
Submitted:
08 November 2025
Posted:
10 November 2025
You are already at the latest version
Abstract
Cyclin-dependent kinase 4/6 inhibitors (CDK4/6i) have transformed the treatment landscape for estrogen receptor-positive (ER+) breast cancer, yet re-sistance remains a major clinical challenge. Although CDK4/6i induce G1 ar-rest and therapy-induced senescence (TIS), the exact nature of this senescent state and its contribution to resistance are not well understood. To explore this, we developed palbociclib- (2PR, 9PR, TPR) and abemaciclib- (2AR, 9AR, TAR) resistant ER+ breast cancer sublines through prolonged drug exposure over six months. Resistant cells demonstrated distinct phenotypic alterations, including cellular senescence, reduced mitochondrial membrane potential, and impaired glycolytic activity. Cytokine profiling and Enzyme-Linked Im-munosorbent Assay (ELISA) validation revealed a non-canonical Senes-cence-Associated Secretory Phenotype (SASP) characterized by elevated Growth/differentiation factor 15 (GDF-15) and serpin E1 (Plasminogen acti-vator inhibitor-1, PAI-1), and absence of classical pro-inflammatory interleu-kins including IL-1α and IL-6. IL-8 levels were significantly elevated, but no association with epithelial-mesenchymal transition (EMT) was observed. Re-sistant cells preserved their epithelial morphology, showed no upregulation of EMT markers, and lacked Aldehyde dehydrogenase 1-positive (ALDH1+) stem-like populations. Additionally, Regulated Upon Activation, Normal T-cell Expressed, and Secreted (RANTES) was strongly upregulated in palbo-ciclib-resistant cells. Together, these findings identify a distinct, non-canonical senescence phenotype associated with CDK4/6i resistance and may provide a foundation for identifying new vulnerabilities in resistant ER+ breast cancers through targeting SASP-related signaling.
Keywords:
breast cancer
; CDK4/6 inhibitors
; resistance to cancer therapies
; therapy-induced senescence
1. Introduction
Cyclin-dependent kinase 4/6 inhibitors (CDK4/6i), palbociclib, ribociclib and abemaciclib, have introduced a new era in the treatment of hormone receptor-positive (HR+), HER2-negative metastatic breast cancer. Landmark clinical trials such as PALOMA, MONALEESA, and MONARCH have demonstrated significant improvements in progression-free survival when these agents are combined with endocrine therapy in both first- and second-line metastatic settings [1,2]. MONARCH-2 trial also demonstrated an overall survival benefit, further reinforcing the role of CDK4/6i as standard of care in advanced disease. These findings have established CDK4/6i as a standard of care in advanced HR+ breast cancer [3].
Mechanistically, CDK4/6i exert their therapeutic effect by blocking CDK4/6 and cyclin D complexes and Rb phosphorylation, leading to a G1 cell cycle arrest [4]. CDK4/6 inhibition also induces a phenotype resembling cellular senescence in vitro, given the key role of the retinoblastoma (RB) protein in the regulation of senescence. This therapy-induced senescence (TIS) represents a cellular state of persistent cell cycle exit with an associated phenotypic alterations [5]. In line with Rb being the main effector of CDK4/6-inhibitor–mediated G1 arrest, Rb status also strongly influences senescence induction upon CDK4/6 inhibition [2,6]. CDK4/6 inhibitors thus cause tumor cells to enter a Rb-dependent senescence program with similarities to conventional senescence.
Tissue context and tumor genetic background (p53 status) can influence senescence. CDK4/6 inhibitors induce a senescent state that is dependent on p53 and lack pro-inflammatory components [7]. Furthermore, active mitogenic signaling appears to set the “depth” of senescence [8,9]. A recent study showed that if growth signaling (mTORC1) is active during CDK4/6 inhibitor-induced cell cycle arrest, then senescence is more pronounced; whereas co-suppression of mTOR attenuates senescence induction [10,11]. In fact, dual mTORC1 inhibition (using everolimus, for example) during palbociclib treatment was reported to prevent “full activation” of senescence [12], consistent with the idea that senescence requires some growth stress (geroconversion) [8,10]. CDK4/6 inhibitors themselves appear to downregulate mTOR activity in breast cancer. As the inflammatory part of the senescent program is controlled by mTOR, this likely impacts the balance between pro- and anti-inflammatory SASP.
TIS is considered a double-edged sword in cancer treatment. On one hand, it acts as a tumor-suppressive mechanism by halting the proliferation of damaged or oncogenic cells in response to treatments such as chemotherapy or radiation. On the other hand, TIS can lead to tumor promotion, reflecting the dual role of senescence in cancer biology through the senescence-associated secretory phenotype (SASP) in a context-dependent manner [13]. The term SASP was first described by Campisi’s group highlighting the complex secretory profile of senescent cells [14]. In response to genotoxic or DNA-damaging stress, senescent tumor cells can secrete a wide array pro-inflammatory cytokines such as IL-6, IL-8, growth factors and proteases [15,16]. By contrast, other forms of non-genotoxic stress have been shown to trigger alternative senescence phenotypes. Campisi’s group described a distinct secretory profile, named as mitochondrial dysfunction-associated senescence (MiDAS). It occurs when mitochondrial function is lost or impaired, still resulting in growth arrest and some features of senescence [17]. This metabolic state defined by lower NAD+/NADH ratios lacks the IL-1-dependent inflammatory arm of the SASP [17,18]. Instead, in MiDAS, low NAD+/NADH ratios activate p53 through AMP-activated protein kinase (AMPK), which then suppresses canonical nuclear factor kappa-light-chain-enhancer (NF-κB)–mediated inflammatory SASP signaling [19]. This distinct secretory profile implicates that senescent cells can be on a spectrum with respect to their secretory phenotypes, ranging from highly tumor-promoting to tumor-suppressive phenotypes.
Despite recent studies exploring the role of CDK4/6 inhibitor-induced senescence, it remains unclear whether senescence directly contributes to therapeutic resistance in the context of CDK4/6 inhibition, as the underlying mechanisms are not yet fully understood. In this study, we developed palbociclib and abemaciclib–resistant ER+ breast cancer cells (LCC2, LCC9, and T47D) through stepwise increasing exposure to the respective CDK4/6 inhibitors to investigate the nature of senescence in resistance to these therapies. Here, we report a non-canonical SASP phenotype that may play an overlooked role in shaping resistance mechanisms. Future studies should explore the therapeutic potential of targeting specific components of this altered SASP to overcome or prevent resistance to CDK4/6 inhibitor-based therapies.
2. Materials and Methods
2.1. Cell Culture and Generation of CDK4/6-Resistant Breast Cancer Cell Lines
Parental breast cancer cell lines (LCC2, LCC9, MCF7 and T47D) were grown in Minimum Essential Medium (MEM) with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin at 37 °C in a humidified 5% CO2 incubator. Culture medium was refreshed every 3 days with fresh complete MEM. LCC2 (tamoxifen-resistant) and LCC9 (fulvestrant-resistant and tamoxifen cross-resistant cell lines were kind gifts from Dr. R. Clarke (Georgetown University Medical School, Washington, DC) [20,21]. MCF7 and T47D cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA). We developed CDK4/6-resistant sublines of these cell lines through a stepwise increase of palbociclib or abemaciclib drug concentration from 50 nM to 2.5 µM. Cells maintained their ability to proliferate in the presence of palbociclib or abemaciclib. After six months of continuous treatment, drug-resistant sublines were established. Palbociclib-resistant cells of LCC2 (2PR), LCC9 (9PR), and T47D (TPR) were continuously cultured in the presence of 2.5 µM palbociclib, and abemaciclib-resistant cells of LCC2 (2AR), LCC9 (9AR), T47D (TAR) were cultured in 2.5 µM abemaciclib and used for the experiments described below. Parental (drug-sensitive) LCC2, LCC9, and T47D cells were cultured in parallel under the same conditions but without drug. All cell lines were routinely confirmed to be mycoplasma-free and used at low passage numbers.
2.2. Cell Proliferation
Cell viability was assessed using the CyQuant Cell Proliferation Assay, following the manufacturer’s instructions (ThermoFisher Scientific). Abemaciclib and palbociclib were obtained from Selleck Chemicals LLC (Houston, TX).
2.3. Senescence Analysis
Senescence-associated β-galactosidase (SA-β-gal) activity was measured using the CellEvent Senescence Green Flow Cytometry Assay (ThermoFisher), according to the manufacturer’s protocol. Flow cytometric analysis was performed using BD FACSuite software. The proportion of senescent cells (green fluorescence) in resistant or parental cell populations was quantified to assess senescence induction by CDK4/6 inhibitors.
