Submitted:
07 November 2025
Posted:
10 November 2025
You are already at the latest version
Abstract
Abstract:
Neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and amyotrophic lateral sclerosis share common molecular mechanisms involving oxidative stress, neuroinflammation, and microvascular dysfunction. These interrelated processes form a self-perpetuating cycle that accelerates neuronal loss and cognitive decline. Oxidative stress triggers mitochondrial impairment and lipid peroxidation, while chronic glial activation amplifies the inflammatory milieu through cytokine release and reactive oxygen species generation. Concurrently, endothelial and pericytic injury disrupts the blood–brain barrier (BBB), compromising neurovascular coupling and metabolic homeostasis. This review summarizes the mechanistic crosstalk between oxidative, inflammatory, and vascular pathways in neurodegeneration, highlighting recent advances in antioxidant, anti-inflammatory, and vasoprotective strategies. We further discuss emerging therapeutic approaches such as nanocarrier-based delivery systems, multitarget drugs, and genetic modulation aimed at restoring redox balance and microvascular integrity. Understanding the molecular intersections of these mechanisms may provide new insights into disease-modifying interventions for neurodegenerative disorders.
Keywords:
oxidative stress
; neuroinflammation
; microvascular damage
; neurodegeneration
; blood–brain barrier
; mitochondria
; therapeutic approaches
1. Introduction
Neurodegenerative diseases (NDs) such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) represent a major global health burden characterized by progressive neuronal loss, cognitive decline, and functional disability [1,2]. Despite the heterogeneity of their clinical manifestations, these disorders share fundamental pathophysiological mechanisms involving oxidative stress, neuroinflammation, and microvascular dysfunction [3,4,5]. The convergence of these processes underlies a self-perpetuating cycle that disrupts neuronal homeostasis, impairs synaptic plasticity, and compromises the blood–brain barrier (BBB), ultimately contributing to neurodegeneration [6,7,8,9].
Oxidative stress, resulting from an imbalance between reactive oxygen species (ROS) generation and antioxidant defense systems, induces lipid peroxidation, mitochondrial dysfunction, and DNA damage [10,11,12]. Chronic activation of glial cells—particularly microglia and astrocytes—drives a sustained neuroinflammatory response, releasing cytokines, chemokines, and reactive nitrogen species that amplify neuronal injury [13,14,15,16]. Simultaneously, endothelial dysfunction, pericyte loss, and tight-junction disruption contribute to BBB breakdown and cerebral hypoperfusion, creating a permissive environment for neurotoxic cascades [17,18,19,20].
In recent years, the neurovascular unit (NVU) has emerged as a central integrative framework for understanding how redox imbalance and inflammation converge at the microvascular level to accelerate neurodegenerative processes [21,22,23]. Growing evidence suggests that restoring vascular integrity, modulating glial activation, and re-establishing redox homeostasis may hold promise as disease-modifying strategies [24,25,26,27].
This review provides a comprehensive synthesis of current mechanistic insights into the oxidative–inflammatory–vascular triad in neurodegeneration and highlights emerging therapeutic interventions targeting these interconnected pathways [28,29,30]. Understanding their crosstalk offers novel opportunities for early intervention and neuroprotection in aging and disease contexts [31,32,33].
2. Molecular Basis of Oxidative Stress in Neurodegeneration
Oxidative stress is a major pathological hallmark across neurodegenerative diseases, arising from an imbalance between reactive oxygen species (ROS) production and the antioxidant defense mechanisms that maintain redox homeostasis [1,2,3]. In the central nervous system (CNS), neurons are particularly vulnerable to oxidative damage due to their high oxygen consumption, abundant polyunsaturated lipids, and limited regenerative capacity [4,5]. Mitochondria, NADPH oxidases, and peroxisomes are the primary sources of ROS, while antioxidant enzymes such as superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx), and peroxiredoxins act as the main defensive systems [6,7,8].
In neurodegenerative conditions like Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS), excessive ROS and reactive nitrogen species (RNS) lead to protein oxidation, lipid peroxidation, and nucleic acid damage, ultimately compromising neuronal survival [9,10,11,12]. Mitochondrial dysfunction—particularly in complex I and complex IV of the electron transport chain—results in increased superoxide anion generation and reduced ATP synthesis [13,14]. The accumulation of oxidized mitochondrial DNA (mtDNA) further triggers apoptotic signaling via cytochrome c release and caspase activation [15,16].
Beyond the mitochondria, aberrant activation of NADPH oxidase (NOX) isoforms, particularly NOX2 and NOX4, has been implicated in microglial ROS production and synaptic injury [17,18]. Elevated NOX activity amplifies oxidative stress through a feed-forward loop that activates NF-κB and other pro-inflammatory transcription factors, linking redox imbalance directly to neuroinflammation [19,20,21]. Moreover, lipid peroxidation products such as 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA) form adducts with neuronal proteins, altering their structure and function, and serving as biomarkers of oxidative injury [22,23].
Dysregulation of antioxidant defenses further exacerbates neuronal vulnerability [24]. Reduced glutathione (GSH) levels and decreased activity of GPx and SOD have been consistently observed in both brain tissue and cerebrospinal fluid of patients with AD and PD [25,26,27]. Additionally, impairment of the nuclear factor erythroid 2–related factor 2 (Nrf2) pathway—a master regulator of antioxidant gene expression—has been shown to aggravate oxidative damage and mitochondrial failure [28,29,30]. Restoration of Nrf2 signaling has therefore become a promising therapeutic strategy, with agents such as sulforaphane, dimethyl fumarate, and curcumin demonstrating neuroprotective effects through activation of Nrf2–ARE (antioxidant response element)–dependent transcription [31,32,33,34].
Recent studies emphasize the bidirectional relationship between oxidative stress and protein aggregation [35]. In AD, oxidative modification of amyloid precursor protein (APP) and tau accelerates plaque and tangle formation, while in PD, oxidation of α-synuclein promotes its misfolding into toxic oligomers [36,37,38]. Conversely, these aggregates further disrupt mitochondrial function and calcium homeostasis, intensifying oxidative injury [39,40]. Thus, oxidative stress is not merely a byproduct but a central driver of neurodegenerative cascades, interwoven with inflammation and vascular dysfunction within the neurovascular unit (NVU) [41,42,43].
Collectively, these findings underscore oxidative stress as a unifying molecular mechanism in neurodegeneration [44,45]. Targeting mitochondrial resilience, NOX inhibition, and Nrf2 activation represents a triad of potential interventions to restore redox balance and attenuate disease progression [46,47,48].
Figure 1.
Molecular Mechanisms of Oxidative Stress and Nrf2 Pathway in Neurodegeneration.
