Submitted:
10 November 2025
Posted:
10 November 2025
You are already at the latest version
Abstract
Non-motor symptoms (NMS) of Parkinson's disease (PD) were recognized by James Parkinson himself about 200 years ago and are now considered to be an integral part of PD, significantly contributing to the worse of patients’ quality of life. Although awareness of NMS is growing, and several scientific societies have now dedicated non-motor study groups, the NMS burden is still the hidden face of PD and in most cases the clinicians’ views are focused on the motor symptoms alone. Literature research has been performed using several databases and scientific journals; this review provides a comprehensive description of the most common NMS in PD, their clinical phenotype, social impact, diagnosis, and therapeutic management. Early recognition of these features may lead to more prompt and effective treatment and may help to better understand patients’ needs.
Keywords:
Parkinson's disease
; non-motor symptoms
; quality of life
; management
Introduction
Parkinson’s disease (PD) is a degenerative disorder of the central nervous system (CNS), typically characterized by a triad of motor symptoms, namely hypokinesia/bradykinesia, rigidity, and tremor, caused by a striatal deficiency of the neurotransmitter dopamine [1]. As such, current treatment of PD is based on the replacement of dopamine. While this can offer good control of motor symptoms, it does not halt the progression of neurodegeneration and consequent disability [1]. Globally, PD affects around six million people, and this number continues to increase due to numerous factors, including an ageing population, environmental factors, and a longer disease duration [2]. PD also presents a significant economic burden; in 2017, the estimated total medical cost attributable to this pathology was just over $25 billion in the United States alone, with $18.9 billion attributed to patients care and another $6.1 billion to unpaid care partners [3]. As well as the cardinal motor symptoms of PD, there are also so-called non-motor symptoms (NMS) of the condition. The NMS include a wide variety of manifestations such as autonomic dysfunctions, hyposmia, sleep problems, neuropsychiatric symptoms, and cognitive impairment [4]. These NMS are consistent with the Braak hypothesis that PD is not restricted to the dopaminergic neurons of the substantia nigra pars compacta, but involves the brain stem, limbic and neocortical structures, along with the peripheral and enteric nervous systems [5]. These symptoms have gained attention over the past decade due to their impact on the quality of life (QoL) for both patients and caregivers, and the overall burden on the healthcare system [6]. Patients often indicate that their NMS are more difficult to manage than their motor problems, and frequent hospitalization as a result of these symptoms is estimated to increase the cost of PD care fourfold [7]. In the UK, The National Institute for Health and Care Excellence (NICE) recognize NMS, and their management, as an important area of unmet need in PD [8]. However, a 2019 cross-sectional study found that up to 72% of people with PD do not report NMS to their healthcare professional [9]. The most commonly reported barriers to seeking help were acceptance of the symptoms, a lack of awareness that the symptom was associated with PD, and embarrassment to discuss some symptoms such as sexual problems or incontinence [9]. Clinicians often fail to identify NMS during consultations, due to lack of time, focusing on motor aspects of PD, and the perception that no effective treatments are available [9]. Despite their frequency and impact, the NMS of PD continue to be under-reported and inadequately treated, and there is a need for more clinical trials considering NMS as outcome measures [5]. The aim of this publication is to inform clinicians and other healthcare professional about the key aspects of NMS in PD, their pathophysiology, burden, economic costs, treatment and the importance of their diagnosis. As this is a narrative review, a systematic literature review was not conducted. No new clinical or pre-clinical studies were performed by the authors, nor patients were recruited. No patient-specific efficacy or safety data were reported. Therefore, institutional review board/ethics approval was not required.
Epidemiology
The term “non-motor symptoms” of PD covers a diverse group of symptoms, encompassing cognitive, emotional, autonomic, and sensory disturbances. NMS are highly prevalent in PD patients: data of >2,500 PD patients using the validated Non-Motor Symptoms Questionnaire indicated that almost all patients (>98 %) reported up to eight different symptoms (Table 1) [5].
NMS are numerous and heterogeneous in their presentation and in the different systems of the body affected (Figure 1).
As a result of their heterogeneity, the division of NMS in one single category is difficult; they reflect a wide range of symptoms, from cognitive to sensory, and can also be classified according to their pathophysiology or drug therapy complications (Table 2).
The heterogeneity of NMS observed in epidemiological studies may be related to the inclusion of patients of different age groups, duration of disease and Hoehn and Yahr (HY) stage. Crosiers et al. revealed that the average of NMS was 6.5 in patients with disease duration <5 years and 10 in those with >15 years [10]. Hallucinations, urological, cardiovascular and gastrointestinal symptoms are more frequent in older patients as well as in patients with HY stages 3-5, while younger patients or patients with HY stage ≤2 reported more frequently sleep problems, pain, anxiety and depression [10]. Also, sex-related differences in the frequency and severity of NMS have been reported by several studies [11]. Anxiety, sadness, depression, fatigue, dysphagia, constipation, and pain are more common in women, while men suffer more from sialorrhea, urinary dysfunction, hypotension, cognitive impairment, sleep behavior disorders, and daytime sleepiness [11]. Regarding impulse control disorders, men show compulsive sexual behavior while women are more likely to develop pathological shopping and “binge eating” [11]. Patients with PD also experience “non-motor fluctuations” (NMF) which often develop in tandem with motor fluctuations, particularly during the “wearing off” periods, and female sex is a major risk factor for the development of NMF [12]. Psychiatric symptoms, and in particular anxiety and depression, fluctuate more frequently and severely than other NMS [13]. There are few studies investigating possible variations in NMS among different ethnic groups; the current evidence, however, suggests that NMS prevalence is high across all ethnic people [14]. An evolving area of research in PD relates to different genetic forms of PD and how they relate to non-motor signs. Although only a minority (5-10%) of PD cases is due to well-defined genetic causes, important clues about this pathology can be garnered from monogenic model diseases [15]. At least 18 mutations in the synuclein alpha (SNCA) gene, one of the PARK family of genes known to be associated with PD, have been described [15]. Analysis of SNCA missense mutations and multiplications, albeit in a relatively small number of PD patients, has suggested that severe cognitive dysfunction is a marker for this genetic form of PD [16]. LRRK2 (leucine-rich repeat kinase 2, another of the PARK gene family) mutations on the other hand are commonly associated with sleep disorders [16]. Variations in the GBA (glucosidase beta acid) gene are commonly seen in PD and seem to be associated with autonomic problems, dementia and hallucinations, and patients often have a high degree of anxiety and depression [17]. Some studies have found a higher prevalence of LRRK2 and GBA mutations among women compared to men; this difference might explain the different NMS manifestation in both sexes [18]. Mutations in PINK1 and DJ-1 genes cause autosomal recessive forms of PD and may have prominent psychiatric symptoms of depression and anxiety [15]. These emerging data on the non-motor markers of genetic subtypes of PD may help refine treatment strategies in the future.
Timing of Symptom Presentation
NMS often pre-date motor symptoms by 10–15 years, most notably depression or apathy, constipation, hyposmia, rapid eye movement (REM) and sleep behavior disorder (RBD) [19]. Other NMS complicate the entire disease course (pain, fatigue) and especially the advanced stages (dementia, dysautonomia, psychiatric symptoms), as shown in the 20 years Sydney multicentre study [20]. There is growing evidence that the degeneration of non-dopaminergic neurons occurs before the dopaminergic neuron loss and the onset of motor symptoms: most of the non-motor features associated with PD are related to the global involvement of the non-dopaminergic systems (i.e. serotoninergic, noradrenergic, cholinergic, glutamatergic, and GABAergic transmissions) [21]. By the time a patient shows the first clinically evident motor symptoms of PD, approximately 50% of the terminals and 30% of the nigral cells are lost, as estimated by postmortem data and using Positron Emission Tomography (PET) imaging in a nonhuman primate model of PD [1]. This indicates that the pathological process begins at the nerve terminals and moves retrogradely, and that there is a substantial number of dopaminergic neurons that are dysfunctional but not lost [21]. This unique timing of symptom presentation in PD has prompted a redefinition of the disease led by the Movement Disorder Society (MDS) Task Force on the Definition of PD, giving rise to three distinct clinical stages [22]:
- a preclinical phase, supported by molecular or imaging markers, but without clinical signs or symptoms of PD
- a premotor phase (or prodromal phase), characterized by NMS such as hyposmia and sleep behavior disorder
- and the motor phase, often including NMS such as pain, fatigue, and dementia
Detecting PD before the appearance of overt motor symptoms could help disease management and provide an early window of opportunity for any future treatments aimed at slowing or even stopping neuronal death and disease progression.
What Causes NMS in PD?
Although PD has been previously considered as primarily associated with dopamine depletion, it is now recognized that multiple neurotransmitter pathways are also impacted, affecting areas that are not directly involved in motor control, as illustrated in Table 3 [23].
Dopamine Pathways Affected in PD:
Dopamine is essential for the control of movement and also plays a key role in the control of systems involved in regulating pleasure, reward, sleep, and cognition. The nigrostriatal, mesocortical and mesolimbic dopaminergic pathways are all affected in PD, contributing to depression, anxiety, pain, sleep disorders, cognitive impairments, and bladder dysfunction [23]. Dopaminergic therapies can also contribute to the development of some NMS, such as hallucinations and daytime sleepiness [24].
Serotonergic Pathways Affected in PD:
Serotonin neurons in the dorsal raphe nuclei project to the basal ganglia, the frontal cortex, and the limbic system [25]. The serotonergic system is thought to be involved in the control of various processes, including mood, emotion and sleep, therefore, serotonergic dysfunction in PD has been directly linked to depression, anxiety, sleep problems, and visual hallucinations [25].
Noradrenergic Pathways Affected in PD:
The locus coeruleus (LC) is the primary site of noradrenergic production in the brain, with neurons projecting out to numerous brain regions including the thalamus, the limbic system, forebrain, and the cortex [26]. Surprisingly, neuronal loss in the LC is greater (83%) than in the substantia nigra (78%) in PD, and the subsequent loss of noradrenergic innervation of the limbic system is associated with cognitive and neurobehavioral problems, including early cognitive dysfunction, dementia, depression, and anxiety [27].
Glutamatergic Pathways Affected in PD:
Loss of nigral dopaminergic neurons and the subsequent striatal depletion of dopamine leads to glutamate hyperactivity in the basal ganglia [28]. Considering its widespread role in important central and peripheral processes, glutamate dysregulation (mainly hyperactivity) is implicated in cognition, depression, anxiety, sleep disorders and pain [28].
Cholinergic Pathways Affected in PD:
Acetylcholine has several distinct functions in the brain, including arousal, attention, memory, and motivation. Cholinergic disruption in PD is known to contribute to cognitive deterioration, psychosis, sleep abnormalities, and altered olfactory function [29]. These symptoms have been specifically attributed to alterations in the cholinergic signaling in the striatum, and to degeneration of cholinergic nuclei, including the nucleus basalis and the pedunculopontine nucleus [30]. Cholinergic neurons also provide parasympathetic autonomic innervation to peripheral organs, and a reduced cholinergic tone contributes to urinary problems [30].
GABAergic Dysfunction Affected in PD:
The γ-amino butyric acid (GABA) system is the main inhibitory neurotransmitter in the central, peripheral, and enteric nervous systems. A deficit of GABA may result in chronic dysfunctions such as sleep disturbances, hyposmia, hallucinations, and anxiety [31]. GABAergic dysregulation has been observed in PD patients in the basal ganglia post-mortem and in vivo with magnetic resonance spectroscopy [31].
Understanding what causes the neuronal degeneration in the different neurotransmitter pathways remains the holy grail of PD research. One promising area of interest focuses on protein misfolding, a common feature of neurodegenerative disease [32]. In PD, misfolded α-synuclein forms insoluble aggregates, and Lewy bodies disrupt the functioning of many organelles, including mitochondria, endoplasmic reticulum (ER), and the Golgi apparatus (GA) [32]. Originally discovered in the substantia nigra, it is now known that Lewy bodies occur across different brain regions; this accumulation of α-synuclein outside of the nigrostriatal pathway is thought to contribute to the early onset of certain NMS [32]. In the prodromal phase of PD, misfolded α-synuclein has been noted in the enteric nervous system and associated with the early symptoms of constipation and abdominal pain [33]. Misfolded and aggregated α-synuclein has also been observed in the olfactory bulb in early PD and subsequently linked to the prodromal symptom of anosmia [33]. These observations led to a theory, spearheaded by Braak and colleagues, that a “bottom-up” pathological spread of misfolded α-synuclein underlies PD [34]. It has been postulated that α-synuclein mediated neurodegeneration spreads in a caudo-rostral direction from the olfactory bulb, the enteric nervous system, the spinal cord, and the dorsal motor nucleus of the vagus nerve into the brain, affecting the different neurotransmitter populations before reaching the substantia nigra [34]. However, this theory may not apply to all patients: in fact, 7–8% of patients have been reported with no α synuclein pathology in the dorsal motor nucleus [33]. Halliday and colleagues have shown that early-onset PD neurodegeneration and Lewy body deposition (predominantly limbic involvement) is different from that seen in later-onset disease (predominantly cortical and superior brainstem involvement) [35]. This leads to segregation into three distinct phenotypes: brainstem dominant, limbic dominant and cortical dominant, each associated with different NMS [36]. Limbic dominant is mostly associated with symptoms of fatigue and pain, cortical dominant with cognitive symptoms and apathy and brainstem dominant with sleep symptoms and dysautonomia [36].
