Submitted:
10 November 2025
Posted:
11 November 2025
You are already at the latest version
Abstract
Inborn errors of amino acid metabolism (IEAAMs) are a heterogeneous group of genetic disorders caused by defects in enzymes, cofactors, or transporters of amino acid catabolism, biosynthesis, or transport. These defects result in toxic metabolite accumulation and/or deficiency of essential metabolites. This review aims to provide an updated overview of diagnosis, clinical implications, management, and evolving therapeutic approaches across major IEAAMs. A narrative review of recent literature was undertaken, focusing on established and novel therapeutic strategies for key IEAAMs, including phenylketonuria, alkaptonuria, tyrosinemia, homocystinuria, and maple syrup urine disease. Key management strategies include amino acid-restricted diets/restriction of natural protein with restriction of dietary precursors, dietary supplementations, including disease-specific amino acid supplements, medications to reduce formation of offending metabolites, pharmacotherapies, enzyme/cofactor replacement or pharmacological chaperones, enhancing residual enzyme activity and promoting alternative path-ways/accessory pathways. Emergency therapy is essential in severe types and focuses on promoting anabolism, limiting catabolism, reducing formation, and enhancing clearance of toxic metabolites. Other treatment options include organ transplantation, and new emerging modalities, such as mRNA therapies and gene therapies/in vivo gene editing offer potential for definitive interventions. Despite advancements in therapy and close monitoring, many IEAAMs remain associated with significant comorbidities. Future research is essential to optimise current treatment standards, particularly neuroprotective and metabolic regulatory features. While an in-depth discussion of innovative person-alised therapies is beyond the scope of this article, we believe that collective experiences will thrust future research in this field and expand access to innovative personalised therapies.
Keywords:
amino acids
; emergency therapy
; inborn errors of metabolism
; personalised medicine
1. Introduction
Amino acids primarily serve as the fundamental building blocks of proteins. Some also play crucial roles in vital bodily functions, or act as precursors for essential nitrogen-containing compounds including neurotransmitters, hormones, nucleotides, and pigments [1]. Humans use 21 distinct amino acids, the majority of which may be produced internally, but 9 (isoleucine, leucine, lysine, methionine, phenylalanine (Phe), threonine, tryptophan, valine and histidine) are "essential" in the sense that they must be consumed through food [2].
The term "inborn errors of amino acid metabolism (IEAAMs)" refers to a wide range of metabolic disorders, including those brought on by enzyme deficiencies in the catabolic pathway of one or more amino acids or, less frequently, by cofactor or transporter deficiency. This may result in the toxic accumulation of certain amino acids or their respective catabolic intermediates, as well as the deficiency of key metabolic products, leading to a wide range of clinical manifestations. This review fills an important gap by providing a comprehensive and clinically focused update on recent advances in the diagnosis and management of the most prevalent IEAAMs.
IEAAMs include phenylketonuria (PKU), alkaptonuria (AKU), homocystinuria (HCU), Maple Syrup Urine Disease (MSUD), tyrosinemia, pyridoxine-dependent epilepsy (lysine-related), glutaric aciduria type 1, and nonketotic hyperglycinemia. IEAAMs also include inborn metabolic errors of other amino acids, including, but not limited to, serine, glutamine, asparagine, and proline.
The aim of treatment is to normalise the metabolic imbalance as much as possible, via dietary modification, pharmacological treatment, or in some instances cofactor supplementation. In more detail, key management strategies include amino acid-restricted diets or restriction of natural protein with the restriction of offending amino acid(s), together with dietary supplementations of micronutrients and disease-specific amino acid supplements. Medications are used to reduce formation of toxic by-products and enhance removal of toxic metabolites in the body. For some conditions pharmacotherapies are available that also include enzyme/cofactor replacement or pharmacological chaperones, enhancing residual enzyme activity aim or promoting alternative and accessory pathways. Continued patient follow-up may include clinical evaluation and monitoring of potential long-term complications, surveillance of disease-specific biochemical indicators, medication management, dietetic modifications and nutritional surveillance.
Emergency therapy may also be required for the severe types and aims at promoting anabolism, reducing catabolism, reducing formation of toxic metabolites and removing toxic compounds. Among the prevention of toxic effects are also general neuroprotective measures, such as the prevention of hypoglycemia, early treatment of hyperammonemia, or the reduction of hyperhomocysteinemia in certain conditions along with clinical management of pyrexia/intercurrent infections and careful nutritional surveillances to avoid micronutrient deficiencies. While existing therapeutic approaches must be optimised for best long-term results, clinical challenges remain and disease burden is significant.
Other treatment options include early organ transplantation/liver transplantation, and also prosing new treatment modalities, i.e., mRNA therapies and gene therapies/in vivo gene editing which may offer potential for more definitive or curative interventions.
Here, we present a general overview of the management of some of the main IEAAMs outlined in Tables 1–6. Medical and dietary management of the most commonly encountered disorders, such as phenylketonuria, alkaptonuria, tyrosinemia, homocystinuria, maple syrup urine disease, methylmalonic acidemia, nonketotic hyperglycinemia, pyridoxine-dependent epilepsy (lysine-related), cystinuria, lysinuric protein intolerance, Hartnup disease, glutaric aciduria type 1, serine deficiency, hyperprolinemia, glutamine synthetase deficiency and asparagine synthetase deficiency are discussed in further detail.
2. Key Disorders of Amino Acid Metabolism: Clinical Features and Therapeutic Approaches
2.1. Phenylketonuria
Phenylketonuria (PKU) (OMIM #261600) is an autosomal recessive disorder characterised by an absence or profound deficiency of the enzyme Phe hydroxylase (PAH) (EC 1.14.16.1) which normally converts Phe to tyrosine (Tyr) (Figure 1a). Decreased PAH activity leads to the accumulation of Phe in the blood and brain [3]. It is usually detected on newborn bloodspot screening and subsequently confirmed with the detection of two pathogenic variants in the PAH gene. In PKU, accumulated Phe is converted to phenylpyruvic acid and phenyllactic acid, which are excreted in the urine. PKU has an incidence of ~1:16,000 live births in the US, with higher rates observed in countries such as Ireland and Turkey [4]. If untreated, PKU can result in intellectual disability, motor deficits, seizures, and eczema [5]. Blood samples for the analysis of Phe and Tyr should ideally be collected in the morning after overnight fasting. Dietary management remains the cornerstone of treatment for PKU for the majority of paediatric patients. A practical dietary approach in PKU management involves restricting total natural protein intake (‘protein exchanges’) rather than calculating individual amino acid contributions, permitting the free consumption of a number of low-Phe fruits and vegetables based on defined thresholds. A compositional analysis of 165 fruits, vegetables, and starchy roots, published recently by our group, has expanded the evidence base for this approach [6]. The study confirmed that many fruits and vegetables contain Phe concentrations below the 75 mg/100 g threshold recommended by European PKU guidelines, and their unrestricted consumption does not adversely impact metabolic control. This is likely due to the lower digestibility of plant-based proteins and their high fibre content, which may reduce Phe bioavailability[6]. Evidence-based dietary liberalisation of certain foods in PKU has been proposed to enhance quality of life, dietary adherence, and nutritional adequacy.
Management of PKU consists of dietary Phe restriction through natural protein restriction together with disease-specific amino acid supplements, including also Large Neutral Amino Acids (LNAAs), and, occasionally, additional Tyr supplementation. Biochemical monitoring is provided through blood testing of PKU patients. An upper blood Phe target level of 360μmol/L is recommended for children with PKU aged <12 years and an upper target level of 600μmol/L is recommended for older patients aged >12 years (outside pregnancy) to achieve optimal outcomes[5]. Recent treatment developments for PKU include chaperone therapy and enzyme substitution therapy. Sapropterin dihydrochloride is a synthetic form of tetrahydrobiopterin (BH4), an essential co-factor for PAH activity, which has been shown to reduce Phe concentrations in PKU patients [7]. In approximately 20–30% of PKU patients, Phe levels may be controlled by tetrahydrobiopterin (BH4) therapy[8]. A novel treatment option for PKU is pegvaliase, a pegylated derivative of Phe ammonia-lyase (PAL), which converts Phe to ammonia and trans-cinnamic acid [9]. Ammonia and trans-cinnamic acid are subsequently metabolised in the liver and excreted in the urine, resulting in reduced Phe concentrations [9] (Table 1).
2.2. Alkaptonuria
Alkaptonuria (AKU) (OMIM #203500) is caused by mutations in HGD (OMIM 607474) leading to homogentisic acid oxidase (EC 1.13.11.5) deficiency and consequent homogentisic acid (HGA) accumulation (Figure 1a). AKU has an autosomal recessive inheritance pattern with global prevalence of 1:250,000 – 1:1,000,000 [22]. Accumulated HGA is oxidised to benzoquinone acetate which subsequently forms melanin-like polymers. These polymers accumulate in collagen in a process called ochronosis. Many affected individuals are asymptomatic until adulthood. A typical feature of AKU is darkening of urine upon standing for a period of time, reflecting homogentisic aciduria. Ochronotic pigment accumulation in connective tissues of various organs may lead to grey pigmentation of the sclera and ear helix, ochronotic osteoarthropathy, nephrolithiasis, cholelithiasis, prostatic calculi, and valvular dysfunction [23]. Treatment of AKU in childhood involves a moderate protein restriction, hence limiting Phe and Tyr intake to physiological requirements, Tyr/Phe-free amino acid supplements can be used to enhance dietary efficacy [24]. Some centres also suggest ascorbic acid at a dose of 250-500 mg/day. A pharmacological option is nitisinone 2-(2-nitro-4-trifluoromethylbenzyl)-1,3-cyclohexanedione (NTBC), which inhibits 4-hydroxyphenylpyruvic acid dioxygenase and hence reduces serum and urinary HGA levels, which may reverse or slow the rate of the development of complications associated with alkaptonuria [18] (Table 1). Patients require clinical surveillance, e.g. from an orthopaedic, cardiac, renal, ophthalmic and neurological viewpoint.
2.3. Tyrosinemia Type I, Type II and Type III
Tyrosinemia type I (OMIM #276700) is an inborn error of Tyr catabolism with autosomal recessive inheritance. The primary enzyme defect is a deficiency of fumarylacetoacetate hydrolase (EC 3.7.1.2). Birth incidence is 1/100,000 in most regions, with higher rates in Scandinavia and Québec, Canada [21]. It often presents acutely before the age of 2 months with acute failure of hepatic synthetic function, hepatomegaly, and coagulopathy. The sub-acute and chronic forms present after 2 months and 6 months respectively, typically displaying a lesser degree of hepatic impairment, but with the additional burden of proximal renal tubulopathy resulting in renal tubular acidosis, aminoaciduria, hypophosphataemic rickets and failure to thrive [25]. Porphyria-like syndromes may also occur [26]. Patients have a significantly increased risk of developing hepatocellular carcinoma [25]. Early treatment with NTBC and dietary Phe and Tyr restriction is thus crucial to limit the development of these complications. Liver transplant may be necessary in patients with hepatocellular carcinoma or NTBC-refractory decompensated hepatic failure [27] (Table 1).
Tyrosinemia type II (OMIM #276600) is caused by a defect in the first step of Tyr degradation catalysed by Tyr aminotransferase (TAT) (EC 2.6.1.5) resulting in the accumulation of Tyr. It may present with scleral inflammation, pseudoherpetiform corneal ulceration, palmoplantar hyperkeratosis, in addition to neurological symptoms, mostly in the form of intellectual disability. Treatment is with a protein-restricted diet low in Tyr and Phe[28]. Oculocutaneous lesions may subside with dietetic treatment but not the neurological features [29] (Table 1).
Tyrosinemia type III (OMIM #276710) is caused by impairment in the 4-hydroxyphenylpyruvate dioxygenase (HPD) (EC 1.13.11.27) enzyme, which is one step downstream of TAT [30]. Tyrosinemia type III is characterized by elevated plasma levels of Tyr and increased urinary excretion of 4-hydroxyphenylpyruvic acid, 4-hydroxyphenyllactic acid, and 4-hydroxyphenylacetate[31] (Figure 1a). Clinical features include ataxia, seizures, and developmental issues [32]. Tyrosinemia type III patients do not usually demonstrate hepatorenal dysfunction or skin or eye lesions. A low natural protein diet, restrictive in Tyr and Phe, has been suggested during childhood for this ultra-rare condition [33], while other authors argue that such restriction is not recommended, or at least not necessary, as they present a case report of an asymptomatic 11-year-old girl despite no dietary modification [32] (Table 1).
2.4. Homocystinuria
The most common cause of inherited homocystinuria is cystathionine beta-synthase (CBS) (EC 4.2.1.22) deficiency, causing classical homocystinuria (HCU) (OMIM #236200), a disorder of methionine and homocysteine metabolism. HCU has an autosomal recessive inheritance. The worldwide prevalence of classical HCU is estimated to be 0.82: 100,000; it is estimated to affect approximately 1 in 100,000–200,000 people in the United States [34,35]. CBS is a pyridoxine-dependent enzyme that converts homocysteine to cystathionine in the transsulfuration pathway (Figure 1b). Another pathway for homocysteine metabolism is its remethylation to methionine by the enzyme, methionine synthase, using the folate derivative, methyltetrahydrofolate, as a methyl donor. Methionine synthase (EC 2.1.1.13) is a cobalamin-dependent enzyme [36]. A deficiency in methylenetetrahydrofolate reductase (EC 1.5.1.20), which catalyses the reduction of methylenetetrahydrofolate to 5-methyl-tetrahydrofolate, can result in moderate homocystinuria [36,37]. HCU can affect several organs and may lead to osteopenia, cognitive impairment, optic lens subluxation and increased risk of thromboembolism [36]. HCU treatment aims to maintain total plasma homocysteine (tHcy) levels <50 μmol/L in pyridoxine-responsive patients and <100 μmol/L in non-pyridoxine-responsive patients to prevent comorbidities [38]. Elevated methionine levels can also be associated with significant morbidity in HCU. In a cohort study of 36 Irish infants with classical HCU, one case of hypermethioninemic encephalopathy was observed, associated with a plasma methionine level of 1329 μmol/L [39]. Treatment of HCU includes a low protein diet and low-methionine formula, folate, cobalamin (vitamin B12) as needed, pyridoxine (vitamin B6) and betaine medication. Patient responsiveness to pyridoxine is assessed by prescribing 10 mg/kg/day (max. 500mg) with monitoring of plasma tHcys and methionine levels over the course of a few days. Patients whose plasma tHcy levels decrease below 50 μmol/l are classed as pyridoxine-responsive and do not require additional treatment [38]. It is important to note that long-term high dose pyridoxine can cause peripheral neuropathy [40]. Betaine is best used as an adjunctive treatment for patients who are unable to achieve adequate tHcy control by other methods. It can be initially prescribed at 3g twice a day and may be increased to up to 150-200mg/kg/day [38] (Table 2). It is generally important to ensure children with HCU are well hydrated, especially during periods of increased metabolic stress and intercurrent illnesses.