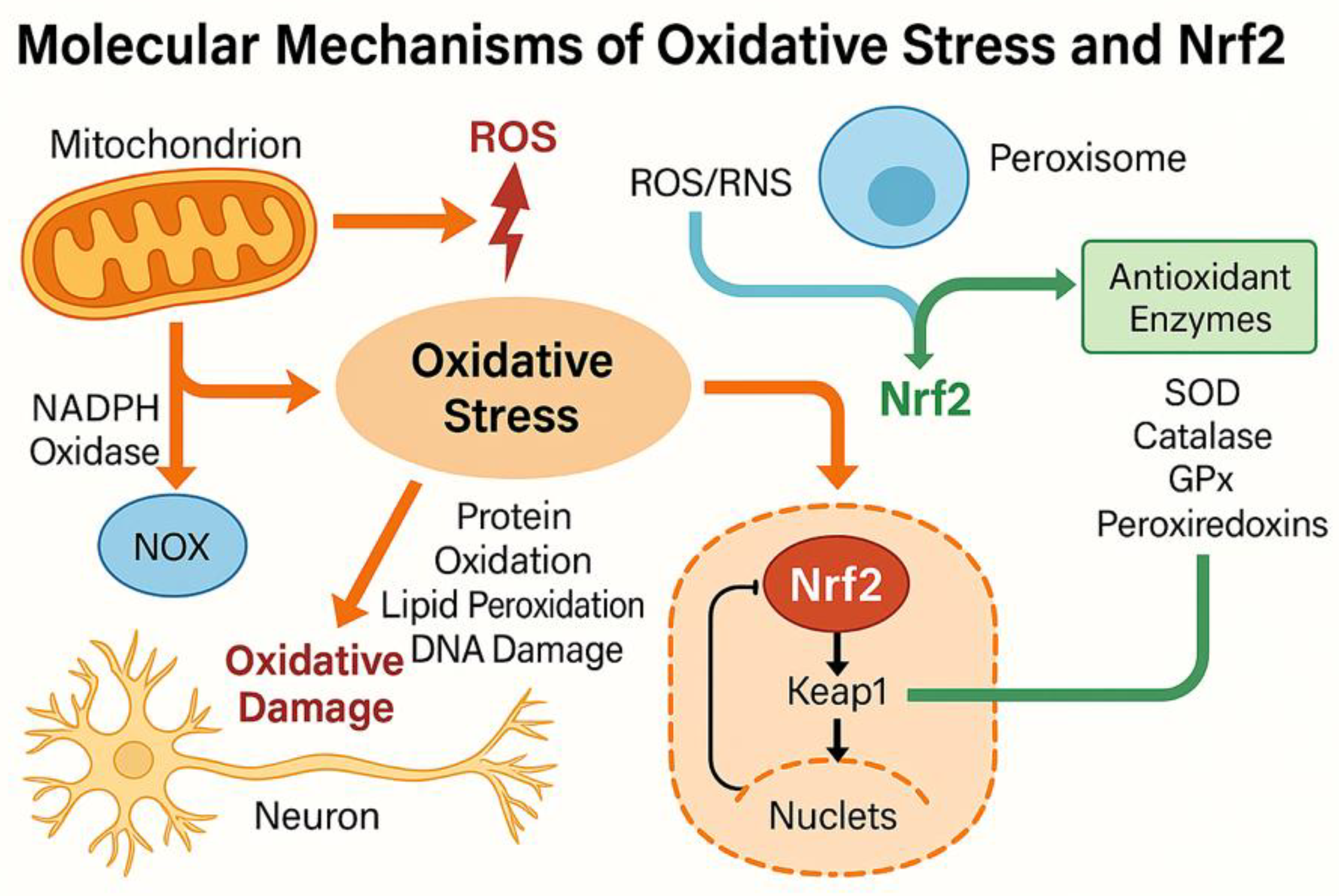
The figure illustrates the main sources and regulatory pathways of oxidative stress in neuronal cells. Mitochondrial dysfunction and NADPH oxidase (NOX) activity generate reactive oxygen species (ROS), leading to protein oxidation, lipid peroxidation, and DNA damage. Under oxidative stress, nuclear factor erythroid 2–related factor 2 (Nrf2) dissociates from its repressor Keap1, translocates into the nucleus, and activates antioxidant response element (ARE)-dependent transcription of detoxifying enzymes, including superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx), and heme oxygenase-1 (HO-1), which restore redox balance and protect neurons from oxidative injury.
3. Neuroinflammatory Pathways and Glial Crosstalk
Neuroinflammation constitutes a second major pathological axis in neurodegenerative deseases (NDs), orchestrated primarily by glial cells—microglia and astrocytes—that serve as the immune sentinels of the central nervous system (CNS) [49,50,51]. While acute inflammatory responses can promote neuroprotection and tissue repair, chronic activation of these glial populations leads to persistent release of pro-inflammatory mediators, oxidative radicals, and excitotoxins that amplify neuronal damage [52,53].
Microglial activation represents the earliest and most dynamic component of neuroinflammation [54]. Under physiological conditions, microglia maintain homeostasis through synaptic pruning, phagocytosis of cellular debris, and neurotrophic factor release [55]. However, in response to chronic stressors such as misfolded proteins (amyloid-β, α-synuclein, or mutant huntingtin), mitochondrial dysfunction, and damaged neurons, microglia shift from a surveillant to an activated phenotype [56,57,58]. This activation involves the engagement of pattern recognition receptors (PRRs), including toll-like receptors (TLR2, TLR4) and NOD-like receptors (NLRP3 inflammasome) [59,60]. The downstream signaling cascade activates NF-κB and MAPK pathways, leading to the transcription of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6, as well as inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) [61,62].
Persistent activation of microglia results in a “cytokine storm” that sustains oxidative and nitrosative stress [63]. The release of nitric oxide (NO) and superoxide anions generates peroxynitrite (ONOO⁻), a potent oxidant capable of inducing lipid peroxidation, protein nitration, and mitochondrial injury [64,65]. Moreover, activated microglia express NADPH oxidase (NOX2), contributing directly to the ROS pool and reinforcing a feed-forward loop between inflammation and oxidative stress [66,67].
Astrocytes, which normally maintain extracellular ion balance, neurotransmitter recycling, and BBB integrity, also undergo reactive transformation (astrogliosis) in response to chronic inflammation [68]. Reactive astrocytes upregulate glial fibrillary acidic protein (GFAP), release cytokines and complement components (C3, C1q), and exhibit altered glutamate uptake through decreased expression of the excitatory amino acid transporter EAAT2 [69,70]. This dysregulation increases extracellular glutamate levels, promoting excitotoxicity and neuronal death [71]. Additionally, astrocyte–microglia communication via cytokines (IL-33, CXCL10, TGF-β) creates a paracrine network that sustains neuroinflammatory signaling [72,73].
Crosstalk between glial cells and endothelial components of the neurovascular unit (NVU) further exacerbates pathology [74]. Inflammatory cytokines disrupt tight-junction proteins such as claudin-5, occludin, and ZO-1, increasing BBB permeability and facilitating infiltration of peripheral immune cells [75]. These infiltrating leukocytes amplify the inflammatory milieu by releasing proteases, ROS, and matrix metalloproteinases (MMP-2, MMP-9), leading to vascular leakage and microvascular degeneration [76,77].
Recent evidence highlights the existence of dual glial phenotypes, with M1-like microglia and A1 astrocytes exhibiting pro-inflammatory, neurotoxic profiles, whereas M2-like microglia and A2 astrocytes promote tissue repair, phagocytosis, and release of anti-inflammatory mediators (IL-10, TGF-β) [78,79]. However, in neurodegeneration, the equilibrium between these phenotypes shifts toward the M1/A1 axis, reinforcing a state of chronic neuroinflammation [80]. Therapeutic modulation of glial polarization—through agents targeting TLR4, PPAR-γ, or Nrf2 pathways—has shown promising neuroprotective outcomes in preclinical studies [81,82,83].
In summary, the interplay between microglia, astrocytes, and endothelial cells defines the inflammatory landscape of neurodegenerative diseases [84]. Persistent glial activation transforms the NVU from a neuroprotective barrier into a source of oxidative and inflammatory stress, thereby bridging neuroinflammation with vascular dysfunction and neuronal loss [85,86,87].
4. Microvascular Dysfunction and Blood–Brain Barrier Breakdown
The integrity of the cerebral microvasculature is essential for maintaining neural homeostasis. The blood–brain barrier (BBB), composed of endothelial cells, pericytes, astrocytic endfeet, and basement membrane, ensures selective permeability and protects the brain from peripheral insults [73,74,75]. However, in neurodegenerative diseases (NDs), chronic oxidative stress and inflammation lead to progressive microvascular dysfunction, compromising BBB structure and function [76,77,78].
