.M.; Antoniou, J.; Mwale, F. Calcification in human intervertebral disc degeneration and scoliosis. J Orthop Res 2011, 29, 1888–1895. [Google Scholar] [CrossRef]
- Wang, Y.; Videman, T.; Battie, M.C. Lumbar vertebral endplate lesions: prevalence, classification, and association with age. Spine (Phila Pa 1976) 2012, 37, 1432–1439. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cisewski, S.E.; Wegner, N.; Zhao, S.; Pellegrini, V.D., Jr.; Slate, E.H.; Yao, H. Region and strain-dependent diffusivities of glucose and lactate in healthy human cartilage endplate. J Biomech 2016, 49, 2756–2762. [Google Scholar] [CrossRef]
- Huang, C.Y.; Gu, W.Y. Effects of mechanical compression on metabolism and distribution of oxygen and lactate in intervertebral disc. J Biomech 2008, 41, 1184–1196. [Google Scholar] [CrossRef]
- Liu, Z.; Zheng, J.; Ding, T.; Chen, H.; Wan, R.; Zhang, X.; Zhang, W. HIF-1alpha protects nucleus pulposus cells from oxidative stress-induced mitochondrial impairment through PDK-1. Free Radic Biol Med 2024, 224, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Shen, J.; Zhang, X.; Hu, Z. LDHA-Mediated Glycolytic Metabolism in Nucleus Pulposus Cells Is a Potential Therapeutic Target for Intervertebral Disc Degeneration. Biomed Res Int 2021, 2021, 9914417. [Google Scholar] [CrossRef]
- Zhao, K.; An, R.; Xiang, Q.; Li, G.; Wang, K.; Song, Y.; Liao, Z.; Li, S.; Hua, W.; Feng, X.; et al. Acid-sensing ion channels regulate nucleus pulposus cell inflammation and pyroptosis via the NLRP3 inflammasome in intervertebral disc degeneration. Cell Prolif 2021, 54, e12941. [Google Scholar] [CrossRef]
- Trone, M.A.R.; Stover, J.D.; Almarza, A.; Bowles, R.D. pH: A major player in degenerative intervertebral disks. JOR Spine 2024, 7, e70025. [Google Scholar] [CrossRef]
- Gilbert, H.T.J.; Hodson, N.; Baird, P.; Richardson, S.M.; Hoyland, J.A. Acidic pH promotes intervertebral disc degeneration: Acid-sensing ion channel -3 as a potential therapeutic target. Sci Rep 2016, 6, 37360. [Google Scholar] [CrossRef]
- Razaq, S.; Wilkins, R.J.; Urban, J.P. The effect of extracellular pH on matrix turnover by cells of the bovine nucleus pulposus. Eur Spine J 2003, 12, 341–349. [Google Scholar] [CrossRef]
- Vadala, G.; Ambrosio, L.; Russo, F.; Papalia, R.; Denaro, V. Interaction between Mesenchymal Stem Cells and Intervertebral Disc Microenvironment: From Cell Therapy to Tissue Engineering. Stem Cells Int 2019, 2019, 2376172. [Google Scholar] [CrossRef] [PubMed]
- Vo, N.V.; Hartman, R.A.; Patil, P.R.; Risbud, M.V.; Kletsas, D.; Iatridis, J.C.; Hoyland, J.A.; Le Maitre, C.L.; Sowa, G.A.; Kang, J.D. Molecular mechanisms of biological aging in intervertebral discs. J Orthop Res 2016, 34, 1289–1306. [Google Scholar] [CrossRef]
- Zhan, J.; Cui, Y.; Zhang, P.; Du, Y.; Hecker, P.; Zhou, S.; Liang, Y.; Zhang, W.; Jin, Z.; Wang, Y.; et al. Cartilage Endplate-Targeted Engineered Exosome Releasing and Acid Neutralizing Hydrogel Reverses Intervertebral Disc Degeneration. Adv Healthc Mater 2025, 14, e2403315. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.H.; Twomey, J.D. Cellular mechanobiology of the intervertebral disc: new directions and approaches. J Biomech 2010, 43, 137–145. [Google Scholar] [CrossRef]
- Erwin, W.M.; Hood, K.E. The cellular and molecular biology of the intervertebral disc: A clinician's primer. J Can Chiropr Assoc 2014, 58, 246–257. [Google Scholar] [PubMed]
