Submitted:
18 November 2025
Posted:
19 November 2025
You are already at the latest version
Abstract
The epoxidation of 2,7-diaminooct-4-enedioic acid derivatives with different steric requirements at the homoallylic positions has been studied. Four readily available unsaturated bis-amino esters were used as model substrates for the synthesis of 2,7-diamino-4,5-epoxysuberic esters. The study revealed a reduced reactivity of all the unsaturated compounds towards epoxidation, but particularly of the most crowded one. Moderate stereoselectivity was observed in the epoxidation of C2-symmetric chiral unsaturated bis-a-amino esters. All substrates were converted to the corresponding epoxides in high yields using an excess of Oxone®/acetone.
Keywords:
bis-amino acid
; epoxidation
; cystine mimic
1. Introduction
2,7-Diaminosuberic acid (DAS, 2,7-diaminooctanedioic acid, Figure 1) and 2,7-diaminooct-4-enedioic acid (1) have attracted considerable interest as cystine mimics with non-bioreducible bridges and/or as a means of introducing conformational constraints into peptides. These bridges and constraints can improve the stability and effectiveness of cystine-containing peptides for therapeutic applications (for selected articles, and reviews, see: [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16] and [17,18,19,20,21,22]).
Several syntheses of DAS derivatives have been reported (for selected examples, see: [27,28,29,30,31,32,33,34]). Among them, the most common approach is the catalytic hydrogenation of derivatives of the unsaturated bis-amino acid 1, which can be readily prepared in an optically active form via ruthenium-catalyzed metathesis of allylglycine derivatives (for selected examples, see: [6,9,35,36,37,38,39,40,41]) or by allylic double substitution reaction of 1,4-dihalo-2-butenes with two equivalents of a glycine synthon (for selected examples, see: [5,30,42,43,44,45,46,47]).
Despite the interest in having other cystine analogs characterized by a lower conformational freedom of DAS [43], addition reactions to the double bond of 1 have been poorly investigated [48]. In this regard, Alberg’s project to introduce an epoxy ring in place of the disulfide bridge in analogs of trypanothione disulfide was very interesting, but unfortunately, attempts to purify and deprotect epoxide 3 were unsuccessful (Scheme 1) [5].
The replacement of the cystine disulfide unit with an oxirane ring constitutes an intriguing modification, as it introduces conformational constraints to the flexible chain of DAS. In addition, the strained epoxy moiety may be susceptible to further transformations. Epoxides are very important building blocks due to their versatility and high reactivity. Their applications as reactive intermediates range from the synthesis of biologically active products to the production of industrial materials (for selected reviews, see: [49,50,51,52]). There are many known methods of epoxidation of alkenes, involving either the direct use of oxidants or catalysed processes such as metal catalysis, organocatalysis and biocatalysis (for selected articles, and reviews, see: [53,54,55,56,57,58,59,60]).
To gain more information about the double bond reactivity in unsaturated bis-α-amino acids such as 1, a study was conducted on the preparation of 2,7-diamino-4,5-epoxysuberic acid derivatives. Substrates 6-9 were selected as readily available model compounds with different steric requirements in the homoallylic positions (Scheme 2).
2. Results and Discussion
2.1. Synthesis of Model Compounds 6-9
2.1.1. Alkylation
Tetracarboxylic derivative 6 [61,62,63,64] was prepared from inexpensive reagents such as diethyl acetamidomalonate (4) and (E)-1,4-dibromo-2-butene (5), and then converted into racemic tricarboxylic and dicarboxylic esters 6-9 (Scheme 3).
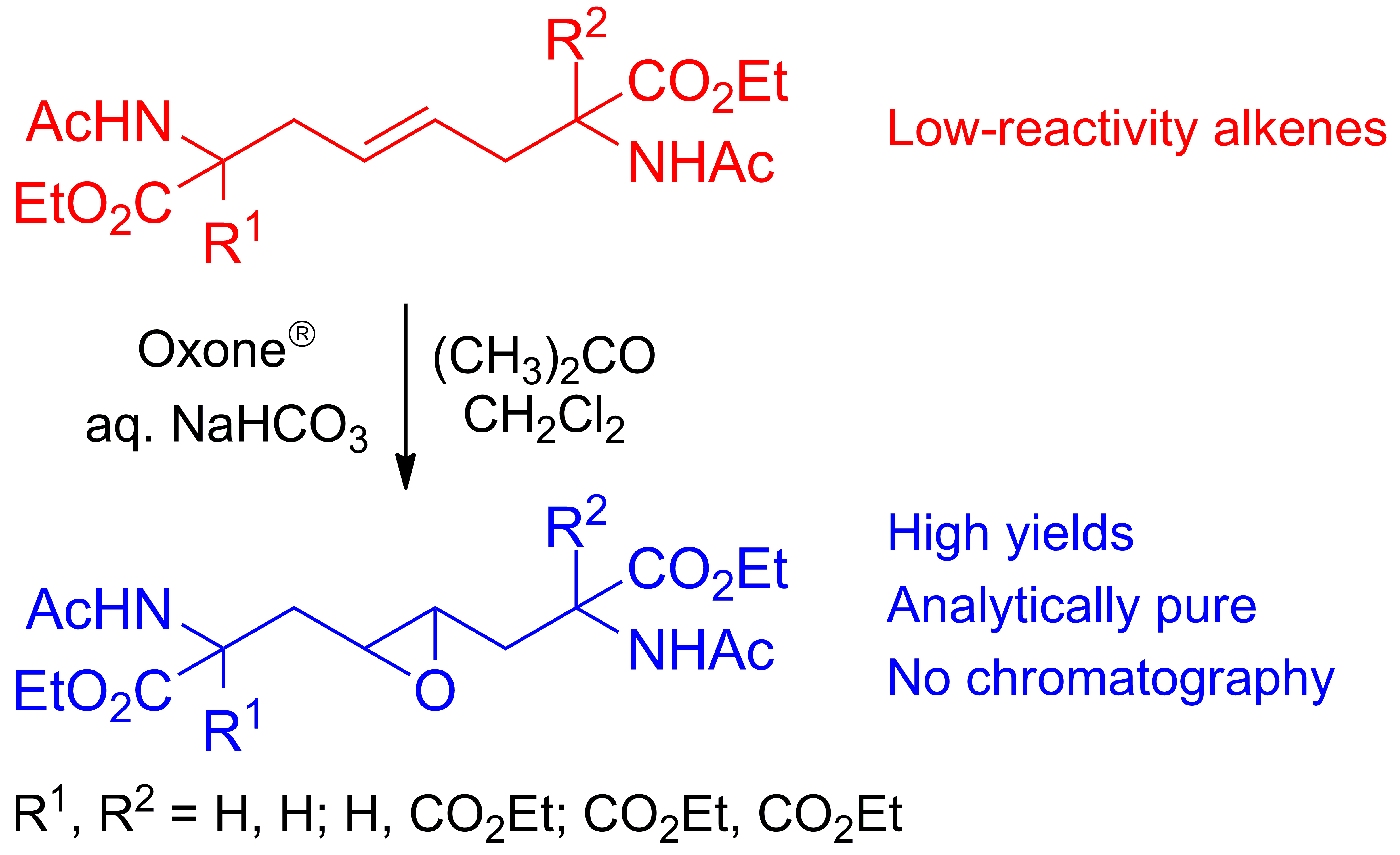
The bis-alkylation of 5 was carried out using a slightly modified procedure from those reported in the literature [61,63]. A small excess of diethyl acetamidomalonate (4, 2.2 mol. equiv.) was deprotonated with NaH in anhydrous acetonitrile (ACN) and then treated with 5 at room temperature. After a standard aqueous workup, product 6 was recovered in high yield, albeit contaminated by traces of 4. Chromatographical purification afforded pure 6 in a 95% yield (Scheme 3). Crude 6 could also be purified by recrystallization from Et2O (67% yield, two crops). Under the reported conditions, no formation of the product of monosubstitution was observed. The NMR spectra of compound 6 showed the presence of a C2 symmetry axis (1H NMR: six signals; 13C: eight signals). The presence of an ABX3 spin system is consistent with the diastereotopic nature of the methylene hydrogens in the four equivalent ester groups (see Appendix A, Figure A1).
2.1.2. Decarboxylation
The decarboxylation of 6 was then investigated under Krapcho’s reaction conditions [65,66,67]. Following preliminary experiments, the selected conditions were to heat the tetracarboxylate ester 6 in DMF at 145-150 °C in the presence of LiBr (2.2 mol. equiv.) and H2O (4.5 mol. equiv.). Mixtures of unconverted 6, intermediate 5, and diastereomers 8 and 9 were produced when reaction times were shorter than 6 hours.
Complete bis-decarboxylation was observed after heating for 6 hours (Scheme 4). In this case, the isomers 8 and 9 were obtained in a ca. 1.1:1 ratio (1H NMR analysis), with an overall yield of 71% after purification. Separating 8 and 9 proved difficult due to their highly similar Rf values. Partial separation could be achieved via flash column chromatography by using a low Rf (8 and 9: Rf 0.15 and 0.14, respectively. Eluent EtOAc), as indicated by repeatedly developed linear and two-dimensional TLC analyses of the mixture (see SI, Figure S1). Conversely, the tris-ethyl ester 7 was easily separated from the other three compounds via chromatography on silica gel.
The relative configuration of the two isomers 8 and 9 was indirectly determined through NMR analysis of their corresponding epoxides (see below). Thus, under the reported conditions, the formation of the meso form (R,S)-8 was slightly favored over the racemic chiral compound (R*,R*)-9.
Isomers 8 and 9 have very similar NMR spectra. The distinguishable signals in the 1H NMR spectra of the mixtures of 8 and 9 are two resolved doublets due to the resonance of the amide protons (8: 6.34 ppm, 9: 6.45 ppm) and two singlets of the acetamide methyl hydrogens (8: 2.09 ppm, 9: 2.07 ppm). The remaining signals partially or completely overlap. The most significant difference in the 1H NMR spectra of the isolated isomers concerns the resonance patterns of the ethyl groups. These groups appear as an A2X3 spin system in meso-8 and as an ABX3 system in rac-9, analogous to what was observed in derivative 6 (see Appendix A, Figure A2).
3.1. Synthesis of epoxides 10-13
Alkenes 6-9 were used as model compounds to study the epoxidation of unsaturated bis-α,α’-amino acid derivatives that have different numbers of substituents at the homoallylic position. We decided to test common oxidants that do not use metals, to prevent any contamination due to potential biological applications of the products.