The Burden of NMS
A body of evidence suggests that the global burden of NMS seems to have a greater impact on patients’ quality of life (QoL) than their motor symptoms; in a large international cross-sectional study of 951 patients with PD, the highest burden was noted in pain, sleep, fatigue, mood, urinary, and gastrointestinal domains [37]. These NMS have a substantial impact on QoL through a direct consequence of symptoms and also indirectly, due to the consequent disability; moreover, as the condition progresses, they become more and more burdensome [38]. In a study of 265 PD patients in the UK, those with more advanced PD listed fluctuations and non-motor aspects as their most troublesome [39]. NMS cause a significative burden not only to patients’ life, but also to caregivers’ life: the majority of care for patients with PD is provided by informal caregivers, not only offering physical and emotional support for patients but also playing a large economic supporting role and preventing early nursing home placement [40]. An online survey of people with PD and their carers found that care partners report that NMS were more challenging than motor symptoms (58% vs. 32%) [41]. NMS commonly associated with caregiver burden are depression, anxiety, cognitive impairment, sleep disturbances, hallucinations and psychosis [40]. Psychosis, in particular, causes greater distress: a systematic review of UK studies found that caregivers had a comorbidity rate nearly five times greater compared to the age-matched population, when the patients cared had psychiatric symptoms, with a comparable reduction in QoL over time affecting their social, psychological, and physical wellbeing [42]. Finally, NMS cause a large financial burden on patients, caregivers and healthcare system and the economic costs are known to double by each Hoehn & Yahr stage [43]. Falls, dementia and hallucinations are the major reasons for admission to hospital and residential care, while mood disorders and fatigue account for hidden costs such as sick leave, loss of productivity and early retirement [43]. In the UK, the annual direct costs for patients living in full-time institutional care was estimated at about five times higher than in the home setting [44]. An Australian study found that the largest economic burden from the household perspective was the cost of informal care, which was estimated at $12,548 per person over 12 months [45]. People with moderate-to-severe disease reported an average of 775 informal care hours annually [45]. The cost of formal care was 3.8 times higher for patients with 10 NMS or more compared to those with 6–9 NMS [46]. Moreover, women with PD receive less social and familiar care then men, and more frequently pay for a professional caregiver [11]. Clinically, the broad symptomatology and potential severity of NMS requires multidisciplinary approaches, often with a dedicated team of around 15 different specialties [47]. Thus, through early identification of NMS, physicians will be able to provide patients with better QoL, limiting the financial impact of PD.
A Focus on PD Nurses
With the convergence of numerous medical professionals for PD diagnosis and treatment, a specialist role was required to bridge the gap between this medical management and the unique personal needs of the patient [48].
In 1989 in the UK, the PD nurse specialist (PDNS) was created to meet this need by:
- ✓ providing information, education, and instruction
- ✓ supporting the patient and caregiver in the promotion of self-management
- ✓ supporting psychosocial care questions
- ✓ specializing in diagnostic strategies and therapeutic nursing interventions
- ✓ promoting multidisciplinary collaboration.
Based on expert opinion from healthcare professionals, it is recommended that every person with PD could benefit from PDNS care, and the role is now common across Europe [48]. For example, in Sweden, the PDNS is the primary link to medical care for PD patients, evaluating changes in medical treatment, and asking the patient for feedback on efficacy and side-effects [49]. Providing emotional support for both patients and caregivers is also an essential part of the work; as such, PDNS are often the first healthcare professionals to become aware about patients NMS [49]. It is therefore important that the nurse is well-informed about the effect of these symptoms and any treatments that may be available [50]. PDNS care may improve patient wellbeing and quality of life, and reduce anxiety and depression, saving money in the long term, due to reductions in consultations with physicians and neurologists [50].
Tools for Assessing NMS
To effectively recognize and treat NMS, clinically validated holistic tools have been developed and are now routinely used by physicians. The Non-Motor Symptoms Screening Questionnaire (NMSQuest) was developed by a multidisciplinary group of international experts in PD [51]. The questionnaire is a 30-items self-completed tool, with a yes/no response format, used to identify those symptoms requiring further evaluation during clinical consultation [51]. However, the questionnaire is only used for screening and does not grade symptom severity, therefore cannot be used to assess treatment effects [51]. The Non-Motor Symptoms Scale (NMSS) is a 30-item rater-based scale that measures the severity and frequency of NMS across nine dimensions in patients with PD at all stages of the condition, thus capturing the global burden for patients [52]. Importantly, the scale can be used in a clinical trial setting [52]. Other available tools include the battery of SCales for Outcomes in PArkinson’s disease (SCOPA) [53], investigating cognition, dysautonomia, sleep disorders and psychiatric symptoms, and the Movement Disorder Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) which integrate motor and non-motor aspects of the disease [54]. There are other scales not specific for the assessment of NMS or grading only some symptoms, such as the Parkinson’s Disease Questionnaire – 39 items (PDQ-39) [55], a self-administered quality of life scale including some questions related to cognition, mood and pain, the King’s Parkinson’s disease Pain Scale (KPPS) [56] for PD chronic pain, the Parkinson’s Disease Sleep Scale (PDSS-2) and the Epworth Sleepiness Scale (ESS) for sleep disturbances [57,58], the Hamilton Depression rating scale (HAMD) and the Beck Inventory for depression [59,60], the Fatigue Severity Scale (FSS) and the Parkinson Fatigue Scale – 16 items (PFS-16) for fatigue [61,62].
The Different NMS in PD
The symptoms can be broadly divided into five categories:
- Pain and other sensory symptoms (olfactory disfunction; changes in visual function)
- Neuropsychiatric symptoms (depression; anxiety; apathy; cognitive impairment and dementia; psychotic symptoms, hallucinations and delusions; compulsive behaviors)
- Sleep disorders (rapid eye movement sleep behavior disorder; insomnia; restless legs syndrome and periodic limb movements; excessive daytime sleepiness)
- Autonomic symptoms (bladder dysfunction; gastrointestinal symptoms; neurogenic orthostatic hypotension; sexual dysfunction)
- Fatigue
Pain and Other Sensory Symptoms
Pain
Pain affects between 40-85% of PD patients, and is considered one of the most bothersome NMS, often more distressing than motor disability [63]. However, it is often underdiagnosed despite its substantial impact on patients’ quality of life; many patients also report poor management, and around 50% do not receive any analgesic prescription [63]. Pain is also one of the earliest symptoms of PD, sometimes present at the preclinical stage [63]. There are various methods of classifying PD pain, the most widely accepted is the Ford classification, which distinguishes five different types of pain: musculoskeletal, dystonic, radicular/neuropathic, primary/central and akathisia; these types also often overlap, with further complicating diagnosis and treatment [64]. Recently, a new classification has been published by a panel of specialists (including movement disorders experts, nurses, physiotherapists, and psychologists), and validated through an international, cross-sectional multicenter study: the Parkinson’s disease Pain Classification System (PD-PCS) [65]. The PD-PCS differentiates between pain related or unrelated to PD, and distinguishes three different types of pain: nociceptive, neuropathic, and nociplastic. The PD-PCS also provides a score that covers pain intensity, effect, and frequency [65]. The neurobiology of pain in PD is complex and appears to involve dopaminergic, serotonergic, noradrenergic, glutamatergic, and GABAergic neurotransmission [66]. In addition to motor functions, the basal ganglia are involved in the processing of nociceptive inputs; thus, nigrostriatal damage may at least partly account for the sensation of pain experienced in PD [66]. Furthermore, the serotonergic raphe nuclei and the noradrenergic locus coeruleus have a role in ‘gain setting’ for pain control and are impacted over the course of PD [66]. Changes in dopaminergic function in PD may also modulate sensory perception; for instance, pain occurs more frequently in patients during ‘OFF’ periods, and pain thresholds are raised by dopaminergic medication [67]. Finally, peripheral sensory information and pain signals are transmitted to the spinal cord via primary afferent neurons, the majority of which are glutamatergic; upon noxious stimulation, glutamate is released from central terminals in the spinal cord [68]. Glutamate has also been implicated in the phenomenon underlying chronic pain, including effects of allodynia and hyperalgesia [69]. Specific demographic and clinical factors have been associated with the variability of chronic pain in PD: age at onset, sex, ethnicity, and the presence of comorbid conditions can all shape the perception and experience of pain [68]. For example, females with PD may experience more severe or more frequent pain than males [70]. At the same time, people with younger-onset PD may report different pain patterns compared to those diagnosed later in life [71]. Genetic factors might contribute to these differences; for instance, the dopamine receptor D2 (DRD2) rs2283265 polymorphism has been associated with an increased risk of PD-related pain in females [72], while the CHRNA4 gene rs1044397 variant may influence PD onset age in females with chronic pain [73]. No drugs are specifically indicated for PD pain. Dopamine agonists can be useful for musculoskeletal pain, as well as non-steroidal anti-inflammatory drugs and intra-articular corticosteroid injections [66]. Deep brain stimulation (DBS), apomorphine or adjustments of levodopa dosage are indicated for dystonic pain, when occurs during wearing off, while botulinum toxin may be useful in localized dystonia and amantadine for dystonic dyskinesia [66]. Antiepileptics (such as pregabalin and gabapentin), tricyclic antidepressants (TCAs) including amitriptyline, and serotonin and norepinephrine reuptake inhibitors (SNRIs) such as duloxetine are recommended for neuropathic pain [66]. Glutamatergic neurotransmission is elevated during neuropathic pain; therefore, the use of glutamate modulators may help to reduce pain symptoms [74]. In a systematic review and meta-analysis, the greatest improvement of pain severity was seen with safinamide [75]. Safinamide reduced by 43.6% the total score of the King’s Parkinson’s disease Pain Scale, with significant improvements observed in in musculoskeletal, fluctuation-related, nocturnal and radicular pain, and with a reduction of concomitant pain treatments by 26% [76]. In severe cases, the opioid drugs tramadol or tapentadol may be prescribed, while lidocaine or capsaicin patches or botulinum injections are used for peripheral neuropathies [77]. Data of 11.466 PD patients obtained from the French System of Health Insurance showed that the prevalence of analgesic drugs prescriptions was higher in these patients compared to the general population (82% versus 77%) [78]. Also, their chronic prescription (more than 90 days) was more prevalent in PD patients (33%) than in the general population (20%) [78]. Some non-pharmacological treatments, in addition to DBS, may improve pain. High-frequency repetitive transcranial magnetic stimulation (rTMS) reduced musculoskeletal pain [79]. Acupuncture alleviate nociceptive pain, physical activity, particularly Nordic walking, may be beneficial for back, hand and legs pain and yoga for lower back pain [80].
Olfactory Disfunction
Hyposmia is clinically recognized as one of the earliest symptoms of PD; a patient with hyposmia has a 10% increased risk of developing PD within 2 years of onset of olfactory dysfunction compared with asymptomatic relatives [81]. Thus, the symptom of hyposmia may represent an opportunity for early diagnosis and potential treatment before the onset of motor symptoms [81]. It is estimated in total that around 75% of people with PD have a poorer sense of smell compared with people of the same age; this includes both an increased olfactory threshold, and a decreased ability to discriminate odors [82]. As well as loss of smell, another recent unexpected finding is that patients with PD also have a unique ‘musky odor’ discernible by so-called ‘super smellers’ [83]. This unique scent results from several compounds, particularly hippuric acid, eicosane, and octadecanal being produced in higher than usual concentrations in the sebum of people with PD and also represents a potential biomarker for early diagnosis [83]. The reason why the sense of smell is impaired in PD is not completely understood; it has been suggested that it is related to early α-synuclein clumping in the olfactory bulb, visible before the loss of dopaminergic neurons in the substantia nigra [84]. According to the six-stage pathological process proposed by Braak and colleagues, Lewy bodies initially form in the olfactory bulb and anterior olfactory nucleus, producing olfactory dysfunction [5]. Hyposmia or anosmia have been reported in other disorders with abnormal synuclein, such as Lewy-body dementia and multiple system atrophy, suggesting a common pathogenic process [85]. Moreover, the non-synucleinopathies, such as vascular parkinsonism, corticobasal degeneration and progressive supranuclear palsy, tend to have intact olfactory function [86]. However, the presence of olfactory dysfunction in later stages of PD may also be linked to cholinergic denervation, cortical processing of olfactory inputs, the onset of cognitive deficits, and dementia [85]. A 2010 study also found that individuals with PD have a loss of mitral cells and cells containing substance P in the olfactory bulb, and a reduction in the level of calcium-binding protein in this region [86]. While dopamine plays an important modulatory role throughout the olfactory system, dopaminergic pharmacotherapy has no discernible impact on smell loss [1]. Olfactory disfunction is an important component of PD, but its pathological basis and relationship to the progression of the disease remains largely undefined.
Changes in Visual Function
Parkinson’s disease is associated with several ocular disturbance, including blurred vision, diplopia, reduction of eye movements, impaired color discrimination, convergence insufficiency and contrast deficits [87]. Reduced blink rates can lead to dry eyes and hypomimia appearance; moreover, between 25% and 40% of patients experience visual hallucinations due to the presence of Lewy bodies in the occipital lobe and in ret inal neurons, and to the loss of dopaminergic amacrine cells and of the regulatory role of dopamine receptors in the eye [87]. Treatment with dopamine agonists is associated with an increased risk of visual hallucinations, implying that dopaminergic signaling is involved in their generation, and visual hallucinations are predictive of cognitive decline in later stages of the disease [88]. All these visual disturbances may negatively impact reading, driving, gait and balance (increasing fall risk) and daily activities [87]. Reduced color discrimination is one of the visual hallmarks of PD and may reflect neurodegenerative changes in thalamus and posterior parietal lobe [89]. Patients show shortened chromatic fusion time, which indicates perceptual acuity for monochromatic contours, especially for light-blue and dark-green contours [89]. Innervation around the fovea (responsible for sharp central vision) are largely dopaminergic: postmortem studies of untreated PD patients showed decreased retinal dopamine concentrations compared with those treated with dopaminergic drugs [89]. The deposition of alpha-synuclein has been described in amacrine and ganglion cells within the inner retina [89]. Many patients report blurred vision, which typically emerges at lower light levels during OFF periods [90]. Dopamine deficiency may reduce retina’s ability to differentiate spatially distinct stimuli, impairing contrast sensitivity, while during levodopa-induced dyskinesia, dopaminergic overstimulation may produce rapid fluctuations in contrast sensitivity, leading to blurred vision [91]. Artificial tears may alleviate dry eyes, botulin toxin injections can be used for blepharospasm, glasses or prisms for diplopia, whereas sustained release formulations of dopaminergic drugs may improve oculomotor symptoms during OFF periods [92]. Anticholinergic drugs and amantadine should be avoided because may produce visual system side effects, such as mydriasis, photophobia, dry eyes, anterior angle closure, blurred vision [92]. Care coordination among optometrists, ophthalmologists, occupational and physical therapists may be required [47].
