
Submitted:
03 November 2025
Posted:
04 November 2025
You are already at the latest version
Abstract
Neurodegenerative diseases pose major clinical challenges partly due to the underappreciation of the brain's vascular and clearance systems. Evidence suggests that neurovascular dysfunction and glymphatic impairment are early contributors to disease onset, preceding established markers such as protein aggregation. This review synthesizes recent advances in understanding how disruption of the neurovascular unit (NVU) and glymphatic pathways contributes to neurodegeneration. We analyzed published literature documenting the temporal relationship between vascular dysfunction, glymphatic clearance impairment, and subsequent neurodegenerative pathology, with a focus on identifying therapeutic targets within this axis. Current research demonstrates that BBB breakdown, pericyte dysfunction, and compromised cerebral perfusion precede protein aggregation in multiple neurodegenerative disorders. Glymphatic dysfunction, characterized by aquaporin-4 (AQP4) depolarization and abnormalities in meningeal lymphatic vessels, impairs the clearance of neurotoxic metabolites. Novel therapeutic opportunities include the preservation of pericyte function, restoration of AQP4 polarity, enhancement of meningeal lymphatic drainage via VEGF-C/VEGFR-3 signaling, and targeted modulation of microRNA and complement pathways that regulate neuroinflammation. By targeting the earliest vascular and glymphatic disruptions, emerging therapeutic strategies may halt or delay disease progression before irreversible neuronal loss occurs. This neurovascular-glymphatic approach represents an unexplored frontier that complements traditional protein-centric therapeutic paradigms, offering new possibilities for early intervention in neurodegenerative disorders.
Keywords:
neurovascular dysfunction
; glymphatic system
; blood-brain barrier
; neurodegeneration
; precision medicine
1. Introduction
The conventional understanding of neurodegenerative disorders has predominantly focused on protein aggregation, neuronal death, and synaptic dysfunction as primary pathogenic mechanisms [1]. However, emerging evidence reveals a critical and underappreciated pathophysiological axis that precedes and potentially drives these classic hallmarks: the neurovascular-glymphatic dysfunction cascade [2]. This review presents a comprehensive analysis of an unexplored therapeutic frontier centered on the intricate relationship between cerebrovascular integrity, glymphatic clearance mechanisms, and the inflammatory cascade that culminates in neurodegeneration.
The BBB, once considered a static protective barrier, is now recognized as a dynamic interface critically involved in the pathogenesis of multiple neurodegenerative conditions [3]. The earliest indicators of multiple neurodegenerative disorders in humans and animal models include impaired BBB stability, regional cerebral blood flow shortfalls, and vascular inflammation associated with BBB dysfunction [4]. Concurrently, the recently discovered glymphatic system represents a fundamental brain waste clearance mechanism whose dysfunction may precede classical pathological changes in Alzheimer's disease and other neurodegenerative disorders [2]. The convergence of BBB dysfunction, glymphatic impairment, and neuroinflammation creates a self-perpetuating cycle that accelerates neurodegeneration through mechanisms that remain largely untargeted by current therapeutic approaches. This neurovascular dysfunction represents one of the earliest detectable changes in neurodegeneration, often preceding classical pathological markers by years or decades[5].
This review identifies three critical knowledge gaps that represent unprecedented therapeutic opportunities: (1) the role of pericyte dysfunction as a primary initiator of neurovascular NVU failure, (2) the therapeutic potential of targeting glymphatic-lymphatic interfaces, and (3) the development of precision medicine approaches that address the vascular-inflammatory axis in neurodegeneration. These interconnected pathways offer novel pharmacological targets that could potentially halt or reverse the neurodegenerative process before irreversible neuronal damage occurs.
2. Pathophysiology of Neurovascular Unit Dysfunction
2.1. The Neurovascular Unit as a Therapeutic Target
The NVU comprises endothelial cells, pericytes, astrocytes, microglia, and neurons, collectively maintaining cerebrovascular homeostasis and BBB integrity [6]. The persistent neurovascular unit dysfunction (NVUD) hypothesis proposes that continuous abnormalities in the NVU following initial insults serve as the pathophysiological substrate yielding chronic neuroinflammation, proteinopathies, and oxidative stress [7]. Figure 1 illustrates the progressive cascade of neurovascular dysfunction, from early pericyte injury and subtle BBB disruption to advanced neurodegeneration characterized by severe vascular damage, protein accumulation, and chronic neuroinflammation, highlighting how these changes precede and potentially drive classical disease manifestations.
This framework suggests that targeting NVUD could provide both treatment and prevention strategies for late-onset neurodegenerative diseases, representing a paradigm shift from protein-centric to vascular-centric therapeutic approaches. The NVU's vulnerability stems from its high metabolic demands and continuous exposure to systemic inflammatory mediators, making it a critical therapeutic target for early intervention strategies [8]. Recent evidence demonstrates that NVUD with BBB hyperpermeability contributes to major depressive disorder and various neurological conditions through oxidative stress and neuroinflammation mechanistically linked to neurovascular dysfunction [9].
Understanding the contribution of neurovascular dysfunction with BBB hyperpermeability to neurodegeneration pathophysiology may help identify novel therapeutic and preventative approaches [10]. The temporal relationship where BBB dysfunction and decreased cerebral blood flow are early pathophysiological changes in neurodegenerative disorders suggests that vascular-targeted therapies could potentially halt disease progression before irreversible neuronal damage occurs [11]. Table 1 summarizes the key biomarkers of neurovascular and glymphatic dysfunction that can be detected in cerebrospinal fluid (CSF), plasma, and brain tissue, providing critical diagnostic and monitoring tools for both clinical assessment and therapeutic development.
Table 1: Key Biomarkers of Neurovascular and Glymphatic Dysfunction in Neurodegeneration.
| Biomarker | Source/Location | Pathophysiological Role | Clinical Significance | Detection Method | Key References |
| sPDGFRβ | CSF, released from injured pericytes | Indicates pericyte injury and BBB breakdown; correlates with neuroinflammation | Elevated in early-stage neurodegenerative disorders; correlates with cognitive decline and BBB dysfunction (QAlb) | ELISA, MSD electrochemiluminescence | [12,13,14,15] |
| CSF/Plasma Albumin Ratio (QAlb) | CSF and plasma | Reflects BBB permeability; increased ratio indicates BBB breakdown | Correlates with age, pericyte damage, and neuroinflammation; elevated in MCI and AD | Nephelometry, ELISA | [12,13,16] |
| C1q | Brain tissue, synapses (microglia-derived) | Tags synapses for complement-mediated elimination; initiates classical complement cascade | Increased and localized to synapses before plaque deposition in AD; associated with early synapse loss | Immunohistochemistry, Western blot | [17,18,19] |
| C3/iC3b | Brain tissue, synapses (astrocyte and microglia-derived) | Opsonizes synapses for microglial phagocytosis via CR3 receptor | Elevated in vulnerable brain regions; C3 deficiency protects against age-related synapse loss | Immunohistochemistry, flow cytometry | [18,19,20] |
| AQP4 Polarization Index | Astrocytic perivascular endfeet | Maintains glymphatic fluid flow; loss of polarization impairs waste clearance | Depolarization correlates with disease progression and impaired Aβ clearance | Immunofluorescence microscopy | [21,22,23] |
| CSF YKL-40 | CSF (astrocyte activation marker) | Indicates astrocytic activation and neuroinflammation | Elevated in AD and correlates with BBB dysfunction and PDGFRβ | ELISA | [24,25] |
| CSF GFAP | CSF (astrocyte marker) | Reflects astrocytic reactivity and glial activation | Increased with age and neuroinflammation; associated with BBB dysfunction | ELISA, Simoa | [26] |
| miR-124 | Plasma, CSF, brain tissue | Anti-inflammatory microRNA; maintains microglial quiescence | Downregulated in neurodegeneration; loss promotes M1 microglial polarization | qRT-PCR, sequencing | [27] |
| miR-155 | Plasma, CSF, brain tissue | Pro-inflammatory microRNA; promotes neuroinflammation | Upregulated in MS and AD; correlates with disease severity | qRT-PCR, sequencing | [28,29] |
| VEGF-C | CSF, brain tissue | Regulates meningeal lymphatic vessel function and lymphangiogenesis | Reduced levels associated with impaired brain clearance; therapeutic target | ELISA, Western blot | [30,31] |
| CSF Fibrinogen | CSF (blood-derived) | BBB leakage marker; promotes neuroinflammation | Elevated in AD; correlates with pericyte loss and reduced oxygenation | ELISA, immunohistochemistry | [12] |
Abbreviations: sPDGFRβ, soluble platelet-derived growth factor receptor-β; CSF, cerebrospinal fluid; BBB, blood-brain barrier; QAlb, albumin quotient; MCI, mild cognitive impairment; AD, Alzheimer’s disease; AQP4, aquaporin-4; Aβ, amyloid-β; GFAP, glial fibrillary acidic protein; MS, multiple sclerosis; VEGF-C, vascular endothelial growth factor-C; MSD, Meso Scale Discovery.
2.2. Pericyte Dysfunction: The Primary Pathogenic Event
Pericytes are contractile cells embedded within the capillary basement membrane that have emerged as central regulators of BBB integrity and cerebral blood flow [32]. Pericyte dysfunction, characterized by the release of soluble platelet-derived growth factor receptor-β (sPDGFRβ), serves as both a biomarker of BBB dysfunction and a potential therapeutic target [15]. The loss of pericytes has been associated with the development and progression of various diseases, such as diabetes, Alzheimer's disease, stroke and traumatic brain injury [5,33]. Recent clinical evidence demonstrates that CSF levels of PDGFRβ are elevated in early-stage neurodegenerative disorders, correlating with neuroinflammation and cognitive decline [34]. BBB alterations may contribute to Alzheimer's disease pathology through various mechanisms, including impaired amyloid-β clearance and neuroinflammation, with soluble PDGFRβ emerging as a potential biomarker for BBB integrity [34].
The PDGF-BB/PDGFRβ signaling pathway maintains pericyte survival and vascular stability through activation of ERK and PI3K pathways [35]. Disruption of this signaling cascade leads to pericyte loss, BBB breakdown, and subsequent neuroinflammation [36]. Notably, pericyte dysfunction appears to be particularly pronounced in APOE4 carriers, where impaired APOE-mediated signaling accelerates pericyte injury and vascular regression. APOE4 promotes the cyclophilin A-nuclear factor B-matrix metalloproteinase 9 complex pathway, which directly increases pericyte injury and impairs the formation of basement membranes [37].
Pericyte loss is one of the earliest characteristics of cerebral amyloid angiopathy, and although pericyte loss correlates with neuronal loss, the molecular mechanisms by which pericyte loss contributes to neurodegeneration remain poorly understood. BBB disruption resulting from pericyte loss serves as an early pathological hallmark in cerebral amyloid angiopathy, promoting amyloid-β accumulation and neurodegeneration via MAPK-dependent pathways [38].
