
Submitted:
03 November 2025
Posted:
04 November 2025
You are already at the latest version
Abstract
5q-associated spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder caused by mutations in the survival of motor neuron-1 (SMN1) gene, leading to progressive muscle weakness, atrophy, and respiratory failure. Although traditionally considered a motor neuron (MN)–specific disease, recent research highlights the in-volvement of astrocytes, particularly in regulating extracellular glutamate levels and promoting motor neuron toxicity. Dysfunctional MN–astrocyte interactions and astrocytic glutamate dysregulation contribute to MN degeneration. We investigated the role of astrocytic gap junctions, focusing on connexin 43 (Cx43), a key protein in astrocytic communication. We employed a combination of in vivo and in vitro approaches, including a mouse model representative of late-onset SMA, human astrocytes derived from induced pluripotent stem cells, and murine astrocyte cultures. We assessed changes in expression and localization using genetic modification, immunostaining, Western blotting, and quantitative PCR. Functional outcomes were further investigated using ex vivo spinal cord slice cultures, Ca2+-imaging, and glutamate release assays. Our experiments revealed a significant upregulation of Cx43 in the spinal cord of late-onset SMA mice and in SMN-deficient murine and human induced astrocyte cultures. Elevated Cx43 expression was associated with increased astrocytic glutamate release and motor neuron (MN) tox-icity. Ca2+-imaging demonstrated that this enhanced glutamate release was mediated via Cx43-dependent mechanisms. Notably, pharmacological inhibition of Cx43 using Gap27, a Cx32 hemichannel blocker, in slice cultures attenuated glutamate release. It reduced MN Ca2+ response, supporting a causal role of Cx43 in mediating astrocyte-driven excitotoxicity in late-onset SMA. These findings suggest that astrocytic Cx43 contributes to gluta-mate-mediated MN toxicity in late-onset SMA. By identifying Cx43 as a contributor to astrocyte dysfunction, our work supports the growing recognition of non-neuronal con-tributions to late-onset SMA pathology.
Keywords:
calcium ion
; excitotoxicity
; Gap27
; gap junction
; motor neuron
; mouse model
; induced pluripotent stem cells
1. Introduction
Spinal Muscular Atrophy (SMA) is an inherited motor neuron disorder (MND) characterized by progressive muscle atrophy caused by the loss of lower motor neurons (MNs) in the ventral horn of the spinal cord, resulting in weakness and wasting of proximal limb and respiratory muscles. The disease is triggered by homozygous loss-of-function variation in the survival of motor neuron 1 (SMN1) gene [1,2], which encodes the SMN protein, a key factor in post-transcriptional RNA processing, including mRNA splicing. Disease severity is strongly modified by the copy number of the paralogous SMN2 gene. Patients with few SMN2 copies typically develop severe, early-onset SMA within the first months of life, while higher copy numbers lead to milder forms with later onset of symptoms [2,3,4,5].
The identification of the genetic basis of SMA has led to the development of therapies that restore SMN expression, with nusinersen becoming the first approved treatment in 2016 [6,7]. While SMN-enhancing drugs have significantly improved survival and motor outcomes, their therapeutic effect is limited when treatment is initiated at advanced disease stages [8,9,10,11,12]. Persistent motor deficits and the high economic burden of treatment highlight the need for complementary, SMN-independent therapeutic strategies.
Astrocytes, the predominant glial cell type in the central nervous system, have emerged as significant contributors to the pathophysiology of MND, including Amyotrophic Lateral Sclerosis (ALS) and SMA. Several studies have shown astrocytic activation preceding MN degeneration [13,14,15,16,17]. In late-onset SMA, astrocytic dysfunction has been implicated in MN death, with evidence pointing to dysregulated glutamate homeostasis as a key driver of neurotoxicity in both a late-onset SMA mouse model and patient-derived samples [16].
Astrocytes communicate through gap junctions composed of connexons, each formed by six connexin proteins [18,19]. Connexin 43 (Cx43), encoded by GJA1, is the predominant connexin expressed in astrocytes and plays a central role in intercellular coupling. Cx43 has been implicated in maintaining central nervous system homeostasis through multiple mechanisms, including buffering extracellular ions, synchronizing astrocytic networks, providing metabolic support to neurons, regulating the blood–brain barrier, and modulating synaptic plasticity [20,21,22]. Importantly, Cx43 has been linked to the regulation of extracellular glutamate, suggesting that astrocytic gap junctions are involved in controlling excitatory neurotransmission [23,24]. Dysregulation of astrocytic Cx43 has been associated with neurodegeneration, and its contribution to MN pathology has already been established in ALS [25].
Building on our recent findings that astrocytic activation and glutamate dysregulation drive MN death in late-onset SMA [16,17], Cx43 emerges as a promising candidate mechanism for contributing to astrocyte-mediated neurotoxicity. In the present study, we investigate the role of astrocytic gap junctions, with a focus on Cx43, in the context of late-onset SMA. To this end, we employ complementary in vitro and ex vivo approaches, including SMN-deficient human induced astrocytes, murine astrocyte cultures, spinal cord slice preparations, and the Taiwanese mild SMA mouse model, which recapitulates the late-onset phenotype [26]. We hypothesize that astrocytic Cx43 contributes to MN degeneration in late-onset SMA and propose that targeting this protein may provide a potential SMN-independent therapeutic strategy.
2. Materials and Methods
2.1. Animals
The Taiwanese mild SMA mouse model reflecting late-onset SMA (FVB.Cg-Smn1<sup>tm1Hung</sup>Tg(SMN2)2Hung/J; Jackson Laboratory #005058) was obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and bred in the Animal Research Facility of the University Hospital Essen (North Rhine-Westphalia, Germany). These mice are double homozygotes carrying a knockout of murine SMN1 together with four copies of the human SMN2 gene. Compared with wild-type FVB/N mice, they are smaller in size and exhibit reduced body weight, decreased grip strength, and hindlimb weakness. In addition, ear and tail necrosis typically occur, resulting in shortened ears and thickened, shortened tails. Previous studies demonstrated MN loss in this model at approximately postnatal day (P) 35, defining this stage as a critical time point to investigate the role of Cx43 [16,17]. For temporal analysis, P20 was defined as the early stage, P35 as the onset of MN loss, and >P100 as the late stage of disease progression.
Wild-type (WT) FVB/N mice were used as controls and for culture experiments. For MN preparation, embryos were harvested at embryonic day (E) 14 from timed pregnant WT females. All animals were maintained under a 12 h light/12 h dark cycle with food and water provided ad libitum.
All animal experiments were performed in accordance with the institutional animal welfare guidelines of the University of Duisburg-Essen, Germany. The use of the late-onset SMA mouse model was approved by the State Office for Consumer Protection and Food Safety (LAVE), North Rhine-Westphalia, Germany (approval number 81–02.04–2020.A335).
2.2. Isolation of the Spinal Cord
To extract the spinal cord, WT mice were euthanized by decapitation, after deep anesthetization with isoflurane. The spine was then extracted, and the spinal cord was obtained by hydraulic extrusion. The meninges were removed, and the cord was ready for use.
2.3. Spinal Cord Slice Preparation
The lumbar part of the spinal cord was placed in embedding medium and snap frozen using liquid nitrogen. The tissue was then cut into 20 µm slices using a cryotome. Every fifth section of each spinal cord was mounted on an independent microscopy slide and stored at -20 °C.
2.4. Culture of Spinal Astrocytes from WT Mice
The spinal cord tissue was finely diced with a razor blade and placed in a 0.25% trypsin/EDTA solution (#25200056, Thermo Fisher Scientific, Dreieich, Germany) for 30 min at 37 °C. Enzymatic digestion was halted by introducing DMEM/F12 (#210410202, Thermo Fisher Scientific, Dreieich, Germany) supplemented with 10% fetal bovine serum (FBS, #16140071, Thermo Fisher Scientific, Dreieich, Germany) to the solution. A homogenized cell solution was then produced by mechanical titration with a pipette. The solution was centrifuged down at 500 G for 5 min. The supernatant was removed, 10 mL of our culture medium (a DMEM/F12 solution containing 10% FBS and 1% penicillin/streptomycin (P/S, #15140122, Thermo Fisher Scientific, Dreieich, Germany), was added and then transferred to a 75 cm2 cell culture flask (T75) and incubated at 37 °C and 5% CO2. The next day and for every 2 days after that, the medium was exchanged with fresh culture medium until a confluency of about 65% was reached at 10-14 days in vitro (DIV). The flask was then placed on an orbital shaker (250 rpm, 37 °C, 5% CO2) overnight for the microglia to detach. Subsequently, the culture medium was exchanged, and cells were detached from the cell culture flask, enumerated, and then seeded with 3500 cells per coverslip onto poly-d-lysine (PDL) (Sigma-Aldrich, Taufkirchen, Germany)-treated glass coverslips in a 24-well plate. The culture medium was again replaced in the same manner.
2.5. Human Tissue Samples
Human skin biopsy was performed by a physician. All participants provided informed consent, and the study was approved by the Ethics Committee of the University of Duisburg-Essen (approval number 19–9011-BO).
2.6. Generation of hiAstrocytes from Skin Fibroblasts
hiAstrocytes were generated from dermal fibroblasts of healthy donors as previously described [27]. In brief, fibroblasts were directly reprogrammed into induced pluripotent stem cells (iPSCs), via retroviral transduction with Oct4, Sox2, Klf4, and c-Myc (#RF101, ALSTEM, USA) in the presence of a neural induction medium. Following transfection, cells were maintained in conversion medium. Conversion medium was a solution of DMEM/F12 supplemented with 1% GlutaMAX, 1% N-2 supplement, 1% B27 supplement, 1% penicillin/streptomycin, 20 ng/mL human fibroblast growth factor (FGF)-basic (#100-18B, PreproTech, Germany), 20 ng/mL human epidermal growth factor (EGF) (#AF-100–15, PreproTech, Germany), and 5 μg/mL heparin). Cells were passaged at a 1:2 ratio after five days to allow further multiplication and then plated after an additional 2–3 days onto PDL-coated glass coverslips.
iPSCs were subsequently cultured in iPSC medium (DMEM/F12 with 1% GlutaMAX, 1% N-2 supplement, 1% B27 supplement, 1% penicillin/streptomycin, and 40 ng/mL human FGF-basic) until confluent. The cells were then enzymatically dissociated using Accutase and seeded into 24-well plates containing PDL-coated coverslips. Differentiation into hiAstrocytes was induced using an astrocyte conversion medium (DMEM high glucose supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 0.2% N-2 supplement) and maintained until approximately 80% confluency was achieved.