- Smith, L.J.; Fazzalari, N.L. The elastic fibre network of the human lumbar anulus fibrosus: architecture, mechanical function and potential role in the progression of intervertebral disc degeneration. Eur Spine J 2009, 18, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Mirzaeipoueinak, M.; Mordechai, H.S.; Bangar, S.S.; Sharabi, M.; Tipper, J.L.; Tavakoli, J. Structure-function characterization of the transition zone in the intervertebral disc. Acta Biomater 2023, 160, 164–175. [Google Scholar] [CrossRef]
- Urban, J.P.; Maroudas, A. Swelling of the intervertebral disc in vitro. Connect Tissue Res 1981, 9, 1–10. [Google Scholar] [CrossRef]
- Urban, J.P.; McMullin, J.F. Swelling pressure of the lumbar intervertebral discs: influence of age, spinal level, composition, and degeneration. Spine (Phila Pa 1976) 1988, 13, 179–187. [Google Scholar] [CrossRef]
- Yao, H.; Justiz, M.A.; Flagler, D.; Gu, W.Y. Effects of swelling pressure and hydraulic permeability on dynamic compressive behavior of lumbar annulus fibrosus. Ann Biomed Eng 2002, 30, 1234–1241. [Google Scholar] [CrossRef]
- Urban, J.P.; Maroudas, A.; Bayliss, M.T.; Dillon, J. Swelling pressures of proteoglycans at the concentrations found in cartilaginous tissues. Biorheology 1979, 16, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Best, B.A.; Guilak, F.; Setton, L.A.; Zhu, W.; Saed-Nejad, F.; Ratcliffe, A.; Weidenbaum, M.; Mow, V.C. Compressive mechanical properties of the human anulus fibrosus and their relationship to biochemical composition. Spine (Phila Pa 1976) 1994, 19, 212–221. [Google Scholar] [CrossRef]
- Gu, W.Y.; Mao, X.G.; Foster, R.J.; Weidenbaum, M.; Mow, V.C.; Rawlins, B.A. The anisotropic hydraulic permeability of human lumbar anulus fibrosus. Influence of age, degeneration, direction, and water content. Spine (Phila Pa 1976) 1999, 24, 2449–2455. [Google Scholar] [CrossRef]
- Perie, D.; Korda, D.; Iatridis, J.C. Confined compression experiments on bovine nucleus pulposus and annulus fibrosus: sensitivity of the experiment in the determination of compressive modulus and hydraulic permeability. J Biomech 2005, 38, 2164–2171. [Google Scholar] [CrossRef] [PubMed]
- Selard, E.; Shirazi-Adl, A.; Urban, J.P. Finite element study of nutrient diffusion in the human intervertebral disc. Spine (Phila Pa 1976) 2003, 28, 1945–1953. [Google Scholar] [CrossRef]
- Soukane, D.M.; Shirazi-Adl, A.; Urban, J.P. Analysis of nonlinear coupled diffusion of oxygen and lactic acid in intervertebral discs. J Biomech Eng 2005, 127, 1121–1126. [Google Scholar] [CrossRef]
- Baltazar, F.; Afonso, J.; Costa, M.; Granja, S. Lactate Beyond a Waste Metabolite: Metabolic Affairs and Signaling in Malignancy. Front Oncol 2020, 10, 231. [Google Scholar] [CrossRef]
- Brooks, G.A. The lactate shuttle during exercise and recovery. Med Sci Sports Exerc 1986, 18, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. Lactate production under fully aerobic conditions: the lactate shuttle during rest and exercise. Fed Proc 1986, 45, 2924–2929. [Google Scholar]
- Mason, S. Lactate Shuttles in Neuroenergetics-Homeostasis, Allostasis and Beyond. Front Neurosci 2017, 11, 43. [Google Scholar] [CrossRef]
- Brooks, G.A. Cell-cell and intracellular lactate shuttles. J Physiol 2009, 587, 5591–5600. [Google Scholar] [CrossRef] [PubMed]
- Karchevskaya, A.E.; Poluektov, Y.M.; Korolishin, V.A. Understanding Intervertebral Disc Degeneration: Background Factors and the Role of Initial Injury. Biomedicines 2023, 11. [Google Scholar] [CrossRef]