2.3. Vascular Endothelial Growth Factor as a Dual-Acting Therapeutic Target
Vascular endothelial growth factor (VEGF) represents a critical mediator of neurovascular coupling and brain clearance mechanisms with established neuroprotective properties. VEGF prevents neurons from death under critical conditions such as hypoxia and glucose deprivation through binding to specific receptors, which are also expressed on the surface of neuronal cells. The neuroprotective actions occur directly through the inhibition of programmed cell death or apoptosis and the stimulation of neurogenesis [39]. VEGF binding to VEGFR-2 receptors triggers the phosphatidylinositol 3-kinase/Akt signal transduction system and, in consequence, leads to the inhibition of programmed cell death by activating antiapoptotic proteins through the transcription factor NF-κB and inhibiting proapoptotic signaling [39]. Recent clinical evidence demonstrates that transcranial radiofrequency wave treatment increases VEGF levels in Alzheimer's disease patients, correlating with enhanced clearance of tau and amyloid-β proteins from the brain through facilitation of meningeal lymphatic vessel flow and toxin clearance [40].
Exogenous application of VEGF can increase the permeability of the BBB without causing brain edema, and pretreatment with VEGF may be a feasible method to facilitate drug delivery into the CNS [41]. VEGF treatment at optimal concentrations significantly reduced brain weight loss and gross brain injury in neonatal hypoxic-ischemic brain injury models. The neuroprotective effects may be related to activation of the Akt/ERK signaling pathway, as VEGF increased phosphorylation of protein kinase B and extracellular-signal regulated kinase 1/2 in the cortex [42]. the temporal aspects of VEGF treatment are critical, as early inhibition of VEGF may have significant potential against cerebral ischemia, partly by regulating the expression of matrix metalloproteinases [43].
2.4. The Glymphatic-Lymphatic Interface
The glymphatic system, a brain-wide network facilitating CSF-interstitial fluid exchange, represents a fundamental mechanism for clearing metabolic waste and pathological proteins [44]. This system functions through perivascular pathways, where AQP4 water channels on astrocytic endfeet facilitate fluid movement [45]. Dysfunction of this system has emerged as an early and predictive marker of neurodegeneration, often preceding amyloid pathology [2]. The glymphatic system was identified as a waste drainage system in the brain that promotes the elimination of amyloid-β and tau protein [46]. Regional variation in glymphatic function dictates tau accumulation in mouse models of Alzheimer's disease tauopathy, with impaired CSF-interstitial fluid exchange and AQP4 polarization observed in affected regions [47]. The central role of AQP4 in the glymphatic clearance of tau from the brain has been established through studies showing marked impaired glymphatic CSF-interstitial fluid exchange and tau protein clearance using novel AQP4 inhibitors [47].
Impaired glymphatic clearance is an important cause of metabolite accumulation in Alzheimer's disease, as the disease is characterized by the abnormal accumulation of amyloid-β protein creating neuritic plaques and hyperphosphorylated tau protein forming neurofibrillary tangles [48]. Multisensory gamma stimulation has been shown to promote glymphatic clearance, as glymphatic transport clears parenchymal metabolites, including pathogenic proteins such as amyloid-β [49].
2.5. Aquaporin-4 Polarity Loss: A Therapeutic Target
The polarized localization of AQP4 at perivascular astrocytic end feet is essential for efficient glymphatic function [45]. In Alzheimer's disease Loss of AQP4 polarity occurs when AQP4 expression is mislocalized within astrocytes, becoming broadly distributed rather than concentrated at the perivascular end feet, impairing its efficiency in fluid transport and waste clearance, which exacerbates the accumulation of amyloid-β, contributing to the progression of Alzheimer's disease pathology. Studies have shown that various factors, such as APOE4 and amyloid-β, influence the structure and function of AQP4, thereby regulating glymphatic system flow and affecting cognitive function. AQP4 holds great potential as a therapeutic target for Alzheimer's disease, with drug development and lifestyle interventions, such as aerobic exercise and dietary regulation, being promising approaches to restore AQP4 polarity and enhance its metabolic waste (i.e.β-amyloid) clearance capacity [50,51].
Recent research identifies calmodulin-dependent phosphorylation of AQP4 as leading to increased expression of AQP4 at the plasma membrane of astrocytes in hypoxia-induced edema. The mechanism involves transient receptor potential vanilloid type 4-facilitated calcium influx that activates calmodulin, leading to cAMP-dependent protein kinase A activation. The phosphorylation of AQP4 at Ser276 causes AQP4 to relocalize to the plasma membrane, and inhibition of calmodulin with trifluoperazine significantly reduced AQP4 translocation, CNS edema, and accelerated functional recovery compared with untreated animals [52]. Alterations in AQP4 expression and polarization occur in neurodegenerative diseases, with depolarized AQP4 expression observed to occur in line with disease progression. AQP4 depolarization may be a pathological factor associated with disease onset and progression, as sustained depolarization of AQP4 impairs the function of maintaining water balance in the spinal cord, leading to swelling and malformation of astrocytes and interfering with neuronal function [53].
The astrocyte AQP4 polarized distribution-mediated glymphatic system is essential for amyloid-β and abnormal tau clearance and represents a potential therapeutic target for Alzheimer's disease. High-intensity interval training has been shown to ameliorate Alzheimer's disease pathology through enhancement of the glymphatic system via restoration of AQP4 polarization [54]. Aerobic exercise improves clearance of amyloid-β via the glymphatic system, as previous studies have suggested that aquaporin-4-mediated glymphatic system is an important pathway to clear β-amyloid in the brain [50].
2.6. Meningeal Lymphatic Vessels: A Novel Drainage Target
The discovery of meningeal lymphatic vessels (mLVs) has revolutionized understanding of brain drainage mechanisms [55]. These vessels, which drain approximately 50% of CSF volume, represent a direct connection between the central nervous system (CNS) and peripheral lymphatic circulation [56]. Dysfunction of mLVs has been implicated in protein accumulation and cognitive decline, making them attractive targets for therapeutic intervention. VEGF-C and VEGFR3 signaling pathways control mLV development and maintenance, and pharmacological enhancement of this signaling can potentially restore drainage capacity in neurodegenerative conditions.
VEGF-C prophylaxis favors lymphatic drainage and improves neurological outcomes after ischemic stroke through enhanced CSF drainage to deep cervical lymph nodes [57]. Age-related changes in meningeal lymphatic function may contribute to the accumulation of neurotoxic proteins and the development of age-related neurodegenerative diseases [58].
3. Neuroinflammation and the Tripartite Synapse
3.1. Microglial Dysfunction and Synaptic Clearance
Microglial cells serve dual functions as brain immune sentinels and regulators of synaptic plasticity [59]. In neurodegenerative conditions, chronically activated microglia produce neurotoxic factors including tumor necrosis factor-α, nitric oxide, and reactive oxygen species, creating a self-perpetuating inflammatory cycle [59]. This chronic activation is maintained through reactive microgliosis, were neuronal damage signals further microglial activation, creating a feed-forward loop of neurodegeneration. Microglia can be categorized into two opposite types: classical (M1) or alternative (M2), though there's a continuum of different intermediate phenotypes between M1 and M2, and microglia can transit from one phenotype to another. M1 microglia release inflammatory mediators and induce inflammation and neurotoxicity, while M2 microglia release anti-inflammatory mediators and induce anti-inflammatory effects and neuroprotection [60]. The balance between M1 (pro-inflammatory) and M2 (anti-inflammatory) microglial phenotypes is critically important for neurological recovery. In neurodegenerative diseases, activated microglia are excessively shifted toward the M1 or neurotoxic phenotype due to microRNA dysregulation, particularly involving miR-124 and miR-155 pathways that control neuroinflammatory processes [61]. M1-type microglia release diverse proinflammatory mediators and free radicals that inhibit brain repair and regeneration. Conversely, microglia of the M2 phenotype improve brain repair and regeneration by enhancing phagocytosis, releasing trophic factors, and reducing brain inflammation. Following stimulation with LPS or IFN-γ, M1 microglia express high levels of inducible nitric oxide synthase and pro-inflammatory cytokines/chemokines such as TNF-α, IL-1β, and CC chemokine ligand 2 [62].
3.2. Complement-Mediated Synaptic Pruning
The complement system, particularly C1q and C3 components, mediates synaptic pruning through microglial phagocytosis [63]. While essential for normal development, excessive complement activation in neurodegenerative conditions leads to pathological synapse loss [20]. In pathological conditions such as Alzheimer's disease, virus infection, or radiation-induced injury, excessive complement-mediated synaptic pruning results in excessive elimination of synapses and is associated with cognitive impairment [63]. C1q localizes predominantly to presynaptic terminals, suggesting that complement-mediated pruning is initiated by presynaptic processes [64]. Recent evidence demonstrates that complement-mediated synaptic loss involves local apoptotic-like mechanisms within synapses, indicating that targeted anti-apoptotic therapies could preserve synaptic integrity [64].
Deletion or blockage of C1q, C3, or CR3 [65] in mouse models of Alzheimer's disease have been shown to protect synapses and prevent cognitive impairments, highlighting the therapeutic potential of complement inhibition strategies [66]. The role of the complement system in synaptic pruning and neurodegeneration presents novel therapeutic opportunities for controlling excessive synaptic elimination [65]. TREM2, a microglial receptor, modulates complement-mediated synaptic pruning by regulating microglial phagocytic capacity and inflammatory responses [67]. The specific mechanism of TREM2 regulation of synaptic clearance involves modulation of microglial activation states and phagocytic function [67].
3.3. MicroRNA-Mediated Inflammation Control
MicroRNAs, particularly miR-124 and miR-155, serve as critical regulators of neuroinflammation and represent promising therapeutic targets [61]. MiR-124 functions as an anti-inflammatory regulator that maintains microglial homeostasis and inhibits microglial activation by repressing C/EBPα, PU.1, and CREB1, thereby reducing TNF-α expression while upregulating ARG-1 and IL-10 expression [68].
MiR-124 is a key player in microglia-mediated neuroinflammation, functioning as a master regulator of microglial quiescence and activation [68]. In multiple sclerosis pathogenesis, miR-124 as an anti-inflammatory marker is significantly downregulated, while miR-155 shows an increase [61]. Conversely, miR-155 acts as a pro-inflammatory mediator that is continuously increased in multiple sclerosis patients and promotes inflammatory processes by suppressing NF-κβ-dependent toll-like receptor signaling pathways [61].
