Submitted:
02 November 2025
Posted:
03 November 2025
You are already at the latest version
Abstract
Diabetes mellitus (DM) is a complex, heterogeneous, chronic metabolic disorder characterized by persistent hyperglycemia. The most common classification of diabetes includes type 1 diabetes mellitus (T1DM), type 2 diabetes mellitus (T2DM), and gestational diabetes. The aim of the study is to investigate the underlying mechanisms of T2DM, its clinical presentation, and complications. The association of the pathogenesis of T2DM and the occurrence of its complications with obesity, chronic inflammation, oxidative stress (OS), insulin resistance (IR), hepatic steatosis, dyslipidemia, and molecular pathways, such as NF-κB and JAK/STAT is also summarized, under the scope of the effects of several antioxidant compounds and minerals in their progression. T2DM is characterized by progressive loss of insulin secretion from pancreatic islet β-cells due to IR, which is a parameter of metabolic syndrome (MetS). Chronic hyperglycemia in patients with TDM in synergy with other metabolic abnormalities causes acute complications such as diabetic ketoacidosis, osmotic diuresis and hyperglycemic diabetic coma, chronic microvascular complications such as diabetic retinopathy, nephropathy and neuropathy and macrovascular complications such as atherosclerotic cardiovascular disease (ASCVD), peripheral artery disease (PAD) and cerebrovascular events, which implicate the formation of reactive species and the promotion of inflammatory pathways. In these events natural or synthetic antioxidants and minerals seem to be ameliorating or co-treatment beneficial options. Obese patients are recommended to be screened for T2DM and to make lifestyle changes including exercise, diet modification, and weight loss, potentially together with anti-obesity pharmacotherapy, and multifunctional supplements with antioxidant and anti-inflammatory potential.
Keywords:
obesity
; oxidative stress
; antioxidants
; natural antioxidants
; vitamins
; catechins
; flavonoids
; adipocytes
; cardiovascular disease
; diabetes
1. Introduction
The term diabetes originates from the Greek verb diabaino, meaning “to pass through” [1]. The ancient physician Aretaeus of Cappadocia used the term diabetes to describe a condition characterized by excessive urination, or polyuria [1]. Since then, a distinction has been made between diabetes mellitus and diabetes insipidus, the latter referring to a condition in which the urine is tasteless and lacks any distinctive quality [2].
Diabetes mellitus is a heterogeneous metabolic disorder characterized by chronic hyperglycemia resulting from impaired insulin secretion, defective insulin action, or a combination of both [3]. The most common classifications include T1DM, T2DM, and gestational diabetes [4]. The disease typically develops through a prediabetic stage, defined by impaired fasting glucose (IFG) and/or impaired glucose tolerance (IGT), or glycated hemoglobin (HbA1c) values ranging from 6.0% to 6.4% [3,5].
T2DM is among the most significant non-communicable chronic diseases, exhibiting high rates of morbidity and mortality [6]. It accounts for approximately 96% of all diabetes cases and is characterized by (a) non-autoimmune, progressive loss of adequate insulin secretion by pancreatic β-cells within the islets of Langerhans, (b) IR, and (c) MetS [7,8]. Furthermore, its strong association with obesity and the involvement of OS in both the onset and complications of T2DM are well-documented. Numerous inflammatory and oxidative mechanisms contribute to this relationship [9,10]. In light of the above, the present study aims to elucidate the pathways implicated in the development and progression of T2DM and its complications, emphasizing their interrelation with obesity and OS-related signaling. Furthermore, it seeks to analyze the impact of antioxidant vitamins, natural compounds, and minerals on these processes, drawing upon data from the international literature, mainly from the last decade, and comparing findings from in cellulo, in vivo, and clinical studies.
2. Pathophysiology of Obesity-Induced Insulin Resistance and Type 2 Diabetes Mellitus
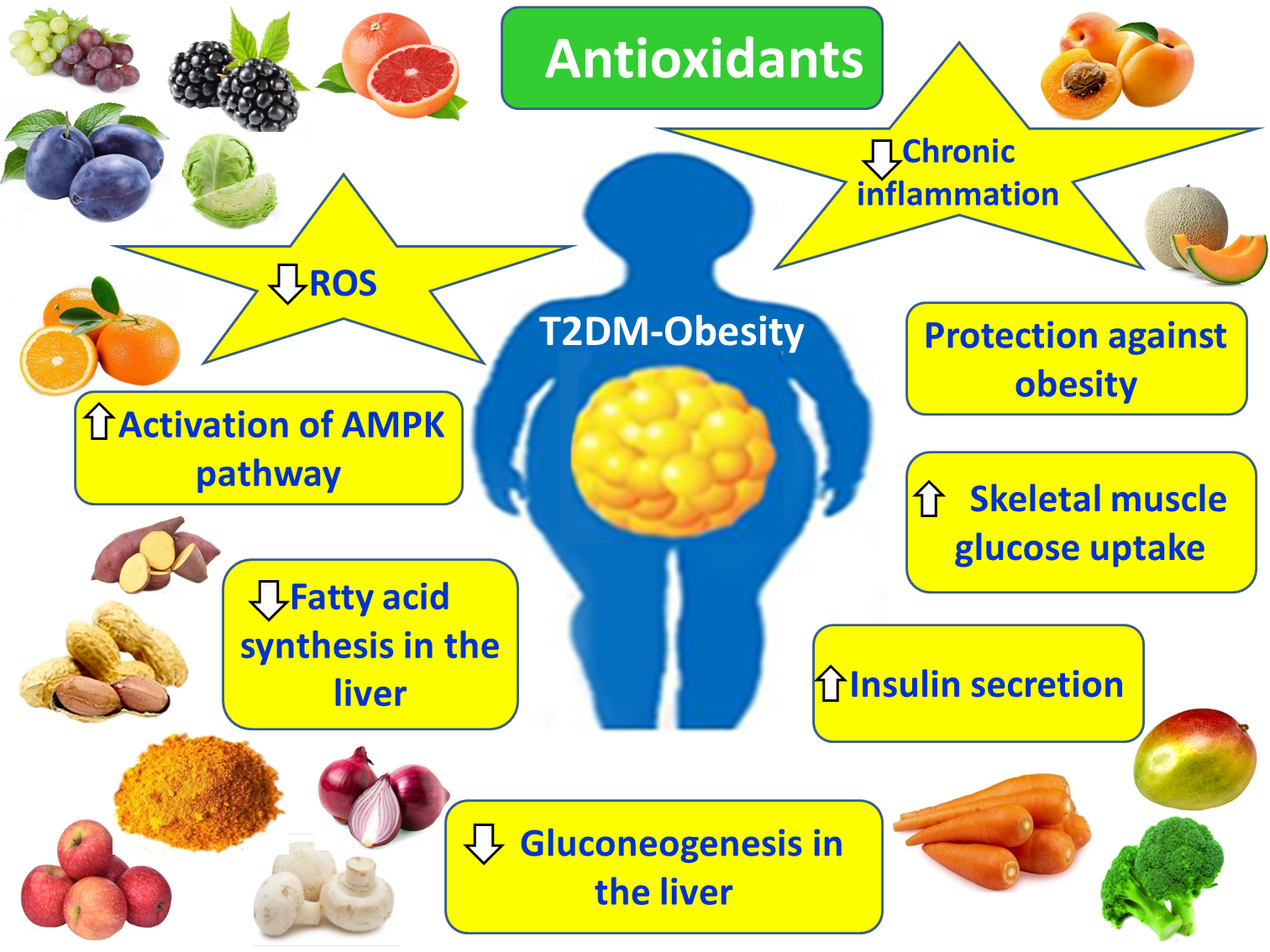
Hyperglycemia in T2DM results from reduced glucose utilization due to non-autoimmune insulinopenia and the excessive production of glucagon [11]. Insulinopenia leads to decreased glucose uptake by muscle and adipose tissues, reduced glycogen synthesis, and diminished lipogenesis [6]. Excessive glucagon secretion, which becomes the dominant hormone in carbohydrate metabolism, promotes increased glycogenolysis and gluconeogenesis in the liver [12]. Among these mechanisms, elevated hepatic gluconeogenesis (mediated by glucagon activity) contributes more significantly to hyperglycemia than impaired glucose utilization [13]. The ensuing consequences of hyperglycemia include glucosuria, osmotic diuresis, dehydration, and diabetic ketoacidosis [14] (Figure 1).
2.1. Insulin Signaling Pathways, IR, and the Pathogenesis of T2DM
Under normal physiological conditions, insulin signaling begins with the phosphorylation of its cytoplasmic receptor and proceeds through two parallel pathways: (a) the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathway and (b) the mitogen-activated protein kinase (MAPK) pathway [15]. Activation of AKT by insulin induces the translocation of the glucose transporter GLUT4 from cytoplasmic vesicles to the plasma membrane, thereby promoting glucose uptake [6]. In addition, AKT activation by insulin phosphorylates the transcription factor FOXO1 (Forkhead box protein O1) in the nucleus and translocates it to the cytoplasm, resulting in its inactivation and the suppression of hepatic gluconeogenesis [6,16]. Furthermore, insulin-induced AKT activation phosphorylates and inactivates glycogen synthase kinase 3 (GSK3), leading to enhanced glycogen synthesis, while it also stimulates mTOR activity, promoting protein synthesis [6]. Under certain conditions, insulin can also activate the MAPK pathway, leading to increased secretion of endothelin-1 from vascular endothelial cells and subsequent vasoconstriction [15].
Insulin resistance (IR) refers to the reduced ability of insulin target tissues to respond to insulin signaling, resulting in compensatory hyperinsulinemia, which is closely associated with MetS and related disorders such as obesity, non-alcoholic fatty liver disease (NAFLD), and hypertension [6]. In the early stages of IR, pancreatic β-cells become hyperfunctional in an attempt to increase insulin secretion and maintain normal plasma glucose levels [17]. As the condition progresses, β-cells can no longer secrete sufficient insulin to compensate for the abnormally high blood glucose concentrations, leading to prediabetes or overt T2DM [18].
In adipose tissue, IR induces enhanced lipolysis, while in the liver it promotes lipogenesis and gluconeogenesis, resulting in compensatory hyperinsulinemia to maintain normoglycemia [15]. In skeletal muscle, IR impairs GLUT4 translocation to the plasma membrane, thereby reducing glucose uptake [15]. Increased lipolysis of visceral fat releases free fatty acids (FFAs) into the liver via the portal circulation, leading to elevated secretion of very-low-density lipoproteins (vLDLs) and triglycerides. The accumulation of triglycerides and FFAs in the liver causes hepatic lipotoxicity and contributes to the development of NAFLD [15]. Moreover, elevated plasma FFAs contribute to increased low-density lipoprotein (LDL) levels and decreased high-density lipoprotein (HDL) concentrations, thus generating an atherogenic dyslipidemia [19]. In parallel, IR elevates serum fibrinogen and plasminogen activator inhibitor-1 (PAI-1) levels, promoting a prothrombotic state in T2DM, thereby increasing the risk of thrombosis and cardiovascular diseases (CVDs) [20].
2.2. Obesity, Chronic Low-Grade Inflammation, OS and the Pathogenesis of T2DM
Obesity is closely associated with chronic low-grade inflammation within adipose tissue. In obesity-related chronic inflammation, there is increased secretion of chemotactic factors, anti-inflammatory and pro-inflammatory adipokines, as well as pro-inflammatory cytokines [15]. This inflammatory process involves a phenotypic shift in adipose tissue macrophages, from anti-inflammatory M2 macrophages, predominant in lean individuals, to pro-inflammatory M1 macrophages, which dominate in overweight and obese subjects [15]. In individuals with normal body weight, adipose tissue macrophages are mainly of the M2 subtype, producing anti-inflammatory adipokines, such as adiponectin and apelin, and anti-inflammatory cytokines including IL-4, IL-10, IL-11, IL-13, and IL-1Ra [15]. Excessive caloric intake and physical inactivity lead to adipocyte hypertrophy, promoting increased secretion of chemotactic factors such as MCP-1 into the circulation, which in turn stimulates the recruitment and activation of pro-inflammatory M1 macrophages [21]. M1 macrophages infiltrate adipose tissue, resulting in the production of (i) pro-inflammatory adipokines such as visfatin, resistin, leptin, and PAI-1; (ii) pro-inflammatory cytokines such as IL-6, TNF-α, and IL-1β; and (iii) inducible nitric oxide synthase (iNOS). The chronic inflammation that ensues leads to reduced adiponectin levels [15]. These pro-inflammatory adipokines and cytokines impair insulin signaling in the liver, adipose tissue, and skeletal muscle, while activating multiple inflammatory signaling cascades, including NF-κB, JAK/STAT, and JNK pathways. This molecular dysregulation promotes the development of MetS and T2DM [6].
In the liver, adipose tissue, and skeletal muscles, chronic hyperinsulinemia, secondary to persistent hyperglycemia, further diminishes insulin signaling. The infiltration of adipose tissue by M1 macrophages enhances the secretion of pro-inflammatory cytokines, perpetuating chronic inflammation and contributing to OS [15]. Moreover, hyperglycemia increases mitochondrial production of nicotinamide adenine dinucleotide phosphate (NADPH), which enhances the generation of reactive oxygen species (ROS). These ROS exceed the capacity of endogenous antioxidant systems, thereby promoting OS [22]. Elevated ROS levels exacerbate OS and downregulate AKT activity, resulting in sustained hyperinsulinemia and IR. The latter further diminishes insulin signaling in the liver, adipose tissue, and skeletal muscles [15]. Concurrently, both increased secretion of pro-inflammatory cytokines such as IL-1β from adipose tissue and chronic hyperinsulinemia contribute to pancreatic β-cell dysfunction, leading to T2DM [15].
OS is also implicated in the pathogenesis of T2DM complications, including renal dysfunction, diabetic retinopathy, and diabetic peripheral neuropathy [6]. In diabetic nephropathy, chronic hyperglycemia induces excessive ROS accumulation within the renal mesangium, triggering activation of Mtor [6], and sirtuin 1 [23], ultimately leading to mesangial cell apoptosis [6,24]. Similarly, in diabetic retinopathy, persistent hyperglycemia causes excessive accumulation of ROS and advanced glycation end products (AGEs) in ocular tissues [25], activating the polyol, hexosamine, and PKC pathways, which culminate in retinal cell apoptosis and disease progression [26]. In diabetic peripheral neuropathy, both hyperglycemia and elevated saturated fatty acids (SFAs) contribute to mitochondrial membrane depolarization and ROS accumulation in Schwann cells (SCs), leading to activation of downstream effectors such as PKCε [27] and JNK [28], and ultimately resulting in mitochondrial dysfunction and Schwann cell apoptosis [29].
2.3. The NF-κB Signaling Pathway in the Pathogenesis of T2DM and Its Complications
Proinflammatory cytokines secreted by adipose tissue in obese individuals with T2DM phosphorylate and degrade the IκB kinase (IKK) complex [30], thereby releasing and activating the nuclear transcription factor κB (NF-κB), which subsequently translocates into the cell nucleus to exert its transcriptional activity [6]. NF-κB regulates immune responses, stress reactions, apoptosis, and cellular differentiation [31]. Specifically, NF-κB inhibits the activation of insulin receptor substrate-1 (IRS-1) by increasing the expression of inducible nitric oxide synthase (iNOS) and the production of nitric oxide (NO) [6]. Moreover, IRS-1 activation is suppressed through serine phosphorylation by IKKβ, a subunit of the IKK complex. Additionally, NF-κB inhibits IRS-1 activation through the stimulation of protein kinase C epsilon (PKCε) via the NLRP3 inflammasome, which itself is activated by NF-κB [6]. Concurrently, NF-κB upregulates the transcription of IL-1β, IL-18, and TNF-α, creating a positive feedback loop that further amplifies its activity and contributes to the development of metabolic syndrome–related comorbidities such as diabetic nephropathy, diabetic foot ulcers, and diabetic retinopathy [6]. In diabetic nephropathy, NF-κB activation and its stimulation of the NLRP3 inflammasome promote renal inflammation and enhance the deposition of fibronectin and collagen in the kidney [32], leading to thickening of the glomerular basement membrane, glomerulosclerosis, podocyte injury, and ultimately renal fibrosis [33]. In diabetic foot ulcers, NF-κB activation induces wound inflammation and increases caspase-3 activity, resulting in apoptosis of vascular endothelial cells and delayed wound healing [34]. During the early stages of diabetic retinopathy, NF-κB activation interacts with ROS to promote pro-apoptotic processes [35]. In the later stages of proliferative diabetic retinopathy (PDR), abnormal neovascularization occurs, accompanied by apoptosis of pericytes in the retinal layer due to elevated levels of the NLRP3 inflammasome and IL-1β driven by NF-κB activity [36].
2.4. The JAK/STAT Signaling Pathway in the Pathogenesis of T2DM and Its Complications
The Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling pathway serves as a key mediator in the downstream signaling of various cytokines, hormones, and growth factors, and plays a crucial role in regulating cellular proliferation, differentiation, apoptosis, and inflammatory responses [6]. In T2DM, elevated levels of proinflammatory cytokines, such as interleukin-6 (IL-6) and interferon-gamma (IFN-γ), activate the JAK2/STAT3 pathway, leading to the upregulation of suppressor of cytokine signaling 3 (SOCS3), which inhibits the activation of insulin receptor substrate-1 (IRS-1) [37,38]. Activation of the JAK/STAT3 pathway by these proinflammatory cytokines has also been found to induce NF-κB activation [37]. Furthermore, JAK/STAT3 signaling is implicated in T2DM-related complications, such as diabetic nephropathy and diabetic foot ulcers. In diabetic nephropathy, increased JAK expression in glomerular podocytes activates the STAT3/NF-κB axis, resulting in persistent low-grade renal inflammation, which promotes fibrosis and progressive loss of renal function [39,40]. In diabetic foot ulcers, elevated proinflammatory cytokines, such as IL-6, enhance STAT3 signaling [41], which impairs immune cell activation, recruitment, and survival, ultimately leading to delayed wound healing [42].