- Wu, R.; Zhao, X.J.; Du, Y.; Dong, Y.; Song, X.; Zhu, Y. Lipid metabolic disorders and their impact on cartilage endplate and nucleus pulposus function in intervertebral disk degeneration. Front Nutr 2025, 12, 1533264. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, Y.; Huang, B.; Liu, L.T.; Liu, M.H.; Wang, J.; Li, C.Q.; Zhang, Z.F.; Chu, T.W.; Xiong, C.J. Utilization of stem cells in alginate for nucleus pulposus tissue engineering. Tissue Eng Part A 2014, 20, 908–920. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, L.; Deng, X.; Shi, D.; Wu, F.; Liang, H.; Huang, D.; Shao, Z. Mesenchymal Stem Cells Protect Nucleus Pulposus Cells from Compression-Induced Apoptosis by Inhibiting the Mitochondrial Pathway. Stem Cells Int 2017, 2017, 9843120. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Deng, G.; Ma, J.; Huang, X.; Yu, J.; Xi, Y.; Ye, X. Transplantation of Hypoxic-Preconditioned Bone Mesenchymal Stem Cells Retards Intervertebral Disc Degeneration via Enhancing Implanted Cell Survival and Migration in Rats. Stem Cells Int 2018, 2018, 7564159. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Chen, H.; Li, Y.; Li, H.; Chen, X.; Fu, H.; Mao, D.; Chen, W.; Lan, L.; Wang, C.; Hu, K.; et al. NBS1 lactylation is required for efficient DNA repair and chemotherapy resistance. Nature 2024, 631, 663–669. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, Y.; Yang, B.; Sun, S.; Zhang, P.; Luo, Z.; Feng, T.; Cui, Z.; Zhu, T.; Li, Y.; et al. Lactylation of METTL16 promotes cuproptosis via m(6)A-modification on FDX1 mRNA in gastric cancer. Nature communications 2023, 14, 6523. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, D.; Singh, P.; Chakraborty, B.; Hennessey, M.; Tannir, A.J.; Byregowda, S.; Natarajan, S.M.; Trujillo-Ocampo, A.; Im, J.S.; Goswami, S. Histone lactylation drives CD8(+) T cell metabolism and function. Nature immunology 2024, 25, 2140–2151. [Google Scholar] [CrossRef]
- Feng, P.; Che, Y.; Gao, C.; Zhu, L.; Gao, J.; Vo, N.V. Immune exposure: how macrophages interact with the nucleus pulposus. Front Immunol 2023, 14, 1155746. [Google Scholar] [CrossRef]
- Shi, Y.; Li, F.; Lin, W.; Han, L.; Wang, J.; Yan, C.; Sun, J.; Ji, C.; Shi, J.; Sun, K. Integrating Bulk RNA and Single-Cell RNA Sequencing Identifies and Validates Lactylation-Related Signatures for Intervertebral Disc Degeneration. J Cell Mol Med 2024, 28, e70262. [Google Scholar] [CrossRef]
- Sheng, L.; Xu, H.; Wang, Y.; Ni, J.; Xiang, T.; Xu, H.; Zhou, X.; Wei, K.; Dai, J. Systematic analysis of lysine lactylation in nucleus pulposus cells. iScience 2024, 27, 111157. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, Z.; Han, W.; Wu, J.; Li, S.; Qin, T.; Zhang, C.; Shi, M.; Han, S.; Gao, B.; et al. Glutamine suppresses senescence and promotes autophagy through glycolysis inhibition-mediated AMPKalpha lactylation in intervertebral disc degeneration. Communications biology 2024, 7, 325. [Google Scholar] [CrossRef]
- Zhou, M.M.; Cole, P.A. Targeting lysine acetylation readers and writers. Nat Rev Drug Discov 2025, 24, 112–133. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.F.; Zhang, H.L.; Li, L.Z.; Lv, Y.; Li, H.; Li, Z.; Yu, F.L.; Jiang, T.; Zhang, T.Y.; Xin, F.; et al. Sirt1 blocks nucleus pulposus and macrophages crosstalk by inhibiting RelA/Lipocalin 2 axis. J Orthop Translat 2025, 50, 30–43. [Google Scholar] [CrossRef]