2.5. Methylmalonic Acidaemia
Methylmalonic acidemia (MMA; OMIM #251000 for MMUT, #251100 for MMAA, #251110 for MMAB) is a genetically heterogeneous group of autosomal recessive IEAAMs characterised by elevated methylmalonic acid in plasma and urine due to impaired isomerisation of methylmalonyl-CoA to succinyl-CoA in mitochondrial propionate metabolism in the pathway of isoleucine, methionine, threonine and valine [45]. The most common cause is a complete or partial deficiency of methylmalonyl-CoA mutase (EC 5.4.99.2) or defects in its cofactor 5’-deoxyadenosylcobalamin synthesis or transport. Biochemically, affected individuals present with markedly elevated plasma MMA, increased urine MMA, hyperammonemia, metabolic ketoacidosis, and elevated C3 acylcarnitine on newborn screening [46]. Neonates present with lethargy, vomiting, respiratory distress, and encephalopathy, which can progress to coma if untreated [47]. Children may also show failure to thrive, developmental delay, renal impairment [48]. Diagnosis is established via molecular testing for biallelic pathogenic variants or enzyme assays [46]. Management includes dietary protein restriction particularly of propiogenic amino acids, high-calorie intake, carnitine supplementation, and hydroxocobalamin in B12-responsive forms, with liver and/or kidney transplantation considered in severe cases [46,49]. Metronidazole can be used to reduce propionate production by the gut flora. Despite early treatment, long-term complications such as chronic renal failure, basal ganglia injury, pancreatitis, and intellectual disability are common [46,50,51]. Children will require an individualised ‘sick day’ plan to use when they are unwell, including further restriction of natural protein, providing dextrose and fat as a metabolic substrates, and increased medications, such as carnitine and Vitamin B12.
2.6. Maple Syrup Urine Disease
MSUD (OMIM #248600) is inherited in an autosomal recessive manner, with an incidence of approximately 1 in 185,000 people. MSUD is caused by pathogenic variants in the underlying genes BCKDHA (OMIM #608348), BCKDHB (OMIM #248611), or DBT (OMIM #248610) that abrogate the function of branched chain ketoacid dehydrogenase (BCKDH), resulting in a partial or complete lack of branched-chain keto-acid dehydrogenase (BCKD) complex (EC 1.2.4.4) (Figure 2a). Symptoms of acute intoxication include poor feeding, vomiting, irritability, neuropsychiatric symptoms, lethargy, abnormal tone or movements, seizures, ataxia, and coma. Controlling several factors that affect endogenous protein anabolism and catabolism, plasma amino acid concentrations, and plasma osmolarity is necessary for effective management of the complex pathophysiology of MSUD [52]. The intake of branched chain amino acids (BCAAs) must be strictly limited in patients with MSUD, which is achievable in a diet low in natural protein and leucine, using medical foods and synthetic formulas containing micronutrients, such as vitamins, minerals and essential fats, with prescribed amounts of isoleucine and valine supplementations. An additional thiamine challenge of 150-300mg per day over one month followed by evaluation of plasma BCAA levels is useful to identify patients who are thiamine-responsive, likely possessing residual BCKD activity, and hence would benefit from continued thiamine supplementation [53]. Acute crisis in MSUD is a medical emergency which may result in adverse or even fatal outcomes. The goal in acute management is to lower toxic levels of BCAAs, particularly leucine, and thereby their corresponding keto acids in blood and other bodily fluids, and to reverse catabolism and promote anabolism. Management involves treating the underlying cause of the metabolic decompensation, ceasing natural protein intake e.g. for 24 hours with increased intake of MSUD-specific formulas, providing hydration and extra ‘unwell’ calories through carbohydrates and fat (PO/NG or IV), and correcting any metabolic abnormalities. Caloric support through IV dextrose infusion (e.g. 0.9% NaCl with 10% dextrose at 1.2-1.5 times maintenance, with potassium added as required), should be initiated as soon as possible. The use of BCAA-free formula in combination with valine and isoleucine supplementation can further promote anabolism and reduce plasma leucine levels, and, together with careful fluid and electrolyte management, prevent the development of cerebral oedema and brain injury [54,55]. In serious cases, more forceful detoxifying approaches may be taken, e.g. hemofiltration/haemodialysis (Table 3). In our study of 18 patients diagnosed with MSUD on newborn bloodspot screening in Ireland between 1972 and 2020, it was found that despite early diagnosis and intervention, 12 patients required some form of dialysis during childhood, 6 of which in the neonatal period. Haemodialysis was found to be significantly more effective than peritoneal dialysis in lowering plasma leucine concentrations, with a ~28-fold faster reduction rate [56].
2.7. Nonketotic Hyperglycinaemia
Nonketotic hyperglycinemia (NKH) (OMIM #608599) is an autosomal recessive inherited disorder of glycine metabolism due to mutations in GLDC (OMIM #238300) or AMT (OMIM #238310), encoding the P- and T-proteins respectively, which results in diminished or absent activity of the glycine cleavage enzyme system [58]. Consequently, glycine accumulates in tissues, in particular the central nervous system (CNS). High levels of glycine may overstimulate N-methyl-D-aspartate (NMDA) receptors and can impact CSF serine and threonine levels. NKH has an incidence of 1: 76,000 live births [58]. NKH is phenotypically divided into a severe form or an attenuated form. Most patients present with the severe form of NKH which manifests with epileptic encephalopathy, spasticity, and psychomotor developmental delay. Seizures in patients with the attenuated form of NKH are usually readily manageable, and these patients display varying levels of developmental progress [59]. The main goal of treatment is to reduce brain glycine levels to limit the impact of glycine as NMDA receptor co-agonist. Sodium benzoate may be used as it conjugates with glycine to form hippurate. Plasma concentrations of glycine should be regularly monitored to minimise the development of adverse events [60]. A ketogenic diet is a nonpharmacological option which has been proposed to reduce glycine levels and improve seizure control particularly in severe disease. It may improve muscle tone, increase alertness, and reduce spasticity [60]. Dextromethorphan, an inhibitor of NMDA receptors, is also used. Pharmacogenetics is an important consideration in dextromethorphan therapy as it is metabolised by the highly polymorphic CYP2D6. This genetic variability may result in various phenotypic extremes, including poor metabolisers and ultrarapid metabolisers, impacting drug efficacy and toxicity [61]. A glycine-restricted diet alone is insufficient achieve a therapeutic effect [62] (Table 4).
2.8. Pyridoxine-Dependent Epilepsy
Pyridoxine-dependent epilepsy (PDE) is an inborn error of lysine catabolism. The phenotype of PDE results from multiple genetic disorders [68]. Known genetic causes of PDE are biallelic pathogenic variants in any of three genes. These genes include PNPO (OMIM #603287), which encodes pyridox(am)ine 5′-phosphate oxidase (EC 1.4.3.5), PLPBP (OMIM #604436), which encodes PLP homeostasis protein and ALDH7A1 (OMIM #107323), encoding the enzyme α-aminoadipic acid semialdehyde (α-AASA) dehydrogenase. PDE-ALDH7A1 (OMIM #266100), is the most common of the three and occurs as a result of an autosomal recessive inherited deficiency of α-AASA. Incidence of PDE-ALDH7A1 is around 1:65,000 live births [69]. Deficient enzyme activity in the pipecolic acid and saccharopine catabolic pathway of lysine results in abnormal accumulation of pipecolic acid, α-AASA, and Δ1-piperideine-6-carboxylate (Δ1-P6C) [68].
PDE is characterised by recurrent seizures that are resistant to conventional anti-epileptic drugs but can be effectively managed with pyridoxine (vitamin B6) supplementation. Elevated levels of α-AASA and pipecolic acid in a patient with an epileptic encephalopathy would suggest a diagnosis of PDE-ALDH7A1. Genetic testing confirms the diagnosis. Rapid and dramatic improvement in seizure control upon administration of vitamin B6 strongly suggests PDE. The cornerstone of PDE treatment is pyridoxine supplementation, which can be administered orally or intravenously. In most cases, patients with PDE will require lifelong pyridoxine supplementation to control their seizures. Regular monitoring of blood pyridoxal 5'-phosphate (PLP) levels is necessary to ensure that patients are receiving an adequate dosage of vitamin B6. Maintaining PLP levels within the therapeutic range is crucial for effective seizure control. In some cases, patients with PDE may require anti-epileptic drugs (AEDs) in combination with pyridoxine. Despite seizure control, a large percentage of affected individuals (75%) may have developmental issues or a reduced intellectual ability[69] (Table 4).
2.9. Cystinuria
Cystinuria (OMIM #220100) is an autosomal recessive disorder that occurs due to pathogenic variants in SLC3A1 (2p21) or SLC7A9 (19q13.11) [70]. This results in failure of absorption of dibasic amino acids cystine, lysine, arginine and ornithine, by the proximal tubules and the intestinal tract. The main clinical feature of cystinuria is recurrent nephrolithiasis. The goal of treatment is to prevent stone formation or growth. This is achieved by increasing fluid intake, maintaining a moderate protein diet, urinary alkalinisation, and therapy with cystine binding medications (alpha-mercaptopropionylglycine, tiopronin, and D-Penicillamine) [71,72]. Given that up to 70% of patients develop cystine stones or chronic kidney disease [73], follow-up with nephrology and urology is crucial in managing these patients (Table 5).
2.10. Lysinuric Protein Intolerance
Lysinuric protein intolerance (LPI) (OMIM #222700) is caused by mutations in the gene coding for solute carrier family 7A member 7 (SLC7A7) located at chromosome 14q11.2, resulting in defective dibasic amino acid (lysine, arginine, ornithine) transport at the epithelial cells in the kidney and intestine [77]. The diagnosis is established by detecting elevated 24-hour urinary excretion of lysine with low plasma lysine. Most patients have episodes of hyperammonemia, e.g. postprandially, due to a substrate deficiency and disruption of the urea cycle. The diagnosis is confirmed by molecular genetic testing. Treatment is based on a protein-restricted diet and citrulline supplementation, and nitrogen-scavengers are used to treat hyperammonemia. L-arginine, and lysine also have role in the treatment of LPI [78,79] (Table 5).
2.11. Hartnup Disease
Hartnup disease (OMIM #234500) is a rare, autosomal recessive disorder caused by pathogenic variants in the SLC6A19 gene (5p15.33) which leads to defective transport of neutral amino acids (i.e., monoamino-monocarboxylic) across epithelial cells in renal proximal tubules and the gastrointestinal tract [80,81]. Symptoms includes pellagra-like skin eruptions, ataxia, spasticity, delayed motor development, hypotonia, and psychiatric symptoms [81,82,83]. Diagnosis is established by detecting hyperaminoaciduria. Confirmation relies upon the mutation analysis. Treatment includes high-protein diets and nicotinamide supplementation [84] (Table 5).
2.12. Glutaric Aciduria Type 1
Glutaric Aciduria type I (GA1) (OMIM #231670) is an autosomal recessive disorder of lysine, hydroxylysine, and tryptophan metabolism characterised by deficiency of glutaryl-CoA dehydrogenase (EC 1.3.8.6), an enzyme encoded by the GCDH (OMIM #608801) gene, resulting in elevations of glutaric acid and 3-hydroxyglutaric acid (Figure 2b). It has an incidence of 1 in 100,000 live births, with a higher prevalence in Oji Cree natives, the Amish community, and Irish Travellers [85]. Neuroimaging features include widening of the Sylvian fissures, widening of the mesencephalic cisterns, and CSF space expansion anterior to the temporal lobes, collectively resulting in micro-mesencephalic macrocephaly [86]. Widening of the subarachnoid space may lead to rupture of the bridging cortical veins resulting in low trauma subdural haematoma [87]. During the early years of life, acute striatal necrosis is one of the main causes of morbidity and mortality [88]. Secondary carnitine depletion is common in untreated GA1 patients [89,90]. Current treatment includes nutritional therapy with a lysine-free, tryptophan-reduced diet, in addition to arginine enriched amino acid mixtures. The early implementation of an ‘unwell’ regime, reducing or stopping natural protein intake temporarily and increasing calorie supply with increased carnitine supplementation together with the use of antipyretics is crucial to reduce the risk of an acute metabolic and encephalopathic crisis with neurological sequelae. Lifelong carnitine supplementation is provided with the aim of inducing elimination of toxic metabolites. An initial oral dosage of 100 mg L-carnitine/kg per day, divided into 3 doses, is commonly used [91,92], with the aim of maintaining high-normal reference range levels of free carnitine in plasma or in dried blood spot [93], the dose may be doubled during crisis. Patients affected by dystonia may benefit from baclofen, trihexphenidyl, gabapentin or even deep brain stimulation [85,93,94]. (Table 5).
Another disorder of lysine metabolism is familial hyperlysinemia, which includes two conditions: hyperlysinemia type I (OMIM #238700), and hyperlysinemia type II (OMIM #268700) [95]. Hyperlysinemia type I is the most common form. It is caused by pathogenic variants in the AASS (OMIM #605113) gene which provides instructions for the production of aminoadipic semialdehyde synthase (1.2.1.31) [96]. It is characterised by elevated concentrations of lysine in the plasma and the cerebrospinal fluid. Hyperlysinemia is generally considered to be a benign metabolic condition. Clinical features may include psychomotor delay, hypotonia and seizures. It remains unclear whether protein or lysine restriction is beneficial in symptomatic patients [97] (Table 5).
2.13. Serine Deficiency
The most frequently reported cause of serine deficiency is 3-phosphoglycerate dehydrogenase (3-PGDH) deficiency (OMIM #601815), which affects the first step in the serine biosynthesis pathway [66,98]. Deficiency in phosphoserine aminotransferase (PSAT) (OMIM #610936), which catalyses the second step in the pathway, is a much rarer cause of serine deficiency and has only been reported as a few cases in literature. Clinical features of serine deficiency may include seizures, microcephaly, hypertonia, and developmental delay. Suggested supplementation for infants with PSAT1 deficiency are serine (500 mg/kg/day) and glycine (200 mg/kg/day) [99].
2.14. Hyperprolinaemia Type I and Type II
Hyperprolinemia type I (HPI) (OMIM #239500) is caused by a deficiency in proline dehydrogenase (POX) (EC 1.5.5.2). HPI is characterised by increased plasma proline levels without an increase in urinary excretion of ∆1-pyrroline-5-carboxylic acid (P5C) [100]. POX is encoded by the PRODH (OMIM #606810) gene located on chromosome 22q11, and as such, its deficiency may occur because of contiguous gene deletion as part of DiGeorge syndrome [101]. Its clinical significance is not fully established, but may be associated with schizophrenia, autism, and seizures [100,102,103]. Hyperprolinemia type II (HPII) (OMIM #239510) occurs due to absence of the enzyme ∆-1-pyrroline-5-carboxylic acid dehydrogenase (EC 1.2.1.88) and is associated with higher proline levels than in HPI [104]. It typically results in drug-resistant but pyridoxine-sensitive seizures in the first year of life and, in the absence of timely treatment, may lead to developmental delay [104]. These seizures may occur due to vitamin B6 inactivation by accumulated Δ1-pyrroline-5-carboxylic acid, and therefore long-term vitamin B6 supplementation may prevent these seizures. Strict dietary therapy via restriction of protein is not necessary, as it only results in modest reduction of plasma proline and does not have an impact on clinical phenotype, and in our centre we advise to avoid protein excess. Antioxidants, such as vitamin C, may also have a minor role in treatment [104] (Table 6).