2.7. Induction of SMN Deficiency in Cultured Spinal Astrocytes
Following the replating process, small interfering (si)RNA experiments were commenced after 7 DIV. A genetic knockdown was achieved by transfecting WT murine astrocyte cultures with mouse-specific SMN1-siRNA (#SR408287, OriGene, USA) to induce SMN deficiency. For hiAstrocytes, human SMN1 (#SR304480 OriGene, USA) was used. Transfection with scrambled(scr)-siRNA-FITC (#sc-36869, Santa Cruz, Dallas, TX, USA) served as our control. For this purpose, 2 h before applying siRNA to the spinal astrocytes, the medium was replaced with FBS-free medium (only DMEM/F12 containing 1% P/S).
Subsequently, 10 nM of the SMN1-siRNA was combined with 200 μM of SilenceMag (#SM11000, OZ Biosciences, Marseille, France) and incubated for 15 min at room temperature (RT). The solution was then added to the cells and incubated for 2 h on a magnetic plate at 37 °C and 5% CO2. The magnetic plate was then removed, and the cells allowed to incubate until the next day, when the siRNA medium was exchanged with fresh medium (DMEM/F12, 10% FBS, 1% P/S) and in part with 200 μM Gap27 (#E0040, Selleck Chemicals, Houston, TX, USA), a Cx43 inhibitor. The cells were maintained in culture for three more days before being used in experiments. The efficiency of this transfection method has been previously evaluated by immunostaining for SMN-positive cells after transfection [17].
2.8. Immunostaining
For immunostaining, cell cultures and tissue were fixed in 4% paraformaldehyde, washed with phosphate-buffered saline (PBS), and permeabilized with 0.1 v/w Triton X-100 in PBS. The tissue was then blocked in a solution of 5% bovine serum albumin (BSA) in PBS, and the cells were treated with 1% BSA in PBS.
Primary antibodies against Cx43 (rabbit, 1:500, #SAB4501175 Sigma Aldrich, Taufkirchen, Germany), βIII-tubulin (TUJ1) (mouse, 1:500, #8578 Sigma Aldrich, Taufkirchen, Germany), non-phosphorylated neurofilament proteins (SMI-32, mouse, 1:500, #801701, BioLegend, San Diego, CA, USA), glial fibrillary acidic protein (GFAP) (mouse, 1:500, #63893, Sigma Aldrich, Taufkirchen, Germany) were diluted in the appropriate blocking agent and incubated over night at 4 °C.
After washing, secondary antibodies (goat anti-rabbit, goat anti-mouse, 1:300, Dianova, Hamburg, Germany) and 4′,6-diamidino-2-phenylindole (DAPI, 1:500, Sigma Aldrich, Taufkirchen, Germany), diluted in blocking solution, were administered to the tissue and cells, incubated for 1.5 h at RT, then washed, covered, dried overnight, and sealed.
Images were captured utilizing a Zeiss Axio Observer.Z1 fluorescence microscope (Zeiss, Jena, Germany). The Zeiss Zen software was employed to visualize the target proteins. To ensure consistency in the analyses, all microscope settings, including laser intensity, exposure time, and contrast, were maintained at identical parameters for each protein. With the use of the ImageJ software (NIH, Bethesda, MD, USA), the immunoreactivity of the tagged protein in the spinal cord tissue slices was measured by marking the target area. The fluorescence intensity was then normalized against the background intensity of each area. ImageJ was used to determine the cell count of MNs, and the quantity of Cx43 particles in the astrocytes.
All analyses were conducted by setting the experiment into the perspective of an appropriate control. WT mice were used as a control for SMA mice. scr.-siRNA transfected astrocytes were used as controls for SMN-deficient astrocytes.
2.9. Western Blots
Western blot (WB) analysis was performed to confirm the protein levels assessed through immunostaining. To achieve this, spinal cord tissue from SMA and WT mice was homogenized using RIPA buffer supplemented with a protease inhibitor cocktail (Roche, Germany). The protein content in these lysates was quantified using a bicinchoninic acid protein assay (BCA).
A total of 10 µm of protein was applied to 4–15% TGX Stain-Free gels (Biorad, Germany); subsequent protein transfer to 0.2 µm nitrocellulose membranes was achieved through a semi-dry blotting technique. Membrane images were captured for comprehensive assessment of total protein. The membranes then underwent a 10-min incubation in fast-blocking solution (Biorad, Germany) with gentle agitation at RT. Following this, the membranes were incubated with primary antibodies diluted 1:8000 in blocking solution, targeting Cx43, at 4 °C overnight.
The secondary anti-rabbit antibody diluted at 1:10000 in blocking solution and coupled to horseradish peroxidase was then introduced to the membranes after thorough washing and incubated for 90 min at RT. After another washout, an enhanced chemiluminescence substrate was added, and immunoreactivity was detected using a WB imaging system (BioRad, Germany). The WB signals were analyzed with Biorad imaging software. Initially, the signal intensity of Cx43 lanes were assessed. Subsequently, the protein signal of the lanes was adjusted relative to its total protein value. Lastly, the determined protein level in SMA mice was normalized against the value observed in age-matched controls. The blots and the total protein are shown in the Additional file.
2.10. Real-Time Quantitative Polymerase Chain Reactions
The total RNA was isolated from spinal cord samples employing Qiazol (Qiagen #79306). One microgram of each RNA sample was used for first-strand complementary DNA synthesis in a 20-μL reaction using the high-capacity cDNA RT Kit (Applied Biosystems #4368814). Expression levels of the Cx43 gene GJA1 (forward primer: GGTGATGAACAGTCTGCCTTTCG, reverse primer: GTGAGCCAAGTACAGGAGTGTG; #MP205239, OriGene, United States) were quantified through real-time quantitative polymerase chain reactions (qPCR) analysis utilizing Power SYBR™ Green PCR Master Mix (#4,367,659, Applied Biosystems, United States). Data were normalized to a β-actin gene (forward primer: CATTGCTGACAGGATGCAGAAGG, reverse primer: TGCTGGAAGGTGGACAGTGAGG; #MP200232, OriGene, United States).
2.11. Isolation and Culture of Organotypic Spinal Cord Slice Cultures from WT and SMA-Mice
To achieve the best resemblance of SMA pathophysiology in the mouse spinal cord and the effect of pharmacology on this tissue with the lowest burden on the animal, slice culture experiments were established. After isolating the spinal cord of SMA mice at P25(±1), the lumbar section was extracted, placed on an appropriately cut agar plate, and introduced to a vibratome filled with artificial cerebrospinal fluid, 125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 25 mM NaHCO3, 1.25 mM NaH2PO4, 25 mM glucose. 350 µm slices were prepared and put on inserts with a semi-permeable membrane into wells filled with a culture medium consisting of Neurobasal-A (#10888022, Thermo Fisher Scientific, Dreieich, Germany) containing 1% P/S and 2% B-27 Plus Supplement (#17504044, Thermo Fisher Scientific, Dreieich, Germany). Age-matched control mice were utilized in a different experiment with the same protocol.
On 1 DIV, the culture media was replaced with fresh medium, and half of the samples were treated with Gap27. The supernatant (SN) was collected at 1, 7, and 14 DIV, snap-frozen with liquid nitrogen, and stored at -80 °C for experimentation purposes.
2.12. Isolation and Culture of Spinal Motor Neurons from Embryonic WT Mice
Embryos at E14 were harvested from the expecting mother, decapitated, fixed to a plate, and had their spinal cord removed under the microscope. The meninges were carefully removed, and the cord was kept in Neurobasal (#21103049, Thermo Fisher Scientific, Dreieich, Germany) on ice. Next, the spinal cords were introduced to our 0.25% trypsin/EDTA solution and allowed to digest at 37 °C.
After enzymatic digestion and mechanical dissociation with a pipette, the tissue was centrifuged and added to a culture medium consisting of DMEM:F12 with 1% P/S, 10% FBS, and 1:800 GlutaMAX (#35050061, Thermo Fisher Scientific, Dreieich, Germany). The cell suspension was then added to a petri dish and incubated at 37 °C and 5% CO2 for 1h. The debris was gently washed off, and the adhering cells were scraped off, added to the culture medium, and plated on PDL-coated wells (5000 cells/well). After incubation at 37 °C and 5% CO2, the wells were flooded with culture medium to a total of 500 µl.
The next day, the medium was removed. A new medium was introduced, consisting of Neurobasal with 2% B-27, 1% P/S, 1:800 GlutaMAX, 1:400 ciliary neurotrophic factor (CNTF, #C-240, Alomone Labs, Jerusalem, Israel), 1:100 glial cell line-derived neurotrophic factor (GDNF, #G-240, Alomone Labs, Jerusalem, Israel), and 1 µl cytosine β-D-arabinofuranoside (AraC, #C6645, Sigma Aldrich, Taufkirchen, Germany). The medium was then changed at 8 DIV, and the culture was usable for further experimentation after 14 DIV.
2.13. Calcium Imaging
To determine whether inhibiting Cx43 in the mouse spinal cord had any impact on spinal MNs, the collected medium of the slice cultures was introduced to the cultured WT MN and incubated overnight at 37 °C and 5% CO2.
The next day, the wells were washed and introduced to 1 µM of the calcium ion (Ca2+) fluorescence dye Fluo-4 AM (#F14201, Thermo Fisher Scientific, Waltham, MA, USA) in FBS-free medium and incubated for 15 min. Using a Zeiss Axio Examiner fluorescent microscope, images were captured under various conditions, and the difference in Ca2+ signaling intensity was measured using ImageJ.
In a separate experiment, Fluo-4 was introduced to WT MNs. The MNs were then examined under the microscope, and a video recording was started. At the one-minute mark (application), the SN of SMA or SMA treated with Gap27 slice cultures was added to the cultures and recorded for an additional four minutes. The spike amplitude of all viable cells was measured as the difference between the highest point and the point of application. Measurements were done using ImageJ Software.
2.14. Glutamate Assay
To measure the glutamate level in the supernatants of slice cultures and transfected astrocyte cultures, a glutamate assay kit (#MAK004, Sigma Aldrich, USA) was used according to the manufacturer’s protocol. The total protein in the supernatants was measured using a BCA and normalized to the measured glutamate level and to their appropriate controls.
2.15. Statistical Analysis
For statistical analyses between two conditions, Student’s t-tests and Mann-Whitney U tests were applied; for analyses between two or more conditions, the one-way ANOVA was used. Normality was verified by applying the D’Agostino & Pearson test. Significances were defined at a value of ⁕ p < 0.05; ⁕⁕ p < 0.01; ⁕⁕⁕ p < 0.001. All values are given as mean ± standard deviation. All statistical analyses and their graphical presentations were made using GraphPad Prism 9. All graphical presentations of methodological approaches were done using biorender.com.