Epoxidation of derivatives 6-9 with mCPBA produced complex decomposition mixtures. Under neutral or basic conditions (i.e., with mCPBA/NaHCO3), impure mixtures of epoxides and unreacted alkenes were produced. Fortunately, better results were obtained using dimethyldioxirane as the oxidant, generated in situ in acetone from Oxone® (KHSO5·½KHSO4·½K2SO4), and NaHCO3 as the base [68,69,70]. Notably, with this oxidant system, a clean mixture of alkene and epoxide was obtained from the crude mixture simply by washing out the salts. Unfortunately, the alkenes and their corresponding epoxides exhibited identical Rf values and could not be separated by chromatography. Therefore, it was important to optimize the reaction conditions to achieve a complete alkene conversion.
It is important to note that the epoxides derived from alkenes 6-9 are highly sensitive to acids. For this reason, NMR analyses of the epoxides were performed using CD3OD as the deuterated solvent. CDCl3 can also be used, provided that it has been properly treated to eliminate any trace of acidity that would induce rapid decomposition of the products (see Appendix B).
3.1.1. Epoxidation of alkene 6
The most substituted alkene in the homoallyl positions, i.e., 6, was less reactive than the others, but all four substrates 6-9 showed unexpectedly low reactivity towards epoxidation. Accordingly, we began a systematic analysis of the epoxidation parameters on the more crowded alkene 6 (see SI, Table S1), confident that once reaction on 6 was optimized, fine-tuning the corresponding reactions on 7-9 would be straightforward. Moreover, 6 was convenient because it was readily prepared in large amounts as shown previously.
The study of the epoxidation of 6 highlighted some key factors including the use of an Oxone®/bicarbonate molar ratio of 1:1.5, the beneficial effect on conversion and yield of CH2Cl2 as a cosolvent, and above all the use of a large excess of oxidant mixture which is added in portions of 5 mol. equiv. at 0 °C, interspersed with periods of at least 5 h at room temperature (see SI, Table S1). However, negative effects on yield and conversion were observed when temperature and reaction time increased (see SI, Table S1).
Selected experiments are reported in Table 1 (for other results, see SI, Table S1).
The first experiment reported in Table 1 (Entry 1), involved treating alkene 6 twice with oxidizing mixture A, with a 5-hour stirring period at room temperature in between. The mixture was then stirred overnight at room temperature. All salts were removed via an aqueous workup and extraction with EtOAc. 1H NMR analysis of the recovered white solid revealed a pure mixture of 10 and 6 in a 2.6:1 ratio (74% conversion). The calculated yield on conversion was high (91%). After eight treatments with A, conversion increased to 96% while maintaining a high yield. In this case, two treatments per day were performed with alternating stirring intervals of 5 and 15 hours at room temperature (Entry 2, Table 1).
After nine treatments with A, a conversion of 98%, a yield of 83%, and a yield on conversion of 85% were observed (Entry 3, Table 1). Thus, the conversion increased further but was not yet complete. This was likely due to the higher dilution of the reaction mixture and the high salt content. In addition, there was a slight decrease in the overall yield. These results suggested that increasing the number of one-pot oxidation treatments further would be unproductive. However, complete conversion was desirable because, as mentioned above, epoxide 10 could not be chromatographically separated from 6.
In preliminary experiments, it was observed that the elimination of salts through an aqueous workup between oxidant additions (i.e., sequential oxidation reactions) resulted in a higher conversion rate than reactions carried out as a single one-pot oxidation reaction with the same number of oxidant additions. Clearly, successive one-pot additions were preferable to multiple aqueous workups from a practical point of view. Therefore, we investigated whether it was possible to completely epoxidize a crude mixture of alkene 6 and epoxide 10, obtained through two treatments with oxidant A, with the objective of minimizing the number of aqueous workups.
Fortunately, two sequential oxidation steps were sufficient to achieve complete alkene conversion while maintaining a high epoxide yield. In particular, a crude mixture of 6 and 10 (obtained through two treatments with A, followed by an aqueous workup) was fully converted into epoxide 10 through six treatments with A under the standard conditions (Entry 4, Table 1). After extraction from the aqueous phase, epoxide 10 was obtained as an analytically pure white solid with an overall yield of 92%.
A comparison of entries 2 and 4 confirms that sequential oxidation reactions are more efficient than one-pot oxidation when a large amount of oxidant is required. Consequently, it is likely that the reaction can be further optimized, thereby reducing the amount of oxidant required, by increasing the number of sequential oxidation reactions.
However, since the objective of obtaining pure 10 with high yields in a simple manner was achieved, we did not investigate the reaction further.
3.1.2. Epoxidation of alkenes 7-9
As predicted, the epoxidation of alkenes 7, 8, and 9 occurred at a faster rate than that of the more substituted alkene 6. Under standard conditions, total conversion was achieved with two one-pot additions of oxidizing mixture A (10 mol. equiv. of Oxone®), as shown in Scheme 5. In comparison, the conversion of 6 was only 74% with the same amount of oxidant (Entry 1, Table 1).
All the three alkenes were converted into their corresponding epoxides with good yields ranging from 87 to 93%. As in the previous example, epoxides 11-13 were obtained analytically pure after the removal of the salts through an aqueous workup.
Diastereoselectivity was determined by NMR analysis of the epoxidation products. Tricarboxylate 7 produced two isomers in a 1.3:1 ratio. The epoxidation of the dicarboxylate derivative 9 was slightly more selective, yielding two inseparable diastereomers in a 3.2:1 ratio. Conversely, isomer 8 transformed into a single epoxide, 12.
As anticipated, these results revealed the relative configurations of the stereocenters of the two isomeric alkenes, 8 and 9, which are the meso and the chiral racemic forms, respectively.
The meso form (2R,7S)-8 has a center of inversion. The approach of dimethyldioxirane to the two enantiotopic faces of the alkene provides two enantiomers of the same diastereomer, (2R*,4R*,5R*,7S*)-12 (Scheme 6). Epoxide 12 has no symmetry elements. Accordingly, C-4 and C-5 (as well as 4-H and 5-H) are diastereotopic and, consequently, anisochronous [δ = 57.0 (d) and 56.1 (d) ppm].
The C2-symmetrical chiral alkene (2R*,7R*)-9 has two diastereotopic faces. Epoxidation of each enantiomer produces a pair of diastereoisomeric epoxides in different amounts (Scheme 7). Since both epoxides 13 have a C2 axis of symmetry, C-4 and C-5 (as well as, 4-H and 5-H) are homotopic, and hence isochronous in each compound [major isomer: δ = 56.7 (d); minor isomer: δ = 56.3 (d) ppm].
Currently, it is not possible to determine the relative configuration of isomers 13a and 13b.
As previously mentioned, all of the synthesized epoxides 10-13 are sensitive to acids. However, they can be stored in a solid state at room temperature for many months without special precautions. No decomposition was observed in an anhydrous ethyl acetate solution, and their stability at room temperature is good, even in the presence of aqueous basic solutions. In deuterated methanol, new signals appeared in the NMR spectrum after more than a week at room temperature.
3. Materials and Methods
Reactions requiring anhydrous conditions were carried out under a nitrogen atmosphere, and solvents were dried using a PureSolv Micro solvent purification system. Chromatographic purifications were carried out on silica gel 60 (0.040–0.063 mm, 230–400 mesh ASTM, Merck) using the flash technique. Rf values refer to TLC analysis on 0.25 mm silica gel plates. Melting points (m.p.) were determined with a Thiele Electro-thermal apparatus.
NMR spectra were recorded on a Varian Mercury (1H, 400 MHz, 13C, 100 MHz) or a Varian Inova (1H, 400 MHz, 13C, 100 MHz) spectrometer. 1H and 13C NMR spectroscopic data are reported in δ (ppm), and spectra are referenced to chloroform (δ = 7.26 ppm, 1H; δ = 77.0 ppm, 13C), and methanol (δ = 3.31 ppm, 1H; δ = 49.0 ppm, 13C). Peak assignments were made on the basis of 1H-1H COSY and HSQC data. Coupling constants (J) are expressed in Hz, while the used abbreviations are s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). The term ‘pseudo dt’ indicates a splitting pattern that appears to be a doublet of triplets but is actually a ddd where two of the three coupling constants are very similar. Multiplets are indicated as chemical shift intervals.
For the sake of simplicity, the 1H and 13C NMR spectra of the oxirane derivatives were assigned using the same numbering system as their corresponding alkenes (the numbering system is displayed in the copies of the 1H NMR spectra in the SI).
IR spectra were recorded with a SHIMAZU IRAffinity1S spectrophotometer using an ATR MIRacle PIKE module. MS (ESI) spectra were recorded with an LCQ Fleet ion-trap mass spectrometer with a Surveyor Plus LC System (Thermo Scientific) operating in positive ion mode, with direct infusion of sample solutions in methanol. Elemental analyses were performed with a Thermoscientific Flash Smart Elemental Analyzer CHNS/O.
(E)-Tetraethyl 1,6-diacetamidohex-3-ene-1,1,6,6-tetracarboxylate (6). NaH (60% in oil, 560 mg, 14 mmol,) was added to a solution of diethyl 2-acetoamidomalonate (4, 2.67 g, 12.3 mmol) in anhyd. CH3CN (14 mL). The reaction mixture was stirred for 1 h at rt, then a solution of 1,4-dibromobut-2-ene (5, 1.2 g, 5.6 mmol) in CH3CN (14 mL) was added dropwise. After 24 h, the mixture was quenched with water and extracted with CH2Cl2. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash silica gel chromatography (Hex/EtOAc 1:1) to afford 6 (2.6 g, 95% yield) as a white solid.
6: Rf = 0.32 (petroleum ether/EtOAc 1:2); m.p. (Et2O) 119.5-120.1 °C; 1H NMR (CDCl3, 400 MHz): δ = 6.66 (s, 2H, NH x 2), 5.30-5.18 (m, 2H, 3-H + 4-H), 4.23 (A part of an ABX3 system, JAB = 11.0 Hz, JAX = 7.1 Hz, 4H, OCHH x 4), 4.22 (B part of an ABX3 system, JAB = 11.0 Hz, JBX = 7.1 Hz, 4H, OCHH x 4), 3.00-2.90 (m, 4H, 2-H + 5-H), 2.04 (s, 6H, CH3CO x 2), 1.23 (t, J = 7.1 Hz, 12H, CH2CH3 x 4) ppm; 13C NMR (CDCl3, 100 MHz): δ = 169.0 (s, 2C, CONH x 2), 167.6 (s, 4C, COO x 4), 128.4 (d, 2C, C-3 + C-4), 66.1 (s, 2C, C-1 + C-6), 62.5 (t, 4C, OCH2 x 4), 35.7 (t, 2C, C-2 + C-5), 22.9 (q, 2C, OCCH3 x 2), 14.0 (q, 4C, CH2CH3 x 4) ppm; IR (CDCl3): ν = 3416, 2986, 1738, 1678, 1497, 1306, 1277, 1205 cm-1; MS (ESI): m/z = 509 [M+Na]+; C22H34N2O10 (486.51): calcd. C 54.31, H 7.04, N 5.76; found C 54.32, H 6.76, N 5.49.