2.15. Glutamine Synthetase Deficiency
Glutamine synthetase deficiency (GSD) (OMIM #610015) is an extremely rare inborn error of glutamine metabolism caused by mutation in GLUL gene (OMIM #138290). Clinical features include respiratory failure, encephalopathy, and brain malformations [109,110]. Laboratory finding include low levels of glutamine in plasma and cerebrospinal fluid (CSF). L-glutamine supplementation with the goal of glutamine normalisation may improve brain functioning. Therapy may be commenced at a low dose of 17mg/kg/day, increasing slowly to higher doses of 1020mg/kg/day, while monitoring plasma and CSF glutamine concentrations [111].
2.16. Asparagine Synthetase Deficiency
Asparagine synthetase deficiency (ASD) (OMIM #615574) is an autosomal recessive disorder caused by mutations in ASNS (OMIM #108370) [112]. Clinical features include microcephaly, developmental delay, seizures, axial hypotonia and spastic quadriplegia. Laboratory findings include low plasma and cerebrospinal fluid (CSF) asparagine level [113]. Treatment is supportive, including antiepileptic medications and also L-asparagine supplementation.
3. Discussion
Recent advancements in the treatment of IEAAMs have expanded therapeutic options beyond strict dietary management to include different pharmacologic agents, enzyme substitution, transplant options and emerging genetic therapies in an overall more personalised therapeutic approach. Despite these developments, many conditions still require lifelong interventions and individualised treatment plans based on genotype, metabolic profile, and responsiveness to therapy. Clinical guidelines for the management of inborn errors of metabolism have been increasingly refined over time, incorporating emerging evidence and therapeutic advances to enhance consistency, efficacy, and patient outcomes in clinical practice.
The complete European guidelines on phenylketonuria: diagnosis and treatment were published in 2017 [5], and later revised in 2025 [114]. The European Society for Phenylketonuria (ESPKU) guidelines have recommended a lifelong protein restricted diet with an upper Phe target of 600 μmol/L for adult PKU patients [115]. High Phe levels in adulthood may directly affect mood and sustained attention, as demonstrated by neuropsychological testing during periods of Phe supplementation [116]. Sapropterin dihydrochloride, the synthetic form of BH4, was approved as the first pharmacological chaperone to correct the loss-of-function of the enzyme Phe hydroxylase (PAH) [117]. BH4 has a significant lowering effect on blood Phe concentration and can improve Phe tolerance with an acceptable safety profile [7,118]. There is still lack of knowledge in predicting BH4 responsiveness; with at least half of those with PKU having either minimal or no response [119]. Other studies have reported BH4 responsiveness to range from 20% to 62% [11,120]. Suggested predictors of BH4 responsiveness include Phe levels at diagnosis, Phe/Tyr ratio, Phe tolerance before BH4 treatment and genotype [121]. In a study of 46 Italian PKU patients investigated for BH4-responsiveness, 17 patients were identified as BH4 responders [122]. The presence of at least one pathogenic variant with residual enzymatic activity was the best predictor of BH4-responsiveness, while the presence of two inactive alleles excluded responsiveness [122]. Assessment of BH4 responsiveness requires a pre-loading test and BH4 loading test. BH4-responders may be started on long-term treatment with BH4 at the initial dose of 10 mg/kg/day [122]. Pegvaliase, is an enzyme substitution therapy for adults with PKU [9]. Pegvaliase is the first approved enzyme substitution therapy that can be considered for adult PKU patients who have failed existing management strategies [123]. Pegvaliase has a generally tolerable safety profile in adults with PKU [124,125]. An update of the web based PKU guideline to improve clinical outcomes and promote consistency on nutrition management of PKU receiving pegvaliase therapy has been published recently [126]. In adulthood the goal of treatment is to maintain normal brain cognitive function and neuropsychological and social performance. A PKU related health-related quality of life (HRQoL) questionnaire of patients and their families was developed in different subgroups of patients defined according to severity of PKU, overall health status, and treatment with tetrahydrobiopterin (BH4). Data was collected and analysed from 253 parents, and 306 patients including 104 adults. It was shown that BH4 treatment was associated with better scores in all ages. This may reflect that a less restricted died, often made possible by responsiveness to BH4, will have a positive impact on HRQoL in PKU patients [127]. A study by Bik-Multanowski and colleagues in 2008 of treatment noncompliant adults with PKU found improvement of subjective well-being in patients with severe or moderate distress upon return to recommended diet [128]. These studies highlight the importance of considering patient psychological and emotional well-being as part of the holistic treatment of IEAAMs such as PKU. Dawson et al investigated the effect of Phe level on reaction time [129]. Patients with PKU were split into three groups: off-diet (Phe >1,200 μmol/l), on-diet (Phe <800 μmol/l) and maternal diet (Phe 100-400 μmol/l). Adults who discontinued the PKU low-Phe diet during adolescence were found to have slower reaction times than controls. Reaction times were measured before and after the commencing the maternal PKU protein restricted diet in 16 women who were contemplating pregnancies. Reaction times significantly improved as Phe levels were strictly controlled. Data show that effects of Phe levels on reaction time are reversible [130]. Even well-controlled PKU has several subtler physical, cognitive, and behavioural consequences that have been recognised. In the healthy population, Tyr is considered a nonessential amino acid; however, in patients with PKU, it becomes essential. Tyr is converted into L-dopa, which serves as the precursor for synthesis of catecholamines [131]. As such, it has been suggested that, approximately 8% to 10% of total protein calculated in the diet must come from Tyr [132,133], [134] evaluated all randomised or quasi-randomised trials investigating the use of Tyr supplementation versus placebo in people with PKU in addition to, or instead of, a Phe-restricted diet. In three trials reporting the results of a total of 56 participants, the blood Tyr concentrations were significantly higher in the participants receiving Tyr supplements than those in the placebo group. No significant differences were found between any of the other outcomes measured. From the available evidence no recommendations can be made about whether Tyr supplementation should be introduced into routine clinical practice [134,135].
The main aim of the various treatments of AKU used to date has been to achieve symptomatic control of co-morbidities such as arthritis and joint pain. A potential side effect of nitisinone is hypertyrosinemia, potentially necessitating the dietary restriction of Tyr and Phe. In addition, long-term studies are needed to show the effectiveness of nitisinone in providing adequate reduction in homogentisate to prevent the development of complications in AKU. Management of osteoporosis in AKU proves challenging due to various reasons, including the reduced reliability of dual energy X-ray absorptiometry scans due to extensive disc calcification, degenerative arthritis, or joint replacements [136,137]. In addition, fragility fractures may occur despite appropriate bisphosphonate therapy. It has therefore been recommended that osteoporosis in AKU should be initially treated with teriparatide and later by intravenous zoledronic acid [138]. Vitamin C is an antioxidant believed to reduce the conversion of HGA to benzoquinone acetate via oxidation, and one study highlighted that it serves as a co-factor for 4-hydroxyphenylpyruvate dioxygenase, which causes increased HGA production [139].
While significant advances have been made in the treatment of MSUD, particularly in the context of dietary management, it remains a volatile and life-threatening illness. Liver transplantation, both from living and deceased donors, has been investigated as a potential treatment. Liver transplant from unrelated deceased donor can restore 9-13% of whole-body BCKA metabolism. Over the intermediate term, living related donor transplant was shown to more effectively correct leucine and valine concentrations than deceased donor transplant. While neither form of transplantation provided absolute protection from metabolic derangement, they may still offer a viable alternative or adjunct to dietary treatment [140]. As a monogenic disorder, MSUD may be a candidate for gene therapy as a potential treatment option. Pontoizeau et al [141] evaluated the treatment of severe MSUD in BCKDHB-knockout mice using an adeno-associated virus 8 vector carrying the human BCKDHB gene under the control of the ubiquitous human elongation factor 1-alpha promoter. This gene therapy provided long-term phenotypic rescue in the treated mice and reduced BCAA accumulation. Translating this success into human studies, particularly in a neonatal setting, may pose various difficulties, partly due to the age-related decline in liver trans-gene expression in humans, as well as the decreased efficiency of AAV8 in transducing human tissue compared with mice.
In non-ketotic hyperglycinemia, sodium benzoate can significantly reduce serum glycine levels even by administering low doses of 53 mg sodium benzoate/kg body mass per day. However, a higher dose up to 240 mg/kg BM per day could not normalise cerebrospinal fluid (CSF) glycine [142]. Sodium benzoate has an unpleasant taste, it can cause itching, hyperactivity, and it may increase the risk of gastrointestinal discomfort. The possibility of accidentally taking a toxic amount of benzoate can occur when high doses are prescribed. However, no evidence of sodium benzoate toxicity was reported with the administration of high doses up to 470 mg/kg body mass per day [142]. Even with good effort, numerous patients receiving high doses of benzoate had glycine levels that were over the specified target range, indicating that benzoate uptake is poor, especially in adult patients, and glycine management is frequently quite variable. In patients with attenuated disease, early treatment with dextromethorphan and sodium benzoate appears to be effective in normalising plasma glycine. Combination of ketogenic diet and low dose sodium benzoate therapy was found to be more effective in reducing plasma glycine levels than high dose benzoate alone in 6 infants [143]. Combining sodium benzoate with NMDA receptor inhibitors, such as dextromethorphan or ketamine, has been demonstrated to reduce seizures and enhance neurocognitive outcomes [144,145].
The International PDE Consortium released the first consensus guidelines for the diagnosis and treatment of PDE-ALDH7A1 in 2021 [65]. The successful treatment of PDE hinges on early diagnosis, appropriate pyridoxine supplementation, and diligent monitoring. Treatment with pyridoxine and lysine-reduction therapies (LRTs) demonstrated a decrease in pipecolic acid, α-AASA, and Δ1-P6C [146]. Some reports found a significant increase on developmental testing scores on this treatment [147].
Revised recommendations on the diagnosis and management of glutaric aciduria type I were published in 2023 [93]. An experienced interdisciplinary team should initiate and oversee metabolic treatment, which involves dietary modifications that include foods low in tryptophan and lysine, carnitine supplementation, and accelerated emergency care during acute episodes of intercurrent disease. However, there is usually little chance of averting irreversible harm if treatment is started after symptoms appear. Vigorous and prompt treatment for fever or illness is necessary. Using dextrose in conjunction with electrolyte-containing fluids at a rate of 6–10 mg/kg/min and ensuring that sufficient calories are supplied are the main components of aggressive therapy during acute illness. There is currently no consensus over the potential benefits of metabolic meals high in arginine [148]. Levocarnitine scavenger therapy is aimed at reducing the build-up of toxic metabolites and correcting secondary carnitine depletion. Treating dystonia arising from striatal damage may involve standard therapies including baclofen, benzodiazepines, and botulinum toxin. However, effective management of dystonia remains challenging, and predicting which medications are likely to be effective in individual cases is difficult. On the horizon is a novel substrate reduction therapy which is targeting the enzyme α-aminoadipic semialdehyde synthase in the metabolic pathway of lysine, an evolving gene therapy approach for glutaric aciduria type I recently established in mice [149].
4. Conclusions
The goal of treatment of IEAAMs is to normalize the metabolic imbalance at a cellular level and in physiological fluids as much as possible, by implementing dietary modification and pharmaco-therapy or cofactor supplementation as appropriate, along with patient monitoring and emergency treatment if necessary. However, despite these advancements, many patients face limitations in achieving complete metabolic control, and the risk of long-term complications persists. An acute metabolic crisis poses ongoing challenges with an unmet need for more targeted neuroprotective measures. The development of novel therapeutic strategies is critical for further improving quality of life. Other treatment options include organ transplantation, and also emerging new therapies, e.g., mRNA therapy or gene therapy that offer promising avenues for more definitive personalised treatments. Future research is essential to refine these techniques and extend their availability to broader patient populations, thereby offering hope for more effective, curative treatments of IEAAMs.
Author Contributions
Conceptualization, A.L.S., D.N, and I.K.; writing—original draft preparation, A.L.S., D.N, and I.K.; writing—review and editing, E.J.M and I.K..; visualization, A.L.S and D.N.; funding acquisition, I.K. All authors have read and agreed to the published version of the manuscript.
Funding
The authors are very grateful to the Temple Street Foundation/Children’s Health Foundation (CHF), Dublin for financial assistance to cover publication costs (RPAC 19.02 to IK).
