Submitted:
01 November 2025
Posted:
04 November 2025
Read the latest preprint version here
Abstract
Inflammatory bowel disease (IBD) arises from the interplay of genetic susceptibility, microbial dysbiosis, and dysregulated innate immunity, leading to chronic intestinal inflammation. Recent advances in single-cell and spatial multi-omics technologies have redefined neutrophils as highly heterogeneous and transcriptionally plastic cells rather than short-lived effectors. In the inflamed gut, cytokine gradients, hypoxia, and microbial metabolites such as short-chain fatty acids dynamically reprogram neutrophil states toward either protective or pathogenic phenotypes. Emerging evidence identifies de novo induction of the NADPH oxidase enzyme DUOX2 in intestinal neutrophils as a key driver of redox signaling, amplifying epithelial and immune activation through NF-κB and p38 MAPK pathways. This redox diversification enhances antimicrobial defense but also perpetuates dysbiosis, barrier dysfunction, and NET-associated thrombo-inflammation. Experimental models demonstrate that myeloid DUOX2 ablation mitigates colitis, supporting therapeutic strategies that target this axis—such as JAK/STAT inhibition, CXCR2 blockade, p38/MK2 modulation, or butyrate-based interventions—to recalibrate neutrophil function while preserving host protection. Integrating single-cell atlases, redox proteomics, and functional imaging will be essential to translate neutrophil plasticity into biomarker-guided, precision therapies for durable mucosal healing in IBD.
Keywords:
neutrophil plasticity
; DUOX2 expression
; IBD pathogenesis
; redox signaling
; precision therapeutics
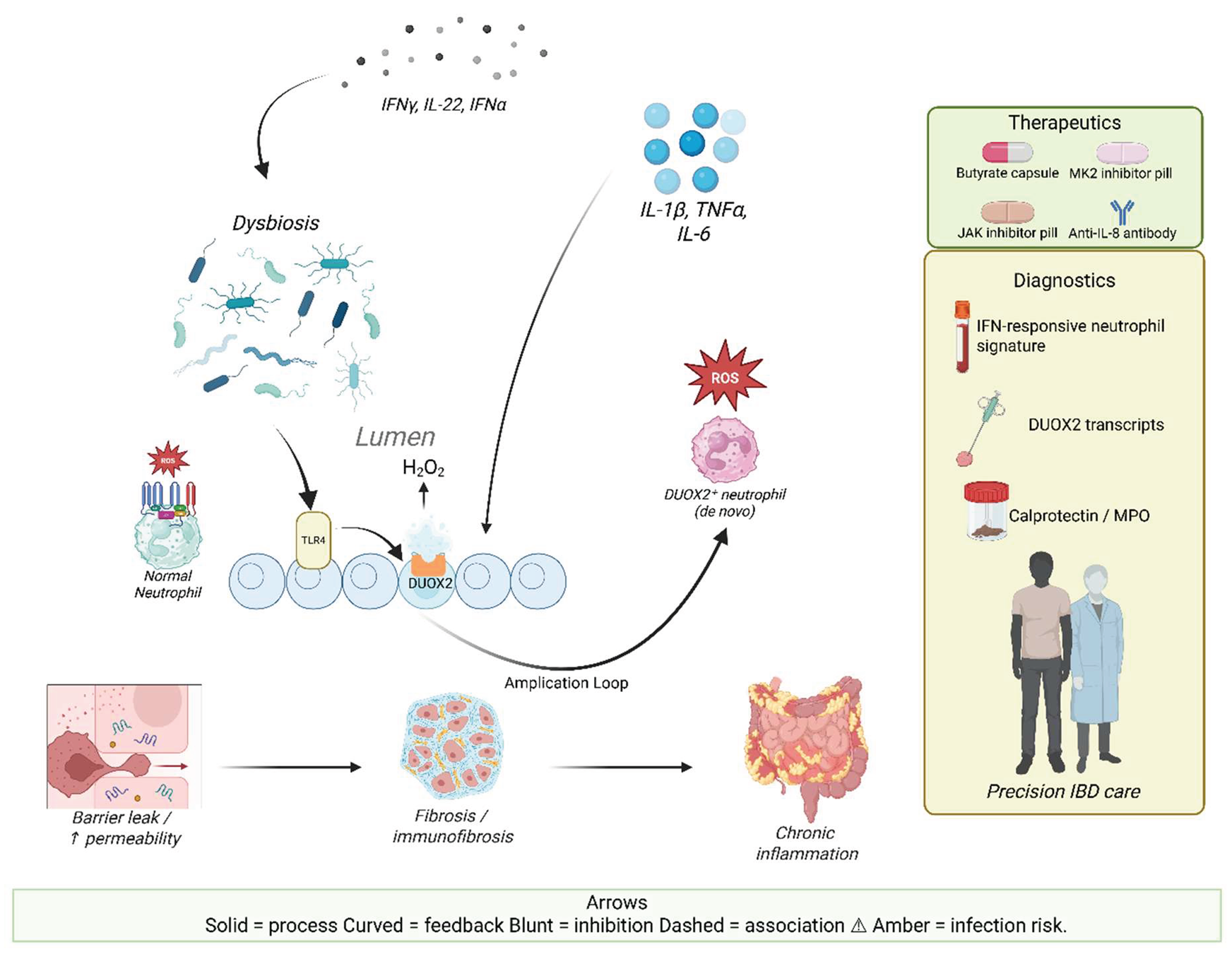
Graphical Abstract: Neutrophil plasticity and DUOX2 in IBD pathogenesis and therapy. Dysbiosis and cytokine signaling (IFNγ, IL-22, IFNα) activate epithelial DUOX2, releasing H₂O₂ into the lumen. This drives neutrophil reprogramming into DUOX2⁺ subsets with enhanced ROS production, creating an amplification loop that sustains epithelial injury. Elevated cytokine release (IL-1β, TNFα, IL-6) further reinforces the epithelial–neutrophil axis. Downstream outcomes include barrier leak and permeability defects, fibrosis/immunofibrosis via NET–fibroblast interactions, and chronic intestinal inflammation. Translational opportunities include therapeutics (butyrate, MK2 inhibitors, JAK inhibitors, anti-IL-8 antibodies) and diagnostic biomarkers (IFN-responsive neutrophil signatures, DUOX2 transcripts, stool calprotectin/MPO, NET proteins), converging on precision IBD care.
1. Introduction
Inflammatory bowel diseases (IBD), encompassing Crohn’s disease (CD) and ulcerative colitis (UC), are chronic, relapsing inflammatory disorders of the gastrointestinal tract arising from the complex interplay of host genetics, environmental exposures, immune dysregulation, and alterations in the intestinal microbiota [1]. CD can affect any part of the gastrointestinal tract and is characterized by transmural inflammation, granuloma formation, and fibrotic complications driven largely by Th1/Th17 cytokines such as interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and interleukin-17 (IL-17) [2]. In contrast, UC is confined to the colonic mucosa and involves continuous superficial ulceration, crypt abscesses, and a Th2-skewed inflammatory profile dominated by cytokines including IL-13 and IL-5 [3]. Genome-wide association studies have identified more than 230 susceptibility loci, including NOD2, ATG16L1, IL23R, and NCF4, many of which converge on microbial recognition, autophagy, and neutrophil function [4,5]. However, genetic predisposition alone cannot explain the rapid rise in global incidence, underscoring the critical contribution of environmental triggers and dysbiosis—marked by reduced microbial diversity, expansion of pathobionts such as adherent-invasive Escherichia coli (AIEC), and diminished butyrate-producing bacteria—which collectively compromise epithelial barrier integrity and amplify aberrant immune activation [6].
Innate immune cells form the first line of defense against microbial invasion, and among them neutrophils are rapidly recruited to inflamed mucosa in IBD [7]. Historically regarded as short-lived, terminally differentiated effectors, neutrophils were long considered simple executors of host defense through phagocytosis, degranulation, reactive oxygen species (ROS) production, and neutrophil extracellular trap (NET) release [8]. In the context of IBD, their massive infiltration leads to hallmark features such as cryptitis and crypt abscesses, while neutrophil granule proteins like calprotectin serve as clinically useful biomarkers of disease activity [9,10]. Yet the functional role of neutrophils has remained controversial due to their apparent duality: on one hand, they are indispensable for containing microbial translocation and facilitating wound healing, while on the other, their excessive or dysregulated activity exacerbates epithelial damage, drives oxidative stress, and perpetuates chronic inflammation [11]. This paradox has fueled a shift in understanding toward the concept of neutrophil plasticity. Advances in single-cell transcriptomics and spatial profiling have revealed that neutrophils are not a homogenous population but exhibit remarkable transcriptional and functional heterogeneity, reprogramming in response to local cytokines, microbial cues, and metabolic signals to acquire context-dependent phenotypes that can be either pro-inflammatory or pro-resolving [12,13].
A novel dimension of this plasticity is the induction of dual oxidase 2 (DUOX2) expression in neutrophils recruited to the inflamed gut. DUOX2, a member of the NADPH oxidase family normally restricted to intestinal epithelial cells, generates hydrogen peroxide (H₂O₂) at mucosal surfaces, contributing to baseline antimicrobial defense and epithelial barrier regulation [14]. In IBD, epithelial DUOX2 is markedly upregulated early in disease in response to dysbiosis, pattern recognition receptor activation, and cytokines such as IFN-γ and IL-22, whereas microbial metabolites like butyrate suppress its expression [15,16]. While physiologic DUOX2 activity maintains mucosal homeostasis, its overexpression drives oxidative stress, epithelial injury, and microbial imbalance [17,18]. Intriguingly, recent evidence demonstrates that neutrophils themselves can undergo transcriptional reprogramming to express DUOX2, thereby expanding their oxidative capacity beyond canonical NOX2 activity [19,20]. This discovery highlights a previously unrecognized neutrophil–epithelial crosstalk axis, wherein DUOX2 serves as both a sentinel of microbial imbalance and a driver of tissue pathology.
This review integrates emerging concepts of neutrophil plasticity and the evolving role of DUOX2 in IBD pathogenesis. We synthesize evidence from single-cell and mechanistic studies to examine how microenvironmental signals orchestrate neutrophil heterogeneity, how de novo DUOX2 expression reshapes neutrophil effector functions, and how this intersection contributes to the amplification or resolution of intestinal inflammation. Finally, we discuss the therapeutic implications of targeting the neutrophil–DUOX2 axis as a novel strategy to restore mucosal homeostasis without compromising host defense. As illustrated in Figure 2, the interplay between DUOX2 expression in epithelial cells and neutrophil recruitment contributes to the amplification of oxidative stress, barrier dysfunction, and chronic inflammation in IBD.
Figure 1.
DUOX2 in IBD pathogenesis. Panel A: microbial and cytokine cues induce epithelial DUOX2. Panel B: excessive DUOX2 drives barrier dysfunction and dysbiosis. Panel C: recruited neutrophils reprogram into DUOX2+ subsets that amplify cytokine and fibrotic loops. Panel D: therapeutic interventions (butyrate, MK2/JAK/IL-8 blockade) and biomarker strategies can modulate this axis.
Figure 1.
DUOX2 in IBD pathogenesis. Panel A: microbial and cytokine cues induce epithelial DUOX2. Panel B: excessive DUOX2 drives barrier dysfunction and dysbiosis. Panel C: recruited neutrophils reprogram into DUOX2+ subsets that amplify cytokine and fibrotic loops. Panel D: therapeutic interventions (butyrate, MK2/JAK/IL-8 blockade) and biomarker strategies can modulate this axis.

2. Neutrophil plasticity in IBD and tissue contexts
2.1. Evidence for neutrophil heterogeneity and plasticity in IBD
The long-standing notion that neutrophils are short-lived, transcriptionally inert effectors has been overturned by single-cell genomics demonstrating discrete, stimulus-tunable neutrophil states in humans. In peripheral blood, reproducible transcriptomic clusters (Nh0–Nh3) span maturation and activation continua, including interferon-responsive states that are invisible to classical surface phenotyping [21]. Within the inflamed human colon, single-cell and spatial multi-omics reveal that neutrophils diversify further into tissue-adapted subpopulations with distinct chemokine, interferon, and redox programs; these subsets are differentially enriched across Crohn’s disease (CD) and ulcerative colitis (UC) and participate in dense crosstalk with inflammatory fibroblasts and other myeloid cells [13,22,23]. Collectively, these data establish neutrophil plasticity as a core feature of IBD pathobiology rather than a peripheral epiphenomenon [13,22,23]. A summary of major neutrophil subsets, their defining markers, tissue contexts, and functional roles in IBD is provided in Table 1.

Table 1.