- Farsetti, A.; Illi, B.; Gaetano, C. How epigenetics impacts on human diseases. Eur J Intern Med 2023, 114, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Che, H.; Li, J.; Li, Y.; Ma, C.; Liu, H.; Qin, J.; Dong, J.; Zhang, Z.; Xian, C.J.; Miao, D.; et al. p16 deficiency attenuates intervertebral disc degeneration by adjusting oxidative stress and nucleus pulposus cell cycle. eLife 2020, 9. [Google Scholar] [CrossRef]
- Ji, M.L.; Jiang, H.; Zhang, X.J.; Shi, P.L.; Li, C.; Wu, H.; Wu, X.T.; Wang, Y.T.; Wang, C.; Lu, J. Preclinical development of a microRNA-based therapy for intervertebral disc degeneration. Nat Commun 2018, 9, 5051. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Lin, X.; Dong, W.; Huang, W.; Jiang, W.; Lin, L.; Qiu, Q.; Zhang, X.; Shen, J.; Song, Z.; et al. SIRT1 alleviates senescence of degenerative human intervertebral disc cartilage endo-plate cells via the p53/p21 pathway. Sci Rep 2016, 6, 22628. [Google Scholar] [CrossRef]
- Ramteke, P.; Watson, B.; Toci, M.; Tran, V.A.; Johnston, S.; Tsingas, M.; Barve, R.A.; Mitra, R.; Loeser, R.F.; Collins, J.A.; et al. Sirt6 deficiency promotes senescence and age-associated intervertebral disc degeneration in mice. Bone Res 2025, 13, 50. [Google Scholar] [CrossRef]
- Zheng, Z.; Bian, Y.; Zhang, Y.; Ren, G.; Li, G. Metformin activates AMPK/SIRT1/NF-kappaB pathway and induces mitochondrial dysfunction to drive caspase3/GSDME-mediated cancer cell pyroptosis. Cell cycle 2020, 19, 1089–1104. [Google Scholar] [CrossRef]
- Yang, W.; Wang, P.; Cao, P.; Wang, S.; Yang, Y.; Su, H.; Nashun, B. Hypoxic in vitro culture reduces histone lactylation and impairs pre-implantation embryonic development in mice. Epigenetics Chromatin 2021, 14, 57. [Google Scholar] [CrossRef]
- Dai, X.; Lv, X.; Thompson, E.W.; Ostrikov, K.K. Histone lactylation: epigenetic mark of glycolytic switch. Trends Genet 2022, 38, 124–127. [Google Scholar] [CrossRef]
- Moreno-Yruela, C.; Zhang, D.; Wei, W.; Baek, M.; Liu, W.; Gao, J.; Dankova, D.; Nielsen, A.L.; Bolding, J.E.; Yang, L.; et al. Class I histone deacetylases (HDAC1-3) are histone lysine delactylases. Sci Adv 2022, 8, eabi6696. [Google Scholar] [CrossRef]
- Yang, K.; Fan, M.; Wang, X.; Xu, J.; Wang, Y.; Tu, F.; Gill, P.S.; Ha, T.; Liu, L.; Williams, D.L.; et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ 2022, 29, 133–146. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Li, W.; Zhou, X. Lactylation, an emerging hallmark of metabolic reprogramming: Current progress and open challenges. Front Cell Dev Biol 2022, 10, 972020. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hu, H.; Liu, M.; Zhou, T.; Cheng, X.; Huang, W.; Cao, L. The role and mechanism of histone lactylation in health and diseases. Frontiers in genetics 2022, 13, 949252. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Chen, Z.; Li, J.; Ding, L. Effects of lactylation on the hallmarks of cancer (Review). Oncol Lett 2025, 30, 492. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef]
- Latham, T.; Mackay, L.; Sproul, D.; Karim, M.; Culley, J.; Harrison, D.J.; Hayward, L.; Langridge-Smith, P.; Gilbert, N.; Ramsahoye, B.H. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic acids research 2012, 40, 4794–4803. [Google Scholar] [CrossRef]
- Gong, H.; Zhong, H.; Cheng, L.; Li, L.P.; Zhang, D.K. Post-translational protein lactylation modification in health and diseases: a double-edged sword. J Transl Med 2024, 22, 41. [Google Scholar] [CrossRef]