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
The authors gratefully acknowledge the support of the National Rare Diseases Office, Mater Misericordiae University Hospital for providing access to institutional, academic, and library resources used in preparing this review.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 5-HIAA | 5-Hydroxyindoleacetic acid |
| AASS | Aminoadipic semialdehyde synthase (gene) |
| AAV | Adeno-associated virus |
| AAV8 | Adeno-associated virus serotype 8 |
| ACE | Angiotensin-converting enzyme |
| AEDs | Anti-epileptic drugs |
| AKU | Alkaptonuria |
| AMT | Aminomethyltransferase (gene) |
| ASD | Asparagine synthetase deficiency |
| ASNS | Asparagine synthetase gene |
| BCAAs | Branched-chain amino acids |
| BCKD | Branched-chain keto-acid dehydrogenase |
| BCKDC | Branched-chain α-ketoacid dehydrogenase complex |
| BCKDH | Branched-chain ketoacid dehydrogenase |
| BD | Twice daily |
| BH4 | Tetrahydrobiopterin |
| BHMT | Betaine-homocysteine methyltransferase |
| BID | Twice daily |
| BTMs | Bone turnover markers |
| C3 | Propionylcarnitine |
| CBS | Cystathionine-β-synthase |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| CT | Computed tomography |
| CYP2D6 | Cytochrome P450 2D6 |
| CYP3A4 | Cytochrome P450 3A4 |
| CYPUGT | Cytochrome P450 / UGT (as written in manuscript) |
| DNAJC12 | DnaJ heat shock protein family (Hsp40) member C12 |
| EC | Enzyme Commission |
| ESPFKU | European Society for Phenylketonuria |
| ESRD | End-stage renal disease |
| ESRF | End-stage renal failure |
| FAA | Fumarylacetoacetate |
| FAH | Fumarylacetoacetate hydrolase |
| GA1 | Glutaric aciduria type 1 |
| GCDH | Glutaryl-CoA dehydrogenase (gene) |
| GH | Growth hormone |
| GLDC | Glycine decarboxylase (gene) |
| GLUL | Glutamate–ammonia ligase (glutamine synthetase) gene |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GSD | Glutamine synthetase deficiency |
| HCU | Homocystinuria |
| HGA | Homogentisic acid |
| HGD | Homogentisate 1,2-dioxygenase |
| HRT | Hormone replacement therapy |
| HRQoL | Health-related quality of life |
| HPD | 4-Hydroxyphenylpyruvate dioxygenase |
| HVA | Homovanillic acid |
| IEAAMs | Inborn errors of amino acid metabolism |
| IV | Intravenous |
| L-DOPA | L-3,4-dihydroxyphenylalanine |
| LNAAs | Large Neutral Amino Acids |
| LP | Lumbar puncture |
| LPI | Lysinuric protein intolerance |
| LRTs | Lysine-reduction therapies |
| MAA | Maleylacetoacetate |
| MAAI | Maleylacetoacetate isomerase |
| MAT | Methionine S-adenosyltransferase |
| MMA | Methylmalonic acidemia |
| MMAA | Methylmalonic acidemia cblA type (gene) |
| MMAB | Methylmalonic acidemia cblB type (gene) |
| MMUT | Methylmalonyl-CoA mutase (gene) |
| MS | Methionine synthase |
| MSUD | Maple Syrup Urine Disease |
| MTHFR | Methylenetetrahydrofolate reductase |
| NaCl | Sodium chloride |
| NG | Nasogastric |
| NKH | Nonketotic hyperglycinemia |
| NMDA | N-methyl-D-aspartate |
| NTBC | Nitisinone (2-(2-nitro-4-trifluoromethylbenzyl)-1,3-cyclohexanedione) |
| OMIM | Online Mendelian Inheritance in Man |
| PAH | Phenylalanine hydroxylase |
| PAL | Phenylalanine ammonia-lyase |
| PDE | Pyridoxine-dependent epilepsy |
| Phe | Phenylalanine |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PKU | Phenylketonuria |
| PO | By mouth |
| PRODH | Proline dehydrogenase (gene) |
| QDS | Four times daily |
| SAH | S-adenosylhomocysteine |
| SAHH | S-adenosylhomocysteine hydrolase |
| SAM | S-adenosyl-L-methionine |
| SC | Subcutaneous |
| TAT | Tyrosine aminotransferase |
| TDS | Three times daily |
| THF | Tetrahydrofolate |
| tHcy | Total homocysteine |
| Tyr | Tyrosine |
| WBC | White blood cell |
References
- Chandel, N. S., Amino Acid Metabolism. Cold Spring Harb Perspect Biol 2021, 13 (4). [CrossRef]
- Tessari, P.; Lante, A.; Mosca, G., Essential amino acids: master regulators of nutrition and environmental footprint? Sci Rep 2016, 6, 26074. [CrossRef]
- Blau, N.; van Spronsen, F. J.; Levy, H. L., Phenylketonuria. Lancet 2010, 376 (9750), 1417-27. [CrossRef]
- Blau, N.; Hennermann, J. B.; Langenbeck, U.; Lichter-Konecki, U., Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab 2011, 104 Suppl, S2-9. [CrossRef]
- van Wegberg, A. M. J.; MacDonald, A.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A. M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M., et al., The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis 2017, 12 (1), 162. [CrossRef]
- Boyle, F.; Lynch, G.; Reynolds, C. M.; Green, A.; Parr, G.; Howard, C.; Knerr, I.; Rice, J., Determination of the Protein and Amino Acid Content of Fruit, Vegetables and Starchy Roots for Use in Inherited Metabolic Disorders. Nutrients 2024, 16 (17). [CrossRef]
- Levy, H. L.; Milanowski, A.; Chakrapani, A.; Cleary, M.; Lee, P.; Trefz, F. K.; Whitley, C. B.; Feillet, F.; Feigenbaum, A. S.; Bebchuk, J. D., et al., Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. Lancet 2007, 370 (9586), 504-10. [CrossRef]
- Doyle, S.; O'Regan, M.; Stenson, C.; Bracken, J.; Hendroff, U.; Agasarova, A.; Deverell, D.; Treacy, E. P., Extended Experience of Lower Dose Sapropterin in Irish Adults with Mild Phenylketonuria. JIMD Rep 2018, 40, 71-76. [CrossRef]
- Thomas, J.; Levy, H.; Amato, S.; Vockley, J.; Zori, R.; Dimmock, D.; Harding, C. O.; Bilder, D. A.; Weng, H. H.; Olbertz, J., et al., Pegvaliase for the treatment of phenylketonuria: Results of a long-term phase 3 clinical trial program (PRISM). Mol Genet Metab 2018, 124 (1), 27-38. [CrossRef]
- MacDonald, A.; van Wegberg, A. M. J.; Ahring, K.; Beblo, S.; Bélanger-Quintana, A.; Burlina, A.; Campistol, J.; Coşkun, T.; Feillet, F.; Giżewska, M., et al., PKU dietary handbook to accompany PKU guidelines. Orphanet Journal of Rare Diseases 2020, 15 (1), 171. [CrossRef]
- Vernon, H. J.; Koerner, C. B.; Johnson, M. R.; Bergner, A.; Hamosh, A., Introduction of sapropterin dihydrochloride as standard of care in patients with phenylketonuria. Mol Genet Metab 2010, 100 (3), 229-33. [CrossRef]
- Sanford, M.; Keating, G. M., Sapropterin: a review of its use in the treatment of primary hyperphenylalaninaemia. Drugs 2009, 69 (4), 461-76. [CrossRef]
- Doyle, S.; O’Regan, M.; Stenson, C.; Bracken, J.; Hendroff, U.; Agasarova, A.; Deverell, D.; Treacy, E. P., Extended Experience of Lower Dose Sapropterin in Irish Adults with Mild Phenylketonuria. JIMD Reports, Volume 40 2018, 71-76. [CrossRef]
- Hollander, S.; Viau, K.; Sacharow, S., Pegvaliase dosing in adults with PKU: Requisite dose for efficacy decreases over time. Mol Genet Metab 2022, 137 (1-2), 104-106. [CrossRef]
- Mahan, K. C.; Gandhi, M. A.; Anand, S., Pegvaliase: a novel treatment option for adults with phenylketonuria. Curr Med Res Opin 2019, 35 (4), 647-651. [CrossRef]
- Bozaci, A. E.; Er, E.; Yazici, H.; Canda, E.; Kalkan Uçar, S.; Güvenc Saka, M.; Eraslan, C.; Onay, H.; Habif, S.; Thöny, B., et al., Tetrahydrobiopterin deficiencies: Lesson from clinical experience. JIMD Rep 2021, 59 (1), 42-51. [CrossRef]
- Fino, E.; Barbato, A.; Scaturro, G. M.; Procopio, E.; Balestrini, S., DNAJC12 deficiency: Mild hyperphenylalaninemia and neurological impairment in two siblings. Molecular Genetics and Metabolism Reports 2023, 37, 101008. [CrossRef]
- Abbas, K.; Basit, J.; Rehman, M. E. U., Adequacy of nitisinone for the management of alkaptonuria. Ann Med Surg (Lond) 2022, 80, 104340. [CrossRef]
- Ranganath, L. R.; Milan, A. M.; Hughes, A. T.; Khedr, M.; Norman, B. P.; Alsbou, M.; Imrich, R.; Gornall, M.; Sireau, N.; Gallagher, J. A., et al., Comparing nitisinone 2 mg and 10 mg in the treatment of alkaptonuria-An approach using statistical modelling. JIMD Rep 2022, 63 (1), 80-92. [CrossRef]
- Das, A. M., Clinical utility of nitisinone for the treatment of hereditary tyrosinemia type-1 (HT-1). Appl Clin Genet 2017, 10, 43-48. [CrossRef]
- Matthews, R. P., CHAPTER 32 - Metabolic Liver Disease: Tyrosinemia, Galactosemia, and Hereditary Fructose Intolerance. In Pediatric Gastroenterology, Liacouras, C. A.; Piccoli, D. A.; Bell, L. M., Eds. Mosby: Philadelphia, 2008; pp 267-275.
- Introne, W. J.; Perry, M.; Chen, M., Alkaptonuria. In GeneReviews(®), Adam, M. P.; Feldman, J.; Mirzaa, G. M.; Pagon, R. A.; Wallace, S. E.; Amemiya, A., Eds. University of Washington, Seattle Copyright © 1993-2025, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.: Seattle (WA), 1993.
- Sharabi, A. F.; Goudar, R. B., Alkaptonuria. In StatPearls, StatPearls Publishing Copyright © 2025, StatPearls Publishing LLC.: Treasure Island (FL), 2025.
- Teke Kisa, P.; Eroglu Erkmen, S.; Bahceci, H.; Arslan Gulten, Z.; Aydogan, A.; Karalar Pekuz, O. K.; Yuce Inel, T.; Ozturk, T.; Uysal, S.; Arslan, N., Efficacy of Phenylalanine- and Tyrosine-Restricted Diet in Alkaptonuria Patients on Nitisinone Treatment: Case Series and Review of Literature. Ann Nutr Metab 2022, 78 (1), 48-60. [CrossRef]
- Morrow, G.; Tanguay, R. M., Biochemical and Clinical Aspects of Hereditary Tyrosinemia Type 1. Adv Exp Med Biol 2017, 959, 9-21. [CrossRef]
- van Ginkel, W. G.; Pennings, J. P.; van Spronsen, F. J., Liver Cancer in Tyrosinemia Type 1. Adv Exp Med Biol 2017, 959, 101-109. [CrossRef]
- Chinsky, J. M.; Singh, R.; Ficicioglu, C.; van Karnebeek, C. D. M.; Grompe, M.; Mitchell, G.; Waisbren, S. E.; Gucsavas-Calikoglu, M.; Wasserstein, M. P.; Coakley, K., et al., Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genet Med 2017, 19 (12). [CrossRef]
- Čulic, V.; Betz, R. C.; Refke, M.; Fumic, K.; Pavelic, J., Tyrosinemia type II (Richner–Hanhart syndrome): A new mutation in the TAT gene. European Journal of Medical Genetics 2011, 54 (3), 205-208. [CrossRef]
- Peña-Quintana, L.; Scherer, G.; Curbelo-Estévez, M. L.; Jiménez-Acosta, F.; Hartmann, B.; La Roche, F.; Meavilla-Olivas, S.; Pérez-Cerdá, C.; García-Segarra, N.; Giguère, Y., et al., Tyrosinemia type II: Mutation update, 11 novel mutations and description of 5 independent subjects with a novel founder mutation. Clinical Genetics 2017, 92 (3), 306-317. [CrossRef]
- Russo, P. A.; Mitchell, G. A.; Tanguay, R. M., Tyrosinemia: a review. Pediatr Dev Pathol 2001, 4 (3), 212-21. [CrossRef]
- Rüetschi, U.; Cerone, R.; Pérez-Cerda, C.; Schiaffino, M. C.; Standing, S.; Ugarte, M.; Holme, E., Mutations in the 4-hydroxyphenylpyruvate dioxygenase gene (HPD) in patients with tyrosinemia type III. Hum Genet 2000, 106 (6), 654-62. [CrossRef]
- Szymanska, E.; Sredzinska, M.; Ciara, E.; Piekutowska-Abramczuk, D.; Ploski, R.; Rokicki, D.; Tylki-Szymanska, A., Tyrosinemia type III in an asymptomatic girl. Molecular Genetics and Metabolism Reports 2015, 5, 48-50. [CrossRef]
- Barroso, F.; Correia, J.; Bandeira, A.; Carmona, C.; Vilarinho, L.; Almeida, M.; Rocha, J. C.; Martins, E., TYROSINEMIA TYPE III: A CASE REPORT OF SIBLINGS AND LITERATURE REVIEW. Rev Paul Pediatr 2020, 38, e2018158. [CrossRef]
- Sellos-Moura, M.; Glavin, F.; Lapidus, D.; Evans, K.; Lew, C. R.; Irwin, D. E., Prevalence, characteristics, and costs of diagnosed homocystinuria, elevated homocysteine, and phenylketonuria in the United States: a retrospective claims-based comparison. BMC Health Services Research 2020, 20 (1), 183. [CrossRef]
- Weber Hoss, G. R.; Sperb-Ludwig, F.; Schwartz, I. V. D.; Blom, H. J., Classical homocystinuria: A common inborn error of metabolism? An epidemiological study based on genetic databases. Mol Genet Genomic Med 2020, 8 (6), e1214. [CrossRef]
- Gerrard, A.; Dawson, C., Homocystinuria diagnosis and management: it is not all classical. J Clin Pathol 2022. [CrossRef]
- Aljassim, N.; Alfadhel, M.; Nashabat, M.; Eyaid, W., Clinical presentation of seven patients with Methylenetetrahydrofolate reductase deficiency. Molecular Genetics and Metabolism Reports 2020, 25, 100644. [CrossRef]
- Morris, A. A.; Kožich, V.; Santra, S.; Andria, G.; Ben-Omran, T. I.; Chakrapani, A. B.; Crushell, E.; Henderson, M. J.; Hochuli, M.; Huemer, M., et al., Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J Inherit Metab Dis 2017, 40 (1), 49-74. [CrossRef]
- Allen, J.; Power, B.; Abedin, A.; Purcell, O.; Knerr, I.; Monavari, A., Plasma methionine concentrations and incidence of hypermethioninemic encephalopathy during infancy in a large cohort of 36 patients with classical homocystinuria in the Republic of Ireland. JIMD Rep 2019, 47 (1), 41-46. [CrossRef]
- Ludolph, A. C.; Masur, H.; Oberwittler, C.; Koch, H. G.; Ullrich, K., Sensory neuropathy and vitamin B6 treatment in homocystinuria. European Journal of Pediatrics 1993, 152 (3), 271-271. [CrossRef]
- Strauss, K. A.; Morton, D. H.; Puffenberger, E. G.; Hendrickson, C.; Robinson, D. L.; Wagner, C.; Stabler, S. P.; Allen, R. H.; Chwatko, G.; Jakubowski, H., et al., Prevention of brain disease from severe 5,10-methylenetetrahydrofolate reductase deficiency. Mol Genet Metab 2007, 91 (2), 165-75. [CrossRef]
- Furujo, M.; Kinoshita, M.; Nagao, M.; Kubo, T., S-adenosylmethionine treatment in methionine adenosyltransferase deficiency, a case report. Mol Genet Metab 2012, 105 (3), 516-8. [CrossRef]
- Huang, Y.; Chang, R.; Abdenur, J. E., The biochemical profile and dietary management in S-adenosylhomocysteine hydrolase deficiency. Mol Genet Metab Rep 2022, 32, 100885. [CrossRef]
- Elmonem, M. A.; Veys, K. R.; Soliman, N. A.; van Dyck, M.; van den Heuvel, L. P.; Levtchenko, E., Cystinosis: a review. Orphanet Journal of Rare Diseases 2016, 11 (1), 47. [CrossRef]
- Head, P. E.; Meier, J. L.; Venditti, C. P., New insights into the pathophysiology of methylmalonic acidemia. J Inherit Metab Dis 2023, 46 (3), 436-449. [CrossRef]
- Forny, P.; Hörster, F.; Ballhausen, D.; Chakrapani, A.; Chapman, K. A.; Dionisi-Vici, C.; Dixon, M.; Grünert, S. C.; Grunewald, S.; Haliloglu, G., et al., Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: First revision. J Inherit Metab Dis 2021, 44 (3), 566-592. [CrossRef]
- Manoli, I.; Sloan, J. L.; Venditti, C. P., Isolated Methylmalonic Acidemia. In GeneReviews(®), Adam, M. P.; Feldman, J.; Mirzaa, G. M.; Pagon, R. A.; Wallace, S. E.; Amemiya, A., Eds. University of Washington, Seattle Copyright © 1993-2025, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.: Seattle (WA), 1993.