Selected neutrophil subsets and phenotypes in health and IBD.
| Neutrophil Subset | Key Markers / Traits | Context / Location | Functional Roles | References |
| Resting circulating neutrophils | CD16^+, CD62L^+, CXCR2^+ | Blood (homeostasis) | Baseline antimicrobial defense; short-lived; enter tissues upon chemokine signaling. | [24] |
| CD177^+ neutrophils | CD177^hi, CD66b^+, FcγRIIIb^+ | Inflamed intestinal mucosa in IBD | Enhanced chemotaxis and bactericidal activity; support barrier defense; also release pro-inflammatory mediators; correlate with IBD severity. | [25,26] |
| CD177^- neutrophils | CD177^-; otherwise phenotypically similar to CD177^+ | Blood, IBD mucosa | Lower recruitment compared to CD177^+; may have less effector potency; role still under investigation. | [27] |
| Low-density neutrophils (LDNs) | Low buoyant density; often immature; heterogeneous; markers vary | Autoimmunity, cancer, severe inflammation, IBD flares | Immunosuppressive (MDSC-like) or proinflammatory; can release ROS and NETs; expansion reported in active IBD. | [28,29] |
| Inflammation-primed neutrophils | Upregulated CXCR1/2, CD11b; downregulated CXCR4; increased activation markers | Inflamed tissue niches (gut mucosa in IBD) | Heightened effector functions (ROS, degranulation, NETs); strong recruitment cascades; IFN-priming can drive fibrotic plasticity in Crohn’s. | [30] |
| DUOX2^+ neutrophils | De novo DUOX2 expression (with NOX2); upregulated chemokines (CXCL1/2), cytokines (IL-1β, TNFα, IL-6) | Inflamed intestine (murine colitis, human IBD) | Expanded oxidative capacity; extracellular H₂O₂ production; amplify cytokine loops; sustain chronic mucosal inflammation and fibrosis. | [31] |
2.2. Microenvironmental cues that drive tissue reprogramming
Tissue entry exposes neutrophils to an IBD-specific milieu—hypoxia, cytokines (e.g., IL-1 family, type I/II interferons), stromal mediators, and microbial metabolites—that together imprint niche-directed phenotypes. Spatial-transcriptomic mapping of deep ulcer beds identifies an IL-1–high stromal niche that programs neutrophils and fibroblasts into a self-amplifying inflammatory circuit associated with therapy non-response [23]. Beyond IL-1, type I interferon can “pre-prime’’ circulating CD neutrophils toward a fibrogenic effector program; mechanistically, IFN-α–conditioned neutrophils generate NET-rich secretomes that activate intestinal fibroblasts, and JAK inhibition interrupts this neutrophil–fibroblast axis in experimental systems [32]. Microbiota signals provide an orthogonal layer of control: in patients with IBD, the short-chain fatty acid butyrate directly restrains neutrophil inflammatory functions—suppressing cytokine/chemokine output, migration, ROS/MPO release, and NET formation—via histone-deacetylase inhibition, and ameliorates neutrophil-dominated colitis in vivo [33]. In parallel, homeostatic and inflammatory trafficking cues re-shape neutrophil life-cycle dynamics; for example, CXCR4–CXCL12 signaling governs marrow retention and the aging (CXCR4^hi) neutrophil phenotype, which is frequently observed in tissues and can mark specialized, niche-adapted states [34].
2.3. Dichotomous effector programs: protection versus pathology
Neutrophils are indispensable for barrier defense when the epithelium is breached—coordinating phagocytosis, oxidative burst, degranulation, and, when required, NET-mediated trapping—to contain microbes and limit dissemination [13,22,23,33]. The same armamentarium becomes pathogenic when chronically sustained or spatially dysregulated: intestinal NETs and associated proteases/oxidants disrupt tight junctions, increase epithelial apoptosis and permeability, and potentiate thrombo-inflammatory cascades; enzymatic dismantling or upstream inhibition of NETosis alleviates intestinal injury in preclinical models, underscoring causal involvement [23,33,35]. Thus, in IBD, neutrophil plasticity resolves into two broad, context-contingent programs—host-protective versus tissue-damaging—determined by local cytokine tone, stromal instruction, microbial composition, and metabolic substrates [13,22,23,32,33,35]. The dual roles of key neutrophil effector mechanisms in IBD are summarized in Table 2.
Table 2.
Dual functions of neutrophil effector mechanisms in IBD.
| Effector Mechanism | Protective Role in IBD | Pathogenic Role in IBD | References |
| Reactive Oxygen Species (ROS) | Potent bactericidal activity that limits microbial dissemination; essential for early host defense. | Excessive ROS disrupts epithelial tight junctions, damages DNA/proteins, and induces apoptosis of intestinal epithelial cells. | [36,37] |
| Neutrophil Extracellular Traps (NETs) | Trap and neutralize pathogens extracellularly, preventing microbial spread; can support wound repair and resolution. | Dysregulated NETosis increases gut permeability, induces IEC apoptosis, and destroys tight junctions, perpetuating inflammation. | [38,39] |
| Proteases (MMPs, elastase) | Release of lytic enzymes clears invading microbes and assists in matrix remodeling during repair. | Overproduction degrades epithelial adherens junctions, weakens barrier integrity, and contributes to mucosal injury. | [40,41] |
| Immune Cell Recruitment | Release of chemokines ensures rapid recruitment of immune cells, enabling pathogen clearance and resolution. | Excessive or chronic recruitment drives uncontrolled inflammation, amplifies cytokine cascades, and worsens epithelial damage. | [30,42] |
2.4. Disease-relevant subsets in the gut niche (including DUOX2+ and CD177+ states)
IBD tissues harbor neutrophil subsets with distinct molecular specializations. Recent multi-omic work demonstrates de novo DUOX2 induction in intestinal neutrophils, expanding their oxidative repertoire beyond NOX2 to epithelial-style H_2O_2 generation; this DUOX2^+ program tracks with inflamed niches and augments chemokine-rich microenvironments, providing a mechanistic route by which neutrophil reprogramming can amplify chronic mucosal inflammation [13]. In contrast, CD177^+ neutrophils represent a functionally activated, barrier-supportive state in IBD: they display enhanced antimicrobial activity and produce IL-22, a cytokine linked to epithelial restitution, with evidence for negative regulation of excessive inflammation and improved mucosal integrity in experimental systems and patient cohorts [11,33]. Together with interferon-responsive and CXCR4^hi tissue-adapted populations identified by single-cell atlases, these findings position neutrophil subset biology—not mere abundance—as a determinant of clinical phenotype (e.g., fibrosis-prone CD, therapy-refractory UC) and a substrate for precision intervention [11,13,22,23,32]. Figure 3 illustrates the de novo expression of DUOX2 in neutrophils during IBD and highlights its role in enhancing oxidative capacity and promoting chronic inflammation. The discovery of this DUOX2+ neutrophil subset provides crucial insights into the dual roles of neutrophils in both inflammation resolution and tissue damage.
Additional subsets include: (i) low-density granulocytes (LDGs), a circulating pro-inflammatory population enriched in active IBD, capable of spontaneous NET release and T-cell modulation [43] ; and (ii) Siglec-9hi neutrophils, recently described as immunoregulatory and enriched in resolving mucosal niches, potentially restraining excessive inflammation [44]. These specialized states underscore that neutrophil heterogeneity extends beyond DUOX2⁺ and CD177⁺ phenotypes, broadening the therapeutic landscape.
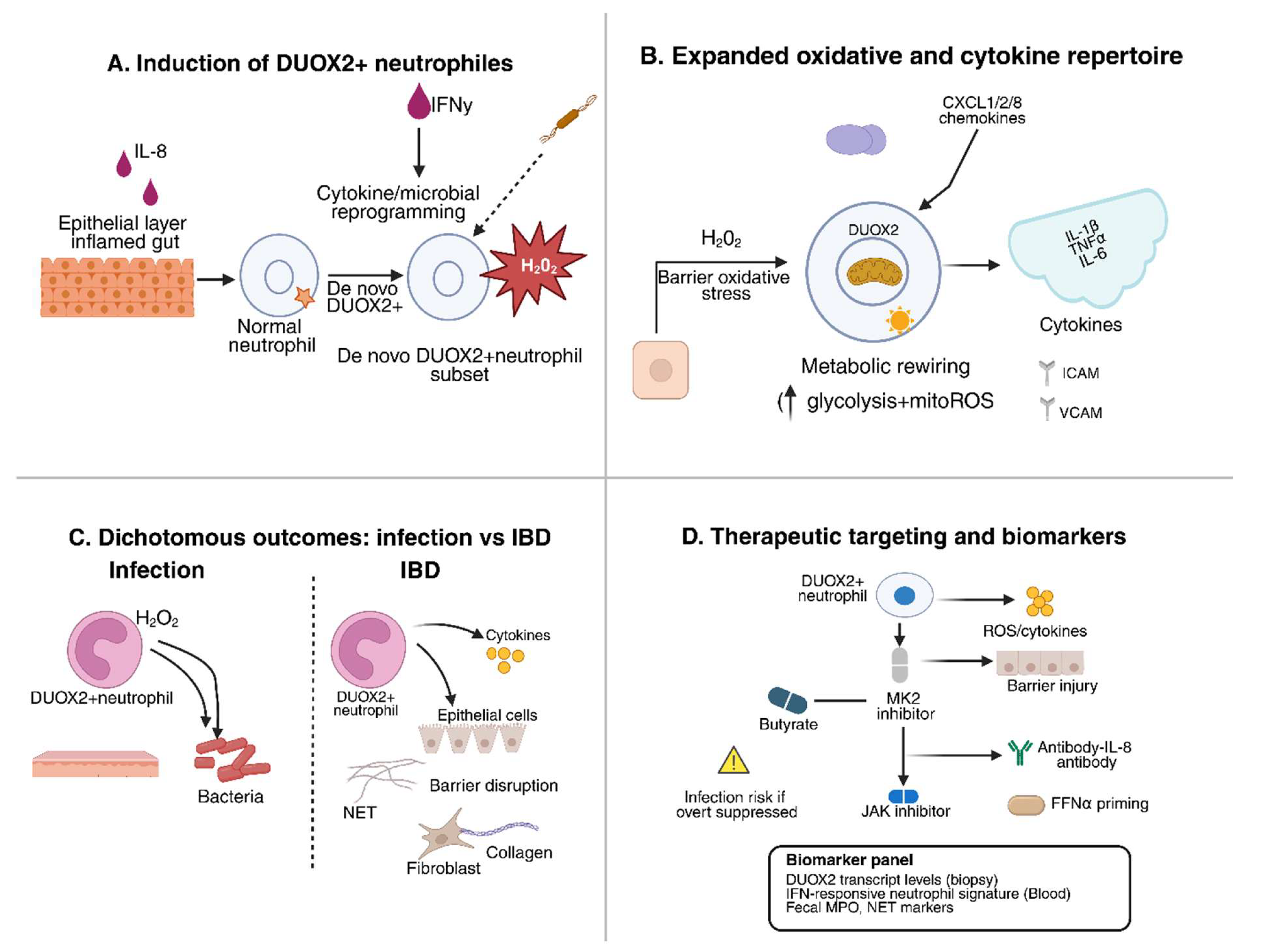
Figure 3.
De novo DUOX2 expression in neutrophils. Panel A: neutrophils recruited into inflamed intestine reprogram to express DUOX2. Panel B: DUOX2+ neutrophils exhibit expanded oxidative and cytokine outputs. Panel C: while protective during infection, DUOX2 neutrophils perpetuate chronic inflammation and fibrosis in IBD. Panel D: therapeutic strategies (butyrate, MK2, JAK, IL-8 blockade) and biomarkers can selectively modulate this axis.
Figure 3.
De novo DUOX2 expression in neutrophils. Panel A: neutrophils recruited into inflamed intestine reprogram to express DUOX2. Panel B: DUOX2+ neutrophils exhibit expanded oxidative and cytokine outputs. Panel C: while protective during infection, DUOX2 neutrophils perpetuate chronic inflammation and fibrosis in IBD. Panel D: therapeutic strategies (butyrate, MK2, JAK, IL-8 blockade) and biomarkers can selectively modulate this axis.
3. DUOX2 in IBD Pathogenesis
3.1. Regulation of epithelial DUOX2 expression
Dual oxidase 2 (DUOX2) is a transmembrane NADPH oxidase located predominantly at the apical membrane of intestinal epithelial cells, where it generates hydrogen peroxide (H₂O₂) as part of mucosal host defense [14]. Under homeostatic conditions, DUOX2 contributes to redox-based signaling and antimicrobial activity, preventing bacterial overgrowth at the mucosal interface. Germ-free mice display markedly reduced intestinal DUOX2 expression, which becomes rapidly upregulated upon colonization with commensal bacteria [15]. Specific pathobionts, such as segmented filamentous bacteria, are potent inducers of epithelial DUOX2 via induction of IL-22 and Th17 responses [45].
Cytokines are central regulators of DUOX2 transcription. IFN-γ, a signature Th1 cytokine enriched in Crohn’s disease (CD), robustly upregulates DUOX2 in human intestinal organoids and epithelial lines [46]. Similarly, IL-22—a cytokine derived from group 3 innate lymphoid cells (ILC3s) and Th17 cells—can synergistically augment DUOX2 induction, enhancing epithelial antimicrobial capacity [45]. On the microbial side, epithelial Toll-like receptor (TLR) signaling provides a second layer of control: TLR4 engagement by bacterial lipopolysaccharide induces DUOX2 activity and H₂O₂ release, which in turn modulates epithelial barrier signaling [37].
Negative regulators of DUOX2 include microbiota-derived metabolites, particularly short-chain fatty acids (SCFAs) such as butyrate. Butyrate downregulates DUOX2 expression at both mRNA and protein levels and attenuates H₂O₂ generation, at least in part via histone deacetylase (HDAC) inhibition [16,33]. Pharmacologic HDAC inhibitors mimic this effect, underscoring the epigenetic sensitivity of DUOX2 transcriptional regulation. Collectively, DUOX2 expression is dynamically tuned by cytokine networks, microbial ligands, and metabolites, positioning it as a central epithelial sensor of microbial imbalance. Key regulators of DUOX2 expression and their pathophysiological consequences are summarized in Table 3.
3.2. DUOX2 as a driver of dysbiosis and barrier dysfunction
Although DUOX2 contributes to epithelial defense, its dysregulated activation in IBD appears pathogenic. Transcriptomic and proteomic profiling of IBD biopsies consistently demonstrate robust DUOX2 upregulation, detectable even in non-inflamed regions or at early disease stages [59]. Elevated DUOX2 correlates with reduced microbial diversity, expansion of Proteobacteria (including adherent-invasive Escherichia coli), and depletion of butyrate-producing Firmicutes [6]. This aligns with data showing that DUOX2-derived H₂O₂ alters the redox landscape of the intestinal lumen, selectively disadvantaging obligate anaerobes while enabling the expansion of facultative pathogens [60].