Endothelial dysfunction is one of the earliest manifestations of vascular compromise [79]. Cerebral endothelial cells are highly sensitive to oxidative and inflammatory stimuli, which impair nitric oxide (NO) bioavailability, induce mitochondrial injury, and up-regulate adhesion molecules such as ICAM-1, VCAM-1, and E-selectin [80,81]. These alterations promote leukocyte adhesion and trans-endothelial migration, triggering local inflammation and microvascular obstruction [82]. Furthermore, ROS-mediated oxidation of tight-junction proteins (claudin-5, occludin, ZO-1) disrupts intercellular cohesion, increasing BBB permeability and facilitating neurotoxic molecule entry from the circulation [83,84].
Pericyte degeneration represents a critical event linking vascular and neuronal pathology [85,86]. Pericytes regulate capillary tone, endothelial proliferation, and BBB stability through PDGFR-β and Ang/Tie2 signaling [87]. In Alzheimer’s disease and other NDs, pericyte loss has been demonstrated in both animal models and human autopsy studies, correlating with cerebral hypoperfusion, micro-aneurysm formation, and accumulation of plasma proteins such as fibrinogen and albumin in the parenchyma [88,89]. These extravasated proteins further exacerbate neuroinflammation by activating astrocytes and microglia, thereby amplifying tissue injury [90].
Astrocytic and glial contributions to BBB disruption are increasingly recognized [91]. Under oxidative and inflammatory stress, astrocytic endfeet retract and lose aquaporin-4 (AQP4) polarization, impairing water and ion homeostasis [92]. This alteration compromises the glymphatic system, a recently described clearance pathway essential for removing metabolic waste and amyloid aggregates [93]. Dysfunctional glymphatic flow leads to toxic metabolite accumulation, perpetuating oxidative stress and neuronal degeneration [94,95].
Senescence of endothelial and glial cells adds a further layer of complexity [96]. Senescent cells exhibit a pro-inflammatory secretory phenotype (SASP) characterized by cytokine and MMP release, which deteriorates the extracellular matrix and basement membrane [97]. This process weakens capillary walls and reduces cerebral blood-flow regulation, promoting micro-infarcts and white-matter lesions observed in aging and dementia [98].
Additionally, vascular rarefaction and hypoxia emerge as key pathological features [99]. Chronic hypoperfusion decreases oxygen and nutrient supply to neurons, activates hypoxia-inducible factor-1α (HIF-1α), and shifts metabolism toward glycolysis [100]. Although initially adaptive, sustained HIF-1α activation induces VEGF over-expression, leading to abnormal angiogenesis and leaky, immature vessels that further compromise BBB integrity [100].
In summary, microvascular dysfunction in neurodegenerative diseases results from the synergistic impact of oxidative stress, endothelial injury, pericyte degeneration, astrocytic dysregulation, and cellular senescence [99,100]. These events collectively transform the BBB from a neuroprotective interface into a source of neurotoxicity. Preservation of microvascular health and BBB stability is therefore central to halting the cascade of neurodegeneration and represents a crucial therapeutic target for future interventions [100].
5. Interconnection Between Oxidative Stress, Neuroinflammation, and Microvascular Damage
The interplay between oxidative stress, neuroinflammation, and microvascular dysfunction forms a self-amplifying pathogenic triad that drives the progression of neurodegenerative diseases (NDs) [85,86,87]. These processes do not act in isolation; rather, they are mechanistically and temporally interconnected through reciprocal signaling within the neurovascular unit (NVU), ultimately leading to neuronal death and cognitive decline [88,89].
Oxidative stress as the initiator: Reactive oxygen and nitrogen species (ROS/RNS) generated by mitochondrial dysfunction, NADPH oxidases, and activated glia initiate redox imbalance [90,91]. Oxidative stress acts as a potent trigger for microglial activation through redox-sensitive transcription factors such as NF-κB and AP-1 [92,93]. The resulting cytokine release (IL-1β, TNF-α, IL-6) recruits additional immune cells and amplifies the inflammatory cascade [94]. Furthermore, ROS directly impair endothelial cell function by oxidizing membrane lipids and disrupting nitric-oxide signaling, thereby contributing to vasoconstriction and BBB permeability [95,96].
Neuroinflammation as the amplifier: Once initiated, the inflammatory response perpetuates oxidative stress through feed-forward mechanisms [97,98]. Activated microglia and astrocytes release large amounts of superoxide, NO, and hydrogen peroxide, overwhelming antioxidant defenses [99]. Cytokines such as IL-1β and TNF-α activate iNOS and NOX enzymes, establishing a vicious cycle that sustains redox imbalance [100]. The chronic presence of these mediators promotes endothelial activation, up-regulating adhesion molecules and metalloproteinases that degrade tight-junction proteins [94,95,96].
Microvascular dysfunction as the facilitator: The breakdown of the BBB and microvascular integrity permits peripheral immune-cell infiltration, introducing additional ROS and inflammatory mediators from the circulation [89,97]. This extravasation exacerbates oxidative and inflammatory signaling within the CNS, creating a bidirectional feedback loop between systemic and central processes [98]. Loss of pericytes and endothelial senescence further impair cerebrovascular autoregulation, resulting in hypoxia and nutrient deprivation, which intensify mitochondrial dysfunction and oxidative injury [99,100].
Cellular and molecular crosstalk within the NVU: Endothelial cells, astrocytes, microglia, and neurons engage in complex intercellular communication mediated by cytokines, chemokines, and oxidative metabolites [92,93]. For example, microglial-derived ROS activate endothelial NF-κB signaling, while endothelial IL-6 and MCP-1 further activate microglia, forming a sustained inflammatory loop [94,95]. Astrocytes, in turn, sense oxidative stress and release glutamate and ATP, propagating excitotoxicity and calcium dysregulation in neurons [96,97,98]. This integrated failure of the NVU transforms a local redox disturbance into widespread neurovascular dysfunction [99,100].
Emerging conceptual model: Current evidence supports the notion that these three mechanisms—oxidative stress, inflammation, and vascular injury—converge into a unified neurodegenerative pathway [90,94,99]. Early oxidative imbalance sets the stage for glial activation, while neuroinflammation accelerates endothelial deterioration and BBB leakage [95,96]. Vascular dysfunction, in turn, fuels hypoxia and metabolic distress, perpetuating redox imbalance [97,98,99,100]. This cyclic interdependence accounts for the progressive and irreversible nature of neuronal loss observed in disorders such as Alzheimer’s, Parkinson’s, and ALS [88,89,90].
Understanding this triadic interplay offers a paradigm shift in neurodegeneration research [91,92]. It suggests that effective therapeutic strategies should adopt multimodal approaches that simultaneously target redox homeostasis, inflammatory modulation, and vascular protection, rather than treating these mechanisms independently [93,94,95].
Figure 2.
Oxidative Stress–Neuroinflammation–Microvascular Dysfunction Cycle.