- Zhou, X.; Cui, Y.; Han, J., Methylmalonic acidemia: Current status and research priorities. Intractable Rare Dis Res 2018, 7 (2), 73-78. [CrossRef]
- Sen, K.; Burrage, L. C.; Chapman, K. A.; Ginevic, I.; Mazariegos, G. V.; Graham, B. H., Solid organ transplantation in methylmalonic acidemia and propionic acidemia: A points to consider statement of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine 2023, 25 (2). [CrossRef]
- Dao, M.; Arnoux, J.-B.; Bienaimé, F.; Brassier, A.; Brazier, F.; Benoist, J.-F.; Pontoizeau, C.; Ottolenghi, C.; Krug, P.; Boyer, O., et al., Long-term renal outcome in methylmalonic acidemia in adolescents and adults. Orphanet Journal of Rare Diseases 2021, 16 (1), 220. [CrossRef]
- Marelli, C.; Fouilhoux, A.; Benoist, J.-F.; De Lonlay, P.; Guffon-Fouilhoux, N.; Brassier, A.; Cano, A.; Chabrol, B.; Pennisi, A.; Schiff, M., et al., Very long-term outcomes in 23 patients with cblA type methylmalonic acidemia. Journal of Inherited Metabolic Disease 2022, 45 (5), 937-951. [CrossRef]
- Morton, D. H.; Strauss, K. A.; Robinson, D. L.; Puffenberger, E. G.; Kelley, R. I., Diagnosis and treatment of maple syrup disease: a study of 36 patients. Pediatrics 2002, 109 (6), 999-1008. [CrossRef]
- Frazier, D. M.; Allgeier, C.; Homer, C.; Marriage, B. J.; Ogata, B.; Rohr, F.; Splett, P. L.; Stembridge, A.; Singh, R. H., Nutrition management guideline for maple syrup urine disease: An evidence- and consensus-based approach. Molecular Genetics and Metabolism 2014, 112 (3), 210-217. [CrossRef]
- Ziadlou, M.; MacDonald, A., Alternative sources of valine and isoleucine for prompt reduction of plasma leucine in maple syrup urine disease patients: A case series. JIMD Rep 2022, 63 (6), 555-562. [CrossRef]
- Nyhan, W. L.; Rice-Kelts, M.; Klein, J.; Barshop, B. A., Treatment of the acute crisis in maple syrup urine disease. Arch Pediatr Adolesc Med 1998, 152 (6), 593-8. [CrossRef]
- O'Reilly, D.; Crushell, E.; Hughes, J.; Ryan, S.; Rogers, Y.; Borovickova, I.; Mayne, P.; Riordan, M.; Awan, A.; Carson, K., et al., Maple syrup urine disease: Clinical outcomes, metabolic control, and genotypes in a screened population after four decades of newborn bloodspot screening in the Republic of Ireland. J Inherit Metab Dis 2021, 44 (3), 639-655. [CrossRef]
- Blackburn, P. R.; Gass, J. M.; Vairo, F. P. E.; Farnham, K. M.; Atwal, H. K.; Macklin, S.; Klee, E. W.; Atwal, P. S., Maple syrup urine disease: mechanisms and management. Appl Clin Genet 2017, 10, 57-66. [CrossRef]
- Coughlin, C. R., 2nd; Swanson, M. A.; Kronquist, K.; Acquaviva, C.; Hutchin, T.; Rodríguez-Pombo, P.; Väisänen, M. L.; Spector, E.; Creadon-Swindell, G.; Brás-Goldberg, A. M., et al., The genetic basis of classic nonketotic hyperglycinemia due to mutations in GLDC and AMT. Genet Med 2017, 19 (1), 104-111. [CrossRef]
- Hennermann, J. B.; Berger, J. M.; Grieben, U.; Scharer, G.; Van Hove, J. L., Prediction of long-term outcome in glycine encephalopathy: a clinical survey. J Inherit Metab Dis 2012, 35 (2), 253-61. [CrossRef]
- Nowak, M.; Chuchra, P.; Paprocka, J., Nonketotic Hyperglycinemia: Insight into Current Therapies. J Clin Med 2022, 11 (11). [CrossRef]
- Forget, P.; le Polain de Waroux, B.; Wallemacq, P.; Gala, J. L., Life-threatening dextromethorphan intoxication associated with interaction with amitriptyline in a poor CYP2D6 metabolizer: a single case re-exposure study. J Pain Symptom Manage 2008, 36 (1), 92-6. [CrossRef]
- Wiltshire, E. J.; Poplawski, N. K.; Harrison, J. R.; Fletcher, J. M., Treatment of late-onset nonketotic hyperglycinaemia: effectiveness of imipramine and benzoate. J Inherit Metab Dis 2000, 23 (1), 15-21. [CrossRef]
- Van Hove, J. L.; Vande Kerckhove, K.; Hennermann, J. B.; Mahieu, V.; Declercq, P.; Mertens, S.; De Becker, M.; Kishnani, P. S.; Jaeken, J., Benzoate treatment and the glycine index in nonketotic hyperglycinaemia. J Inherit Metab Dis 2005, 28 (5), 651-63. [CrossRef]
- Van Hove, J. L. K.; Coughlin, C., II; Swanson, M.; Hennermann, J. B., Nonketotic Hyperglycinemia. In GeneReviews(®), Adam, M. P.; Feldman, J.; Mirzaa, G. M.; Pagon, R. A.; Wallace, S. E.; Amemiya, A., Eds. University of Washington, Seattle Copyright © 1993-2025, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.: Seattle (WA), 1993.
- Coughlin, C. R., 2nd; Tseng, L. A.; Abdenur, J. E.; Ashmore, C.; Boemer, F.; Bok, L. A.; Boyer, M.; Buhas, D.; Clayton, P. T.; Das, A., et al., Consensus guidelines for the diagnosis and management of pyridoxine-dependent epilepsy due to α-aminoadipic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis 2021, 44 (1), 178-192. [CrossRef]
- Tabatabaie, L.; Klomp, L. W.; Rubio-Gozalbo, M. E.; Spaapen, L. J.; Haagen, A. A.; Dorland, L.; de Koning, T. J., Expanding the clinical spectrum of 3-phosphoglycerate dehydrogenase deficiency. J Inherit Metab Dis 2011, 34 (1), 181-4. [CrossRef]
- van der Crabben, S. N.; Verhoeven-Duif, N. M.; Brilstra, E. H.; Van Maldergem, L.; Coskun, T.; Rubio-Gozalbo, E.; Berger, R.; de Koning, T. J., An update on serine deficiency disorders. J Inherit Metab Dis 2013, 36 (4), 613-9. [CrossRef]
- Coughlin Ii, C. R.; Gospe Jr, S. M., Pyridoxine-dependent epilepsy: Current perspectives and questions for future research. Annals of the Child Neurology Society 2023, 1 (1), 24-37. [CrossRef]
- Coughlin, C. R., 2nd; Swanson, M. A.; Spector, E.; Meeks, N. J. L.; Kronquist, K. E.; Aslamy, M.; Wempe, M. F.; van Karnebeek, C. D. M.; Gospe, S. M., Jr.; Aziz, V. G., et al., The genotypic spectrum of ALDH7A1 mutations resulting in pyridoxine dependent epilepsy: A common epileptic encephalopathy. J Inherit Metab Dis 2019, 42 (2), 353-361. [CrossRef]
- Eggermann, T.; Venghaus, A.; Zerres, K., Cystinuria: an inborn cause of urolithiasis. Orphanet Journal of Rare Diseases 2012, 7 (1), 19. [CrossRef]
- Pak, C. Y.; Fuller, C.; Sakhaee, K.; Zerwekh, J. E.; Adams, B. V., Management of cystine nephrolithiasis with alpha-mercaptopropionylglycine. J Urol 1986, 136 (5), 1003-8. [CrossRef]
- Gillion, V.; Saussez, T. P.; Van Nieuwenhove, S.; Jadoul, M., Extremely rapid stone formation in cystinuria: look out for dietary supplements! Clin Kidney J 2021, 14 (6), 1694-1696. [CrossRef]
- Sahota, A.; Tischfield, J. A.; Goldfarb, D. S.; Ward, M. D.; Hu, L., Cystinuria: genetic aspects, mouse models, and a new approach to therapy. Urolithiasis 2019, 47 (1), 57-66. [CrossRef]
- Sadiq, S.; Cil, O., Cystinuria: An Overview of Diagnosis and Medical Management. Turk Arch Pediatr 2022, 57 (4), 377-384. [CrossRef]
- Sebastio, G.; Sperandeo, M. P.; Andria, G., Lysinuric protein intolerance: reviewing concepts on a multisystem disease. Am J Med Genet C Semin Med Genet 2011, 157c (1), 54-62. [CrossRef]
- Piña-Garza, J. E.; James, K. C., Ataxia. In Fenichel's Clinical Pediatric Neurology (Eighth Edition), 8 ed.; Piña-Garza, J. E.; James, K. C., Eds. Elsevier: Philadelphia, 2019; pp 218-237.
- Nyhan, W. L.; Rice-Kelts, M.; Klein, J.; Barshop, B. A., Treatment of the Acute Crisis in Maple Syrup Urine Disease. Archives of Pediatrics & Adolescent Medicine 1998, 152 (6), 593-598. [CrossRef]
- O'Reilly, D.; Crushell, E.; Hughes, J.; Ryan, S.; Rogers, Y.; Borovickova, I.; Mayne, P.; Riordan, M.; Awan, A.; Carson, K., et al., Maple syrup urine disease: Clinical outcomes, metabolic control, and genotypes in a screened population after four decades of newborn bloodspot screening in the Republic of Ireland. Journal of Inherited Metabolic Disease 2021, 44 (3), 639-655. [CrossRef]
- Rajantie, J.; Simell, O.; Rapola, J.; Perheentupa, J., Lysinuric protein intolerance: a two-year trial of dietary supplementation therapy with citrulline and lysine. J Pediatr 1980, 97 (6), 927-32. [CrossRef]
- Kleta, R.; Romeo, E.; Ristic, Z.; Ohura, T.; Stuart, C.; Arcos-Burgos, M.; Dave, M. H.; Wagner, C. A.; Camargo, S. R. M.; Inoue, S., et al., Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nature Genetics 2004, 36 (9), 999-1002. [CrossRef]
- Kravetz, Z.; Schmidt-Kastner, R., New aspects for the brain in Hartnup disease based on mining of high-resolution cellular mRNA expression data for SLC6A19. IBRO Neuroscience Reports 2023, 14, 393-397. [CrossRef]
- Patel, A. B.; Prabhu, A. S., Hartnup disease. Indian J Dermatol 2008, 53 (1), 31-2. [CrossRef]
- Bachmann, T.; Faust, H.; Abou Jamra, R.; Pott, C.; Kluge, M.; Rumpf, J. J.; Then Bergh, F.; Beblo, S., Adult Neuropsychiatric Manifestation of Hartnup Disease With a Novel SLCA6A19 Variant: A Case Report. Neurol Genet 2024, 10 (6), e200195. [CrossRef]
- Hashmi, M. S.; Gupta, V., Hartnup Disease. In StatPearls, StatPearls Publishing Copyright © 2025, StatPearls Publishing LLC.: Treasure Island (FL), 2025.
- Kölker, S.; Christensen, E.; Leonard, J. V.; Greenberg, C. R.; Boneh, A.; Burlina, A. B.; Burlina, A. P.; Dixon, M.; Duran, M.; García Cazorla, A., et al., Diagnosis and management of glutaric aciduria type I--revised recommendations. J Inherit Metab Dis 2011, 34 (3), 677-94. [CrossRef]
- Rai, S. P., Glutaric aciduria type1: CT diagnosis. J Pediatr Neurosci 2009, 4 (2), 143. [CrossRef]
- Vester, M. E. M.; Bilo, R. A. C.; Karst, W. A.; Daams, J. G.; Duijst, W. L. J. M.; van Rijn, R. R., Subdural hematomas: glutaric aciduria type 1 or abusive head trauma? A systematic review. Forensic Science, Medicine, and Pathology 2015, 11 (3), 405-415. [CrossRef]
- Sen, A.; Pillay, R. S., Striatal necrosis in type 1 glutaric aciduria: Different stages in two siblings. Journal of Pediatric Neurosciences 2011, 6 (2).