Mechanistically, chronic DUOX2 activation perturbs epithelial junctional complexes. Persistent H₂O₂ generation damages tight junction proteins such as claudins and zonula occludens-1 (ZO-1), resulting in increased permeability (“leaky gut”) and translocation of luminal bacteria (18) . DUOX2-driven oxidative stress also compromises epithelial stem cell niches, impairing regenerative capacity and sustaining low-grade inflammation (33). Preclinical studies reveal that DUOX2 hyperactivation predisposes mice to barrier breakdown and spontaneous colitis, whereas DUOX2 deficiency renders epithelia more permissive to bacterial overgrowth but protects against inflammation-driven dysbiosis [6,15].
Importantly, DUOX2 does not act in isolation but establishes feed-forward loops with neutrophils and other innate effectors. Excess H₂O₂ triggers epithelial release of chemokines such as IL-8 (CXCL8), which recruit neutrophils to the lamina propria [23]. These neutrophils, in turn, can undergo transcriptional reprogramming to express DUOX2 themselves, amplifying local oxidative stress and perpetuating mucosal injury [31]. Thus, DUOX2 overexpression becomes both a marker and a driver of the self-sustaining inflammatory cycle characteristic of IBD.
3.3. Crosstalk between epithelial DUOX2 and immune-mediated pathology
A critical insight into IBD pathogenesis is the recognition of epithelial DUOX2 as a nodal point connecting microbial dysbiosis, barrier dysfunction, and immune cell recruitment. Spatial transcriptomic analyses of ulcerated IBD tissues show DUOX2 enrichment in epithelial zones adjacent to neutrophil-rich infiltrates, suggesting local oxidative “hot spots” of neutrophil–epithelial crosstalk [22]. In these niches, DUOX2-derived ROS activate NF-κB and p38 MAPK pathways, further inducing epithelial production of TNF, IL-6, and IL-1β, which amplify neutrophil recruitment [37].
Recent work in Crohn’s strictures has implicated DUOX2-driven neutrophil activation in immunofibrosis. IFN-α–primed neutrophils release extracellular traps (NETs) rich in oxidants and proteases that activate intestinal fibroblasts into collagen-producing, pro-fibrotic phenotypes [61]. These fibroblasts, in turn, secrete IL-8, sustaining neutrophil influx and perpetuating the fibrotic loop. DUOX2 induction in neutrophils enhances the oxidative burden within these fibro-inflammatory circuits, linking epithelial redox imbalance to tissue remodeling and fibrosis.
In addition to fibrosis, DUOX2 contributes to carcinogenesis. In mouse models, chronic DUOX2 upregulation increases susceptibility to colitis-associated colorectal cancer by inducing DNA strand breaks, mutagenesis, and epithelial senescence [62]. Human genomic data reveal DUOX2 polymorphisms and rare DUOXA2 mutations in very-early-onset IBD, underscoring its role as a genetic and functional contributor to epithelial pathophysiology [63].
Therapeutically, targeting the DUOX2 axis offers multiple opportunities. Dietary supplementation with butyrate or pharmacologic HDAC inhibitors restores barrier integrity and reduces inflammation via DUOX2 suppression [16,33]. Inhibition of JAK/STAT signaling (e.g., with baricitinib) indirectly dampens IFN-γ/IL-22–driven DUOX2 induction [61]. Small-molecule NADPH oxidase inhibitors are being explored in preclinical colitis models as a means to selectively constrain epithelial oxidative output while preserving basal antimicrobial activity [64].
Collectively, these findings position DUOX2 not merely as an antimicrobial enzyme but as a pathogenic amplifier of intestinal inflammation when dysregulated. By linking microbial cues, epithelial signaling, and immune effector recruitment, DUOX2 creates a vicious cycle that sustains dysbiosis, barrier injury, and chronic inflammation. Deciphering and therapeutically modulating this DUOX2-centered network represents a promising frontier for restoring mucosal homeostasis in IBD.
4. De novo DUOX2 expression in neutrophils
The canonical view of neutrophil redox biology has long centered on the phagocytic NADPH oxidase complex NOX2, which mediates the respiratory burst and intracellular killing of engulfed microbes. However, recent single-cell and bulk transcriptomic analyses have fundamentally challenged this paradigm by demonstrating that neutrophils infiltrating the inflamed intestine can undergo transcriptional reprogramming to express dual oxidase 2 (DUOX2), a NADPH oxidase previously thought to be restricted to epithelial cells [31]. In murine models of dextran sulfate sodium (DSS)-induced colitis, Citrobacter rodentium infection, and in neutrophils isolated from patients with idiopathic inflammatory bowel disease (IBD) or chronic granulomatous disease (CGD), DUOX2 transcripts and protein are robustly induced in subsets of infiltrating neutrophils [31]. These DUOX2^+ neutrophils co-express the maturation factors DUOXA1/2, enabling functional assembly of the DUOX2 complex at the plasma membrane. Functionally, this confers the capacity to generate extracellular hydrogen peroxide (H₂O₂) in addition to the classical phagosomal ROS burst mediated by NOX2 [65,66].
4.1. Functional consequences of neutrophil DUOX2
The emergence of DUOX2^+ neutrophils significantly alters the inflammatory microenvironment. Compared to DUOX2-negative counterparts, DUOX2^+ neutrophils secrete higher levels of chemokines (CXCL1, CXCL2, CXCL8) and cytokines (IL-1β, TNF-α, IL-6), establishing a pro-inflammatory circuit that recruits additional neutrophils and monocytes [31,60,67]. Spatial transcriptomics reveals co-localization of DUOX2^+ neutrophils with epithelial DUOX2 expression, creating oxidative “hot spots” in the mucosa where extracellular H₂O₂ modifies redox signaling, enhances epithelial stress responses, and amplifies NF-κB activation [22].
Genetic dissection underscores the dichotomous impact of this pathway. Mice engineered with conditional myeloid-specific deletion of DUOX2 exhibit attenuated colitis severity, reduced mucosal neutrophil infiltration, and decreased cytokine burden in DSS colitis models [31]. Conversely, during enteric infection, DUOX2-deficient neutrophils are impaired in bacterial clearance, with delayed pathogen elimination and higher microbial loads [31,68]. These findings highlight the dual role of neutrophil DUOX2: beneficial in acute host defense but deleterious in the context of chronic inflammation.
4.2. Mechanistic insights: redox diversification
Mechanistically, DUOX2 expression diversifies the neutrophil redox arsenal beyond the NOX2-driven oxidative burst. Unlike phagosomal ROS, DUOX2-derived H₂O₂ is predominantly extracellular, enabling paracrine modification of surrounding epithelial and stromal cells [37]. This extracellular H₂O₂ promotes neutrophil adhesion by oxidizing integrin thiols, enhances NETosis, and alters epithelial redox tone. In fibro-inflammatory niches, DUOX2 activity supports immunofibrosis by amplifying neutrophil–fibroblast cross-talk, a process dependent on oxidant-driven NET release [32].
DUOX2 also modulates neutrophil metabolism. Single-cell metabolic profiling suggests that DUOX2 induction correlates with increased glycolytic flux and elevated mitochondrial ROS, pointing to a feed-forward redox circuit that stabilizes hypoxia-inducible factor-1α (HIF-1α) and sustains inflammatory transcriptional programs [69,70]. The evolutionary implication is that DUOX2 provides neutrophils with a flexible oxidative strategy, enabling both antimicrobial activity and niche-adapted inflammatory persistence.
4.3. Therapeutic implications
The discovery of DUOX2^+ neutrophils reframes therapeutic strategies in IBD. Global inhibition of ROS is neither feasible nor desirable, given the indispensable role of NOX2 in host defense. However, selective DUOX2 targeting represents a promising approach to attenuate chronic intestinal inflammation without compromising antimicrobial function. Preclinical data show that small-molecule NADPH oxidase inhibitors can preferentially constrain DUOX2-mediated H₂O₂ output, dampen chemokine circuits, and reduce mucosal injury [37,64].
At the translational level, DUOX2^+ neutrophils may serve as biomarkers of disease activity. Their presence in mucosal biopsies correlates with treatment-refractory inflammation, and ongoing efforts are directed at validating DUOX2 transcript and protein expression in large patient cohorts [31,71,72]. Combining DUOX2 inhibition with microbiota-directed therapies (e.g., SCFA supplementation) could synergistically restore mucosal redox balance and barrier function.
In sum, the recognition of DUOX2 as an inducible neutrophil oxidase represents a paradigm shift in our understanding of innate immunity in IBD. By extending neutrophil oxidative capacity beyond NOX2, DUOX2 reprograms neutrophils into context-specific inflammatory effectors that both defend against pathogens and propagate chronic inflammation. Targeting this axis offers new opportunities for therapeutic intervention in IBD. Current and emerging therapeutic strategies that target neutrophil–DUOX2 signaling in IBD are summarized in Table 4.
5. Targeting neutrophils in IBD: therapeutic and diagnostic frontiers
5.1. Why current therapies leave a neutrophil-shaped gap
Biologic agents that modulate adaptive immunity (anti-TNF, anti-IL-12/23) or prevent lymphocyte trafficking (anti-α4β7) have transformed IBD care, yet primary non-response and secondary loss of response remain common. Single-cell and spatial analyses of inflamed colon identify deep-ulcer “pathotypes” dominated by IL-1–driven stromal–neutrophil circuits that persist despite standard therapies and correlate with treatment refractoriness [23]. In these niches, activated fibroblasts and endothelial cells produce IL-1 family cytokines and neutrophil chemokines (e.g., CXCL1/2/8), which recruit and retain neutrophils; in turn, neutrophil proteases, ROS and NETs amplify stromal activation and epithelial stress, creating a self-sustaining loop [23]. Parallel epithelial programs (including DUOX2 upregulation) further fuel neutrophil recruitment and redox stress at the mucosal surface [6,15,37]. Collectively, these mechanistic data rationalize a precision shift toward neutrophil-centric interventions—attenuating pathogenic neutrophil programs, dismantling stromal-neutrophil crosstalk, and restoring epithelial redox–barrier homeostasis—while preserving host defense.
5.2. Drugging neutrophil pathways: from tractable targets to rational combinations
DUOX2 / epithelial–neutrophil redox axis: Epithelial DUOX2 is inducible by dysbiosis, TLR signaling, IFN-γ and IL-22; its overexpression perturbs the luminal redox milieu, weakens tight junctions, and selects for Proteobacteria expansion [6,15,37,45,61]. Although selective DUOX2 inhibitors are not yet available clinically, two feasible routes exist: (i) upstream transcriptional control via cytokine blockade (e.g., JAK inhibition to blunt IFN-driven DUOX2 induction) and epigenetic repression (butyrate/HDAC inhibition) [88,89]; and (ii) downstream oxidase modulation using NADPH oxidase–directed small molecules (noting current agents favor NOX1/4 selectivity and off-target profiles) [64]. Given the emerging evidence that neutrophils can acquire DUOX2 in the IBD niche (and co-localize with DUOX2-high epithelium), strategies that restrain epithelial induction plus neutrophil reprogramming could break redox-chemokine amplification loops [21,70,88].
Chemotaxis and recruitment (IL-8/CXCR2). CXCR2 signaling governs neutrophil trafficking into inflamed mucosa. Genetic and pharmacologic blockade of CXCR2 reduces experimental colitis severity and dampens neutrophil infiltration [90,91]. Human data implicate heightened IL-8/CXCR2 signaling in UC mucosa and peripheral myeloid populations [90].
Translational path: gut-restricted or topically delivered CXCR2 antagonists (to minimize systemic infection risk), potentially in short induction pulses paired with mucosal healing regimens. As fibroblast-derived IL-8 sustains immunofibrosis in CD, CXCR2 antagonism could synergize with anti-fibrotic strategies (see IFN axis below) [32,92].
Type I/II interferon and JAK–STAT signaling: IFN-γ drives epithelial DUOX2; IFN-α primes neutrophils toward pro-fibrotic, NET-competent states that activate intestinal fibroblasts [15,68,88,93]. JAK inhibition (e.g., tofacitinib in UC; upadacitinib in CD) achieves clinical and endoscopic efficacy by intercepting multiple cytokine nodes, plausibly including interferon-dependent priming of neutrophils and stromal compartments [77,78,94]. Precision escalation—early JAK inhibition in patients with interferon-high, neutrophil-enriched pathotypes—merits formal testing [95,96].
Neutrophil ROS generation (MK2/p38 axis) and effector enzymes: MK2 is required for neutrophil NADPH-oxidase activation; myeloid MK2 deletion reduces ROS, MPO, cytokines and histologic injury in DSS colitis [82]. This positions MK2 or upstream p38 as tractable anti-injury nodes. Downstream, myeloperoxidase (MPO) inhibitors (e.g., AZD3241) ameliorate DSS colitis and reduce oxidative tissue damage in preclinical models [97]. Given MPO’s central role in NET formation and chlorinating oxidants, MPO inhibition may couple anti-NET and anti-oxidant benefits.
NETosis and PAD4. NETs disrupt epithelial barriers and propagate thrombo-inflammation. In colitis models, pharmacologic PAD4 inhibition reduces mucosal NET burden; while a 2025 study showed NET reduction without full biomarker normalization, the consistent histologic and barrier improvements across NET-targeting studies support PAD4 (and NET dismantling approaches) as adjunct targets—best deployed in combination rather than monotherapy [98,98].