2.4. NAD+/NADH Quantification
Intracellular levels of NAD+ and NADH were measured using a fluorometric cycling assay (NAD+/NADH Assay Kit, Cell Biolabs, Cat# MET-5030) per manufacturer’s protocol. Briefly, 4 × 106 cells per sample were harvested and washed with cold PBS before extraction in 0.5 mL of Extraction Buffer (included in the kit) on ice. Lysates were clarified by centrifugation (5 min, 14,000 × g, 4 °C) and deproteinated with a 10 kDa molecular-weight cutoff spin filter to remove any enzymes that could degrade NAD. Two aliquots of each sample were prepared and treated differently to distinguish between oxidized and reduced nucleotides. One aliquot was incubated in 0.1 N HCl at 80 °C for 60 min to degrade NAD+ (reporting NADH levels), while the other was incubated in 0.1 N NaOH at 80 °C for 60 min to degrade NADH (reporting NAD+ levels). Following neutralization and cooling, treated extracts were combined with cycling enzyme, NAD+ substrate, and the kit’s fluorescent probe in a black 96-well plate. The enzyme cycling reaction converts NAD+ to NADH, which then reacts with the fluorometric probe to generate a fluorescent signal proportional to the NAD concentration. Fluorescence intensity was measured on a plate reader using 530 nm excitation and 590 nm emission settings. Absolute concentrations of NAD+ and NADH were determined by comparing to a NAD+ standard curve generated in parallel, and NAD+/NADH ratio was calculated for each sample. At least triplicate independent samples were assessed for each condition.
2.5. Mitochondrial Membrane Potential Assay (TMRE)
Mitochondrial membrane potential was measured using a tetramethylrhodamine ethyl ester (TMRE) fluorescence assay (TMRE-Mitochondrial Membrane Potential Assay Kit, Abcam, Cat# ab113852). Cells were plated into black-walled 96-well plates (clear bottom) at appropriate densities and allowed to attach overnight. On the day of the assay, cells were incubated with TMRE working solution (200 nM final concentration in culture medium) for 30 min at 37 °C, in the dark. After incubation, the TMRE-containing medium was carefully removed, and cells were washed twice with warm PBS with 0.2% BSA to reduce background fluorescence. Fluorescence was then measured immediately in a microplate reader (bottom-read mode) with 549 nm excitation/575 nm emission filters, which detects the accumulation of TMRE in polarized mitochondria. A subset of cells was pre-treated with the protonophore carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (20 µM, added 10 min before TMRE incubation) in each experiment as a positive control to depolarize the mitochondrial membrane, which is expected to result in loss of TMRE signal. Unstained control wells (containing no TMRE dye) were included in all experiments to account for background autofluorescence. Triplicate wells were measured for each condition and the assay repeated in at least three independent experiments.
2.6. Agilent Seahorse XFp Cell Energy Phenotype Assays
Mitochondrial respiration, and glycolysis were assessed using Agilent Seahorse XF Cell Mito Stress test and Agilent Seahorse XF Glycolysis Stress test, respectively, as previously described [22]. Cells were seeded in XFp miniplates and incubated overnight. The test involved sequential treatment with oligomycin (an ATP synthase inhibitor) and FCCP (a mitochondrial uncoupler) to induce metabolic stress and evaluate cellular energy metabolism. These perturbations enabled the assessment of baseline and stressed energy phenotypes, as well as the calculation of metabolic potential by measuring changes in oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). To fully inhibit mitochondrial respiration and confirm non-mitochondrial oxygen consumption, rotenone (a complex I inhibitor) and antimycin A (a complex III inhibitor) were subsequently injected. This final step ensures accurate quantification of mitochondrial-dependent respiration by subtracting non-mitochondrial OCR from total OCR values. The results presented are the combination of three independent assays, and two-way ANOVA analyses were performed using GraphPad software (P < 0.05, statistically significant).
2.7. Cytokine Array
The cytokine array was performed using Proteome Profiler Array Human XL Cytokine Array Kit (Bio-Techne/R&D Systems, #ARY022B), according to the manufacturer’s instructions. Imaging and quantification were performed using Amersham Imager 600 software (GE Healthcare Life Sciences). The signal intensities of six reference spots in each membrane were measured and defined as 100%.
2.8. Cytokine Quantification by ELISA
Interleukin-6 (IL-6), Interleukin-8 (IL-8), Interleukin 1 alpha (IL-1α), Growth/differentiation factor 15 (GDF-15), High Mobility Group Box 1 (HMGB1), Regulated Upon Activation, Normal T-cell Ex-pressed, and Secreted (RANTES), Angiogenin, Vascular endothelial growth factor (VEGF-A), Platelet-derived growth factor-AA (PDGF-AA) and serpin E1 (Plasminogen Activator Inhibitor-1, PAI-1) in cell culture supernatants were measured by sandwich ELISA using pre-coated 96-well plate kits (Invitrogen, Thermo Fisher Scientific, USA). For each assay, cell culture supernatants were collected after 72 h of incubation (at ~80% cell confluence) and clarified by centrifugation to remove cells and debris. Samples and provided standards were added to the antibody-coated wells and incubated according to manufacturer’s instructions, followed by addition of biotinylated detection antibodies and horseradish peroxidase (HRP)-conjugated streptavidin. After washing, TMB substrate was added and allowed to develop for the recommended time before reaction was stopped. Absorbance at 450 nm (reference 540–570 nm) was measured on a microplate reader. Cytokine concentrations in each sample were determined by comparison to the standard curve generated on the same plate. All samples were analyzed in duplicate wells, and each experiment was performed at least in duplicate.
2.9. Morphology Assessment
Cells were grown in 8-well slide chambers (MatTek) in CSS media for 4 days. The slides were then fixed in 60% ethanol and stained with hematoxylin and eosin (H&E) stains. The slides were examined under an Olympus BX41 microscope, and images were obtained using a DP-72 camera and CellSensTM software at 40× magnification.
2.10. Western Blot Analysis
Protein lysates from control and CDK4/6 resistant cell lines were prepared and quantified using the Bio-Rad DC Protein Assay kit (Bio-Rad, Hercules, CA, USA). Equal protein amounts were analyzed by Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS–PAGE) and Western blot as previously described [22]. Briefly, membranes were probed with primary antibodies against Snail (SNAIL), Slug (SNAI2), TWIST1 and Vimentin (Cell Signaling Technology, Danvers, MA). β-actin (Sigma, St. Louis, MO) served as a loading control. Detection was performed using the SuperSignal™ West Pico PLUS Chemiluminescent Substrate kit (Amersham, Piscataway, NJ), and images were captured with the Amersham Imager 600 system (GE Healthcare Life Sciences, Pittsburgh, PA). Results shown are representative of three independent experiments.
3. Results
3.1. Prolonged Exposure to Palbociclib or Abemaciclib Confers Resistance in ER+ Breast Cancer Cells
The sensitivity to palbociclib and abemaciclib in both parental (LCC2, LCC9, and T47D) and resistant sublines (2PR, 9PR, TPR, 2AR, 9AR and TAR) was assessed by measuring cell survival rates. Figure 1 shows the comparison of IC50 values, representing the concentrations required to reduce cell viability by 50%, for both parental control and resistant sublines. The 2PR, 9PR and TPR sublines exhibited 645-fold, 57-fold and 28-fold increases to palbociclib in IC50 values compared to their parental sensitive counterparts, respectively. For abemaciclib, the 2AR, 9AR and TAR sublines displayed 76-fold, 88-fold and 18-fold increases compared to their parental sensitive counterparts, respectively. This differential response demonstrates that the resistant sublines successfully acquired resistance to each drug.
3.2. Palbociclib and Abemaciclib-Resistant Cells Induces Senescence and Exhibit Markers of Therapy-Induced Senescence
Given the link between CDK4/6 inhibition and therapy-induced senescence (TIS), we evaluated whether the resistant cells had adopted a senescent phenotype. To assess this, we measured senescence-associated β-galactosidase (SA-β-Gal) activity, a widely used surrogate marker of cellular senescence. The fluorescence intensity of resistant sublines (2PR, 2AR, 9PR, 9AR, and TAR) exhibited a significant rightward shift compared to their sensitive counterparts, resulting in increased senescence phenotype. whereas T47D palbociclib-subline (TPR) did not exhibit senescence induction, suggesting the differences among the CDK4/6 inhibitors (Figure 2).
Quantification of their intensities reveals that the senescent fraction of 2PR cells is 48.7%, while 2AR cells display 40.8% positivity for SA-β-Gal, in contrast to the parental LCC2 cells at 10.6% (p < 0.0001). Specifically, 9PR and 9AR cells exhibited 56% and 39% SA-β-Gal positivity, respectively, versus 5% in parental LCC9 cells, while TPR and TAR cells displayed 2.5% and 31.5% positivity compared with 4.9% in parental T47D cells (p < 0.01).
To further explore senescence in CDK4/6i-resistant cells, we first examined the protein expression of established markers, lamin B1 and p21, linked with therapy-induced senescence (TIS) in cancer. Lamin B1, a structural nuclear envelope protein typically downregulated in senescent cells and considered a negative marker of TIS, was significantly reduced in PR sublines (2PR; p=0.0015, 9PR; p= 0.0141 and TPR; p= 0.01820 and abemaciclib TAR subline (p=0.0474), indicating significant senescence-associated changes (Supplementary Figure S1A-B). p21(Cip1/Waf1), a cyclin-dependent kinase inhibitor regulated by p53 and a key mediator of senescence-associated growth arrest, showed a significant decrease in 2PR (p=0.0036), while it was significantly upregulated in both TPR (p = 0.0372) and TAR (p =0.0011) sublines, highlighting the heterogeneity of the senescence markers expression across different sublines.