3. Pathophysiology of T2DM Complications
Patients with diabetes mellitus frequently present with excessive thirst (polydipsia), extreme hunger (polyphagia), increased urination (polyuria), lack of energy and fatigue, bacterial and fungal infections of the skin and genital area, dry mouth, xerosis, nausea, vomiting, abdominal pain, headache, facial flushing, dehydration, and delayed wound healing [43]. Some individuals may also report numbness or tingling in the hands and feet, or blurred vision [43]. Persistent hyperglycemia in uncontrolled diabetes can lead to both acute and chronic complications. Acute complications include hypoglycemia, diabetic ketoacidosis, hyperglycemic hyperosmolar state, and hyperglycemic diabetic coma [43].
Chronic microvascular complications include nephropathy, neuropathy, and diabetic retinopathy, whereas chronic macrovascular complications include atherosclerotic cardiovascular disease (ASCVD), peripheral artery disease (PAD), and cerebrovascular events [3,6,43]. It is estimated that 1.4-4.7% of middle-aged individuals with diabetes experience a cardiovascular event annually [44,45]. Obese individuals with T2DM exhibit elevated circulating cholesterol levels and are at higher risk for severe arteriosclerosis, characterized by the formation of atherosclerotic plaques within the arterial wall, which narrow the vessel lumen [46]. The resulting restriction in blood flow increases flow velocity and turbulence, further damaging the vascular wall and promoting thrombosis, events that underlie myocardial infarction and stroke [47,48].
Diabetic foot ulcers arise from poor peripheral circulation [49]. Early symptoms of diabetic peripheral neuropathy include hypoesthesia, numbness, and tingling in the lower extremities [50]. Due to sensory loss, injuries to the foot may occur and, because of impaired local circulation, these wounds often fail to heal properly, potentially leading to lower-limb amputation [51].
3.1. Pathophysiological Mechanisms Underlying Diabetic Ketoacidosis
Diabetic ketoacidosis (DKA) is a medical emergency in diabetic patients characterized by plasma glucose levels exceeding 240 mg/dL, resulting from the inhibition of lipogenesis [52]. Consequently, fatty acids fail to enter the citric acid (Krebs) cycle and instead undergo mitochondrial β-oxidation, leading to the formation of ketone bodies [53]. The two primary ketone bodies are acetoacetic acid and β-hydroxybutyric acid, with acetone being the third and least abundant [54]. Ketone bodies are weak acids; however, their excessive accumulation may exceed the compensatory capacity of the body’s acid–base homeostatic mechanisms, leading to severe metabolic acidosis, known as diabetic ketoacidosis, defined by blood pH less than 7.35 and bicarbonate less than 18 mmol/L [55,56]. DKA is further exacerbated by dehydration resulting from osmotic diuresis [55]. If not treated promptly with high-dose insulin therapy and adequate rehydration, DKA can progress to diabetic coma and ultimately death [57]. To compensate for the acidosis, respiration becomes rapid and deep (Kussmaul respiration), promoting increased elimination of carbon dioxide [55]. Additionally, the kidneys contribute to compensatory mechanisms by reabsorbing bicarbonate from the extracellular fluid and synthesizing new bicarbonate ions [58]. However, this renal buffering depletes extracellular bicarbonate reserves [59,60]. Severe DKA develops primarily in cases of poorly controlled diabetes mellitus [61]. When blood pH drops below 7.0, diabetic coma and death may ensue within hours [61]. The clinical signs and symptoms of diabetic ketoacidosis include: (1) elevated blood glucose levels (>240 mg/dL), (2) ketonuria, (3) polydipsia and polyuria, (4) nausea, vomiting, and abdominal pain, (5) dyspnea or labored breathing, (6) a fruity or acetone odor on the breath, (7) flushing, (8) fatigue, (9) dehydration, and (10) loss of consciousness [14,55,60,61].
3.2. Pathophysiological Mechanisms of Osmotic Diuresis and Muscle Mass Reduction in T2DM
Under normal physiological conditions, glucose from the renal filtrate is completely reabsorbed by the renal tubules into the peritubular capillaries and reenters systemic circulation [62]. The plasma glucose concentration threshold of 180 mg/dL, known as the renal glucose threshold, represents the saturation point of the glucose transporters that reabsorb glucose from the filtrate into the bloodstream [63]. When plasma glucose levels exceed 180 mg/dL, the filtration of glucose into the renal tubules surpasses the reabsorptive capacity of these transporters, resulting in the excretion of excess glucose in the urine [64]. The elevated glucose concentration within the renal tubules increases the osmotic pressure, thereby reducing tubular water reabsorption by the peritubular capillaries [65]. Consequently, large volumes of water are lost through the urine, leading to dehydration of the extracellular fluid, followed by compensatory dehydration of the intracellular compartment, ultimately causing widespread cellular dehydration [64].
In uncontrolled T2DM, this mechanism manifests clinically as polyuria and intense thirst, as the body attempts to compensate for the excessive water loss [66]. The pathophysiology of T2DM also involves inappropriate hypersecretion of glucagon from pancreatic α-cells under hyperglycemic conditions, further elevating plasma glucose levels via enhanced hepatic gluconeogenesis [67]. Simultaneously, glucagon promotes protein catabolism [68]. This reduction in total body protein stores in T2DM patients leads to loss of muscle mass and progressive weakness [68]. Severe muscle wasting may result in quadriplegia and ultimately death [69].
4. Antioxidants in T2DM and the Amelioration of Obesity-Associated T2DM
4.1. Vitamin E
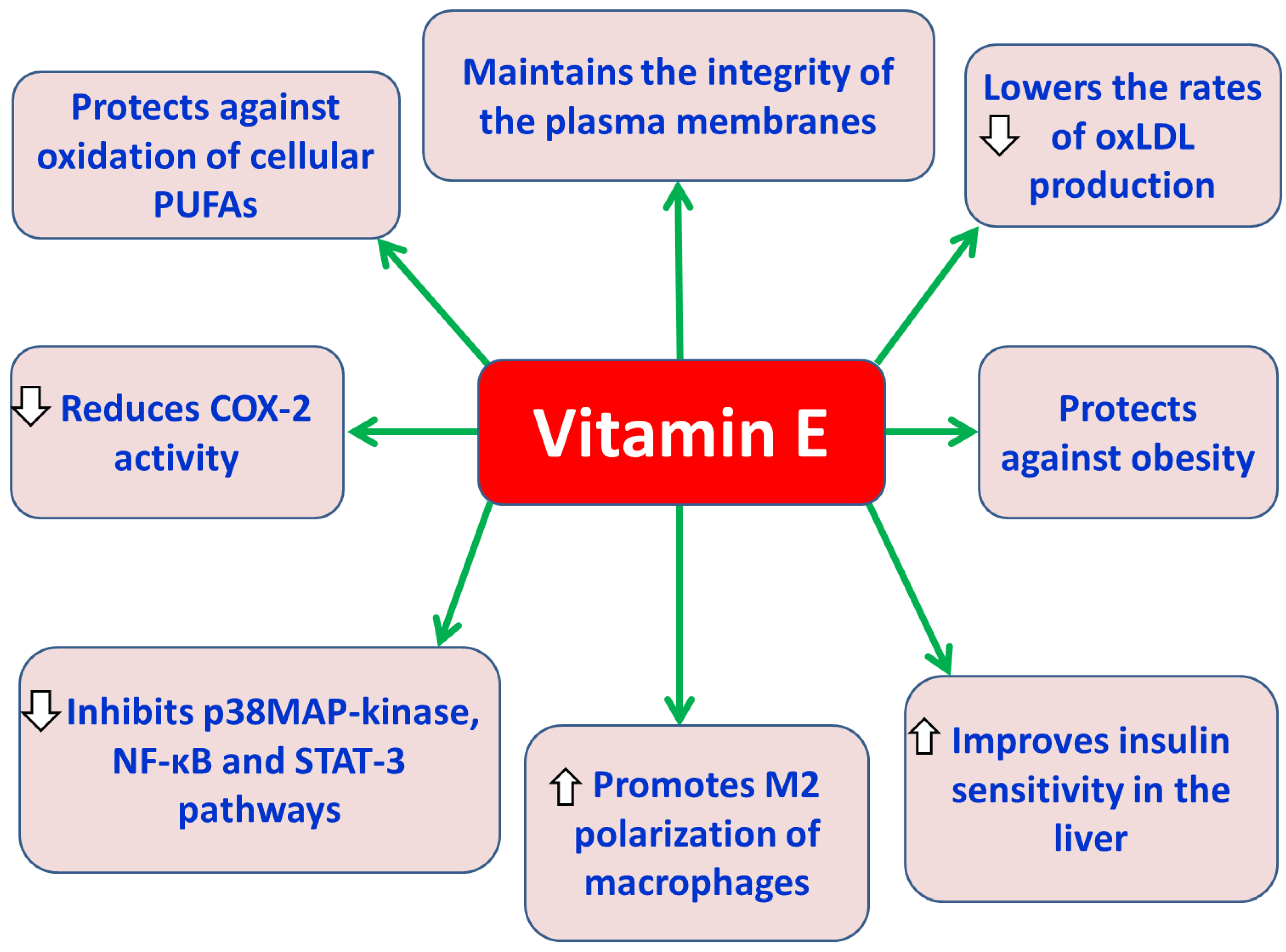
Vitamin E is a group of lipid-soluble substances (tocols) including tocopherols (saturated) and tocotrienols (unsaturated) each with four natural isomers (α-, β-, γ-, and δ-) depending on the presence and position of the methyl group(s) as side chains on their aromatic chromanol ring [70,71]. Vitamin E is exclusively obtained from various staple foods, such as vegetable oils, palm oils, rice bran, wheat germ, olive, barley, soybean, nuts and grains [70,72]. Vitamin E is an important cell membrane component of all tissues and is transported in the body via plasma lipoproteins [73]. All eight forms of vitamin E act as antioxidants when fat undergoes oxidation. In particular, tocopherols and tocotrienols have a hydroxyl (–OH) group on their chromanol ring, which allows them to donate hydrogen atoms to free radicals to prevent the oxidation of tissue polyunsaturated fatty acids (PUFAs) in tissues [74]. Alpha-tocopherol is recognized as the most potent natural source of vitamin E, and it acts as chain-breaking antioxidant in lipoproteins preventing lipid peroxidation and maintaining plasma membrane integrity [75]. Also, alpha-tocopherol reduces LDL oxidation preventing the the formation of foam cells and the development of atherosclerosis [74]. Vitamin E supplementation reduces the inflammatory response by reducing the expression of IL-6, TNF-α and C reactive protein [76]. Vitamin E has been found to inhibit the signaling pathways of p38 MAP-kinases [77], NF-κB and STAT-3 [78], and to reduce the activity of cyclooxygenase-2 (COX-2) and the production of eicosanoids [79]. Vitamin E has also shown antidiabetic properties, since it improves insulin sensitivity (alpha-tocopherol) and IR in the liver [80]. The ability of alpha-tocopherol to act as a scavenger of oxidized low-density lipoprotein cholesterol (oxLDL) seems to be beneficial for the health of patients with metabolic syndrome by assisting to the reduction of the risk of development of T2DM and the prevention of atherosclerosis associated with obesity [80]. Vitatin E has been found in obese animal models to have anti-obesity effects [81]. Also, in an experimental model of diabetic rats, it was observed that the administration of tocotrienol-rich supplements (200 mg/kg/day) reduced fasting blood glucose levels and levels of oxidative stress markers and improved dyslipidemia [82]. Additionally, in another experimental model of obese mice, it was found that oral administration of γ-tocotrienol supplements (50 mg/kg) significantly reduced fasting blood glucose levels, insulin levels, and proinflammatory cytokine secretion, and enhanced insulin signaling in adipose tissue [83]. At the same time, it has been found that the administration of alpha-tocopherol to obese individuals caused polarization of macrophages towards the anti-inflammatory (M2) phenotype [84] and reduced oxidative damage caused by ROS in T2DM [84].
Figure 2.
Protective effects of vitamin E against obesity-associated T2DM. Abbreviations: COX-2: cyclooxygenase-2; NF-κB: nuclear factor kappa B; oxLDL: oxidized low-density lipoprotein cholesterol; p38 MAP-kinases: p38 mitogen-activated protein kinases; PUFAs: polyunsaturated fatty acids; STAT-3: signal transducer and activator of transcription 3.
Figure 2.
Protective effects of vitamin E against obesity-associated T2DM. Abbreviations: COX-2: cyclooxygenase-2; NF-κB: nuclear factor kappa B; oxLDL: oxidized low-density lipoprotein cholesterol; p38 MAP-kinases: p38 mitogen-activated protein kinases; PUFAs: polyunsaturated fatty acids; STAT-3: signal transducer and activator of transcription 3.
4.2. Vitamin C
Vitamin C (ascorbic acid) is a naturally occurring organic compound found both in animals and plants, and functions as a cofactor in many enzymatic reactions, as an enzyme component and as a powerful antioxidant, affecting also other antioxidant enzymes and compounds like vitamin E [85]. Human body lacks the ability to synthesize vitamin C on its own and must obtain it through diet, primarily from fresh fruits and vegetables or from supplements [85]. Several studies have examined the effects of vitamin C supplementation in T2DM, with some showing potential benefits, particularly at higher doses, in improving fasting blood glucose and HbA1c levels and reducing IR and OS [86]. Naziroglu et al. evaluated the effects of hormone replacement therapy (HRT) and vitamin C and E supplementation in postmenopausal women with T2DM, compared to non-diabetic postmenopausal women and found that the therapy decreased lipid peroxidation and plasma levels of total cholesterol, LDL-cholesterol and triglycerides. Also, there was a significant increase in plasma levels of β-carotene catalase, glutathione peroxidase (GPx) and reduced glutathione, in postmenopausal women with T2DM, treated with HRT and vitamin C and E combination [87]. Also, Harding et al., in a long-term study, examined whether the consumption of fruits and vegetables and the plasma vitamin C levels were negatively associated with the risk of T2DM development [88]. The researchers found a significant association between the higher plasma vitamin C levels and the decreased risk of T2DM development, suggesting that the high consumption of fruits and vegetables prevents T2DM development [88]. In addition, Rafighi et al. evaluated the outcome of vitamin C and E supplementation for 3 months on T2DM patients, aged 30-60 years with BMI>25 and found that the consumption of vitamin C, E and C plus E significantly reduced the levels of fasting plasma glucose (FBG) and glycated hemoglobin (HbA1c) and increased the levels of superoxide dismutase (SOD) and glutathione (GSH) in T2DM patients compared to the placebo group [86].
4.3. N-Acetylcysteine (NAC)
NAC is an N-acetylated form of the amino acid L-cysteine, and it is deacetylated into cysteine, which is then used for the formation of glutathione (GSH) [89]. NAC possesses antioxidant, cytoprotective, anti-inflammatory, antimicrobial and mucolytic properties, making it a promising therapeutic agent for a wide range of diseases in humans [89]. NAC reduces ROS through both direct and indirect antioxidant effects [90]. It directly scavenges some free radicals like hydroxyl radicals (•OH), nitrogen dioxide radicals (•NO2) and carbon trioxide radicals (CO3•−) [91]. In addition, it indirectly boosts intracellular antioxidant defenses by serving as a precursor for glutathione (GSH). The indirect pathway involves the synthesis of GSH, which detoxifies various oxidants and replenishes cellular thiol stores, contributing to redox balance overally [92]. However, the oral bioavailability of GSH is low due to its rapid elimination in the gastrointestinal tract, leading to poor absorption and low blood levels, making traditional oral forms ineffective for therapeutic purposes [93]. Alternative delivery methods using nanoformulations such as solid lipid nanoparticles (SLNs), liposomes and nanoemulsions are being developed for NAC to enhance GSH absorption and efficacy. These lipid-based nanosystems protect NAC from degradation, improve its solubility in the body and facilitate targeted delivery across biological barriers by providing a protective barrier and controlled release. This leads to increased bioavailability, extended half-life, and reduced side effects, improving the therapeutic benefit of NAC for various conditions [94]. Many studies support that NAC treatment in obese mice shows reduced fat mass, increased insulin sensitivity, and improved glucose and lipid metabolism [95]. Daily NAC treatment for 11 weeks in high-carbohydrate diet-fed rats significantly reduced serum insulin levels and improved insulin sensitivity, preventing the development of IR [96]. Also, NAC treatment was found to increase the phosphorylation of the PI3K/AKT and JNK2/STAT3 signaling pathways [97]. Moreover, NAC has been shown to inhibit lipid accumulation in cultured adipocytes, and to reduce the expression of proteins like heat shock protein 70 (HSP70), aminoacylase-1 (ACY-1), and transketolase [98]. NAC reduces the release of inflammatory cytokines like TNF-α, IL-6, IL-8, and IL-1β by suppressing NF-κB activation through several mechanisms, including scavenging ROS, inhibiting the IKKβ/NF-κB signaling pathway, and preventing the nuclear translocation of NF-κB [99]. In addition, NAC can reduce triglyceride accumulation in cells by inhibiting adipogenic transcription factors like PPARγ and SREBP-1, as demonstrated in various preclinical studies [100]. Furthermore, research indicates that NAC can enhance insulin-induced glucose uptake in adipocytes by influencing the insulin signaling pathway, specifically by modulating the activity of PI3K and IRS-1 [95]. Findings suggest that NAC may prevent obesity-related liver issues, like nonalcoholic steatohepatitis (NASH) by normalizing malondialdehyde (MDA) and improving the function of antioxidant enzymes such as SOD [101]. NAC reduces the increased thrombotic tendency in T2DM, boosting platelet GSH levels, which enhances their antioxidant defenses and reduces ROS production. This increased antioxidant capacity helps to counteract the platelet hyperaggregability characteristic of T2DM, potentially making NAC a beneficial therapy for these patients [102]. A growing body of research indicates that NAC supplementation may protect the heart from oxidative damage caused by hyperglycemia, a key factor in diabetic cardiomyopathy. By acting as a potent antioxidant and GSH precursor, NAC helps to reduce ROS in cardiac cells, which in turn can attenuate heart remodeling, improve cardiac function, and potentially prevent myocardial injury [95].