Trafficking checkpoints (integrins/addressins) with myeloid focus: While vedolizumab primarily limits α4β7^+ lymphocyte homing, anti-trafficking paradigms are expanding to innate compartments. Endothelial MAdCAM-1/PNAd remodeling accompanies UC activity and could be harnessed to reduce myeloid ingress or to deliver drugs selectively to inflamed venules [99,100,101]. This suggests ligand-guided biologics or nanoparticles that exploit gut addressins for myeloid-biased delivery, minimizing systemic exposure.
Because neutrophil pathogenicity is emergent from networked cues (interferons → epithelial DUOX2 → IL-8 → neutrophil influx → NET/ROS → fibroblast activation), two- to three-node combinations may be required. Examples include: JAK inhibition + CXCR2 antagonism (cut interferon priming and recruitment); butyrate/HDAC modulation + MPO inhibition (restore epithelial redox tone and blunt neutrophil oxidants); or NETosis blockade + anti-IL-1 strategies in the IL-1 stromal pathotype [3,82,95,96,97,98,102,103,104,105,106]. Figure 4 provides an overview of the various therapeutic strategies targeting neutrophil pathways in IBD, including JAK inhibition, CXCR2 antagonism, and microbiota modulation. These strategies aim to modulate neutrophil function and restore immune homeostasis without compromising host defense.
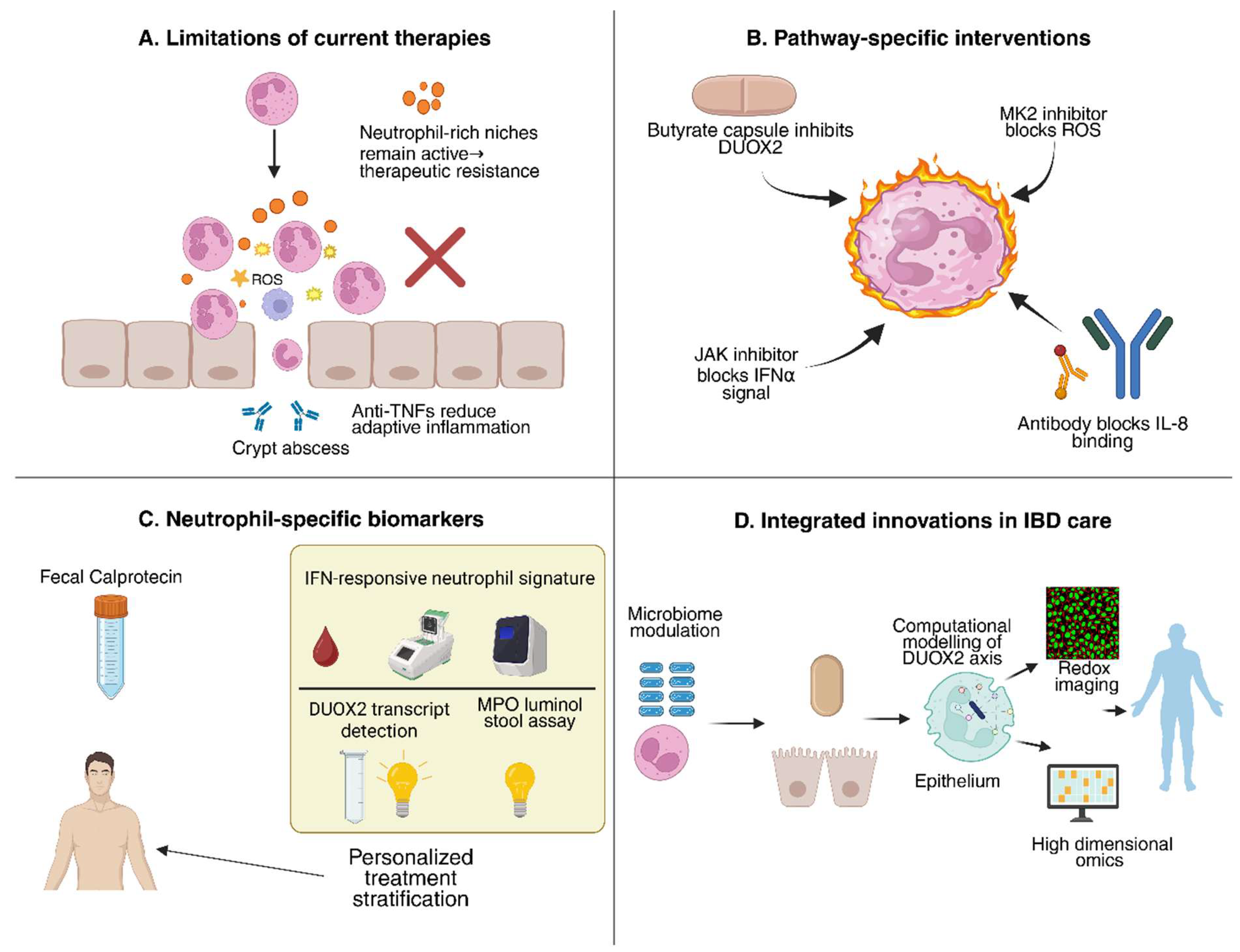
Figure 3.
Therapeutic and diagnostic targeting of neutrophils in IBD. Panel A: limitations of current therapies that fail to resolve neutrophil-driven inflammation. Panel B: pathway-specific interventions targeting DUOX2, MK2, IFNα, and IL-8. Panel C: emerging neutrophil biomarkers enabling personalized patient stratification. Panel D: integrated innovations—including microbiome modulation, systems biology modeling, redox imaging, and omics-based diagnostics—chart a roadmap toward precision IBD care.
Figure 3.
Therapeutic and diagnostic targeting of neutrophils in IBD. Panel A: limitations of current therapies that fail to resolve neutrophil-driven inflammation. Panel B: pathway-specific interventions targeting DUOX2, MK2, IFNα, and IL-8. Panel C: emerging neutrophil biomarkers enabling personalized patient stratification. Panel D: integrated innovations—including microbiome modulation, systems biology modeling, redox imaging, and omics-based diagnostics—chart a roadmap toward precision IBD care.
5.3. Diagnostics and monitoring: beyond calprotectin toward functional neutrophil readouts
Established stool markers: Fecal calprotectin (S100A8/A9) remains a non-invasive mainstay for activity assessment and treat-to-target monitoring; S100A12 and lactoferrin provide complementary inflammatory readouts and help predict relapse and postoperative recurrence [10,107,108]. However, these bulk neutrophil load proxies do not resolve qualitative neutrophil states (e.g., interferon-primed, DUOX2^+, NET-active).
Molecular signatures of neutrophil states: Single-cell atlases and spatial maps supply gene signatures for tissue-adapted neutrophils and IL-1 stromal niches [67,95]. Translationally, blood-based interferon-response signatures in neutrophils could stratify CD patients at risk for immunofibrosis and identify likely JAK-responsive phenotypes [78,93,94]. Likewise, tissue DUOX2^high signatures (epithelium ± neutrophils) might enrich for patients who benefit from redox-modulating regimens [22,88].
NET-related biomarkers: Circulating and fecal MPO–DNA complexes, citrullinated histone H3, and cell-free DNA quantify NET activity and associate with disease activity; NETs also impair barrier function through cGAS–STING activation, providing a mechanistic link to permeability that can be tracked with biomarker panels [98,109]. Incorporating NET assays into monitoring could flag patients likely to respond to PAD4/MPO-targeted adjuncts.
Functional imaging of innate inflammation: PET tracers targeting innate cytokines and myeloid markers (e.g., ^89Zr-anti-IL-1β, ^89Zr-anti-CD11b) detect colonic inflammation with higher sensitivity than MRI in preclinical models and offer lesion-wise quantification of innate activity [110,111]. More broadly, dedicated IBD PET reviews highlight a maturing pipeline (e.g., tracers for MPO activity, IL-1β, VCAM-1) that could complement endoscopy, especially where deep-ulcer myeloid niches drive pathology [112,113]. The near-term translational concept is “innate PET phenotyping” to select patients for neutrophil-targeted therapies and to monitor on-target suppression non-invasively.
Stratified treat-to-target frameworks: Contemporary treat-to-target emphasizes biomarker-guided escalation; embedding neutrophil state metrics (interferon signature, NET biomarkers) alongside calprotectin may refine early decision points and reduce therapeutic drift in non-responders [107]. A practical algorithm is: (i) screen with calprotectin/CRP; (ii) if active disease, phenotype blood/tissue for interferon-neutrophil and DUOX2-redox signatures; (iii) assign add-on targeting (e.g., JAK, CXCR2, NET/MPO, redox modulators) accordingly; (iv) monitor with signature-matched biomarkers and innate-focused PET in complex cases [15,33,78,82,94,95,110,111,112].
5.4. Translational roadmap
Targeted delivery to the myeloid niche: To balance efficacy and safety, gut-selective delivery is paramount. Two concrete modalities: (i) α4β7/MAdCAM-directed vehicles (antibody fragments, peptides) that ferry small-molecule MPO or MK2 inhibitors to inflamed venules and lamina propria [99,100,101,114]; and (ii) pH-triggered oral formulations that release butyrate pro-drugs or HDAC inhibitors at the distal ileum/colon to downregulate epithelial/neutrophil DUOX2 without systemic exposure [64,103].
Network-aware combinations and adaptive dosing: Systems reasoning favors short, induction-phase use of recruitment or NET blockers (CXCR2, PAD4) to blunt the peak influx/NETosis, followed by maintenance with epithelial redox restoration (butyrate/HDAC) and background cytokine control (JAK), with biomarker-based tapering to minimize infection risk [78,82,93,94,95,97,98,103,104,115,116].
Prospective biomarker validation: Multi-center observational cohorts should pre-specify neutrophil pathotypes (interferon-high, DUOX2-redox-high, NET-high) and embed matched interventions to test response enrichment. Parallel development of standardized NET assays and innate PET protocols will enable rigorous pharmacodynamic readouts and dose-finding [98,110,111,112].
Safety guardrails: Because neutrophils are essential for pathogen control, all neutrophil-directed strategies should include infection surveillance, vaccine optimization, and stop rules keyed to biomarker oversuppression (e.g., precipitous falls in NET markers during active infection). The aim is re-balancing—not blanket suppression—of neutrophil functions integral to mucosal defense.
6. Future Directions and Research Gaps
The recognition that neutrophils are not merely terminally differentiated effectors but plastic, transcriptionally adaptable cells has transformed our understanding of their role in inflammatory bowel disease (IBD). The recent discovery that subsets of neutrophils can undergo de novo expression of dual oxidase 2 (DUOX2), a hydrogen peroxide–generating NADPH oxidase previously thought to be restricted to epithelia, has reframed the neutrophil as a redox-modifying cell capable of reshaping the inflammatory tissue niche. This conceptual shift raises pressing questions about prevalence, mechanisms, and clinical implications, which define a broad research agenda for the coming decade. One immediate gap is the need for comprehensive human single-cell profiling. While murine models and small patient cohorts have defined DUOX2+ neutrophils, the true distribution, frequency, and functional heterogeneity of these subsets across Crohn’s disease and ulcerative colitis remain unknown. State-of-the-art multimodal single-cell pipelines now allow simultaneous capture of RNA, protein, chromatin accessibility, and TCR/BCR clonotypes, and when coupled with high-plex spatial transcriptomics and imaging, these can resolve neutrophil states in situ with their tissue and microbial partners [22,117,118,119,120]. Importantly, the largest between-patient variability in inflamed intestine appears to localize to myeloid and neutrophil compartments [22], underscoring the translational importance of such mapping. To be clinically impactful, these efforts must extend beyond cross-sectional atlases to longitudinal cohorts, quantifying whether the appearance of DUOX2+ neutrophils predicts flare onset, therapeutic resistance, or fibrotic complications. Pre-flare or pre-treatment samples that integrate single-cell maps with stool and plasma biomarkers may enable predictive signatures for precision stratification (83,85–87).
At the mechanistic level, DUOX2 biology in neutrophils remains largely unexplored. Unlike the classical NOX2 oxidase, which generates superoxide within phagosomes, DUOX2 localizes to the plasma membrane and produces extracellular hydrogen peroxide, thereby altering the redox microenvironment. Mapping the downstream signaling pathways engaged by this extracellular ROS is critical to understanding its dualistic roles in antimicrobial defense and tissue injury. Redox proteomics now enables site-resolved detection of cysteine sulfenylation across thousands of proteins, revealing how localized peroxide flux rewires kinase cascades, cytoskeletal dynamics, adhesion complexes, and transcription factor activity [121,122,123,124,125]. Applying these workflows to DUOX2+ neutrophils from human IBD biopsies could uncover neutrophil-specific peroxide targets distinct from epithelial ones, such as thiol switches on integrins that regulate chemotaxis, or sulfenylation of inflammasome adaptors that amplify IL-1β release. Comparative redoxomics of neutrophils versus epithelium may therefore delineate cell-type-specific networks and explain how DUOX2-derived oxidants differentially influence epithelial barrier breakdown, stromal activation, or microbial control [126,127]. These mechanistic insights should be coupled with computational approaches: systems models that integrate cytokine networks, epithelial permeability, interferon signaling, and ROS flux can reveal whether DUOX2 activity acts as a bistable switch, pushing mucosal niches into chronic inflammatory attractor states once oxidative thresholds are exceeded [128]. Such models could guide rational design of combination therapies, predicting which nodes must be simultaneously perturbed to reset the system toward homeostasis.