The therapeutic manipulation of these microRNA pathways has shown promise, with miR-155 deletion demonstrating neuroprotective effects and improved histological and functional outcomes in experimental spinal cord injury models [69]. MiR-124 mediates cholinergic anti-inflammatory pathways, suggesting that modulation of these microRNA networks could provide novel therapeutic approaches for controlling neuroinflammation [70]. CD200-Fc treatment of lipopolysaccharide-triggered rat macrophages upregulates M2 cells while downregulating the M1 subtype and proinflammatory cytokines [71]. The modulation of microglial polarization from M1 to M2 phenotype represents a significant therapeutic strategy, as M1 microglia secrete pro-inflammatory factors and neurotoxic substances to promote neuroinflammation and nerve fiber injury, while M2 microglia promote tissue repair and neuroprotection [72].
4. Discussion
4.1. Inadequacy of Protein-Centric Approaches
Current therapeutic strategies for neurodegenerative disorders have predominantly focused on reducing pathological protein accumulation, particularly amyloid-β and tau in Alzheimer's disease [11]. The vascular hypothesis of Alzheimer's disease proposes that vascular risk factors result in dysregulation of the NVU and hypoxia, which may reduce amyloid-β clearance from the brain and increase its production, leading to both parenchymal and vascular accumulation [11]. Several protein-centric approaches have either been associated with inappropriate immune responses triggering inflammation or have failed to improve cognition, highlighting the need for alternative therapeutic targets beyond protein aggregation [11]. The failure of numerous clinical trials targeting amyloid-β underscores the complexity of neurodegeneration and suggests that therapeutic interventions must address multiple pathophysiological mechanisms simultaneously [11].
The neurovascular dysfunction hypothesis provides a framework for understanding how vascular pathology precedes and potentially drives protein aggregation, offering new avenues for early intervention [11]. Understanding the contribution of neurovascular dysfunction with BBB hyperpermeability to neurodegeneration pathophysiology may help identify novel therapeutic and preventative approaches [9].
4.2. Blood-Brain Barrier Permeability as an Overlooked Target
This temporal relationship suggests that vascular-targeted therapies could potentially halt disease progression before irreversible neuronal damage occurs [5]. Blood-based biomarkers are quantitative, non-invasive diagnostic tools that can identify candidate biomarkers for Alzheimer's disease using the hypothesis that with BBB dysfunction, brain-synthesized proteins can leak into plasma for detection [73]. Pericytes in Alzheimer's disease are key players in disease pathogenesis, and transplanted neural stem cells have been shown to alleviate Alzheimer's disease pathology and cognitive decline, partly by replenishing pericytes [74].
4.3. Inflammation-Mediated Neurovascular Damage
The majority of vascular transcriptional changes occur in pericytes, with SMAD3 upregulated in Alzheimer's disease pericytes having the highest number of ligands including VEGFA, which is downregulated in Alzheimer's disease astrocytes [75]. Microglia-mediated neuroinflammation is considered a double-edged sword, performing both harmful and helpful effects in neurodegenerative diseases [60]. Balancing microglia M1/M2 polarization has a promising therapeutic prospect in neurodegenerative diseases [60].
4.4. Precision Medicine Approaches to Neurovascular Dysfunction
Biomarker-guided approaches utilizing vascular dysfunction indicators such as CSF PDGFRβ levels could enable early identification of at-risk individuals before classical pathological changes occur [15]. As outlined in Table 2, these findings have informed the development of multiple promising therapeutic targets addressing various aspects of neurovascular and glymphatic dysfunction, each with distinct mechanisms of action and potential clinical applications based on preclinical evidence.
Models of precision medicine for neurodegeneration focus on developing personalized therapeutic strategies based on individual pathophysiological profiles [1]. The integration of multi-modal biomarker approaches including neuroimaging, fluid biomarkers, and genetic profiling could enable personalized therapeutic strategies targeting specific aspects of neurovascular dysfunction [76]. Biomarker discovery in Alzheimer's and neurodegenerative diseases focuses on identifying novel targets for early intervention and personalized treatment approaches [77].
4.5. Molecular Pathway-Based Therapeutic Targets
Clinical translation of VEGF-C therapies could involve various delivery approaches, including intrathecal administration or systemic delivery with brain-targeting strategies [57].
The prophylactic administration of VEGF-C has shown particular promise, promoting multiple vascular, immune, and neural responses that culminate in protection against neurological damage in acute ischemic stroke models [57]. Therapeutic approaches to CNS diseases via the meningeal lymphatic system represent a novel frontier in neurodegenerative disease treatment [95]. The meningeal lymphatic drainage provides novel insights into CNS clearance mechanisms and offers new therapeutic targets [56].
Therapeutic targeting of the complement system represents a promising approach for controlling excessive synaptic pruning in neurodegenerative conditions [63]. The complement system in neurodegenerative and neuroinflammatory diseases presents novel therapeutic opportunities for controlling pathological complement activation [96]. Several preclinical complement-targeted therapeutics are in development, focusing on selective inhibition of complement components to preserve beneficial synaptic refinement while preventing pathological synapse loss [63].
MiR-124 and miR-155 serve as therapeutic targets in microglia-mediated neuroinflammation, offering novel approaches for modulating microglial polarization. Therapeutic strategies could involve miR-124 replacement therapy to restore anti-inflammatory signaling or miR-155 inhibition to reduce pro-inflammatory responses. These approaches could be delivered using various platforms including lipid nanoparticles, viral vectors, or conjugated oligonucleotides [61]. MicroRNA regulation in Parkinson's disease and their potential therapeutic applications demonstrate the broad applicability of microRNA-based therapies [97].
4.6. Future Directions and Research Priorities
Neurovascular Unit-Targeted Drug Delivery: Future therapeutic development should focus on NVU-targeted delivery systems including BBB-crossing technologies, nanoparticles, and localized delivery methods [98]. Theranostic platforms combining imaging capabilities with therapeutics could enable real-time treatment monitoring [8]. Non-invasive characterization of pericyte dysfunction represents a critical advancement [99], while integration of controlled-release technologies with biomarker-guided dosing could optimize therapeutic windows [8]. Recent advances in BBB tissue repair after stroke provide additional therapeutic insights [100].
Combination Therapy Approaches: The complex nature of neurovascular dysfunction, glymphatic impairment, and neuroinflammation necessitates combination therapeutic strategies [101]. Potential combinations include VEGF-C enhancement with complement modulation, AQP4 activation with microRNA-based anti-inflammatory therapy, or pericyte protection with glymphatic enhancement [102]. The microbiota-gut-brain axis [102] and gut-brain vascular axis [103] represent additional therapeutic opportunities. Novel pharmacological targets require rational combination design considering the temporal sequence of pathophysiological events [104].
Translational Challenges and Biomarker Development: Translation to clinical applications faces challenges including development of appropriate biomarkers, establishing relevant trial endpoints, and addressing species differences in neurovascular anatomy [105]. The Global Neurodegeneration Proteomics Consortium represents an important initiative for addressing these challenges [105]. Future clinical trials should incorporate adaptive approaches allowing biomarker-guided dose optimization and patient stratification [106]. Advanced biomarkers for BBB dysfunction [15,73] are critical for precision medicine, with biofluid markers for Alzheimer's disease focusing on vascular and inflammatory indicators [107]. MiRNA neuroinflammatory biomarkers offer novel approaches to monitoring therapeutic responses [108], while reference ranges for CSF PDGFRβ provide clinical assessment standards for pericyte dysfunction [109]. Associations between CSF PDGFRβ, aging, BBB dysfunction, and neuroinflammation provide mechanistic insights into vascular contributions to neurodegeneration [110].
5. Conclusion
Neurovascular dysfunction and glymphatic impairment constitute foundational yet underappreciated mechanisms driving the pathogenesis of neurodegenerative diseases. Therapeutic approaches that exclusively target protein aggregation have not sufficiently addressed the complex cascade of vascular and clearance deficits that initiate and perpetuate neuronal injury. This review highlights the critical importance of maintaining NVU integrity, preserving pericyte function, restoring AQP4 polarization, and enhancing meningeal lymphatic drainage as integral strategies to interrupt neurodegenerative progression. Furthermore, modulation of microglial inflammatory phenotypes and complement-mediated synaptic pruning offers additional avenues to mitigate neuroinflammation and synaptic loss. The integration of advanced biomarkers reflecting vascular and glymphatic dysfunction with precision medicine approaches promises to refine early diagnosis and enable tailored interventions. Fostering research and clinical translation targeting these interconnected vascular-inflammatory pathways holds substantial potential to transform therapeutic paradigms and improve outcomes for patients with Alzheimer’s disease and related neurodegenerative disorders.
| Aβ | Amyloid-β |
| Akt | Protein Kinase B |
| APOE4 | Apolipoprotein Epsilon 4 |
| AQP4 | Aquaporin-4 |
| ARG-1 | Arginase 1 |
| BBB | Blood Brain Barrier |
| C/EBPα | CCAAT/Enhancer-Binding Protein alpha |
| C1q | Complement component 1q |
| C3 | Complement component 3 |
| C5aR1 | C5a Receptor 1 |
| CD200-CD200R | CD200-CD200 Receptor |
| CR3 | Complement Receptor 3 |
| CREB1 | cAMP Response Element Binding Protein 1 |
| CSF | Cerebrospinal Fluid |
| ERK | Extracellular signal-Regulated Kinase |
| GFAP | Glial Fibrillary Acidic Protein |
| IL-10 | Interleukin 10 |
| IL-1β | Interleukin 1 beta |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-Activated Protein Kinase |
| miR-124 | microRNA 124 |
| miR-155 | microRNA 155 |
| NF-κB | Nuclear Factor kappa B |
| PDGF-BB | Platelet-Derived Growth Factor-BB |
| PDGFRβ | Platelet-Derived Growth Factor Receptor-β |
| PI3K | Phosphatidylinositol 3-Kinase |
| PU.1 | PU.1 (also known as SPI1) |
| Qalb | CSF/Plasma Albumin Ratio |
| Ser276 | Serine at position 276 |
| sPDGFRβ | soluble Platelet-Derived Growth Factor Receptor-β |
| TLR | Toll-Like Receptor |
| TNF-α | Tumor Necrosis Factor-α |
| TREM2 | Triggering Receptor Expressed on Myeloid cells 2 |
| VEGF | Vascular Endothelial Growth Factor |
Author Contributions
G.K.M: Investigation, Methodology, Resources, Formal analysis, Writing – original draft. OB: Conceptualization, Formal analysis, Methodology, Validation, Writing – original draft, Writing – review and editing. A.W.H: Investigation, Methodology, Resources, Formal analysis, Writing – original draft, VM: Conceptualization, Methodology, Visualization, Resources, Validation, Writing – original draft, Writing – review and editing.
Funding
This research received no external funding.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
References
- Espay, A.J. Models of Precision Medicine for Neurodegeneration. Handb Clin Neurol 2023, 192, 21–34, doi:10.1016/B978-0-323-85538-9.00009-2.