3.3. Loss of Mitochondrial Membrane Potential in Resistant Cells
To better define the nature of senescence in palbociclib and abemaciclib-resistant cells, we next examined mitochondrial dysfunction-associated senescence (MiDAS), as described first by Campisi group [17]. As an indication of MiDAS state, we first measured NAD+/NADH ratio in parental LCC2 cells and their palbociclib- and abemaciclib-resistant derivatives, 2PR and 2AR. Following normalization to LCC2, the mean (±SD) NAD+ and the NADH values for 2PR and 2AR were not significantly altered (Figure 3A). Further analysis of the NAD+/NADH ratio also revealed no significant alterations between the resistant sublines and their sensitive counterpart, as determined by one-way ANOVA. Consistent outcomes were observed in parallel assays using LCC9 and T47D parental cell lines and their resistant derivatives (9PR, TPR, 9AR, and TAR), further supporting these observations (Supplementary Figure S2A). These results suggest that there is no detectable change in the redox balance in the resistant cells, indicating a lack of substantial mitochondrial dysfunction or metabolic stress. To further evaluate mitochondrial health, we measured mitochondrial membrane potential (ΔΨm) using a TMRE-based assay. This analysis revealed a marked and statistically significant reduction in TMRE intensity in both resistant cell lines compared to LCC2 (Figure 3B), indicating a loss of mitochondrial membrane potential (ΔΨm). WE verified these results using LCC9 and T47D parental cell lines and their resistant derivatives (9PR, TPR, 9AR, and TAR), further supporting these observations (Supplementary Figure S2B). These data suggest that the absence of canonical MiDAS, but altered mitochondrial membrane potential in CDK4/6-resistant cells do manifest a different metabolic phenotype, marked by mitochondrial activity impairment.
To further assess the metabolic profile of palbociclib (2PR)- and abemaciclib (2AR)-resistant cells, we have performed Seahorse assays to measure key metabolic parameters, including the extracellular acidification rate (ECAR) and the oxygen consumption rate (OCR)(Supplementary S3A, B). Both 2PR and 2AR cells exhibited a marked decrease in ECAR compared to the parental LCC2 cells, suggesting a metabolic shift toward reduced glycolytic capacity and reserve. In addition, 2PR and 2AR cells exhibit significantly reduced mitochondrial function compared to LCC2 cells, as evidenced by lower basal respiration, spare respiratory capacity, and ATP production. Proton leak remains unchanged across comparisons, suggesting that mitochondrial membrane integrity is not notably affected. Similarly, LCC9- and T47D-derived resistant sublines (9PR, 9AR, TPR, TAR) exhibited reduced glycolytic capacity and reserve relative to parental counterparts. Although LCC9-derived resistant cells demonstrated moderate reductions in spare respiratory capacity, they maintained their total mitochondrial function. In contrast, T47D-resistant sublines did not significantly alter any OCR parameters. These observations point to a potential difference in cellular metabolism in the resistant lines, and alterations in energy production pathways. The decrease in ECAR further supports the idea that the resistant cells may rely more on oxidative phosphorylation or other alternative metabolic pathways, as opposed to glycolysis, to meet their energy demands.
3.4. SASP Changes in Abemaciclib and Palbociclib-Resistant Breast Cancer Cells
Senescent cells secrete molecules known as the senescence-associated secretory phenotype (SASP), which includes pro-inflammatory cytokines, proteases, and growth and angiogenesis factors [23]. These factors can contribute to therapy resistance. To better understand the role of senescence in 2PR and 2AR-resistant cells compared to their parental sensitive counterpart (LCC2), we next performed a human cytokine array consisting of key SASP components. Among these factors analyzed, we observed a significant increase in growth/differentiation factor 15 (GDF-15) levels in conditioned media of 2AR and 2PR cells, while angiogenin was dramatically decreased in both resistant cells compared to parental cells (Figure 4; Supplementary Figure S4A–C). Additionally, elevated secretion of VEGF, PDGF-AA, RANTES and serpin E1 significantly elevated in 2PR cells. Insulin Like Growth Factor Binding Protein 2 (IGFBP-2) levels were significantly decreased in 2PR an 2AR, signals for IL-6 and IL-1α remain unchanged between resistant and parental cells (Supplementary Figure S4A–C), These results suggest that resistant cells adopt an altered SASP profile with unique inflammatory and pro-tumorigenic secretions.
3.5. ELISA Assays Confirmed Alterations in Cytokine Levels Within the SASP Profiles of Palbociclib and Abemaciclib-Resistant Breast Cancer Cells
To validate the array results, we quantitatively measured the levels of key SASP factors in the conditioned medium of 2PR and 2AR sublines compared to parental LCC2 cells by ELISA (Figure 5). These analyses confirmed that that IL-8 (2PR, p < 0.0001; 2AR, p < 0.001), GDF-15 (2PR, p < 0.0001; 2AR, p < 0.0001), and serpin E1 (PAI-1)( 2PR, p < 0.01) and 2AR, p < 0.001) were significantly upregulated in both 2PR and 2AR conditioned media compared to LCC2. RANTES was upregulated only in 2PR cells (p < 0.0001). In contrast, secretion of IL-6 or IL-1α was not significantly changed in the resistant cells. Similarly, angiogenin, VEGF, and PDGF-AA levels were not significantly altered in 2PR. In contrast, they are significantly decreased in 2AR cells (angiogenin, p < 0.01; VEGF, p < 0.01 and PDGF-AA, p < 0.0001). We also observed similar alterations of these cytokines/markers in palbociclib and abemaciclib-resistant derivatives of LCC9 and T47D cells. (Supplementary Figure S5A,B).
We also measured other SASP markers that have been reported by the Campisi group to contribute to the senescence-associated arrest including High-Mobility Group Box 1 (HMGB1), AMP-activated protein kinase (AMPK), nuclear factor kappa-light-chain-enhancer of activated B (NF-κB) and p53 [17]. HGMB1 secretion was significantly reduced in the 2PR (p < 0.0001) and 2AR (p < 0.0001) conditioned media, supporting that this pro-inflammatory nuclear protein is not involved. In addition, we did not observe any change in protein expression of p53, AMPK, and phospho-AMPK (Supplementary Figure S6A,B). Overall, these results demonstrate that LCC2 cells selected for CDK4/6 inhibitor resistance acquire a distinct SASP-like secretory phenotype, which is characterized by the marked upregulation of some pro-inflammatory cytokines (IL-8), but not the classical inflammatory cytokines including IL1-α and IL-6. Consistent with these results, NF-κB levels, the major pathway activated by IL1-α, were significantly downregulated in 2PR and not altered in any other resistant sublines compared to their sensitive counterparts. Other markers, including AMPK, phospho-AMPK, and p16, were also unaltered in the resistant cells. Moreover, p53, another factor commonly associated with senescence, was not involved in these resistant cell lines, suggesting that the mechanism underlying resistance is p53-independent. We also observed similar alterations of these cytokines/markers in palbociclib and abemaciclib-resistant derivatives of LCC9 and T47D cells. Taken together, these studies indicate that CDK4/6 inhibitor–resistant ER-positive breast cancer cells are characterized by a non-canonical inflammatory secretome rather than a classical pro-inflammatory cytokine profile.
3.6. Resistance to CDK4/6 Inhibitors Does Not Promote EMT Phenotype in ER+ Breast Cancer Models
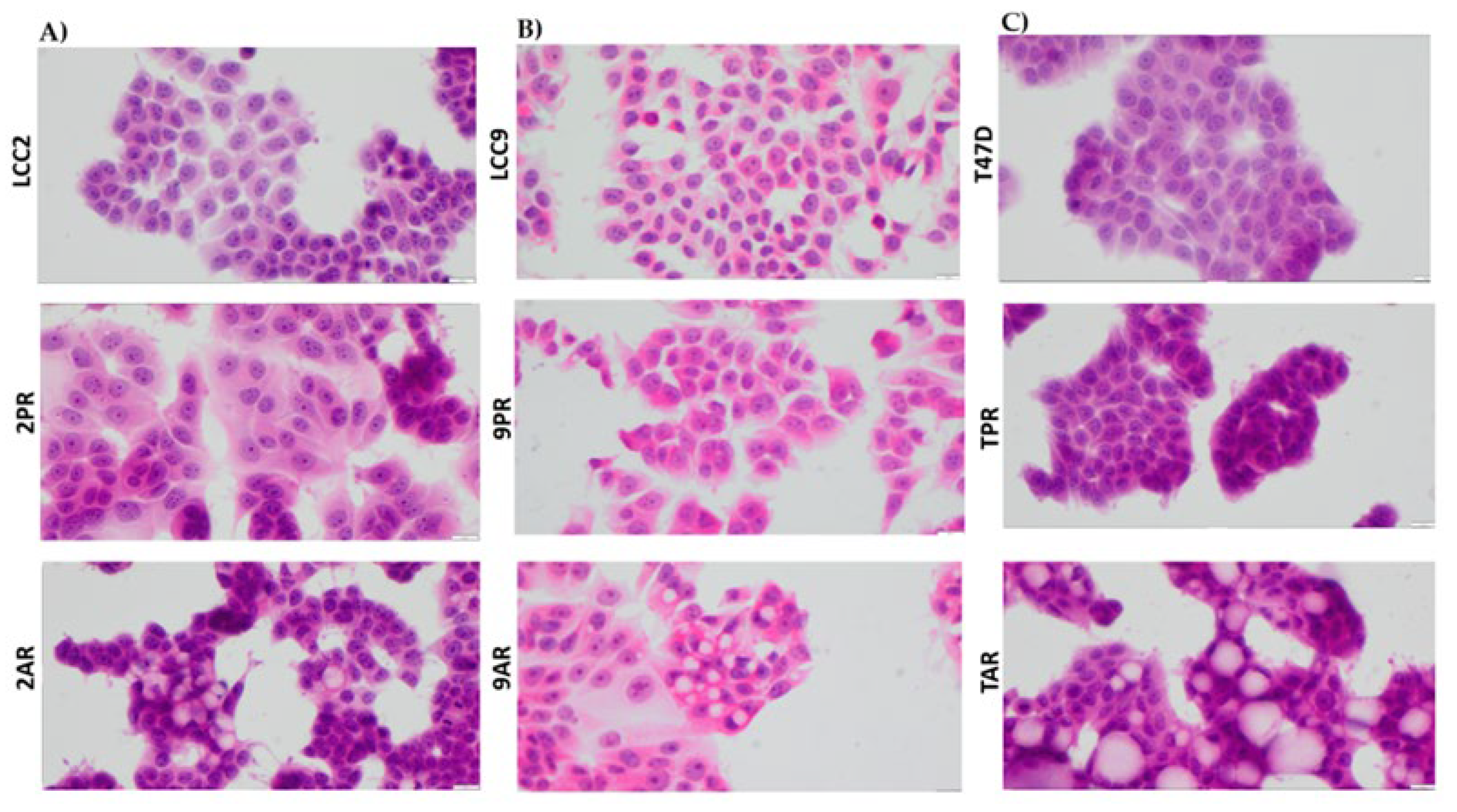
IL-8 has been implicated in promoting epithelial-to-mesenchymal transition (EMT) in various tumor types, and resistance to therapies is often associated with mesenchymal-like characteristics. To assess whether EMT contributes to resistance in CDK4/6 inhibitor (CDK4/6i) models, we examined the morphology of palbociclib- and abemaciclib-resistant breast cancer cells. All resistant models (2PR, 9PR, TPR, 2AR, 9AR and TAR) maintained an epithelial morphology, with no evidence of mesenchymal-like changes (Figure 6).