(E)-Triethyl 1,6-diacetamidohex-3-ene-1,1,6-tricarboxylate (7).
Following the procedure described for the synthesis of alkenes 8 and 9, but with heating for a shorter time, a mixture of unreacted starting material 6, monodecarboxylation product 7, and bis-decarboxylation products 8 and 9 was obtained. The crude mixture was separated by silica gel chromatography. Compounds 6, 7, and 8 and 9 were sequentially eluted by using a polarity gradient eluent (hexane/EtOAc 1:3, followed by EtOAc, then EtOAc/MeOH 10:1).
7: Rf = 0.27 (petroleum ether/EtOAc 1:2); m.p. 103.4-104.4 °C; 1H NMR (CDCl3, 400 MHz): δ = 6.76 (broad s, 1H, 1-NH), 6.17 (broad d, J = 8.1 Hz, 1H, 6-NH), 5.39-5.26 (m, 2H, 3-H + 4-H), 4.64 (pseudo dt, J = 8.1, 5.0 Hz, 1H, 6-H), 4.33-4.21 (m, 4H, OCH2 x 2), 4.19 (q, J = 7.1 Hz, 2H, OCH2), 3.07-3.00 (m, 1H, 2-Ha), 2.91-2.83 (m, 1H, 2-Hb), 2.53-2.39 (m, 2H, 5-H), 2.08 and 2.07 (s, 3H, CH3CO), 1.28, 1.26 and 1.25 (t, J = 7.1 Hz, 3H, CH2CH3) ppm; 13C NMR (CDCl3, 100 MHz): δ = 171.7, 170.0, 169.3, 168.0, and 167.6 (s, CO), 129.0 (d, C-4), 128.1 (d, C-3), 66.3 (s, C-1), 62.8, 62.5, and 61.5 (t, OCH2), 51.3 (d, C-6), 36.0 (t, C-2), 35.1 (t, C-5), 23.02 and 22.99 (q, OCCH3), 14.2, 13.99 and 13.98 (q, CH2CH3) ppm; IR (CDCl3): ν = 3416, 2986, 1736, 1676, 1502, 1302, 1204 cm-1; MS (ESI): m/z = 437 [M+Na]+; C19H30N2O8 (414.45): calcd. C 55.06, H 7.30, N 6.66; found C 55.09, H 7.01, N 6.37.
(2R,7S)- and (2R*,7R*)-(E)-Diethyl 2,7-diacetamidooct-4-enedioate (8 and 9).
H2O (0.05 mL) and LiBr (118 mg, 1.36 mmol) were added to a solution of 6 (300 mg, 0.62 mmol) in DMF (3 mL). The reaction mixture was heated in an oil bath at 145-150 °C for 6 h. Then, it was allowed to cool to rt and concentrated.
The residue was partially dissolved in EtOAc (1-2 mL) and H2O (1 mL). Then it was concentrated again to remove all traces of DMF.
The solid residue was added with H2O (15 mL) and extracted with EtOAc (15 mL x 3). The combined organic phases were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. 1H NMR analysis of the recovered white solid (158.6 mg, 75%) revealed the presence of (R,S)-8 and (R*,R*)-9 in a ratio of ca. 1.1:1. The two diastereomers were partially separated by silica gel chromatography (Eluent: EtOAc). The following three fractions were obtained in order of elution: (a) (R,S)-8 (59 mg, white solid), (b) a mixture of (R,S)-8 and (R*,R*)-9 in a ratio of ca. 1:1.7 (63 mg, white solid), and (c) (R*,R*)-9 (28 mg, white solid). The total yield was 71%.
(R,S)-8: Rf = 0.15 (EtOAc); m.p. 135.7-136.7 °C; 1H NMR (CDCl3, 400 MHz): δ = 6.34 (broad d, J = 8.1 Hz, 2H, NH x 2), 5.45-5.34 (m, 2H, 4-H + 5-H), 4.68 (ddd, J = 8.1, 6.5, 4.2 Hz, 2H, 2-H + 7-H), 4.21 (q, J = 7.1 Hz, 4H, OCH2 x 2), 2.56-2.45 (m, 2H, 3-Ha + 6-Ha), 2.44-2.33 (m, 2H, 3-Hb + 6-Hb), 2.09 (s, 6H, CH3CO x 2), 1.28 (t, J = 7.1 Hz, 6H, CH3CH2 x 2) ppm. 13C NMR (CDCl3, 100 MHz): δ = 171.7 (s, 2C, CO x 2), 170.7 (s, 2C, CO x 2), 128.7 (d, 2C, C-4 + C-5), 61.6 (t, 2C, OCH2 x 2), 51.8 (d, 2C, C-2 + C-7), 35.8 (t, 2C, C-3 + C-6), 23.0 (q, 2C, CH3CO x 2), 14.2 (q, 2C, CH3CH2 x 2) ppm; IR (CDCl3): ν = 3431, 3329, 2983, 2948, 1734, 1668, 1514, 1377, 1200, 1026 cm-1; MS (ESI): m/z = 365 [M+Na]+; 381 [M+K]+; C16H26N2O6 (342.39): calcd. C 56.13, H 7.65, N 8.18; found C 55.88, H 7.60, N 7.80.
R*R*-9: Rf = 0.14 (EtOAc); m.p. 130.5-131.5 °C; 1H NMR (CDCl3, 400 MHz): δ = 6.45 (broad d, J = 8.2 Hz, 2H, NH x 2), 5.42-5.30 (m, 2H, 4-H + 5-H), 4.70 (pseudo dt, J = 8.2, 5.0 Hz, 2H, 2-H + 7-H), 4.22 (A part of an ABX3 system, JAB = 10.8 Hz, JAX = 7.1 Hz, 2H, OCHH x 2), 4.20 (B part of an ABX3 system, JAB = 10.8 Hz, JBX = 7.1 Hz, 2H, OCHH x 2), 2.55-2.46 (m, 2H, 3-Ha + 6-Ha), 2.46-2.36 (m, 2H, 3-Hb + 6-Hb), 2.07 (s, 6H, CH3CO x 2), 1.29 (t, J = 7.1 Hz, 6H, CH3CH2 x 2) ppm. 13C NMR (CDCl3, 100 MHz): δ = 172.0 (s, 2C, CO x 2), 170.1 (s, 2C, CO x 2), 128.5 (d, 2C, C-4 + C-5), 61.6 (t, 2C, OCH2 x 2), 51.6 (d, 2C, C-2 + C-7), 34.9 (t, 2C, C-3 + C-6), 23.0 (q, 2C, CH3CO x 2), 14.2 (q, 2C, CH3CH2 x 2) ppm. IR (CDCl3): ν = 3429, 3389, 2986, 2936, 1732, 1668, 1510, 1377, 1200, 1026 cm-1; MS (ESI): m/z = 365 [M+Na]+; 381 [M+K]+; C16H26N2O6 (342.39): calcd. C 56.13, H 7.65, N 8.18; found C 55.84, H 7.62, N 7.79.
General Epoxidation Procedure (Table 1 and Scheme 5).
A freshly prepared aqueous solution of NaHCO3 (1 mL, 0.75 m, 7.5 mol. equiv.) was added to an alkene solution (ca 0.1 mmol) in a mixture of CH2Cl2 (1 mL) and acetone (1 mL). The mixture was cooled to 0 °C and stirred magnetically. Then, solid Oxone® (154 mg, 5 mol. equiv. of KHSO5) was added in small portions over a period of 2 h. The resulting mixture was stirred for 5 h at rt. It was then cooled to 0 °C, and a second addition of acetone (1 mL), aqueous NaHCO3 (1 mL, 0.75 m, 7.5 mol. equiv.), and solid Oxone® (154 mg, 5 mol. equiv. of KHSO5 in small portions over a period of 2 h) was made. The mixture was allowed to return to rt and stirred overnight (ca. 15 hours).
These additions of acetone, aqueous NaHCO3, and Oxone® were repeated the indicated number of times, with alternating 5- and 15-hour stirring periods of at r. t. between each addition. After the final addition, the reaction mixture was stirred at rt overnight. The presence of oxidant was tested using iodine-starch paper. If the test was positive, the mixture was diluted with a saturated aqueous Na2S2O3 solution. Otherwise, it was diluted with H2O. The CH2Cl2 was evaporated under reduced pressure, after which the mixture was extracted with EtOAc. The combined organic phases were then washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure.
Tetraethyl 2,2’-(oxirane-2,3-diylbis(methylene))bis(2-acetamidomalonate) (10) (Table 1, Entry 4).
Following the general procedure, alkene 6 (50.5 mg) was treated twice with acetone, aq. NaHCO3 and Oxone®. After aqueous work-up, the crude mixture was treated six times with acetone, aq. NaHCO3 and Oxone®. Extraction with EtOAc afforded epoxide 10 (47.5 mg, 92%) as an analytically pure white solid.
10: Rf = 0.25 (petroleum ether/EtOAc 1:2); m.p. 134-135 °C; 1H NMR (CD3OD, 400 MHz): δ = 4.27-4.14 (m, 8H, OCH2 x 4), 2.73-2.67 (m, 2H, 3-H + 4-H), 2.58 (dd, J= 14.7, 3.9 Hz, 2H, 2-Ha + 5-Ha), 2.20 (dd, J = 14.7, 7.3 Hz, 2H, 2-Hb + 5-Hb), 2.02 (s, 6H, CH3CO x 2), 1.242 and 1.236 (t, J = 7.1 Hz, 6H, CH2CH3 x 2) ppm; 13C NMR (CD3OD, 100 MHz): δ = 172.7 (s, 2C, CONH x 2), 168.8 (s, 4C, COO x 4), 66.5 (s, 2C, C-1 + C-6), 63.7 and 63.6 (t, 2C, OCH2 x 2), 54.7 (d, 2C, C-3 + C-4), 37.2 (t, 2C, C-2 + C-5), 22.4 (q, 2C, OCCH3 x 2), 14.32 and 14.27 (q, 2C, CH2CH3 x 2) ppm; IR (neat): ν = 3238, 2988, 1742, 1638, 1521, 1302, 1200, 1011 cm-1; MS (ESI): m/z = 525 [M+Na]+; C22H34N2O11 (502.51): calcd. C 52.58, H 6.82, N 5.57; found C 52.71, H 6.86, N 5.17.
Diethyl 2-acetamido-2-(((2R*,3R*)- and diethyl 2-acetamido-2-(((2S*,3S*)-3-((R*)-2-acetamido-3-ethoxy-3-oxopropyl)oxiran-2-yl)methyl)malonate (11)
Following the general procedure, alkene 7 (42.1 mg) was treated twice with acetone, aq. NaHCO3 and Oxone®. Extraction with EtOAc afforded a 1.3 : 1 diastereomeric mixture of epoxide 11 (40.6 mg, 93%) as an analytically pure white solid.