- Xin, Q.; Wang, H.; Li, Q.; Liu, S.; Qu, K.; Liu, C.; Zhang, J. Lactylation: a Passing Fad or the Future of Posttranslational Modification. Inflammation 2022, 45, 1419–1429. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X. Virus-Induced Histone Lactylation Promotes Virus Infection in Crustacean. Advanced science 2024, 11, e2401017. [Google Scholar] [CrossRef]
- Jennings, E.Q.; Ray, J.D.; Zerio, C.J.; Trujillo, M.N.; McDonald, D.M.; Chapman, E.; Spiegel, D.A.; Galligan, J.J. Sirtuin 2 Regulates Protein LactoylLys Modifications. Chembiochem 2021, 22, 2102–2106. [Google Scholar] [CrossRef] [PubMed]
- Zu, H.; Li, C.; Dai, C.; Pan, Y.; Ding, C.; Sun, H.; Zhang, X.; Yao, X.; Zang, J.; Mo, X. SIRT2 functions as a histone delactylase and inhibits the proliferation and migration of neuroblastoma cells. Cell Discov 2022, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Ran, R.; Wu, Y.; Zhang, H.H. Acid-sensing Ion Channels: Implications for Intervertebral Disc Degeneration. Curr Pharm Biotechnol 2023, 24, 1343–1350. [Google Scholar] [CrossRef]
- Grunder, S.; Vanek, J.; Pissas, K.P. Acid-sensing ion channels and downstream signalling in cancer cells: is there a mechanistic link? Pflugers Arch 2024, 476, 659–672. [Google Scholar] [CrossRef]
- Ding, J.; Zhang, R.; Li, H.; Ji, Q.; Cheng, X.; Thorne, R.F.; Hondermarck, H.; Liu, X.; Shen, C. ASIC1 and ASIC3 mediate cellular senescence of human nucleus pulposus mesenchymal stem cells during intervertebral disc degeneration. Aging (Albany NY) 2021, 13, 10703–10723. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, F.R.; Xu, R.S.; Hu, W.; Jiang, D.L.; Ji, C.; Chen, F.H.; Yuan, F.L. Acid-sensing ion channel 1a-mediated calcium influx regulates apoptosis of endplate chondrocytes in intervertebral discs. Expert Opin Ther Targets 2014, 18, 1–14. [Google Scholar] [CrossRef]
- Llibre, A.; Kucuk, S.; Gope, A.; Certo, M.; Mauro, C. Lactate: A key regulator of the immune response. Immunity 2025, 58, 535–554. [Google Scholar] [CrossRef]
- Liu, C.; Wu, J.; Zhu, J.; Kuei, C.; Yu, J.; Shelton, J.; Sutton, S.W.; Li, X.; Yun, S.J.; Mirzadegan, T.; et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem 2009, 284, 2811–2822. [Google Scholar] [CrossRef]
- Langin, D. Adipose tissue lipolysis revisited (again!): lactate involvement in insulin antilipolytic action. Cell Metab 2010, 11, 242–243. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain lactate metabolism: the discoveries and the controversies. J Cereb Blood Flow Metab 2012, 32, 1107–1138. [Google Scholar] [CrossRef]
- Harun-Or-Rashid, M.; Inman, D.M. Reduced AMPK activation and increased HCAR activation drive anti-inflammatory response and neuroprotection in glaucoma. J Neuroinflammation 2018, 15, 313. [Google Scholar] [CrossRef]
- Zou, Y.; Zeng, S.; Huang, M.; Qiu, Q.; Xiao, Y.; Shi, M.; Zhan, Z.; Liang, L.; Yang, X.; Xu, H. Inhibition of 6-phosphofructo-2-kinase suppresses fibroblast-like synoviocytes-mediated synovial inflammation and joint destruction in rheumatoid arthritis. Br J Pharmacol 2017, 174, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Abebayehu, D.; Spence, A.J.; Qayum, A.A.; Taruselli, M.T.; McLeod, J.J.; Caslin, H.L.; Chumanevich, A.P.; Kolawole, E.M.; Paranjape, A.; Baker, B.; et al. Lactic Acid Suppresses IL-33-Mediated Mast Cell Inflammatory Responses via Hypoxia-Inducible Factor-1alpha-Dependent miR-155 Suppression. J Immunol 2016, 197, 2909–2917. [Google Scholar] [CrossRef] [PubMed]