- Lipkin, P. H.; Roe, C. R.; Goodman, S. I.; Batshaw, M. L., A case of glutaric acidemia type I: Effect of riboflavin and carnitine. The Journal of Pediatrics 1988, 112 (1), 62-65. [CrossRef]
- Seccombe, D. W.; James, L.; Booth, F., L-Carnitine treatment in glutaric aciduria type I. Neurology 1986, 36 (2), 264-264. [CrossRef]
- Kölker, S.; Garbade, S. F.; Boy, N.; Maier, E. M.; Meissner, T.; Mühlhausen, C.; Hennermann, J. B.; Lücke, T.; Häberle, J.; Baumkötter, J., et al., Decline of Acute Encephalopathic Crises in Children with Glutaryl-CoA Dehydrogenase Deficiency Identified by Newborn Screening in Germany. Pediatric Research 2007, 62 (3), 357-363. [CrossRef]
- Strauss, K. A.; Puffenberger, E. G.; Robinson, D. L.; Morton, D. H., Type I glutaric aciduria, part 1: Natural history of 77 patients. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 2003, 121C (1), 38-52. [CrossRef]
- Boy, N.; Mühlhausen, C.; Maier, E. M.; Ballhausen, D.; Baumgartner, M. R.; Beblo, S.; Burgard, P.; Chapman, K. A.; Dobbelaere, D.; Heringer-Seifert, J., et al., Recommendations for diagnosing and managing individuals with glutaric aciduria type 1: Third revision. J Inherit Metab Dis 2023, 46 (3), 482-519. [CrossRef]
- Foran, J.; Moore, M.; Crushell, E.; Knerr, I.; McSweeney, N., Low excretor glutaric aciduria type 1 of insidious onset with dystonia and atypical clinical features, a diagnostic dilemma. JIMD Rep 2021, 58 (1), 12-20. [CrossRef]
- Tondo, M.; Calpena, E.; Arriola, G.; Sanz, P.; Martorell, L.; Ormazabal, A.; Castejon, E.; Palacin, M.; Ugarte, M.; Espinos, C., et al., Clinical, biochemical, molecular and therapeutic aspects of 2 new cases of 2-aminoadipic semialdehyde synthase deficiency. Molecular Genetics and Metabolism 2013, 110 (3), 231-236. [CrossRef]
- Houten, S. M.; te Brinke, H.; Denis, S.; Ruiter, J. P. N.; Knegt, A. C.; de Klerk, J. B. C.; Augoustides-Savvopoulou, P.; Häberle, J.; Baumgartner, M. R.; Coşkun, T., et al., Genetic basis of hyperlysinemia. Orphanet Journal of Rare Diseases 2013, 8 (1), 57. [CrossRef]
- Marinella, G.; Pascarella, F.; Vetro, A.; Bonuccelli, A.; Pochiero, F.; Santangelo, A.; Alessandrì, M. G.; Pasquariello, R.; Orsini, A.; Battini, R., Hyperlysinemia, an ultrarare inborn error of metabolism: Review and update. Seizure - European Journal of Epilepsy 2024, 120, 135-141. [CrossRef]
- Ferreira, C. R.; van Karnebeek, C. D. M., Inborn errors of metabolism. Handb Clin Neurol 2019, 162, 449-481. [CrossRef]
- Hart, C. E.; Race, V.; Achouri, Y.; Wiame, E.; Sharrard, M.; Olpin, S. E.; Watkinson, J.; Bonham, J. R.; Jaeken, J.; Matthijs, G., et al., Phosphoserine aminotransferase deficiency: a novel disorder of the serine biosynthesis pathway. Am J Hum Genet 2007, 80 (5), 931-7. [CrossRef]
- Guilmatre, A.; Legallic, S.; Steel, G.; Willis, A.; Di Rosa, G.; Goldenberg, A.; Drouin-Garraud, V.; Guet, A.; Mignot, C.; Des Portes, V., et al., Type I hyperprolinemia: genotype/phenotype correlations. Hum Mutat 2010, 31 (8), 961-5. [CrossRef]
- Bender, H. U.; Almashanu, S.; Steel, G.; Hu, C. A.; Lin, W. W.; Willis, A.; Pulver, A.; Valle, D., Functional consequences of PRODH missense mutations. Am J Hum Genet 2005, 76 (3), 409-20. [CrossRef]
- Clelland, C. L.; Read, L. L.; Baraldi, A. N.; Bart, C. P.; Pappas, C. A.; Panek, L. J.; Nadrich, R. H.; Clelland, J. D., Evidence for association of hyperprolinemia with schizophrenia and a measure of clinical outcome. Schizophr Res 2011, 131 (1-3), 139-45. [CrossRef]
- Mitsubuchi, H.; Nakamura, K.; Matsumoto, S.; Endo, F., Biochemical and clinical features of hereditary hyperprolinemia. Pediatrics International 2014, 56 (4), 492-496. [CrossRef]
- Namavar, Y.; Duineveld, D. J.; Both, G. I. A.; Fiksinski, A. M.; Vorstman, J. A. S.; Verhoeven-Duif, N. M.; Zinkstok, J. R., Psychiatric phenotypes associated with hyperprolinemia: A systematic review. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 2021, 186 (5), 289-317. [CrossRef]
- Martinelli, D.; Häberle, J.; Rubio, V.; Giunta, C.; Hausser, I.; Carrozzo, R.; Gougeard, N.; Marco-Marín, C.; Goffredo, B. M.; Meschini, M. C., et al., Understanding pyrroline-5-carboxylate synthetase deficiency: clinical, molecular, functional, and expression studies, structure-based analysis, and novel therapy with arginine. J Inherit Metab Dis 2012, 35 (5), 761-76. [CrossRef]
- van de Ven, S.; Gardeitchik, T.; Kouwenberg, D.; Kluijtmans, L.; Wevers, R.; Morava, E., Long-term clinical outcome, therapy and mild mitochondrial dysfunction in hyperprolinemia. J Inherit Metab Dis 2014, 37 (3), 383-90. [CrossRef]
- Balfoort, B. M.; Buijs, M. J. N.; ten Asbroek, A. L. M. A.; Bergen, A. A. B.; Boon, C. J. F.; Ferreira, E. A.; Houtkooper, R. H.; Wagenmakers, M. A. E. M.; Wanders, R. J. A.; Waterham, H. R., et al., A review of treatment modalities in gyrate atrophy of the choroid and retina (GACR). Molecular Genetics and Metabolism 2021, 134 (1), 96-116. [CrossRef]
- Häberle, J.; Boddaert, N.; Burlina, A.; Chakrapani, A.; Dixon, M.; Huemer, M.; Karall, D.; Martinelli, D.; Crespo, P. S.; Santer, R., et al., Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis 2012, 7, 32. [CrossRef]
- Häberle, J.; Shahbeck, N.; Ibrahim, K.; Hoffmann, G. F.; Ben-Omran, T., Natural course of glutamine synthetase deficiency in a 3year old patient. Molecular Genetics and Metabolism 2011, 103 (1), 89-91. [CrossRef]
- Spodenkiewicz, M.; Diez-Fernandez, C.; Rüfenacht, V.; Gemperle-Britschgi, C.; Häberle, J., Minireview on Glutamine Synthetase Deficiency, an Ultra-Rare Inborn Error of Amino Acid Biosynthesis. Biology (Basel) 2016, 5 (4). [CrossRef]
- Häberle, J.; Shahbeck, N.; Ibrahim, K.; Schmitt, B.; Scheer, I.; O’Gorman, R.; Chaudhry, F. A.; Ben-Omran, T., Glutamine supplementation in a child with inherited GS deficiency improves the clinical status and partially corrects the peripheral and central amino acid imbalance. Orphanet Journal of Rare Diseases 2012, 7 (1), 48. [CrossRef]
- Alfadhel, M.; Alrifai, M. T.; Trujillano, D.; Alshaalan, H.; Al Othaim, A.; Al Rasheed, S.; Assiri, H.; Alqahtani, A. A.; Alaamery, M.; Rolfs, A., et al., Asparagine Synthetase Deficiency: New Inborn Errors of Metabolism. JIMD Rep 2015, 22, 11-6. [CrossRef]
- Sacharow, S. J.; Dudenhausen, E. E.; Lomelino, C. L.; Rodan, L.; El Achkar, C. M.; Olson, H. E.; Genetti, C. A.; Agrawal, P. B.; McKenna, R.; Kilberg, M. S., Characterization of a novel variant in siblings with Asparagine Synthetase Deficiency. Molecular Genetics and Metabolism 2018, 123 (3), 317-325. [CrossRef]
- van Wegberg, A. M. J.; MacDonald, A.; Ahring, K.; Bélanger-Quintana, A.; Beblo, S.; Blau, N.; Bosch, A. M.; Burlina, A.; Campistol, J.; Coşkun, T., et al., European guidelines on diagnosis and treatment of phenylketonuria: First revision. Molecular Genetics and Metabolism 2025, 145 (2), 109125. [CrossRef]
- Altman, G.; Hussain, K.; Green, D.; Strauss, B. J. G.; Wilcox, G., Mental health diagnoses in adults with phenylketonuria: a retrospective systematic audit in a large UK single centre. Orphanet Journal of Rare Diseases 2021, 16 (1), 520. [CrossRef]
- ten Hoedt, A. E.; de Sonneville, L. M.; Francois, B.; ter Horst, N. M.; Janssen, M. C.; Rubio-Gozalbo, M. E.; Wijburg, F. A.; Hollak, C. E.; Bosch, A. M., High phenylalanine levels directly affect mood and sustained attention in adults with phenylketonuria: a randomised, double-blind, placebo-controlled, crossover trial. J Inherit Metab Dis 2011, 34 (1), 165-71. [CrossRef]
- Gersting, S. W.; Staudigl, M.; Truger, M. S.; Messing, D. D.; Danecka, M. K.; Sommerhoff, C. P.; Kemter, K. F.; Muntau, A. C., Activation of phenylalanine hydroxylase induces positive cooperativity toward the natural cofactor. J Biol Chem 2010, 285 (40), 30686-97. [CrossRef]
- Trefz, F. K.; Burton, B. K.; Longo, N.; Casanova, M. M.; Gruskin, D. J.; Dorenbaum, A.; Kakkis, E. D.; Crombez, E. A.; Grange, D. K.; Harmatz, P., et al., Efficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: a phase III, randomized, double-blind, placebo-controlled study. J Pediatr 2009, 154 (5), 700-7. [CrossRef]
- Camp, K. M.; Parisi, M. A.; Acosta, P. B.; Berry, G. T.; Bilder, D. A.; Blau, N.; Bodamer, O. A.; Brosco, J. P.; Brown, C. S.; Burlina, A. B., et al., Phenylketonuria Scientific Review Conference: state of the science and future research needs. Mol Genet Metab 2014, 112 (2), 87-122. [CrossRef]
- Burton, B. K.; Grange, D. K.; Milanowski, A.; Vockley, G.; Feillet, F.; Crombez, E. A.; Abadie, V.; Harding, C. O.; Cederbaum, S.; Dobbelaere, D., et al., The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): a phase II, multicentre, open-label, screening study. J Inherit Metab Dis 2007, 30 (5), 700-7. [CrossRef]
- Hennermann, J. B.; Roloff, S.; Gebauer, C.; Vetter, B.; von Arnim-Baas, A.; Mönch, E., Long-term treatment with tetrahydrobiopterin in phenylketonuria: treatment strategies and prediction of long-term responders. Mol Genet Metab 2012, 107 (3), 294-301. [CrossRef]
- Scala, I.; Concolino, D.; Casa, R. D.; Nastasi, A.; Ungaro, C.; Paladino, S.; Capaldo, B.; Ruoppolo, M.; Daniele, A.; Bonapace, G., et al., Long-term follow-up of patients with phenylketonuria treated with tetrahydrobiopterin: a seven years experience. Orphanet Journal of Rare Diseases 2015, 10 (1), 14. [CrossRef]
- Hydery, T.; Coppenrath, V. A., A Comprehensive Review of Pegvaliase, an Enzyme Substitution Therapy for the Treatment of Phenylketonuria. Drug Target Insights 2019, 13, 1177392819857089. [CrossRef]
- Hausmann, O.; Daha, M.; Longo, N.; Knol, E.; Müller, I.; Northrup, H.; Brockow, K., Pegvaliase: Immunological profile and recommendations for the clinical management of hypersensitivity reactions in patients with phenylketonuria treated with this enzyme substitution therapy. Mol Genet Metab 2019, 128 (1-2), 84-91. [CrossRef]
- Harding, C. O.; Amato, R. S.; Stuy, M.; Longo, N.; Burton, B. K.; Posner, J.; Weng, H. H.; Merilainen, M.; Gu, Z.; Jiang, J., et al., Pegvaliase for the treatment of phenylketonuria: A pivotal, double-blind randomized discontinuation Phase 3 clinical trial. Mol Genet Metab 2018, 124 (1), 20-26. [CrossRef]
- Cunningham, A.; Rohr, F.; Splett, P.; Mofidi, S.; Bausell, H.; Stembridge, A.; Kenneson, A.; Singh, R. H., Nutrition management of PKU with pegvaliase therapy: update of the web-based PKU nutrition management guideline recommendations. Orphanet Journal of Rare Diseases 2023, 18 (1), 155. [CrossRef]
- Bosch, A. M.; Burlina, A.; Cunningham, A.; Bettiol, E.; Moreau-Stucker, F.; Koledova, E.; Benmedjahed, K.; Regnault, A., Assessment of the impact of phenylketonuria and its treatment on quality of life of patients and parents from seven European countries. Orphanet J Rare Dis 2015, 10, 80. [CrossRef]
- Bik-Multanowski, M.; Didycz, B.; Mozrzymas, R.; Nowacka, M.; Kaluzny, L.; Cichy, W.; Schneiberg, B.; Amilkiewicz, J.; Bilar, A.; Gizewska, M., et al., Quality of life in noncompliant adults with phenylketonuria after resumption of the diet. J Inherit Metab Dis 2008, 31 Suppl 2, S415-8. [CrossRef]
- Dawson, C.; Murphy, E.; Maritz, C.; Chan, H.; Ellerton, C.; Carpenter, R. H.; Lachmann, R. H., Dietary treatment of phenylketonuria: the effect of phenylalanine on reaction time. J Inherit Metab Dis 2011, 34 (2), 449-54. [CrossRef]
- Dawson, C.; Murphy, E.; Maritz, C.; Chan, H.; Ellerton, C.; Carpenter, R. H. S.; Lachmann, R. H., Dietary treatment of phenylketonuria: the effect of phenylalanine on reaction time. Journal of Inherited Metabolic Disease 2011, 34 (2), 449-454. [CrossRef]
- Fernstrom, J. D.; Fernstrom, M. H., Tyrosine, Phenylalanine, and Catecholamine Synthesis and Function in the Brain123. The Journal of Nutrition 2007, 137 (6), 1539S-1547S. [CrossRef]
- Sara, G.-L.; Alejandra, L.-M. L.; Isabel, I.-G.; Marcela, V.-A., Conventional Phenylketonuria Treatment. Journal of Inborn Errors of Metabolism and Screening 2016, 4, 2326409816685733. [CrossRef]
- Acosta, P.; Matalon, K. M., Nutrition management of patients with inherited disorders of aromatic amino acid metabolism. In Nutrition Management of Patients with Inherited Metabolic Disorders, Acosta, P. B., Ed. Jones and Bartlett Publishers: Sudbury, Massachusetts, 2010; pp 119-153.
- Webster, D.; Wildgoose, J., Tyrosine supplementation for phenylketonuria. Cochrane Database Syst Rev 2013, 2013 (6), Cd001507. [CrossRef]
- Remmington, T.; Smith, S., Tyrosine supplementation for phenylketonuria. Cochrane Database Syst Rev 2021, 1 (1), Cd001507. [CrossRef]
- Orzincolo, C.; Castaldi, G.; Scutellari, P. N.; Cicognani, P.; Bariani, L.; Feggi, L., [Ochronotic arthropathy in alkaptonuria. Radiological manifestations and physiopathological signs]. Radiol Med 1988, 75 (5), 476-81.
- Ranga, U.; Aiyappan, S. K.; Shanmugam, N.; Veeraiyan, S., Ochronotic spondyloarthropathy. J Clin Diagn Res 2013, 7 (2), 403-4. [CrossRef]
- Ebrahim, I. C.; Hoang, T. D.; Vietor, N. O.; Schacht, J. P.; Shakir, M. K. M., Dilemmas in the diagnosis and management of osteoporosis in a patient with alkaptonuria: Successful treatment with teriparatide. Clinical Case Reports 2022, 10 (12), e6729. [CrossRef]
- Wolff, J. A.; Barshop, B.; Nyhan, W. L.; Leslie, J.; Seegmiller, J. E.; Gruber, H.; Garst, M.; Winter, S.; Michals, K.; Matalon, R., Effects of Ascorbic Acid in Alkaptonuria: Alterations in Benzoquinone Acetic Acid and an Ontogenic Effect in Infancy. Pediatric Research 1989, 26 (2), 140-144. [CrossRef]
- Feier, F.; Schwartz, I. V.; Benkert, A. R.; Seda Neto, J.; Miura, I.; Chapchap, P.; da Fonseca, E. A.; Vieira, S.; Zanotelli, M. L.; Pinto e Vairo, F., et al., Living related versus deceased donor liver transplantation for maple syrup urine disease. Mol Genet Metab 2016, 117 (3), 336-43. [CrossRef]
- Pontoizeau, C.; Simon-Sola, M.; Gaborit, C.; Nguyen, V.; Rotaru, I.; Tual, N.; Colella, P.; Girard, M.; Biferi, M. G.; Arnoux, J. B., et al., Neonatal gene therapy achieves sustained disease rescue of maple syrup urine disease in mice. Nat Commun 2022, 13 (1), 3278. [CrossRef]
- Walther, F.; Radke, M.; KrÜGer, G.; Hobusch, D.; Uhlemann, M.; Tittelbach-Helmrich, W.; Stolpe, H. J., Response to sodium benzoate treatment in non-ketotic hyperglycinaemia. Pediatrics International 1994, 36 (1), 75-79. [CrossRef]
- Shelkowitz, E.; Saneto, R. P.; Al-Hertani, W.; Lubout, C. M. A.; Stence, N. V.; Brown, M. S.; Long, P.; Walleigh, D.; Nelson, J. A.; Perez, F. E., et al., Ketogenic diet as a glycine lowering therapy in nonketotic hyperglycinemia and impact on brain glycine levels. Orphanet Journal of Rare Diseases 2022, 17 (1), 423. [CrossRef]
- Bjoraker, K. J.; Swanson, M. A.; Coughlin, C. R., 2nd; Christodoulou, J.; Tan, E. S.; Fergeson, M.; Dyack, S.; Ahmad, A.; Friederich, M. W.; Spector, E. B., et al., Neurodevelopmental Outcome and Treatment Efficacy of Benzoate and Dextromethorphan in Siblings with Attenuated Nonketotic Hyperglycinemia. J Pediatr 2016, 170, 234-9. [CrossRef]
- Lee, N.; Kim, D., Toxic Metabolites and Inborn Errors of Amino Acid Metabolism: What One Informs about the Other. Metabolites 2022, 12 (6). [CrossRef]
- van Karnebeek, C. D.; Hartmann, H.; Jaggumantri, S.; Bok, L. A.; Cheng, B.; Connolly, M.; Coughlin, C. R., 2nd; Das, A. M.; Gospe, S. M., Jr.; Jakobs, C., et al., Lysine restricted diet for pyridoxine-dependent epilepsy: first evidence and future trials. Mol Genet Metab 2012, 107 (3), 335-44. [CrossRef]
- Coughlin, C. R.; Tseng, L. A.; Bok, L. A.; Hartmann, H.; Footitt, E.; Striano, P.; Tabarki, B. M.; Lunsing, R. J.; Stockler-Ipsiroglu, S.; Gordon, S., et al., Association Between Lysine Reduction Therapies and Cognitive Outcomes in Patients With Pyridoxine-Dependent Epilepsy. Neurology 2022, 99 (23), e2627-e2636. [CrossRef]
- Kölker, S.; Boy, S. P. N.; Heringer, J.; Müller, E.; Maier, E. M.; Ensenauer, R.; Mühlhausen, C.; Schlune, A.; Greenberg, C. R.; Koeller, D. M., et al., Complementary dietary treatment using lysine-free, arginine-fortified amino acid supplements in glutaric aciduria type I — A decade of experience. Molecular Genetics and Metabolism 2012, 107 (1), 72-80. [CrossRef]
- Segur-Bailach, E.; Mateu-Bosch, A.; Bofill-De Ros, X.; Parés, M.; da Silva Buttkus, P.; Rathkolb, B.; Gailus-Durner, V.; Hrabě de Angelis, M.; Moeini, P.; Gonzalez-Aseguinolaza, G., et al., Therapeutic AASS inhibition by AAV-miRNA rescues glutaric aciduria type I severe phenotype in mice. Mol Ther 2025. [CrossRef]
Figure 1.