Neuropsychiatric Symptoms
Depression
Depression is a frequent neuropsychiatric symptom of PD, affecting up to 45% of patients with a substantial negative impact on their QoL, and can appear several years before the motor impairment [93]. Diagnosis can be difficult since some typical symptoms such as lack of expression, apathy, sleep disorders, loss of appetite, weight loss, lack of energy, and asthenia are also part of the clinical picture of PD [93]. It is a complex phenomenon that changes over the disease duration and may be a pathological consequence, a reaction to the associated disability, an independent separate comorbidity, or a combination of all three [93]. It has been hypothesized that depression in PD arises from degeneration of the mesocortical and mesolimbic dopaminergic neurons, disrupting serotonergic neurons in the midbrain and leading to dysfunction of the depression-related-thalamic circuits [94]. Catecholamines may be also involved: depressed patients have a reduction in both dopaminergic and noradrenergic innervation in the locus coeruleus, thalamus, and in the limbic system [94]. The risk of suicidal behaviour in PD patients is twice than controls [95]. Selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine, sertraline, citalopram, and paroxetine remain the first treatment of choice; a large clinical trial demonstrated the efficacy of the serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine [96]. Tricyclic antidepressant drugs affecting noradrenaline reuptake, such as amitriptyline and nortriptyline, may be effective in the treatment of PD related depression; however, their use may be limited by side effects such as cognitive impairment [96]. Pramipexole, a dopamine-agonist, has been shown to improve depressive symptoms in PD patients [97]. Inhibition of monoamine oxidase enzyme increases the levels of biogenic amines (in particular dopamine) in the synaptic cleft and may enhance mood and motivation; nevertheless, rasagiline has shown no significant effects on depression in PD patients [98]. Growing evidence suggests that the glutamatergic system is directly involved in mood disorders in PD [99]. Glutamate levels are regulated by sodium-dependent glutamate transporter: mood stabilizing agents, like valproic acid and lamotrigine, modulate glutamate neurotransmission by blocking sodium channels [99]. Safinamide, a reversible MAO-B inhibitor and glutamate modulator, showed clinical benefits on mood fluctuations in patients with PD and improved emotional well-being [100]. However, robust evidence for a good anti-depressant is lacking; the current data does not indicate towards one specific class of drugs, and the heterogeneity of contributory factors for PD depression should be considered by clinicians when prescribing a treatment [98]. The approach to the treatment of depression should be therefore multidimensional, comprising medications as appropriate, education about mood disturbances in PD, and emotional support [47]. Regarding non-pharmacological treatments, some beneficial effects on depression outcome measures have been reported with repetitive transcranial magnetic stimulation (rTMS), cognitive behavioral therapy (CBT) and deep brain stimulation (DBS) [101]. CBT in particular may be first-line or add-on treatment for patients with mild depression. In controlled, randomized clinical trials of CBT for PD depression, symptom severity improved by 56% compared with 8% for the control group [102]. Electroconvulsive therapy (ECT) may be considered for refractory depression, with caveats of side effects, limited trial evidence and contraindication after DBS treatment [103]. Physical exercise, tai-chi, yoga, acupuncture and bright light therapy have shown beneficial effects with high variability, and the results should be confirmed by further studies [101].
Anxiety
Anxiety is very common in PD (prevalence 25-60%), but do not fit the usual criteria for anxiety disorders, leading to a diagnosis of PD-specific anxiety whose manifestations are panic attacks, specific phobias (e.g. phobia of falling), social phobia, obsessive-compulsive disorder or generalized anxiety disorder [104]. Anxiety is more frequent in females than in males and somatic symptoms include palpitations, shortness of breath, sweating, and dizziness. [11]. Many symptoms can occur as a dopamine-deficit event related to ‘OFF’ periods and improve with an appropriate dopaminergic therapy [104]. On the other hand, there are forms of anxiety, such as the fear of dying or going insane, that are independent of dopaminergic state and do not respond to dopaminergic drugs; these are more likely a reactive symptom to the diagnosis and progression of PD [104]. Anxiety can impact on the severity of the physical symptoms of PD: for example, patients report that episodes of anxiety can worsen pre-existing tremor or dyskinesia [105]. Interestingly, epidemiologic observations have found that patients with PD are at greater risk of developing anxiety before their PD diagnosis compared with age-matched controls, suggesting that anxiety may be an early non-motor marker of the condition [106]. Degeneration of subcortical nuclei and ascending dopamine, norepinephrine, and serotonin pathways within the basal ganglia–frontal circuits may be responsible for anxiety [106]. Positron emission tomography (PET) analysis has shown that the severity of anxiety in patients with PD is also inversely correlated with dopamine and noradrenaline-transporter binding in the amygdala, locus coeruleus, and thalamus [107]. These results suggest that anxiety in PD might be associated with a specific loss of dopaminergic and noradrenergic innervation in the locus coeruleus and the limbic system [107]. The first step to treat anxiety is to optimize the dopaminergic therapy, followed by the use of SSRIs, SNRIs and tricyclics [108]. Benzodiazepines, buspirone and pregabalin are sometimes prescribed, but with a limited efficacy [108]. CBT, acupuncture and Qigong may reduce anxiety symptoms [101]. CBT has large effects, maintaining improvements in situational anxiety, social anxiety and avoidance behavior up to six months and reducing caregiver burden [109]. The benefit of physical exercise for the treatment of anxiety in PD is much less convincing than for depression [101]. More research is needed to tailor treatment to the individuals’ needs and combined interventions may provide synergistic effects.
Apathy
Apathy is is one of the more common and debilitating neuropsychiatric disturbances in PD, present in 17-70% of patients, and consists of a loss of motivation, emotional indifference, and reduced desire to engage in activities [110]. The high heterogeneity in estimations of prevalence arises from the difficulty in distinguish apathy from other symptoms of PD such as bradyphrenia, depression, anxiety, cognitive impairment or fatigue [110]. To address this difficulty, a more operational definition of apathy has recently been adopted: a diagnosis of apathy should be made if a quantitative reduction of goal-directed activity either in behavioral, cognitive, emotional, or social dimensions is observed in comparison to the patient’s previous level of functioning in these areas [111]. Apathy contributes to reduction in quality of life and increase caregiver distress and care dependency; moreover, apathy increases the risk of developing dementia, differentiating patients with intact cognition from those with cognitive impairment [110]. Motivation depends on subcortical structures linking the prefrontal cortex with the limbic system; disruption of these circuits through mesolimbic dopaminergic denervation, depletion of cholinergic neurons and global serotonergic dysfunction, results in feelings of apathy [112]. Decreased activity levels in the supplementary motor area and lesions in the basal ganglia and thalamus, have also been implicated as underlying mechanisms of apathy [112]. Apathy and impulse control disorders have been considered as two faces of the same coin, caused respectively by hypo- and hyper-dopaminergic behaviors [113]. Dopaminergic contribution to apathy is supported by its onset after bilateral DBS, however, dopaminergic replacement therapy generally has a partial or no effect on apathy in PD [114]. The dopamine agonist piribedil showed some preliminary positive results, while rotigotine was ineffective; therefore, the role of other neurotransmitter systems involved in the normal functioning of the basal ganglia should be considered [115]. Substantial evidence implicates glutamine-to-glutamate ratio in the nucleus accumbens on the prediction of specific components of motivated behavior [116]. Safinamide, a glutamate modulator, reduced significantly scores of the Apathy Evaluation Scale and of the 14-item Apathy Scale score in 46.6% of patients clinically apathetic [117]. The cholinergic system may have also a role in apathy; a multicenter, placebo-controlled trial with the anticholinesterase agent rivastigmine, administered via a transdermal patch, reported some improvements in apathy [118]. The use of antidepressants for apathy is controversial: SSRIs have been related to increased apathy in PD, while the norepinephrine-dopamine reuptake inhibitor bupropion improved motivation [115]. One study using 5-hydroxytryptophan observed positive effect on depression but not on apathy [116]. Physical exercise (aerobic, dance, nordic walking) and mindfulness may be useful; exercise improves apathy through goal-directed behaviour change and engagement in social interactions, mindfulness by targeting the emotional and cognitive domains [119]. There are no guidelines currently managing apathy in PD, hence, there is an urgent unmet need to adequately explore treatments to improve this NMS.
Cognitive Impairment and Dementia
Analysis of NMS Questionnaire data revealed that 46% of patients with PD experience cognitive problems with preserved function, known as mild cognitive impairment (MCI), at time of diagnosis, and 70–80% of patients will progress to PD dementia (PDD) within 15–20 years [120]. PD patients have a fivefold increased risk of developing dementia than their equivalent healthy age group [120]. People with MCI may experience difficulties with memory, language, thinking, or judgement that are greater than the cognitive changes expected with normal aging, whereas PDD is characterized by impairments in attention, executive, memory (especially retrieval rather than recognition) and visuo-spatial functions [121]. In a meta-analysis of 32 cohort studies, nine risk factors were found for PD cognitive impairment, including hallucinations, orthostatic hypotension, and diabetes mellitus [122]. Cognitive decline is associated with significant impairments in quality of life, loss of independence, caregiver burden and increased healthcare costs [121]. Given the heterogeneous neuropathological and neuropsychological nature of cognitive deficits, it has been hypothesized that there are two independents, partially overlapping syndromes in PD: a “frontal-striatal” network dysfunction present at the early stage of the disease, dopamine-modulated, leading to deficits in executive functions, working memory, attention, planning, and response inhibition; a “posterior cortical” degeneration, associated with Lewy body formation and cholinergic loss, leading to dementia, characterized by visuospatial and memory deficits [123]. As such, dopaminergic drugs have little effect and can worsen psychosis and hallucinations associated with this dementia [123]. Loss of noradrenergic input from the locus coeruleus to cortical regions has also been noted in more cases of people with PD-related dementia compared with those without [124]. A multidisciplinary approach integrating pharmacological, non-pharmacological and psychosocial strategies is critical [47]. Few studies have addressed the treatment of PD cognitive impairment. The first step should be to discontinue drugs that can aggravate cognitive deficits such as tricyclic antidepressant and benzodiazepines [125]. Cholinesterase inhibitors such as rivastigmine and donepezil are generally prescribed in common clinical practice, however, this class of drugs is associated with a higher rate of discontinuation due to adverse events [125]. Memantine and amantadine, two glutamate NMDA receptor antagonists, slightly improves cognitive dysfunction [125]. Finally, despite a small, open-label pilot study of the SNRI atomoxetine found an improvement in cognitive function measures, these results have not been confirmed in other trials [126]. Non-pharmacological strategies include transcranial direct-current stimulation, physical exercise (aerobic or dance), Tai Chi or Qigong, cognitive training [127].
Psychotic Symptoms, Hallucinations and Delusions
Psychosis is one of the major reasons for admitting PD patients into nursing homes and long-term care facilities, and has been associated with worsened quality of life, caregiver burden and increased mortality [128]. Prevalence of psychosis varies widely, being rarely reported in early stage of PD and ranging from 20–70% in advanced stages [128]. Manifestation of psychosis ranges from minor signs (mild illusions, vivid dreams) to more severe symptoms (visual hallucinations, paranoid delusions and delirium) [128]. The visual hallucinations range from passing shadows in the periphery to recognizable people or animals and often precede or accompany cognitive decline; as they become more vivid, the patient may lose insight into what is happening and start acting upon the hallucinations [129]. Occasionally, auditory hallucinations also appear [129]. Paranoid delusions, though less common than psychosis (occurring in 5% to 10% of patients with PD), can be extremely distressing for both the patient and relatives, with common recurring themes of spousal infidelity or abandonment, intent of harm by strangers, or even by caregivers [130]. As with the other neuropsychiatric symptoms, psychosis in PD remains poorly understood. The pathophysiological processes underlying PD psychosis can be subdivided into intrinsic (derived from neurotransmitter dysfunction) and extrinsic causes (a result of the use of pharmacological agents) [131]. Intrinsic psychosis is thought to be caused by alterations in dopamine, serotonin, and acetylcholine systems involving subcortical projections, as well as synaptic and neuronal changes in limbic and cortical structures [131]. Visual hallucinations have been linked to the presence of Lewy bodies in the occipital lobe and in retinal neurons, and to the loss of dopaminergic amacrine cells and the regulatory role of D1 and D2 dopamine receptors in the eye [131]. Sleep disturbances commonly preceded psychosis in PD. Acetylcholine is an important neurotransmitter in the control of REM sleep and some researchers have suggested a link between hallucinations and REM sleep behavior disorder [132]. Various risk factors have been associated with the development of PD psychosis, including the use of dopamine agonists and a polymorphism in the cholecystokinin gene [133].