3. Results
3.1. Spinal Cord Tissue Exhibited Increased Expression of Cx43 in Late-Onset SMA Mice
To examine Cx43 expression in situ, lumbar spinal cord sections from late-onset SMA and WT mice were immunostained and analyzed (Figure 1A). Quantification of total Cx43 immunoreactivity within the ventral horn, normalized to WT levels, revealed significantly increased expression in late-onset SMA tissue at P20 (1.25-fold, p = 0.0035), P35 (1.59-fold, p = 0.0094), and P>100 (2.3-fold, p = 0.0071) (Figure 1B-D).
These findings were confirmed by western blot analysis (Figures 1E-H and S1A–D).
3.2. Cx43 mRNA Expression Is Post-Transcriptionally Modulated
To investigate whether Cx43 expression is transcriptionally regulated in late-onset SMA pathology, qPCR analysis was performed on lumbar spinal cord tissue. Cx43 mRNA levels were significantly elevated 1.8-fold in the spinal cord tissue of late-onset SMA mice compared to WT at P20 (p = 0.029) (Figure 2A). In contrast, no significant differences were observed at P35 (p = 0.9485) or P>100 (p = 0.0542) (Figure 2B&C).
3.3. SMN Deficiency Increased Astrocytic Cx43 Expression
To assess whether SMN deficiency affects Cx43 expression in spinal astrocytes, primary WT astrocyte cultures were transfected with SMN-targeting siRNA, while cultures transfected with scr-siRNA served as controls (Figure 3A).
The astrocytic purity of the cultures was verified by GFAP immunostaining, revealing that > 98% of all DAPI⁺ cells were GFAP⁺ (Figure 3B&C). Immunolabeling for Cx43 followed by quantitative particle analysis demonstrated a 1.6-fold increase in Cx43 expression in SMN-deficient astrocytes compared to control cells (p = 0.0298) (Figure 3D&E).
3.4. Translational Validation of Cx43 Dysregulation Using a Human iPSC-Based Model
To further explore the contribution of Cx43 dysregulation in late-onset SMA, we employed an iPSC-based human model. Human iPSC-derived astrocytes (hiAstrocytes) were generated and transfected with SMN1-specific siRNA to mimic SMN deficiency observed in late-onset SMA mice.
Astrocytic identity and culture purity were confirmed by GFAP immunostaining, showing that > 98% of all DAPI⁺ cells were GFAP⁺ (Figure 4A&B).
Transfection efficiency was verified by SMN immunostaining, revealing a significant reduction in SMN expression in SMN-siRNA-treated cells compared to scrambled siRNA controls (0.82-fold, p = 0.006) (Figure 4C&D).
SMN-deficient hiAstrocytes exhibited a 1.7-fold increase in Cx43 expression relative to controls (p = 0.02), which was reduced upon treatment with the Cx43 inhibitor GAP27 (0.5-fold, p = 0.007) (Figure 4E&F).
3.5. Cx43 Inhibition Reduces SMA-Associated Calcium Levels in MNs
To investigate the functional impact of pathological Cx43 expression on spinal MNs, organotypic spinal cord slice cultures and primary MN cultures were prepared. MNs were subsequently incubated with supernatants (SN) collected from treated and untreated spinal cord slice cultures of late-onset SMA mice and untreated WT mice (Figure 5).
Immunocytochemical characterization revealed that > 97% of the cultured cells were TUJ1⁺ neurons, of which > 93% were SMI-32⁺ MNs, confirming a motor neuron purity of > 95% (Figure 6A-C).
After overnight incubation of MNs with SN collected from 7 DIV spinal cord slice cultures of WT, late-onset SMA, and SMA treated with the Cx43 inhibitor Gap27 (SMA + Gap27), intracellular Ca²⁺ levels were assessed using Fluo-4 staining.
Microscopic analysis revealed a 2.8-fold increase in Ca²⁺ response in MNs incubated with SMA SN compared to WT SN (p = 0.0076). Treatment of SMA slice cultures with Gap27 significantly reduced the Ca²⁺ response in MNs by 0.29-fold compared to SMA SN (p = 0.002), restoring levels comparable to those observed in MNs exposed to WT SN (p = 0.82) (Figure 7A&B).
In addition, short-term exposure of Fluo-4–loaded MNs to SN from SMA or SMA + Gap27 slice cultures, followed by Ca²⁺ imaging, revealed a significant 0.76-fold reduction in spontaneous spike amplitude in MNs treated with SMA + Gap27 SN compared to SMA SN (p = 0.0156) (Figure 7C&D).
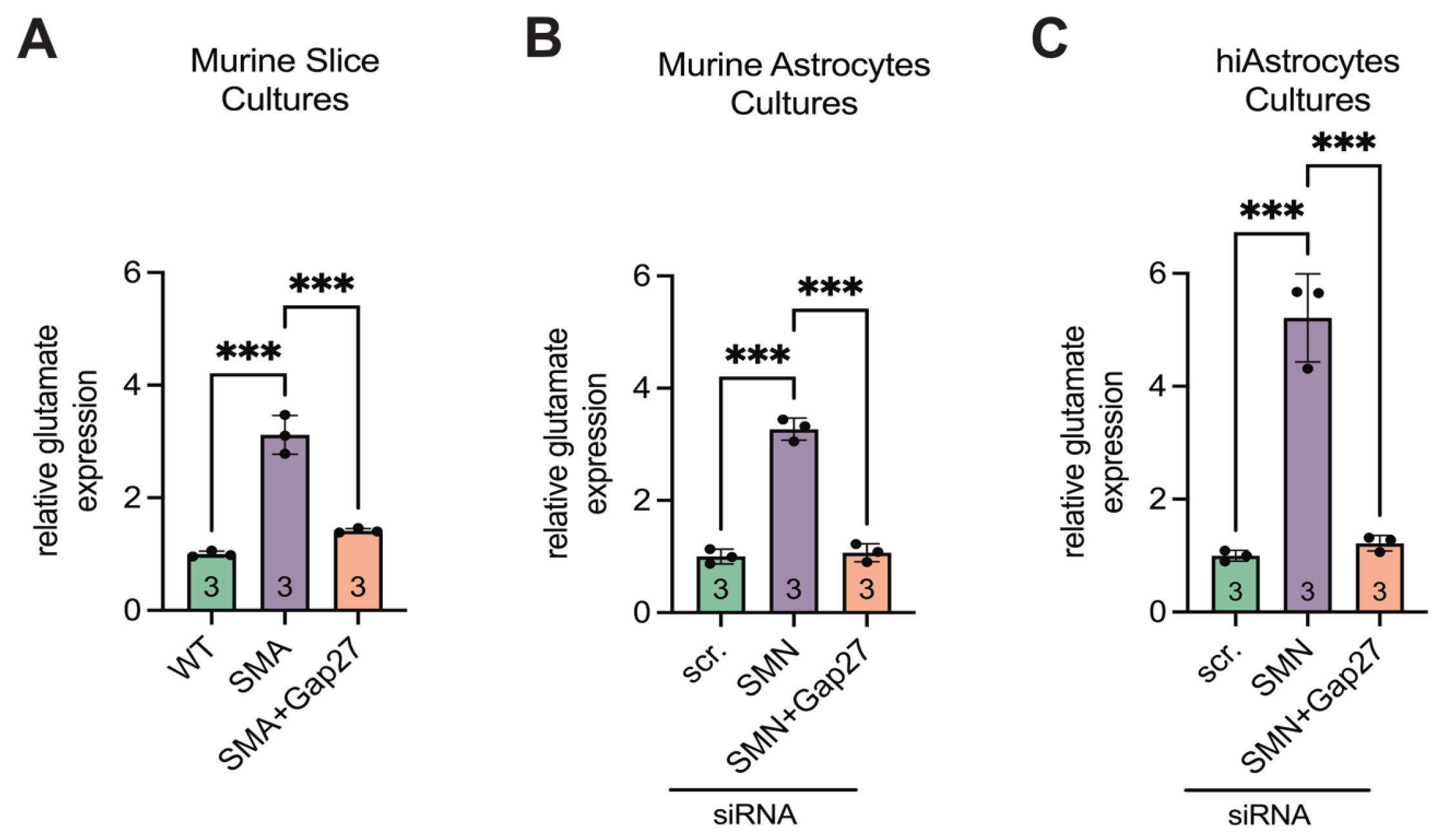
3.6. SMA Cultures Showed Elevated Glutamate Levels, Reversed by Inhibiting Cx43
Glutamate assay analysis revealed a 3.1-fold increase in extracellular glutamate levels in late-onset SMA spinal cord slice cultures compared to WT (p < 0.001). Treatment of SMA slice cultures with the Cx43 inhibitor Gap27 significantly reduced glutamate levels (p < 0.001), restoring them to values comparable to WT (p = 0.1) (Figure 8A).
Supernatants from siRNA-transfected murine astrocyte cultures showed consistent results. SMN-deficient astrocytes exhibited a 3.3-fold increase in glutamate levels compared to scr-siRNA controls (p < 0.001), whereas Gap27 treatment normalized glutamate concentrations to control levels (p < 0.001 vs. SMA; p = 0.85 vs. scr-siRNA) (Figure 8B).
Similarly, in hiAstrocyte cultures, siRNA-mediated SMN knockdown led to a 5.5-fold increase in glutamate levels (p < 0.001). Gap27 treatment significantly reduced glutamate release (p < 0.001), restoring levels to those observed in scr-siRNA-treated hiAstrocytes (p = 0.92) (Figure 8C).
4. Discussion
This study identifies astrocytic Cx43 as a contributor of glutamate-driven MN loss in late-onset SMA. We demonstrate that SMN deficiency leads to a pronounced upregulation of Cx43 in spinal astrocytes, both in a murine and in human iPSC-derived model. Elevated Cx43 expression was accompanied by increased extracellular glutamate and abnormal MN Ca²⁺ responses, indicating enhanced excitotoxic stress. Pharmacological inhibition of Cx43 with the hemichannel blocker Gap27 effectively normalized glutamate levels and restored MN Ca²⁺ signaling to control values, establishing a direct functional link between astrocytic Cx43 and MN excitotoxicity in SMA.
Our findings highlight the importance of non-neuronal mechanisms in SMA pathogenesis. Although SMA is considered an MND caused by loss of SMN protein, growing evidence indicates that astrocytes critically modulate disease progression. Previous work has shown astrocytic activation and glutamate dysregulation preceding MN degeneration in late-onset SMA mice [16,17]. The present data extend these findings by identifying Cx43 as a central molecular contributor to this glial dysfunction.
Cx43 is the predominant connexin in astrocytes and forms both gap junctions and hemichannels that regulate intercellular signaling, ion homeostasis, and neurotransmitter clearance [22,24]. Under pathological conditions, excessive opening of Cx43 hemichannels results in uncontrolled release of glutamate, ATP, and other neuroactive molecules, thereby amplifying excitotoxicity and inflammation [28]. Our results indicate that late-onset SMA condition promotes such aberrant Cx43 activity, leading to impaired glutamate buffering and excitotoxic stress on MNs. The reversal of these effects by Gap27 demonstrates that Cx43 hemichannels are functionally involved in the astrocyte-mediated toxicity observed in late-onset SMA.