- Caslin, H.L.; Abebayehu, D.; Abdul Qayum, A.; Haque, T.T.; Taruselli, M.T.; Paez, P.A.; Pondicherry, N.; Barnstein, B.O.; Hoeferlin, L.A.; Chalfant, C.E.; et al. Lactic Acid Inhibits Lipopolysaccharide-Induced Mast Cell Function by Limiting Glycolysis and ATP Availability. J Immunol 2019, 203, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, X.; Chen, X.; Jiang, Y.; Zeng, Y.; Ren, R.; Nie, M.; Zhang, Z.; Bao, Y.; Kang, H. Genkwanin alleviates intervertebral disc degeneration via regulating ITGA2/PI3K/AKT pathway and inhibiting apoptosis and senescence. Int Immunopharmacol 2024, 133, 112101. [Google Scholar] [CrossRef]
- Liang, Z.H.; Song, J.; Shangguan, W.J.; Zhang, Q.Q.; Shao, J.; Zhang, Y.H. Melatonin mitigates matrix stiffness-induced intervertebral disk degeneration by inhibiting reactive oxygen species and melatonin receptors mediated PI3K/AKT/NF-kappaB pathway. Am J Physiol Cell Physiol 2024, 327, C1236–C1248. [Google Scholar] [CrossRef]
- Qiu, X.; Ma, C.; Luo, Z.; Zhang, Y.; Kang, J.; Zhu, D.; Wang, Z.; Li, L.; Wei, Z.; Wang, Z.; et al. Bradykinin protects nucleus pulposus cells from tert-butyl hydroperoxide-induced damage and delays intervertebral disc degeneration. Int Immunopharmacol 2024, 134, 112161. [Google Scholar] [CrossRef]
- Risbud, M.V.; Fertala, J.; Vresilovic, E.J.; Albert, T.J.; Shapiro, I.M. Nucleus pulposus cells upregulate PI3K/Akt and MEK/ERK signaling pathways under hypoxic conditions and resist apoptosis induced by serum withdrawal. Spine (Phila Pa 1976) 2005, 30, 882–889. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Qi, Y.; Lou, J.; Chen, Y.; Liu, C.; Li, H.; Chang, X.; Hu, Z.; Li, Y.; et al. Lactic acid promotes nucleus pulposus cell senescence and corresponding intervertebral disc degeneration via interacting with Akt. Cell Mol Life Sci 2024, 81, 24. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, X.; Alexander, P.G.; Feng, P.; Zhang, J. An Analysis of AMPK and Ferroptosis in Cancer: A Potential Regulatory Axis. Front Biosci (Landmark Ed) 2025, 30, 36618. [Google Scholar] [CrossRef]
- Chen, J.; Yang, X.; Feng, Y.; Li, Q.; Ma, J.; Wang, L.; Quan, Z. Targeting Ferroptosis Holds Potential for Intervertebral Disc Degeneration Therapy. Cells 2022, 11. [Google Scholar] [CrossRef]
- Xiang, Q.; Zhao, Y.; Li, W. Identification and validation of ferroptosis-related gene signature in intervertebral disc degeneration. Front Endocrinol (Lausanne) 2023, 14, 1089796. [Google Scholar] [CrossRef]
- Li, Q.; Peng, J.; Ding, F. 1,25(OH)(2)D(3) inhibits ferroptosis in nucleus pulposus cells via VDR signaling to mitigate lumbar intervertebral disc degeneration. Sci Rep 2025, 15, 7968. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Song, Y.; Luo, R.; Li, S.; Li, G.; Wang, K.; Liao, Z.; Wang, B.; Ke, W.; Xiang, Q.; et al. Ferroportin-Dependent Iron Homeostasis Protects against Oxidative Stress-Induced Nucleus Pulposus Cell Ferroptosis and Ameliorates Intervertebral Disc Degeneration In Vivo. Oxid Med Cell Longev 2021, 2021, 6670497. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Shi, Y.; Yan, C.; Wang, S.; Han, L.; Li, F.; Xu, X.; Wang, Y.; Sun, J.; Kang, Z.; et al. Glycolysis-Derived Lactate Induces ACSL4 Expression and Lactylation to Activate Ferroptosis during Intervertebral Disc Degeneration. Advanced science 2025, 12, e2416149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, Z.; Xie, Z.; Chen, Y.; Zheng, Z.; Wei, X.; Huang, B.; Shan, Z.; Liu, J.; Fan, S.; et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic Biol Med 2020, 160, 552–565. [Google Scholar] [CrossRef]
Figure 1.