Biomarker discovery represents a parallel frontier. Current non-invasive tools such as fecal calprotectin reflect bulk neutrophil load but fail to distinguish protective from pathogenic states [10,107,108]. The identification of interferon-primed neutrophil signatures in Crohn’s disease, DUOX2+ neutrophils in active colitis, and NET-associated proteomic footprints provides opportunities to design next-generation biomarkers [32,109]. Candidate assays include blood-based IFN-responsive gene panels, stool DUOX2 transcripts or oxidized-protein surrogates, and circulating MPO–DNA complexes as NET proxies. Moreover, device-ready luminol chemistries for stool myeloperoxidase have recently been validated as rapid inflammation readouts [129], opening the door to point-of-care neutrophil function testing. Embedding these markers into longitudinal treat-to-target frameworks (STRIDE-II) [130] could allow earlier escalation in patients with emerging DUOX2/neutrophil pathotypes, rather than waiting for clinical relapse or gross mucosal damage. Equally, integrating biomarker endpoints into clinical trials of novel therapies would accelerate mechanism-of-action understanding and de-risk development.
Interventional studies are urgently needed to translate these mechanistic insights into therapeutic practice. Several rational strategies are already in view. Butyrate, a microbiota-derived short-chain fatty acid reduced in IBD, downregulates epithelial and neutrophil DUOX2 expression via histone deacetylase inhibition and restores barrier function [15,77,102,131,132,133]. While early clinical trials of butyrate enemas yielded inconsistent results, newer oral formulations with colonic-release profiles and microbiome-based delivery may prove more effective, particularly when stratified to patients with DUOX2-high pathotypes. Similarly, inhibitors of the MAPK-activated protein kinase 2 (MK2), required for neutrophil-derived ROS production, ameliorate colitis in preclinical models [81,82,105,134,135]. Repurposing MK2 inhibitors or peptides could therefore attenuate neutrophil-mediated tissue damage without compromising antimicrobial phagocytosis. For Crohn’s disease with fibrotic complications, disrupting the IFNα–neutrophil–fibroblast axis represents a compelling anti-fibrotic strategy, potentially through JAK inhibition or direct IFNAR blockade, informed by safety precedents in systemic lupus erythematosus [32,136]. Furthermore, targeting NETosis through PAD4 inhibition has demonstrated reduction in mucosal NET burden in colitis models [98,104], though normalizing inflammatory biomarkers requires combination with upstream cytokine or chemokine blockade. A unifying principle is that neutrophil-targeted interventions should be network-aware, deployed in short induction pulses to suppress acute amplification loops, then transitioned to maintenance regimens that restore epithelial and microbial homeostasis.
Safety remains a non-trivial challenge. Neutrophils and their oxidases are indispensable for host defense, as evidenced by the susceptibility to infection in chronic granulomatous disease. Any DUOX2-directed therapy must therefore balance suppression of pathogenic overactivity against preservation of antimicrobial capacity. This argues for gut-selective delivery systems—oral pH-triggered release, ligand-directed nanoparticles, or α4β7/MAdCAM-targeted vehicles that deposit drugs specifically in inflamed mucosa [99,100,101,114]. Equally, biomarker-driven dosing and infection surveillance will be necessary guardrails, ensuring that neutrophil suppression does not cross thresholds of impaired pathogen clearance. Prospective cohort studies should embed infection outcomes and vaccine optimization as integral endpoints when trialing neutrophil- or DUOX2-targeted agents.
Finally, it is important to situate these discoveries within the broader landscape of immune-mediated disease. DUOX2+ neutrophils may not be unique to IBD; analogous plasticity may occur in other neutrophil-rich niches such as rheumatoid arthritis synovium, psoriasis plaques, or vasculitic lesions. Comparative profiling across diseases could reveal conserved versus gut-specific programs, distinguishing features driven by microbial exposure from those induced by sterile inflammatory signals. If DUOX2+ neutrophils prove to be a generalizable phenomenon of chronic inflammation, therapeutic strategies developed in IBD may have far wider applicability. Conversely, if they are gut-specific, they provide a compelling explanation for the unique chronicity and microbial dependence of intestinal inflammation. Either outcome would fundamentally reshape our understanding of neutrophil biology.
The emerging neutrophil–DUOX2 axis defines a fertile ground for translational discovery in IBD. The next wave of research must integrate high-dimensional human mapping with mechanistic redox proteomics, embed neutrophil-centric biomarkers into longitudinal treat-to-target designs, and test targeted interventions in biomarker-stratified cohorts. Computational modeling should accompany empirical studies to predict network dynamics and guide rational combinations. Above all, translation must proceed with infection-conscious safeguards, leveraging gut-selective delivery and adaptive dosing. By closing these gaps, the field can move beyond descriptive immunology toward actionable, precision interventions that recalibrate neutrophil plasticity and redox balance, offering patients durable remission and protection from irreversible complications. Figure 5 illustrates the future directions and research gaps in neutrophil–DUOX2 biology in IBD. It highlights key areas of exploration, such as human single-cell profiling, mechanistic mapping of DUOX2 signaling, longitudinal studies in patient cohorts, and innovative interventional trials to validate these findings.
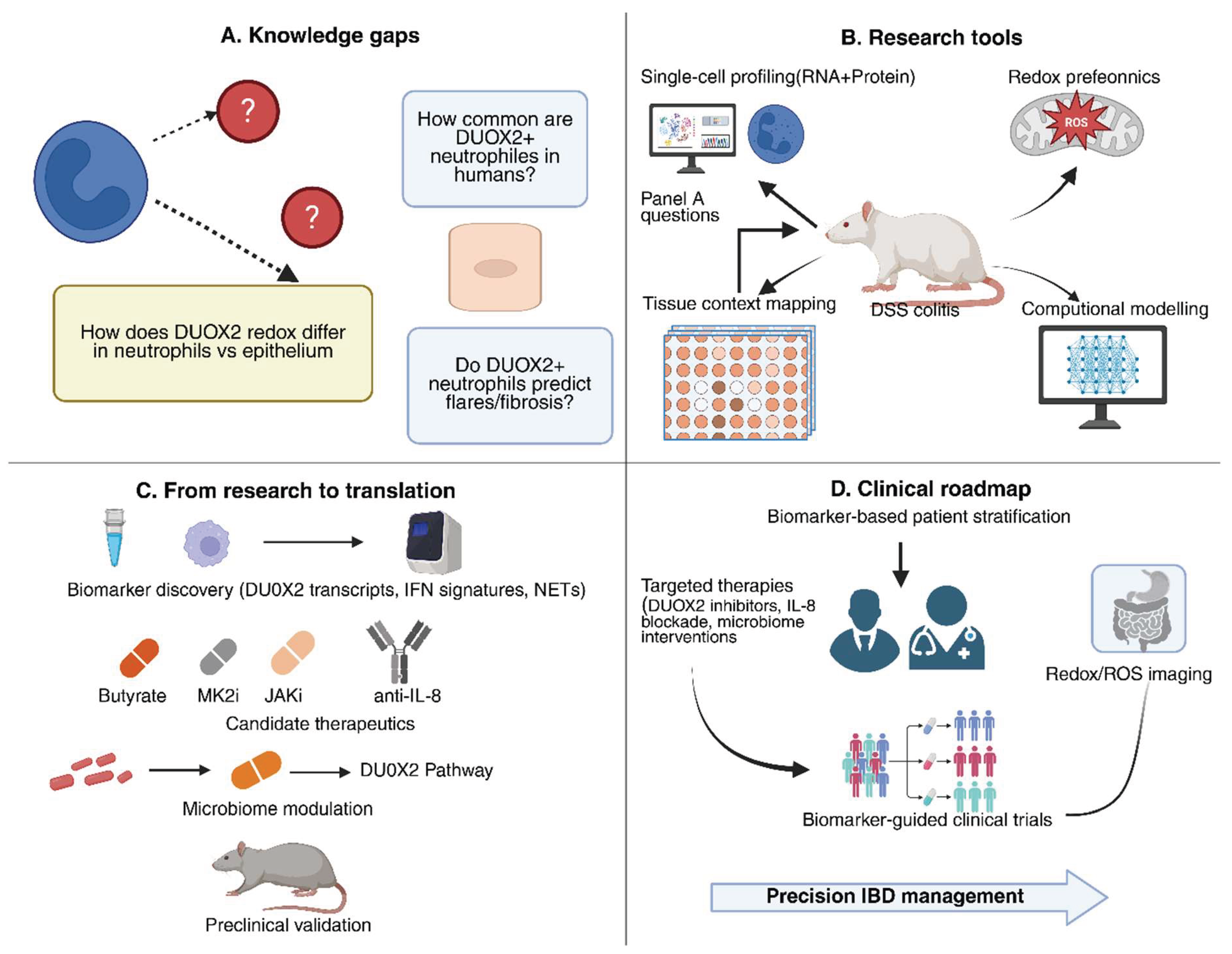
Figure 4.
Future directions and research gaps. Panel A: key unanswered questions regarding DUOX2+ neutrophils in IBD. Panel B: experimental tools—single-cell profiling, redox proteomics, spatial transcriptomics, computational modeling—to address these questions. Panel C: translation of discoveries into biomarkers and therapeutic strategies. Panel D: integration into clinical practice via patient stratification, targeted therapy, imaging, and biomarker-guided trials toward precision IBD management.
Figure 4.
Future directions and research gaps. Panel A: key unanswered questions regarding DUOX2+ neutrophils in IBD. Panel B: experimental tools—single-cell profiling, redox proteomics, spatial transcriptomics, computational modeling—to address these questions. Panel C: translation of discoveries into biomarkers and therapeutic strategies. Panel D: integration into clinical practice via patient stratification, targeted therapy, imaging, and biomarker-guided trials toward precision IBD management.
7. Conclusions
The recognition that neutrophils are not static effectors but highly plastic cells capable of de novo DUOX2 expression marks a paradigm shift in our understanding of IBD pathogenesis. This axis integrates innate immunity, epithelial redox biology, and microbial ecology into a self-perpetuating loop that fuels chronic inflammation, barrier dysfunction, and fibrotic remodeling. By reframing neutrophils as context-dependent regulators—protective in host defense yet pathogenic when maladapted—this perspective highlights both the complexity and therapeutic potential of the neutrophil–DUOX2 circuit.
Translational opportunities are already in sight. Preclinical data suggest that modulating DUOX2 activity, intercepting IFN- and IL-8–driven neutrophil programs, or restoring microbiota-derived metabolites such as butyrate can attenuate mucosal damage without abolishing essential antimicrobial functions. The challenge ahead is to translate these mechanistic insights into therapies that recalibrate, rather than suppress, neutrophil responses. Equally critical is the development of biomarkers that capture neutrophil states and DUOX2 activity, enabling precise patient stratification and treat-to-target frameworks.
Ultimately, future progress will depend on integrating high-dimensional human profiling, redox proteomics, and systems modeling with biomarker-guided clinical trials. Such efforts can define when and how DUOX2+ neutrophils emerge, how they shape tissue outcomes, and how best to intervene without compromising immunity. If realized, this paradigm may shift IBD management from broad immunosuppression to tailored modulation of innate immune plasticity, offering more durable remission and reduced complications. By harnessing, rather than fearing, neutrophil adaptability, the field has the opportunity to transform chronic gut inflammation into a tractable and preventable disorder.
Funding
The author(s) acknowledge the funds received by Uddalak Das from the Department of Biotechnology (Grant No: DBTHRDPMU/JRF/BET-24/I/2024-25/376), the Council of Scientific and Industrial Research (Grant No: 24J/01/00130), and the Indian Council of Medical Research (Grant No: 3/1/3/BRET-2024/HRD (L1)).
Biotechnology
(Grant No: DBTHRDPMU/JRF/BET-24/I/2024-25/376), the Council of Scientific and Industrial Research (Grant No: 24J/01/00130), and the Indian Council of Medical Research (Grant No: 3/1/3/BRET-2024/HRD (L1)).
Industrial Research
(Grant No: 24J/01/00130), and the Indian Council of Medical Research (Grant No: 3/1/3/BRET-2024/HRD (L1)).
Authorship Contribution Statement
IS.T. Gopukumar: Writing - Review & Editing; Dyumn Dwivedi: Writing - Original Draft, Writing - Review & Editing, Visualization; S. Muthumareeswari: Writing - Review & Editing; Prabhjot Kaur: Writing - Review & Editing, Referencing; Randhir Singh: Visualization, Referencing; Karpakavalli Meenakshi Sundaram: Writing - Review & Editing; Uddalak Das: Conceptualization, Project Administration, Supervision, Funding Acquisition, Review & Editing. All authors critically revised the paper and approved the final version
Conflicts of Interest
The author(s) report no conflict of interest.
Declaration of generative AI and AI-assisted technologies in the writing process
The writing of this review paper involved the use of generative AI and AI-assisted technologies only to enhance the clarity, coherence, and overall quality of the manuscript. The authors acknowledge the contributions of AI in the writing process while ensuring that the final content reflects the author's own insights and interpretations of the literature. All interpretations and conclusions drawn in this manuscript are the sole responsibility of the author.
Data and Materials Availability
Not applicable.
References
- Kaplan GG, Windsor JW. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2021 Jan;18(1):56–66.
- Neurath MF. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat Immunol. 2019 Aug;20(8):970–9.
- Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012 Nov 3;380(9853):1606–19.
- Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, Abedian S, Cheon JH, Cho J, Dayani NE, Franke L, Fuyuno Y, Hart A, Juyal RC, Juyal G, Kim WH, Morris AP, Poustchi H, Newman WG, Midha V, Orchard TR, Vahedi H, Sood A, Sung JY, Malekzadeh R, Westra HJ, Yamazaki K, Yang SK, International Multiple Sclerosis Genetics Consortium, International IBD Genetics Consortium, Barrett JC, Alizadeh BZ, Parkes M, Bk T, Daly MJ, Kubo M, Anderson CA, Weersma RK. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015 Sep;47(9):979–86.