(a) Pathways of phenylalanine and tyrosine metabolism. “PAH” denotes phenylalanine hydroxylase; tyrosine aminotransferase (TAT); 4-hydroxyphenylpyruvate dioxygenase (HPD); homogentisate 1,2-dioxygenase (HGD); maleylacetoacetate isomerase (MAAI), which converts maleylacetoacetate (MAA) to fumarylacetoacetate (FAA); and fumarylacetoacetate hydrolase (FAH). (b) Pathways of sulphur-containing amino acid metabolism. “MS” denotes methionine synthase; cystathionine-β-synthase (CBS); tetrahydrofolate (THF); methylene tetrahydrofolate reductase (MTHFR); methionine S-adenosyltransferase (MAT); and S-adenosylhomocysteine hydrolase (SAHH).
Figure 1.
(a) Pathways of phenylalanine and tyrosine metabolism. “PAH” denotes phenylalanine hydroxylase; tyrosine aminotransferase (TAT); 4-hydroxyphenylpyruvate dioxygenase (HPD); homogentisate 1,2-dioxygenase (HGD); maleylacetoacetate isomerase (MAAI), which converts maleylacetoacetate (MAA) to fumarylacetoacetate (FAA); and fumarylacetoacetate hydrolase (FAH). (b) Pathways of sulphur-containing amino acid metabolism. “MS” denotes methionine synthase; cystathionine-β-synthase (CBS); tetrahydrofolate (THF); methylene tetrahydrofolate reductase (MTHFR); methionine S-adenosyltransferase (MAT); and S-adenosylhomocysteine hydrolase (SAHH).
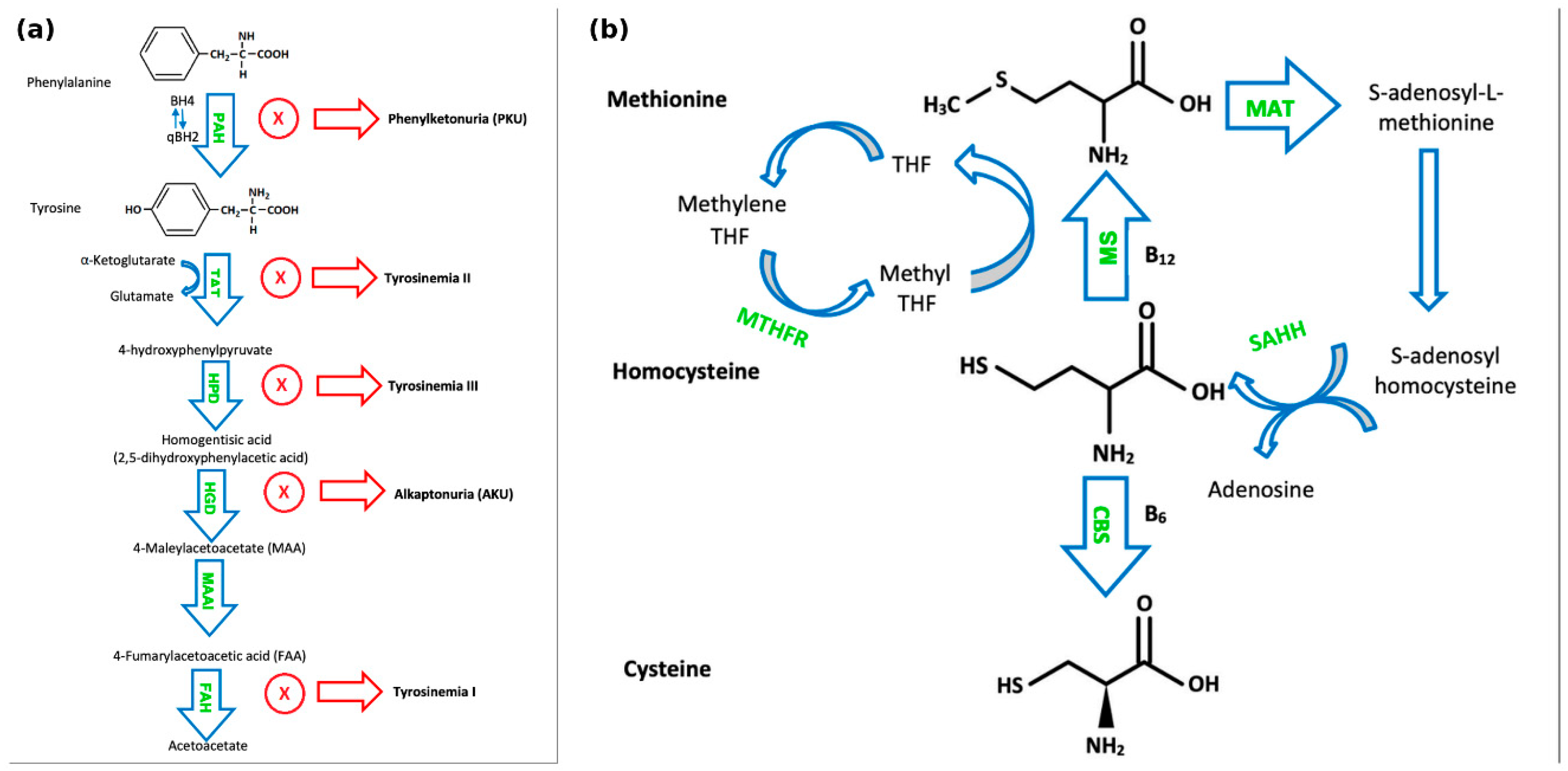
Figure 2.
(a) Pathways of branched-chain amino acid metabolism. “BCKDC” denotes branched-chain α-keto acid dehydrogenase complex. (b) Pathways of lysine, hydroxylysine, and tryptophan metabolism. “GCDH” denotes glutaryl-CoA dehydrogenase.
Figure 2.
(a) Pathways of branched-chain amino acid metabolism. “BCKDC” denotes branched-chain α-keto acid dehydrogenase complex. (b) Pathways of lysine, hydroxylysine, and tryptophan metabolism. “GCDH” denotes glutaryl-CoA dehydrogenase.
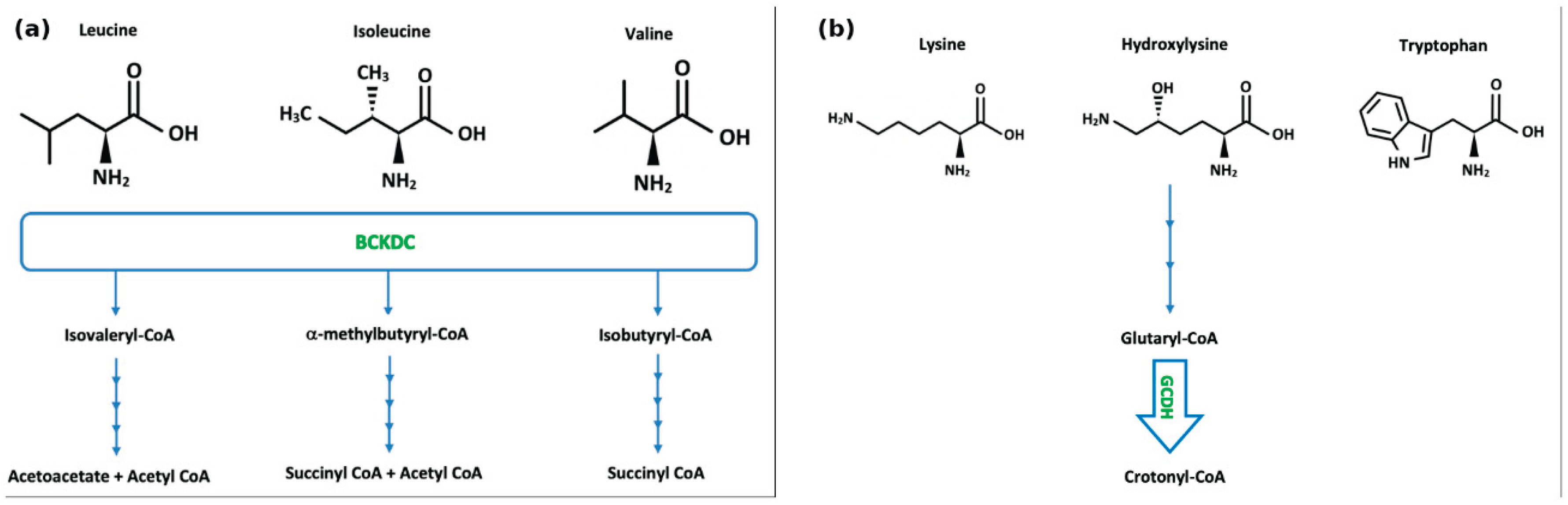

Table 1.
Medical and dietetic management of Phenylalanine and Tyrosine disorders.
| Disorder | Treatment | Rationale/ Mechanism |
Dose | Biochemical Monitoring |
|---|---|---|---|---|
| Phenylketonuria (PKU) | Phenylalanine-free amino acid supplements Dietary restriction of phenylalanine |
To limit intake of offending amino acid | Small, frequent doses (3-4) spaced evenly across day [10]. | Blood phenylalanine levels |
| Sapropterin dihydrochloride (Kuvan®) | Synthetic form of cofactor tetrahydrobiopterin (BH4). | Recommended starting dose in patients is 10 mg/kg body weight/day. Dose is adjusted, usually between 5-20 mg/kg/day, to achieve and maintain blood Phe control [11,12,13]. | Blood phenylalanine levels |
|
| Pegvaliase (Palynziq®) | Recombinant phenylalanine ammonia lyase (PAL) enzyme (patients ≥ 16 yrs) | Recommended starting dose is 2.5mg once per week for 4 weeks. Dose escalated gradually based on tolerability to daily maintenance dose needed to achieve blood Phe control. Maintenance dose is individualised to achieve blood Phe control [9,14,15]. | Blood phenylalanine levels |
|
|
Biopterin defects causing hyperphenylalaninemia [101] |
Dietary restriction of phenylalanine (GTPCH and DHPR deficiency patients). Phenylalanine-free amino acid supplements. | To limit intake of offending amino acid | Small, frequent doses (3-4) spaced evenly across day [10]. | Blood phenylalanine levels |
| Sapropterin dihydrochloride (Kuvan®) (All BH4 deficiency patients) | Synthetic form of cofactor tetrahydrobiopterin (BH4). | Recommended starting dose in adult patients is 10 mg/kg body weight/day. Dose is adjusted, usually between 5-20 mg/kg/day, to achieve and maintain blood phenylalanine control [8,11,12]. | Blood phenylalanine levels |
|
| l-3,4- dihydroxyphenylalanine/carbidopa (L-DOPA) and 5-OH-Tryptophan |
For neurotransmitter related movement disorders | L-DOPA in 4 divided doses with similar dosing for 5-OH-Tryptophan [16], age-dependent. | LP for CSF neurotransmitters measurement (HVA, 5-HIAA); prolactin levels. | |
| Folinic acid | For movement disorders, to prevent cerebral folate deficiency | 10-15mg/day [16] | Monitoring of CSF folate and folinic acid status | |
|
Hyperphenylalaninemia due to DNAJC12 |
Dietary restriction of phenylalanine. Phenylalanine-free amino acid supplements | To limit intake of offending amino acid | Small, frequent doses (3-4) spaced evenly across day [10]. | Blood phenylalanine levels |
| Sapropterin dihydrochloride (Kuvan®) | Synthetic form of cofactor tetrahydrobiopterin (BH4). | Recommended starting dose in adult patients is 10 mg/kg body weight/day. Dose is adjusted, usually between 520 mg/kg/day, to achieve and maintain blood Phe control 8, 95, 126. | Blood phenylalanine levels |
|
| L-DOPA and Tryptophan | For neurotransmitter related movement disorder | Starting dose of 2.5mg/kg/day (can be increased to 6mg/kg/day) [17]. |
LP for CSF neurotransmitters measurement (HVA, 5-HIAA) | |
|
Alkaptonuria (AKU) |
Dietary restriction of phenylalanine. Tyrosine/phenylalanine-free amino acid supplements | To limit intake of offending amino acid. | Moderate restriction of natural protein | Plasma amino acids (phenylalanine, tyrosine) |
| Nitisinone (currently Nitisinone is approved for alkaptonuria treatment in adults only) |
Inhibits 4- hydroxyphenylpyruvic acid dioxygenase |
The recommended dose in the adult AKU population is 10 mg once daily [18,19]. | Plasma amino acids (phenylalanine, tyrosine) | |
| Bisphosphonate | Inhibit bone resorption by preventing hydroxyapatite breakdown | As clinically indicated | Bone turnover markers (BTMs) |
|
| Teriparatide | Activates PKA- and PKC-dependent signaling pathways | 20mcg/day SC (approved in adults) | BTMs, plasma calcium levels | |
|
Tyrosinemia type I |
Dietary restriction of phenylalanine and tyrosine. Tyrosine/phenylalanine-free amino acid supplements | To limit intake of offending amino acids | Plasma amino acids (phenylalanine, tyrosine, methionine), liver function; blood/urine succinylacetone |
|
| Nitisinone (Nitisinone is approved for tyrosinemia type I treatment in children) |
Inhibits 4- hydroxyphenylpyruvic acid dioxygenase |
Recommended starting dose in adult patients is 1 mg/kg body weight/day. Dose should be adjusted individually. Maximum of dose of 2 mg/kg body weight/day [20,21]. |
Blood tyrosine levels, blood/urine succinylacetone; NTBC drug levels, liver function, alpha-fetoprotein |
|
| Liver transplant | If end-stage liver disease, liver failure, or hepatocellular carcinoma develops | |||
| Tyrosinemia type II | Dietary restriction of phenylalanine and tyrosine. Tyrosine/phenylalanine-free amino acid supplements | To limit intake of offending amino acids | Blood tyrosine and phenylalanine levels | |
| Tyrosinemia type III | A restrictive tyrosine and phenylalanine diet has been suggested during childhood [20], while other authors argue that such restriction is not recommended |
Table 2.