At the molecular level, the early but transient increase in Cx43 mRNA and sustained protein overexpression suggest post-transcriptional dysregulation. This pattern aligns with the established function of SMN in RNA processing and transport [29,30] and supports the hypothesis that SMN deficiency interferes with the post-transcriptional regulation of astrocytic genes. Such mechanisms may contribute to the persistent elevation of Cx43 protein seen at symptomatic and late disease stages.
Notably, the upregulation of astrocytic Cx43 is not unique to SMA and has been described in other neurodegenerative and neuromuscular disorders, including ALS, Duchenne Muscular Dystrophy, and Multiple Sclerosis [15,25,31]. In ALS, Cx43-dependent hemichannel opening has been shown to promote MN degeneration, and its inhibition mitigates excitotoxic damage [25]. Our study provides converging evidence that similar mechanisms operate in late-onset SMA, reinforcing the concept that Cx43-mediated astrocytic dysfunction is a common pathway contributing to MN loss across distinct MNDs.
Importantly, our experiments using human SMN-deficient astrocytes reproduced the findings obtained in mouse models, underscoring the translational relevance of this mechanism. Gap27 treatment normalized glutamate release in both systems, suggesting that pharmacological modulation of Cx43 may represent a potential therapeutic strategy. Such approaches could be especially valuable for late-onset SMA patients who are diagnosed at advanced stages, when SMN-enhancing therapies alone show limited efficacy [32].
Nevertheless, several limitations must be acknowledged. The study relies primarily on ex vivo and in vitro systems, which, while allowing controlled mechanistic analysis, may not fully capture the complexity of the in vivo spinal environment. In addition, although Gap27 efficiently inhibited pathological Cx43 activity in culture, its limited bioavailability and tissue penetration pose challenges for clinical application [33]. Future work should therefore employ conditional, astrocyte-specific Cx43 knockout models and evaluate clinically tested gap junction modulators such as Tonabersat [34,35,36] in different phenotype-related SMA models to determine their therapeutic potential.
Beyond glutamate regulation, Cx43 hemichannels also control the release of ATP and cytokines [37,38], which may further exacerbate neuroinflammation and MN injury. Thus, astrocytic Cx43 likely contributes to late-onset SMA pathology through multiple converging mechanisms that warrant deeper investigation.
5. Conclusions
This study identifies astrocytic Cx43 as a regulator of glutamate-mediated MN stress in late-onset SMA and demonstrates that its inhibition mitigates excitotoxicity. These findings expand current concepts of late-onset SMA pathogenesis beyond SMN deficiency and underscore the therapeutic potential of targeting astrocytic dysfunction as a complementary strategy to existing SMN-enhancing treatments.
This section is not mandatory but can be added to the manuscript if the discussion is unusually long or complex.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org.
Author Contributions
Conceptualization: ML; methodology: SS, L-IS, ML, KCL, SH, AR; formal analysis and investigation: SS, L-IS; writing and original draft preparation: SS; writing—review and editing: CK, L-IS, SH, AR, USS, ML, TH; funding acquisition: TH; resources: CK, TH; supervision: ML, TH. All authors read and approved the final manuscript.
Funding
No fundings were received for this project.
Institutional Review Board Statement
The study was performed in line with the principles of the Declaration of Helsinki. The study was approved by the Bioethic Committee of the University of Duisburg-Essen (approval number 18–8285-BO; 19–9011-BO). All experiments were conducted under the animal welfare guidelines of the University of Duisburg-Essen. Furthermore, the SMA mouse model used was approved by the State Office for Consumer Protection and Food Safety (LAVE) in NorthRhine-Westphalia, Germany (reference number 81–02.04–2020.A335).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors have no competing interests to declare that are relevant to this article.
Abbreviations
The following abbreviations are used in this manuscript:
| ALS | amyotrophic lateral sclerosis |
| ATP | adenosine triphosphate |
| BCA | bicinchoninic acid protein assay |
| Ca2+ | calcium ion |
| Cx43 | connexin 43 |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DIV | days in vitro |
| DMD | Duchenne muscular dystrophy |
| E | embryonal day |
| EGF | epidermal growth factor |
| ELISA | enzyme-linked immunosorbent assay |
| FBS | fetal bovine serum |
| FGF | fibroblast growth factor |
| iPSC | induced pluripotent stem cells |
| GFAP | glial fibrillary acidic protein |
| GJA1 | gap junction alpha-1 |
| hiAstrocytes | human induced astrocytes |
| late-onset SMA | late-onset spinal muscular atrophy |
| P | postnatal day |
| PBS | phosphate-buffered saline |
| PDL | poly-d-lysine |
| qPCR | quantitative polymerase chain reactions |
| RT | room temperature |
| scr | scrambled |
| SMA | spinal muscular atrophy |
| SMI-32 | non-phosphorylated neurofilament H |
| SMN1 | survival of motor neuron-1 |
| SMN2 | survival of motor neuron-2 |
| SMN | survival of motor neuron |
| TUJ1 | βIII-tubulin |
| WB | Western blot |
| WT | wild-type |
References
- Crawford, T. O. and C. A. Pardo. “The neurobiology of childhood spinal muscular atrophy.” Neurobiology of Disease 3 (1996): 97-110. [CrossRef]
- Lefebvre, S., L. Bürglen, S. Reboullet, O. Clermont, P. Burlet, L. Viollet, B. Benichou, C. Cruaud, P. Millasseau, M. Zeviani, et al. “Identification and characterization of a spinal muscular atrophy-determining gene.” Cell 80 (1995): 155-65. [CrossRef]
- Monani, U. R. “The human centromeric survival motor neuron gene (smn2) rescues embryonic lethality in smn-/- mice and results in a mouse with spinal muscular atrophy.” Human Molecular Genetics 9 (2000): 333-39. [CrossRef]
- Feldkötter, M., V. Schwarzer, R. Wirth, T. F. Wienker and B. Wirth. “Quantitative analyses of smn1 and smn2 based on real-time lightcycler pcr: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy.” The American Journal of Human Genetics 70 (2002): 358-68. [CrossRef]
- McAndrew, P. E., D. W. Parsons, L. R. Simard, C. Rochette, P. N. Ray, J. R. Mendell, T. W. Prior and A. H. M. Burghes. “Identification of proximal spinal muscular atrophy carriers and patients by analysis of smnt and smnc gene copy number.” The American Journal of Human Genetics 60 (1997): 1411-22. [CrossRef]
- Chaytow, H., K. M. E. Faller, Y.-T. Huang and T. H. Gillingwater. “Spinal muscular atrophy: From approved therapies to future therapeutic targets for personalized medicine.” Cell Reports Medicine 2 (2021): 10.1016/j.xcrm.2021.100346.
- Finkel, R. S., E. Mercuri, B. T. Darras, A. M. Connolly, N. L. Kuntz, J. Kirschner, C. A. Chiriboga, K. Saito, L. Servais, E. Tizzano, et al. “Nusinersen versus sham control in infantile-onset spinal muscular atrophy.” N Engl J Med 377 (2017): 1723-32. https://www.ncbi.nlm.nih.gov/pubmed/29091570. [CrossRef]
- Hagenacker, T., C. D. Wurster, R. Günther, O. Schreiber-Katz, A. Osmanovic, S. Petri, M. Weiler, A. Ziegler, J. Kuttler, J. C. Koch, et al. “Nusinersen in adults with 5q spinal muscular atrophy: A non-interventional, multicentre, observational cohort study.” The Lancet Neurology 19 (2020): 317-25. [CrossRef]
- Chiriboga, C. A., K. J. Swoboda, B. T. Darras, S. T. Iannaccone, J. Montes, D. C. De Vivo, D. A. Norris, C. F. Bennett and K. M. Bishop. “Results from a phase 1 study of nusinersen (isis-smn rx) in children with spinal muscular atrophy.” Neurology 86 (2016): 890-97. [CrossRef]
- Mercuri, E., B. T. Darras, C. A. Chiriboga, J. W. Day, C. Campbell, A. M. Connolly, S. T. Iannaccone, J. Kirschner, N. L. Kuntz, K. Saito, et al. “Nusinersen versus sham control in later-onset spinal muscular atrophy.” New England Journal of Medicine 378 (2018): 625-35. [CrossRef]
- Mercuri, E., F. Muntoni, G. Baranello, R. Masson, O. Boespflug-Tanguy, C. Bruno, S. Corti, A. Daron, N. Deconinck, L. Servais, et al. “Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (str1ve-eu): An open-label, single-arm, multicentre, phase 3 trial.” The Lancet Neurology 20 (2021): 832-41. [CrossRef]
- Mercuri, E., N. Deconinck, E. S. Mazzone, A. Nascimento, M. Oskoui, K. Saito, C. Vuillerot, G. Baranello, O. Boespflug-Tanguy, N. Goemans, et al. “Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (sunfish part 2): A phase 3, double-blind, randomised, placebo-controlled trial.” The Lancet Neurology 21 (2022): 42-52. [CrossRef]
- Ponath, G., S. Ramanan, M. Mubarak, W. Housley, S. Lee, F. R. Sahinkaya, A. Vortmeyer, C. S. Raine and D. Pitt. “Myelin phagocytosis by astrocytes after myelin damage promotes lesion pathology.” Brain 140 (2017): 399-413. [CrossRef]
- Yamanaka, K., S. J. Chun, S. Boillee, N. Fujimori-Tonou, H. Yamashita, D. H. Gutmann, R. Takahashi, H. Misawa and D. W. Cleveland. “Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis.” Nature Neuroscience 11 (2008): 251-53. [CrossRef]
- McGivern, J. V., T. N. Patitucci, J. A. Nord, M. A. Barabas, C. L. Stucky and A. D. Ebert. “Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production.” Glia 61 (2013): 1418-28. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3941074/pdf/nihms-561874.pdf. [CrossRef]
- Schmitt, L. I., C. David, R. Steffen, S. Hezel, A. Roos, U. Schara-Schmidt, C. Kleinschnitz, M. Leo and T. Hagenacker. “Spinal astrocyte dysfunction drives motor neuron loss in late-onset spinal muscular atrophy.” Acta Neuropathologica 145 (2023): 611-35. https://www.ncbi.nlm.nih.gov/pubmed/36930296. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10119066/pdf/401_2023_Article_2554.pdf. [CrossRef]
- Leo, M., L. I. Schmitt, M. Fleischer, R. Steffen, C. Osswald, C. Kleinschnitz and T. Hagenacker. “Induction of survival of motor neuron (smn) protein deficiency in spinal astrocytes by small interfering rna as an in vitro model of spinal muscular atrophy.” Cells 11 (2022): https://mdpi-res.com/d_attachment/cells/cells-11-00558/article_deploy/cells-11-00558-v2.pdf?version=1644496536. [CrossRef]
- Konietzko, U. and C. M. Müller. “Astrocytic dye coupling in rat hippocampus: Topography, developmental onset, and modulation by protein kinase c.” Hippocampus 4 (2004): 297-306. [CrossRef]
- Musil, L. S. and D. A. Goodenough. “Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques.” The Journal of cell biology 115 (1991): 1357-74. [CrossRef]
- Soares, A. R., T. Martins-Marques, T. Ribeiro-Rodrigues, J. V. Ferreira, S. Catarino, M. J. Pinho, M. Zuzarte, S. Isabel Anjo, B. Manadas, J. P.G. Sluijter, et al. “Gap junctional protein cx43 is involved in the communication between extracellular vesicles and mammalian cells.” Scientific Reports 5 (2015): 10.1038/srep13243.
