
Submitted:
01 December 2025
Posted:
03 December 2025
You are already at the latest version
Abstract
Acute myeloid leukemia (AML) and myelodysplastic neoplasms (MDS) are clonal hematopoietic malignancies in which next-generation sequencing (NGS) has become integral for diagnosis, classification, risk stratification, and measurable residual disease (MRD) monitoring. Traditional cytogenetic and PCR-based assays remain useful, but targeted NGS panels now represent the standard of care, providing rapid and sensitive detection of recurrent gene mutations, structural variants, and gene fusions. Whole-genome, whole-exome, RNA sequencing, and long-read platforms expand the spectrum of detectable alterations, though targeted panels remain most practical for routine diagnostics. Bioinformatic pipelines and quality metrics-including read length, sequencing depth, and coverage-are critical for accurate variant calling, with validation often required for variants of uncertain significance or those near detection thresholds. NGS is now embedded in diagnostic frameworks, including the WHO 2022 and ICC classifications, which incorporate recurrently mutated genes such as TP53, ASXL1, RUNX1, and FLT3. These data inform prognostic models, with ELN-2022 defining adverse-risk AML subgroups for patients treated with intensive chemotherapy, ELN-2024 AML patients treated with less-intensive therapies, and the IPSS-M refining MDS risk categories by integrating mutational data. NGS also enables MRD monitoring, with gene panels and PCR-NGS hybrid approaches (e.g., for FLT3-ITD) showing increasing clinical utility, though standardization is still lacking. Furthermore, diagnostic NGS frequently uncovers germline predisposition syndromes (e.g., DDX41, GATA2), with significant implications for treatment decisions and donor selection in transplantation. In this manuscript, we review the advantages, limitations, and future perspectives of NGS in the clinical management of AML and MDS with a particular emphasis on the biological and technical principles underlying its use in these diseases. Furthermore, we discuss how NGS findings may influence diagnosis, prognostic classification and therapeutic decision-making within current clinical frameworks. Our aim is to provide a comprehensive overview of NGS fundamentals to support clinicians in navigating the increasing complexity of molecular data in daily practice.