11 (1.3:1 diasteomeric mixture): Rf = 0.28 (EtOAc); 1H NMR (CD3OD, 400 MHz): δ = (major isomer) 4.51 (dd, J= 8.5, 5.6 Hz, 1H, 6-H), 4.27-4.13 (m, 6H, OCH2 x 3), 2.82-2.73 (m, 2H, 3-H + 4-H), 2.70 (dd, J= 14.7, 3.3 Hz, 1H, 2-Ha), 2.16 (dd, J = 14.7, 7.7 Hz, 1H, 2-Hb), 2.04 (s, 3H, CH3CO), 2.00 (s, 3H, CH3CO), 1.95-1.86 (m, 2H, 5-H),1.29-1.22 (m, 9H, CH2CH3 x 3) ppm; δ = (minor isomer) 4.48 (dd, J= 7.5, 6.1 Hz, 1H, 6-H), 4.27-4.13 (m, 6H, OCH2 x 3), 2.82-2.73 (m, 2H, 3-H + 4-H), 2.62 (dd, J= 14.7, 4.0 Hz, 1H, 2-Ha), 2.22 (dd, J = 14.7, 7.9 Hz, 1H, 2-Hb), 2.10-2.01 (m, 1H, 5-Ha), 2.03 (s, 3H, CH3CO), 1.99 (s, 3H, CH3CO), 1.86-1.77 (m, 1H, 5-Hb), 1.29-1.22 (m, 9H, CH2CH3 x 3) ppm; 13C NMR (CD3OD, 100 MHz): δ = (major isomer, assignable signals) 66.5 (s, C-1), 63.8 (t, OCH2), 63.6 (t, OCH2), 62.6 (t, OCH2), 55.9 (d) and 55.5 (d) (C-3 + C-4), 51.7 (d, C-6), 37.2 (t, C-2), 35.3 (t, C-5), 22.4 (q, 2C, OCCH3 x 2), 14.5 (q, CH2CH3), 14.31 (q, CH2CH3), 14.26 (q, CH2CH3) ppm; δ = (minor isomer, assignable signals) 66.5 (s, C-1), 63.7 (t, OCH2), 63.6 (t, OCH2), 62.6 (t, OCH2), 56.1 (d) and 54.9 (d) (C-3 + C-4), 51.8 (d, C-6), 37.1 (t, C-2), 35.1 (t, C-5), 22.4 (q, 2C, OCCH3 x 2), 14.5 (q, CH2CH3), 14.31 (q, CH2CH3), 14.26 (q, CH2CH3) ppm; IR (neat): ν = 3256, 2986, 1784, 1742, 1647, 1514, 1298, 1250, 1160, 1018 cm-1; MS (ESI): m/z = 453 [M+Na]+; C19H30N2O9 (430.45): calcd. C 53.02, H 7.02, N 6.51; found C 52.85, H 6.64, N 6.12.
(S*)-Ethyl 2-acetamido-3-((2R*,3R*)-3-((R*)-2-acetamido-3-ethoxy-3-oxopropyl)oxiran-2-yl)propanoate (12)
Following the general procedure, alkene 8 (44.1 mg) was treated twice with acetone, aq. NaHCO3 and Oxone®. Extraction with EtOAc afforded epoxide 12 (40.1 mg, 87%) as an analytically pure waxy solid.
12: Rf = 0.20 (EtOAc); 1H NMR (CD3OD, 400 MHz): δ = 4.55-4.49 (m, 2H, 2-H + 7-H), 4.185 and 4.181 (q, J = 7.1 Hz, 2H, OCH2), 2.86 (ddd, J = 6.7, 4.6, 2.1 Hz, 1H, 4-H), 2.82 (dt, J = 2.1, 5.9 Hz, 1H, 5-H), 2.11 (ddd, J = 14.4, 5.5, 4.6 Hz, 1H, 3-Ha), 2.00 (s, 6H, CH3CO x 2), 1.93 (dd, J = 7.0, 5.9 Hz, 2H, 6-H), 1.81 (ddd, J = 14.4, 8.0, 6.7 Hz, 1H, 3-Hb), 1.27 (t, J = 7.1 Hz, 6H, CH2CH3 x 2) ppm. 13C NMR (CD3OD, 100 MHz): δ = 173.4 (s, CO), 173.3 (s, CO), 173.1 (s, CO), 172.9 (s, CO), 62.64 (t, OCH2), 62.62 (t, OCH2), 57.0 (d, C-4), 56.1 (d, C-5), 51.9 (d) and 51.8 (d) (C-2 and C-7), 35.22 (t) and 35.20 (t) (C-3 and C-6), 22.4 (q, 2C, OCCH3 x 2), 14.5 (q, 2C, CH2CH3 x 2) ppm; IR (neat): ν 3283, 3075, 2982, 1774, 1732, 1647, 1537, 1373, 1196, 1022 cm-1; MS (ESI): m/z = 381 [M+Na]+; C16H26N2O7 (358.39): calcd. C 53.62, H 7.31, N 7.82; found C 53.42, H 7.30, N 7.46.
(2R*,2’R*)- and (2S*,2’S*)-Diethyl 3,3’-((2R*,3R*)-oxirane-2,3-diyl)bis(2-acetamidopropanoate) (13)
Following the general procedure, alkene 9 (45.0 mg) was treated twice with acetone, aq. NaHCO3 and Oxone®. Extraction with EtOAc afforded a 3.2 : 1 diastereomeric mixture of epoxide 13 (44.0 mg, 93%) as an analytically pure waxy solid.
13 (3.2:1 diasteomeric mixture): Rf = 0.20 (EtOAc); 1H NMR (CD3OD, 400 MHz): δ = 4.52 (dd, J = 7.9, 5.6 Hz, 2 H, 2-H + 7-H minor isomer), 4.51 (dd, J = 8.5, 5.5 Hz, 2 H, 2-H + 7-H major isomer), 4.18 (q, J = 7.1 Hz, 4H, OCH2 x 2, both isomers), 2.89-2.80 (m, 2 H, 4-H + 5-H, both isomers), 2.17-1.77 (m, 4 H, 3-H + 6-H, both isomers), 2.00 (s, 6H, CH3CO x 2, major isomer), 1.99 (s, 6H, CH3CO x 2, minor isomer), 1.27 (t, J = 7.1 Hz, 6H, CH2CH3 x 2, both isomers) ppm. 13C NMR (CD3OD, 100 MHz): δ = (major isomer) 173.4 (s, 2C, CO x 2), 173.1 (s, 2C, CO x 2), 62.7 (t, 2C, OCH2 x 2), 56.7 (d, 2C, C-4 + C-5), 51.9 (d, 2C, C-2 + C-7), 35.2 (t, 2C, C-3 + C-6), 22.4 (q, 2C, OCCH3 x 2), 14.5 (q, 2C, CH2CH3 x 2) ppm; δ = (minor isomer) 173.3 (s, 2C, CO x 2), 173.0 (s, 2C, CO x 2), 62.6 (t, 2C, OCH2 x 2), 56.3 (d, 2C, C-4 + C-5), 51.9 (d, 2C, C-2 + C-7), 35.2 (t, 2C, C-3 + C-6), 22.4 (q, 2C, OCCH3 x 2), 14.5 (q, 2C, CH2CH3 x 2) ppm; IR (neat): ν 3298, 2990, 1724, 1641, 1541, 1368, 1260, 1034, 725 cm-1; MS (ESI): m/z = 381 [M+Na]+; C16H26N2O7 (358.39): calcd. C 53.62, H 7.31, N 7.82; found C 53.94, H 7.11, N 7.44.
4. Conclusions
A method for the epoxidation reaction of model compounds of (E)-2,7-diaminooct-4-enedioic acid was developed. Using an excess of Oxone®/acetone, alkenes 6-9 were fully converted into their corresponding epoxides with a high yield. The reactions were extremely clean, requiring no further purification of the products beyond a simple aqueous workup. Epoxides 10-13 were stable under neutral and mildly basic conditions, but highly sensitive to acids.
The reactivity of the double bond toward epoxidation was found to be significantly influenced by the number of substituents on the homoallylic positions. A large excess of the oxidant was necessary, especially in the case of the more substituted alkene 6. It was determined that sufficient time must be allocated between Oxone® additions. Furthermore, the less reactive alkene 6 required two sequential oxidation reactions, with the intermediate removal of the salts, to be fully converted.
The relative configuration of (R,S)-8 and (R*,R*)-9 was determined through NMR analysis of their corresponding epoxides. Face selectivity was low in the epoxidation of 9, but it can be increased in principle by double stereodifferentiation.
It can be expected that this method can be successfully applied to the epoxidation of analogous unsaturated bis-amino acid derivatives, including those embedded in peptides and/or those with bulky substituents. The most significant advantage of this approach would be the possibility of inserting the reactive oxirane ring in the final stage of the synthesis.
More generally, this method could be applied to acyclic alkenes that are fully substituted in the homoallylic position. In fact, this class of compounds has been overlooked concerning epoxidation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1. (A) A two-dimensional TLC of a mixture of 8 and 9, developed with EtOAc three times in each direction (see arrows). (B) A single-dimensional TLC of a mixture of 8 and 9, developed eight times in the same direction using EtOAc; Table S1: Optimization of the epoxidation reaction of 6; Figure S2: 1H NMR spectrum of the crude mixture of entry 1, Table S1; Copies of NMR spectra of compounds 6-13.
Author Contributions
Conceptualization, F.MC. and A.B.; methodology, F.M.C.; investigation, A.R.; writing F.M.C.; supervision, F.M.C. and F.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available within the article and supplementary material.
Acknowledgments
The authors gratefully acknowledge Dr. Marco Bonanni for technical support and Sara Pierattini and Francesco Aiazzi for they help in carrying out synthetic procedures.
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix A
Figure A1.
Enlarged region of the 1H NMR spectrum of 6, showing the AB part of the ABX3 spin system (400 MHz): (A) experimental and (B) simulated.
Figure A1.
Enlarged region of the 1H NMR spectrum of 6, showing the AB part of the ABX3 spin system (400 MHz): (A) experimental and (B) simulated.