- Wang, X., M. L. Veruki, N. V. Bukoreshtliev, E. Hartveit and H.-H. Gerdes. “Animal cells connected by nanotubes can be electrically coupled through interposed gap-junction channels.” Proceedings of the National Academy of Sciences 107 (2010): 17194-99. [CrossRef]
- Giaume, C. and X. Liu. “From a glial syncytium to a more restricted and specific glial networking.” Journal of Physiology-Paris 106 (2012): 34-39. [CrossRef]
- Cheung, G., O. Chever, A. Rollenhagen, N. Quenech’du, P. Ezan, J. H. R. Lübke and N. Rouach. “Astroglial connexin 43 regulates synaptic vesicle release at hippocampal synapses.” Cells 12 (2023): 10.3390/cells12081133.
- Jiang, S., H. Yuan, L. Duan, R. Cao, B. Gao, Y.-F. Xiong and Z.-R. Rao. “Glutamate release through connexin 43 by cultured astrocytes in a stimulated hypertonicity model.” Brain Research 1392 (2011): 8-15. [CrossRef]
- Almad, A. A., A. Doreswamy, S. K. Gross, J. P. Richard, Y. Huo, N. Haughey and N. J. Maragakis. “Connexin 43 in astrocytes contributes to motor neuron toxicity in amyotrophic lateral sclerosis.” Glia 64 (2016): 1154-69. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5635605/pdf/nihms908168.pdf. [CrossRef]
- Hsieh-Li, H. M., J. G. Chang, Y. J. Jong, M. H. Wu, N. M. Wang, C. H. Tsai and H. Li. “A mouse model for spinal muscular atrophy.” Nature Genetics 24 (2000): 66-70. https://www.ncbi.nlm.nih.gov/pubmed/10615130. https://www.nature.com/articles/ng0100_66. [CrossRef]
- Meyer, K., Y. Rodriguez, F. S. Roussel, A. M. Hartlaub, S. S. Ray, J. A. Sierra-Delgado and C. N. Dennys. “In vitro modeling for neurological diseases using direct conversion from fibroblasts to neuronal progenitor cells and differentiation into astrocytes.” Journal of Visualized Experiments (2021): 10.3791/62016.
- Orellana, J. A., D. E. Hernández, P. Ezan, V. Velarde, M. V. L. Bennett, C. Giaume and J. C. Sáez. “Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels.” Glia 58 (2009): 329-43. [CrossRef]
- Baker, K. E. and J. Coller. “The many routes to regulating mrna translation.” Genome Biology 7 (2006): 10.1186/gb-2006-7-12-332.
- Pellizzoni, L., N. Kataoka, B. Charroux and G. Dreyfuss. “A novel function for smn, the spinal muscular atrophy disease gene product, in pre-mrna splicing.” Cell 95 (1998): 615-24. [CrossRef]
- Markoullis, K., I. Sargiannidou, N. Schiza, A. Hadjisavvas, F. Roncaroli, R. Reynolds and K. A. Kleopa. “Gap junction pathology in multiple sclerosis lesions and normal-appearing white matter.” Acta Neuropathologica 123 (2012): 873-86. [CrossRef]
- Mercuri, E., M. C. Pera, M. Scoto, R. Finkel and F. Muntoni. “Spinal muscular atrophy — insights and challenges in the treatment era.” Nature Reviews Neurology 16 (2020): 706-15. [CrossRef]
- Abudara, V. n., J. Bechberger, M. Freitas-Andrade, M. De Bock, N. Wang, G. Bultynck, C. C. Naus, L. Leybaert and C. Giaume. “The connexin43 mimetic peptide gap19 inhibits hemichannels without altering gap junctional communication in astrocytes.” Frontiers in Cellular Neuroscience 8 (2014): 10.3389/fncel.2014.00306.
- Grek, C. L., G. M. Prasad, V. Viswanathan, D. G. Armstrong, R. G. Gourdie and G. S. Ghatnekar. “Topical administration of a connexin43--based peptide augments healing of chronic neuropathic diabetic foot ulcers: A multicenter, randomized trial.” Wound Repair and Regeneration 23 (2015): 203-12. [CrossRef]
- Ghatnekar, G. S., C. L. Grek, D. G. Armstrong, S. C. Desai and R. G. Gourdie. “The effect of a connexin43-based peptide on the healing of chronic venous leg ulcers: A multicenter, randomized trial.” Journal of Investigative Dermatology 135 (2015): 289-98. [CrossRef]
- Kwakowsky, A., B. Chawdhary, A. de Souza, E. Meyer, A. H. Kaye, C. R. Green, S. S. Stylli and H. Danesh-Meyer. “Tonabersat significantly reduces disease progression in an experimental mouse model of multiple sclerosis.” International Journal of Molecular Sciences 24 (2023): 10.3390/ijms242417454. https://mdpi-res.com/d_attachment/ijms/ijms-24-17454/article_deploy/ijms-24-17454-v2.pdf?version=1702716205.
- Wei, H., F. Deng, Y. Chen, Y. Qin, Y. Hao and X. Guo. “Ultrafine carbon black induces glutamate and atp release by activating connexin and pannexin hemichannels in cultured astrocytes.” Toxicology 323 (2014): 32-41. [CrossRef]
- Mugisho, O. O., C. R. Green, D. T. Kho, J. Zhang, E. S. Graham, M. L. Acosta and I. D. Rupenthal. “The inflammasome pathway is amplified and perpetuated in an autocrine manner through connexin43 hemichannel mediated atp release.” Biochimica et Biophysica Acta (BBA) - General Subjects 1862 (2018): 385-93. [CrossRef]
Figure 1.
SMA spinal cord analyses showed an increased expression of Cx43 compared to WT. (A) Spinal cord slices of SMA and WT mice at ages P20, P35, and P>100 (n = 4-5 animals) were stained for Cx43 (green). Imaging studies showed an increased expression of Cx43 in SMA at P20 (** p = 0.0035). This increased expression was also visible at P35 and P>100 (** p = 0.0094 and p = 0.0071, respectively). Higher accumulation of Cx43 was evident around motor neurons, particularly in SMA, pointed out in the SMA P35 image. Scale bar 20 μm. (B) For WB studies, lumbar spinal cord tissue of n = 3-4 animals was used. Results were then adjusted to the total protein value. At P35 (* p = 0.0309) and P>100 (** p = 0.0037), Cx43 expression was increased in SMA compared to WT. At P20, we identified no difference in the Cx43 expression (p = 0.1115). Analysis was done using unpaired Student’s t-tests. Full blots are shown in the Additional file. Abbreviations: SMA, spinal muscular atrophy; WB, Western blot; WT, wild-type; P, postnatal day.
Figure 1.
SMA spinal cord analyses showed an increased expression of Cx43 compared to WT. (A) Spinal cord slices of SMA and WT mice at ages P20, P35, and P>100 (n = 4-5 animals) were stained for Cx43 (green). Imaging studies showed an increased expression of Cx43 in SMA at P20 (** p = 0.0035). This increased expression was also visible at P35 and P>100 (** p = 0.0094 and p = 0.0071, respectively). Higher accumulation of Cx43 was evident around motor neurons, particularly in SMA, pointed out in the SMA P35 image. Scale bar 20 μm. (B) For WB studies, lumbar spinal cord tissue of n = 3-4 animals was used. Results were then adjusted to the total protein value. At P35 (* p = 0.0309) and P>100 (** p = 0.0037), Cx43 expression was increased in SMA compared to WT. At P20, we identified no difference in the Cx43 expression (p = 0.1115). Analysis was done using unpaired Student’s t-tests. Full blots are shown in the Additional file. Abbreviations: SMA, spinal muscular atrophy; WB, Western blot; WT, wild-type; P, postnatal day.
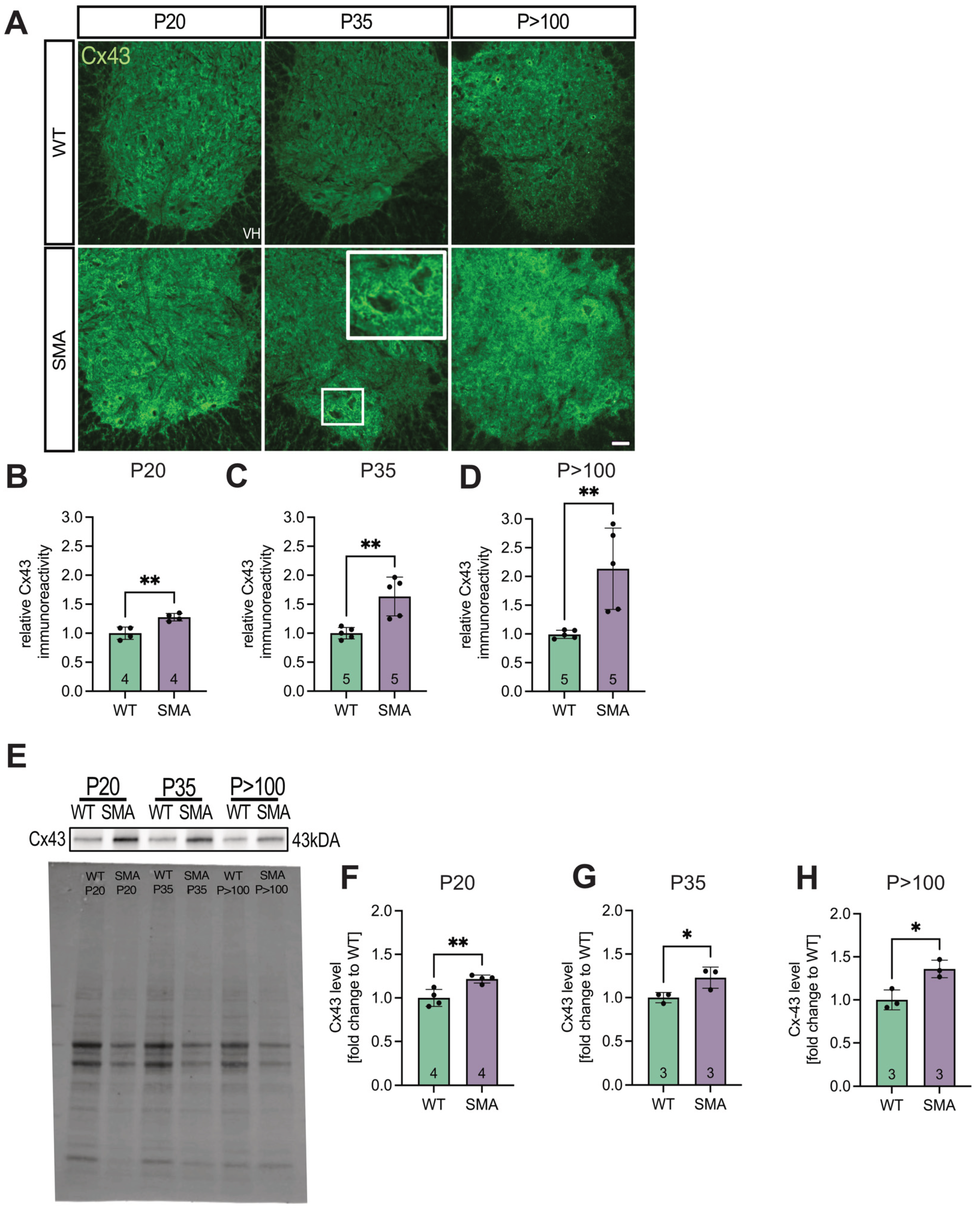
Figure 2.