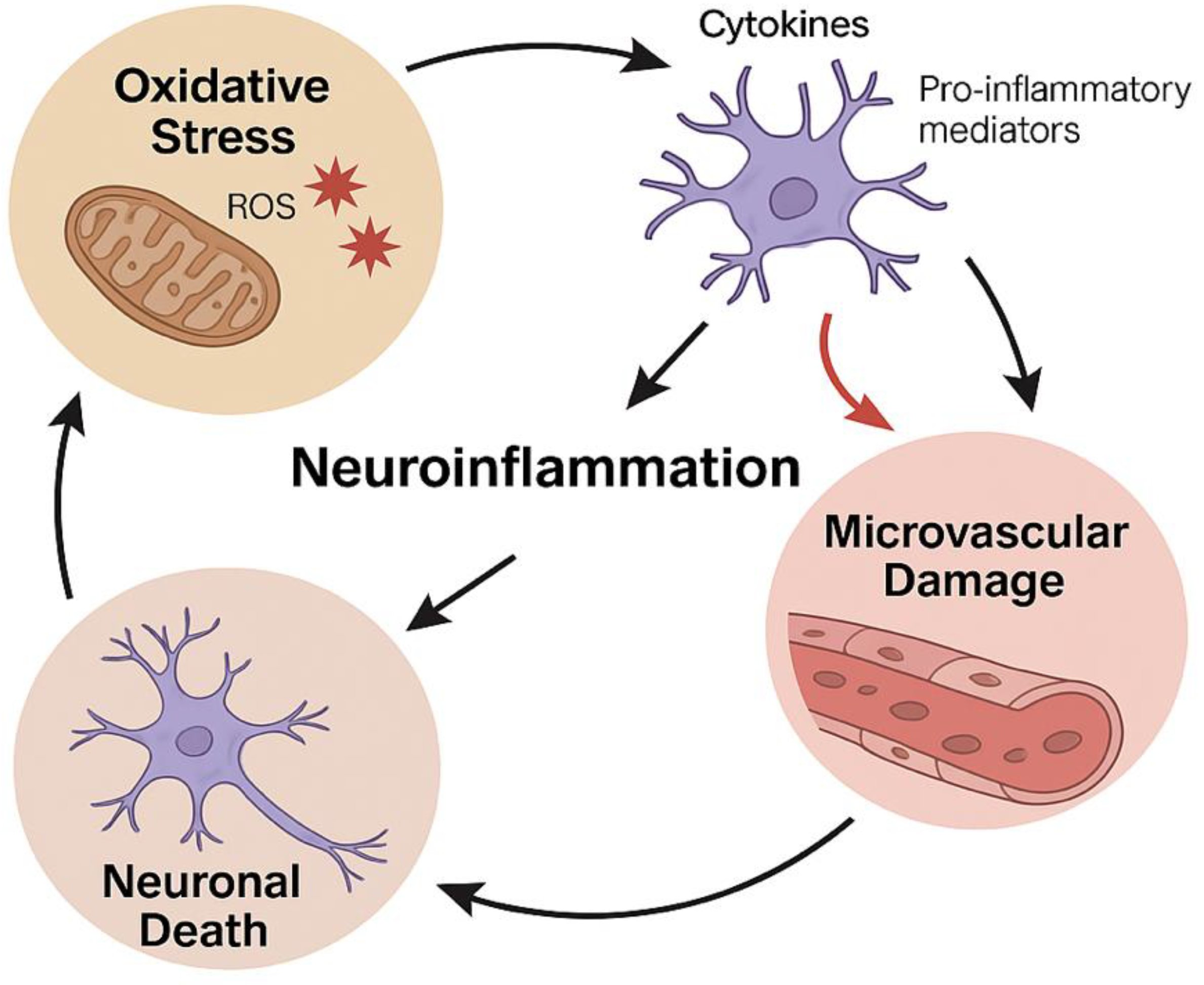
The diagram illustrates the self-perpetuating cycle linking oxidative stress, neuroinflammation, and microvascular damage in neurodegenerative diseases. Excessive production of reactive oxygen species (ROS) promotes glial activation and the release of pro-inflammatory mediators, which in turn exacerbate endothelial dysfunction and blood–brain barrier breakdown. These events contribute to neuronal death and perpetuate the oxidative–inflammatory–vascular feedback loop within the neurovascular unit (NVU).
6. Therapeutic and Preventive Strategies
The intricate interdependence between oxidative stress, neuroinflammation, and microvascular dysfunction highlights the necessity for multitarget therapeutic approaches capable of restoring redox balance, modulating inflammatory signaling, and preserving neurovascular integrity [91,92,93]. Although no current pharmacological intervention fully halts neurodegeneration, numerous preclinical and clinical studies suggest that combined strategies acting on these interconnected mechanisms may significantly slow disease progression [94,95,96].
6.1. Antioxidant-Based Therapies
Restoration of redox equilibrium remains a cornerstone of neuroprotective strategies [97]. Traditional antioxidants such as vitamin E, vitamin C, and coenzyme Q10 have shown limited clinical efficacy due to poor bioavailability and inability to cross the BBB [98]. Consequently, research has shifted toward mitochondria-targeted antioxidants such as MitoQ, SkQ1, and SS-31 (elamipretide), which directly localize within mitochondria to prevent ROS overproduction and maintain ATP synthesis [99,100].
Polyphenolic compounds—including resveratrol, curcumin, quercetin, and epigallocatechin gallate (EGCG)—have demonstrated robust neuroprotective effects in experimental models [83,84]. Their mechanisms extend beyond direct ROS scavenging to activation of the Nrf2–ARE pathway, which enhances the expression of endogenous antioxidants (SOD, catalase, GPx, HO-1) [85,86]. Moreover, melatonin exhibits dual antioxidant and mitochondrial-protective functions, modulating Bcl-2 family proteins and preventing cytochrome c release [87].
6.2. Anti-Inflammatory and Glial-Modulating Approaches
Targeting chronic glial activation is critical for mitigating the inflammatory amplification loop [88]. Nonsteroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen and indomethacin have shown partial protection in epidemiological studies but carry risks of systemic side effects [89]. Modern approaches aim to achieve selective immunomodulation without global suppression [90].
Agents such as minocycline and pioglitazone (a PPAR-γ agonist) have been shown to inhibit microglial activation, suppress NF-κB signaling, and shift microglia toward the anti-inflammatory M2 phenotype [91,92]. Similarly, dimethyl fumarate (approved for multiple sclerosis) exerts combined anti-inflammatory and antioxidant effects through activation of Nrf2 and inhibition of pro-inflammatory cytokines [93]. Novel small molecules targeting NLRP3 inflammasome assembly (e.g., MCC950) or TLR4 antagonists (e.g., TAK-242) show promise in restoring glial homeostasis [94,95,96].
Astrocyte-directed therapies are gaining attention as regulators of BBB integrity and excitotoxicity [97,98]. Ceftriaxone, an FDA-approved β-lactam antibiotic, upregulates the glutamate transporter EAAT2, reducing glutamate-induced neuronal injury [99]. Similarly, adenosine A2A receptor antagonists modulate astrocyte–microglia communication and have demonstrated cognitive benefits in early clinical trials for Parkinson’s disease [100].
6.3. Vascular and BBB-Targeted Interventions
Protection and restoration of microvascular function represent a growing therapeutic frontier [95,96]. Angiotensin II receptor blockers (ARBs) and ACE inhibitors have demonstrated neurovascular protective effects independent of blood pressure regulation, partly by enhancing endothelial NO availability and reducing oxidative stress [97]. Statins, through pleiotropic antioxidant and anti-inflammatory properties, improve endothelial function and reduce amyloid-β deposition [98,99].
Experimental compounds that stabilize the tight-junction architecture (e.g., claudin-5 mimetics, MMP inhibitors) and enhance pericyte survival (via PDGF-BB or Tie2 agonists) have shown promise in preclinical neurodegeneration models [100]. Vasoprotective peptides, such as angiopoietin analogues and VEGF modulators, are under active investigation to repair BBB leakage while preventing aberrant angiogenesis [99,100].
6.4. Emerging and Multimodal Strategies
Given the multifactorial nature of neurodegeneration, combined or multitarget therapies are increasingly favored [93,94]. Nanocarrier-based delivery systems improve drug penetration across the BBB and allow co-delivery of antioxidants and anti-inflammatory agents [95,96]. Lipid nanoparticles, polymeric micelles, and exosome-based carriers are being engineered to transport Nrf2 activators, siRNAs, or mitochondria-protective compounds with enhanced precision [97,98].