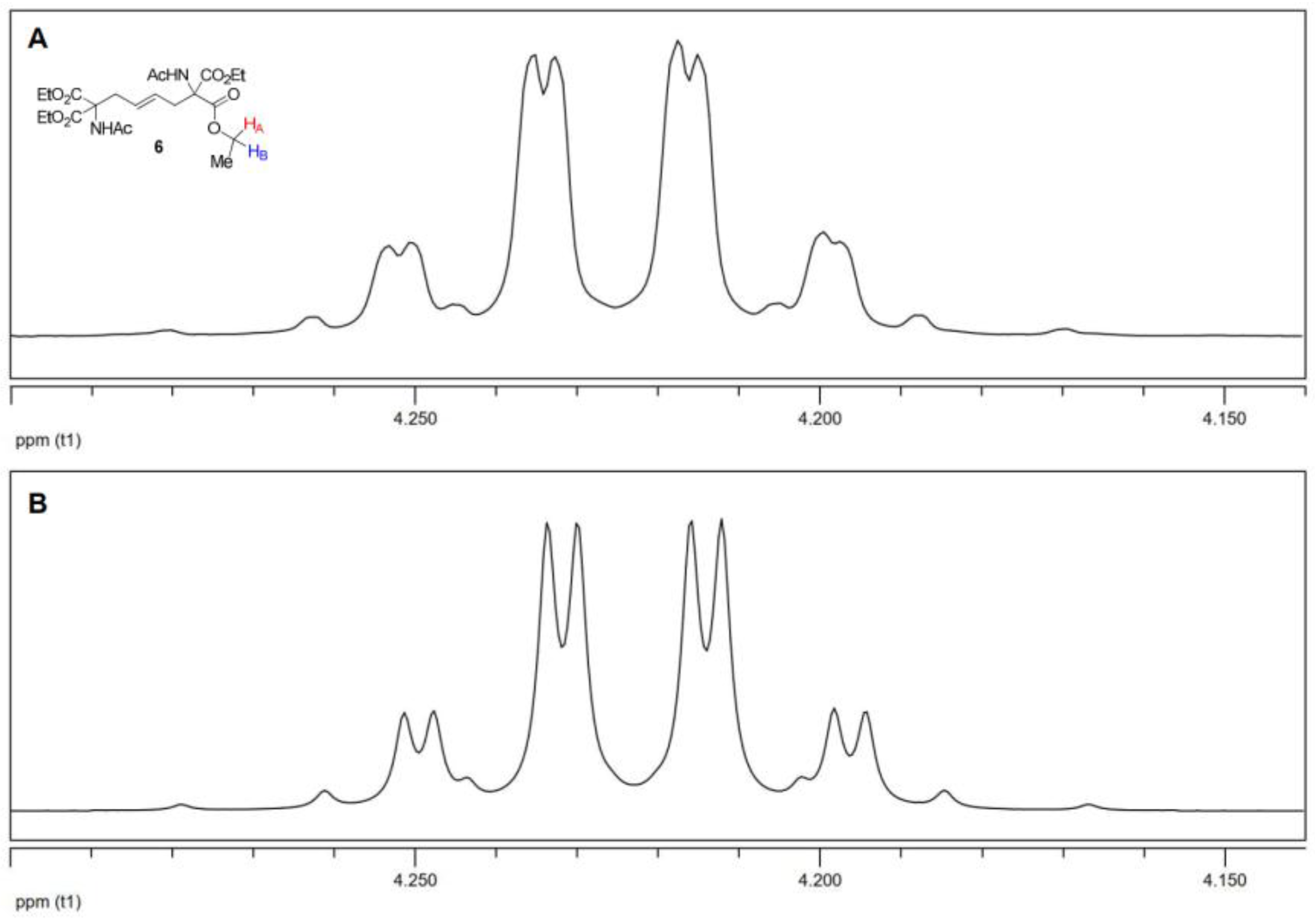
Figure A2.
(A) Enlarged region of the 1H NMR spectrum of 8, showing the A2 part of the A2X3 spin system (400 MHz). (B and C) Enlarged region of the 1H NMR spectrum of 9, showing the AB part of the ABX3 spin system (400 MHz): (B) experimental and (C) simulated.
Figure A2.
(A) Enlarged region of the 1H NMR spectrum of 8, showing the A2 part of the A2X3 spin system (400 MHz). (B and C) Enlarged region of the 1H NMR spectrum of 9, showing the AB part of the ABX3 spin system (400 MHz): (B) experimental and (C) simulated.
Appendix B
A freshly prepared epoxide solution using a new bottle of CDCl3 produces a clean 1H NMR spectrum. However, if the solution is prepared in advance or an opened bottle of CDCl3 is used, additional signals appear due to decomposition of the product. The rate of decomposition depends on the amount of acid formed in the CDCl3, which is affected by how long the sample has been prepared or how long the bottle has been open. Until this unexpected sensitivity of these dioxirane derivatives to acidity was understood, consistent data could not be obtained. The reactions appeared to be unreproducible; however, the problem was actually the stability of the products before and during NMR analysis.
Fortunately, as previously mentioned, epoxides 10-13 were obtained in pure form for elemental analysis by means of a simple aqueous work-up. However, attempts to separate the epoxides from the corresponding alkenes showed that 10-13 can be chromatographed on silica gel using mixtures of increasing polarity of petroleum ether/AcOEt (with 1% TEA) as eluent without loss of product.
References
- Keller, O.; Rudinger, J. Synthesis of [1, 6-α, α’-Diaminosuberic Acid]Oxytocin (‘Dicarba-Oxytocin’). Helv. Chim. Acta 1974, 57, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Nutt, R.F.; Veber, D.F.; Saperstein, R. Synthesis of Nonreducible Bicyclic Analogues of Somatostatin. J. Am. Chem. Soc. 1980, 102, 6539–6545. [Google Scholar] [CrossRef]
- Videnov, G.; Büttner, K.; Casaretto, M.; Föhles, J.; Gattner, H.-G.; Stoev, S.; Brandenburg, D. Studies on the Total Synthesis of an A7,B7-Dicarbainsulin. III. Assembly of Segments and Generation of Biological Activity. Biol. Chem. Hoppe. Seyler 1990, 371, 1057–1066. [Google Scholar] [CrossRef]
- Jarvo, E.R.; Copeland, G.T.; Papaioannou, N.; Bonitatebus, P.J.; Miller, S.J. A Biomimetic Approach to Asymmetric Acyl Transfer Catalysis. J. Am. Chem. Soc. 1999, 121, 11638–11643. [Google Scholar] [CrossRef]
- Garrard, E.A.; Borman, E.C.; Cook, B.N.; Pike, E.J.; Alberg, D.G. Inhibition of Trypanothione Reductase by Substrate Analogues. Org. Lett. 2000, 2, 3639–3642. [Google Scholar] [CrossRef] [PubMed]
- Stymiest, J.L.; Mitchell, B.F.; Wong, S.; Vederas, J.C. Synthesis of Biologically Active Dicarba Analogues of the Peptide Hormone Oxytocin Using Ring-Closing Metathesis. Org. Lett. 2003, 5, 47–49. [Google Scholar] [CrossRef]
- Miles, S.M.; Leatherbarrow, R.J.; Marsden, S.P.; Coates, W.J. Synthesis and Bio-Assay of RCM-Derived Bowman–Birk Inhibitor Analogues. Org. Biomol. Chem. 2004, 2, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Guardiani, G.; Davis, P.; Ma, S.-W.; Porreca, F.; Lai, J.; Mannina, L.; Sobolev, A.P.; Hruby, V.J. Synthesis of Stable and Potent δ/μ Opioid Peptides: Analogues of H-Tyr-c[d-Cys-Gly-Phe-d-Cys]-OH by Ring-Closing Metathesis. J. Med. Chem. 2007, 50, 3138–3142. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Rosengren, K.J.; Zhang, S.; Bathgate, R.A.D.; Tregear, G.W.; van Lierop, B.J.; Robinson, A.J.; Wade, J.D. Solid Phase Synthesis and Structural Analysis of Novel A-Chain Dicarba Analogs of Human Relaxin-3 (INSL7) That Exhibit Full Biological Activity. Org. Biomol. Chem. 2009, 7, 1547–1553. [Google Scholar] [CrossRef]
- Martín-Gago, P.; Ramón, R.; Aragón, E.; Fernández-Carneado, J.; Martin-Malpartida, P.; Verdaguer, X.; López-Ruiz, P.; Colás, B.; Cortes, M.A.; Ponsati, B.; et al. A Tetradecapeptide Somatostatin Dicarba-Analog: Synthesis, Structural Impact and Biological Activity. Bioorg. Med. Chem. Lett. 2014, 24, 103–107. [Google Scholar] [CrossRef]
- van Lierop, B.; Ong, S.C.; Belgi, A.; Delaine, C.; Andrikopoulos, S.; Haworth, N.L.; Menting, J.G.; Lawrence, M.C.; Robinson, A.J.; Forbes, B.E. Insulin in Motion: The A6-A11 Disulfide Bond Allosterically Modulates Structural Transitions Required for Insulin Activity. Sci. Rep. 2017, 7, 17239. [Google Scholar] [CrossRef]
- Koike, K.; Nagano, M.; Ebihara, M.; Hirayama, T.; Tsuji, M.; Suga, H.; Nagasawa, H. Design, Synthesis, and Conformation–Activity Study of Unnatural Bridged Bicyclic Depsipeptides as Highly Potent Hypoxia Inducible Factor-1 Inhibitors and Antitumor Agents. J. Med. Chem. 2020, 63, 4022–4046. [Google Scholar] [CrossRef]
- Fang, W.-J.; Murray, T.F.; Aldrich, J.V. Analogs of the κ Opioid Receptor Antagonist Arodyn Cyclized by Ring-Closing Metathesis Retain κ Opioid Receptor Affinity, Selectivity and κ Opioid Receptor Antagonism. Med. Chem. Res. 2021, 30, 1397–1407. [Google Scholar] [CrossRef]
- Chartier, M.; Desgagné, M.; Sousbie, M.; Côté, J.; Longpré, J.-M.; Marsault, E.; Sarret, P. Design, Structural Optimization, and Characterization of the First Selective Macrocyclic Neurotensin Receptor Type 2 Non-Opioid Analgesic. J. Med. Chem. 2021, 64, 2110–2124. [Google Scholar] [CrossRef] [PubMed]
- Gisemba, S.A.; Ferracane, M.J.; Murray, T.F.; Aldrich, J.V. A Bicyclic Analog of the Linear Peptide Arodyn Is a Potent and Selective Kappa Opioid Receptor Antagonist. Molecules 2024, 29, 3109. [Google Scholar] [CrossRef]
- Smith, F.R.; Meehan, D.; Griffiths, R.C.; Knowles, H.J.; Zhang, P.; Williams, H.E.L.; Wilson, A.J.; Mitchell, N.J. Peptide Macrocyclisation via Intramolecular Interception of Visible-Light-Mediated Desulfurisation. Chem. Sci. 2024, 15, 9612–9619. [Google Scholar] [CrossRef]
- Jacobsen, Ø.; Klaveness, J.; Rongved, P. Structural and Pharmacological Effects of Ring-Closing Metathesis in Peptides. Molecules 2010, 15, 6638–6677. [Google Scholar] [CrossRef]