Keywords:
1. Introduction
2. NGS Techniques
2.1. NGS Workflow
2.2. NGS Types in Clinical Practice
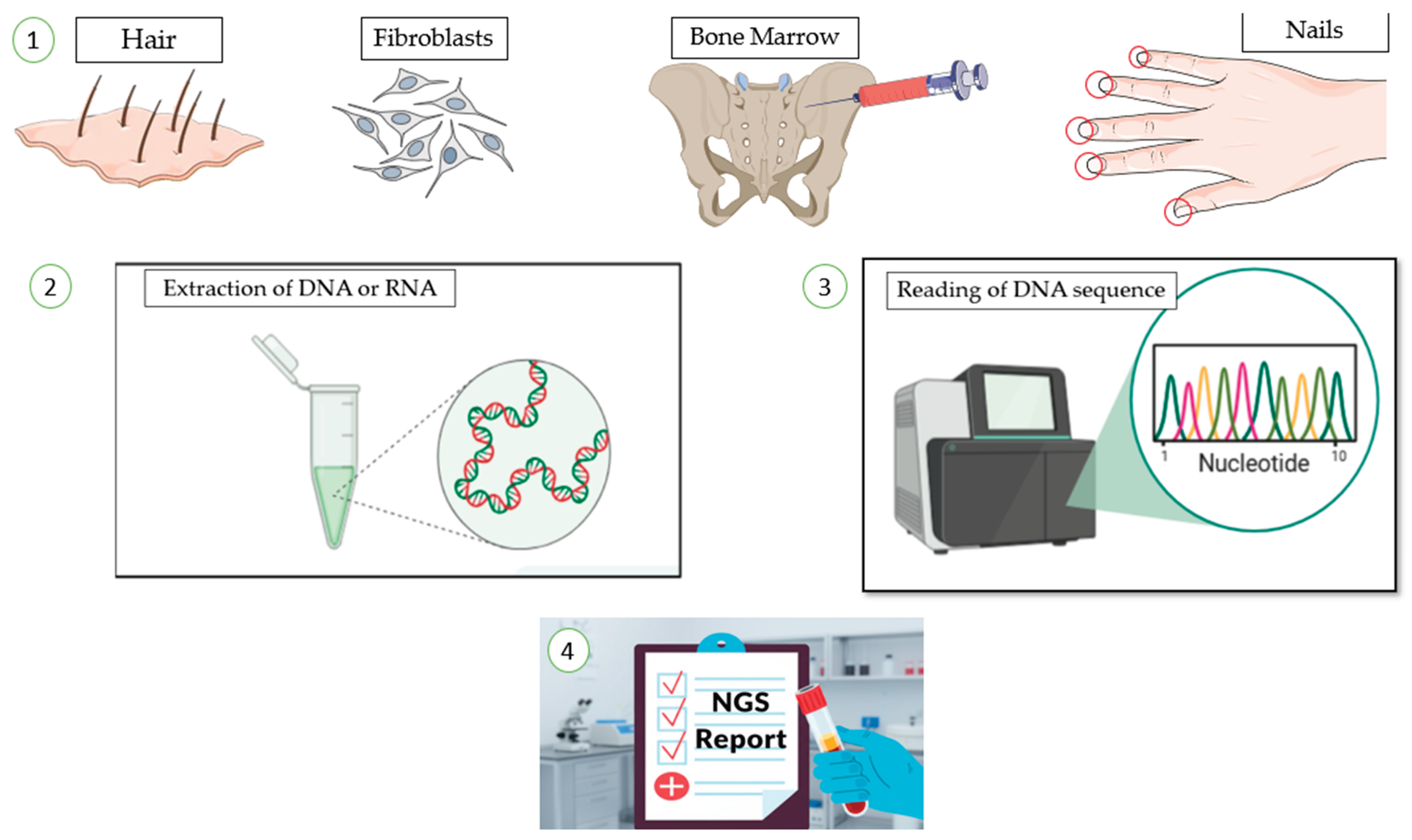
2.3. Sample Type and Quantity
2.4. Results Validation
2.5. Standardization
2.6. Type of Alterations
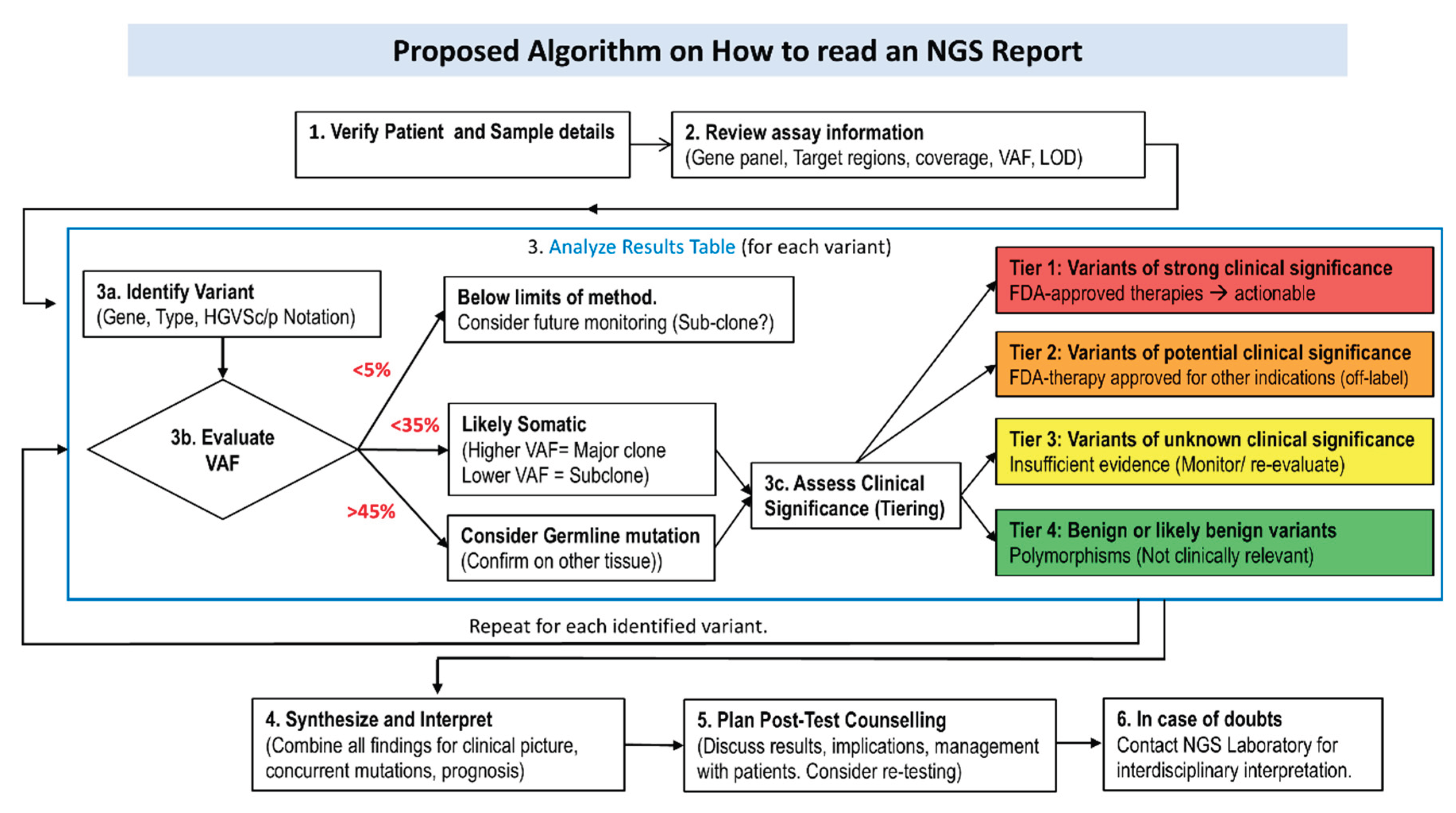
2.7. Result Interpretation
- − Primary Analysis:
- Fluorescent signals generated during sequencing are converted into text files (FASTQ) representing the sequence of individual bases.
- − Secondary Analysis
- Sequences from multiple samples run together are separated according to their indices in a process called demultiplexing. The reads are then aligned to a reference genome, and differences are detected in a step known as variant calling.
- − Tertiary Analysis
- The final stage involves interpreting the identified variants in the clinical context, evaluating their potential relevance and pathogenicity, and determining possible causal relationship with the patient’s disease.