Management of psychosis is based as first step on reduction or discontinuation of dopaminergic medications, in particular dopamine agonists, and of drugs that may have negative effects such as anticholinergics, benzodiazepines, phenothiazines and uro-spasmolytics [134]. Antipsychotics with dopamine antagonism, such as haloperidol, risperidone, olanzapine, should not be given because can worsen motor symptoms, while atypical antipsychotics blocking serotonin may be useful, in particular clozapine which is the most efficacious, but requires blood monitoring for the risk of agranulocytosis [134]. A further option is to use cholinesterase inhibitors such as rivastigmine or donepezil [134]. Pimavanserin, a serotonin 2A receptor inverse agonist, obtained regulatory approval in the US for PD psychosis [135]. Psychosocial support for patients and caregivers are essential, visual aids and art therapy may also be helpful [136]. More research is needed in this area to determine what is the most effective treatment for managing this problem.
Compulsive Behaviors
Compulsive behaviors or impulse control disorders (ICDs) include a variety of repetitive, often rewarding, actions, from pathological shopping, eating, gambling, to hypersexuality and medication abuse [137]. These behaviors are problematic not only for the patients but also for the caregiver and are often not declared because they may be socially unacceptable, embarrassing and a source of financial distress; therefore, physicians must specifically ask for their presence during patients’ visits [137]. ICDs occur in 14% of PD patients, sometimes having more than one compulsive behavior, with pathological gambling ranging between 3% and 8%, hypersexuality occurring in 2-8% and compulsive medication use in 3-4% Therefore, physicians must specifically ask for their presence during patients’ visits [138]. Dopamine agonist use is one of the risk factors, even if they can occur also in untreated subjects, suggesting that other factors related to cognitive process, personality trait and psychiatric problems may play a role [137]. Levodopa is mainly implicated in dopamine dysregulation syndrome, associated with medication overuse [138]. The proposed pathophysiology is alterations in dopaminergic functions within the nucleus accumbens and ventral striatum and altered connections between striatal and associative/limbic cortical regions [139]. A balance of dopamine agonists dosage or a gradual reduction is recommended rather than a sudden or rapid discontinuation that may cause a withdrawal syndrome with panic attacks, dysphoria, dysautonomia, sleep disturbances and, in rare cases, symptoms resembling the neuroleptic malignant syndrome [138]. Subthalamic DBS may help manage ICDs, naltrexone and clonidine have insufficient evidence [138].
Sleep Disorders
Nearly all PD patients have sleep disturbances that usually start early with a prevalence of 60-98% [140]. The more common are rapid eye movement (REM) sleep behavior disorder (RBD); insomnia; restless legs syndrome (RLS); excessive daytime sleepiness (EDS) and sleep-disordered breathing [140]. The pathogenesis of sleep disorders is multifactorial; but degeneration of central sleep regulation centers in the brainstem and thalamocortical pathways with involvement of dopamine; serotonin; noradrenaline and hypocretin is the most important [140]. Other factors that may contribute to sleep disruption include motor symptoms; nocturia; anxiety; depression and dopaminergic treatments. Sleep disturbances greatly affect the quality of life of both patients and caregivers [141].
Rapid Eye Movement Sleep Behavior Disorder (RBD)
RBD is a parasomnia characterized by vivid and frightening dreams associated with elaborate motor activity, as the patients “act out” their dreams, and loss of muscle atonia during REM sleep [142]. Bed partners report on vocalizations and abnormal and sometimes violent movements that may cause injuries such as lacerations, ecchymosis and dislocations [142]. Diagnosis of RBD should be made via single-night PSG testing because clinical criteria alone have been shown to be only 33% sensitive [143]. RBD is thought to precede the onset of motor symptoms in over 40% of PD patients and RBD is a risk factor for cognitive impairment and psychosis in PD [142]. The pathological basis of RBD is still unclear: a hypothesis is that RBD arises because of degeneration of lower brainstem nuclei, consistent with Braak stages 1 and 2 [144]. Degeneration of the sublaterodorsal nucleus, with its direct and indirect projections to the spinal interneurons, has been implicated as well as involvement of the laterodorsal tegmental nucleus, perilocus coerulus region, nucleus reticularis magnocellularis, pedunculopontine nucleus and ventrolateral reticulospinal tracts [144]. Sleep fragmentation may result, in part, due to night-time motor fluctuations; this can be mitigated by continuous drug delivery or by controlled-release formulations of L-dopa or dopamine agonists [145]. Use of clonazepam , melatonin or rivastigmine and sodium oxybate may reduce involuntary nocturnal movements during sleep [145]. In a recent study, safinamide had a beneficial effect on patients’ sleep, as assessed by both clinical scales and PSG recordings, while rasagiline did not improve PSG sleep parameters nor clinical questionnaires [146].
Insomnia
Difficulty falling asleep and difficulties maintaining sleep are both common in PD and risk factors include advanced disease and female gender [11]. Sleep maintenance problems and consequent sleep fragmentation can be caused by many different problems, such as nighttime akinesia, dystonia and non-motor problems (nocturia, mood disorders, obstructive sleep apnea), and may be associated to daytime sleepiness [147]. Management of insomnia thus depends on a precise analysis of the cause: if the reason is mainly due to nocturnal PD symptoms, additional nighttime dopaminergic medications doses, preferably long-acting formulations, may be helpful [147]. For example, transdermal delivery of rotigotine can be effective in improving nocturnal motor disability, as rotigotine does not share the high affinity D3 receptor profile of other dopamine agonists [147]. When there is the suspect that insomnia could be a side effect of parkinsonian drugs, such as selegeline or dopamine agonists, it might be advisable to reduce their dosages or to change the treatment [148]. Antidepressants, anxiolitics and melatonin have weak evidence for sleep insomnia [148]. Regarding non-pharmacological treatments, basic practices of sleep hygiene are part of standard management [127]. CBT and bright light therapy may also improve sleep efficiency [127].
Restless Legs Syndrome and Periodic Limb Movements
Restless legs syndrome (RLS) and periodic limb movements are closely linked and are sensitive to dopaminergic treatments: 61% of PD patients report a correlation between RLS and nighttime “wearing off” [149]. RLS is defined as an unpleasant feeling in a limb that appears or worsen when the patients is sitting or lying, mainly in the evening or night, and may cause uncontrolled and sometimes potentially dangerous movements [149]. The relationship between PD and RLS is not completely clear, and their symptomatology may overlap [149]. RLS and periodic limb movements are another frequent cause of sleep disruption. The prevalence of RLS is estimated at 20% in PD patients, with an onset around 4.5 years after PD onset [150]. The pathophysiology of RLS and periodic limb movements is thought to be related to changes in mesocortical dopamine [150]. Continuous infusion of apomorphine resulted in significant reduction of nocturnal discomfort and decreased leg movements [151]. Long-acting levodopa or dopamine-agonists, gabapentin, pregabalin, intravenous iron can be considered while medications that can exacerbate symptoms, such as antidepressant, anticholinergics and antihistaminergics, should be avoided [151].
Excessive Daytime Sleepiness (EDS)
Excessive daytime sleepiness (EDS), defined as a debilitating trend to drift off to sleep, or rapid-onset sleep without any prior drowsiness, affects up to 60% of PD patients and can profoundly influence quality of life [152]. EDS can occur early in disease development, often predating diagnosis, and may be linked to cognitive impairment in the late stage of the disease [152]. Daytime somnolence is associated with poor concentration and memory and may lead to driving and/or occupational accidents [152]. The causes are multifactorial and include the disease process itself, nighttime sleep disruption, depression and drug therapy with agents including antihistamines, dopaminergic therapies, anxiolytics and selective serotonin reuptake inhibitors [153]. Haq et al. have suggested a secondary narcoleptic phenotype in PD connected to degeneration of hypocretin-containing neurons in the hypothalamus [154]. Saper et al. proposed that a “flip-flop” switch is responsible for the sleep-wake cycle in which wake- and sleep-promoting neurons inhibit each other, resulting in stable wakefulness and sleep; disruption of wake- or sleep-promoting pathways results in behavioral state instability [155]. Dopaminergic dysfunction and neural degeneration have been suggested to destabilize the switch and its regulators, promoting rapid transitions to sleep; dopamine agonists and levodopa could be involved [155]. EDS in PD patients is also associated with reduced spontaneous neural activity in the left angular gyrus and with reduced functional connectivity between the left angular gyrus and cerebellum [156]. EDS and sudden onset of sleep can increase the risk of serious injury, particularly if the patients drive a vehicle or operate machinery [152]. To improve these symptoms, exclusion/substitution of the suspected drugs can be tried. Treating patients with stimulants such as modafinil or caffeine is only partially efficacious, while anti-H3-receptor drugs, solriamfetol and sodium oxybate seem more promising [153]. Sleep-hygiene education, and exposure to bright light may be helpful [127].
Autonomic Symptoms
Autonomic dysfunction in PD is common and debilitating, encompassing bladder, gastrointestinal, cardiovascular and sexual problems and may precede motor features [157].
Bladder Dysfunction
Bladder dysfunction is a common complaint in PD and is associated with poorer QoL, alteration of daily routine, falls, and admission to care, with a greater effect on emotional and social wellbeing on men than in women [158]. Urinary symptoms that predominate are storage symptoms (urinary urgency, frequency, and nocturia), suggestive of detrusor hyperreflexia (hyperactive bladder due to detrusor overactivity), classified as “irritative”, and voiding symptoms (incomplete emptying, slow and/or interrupted stream, terminal dribble, hesitancy, and straining), classified as “obstructive” [158]. Over 80% of PD patients experience these problems during the course of their illness [158]. In the PRIAMO study, the urinary dysfunction was reported in 57.3% of patients ranging from 37.7% in early PD (Hoehn & Yahr stage 1) to 89.8% in late stage (Hoehn & Yahr stage 4-5). Nocturia is the most prevalent (up to 86% of patients) followed by urgency (33-71%) and frequency (16-68%) [159]. These symptoms may lead to urinary incontinence and cause sleep disturbances [158]. Dopaminergic mechanisms play an important role in normal micturition control: dopaminergic neurons have both inhibitory and stimulatory effects in micturition acting via D1 and D2-receptors, respectively [160]. Such neurons are particular abundant in the substantia nigra pars compacta (SNC) and the ventral tegmental area (VTA) of the midbrain and dysfunction of these mechanisms may lead to detrusor overactivity (DO) [161]. The most widely accepted theory is that basal ganglia inhibit the micturition reflex in the normal situation via D1 receptors and cell depletion in SNC in idiopathic PD result in loss of D1 receptor-mediated inhibition and consequently DO [161]. Severity of urinary dysfunction may also correlate with the relative degeneration of the caudate nucleus, which receives dopamine-rich innervations from the SNC and VTA [161]. When treating urinary dysfunction, it is important to establish if the symptoms are related to fluctuation; if so, adjustment of dopaminergic medication should be considered [161]. Levodopa showed both aggravation and alleviation of bladder symptoms, worsening detrusor overactivity during bladder filling, but facilitating voiding through an effect on detrusor contractility; apomorphine improved voiding efficiency by increasing urine flow and reducing post-void residual urine volume, although its effect on detrusor muscle may vary [161]. Antimuscarinics are the most routinely used class of drugs to treat overactive bladder symptoms in PD, in particular trihexyphenidyl and oxybutynin. However, these treatments should be used with caution in patients experiencing hallucinations and cognitive decline [161]. Moreover, blocking M2 and M3 receptors can lead to dry mouth and constipation. The beta-3 adrenergic agonist, mirabegron, and intravesical botulinum toxin injections can be also considered [162]. In a retrospective study, safinamide improved urinary symptoms (measured through the SCOPA Autonomic-Urinary scale), as well as urgency, incontinence, frequency and nocturia [163]. Non-pharmacological treatments include intermittent catheterization and pelvic floor therapy [164]. Finally, patients should be referred to a urologist for further investigation if persistent urinary symptoms are suspected to be related to bladder outlet obstruction.
Gastrointestinal Symptoms
Gastrointestinal disorders are present in more than half of patients with PD, including salivary excess (sialorrhea), dysphagia, nausea/gastroparesis, bowel dysfunction (constipation, defecatory dysfunction) and can cause serious complications, such as weight loss [165]. Gastrointestinal function is impaired primarily due to disruption to both extrinsic and intrinsic innervation of the gut, partly caused by accumulation of Lewy bodies and alpha-synuclein deposits in the dorsal motor nucleus of the vagus nerve, sacral parasympathetic nuclei, sympathetic ganglia, and the enteric nervous system, with consequent microbiome changes and inflammation [166]. Sialorrhea affects 70–80% of patients and is generally observed in the late stages of the disease and during ‘OFF’ states. Saliva accumulation is caused by poor bolus formation and reduced frequency and efficiency of swallowing [167]. Drooling can affect eating and speech and cause social embarrassment. Locally administered anticholinergic drugs may be helpful, but their use should be limited to short periods because they can aggravate constipation or cause urinary retention and cognitive impairment [168]. Sublingual atropine drops, ipratropium spray and botulinum toxin injections into salivary glands (except if there is esophageal dysmotility) may decrease saliva production without systemic adverse events [168]. Dysphagia affects from 30% to 80% of patients [169]. The oral, pharyngeal and esophageal stages of swallowing may be affected; pharyngeal dysmotility can lead to increased risk of aspiration [169]. There is no standard drug treatment for dysphagia; based on the literature, it is recommended to optimize the timing of administration of dopaminergic therapies to allow patients to consume meals in their best ON state [170]. Among pharmacological options, botulinum toxin injections in the cricopharyngeal muscle may improve upper oesophageal sphincter relaxation during swallowing [170]. Non-pharmacological methods include chin-down swallowing, encouraging patients to eat slowly and use of thickened liquids [170]. Continuous monitoring of patients’ diet and nutritional care by clinical nutritionists to reduce the risk of dehydration and malnutrition [170]. Nausea often is caused by dopaminergic medications but may develop without antiparkinsonian drugs due to impaired or delayed gastric emptying (gastroparesis), corresponding to decreased stomach motility, which can affect gut transit [165]. In addition to nausea, chronic problems with gastric emptying can cause a sensation of fullness, abdominal pain, and bloating [165]. Gastroparesis may interfere with drug absorption and bioavailability [171]. Oral levodopa is absorbed in the proximal small intestine; slow gastric emptying delays levodopa delivery to the small intestine, increasing pre-systemic decarboxylation and resulting in reduced absorption and efficacy [171]. This may in turn be the cause of motor fluctuations; various formulations of levodopa and other anti-Parkinson drugs have been proposed to overcome this problem, such as controlled-release, subcutaneous, liquid, patch and inhaled formulations [171]. Domperidone, a dopamine-2-receptor antagonist non crossing the blood-brain barrier, improve gastric emptying and drug absorption [172]. Serotonin 5-HT4 receptor agonist stimulating acetylcholine release such as cisapride and mosapride, have prokinetic properties, but their use is limited by their cardiotoxicity [172]. Constipation is the most common bowel dysfunction, affects around 50% of patients, and may precede the appearance of motor symptoms by several years [173]. The pathophysiologic basis for reduced bowel movement frequency is decreased colonic smooth muscle and phasic rectal contractions, with consequent prolonged colon transit time [173]. Management includes lifestyle and dietary modifications such as increasing physical activity and the consumption of fiber (e.g. psyllium) and fluids [174]. A stool softener such as docusate may also be helpful as well as osmotic laxatives (for short-term use), prokinetic agents, lubiprostone (a chloride channel activator) and probiotics [174]. Defecatory disfunction may be present in up to 70% of PD patients, resulting in pain, excessive straining and incomplete elimination [175]. There are no specific treatments; possible therapies include biofeedback training to relax muscles and botulinum toxin injections into the puberorectalis muscle [175].