4.4. Zinc
Zinc is an essential trace element of living organisms that serves as a cofactor for over 300 enzymes and 2000 transcription factors contributing to DNA and protein synthesis and cellular metabolism and signaling pathways [103]. Zinc also provides molecular structural stability of the cellular membrane [104]. Under normal conditions, zinc binds to metallothioneins (MTs) in mammals with low affinity, while in oxidative stress zinc is released from its complex with metallothionein (MT) to exert antioxidant activity [105]. Zinc is a structural component of the cytoplasmic enzyme, superoxide dismutase [106], which promotes the conversion of two superoxide radicals (O-2) into ordinary molecular oxygen (O2) and hydrogen peroxide (H2O2) and it also reduces ROS production and propagation [107]. Moreover, zinc regulates the expression of glutamate-cysteine ligase (GCL), which is the rate-limiting enzyme for de novo cytoplasmic synthesis of glutathione (GSH), an effective antioxidant in the body [107]. Zinc supplementation enhances insulin sensitivity by inactivating protein tyrosine phosphatase 1B (PTP1B), thereby preventing dephosphorylation of insulin receptor (IR) [108]. Therefore, zinc improves fasting and postprandial plasma glucose levels [108,109]. Under hyperglycemic conditions, zinc ions increase insulin secretion from pancreatic β-cells, insulin storage capacity, and insulin structural stability, reducing chronic hyperglycemia [109,110]. Thus, patients who cannot be adequately controlled by antidiabetic drugs may benefit from zinc supplementation. Patients with T2DM also experience zinc deficiency due to hyperglycemic osmotic diuresis, which causes increased urinary zinc excretion [111]. At the same time, chronic low zinc intake is a risk factor for the development of obesity and T2DM, while zinc supplementation appears to prevent the onset of metabolic syndrome and T2DM [109]. Wang et al. in a meta-analysis showed that zinc supplementation improves the glycemic profile of diabetic patients and individuals at high risk of progression to T2DM [112]. It has also been found that insufficient concentrations of zinc promote the production of pro-inflammatory cytokines, such as IL-1β, IL-2, IL-6 and TNF-α, resulting in oxidative stress [113]. Furthermore, Zn deficiency disrupts cell growth, attenuates intracellular signaling pathways, and enhances the p53 signaling pathway, resulting in apoptosis initiation [114]. However, excessive zinc intake causes copper deficiency and affects the expression of copper-dependent enzymes, such as superoxide dismutase and ceruloplasmin, which are important antioxidant enzymes [115]. Therefore, the ideal therapeutic regimen of dose and duration of zinc supplementation for the prevention and treatment of T2DM should be determined in adult obese patients [112].
4.5. Alpha-Lipoic Acid
Alpha-lipoic acid (also known as thioctic acid) is an eight-carbon saturated fatty acid [116], which is found in certain foods at low content, such as red meat, spinach, broccoli, potatoes, sweet potatoes, carrots, beets, and yeast [117]. Alpha-lipoic acid is also de novo sythesized in small amounts by the mitochondria of the human body and is an essential coenzyme for several enzymes, such as the 2-oxoglutarate dehydrogenase complex, the branched-chain oxoacetate dehydrogenase complex, and the acetoin dehydrogenase complex [117]. It has a short half-life and bioavailability primarily due to poor solubility, hepatic degradation and instability in the stomach [118]. Alpha-lipoic acid is considered a potent antioxidant by modulating various signaling pathways related to inflammation and oxidative stress, scavenging ROS and regenerating other antioxidants such as glutathione (GSH), vitamin C, vitamin E and coenzyme Q10 [119,120]. Alpha-lipoic acid supplementation in diabetic patients seems to reduce significantly the risk of obesity and its complications, exerting its effects by inhibiting hepatic gluconeogenesis and enhancing peripheral glucose uptake into peripheral cells [121]. Ansar et al., in an eight-week randomized double-blind, placebo-controlled trial found that daily administration of 300 mg alpha-lipoic acid, in T2DM patients, significantly reduced fasting and postprandial blood glucose levels and IR [122]. Also, Zhang et al. in a 2-week randomized, double-blind, placebo-controlled trial in 22 obese patients found that the intravenous administration of 600 mg alpha-lipoic acid improved insulin sensitivity and plasma lipid profile possibly by ameliorating OS and chronic inflammation [123]. Moreover, in a 20-week double-blind, placebo-controlled trial, in 360 obese patients, was found that per os administration of 1800 mg/day alpha-lipoic acid led to a modest weight loss compared to placebo group [124]. The positive effects of α-lipoic acid supplementation in the regulation of glucose and lipid metabolism, the reduction in energy intake, the increase in energy expenditure and the loss of body weight possibly seem to be associated with the activation of AMPK [125]. Haghighatdoost et al. in a meta-analysis of randomized controlled trials showed that alpha-lipoic acid supplementation in obese patients for more than 8 weeks appeared to be associated with a decrease in leptin levels and an increase in adiponectin levels [126]. The increased levels of adiponectin after alpha-lipoic acid administration seem to be, at least partly, due to the activation of AMPK and PPAR-γ [127].
4.6. L-Carnitine
Carnitine occurs as two isomeric forms (D and L). D-carnitine is not naturally found in the body, but is only present in some synthetic preparations [128]. In animal models, D-carnitine supplementation has been linked to liver inflammation, OS, and apoptosis [129]. L-carnitine (levo-carnitine) is the biologically active form of carnitines. It is a water-soluble quaternary amine that is vital for energy production, as it transports FFAs into the mitochondria for their conversion to acetyl-CoA through a process called β-oxidation. Then, acetyl-CoA enters the Krebs cycle (also known as the citric acid cycle) and through a series of reactions leads to the production of ATP, which provides energy to the cells [117]. Dietary sources of L-carnitine include red meat, fish, poultry, eggs, dairy products, soy, nuts, and seeds [117]. However, vegetables, fruits and grains contain very low amounts of L-carnitine [129]. L-carnitine supplementation has been shown to significantly reduce OS and increase the activity of antioxidant enzymes [117]. A meta-analysis by Fathizadeh et al. revealed that L-carnitine supplementation significantly reduced levels of CRP, IL-6, TNF-α, and MDA, and increased levels of SOD [130]. In addition, L-carnitine supplementation significantly reduces body weight [131]. Also, L-carnitine induces beneficial changes in the glycemic and lipid profiles of patients with type II diabetes mellitus who follow a Mediterranean diet and receive their recommended treatment [132].
4.7. Coenzyme Q10 (CoQ10)
CoQ10, or ubiquinone, is a fat-soluble benzoquinone with a side chain of 10 isoprenoid units, found in almost all human and animal cells, synthesized endogenously from precursors derived from both phenylalanine (for the benzoquinone ring) and mevalonic acid (for the isoprenoid side chain) [133]. Coenzyme Q10 exists in 2 forms: the oxidized form (ubiquinones, oCoQ10) and the reduced form (ubiquinol, rCoQ10) [133]. Within cells, ubiquinol and ubiquinone are constantly interconverted between oxidized and reduced forms [134]. CoQ10 is involved in the mitochondrial electron transport chain, which is essential for the synthesis of adenosine triphosphate (ATP) and therefore has a key role in cellular energy supply [135]. A small increase in the concentration of coenzyme Q10 in mitochondrial membranes can increase the mitochondrial respiration [136]. Primary dietary sources of CoQ10 include beef and pork, organ meats (such as heart, kidneys and liver), poultry, oily fish (such as salmon and tuna), olive oil, soybeans, grape seeds, whole grains, nuts and most dairy products (in smaller amounts) [137]. CoQ10 absorption from the diet occurs mainly in the small intestine and is best absorbed by taking a meal containing oil or foods rich in fat and is then transported to the liver to form a lipoprotein complex [138]. Ubiquinol supplements offer superior bioavailability compared to ubiquinone probably due to more efficient absorption and incorporation into mixed micelles in the intestine [139]. Also, intestinal absorption of CoQ10 varies significantly between individuals [134]. CoQ10 as a dietary supplement is relatively safe, but approximately 1% of the population may experience side effects such as headache, stomach ache, diarrhea, loss of appetite, dizziness, irritability, itching, skin rash, and nausea [140]. CoQ10 possesses significant antioxidant and anti-inflammatory properties [141], protecting cellular components like lipids, proteins, and DNA from oxidative damage [142], and also modulating inflammatory gene expression to reduce pro-inflammatory cytokines, such as TNF-α and IL-6 [143]. However, CoQ10 supplementation does not appear to improve body weight, BMI and waist circumference [144], but seems to have beneficial effects on glycemic control, lipid profile and blood pressure in patients with T2DM [145]. The beneficial effects of CoQ10 on lipid metabolism may be due to increased expression of PPAR-γ gene, which produces a nuclear receptor protein that regulates lipid metabolism and inflammation [146,147]. In addition, CoQ10 supplementation regulates hepatic lipid metabolism by the activation of the AMP-activated protein kinase (AMPK) pathway [148]. Moreover, CoQ10 supplementation suppresses the LOX-1 receptor and reduces the ROS production by inhibiting the activation of NADPH oxidation and other inflammatory pathways [149].
4.8. Superoxide Dismutases (SODs)
Superoxide dismutases (SODs) are crucial endogenous metalloenzymes that protects cells by converting superoxide radicals (O2-) into less harmful hydrogen peroxide (H2O2) and molecular oxygen (O2) [142]. Then, other endogenous antioxidant enzymes such as catalase, glutathione peroxidase (GPx) and peroxiredoxines (Prx) eliminate hydrogen peroxide (H2O2) reducing it to water (H2O) [142]. On the other hand, under specific conditions SODs can indirectly act as prooxidant by accumulating hydrogen peroxide (H2O2), which in the presence of ferrous ions (Fe2+) undergoes the Fenton reaction. This reaction produces high reactive hydroxyl radicals (•OH), which are toxic ROS that cause cellular damage [142]. In mammals, the three SOD isoforms are SOD1 (copper-zinc SOD) located in the cytoplasm; SOD2 (manganese SOD) located in mitochondria; and SOD3 (extracellular cooper-zinc SOD), located in the extracellular space. Each isoform is produced by a distinct gene, but requires different metal cofactors for its catalytic activity produced by distinct genes and characterized according to the location they are located and the metal they catalyze [150]. TNF-α is a potent activator of SOD2 contributing to its immune-modulatory effects [151]. The inflammatory mediators like IL-1, IL-4, IL-6, and TNF-α activate SOD2, which is induced by its binding sites for transcription factors such as NF-κB, NF-1, and C/EBPs within the SOD2 gene [151]. Evidence indicates that SODs and their mimetics can improve the obesity phenotype by reducing OS and inflammation, which are key features of obesity [152] An in vivo study in mice fed a high-fat diet and supplemented with the antioxidant Tempol experienced significantly greater body weight loss and reduced lipid accumulation compared to control mice, which was linked to alterations in the gut microbiome and bile acid composition [153]. Mice on high-fat diets (HFDs) receiving hydrodynamic injections of SOD3 plasmids showed improved metabolic health markers, including reduced body weight, blood levels of triglycerides and cholesterol, along with better glucose tolerance and insulin sensitivity, compared to control mice. Additionally, the metabolic improvement was associated with higher mRNA levels of key genes involved in energy expenditure and breakdown, such as CPT1α, CPT1β, PGC1α, PGC1β, and UCP2 [154]. Also, an in vivo study using nanoparticulated SOD on mice with a high-fat diet (HFD) demonstrated improvements in lipid metabolism, specifically by reducing serum triglyceride levels, decreasing liver triglyceride content, and mitigating hepatic lipid accumulation, which are key indicators of metabolic dysfunction associated with obesity [155]. Coudriet et al. found that treating T2DM-induced HFD in mice with a manganese porphyrin SOD mimetic improved mitochondrial function, glucose tolerance and IR by stabilizing mitochondrial membranes and enhancing aconitase activity. Additionally, the MnP treatment improved liver function and reduced hepatic steatosis [156].
4.9. Catechins
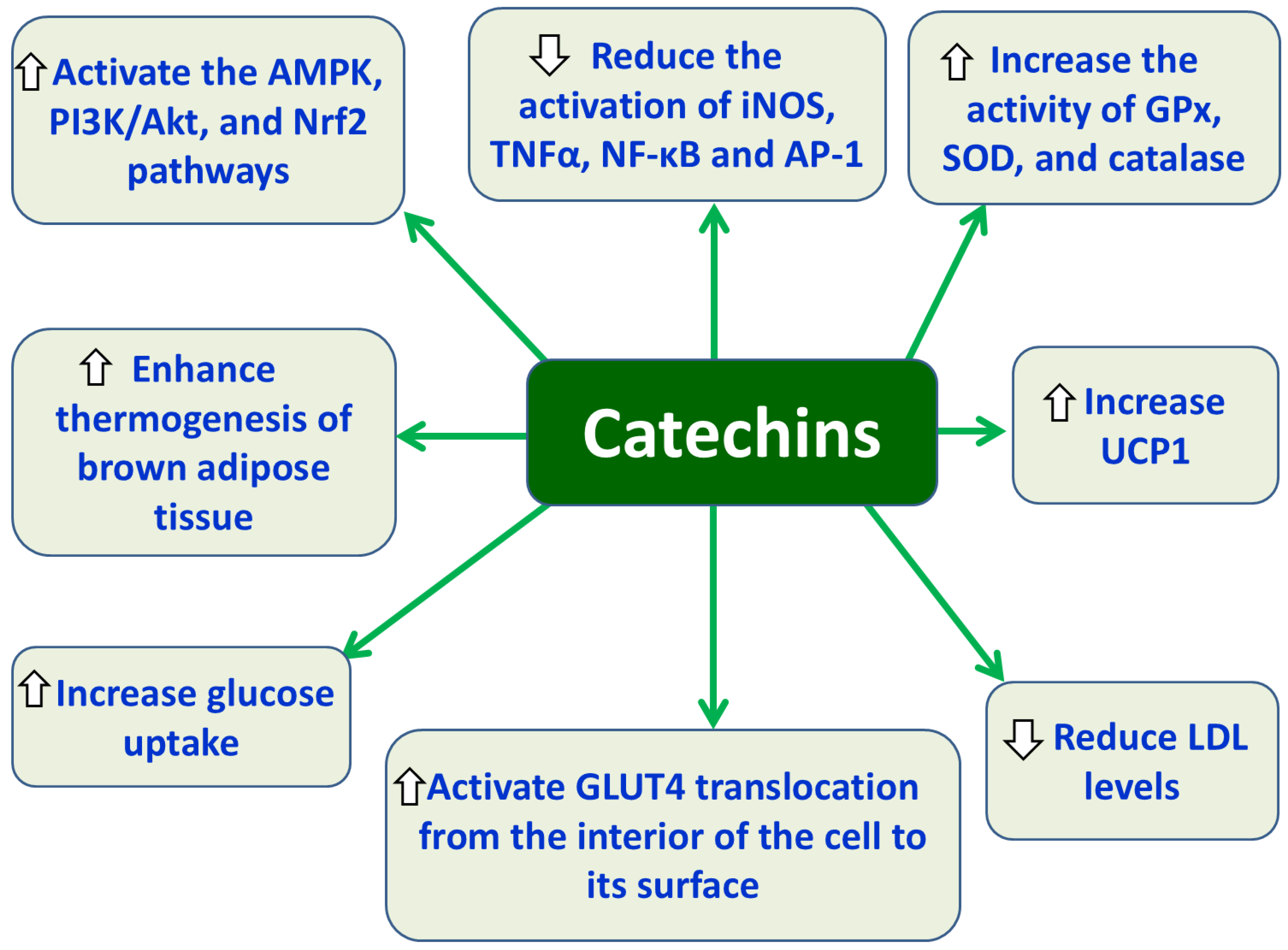
Catechins, including epigallocatechin gallate (EGCG), are plant compounds classified as flavanols, a subgroup of flavonoids, known for their potent antioxidant, anti-inflammatory, and therapeutic properties. Found primarily in green tea, cocoa, red wine, apples, and berries, catechins act by scavenging free radicals, inhibiting OS, and modulating signaling pathways [157]. EGCG exhibits antioxidant effects by increasing the activity of the antioxidant enzymes GPx, SOD, catalase and anti-inflammatory effects, by decreasing the activation of inflammatory markers like inducible nitric oxide synthase (iNOS), TNFα, NF-κB, and AP-1, which contribute to cellular damage and inflammatory response [157,158]. Moreover, EGCG activates the Nrf2 pathway by stimulating the PI3K signaling cascade, which leads to beneficial effects in ameliorating various diabetic complications, such as diabetic nephropathy through the reduction of oxidative damage, inflammation, and fibrosis [159]. Furthermore, EGCG acts like insulin promoting glucose uptake by triggering the translocation of GLUT4 from inside the cell to its surface, a process that requires PI3K/Akt pathway and activation of AMPK [160]. In parallel, EGCG supplementation enhances brown adipose tissue (BAT) thermogenesis by activating AMPK pathway and increasing uncoupling protein 1 (UCP1), thereby promoting heat production, a process critical for energy expenditure and anti-obesity effects [161]. Accumulating evidence suggests that EGCG supplementation, often in the form of green tea extract, improves endothelial function and has significant beneficial effects in reducing systolic blood pressure (SBP) by reducing the secretion of vasoconstrictor substances like endothelin-1 (ET-1) and increasing the production of nitric oxide (NO) [162]. Also, consumption of green tea EGCG can reduce LDL cholesterol levels [163], accentuating the potential positive effects of EGCG on metabolic imbalances provoked by oxidative stress and inflammatory conditions.