- Huang, S.-Y.; Zhang, Y.-R.; Guo, Y.; Du, J.; Ren, P.; Wu, B.-S.; Feng, J.-F.; Cheng, W.; Yu, J.-T. Glymphatic System Dysfunction Predicts Amyloid Deposition, Neurodegeneration, and Clinical Progression in Alzheimer’s Disease. Alzheimers Dement 2024, 20, 3251–3269, doi:10.1002/alz.13789.
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-Brain Barrier Breakdown in Alzheimer Disease and Other Neurodegenerative Disorders. Nat Rev Neurol 2018, 14, 133–150, doi:10.1038/nrneurol.2017.188.
- Chen, T.; Dai, Y.; Hu, C.; Lin, Z.; Wang, S.; Yang, J.; Zeng, L.; Li, S.; Li, W. Cellular and Molecular Mechanisms of the Blood-Brain Barrier Dysfunction in Neurodegenerative Diseases. Fluids Barriers CNS 2024, 21, 60, doi:10.1186/s12987-024-00557-1.
- Yu, X.; Ji, C.; Shao, A. Neurovascular Unit Dysfunction and Neurodegenerative Disorders. Front Neurosci 2020, 14, 334, doi:10.3389/fnins.2020.00334.
- Kugler, E.C.; Greenwood, J.; MacDonald, R.B. The “Neuro-Glial-Vascular” Unit: The Role of Glia in Neurovascular Unit Formation and Dysfunction. Front Cell Dev Biol 2021, 9, 732820, doi:10.3389/fcell.2021.732820.
- Zhou, Y.; Chen, Q.; Wang, Y.; Wu, H.; Xu, W.; Pan, Y.; Gao, S.; Dong, X.; Zhang, J.H.; Shao, A. Persistent Neurovascular Unit Dysfunction: Pathophysiological Substrate and Trigger for Late-Onset Neurodegeneration After Traumatic Brain Injury. Front Neurosci 2020, 14, 581, doi:10.3389/fnins.2020.00581.
- van Vliet, E.A.; Marchi, N. Neurovascular Unit Dysfunction as a Mechanism of Seizures and Epilepsy during Aging. Epilepsia 2022, 63, 1297–1313, doi:10.1111/epi.17210.
- Najjar, S.; Pearlman, D.M.; Devinsky, O.; Najjar, A.; Zagzag, D. Neurovascular Unit Dysfunction with Blood-Brain Barrier Hyperpermeability Contributes to Major Depressive Disorder: A Review of Clinical and Experimental Evidence. J Neuroinflammation 2013, 10, 142, doi:10.1186/1742-2094-10-142.
- Najjar, S.; Pahlajani, S.; De Sanctis, V.; Stern, J.N.H.; Najjar, A.; Chong, D. Neurovascular Unit Dysfunction and Blood-Brain Barrier Hyperpermeability Contribute to Schizophrenia Neurobiology: A Theoretical Integration of Clinical and Experimental Evidence. Front Psychiatry 2017, 8, 83, doi:10.3389/fpsyt.2017.00083.
- Soto-Rojas, L.O.; Pacheco-Herrero, M.; Martínez-Gómez, P.A.; Campa-Córdoba, B.B.; Apátiga-Pérez, R.; Villegas-Rojas, M.M.; Harrington, C.R.; de la Cruz, F.; Garcés-Ramírez, L.; Luna-Muñoz, J. The Neurovascular Unit Dysfunction in Alzheimer’s Disease. Int J Mol Sci 2021, 22, 2022, doi:10.3390/ijms22042022.
- Miners, J.S.; Kehoe, P.G.; Love, S.; Zetterberg, H.; Blennow, K. CSF Evidence of Pericyte Damage in Alzheimer’s Disease Is Associated with Markers of Blood-Brain Barrier Dysfunction and Disease Pathology. Alzheimers Res Ther 2019, 11, 81, doi:10.1186/s13195-019-0534-8.
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-Brain Barrier Breakdown Is an Early Biomarker of Human Cognitive Dysfunction. Nat Med 2019, 25, 270–276, doi:10.1038/s41591-018-0297-y.
- Sweeney, M.D.; Sagare, A.P.; Pachicano, M.; Harrington, M.G.; Joe, E.; Chui, H.C.; Schneider, L.S.; Montagne, A.; Ringman, J.M.; Fagan, A.M.; et al. A Novel Sensitive Assay for Detection of a Biomarker of Pericyte Injury in Cerebrospinal Fluid. Alzheimers Dement 2020, 16, 821–830, doi:10.1002/alz.12061.
- Vrillon, A.; Ashton, N.J.; Bouaziz-Amar, E.; Mouton-Liger, F.; Cognat, E.; Dumurgier, J.; Lilamand, M.; Karikari, T.K.; Prevot, V.; Zetterberg, H.; et al. Dissection of Blood-Brain Barrier Dysfunction through CSF PDGFRβ and Amyloid, Tau, Neuroinflammation, and Synaptic CSF Biomarkers in Neurodegenerative Disorders. EBioMedicine 2025, 115, 105694, doi:10.1016/j.ebiom.2025.105694.
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302, doi:10.1016/j.neuron.2014.12.032.
- Stephan, A.H.; Madison, D.V.; Mateos, J.M.; Fraser, D.A.; Lovelett, E.A.; Coutellier, L.; Kim, L.; Tsai, H.-H.; Huang, E.J.; Rowitch, D.H.; et al. A Dramatic Increase of C1q Protein in the CNS during Normal Aging. J Neurosci 2013, 33, 13460–13474, doi:10.1523/JNEUROSCI.1333-13.2013.
- Shi, Q.; Colodner, K.J.; Matousek, S.B.; Merry, K.; Hong, S.; Kenison, J.E.; Frost, J.L.; Le, K.X.; Li, S.; Dodart, J.-C.; et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J Neurosci 2015, 35, 13029–13042, doi:10.1523/JNEUROSCI.1698-15.2015.
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and Microglia Mediate Early Synapse Loss in Alzheimer Mouse Models. Science 2016, 352, 712–716, doi:10.1126/science.aad8373.
- Presumey, J.; Bialas, A.R.; Carroll, M.C. Complement System in Neural Synapse Elimination in Development and Disease. Adv Immunol 2017, 135, 53–79, doi:10.1016/bs.ai.2017.06.004.
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci Transl Med 2012, 4, 147ra111, doi:10.1126/scitranslmed.3003748.
- Smith, A.J.; Yao, X.; Dix, J.A.; Jin, B.-J.; Verkman, A.S. Test of the “glymphatic” Hypothesis Demonstrates Diffusive and Aquaporin-4-Independent Solute Transport in Rodent Brain Parenchyma. Elife 2017, 6, e27679, doi:10.7554/eLife.27679.
- Mestre, H.; Hablitz, L.M.; Xavier, A.L.; Feng, W.; Zou, W.; Pu, T.; Monai, H.; Murlidharan, G.; Castellanos Rivera, R.M.; Simon, M.J.; et al. Aquaporin-4-Dependent Glymphatic Solute Transport in the Rodent Brain. Elife 2018, 7, e40070, doi:10.7554/eLife.40070.
- Craig-Schapiro, R.; Perrin, R.J.; Roe, C.M.; Xiong, C.; Carter, D.; Cairns, N.J.; Mintun, M.A.; Peskind, E.R.; Li, G.; Galasko, D.R.; et al. YKL-40: A Novel Prognostic Fluid Biomarker for Preclinical Alzheimer’s Disease. Biol Psychiatry 2010, 68, 903–912, doi:10.1016/j.biopsych.2010.08.025.
- Janelidze, S.; Mattsson, N.; Stomrud, E.; Lindberg, O.; Palmqvist, S.; Zetterberg, H.; Blennow, K.; Hansson, O. CSF Biomarkers of Neuroinflammation and Cerebrovascular Dysfunction in Early Alzheimer Disease. Neurology 2018, 91, e867–e877, doi:10.1212/WNL.0000000000006082.
- Benedet, A.L.; Milà-Alomà, M.; Vrillon, A.; Ashton, N.J.; Pascoal, T.A.; Lussier, F.; Karikari, T.K.; Hourregue, C.; Cognat, E.; Dumurgier, J.; et al. Differences Between Plasma and Cerebrospinal Fluid Glial Fibrillary Acidic Protein Levels Across the Alzheimer Disease Continuum. JAMA Neurol 2021, 78, 1471–1483, doi:10.1001/jamaneurol.2021.3671.
- Ponomarev, E.D.; Veremeyko, T.; Barteneva, N.; Krichevsky, A.M.; Weiner, H.L. MicroRNA-124 Promotes Microglia Quiescence and Suppresses EAE by Deactivating Macrophages via the C/EBP-α-PU.1 Pathway. Nat Med 2011, 17, 64–70, doi:10.1038/nm.2266.
- Guedes, J.R.; Custódia, C.M.; Silva, R.J.; de Almeida, L.P.; Pedroso de Lima, M.C.; Cardoso, A.L. Early miR-155 Upregulation Contributes to Neuroinflammation in Alzheimer’s Disease Triple Transgenic Mouse Model. Hum Mol Genet 2014, 23, 6286–6301, doi:10.1093/hmg/ddu348.
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 Restores Abnormal Microglia and Attenuates Disease in SOD1 Mice. Ann Neurol 2015, 77, 75–99. Erratum in. Ann Neurol. 2015, 77: 1085. doi:10.1002/ana.24304.
- Da Mesquita, S.; Louveau, A.; Vaccari, A.; Smirnov, I.; Cornelison, R.C.; Kingsmore, K.M.; Contarino, C.; Onengut-Gumuscu, S.; Farber, E.; Raper, D.; et al. Functional Aspects of Meningeal Lymphatics in Ageing and Alzheimer’s Disease. Nature 2018, 560, 185–191. Erratum in. Nature. 2018, 564: E7. doi:10.1038/s41586-018-0368-8.
- Ahn, J.H.; Cho, H.; Kim, J.-H.; Kim, S.H.; Ham, J.-S.; Park, I.; Suh, S.H.; Hong, S.P.; Song, J.-H.; Hong, Y.-K.; et al. Meningeal Lymphatic Vessels at the Skull Base Drain Cerebrospinal Fluid. Nature 2019, 572, 62–66, doi:10.1038/s41586-019-1419-5.
- Brown, L.S.; Foster, C.G.; Courtney, J.-M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front Cell Neurosci 2019, 13, 282, doi:10.3389/fncel.2019.00282.
- Li, P.; Fan, H. Pericyte Loss in Diseases. Cells 2023, 12, 1931, doi:10.3390/cells12151931.