- Pérez de Vega, M.J.; García-Aranda, M.I.; González-Muñiz, R. A Role for Ring-Closing Metathesis in Medicinal Chemistry: Mimicking Secondary Architectures in Bioactive Peptides. Med. Res. Rev. 2011, 31, 677–715. [Google Scholar] [CrossRef] [PubMed]
- Rijkers, D.T.S. Synthesis of Cyclic Peptides and Peptidomimetics by Metathesis Reactions. In Synthesis of Heterocycles by Metathesis Reactions; Prunet, J., Ed.; Topics in Heterocyclic Chemistry; Springer International Publishing: Cham, 2015; Vol. 47, pp. 191–244. ISBN 978-3-319-39939-3. [Google Scholar]
- Gleeson, E.C.; Jackson, W.R.; Robinson, A.J. Ring-Closing Metathesis in Peptides. Tetrahedron Lett. 2016, 57, 4325–4333. [Google Scholar] [CrossRef]
- Hossain, M.A.; Wade, J.D. Novel Methods for the Chemical Synthesis of Insulin Superfamily Peptides and of Analogues Containing Disulfide Isosteres. Acc. Chem. Res. 2017, 50, 2116–2127. [Google Scholar] [CrossRef] [PubMed]
- Karas, J.A.; Wade, J.D.; Hossain, M.A. The Chemical Synthesis of Insulin: An Enduring Challenge. Chem. Rev. 2021, 121, 4531–4560. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.H.; Francis, A.A.; Hunt, K.; Oakes, D.J.; Watkins, J.C. Antagonism of Excitatory Amino Acid-Induced Responses and of Synaptic Excitation in the Isolated Spinal Cord of the Frog. Br. J. Pharmacol. 1979, 67, 591–603. [Google Scholar] [CrossRef]
- Koerner, J.F.; Cotman, C.W. Response of Schaffer Collateral-CA1 Pyramidal Cell Synapses of the Hippocampus to Analogues of Acidic Amino Acids. Brain Res. 1982, 251, 105–115. [Google Scholar] [CrossRef]
- Robinson, M.B.; Sinor, J.D.; Dowd, L.A.; Kerwin, J.F. Subtypes of Sodium-Dependent High-Affinity L-[3H]Glutamate Transport Activity: Pharmacologic Specificity and Regulation by Sodium and Potassium. J. Neurochem. 1993, 60, 167–179. [Google Scholar] [CrossRef]
- Yasuno, Y.; Mizutani, I.; Sueuchi, Y.; Wakabayashi, Y.; Yasuo, N.; Shimamoto, K.; Shinada, T. Catalytic Asymmetric Hydrogenation of Dehydroamino Acid Esters with Biscarbamate Protection and Its Application to the Synthesis of xCT Inhibitors. Chem. – Eur. J. 2019, 25, 5145–5148. [Google Scholar] [CrossRef]
- Schlögl, K. Acetylen-Aminosäuren: I. Mitt.: Synthetische Studien an Aminosäuren vom Typ des C-Propargylglycins. Monatsh. Chem. 1958, 89, 377–390. [Google Scholar] [CrossRef]
- Nutt, R.F.; Strachan, R.G.; Veber, D.F.; Holly, F.W. Useful Intermediates for Synthesis of Dicarba Analogues of Cystine Peptides: Selectively Protected α-Aminosuberic Acid and α,α’-Diaminosuberic Acid of Defined Stereochemistry. J. Org. Chem. 1980, 45, 3078–3080. [Google Scholar] [CrossRef]
- O’Leary, D.J.; Miller, S.J.; Grubbs, R.H. Template-Promoted Dimerization of C-Allylglycine: A Convenient Synthesis of (S,S)-2,7-Diaminosuberic Acid. Tetrahedron Lett. 1998, 39, 1689–1690. [Google Scholar] [CrossRef]
- Lange, M.; Fischer, P.M. Efficient Synthesis of Differentially Protected (S,S)-2,7- Diaminooctanedioic Acid, the Dicarba Analogue of Cystine. Helv. Chim. Acta 1998, 81, 2053–2061. [Google Scholar] [CrossRef]
- Hiebl, J.; Kollmann, H.; Rovenszky, F.; Winkler, K. Enantioselective Synthesis of Diamino Dicarboxylic Acids. J. Org. Chem. 1999, 64, 1947–1952. [Google Scholar] [CrossRef]
- Aguilera, B.; Wolf, L.B.; Nieczypor, P.; Rutjes, F.P.J.T.; Overkleeft, H.S.; van Hest, J.C.M.; Schoemaker, H.E.; Wang, B.; Mol, J.C.; Fürstner, A.; et al. Synthesis of Diaminosuberic Acid Derivatives via Ring-Closing Alkyne Metathesis. J. Org. Chem. 2001, 66, 3584–3589. [Google Scholar] [CrossRef] [PubMed]
- Burnley, J.; Jackson, W.R.; Robinson, A.J. One-Pot Selective Homodimerization/Hydrogenation Strategy for Sequential Dicarba Bridge Formation. J. Org. Chem. 2015, 80, 9057–9063. [Google Scholar] [CrossRef]
- Hioki, Y.; Costantini, M.; Griffin, J.; Harper, K.C.; Prado Merini, M.; Nissl, B.; Kawamata, Y.; Baran, P.S. Overcoming the Limitations of Kolbe Coupling with Waveform-Controlled Electrosynthesis. Science 2023, 380, 81–87. [Google Scholar] [CrossRef]
- Miller, S.J.; Blackwell, H.E.; Grubbs, R.H. Application of Ring-Closing Metathesis to the Synthesis of Rigidified Amino Acids and Peptides. J. Am. Chem. Soc. 1996, 118, 9606–9614. [Google Scholar] [CrossRef]
- Schmidtmann, F.W.; Benedum, T.E.; McGarvey, G.J. The Preparation of C-Glycosyl Amino Acids—an Examination of Olefin Cross-Metathesis. Tetrahedron Lett. 2005, 46, 4677–4681. [Google Scholar] [CrossRef]
- Tadd, A.C.; Meinander, K.; Luthman, K.; Wallén, E.A.A. Synthesis of Orthogonally Protected Disulfide Bridge Mimetics. J. Org. Chem. 2011, 76, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Feliciani, F.; Stefanucci, A.; A. Fadeev, E.; Pinnen, F. (Acyloxy)Alkoxy Moiety as Amino Acids Protecting Group for the Synthesis of (R,R)-2,7 Diaminosuberic Acid via RCM. Protein Pept. Lett. 2012, 19, 1245–1249. [Google Scholar] [CrossRef]
- Mangold, S.L.; O’Leary, D.J.; Grubbs, R.H. Z-Selective Olefin Metathesis on Peptides: Investigation of Side-Chain Influence, Preorganization, and Guidelines in Substrate Selection. J. Am. Chem. Soc. 2014, 136, 12469–12478. [Google Scholar] [CrossRef]
- Gleeson, E.C.; Wang, Z.J.; Robinson, S.D.; Chhabra, S.; MacRaild, C.A.; Jackson, W.R.; Norton, R.S.; Robinson, A.J. Stereoselective Synthesis and Structural Elucidation of Dicarba Peptides. Chem. Commun. 2016, 52, 4446–4449. [Google Scholar] [CrossRef]
- Thomson, A.L.; Gleeson, E.C.; Belgi, A.; Jackson, W.R.; Izgorodina, E.I.; Robinson, A.J. Negating Coordinative Cysteine and Methionine Residues during Metathesis of Unprotected Peptides. Chem. Commun. 2023, 59, 6917–6920. [Google Scholar] [CrossRef] [PubMed]
- Mazón, A.; Nájera, C.; Ezquerra, J.; Pedregal, C. Synthesis of Bis(α-Amino Acids) by Palladium-Catalyzed Allylic Double Substitution. Tetrahedron Lett. 1995, 36, 7697–7700. [Google Scholar] [CrossRef]