Neurogenic Orthostatic Hypotension
Blood pressure dysregulation is the most frequent cardiovascular symptom in PD, particularly neurogenic orthostatic hypotension (OH) and neurogenic hypertension in the supine position (SH) [176]. Management of symptomatic OH and SH can be challenging as treating one usually aggravates the other [176]. OH is the most common manifestation with a reported frequency of 30–58%; this prevalence increases with age and disease duration [176]. OH is highly disabling, increasing fall risk and decreasing independence [177]. Light headedness, blurry vision, dizziness and faint feeling are easily recognized as caused by OH; other less specific symptoms include tiredness, cognitive impairment, dyspnea, neck and shoulder discomfort, or angina [177]. Symptoms are due to tissue hypoperfusion as a result of OH, defined as a sustained fall in blood pressure of ≥20 mmHg systolic or 10 mmHg diastolic when moving from supine to standing [177]. Normally, vasoconstriction maintains blood pressure in the standing position: standing up unloads the baroreceptors, which triggers norepinephrine release from sympathetic post-ganglionic nerves causing vasoconstriction [178]. In patients with PD, this compensatory mechanism is absent or attenuated [178]. Plasma norepinephrine, a marker of sympathetic neuronal integrity, is lower in PD patients with OH; imaging and neuropathological data show that post-ganglionic peripheral sympathetic neurons innervating the myocardium and sympathetic fibers innervating blood vessels are functionally affected due to α-synuclein deposits and fiber loss [178]. This results in impaired norepinephrine release and defective vasoconstriction upon standing, causing the fall in blood pressure [178]. Consensus guidelines for the treatment of OH state that the goal is not to normalize standing blood pressure, but to reduce symptom burden, improve QoL, and reduce morbidity and mortality [179]. The first step to manage OH is to correct aggravating factors (eliminating if not necessary diuretics, vasodilators, and drugs that block norepinephrine release/activity at the neurovascular junction), investigating and treating anemia, adjust the doses of levodopa and/or dopamine agonists [179]. Non-pharmacological strategies include reducing or stopping caffeine and alcohol intake, due to their diuretic and vasodilator effect, avoid sugar beverages, increase fluid intake, physical exercise and salt intake (sodium-enriched diet), eating smaller, more frequent meals, and reduce carbohydrates in case of postprandial hypotension, elevate head position during sleep, use support stocking or abdominal binders [179]. Drug therapies include fludrocortisone or increase peripheral vascular resistance with midodrine, droxidopa, or norepinephrine transporter inhibitors (atomoxetine) [179].
Sexual Dysfunction
Sexual dysfunction is frequently experienced by PD patients, with a prevalence from 36% to 80%, and is often poorly discussed or investigated in clinical evaluations [180]. Over two-thirds of male patients have erectile dysfunction (ED) while 70% of female patients have decreased libido, moreover women with PD are more likely to have anxiety or inhibition during sex, vaginal tightness, and involuntary urination [180]. The use of SSRIs for comorbid depression or advanced disease stage may contribute to the development of sexual dysfunction [180]. The exact pathophysiology of sexual dysfunction is not fully known; it has been hypothesized that disturbances of the mesocortical and mesolimbic dopaminergic pathways may be involved [181]. Libido and erection are thought to be regulated by the hypothalamus, particularly the medial preoptic area [181]. Dopamine may help to facilitate sexual behavior; in the paraventricular nucleus, dopamine activates oxytogeneric neurons that project to the hippocampus, medulla oblungata, and spinal cord, all of which play important roles in sexual motivation and reward [181]. Dopamine agonists such as apomorphine, ropinirole and pergolide mesylate may be helpful in inducing erection; however, dopamine agonists may induce impulse control disorders, including hypersexuality [182]. The phosphodiesterase-5 inhibitor sildenafil is effective for treating erectile dysfunction but standing and lying blood pressure must be measured before prescribing this agent for potential orthostatic hypotension induction or deterioration [183]. Moreover, sildenafil cannot be given with nitrate therapy for cardiac disorders [183]. Lubrication agents and systemic or local hormonal replacement therapy may help women with decreased libido; sex and behavioral therapy may also be helpful for addressing the psychosocial and relationship aspects of sexual dysfunction [182].
Fatigue
Fatigue in PD is a subjective sensation of profound tiredness, lack of energy and exhaustion, contributing to restriction in daily activities and social activities and associated with nonrestorative rest [184]. Even if fatigue often coexists with apathy, anxiety, depression, and sleep disorders, it is an independent symptom [184]. Up to 70% of the PD population experience fatigue, which may emerge at the early stages of the disease or in the prodromal phase before motor symptoms and can be divided into mental or physical fatigue [185]. Physical fatigue is a lack of energy to perform physical tasks despite personal ability and motivation. Mental fatigue refers to the cognitive effects experienced during and after prolonged periods of activities requiring sustained concentration, such as driving in heavy traffic [185]. Despite its relevance, its pathophysiological mechanisms are still not fully understood. Several hypotheses suggest a multifactorial aetiology involving dysfunction in dopaminergic and non-dopaminergic pathways, neuroinflammation, genetic predispositions, and metabolic dysregulation [186]. Patients with fatigue have abnormalities in the prefrontal cortex, reduced perfusion in the frontal lobe and dysfunction in the connections between the hypothalamic-pituitary axis and the basal ganglia [186]. Dopamine is only partially involved in the pathogenesis of fatigue; other neurotransmitter systems should be considered, in particular serotonin and glutamate [187]. A study published by Pavese et al. showed a significant reduction in serotonin reuptake transporter binding in the caudate nucleus, putamen, ventral striatum, thalamus, cingu late and amygdala of PD patients with fatigue compared to those without [188]. Glutamatergic dysregulation is another key factor. Glutamate hyperactivity is implicated in excitotoxicity, oxidative stress, and neuronal death and contributes to the development of chronic inflammation, triggering the release of pro-inflammatory cytokines, such as interleukin-6 and tumour necrosis factor-alpha [189]. High levels of these cytokines have been observed in patients with severe fatigue; therefore, targeting glutamatergic pathways to mitigate neuroinflammation may represent a promising therapeutic approach for alleviating symptoms [189]. Recent studies showed a significant association between elevated levels of cerebrospinal fluid alpha-synuclein and fatigue in PD patients, probably due the activation of pro-inflammatory cytokines released in the microglial cells mediated by alpha-synuclein [190]. Versace et al. reported an increase in fatigue due to a reduction in GABAergic inhibition in the primary motor cortex [191]. Genetic factors also affect the fatigue mechanism, as several mutations have been linked with fatigue in patients with PD, such as glucocerebrosidase beta mutations [192] and the leucine-rich repeat kinase 2 (LRRK2) G2385R variant [193]. Management of fatigue requires a complex approach, including pharmacological, non-pharmacological, and lifestyle interventions [194]. Optimization of pharmacotherapy may alleviate fatigue. Levodopa and intrajejunal levodopa infusion may improve physical fatigue in PD patients; dopamine agonists, apomorphine and selegiline have controversial effects in fatigue management, while rasagiline was found to decrease the score of the fatigue scale in PD patients [195]. Some studies support safinamide’s role in reducing PD-related fatigue, possibly due to its effect on glutamate regulation [196]. Modafinil and methylphenidate, two stimulant agents, may reduce physical fatigue, doxepin (a tricyclic antidepressant) was associated with improvement in general fatigue (both physical and mental), while caffeine and memantine have no effect [195]. Bilateral subthalamic deep brain stimulation has shown potential benefits for some non-motor symptoms including fatigue [197]. Non-pharmacological strategies, such as structured exercise programs (in particular Nordic walking), CBT, dance therapy and sleep optimization, have shown benefits in reducing fatigue severity [198]. Additionally, lifestyle modifications, including maintaining a balanced diet, managing stress, and ensuring proper hydration, can help improve energy levels [199].
Conclusions
In addition to motor symptoms, NMS are now recognized as extremely frequent and important components of PD that can appear during all stages of the disorder, including the premotor stage. NMS are a major determinant of health-related quality of life and progression of overall disability, emphasizing the need for early detection and awareness. NMS substantially increase the cost of care, requiring frequent hospitalization, nursing home assistance and specific treatments, and pose a major challenge to healthcare professionals. With the necessary knowledge and expertise, clinicians should be able to timely and accurately detect NMS in PD and implement appropriate interventions to mitigate their impact.
Authors Contribution
All authors contributed to writing and reviewing the paper, collecting, and analysing the literature, designing the tables/figure and approved the final manuscript.
Ethical Approval
No new clinical or pre-clinical studies were performed by the authors, nor patients were recruited for this narrative review. No patient-specific efficacy or safety data were reported; therefore, institutional review board/ethics approval was not required.
Funding
Financial support was provided by Zambon SpA, however Zambon SpA was not involved in writing of this article or the decision to submit it for publication. The opinions expressed in this article are those of the authors who did not receive any honorarium/fee or other form of financial support related to the development of this publication.
<i>Acknowledgments</i>
None.
Conflicts of Interest
C. Cattaneo is an employee of Zambon SpA, G. Fabbrini has received compensation for speaker related activities from Zambon SpA., A. Fabbrini, D. Belvisi D, F. Aiello, F. Marchet do not received any compensation from Zambon SpA nor had any previous relationship with Zambon SpA., All authors declare no other competing interests or financial disclosures for this review.
References
- Tanner CM, Ostrem JL. Parkinson’s disease. N Engl J Med 2024; 391(5): 442-452.
- Su D, Cui Y, He C et al. Projections for prevalence of Parkinson’s disease and its driving factor in 195 countries and territories to 2050: modelling study of Global Burden of Disease Study 2021; BMJ 2025; 388: e080952.
- Dahodwala N, Li P, Jahnke J et al. Burden of Parkinson’s disease by severity: health care costs in the US Medicare population. Mov Disord 2021; 36(1): 133-142.
- Goldman, JG. Non-motor symptoms and treatments in Parkinson’s disease. Neurol Clin 2025; 43(2): 291-317.
- Rietdijk CD, Perez-Pardo P, Garssen J et al. Exploring Braak’s hypothesis of Parkinson’s disease. Front Neurol 2017; 8: 37.
- Van der Meer F, Jorgensen H, Hiligsmann M. Burden of non-motor symptoms of Parkinson’s disease: cost-of-illness and quality-of-life estimates through a scoping review. Expert Rev Pharmacoecon Outcomes Res 2025; 25(1): 17-27.
- Rukavina K, Batzu L, Boogers A et al. Non-motor complications in late-stage Parkinson’s disease: recognition, management and unmet needs. Expert Rev Neurother 2021; 21(3): 335-352.
- Lewitt PA, Chaudhury KR. Unmet needs in Parkinsons disease: motor and non-motor. Parkinsonism Relat Disord 2020; 80(Suppl 1): S7-S12.
- Hurt CS, Rixon L, Chaudhuri KR et al. Barriers to reporting non-motor symptoms to health-care providers in people with Parkinson’s. Parkinsonism Relat Disord 2019; 64: 220–225.
- Crosiers D, Pickut B, Theuns J et al. Non-motor symptoms in a Flanders-Belgian population of 215 Parkinson’s disease patients as assessed by the non-motor symptoms questionnaire. American Journal of Neurodegenerative Disease 2012; 1(2): 160-167.
- Cattaneo C, Pagonabarraga J. Sex differences in Parkinson’s disease: a narrative review. Neurol Ther 2025; 14(1): 57-70.
- Brun L, Lefaucheur R, Fetter D et al. Non-motor fluctuations in Parkinson’s disease: prevalence, characteristics and management in a large cohort of parkinsonian outpatients. Clin Neurol Neurosurg 2014; 127: 93–96.
- Batzu L, Podlewska A, Gibson L et al. A general clinical overview of the non-motor symptoms in Parkinson’s disease: neuropsychiatric symptoms. Int Rev Neurobiol 2024; 174: 59-97.
- Sauerbier A, Lenka A, Aris A, Pal PK. Nonmotor symptoms in Parkinson's disease: gender and ethnic differences. Int Rev Neurobiol 2017; 133: 417-446.
- Jercic KG, Blazekovic A, Borovecki S, Borovecki F. Non-motor symptoms of Parkinson’s disease - insights from genetics. J Neural Transm 2024; 131(11): 1277-1284.