To further evaluate EMT status, we analyzed the expression of canonical EMT transcription factors using Western blotting (Supplementary Figure S7A,B). Expression levels of the EMT inducers Slug (SNAI2), Snail (SNAI1), and TWIST1 were not significantly altered in either resistance model. Vimentin, a key mesenchymal cytoskeletal marker, was undetectable in both palbociclib- and abemaciclib-resistant cells. These findings indicate that resistance to CDK4/6 inhibitors does not involve EMT, and resistant cells do not acquire a mesenchymal phenotype. To assess whether stem-like properties contribute to resistance, we also evaluated ALDH1 activity—a recognized marker of breast cancer stem cells—using the ALDEFLUOR assay. No ALDH1 activity was detected in CDK4/6 inhibitor-resistant cells (Supplementary Figure S8), providing no evidence to support a role for ALDH1-positive stem-like cells in resistance.
4. Discussion
We show that CDK4/6 inhibitor-resistant ER+ breast cancer cells display a non-canonical, metabolically altered senescence phenotype marked by loss of mitochondrial membrane potential, and a selectively altered SASP profile. This distinct phenotype underscores the atypical nature of these resistant cells and calls into question conventional definitions of senescence. Conventionally, therapy-induced senescence (TIS) in cancer cells has been characterized by permanent growth arrest, coupled with a pro-inflammatory senescence-associated secretory phenotype (SASP) enriched in key cytokines like IL-6 and IL-8, as well as various growth factors [24]. IL-1α is generally thought to be required for the amplification of the SASP via the NF-κB pathway [27], and its absence here could indicate a selective suppression or rewiring of upstream regulators in the resistant cells. Besides the IL-1/NF-κB–dependent secretome, Campisi group further categorized senescent phenotypes into mitochondrial dysfunction-associated senescence (MiDAS) secretome using a fibroblast model system. They also demonstrated MiDAS as another form of cellular senescence that may play a role in age-related diseases, where it was observed in mouse models of progeria, and affecting metabolism [17]. In our study, the CDK4/6 inhibitor–resistant breast cancer models diverged significantly from these senescence programs. Specifically, the lack of alteration of key SASP components such as IL-6, IL-1α and NF-κB in these resistant cells point to a senescent phenotype that is distinct from this pro-inflammatory senescence arrest. In addition, we did not observe the expected decrease in the NAD+/NADH ratio typically associated with MiDAS. Instead, our study revealed an altered cellular metabolism marked by significant loss of mitochondrial membrane potential and decrease in ATP production via oxidative phosphorylation (OXPHOS) in resistant sublines indicating a distinct altered cellular metabolism. These findings together suggest the presence of mitochondrial stress, and reflects an adaptive mechanism by shifting energy balance or altering signaling pathways to bypass drug-induced cell cycle arrest. This state could signal a non-canonical senescence phenotype, where cells remain metabolically active, but do not conform to traditional senescence markers including p16 and p53 or AMPK. HMGB1 functions as a key mediator of senescence-associated inflammation, dependent p53 activity to the SASP [25]. Significant downregulation of secreted HMGB1 rules out inflammatory signaling in our models.
To further understand the SASP in resistant cells, we conducted a detailed analysis of other secretory profiles in resistant cells compared to their sensitive counterparts. The upregulation of SASP factors including serpin E1 (PAI-1), IL-8, GDF-15, [26,27,28], but not IL1-α and IL-6 dependent signaling suggests a non-canonical senescence phenotype characterized by distinct SASP features. An increased level of serpin E1/PAI-1, a multifunctional secreted protein, has been previously implicated in metastasis and stromal remodeling [29]. uPA/PAI-1 is a Level 1 prognostic biomarker in early-stage breast cancer; high levels are being associated with poor progression free and overall survival [30,31]. PAI-1 is linked to enhanced tumor cell survival, invasion, and metastasis, particularly through its interaction with the urokinase plasminogen activator (uPA) system [32]. High PAI-1 mRNA levels predict poor outcomes and shorter overall survival [33,34]. Elevated plasma PAI-1 correlates with higher relapse risk and worse overall survival [35]. Significant upregulation of PAI-1 was also observed in other therapies- such as Alisertib, an Aurora kinase inhibitor [36]. These findings support that PAI-1 is not only a marker of senescence, but also as a maintainer of the senescent phenotype under therapeutic stress. Its selective induction in aggressive breast cancer models following senescence activation further suggests a functional role in therapy-induced senescence.
We also observed an increased level of GDF-15, a member of the transforming growth factor-beta (TGF-β) superfamily that has been upregulated in response to cellular stress, including mitochondrial dysfunction, oxidative stress, and inflammation [37]. Other studies reported that GDF-15 overexpression is linked to aggressive phenotypes, radio-resistance, poor response to chemotherapy, and failure of immune checkpoint inhibitors (ICIs) in cancer including head and neck squamous cell carcinoma and breast cancer [38,39]. GDF-15 is upregulated in breast cancer tissues, especially in drug-tolerant persister (DTP) cells, which are linked to treatment resistance and recurrence [40].
IL-8 (CXCL8), another upregulated chemokine in our CDK4/6 inhibitor-resistant sublines, plays a critical role in tumor angiogenesis, metastasis, and chemoresistance, contributing to cancer progression [41]. Elevated IL-8 levels at baseline are linked to poor overall survival in breast cancer patients undergoing chemotherapy. Notably, the observed upregulation of IL-8 without corresponding upregulation of IL-1α is intriguing. A remodeled SASP may represent a key element of the senescence phenotype that allows for tumor progression.
The non-canonical senescence state we have described here also opens the possibility that senescence is not an end-stage process in cancer and can persist in tumors. Several recent reports show that senescent tumor cells can acquire stem-like properties and re-enter the cell cycle, contributing to tumor recurrence [42]. The secretory phenotype of these drug-resistant cells, and their dependence on the selective secretion of IL-8 and other inflammatory mediators, could facilitate such plasticity in adjacent cells in a manner consistent with the senescence-associated stemness phenotype recently described in lymphoma and colorectal cancer models [43]. Despite the upregulation of IL-8, we do not see epithelial-mesenchymal transition (EMT) phenotype or any upregulation EMT-TFs as well as ALDH activity, a stem cell marker in breast cancer.
5. Conclusions
This study revealed a distinct, non-canonical senescence mechanism employed by CDK4/6 inhibitor-resistant breast cancer cells to maintain therapeutic resistance. This mechanism is marked by altered metabolic functions and an atypical SASP profile. From a clinical perspective, the drug-persistent senescent state we have identified offers a promising avenue for patient stratification and the development of targeted treatment strategies. These represent novel therapeutic vulnerabilities that could enhance the efficacy of CDK4/6 inhibitors and guide the rational design of combination therapies aimed at either eradicating these resilient cells or reversing their senescent phenotype.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
“Conceptualization, S.S.B. and Y.G-P.; methodology, A.M. Y.G. and L.R.; validation, A.M. Y.G. and L.R. writing—original draft preparation, A.M., S.S.B., and Y.G-P.; writing—review and editing, A.M., S.S.B., and Y.G-P; funding acquisition, S.S.B, and Y.G-P. All authors have read and agreed to the published version of the manuscript.”.