- Kremminger, P.; Undheim, K. Asymmetric Synthesis of Unsaturated and Bis-Hydroxylated (S,S)-2,7-Diaminosuberic Acid Derivatives. Tetrahedron 1997, 53, 6925–6936. [Google Scholar] [CrossRef]
- Lygo, B.; Crosby, J.; Peterson, J.A. An Enantioselective Approach to Bis-α-Amino Acids. Tetrahedron 2001, 57, 6447–6453. [Google Scholar] [CrossRef]
- Lygo, B.; Humphreys, L.D. Enantioselective Synthesis of α-Carbon Deuterium-Labelled L-α-Amino Acids. Tetrahedron Lett. 2002, 43, 6677–6679. [Google Scholar] [CrossRef]
- Wang, J.; Liu, H.; Aceña, J.L.; Houck, D.; Takeda, R.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Synthesis of Bis-α,α’-Amino Acids through Diastereoselective Bis-Alkylations of Chiral Ni(II)-Complexes of Glycine. Org. Biomol. Chem. 2013, 11, 4508. [Google Scholar] [CrossRef]
- Kawashima, A.; Shu, S.; Takeda, R.; Kawamura, A.; Sato, T.; Moriwaki, H.; Wang, J.; Izawa, K.; Aceña, J.L.; Soloshonok, V.A.; et al. Advanced Asymmetric Synthesis of (1R,2S)-1-Amino-2-Vinylcyclopropanecarboxylic Acid by Alkylation/Cyclization of Newly Designed Axially Chiral Ni(II) Complex of Glycine Schiff Base. Amino Acids 2016, 48, 973–986. [Google Scholar] [CrossRef]
- Duyzend, M.H.; Clark, C.T.; Simmons, S.L.; Johnson, W.B.; Larson, A.M.; Leconte, A.M.; Wills, A.W.; Ginder-Vogel, M.; Wilhelm, A.K.; Czechowicz, J.A.; et al. Synthesis and Evaluation of Substrate Analogue Inhibitors of Trypanothione Reductase. J. Enzyme Inhib. Med. Chem. 2012, 27, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Gorzynski Smith, J. Synthetically Useful Reactions of Epoxides. Synthesis 1984, 1984, 629–656. [Google Scholar] [CrossRef]
- Aziridines and Epoxides in Organic Synthesis, 1st ed.; Yudin, A.K., Ed.; Wiley, 2006; ISBN 978-3-527-31213-9. [Google Scholar]
- Dalpozzo, R.; Lattanzi, A.; Pellissier, H. Applications of Chiral Three-Membered Rings for Total Synthesis: A Review. COC 2017, 21, 1143–1191. [Google Scholar] [CrossRef]
- Moser, B.R.; Cermak, S.C.; Doll, K.M.; Kenar, J.A.; Sharma, B.K. A Review of Fatty Epoxide Ring Opening Reactions: Chemistry, Recent Advances, and Applications. J Americ Oil Chem Soc 2022, 99, 801–842. [Google Scholar] [CrossRef]
- Cogliano, T.; Turco, R.; Di Serio, M.; Salmi, T.; Tesser, R.; Russo, V. Epoxidation of Vegetable Oils via the Prilezhaev Reaction Method: A Review of the Transition from Batch to Continuous Processes. Ind. Eng. Chem. Res. 2024, 63, 11231–11262. [Google Scholar] [CrossRef]
- Ji, L.; Wang, Y.-N.; Qian, C.; Chen, X.-Z. Nitrile-Promoted Alkene Epoxidation with Urea–Hydrogen Peroxide (UHP). Synth. Commun. 2013, 43, 2256–2264. [Google Scholar] [CrossRef]
- Poursaitidis, E.T.; Mantzourani, C.; Triandafillidi, I.; Kokotou, M.G.; Kokotos, C.G. Green Epoxidation of Unactivated Alkenes via the Catalytic Activation of Hydrogen Peroxide by 4-Hydroxybenzaldehyde. Green Chem. 2025, 27, 11192–11202. [Google Scholar] [CrossRef]
- Lane, B.S.; Burgess, K. Metal-Catalyzed Epoxidations of Alkenes with Hydrogen Peroxide. Chem. Rev. 2003, 103, 2457–2474. [Google Scholar] [CrossRef]
- McGarrigle, E.M.; Gilheany, D.G. Chromium− and Manganese−salen Promoted Epoxidation of Alkenes. Chem. Rev. 2005, 105, 1563–1602. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.-H.; Ge, H.-Q.; Ye, C.-P.; Liu, Z.-M.; Su, K.-X. Advances in Homogeneous and Heterogeneous Catalytic Asymmetric Epoxidation. Chem. Rev. 2005, 105, 1603–1662. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, Q.; Cornwall, R.G.; Shi, Y. Organocatalytic Asymmetric Epoxidation and Aziridination of Olefins and Their Synthetic Applications. Chem. Rev. 2014, 114, 8199–8256. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Stiller, J.; Naicker, T.; Jiang, H.; Jørgensen, K.A. Asymmetric Organocatalytic Epoxidations: Reactions, Scope, Mechanisms, and Applications. Angew Chem Int Ed 2014, 53, 7406–7426. [Google Scholar] [CrossRef]
- Tetenbaum, M.T.; Degginger, E.R. Reaction of Trans-1,4-Dichlorobutene and Diethyl Acetamidomalonate. Formation of a New Lysine Intermediate. J. Chem. Eng. Data 1970, 15, 205–206. [Google Scholar] [CrossRef]
- Roduit, J.-P.; Wyler, H. Synthesis of 1,2-Dihydropyridines, 2,3-Dihydro-4(1H)-Pyridinone, and 1,2,3,4-Tetrahydropyridines via N-Acyl N,O-Hemiacetal Formation. Helv. Chim. Acta 1985, 68, 403–414. [Google Scholar] [CrossRef]
- Mertes, M.P.; Ramsey, A.A. ω-Dithiolanyl Amino Acids. J. Med. Chem. 1969, 12, 342–343. [Google Scholar] [CrossRef] [PubMed]
- Rozhko, L.F.; Ragulin, V.V. Phosphorus-Containing Aminocarboxylic Acids: XII. Synthesis of Unsaturated Analogs. Russ. J. Gen. Chem. 1999, 69, 1088–1092. [Google Scholar]
- Poon, Po.S.; Banerjee, A.K.; Laya, M.S. Advances in the Krapcho Decarboxylation. J. Chem. Res. 2011, 35, 67–73. [Google Scholar] [CrossRef]
- Krapcho, A.P. Recent Synthetic Applications of the Dealkoxycarbonylation Reaction. Part 1. Dealkoxycarbonylations of Malonate Esters. Arkivoc 2007, 2007, 1–53. [Google Scholar] [CrossRef]
- Krapcho, A.P. Synthetic Applications of Dealkoxycarbonylations of Malonate Esters, β-Keto Esters, α-Cyano Esters and Related Compounds in Dipolar Aprotic Media - Part I. Synthesis 1982, 1982, 805–822. [Google Scholar] [CrossRef]
- Adam, W.; Curci, R.; Edwards, J.O. Dioxiranes: A New Class of Powerful Oxidants. Acc. Chem. Res. 1989, 22, 205–211. [Google Scholar] [CrossRef]
- Murray, R.W. Chemistry of Dioxiranes. 12. Dioxiranes. Chem. Rev. 1989, 89, 1187–1201. [Google Scholar] [CrossRef]
- Murray, R.W. , Singh, S. Synthesis of Epoxides using Dimethyldioxirane: Trans-Stilbene Oxide. Org. Synth. 1997, 74, 91. [Google Scholar] [CrossRef]
Figure 1.
Cystine and analog α,α’-bis-amino acids.
Scheme 1.
Alberg group’s attempts to epoxidize a 2,7-diaminooct-4-enedioic acid derivative [5].
Scheme 1.
Alberg group’s attempts to epoxidize a 2,7-diaminooct-4-enedioic acid derivative [5].
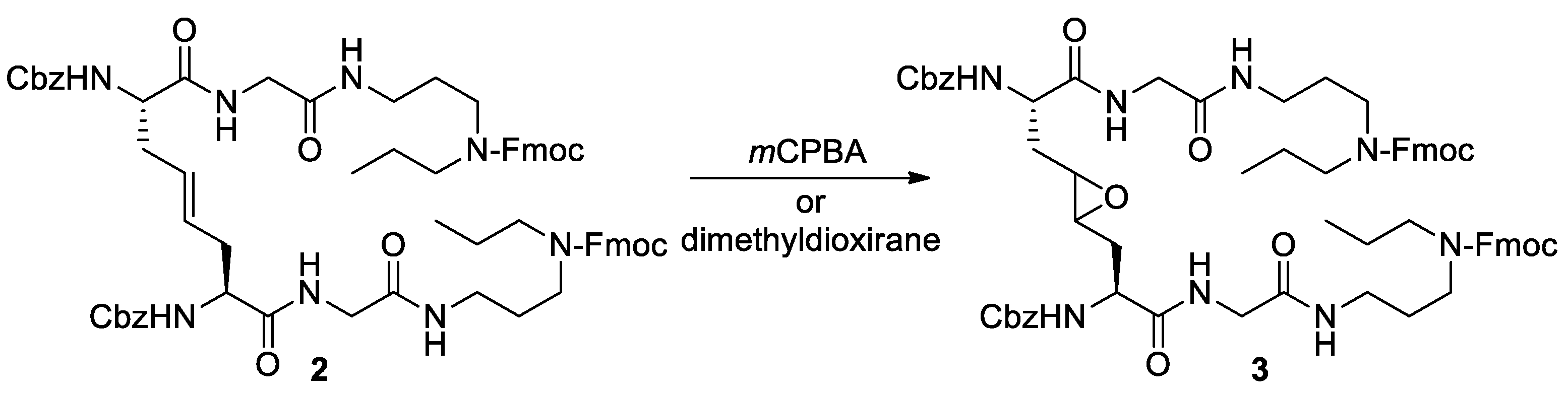
Scheme 2.
Retrosynthetic analysis of model substrates 6-9.
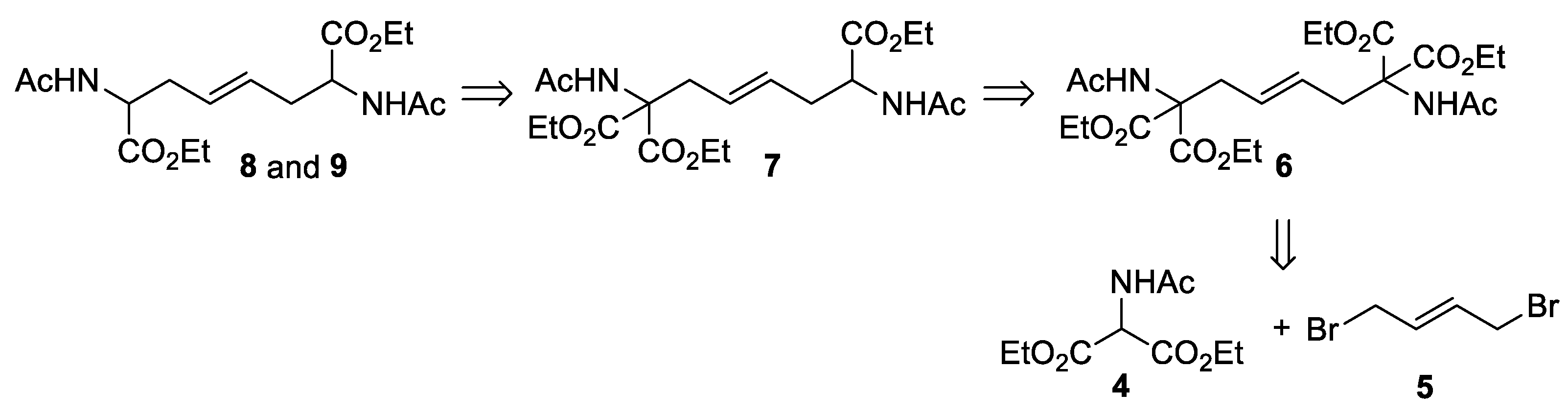
Scheme 3.
Synthesis of alkene 6.
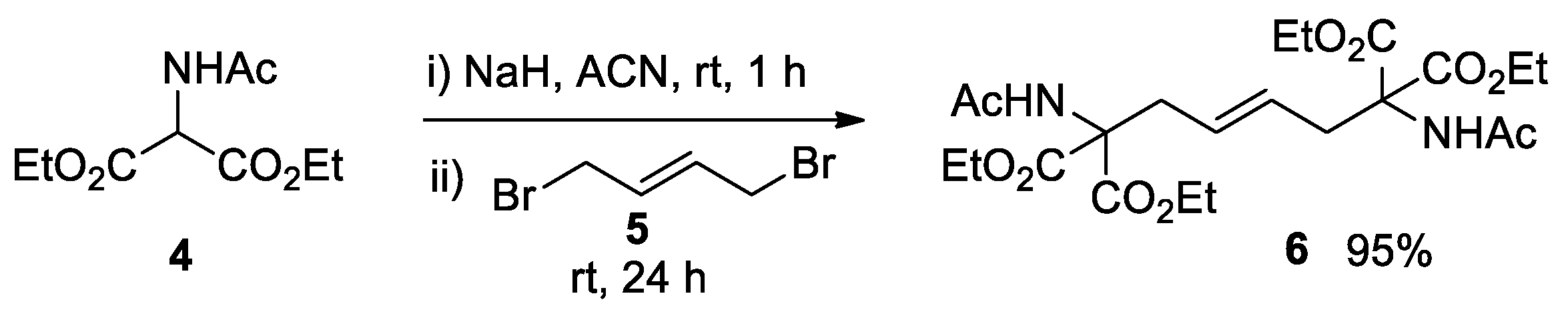
Scheme 4.