Figure 3.
Protective effects of catechins against obesity-associated T2DM. Abbreviations: AMPK: AMP-activated protein kinase; PI3K: phosphoinositide 3-kinase; Nrf2: nuclear factor erythroid 2-related factor 2; iNOS: inducible nitric oxide synthase; TNF-α: tumor necrosis factor-alpha; NF-κB: nuclear factor kappa B; AP-1: activator protein-1; GPx: glutathione peroxidase; SOX: sulfite oxidase; GLUT4: glucose transport type 4; UCP1: uncoupling protein 1; LDL: low-density lipoprotein.
Figure 3.
Protective effects of catechins against obesity-associated T2DM. Abbreviations: AMPK: AMP-activated protein kinase; PI3K: phosphoinositide 3-kinase; Nrf2: nuclear factor erythroid 2-related factor 2; iNOS: inducible nitric oxide synthase; TNF-α: tumor necrosis factor-alpha; NF-κB: nuclear factor kappa B; AP-1: activator protein-1; GPx: glutathione peroxidase; SOX: sulfite oxidase; GLUT4: glucose transport type 4; UCP1: uncoupling protein 1; LDL: low-density lipoprotein.
4.10. Quercetin
Quercetin (3,5,7,3′,4′-pentahydroxyflavone) is a natural yellow flavonol [164]. Quercetin is found in many fruits such as cherries, berries and apples, vegetables such as onions, capers, cabbage, red lettuce leaves, broccoli, nuts and beverages such as green tea, coffee and red wine [165]. Also, quercetin is found in various medicinal plants such as Acanthopanax seticosus (Siberian ginseng), momordica charantia (bitter melon), and brassica rapa (turnip) and has been used in traditional medicine [164]. It is completely soluble in lipids and alcohol, but not water, which limits its absorption from the digestive system [164]. Also, quercetin has low oral bioavailability because it is extensively metabolized into inactive forms that are quickly excreted [166]. This low oral bioavailability is a major challenge for its clinical use, but methods like nanoformulations are being explored to help improve its uptake and delivery to the target site [167]. Another factor contributing to its low bioavailability includes its rapid clearance from the body [165]. Quercetin is absorbed primarily in the small intestine, with some evidence suggesting that certain forms, like quercetin glucosides, can be transported by the sodium-glucose cotransporter 1 (SGLT1) [166]. Absorption in the stomach is limited, and its poor solubility can hinder its absorption without fat parallel intake [166]. Quercetin absorbed by enterocytes is rapidly conjugated with glucuronic acid and/or sulfate by enzymes during first-pass metabolism, which significantly reduces the amount of free quercetin available in the bloodstream. These reactions convert quercetin into more water-soluble forms like quercetin-3-O-glucuronide and quercetin-3′-O-sulfate [168]. During phase II metabolism, quercetin in the intestine, kidneys and liver undergoes glucuronidation, sulfation, and methylation, leading to excretion in bile, urine and feces. During this process, enzymes like UDP-glucuronyltransferases (UGTs), sulfotransferases (SULTs), and catechol-O-methyltransferases (COMTs) add hydrophilic groups to quercetin, making it more water-soluble and easier to be excreted from the body [166]. Quercetin has a wide range of pharmacological properties, including antioxidant, anti-inflammatory, antihypertensive, and vasodilatory effects, which contribute to its potential application in treating conditions like obesity, diabetes and cardiovascular diseases. It is also being studied for its anticancer and neuroprotective effects [169]. Quercetin’s antioxidant activity involves activating the KEAP1-Nrf2 pathway. Quercetin assists to the disruption of the KEAP1-NRF2 interaction, allowing NRF2 to translocate to the nucleus and activate this protective antioxidant response [170]. In addition, quercetin can reduce serum levels of MDA, increasing the antioxidant potential via the reduction of lipid peroxidation [171]. Furthermore, quercetin reduces inflammation by suppressing the release of pro-inflammatory cytokines like IL-1β, IL-6, and TNF-α, and by lowering the expression of chemokines such as MCP-1 by inhibiting the NF-κB pathway [172]. Data suggests that quercetin prevents lipogenesis by inhibiting the production of new fat molecules and promoting their breakdown. This is achieved by suppressing key enzymes involved in fat synthesis, such as acetyl-CoA carboxylase (ACACA) and fatty acid synthase (FASN), and transcription factors like SREBP-1c as well. It also simultaneously promotes fat breakdown by up-regulating enzymes involved in fatty acid oxidation. This dual action leads to a reduction in overall body fat accumulation [173]. Also, quercetin can modulate the insulin signaling pathway to further decrease fat and triglyceride accumulation in hepatic cells, protecting therefore from nonalcoholic fatty liver disease (NAFLD) development [174]. Quercetin has shown significant antidiabetic potential by inhibiting intestinal glucose absorption, improving insulin secretion and sensitivity, and boosting glucose uptake in tissues like the liver and skeletal muscle. It also protects pancreatic cells from damage and reduces inflammation, although its low bioavailability is a hurdle for effective human application [164]. Research shows that quercetin, in rats with diabetes, induced by streptozotocin (STZ), lowered plasma glucose levels and improved glucose tolerance by regenerating pancreatic islets, increasing insulin secretion and improving antioxidant defense. It also enhances glucose uptake by activating insulin signaling molecules, such as phosphatidylinositol kinase 3 (PI3K) and insulin receptor substrate-1 (IRS-1), which leads to a significant reduction in plasma glucose levels and improved glucose tolerance [164]. In addition, a 12-week treatment of quercetin in STZ-induced diabetic rats does promote hepatic glycogen synthesis and lower blood glucose levels by activating the AKT and SIRT1 signaling pathways [175]. This is achieved because quercetin activation of SIRT1 leads to subsequent activation of the AKT pathway, which can improve glucose and lipid metabolism [175]. In hepatocytes, quercetin activates AMPK, which in turn inhibits the expression of gluconeogenic enzymes like glucose-6-phosphatase [176]. Moreover, in skeletal muscle cells, quercetin activates signaling pathways, like the PI3K/Akt and AMPK pathways, which promotes the translocation of the GLUT4 transporter to the cell membrane, increasing glucose uptake. This mechanism is similar to some anti-diabetic drugs and suggests that quercetin enhances glucose metabolism by improving also insulin sensitivity [177]. However, daily oral quercetin supplementation at 250 mg for 8 weeks did not significantly alter glycemic control or lipid profiles, but it improved their antioxidant status [178]. However, quercetin intake at daily doses of 500 mg or more for a period of at least 8 weeks significantly reduced fasting plasma glucose levels in individuals with metabolic disorders. This effect is likely due to quercetin’s ability to improve insulin sensitivity and stimulate glucose uptake [179]. Quercetin also protects against complications of diabetes mellitus by lowering blood glucose, reducing oxidative stress, and exerting anti-inflammatory effects [180]. A dose of 150 mg/kg of quercetin has been shown to improve diabetic retinopathy in streptozotocin-induced rats by activating the heme oxygenase-1 (HO-1) pathway, which helps counteract OS, inflammation and neovascularization associated with the disease [181]. In addition, quercetin inhibits the SphK1-S1P pathway, which is involved in development of renal fibrosis in cases of diabetic kidneys. This inhibition can protect against diabetic complications, leading to reduced renal fibrosis [182]. Furthermore, research shows that quercetin can increase cardioprotection in rats with streptozotocin-induced diabetes by improving endothelial function. This is achieved by reducing OS, which in turn increases the production of nitric oxide (NO) and protects against aortic damage caused by the diabetes [183]. Also, it has been found that quercetin supplementation in T2DM women significantly reduces systolic blood pressure [184].
4.11. Curcumin
Curcumin is the main active compound from the turmeric plant (Curcuma longa) and is a pleiotropic polyphenol, including strong antioxidant and anti-inflammatory properties [185]. The low bioavailability of curcumin is due to its low solubility in water, poor permeability across the intestinal lining, and rapid metabolism and clearance from the body [186]. Curcumin reduces inflammation by inhibiting key signaling pathways like NF-κB, MAPK, and JAK/STAT, which decreases the production of pro-inflammatory cytokines such as TNF-α and INF-γ, and chemokines like RANTES [185]. At the same time, curcumin functions as a metal chelator and directly scavenges ROS. It also indirectly boosts the body’s own antioxidant defenses by increasing the activity of enzymes like SOD and catalase, which reduces OS markers like malondialdehyde (MDA) and increases total antioxidant capacity (TAC) [185]. Curcumin improves insulin sensitivity by inhibiting pro-inflammatory signals like ERK/JNK phosphorylation and activating PI3K-Akt-GSK3B pathway, which promotes glucose uptake [187]. Also, curcumin promotes insulin sensitivity and redox homeostasis by regulating Keap1 expression to activate Nrf2 [185]. In addition, curcumin improves hyperglycemia [188] and hyperlipidemia by reducing triglycerides, total cholesterol, and LDL-c levels, and increasing HDL-c levels [189]. Moreover, it enhances weight loss through its anti-inflammatory, antioxidant, and metabolic regulatory properties [188,190]. Ιn vitro data suggest that curcumin can promote the “browning” of white adipocytes, leading to enhanced lipolysis, by increasing the levels of hormone-sensitive lipase and p-acyl-CoA carboxylase [190]. Accumulating evidence suggests that curcumin can modulate targets involved in metabolic diseases by suppressing the inflammatory pathways NF-κB and its regulators, IKK and JNK [188,191]. Curcumin regulates signaling pathways that target inflammatory mediators in diabetes mellitus, improves beta-cell function, and reduces IR through its antioxidant and anti-inflammatory properties [192]. In addition, curcumin can inhibit enzymes such as α-glucosidase and aldose reductase and aldose reductase inhibitors, which are linked to diabetes complications [187]. Panahi et al. found that providing 1000 mg of curcuminoids daily for 12 weeks to patients with type 2 diabetes promotes total serum antioxidant activity (TAC) and SOD activity and reduces malondialdehyde (MDA) levels [193]. A systematic review by Matron et al., found that curcumin supplementation significantly reduced BMI, HbA1c and fasting blood glucose levels [194], whilst curcumin-based nanoparticles are also being investigated for their potential therapeutic properties in T2DM, in order to enhance its solubility, bioavailability and transport across biological membranes [185].
4.12. Resveratrol (RSV)
RSV (3,4′,5-trihydroxystilbene) is a polyphenol found in foods like red wine, red grapes, berries, apples, plums, blueberries, peanuts, and chocolate [195]. RSV is associated with a range of health benefits, such as anti-inflammatory, antioxidant, and anti-aging effects, which may help in the prevention or management of conditions like T2DM, CVDs, certain cancers, and neurodegenerative disorders [185]. However, RSV’s rapid metabolism in the small intestine and by the gut bacteria, along with its poor water solubility, leads to low plasma bioavailability, which can limit its effectiveness [196]. RSV modifies the gut microbiota by increasing beneficial bacteria like Bacteroidetes and Bifidobacterium, while reducing opportunistic pathogens like Escherichia coli. This modulation of the gut microbiome is associated with benefits for metabolic and cardiovascular health, including a reduction in levels of trimethylamine-N-oxide (TMAO) [197]. The mechanism of RSV action involves the inhibition of PPARγ and activation of SIRT1 leading to a decrease in BMI by reducing lipogenesis and increasing lipolysis [198]. RSV provides potent antioxidant effects in diabetic models by activating the Nrf2/Keap1 pathway, increasing Nrf2 protein levels and the expression of antioxidant genes such as HO-1 and GST. This activation process reduces accumulated ROS and is a key mechanism by which RSV protects against OS and other diabetes-associated complications [198,199]. Moreover, RSV has demonstrated anti-inflammatory effects in diabetic models, primarily by inhibiting the NF-κB pathway. This action reduces pro-inflammatory cytokines like TNF-α, IL-1β, and IL-6, which in turn improves insulin signaling and protects insulin-producing cells from damage in T2DM [199]. The anti-inflammatory effects of RSV are, in part, mediated by its activation of the SIRT1 enzyme, which then deactivates NF-κB by binding to its p65 subunit [200]. Inactivating the transcription factor NF-κB halts the transcription of genes for inflammatory molecules like the adhesion molecules ICAM-1 and VCAM-1 [201]. In parallel, RSV protects pancreatic β-cells from stress by down-regulating the NF-κB signaling cascade and pro-apoptotic signals, which improves β-cell survival [199]. In addition, RSV has antidiabetic properties by activating the AMPK/SIRT1 pathway to improve insulin sensitivity in tissues like the liver and muscle. This activation enhances insulin signaling, leading to improved glucose uptake and metabolism [199]. The activation of SIRT1 and AMPK by the resveratrol, in turn activates FOXO1 (Forkhead Box O1) and PGC-1α (Peroxisome Proliferator- Activated Receptor Gamma Coactivator-1α). This activation leads to improved mitochondrial function and protects against metabolic disorders like T2DM [199]. At the same time, RSV suppresses the negative regulator PTP1B (protein tyrosine phosphatase 1B), which allows the IRS-1/PI3K/AKT pathway to remain active, thereby promoting insulin sensitivity and glucose uptake [199]. RSV can improve glucose metabolism in muscle cells through the promotion of the Akt pathway, which triggers the translocation of the GLUT4 protein to the cell membrane [199]. A meta-analysis by Abdelhaleem et al. of 17 RCTs with 871 patients with T2DM concluded that supplementation of RSV at doses of 500 mg or more significantly improved glycemic control and cardiometabolic parameters compared to placebo. Specifically, it led to significant improvements in fasting blood glucose (FBG) and HbA1c levels, and total cholesterol (TC) levels and reduced systolic blood pressure [202]. Zeraattalab-Motlagh et al. found that RSV supplementation showed, at least partly, beneficial effects in patients with T2DM, MetS, and NAFLD, such as improvement of lipid profiles, glycemic control, and inflammatory markers. However, the study concluded that the overall evidence does not support the usage of RSV for the management of cardiometabolic risk factors in these patients groups due to the trivial magnitude of effects, low certainty of evidence, and limited trial numbers for most outcomes. A specific exception was the short-term (<12 weeks) improvement in HbA1c, which was clinically significant but requires further research given the limitations [203].
4.13. Lycopene
Carotenoids are a group of over 700 naturally occurring compounds synthesized by plants, algae and microorganisms like fungi, and bacteria that provide yellow, orange, and red colors [204,205]. Approximately 90% of carotenoids in both diet and human body are α-carotene, β-carotene, lycopene, lutein and cryptoxanthin [204]. Lycopene, also known as psi-carotene, is a fat-soluble pigment primarily found in tomatoes [206]. Also, other foods like red guava, papaya, watermelon, pink grapefruit, red grapes, apricots, rose hips, eggplant and even potatoes contain lycopene [204]. Moreover, lycopene can be found in certain algae, and fungi [204]. Lycopene has both antioxidant and anti-inflammatory properties that protect the body from cellular damage [207]. Its antioxidant role involves neutralizing of harmful ROS via electron transfer, where lycopene acts as an electron donor [208,209]. The anti-inflammatory lycopene effects come from its ability to inhibit pro-inflammatory cytokines like TNF-α, IL-1β and IL-6, and block signaling pathways like the NF-κB pathway, which decreases inflammation in both adipocytes and macrophages [210,211]. Lycopene seems to prevent obesity-related inflammation by shifting adipose tissue macrophages (ATM) from a pro-inflammatory M1 state to an anti-inflammatory M2 state [212]. Lycopene consumption contributes to obesity in animal studies. Particularly, in animal models, lycopene has been shown to reduce blood lipid levels, prevent weight gain, and decrease liver fat accumulation by improving overall lipid metabolism [210]. Also, a 12-week study on Swiss albino mice showed that lycopene counteracts the effects of a high-fat diet (HFD) by preventing weight gain, reducing fat in adipose tissue, and improving IR. These are achieved by decreasing total triglyceride levels, improving liver function related to glucose and lipids, and boosting glucose clearance and insulin sensitivity [213]. In addition, a study on C57BL/6J mice found that lycopene improved metabolic health by promoting lipolysis and improving glucose control, while also reducing fat-related markers like triglycerides, NEFA, and HOMA-IR. Lycopene also reduces inflammation and adipocyte hypertrophy and inhibites genes linked to lipid storage, suggesting a multifaceted beneficial effect on obesity-related metabolic dysfunction [214]. Moreover, a study on mice showed that lycopene reduces fat accumulation by inhibiting lipogenesis genes (Fas, Acaca, Pparγ) and promoting lipolysis via thermogenetic genes (Pgc1α, Prdm16, Ucps) and mitochondrial functional genes (Cox5b, Cox8b, CoxII, Cycs, Sirt1). It also prevents lipid buildup caused by autophagy by down-regulating autophagy-related genes (Atg7, Atg14, P62, Lc3, Beclin) [215]. Furthermore, a study on Wister rats demonstrated that lycopene could inhibit obesity-related complications by preventing body and liver weight gain. It also improved blood lipid profile by lowering serum levels of cholesterol, LDL, TGs, and Apo-B, while raising HDL serum levels. Also, lycopene can mitigate liver damage from obesity by reducing OS, inflammation, and fibrosis through antioxidant and anti-inflammatory actions. At the same time, lycopene can prevent cardiac complications by reducing LDH and creatine kinase (CK) levels [216].