qPCR analyses showed an increase in Cx43 mRNA expression early in the pathogenesis. (A) Whole lumbar spinal cord tissue of SMA and WT mice at P20, P35, and P>100 were prepared, and qPCR analyses were performed. The results suggested an increase in mRNA expression at P20 in SMA compared to WT (* p = 0.029). (B&C) No significant difference was visible between SMA and WT at later stages (P35, p = 0.9485; P>100, p = 0.0542). n = 3 individual animals, analysis was done using unpaired Student’s t-tests. Abbreviations: SMA, spinal muscular atrophy; qPCR, quantitative polymerase chain reaction; WT, wild-type; P, postnatal day.
Figure 2.
qPCR analyses showed an increase in Cx43 mRNA expression early in the pathogenesis. (A) Whole lumbar spinal cord tissue of SMA and WT mice at P20, P35, and P>100 were prepared, and qPCR analyses were performed. The results suggested an increase in mRNA expression at P20 in SMA compared to WT (* p = 0.029). (B&C) No significant difference was visible between SMA and WT at later stages (P35, p = 0.9485; P>100, p = 0.0542). n = 3 individual animals, analysis was done using unpaired Student’s t-tests. Abbreviations: SMA, spinal muscular atrophy; qPCR, quantitative polymerase chain reaction; WT, wild-type; P, postnatal day.
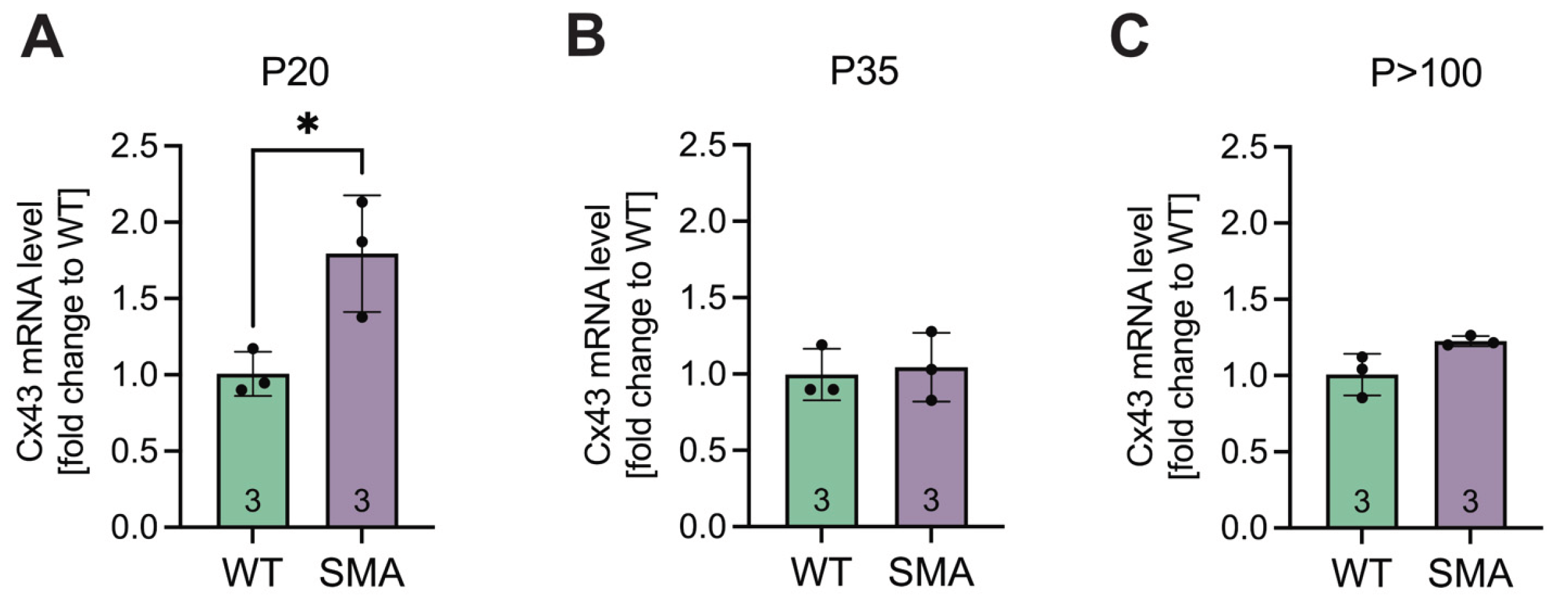
Figure 3.
Overexpression of Cx43 in a model of in vitro-induced SMN-deficiency in astrocytes. (A) Astrocytic cultures were prepared from the lumbar part of the spinal cord of WT mice and transfected with SMN-siRNA to induce SMN-deficiency. Appropriate control cultures were transfected with scr-siRNA and fluorescence imaging was conducted. (B&C) GFAP (red) and DAPI (blue) staining of the same cultures demonstrated an astrocyte proportion of >98% compared to all viable cells; n = 4 individual cultures for each of the three animals, scale bar 50 μm. (D&E) Immunostaining of Cx43 (green) in transfected astrocytes. Analysis showed a 1.6-fold increase in Cx43 expression in SMN-deficient cells compared to control (* p = 0.03; n = 3 independent experiments for each of the three individual animals, analysis was done using unpaired Student’s t-tests, scale bar 50 μm. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; GFAP, glial fibrillary acidic protein; siRNA, small interfering RNA; SMN, survival of motor neuron; WT, wild-type.
Figure 3.
Overexpression of Cx43 in a model of in vitro-induced SMN-deficiency in astrocytes. (A) Astrocytic cultures were prepared from the lumbar part of the spinal cord of WT mice and transfected with SMN-siRNA to induce SMN-deficiency. Appropriate control cultures were transfected with scr-siRNA and fluorescence imaging was conducted. (B&C) GFAP (red) and DAPI (blue) staining of the same cultures demonstrated an astrocyte proportion of >98% compared to all viable cells; n = 4 individual cultures for each of the three animals, scale bar 50 μm. (D&E) Immunostaining of Cx43 (green) in transfected astrocytes. Analysis showed a 1.6-fold increase in Cx43 expression in SMN-deficient cells compared to control (* p = 0.03; n = 3 independent experiments for each of the three individual animals, analysis was done using unpaired Student’s t-tests, scale bar 50 μm. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; GFAP, glial fibrillary acidic protein; siRNA, small interfering RNA; SMN, survival of motor neuron; WT, wild-type.
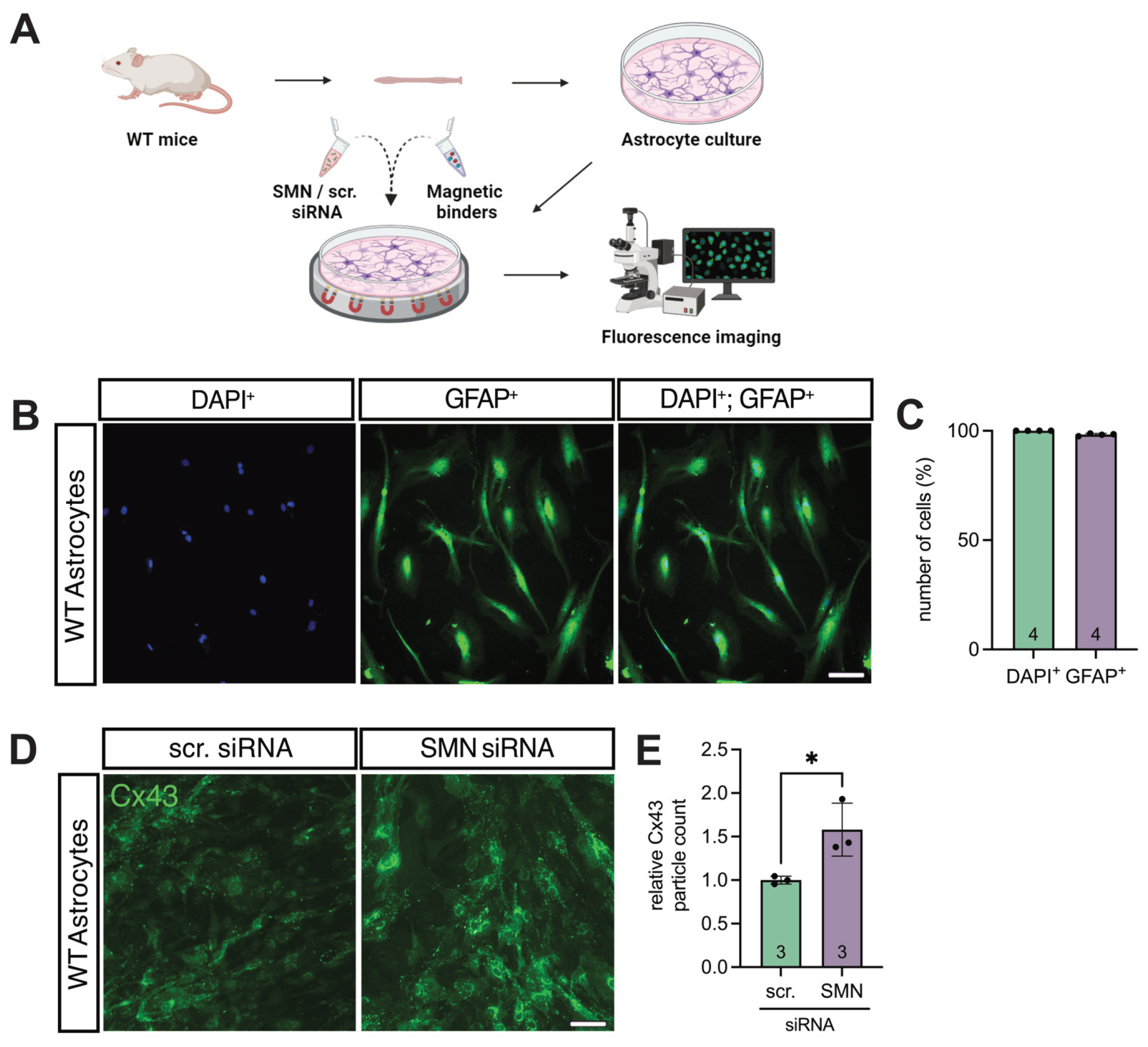
Figure 4.