Gene and cell-based therapies are also entering the translational stage [99]. Overexpression of antioxidant enzymes (e.g., SOD2, GPx1) via viral vectors reduces ROS burden and protects against neuronal loss [100]. Mesenchymal stem cell (MSC)-derived exosomes exert potent anti-inflammatory and vasoprotective effects by transferring microRNAs that regulate Nrf2 and NF-κB pathways [95,96,97].
Additionally, lifestyle and metabolic interventions—including physical exercise, caloric restriction, and ketogenic diets—modulate mitochondrial function and vascular health through activation of sirtuins and AMP-activated protein kinase (AMPK) [98,99]. These non-pharmacological strategies have demonstrated synergistic effects when combined with pharmacological treatments [100].
6.5. Integrative Perspective
Collectively, these therapeutic approaches underscore the importance of addressing neurodegeneration as a multisystem disorder encompassing oxidative, inflammatory, and vascular dimensions [99,100]. Future research should prioritize the development of multimodal therapies that integrate antioxidant and anti-inflammatory actions with microvascular protection, ideally delivered through advanced targeted systems capable of crossing the BBB [95,96]. Precision-medicine approaches—guided by patient-specific redox and vascular biomarkers—may enable early intervention and improved clinical outcomes in neurodegenerative disorders [99,100].
Figure 3.
Multimodal Therapeutic Strategies Targeting the Oxidative–Inflammatory–Vascular Axis in Neurodegenerative Diseases.
Figure 3.
Multimodal Therapeutic Strategies Targeting the Oxidative–Inflammatory–Vascular Axis in Neurodegenerative Diseases.
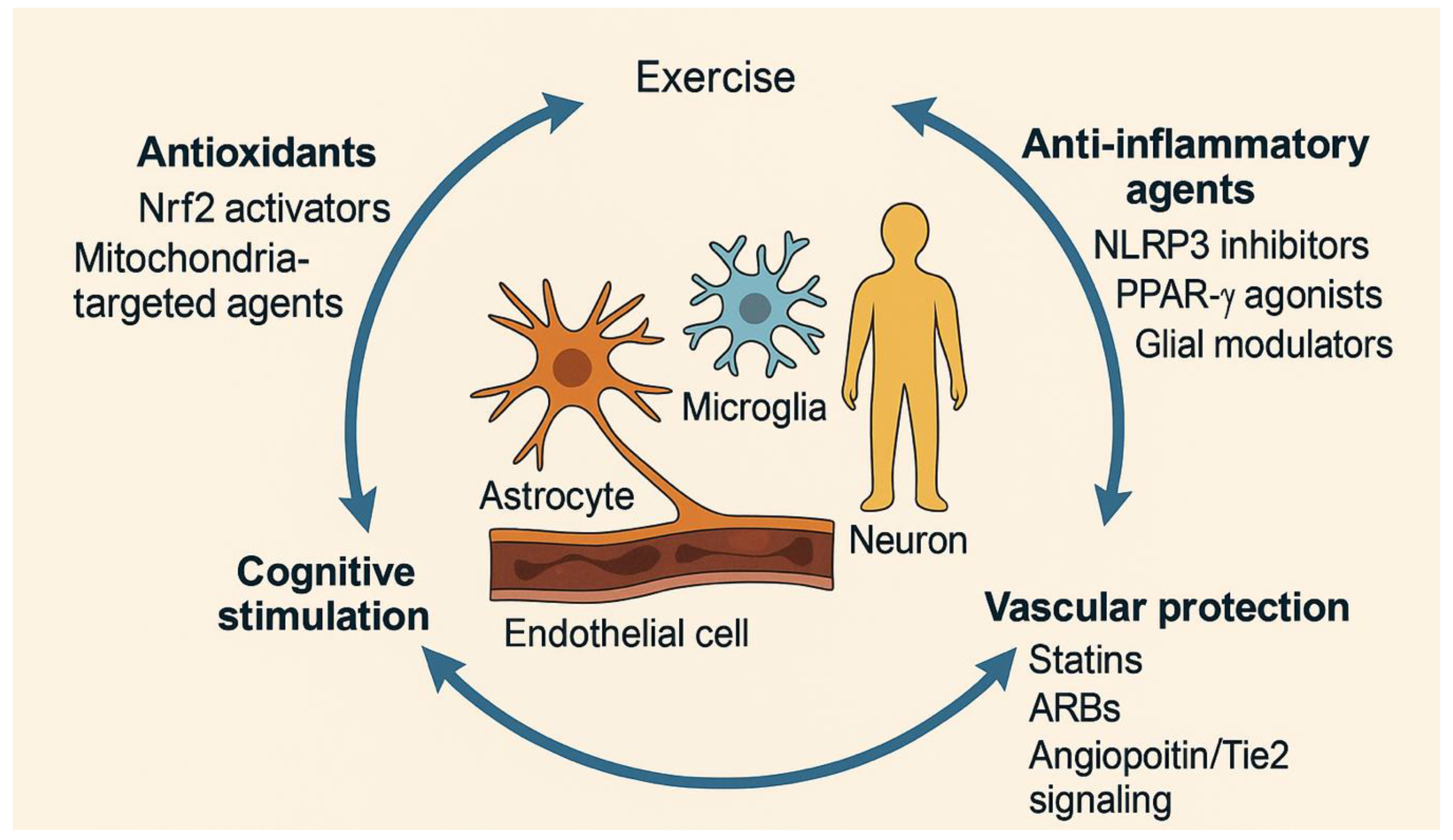
The diagram summarizes integrative therapeutic approaches aimed at restoring redox and neurovascular homeostasis. Antioxidant therapies (e.g., Nrf2 activators, mitochondria-targeted agents) reduce oxidative stress; anti-inflammatory agents (PPAR-γ agonists, NLRP3 inhibitors, glial modulators) attenuate chronic neuroinflammation; and vascular-protective compounds (statins, ARBs, Angiopoietin/Tie2 signaling modulators) maintain endothelial and BBB integrity. Lifestyle interventions such as exercise, diet optimization, and cognitive stimulation further enhance neuroprotection and functional recovery.
7. Future Perspectives and Conclusions
The convergence of oxidative stress, neuroinflammation, and microvascular dysfunction represents a unified mechanistic framework underlying the onset and progression of neurodegenerative diseases [94,95,96]. Rather than independent pathological entities, these processes form an interdependent network that accelerates neuronal damage through reciprocal amplification [97,98]. Mitochondrial dysfunction, glial activation, and vascular breakdown together compromise the structural and functional integrity of the neurovascular unit (NVU), transforming a once-protective environment into a pro-degenerative niche [99,100].
Future research directions should focus on dissecting this triadic interplay at cellular and molecular levels using high-resolution imaging, multi-omics approaches, and integrative computational models [95,96,97]. Systems biology and single-cell transcriptomics are particularly promising for identifying novel biomarkers that capture the dynamic crosstalk between redox, inflammatory, and vascular pathways [98,99]. The discovery of early circulating biomarkers—such as oxidative metabolites, cytokine profiles, or endothelial-derived extracellular vesicles—could revolutionize diagnosis, enabling pre-symptomatic detection and therapeutic intervention at reversible stages [100].
Translationally, there is growing consensus that monotherapy approaches targeting a single mechanism have limited efficacy in complex disorders like Alzheimer’s or Parkinson’s disease [93,94,95]. Consequently, multimodal interventions that simultaneously modulate oxidative, inflammatory, and vascular targets are likely to yield more durable benefits [96,97,98]. Advances in nanomedicine, gene therapy, and stem cell-derived exosomes open new avenues for delivering neuroprotective agents across the BBB with higher specificity and safety [99,100]. Furthermore, the integration of lifestyle interventions—including exercise, diet modulation, and cognitive stimulation—may potentiate pharmacological treatments by enhancing mitochondrial resilience and vascular perfusion [97,98,99].