- Song T, Zhou X, Wang C et al. Clinical features and progression of Parkinson’s disease with LLRK2 variants: a prospective study. Ann Clin Transl Neurol 2025; 12(1): 34-42.
- Brockmann K, Srulijes K, Pflederer S et al. GBA-associated Parkinsons’ disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 2015; 30(3): 407-411.
- Li Q, Jing Y, Lun P et al. Association of gender and age at onset with glucocerebrosidase associated Parkinson’s disease: a systematic review and meta-analysis. Neurol Sci 2021; 42(6): 2261–71.
- Chen H, Burton EA, Ross GW et al., Research on the premotor symptoms of Parkinson’s disease: clinical and etiological implications. Environ Health Perspect 2013; 121: 1245–1252.
- Hely MA, Reid WG, Adena M, et al. The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov Disord 2008; 23: 837–844.
- Morris HR, Spillantini MG, Sue CM, Williams-Gray CH. The pathogenesis of Parkinson’s disease. Lancet 2024; 403(10423): 293–304.
- Berg D, Postuma RB, Bloem B et al. Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson’s disease. Mov Disord 2014; 29: 454–62.
- Schapira AHV, Chaudhuri KR and Jenner P. Non-motor features of Parkinson disease. Nat Rev Neurosci 2017; 18: 435–450.
- Park A and Stacy, M. Dopamine-induced nonmotor symptoms of Parkinson’s disease. Parkinsons Dis 2011: 485063.
- Politis M and Loane, C. Serotonergic dysfunction in Parkinson’s disease and its relevance to disability. ScientificWorldJournal 2011; 11: 1726–1734.
- Delaville C, Deurwaerdère PD and Benazzouz A. Noradrenaline and Parkinson’s disease. Front Syst Neurosci 2011; 5: 31.
- Remy P, Doder M, Lees A et al. Depression in Parkinson’s disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain 2005; 128: 1314–1322.
- Jenner P and Caccia, C. The role of glutamate in the healthy brain and in the pathophysiology of Parkinson’s disease. Eur Neurol Rev 2019; 14: 2–12.
- Müller MLTM, Bohnen NI. Cholinergic dysfunction in Parkinson’s disease. Curr Neurol Neurosci Rep 2013; 13(9): 377.
- Pasquini J, Brooks DJ, Pavese N. The involvement of the cholinergic system in Parkinson disease. Handb Clin Neurol 2025; 211: 215-229.
- Alharbi B, Al-Kuraishy HM, Al-Gareeb AL et al. Role of GABA pathway in motor and non-motor symptoms in Parkinson’s disease: a bidirectional circuit. Eur J Med Res 2024; 29(1): 205.
- Mehra S, Sahay S, Maji SK. Alpha-synuclein misfolding and aggregation: implications in Parkinson’s disease pathogenesis. Biochim Biophys Acta Proteins Proteom 2019; 1867(10): 890-908.
- Del Tredici K, Braak H. Idiopathic Parkinson’s disease: staging an α -synucleinopathy with a predictable pathoanatomy. In: Kahle P, Haass C, eds. Molecular mechanisms in Parkinson’s disease. Georgetown, TX: Landes Bioscience, 2004; 1–32.
- Braak H, Del Tredici K, Rüb U et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24: 197–211.
- Halliday G, Lees A, Stern M. Milestones in Parkinson’s disease – clinical and pathologic features. Mov Disord 2011; 26(6): 1015–1021.
- Kulcsarova K, Skorvanek M, Postuma RB, Berg D. Defining Parkinson’s disease: past and future. J Parkinsons Dis 2024; 14(s2): S257-S271.
- Global Parkinson’s Disease Survey Steering Committee. Factors impacting on quality of life in Parkinson’s disease: results from an international survey. Mov Disord 2022; 17: 60-67.
- Wu DD; Su W, He J et al. Nonmotor symptoms and quality of life in Parkinson’s disease with different motor subtypes. Z Gerontol Geriatr 2022; 55(6): 496-501.
- Candel-Parra E, Corcoles-Jimenez MP, Delicado-Useros V et al. Evolution of quality of life in persons with Parkinson’s disease: a prospective cohort study. J Clin Med 2021; 10(9): 1824.
- Martinez-Martin P, Macaulay D, Jalundhwala YJ et al. The long- term direct and indirect economic burden among Parkinson’s disease caregivers in the United States. Mov Disord 2019; 34(2): 236–245.
- Hermanowicz N, Jones SA and Hauser RA. Impact of non-motor symptoms in Parkinson’s disease: a PMDAlliance survey. Neuropsychi atr Dis Treat 2019; 15: 2205–2212.
- Gumber A, Ramaswamy B, Thongchundee O. Effects of Parkinson’s on employment, cost of care, and quality of life of people with condition and family caregivers in the UK: a systematic literature review. Patient Relat Outcome Meas 2019; 10: 321–333.
- Yang W, Hamilton JL, Kopil C et al. Current and projected future economic burden of Parkinson’s disease in the U.S. NPJ Parkinsons Dis 2020; 6: 15.
- Findley L, Aujla M, Bain PG et al. Direct economic impact of Parkinson’s disease: a research survey in the United Kingdom. Mov Disord 2003; 18: 1139-1145.
- Mudiyanselage SB, Watts JJ, Abimanyi-Ochom J et al. Cost of living with Parkinson’s disease over 12 months in Australia: a prospective cohort study. Parkinsons Dis 2017; 2017: 5932675.
- Gustafsson A, Hjalte F, Norlin J et al. The association between non-motor symptoms and cost in Parkinson’s disease. J Neurol 2025; 272(4): 297.
- Weise D, Claus I, Dresel C et al. Multidisciplinary care in Parkinson’s disease. J Neural Transm 2024; 131(10): 1217–1227.
- Radder DLM, Lennaerts HH, Vermeulen H et al. The cost-effectiveness of specialized nursing interventions for people with Parkinson’s disease: the NICE-PD study protocol for a randomized controlled clinical trial. Trials 2020; 21(1): 88.
- Hellqvist C, Berterö C. Support supplied by Parkinson’s disease specialist nurses to Parkinson’s disease patients and their spouses. Appl Nurs Res 2015; 28(2): 86–91.
- Fujita T, Iwaki M, Hatono Y. The role of nurses for patients with Parkinson’s disease at home: a scoping review. BMS Nurs 2024; 23(1): 318.
- Chaudhuri KR, Martinez-Martin P, Schapira AH et al. International multicenter pilot study of the first comprehensive self-completed nonmotor symptoms questionnaire for Parkinson’s disease: the NMSQuest study. Mov Disord 2006; 21: 916-923.
- Chaudhuri KR, Martinez-Martin P, Brown RG et al. The metric properties of a novel non-motor symptoms scale for Parkinson’s disease: Results from an international pilot study. Mov Disord 2007; 22: 1901–1911.
- Marinus J, Visser M, Stiggelbout AM et al. A short scale for the assessment of motor impairments and disabilities in Parkinson's disease: the SPES/SCOPA. J Neurol Neurosurg Psychiatry 2004; 75(3): 388-95.
- Goetz CG, Tilley BC, Shaftman SR et al. Movement Disorder Society -sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord 2008; 23: 2129-2170.
- Peto V, Jenkinson C, Fitzpatrick R et al. The development and validation of a short measure of functioning and well-being for individuals with Parkinson’s disease. Qual Lif Res 1995; 4: 241-248.
- Chaudhuri KR, Rizos A, Trenkwalder C et al. King´s Parkinson´s disease pain scale, the first scale for pain in PD: an international validation. Mov Disord 2015; 30: 1623–163.
- Trenkwalder C, Kohnen R, Högl B et al. Parkinson’s disease sleep scale validation of the revised version PDSS-2. Mov. Disord 2011; 26(4): 644-652.
- Johns, MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 1991; 14(6): 540-545.
- Hamilton, M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960: 23: 56–62.
- Beck AJ, Ward CH, Mendelson M et al. An inventory for measuring depression. Arch Gen Psychiatry 1961; 4: 561–71.
- Krupp LB, LaRocca NG, Muir-Nash J et al. The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Arch Neurol 1989; 46(10): 1121-1123.
- Brown RG, Dittner A, Findley L et al. The Parkinson fatigue scale. Parkinsonism Relat Disord 2005; 11(1): 49–55.
- Antonini A, Tinazzi M, Abbruzzese G et al. Pain in Parkinson’s disease: facts and uncertainties. Eur J Neurol 2018; 25: 917-e69.
- Ford, B. Pain in Parkinson’s disease. Clin Neurosci 1998; 5(2): 63–72.
- Mylius V, Perez Lloret S, Cury RG et al. The Parkinson disease pain classification system: results from an international mechanism-based classification approach. Pain 2021; 162: 1201–1210.
- Cattaneo C, Jost WH. Pain in Parkinson’s disease: pathophysiology, classification and treatment. J Integr Neurosci 2023; 22(5): 132.
- Storch A, Bremer A, Gandor F et al. Pain fluctuations in Parkinson’s disease and their association with motor and non-motor fluctuations. J Parkinsons Dis 2024; 14(7): 1451-1468.
- Tinazzi M, Gandolfi M, Artusi CA et al. Advances in diagnosis, classification and management of pain in Parkinson’s disease. Lancet Neurol 2025; 24(4): 331-347.
- Pagonabarraga J, Tinazzi M, Caccia C, Jost WH. The role of glutamatergic neurotransmission in the motor and non-motor symptoms in Parkinson’s disease: clinical cases and a review of the literature. J Clin Neurosci 2021; 90: 178–183.
- Gao L, Yang Y, Cai L, Xiong Y. Gender differences in pain subtypes among patients with Parkinson’s disease. J Integr Neurosci 2022; 21(4): 120.
- Mehanna R, Jankovic J. Young-onset Parkinson’s disease: Its unique features and their impact on quality of life. Parkinsonism Relat Disord 2019; 65: 39–48.
- Eryilmaz IE, Erer S, Zarifoglu M et al. Contribution of functional dopamine D2 and D3 receptor variants to motor and non-motor symptoms of early onset Parkinson’s disease. Clin Neurol Neurosurg 2020; 199: 106257.
- Zhang LM, Zhang XP, Chen YQ, Ye W. Association of CHRNA4 gene rs1044396 and rs1044397 polymorphisms with Parkinson’s disease symptoms and smoking. Genet Mol Res 2015; 14(2): 5112–5122.
- Osikowicz M, Mika J, Przewlocka B. The glutamatergic system as a target for neuropathic pain relief. Exp Physiol 2013; 98(2): 372–384.
- Qureshi AR, Rana AQ, Malik SH et al. Comprehensive examination of therapies for pain in Parkinson’s disease: a systematic review and meta-analysis. Neuroepidemiology 2018; 51(3-4): 190–206.
- Santos García D, Yáñez Baña R, Labandeira Guerra C et al. Pain improvement in Parkinson’s disease patients treated with safinamide: results from the SAFINONMOTOR study. J Pers Med 2021; 11(8): 798.
- Edinoff A, Sathivadivel N, McBride T et al. Chronic pain treatment strategies in Parkinsons’ disease. Neurol Int 2020; 12(3): 61-76.
- Brefel-Courbon C, Grolleau S, Thalamas C et al. Comparison of chronic analgesic drugs prevalence in Parkinson’s disease, other chronic diseases and the general population. Pain 2009; 141(1-2): 14-18.
- Li J, Mi TM, Zhu BF et al. High-frequency repetitive transcranial magnetic stimulation over the primary motor cortex relieves musculoskeletal pain in patients with Parkinson’s disease: a randomized controlled trial. Parkinsonism Relat Disord 2020; 80: 113–119.
- Huissoud M, Boussac M, Joineau K et al. The effectiveness and safety of non-pharmacological intervention for pain management in Parkinson’s disease: a systematic review. Rev Neurol 2024; 180(8): 715–735.
- Ponsen MM, Stoffers D, Booij J et al. Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann Neurol 2004; 56: 173–181.
- Haehner A, Hummel T and Reichmann H. Olfactory loss in Parkinson’s disease. Parkinson’s Disease 2011; 2011: 450939.
- Mitchell E, Mattjie C, Bestwick JP et al. Hyposmia in Parkinson’s disease; exploring selective odour loss. NPJ Parkinsons Dis 2025; 11(1): 67.
- Marek K, Russel DS, Concha-Marambio L et al. Evidence of alpha-synuclein aggregation in older individuals with hyposmia: a cross-sectional study. EBioMedicine 2025; 112: 105567.
- Hawkes, C. Olfaction in neurodegenerative disorders. Mov Disord 2003; 18(4): 364–372.
- Ubeda-Bañon I, Saiz-Sanchez D, de la Rosa-Prieto C et al. Alpha-synucle inopathy in the human olfactory system in Parkinson’s disease: involvement of calcium-binding protein- and substance P-positive cells. Acta Neuropathol 2010; 119: 723–735.
- Nieto-Esamez F, Obrero-Gaitan E, Cortés-Pérez I. Visual dysfunction in Parkinson’s disease. Brain Sci 2023; 13(8): 1173.
- Gryc W, Roberts KA, Zabetian CP et al. Hallucinations and development of dementia in Parkinson’s disease. J Parkinsons Dis 2020; 10(4): 1643-1648.
- Tran KKN, Lee PY, Finkelstein DI et al. Altered outer retinal structure, electrophysiology and visual perception in Parkinson’s disease. J Parkinsons Dis 2024; 14(1): 167-180.
- Borm CDJM, Bloem BR, Hoyng C et al. The many faces of blurry vision in Parkinson’s disease: an illustrative case series. Case Rep Neurol 2022; 14(1): 173-178.