Pathways of lactate metabolism within the IVD.
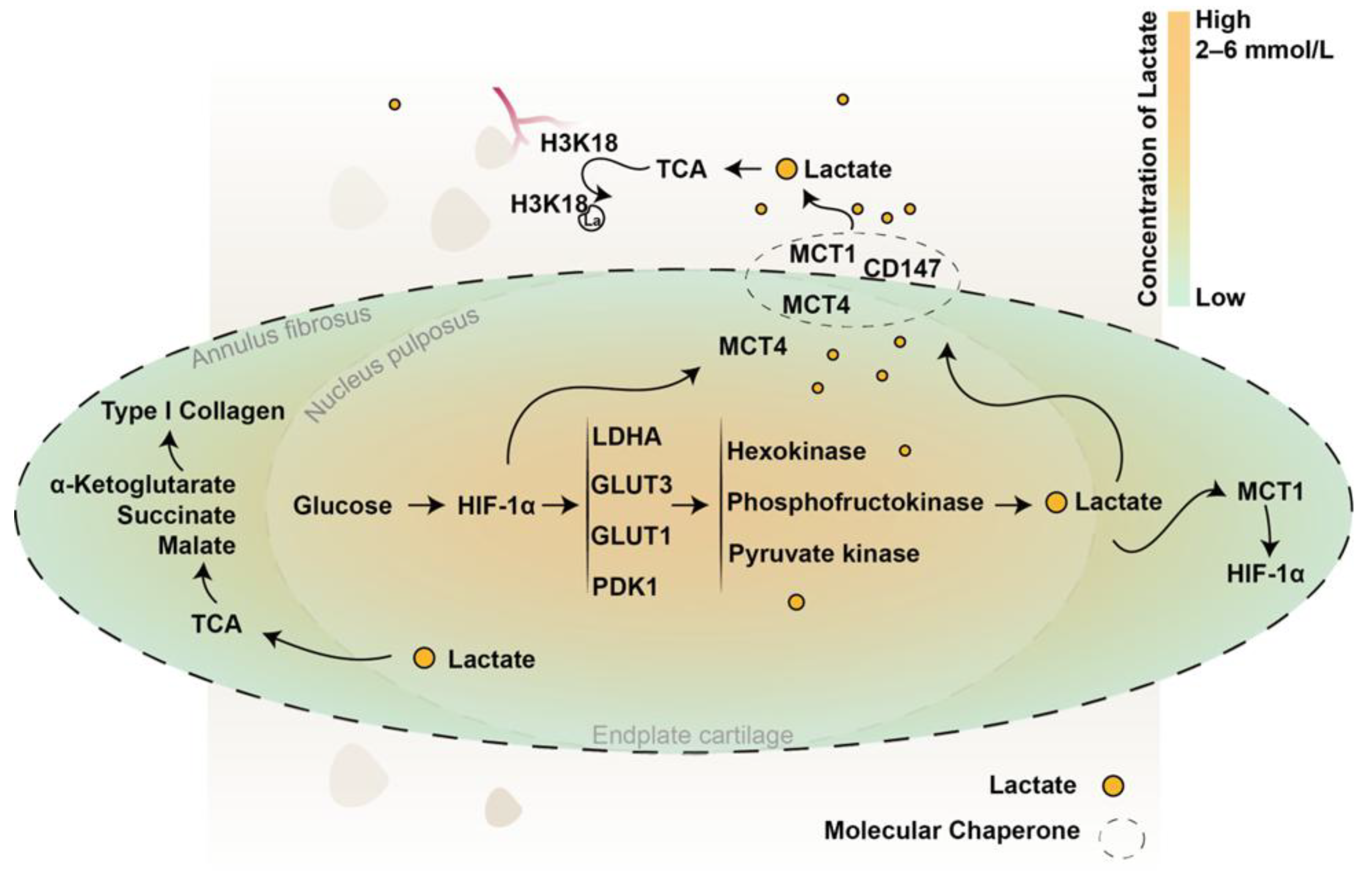
Figure 2.