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Büning C, Cohain A, Cichon S, D’Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H, International IBD Genetics Consortium (IIBDGC), Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A, Parkes M, Vermeire S, Barrett JC, Cho JH. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012 Nov 1;491(7422):119–24.
- Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol. 2018 Feb;11(1):1–10.
- Mitsialis V, Wall S, Liu P, Ordovas-Montanes J, Parmet T, Vukovic M, Spencer D, Field M, McCourt C, Toothaker J, Bousvaros A, Boston Children’s Hospital Inflammatory Bowel Disease Center, Brigham and Women’s Hospital Crohn’s and Colitis Center, Shalek AK, Kean L, Horwitz B, Goldsmith J, Tseng G, Snapper SB, Konnikova L. Single-Cell Analyses of Colon and Blood Reveal Distinct Immune Cell Signatures of Ulcerative Colitis and Crohn’s Disease. Gastroenterology. 2020 Aug;159(2):591-608.e10.
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013 Mar;13(3):159–75.
- Sidler MA, Leach ST, Day AS. Fecal S100A12 and fecal calprotectin as noninvasive markers for inflammatory bowel disease in children. Inflamm Bowel Dis. 2008 Mar;14(3):359–66.
- Ishida N, Onoue S, Takebe T, Takahashi K, Asai Y, Tamura S, Matsuura T, Yamade M, Iwaizumi M, Hamaya Y, Yamada T, Osawa S, Sugimoto K. Fecal Calprotectin as a Biomarker of Crohn’s Disease in Patients With Short Disease Durations: A Prospective, Single-Center, Cross-Sectional Study. [cited 2025 Sep 4]; Available from: https://onlinelibrary.wiley.com/doi/10.1155/grp/9984055.
- Zhou G, Yu L, Fang L, Yang W, Yu T, Miao Y, Chen M, Wu K, Chen F, Cong Y, Liu Z. CD177+ neutrophils as functionally activated neutrophils negatively regulate IBD. Gut. 2018 Jun;67(6):1052–63.
- Xie X, Shi Q, Wu P, Zhang X, Kambara H, Su J, Yu H, Park SY, Guo R, Ren Q, Zhang S, Xu Y, Silberstein LE, Cheng T, Ma F, Li C, Luo HR. Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat Immunol. 2020 Sep;21(9):1119–33.
- Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J, Sud M, Andrews E, Velonias G, Haber AL, Jagadeesh K, Vickovic S, Yao J, Stevens C, Dionne D, Nguyen LT, Villani AC, Hofree M, Creasey EA, Huang H, Rozenblatt-Rosen O, Garber JJ, Khalili H, Desch AN, Daly MJ, Ananthakrishnan AN, Shalek AK, Xavier RJ, Regev A. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell. 2019 Jul 25;178(3):714-730.e22.
- Donkó A, Péterfi Z, Sum A, Leto T, Geiszt M. Dual oxidases. Philos Trans R Soc Lond B Biol Sci. 2005 Dec 29;360(1464):2301–8.
- Grasberger H, Gao J, Nagao-Kitamoto H, Kitamoto S, Zhang M, Kamada N, Eaton KA, El-Zaatari M, Shreiner AB, Merchant JL, Owyang C, Kao JY. Increased Expression of DUOX2 Is an Epithelial Response to Mucosal Dysbiosis Required for Immune Homeostasis in Mouse Intestine. Gastroenterology. 2015 Dec;149(7):1849–59.
- Aguilar EC, Santos LCD, Leonel AJ, de Oliveira JS, Santos EA, Navia-Pelaez JM, da Silva JF, Mendes BP, Capettini LSA, Teixeira LG, Lemos VS, Alvarez-Leite JI. Oral butyrate reduces oxidative stress in atherosclerotic lesion sites by a mechanism involving NADPH oxidase down-regulation in endothelial cells. J Nutr Biochem. 2016 Aug;34:99–105.
- Wen X, Tang L, Zhong R, Liu L, Chen L, Zhang H. Role of Mitophagy in Regulating Intestinal Oxidative Damage. Antioxidants (Basel). 2023 Feb 14;12(2):480.
- Haque PS, Kapur N, Barrett TA, Theiss AL. Mitochondrial function and gastrointestinal diseases. Nat Rev Gastroenterol Hepatol. 2024 Aug;21(8):537–55.
- Filep JG. Targeting Neutrophils for Promoting the Resolution of Inflammation. Front Immunol [Internet]. 2022 Mar 16 [cited 2025 Sep 13];13. Available from: https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.866747/full.
- Byun MJ, Nakasone ES, Shin HE, Lee H, Park JC, Lee W, Park W, Park CG, Park J, Kim SN. Advanced Nanoparticle Therapeutics for Targeting Neutrophils in Inflammatory Diseases. Adv Healthc Mater. 2025 Jul 22;e2502092.
- Wigerblad G, Cao Q, Brooks S, Naz F, Gadkari M, Jiang K, Gupta S, O’Neil L, Dell’Orso S, Kaplan MJ, Franco LM. Single-Cell Analysis Reveals the Range of Transcriptional States of Circulating Human Neutrophils. J Immunol. 2022 Aug 15;209(4):772–82.
- Garrido-Trigo A, Corraliza AM, Veny M, Dotti I, Melón-Ardanaz E, Rill A, Crowell HL, Corbí Á, Gudiño V, Esteller M, Álvarez-Teubel I, Aguilar D, Masamunt MC, Killingbeck E, Kim Y, Leon M, Visvanathan S, Marchese D, Caratù G, Martin-Cardona A, Esteve M, Ordás I, Panés J, Ricart E, Mereu E, Heyn H, Salas A. Macrophage and neutrophil heterogeneity at single-cell spatial resolution in human inflammatory bowel disease. Nat Commun. 2023 Jul 26;14(1):4506.
- Friedrich M, Pohin M, Jackson MA, Korsunsky I, Bullers SJ, Rue-Albrecht K, Christoforidou Z, Sathananthan D, Thomas T, Ravindran R, Tandon R, Peres RS, Sharpe H, Wei K, Watts GFM, Mann EH, Geremia A, Attar M, Oxford IBD Cohort Investigators, Roche Fibroblast Network Consortium, McCuaig S, Thomas L, Collantes E, Uhlig HH, Sansom SN, Easton A, Raychaudhuri S, Travis SP, Powrie FM. IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat Med. 2021 Nov;27(11):1970–81.
- Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. 2010 Aug;31(8):318–24.
- Xu H, Zhan M, Wu Z, Chen J, Zhao Y, Feng F, Wang F, Li Y, Zhang S, Liu Y. Aberrant expansion of CD177+ neutrophils promotes endothelial dysfunction in systemic lupus erythematosus via neutrophil extracellular traps. J Autoimmun. 2025 Mar;152:103399.
- Bai M, Grieshaber-Bouyer R, Wang J, Schmider AB, Wilson ZS, Zeng L, Halyabar O, Godin MD, Nguyen HN, Levescot A, Cunin P, Lefort CT, Soberman RJ, Nigrovic PA. CD177 modulates human neutrophil migration through activation-mediated integrin and chemoreceptor regulation. Blood. 2017 Nov 9;130(19):2092–100.
- von Vietinghoff S, Tunnemann G, Eulenberg C, Wellner M, Cristina Cardoso M, Luft FC, Kettritz R. NB1 mediates surface expression of the ANCA antigen proteinase 3 on human neutrophils. Blood. 2007 May 15;109(10):4487–9.
- Hacbarth E, Kajdacsy-Balla A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum. 1986 Nov;29(11):1334–42.
- Blanco-Camarillo C, Alemán OR, Rosales C. Low-Density Neutrophils in Healthy Individuals Display a Mature Primed Phenotype. Front Immunol. 2021;12:672520.
- Neutrophils in cancer: heterogeneous and multifaceted | Request PDF. ResearchGate [Internet]. [cited 2025 Sep 14]; Available from: https://www.researchgate.net/publication/353047575_Neutrophils_in_cancer_heterogeneous_and_multifaceted.
- Singh AK, Ainciburu M, Wynne K, Bhat SA, Blanco A, Tzani I, Akiba Y, Lalor SJ, Kaunitz J, Bourke B, Kelly VP, Doherty GA, Zerbe CS, Clarke C, Hussey S, Knaus UG. De novo DUOX2 expression in neutrophil subsets shapes the pathogenesis of intestinal disease. Proc Natl Acad Sci U S A. 2025 May 13;122(19):e2421747122.
- Gavriilidis E, Divolis G, Natsi AM, Kafalis N, Kogias D, Antoniadou C, Synolaki E, Pavlos E, Koutsi MA, Didaskalou S, Papadimitriou E, Tsironidou V, Gavriil A, Papadopoulos V, Agelopoulos M, Tsilingiris D, Koffa M, Giatromanolaki A, Kouklakis G, Ritis K, Skendros P. Neutrophil-fibroblast crosstalk drives immunofibrosis in Crohn’s disease through IFNα pathway. Front Immunol. 2024 Sep 13;15:1447608.
- Li G, Lin J, Zhang C, Gao H, Lu H, Gao X, Zhu R, Li Z, Li M, Liu Z. Microbiota metabolite butyrate constrains neutrophil functions and ameliorates mucosal inflammation in inflammatory bowel disease. Gut Microbes. 2021;13(1):1968257.
- De Filippo K, Rankin SM. CXCR4, the master regulator of neutrophil trafficking in homeostasis and disease. Eur J Clin Invest. 2018 Nov;48 Suppl 2(Suppl Suppl 2):e12949.
- Chen F, Liu Y, Shi Y, Zhang J, Liu X, Liu Z, Lv J, Leng Y. The emerging role of neutrophilic extracellular traps in intestinal disease. Gut Pathog. 2022 Jun 22;14(1):27.
- Segal, AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223.
- Aviello G, Knaus UG. NADPH oxidases and ROS signaling in the gastrointestinal tract. Mucosal Immunol. 2018 Jul;11(4):1011–23.
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004 Mar 5;303(5663):1532–5.
- Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol. 2012 Sep 15;189(6):2689–95.
- Pham CTN. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006 Jul;6(7):541–50.
- Kabouridis PS, Lasrado R, McCallum S, Chng SH, Snippert HJ, Clevers H, Pettersson S, Pachnis V. The gut microbiota keeps enteric glial cells on the move; prospective roles of the gut epithelium and immune system. Gut Microbes. 2015 Nov 11;6(6):398–403.
- Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol. 2011 Oct;32(10):452–60.
- Zhu N, Zhu J, Mei Q. Low-density Granulocytes as a Novel Biomarkers of Disease Activity in IBD. Inflammatory Bowel Diseases. 2023 Aug 1;29(8):e31–e31.
- Delaveris C, Wilk A, Riley N, Stark J, Yang S, Rogers A, Ranganath T, Nadeau K, Blish C, Bertozzi C. Synthetic Siglec-9 Agonists Inhibit Neutrophil Activation Associated with COVID-19 [Internet]. Chemistry; 2020 [cited 2025 Sep 14]. Available from: https://chemrxiv.org/engage/chemrxiv/article-details/60c75305469df463f7f44ca1.
- Sonnenberg GF, Fouser LA, Artis D. Functional biology of the IL-22-IL-22R pathway in regulating immunity and inflammation at barrier surfaces. Adv Immunol. 2010;107:1–29.
- Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004 Feb;75(2):163–89.
- Ha EM, Oh CT, Bae YS, Lee WJ. A direct role for dual oxidase in Drosophila gut immunity. Science. 2005 Nov 4;310(5749):847–50.
- Grasberger H, Magis AT, Sheng E, Conomos MP, Zhang M, Garzotto LS, Hou G, Bishu S, Nagao-Kitamoto H, El-Zaatari M, Kitamoto S, Kamada N, Stidham RW, Akiba Y, Kaunitz J, Haberman Y, Kugathasan S, Denson LA, Omenn GS, Kao JY. DUOX2 variants associate with preclinical disturbances in microbiota-immune homeostasis and increased inflammatory bowel disease risk. J Clin Invest. 2021 May 3;131(9):e141676, 141676.
- Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol. 2011 Jan;4(1):31–42.
- Sun X, Chalmers L, Fu X, Zhao M. A Molecular Link Between Interleukin 22 and Intestinal Mucosal Wound Healing. Adv Wound Care (New Rochelle). 2012 Dec 1;1(6):231–7.
- Lamas B, Natividad JM, Sokol H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol. 2018 Jul;11(4):1024–38.
- Small CL, Xing L, McPhee JB, Law HT, Coombes BK. Acute Infectious Gastroenteritis Potentiates a Crohn’s Disease Pathobiont to Fuel Ongoing Inflammation in the Post-Infectious Period. PLOS Pathogens. 2016 Oct 6;12(10):e1005907.
- Ragland SA, Criss AK. From bacterial killing to immune modulation: Recent insights into the functions of lysozyme. PLoS Pathog. 2017 Sep;13(9):e1006512.
- Bots S, Nylund K, Löwenberg M, Gecse K, D’Haens G. Intestinal Ultrasound to Assess Disease Activity in Ulcerative Colitis: Development of a novel UC-Ultrasound Index. J Crohns Colitis. 2021 Aug 2;15(8):1264–71.
- Shang J, Ding M, Zhou X. Recent advances in S-palmitoylation and its emerging roles in human diseases. J Hematol Oncol. 2025 Sep 1;18:83.
- Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol. 2011 Mar 28;17(12):1519–28.
- Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. 2003 Jul;133(7 Suppl):2485S-2493S.
- Jarade A, Garcia Z, Marie S, Demera A, Prinz I, Bousso P, Di Santo JP, Serafini N. Inflammation triggers ILC3 patrolling of the intestinal barrier. Nat Immunol. 2022;23(9):1317–23.
- Ostrowski J, Dabrowska M, Lazowska I, Paziewska A, Balabas A, Kluska A, Kulecka M, Karczmarski J, Ambrozkiewicz F, Piatkowska M, Goryca K, Zeber-Lubecka N, Kierkus J, Socha P, Lodyga M, Klopocka M, Iwanczak B, Bak-Drabik K, Walkowiak J, Radwan P, Grzybowska-Chlebowczyk U, Korczowski B, Starzynska T, Mikula M. Redefining the Practical Utility of Blood Transcriptome Biomarkers in Inflammatory Bowel Diseases. J Crohns Colitis. 2019 Apr 26;13(5):626–33.
- Hegazy AN, West NR, Stubbington MJT, Wendt E, Suijker KIM, Datsi A, This S, Danne C, Campion S, Duncan SH, Owens BMJ, Uhlig HH, McMichael A, Oxford IBD Cohort Investigators, Bergthaler A, Teichmann SA, Keshav S, Powrie F. Circulating and Tissue-Resident CD4+ T Cells With Reactivity to Intestinal Microbiota Are Abundant in Healthy Individuals and Function Is Altered During Inflammation. Gastroenterology. 2017 Nov;153(5):1320-1337.e16.
- Li JY, Xiao J, Gao M, Zhou HF, Fan H, Sun F, Cui DD. IRF/Type I IFN signaling serves as a valuable therapeutic target in the pathogenesis of inflammatory bowel disease. International Immunopharmacology. 2021 Mar 1;92:107350.
- Westbrook AM, Szakmary A, Schiestl RH. Mechanisms of intestinal inflammation and development of associated cancers: lessons learned from mouse models. Mutat Res. 2010;705(1):40–59.
- Parlato M, Charbit-Henrion F, Hayes P, Tiberti A, Aloi M, Cucchiara S, Bègue B, Bras M, Pouliet A, Rakotobe S, Ruemmele F, Knaus UG, Cerf-Bensussan N. First Identification of Biallelic Inherited DUOX2 Inactivating Mutations as a Cause of Very Early Onset Inflammatory Bowel Disease. Gastroenterology. 2017 Aug;153(2):609-611.e3.
- Altenhöfer S, Radermacher KA, Kleikers PWM, Wingler K, Schmidt HHHW. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid Redox Signal. 2015 Aug 10;23(5):406–27.
- Donkó A, Péterfi Z, Sum A, Leto T, Geiszt M. Dual oxidases. Philos Trans R Soc Lond B Biol Sci. 2005 Dec 29;360(1464):2301–8.
- Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 2003 Aug;17(11):1502–4.
- Staicu I. Neutrophil subsets. Nat Immunol. 2024 Apr;25(4):583–583.
- DUOX2 activation drives bacterial translocation and subclinical inflammation in IBD-associated dysbiosis | Gut [Internet]. [cited 2025 Sep 13]. Available from: https://gut.bmj.com/content/74/10/1589.
- Ortega-Zapero M, Gomez-Bris R, Pascual-Laguna I, Saez A, Gonzalez-Granado JM. Neutrophils and NETs in Pathophysiology and Treatment of Inflammatory Bowel Disease. Int J Mol Sci. 2025 Jul 23;26(15):7098.
- Zaiatz Bittencourt V, Jones F, Doherty G, Ryan EJ. Targeting Immune Cell Metabolism in the Treatment of Inflammatory Bowel Disease. Inflamm Bowel Dis. 2021 Mar 10;27(10):1684–93.
- Taman H, Fenton CG, Hensel IV, Anderssen E, Florholmen J, Paulssen RH. Transcriptomic Landscape of Treatment-Naïve Ulcerative Colitis. J Crohns Colitis. 2018 Feb 28;12(3):327–36.
- Nowak JK, Adams AT, Kalla R, Lindstrøm JC, Vatn S, Bergemalm D, Keita ÅV, Gomollón F, Jahnsen J, Vatn MH, Ricanek P, Ostrowski J, Walkowiak J, Halfvarson J, Satsangi J, IBD Character Consortium. Characterisation of the Circulating Transcriptomic Landscape in Inflammatory Bowel Disease Provides Evidence for Dysregulation of Multiple Transcription Factors Including NFE2, SPI1, CEBPB, and IRF2. J Crohns Colitis. 2022 Aug 1;16(8):1255–68.
- Grasberger H, Magis AT, Sheng E, Conomos MP, Zhang M, Garzotto LS, Hou G, Bishu S, Nagao-Kitamoto H, El-Zaatari M, Kitamoto S, Kamada N, Stidham RW, Akiba Y, Kaunitz J, Haberman Y, Kugathasan S, Denson LA, Omenn GS, Kao JY. DUOX2 variants associate with preclinical disturbances in microbiota-immune homeostasis and increased inflammatory bowel disease risk. J Clin Invest. 2021 May 3;131(9):e141676, 141676.
- Ota S, Sakuraba H. Uptake and Advanced Therapy of Butyrate in Inflammatory Bowel Disease. Immuno. 2022 Dec;2(4):692–702.
- Shin Y, Han S, Kwon J, Ju S, Choi TG, Kang I, Kim SS. Roles of Short-Chain Fatty Acids in Inflammatory Bowel Disease. Nutrients. 2023 Oct 21;15(20):4466.
- Conder E, Shay HC, Vekaria H, Erinkitola I, Bhogoju S, Goretsky T, Sullivan P, Barrett T, Kapur N. BUTYRATE-INDUCED MITOCHONDRIAL FUNCTION IMPROVES BARRIER FUNCTION IN INFLAMMATORY BOWEL DISEASE (IBD). Inflamm Bowel Dis. 2023 Feb 1;29(Supplement_1):S71–2.
- Sandborn, WJ. Are short-chain fatty acid enemas effective for left-sided ulcerative colitis? Gastroenterology. 1998 Jan;114(1):218–9.
- Sandborn WJ, Su C, Sands BE, D’Haens GR, Vermeire S, Schreiber S, Danese S, Feagan BG, Reinisch W, Niezychowski W, Friedman G, Lawendy N, Yu D, Woodworth D, Mukherjee A, Zhang H, Healey P, Panés J, OCTAVE Induction 1, OCTAVE Induction 2, and OCTAVE Sustain Investigators. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2017 May 4;376(18):1723–36.
- Zhang X, Guo R, Kambara H, Ma F, Luo HR. The role of CXCR2 in acute inflammatory responses and its antagonists as anti-inflammatory therapeutics. Curr Opin Hematol. 2019 Jan;26(1):28–33.
- Sitaru S, Budke A, Bertini R, Sperandio M. Therapeutic inhibition of CXCR1/2: where do we stand? Intern Emerg Med. 2023 May 30;1–18.
- Sun L, Wu Q, Nie Y, Cheng N, Wang R, Wang G, Zhang D, He H, Ye RD, Qian F. A Role for MK2 in Enhancing Neutrophil-Derived ROS Production and Aggravating Liver Ischemia/Reperfusion Injury. Front Immunol. 2018;9:2610.
- Zhang T, Jiang J, Liu J, Xu L, Duan S, Sun L, Zhao W, Qian F. MK2 Is Required for Neutrophil-Derived ROS Production and Inflammatory Bowel Disease. Front Med (Lausanne). 2020;7:207.
- Tam JSY, Coller JK, Hughes PA, Prestidge CA, Bowen JM. Toll-like receptor 4 (TLR4) antagonists as potential therapeutics for intestinal inflammation. Indian J Gastroenterol. 2021 Feb 1;40(1):5–21.
- Kuzmich NN, Sivak KV, Chubarev VN, Porozov YB, Savateeva-Lyubimova TN, Peri F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines (Basel). 2017 Oct 4;5(4):34.
- Vaure C, Liu Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol. 2014;5:316.
- Paramsothy S, Kamm MA, Kaakoush NO, Walsh AJ, van den Bogaerde J, Samuel D, Leong RWL, Connor S, Ng W, Paramsothy R, Xuan W, Lin E, Mitchell HM, Borody TJ. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet. 2017 Mar 25;389(10075):1218–28.
- Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front Cell Infect Microbiol. 2017;7:373.
- Castrillón-Betancur JC, López-Agudelo VA, Sommer N, Cleeves S, Bernardes JP, Weber-Stiehl S, Rosenstiel P, Sommer F. Epithelial Dual Oxidase 2 Shapes the Mucosal Microbiome and Contributes to Inflammatory Susceptibility. Antioxidants (Basel). 2023 Oct 21;12(10):1889.
- Sommer F, Bäckhed F. The gut microbiota engages different signaling pathways to induce Duox2 expression in the ileum and colon epithelium. Mucosal Immunol. 2015 Mar;8(2):372–9.
- Bento AF, Leite DFP, Claudino RF, Hara DB, Leal PC, Calixto JB. The selective nonpeptide CXCR2 antagonist SB225002 ameliorates acute experimental colitis in mice. J Leukoc Biol. 2008 Oct;84(4):1213–21.
- Zhu F, He H, Fan L, Ma C, Xu Z, Xue Y, Wang Y, Zhang C, Zhou G. Blockade of CXCR2 suppresses proinflammatory activities of neutrophils in ulcerative colitis. Am J Transl Res. 2020;12(9):5237–51.
- Xie Y, Kuang W, Wang D, Yuan K, Yang P. Expanding role of CXCR2 and therapeutic potential of CXCR2 antagonists in inflammatory diseases and cancers. European Journal of Medicinal Chemistry. 2023 Mar 15;250:115175.
- Li JY, Xiao J, Gao M, Zhou HF, Fan H, Sun F, Cui DD. IRF/Type I IFN signaling serves as a valuable therapeutic target in the pathogenesis of inflammatory bowel disease. International Immunopharmacology. 2021 Mar 1;92:107350.
- Loftus EV, Panés J, Lacerda AP, Peyrin-Biroulet L, D’Haens G, Panaccione R, Reinisch W, Louis E, Chen M, Nakase H, Begun J, Boland BS, Phillips C, Mohamed MEF, Liu J, Geng Z, Feng T, Dubcenco E, Colombel JF. Upadacitinib Induction and Maintenance Therapy for Crohn’s Disease. N Engl J Med. 2023 May 25;388(21):1966–80.
- A E, M F, F P. IL-1-mediated remodelling of the stromal-neutrophil landscape is associated with non-response to therapy in a subset of inflammatory bowel disease patients. Cell [Internet]. 2020 Jul 3 [cited 2025 Sep 13]; Available from: https://www.oncology.ox.ac.uk/publications/1116098.
- (PDF) IL-1-driven stromal-neutrophil interaction in deep ulcers defines a pathotype of therapy non-responsive inflammatory bowel disease [Internet]. ResearchGate. [cited 2025 Sep 13]. Available from: https://www.researchgate.net/publication/349100219_IL-1-driven_stromal-neutrophil_interaction_in_deep_ulcers_defines_a_pathotype_of_therapy_non-responsive_inflammatory_bowel_disease.
- Ahmad G, Chami B, Liu Y, Schroder AL, San Gabriel PT, Gao A, Fong G, Wang X, Witting PK. The Synthetic Myeloperoxidase Inhibitor AZD3241 Ameliorates Dextran Sodium Sulfate Stimulated Experimental Colitis. Front Pharmacol. 2020;11:556020.
- Xie K, Hunter J, Lee A, Ahmad G, Witting PK, Ortiz-Cerda T. The PAD4 inhibitor GSK484 diminishes neutrophil extracellular trap in the colon mucosa but fails to improve inflammatory biomarkers in experimental colitis. Biosci Rep. 2025 May 20;45(6):375–97.
- Mehandru S, Colombel JF, Juarez J, Bugni J, Lindsay JO. Understanding the molecular mechanisms of anti-trafficking therapies and their clinical relevance in inflammatory bowel disease. Mucosal Immunology. 2023 Dec;16(6):859–70.
- Soler D, Chapman T, Yang LL, Wyant T, Egan R, Fedyk ER. The binding specificity and selective antagonism of vedolizumab, an anti-alpha4beta7 integrin therapeutic antibody in development for inflammatory bowel diseases. J Pharmacol Exp Ther. 2009 Sep;330(3):864–75.
- Roosenboom B, Lochem EG van, Meijer J, Smids C, Nierkens S, Brand EC, Erp LW van, Kemperman LGJM, Groenen MJM, Horje CSHT, Wahab PJ. Development of Mucosal PNAd+ and MAdCAM-1+ Venules during Disease Course in Ulcerative Colitis. Cells. 2020 Apr 6;9(4):891.
- Facchin S, Vitulo N, Calgaro M, Buda A, Romualdi C, Pohl D, Perini B, Lorenzon G, Marinelli C, D’Incà R, Sturniolo GC, Savarino EV. Microbiota changes induced by microencapsulated sodium butyrate in patients with inflammatory bowel disease. Neurogastroenterol Motil. 2020 Oct;32(10):e13914.
- Hodgkinson K, El Abbar F, Dobranowski P, Manoogian J, Butcher J, Figeys D, Mack D, Stintzi A. Butyrate’s role in human health and the current progress towards its clinical application to treat gastrointestinal disease. Clin Nutr. 2023 Feb;42(2):61–75.
- Chiang HS. PAD4-dependent neutrophil extracellular traps dagame intestinal barrier integrity through histone protein components. J Immunol. 2023 May 1;210(Supplement_1):61.13.