- Van der Lijn I, de Haan GA, Huizinga F et al. Self-reported visual complaints in people with Parkinson’s disease: a systematic review. J Parkinsons Dis 2022; 12(3): 785-806.
- Kedar, S. Symptomatic treatment of neuro-ophthalmic visual disturbances. Continuum 2025; 31(2): 566-582.
- Cong S, Xiang C, Zhang S et al. Prevalence and clinical aspects of depression in Parkinson’s disease: a systematic review and meta-analysis of 129 studies. Neurosci Biobehav Rev. 2022; 141: 104749.
- Dujardin K, Sgambato V. Neuropsychiatric disorders in Parkinson’s disease: what do we know about the role of dopaminergic and non-dopaminergic systems? Front Neurosci 2020; 14: 25.
- Mai AS, Chao Y, Xiao B et al. Risk of suicidal ideation and behaviour in individuals with Parkinson disease: a systematic review and meta-analysis. JAMA Neurol 2024; 81(1): 10-18.
- Mills KA; Greene MC, Dezube R et al. Efficacy and tolerability of antidepressants in Parkinson’s disease: a systematic review and network meta-analysis. Int J Geriatr Psychiatry 2018; 33(4): 642-651.
- Barone P, Poewe W, Albrecht S et al. Pramipexole for the treatment of depressive symptoms in patients with Parkinson’s disease: a randomized, double-bind, placebo-controlled trial. Lancet Neurol 2010; 22: 573-580.
- Barone P, Santangelo G, Morgante L et al. A randomized clinical trial to evaluate the effects of rasagiline on depressive symptoms in non-demented Parkinson’s disease patients. Eur J Neurol 2015; 22: 1184-1191.
- Miladinovic T, Nashed MG, Singh G. Overview of glutamatergic dysregulation in central pathologies. Biomolecules 2015; 5: 3122-3141.
- Peña E, Borrué C, Mata M et al. Impact of SAfinamide on Depressive symptoms in Parkinson’s Disease patients (SADness-PD study): a multicenter retrospective study. Brain Sci 2021; 11(2): 232.
- Wamelen DJV, Rukavina K, Podlewska AM, Chaudhuri KR. Advances in the pharmacological and non-pharmacological management of non-motor symptoms in Parkinson’s disease: an update since 2017. Cur Neuropharmacol 2023; 21(8): 1786-1805.
- Dobkin RD, Mann SL, Interian A et al. Cognitive behavioral therapy improves diverse profiles of depressive symptoms in Parkinson’s disease. Int J Geriatr Psychiatry 2019; 34(5): 722–729.
- Takamiya A, Seki M, Kudo S et al. Electroconvulsive therapy for Parkinson’s disease: a systematic review and meta-analysis. Mov Disord 2021; 36(1): 50-58.
- Dissanayaka NN, Forbes EJ Perepezko K et al. Phenomenology of atypical anxiety disorders in Parkinson’s disease: a systematic review. Am J Geriatr Psychiatry. 2022; 30: 1026–1050.
- Wang Z, Wei H, Xin Y, Qin W. Advances in the study of depression and anxiety in Parkinson’s disease: a review. Medicine 2025; 104(10): e41674.
- Lintel H, Corpuz T, Paracha SU, Grossberg GT. Mood disorders and anxiety in Parkinson’s disease: current concepts. J Geriatr Psychiatry Neurol 2021; 34:280–288.
- Perepezko K, Naaz F, Wagandt C et al. Anxiety in Parkinson’s disease: a systematic review of neuroimaging studies. J Neuropsychiatry Clin Neurosci 2021; 33(4): 280-294.
- Pontone GM, Mills KA. Optimal treatment of depression and anxiety in Parkinson’s disease. Am J Geriatr Psychiatry 2021; 29(6): 530-540.
- Moonen AJH, Mulders AEP, Defebvre L et al. Cognitive behavioral therapy for anxiety in Parkinson’s disease: a randomized controlled trial. Mov Disord 2021; 36(11): 2539-2548.
- Bereau M, Van Waes V, Servant M et al. Apathy in Parkinson’s disease: clinical patterns and neurobiological basis. Cells 2023; 12(12): 1599.
- Miller DS, Robert P, Ereshefsky L et al. Diagnostic criteria for apathy in neurocognitive disorders. Alzheimers Dement 2021; 17(12): 1892-1904.
- Yang C, Hu Y, Talishinsky AD et al. Medial prefrontal cortex and anteromedial thalamus interaction regulates goal-directed behavior and dopaminergic neuron activity. Nat Commun 2022; 13: 1386.
- Maggi G, Loayza F, Vitale C et al. Anatomical correlates of apathy and impulsivity co-occurrence in early Parkinson’s disease. J Neurol 2024; 271(5): 2798-2809.
- Vachez YM, Bahout M, Magnard R et al. Unilateral and Bilateral Subthalamic Deep Brain Stimulation Differently Favour Apathy in Parkinson's Disease. Eur J Neurosci 2025; 61(4): e70019.
- Mele B, Van S, Holroyd-Leduc J et al. Diagnosis, treatment and management of apathy in Parkinson’s disease: a scoping review. BMJ Open 2020; 10(9): e037632.
- Strasser A, Luksys G, Xin L et al. Glutamine-to-Glutamate ratio in the nucleus accumbens predicts effort based motivated performance in humans. Neuropsychopharmacology 2020; 45: 2048–57.
- Kulisevsky J, Martínez-Horta S, Campolongo A et al. A randomized clinical trial to evaluate the effects of safinamide on apathetic non-demented patients with Parkinson’s disease. Front Neurol 2022; 13:866502.
- Devos D, Moreau C, Maltete D et al. Rivastigmine in apathetic but dementia and depression-free patients with Parkinson’s disease: a double-blind, placebo-controlled, randomized clinical trial. J Neurol Neurosurg Psychiatry 2014; 85(6): 668–674.
- Mele B, Ismail Z, Goodarzi Z et al. Non-pharmacologic interventions to treat apathy in Parkinson's disease: a realist review. Clin Park Relat Disord 2021; 4: 100096.
- Aarsland D, Batzu L, Halliday GM et al. Parkinson disease-associated cognitive impairment. Nat Rev Dis Primers 2021; 7(1): 47.
- Goldman JG, Sieg E. Cognitive impairment and dementia in Parkinson’s disease. Clin Geriatr Med 2020; 36(2): 365-377.
- Guo Y, Xu W, Liu FT et al. Modifiable risk factors for cognitive impairment in Parkinson’s disease: a systematic review and meta-analysis of prospective cohort studies. Mov Disord 2019; 34: 876–883.
- Pourzinal D, Yang J, Lawson RA et al. Systematic review of data-driven cognitive subtypes in Parkinson disease. Eur J Neurol 2022; 29(11): 3395-3417.
- Singh, S. Noradrenergic pathways of locus coeruleus in Parkinson's and Alzheimer's pathology. Int J Neurosci 2020; 130(3): 251-261.
- Sun C, Armstrong MJ. Treatment of Parkinson's disease with cognitive impairment: current approaches and future directions. Behav Sci 2021; 11(4): 54.
- Ghosh A, Das S, Behera SK et al. Atomoxetine does not improve complex attention in idiopathic Parkinson’s disease patients with cognitive deficits: a meta-analysis. Parkinsons Dis 2020; 2020: 4853590.
- Zhang Y, Liu S, Xu K et al. Non-pharmacological therapies for treating non-motor symptoms in patients with Parkinson’s disease: a systematic review and meta-analysis. Front Aging Neurosci 2024; 16: 1363115.
- Chendo I, Silva C, Duarte GS et al. Frequency and characteristics of psychosis in Parkinson's disease: a systematic review and meta-analysis. J Parkinsons Dis. 2022;12(1):85-94.
- Pagonabarraga J, Bejr-Kasem H, Martinez-Horta S, Kulisevsky J. Parkinson disease psychosis: from phenomenology to neurobiological mechanisms. Nat Rev Neurol 2024; 20(3): 135-150.
- Brown P, Freeman D, Loe BS et al. Paranoid and unusual sensory experiences in Parkinson’s disease. Aging Ment Health 2025; 29(5): 935-950.
- Pisani S, Gunasekera B, Lu Y et al. Functional and connectivity correlates associated with Parkinson’s disease psychosis: a systematic review. Brain Commun 2024; 6(6): fcae358.
- Komagamine T, Suzuki K, Kokubun N et al. Sleep-related hallucinations in patients with Parkinson’s disease. PLoS One 2022; 17(10): e0276736.
- Goldman JG, Goetz CG, Berry-Kravis E et al. Genetic polymorphisms in Parkinson’s disease subjects with and without hallucinations: an analysis of the cholecystokinin system. Arch Neurol 2004; 61(8): 1280-1284.
- Segal GS, Xie SJ, Paracha SU, Grossberg GT. Psychosis in Parkinson's disease: current treatment options and impact on patients and caregivers. J Geriatr Psychiatry Neurol 2021; 34(4): 274-279.
- Tampi RR, Tampi DJ, Young JJ et al. Evidence for using pimavanserin for the treatment of Parkinson’s disease psychosis. World J Psychiatry 2019; 9(3): 47-54.
- Rose O, Huber S, Trinka E et al. Treatment of Parkinson’s disease psychosis – a systematic review and multi-methods approach. Biomedicines 2024; 12(10) :2317.
- Jellinger, KA. Behavioral disorders in Parkinson’s disease: current view. J Neural Transm 2025; 132(2): 169-201.
- Zhang JF, Wang XX, Feng Y et al. Impulse control disorders in Parkinson’s disease: epidemiology, pathogenesis and therapeutic strategies. Front Psychiatry 2021; 12: 635494.
- Ansari MF, Prasad S, Bhardwaj S et al. Morphometric alterations of the mesocorticolimbic network in Parkinson’s disease with impulse control disorders. J Neural Transm 2024; 131: 229–237.
- Gros P, Videnovic A. Overview of sleep and circadian rhythm disorders in Parkinson disease. Clin Geriatr Med 2020; 36(1): 119–130.
- Liguori C, De Franco V, Cerroni R et al. Sleep problems affect quality of life in Parkinson's disease along disease progression. Sleep Med 2021; 81: 307-311.
- Jones BM, McCarter SJ. Rapid eye movement sleep behavior disorder: clinical presentation and diagnostic criteria. Sleep Med Clin 2024; 19(1): 71-81.
- Gnarra O, Wulf MA, Schäfer C et al. Rapid eye movement sleep behavior disorder: a narrative review from a technological perspective. Sleep 2023; 46(6): zsad030.
- Kunz D, Oster H, Rawashdeh O et al. Sleep and circadian rhythms in α-synucleinopathies-Perspectives for disease modification. Acta Physiol 2023; 238(1): e13966.
- Howell M, Avidan AY, Foldvary-Schaefer N et al. Management of REM sleep behavior disorder: an American Academy of Sleep Medicine clinical practice guideline. J Clin Sleep Med. 2023; 19(4): 759-768.
- Bovenzi R, Conti M, Pierantozzi M et al. Safinamide effect on sleep architecture of motor fluctuating Parkinson’s disease patients: a polysomnographic rasagiline-controlled study. Parkinsonism Relat Disord 2024; 127: 107103.
- Arias-Carrion O, Ortega-Robles E, Ortuno-Sahagun D et al. Sleep-related disorders in Parkinson's disease: mechanisms, diagnosis, and therapeutic approaches. CNS Neurol Disord Drug Targets 2025; 24(2): 132-143.
- Suzuki K, Fujita H, Kobayashi S. Managing sleep issues in Parkinson's disease: an up-to-date review. Expert Rev Neurother 2025; 25(2): 211-226.
- Bliwise DL, Karroum EG, Greer SA et al. Restless legs symptoms and periodic leg movements in sleep among patients with Parkinson's disease. J Parkinsons Dis 2022; 12(4): 1339-1344.
- Maggi G, Barone A, Mastromarino C et al. Prevalence and clinical profile of patients with restless legs syndrome in Parkinson's disease: a meta-analysis. Sleep Med 2024; 121: 275-286.
- Cochen De Cock, V. Therapies for restless legs in Parkinson’s disease. Curr Treat Options Neurol 2019; 21(11): 56.
- Maggi G, Vitale C, Cerciello F, Santangelo G. Sleep and wakefulness disturbances in Parkinson's disease: a meta-analysis on prevalence and clinical aspects of REM sleep behavior disorder, excessive daytime sleepiness and insomnia. Sleep Med Rev 2023; 68: 101759.
- Arias-Carrion O, Ortega-Robles E, Ortuno-Sahagun D et al. Sleep-related disorders in Parkinson's disease: mechanisms, diagnosis, and therapeutic approaches. CNS Neurol Disord Drug Targets 2025; 24(2): 132-143.
- Haq IZ, Naidu Y, Reddy P, Chaudhuri KR. Narcolepsy in Parkinson’s disease. Expert Rev Neurother 2010; 10(6): 879-884.
- Saper CB, Fuller PM, Pedersen NP et al. Sleep state switching. Neuron 2010; 68(6):1023-1042.
- Zheng JH, Ma JJ, Sun WH et al. Excessive daytime sleepiness in Parkinson's disease is related to functional abnormalities in the left angular gyrus. 1: Clin Neuroradiol 2023; 33(1), 2023.
- Fujita H, Ogaki K, Shiina T et al. Impact of autonomic symptoms on the clinical course of Parkinson's disease. Neurol Sci 2024; 45(8): 3799-3807.
- Li FF, Cui YS, Yan R et al. Prevalence of lower urinary tract symptoms, urinary incontinence and retention in Parkinson's disease: a systematic review and meta-analysis. Front Aging Neurosci 2022; 14: 977572).
- Barone P, Antonini A, Colosimo C et al. The PRIAMO study: a multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson's disease. Mov Disord 2009; 24(11): 1641-1649.