Lactate-mediated cellular signaling pathways in the IVD.
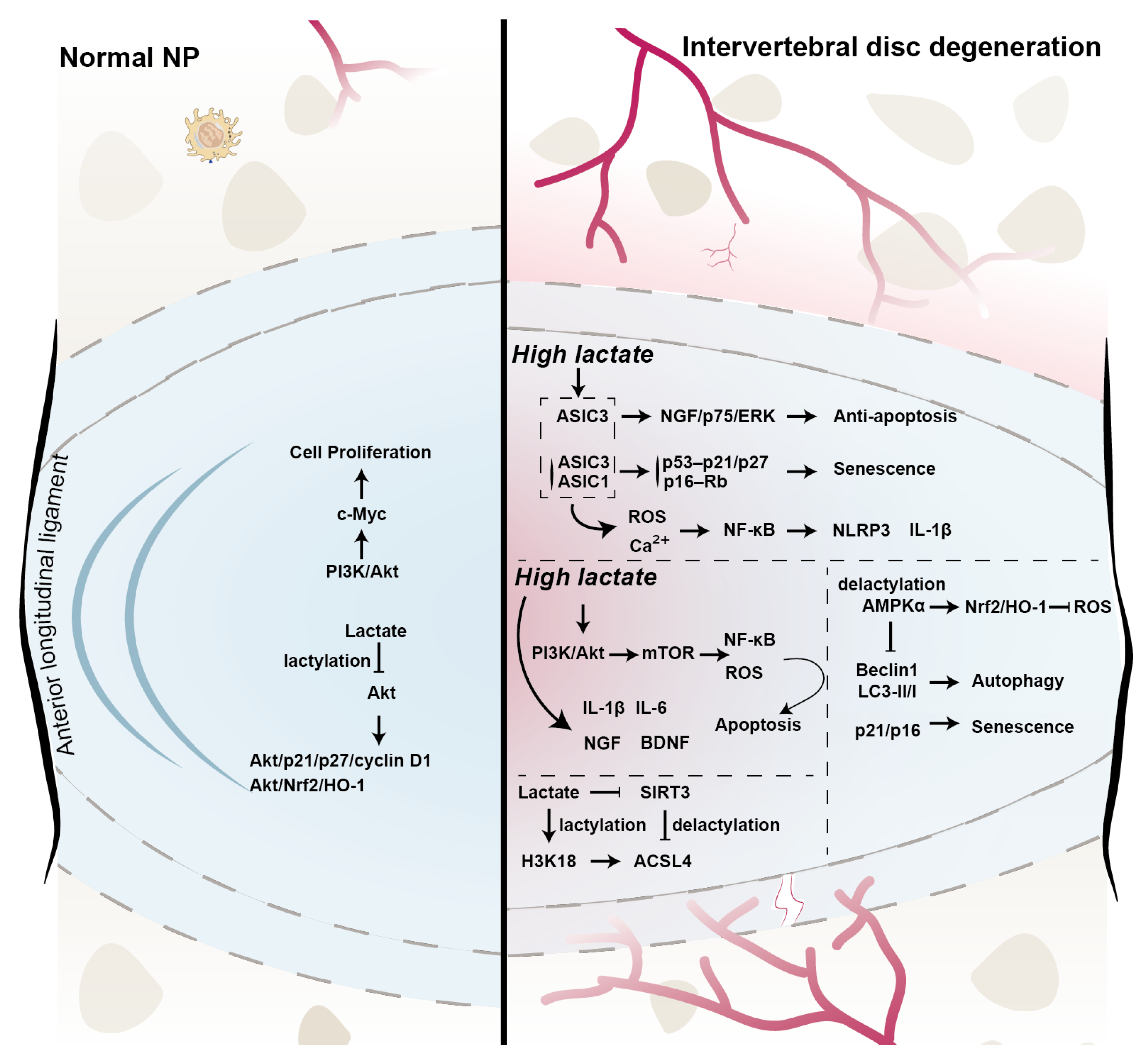
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