Funding
This study was supported partly by the Susan G. Komen for the Cure to Sunil Badve (SAC220219) and Winship Invest$$ pilot grant to Yesim Gokmen-Polar. In addition, Startup funds from Emory University were also utilized. Sunil Badve is a Komen scholar.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is available upon request.
Acknowledgments
Research reported in this publication was supported in part by the Pediatrics/Winship Flow Cytometry Core of Winship Cancer Institute of Emory University, Children’s Healthcare of Atlanta (NIH/NCI) under award number P30CA138292. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institute of Health.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ALDH1 | Aldehyde dehydrogenase 1 |
| AMPK | AMP-activated protein kinase (AMPK) |
| CDK4/6i | CDK4/6 inhibitors |
| ECAR | Extracellular Acidification Rate |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| EMT | Epithelial-mesenchymal transition |
| ER+ | Estrogen Receptor-positive |
| FBS | Fetal Bovine Serum |
| FCCP | Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone |
| GDF-15 | Growth/differentiation factor 15 |
| H&E | Hematoxylin and Eosin |
| HMGB1 | High Mobility Group Box 1 protein |
| IGFBP-2 | Insulin Like Growth Factor Binding Protein 2 |
| IL-1α | Interleukin -1alpha |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| MEM | Minimum Essential Medium |
| MiDAS | Mitochondrial dysfunction associated senescence |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B |
| OCR | Oxygen Consumption Rate |
| OXPHOS | Oxidative phosphorylation |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PDGF-AA | Platelet -derived growth factor AA |
| RANTES | Regulated Upon Activation, Normal T-cell Expressed, and Secreted |
| SA-β-Gal | Senescence-associated β-galactosidase |
| SASP | Senescence-Associated Secretory Phenotype |
| SDS-PAGE | Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis |
| TIS | Therapy-induced Senescence |
| TMRE | Tetramethylrhodamine ethyl ester |
| VEGF | Vascular endothelial growth factor |
| 2PR | Palbociclib-resistant LCC2 subline |
| 9PR | Palbociclib-resistant LCC9 subline |
| TPR | Palbociclib-resistant T47D subline |
| 2AR | Abemaciclib-resistant LCC2 subline |
| 9AR | Abemaciclib-resistant LCC9 subline |
| TAR | Abemaciclib-resistant T47D subline |
References
- Morrison L, Loibl S, Turner NC. The CDK4/6 inhibitor revolution - a game-changing era for breast cancer treatment. Nat Rev Clin Oncol. 2024;21(2):89-105.
- Watt AC, Goel S. Cellular mechanisms underlying response and resistance to CDK4/6 inhibitors in the treatment of hormone receptor-positive breast cancer. Breast Cancer Res. 2022;24(1):17.
- Sledge GW, Jr., Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. The Effect of Abemaciclib Plus Fulvestrant on Overall Survival in Hormone Receptor-Positive, ERBB2-Negative Breast Cancer That Progressed on Endocrine Therapy-MONARCH 2: A Randomized Clinical Trial. JAMA Oncol. 2020;6(1):116-24.
- Glaviano A, Wander SA, Baird RD, Yap KC, Lam HY, Toi M, et al. Mechanisms of sensitivity and resistance to CDK4/CDK6 inhibitors in hormone receptor-positive breast cancer treatment. Drug Resist Updat. 2024;76:101103.
- Wang L, Lankhorst L, Bernards R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer. 2022;22(6):340-55.
- Wagner V, Gil J. Senescence as a therapeutically relevant response to CDK4/6 inhibitors. Oncogene. 2020;39(29):5165-76.
- Wang B, Varela-Eirin M, Brandenburg SM, Hernandez-Segura A, van Vliet T, Jongbloed EM, et al. Pharmacological CDK4/6 inhibition reveals a p53-dependent senescent state with restricted toxicity. EMBO J. 2022;41(6):e108946.
- Blagosklonny MV. Rapamycin, proliferation and geroconversion to senescence. Cell Cycle. 2018;17(24):2655-65.
- Crozier L, Foy R, Adib R, Kar A, Holt JA, Pareri AU, et al. CDK4/6 inhibitor-mediated cell overgrowth triggers osmotic and replication stress to promote senescence. Mol Cell. 2023;83(22):4062-77 e5.
- Maskey RS, Wang F, Lehman E, Wang Y, Emmanuel N, Zhong W, et al. Sustained mTORC1 activity during palbociclib-induced growth arrest triggers senescence in ER+ breast cancer cells. Cell Cycle. 2021;20(1):65-80.
- Zhou FH, Downton T, Freelander A, Hurwitz J, Caldon CE, Lim E. CDK4/6 inhibitor resistance in estrogen receptor positive breast cancer, a 2023 perspective. Front Cell Dev Biol. 2023;11:1148792.
- Michaloglou C, Crafter C, Siersbaek R, Delpuech O, Curwen JO, Carnevalli LS, et al. Combined Inhibition of mTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-term Growth Inhibition in Estrogen Receptor-positive Breast Cancer. Molecular Cancer Therapeutics. 2018;17(5):908-20.
- Schmitt CA, Wang B, Demaria M. Senescence and cancer - role and therapeutic opportunities. Nat Rev Clin Oncol. 2022;19(10):619-36.
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853-68.
- Song KX, Wang JX, Huang D. Therapy-induced senescent tumor cells in cancer relapse. J Natl Cancer Cent. 2023;3(4):273-8.
- Ohtani N. The roles and mechanisms of senescence-associated secretory phenotype (SASP): can it be controlled by senolysis? Inflamm Regen. 2022;42(1):11.
- Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016;23(2):303-14.
- Miwa S, Kashyap S, Chini E, von Zglinicki T. Mitochondrial dysfunction in cell senescence and aging. J Clin Invest. 2022;132(13).
- Martini H, Passos JF. Cellular senescence: all roads lead to mitochondria. FEBS J. 2023;290(5):1186-202.
- Brunner N, Frandsen TL, Holst-Hansen C, Bei M, Thompson EW, Wakeling AE, et al. MCF7/LCC2: a 4-hydroxytamoxifen resistant human breast cancer variant that retains sensitivity to the steroidal antiestrogen ICI 182,780. Cancer Res. 1993;53(14):3229-32.
- Brunner N, Boysen B, Jirus S, Skaar TC, Holst-Hansen C, Lippman J, et al. MCF7/LCC9: an antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross-resistance to the nonsteroidal antiestrogen tamoxifen. Cancer Res. 1997;57(16):3486-93.
- Gokmen-Polar Y, Neelamraju Y, Goswami CP, Gu Y, Gu X, Nallamothu G, et al. Splicing factor ESRP1 controls ER-positive breast cancer by altering metabolic pathways. EMBO Rep. 2019;20(2).
- Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99-118.
- Fitsiou E, Soto-Gamez A, Demaria M. Biological functions of therapy-induced senescence in cancer. Semin Cancer Biol. 2022;81:5-13.
- Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013;201(4):613-29.
- Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020;18(1):e3000599.
- Haake M, Haack B, Schafer T, Harter PN, Mattavelli G, Eiring P, et al. Tumor-derived GDF-15 blocks LFA-1 dependent T cell recruitment and suppresses responses to anti-PD-1 treatment. Nat Commun. 2023;14(1):4253.
- Kaur G, Roy B. Decoding Tumor Angiogenesis for Therapeutic Advancements: Mechanistic Insights. Biomedicines. 2024;12(4).
- Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8(8):877-84.
- Harbeck N, Schmitt M, Meisner C, Friedel C, Untch M, Schmidt M, et al. Ten-year analysis of the prospective multicentre Chemo-N0 trial validates American Society of Clinical Oncology (ASCO)-recommended biomarkers uPA and PAI-1 for therapy decision making in node-negative breast cancer patients. Eur J Cancer. 2013;49(8):1825-35.
- Harris L, Fritsche H, Mennel R, Norton L, Ravdin P, Taube S, et al. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J Clin Oncol. 2007;25(33):5287-312.
- Melzer C, von der Ohe J, Otterbein H, Ungefroren H, Hass R. Changes in uPA, PAI-1, and TGF-beta Production during Breast Cancer Cell Interaction with Human Mesenchymal Stroma/Stem-Like Cells (MSC). Int J Mol Sci. 2019;20(11).
- Sternlicht MD, Dunning AM, Moore DH, Pharoah PD, Ginzinger DG, Chin K, et al. Prognostic value of PAI1 in invasive breast cancer: evidence that tumor-specific factors are more important than genetic variation in regulating PAI1 expression. Cancer Epidemiol Biomarkers Prev. 2006;15(11):2107-14.
- Duffy MJ, McGowan PM, Harbeck N, Thomssen C, Schmitt M. uPA and PAI-1 as biomarkers in breast cancer: validated for clinical use in level-of-evidence-1 studies. Breast Cancer Res. 2014;16(4):428.
- Ferroni P, Roselli M, Portarena I, Formica V, Riondino S, F LAF, et al. Plasma plasminogen activator inhibitor-1 (PAI-1) levels in breast cancer - relationship with clinical outcome. Anticancer Res. 2014;34(3):1153-61.
- Niro F, Pecoraro G, Balestrieri A, Soricelli A, D’Aiuto M, Mossetti G, et al. Cellular senescence as a prognostic marker for predicting breast cancer progression in 2D and 3D organoid models. Biomed Pharmacother. 2025;189:118324.