- Preis, L.; Villringer, K.; Brosseron, F.; Düzel, E.; Jessen, F.; Petzold, G.C.; Ramirez, A.; Spottke, A.; Fiebach, J.B.; Peters, O. Assessing Blood-Brain Barrier Dysfunction and Its Association with Alzheimer’s Pathology, Cognitive Impairment and Neuroinflammation. Alzheimers Res Ther 2024, 16, 172, doi:10.1186/s13195-024-01529-1.
- Smyth, L.C.D.; Highet, B.; Jansson, D.; Wu, J.; Rustenhoven, J.; Aalderink, M.; Tan, A.; Li, S.; Johnson, R.; Coppieters, N.; et al. Characterisation of PDGF-BB:PDGFRβ Signalling Pathways in Human Brain Pericytes: Evidence of Disruption in Alzheimer’s Disease. Commun Biol 2022, 5, 235, doi:10.1038/s42003-022-03180-8.
- Shen, J.; Xu, G.; Zhu, R.; Yuan, J.; Ishii, Y.; Hamashima, T.; Matsushima, T.; Yamamoto, S.; Takatsuru, Y.; Nabekura, J.; et al. PDGFR-β Restores Blood-Brain Barrier Functions in a Mouse Model of Focal Cerebral Ischemia. J Cereb Blood Flow Metab 2019, 39, 1501–1515, doi:10.1177/0271678X18769515.
- Cercy, S.P. Pericytes and the Neurovascular Unit: The Critical Nexus of Alzheimer Disease Pathogenesis? Exploratory Research and Hypothesis in Medicine 2021, 6, 125–134, doi:10.14218/ERHM.2020.00062.
- Liu, T.; Guo, W.; Gong, M.; Zhu, L.; Cao, T.; Huang, Y.; Yang, Y.; Chen, J.; Yang, C.; Zhang, S.; et al. Pericyte Loss: A Key Factor Inducing Brain Aβ40 Accumulation and Neuronal Degeneration in Cerebral Amyloid Angiopathy. Exp Brain Res 2025, 243, 191, doi:10.1007/s00221-025-07134-4.
- Góra-Kupilas, K.; Jośko, J. The Neuroprotective Function of Vascular Endothelial Growth Factor (VEGF). Folia Neuropathologica 2005, 43, 31–39.
- Arendash, G.W.; Lin, X.; Cao, C. Enhanced Brain Clearance of Tau and Amyloid-β in Alzheimer’s Disease Patients by Transcranial Radiofrequency Wave Treatment: A Central Role of Vascular Endothelial Growth Factor (VEGF). J Alzheimers Dis 2024, 100, S223–S241, doi:10.3233/JAD-240600.
- Jiang, S.; Xia, R.; Jiang, Y.; Wang, L.; Gao, F. Vascular Endothelial Growth Factors Enhance the Permeability of the Mouse Blood-Brain Barrier. PLoS One 2014, 9, e86407, doi:10.1371/journal.pone.0086407.
- Feng, Y.; Rhodes, P.G.; Bhatt, A.J. Neuroprotective Effects of Vascular Endothelial Growth Factor Following Hypoxic Ischemic Brain Injury in Neonatal Rats. Pediatr Res 2008, 64, 370–374, doi:10.1203/PDR.0b013e318180ebe6.
- Zhang, H.-T.; Zhang, P.; Gao, Y.; Li, C.-L.; Wang, H.-J.; Chen, L.-C.; Feng, Y.; Li, R.-Y.; Li, Y.-L.; Jiang, C.-L. Early VEGF Inhibition Attenuates Blood-Brain Barrier Disruption in Ischemic Rat Brains by Regulating the Expression of MMPs. Mol Med Rep 2017, 15, 57–64, doi:10.3892/mmr.2016.5974.
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance Systems in the Brain-Implications for Alzheimer Disease. Nat Rev Neurol 2015, 11, 457–470. Erratum in. Nat Rev Neurol. 2016, 12: 248. doi:10.1038/nrneurol.2015.119.
- Gomolka, R.S.; Hablitz, L.M.; Mestre, H.; Giannetto, M.; Du, T.; Hauglund, N.L.; Xie, L.; Peng, W.; Martinez, P.M.; Nedergaard, M.; et al. Loss of Aquaporin-4 Results in Glymphatic System Dysfunction via Brain-Wide Interstitial Fluid Stagnation. Elife 2023, 12, e82232, doi:10.7554/eLife.82232.
- Ota, M.; Sato, N.; Nakaya, M.; Shigemoto, Y.; Kimura, Y.; Chiba, E.; Yokoi, Y.; Tsukamoto, T.; Matsuda, H. Relationships Between the Deposition of Amyloid-β and Tau Protein and Glymphatic System Activity in Alzheimer’s Disease: Diffusion Tensor Image Study. J Alzheimers Dis 2022, 90, 295–303, doi:10.3233/JAD-220534.
- Harrison, I.F.; Ismail, O.; Machhada, A.; Colgan, N.; Ohene, Y.; Nahavandi, P.; Ahmed, Z.; Fisher, A.; Meftah, S.; Murray, T.K.; et al. Impaired Glymphatic Function and Clearance of Tau in an Alzheimer’s Disease Model. Brain 2020, 143, 2576–2593, doi:10.1093/brain/awaa179.
- Hazzard, I.; Batiste, M.; Luo, T.; Cheung, C.; Lui, F. Impaired Glymphatic Clearance Is an Important Cause of Alzheimer’s Disease. Exploration of Neuroprotective Therapy 2024, 4, 401–410, doi:10.37349/ent.2024.00091.
- Murdock, M.H.; Yang, C.-Y.; Sun, N.; Pao, P.-C.; Blanco-Duque, C.; Kahn, M.C.; Kim, T.; Lavoie, N.S.; Victor, M.B.; Islam, M.R.; et al. Multisensory Gamma Stimulation Promotes Glymphatic Clearance of Amyloid. Nature 2024, 627, 149–156, doi:10.1038/s41586-024-07132-6.
- Liang, S.; Liu, H.; Wang, X.; Lin, H.; Zheng, L.; Zhang, Y.; Peng, L.; Huang, S.; Chen, L. Aerobic Exercise Improves Clearance of Amyloid-β via the Glymphatic System in a Mouse Model of Alzheimer’s Disease. Brain Res Bull 2025, 222, 111263, doi:10.1016/j.brainresbull.2025.111263.
- Qianqian, Y.; Li, H.; Liu, H.; Ye, Y.; Yu, C. Factors Affecting Aquaporin-4 and Its Regulatory Mechanisms in Alzheimer’s Disease. Neurology Asia 2025, 30, 361–367, doi:10.54029/2025kfk.
- Patabendige, A.; Chen, R. Astrocytic Aquaporin 4 Subcellular Translocation as a Therapeutic Target for Cytotoxic Edema in Ischemic Stroke. Neural Regen Res 2022, 17, 2666–2668, doi:10.4103/1673-5374.339481.
- Dai, J.; Lin, W.; Zheng, M.; Liu, Q.; He, B.; Luo, C.; Lu, X.; Pei, Z.; Su, H.; Yao, X. Alterations in AQP4 Expression and Polarization in the Course of Motor Neuron Degeneration in SOD1G93A Mice. Mol Med Rep 2017, 16, 1739–1746, doi:10.3892/mmr.2017.6786.
- Feng, S.; Wu, C.; Zou, P.; Deng, Q.; Chen, Z.; Li, M.; Zhu, L.; Li, F.; Liu, T.C.-Y.; Duan, R.; et al. High-Intensity Interval Training Ameliorates Alzheimer’s Disease-like Pathology by Regulating Astrocyte Phenotype-Associated AQP4 Polarization. Theranostics 2023, 13, 3434–3450, doi:10.7150/thno.81951.
- Li, G.; Cao, Y.; Tang, X.; Huang, J.; Cai, L.; Zhou, L. The Meningeal Lymphatic Vessels and the Glymphatic System: Potential Therapeutic Targets in Neurological Disorders. J Cereb Blood Flow Metab 2022, 42, 1364–1382, doi:10.1177/0271678X221098145.
- Zhang, Q.; Niu, Y.; Li, Y.; Xia, C.; Chen, Z.; Chen, Y.; Feng, H. Meningeal Lymphatic Drainage: Novel Insights into Central Nervous System Disease. Signal Transduction and Targeted Therapy 2025, 10, 142, doi:10.1038/s41392-025-02177-z.
- Boisserand, L.S.B.; Geraldo, L.H.; Bouchart, J.; El Kamouh, M.-R.; Lee, S.; Sanganahalli, B.G.; Spajer, M.; Zhang, S.; Lee, S.; Parent, M.; et al. VEGF-C Prophylaxis Favors Lymphatic Drainage and Modulates Neuroinflammation in a Stroke Model. J Exp Med 2024, 221, e20221983, doi:10.1084/jem.20221983.
- Liu, Q.; Wu, C.; Ding, Q.; Liu, X.-Y.; Zhang, N.; Shen, J.-H.; Ou, Z.-T.; Lin, T.; Zhu, H.-X.; Lan, Y.; et al. Age-Related Changes in Meningeal Lymphatic Function Are Closely Associated with Vascular Endothelial Growth Factor-C Expression. Brain Res 2024, 1833, 148868, doi:10.1016/j.brainres.2024.148868.
- Lull, M.E.; Block, M.L. Microglial Activation and Chronic Neurodegeneration. Neurotherapeutics 2010, 7, 354–365, doi:10.1016/j.nurt.2010.05.014.
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front Aging Neurosci 2022, 14, 815347, doi:10.3389/fnagi.2022.815347.
- Asl, E.R.; Hosseini, S.E.; Tahmasebi, F.; Bolandi, N.; Barati, S. MiR-124 and MiR-155 as Therapeutic Targets in Microglia-Mediated Inflammation in Multiple Sclerosis. Cell Mol Neurobiol 2025, 45, 63, doi:10.1007/s10571-025-01578-6.
- Song, G.J.; Suk, K. Pharmacological Modulation of Functional Phenotypes of Microglia in Neurodegenerative Diseases. Front Aging Neurosci 2017, 9, 139, doi:10.3389/fnagi.2017.00139.
- Gomez-Arboledas, A.; Acharya, M.M.; Tenner, A.J. The Role of Complement in Synaptic Pruning and Neurodegeneration. Immunotargets Ther 2021, 10, 373–386, doi:10.2147/ITT.S305420.
- Györffy, B.A.; Kun, J.; Török, G.; Bulyáki, É.; Borhegyi, Z.; Gulyássy, P.; Kis, V.; Szocsics, P.; Micsonai, A.; Matkó, J.; et al. Local Apoptotic-like Mechanisms Underlie Complement-Mediated Synaptic Pruning. Proc Natl Acad Sci U S A 2018, 115, 6303–6308, doi:10.1073/pnas.1722613115.