Lycopene acts against diabetes and its complications through multiple mechanisms, including improving antioxidant defenses, reducing oxidative stress markers like MDA, and inflammatory markers like CRP. It also improves glucose control by reducing markers such as glycated hemoglobin (HbA1c) and positively influences cell survival pathways by modulating proteins involved in apoptosis and cell death like RAGE, NF-қB, Bax, Bcl-Xl, and Bcl-2 [217]. It has been found that the long-term intake of cooked tomatoes (around 200g per day) can improve antioxidant status in patients with T2DM by increasing antioxidant enzymes levels like SOD, GSH, GPx, GR and reducing lipid peroxidation (MDA levels) after 30 days [218]. Moreover, Shidfar et al. found that 32 patients who received 200 g raw tomato daily for 8 weeks showed significant decreases in both systolic and diastolic pressure, along with a significant increase in ApoA-1. These findings suggest that daily tomato consumption may be beneficial for the managements of some cardiovascular risk factors in patients with T2DM [219]. Also, Singh et al. conducted a 3-month study on subjects with T2DM and found that daily lycopene supplementation of 4 mg improved their oxidative-antioxidant status. Participants receiving lycopene showed significant increases in antioxidant enzymes (SOD, GPx, GR) and reduced levels of MDA, along with reduced XOD (xanthine oxidase) levels, compared to the non-lycopene group [220]. In parallel, Guo et al. showed that lycopene increases the expression of the enzyme heme oxygenase-1 (HO-1) in the kidneys of diabetic individuals, protecting against diabetic kidney damage, by reducing oxidative stress and inflammation. This increase in HO-1 expression maintains renal metabolic homeostasis by mitigating damage from conditions like diabetic nephropathy [221]. In addition, Ozmen et al. showed that lycopene treatment in STZ-induced diabetic rats can alleviate diabetes-related pancreatic damage, reduce blood and urine glucose levels, and increase serum insulin levels [222]. Studies on albino rats show that lycopene has potential to prevent T2DM and improve diabetic neuropathy by acting as an antioxidant and improving metabolic health. It reduced OS, as seen by the increased levels of antioxidant enzymes like SOD and GSH-Px and decreased MDA levels. Lycopene also improves glycolipid metabolism by lowering blood glucose, TG, TC, LDL, and GHb, while raising HDL and insulin levels. Furthermore, it can inhibit inflammation by decreasing TNF-α and NO generation [223]. Furthermore, Gao et al. found in 1978 pregnant women a statistically significant inverse relationship between lycopene consumption and the risk of gestational diabetes mellitus (GDM), especially in the first trimester of the pregnancy [224].
5. Conclusions and Perspectives
T2DM can be prevented by maintaining body weight within normal limits and adopting a combination of a healthy diet and regular physical activity. In cases where the prevention of T2DM onset fails, early diagnosis is essential to avoid the development of its complications. Obesity plays a central role in the pathogenesis of T2DM, as it induces a series of disturbances such as extensive expansion of adipose tissue, chronic low-grade inflammation, OS, due to the excessive presence of ROS and nitrogen species (RNS), dysfunction of the liver and skeletal muscles, pancreatic toxicity resulting in a decreased number of functional β-cells, and dysregulation of the microbiota–gut–brain axis.
Given these interconnections between obesity and T2DM, pharmacotherapy of obesity using antioxidant compounds, with anti-inflammatory and antioxidant potential, may reduce inflammation and OS and improve metabolic diseases, such as T2DM. In this context, plant-derived antioxidants, minerals and vitamins could act as adjunct agents through their involvement in multiple mechanisms highlighted in the present study, ultimately improving the outcomes of T2DM and its complications. Nevertheless, due to the limited clinical data and the poor pharmacokinetic profiles of many of these compounds, further studies are required, both at the clinical level and in the development of pharmaceutical formulations, with improved pharmacokinetic characteristics, despite their highly promising potential.
Author’s Contributions
The authors confirm their contribution to the paper as follows: study conception and study design: M.V., P.T.N.; literature review F.N.V., V.K.V.; draft manuscript: F.N.V., M.V., V.K.V., P.T.N.; editing and critical revision of the manuscript: F.N.V., M.V., P.T.N. All authors reviewed and approved the final version of the manuscript.
Funding
This review did not receive any financial support.
Conflict of Interest
The authors declare no conflict of interest.
References
- Karamanou, M.; Protogerou, A.; Tsoukalas, G.; Androutsos, G.; Poulakou-Rebelakou, E. Milestones in the history of diabetes mellitus: The main contributors. World J. Diabetes 2016, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Priya, G.; Kalra, S.; Dasgupta, A.; Grewal, E. Diabetes Insipidus: A Pragmatic Approach to Management. Cureus 2021, 13, e12498. [Google Scholar] [CrossRef]
- Punthakee, Z.; Goldenberg, R.; Katz, P. 2018 Clinical Practice Guidelines Definition, Classification and Diagnosis of Diabetes, Prediabetes and Metabolic Syndrome Diabetes Canada Clinical Practice Guidelines Expert Committee. Can. J. Diabetes 2018, 42, S10–S15. https://www.diabetes.ca/health-care-providers/clinical-practice-guidelines/chapter-3#panel-tab_FullText. [CrossRef] [PubMed]
- Solis-Herrera, C.; Triplitt, C.; Reasner, C.; DeFronzo, R.A.; Cersosimo, E. Classification of Diabetes Mellitus. Endotext [Internet]. Feingold, K.R., Ahmed, S.F., Anawalt, B., Eds.; MDText.com, Inc: South Dartmouth (MA), 2018; Available from: https://www.ncbi.nlm.nih.gov/books/NBK279119/.
- Yudkin, J.S. “Prediabetes”: Are There Problems With This Label? Yes, the Label Creates Further Problems! Diabetes Care 2016, 39, 1468–1471. [Google Scholar] [CrossRef]
- Lu, X.; Xie, Q.; Pan, X.; Zhang, R.; Zhang, X.; Peng, G.; et al. Type 2 diabetes mellitus in adults: pathogenesis, prevention and therapy. Signal Transduct. Targeted. Ther. 2024, 9, 262. [Google Scholar] [CrossRef]
- Arroyave, F.; Montaño, D.; Lizcano, F. Diabetes Mellitus Is a Chronic Disease that Can Benefit from Therapy with Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2020, 21, 8685. [Google Scholar] [CrossRef]
- Dilworth, L.; Facey, A.; Omoruyi, F. Diabetes Mellitus and Its Metabolic Complications: The Role of Adipose Tissues. Int. J. Mol. Sci. 2021, 22, 7644. [Google Scholar] [CrossRef]
- Theodosis-Nobelos, P.; Rekka, E.A. The Antioxidant Potential of Vitamins and Their Implication in Metabolic Abnormalities. Nutrients 2024, 16, 2740. [Google Scholar] [CrossRef]
- Caturano, A.; Rocco, M.; Tagliaferri, G.; Piacevole, A.; Nilo, D.; Di Lorenzo, G.; Iadicicco, I.; Donnarumma, M.; Galiero, R.; Acierno, C.; et al. Oxidative Stress and Cardiovascular Complications in Type 2 Diabetes: From Pathophysiology to Lifestyle Modifications. Antioxidants 2025, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, J.G.; Hamilton, A.; Ramracheya, R.; Tarasov, A.I.; Brereton, M.; Haythorne, E.; et al. Dysregulation of Glucagon Secretion by Hyperglycemia-Induced Sodium-Dependent Reduction of ATP Production. Cell Metab. 2019, 29, 430–442.e4. [Google Scholar] [CrossRef]
- Hædersdal, S.; Lund, A.; Knop, F.K.; Vilsbøll, T. The Role of Glucagon in the Pathophysiology and Treatment of Type 2 Diabetes. Mayo Clin. Proc. 2018, 93, 217–239. [Google Scholar] [CrossRef]
- Mouri, M.; Badireddy, M. Hyperglycemia. StatPearls [Internet]. StatPearls Publishing: Treasure Island (FL), 2023; Available from: https://www.ncbi.nlm.nih.gov/books/NBK4309.
- Gosmanov, A.R.; Gosmanova, E.O.; Kitabchi, A.E. Hyperglycemic Crises: Diabetic Ketoacidosis and Hyperglycemic Hyperosmolar State. Endotext [Internet]. Feingold, K.R., Ahmed, S.F., Anawalt, B., et al., Eds.; MD Text.com, Inc: South Dartmouth (MA), 2021; Available from: https://www.ncbi.nlm.nih.gov/books/NBK279052/.
- Varra, F.-N.; Varras, M.; Varra, V.-K.; Theodosis-Nobelos, P. Molecular and pathophysiological relationship between obesity and chronic inflammation in the manifestation of metabolic dysfunctions and their inflammation-mediating treatment options (Review). Mol. Med. Rep. 2024, 29, 95. [Google Scholar] [CrossRef] [PubMed]
- He, L. Alterations of gut microbiota by overnutrition impact gluconeogenic gene expression and insulin signaling. Int. J. Mol. Sci. 2021, 22, 2121. [Google Scholar] [CrossRef]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: fuel excess and β-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef]
- Weir, G.C.; Bonner-Weir, S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004, 53, S16–S21. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gong, M.; Wu, N. Research progress on the relationship between free fatty acid profile and type 2 diabetes complicated by coronary heart disease. Front. Endocrinol. (Lausanne) 2024, 15, 1503704. [Google Scholar] [CrossRef]
- Altalhi, R.; Pechlivani, N.; Ajjan, R.A. PAI-1 in Diabetes: Pathophysiology and Role as a Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 3170. [Google Scholar] [CrossRef] [PubMed]
- Guria, S.; Hoory, A.; Das, S.; Chattopadhyay, D.; Mukherjee, S. Adipose tissue macrophages and their role in obesity-associated insulin resistance: an overview of the complex dynamics at play. Biosci. Rep. 2023, 43, BSR20220200. [Google Scholar] [CrossRef]
- Gupta, D.; Kono, T.; Evans-Molina, C. The role of peroxisome proliferatoractivated receptor γ in pancreatic β cell function and survival: therapeutic implications for the treatment of type 2 diabetes mellitus. Diabetes Obes. Metab. 2010, 12, 1036–1047. [Google Scholar] [CrossRef]
- Samadi, M.; Aziz, S.G.-G.; Nade, R. The effect of tropisetron on oxidative stress, SIRT1, FOXO3a, and claudin-1 in the renal tissue of STZ-induced diabetic rats. Cell Stress Chaperones 2021, 26, 217–227. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, C.; Fu, Y.; Xie, L.; Kong, Y.; Yang, X. Inflammation, Apoptosis, and Fibrosis in Diabetic Nephropathy: Molecular Crosstalk in Proximal Tubular Epithelial Cells and Therapeutic Implications. Curr. Issues Mol. Biol. 2025, 47, 885. [Google Scholar] [CrossRef]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef] [PubMed]
- Domingueti, C.P.; Dusse, M.S.; Carvalho, M.G.; de Sousa, L.P.; Gomes, K.B.; Fernandes, A.P. Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J. Diabetes Complications 2016, 30, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, S.; Wang, J.; Wang, J.; Yan, Y.; Zhu, M.; et al. Oxidative stress induced by NOX2 contributes to neuropathic pain via plasma membrane translocation of PKCε in rat dorsal root ganglion neurons. J. Neuroinflammation 2021, 18, 106. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhang, Y.-H.; Wang, C.-Y.; Yu, Y.; Li, Y.-Z.; Cui, W.; et al. MicroRNA-7a-5p ameliorates diabetic peripheral neuropathy by regulating VDAC1/JNK/c-JUN pathway. Diabet. Med. 2023, 40, e14890. [Google Scholar] [CrossRef]
- Eid, S.A.; Massry, M.E.I.; Hichor, M.; Haddad, M.; Grenier, J.; Dia, B.; et al. Targeting the NADPH oxidase-4 and liver X receptor pathway preserves Schwann cell integrity in diabetic mice. Diabetes 2020, 69, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Labra, R.; Subiabre, M.; Toledo, F.; Pardo, F.; Sobrevia, L. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol. Asp. Med. 2019, 66, 49–61. [Google Scholar] [CrossRef]
- Liu, P.; Peng, J.; Han, G.-H.; Ding, X.; Wei, S.; Gao, G.; et al. Role of macrophages in peripheral nerve injury and repair. Neural Regen. Res. 2019, 14, 1335–1342. [Google Scholar] [CrossRef]
- Fu, Y.; Wu, N.; Zhao, D. Function of NLRP3 in the Pathogenesis and Development of Diabetic Nephropathy. Med. Sci. Monit. 2017, 23, 3878–3884. [Google Scholar] [CrossRef]
- Matoba, K.; Takeda, Y.; Nagai, Y.; Kawanami, D.; Utsunomiya, K.; Nishimura, R. Unraveling the role of inflammation in the pathogenesis of diabetic kidney disease. Int. J. Mol. Sci. 2019, 20, 3393–2019. [Google Scholar] [CrossRef]
- Sheu, M.L.; Ho, F.M.; Yang, R.S.; Chao, K.F.; Lin, W.W.; Lin-Shiau, S.Y.; Liu, S.-H. High glucose induces human endothelial cell apoptosis through a phosphoinositide 3-kinase-regulated cyclooxygenase-2 pathway. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 539–545. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Yiang, G.-T.; Lai, T.-T.; Li, C.-J. The oxidative stress and mitochondrial dysfunction during the pathogenesis of diabetic retinopathy. Oxid. Med. Cell Longev. 2018, 2018, 3420187. [Google Scholar] [CrossRef]
- Chaurasia, S.S.; Lin, R.R.; Parikh, B.H.; Wey, Y.S.; Tun, B.B.; Wong, T.Y.; et al. The NLRP3 inflammasome may contribute to pathologic neovascularization in the advanced stages of diabetic retinopathy. Sci. Rep. 2018, 8, 2847. [Google Scholar] [CrossRef]
- Tian, S.; Zhao, H.; Song, H. Shared signaling pathways and targeted therapy by natural bioactive compounds for obesity and type 2 diabetes. Crit. Rev. Food Sci. Nutr. 2022, 64, 5039–5056. [Google Scholar] [CrossRef]
- Bako, H.Y.; Ibrahim, M.A.; Isah, M.S.; Ibrahim, S. Inhibition of JAK-STAT and NF-κB signalling systems could be a novel therapeutic target against insulin resistance and type 2 diabetes. Life Sci. 2019, 239, 117045. [Google Scholar] [CrossRef]
- Zhang, H.; Nair, V.; Saha, J.; Atkins, K.B.; Hodgin, J.B.; et al. Podocyte-specific JAK2 overexpression worsens diabetic kidney disease in mice. Kidney Int 2017, 92, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Han, Y.; Zhao, F.; Zhao, Z.; Tian, J.; Jia, K. Nobiletin suppresses high-glucose-induced inflammation and ECM accumulation in human mesangial cells through STAT3/NF-κB pathway. J. Cell Biochem. 2019, 120, 3467–3473. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.G.; Luckett-Chastain, L.R.; Calhoun, K.N.; Frempah, B.; Bastian, A.; Gallucci, R.M. Interleukin 6 function in the skin and isolated keratinocytes is modulated by hyperglycemia. J. Immunol. Res. 2019, 2019, 5087847. [Google Scholar] [CrossRef]
- Sawaya, A.P.; Stone, R.C.; Brooks, S.R.; Pastar, I.; Jozic, I.; Hasneen, K.; et al. Deregulated immune cell recruitment orchestrated by FOXM1 impairs human diabetic wound healing. Nat. Commun. 2020, 11, 4678. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.; Singhal, M.; Jialal, I. Type 2 Diabetes. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK513253/.
- Liakopoulos, V.; Franzén, S.; Svensson, A.M.; Miftaraj, M.; Ottosson, J.; Näslund, I.; Gudbjörnsdottir, S.; Eliasson, B. Pros and cons of gastric bypass surgery in individuals with obesity and type 2 diabetes: nationwide, matched, observational cohort study. BMJ Open 2019, 9, e023882. [Google Scholar] [CrossRef]
- Patoulias, D.; Papadopoulos, C.; Stavropoulos, K.; Zografou, I.; Doumas, M.; Karagiannis, A. Prognostic value of arterial stiffness measurements in cardiovascular disease, diabetes, and its complications: The potential role of sodium-glucose co-transporter-2 inhibitors. J. Clin. Hypertens. (Greenwich) 2020, 22, 562–571. [Google Scholar] [CrossRef]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus–Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef]
- Kushner, A.; West, W.P.; Suheb, M.Z.K.; Pillarisetty, L.S. Virchow Triad. StatPearls [Internet].Treasure Island (FL): StatPearls Publishing. 2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539697/.
- Chiu, J.-J.; Chien, S. Effects of Disturbed Flow on Vascular Endothelium: Pathophysiological Basis and Clinical Perspectives. Physiol. Rev. 2011, 91. [Google Scholar] [CrossRef]
- Yachmaneni, A. Jr.; Jajoo, S.; Mahakalkar, C.; Kshirsagar, S.; Dhole, S. A Comprehensive Review of the Vascular Consequences of Diabetes in the Lower Extremities: Current Approaches to Management and Evaluation of Clinical Outcomes. Cureus 2023, 15, e47525. [Google Scholar] [CrossRef] [PubMed]
- Bodman, M.A.; Dreyer, M.A.; Varacallo, M.A. Diabetic Peripheral Neuropathy. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. 2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK442009/.