Clinically, the implementation of personalized therapeutic algorithms based on patient-specific molecular signatures will be pivotal [95,96]. Stratifying patients according to oxidative, inflammatory, and vascular biomarkers could improve treatment responsiveness and reduce heterogeneity in clinical outcomes [97,98].
In conclusion, the oxidative–inflammatory–vascular axis provides a comprehensive lens through which neurodegeneration can be reinterpreted not as an isolated neuronal disorder, but as a systemic failure of the neurovascular unit [99,100]. Targeting this triad holds transformative potential for developing disease-modifying therapies. Future efforts must bridge mechanistic insights with clinical translation, ensuring that therapeutic advances move from bench to bedside with precision and efficacy [99,100].
Author Contributions
Conceptualization, J.M.N.C.; methodology, J.M.N.C. and M.V.V.D.; investigation, J.M.N.C., G.G.R.M., and K.A.R.V.; resources, A.E.S. and N.G.Q.; writing—original draft preparation, J.M.N.C.; writing—review and editing, J.M.N.C. and M.V.V.D.; supervision, M.V.V.D. and G.G.R.M.; project administration, J.M.N.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors express their sincere gratitude to the Biomedicines Editorial Office (MDPI) for the kind invitation to contribute this manuscript. The authors also wish to thank the Universidad Autónoma de Guadalajara (Facultad de Medicina) for its academic support and collaboration in the development of this work, as well as the valuable contributions of colleagues from partner institutions who participated in the literature review and scientific discussion.
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
- Buga, A.-M.; Oancea, C.-N. Oxidative Stress-Induced Neurodegeneration and Antioxidative Strategies: Current Stage and Future Perspectives. Antioxidants 2023, 12, 1762. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, M.-C.; Jurcau, A.; Diaconu, R.-G. Oxidative Stress in the Pathogenesis of Neurodegenerative Diseases. Stresses 2024, 4, 827–849. [Google Scholar] [CrossRef]
- Pfau, S.J.; Langen, U.H.; Fisher, T.M.; et al. Characteristics of blood–brain barrier heterogeneity between brain regions revealed by profiling vascular and perivascular cells. Nat Neurosci 2024, 27, 1892–1903. [Google Scholar] [CrossRef] [PubMed]
- Konig, S.; Jayarajan, V.; Wray, S.; Kamm, R.; Moeendarbary, E.; et al. Mechanobiology of the blood-brain barrier during development, disease and ageing. Nat Commun 2025, 16, 7233. [Google Scholar] [CrossRef]
- Zhang, X.; Fu, M.; Wang, Y.; Wu, T. Strategies for delivering drugs across the blood-brain barrier for the treatment of neurodegenerative diseases. Front Drug Deliv 2025, 5, 1644633. [Google Scholar] [CrossRef]
- Wang, J. (Review). How endothelial cell metabolism shapes blood–brain barrier function in neurodegenerative disease. Front Mol Neurosci 2025, 18, 1623321. [Google Scholar] [CrossRef] [PubMed]
- Zierfuss, B.; Larochelle, C.; Prat, A. Blood–brain barrier dysfunction in multiple sclerosis: causes, consequences, and potential effects of therapies. Lancet Neurol 2024, 23, 95–109. [Google Scholar] [CrossRef]
- Navarro, E. Multitarget Effects of Nrf2 Signalling in the Brain. Antioxidants 2024, 13, 1502. [Google Scholar] [CrossRef]
- Dustin, CM. NOX2 in Alzheimer’s and Parkinson’s disease. Redox Biol 2024, 76, 103931. [Google Scholar] [CrossRef]
- Zhang, Y. Pericytes in Alzheimer’s disease: Key players and therapeutic targets. Exp Eye Res 2024, 241, 109877. [Google Scholar] [CrossRef]
- Wang, Y. Unveiling the glymphatic system’s impact on neurodegenerative diseases: a bibliometric and knowledge-map analysis. Front Aging Neurosci 2025, 17, 1598608. [Google Scholar] [CrossRef]
- Íñigo-Catalina, L. Melatonin-Mediated Nrf2 Activation as a Potential Therapeutic Strategy in Neurodegeneration. Antioxidants 2025, 14, 1190. [Google Scholar] [CrossRef]
- Tu, D. Activation of neuronal NADPH oxidase NOX2 promotes oxidative stress and neurodegeneration. Signal Transduct Target Ther 2023, 8. [Google Scholar] [CrossRef]
- El-Ghazawi, K. Brain Microvascular Pericyte Pathology Linking Alzheimer’s Disease and Type-2 Diabetes. Microcirculation 2024, 31, e12877. [Google Scholar] [CrossRef]
- Sebghatollahi, Z. Signaling Pathways in Oxidative Stress-Induced Neurodegeneration: Focus on NOX and Mitochondrial Dysregulation. Antioxidants 2025, 14, 457. [Google Scholar] [CrossRef] [PubMed]
- Fletcher-Lloyd, N. 3BTRON: A Blood-Brain Barrier Recognition Network for ultrastructural ageing analysis. Commun Biol 2025, 8, —. [Google Scholar] [CrossRef]
- Korte, N. Inhibiting Ca²⁺ channels in an Alzheimer’s model reverses pericyte-mediated capillary constriction and improves flow. Nat Neurosci 2024, 27, —. [Google Scholar] [CrossRef]
- Rafati, N. Nanosystems for targeted drug delivery: innovations and challenges in overcoming the blood-brain barrier. Int J Pharm 2024, 666, 124800. [Google Scholar] [CrossRef] [PubMed]
- Toader, C. Decoding Neurodegeneration: A Review of Molecular Circuits and Therapeutic Nodes. Int J Mol Sci 2024, 25, 12613. [Google Scholar] [CrossRef]
- Fiadeiro, MB. NADPH Oxidases in Neurodegenerative Disorders: Pathogenic Roles and Therapeutic Targeting. Antioxid Redox Signal. [CrossRef]
- Jurcau, A.; Jurcau, M.-C. Neuroinflammation and Oxidative Stress in Alzheimer’s Disease: Targeting Glial Pathophysiology. Cells 2024, 13, 721. [Google Scholar] [CrossRef]
- Singh, A.; Tripathi, R.; Kumar, S. Crosstalk Between Oxidative Stress and Microglial Activation in Neurodegenerative Diseases. Free Radic. Biol. Med. 2025, 215, 120–134. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Oxidative Stress in Alzheimer’s Disease: Molecular Insights and Therapeutic Perspectives. Antioxidants 2024, 13, 267. [Google Scholar] [CrossRef]
- Alasmari, F.; et al. Cerebral Microvascular Pathology in Alzheimer’s Disease: Mechanisms and Clinical Implications. Biomedicines 2024, 12, 512. [Google Scholar] [CrossRef]
- Yamada, M.; et al. Blood–Brain Barrier Dysfunction and Neuroinflammation in Aging and Dementia. Front. Aging Neurosci. 2024, 16, 1398231. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V. Glial Redox Homeostasis in Aging and Neurodegeneration. Cell Calcium 2023, 113, 102729. [Google Scholar] [CrossRef]