- Liu, H.; Jiang, M.; Chen, Z.; Li, C.; Yin, X.; Zhang, X.; Wu, M. The Role of the Complement System in Synaptic Pruning after Stroke. Aging Dis 2024, 16, 1452–1470, doi:10.14336/AD.2024.0373.
- Cho, K. Emerging Roles of Complement Protein C1q in Neurodegeneration. Aging Dis 2019, 10, 652–663, doi:10.14336/AD.2019.0118.
- Qin, Q.; Wang, M.; Yin, Y.; Tang, Y. The Specific Mechanism of TREM2 Regulation of Synaptic Clearance in Alzheimer’s Disease. Front Immunol 2022, 13, 845897, doi:10.3389/fimmu.2022.845897.
- Zhao, J.; He, Z.; Wang, J. MicroRNA-124: A Key Player in Microglia-Mediated Inflammation in Neurological Diseases. Front Cell Neurosci 2021, 15, 771898, doi:10.3389/fncel.2021.771898.
- Gaudet, A.D.; Fonken, L.K.; Watkins, L.R.; Nelson, R.J.; Popovich, P.G. MicroRNAs: Roles in Regulating Neuroinflammation. Neuroscientist 2018, 24, 221–245, doi:10.1177/1073858417721150.
- Sun, Y.; Li, Q.; Gui, H.; Xu, D.-P.; Yang, Y.-L.; Su, D.-F.; Liu, X. MicroRNA-124 Mediates the Cholinergic Anti-Inflammatory Action through Inhibiting the Production of pro-Inflammatory Cytokines. Cell Res 2013, 23, 1270–1283, doi:10.1038/cr.2013.116.
- Nagy, E.E.; Frigy, A.; Szász, J.A.; Horváth, E. Neuroinflammation and Microglia/Macrophage Phenotype Modulate the Molecular Background of Post-Stroke Depression: A Literature Review. Exp Ther Med 2020, 20, 2510–2523, doi:10.3892/etm.2020.8933.
- Long, Y.; Li, X.-Q.; Deng, J.; Ye, Q.-B.; Li, D.; Ma, Y.; Wu, Y.-Y.; Hu, Y.; He, X.-F.; Wen, J.; et al. Modulating the Polarization Phenotype of Microglia - A Valuable Strategy for Central Nervous System Diseases. Ageing Res Rev 2024, 93, 102160, doi:10.1016/j.arr.2023.102160.
- Dan, Y.R.; Chiam, K.-H. Discovery of Plasma Biomarkers Related to Blood-Brain Barrier Dysregulation in Alzheimer’s Disease. Front Bioinform 2024, 4, 1463001, doi:10.3389/fbinf.2024.1463001.
- Zhang, Y.; Mu, B.-R.; Ran, Z.; Zhu, T.; Huang, X.; Yang, X.; Wang, D.-M.; Ma, Q.-H.; Lu, M.-H. Pericytes in Alzheimer’s Disease: Key Players and Therapeutic Targets. Exp Neurol 2024, 379, 114825, doi:10.1016/j.expneurol.2024.114825.
- İş, Ö.; Wang, X.; Reddy, J.S.; Min, Y.; Yilmaz, E.; Bhattarai, P.; Patel, T.; Bergman, J.; Quicksall, Z.; Heckman, M.G.; et al. Gliovascular Transcriptional Perturbations in Alzheimer’s Disease Reveal Molecular Mechanisms of Blood Brain Barrier Dysfunction. Nat Commun 2024, 15, 4758, doi:10.1038/s41467-024-48926-6.
- Niotis, K.; Janney, C.; Helfman, S.; Hristov, H.; Clute-Reinig, N.; Angerbauer, D.; Saperia, C.; Murray, S.; Westine, J.; Seifan, A.; et al. A Blood Biomarker-Guided Precision Medicine Approach for Individualized Neurodegenerative Disease Risk Reduction and Treatment: The Future of Preventive Neurology? (P7-3.016). Neurology 2025, 104, 201, doi:10.1212/WNL.0000000000208443.
- Ashton, N.J.; Benedet, A.L.; Di Molfetta, G.; Pola, I.; Anastasi, F.; Fernández-Lebrero, A.; Puig-Pijoan, A.; Keshavan, A.; Schott, J.; Tan, K.; et al. Biomarker Discovery in Alzheimer’s and Neurodegenerative Diseases Using Nucleic Acid-Linked Immuno-Sandwich Assay. medRxiv 2024, 2024.07.29.24311079, doi:10.1101/2024.07.29.24311079.
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes Regulate the Blood-Brain Barrier. Nature 2010, 468, 557–561, doi:10.1038/nature09522.
- Kitchen, P.; Day, R.E.; Taylor, L.H.J.; Salman, M.M.; Bill, R.M.; Conner, M.T.; Conner, A.C. Identification and Molecular Mechanisms of the Rapid Tonicity-Induced Relocalization of the Aquaporin 4 Channel. J Biol Chem 2015, 290, 16873–16881, doi:10.1074/jbc.M115.646034.
- Wang, M.; Ding, F.; Deng, S.; Guo, X.; Wang, W.; Iliff, J.J.; Nedergaard, M. Focal Solute Trapping and Global Glymphatic Pathway Impairment in a Murine Model of Multiple Microinfarcts. J Neurosci 2017, 37, 2870–2877, doi:10.1523/JNEUROSCI.2112-16.2017.
- Ding, X.-B.; Wang, X.-X.; Xia, D.-H.; Liu, H.; Tian, H.-Y.; Fu, Y.; Chen, Y.-K.; Qin, C.; Wang, J.-Q.; Xiang, Z.; et al. Impaired Meningeal Lymphatic Drainage in Patients with Idiopathic Parkinson’s Disease. Nat Med 2021, 27, 411–418, doi:10.1038/s41591-020-01198-1.
- Fonseca, M.I.; Chu, S.-H.; Hernandez, M.X.; Fang, M.J.; Modarresi, L.; Selvan, P.; MacGregor, G.R.; Tenner, A.J. Cell-Specific Deletion of C1qa Identifies Microglia as the Dominant Source of C1q in Mouse Brain. J Neuroinflammation 2017, 14, 48, doi:10.1186/s12974-017-0814-9.
- Lansita, J.A.; Mease, K.M.; Qiu, H.; Yednock, T.; Sankaranarayanan, S.; Kramer, S. Nonclinical Development of ANX005: A Humanized Anti-C1q Antibody for Treatment of Autoimmune and Neurodegenerative Diseases. Int J Toxicol 2017, 36, 449–462, doi:10.1177/1091581817740873.
- Wyss-Coray, T.; Yan, F.; Lin, A.H.-T.; Lambris, J.D.; Alexander, J.J.; Quigg, R.J.; Masliah, E. Prominent Neurodegeneration and Increased Plaque Formation in Complement-Inhibited Alzheimer’s Mice. Proc Natl Acad Sci U S A 2002, 99, 10837–10842, doi:10.1073/pnas.162350199.
- Litvinchuk, A.; Wan, Y.-W.; Swartzlander, D.B.; Chen, F.; Cole, A.; Propson, N.E.; Wang, Q.; Zhang, B.; Liu, Z.; Zheng, H. Complement C3aR Inactivation Attenuates Tau Pathology and Reverses an Immune Network Deregulated in Tauopathy Models and Alzheimer’s Disease. Neuron 2018, 100, 1337-1353.e5, doi:10.1016/j.neuron.2018.10.031.
- Fonseca, M.I.; Ager, R.R.; Chu, S.-H.; Yazan, O.; Sanderson, S.D.; LaFerla, F.M.; Taylor, S.M.; Woodruff, T.M.; Tenner, A.J. Treatment with a C5aR Antagonist Decreases Pathology and Enhances Behavioral Performance in Murine Models of Alzheimer’s Disease. J Immunol 2009, 183, 1375–1383, doi:10.4049/jimmunol.0901005.
- Hernandez, M.X.; Jiang, S.; Cole, T.A.; Chu, S.-H.; Fonseca, M.I.; Fang, M.J.; Hohsfield, L.A.; Torres, M.D.; Green, K.N.; Wetsel, R.A.; et al. Prevention of C5aR1 Signaling Delays Microglial Inflammatory Polarization, Favors Clearance Pathways and Suppresses Cognitive Loss. Mol Neurodegener 2017, 12, 66, doi:10.1186/s13024-017-0210-z.
- Propson, N.E.; Roy, E.R.; Litvinchuk, A.; Köhl, J.; Zheng, H. Endothelial C3a Receptor Mediates Vascular Inflammation and Blood-Brain Barrier Permeability during Aging. J Clin Invest 2021, 131, 140966, doi:10.1172/JCI140966.
- Lopez-Ramirez, M.A.; Wu, D.; Pryce, G.; Simpson, J.E.; Reijerkerk, A.; King-Robson, J.; Kay, O.; de Vries, H.E.; Hirst, M.C.; Sharrack, B.; et al. MicroRNA-155 Negatively Affects Blood-Brain Barrier Function during Neuroinflammation. FASEB J 2014, 28, 2551–2565, doi:10.1096/fj.13-248880.
- Hsu, M.; Rayasam, A.; Kijak, J.A.; Choi, Y.H.; Harding, J.S.; Marcus, S.A.; Karpus, W.J.; Sandor, M.; Fabry, Z. Neuroinflammation-Induced Lymphangiogenesis near the Cribriform Plate Contributes to Drainage of CNS-Derived Antigens and Immune Cells. Nat Commun 2019, 10, 229, doi:10.1038/s41467-018-08163-0.
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 2015, 160, 1061–1071, doi:10.1016/j.cell.2015.01.049.
- Bemiller, S.M.; McCray, T.J.; Allan, K.; Formica, S.V.; Xu, G.; Wilson, G.; Kokiko-Cochran, O.N.; Crish, S.D.; Lasagna-Reeves, C.A.; Ransohoff, R.M.; et al. TREM2 Deficiency Exacerbates Tau Pathology through Dysregulated Kinase Signaling in a Mouse Model of Tauopathy. Mol Neurodegener 2017, 12, 74, doi:10.1186/s13024-017-0216-6.
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979-991.e8, doi:10.1016/j.immuni.2018.04.016.
- Walker, D.G.; McGeer, P.L. Complement Gene Expression in Human Brain: Comparison between Normal and Alzheimer Disease Cases. Brain Res Mol Brain Res 1992, 14, 109–116, doi:10.1016/0169-328x(92)90017-6.
- Zhang, R.; Li, J.; Li, X.; Zhang, S. Therapeutic Approaches to CNS Diseases via the Meningeal Lymphatic and Glymphatic System: Prospects and Challenges. Front Cell Dev Biol 2024, 12, 1467085, doi:10.3389/fcell.2024.1467085.
- Negro-Demontel, L.; Maleki, A.F.; Reich, D.S.; Kemper, C. The Complement System in Neurodegenerative and Inflammatory Diseases of the Central Nervous System. Front Neurol 2024, 15, 1396520, doi:10.3389/fneur.2024.1396520.