- Akkus, G.; Sert, M. Diabetic foot ulcers: A devastating complication of diabetes mellitus continues non-stop in spite of new medical treatment modalities. World J. Diabetes 2022, 13, 1106–1121. [Google Scholar] [CrossRef]
- Koyama, K.; Anno, T.; Kimura, Y.; Kawasaki, F.; Kaku, K.; Tomoda, K.; Kaneto, H. Pathology of Ketoacidosis in Emergency of Diabetic Ketoacidosis and Alcoholic Ketoacidosis: A Retrospective Study. J. Diabetes Res. 2024, 2024, 8889415. [Google Scholar] [CrossRef]
- Talley, J.T.; Mohiuddin, S.S. Biochemistry, Fatty Acid Oxidation StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK556002/.
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone bodies: from enemy to friend and guardian angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Lizzo, J.M.; Goyal, A.; Gupta, V. Adult Diabetic Ketoacidosis. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560723/.
- Ghimire, P.; Dhamoon, A.S. Ketoacidosis. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK534848/.
- Self, W.H.; Evans, C.S.; Jenkins, C.A.; Brown, R.M.; Casey, J.D.; Collins, S.P. Clinical Effects of Balanced Crystalloids vs Saline in Adults With Diabetic Ketoacidosis: A Subgroup Analysis of Cluster Randomized Clinical Trials. JAMA Netw. Open 2020, 3, e2024596. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F.; Clegg, D.J. Electrolyte and Acid–Base Disturbances in Patients with Diabetes Mellitus. N. Engl. J. Med. 2015, 373, 548–59. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.H.I.; Mohebbi, N. Kidney metabolism and acid–base control: back to the basics. Pflugers Arch. 2022, 474, 919–934. [Google Scholar] [CrossRef]
- Sharma, S.; Hashmi, M.F.; Aggarwal, S. Hyperchloremic Acidosis. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482340/.
- Elendu, C.; David, J.A.; Udoyen, A.-O.; Egbunu, E.O.; Ogbuiyi-Chima, I.C.; Unakalamba, L.O.; et al. Comprehensive review of diabetic ketoacidosis: an update. Ann Med Surg (Lond) 2023, 85, 2802–2807. [Google Scholar] [CrossRef]
- Liman, M.N.P.; Jialal, I. Physiology, Glycosuria. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557441/.
- Hieshima, K.; Sugiyama, S.; Yoshida, A.; Kurinami, N.; Suzuki, T.; Ijima, H.; et al. Elevation of the renal threshold for glucose is associated with insulin resistance and higher glycated hemoglobin levels. J. Diabetes Investig. 2020, 11, 617–625. [Google Scholar] [CrossRef]
- Hans, F. Polyuria and Diabetes: Understanding the Connection and Implications. Afr. J. Diabetes Med. 2024, 32, 4. [Google Scholar]
- Vallon, V. Glucose transporters in the kidney in health and disease. Pflugers Arch 2020, 472, 1345–1370. [Google Scholar] [CrossRef]
- Gubbi, S.; Hannah-Shmouni, F.; Koch, C.A.; Joseph, G.; Verbalis, J.G. Diagnostic Testing for Diabetes Insipidus. Endotext [Internet]; Feingold, K.R., Ahmed, S.F., Anawalt, B., et al., Eds.; MDText.com, Inc: South Dartmouth (MA), 2022; Available from: https://www.ncbi.nlm.nih.gov/books/NBK537591/.
- Wendt, A.; Eliasson, L. Pancreatic alpha cells and glucagon secretion: Novel functions and targets in glucose homeostasis. Curr. Opin. Pharmacol. 2022, 63, 102199. [Google Scholar] [CrossRef]
- RR; Corriere, M.; Ferrucci, L. Age-related and disease-related muscle loss: the effect of diabetes, obesity, and other diseases. Lancet Diabetes Endocrinol. 2014, 2, 819–829. [Google Scholar]
- Shen, Y.; Li, M.; Wang, K.; Qi, G.; Liu, H.; Wang, W.; et al. Diabetic Muscular Atrophy: Molecular Mechanisms and Promising Therapies. Front. Endocrinol. (Lausanne) 2022, 13, 917113. [Google Scholar] [CrossRef]
- Zaffarin, A.S.; Ng, S.-F.; Ng, M.H.; Hassan, H.; Alias, E. Pharmacology and pharmacokinetics of vitamin E: Nanoformulations to enhance bioavailability. Int. J. Nanomedicine 2020, 15, 9961–9974. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, K.; Chojnacka, A.; Górnicka, M. Tocopherols and tocotrienols – bioactive dietary compounds. What is certain, what is doubt? Int. J. Mol. Sci. 2021, 22, 6222. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Omage, S.O.; Börmel, L.; Kluge, S.; Schubert, M.; Wallert, M.; Lorkowski, S. Vitamin E and metabolic health: relevance of interactions with other micronutrients. Antioxidants 2022, 11, 1785. [Google Scholar] [CrossRef]
- Riccioni, G.; Bucciarelli, T.; Mancini, B.; Corradi, F.; Di Ilio, C.; Mattei, P.A.; D’Orazio, N. Antioxidant vitamin suppementation in cardiovascular diseases. Ann. Clin. Lab. Sci. 2007, 37, 89–95. [Google Scholar]
- Theodosis-Nobelos, P.; Papagiouvannis, G.; Rekka, E.A. A Review on Vitamin E Natural Analogues and on the Design of Synthetic Vitamin E Derivatives as Cytoprotective Agents. Mini Rev Med Chem. 2021, 21, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Galmes, S.; Serra, F.; Paou, A. Vitamin E metabolic effects and genetic variants: A challenge for precision nutrition in obesity and associated disturbances. Nutrients 2018, 10, 1919. [Google Scholar] [CrossRef]
- Lewis, E.D.; Meydani, S.N.; Wu, D. Regulatory role of vitamin E in the immune system and inflammation. IUBMB Life 2019, 71, 487–494. [Google Scholar] [CrossRef]
- Donohoe, F.; Wilkinson, M.; Baxter, E.; Brennan, D.J. Mitogen-activated protein kinase (MAPK) and obesity-related cancer. Int. J. Mol. Sci. 2020, 21, 1241. [Google Scholar] [CrossRef]
- Jiang, Q. Natural forms of vitamin E: metabolism, antioxidant and anti-inflammatory activities and the role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef]
- Kanchi, M.M.; Shanmugam, M.K.; Rane, G.; Sethi, G.; Kumar, A.P. Tocotrienols: the unsaturated sidekick shifting new paradigms in vitamin E therapeutics. Drug Discov. Today 2017, 22, 1765–1781. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Chin, K.-Y.; Suhaimi, F.H.; Ahmad, F.; Ima-Nirwana, S. Vitamin E as a potential interventional treatment for metabolic syndrome: evidence from animal and human studies. Front. Pharmacol. 2017, 8, 444. [Google Scholar] [CrossRef]
- Alcalá, M.; Sanchez-Vera, I.; Sevillano, J.; Herrero, L.; Serra, D.; Ramos, M.P.; Viana, M. Vitamin E reduces adipose tissue fibrosis, inflammation, and oxidative stress and improves metabolic profile in obesity. Obesity (Silver Spring) 2015, 23, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Budin, S.B.; Othman, F.; Louis, S.R.; Bakar, M.A.; Das, S.; Mohamed, J. The effects of palm oil tocotrienol-rich fraction supplementation on biochemical parameters, oxidative stress and the vascular wall of streptozotocin-induced diabetic rats. Clinics 2009, 64, 235–244. [Google Scholar] [CrossRef]
- Zhao, L.; Kang, I.; Fang, X.; Wang, W.; Lee, M.A.; Hollins, R.R.; Marshall, M.R.; Chung, S. Gamma-tocotrienol attenuates high-fat diet-induced obesity and insulin resistance by inhibiting adipose inflammation and M1 macrophage recruitment. Int J Obes (Lond) 2015, 39, 438–446. [Google Scholar] [CrossRef]
- Tun, S.; Spainhower, C.J.; Cottrill, C.L.; Lakhani, H.V.; Pillai, S.S.; Dilip, A.; Chaudhry, H.; Shapiro, J.I.; Sodhi, K. Therapeutic Efficacy of Antioxidants in Ameliorating Obesity Phenotype and Associated Comorbidities. Front. Pharmacol. 2020, 11, 1234. [Google Scholar] [CrossRef]
- Abdullah, M.; Jamil, R.J.; Attia, J.F. Vitamin C (ascorbic acid). In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023; Available from: https://www.ncbi.nlm.nih.gov/books/NBK499877/.
- Rafighi, Z.; Shiva, A.; Arab, S.; Mohd, Y.R. Association of dietary vitamin C and E intake and antioxidant enzymes in type 2 diabetes mellitus patients. Glob. J. Health Sci. 2013, 5, 183–197. [Google Scholar] [CrossRef]
- Naziroğlu, M.; Simşek, M.; Simşek, H.; Aydilek, N.; Ozcan, Z.; Atilgan, R. The effects of hormone replace therapy combined with vitamins C and E on antioxidants levels and lipid profiles in postmenopausal women with type 2 diabetes. Clin Chim Acta 2004, 344, 63–71. [Google Scholar] [CrossRef]
- Harding, A.H.; Wareham, N.J.; Binham, S.A.; Khaw, K.T.; Luben, R.; Welch, A.; et al. Plasma vitamin C level, fruit and vegetabel consumption, and the risk of new-onset type 2 diabetes mellitus: the European prospective investigation of cancer – Norfolk prospective study. Arch. Intern. Med. 2008, 168, 1493–1499. [Google Scholar] [CrossRef]
- Guerini, M.; Gondrò, G.; Friuli, V.; Maggi, L.; Perugini, P. N-acetylcystein (NAC) and its role in clinical practice management of cystic fibrosis (CF): a review. Pharmaceuticals (Basel) 2022, 15, 217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ju, Y.; Ma, Y.; Wang, T. N-acetylcysteine improves oxidative stress and inflammatory response in patients with community acquired pneumonia: a randomized controlled trial. Medicina (Baltimore) 2018, 97, e13087. [Google Scholar] [CrossRef]
- Pei, Y.; Liu, H.; Yang, Y.; Yang, Y.; Jiao, Y.; Tay, F.R. Biological activities and potential oral applications of N-acetylcystein: Progress and prospects. Oxid. Med. Cell. Longev. 2018, 2018, 2835787. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; O’Keefe, J.H.; DiNicolantonio, J.J. Dietary Glycine Is Rate-Limiting for Glutathione Synthesis and May Have Broad Potential for Health Protection. Ochsner J. 2018, 18, 81–87. [Google Scholar] [PubMed]
- Schmitt, B.; Vicenzi, M.; Garrel, C.; Denis, F.M. Effects of N-acetylcysteine, oral glutathione (GSH) and a novel sublingual form of GSH on oxidative stress markers: A comparative crossover study. Redox Biol. 2015, 6, 198–205. [Google Scholar] [CrossRef]
- Mondal, S.; Hazra, A.; Paul, P.; Saha, B.; Roy, S.; Bhowmick, P.; Bhowmick, M. Formulation and evaluation of n-acetyl cysteine loaded bi-polymeric physically crosslinked hydrogel with antibacterial and antioxidant activity for diabetic wound dressing. Int. J. Biol. Macromol. 2024, 279, 135418. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Mazibuko-Mbeje, S.E.; Nyambuya, T.M.; Mxinwa, V.; Tiano, L.; Marcheggiani, F.; et al. The beneficial effects of N-acetyl cystein (NAC) against obesity associated complications: A systematic review of pre-clinical studies. Pharmacol. Res. 2019, 146, 104332. [Google Scholar] [CrossRef] [PubMed]
- Ismael, M.A.; Talbot, S.; Carbonneau, C.L.; Beauséjour, C.M.; Couture, R. Blockade of sensory abnormalities and kinin B1 receptor expression by N-Acetyl-L-Cystein and ramipril in a rat model of insulin resistance. Eur. J. Pharmacol. 2008, 589, 66–72. [Google Scholar] [CrossRef]
- Wang, T.; Mao, X.; Li, H.; Qiao, S.; Xu, A.; Wang, J.; et al. N-Acetylcysteine and allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via adiponectin and attenuated myocardial postischemic injury in diabetes. Free Radic. Biol. Med. 2013, 63, 291–303. [Google Scholar] [CrossRef]
- Kadota, Y.; Toriuchi, Y.; Aki, Y.; Mizuno, Y.; Kawakami, T.; Nakaya, T.; et al. Metallothioneins regulate the adipoogenic defferentation of 3T3-L1 cells via the insulin signaling pathway. PLoS One 2017, 12, e0176079. [Google Scholar] [CrossRef]
- Sun, J.; Song, P.; Wang, Y.; Chen, Y. Clinical efficacy of acetylcysteine combined with tetrandrine tablets in the treatment of silicosis and the effect on serum IL-6 and TNF-alpha. Exp. Ther. Med. 2019, 18, 3383–3388. [Google Scholar] [CrossRef]
- Hang, W.; Shu, H.; Wen, Z.; Liu, J.; Jin, Z.; Shi, Z.; et al. N-acetyl cysteine ameliorates high-fat diet-induced nonalcoholic fatty liver disease and intracellular triglyceride accumulation by preserving mitochondral function. Front Pharmacol. 2021, 12, 636204. [Google Scholar] [CrossRef]
- Korou, L.M.; Agrogiannis, G.; Koros, C.; Kitraki, E.; Vlachos, I.S.; Tzanetakou, I.; et al. Impact of N-acetylcysteine and sesame oil on lipid metabolism and hypothalamic-pituitary-adrenal axis homeostasis in middle-aged hypercholesterolemic mice. Sci. Rep. 2014, 4, 6806. [Google Scholar] [CrossRef]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; et al. ROS in platelet biology: functional aspects and methodological insights. Int. J. Mol. Sci. 2020, 2, 4866. [Google Scholar] [CrossRef] [PubMed]
- Patil, R.J.; Sontakke, T.; Biradar, A.; Nalage, D. Zinc: an essential trace element for human health and beyond. Food Health 2023, 5, 13. [Google Scholar] [CrossRef]
- Fung, E.B.; Gildergorin, G. Zinc status affects glucose homeostasis and insulin secretion in patients with thalassemia. Nutrients 2015, 7, 4296–4307. [Google Scholar] [CrossRef] [PubMed]
- Ruttkay-Nedecky, B.; Nejdl, L.; Gumulec, J.; Zitka, O.; Masarik, M.; Eckschlager, T.; et al. The role of metallothionein in oxidative stress. Int. J. Mol. Sci. 2013, 14, 6044–6066. [Google Scholar] [CrossRef]
- Muñoz, I.G.; Moran, J.F.; Becana, M.; Montoya, G. The crystal structure of an eukaryotic iron superoxide dismutase suggests intersubunit cooperation during catalysis. Protein Sci. 2005, 14, 387–394. [Google Scholar] [CrossRef] [PubMed]
- do Nascimento Marreiro, D.; Clímaco Cruz, K.J.; Silva Morais, J.B.; Batista Beserra, J.; Soares Severo, J.; de Oliveira, A.R.S. Zinc and oxidative stress: current mechanisms. Antioxidants (Basel) 2017, 6, 24. [Google Scholar]
- Maret, W. Zinc in cellular regulation: The nature and significance of “zinc signals”. Int. J. Mol. Sci. 2017, 18, 2285. [Google Scholar] [CrossRef]
- Fukunaka, A.; Fujitani, Y. Role of zinc homeostasis in the pathogenesis of diabetes and obesity. Int J Mol Sci 2018, 19, 476. [Google Scholar] [CrossRef]
- Anton, I.C.; Mititelu-Tartau, L.; Popa, E.G.; Poroch, M.; Poroch, V.; Pelin, A.M.; et al. Zinc chloride endances the antioxidant status, improving the functional and structural organic disturbances in streptozotocin-induced diabetes in rats. Medicina 2022, 58, 1620. [Google Scholar] [CrossRef]
- He, Z.; Yin, G.; Li, Q.Q.; Zeng, Q.; Duan, J. Diabetes mellitus causes male reproductive dysfunction: a review of the evidence and mechanisms. In vivo 2021, 35, 2503–2511. [Google Scholar] [CrossRef]