- Salminen A. GDF15/MIC-1: a stress-induced immunosuppressive factor which promotes the aging process. Biogerontology. 2024;26(1):19.
- He Y, Zhang X, Zhang Y, Luo W, Zhu Z, Song K, et al. Growth differentiation factor 15 is required for triple-negative breast cancer cell growth and chemoresistance. Anticancer Drugs. 2023;34(3):351-60.
- Li YL, Chang JT, Lee LY, Fan KH, Lu YC, Li YC, et al. GDF15 contributes to radioresistance and cancer stemness of head and neck cancer by regulating cellular reactive oxygen species via a SMAD-associated signaling pathway. Oncotarget. 2017;8(1):1508-28.
- Bellio C, Emperador M, Castellano P, Gris-Oliver A, Canals F, Sanchez-Pla A, et al. GDF15 Is an Eribulin Response Biomarker also Required for Survival of DTP Breast Cancer Cells. Cancers (Basel). 2022;14(10).
- Zhao Z, Wang S, Lin Y, Miao Y, Zeng Y, Nie Y, et al. Epithelial-mesenchymal transition in cancer: Role of the IL-8/IL-8R axis. Oncol Lett. 2017;13(6):4577-84.
- Lee S, Schmitt CA. The dynamic nature of senescence in cancer. Nat Cell Biol. 2019;21(1):94-101.
- Milanovic M, Fan DNY, Belenki D, Dabritz JHM, Zhao Z, Yu Y, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018;553(7686):96-100.
Figure 1.
Resistance profile of cells selected in prolonged exposure to palbociclib and abemaciclib. Parental (LCC2, LCC9 and T47D), palbociclib-resistant (2PR, 9PR and TPR), and abemaciclib-resistant (2AR, 9AR and TAR) cells were treated for 72 h with increasing concentrations of palbociclib or abemaciclib (MTT cell viability assays).
Figure 1.
Resistance profile of cells selected in prolonged exposure to palbociclib and abemaciclib. Parental (LCC2, LCC9 and T47D), palbociclib-resistant (2PR, 9PR and TPR), and abemaciclib-resistant (2AR, 9AR and TAR) cells were treated for 72 h with increasing concentrations of palbociclib or abemaciclib (MTT cell viability assays).
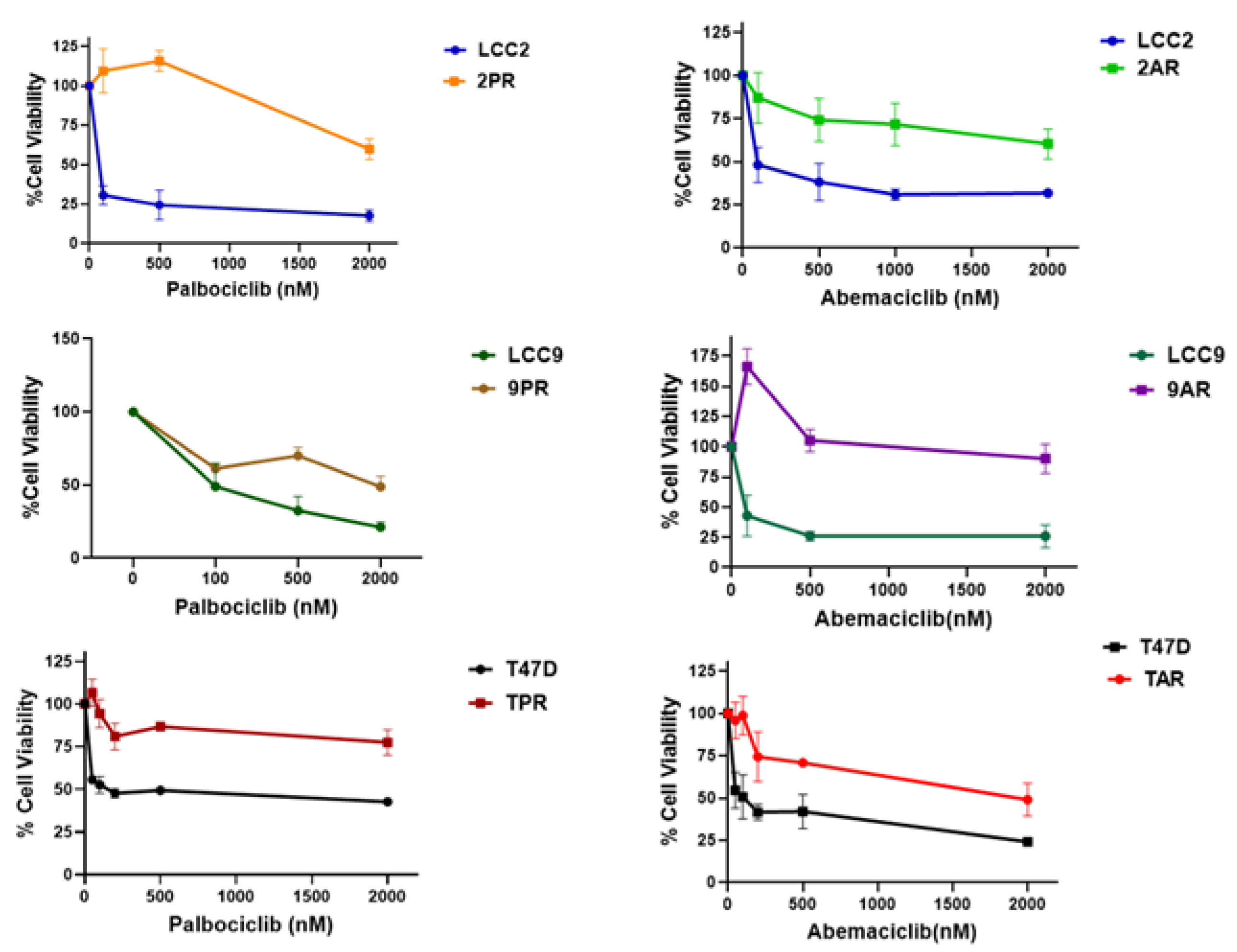
Figure 2.
Palbociclib (PR) and Abemaciclib (AR)-resistant cells exhibit increased senescence. Senescence-associated β-galactosidase (SA-β-Gal) activity was measured using flow cytometry with CellEvent™ Senescence Green Kit (Cat# C10840). Bar plot quantification of the percent of SA-β-Gal–positive cells per group is shown (Mean of n = 3 of biological replicates). One-way ANOVA test was used to determine significance; *p < 0.05.
Figure 2.
Palbociclib (PR) and Abemaciclib (AR)-resistant cells exhibit increased senescence. Senescence-associated β-galactosidase (SA-β-Gal) activity was measured using flow cytometry with CellEvent™ Senescence Green Kit (Cat# C10840). Bar plot quantification of the percent of SA-β-Gal–positive cells per group is shown (Mean of n = 3 of biological replicates). One-way ANOVA test was used to determine significance; *p < 0.05.
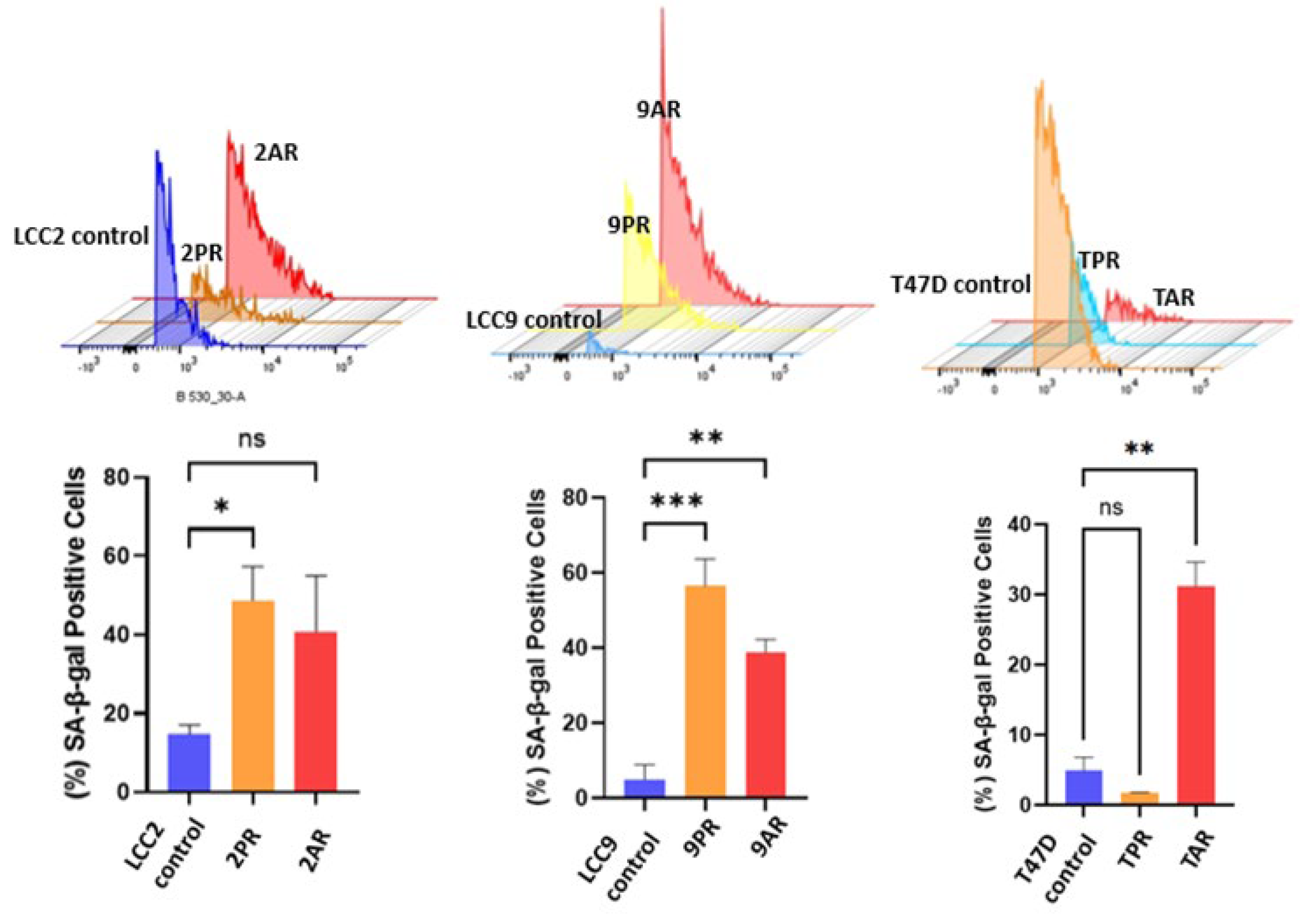
Figure 3.