- Shaheen, N.; Shaheen, A.; Osama, M.; Nashwan, A.J.; Bharmauria, V.; Flouty, O. MicroRNAs Regulation in Parkinson’s Disease, and Their Potential Role as Diagnostic and Therapeutic Targets. NPJ Parkinsons Dis 2024, 10, 186, doi:10.1038/s41531-024-00791-2.
- He, Z.; Sun, J. The Role of the Neurovascular Unit in Vascular Cognitive Impairment: Current Evidence and Future Perspectives. Neurobiol Dis 2025, 204, 106772, doi:10.1016/j.nbd.2024.106772.
- Thalgott, J.H.; Zucker, N.; Deffieux, T.; Koopman, M.S.; Dizeux, A.; Avramut, C.M.; Koning, R.I.; Mager, H.-J.; Rabelink, T.J.; Tanter, M.; et al. Non-Invasive Characterization of Pericyte Dysfunction in Mouse Brain Using Functional Ultrasound Localization Microscopy. Nat Biomed Eng 2025. Erratum in. Nat Biomed Eng. 2025. doi:10.1038/s41551-025-01465-x.
- Qi, L.; Wang, F.; Sun, X.; Li, H.; Zhang, K.; Li, J. Recent Advances in Tissue Repair of the Blood-Brain Barrier after Stroke. J Tissue Eng 2024, 15, 20417314241226551, doi:10.1177/20417314241226551.
- Zhao, K.; Li, Z.; Sun, T.; Liu, Q.; Cheng, Y.; Barreto, G.; Li, Z.; Liu, R. Editorial: Novel Therapeutic Target and Drug Discovery for Neurological Diseases, Volume II. Front Pharmacol 2025, 16, 1566950, doi:10.3389/fphar.2025.1566950.
- Loh, J.S.; Mak, W.Q.; Tan, L.K.S.; Ng, C.X.; Chan, H.H.; Yeow, S.H.; Foo, J.B.; Ong, Y.S.; How, C.W.; Khaw, K.Y. Microbiota-Gut-Brain Axis and Its Therapeutic Applications in Neurodegenerative Diseases. Signal Transduct Target Ther 2024, 9, 37, doi:10.1038/s41392-024-01743-1.
- Carloni, S.; Rescigno, M. The Gut-Brain Vascular Axis in Neuroinflammation. Semin Immunol 2023, 69, 101802, doi:10.1016/j.smim.2023.101802.
- Wasan, K.M.; Iqtadar, S.; Mudogo, C.N.; Chávez-Fumagalli, M.A. Editorial: Novel Pharmacological Targets and Strategies to Treat Neglected Global Diseases (NGDs): An LMIC Perspective. Frontiers in Pharmacology 2025, Volume 15-2024. doi: 10.3389/fphar.2024.1527705.
- Imam, F.; Saloner, R.; Vogel, J.W.; Krish, V.; Abdel-Azim, G.; Ali, M.; An, L.; Anastasi, F.; Bennett, D.; Pichet Binette, A.; et al. The Global Neurodegeneration Proteomics Consortium: Biomarker and Drug Target Discovery for Common Neurodegenerative Diseases and Aging. Nat Med 2025, 31, 2556–2566, doi:10.1038/s41591-025-03834-0.
- Antoniou, M.; Jorgensen, A.L.; Kolamunnage-Dona, R. Biomarker-Guided Adaptive Trial Designs in Phase II and Phase III: A Methodological Review. PLoS One 2016, 11, e0149803, doi:10.1371/journal.pone.0149803.
- Wang, S.; Xie, S.; Zheng, Q.; Zhang, Z.; Wang, T.; Zhang, G. Biofluid Biomarkers for Alzheimer’s Disease. Front Aging Neurosci 2024, 16, 1380237, doi:10.3389/fnagi.2024.1380237.
- Keikha, R.; Hashemi-Shahri, S.M.; Jebali, A. The miRNA Neuroinflammatory Biomarkers in COVID-19 Patients with Different Severity of Illness. Neurologia 2023, 38, e41–e51, doi:10.1016/j.nrl.2021.06.005.
- De Kort, A.M.; Kuiperij, H.B.; Kersten, I.; Versleijen, A.A.M.; Schreuder, F.H.B.M.; Van Nostrand, W.E.; Greenberg, S.M.; Klijn, C.J.M.; Claassen, J.A.H.R.; Verbeek, M.M. Normal Cerebrospinal Fluid Concentrations of PDGFRβ in Patients with Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Alzheimers Dement 2022, 18, 1788–1796, doi:10.1002/alz.12506.
- Cicognola, C.; Mattsson-Carlgren, N.; van Westen, D.; Zetterberg, H.; Blennow, K.; Palmqvist, S.; Ahmadi, K.; Strandberg, O.; Stomrud, E.; Janelidze, S.; et al. Associations of CSF PDGFRβ With Aging, Blood-Brain Barrier Damage, Neuroinflammation, and Alzheimer Disease Pathologic Changes. Neurology 2023, 101, e30–e39, doi:10.1212/WNL.0000000000207358.
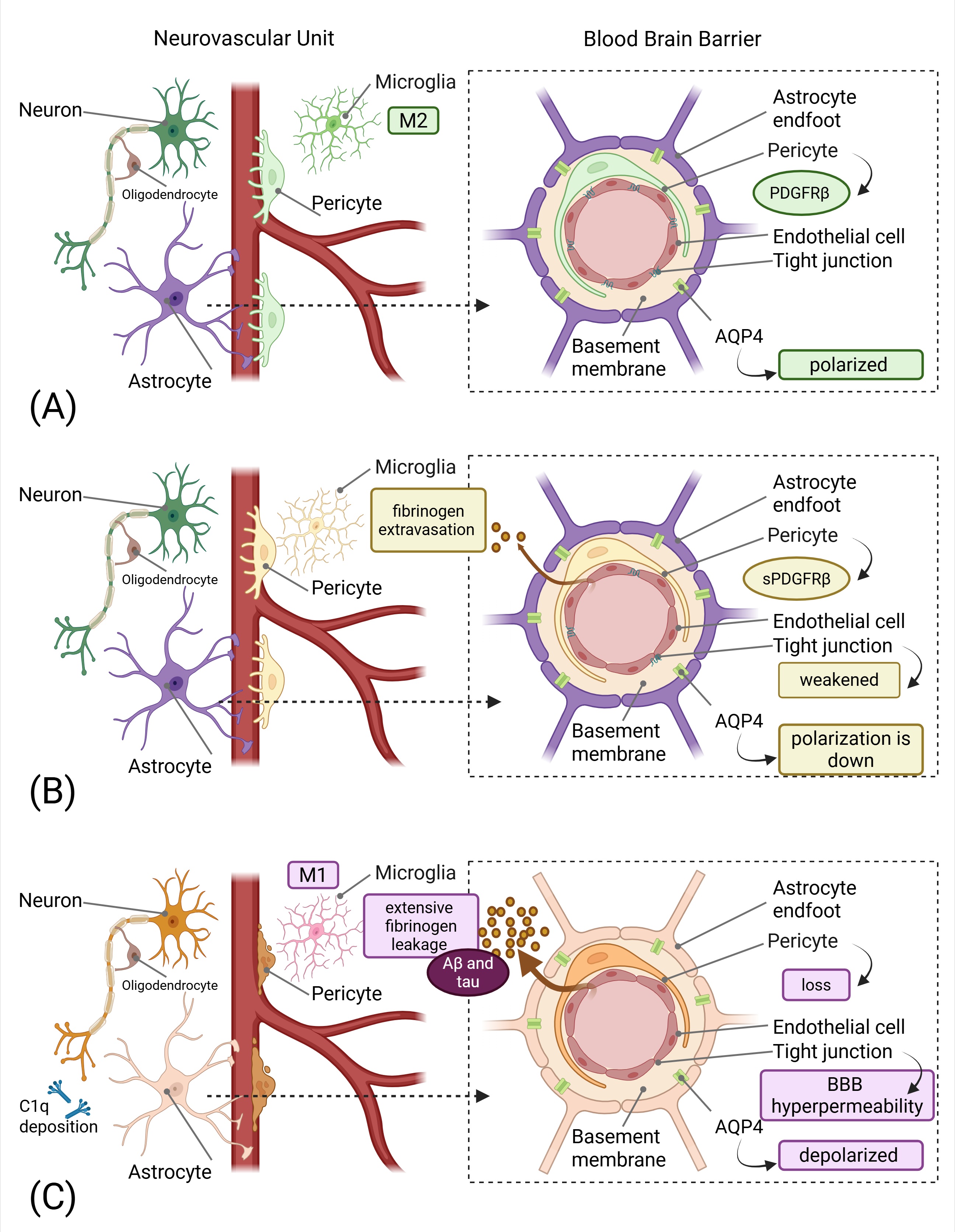
Figure 1.
The Neurovascular-Glymphatic Dysfunction Cascade in Neurodegeneration. Created in BioRender. Mavrych, V. (2026) https://BioRender.com/e97uydy(A) Healthy NVU showing intact BBB with functional pericytes expressing PDGFRβ, endothelial cells with tight junctions, astrocytic endfeet with polarized AQP4, and resting M2 microglia. Normal cerebral blood flow and efficient glymphatic clearance are maintained.(B) Early neurovascular dysfunction characterized by pericyte injury with soluble PDGFRβ (sPDGFRβ) release, initial BBB breakdown indicated by weakened tight junctions and fibrinogen extravasation and beginning loss of AQP4 polarization. Early microglial activation is evident. These changes occur years before clinical symptoms.(C) Progressive pathology showing significant pericyte loss, BBB hyperpermeability with extensive fibrinogen leakage, impaired glymphatic clearance with protein accumulation (Aβ and tau), activated M1 microglia, complement component C1q deposition on synapses, and reactive astrocytes.
Figure 1.
The Neurovascular-Glymphatic Dysfunction Cascade in Neurodegeneration. Created in BioRender. Mavrych, V. (2026) https://BioRender.com/e97uydy(A) Healthy NVU showing intact BBB with functional pericytes expressing PDGFRβ, endothelial cells with tight junctions, astrocytic endfeet with polarized AQP4, and resting M2 microglia. Normal cerebral blood flow and efficient glymphatic clearance are maintained.(B) Early neurovascular dysfunction characterized by pericyte injury with soluble PDGFRβ (sPDGFRβ) release, initial BBB breakdown indicated by weakened tight junctions and fibrinogen extravasation and beginning loss of AQP4 polarization. Early microglial activation is evident. These changes occur years before clinical symptoms.(C) Progressive pathology showing significant pericyte loss, BBB hyperpermeability with extensive fibrinogen leakage, impaired glymphatic clearance with protein accumulation (Aβ and tau), activated M1 microglia, complement component C1q deposition on synapses, and reactive astrocytes.