- Guo, L.; et al. Mitochondrial Dysfunction and Reactive Oxygen Species in Neurodegenerative Diseases: Therapeutic Targeting. Int. J. Mol. Sci. 2023, 24, 11876. [Google Scholar] [CrossRef]
- Cui, Y.; et al. Role of the NLRP3 Inflammasome in Neurovascular Dysfunction. Cells 2025, 14, 33. [Google Scholar] [CrossRef]
- Fernandez, R.; et al. BBB Integrity and Pericyte Loss in Neurodegenerative Conditions. Neurobiol. Dis. 2025, 195, 107058. [Google Scholar] [CrossRef]
- Cencioni, C.; et al. Vascular Senescence and Its Role in Neurodegenerative Disease. Aging Cell 2024, 23, e14024. [Google Scholar] [CrossRef]
- Kirk, R.; et al. Targeting Mitochondrial ROS in Neurodegeneration: Therapeutic Potential of Mitochondria-Targeted Antioxidants. Antioxid. Redox Signal. 2025, 42, 611–628. [Google Scholar] [CrossRef]
- Mishra, R.; et al. Endothelial Senescence and BBB Breakdown in Alzheimer’s Disease: Mechanisms and Interventions. Mol. Neurobiol. 2024, 61, 785–799. [Google Scholar] [CrossRef]
- Chakraborty, A.; et al. Dual Role of Astrocytes in Neurodegeneration: Oxidative Stress and Neuroinflammation. Neurosci. Biobehav. Rev. 2025, 163, 105418. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Paul, S. MMPs and BBB Permeability in Neurodegenerative Diseases: Pathophysiology and Therapeutic Outlook. Front. Cell. Neurosci. 2023, 17, 1170456. [Google Scholar] [CrossRef]
- Liu, Q.; et al. Targeting the Nrf2 Pathway in Neurodegeneration: From Mechanisms to Clinical Translation. Antioxidants 2024, 13, 1497. [Google Scholar] [CrossRef]
- Schiavone, S.; Trabace, L. Redox Imbalance in the Central Nervous System: Implications for Neurodegenerative and Psychiatric Disorders. Oxid. Med. Cell. Longev. 2023, 8810634. [Google Scholar] [CrossRef]
- Rao, P.; et al. Nanocarriers for Brain Delivery of Antioxidants: Advances and Challenges. Pharmaceutics 2025, 17, 210. [Google Scholar] [CrossRef]
- Liu, Z.; et al. Glutathione Depletion and Mitochondrial Redox Dysregulation in Alzheimer’s Disease. Free Radic. Res. [CrossRef]
- Ahmed, S.; et al. BBB Repair Strategies and Vasoprotective Approaches in Neurodegenerative Disorders. Biomedicines 2024, 12, 1432. [Google Scholar] [CrossRef]
- Sun, J.; et al. Crosstalk Between the Gut Microbiome, Neuroinflammation, and Oxidative Stress in Aging. Front. Aging 2025, 6, 1672011. [Google Scholar] [CrossRef]
- Zhao, Y.; et al. Crosstalk Between Oxidative Stress and Autophagy in Alzheimer’s Disease. Front. Aging Neurosci. 2024, 16, 1422019. [Google Scholar] [CrossRef]
- Bhat, A.H.; et al. Oxidative Stress and Neurodegeneration: Mechanisms and Therapeutic Strategies. Life Sci. 2023, 334, 122678. [Google Scholar] [CrossRef]
- Li, W.; et al. Role of Mitochondrial Dynamics and Mitophagy in Oxidative-Stress-Induced Neuronal Death. Int. J. Mol. Sci. 2024, 25, 3920. [Google Scholar] [CrossRef]
- Liu, H.; et al. Microglia–Endothelial Interactions Mediate Neuroinflammation-Induced BBB Disruption. J. Neuroinflammation 2025, 22, 34. [Google Scholar] [CrossRef]
- Wang, T.; et al. Cellular Senescence at the Neurovascular Unit in Aging and Neurodegenerative Disease. Brain Pathol. 2024, 34, e13176. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes and Their Roles in Neurodegeneration. Nat. Rev. Neurosci. 2023, 24, 727–743. [Google Scholar] [CrossRef]
- Li, S.; et al. Targeting the NLRP3 Inflammasome in Neurodegenerative Diseases: Mechanisms and Therapeutic Potential. Pharmacol. Ther. 2024, 257, 108006. [Google Scholar] [CrossRef]
- Ghosh, R.; et al. Role of the NF-κB Pathway in Neuroinflammation and Oxidative Stress: Molecular Targets for Intervention. Antioxidants 2024, 13, 1194. [Google Scholar] [CrossRef]
- Zhu, Q.; et al. Brain Microvascular Endothelial Dysfunction and Its Role in Cognitive Decline. Neurosci. Lett. 2025, 828, 137243. [Google Scholar] [CrossRef]
- Rizvi, S.; et al. Cerebral Hypoperfusion and Oxidative Stress: Interconnected Mechanisms in Neurodegenerative Disorders. Neuroscientist 2024, 30, 186–198. [Google Scholar] [CrossRef]
- Martínez-Hernández, A.; et al. Pericyte Dysfunction as a Driver of Neurovascular Pathology in Aging. Acta Neuropathol. 2025, 149, 255–272. [Google Scholar] [CrossRef]
- Costa, R.; et al. Oxidative Stress Biomarkers in Alzheimer’s Disease: From Mechanistic Insight to Diagnostic Utility. Diagnostics 2024, 14, 571. [Google Scholar] [CrossRef]
- Roy, S.; et al. Microglial Polarization and the M1/M2 Paradigm in Neurodegenerative Diseases. Cells 2024, 13, 1574. [Google Scholar] [CrossRef]
- Hernández-Guillén, J.; et al. Nitrosative Stress and Mitochondrial Dysfunction in Neurodegeneration. Mol. Neurobiol. 2023, 60, 5648–5663. [Google Scholar] [CrossRef]
- Wu, K.; et al. Dysregulation of Tight Junction Proteins in Neurodegenerative Diseases: Mechanisms and Therapies. Front. Neurosci. 2024, 18, 1460009. [Google Scholar] [CrossRef]
- Zhou, M.; et al. Emerging Role of the Glymphatic System in Neurodegenerative Diseases: Redox and Vascular Perspectives. Front. Neurol. 2024, 15, 1449281. [Google Scholar] [CrossRef]
- Rahman, S.; et al. Blood–Brain Barrier Integrity in Neuroinflammation and Neurodegeneration: Molecular Insights. Brain Res. Bull. 2024, 210, 186–199. [Google Scholar] [CrossRef]
- Chiu, C.; et al. Therapeutic Targeting of the Nrf2–Keap1 Pathway in Neurodegenerative Disorders. Biomedicines 2024, 12, 1733. [Google Scholar] [CrossRef]
- Medeiros, R.; et al. Crosstalk Between Microglia and Endothelium in Alzheimer’s Disease. Trends Mol. Med. 2025, 31, 211–226. [Google Scholar] [CrossRef]
- Qin, Y.; et al. Nanotechnology-Based Drug Delivery for Antioxidant Therapy in Neurodegenerative Diseases. Front. Pharmacol. 2025, 16, 1659122. [Google Scholar] [CrossRef]
- Shi, J.; et al. Crosstalk Between the Nrf2 and NF-κB Pathways in Neurodegeneration. Antioxidants 2025, 14, 341. [Google Scholar] [CrossRef]
- Pan, Y.; et al. Pharmacological Activation of Nrf2 for Neuroprotection in Neurodegenerative Diseases. Free Radic. Biol. Med. 2024, 213, 110–128. [Google Scholar] [CrossRef]
- Xu, L.; et al. Pericyte–Endothelial Interactions Regulate BBB Integrity During Oxidative Stress. Front. Cell. Neurosci. 2025, 19, 1683042. [Google Scholar] [CrossRef]