Senescence-related changes in cellular NAD+/NADH ratio and mitochondrial membrane potential. (A) NAD+ and NADH levels were quantified and expressed as % of total NAD+/NADH in LCC2 parental cells, palbociclib-resistant (2PR), and abemaciclib-resistant (2AR) cells. (B) Mitochondrial membrane potential was assessed by TMRE fluorescence in the same cell lines. Data are shown as a normalized percentage relative to LCC2 cells. Data represents mean ± SD of at least n = 3 independent experiments. Two-way ANOVA (A) and one-way ANOVA (B) were used to assess statistical significance followed by post hoc tests. **p < 0.01; ****p < 0.0001; ns: not significant.
Figure 3.
Senescence-related changes in cellular NAD+/NADH ratio and mitochondrial membrane potential. (A) NAD+ and NADH levels were quantified and expressed as % of total NAD+/NADH in LCC2 parental cells, palbociclib-resistant (2PR), and abemaciclib-resistant (2AR) cells. (B) Mitochondrial membrane potential was assessed by TMRE fluorescence in the same cell lines. Data are shown as a normalized percentage relative to LCC2 cells. Data represents mean ± SD of at least n = 3 independent experiments. Two-way ANOVA (A) and one-way ANOVA (B) were used to assess statistical significance followed by post hoc tests. **p < 0.01; ****p < 0.0001; ns: not significant.
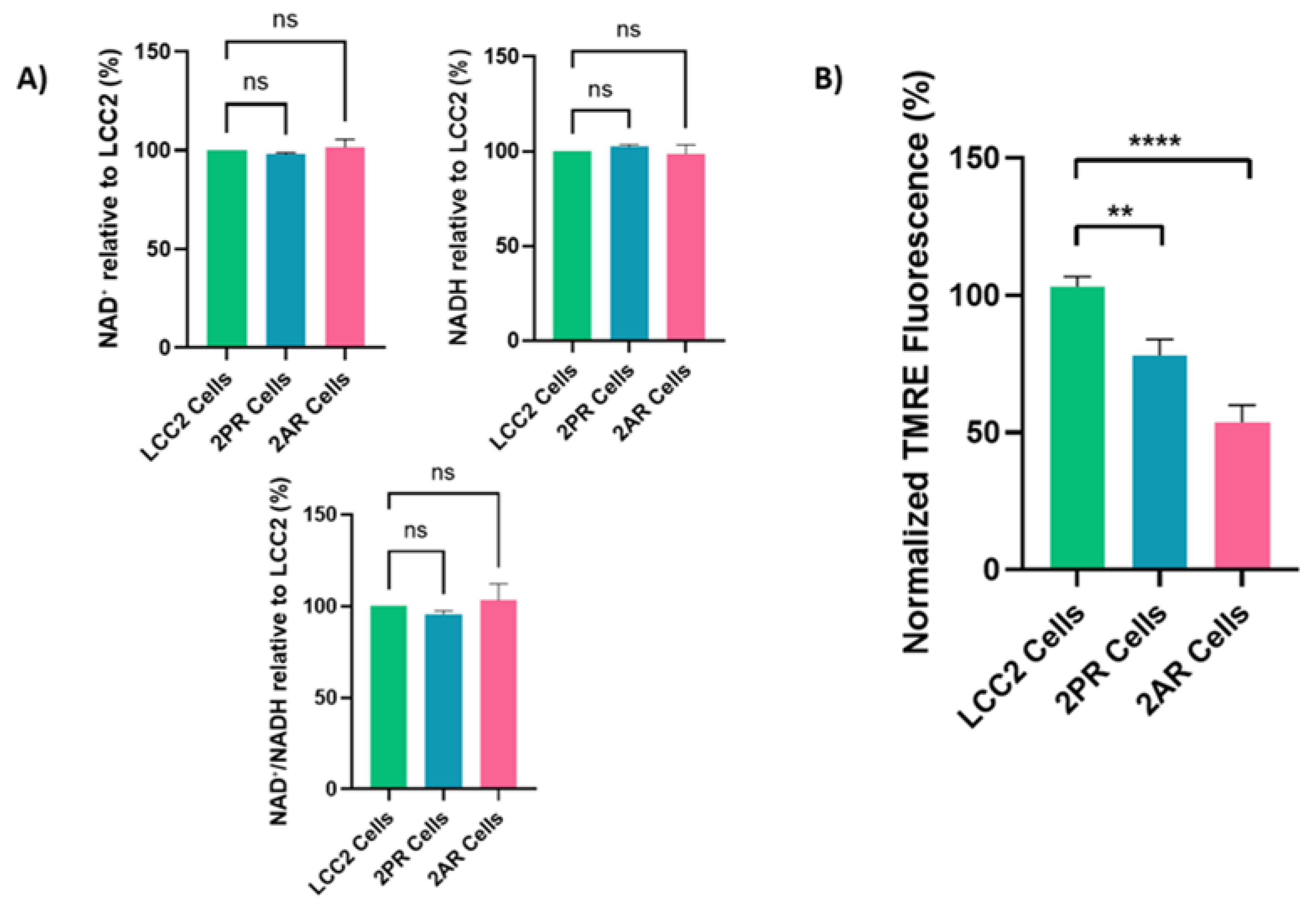
Figure 4.
Quantification of significant cytokines/markers using Proteome Profiler Array Human XL Cytokine Array. Conditioned media collected from parental LCC2 cells and CDK4/6 inhibitor–resistant sublines (2PR, resistant to palbociclib, and 2AR, resistant to abemaciclib) were analyzed using a human cytokine antibody array to profile secreted SASP factors.
Figure 4.
Quantification of significant cytokines/markers using Proteome Profiler Array Human XL Cytokine Array. Conditioned media collected from parental LCC2 cells and CDK4/6 inhibitor–resistant sublines (2PR, resistant to palbociclib, and 2AR, resistant to abemaciclib) were analyzed using a human cytokine antibody array to profile secreted SASP factors.