- Wang P, Liu D, Zhou Z, Liu F, Shen Y, You Q, Lu S, Wu J. The role of protein arginine deiminase 4-dependent neutrophil extracellular traps formation in ulcerative colitis. Front Immunol. 2023;14:1144976.
- Long D, Mao C, Xu Y, Zhu Y. The emerging role of neutrophil extracellular traps in ulcerative colitis. Front Immunol [Internet]. 2024 Aug 7 [cited 2025 Sep 13];15. Available from: https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1425251/full.
- Lutz M, Horst S, Caldera F. Applying Biomarkers in Treat-to-target Approach for IBD. Curr Gastroenterol Rep. 2025;27(1):41.
- Liu F, Lee SA, Riordan SM, Zhang L, Zhu L. Global Studies of Using Fecal Biomarkers in Predicting Relapse in Inflammatory Bowel Disease. Front Med (Lausanne). 2020;7:580803.
- Sun T, Wang P, Zhai X, Wang Z, Miao X, Yang Y, Wu J. Neutrophil extracellular traps induce barrier dysfunction in DSS-induced ulcerative colitis via the cGAS-STING pathway. International Immunopharmacology. 2024 Dec 25;143:113358.
- Dmochowska N, Tieu W, Keller MD, Wardill HR, Mavrangelos C, Campaniello MA, Takhar P, Hughes PA. Immuno-PET of Innate Immune Markers CD11b and IL-1β Detects Inflammation in Murine Colitis. J Nucl Med. 2019 Jun;60(6):858–63.
- Bettenworth D, Reuter S, Hermann S, Weckesser M, Kerstiens L, Stratis A, Nowacki TM, Ross M, Lenze F, Edemir B, Maaser C, Pap T, Koschmieder S, Heidemann J, Schäfers M, Lügering A. Translational 18F-FDG PET/CT imaging to monitor lesion activity in intestinal inflammation. J Nucl Med. 2013 May;54(5):748–55.
- Rezazadeh F, Kilcline AP, Viola NT. Imaging Agents for PET of Inflammatory Bowel Disease: A Review. J Nucl Med. 2023 Dec 1;64(12):1858–64.
- Pickett JR, Wu Y, Ta HT. VCAM-1 as a common biomarker in inflammatory bowel disease and colorectal cancer: unveiling the dual anti-inflammatory and anti-cancer capacities of anti-VCAM-1 therapies. Cancer Metastasis Rev. 2025 Mar 17;44(2):40.
- Ley K, Burns C. Adhesion molecules in lymphocyte trafficking and colitis. Gastroenterology. 2001 Oct 1;121(4):1008–10.
- Zeng MY, Miralda I, Armstrong CL, Uriarte SM, Bagaitkar J. The roles of NADPH oxidase in modulating neutrophil effector responses. Mol Oral Microbiol. 2019 Apr;34(2):27–38.
- Winterbourn CC, Kettle AJ, Hampton MB. Reactive Oxygen Species and Neutrophil Function. Annu Rev Biochem. 2016 Jun 2;85:765–92.
- Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol. 2019 Jun 19;15(6):e8746.
- Friedrich M, Pohin M, Powrie F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity. 2019 Apr 16;50(4):992–1006.
- Chen ML, Sundrud MS. Cytokine networks and T cell subsets in inflammatory bowel diseases. Inflamm Bowel Dis. 2016 May;22(5):1157–67.
- Mennillo E, Kim YJ, Lee G, Rusu I, Patel RK, Dorman LC, Flynn E, Li S, Bain JL, Andersen C, Rao A, Tamaki S, Tsui J, Shen A, Lotstein ML, Rahim M, Naser M, Bernard-Vazquez F, Eckalbar W, Cho SJ, Beck K, El-Nachef N, Lewin S, Selvig DR, Terdiman JP, Mahadevan U, Oh DY, Fragiadakis GK, Pisco A, Combes AJ, Kattah MG. Single-cell and spatial multi-omics highlight effects of anti-integrin therapy across cellular compartments in ulcerative colitis. Nat Commun. 2024 Feb 19;15(1):1493.
- Lismont C, Revenco I, Li H, Costa CF, Lenaerts L, Hussein MAF, De Bie J, Knoops B, Van Veldhoven PP, Derua R, Fransen M. Peroxisome-Derived Hydrogen Peroxide Modulates the Sulfenylation Profiles of Key Redox Signaling Proteins in Flp-In T-REx 293 Cells. Front Cell Dev Biol. 2022;10:888873.
- Day NJ, Zhang T, Gaffrey MJ, Zhao R, Fillmore TL, Moore RJ, Rodney GG, Qian WJ. A deep redox proteome profiling workflow and its application to skeletal muscle of a Duchenne Muscular Dystrophy model. Free Radical Biology and Medicine. 2022 Nov 20;193:373–84.
- Global redox proteome and phosphoproteome analysis reveals redox switch in Akt | Nature Communications [Internet]. [cited 2025 Sep 4]. Available from: https://www.nature.com/articles/s41467-019-13114-4.
- Li X, Gluth A, Zhang T, Qian WJ. Thiol redox proteomics: Characterization of thiol-based post-translational modifications. Proteomics. 2023 Jul;23(13–14):e2200194.
- Zhang T, Gaffrey MJ, Li X, Qian WJ. Characterization of cellular oxidative stress response by stoichiometric redox proteomics. Am J Physiol Cell Physiol. 2021 Feb 1;320(2):C182–94.
- Global redox proteome and phosphoproteome analysis reveals redox switch in Akt | Nature Communications [Internet]. [cited 2025 Sep 13]. Available from: https://www.nature.com/articles/s41467-019-13114-4.
- Su Z, Burchfield JG, Yang P, Humphrey SJ, Yang G, Francis D, Yasmin S, Shin SY, Norris DM, Kearney AL, Astore MA, Scavuzzo J, Fisher-Wellman KH, Wang QP, Parker BL, Neely GG, Vafaee F, Chiu J, Yeo R, Hogg PJ, Fazakerley DJ, Nguyen LK, Kuyucak S, James DE. Global redox proteome and phosphoproteome analysis reveals redox switch in Akt. Nat Commun. 2019 Dec 2;10(1):5486.
- Ruiz Castro PA, Yepiskoposyan H, Gubian S, Calvino-Martin F, Kogel U, Renggli K, Peitsch MC, Hoeng J, Talikka M. Systems biology approach highlights mechanistic differences between Crohn’s disease and ulcerative colitis. Sci Rep. 2021 Jun 1;11(1):11519.
- Kariyawasam V, Davis T, Ghali M, Tejcek D, R O’Neil T, Verley E, Griffiths M, Campos M, Bonney I, Bulmer AC, Ramaswamy Y, Mitrev N, Corte C, Witting PK, Chami B. Myeloperoxidase Luminol Reaction - A Novel Faecal Assay for Predicting Colonoscopy Findings in Patients with Ulcerative Colitis: A Pilot Cross-Sectional Clinical Study. Adv Healthc Mater. 2025 Aug 21;e01825.
- Turner D, Ricciuto A, Lewis A, D’Amico F, Dhaliwal J, Griffiths AM, Bettenworth D, Sandborn WJ, Sands BE, Reinisch W, Schölmerich J, Bemelman W, Danese S, Mary JY, Rubin D, Colombel JF, Peyrin-Biroulet L, Dotan I, Abreu MT, Dignass A, International Organization for the Study of IBD. STRIDE-II: An Update on the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) Initiative of the International Organization for the Study of IBD (IOIBD): Determining Therapeutic Goals for Treat-to-Target strategies in IBD. Gastroenterology. 2021 Apr;160(5):1570–83.
- Scheppach W, Sommer H, Kirchner T, Paganelli GM, Bartram P, Christl S, Richter F, Dusel G, Kasper H. Effect of butyrate enemas on the colonic mucosa in distal ulcerative colitis. Gastroenterology. 1992 Jul;103(1):51–6.
- Breuer RI, Soergel KH, Lashner BA, Christ ML, Hanauer SB, Vanagunas A, Harig JM, Keshavarzian A, Robinson M, Sellin JH, Weinberg D, Vidican DE, Flemal KL, Rademaker AW. Short chain fatty acid rectal irrigation for left-sided ulcerative colitis: a randomised, placebo controlled trial. Gut. 1997 Apr;40(4):485–91.
- Luceri C, Femia AP, Fazi M, Di Martino C, Zolfanelli F, Dolara P, Tonelli F. Effect of butyrate enemas on gene expression profiles and endoscopic/histopathological scores of diverted colorectal mucosa: A randomized trial. Dig Liver Dis. 2016 Jan;48(1):27–33.
- Chulkina M, Rohmer C, McAninch S, Panganiban RP, Villéger R, Portolese A, Ciocirlan J, Yang W, Cohen C, Koltun W, Valentine JF, Cong Y, Yochum G, Beswick EJ, Pinchuk IV. Increased Activity of MAPKAPK2 within Mesenchymal Cells as a Target for Inflammation-Associated Fibrosis in Crohn’s Disease. J Crohns Colitis. 2024 Aug 6;18(7):1147–61.
- Wang Z, Liang XY, Chang X, Nie YY, Guo C, Jiang JH, Chang M. MMI-0100 Ameliorates Dextran Sulfate Sodium-Induced Colitis in Mice through Targeting MK2 Pathway. Molecules. 2019 Aug 3;24(15):2832.
- Deeks ED. Anifrolumab: First Approval. Drugs. 2021 Oct;81(15):1795–802.
Table 3.
Regulation of DUOX2 expression and H₂O₂ production in gut cells.
| Stimulus / Factor | Target Cell Type | Effect on DUOX2 / H₂O₂ | Pathophysiological Consequence | References |
| IFN-γ (Th1 cytokine) | Intestinal epithelium | Strong upregulation of DUOX2 and H₂O₂ | Drives epithelial oxidative burst; promotes chronic inflammation in IBD. | [47,48] |
| IL-22 (Type 17 cytokine) | Intestinal epithelium | Increases DUOX2 expression | Enhances epithelial host defense; may aid barrier repair and antimicrobial protection. | [49,50] |
| TLR4 agonists (LPS) | Intestinal epithelium | Upregulates DUOX2 and H₂O₂ | Couples bacterial sensing to ROS output; implicated in colitis-associated tumorigenesis. | [37,51] |
| Adherent-invasive E. coli | Intestinal epithelium | Potently induces DUOX2 | Amplifies H₂O₂ release during dysbiosis; promotes mucosal inflammation. | [52,53] |
| Dysbiotic microbiota | Intestinal epithelium | Broad activation of DUOX2 | Marker of disrupted homeostasis; correlates with early preclinical IBD changes. | [54,55] |
| Short-chain fatty acids (butyrate) | Intestinal epithelium | Downregulates DUOX2 | Restores barrier integrity; dampens inflammation by lowering epithelial H₂O₂ output. | [56] |
| HDAC inhibitors | Intestinal epithelium | Mimic butyrate effect, suppress DUOX2 | Potential therapeutic avenue to control DUOX2-mediated oxidative stress. | [57] |
| Inflammatory milieu (IL-8, TNF, IFNα) | Neutrophils (new finding) | Induces de novo DUOX2 expression | Neutrophils gain extra oxidative capacity; amplify cytokine circuits and tissue inflammation in IBD. | [57,58] |
Table 4.
Therapeutic strategies targeting the neutrophil–DUOX2 pathway in IBD.
| Strategy / Target | Mechanism of Action | Expected Effect on IBD | References |
| Neutrophil DUOX2 inhibition | Genetic silencing (e.g., conditional knockout) or small-molecule inhibition of DUOX2 | Reduces neutrophil H₂O₂ and cytokine output; suppresses mucosal inflammation; improves colitis in models. ⚠ Risk: impaired pathogen defense. | [31,73] |
| Butyrate / SCFA supplementation | Microbial metabolite; inhibits histone deacetylases (HDACs) | Downregulates epithelial and neutrophil DUOX2; reduces cytokine release and NETosis; enhances barrier integrity. | [74,75,76] |
| JAK inhibitors (e.g., baricitinib, tofacitinib) | Block IFN-α/γ and other cytokine signaling via JAK–STAT pathway | Prevent IFNα-driven neutrophil–fibroblast immunofibrosis; reduce IL-8–mediated recruitment; established anti-inflammatory effect in IBD. | [77,78] |
| CXCR1/2 antagonists (e.g., reparixin) | Block neutrophil chemokine receptors for IL-8/CXCL1/2 | Reduces neutrophil migration into gut mucosa; may lower neutrophil burden and tissue injury. | [79,80] |
| MK2 (p38 MAPK) inhibitors | Inhibit MAPK-activated protein kinase 2 (MK2), required for NADPH oxidase assembly/ROS | Decreases neutrophil-derived ROS and cytokine output; protects against DSS colitis in preclinical studies. | [81,82] |
| TLR4 antagonists (e.g., eritoran) | Block LPS binding to TLR4 on epithelial cells | Prevent LPS-induced DUOX2 overactivation; reduces epithelial oxidative stress and inflammation. | [83,84,85] |
| Probiotics / Fecal microbiota transplantation (FMT) | Restore commensals and SCFA-producers (e.g., Clostridium clusters) | Reduce dysbiosis-driven DUOX2 activation; increase butyrate production; rebalance immune–microbiota crosstalk. | [86] |
| Antioxidants (e.g., N-acetylcysteine, vitamins C/E) | Scavenge reactive oxygen species (ROS) | Neutralize excessive H₂O₂ from DUOX2/NOX; may reduce epithelial oxidative injury, but efficacy is inconsistent. | [15,73,87] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