- Wang, X.; Wu, W.; Zheng, W.; Fang, X.; Chen, L.; Rink, L.; et al. Zinc supplementation improves glycemic control for diabetes prevention and management: a systemic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2019, 110, 76–90. [Google Scholar] [CrossRef]
- Foster, M.; Samman, S. Zinc and regulation of inflammatory cytokines: implications for cardiometabolic disease. Nutrients 2012, 4, 676–694. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Jiang, W.; Wang, X.; Shahid, S.; Saba, N.; Ahmad, M.; et al. Mechanistic impact of zinc deficiency in human development. Front Nutr 2022, 9, 717064. [Google Scholar] [CrossRef]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. (Qassim) 2018, 12, 88–93. [Google Scholar]
- Dajnowicz-Brzezik, P.; Żebrowska, E.; Maciejczyk, M.; Zalewska, A.; Chabowski, A. α -lipoic acid supplementation reduces oxidative stress and inflammation in red skeletal muscle of insulin-resistant rats. Biochem. Biophys. Res. Commun. 2025, 742, 151107. [Google Scholar] [CrossRef]
- Abdali, D.; Samson, S.; Grover, A.K. How effective are antioxidant supplements in obesity and diabetes? Med. Princ. Pract. 2015, 24, 201–15. [Google Scholar] [CrossRef]
- Salehi, B.; Yilmaz, Y.B.; Antika, G.; Tumer, T.B.; Mahomoodally, M.F.; Lobine, D.; et al. Insights on the use of α-lipoic acid for therapeutic purposes. Biomolecules 2019, 9, 356. [Google Scholar] [CrossRef] [PubMed]
- Superti, F.; Russo, R. Alpha-Lipoic Acid: Biological Mechanisms and Health Benefits. Antioxidants (Basel) 2024, 13, 1228. [Google Scholar] [CrossRef] [PubMed]
- Shahid, A.; Nasir, K.; Bhatia, M. Therapeutic Potential of Alpha-Lipoic Acid: Unraveling Its Role in Oxidative Stress and Inflammatory Conditions. Curr. Issues Mol. Biol. 2025, 47, 322. [Google Scholar] [CrossRef] [PubMed]
- Amrousy, D.E.; El-Afify, D. Effects of alpha lipoic acid as a supplement in obese children and adolescents. Cytokine 2020, 130, 155084. [Google Scholar] [CrossRef]
- Ansar, H.; Mazloom, Z.; Kazemi, F.; Hejazi, N. Effect of α-lipoic acid on blood glucose, insulin resistance and glutathione peroxidase of type 2 diabetic patients. Saudi Med. J. 2011, 32, 584–588. [Google Scholar]
- Zhang, Y.; Han, P.; Wu, N.; He, B.; Lu, Y.; Li, S.; et al. Amelioration of lipid abnormalities by α-lipoic acid through antioxidative and anti-inflammatory effects. Obesity (Silver Spring) 2011, 19, 1647–1653. [Google Scholar] [CrossRef]
- Koh, E.H.; Lee, W.J.; Lee, S.A.; Kim, E.H.; Cho, E.H.; Jeong, E.J.; et al. Effects of α-lipoic acid on body weight in obese subjects. Am J. Med. 2011, 124, 85–88. [Google Scholar]
- Capece, U.; Moffa, S.; Improta, I.; Di Giuseppe, G.; Nista, E.C.; Cefalo, C.M.A.; et al. Alpha-lipoic acid and glucose metabolism: a comprehensive update on biochemical and therapeutic features. Nutrients 2023, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Haghighatdoost, F.; Gholami, A.; Hariri, M. Alpha-lipoic acid effect on leptin and adiponectin concentrations: a systematic review and meta-analysis of randomized controlled trials. Eur. J. Clin. Pharmacol. 2020, 76, 649–657. [Google Scholar] [CrossRef]
- Pershadsingh, H.A. Alpha-lipoic acid: physiologic mechanisms and indications for the treatment of metabolic syndrome. Expert. Opin. Investig. Drugs 2007, 16, 291–302. [Google Scholar] [CrossRef]
- Cha, Y.S. Effects of L-carnitine on obesity, diabetes, and as an ergogenic aid. Asia Pac. J. Clin. Nutr. 2008, 17, 306–308. [Google Scholar]
- Li, N.; Zhao, H. Role of carnitine in non-alcoholic fatty liver disease and other related diseases: an update. Front Med. (Lausanne) 2021, 8, 689042. [Google Scholar] [CrossRef]
- Durazzo, A.; Lucarini, M.; Nazhand, A.; Bouto, S.B.; Silva, A.M.; et al. The nutraceutical value of carnitine and its use in dietary supplements. Molecules 2020, 25, 2127. [Google Scholar] [CrossRef] [PubMed]
- Fathizadeh, H.; Milajerdi, A.; Reiner, Ž.; Amirani, E.; Asemi, Z.; Mansournia, M.A.; et al. The effects of L-carnitine supplementation on indicators of inflammation and oxidative stress: a systematic review and meta-analysis of randomized controlled trials. J. Diabetes Metab. Disord. 2020, 19, 1879–1894. [Google Scholar] [CrossRef] [PubMed]
- Askarpour, M.; Hadi, A.; Miraghajani, M.; Symonds, M.E.; Sheikhi, A.; Ghaedi, E. Beneficial effects of l-carnitine supplementation for weight management in overweight and obese adults: An updated systemic review and dose-response meta-analysis of randomized controlled trials. Pharmacol. Res. 2020, 151, 104554. [Google Scholar] [CrossRef]
- Karalis, D.T.; Karalis, T.; Karalis, S.; Kleisiari, A.S. L-Carnitine as a Diet Supplement in Patients With Type II Diabetes. Cureus 2020, 12, e7982. [Google Scholar] [CrossRef]
- Zozina, V.I.; Covantev, S.; Goroshko, O.A.; Krasnykh, L.M.; Kukes, V.G. Coenzyme Q10 in cardiovascular and metabolic diseases: current state and the problem. Curr. Cardiol. Rev. 2018, 14, 164–174. [Google Scholar] [CrossRef]
- Mantle, D.; Dybring, A. Bioavailability of coenzyme Q10: An overview of the absorption process and subsequent metabolism. Antioxidants 2020, 9, 386. [Google Scholar] [CrossRef]
- Mantle, D.; Heaton, R.A.; Hargreaves, I.P. Coenzyme Q10 and immune function: an overview. Antioxidants (Basel) 2021, 10, 579. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Diabetes, oxidative stress and thera- peutic strategies. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2709–2729. [Google Scholar] [CrossRef]
- Alam, A.; Rahman, M. Mitochondrial dysfunction in obesity: potential benefit and mechanism of co-enzyme Q10 supplementation in metabolic syndrome. J. Diabetes Metab. Disord. 2014, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, D.; Waid, P.H. Jordão AAJr Mechanisms of action effects of the administration of Coenzyme Q10 on metabolic syndrome. J. Nutr. Intermed. Metab. 2018, 13, 26–32. [Google Scholar] [CrossRef]
- Failla, M.L.; Chitchumroonchokchai, C.; Aoki, F. Increased bioavailability of ubiquinol compared to that of ubiquinone is due to more efficient micellarization during digestion and greater GSH-dependent uptake and basolateral secretion by Caco-2 cells. J. Agric. Food Chem. 2014, 62, 7174–82. [Google Scholar] [CrossRef]
- Williams, L.; Bradford, J. Should I take coenzyme Q10 with a statin? JAAPA 2007, 19, 61–2. [Google Scholar] [CrossRef]
- Sifuentes-Franco, S.; Sánchez-Macías, D.C.; Carrilo-Ibarra, S.; Rivera-Valdés, J.J.; Zuñiga, L.Y.; Sánchez-López, V.A. Antioxidant and Anti-Inflammatory Effects of Coenzyme Q10 Supplementation on Infectious Diseases. Healthcare (Basel) 2022, 10, 487. [Google Scholar] [CrossRef]
- Vona, R.; Pallotta, L.; Cappelletti, M.; Severi, C.; Matarrese, P. The Impact of Oxidative Stress in Human Pathology: Focus on Gastrointestinal Disorders. Antioxidants 2021, 10, 201. [Google Scholar] [CrossRef]
- McRae, M.P. Coenzyme Q10 Supplementation in Reducing Inflammation: An Umbrella Review. J. Chiropr. Med. 2022, 22, 131–137. [Google Scholar] [CrossRef]
- Mantle, D.; Kozhevnikova, S.; Larsen, S. Coenzyme Q10 and Obesity: An Overview. Antioxidants 2025, 14, 871. [Google Scholar] [CrossRef]
- Li, Y.; El-Sehrawy, A.A.M.A.; Shankar, A.; Mohammed, J.S.; Hjazi, A.; et al. Effects of Coenzyme Q10 Supplementation on Metabolic Indicators in Patients with Type 2 Diabetes: A Systematic Review and Meta-Analysis. Clin. Ther. 47, 235–243. [CrossRef]
- Gholami, M.; Khosrowbeygi, A. Review on the mechanisms of the effects of coenzyme Q10 supplementation on serum levels of leptin and adiponectin. Rom. J. Diabetes Nutr. Metab. Dis. 2023, 30, 124–131. [Google Scholar]
- Nie, X.; Dong, X.; Hu, Y.; Xu, F.; Hu, C.; Shu, C. Coenzyme Q10 Stimulate Reproductive Vatality. Drug Des. Devel. Ther. 2023, 17, 2623–2637. [Google Scholar] [CrossRef]
- Ardekani, A.; Tabrizi, R.; Maleki, E.; Lankarani, K.B.; Heydari, S.T.; Moradinazar, M.; et al. Effects of coenzyme Q10 supplementation on lipid profiles and liver enzymes of nonalcoholic fatty liver disease (NAFLD) patients: A systematic review and meta-analysis of randomized controlled trials. Food Sci. Nutr. 2023, 11, 2580–2588. [Google Scholar] [CrossRef]
- Tsai, K.L.; Chen, L.H.; Chiou, S.H.; Chiou, G.Y.; Chen, Y.C.; Chou, H.Y.; et al. Conzyme Q10 suppresses oxLDL-induced endothelial oxidative injuries by the modulation of LOX-1-mediated ROS generation via the AMPK/PKC/NADPH oxidase signaling pathway. Mol. Nutr. Food Res. 2011, 55, S227–S240. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Kukai, M. Superoxide dismutase: role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Kiningham, K.K.; XU, Y.; Daosukho, C.; Popova, B.; Clair, D.K.S. Nuclear factor κB-dependent mechanisms coordinate the synergistic effect of PMA and cytokines on the induction of superoxide dismutase 2. Biochem. J. 2001, 353, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Tun, S.; Spainhower, C.J.; Cottrill, C.L.; Lakhani, H.V.; Pillai, S.S.; Dilip, A.; et al. Therapeutic Efficacy of Antioxidants in Ameliorating Obesity Phenotype and Associated Comorbidities. Front. Pharmacol. 2020, 11, 1234. [Google Scholar] [CrossRef] [PubMed]
- Samuni, Y.; Cook, J. A.; Choudhuri, R.; Degraff, W.; Sowers, A. L.; Krishna, M.C.; et al. Inhibition of adipogenesis by Tempol in 3T3-L1 cells. Free Radic. Biol. Med. 2010, 49, 667–673. [Google Scholar] [CrossRef]
- Cui, R.; Gao, M.; Qu, S.; Liu, D. Overexpression of superoxide dismutase 3 gene blocks high-fat diet-induced obesity, fatty liver and insulin resistance. Gene Ther. 2014, 21, 840–848. [Google Scholar] [CrossRef]
- Perriotte-Olson, C.; Adi, N.; Manickam, D.S.; Westwood, R.A.; Desouza, C.V.; Natarajan, G.; et al. Nanoformulated copper/zinc superoxide dismutase reduces adipose inflammation in obesity. Obesity (Silver Spring) 2016, 24, 148–156. [Google Scholar] [CrossRef] [PubMed]
- 156 Coudriet, G.M.; Delmastro-Greenwood, M.M.; Previte, D.M.; Marré, M.L.; O’Connor, E.C.; Novak, E.A.; et al. Treatment with a Catalytic Superoxide Dismutase (SOD) Mimetic Improves Liver Steatosis, Insulin Sensitivity, and Inflammation in Obesity-Induced Type 2 Diabetes. Antioxidants (Basel) 2017, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torres, I.; Gastrejón-Téllez, V.; Soto, M.E.; Rudio-Ruiz, M.E.; Manzano-Pech, L.; Guarner-Lans, V. Oxidative stress, plant natural antioxidants, and obesity. Int. J. Mol. Sci. 2021, 22, 1786. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Heo, H.J. The roles of catechins in regulation of systemic inflammation. Food Sci. Biotechnol. 2022, 31, 957–970. [Google Scholar] [CrossRef]
- Pan, C.; Zhou, S.; Wu, J.; Liu, L.; Song, Y.; Li, T.; et al. NRF2 plays a critical role in both self and EGCG protection against diabetic testicular damage. Oxid. Med. Cell Longev. 2017, 2017, 3172692. [Google Scholar] [CrossRef]
- Wen, L.; Wu, D.; Tan, X.; Zhong, M.; Xing, J.; Li, W.; Li, D.; Cao, F. The role of catechins in regulating diabetes: an update review. Nutrients 2022, 14, 4681. [Google Scholar] [CrossRef]
- Leiherer, A.; Mündlein, A.; Drexel, H. Phytochemicals and their impact on adipose tissue inflammation and diabetes. Vascul. Pharmacol. 2013, 58, 3–20. [Google Scholar] [CrossRef]
- Potenza, M.A.; Marasciulo, F.L.; Tarquinio, M.; Tiravanti, E.; Colantuono, G.; Federici, A.; et al. EGCG, a green tea polyphenol, improves endothelial function and insulin sensitivity, reduces blood pressure, and protects against myocardial I/R injury in SHR. Am. J. Physiol. Endocrinol. Metab. 2007, 292, 1378–1387. [Google Scholar] [CrossRef]
- Onakpoya, I.; Spencer, E.; Heneghan, C.; Tompson, M. The effect of green tea on blood pressure and lipid profile: a systematic review and meta-analysis of randomized clinical trials. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Ansari, P.; Choudhury, S.T.; Seidel, V.; Rahman, A.B.; Aziz, M.A.; Richi, A.E.; et al. Therapeutic potential of quercetin in the management of type-2 diabetes mellitus. Life (Basel) 2022, 12, 1146. [Google Scholar] [CrossRef]
- Deepika, *!!! REPLACE !!!*; Maurya, P.K. Health Benefits of Quercetin in Age-Related Diseases. Molecules 2022, 27, 2498. [Google Scholar] [CrossRef]
- Papakyriakopoulou, P.; Veldakis, N.; Khattab, E.; Valsami, G.; Korakianitis, I.; Kadoglou, N.P.E. Potential pharmaceutical applications of quercetin in cardiovascular diseases. Pharmaceuticals 2022, 15, 1019. [Google Scholar] [CrossRef]
- Tomou, Ε.-Μ.; Papakyriakopoulou, P.; Saitani, E.-M.; Valsami, G.; Pippa, N.; Skaltsa, H. Recent Advances in Nanoformulations for Quercetin Delivery. Pharmaceutics 2023, 15, 1656. [Google Scholar] [CrossRef]
- Yang, D.K.; Kang, H.S. Anti-diabetic effect of cotreatment with quercetin and resveratrol in streptozotocin-induced diabetic rats. Biomol. Ther. 2018, 26, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, A.; Razavi, B.M.; Banch, M.; Hosseinzadeh, H. Quercetin and metabolic syndrome: a review. Phytother. Res. 2021, 35, 5352–5364. [Google Scholar] [CrossRef]
- Luo, X.; Weng, X.; Bao, X.; Bai, X.; Lv, Y.; Zhang, S.; et al. A novel anti-atherosclerotic mechanism of quercetin: competitive binding to KEAP1 via Arg483 to inhibit macrophage pyroptosis. Redox Biol. 2022, 57, 102511. [Google Scholar] [CrossRef]
- Jakubczyk, K.; Kruzga, A.; Katarzyna, J.; Skonieczna-Zydecka, K. Antioxidant potential of curcumin – a meta-analysis of randomized clinical trials. Antioxidants (Basel) 2020, 9, 1092. [Google Scholar] [CrossRef]
- D’Andrea, G. Quercetin: a flavonol with multifaceted therapeutic applications? Fitoterapia 2015, 106, 256–271. [Google Scholar] [CrossRef] [PubMed]
- 173 Wang, Y.; Li, Z.; He, J.; Zhao, Y. Quercetin Regulates Lipid Metabolism and Fat Accumulation by Regulating Inflammatory Responses and Glycometabolism Pathways: A Review. Nutrients 2024, 16, 1102. [Google Scholar] [CrossRef]
- Damiano, F.; Giannotti, L.; Gnoni, G.V.; Siculella, L.; Gnoni, A. Quercetin inhibition of SREBPs and ChREBP expression results in reduced cholesterol and fatty acid synthesis in C6 glioma cells. Int J Biochem Cell Biol. 2019, 117, 105618. [Google Scholar] [CrossRef]