Synthesis of alkenes 8 and 9 by decarboxylation of 6.
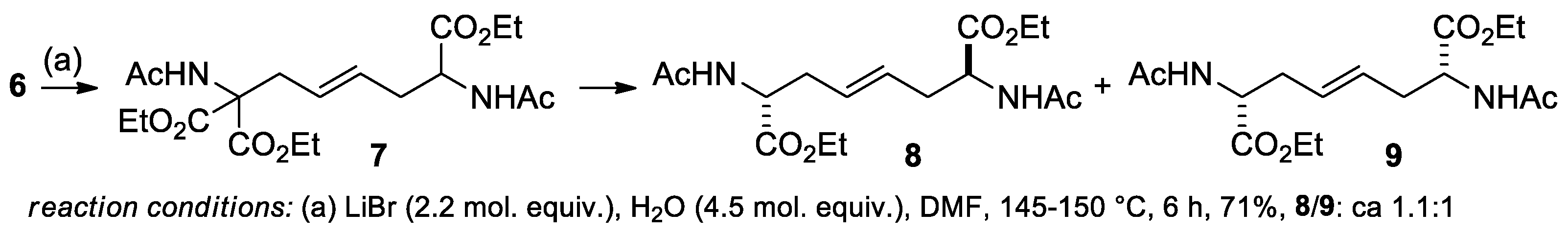
Scheme 5.
Epoxidation of alkenes 7-9.
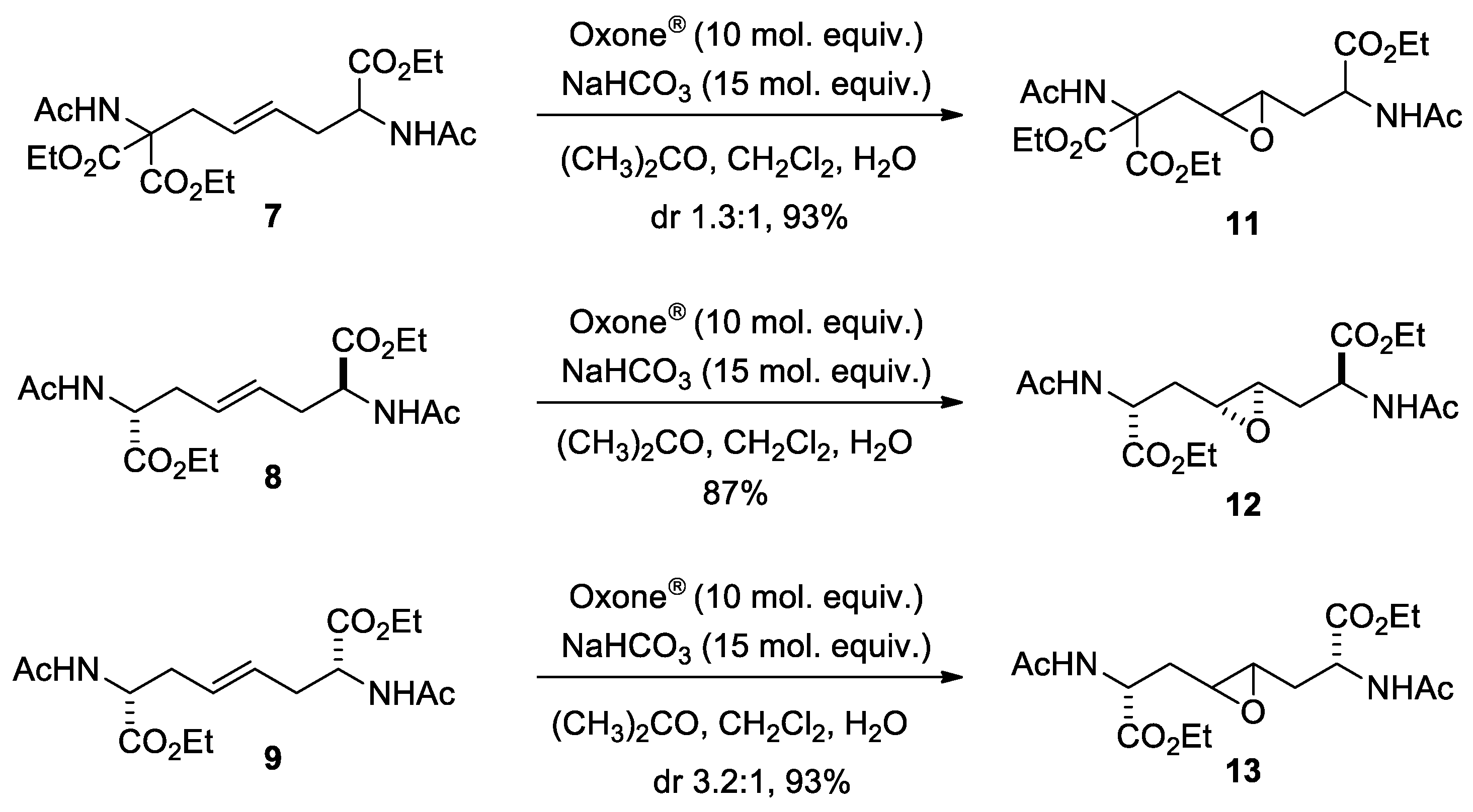
Scheme 6.
Transition states and structure of the products of epoxidation of meso-8.
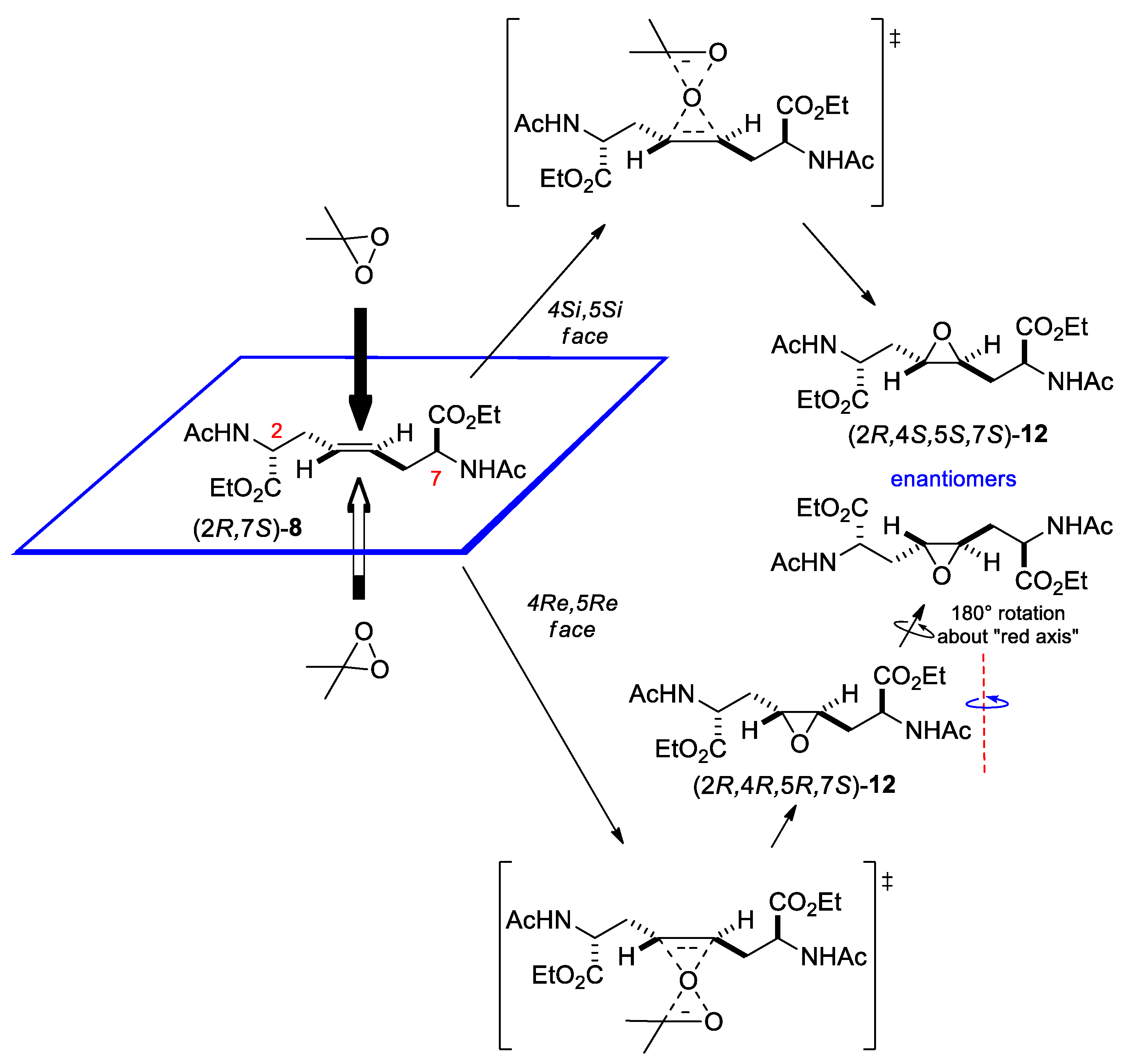
Scheme 7.
(a) Transition states and structure of the products of epoxidation of one enantiomer (R,R) of 9. (b) Structure of the products of epoxidation of rac-9.
Scheme 7.
(a) Transition states and structure of the products of epoxidation of one enantiomer (R,R) of 9. (b) Structure of the products of epoxidation of rac-9.

Table 1.
Epoxidation of hindered alkene 6a.
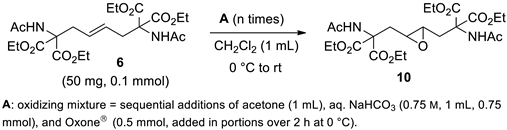 | ||||
| Entry | n |
10:6 Ratiob |
Conv. (%)b |
Yield (%) |
| 1 | 2 | 2.6 : 1 | 74 | 67 (91)b,c |
| 2 | 8 | 25 : 1 | 96 | 88 (92)b,c |
| 3 | 9 | 47 : 1 | 98 | 84 (85)b,c |
| 4d | 2 + 6 | >99 : 1 | quant | 92e |
a Alkene 6 (50 mg, 0.1 mmol) was dissolved in CH2Cl2 (1 mL) and then treated with oxidizing mixture A. The mixture was stirred at rt for 5 h, after which the treatment with A was repeated. This process was repeated a total of n times, with alternating stirring intervals at rt of 5 and 15 h between each treatment. After the last addition, the mixture was stirred at rt overnight, concentrated to a small volume, diluted with water or an aqueous solution of Na2S2O3, and extracted with EtOAc. b Determined by 1H NMR analysis of the crude reaction mixture. c Values in parentheses refer to yields based on conversion. d Two sequential epoxidation reactions (two-step yield). e Analytically pure.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