- Seki S, Igawa Y, Kaidoh K. et al. Role of dopamine D1 and D2 receptors in the micturition reflex in conscious rats. Neurourol Urodyn 2001; 20(1): 105-113.
- Chen Z, Li G, Liu J. Autonomic dysfunction in Parkinson's disease: implications for pathophysiology, diagnosis, and treatment. Neurobiol Dis 2020; 134: 104700.
- Hu JC, Hsu LN, Lee WC et al. Role of urological botulinum toxin-A injection for overactive bladder and voiding dysfunction in patients with Parkinson’s disease or post-stroke. Toxins 2023; 15(2): 166.
- Gómez-López A, Sánchez-Sánchez A, Natera-Villalba E et al. SURINPARK: safinamide for urinary symptoms in Parkinson's disease. Brain Sci 2021 6; 11(1): 57.
- Palma JA, Thijs RD. Non-pharmacological treatment of autonomic dysfunction in Parkinson's disease and other synucleinopathies. J Parkinsons Dis 2024; 14(s1): S81-S92.
- Safarpour D, Sharzehi K, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Drugs 2022; 82(2): 169-197.
- Dogra N, Mani RJ, Katare DP. The gut-brain axis: two ways signaling in Parkinson's disease. Cell Mol Neurobiol 2022; 42(2): 315-332.
- Isaacson J, Patel S, Torres-Yaghi Y, Pagán F. Sialorrhea in Parkinson’s disease. Toxins 2020; 12(11): 691.
- Santos Junior LC, Santos JR, Reis A et al. Effectiveness of the pharmacological treatments for sialorrhea in patients with Parkinson's disease: a systematic review and network meta-analysis. Clin Oral Investig 2023; 27(6): 2449-2463.
- Umemoto G, Furuya H. Management of dysphagia in patients with Parkinson's disease and related disorders. Intern Med. 2020; 59(1): 7-14.
- Schindler A, Pizzorni N, Cereda E et al. Consensus on the treatment of dysphagia in Parkinson's disease. J Neurol Sci 2021; 430: 120008.
- Pfeiffer RF, Isaacson SH, Pahwa R. Clinical implications of gastric complications on levodopa treatment in Parkinson's disease. Parkinsonism Relat Disord 2020; 76: 63-71.
- Soliman H, Coffin B, Gourcerol G. Gastroparesis in Parkinson disease: pathophysiology, and clinical management. Brain Sci 2021; 11(7): 831.
- Yao L, Liang W, Chen J, Wang Q, Huang X. Constipation in Parkinson's disease: a systematic review and meta-analysis. Eur Neurol 2023; 86(1): 34-44.
- Cheesman M, Ho H, Bishop K, Sin MK. Constipation management in Parkinson disease. J Neurosci Nurs 2021; 53(6): 262-266.
- Ramu SK, Oblizajek NR, Savica R et al. Defecatory disorders are a common cause of chronic constipation in Parkinson disease. Neurogastroenterol Motil 2024; 36(5): e14767.
- Palma JA, Kaufmann H. Orthostatic hypotension in Parkinson disease. Clin Geriatr Med 2020; 36(1): 53-67.
- Grosu C, Noea O, Maștaleru A et al. Neurogenic orthostatic hypotension in Parkinson disease-a narrative review of diagnosis and management. J Clin Med 2025; 14(2): 630.
- Kaufmann H, Norcliffe-Kaufmann L, Palma JA. Baroreflex dysfunction. N Engl J Med. 2020; 382(2): 163-178.
- Fanciulli A, Leys F, Falup-Pecurariu C et al. Management of orthostatic hypotension in Parkinson’s disease. J Parkinsons Dis 2020; 10(s1): S57-S64.
- Buhmann, C. Prevalence, clinical presentations and impact on relationship of sexual dysfunction in Parkinson's Disease. Int Rev Neurobiol 2022; 162: 1-19.
- Batzu L, Titova N, Bhattacharyya KB, Chaudhuri KR. The pathophysiology of sexual dysfunction in Parkinson's disease: an overview. Int Rev Neurobiol 2022; 162: 21-34.
- Urso D, Leta V, Rukavina K. Management strategies of sexual dysfunctions in Parkinson's disease. Int Rev Neurobiol 2022; 162: 97-116.
- Bernard BA, Metman LV, Levine L et al. Sildenafil in the treatment of erectile dysfunction in Parkinson's Disease. Mov Disord Clin Pract 2016; 4(3): 412-415.
- Pitton Rissardo J, Jayasinghe M, Rashidi M et al. Exploring fatigue in Parkinson's Disease: a comprehensive literature review. Cureus 2025; 17(3): e81129.
- Lou, JS. Physical and mental fatigue in Parkinson’s disease: epidemiology, pathophysiology and treatment. Drugs Aging 2009; 26(3): 195-208.
- Kang SY, Bang M, Hong JY et al. Neural and dopaminergic correlates of fatigue in Parkinson's disease. J Neural Transm 2020; 127(3): 301-309.
- Di Vico IA, Moretto M, Tamanti A et al. Molecular-informed network analysis unveils fatigue-related functional connectivity in Parkinson's disease. 2025. [CrossRef]
- Pavese N, Metta V, Bose SK et al. Fatigue in Parkinson’s disease is linked to striatal and limbic serotonergic dysfunction. Brain 2010; 133(11): 3434–3443.
- Wang H, Liu Y, Zhao J, Guo X et al. Possible inflammatory mechanisms and predictors of Parkinson's disease patients with fatigue (brief review). Clin Neurol Neurosurg 2021; 208: 106844.
- Wang L, Yi H, Liang X et al.: Plasma TNF-α and phosphorylated α-syn are associated with fatigue in patients with Parkinson's disease. J Neuroimmunol 2023; 385: 578222.
- Versace V, Sebastianelli L, Ferrazzoli D et al. Intracortical GABAergic dysfunction in patients with fatigue and dysexecutive syndrome after COVID-19. Clin Neurophysiol 2021, 132(5): 1138-1143.
- McNeill A, Duran R, Hughes DA et al. A clinical and family history study of Parkinson's disease in heterozygous glucocerebrosidase mutation carriers. J Neurol Neurosurg Psychiatry 2012; 83(8): 853-854.
- Fu R, Cui SS, Du JJ et al. Fatigue correlates with LRRK2 G2385R variant in Chinese Parkinson's disease patients. Parkinsonism Relat Disord 2017; 44: 101-105.
- [194] Panigrahi B, Pillai KS, Radhakrishnan DM, Rajan R, Srivastava AK: Fatigue in Parkinson’s disease—a narrative review. Ann Mov Disord 2024, 7:157-71.
- Tinazzi M, Geroin C, Siciliano M et al. Pain and fatigue in Parkinson's disease: advances in diagnosis and management. Neurol Sci 2025; 46(6): 2437-2454.
- Pauletti C, Locuratolo N, Mannarelli D et al. Fatigue in fluctuating Parkinson’s disease patients: possible impact of safinamide. J Neural Transm 2023; 130(7): 915-923.
- Kluger BM, Parra V, Jacobson C et al. The prevalence of fatigue following deep brain stimulation surgery in Parkinson's disease and association with quality of life. Parkinsons Dis 2012; 2012:769506.
- Folkerts AK, Nielsen J, Gollan R et al. Physical exercise as a potential treatment for fatigue in Parkinson's disease? A systematic review and meta-analysis of pharmacological and non-pharmacological interventions. J Parkinsons Dis 2023; 13(5): 659-679.
- Lawrie S, Coe S, Mansoubi M et al. Dietary patterns and nonmotor symptoms in Parkinson's disease: a cross-sectional analysis. J Am Nutr Assoc 2023; 42(4): 393-402.
Figure 1.
The different non-motor symptoms of Parkinson’s disease.
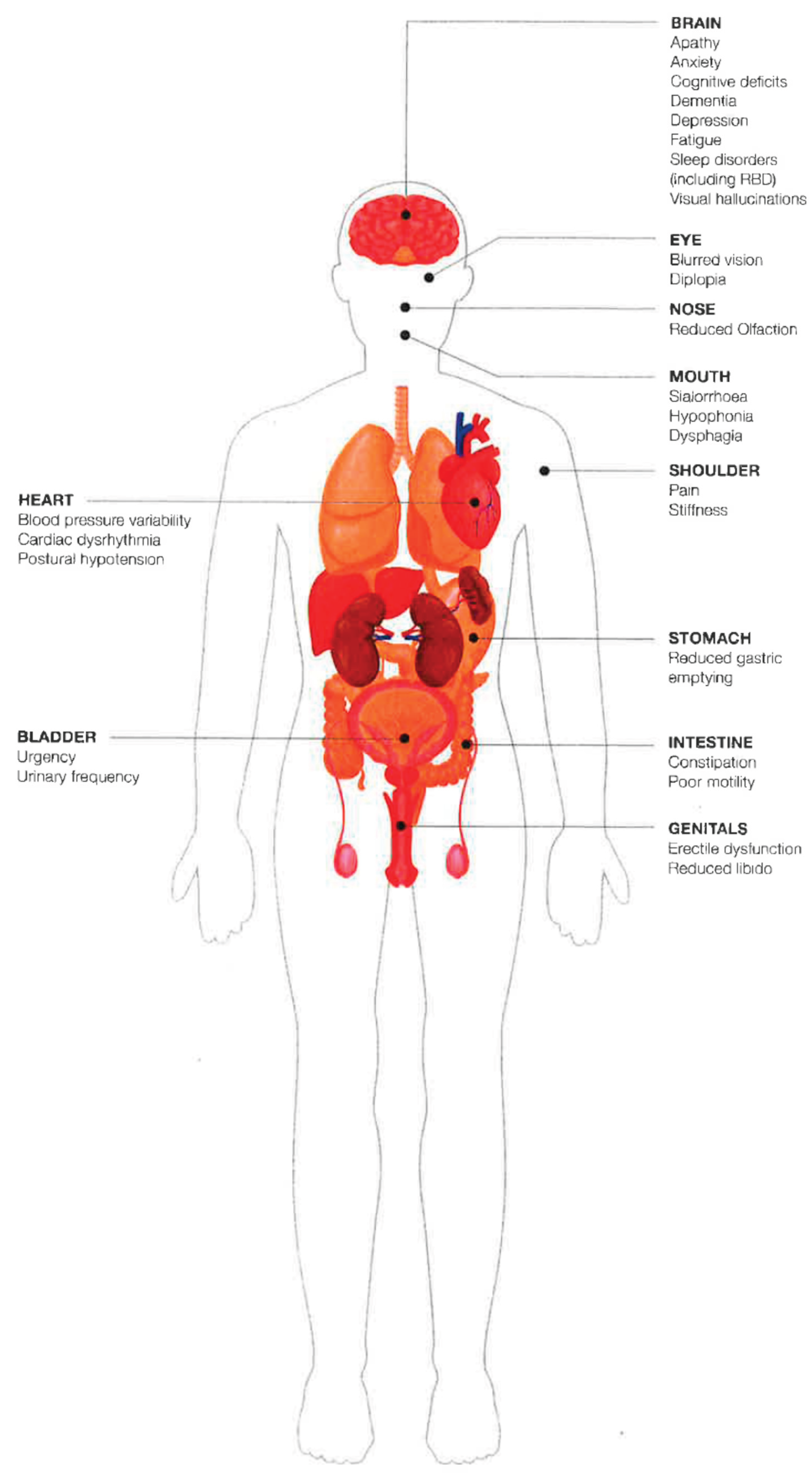

Table 1.
Frequency of occurrence of the eight commonest non-motor symptoms.
| Non-motor symptoms | Mean (%) |
|---|---|
| Urinary problems | 74% |
| Pain | 65% |
| Fatigue | 64% |
| Sleep problems | 62% |
| Constipation | 48% |
| Cognitive impairment | 46% |
| Depression/apathy | 43% |
| Excessive drooling | 35% |
Table 2.
Clinical classifications of NMS of PD.
| Related to the disease process or pathophysiology | Dopaminergic |
|---|---|
| Non-dopaminergic (cholinergic, serotonergic, noradrenergic, glutamatergic, and mixed) | |
| Related to non-motor fluctuations | Present only during OFF periods |
| Present during ON periods and worse in OFF | |
| Related to drug therapy for PD | e.g. hallucinations, impulse control disorders, excessive daytime sleepiness |
| Genetically determined | Dementia or MCI in patients with glucocerebrosidase mutation |
| Depression and sleep disorders in patients with LRRK-2 mutation |
NMS: non-motor symptoms; PD: Parkinson’s disease; MCI: mild cognitive impairment; LRRK-2: leucine-rich repeat kinase-2.
Table 3.
Brain regions and neurotransmitters implicated in NMS.
| NMS | Brain region | Neurotransmitter | |||||
|---|---|---|---|---|---|---|---|
| DA | SE | NA | GLU | ACE | GABA | ||
| Hyposmia | Olfactory bulb & amigdala | √ | √ | ||||
| Hallucinations | Occipital cortex | √ | √ | √ | √ | ||
| Pain | Basal ganglia, locus coeruleus, raphe nucleus, amygdala, thalamus | √ | √ | ||||
| Anxiety | Limbic area including amygdala | √ | √ | √ | √ | √ | |
| Depression | Limbic & cortical areas | √ | √ | √ | √ | ||
| Early cognitive dysfunction | Frontal cortex | √ | √ | √ | √ | ||
| Dementia | Temporal, parietal & occipital lobes | √ | √ | ||||
| Sleep disturbances | Hypothalamus & reticular formation | √ | √ | √ | √ | √ | |
| Bladder hyperreflexia | Basal ganglia | √ | √ | ||||
NMS: non motor symptom; DA: dopamine; SE: serotonine, NA: noradrenaline; GLU: glutamate; ACE: acetylcholine; GABA: γ-amino butyric acid.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