- Peng, J.; Li, Q.; Li, K.; Zhu, L.; Lin, X.; Lin, X.; et al. Quercetin improves glucose and lipid metabolism of diabetic rats: involvement of Akt signaling and SIRT1. J. Diabetes Res. 2017, 2017, 3417306. [Google Scholar] [CrossRef]
- Eid, H.M.; Nachar, A.; Thong, F.; Sweeney, G.; Haddad, P.S. The molecular basis of the antidiabetic action of quercetin in cultured skeletal muscle cells and hepatocytes. Pharmacogn. Mag. 2015, 11, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Dhanya, R.; Kartha, C.C. Quercetin improves oxidative stress-induced pancreatic beta cell alterations via MTOR-signaling. Mol. Cell Biochem. 2021, 476, 3879–3887. [Google Scholar] [CrossRef] [PubMed]
- Mazloom, Z.; Abdollahzadeh, S.M.; Dabbaghmanesh, M.-H.; Rezaianzadeh, A. The effect of quercetin supplementation on oxidative stress, glycemic control, lipid profile and insulin resistance in type 2 diabetes: a randomized clinical trial. J. Heal. Sci. Surveill. Syst. 2014, 2, 8–14. [Google Scholar]
- Ostadmohammadi, V.; Milajerdi, A.; Ayati, E.; Kolahdooz, F.; Asemi, Z. Effects of quercetin supplementation on glycemic control among patients with metabolic syndrome and related disorders: a systematic review and meta-analysis of randomized controlled trials, Phytother. Res. 2019, 33, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.-J.; Li, Y.; Cao, Q.-H.; Wu, H.-X.; Tang, X.-Y.; Gao, X.-H.; et al. In vitro and in vivo evidence that quercetin protects against diabetes and its complications: A systematic review of the literature. Biomed. Pharmacother. 2019, 109, 1085–1099. [Google Scholar] [CrossRef]
- Chai, G.-R.; Liu, S.; Yang, H.-W.; Chen, X.-L. Quercetin protects against diabetic retinopathy in rats by inducing heme oxygenase-1 expression. Neural Regen. Res. 2020, 16, 1344–1350. [Google Scholar] [CrossRef]
- Chen, P.; Chen, J.; Zheng, Q.; Chen, W.; Wang, Y.; Xu, X. Pioglitazone, Extract of Compound Danshen Dripping Pill, and Quercetin Ameliorate Diabetic Nephropathy in Diabetic Rats. J. Endocrinol. Investig. 2013, 36, 422–427. [Google Scholar]
- Mustafa, N.H.; Siti, H.N.; Kamisah, Y. Role of Quercetin in Diabetic Cardiomyopathy. Plants 2025, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Zahedi, M.; Ghiasvand, R.; Feizi, A.; Asgari, G.; Darvish, L. Does Quercetin Improve Cardiovascular Risk Factors and Inflammatory Biomarkers inWomen with Type 2 Diabetes: A Double-Blind Randomized Controlled Clinical Trial. Int. J. Prev. Med. 2013, 4, 777–785. [Google Scholar]
- Fatima, M.T.; Bhat, A.A.; Nisar, S.; Fakhro, K.A.; Akil, A.S.A.-S. The role of dietary antioxidants in type 2 diabetes and neurodegenerative disorders: An assessment of the benefit profile. Heliyon 2022, 9, e12698. [Google Scholar] [CrossRef]
- Yang, J.; Miao, X.; Yang, F.-J.; Cao, J.-F.; Liu, X.; Fu, J.-L.; Su, G.-F. Therapeutic potential of curcumin in diabetic retinopathy (Review). Int. J. Mol. Med. 2021, 47, 75. [Google Scholar] [CrossRef]
- Hussain, Y.; Khan, H.; Alotaibi, G.; Khan, F.; Alam, W.; Aschner, M.; et al. How curcumin targets inflammatory mediators in diabetes: therapeutic insights and possible solutions. Molecules 2022, 27, 4058. [Google Scholar] [CrossRef]
- Aggarwal, B.B. Targeting inflammation-induced obesity and metabolic diseases by curcumin and other nutraceuticals Annu. Rev. Nutr. 2010, 30, 173–199. [Google Scholar] [CrossRef] [PubMed]
- Dahza, M.J.; Ghalandari, H.; Mohammad, H.G.; Amini, M.R.; Askarpour, M. Effects of curcumin/turmeric supplementation on lipid profile: A GRADE-assessed systematic review and dose-response meta-analysis of randomized controlled trials. Complement Ther. Med. 2023, 75, 102955. [Google Scholar] [CrossRef]
- Lone, J.; Choi, J.H.; Kim, S.W.; Yun, J.W. Curcumin induces brown fat-like phenotype in 3T3-L1 and primary white adipocytes. J. Nutr. Biochem. 2016, 27, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.L.; Li, Y.; Wen, Y.; Chen, Y.F.; Na, L.X.; Li, S.T.; et al. Curcumin, a potential inhibitor of up-regulation of TNF-alpha and IL-6 induced by palmitate in 3t3-L1 adipocytes through NF-kappaB and JNK pathway. BioMed Environ. Sci. 2009, 22, 32–39. [Google Scholar] [CrossRef]
- Pivari, F.; Mingione, A.; Brasacchio, C.; Soldati, L. Curcumin and type 2 diabetes mellitus: prevention and treatment. Nutrients 2019, 11, 1837. [Google Scholar] [CrossRef]
- Panahi, Y.; Khalili, N.; Sahebi, E.; Namazi, S.; Karimian, M.S.; Majeed, M.; et al. Antioxidant effects of curcuminoids in patients with type 2 diabetes mellitus: a randomized controlled trial. Inflammopharmacology 2017, 25, 25–31. [Google Scholar] [CrossRef]
- Marton, L.T.; Pescinini-E-Salzedas, L.M.; Camargo, M.E.C.; Barbalho, S.M.; Dos Santos Haber, J.F.; Sinatora, R.V.; et al. The effects of curcumin on diabetes mellitus: a systematic review. Front. Endocrinol. 2021, 12, 66948. [Google Scholar] [CrossRef]
- Koushki, M.; Amiri-Dashatan, N.; Ahmadi, N.; Abbaszadeh, H.-A.; Rezaei-Tavirani, M. Resveratrol: A miraculous natural compound for diseases treatment. Food Sci. Nutr. 2018, 6, 2473–2490. [Google Scholar] [CrossRef]
- Galiniak, S.; Aebisher, D.; Bartusik-Aebisher, D. Health benefits of resveratrol administration. Acta Biochim. Pol. 2019, 66, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Schugar, R.C.; Shih, D.M.; Warrier, M.; Helsley, R.N.; Burrows, A.; Ferguson, D.; et al. The TMAO-Producing Enzyme Flavin-Containing Monooxygenase 3 Regulates Obesity and the Beiging of White Adipose Tissue. Cell Rep. 2017, 19, 2451–2461. [Google Scholar] [CrossRef] [PubMed]
- 198 Springer, M.; Moco, S. Resveratrol and Its Human Metabolites. Effects on Metabolic Health and Obesity. Nutrients 2019, 11, 143. [Google Scholar] [CrossRef]
- Clemente-Suarez, V.J.; Beltran-Velasco, A.I.; Redondo-Florez, L.; Martin-Rodriguez, A.; Tornero-Aguilera, J.F. Global Impacts of Western Diet and Its Effects on Metabolism and Health: A Narrative Review. Nutrients 2023, 15, 2749. [Google Scholar] [CrossRef] [PubMed]
- Ren Z-q Zgeng S-y Sun, Z.; Luo, Y.; Wang, Y.; Yi, P.; et al. Resveratrol: Molecular Mechanisms, Health Benefits, and Potential Adverse Effects. MedComm. (2020) 2025, 6, e70252. [Google Scholar] [CrossRef]
- Pinto, D.S.; Skolnick, A.H.; Kirtane, A.J.; Murphy, S.A.; Barron, H.V.; Giugliano, R.P.; et al. U-Shaped Relationship of Blood Glucose with Adverse Outcomes among Patients with ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2005, 46, 178–180. [Google Scholar] [CrossRef]
- Abdelhaleem, I.A.; Brakat, A.M.; Adayel, H.M.; Asla, M.M.; Rizk, M.A.; Aboalfetoh, A.Y. The Effects of Resveratrol on Glycemic Control and Cardiometabolic Parameters in Patients with T2DM: A Systematic Review and Meta-Analysis. Med. Clin. 2022, 158, 576–585. [Google Scholar] [CrossRef]
- Zeraattalab-Motlagh, S.; Jayedi, A.; Shab-Bidar, S. The Effects of Resveratrol Supplementation in Patients with Type 2 Diabetes, Metabolic Syndrome, and Nonalcoholic Fatty Liver Disease: An Umbrella Review of Meta-Analyses of Randomized Controlled Trials. Am. J. Clin. Nutr. 2021, 114, 1675–1685. [Google Scholar] [CrossRef]
- Leh, H.E.; Lee, L.K. Lycopene: A Potent Antioxidant for the Amelioration of Type II Diabetes Mellitus. Molecules 2022, 27, 2335. [Google Scholar] [CrossRef]
- Sharma, I.; Khare, N.; Rai, A. Carotenoids: Sources, Bioavailability and Their Role in Human Nutrition [Internet]. Physiology. IntechOpen 2024. [Google Scholar] [CrossRef]
- Joshi, S.; Dobhal, A.; Kaur, R.; Rawat, M.; Bhati, D.; Wilson, I. Enhancing lycopene stability and extraction: Sustainable techniques and role of nanotechnology. J Food Compos Anal. 2025, 148, 108192. [Google Scholar] [CrossRef]
- Campos, K.K.D.; Araújo, G.R.; Martins, T.L.; Bandeira, A.C.B.; de Paula Costa, G.; Talvani, A.; et al. The antioxidant and anti-inflammatory properties of lycopene in mice lungs exposed to cigarette smoke. J. Nutr. Biochem. 2017, 48, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Shafras, M.; Sabaragamuwa, R.; Suwair, M. Role of dietary antioxidants in diabetes: An overview. Food Chemistry Advances 2024, 4, 100666. [Google Scholar] [CrossRef]
- Tafail, T.; Ain, H.B.U.; Noreen, S.; Ikram, A.; Arshad, M.T.; Abdullahi, M.A. Nutritional Benefits of Lycopene and Beta-Carotene: A Comprehensive Overview. Food Sci. Nutr. 2024 12, 8715–8741. [CrossRef]
- Abir, M.H.; Mahamud, A.G.M.S.U.; Tonny, S.H.; Anu, M.S.; Hossain, K.H.S.; et al. Pharmacological potentials of lycopene against aging and aging-related disorders: A review. Food Sci. Nutr. 2023, 11, 5701–5735. [Google Scholar] [CrossRef]
- Karaköy, Z.; Cadirci, E.; Dincer, B. A New Target in Inflammatory Diseases: Lycopene. Eurasian J. Med. 2022, 54, S23–S28. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ni, Y.; Nagata, N.; Zhuge, F.; Xu, L.; Nagashimada, M.; et al. Lycopene alleviates obesity-induced Inflammation and insulin resistance by regulating M1/M2 status of macrophages. Mol. Nutr. Food Res. 2019, 63, 1900602. [Google Scholar] [CrossRef]
- Singh, D.P.; Khare, P.; Zhu, J.; Kondepudi, K.K.; Singh, J.; Baboota, R.K.; et al. A novel cobiotic-based preventive approach against high-fat diet-induced adiposity, nonalcoholic fatty liver and gut derangement in mice. Int. J. Obes. 2016, 40, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Fenni, S.; Hammou, H.; Astier, J.; Bonnet, L.; Karkeni, E.; Couturier, C.; et al. Lycopene and tomato powder supplementation similarly inhibit high-fat diet induced obesity, inflammatory response, and associated metabolic disorders. Mol. Nutr. Food Res. 2017, 61, 1601083. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Suo, Y.; Zhang, J.; Zou, Q.; Tan, X.; Yuan, T.; Liu, Z.; Liu, X. Lycopene supplementation attenuates western diet-induced body weight gain through increasing the expressions of thermogenic/mitochondrial functional genes and improving insulin resistance in the adipose tissue of obese mice. J. Nutr. Biochem. 2019, 69, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Albrahim, T.; Alonazi, M.A. Lycopene corrects metabolic syndrome and liver injury induced by high fat diet in obese rats through antioxidant, anti-inflammatory, antifibrotic pathways. Biomed. Pharmacother. 2021, 141, 111831. [Google Scholar] [CrossRef]
- Imran, M.; Ghorat, F.; Ul-Haq, I.; Ur-Rehman, H.; Aslam, F.; Heydari, M.; et al. Lycopene as a natural antioxidant used to prevent human health disorders. Antioxidants 2020, 9, 706. [Google Scholar] [CrossRef]
- Bose, K.; Agrawal, B. Effect of long term supplementation of tomatoes (cooked) on levels of antioxidant enzymes, lipid peroxidation rate, lipid profile and glycated haemoglobin in Type 2 diabetes mellitus. W. Indian Med. J. 2006, 55, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Shidfar, F.; Froghifar, N.; Vafa, M.; Rajab, A.; Hosseini, S.; Shidfar, S.; Gohari, M. The effects of tomato consumption on serum glucose, apolipoprotein B, apolipoprotein A-I, homocysteine and blood pressure in type 2 diabetic patients. Int. J. Food Sci. Nutr. 2011, 62, 289–94. [Google Scholar] [CrossRef]
- Singh, K.; Bal, B.S.; Chopra, S.; Singh, S.; Malhotra, N. Ameliorative effect of lycopene on lipid peroxidation and certain antioxidant enzymes in diabetic patients. J. Diabetes Metab. 2012, 3, 1–5. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, Y.; Wang, Y. Beneficial effect of lycopene on anti-diabetic nephropathy through diminishing inflammatory response and oxidative stress. Food Funct. 2015, 6, 1150–1156. [Google Scholar] [CrossRef]
- Ozmen, O.; Topsakal, S.; Haligur, M.; Aydogan, A.; Dincoglu, D. Effects of caffeine and lycopene in experimentally induced diabetes mellitus. Pancreas 2016, 45, 579–583. [Google Scholar] [CrossRef]
- Yin, Y.; Zheng, Z.; Jiang, Z. Effects of lycopene on metabolism of glycolipid in type 2 diabetic rats. Biomed. Pharmacother. 2019, 109, 2070–2077. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Zhong, C.; Zhou, X.; Chen, R.; Xiong, T.; Hong, M.; et al. The association between intake of dietary lycopene and other carotenoids and gestational diabetes mellitus risk during mid-trimester: a cross-sectional study. Br. J. Nutr. 2019, 121, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Illustration of the pathophysiologic mechanisms of obesity-induced IR and T2DM development: Obesity can cause non-autoimmune insulinopenia through chronic low-grade inflammation, adipose tissue hypoxia, and OS, which lead to hyperglycemia, disruption of insulin signaling, IR, pancreatic β-cell dysfunction and T2DM development. At the same time, obesity can lead to overproduction of glucagon, which causes hyperglycemia and contributes to IR and T2DM development. Hyperglycemia in T2DM leads to glycosuria, which causes osmotic diuresis and dehydration. Also, T2DM can lead to the breakdown of fat for energy production, generating ketone bodies and potentially causing diabetic ketoacidosis. Abbreviations: IR: insulin resistance; T2DM: type 2 diabetes mellitus.
Figure 1.
Illustration of the pathophysiologic mechanisms of obesity-induced IR and T2DM development: Obesity can cause non-autoimmune insulinopenia through chronic low-grade inflammation, adipose tissue hypoxia, and OS, which lead to hyperglycemia, disruption of insulin signaling, IR, pancreatic β-cell dysfunction and T2DM development. At the same time, obesity can lead to overproduction of glucagon, which causes hyperglycemia and contributes to IR and T2DM development. Hyperglycemia in T2DM leads to glycosuria, which causes osmotic diuresis and dehydration. Also, T2DM can lead to the breakdown of fat for energy production, generating ketone bodies and potentially causing diabetic ketoacidosis. Abbreviations: IR: insulin resistance; T2DM: type 2 diabetes mellitus.
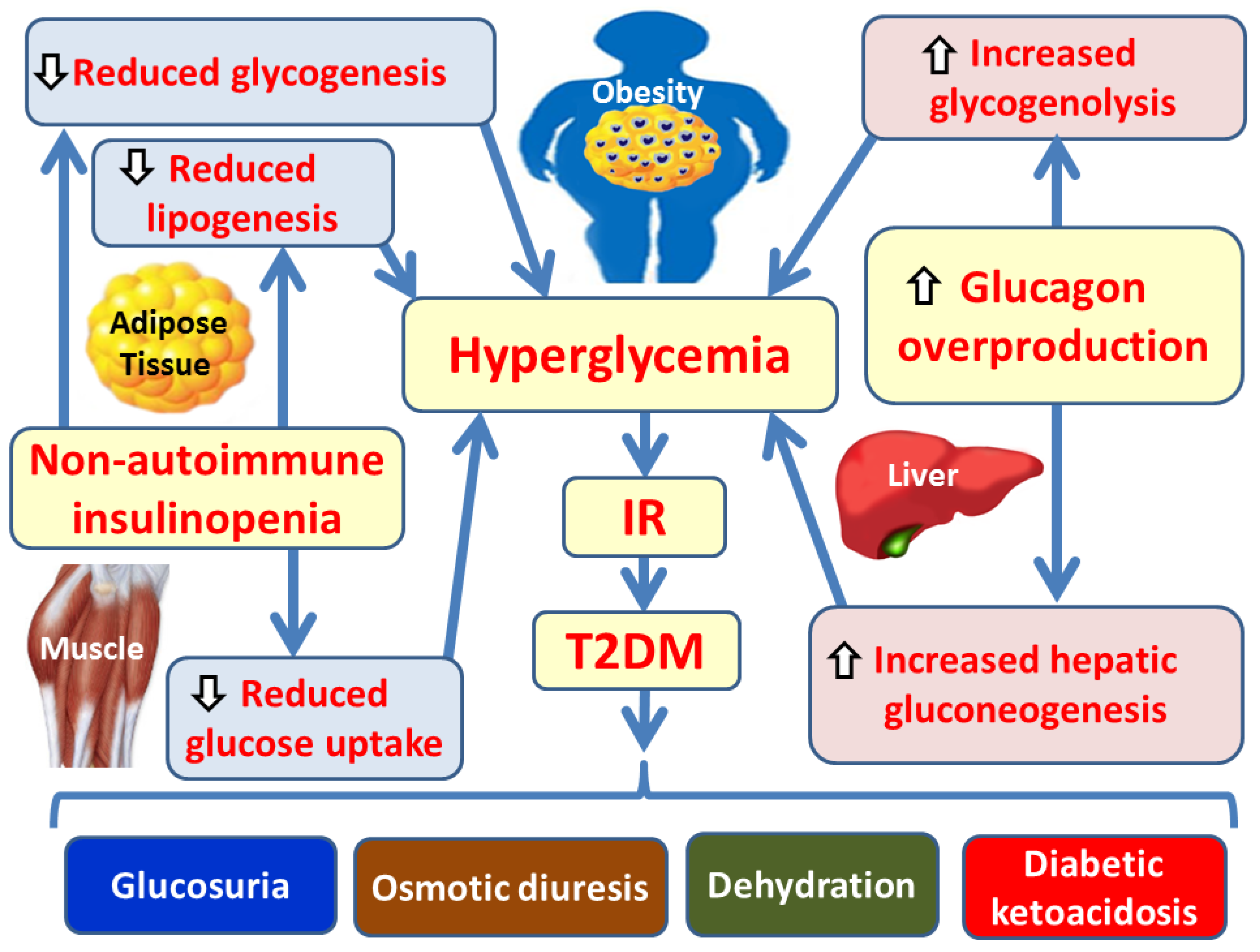
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