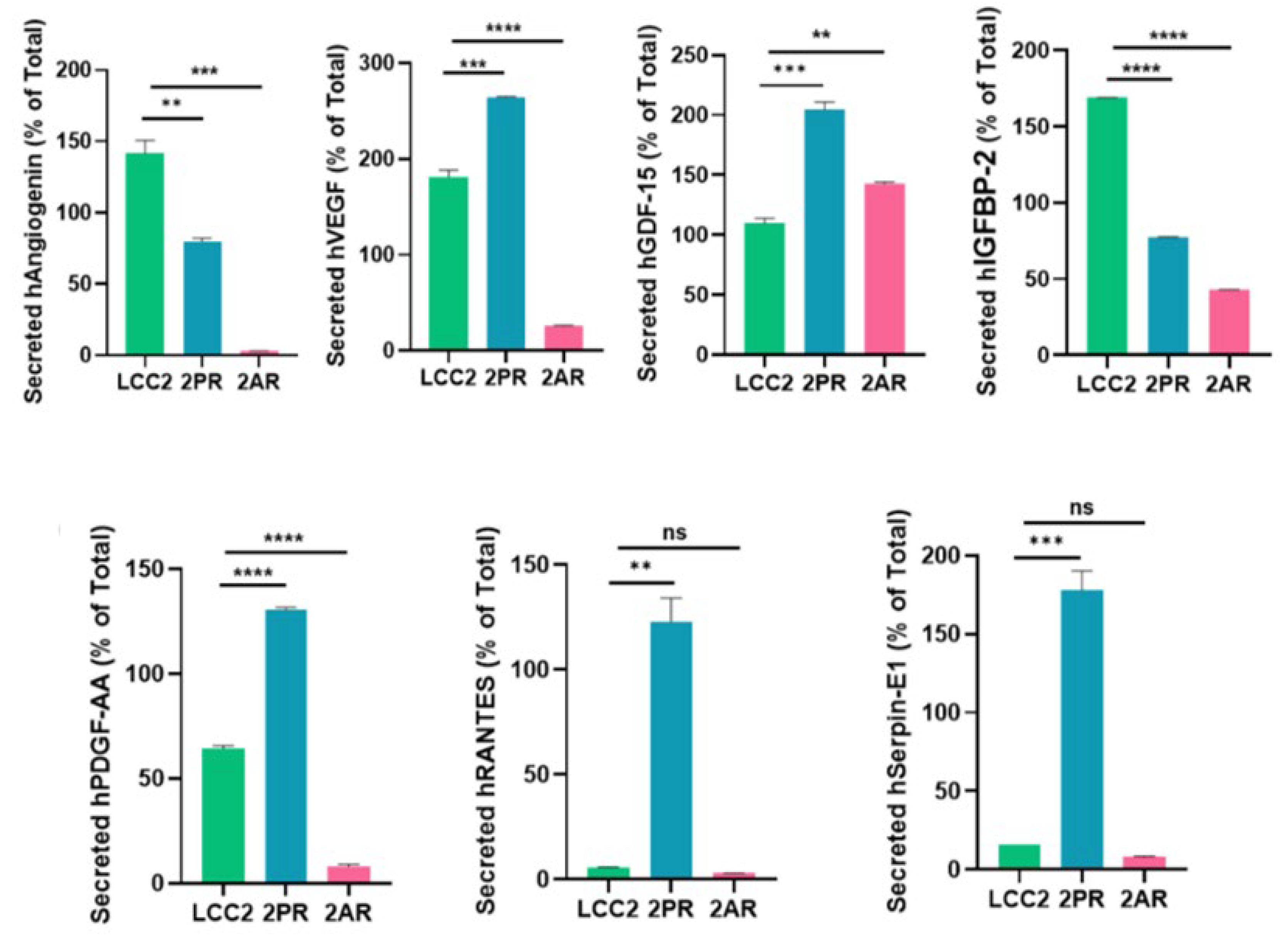
Figure 5.
Quantification of altered SASP factors in palbociclib and abemaciclib resistant LCC2 sublines compared to their parental cells using ELISA. Supernatant from parental LCC2 (green), LCC2-2PR palbociclib-resistant (red), and LCC2-2AR Abemaciclib-resistant (blue) cells were collected and subjected to enzyme-linked immunosorbent assay (ELISA) for a panel of key SASP-associated factors. Ten secreted proteins were quantified: IL-8, IL-6, IL-1α, GDF-15, HMGB1 (high mobility group box 1), RANTES (CCL5), angiogenin, VEGF, PDGF-AA, and serpin E1 (PAI-1). Bar graphs present the mean levels of each factor in the media of the three cell lines (expressed as a percentage of the total level measured for that factor, see Methods), with error bars indicating mean ± SD of n = 3 independent experiments (triplicates each). Significance was determined by one-way ANOVA and post hoc tests: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Figure 5.
Quantification of altered SASP factors in palbociclib and abemaciclib resistant LCC2 sublines compared to their parental cells using ELISA. Supernatant from parental LCC2 (green), LCC2-2PR palbociclib-resistant (red), and LCC2-2AR Abemaciclib-resistant (blue) cells were collected and subjected to enzyme-linked immunosorbent assay (ELISA) for a panel of key SASP-associated factors. Ten secreted proteins were quantified: IL-8, IL-6, IL-1α, GDF-15, HMGB1 (high mobility group box 1), RANTES (CCL5), angiogenin, VEGF, PDGF-AA, and serpin E1 (PAI-1). Bar graphs present the mean levels of each factor in the media of the three cell lines (expressed as a percentage of the total level measured for that factor, see Methods), with error bars indicating mean ± SD of n = 3 independent experiments (triplicates each). Significance was determined by one-way ANOVA and post hoc tests: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
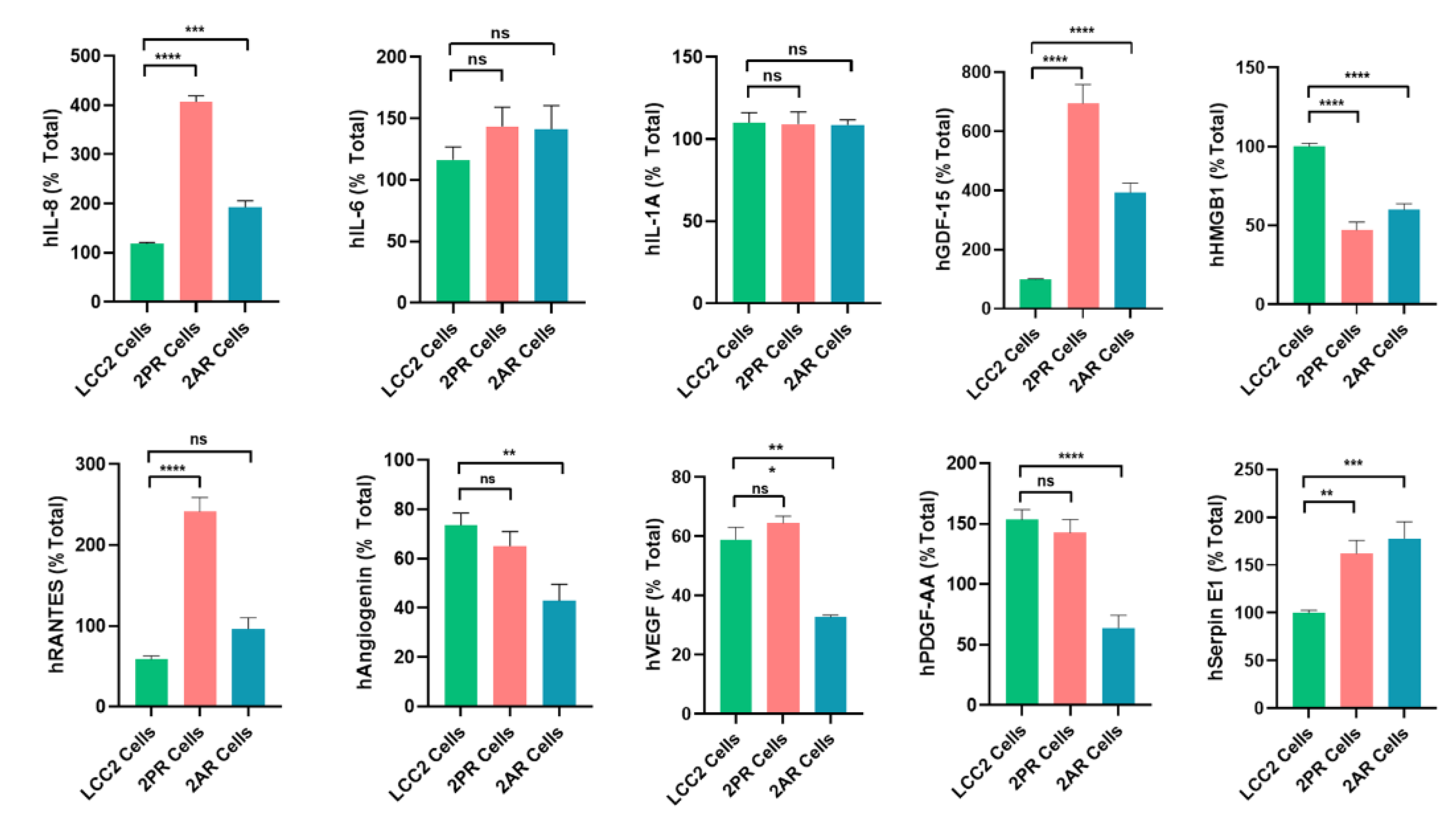
Figure 6.
Palbociclib and abemaciclib resistant cells do not exhibit morphological features characteristic of EMT. Cell morphology using high-power magnification (40x) of the cultured cell lines under a microscope in (A) LCC2 model; 2PR (palbociclib resistant subline of LCC2), 2AR (abemaciclib resistant subline of LCC2), and LCC2 (CDK4/6i sensitive parental cell line). Scale bar: 20 µm. (B) LCC9 model; 2PR (palbociclib resistant subline of LCC2, abemaciclib resistant subline of LCC2) and LCC9 (CDK4/6i sensitive parental cell line). Scale bar: 20 µm. (C) T47D model; TPR (palbociclib resistant subline of LCC2), TAR (abemaciclib resistant subline of LCC2), and T47D (CDK4/6i sensitive parental cell line). Scale bar: 20 µm.
Figure 6.
Palbociclib and abemaciclib resistant cells do not exhibit morphological features characteristic of EMT. Cell morphology using high-power magnification (40x) of the cultured cell lines under a microscope in (A) LCC2 model; 2PR (palbociclib resistant subline of LCC2), 2AR (abemaciclib resistant subline of LCC2), and LCC2 (CDK4/6i sensitive parental cell line). Scale bar: 20 µm. (B) LCC9 model; 2PR (palbociclib resistant subline of LCC2, abemaciclib resistant subline of LCC2) and LCC9 (CDK4/6i sensitive parental cell line). Scale bar: 20 µm. (C) T47D model; TPR (palbociclib resistant subline of LCC2), TAR (abemaciclib resistant subline of LCC2), and T47D (CDK4/6i sensitive parental cell line). Scale bar: 20 µm.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