SMN deficient hiAstrocytes showed a higher Cx43 expression compared to control, reversible by Gap27. (A&B) Culture clarity and iPSC induction success were proven by GFAP staining, showing >98% astrocytes in the cultures. (C&D) hiAstrocytes were transfected with SMN-siRNA to induce SMN deficiency. Appropriate controls were transfected with scr-siRNA. Staining for SMN showed an effective decrease compared to control (p = 0.006). (E&F) Staining for Cx43 showed an increased expression after SMN-knockdown (p = 0.02), which was reduced after treatment with the Cx43 inhibitor Gap27 (p = 0.007). n = 3-7 independent experiments, analysis was done using unpaired Student’s t-tests and ANOVA, scale bar 50 μm. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; E, embryonal day; SMA, spinal muscular atrophy; WT, wild-type.
Figure 4.
SMN deficient hiAstrocytes showed a higher Cx43 expression compared to control, reversible by Gap27. (A&B) Culture clarity and iPSC induction success were proven by GFAP staining, showing >98% astrocytes in the cultures. (C&D) hiAstrocytes were transfected with SMN-siRNA to induce SMN deficiency. Appropriate controls were transfected with scr-siRNA. Staining for SMN showed an effective decrease compared to control (p = 0.006). (E&F) Staining for Cx43 showed an increased expression after SMN-knockdown (p = 0.02), which was reduced after treatment with the Cx43 inhibitor Gap27 (p = 0.007). n = 3-7 independent experiments, analysis was done using unpaired Student’s t-tests and ANOVA, scale bar 50 μm. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; E, embryonal day; SMA, spinal muscular atrophy; WT, wild-type.
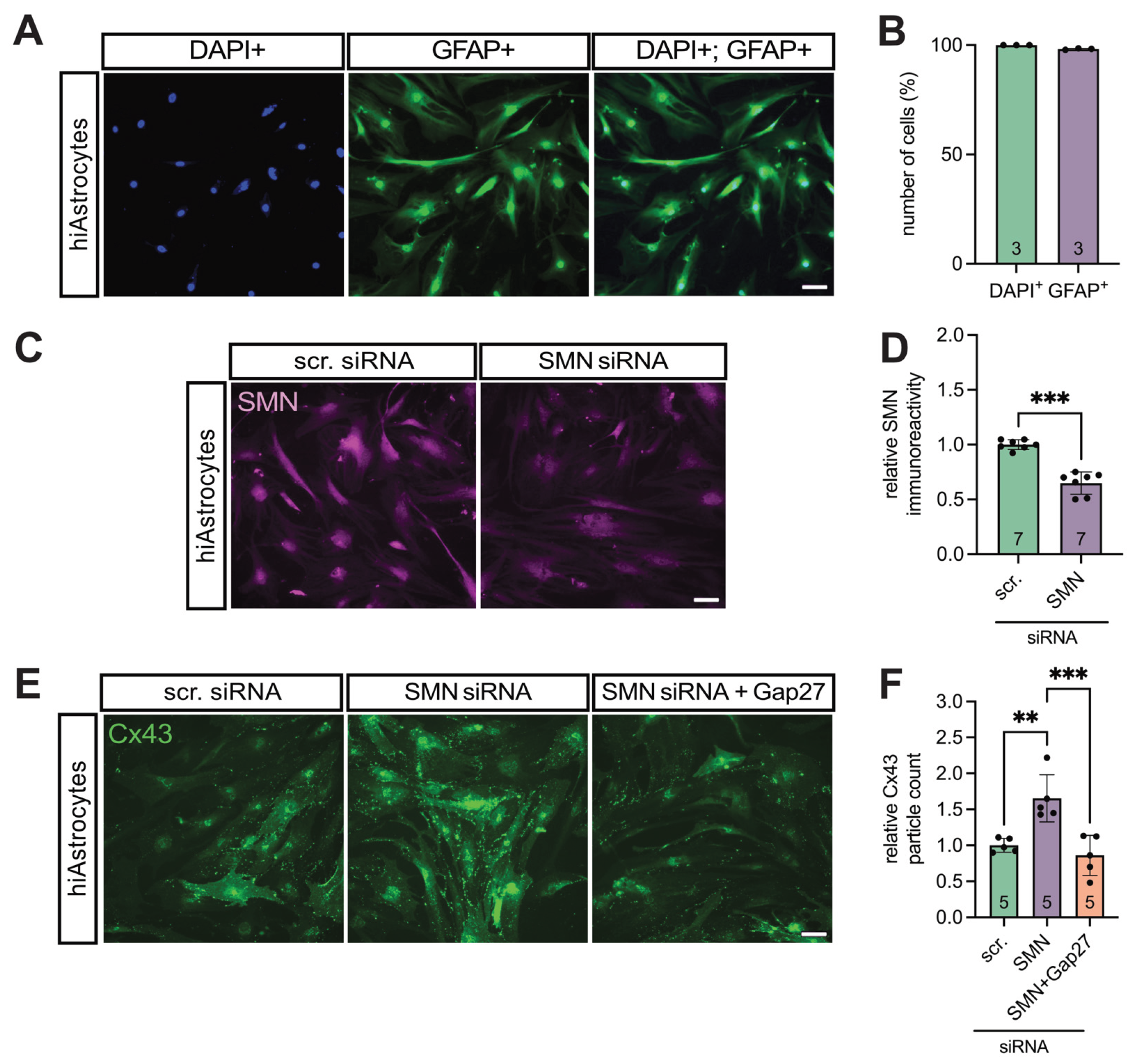
Figure 5.
Preparation of organotypic spinal cord slice cultures as an in vitro model of SMA. Slice cultures from WT and SMA mice were prepared. Supernatant was removed at 1 DIV, and half of the SMA cultures were treated with Gap27. Supernatant was again removed at 7 DIV and at 14 DIV. Embryonal motor neuronal cultures were prepared and, after maturation, the 7 DIV supernatant was introduced to the cultures. After 24-h incubation, the Ca2+ intensity was measured. In another experiment, the Ca2+ spike amplitude was measured immediately after applilacation of 7 DIV supernatants. All supernatants were assayed for glutamate with glutamate assays. Abbreviations: DIV, days in vitro; SMA, spinal muscular atrophy; E, embryonal day; WT, wild-type.
Figure 5.
Preparation of organotypic spinal cord slice cultures as an in vitro model of SMA. Slice cultures from WT and SMA mice were prepared. Supernatant was removed at 1 DIV, and half of the SMA cultures were treated with Gap27. Supernatant was again removed at 7 DIV and at 14 DIV. Embryonal motor neuronal cultures were prepared and, after maturation, the 7 DIV supernatant was introduced to the cultures. After 24-h incubation, the Ca2+ intensity was measured. In another experiment, the Ca2+ spike amplitude was measured immediately after applilacation of 7 DIV supernatants. All supernatants were assayed for glutamate with glutamate assays. Abbreviations: DIV, days in vitro; SMA, spinal muscular atrophy; E, embryonal day; WT, wild-type.
Figure 6.
Clarity of motor neuron cultures were tested using TUJ1 and SMI-32 staining. (A-C) DAPI (blue), TUJ1 (green) for neurons, and SMI-32 (magenta) for MN staining of the neuronal cultures demonstrated that all viable cells in the culture were comprised of neurons (> 97%) and motor neurons (> 93%). MNs made up > 95% of all neurons in the culture. n = 6 independent experiments from six individual cultures, each prepared from 8-9 WT E14 embryos, scale bar 100 μm. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; E, embryonal day.
Figure 6.
Clarity of motor neuron cultures were tested using TUJ1 and SMI-32 staining. (A-C) DAPI (blue), TUJ1 (green) for neurons, and SMI-32 (magenta) for MN staining of the neuronal cultures demonstrated that all viable cells in the culture were comprised of neurons (> 97%) and motor neurons (> 93%). MNs made up > 95% of all neurons in the culture. n = 6 independent experiments from six individual cultures, each prepared from 8-9 WT E14 embryos, scale bar 100 μm. Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; E, embryonal day.