- Aliev, G.; et al. Role of Endothelial Mitochondria in Neurodegenerative Disorders. Antioxid. Redox Signal. 2024, 40, 410–427. [Google Scholar] [CrossRef]
- Chen, H.; et al. Interplay of Oxidative Stress and Mitochondrial Dysfunction in Parkinson’s Disease: Therapeutic Implications. Cells 2024, 13, 1149. [Google Scholar] [CrossRef]
- Lee, S.; et al. Redox Signaling and Mitochondrial Dysfunction in Astrocyte Reactivity. Front. Mol. Neurosci. 2024, 17, 1458873. [Google Scholar] [CrossRef]
- Pires, C.; et al. Antioxidant and Anti-Inflammatory Effects of Polyphenols in Neurodegenerative Diseases. Nutrients 2024, 16, 276. [Google Scholar] [CrossRef]
- Vidal, R.; et al. Therapeutic Modulation of Microglial Polarization: Emerging Strategies for Neuroprotection. Biomedicines 2024, 12, 1528. [Google Scholar] [CrossRef]
- Kim, H.J.; et al. Blood–Brain Barrier Repair as a Therapeutic Target in Alzheimer’s Disease. Int. J. Mol. Sci. 2025, 26, 1189. [Google Scholar] [CrossRef]
- Deng, X.; et al. Crosstalk Between Hypoxia, ROS, and Neuroinflammation in Aging Brains. Exp. Gerontol. 2024, 188, 112485. [Google Scholar] [CrossRef]
- Hao, J.; et al. The Role of Endothelial Senescence in Neurovascular Aging. Aging Dis. 2025, 16, 65–80. [Google Scholar] [CrossRef]
- Galea, E.; et al. The Neurovascular Unit in Aging and Neurodegenerative Disorders. Nat. Rev. Neurosci. 2023, 24, 563–580. [Google Scholar] [CrossRef]
- Tian, J.; et al. Endothelial Dysfunction and Neurovascular Coupling in Alzheimer’s Disease. Brain Res. Rev. 2025, 188, 103265. [Google Scholar] [CrossRef]
- Rahman, K.; et al. Therapeutic Strategies Targeting Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Disorders. Antioxidants 2024, 13, 1988. [Google Scholar] [CrossRef]
- Orellana, J.A.; et al. Astrocyte–Endothelial Gap Junctions and BBB Homeostasis in Aging and Disease. Front. Neurosci. 2024, 18, 1457234. [Google Scholar] [CrossRef]
- Zhou, Y.; et al. Nanomedicine-Based Therapeutic Strategies for Alzheimer’s Disease. Acta Pharm. Sin. B 2024, 14, 2891–2907. [Google Scholar] [CrossRef]
- Wang, Q.; et al. Crosstalk Between Peripheral Inflammation and the Brain Microvasculature in Neurodegenerative Diseases. Front. Immunol. 2024, 15, 1416332. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; et al. Neurovascular Protection in Ischemic and Degenerative Brain Diseases: Emerging Therapeutic Targets. Stroke 2025, 56, 245–256. [Google Scholar] [CrossRef]
- Ghosh, A.; et al. Advances in Targeting Oxidative Stress for Neurodegenerative Therapy: From Bench to Bedside. Int. J. Biochem. Cell Biol. 2024, 165, 107358. [Google Scholar] [CrossRef]
- Leng, F.; et al. Systemic Inflammation and BBB Breakdown in Alzheimer’s Disease: Mechanistic Insights and Therapeutic Targets. Cells 2025, 14, 873. [Google Scholar] [CrossRef]
- Wang, Z.; et al. Crosstalk Between Oxidative Stress and Mitochondrial Biogenesis in Alzheimer’s Disease. Mol. Neurobiol. 2024, 61, 5248–5263. [Google Scholar] [CrossRef]
- Cheng, J.; et al. Reactive Oxygen Species and Mitochondrial Dysfunction as Common Pathways in Neurodegenerative Diseases. Antioxidants 2024, 13, 1132. [Google Scholar] [CrossRef]
- Yoon, S.Y.; et al. Role of Calcium Dysregulation and Oxidative Stress in Neuronal Injury. Front. Cell. Neurosci. 2025, 19, 1692097. [Google Scholar] [CrossRef]
- Caruso, G.; et al. Antioxidants as Multifunctional Agents in Neurodegenerative Disorders: From Preclinical Evidence to Clinical Practice. Antioxidants 2025, 14, 1349. [Google Scholar] [CrossRef]
- Tansey, M.G.; et al. Microglial Dysfunction in Neurodegenerative Disease: Pathophysiology and Therapeutic Targeting. Nat. Rev. Neurosci. 2024, 25, 101–120. [Google Scholar] [CrossRef]
- Zhou, R.; et al. Hypoxia-Induced BBB Leakage and Neuroinflammation: Mechanistic Insights. Neurosci. Bull. 2024, 40, 541–554. [Google Scholar] [CrossRef]
- Kwon, S.; et al. Crosstalk Between Metabolic Dysfunction and Oxidative Stress in Neurovascular Aging. Aging Cell 2025, 24, e14212. [Google Scholar] [CrossRef]
- Papandreou, M.; et al. Targeting Redox Homeostasis for Neuroprotection: A Translational Perspective. Free Radic. Biol. Med. 2024, 215, 98–113. [Google Scholar] [CrossRef]
- Ko, Y.; et al. Pathological Role of Reactive Astrocytes in BBB Breakdown and Neurodegeneration. Cells 2024, 13, 1684. [Google Scholar] [CrossRef]
- Tian, J.; et al. Vascular Endothelial Growth Factor Signaling in Neurodegeneration and Neurorepair. Front. Neurosci. 2025, 19, 1701150. [Google Scholar] [CrossRef]
- Batiuk, M.Y.; et al. Single-Cell Transcriptomics Reveals Astrocyte Diversity and BBB Regulatory Networks. Nat. Neurosci. 2023, 26, 1535–1549. [Google Scholar] [CrossRef]
- Rahimi, M.; et al. Chronic Inflammation and Redox Imbalance as Triggers of Neuronal Aging. Front. Aging 2025, 6, 1683520. [Google Scholar] [CrossRef]
- Martínez, L.; et al. Role of Endothelial Glycocalyx Damage in Cognitive Impairment and Dementia. Cells 2024, 13, 1961. [Google Scholar] [CrossRef]
- Venkataraman, A.; et al. Lipid Peroxidation and Ferroptosis in Neurodegenerative Diseases. Trends Neurosci. 2025, 48, 55–68. [Google Scholar] [CrossRef]
- Morales, I.; et al. Peripheral Inflammation and Microvascular Dysfunction in Parkinson’s Disease. Brain Res. Bull. 2024, 208, 193–208. [Google Scholar] [CrossRef]
- Jeong, D.; et al. Epigenetic Regulation of Nrf2 in the Aging Brain: Implications for Neuroprotection. Biomedicines 2025, 13, 348. [Google Scholar] [CrossRef]
- Navarro, M.; et al. Endothelial Repair and Angiogenic Signaling in the Aging Brain: The Double-Edged Sword. Cells 2025, 14, 493. [Google Scholar] [CrossRef]
- Han, D.; et al. Therapeutic Targeting of Pericyte Dysfunction to Restore BBB Integrity. J. Cereb. Blood Flow Metab. 2025, 45, 789–802. [Google Scholar] [CrossRef]
- Bourdenx, M.; et al. Endolysosomal and Mitochondrial Crosstalk in Neurodegenerative Diseases. Nat. Rev. Neurosci. 2024, 25, 251–266. [Google Scholar] [CrossRef]
- García-López, P.; et al. Therapeutic Advances in BBB-Targeting Nanomedicine for Neurodegenerative Disorders. Pharmaceutics 2025, 17, 745. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