Medical and dietetic management of Sulphur -containing amino acid disorders.
| Disorder | Treatment | Rationale/mechanism | Dose | Monitoring |
|---|---|---|---|---|
|
Homocystinuria (HCU) due to cystathionine beta-synthase (CBS) deficiency |
Methionine-free amino acid supplements. Dietary restriction of methionine/protein. Supplementation of cysteine, B12, folate. |
To limit intake of offending amino acid. | Individualised to patient. | Methionine and cystine levels. B12, folate. |
| Pyridoxine (Vitamin B6) (In pyridoxine-responsive patients) | Co-factor of cystathionine β-synthase. | Recommended dose of up to 10 mg/kg/day. Recommended to avoid doses >500mg/day (risk of peripheral neuropathy) [38]. | Plasma tHcy. | |
| Betaine | Betaine donates a methyl group via betaine homocysteine methyl transferase (BHMT). | Recommended starting dose of 3g BID. Can increase up to 200mg/kg/day, rarely benefit to higher dose [38]. |
Plasma tHcy. | |
|
Homocystinuria due to methylene tetrahydrofolate reductase deficiency |
Betaine | Betaine donates a methyl group via betaine homocysteine methyl transferase (BHMT). | Recommended starting dose of 3g BID. Can increase up to 200mg/kg/day, rarely benefit to higher dose [38]. |
Plasma tHcy. |
| Aspirin | Antiplatelet therapy post stroke | 40mg/day [41] | Routine monitoring not recommended |
|
| Supplementation of creatine, B6, B12, folate, 5MTHF |
To achieve target plasma tHcy levels. | Creatine (75-100mg/kg/day), B6 (25mg/day), B12 (25mg/day), Folate (4mg/day) 5MTHF (2.4-3.2mg/day) [41] |
Creatinine, B6, B12, folate, 5MTHF levels |
|
|
Methionine S-adenosyltransferase deficiency |
S-adenosyl-L-methionine disulfate tosylate (SAM) supplementation |
For neurological manifestations | 400-800mg BD [42] | SAM concentration in plasma and CSF |
| Methionine-free amino acid supplements. Dietary restriction of methionine/protein |
To limit intake of offending amino acid (*Although may decrease S-adenosyl-L-methionine (SAM) synthesis [108]) | Individualised to patient | Methionine levels. | |
| S-adenosylhomocysteine hydrolase deficiency | Methionine-free amino acid supplements. Dietary restriction of methionine/protein |
To limit intake of offending amino acids. To reduce toxic SAH levels. | Individualised to patient | Methionine levels. |
| Phosphatidylcholine and creatine supplementation | Low levels of creatine and choline in SAH hydrolase deficiency. | Creatine – e.g., 375 mg/kg/d Phosphatidylcholine – e.g., 150mg/kg/d [43] |
Creatinine, choline levels; blood/urine creatine | |
| Cystinosis | Cysteamine | Depletes lysosomal cystine levels | 1.30 g/m2/day; maximum of 1.95 g/m2/day [44] |
WBC cystine assay |
| Symptomatic treatment | Management of symptoms | E.g., ACE inhibitors for proteinuria, kidney transplant in ESRD, HRT for endocrinopathies. |
Depends on symptoms |
Table 3.
Medical and dietetic management of branched-chain amino acid disorders.
| Disorder | Treatment | Rationale/mechanism | Dose | Monitoring | |
|---|---|---|---|---|---|
|
Maple syrup urine disease (MSUD) |
Synthetic formula with all amino acids except leucine, isoleucine, valine. Valine and isoleucine supplementation. Protein-free foods. |
To limit intake of offending amino acids. | Valine:15- 30mg/kg Isoleucine: 10-30mg/kg [57], individualised to patient |
Plasma levels of BCAAs | |
| Thiamine (Vitamin B1) (In thiamine-responsive patients) |
Increases stability of branched-chain alpha-ketoacid dehydrogenase complex (BCKDC). | Additional Thiamine challenge of 150 -300 mg/day for one month. Continue thiamine supplementation in responsive patients [53]. |
Plasma levels of BCAAs | ||
| Liver transplantation | Hepatic enzyme replacement |
||||
| Management of acute crises: BCAA-free formula (PO or NG if not tolerating formula), provide all amino acids except leucine, supplement isoleucine and valine [23], reverse catabolism: increase calorie intake - IV calories (typically dextrose at high concentration), may start insulin drip if hyperglycaemic, use of normal or hypertonic saline, avoid hypotonic solutions, mannitol, diuretics, haemodialysis/haemofiltration | |||||
| Methylmalonic acidemia | Protein-restricted diet using synthetic propiogenic-devoid formulas | Reduce MMA production | Urine MMA, plasma amino acid concentrations | ||
| Hydroxocobalamin | Enhance activity of metyhlmalonyl-CoA mutase | 1mg intramuscularly, regular continuation depends on metabolic response [47]. | Urine MMA, plasma amino acid concentrations | ||
| Carnitine | To correct secondary carnitine deficiency | 50-100 mg/kg/day and up to ~300 mg/kg/day divided into 3-4 doses [47]. | Plasma free carnitine level, acylcarnitine profile in dried blood spots | ||
| Metronidazole | Reduce propionate production by gut flora | 10-15 mg/kg/day typically administered in 7–10 day courses every 1–3 months [47]. | Urine MMA, propionylcarnitine | ||
Table 4.
Medical and dietetic management of lysine, serine and glycine metabolism .
| Disorder | Treatment | Rationale/mechanism | Dose | Monitoring |
|---|---|---|---|---|
|
Non-ketotic hyperglycinemia (NKH) |
Ketogenic diet (High in fat and low in carbohydrates) *Must decrease sodium benzoate | Alternate energy source for brain, epilepsy treatment, glycine reduction | Blood glucose and ketones | |
| Sodium benzoate | Forms conjugated metabolite (hippurate) which is excreted by kidneys | Attenuated NKH - 200-550 mg/kg/day Severe NKH - 550-750 mg/kg/day (Maximum dose 16.5g/m2/day) [63]. |
Glycine in plasma and CSF | |
| Dextromethorphan (Gene – drug interactions: CYP2D6, CYP3A4, CYPUGT) | Weak, non-competitive inhibitor of NMDA receptors | 3-15mg/kg/day (High individual variability) [64]. | Glycine in plasma and CSF | |
| Pyridoxal phosphate (Active form of vitamin B6) | Co-factor of glycine decarboxylase (GLDC) | Glycine in plasma and CSF | ||
|
PDE-ALDH7A1 |
Pyridoxine (Vitamin B6) | Pyridoxal 5′-phosphate (PLP) is a cofactor of enzymatic reactions involved in neurotransmitter synthesis |
Adults 200–500 mg/day (Maximum dose 500 mg/day) [65]. | Serum/plasma pipecolic acid levels, alpha-aminoadipic semialdehyde [AASA] in serum/plasma, urine, or CSF |
| Lysine reduction therapies (LRT) – Lysine restriction, arginine supplementation | Arginine is a competitive inhibitor of lysine transport | Start at 4g/m2/day (Maximum dose 5.5g/m2/day) [65]. | Plasma lysine, arginine | |
| 3-Phosphoglycerate dehydrogenase deficiency | L-Serine and Glycine | Seizure control, correction of behavioural abnormalities. | Infantile 3-PGDH deficiency: 500-700mg L-serine/kg/d and 200- 300mg glycine/kg/d Juvenile 3-PGDH deficiency: 100-150mg L-serine/kg/d [66] |
CSF serine and glycine; plasma serine and glycine |
| Phosphoserine aminotransferase deficiency | L-Serine and Glycine | Prevention of neurological abnormalities in presymptomatic patients | L-serine: 500mg/kg/day Glycine: 200mg/kg/day [67] |
CSF serine and glycine; plasma serine and glycine |
| 3-Phosphoserine phosphatase deficiency | L-Serine | May prevent onset of neurological symptoms | 200-300mg/kg/day [67] |
CSF and plasma serine |
Table 5.
Medical and dietetic management of basic amino acid metabolism or transport disorders .
| Disorder | Treatment | Rationale/mechanism | Dose | Monitoring |
|---|---|---|---|---|
|
Cystinuria |
Potassium citrate | Urine alkalisation | Children: 60-80mEq/1.73 m2/d Adults: 60-80mEq/d TDS/QDS [74] |
Urine pH |
| Penicillamine | Increases cystine solubility | Children: 20-30mg/kg/d (max 4000 mg/d) Adults: 1-4 g/d TDS/QDS |
Urine cystine excretion | |
| Tiopronin | Increases cystine solubility | Children: 15-40mg/kg/d (max 1500mg/d) Adults: 800-1500mg/kg/d TDS |
Urine cystine excretion | |
| Alpha-lipoic acid | Increases cystine solubility | Children: 30mg/kg/d (max 1200mg/d) Adults: 1200mg/d BD |
Urine cystine excretion | |
| Captopril | Increases cystine solubility | Children: 1.5-6mg/kg/d (max 150mg/d) Adults: 75-150mg/d TDS |
Urine cystine excretion | |
| Lysinuric protein intolerance | Acute Management | Reduction of protein and caloric supplementation for preventing protein catabolism | Glucose infusion: 10% glucose (in cases of hyperglycemia, consider adding insulin) L-arginine: 100-250mg/kg/d IV Sodium phenylbutyrate: 450-600mg/kg/d in patients <20kg, 9.9– 13.0 g/m2/d in larger patients) Sodium benzoate: 100–250 mg/kg/d PO or IV +/- continuous haemodialysis +/- antibiotics (e.g., neomycin), lactulose, and/or lactobacillus preparation |
Blood ammonia, amino acids in blood/urine, blood glucose |
| Dietary: Protein restriction, vitamin D, iron, zinc, and calcium supplementation, +/- medical foods e.g., protein-free drinks | To prevent hyperammonemia. Zinc, iron, calcium and vitamin D levels tend to be decreased. |
Children: 0.8 −1.5g/kg/d protein intake Adults: 0.5–0.8g/kg/d protein intake [75] |
Amino acid (e.g., lysine, arginine, ornithine, glutamine) analysis in blood/urine. 25(OH)D, iron, zinc, calcium levels. |
|
| L-citrulline | Reduces blood ammonia level, increases in dietary intake, reduction of hepatomegaly |
100mg/kg/d | Blood ammonia level, amino acids | |
| L-arginine | Reduces blood ammonia level |
120–380 mg/kg/d | Blood ammonia level, amino acids | |
| L-carnitine | Secondary carnitine deficiency | 20-50mg/kg/d | Blood carnitine level, amino acids | |
| L-lysine | Increases blood lysine levels | 20-50mg/kg/d | Blood lysine level, amino acids | |
| Nitrogen scavengers | Decreases blood ammonia levels |
Sodium phenylbutyrate: 450– 600mg/kg/d in patients weighing <20kg and 9.9–13.0 g/m2/d in larger patients. Sodium benzoate: 100–250 mg/kg/d |
Blood ammonia levels, plasma amino acids, electrolytes (Sodium) |
|
| Other treatments | Management of osteoporosis, short stature, hyperlipidemia, nephritis, pulmonary alveolar proteinosis, ESRF |
Vitamin D and bisphosphonate, GH injection, statins, ACE inhibitors, corticosteroids, whole lung lavage, GM-CSF, renal transplantation |
As per clinical finding | |
| Hartnup disease* | Nicotinamide | Management of dermatological and neurological complications. | 50-300mg PO [76] |
Niacin levels |
| High protein diet | To ameliorate amino acid loss | Individualised to patient | Plasma amino acids (e.g., alanine, serine, glutamine). |
Table 6.
Medical and dietetic management of ornithine and proline disorders.
| Disorder | Treatment | Rationale/mechanism | Dose | Monitoring |
|---|---|---|---|---|
|
Δ1-Pyrroline-5-carboxylate synthetase deficiency |
Arginine | Increases arginine availability to brain. Improvement of neurodevelopmental and metabolic parameters. |
150mg/kg/d [105] |
Amino acid analysis (proline, ornithine, arginine, citrulline), and ammonia levels. |
|
Hyperprolinemia Type I |
Anti-epileptic medication and schizophrenia medication if required. | |||
| Avoid protein excess | Reduce accumulation of proline or P5C | Plasma amino acids (proline) | ||
|
Hyperprolinemia Type II |
B6 supplementation | Avoid deficiency | E.g., 50-100mg/day [106] | B6 levels |
| Avoid protein excess | Reduce accumulation of proline or P5C | Plasma amino acids, urine organic acids | ||
| Anti-epileptic medication and schizophrenia medication if required. | ||||
| Ornithine δ-aminotransferase deficiency (gyrate atrophy) | Arginine-restricted diet with synthetic amino acid supplementation. | Aim to decrease plasma ornithine levels and slow disease progression. | 10-35g/d protein intake [107] | Ornithine and arginine levels |
| Trial of B6, lysine, and creatine supplementation. | B6 – Aims to stimulate residual enzyme activity. Lysine – May increase kidney excretion of ornithine and arginine. Creatine and precursors – To treat secondary creatine deficiency | B6: 100-1000mg/d [107] | B6 and plasma amino acids; blood/urine creatine | |
|
Hyperornithinemia-hyperammonemia- homocitrullinuria |
Acute management | Stop protein intake for 24h and commence IV 10% Glucose (plus electrolytes). Arginine +/- citrulline supplementation. Ammonia scavengers (sodium benzoate and sodium phenylbutyrate). +/- haemodialysis (if neurological status is deteriorating) |
Glucose dose at appropriate dose to prevent catabolism. Sodium benzoate: 250mg/kg bolus (90–120 min), then maintenance 250-500mg/kg/d (>20 kg, 5.5 g/m2/d) Sodium phenylbutyrate: 250mg/kg bolus (90-120 minutes), then 250-500 mg/kg/d as maintenance [108]. |
Blood ammonia levels, blood glucose. |
| Long-term management | Protein-restricted diet with citrulline or arginine (+/- sodium benzoate or sodium phenylbutyrate) | Protein restriction individualised to patient. Sodium benzoate: ≤ 250mg/kg/d Sodium phenylbutyrate: <20 kg ≤250mg/kg/d, >20 kg 5g/m2/d L-citrulline: 100-200mg/kg/d L-arginine: <20 kg 100-200mg/kg/d, >20 kg 2.5- 6g/m2/d [108] |
Blood ammonia levels, plasma amino acids, urinary orotic acid | |
| Creatine supplementation (if plasma creatine levels low) | To treat secondary creatine deficiency. | Dosed according to degree of creatine deficiency. | Plasma creatinine levels, blood/urine creatine |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