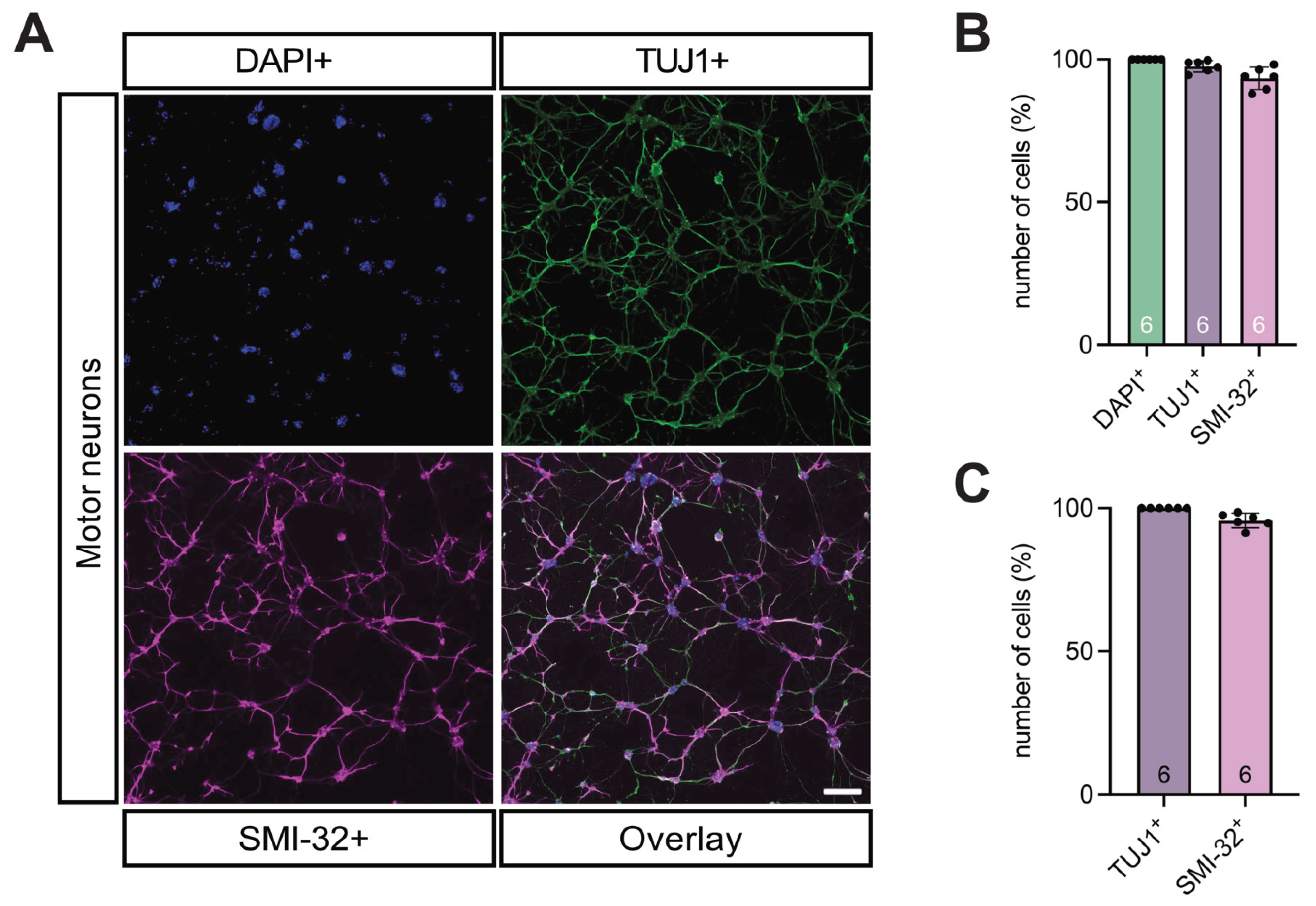
Figure 7.
Inhibition of Cx43 in SMA led to significantly lower WT-like Ca2+ response in MN. (A&B) 24h Incubation of WT MNs with the supernatant from SMA slice cultures (SMA) caused a significant increase in the Ca2+ response compared to incubation with the supernatant from WT mice slice culture (Control) (** p = 0.0076), measured by Fluo-4 staining to visualize Ca2+ processes (green). MNs incubated with SMA slice culture supernatant that had Cx43 inhibited (SMA + Gap27) caused a significant decrease in the Ca2+ response compared to incubation with the untreated SMA supernatant (** p = 0.002). SMA + Gap27 SN showed a result comparable to control (p = 0.82). n = three independent experiments, scale bar 100 μm. (C&D) After acquiring the results from (A), the change in the spontaneous Ca2+ response in MNs to SN of SMA or SMA + Gap27 slice cultures was measured. Acute introduction of SMA + Gap27 slice culture supernatant to WT MNs (Application, at 500 frames = 1min) showed a significant decrease in Ca2+ spike amplitude compared to treatment with SMA supernatant, visualized with Fluo-4 staining in all vial cells (* p < 0.05). n = 3 independent experiments, scale bar 10 μm, analysis was done using unpaired Student’s t-tests and ANOVA. Abbreviations: SMA, spinal muscular atrophy; WT, wild-type.
Figure 7.
Inhibition of Cx43 in SMA led to significantly lower WT-like Ca2+ response in MN. (A&B) 24h Incubation of WT MNs with the supernatant from SMA slice cultures (SMA) caused a significant increase in the Ca2+ response compared to incubation with the supernatant from WT mice slice culture (Control) (** p = 0.0076), measured by Fluo-4 staining to visualize Ca2+ processes (green). MNs incubated with SMA slice culture supernatant that had Cx43 inhibited (SMA + Gap27) caused a significant decrease in the Ca2+ response compared to incubation with the untreated SMA supernatant (** p = 0.002). SMA + Gap27 SN showed a result comparable to control (p = 0.82). n = three independent experiments, scale bar 100 μm. (C&D) After acquiring the results from (A), the change in the spontaneous Ca2+ response in MNs to SN of SMA or SMA + Gap27 slice cultures was measured. Acute introduction of SMA + Gap27 slice culture supernatant to WT MNs (Application, at 500 frames = 1min) showed a significant decrease in Ca2+ spike amplitude compared to treatment with SMA supernatant, visualized with Fluo-4 staining in all vial cells (* p < 0.05). n = 3 independent experiments, scale bar 10 μm, analysis was done using unpaired Student’s t-tests and ANOVA. Abbreviations: SMA, spinal muscular atrophy; WT, wild-type.
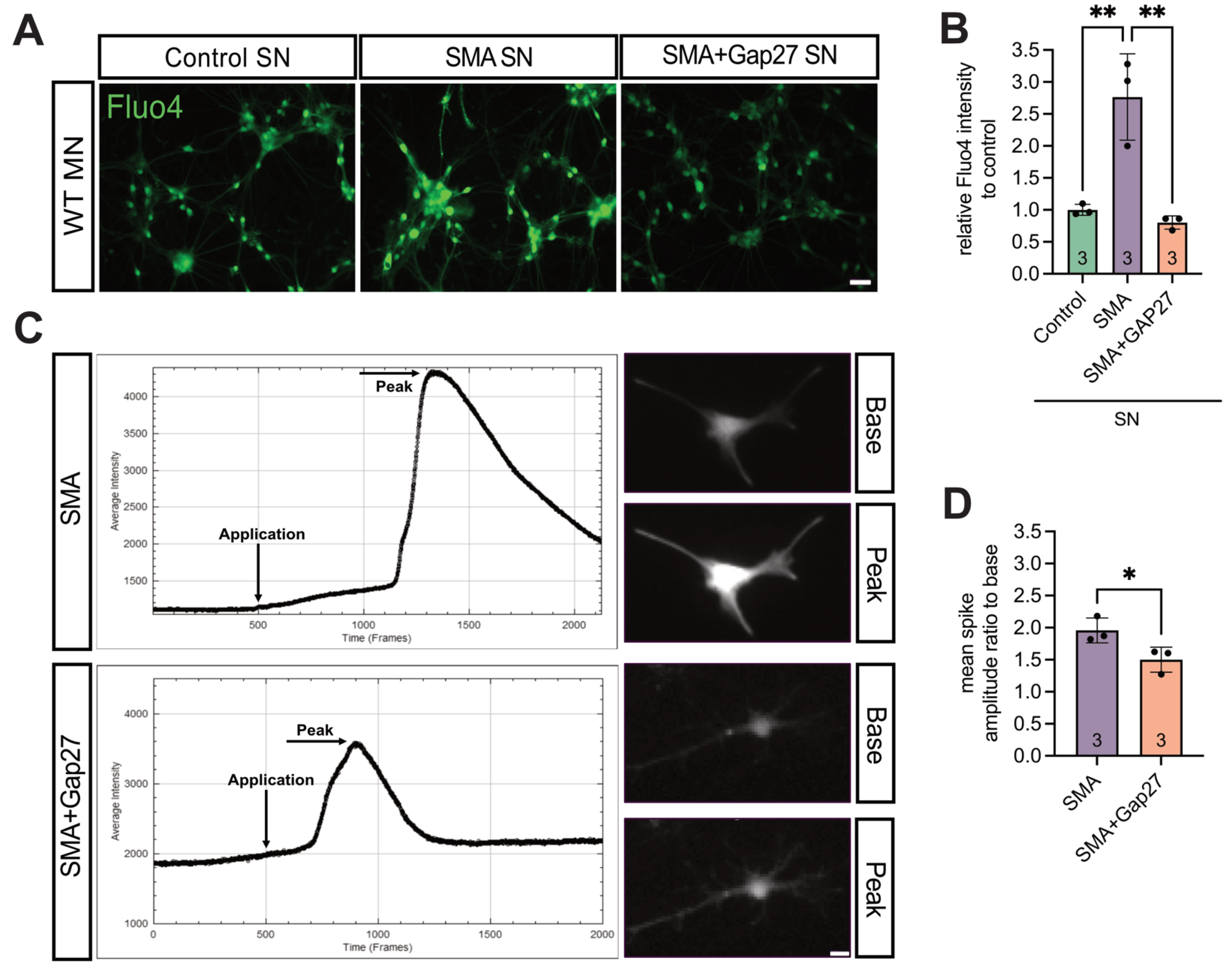
Figure 8.
Glutamate assay showed increased expression in SMA, reversible by Inhibiting Cx43. (A) Slice cultures showed higher glutamate levels in SMA compared to WT (*** p < 0.001). Gap27 treatment in SMA resulted in a decrease in glutamate levels (*** p < 0.001) to the level of WT (p = 0.1). (B) Murine cell cultures also showed higher glutamate levels when SMN was knocked down (*** p < 0.001), with the levels decreasing with Gap27 (*** p < 0.001) to the level of WT (p = 0.85). (C) Similarly, hiAstrocytes showed increased glutamate levels when SMN was knocked down (*** p < 0.001). This significantly decreased after treatment with Gap27 (p < 0.001) to the level of control (p = 0.92). Analysis was done using unpaired Student’s t-tests. n = 3 independent experiments. Abbreviations: SMA, spinal muscular atrophy; SMN, survival of motor neuron; WT, wild-type. hiAstrocytes, human induced astrocytes.
Figure 8.
Glutamate assay showed increased expression in SMA, reversible by Inhibiting Cx43. (A) Slice cultures showed higher glutamate levels in SMA compared to WT (*** p < 0.001). Gap27 treatment in SMA resulted in a decrease in glutamate levels (*** p < 0.001) to the level of WT (p = 0.1). (B) Murine cell cultures also showed higher glutamate levels when SMN was knocked down (*** p < 0.001), with the levels decreasing with Gap27 (*** p < 0.001) to the level of WT (p = 0.85). (C) Similarly, hiAstrocytes showed increased glutamate levels when SMN was knocked down (*** p < 0.001). This significantly decreased after treatment with Gap27 (p < 0.001) to the level of control (p = 0.92). Analysis was done using unpaired Student’s t-tests. n = 3 independent experiments. Abbreviations: SMA, spinal muscular atrophy; SMN, survival of motor neuron; WT, wild-type. hiAstrocytes, human induced astrocytes.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