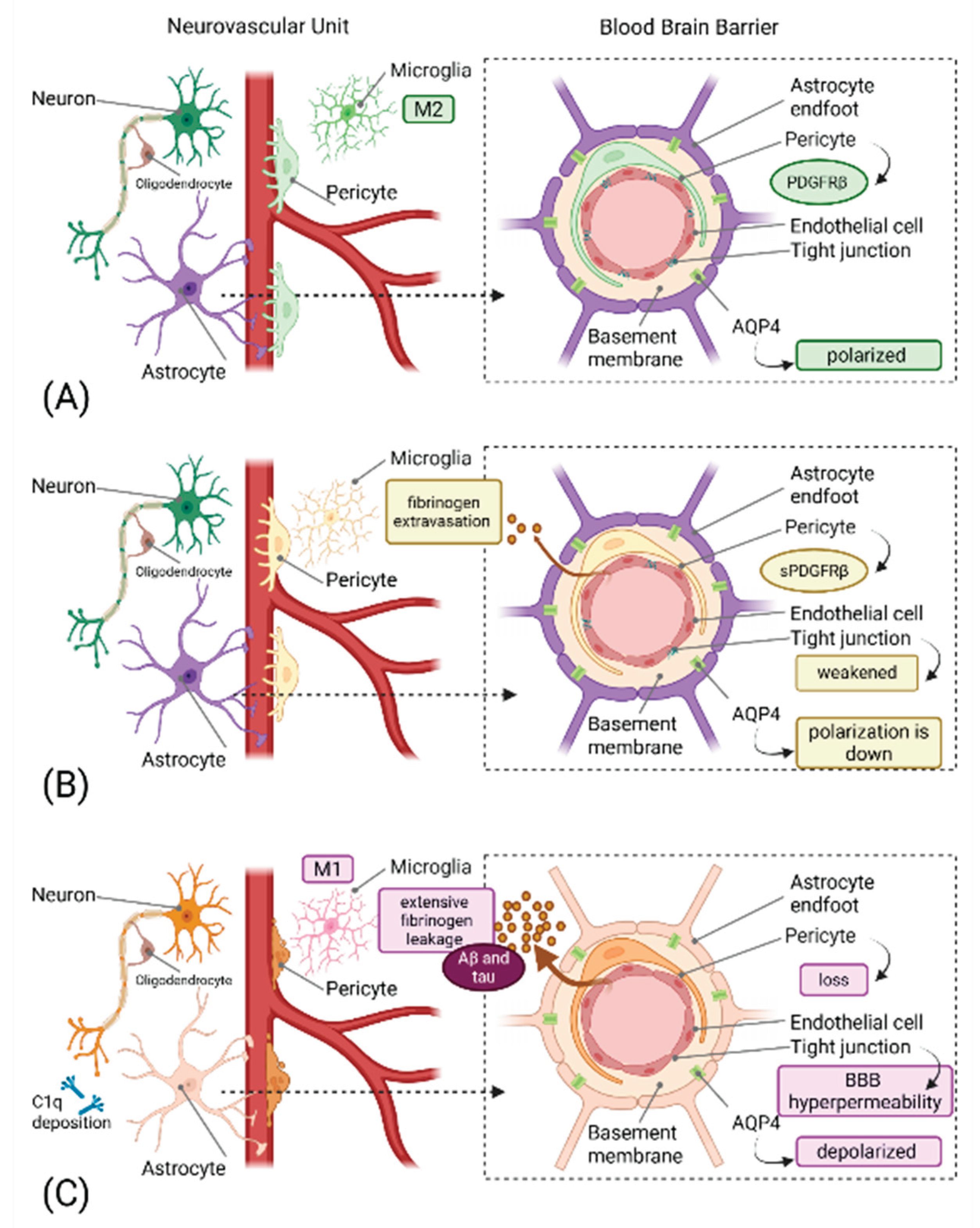

Table 2.
Emerging Therapeutic Targets for Neurovascular-Glymphatic Dysfunction.
| Therapeutic Target | Mechanism of Action | Preclinical Evidence | Proposed Therapeutic Approach | Potential Benefits | Challenges/Considerations | Key References |
| PDGF-BB/PDGFRβ Signaling | Maintains pericyte survival and BBB integrity via ERK and PI3K pathways | PDGFRβ+/- mice show accelerated BBB breakdown and neurodegeneration; restoration protects against vascular damage | PDGF-BB supplementation; prevention of PDGFRβ shedding; APOE4-targeted interventions | Preserves pericyte coverage; maintains BBB integrity; prevents early vascular damage | Timing critical; systemic effects; optimal dosing unclear | [35,78] |
| VEGF-C/VEGFR-3 Signaling | Enhances meningeal lymphatic vessel function and promotes lymphangiogenesis for brain waste clearance | VEGF-C administration in AD mice increases mLV diameter, reduces CSF and brain Aβ, restores cognition | Recombinant VEGF-C (intrathecal or systemic); VEGFR-3 agonists; transcranial radiofrequency stimulation | Enhances protein clearance; reduces tau and Aβ accumulation; improves cognitive function | Delivery route optimization; potential angiogenic effects; dose-finding needed | [30,31] |
| AQP4 Polarization Restoration | Restores proper localization of AQP4 at perivascular astrocytic endfeet to enhance glymphatic flow | Exercise and calmodulin inhibition restore AQP4 polarization and improve Aβ clearance in AD models | High-intensity interval training; aerobic exercise; calmodulin inhibitors (trifluoperazine); pharmacological AQP4 modulators | Enhances glymphatic clearance; reduces protein accumulation; improves waste removal | Exercise compliance; pharmacological specificity; avoiding edema | [79,80,81] |
| Complement C1q Inhibition | Blocks initiation of classical complement cascade; prevents C1q tagging of synapses for elimination | C1q deletion or neutralizing antibodies protect synapses and improve cognition in AD mouse models | Anti-C1q monoclonal antibodies; C1q inhibitor peptides; selective C1q blockers | Prevents excessive synaptic pruning; preserves cognitive function; reduces neuroinflammation | Balancing physiological vs pathological complement; immune surveillance concerns | [19,82,83] |
| Complement C3 Modulation | Prevents C3 cleavage and iC3b-mediated synaptic tagging; blocks complement amplification | C3 deficiency prevents age-related synapse loss and improves LTP in aged mice; protects against AD pathology | C3 inhibitors (compstatin analogs); C3 convertase inhibitors | Reduces synaptic loss; improves cognitive outcomes; maintains neuronal networks | Timing of intervention; systemic complement functions; infection risk | [18,84,85] |
| CR3 (CD11b/CD18) Blockade | Prevents microglial engulfment of iC3b-tagged synapses | CR3 knockout mice protected from Aβ-induced synapse loss; reduced microglial phagocytosis | CR3 antagonists; CD11b-blocking antibodies; small molecule inhibitors | Preserves synapses; reduces microglial-mediated damage; maintains circuit function | Microglial function preservation; specificity for pathological pruning | [19,20] |
| C5aR1 (C5a Receptor) Antagonism | Blocks C5a-mediated microglial activation; reduces excessive synaptic pruning | C5aR1 deletion or PMX205 treatment reduces synapse loss and improves cognition in multiple AD models | PMX205 or PMX53 (C5aR1 antagonists); small molecule C5aR1 inhibitors | Reduces synaptic loss; improves behavior; modulates neuroinflammation without blocking upstream complement | Better therapeutic window than C1q/C3 inhibition; preserves beneficial complement functions | [86,87,88] |
| miR-124 Replacement Therapy | Restores anti-inflammatory signaling; promotes M2 microglial polarization; inhibits inflammatory mediators | miR-124 overexpression reduces neuroinflammation and promotes neuroprotection in injury models | Lipid nanoparticle-encapsulated miR-124; viral vector delivery; synthetic miR-124 mimics | Shifts microglia to anti-inflammatory phenotype; reduces TNF-α; increases IL-10 | Delivery to CNS; off-target effects; stability of miRNA therapeutics | [27] |
| miR-155 Inhibition | Reduces pro-inflammatory signaling; decreases NF-κB activation; attenuates M1 microglial responses | miR-155 deletion improves outcomes in spinal cord injury and reduces neuroinflammation in MS models | AntagomiR-155; locked nucleic acid (LNA) anti-miR-155; GapmeR inhibitors | Reduces neuroinflammation; improves functional recovery; modulates TLR signaling | Delivery challenges; dosing optimization; potential immune effects | [29,89] |
| Meningeal Lymphatic Enhancement | Physical or pharmacological enhancement of mLV structure and function | Exercise enhances mLV flow; VEGF-C expands mLV diameter and improves clearance in aged mice | Aerobic exercise protocols; VEGF-C administration; minimally invasive mLV stimulation | Enhances brain-to-cervical lymph node drainage; improves clearance of proteins and immune cells | Age-related mLV degeneration; non-invasive enhancement methods needed | [30,90] |
| TREM2 Modulation | Regulates microglial phagocytic capacity and metabolic state; modulates complement-mediated pruning | TREM2 deficiency alters microglial response to plaques; affects synaptic engulfment | TREM2 agonistic antibodies; TREM2 activity enhancers (context-dependent) | Modulates microglial function; may enhance beneficial phagocytosis while reducing excessive pruning | Complex role (protective vs detrimental); stage-dependent effects | [91,92,93] |
| CD200-CD200R Axis Enhancement | Maintains microglial quiescence; promotes M2 polarization; reduces inflammatory activation | CD200-Fc treatment shifts macrophages/microglia from M1 to M2; reduces pro-inflammatory cytokines | CD200-Fc fusion protein; CD200R agonists | Reduces neuroinflammation; promotes neuroprotective microglial phenotype; decreases oxidative stress | Systemic delivery; CNS penetration; long-term safety | [94] |
Abbreviations: PDGF-BB, platelet-derived growth factor-BB; PDGFRβ, platelet-derived growth factor receptor-β; BBB, blood-brain barrier; ERK, extracellular signal-regulated kinase; PI3K, phosphatidylinositol 3-kinase; VEGF-C, vascular endothelial growth factor-C; VEGFR-3, vascular endothelial growth factor receptor-3; mLV, meningeal lymphatic vessels; Aβ, amyloid-β; AD, Alzheimer's disease; AQP4, aquaporin-4; CR3, complement receptor 3; C5aR1, C5a receptor 1; miR, microRNA; TNF-α, tumor necrosis factor-α; IL-10, interleukin-10; CNS, central nervous system; MS, multiple sclerosis; NF-κB, nuclear factor kappa B; TLR, toll-like receptor; TREM2, triggering receptor expressed on myeloid cells 2; LTP, long-term potentiation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