2.8. Useful Database
2.9. Report Structure
2.10. NGS vs. Quantitative-PCR vs. Digital-PCR
3. NGS and Impact on Diagnosis and Classification
3.1. NGS Role in MDS: From Diagnosis to Classification
3.2. CHIP and CCUS, the Role of NGS
3.3. Novel Genetically-Based Categories in MDS
3.3.1. SF3B1 in MDS
3.3.2. TP53 in MDS
3.4. Impact of NGS in the Definition of AML, Novel Genetic Classifications
3.4.1. TP53 Mutations in AML [56]
3.4.2. MDS-Related Mutations
3.4.3. RUNX1
3.4.4. AML-Defining Mutations
3.5. AML Mutations Driving Target Therapies (IDH1/2 and FLT3)
4. NGS and Impact on Risk Stratification
4.1. Myelodysplastic Syndromes: Prognostic Impact of Molecular Data
4.2. Acute Myeloid Leukemia
4.2.1. Stratification of AML for Patients Treated with Less-Intensive Therapy
5. The Role of NGS in MRD Measurement and Monitoring
6. The Role of NGS in the Detection of Germline Predisposition [140]
6.1. DDX41
6.2. TP53
6.3. GATA2
6.4. RUNX1
6.5. ETV6
7. The Impact of NGS in the Detection of Druggable Mutations at Relapse
8. Discussion
9. Conclusions
10. Future Directions.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| AMP | Association for Molecular Pathology |
| CAP | College of American Pathologists |
| CCUS | Clonal cytopenia of undetermined significance |
| CHIP | Clonal Hematopoiesis of Indeterminate Potential |
| cnLOH | Copy neutral loss of heterozygosity |
| CMML | Chronic myelomonocytic leukemia |
| HMA | Hypomethylating agents |
| HR | Hazard ratio |
| HSCT | Hematopoietic stem cell transplantation |
| ICC | International Consensus Conference |
| LOH | Loss of heterozygosity |
| MDS | Myelodysplastic neoplasms |
| MN | Myeloid neoplasms |
| MRD | Measurable residual disease |
| NGS | Next generation sequencing |
| OS | Overall survival |
| RFS | Relapse free survival |
| SBS | Sequencing by synthesis |
| VAF | Variant allele frequencies |
| VUS | Variants of uncertain significance |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
| WHO | World Health Organization |
References
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. New England Journal of Medicine 2016, 374, 2209–2221. [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 Recommendations from an International Expert Panel on Behalf of the ELN. Blood 2022, 140, 1345–1377. [CrossRef]
- Gooptu, M.; Murdock, H.M.; Soiffer, R.J. How I Treat AML Relapse after Allogeneic HSCT. Blood 2025, 145, 2128–2137. [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [CrossRef]
- Schmid, S.; Jochum, W.; Padberg, B.; Demmer, I.; Mertz, K.D.; Joerger, M.; Britschgi, C.; Matter, M.S.; Rothschild, S.I.; Omlin, A. How to Read a Next-Generation Sequencing Report—What Oncologists Need to Know. ESMO Open 2022, 7, 100570. [CrossRef]
- Mosele, M.F.; Westphalen, C.B.; Stenzinger, A.; Barlesi, F.; Bayle, A.; Bièche, I.; Bonastre, J.; Castro, E.; Dienstmann, R.; Krämer, A.; et al. Recommendations for the Use of Next-Generation Sequencing (NGS) for Patients with Advanced Cancer in 2024: A Report from the ESMO Precision Medicine Working Group. Annals of Oncology 2024, 35, 588–606. [CrossRef]
- Kim, Y.J.; Takahashi, K. Emerging Technologies of Single-Cell Multi-Omics. Haematologica 2025, 110, 1269–1277. [CrossRef]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J Mol Diagn 2017, 19, 341–365. [CrossRef]
- Duncavage, E.J.; Bagg, A.; Hasserjian, R.P.; DiNardo, C.D.; Godley, L.A.; Iacobucci, I.; Jaiswal, S.; Malcovati, L.; Vannucchi, A.M.; Patel, K.P.; et al. Genomic Profiling for Clinical Decision Making in Myeloid Neoplasms and Acute Leukemia. Blood 2022, 140, 2228–2247. [CrossRef]
- Head, S.R.; Komori, H.K.; LaMere, S.A.; Whisenant, T.; Van Nieuwerburgh, F.; Salomon, D.R.; Ordoukhanian, P. Library Construction for Next-Generation Sequencing: Overviews and Challenges. Biotechniques 2014, 56, 61–64, 66, 68, passim. [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [CrossRef]
- Lee, J.Y. The Principles and Applications of High-Throughput Sequencing Technologies. Dev Reprod 2023, 27, 9–24. [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of Age: Ten Years of next-Generation Sequencing Technologies. Nat Rev Genet 2016, 17, 333–351. [CrossRef]
- Peroni, E.; Randi, M.L.; Rosato, A.; Cagnin, S. Acute Myeloid Leukemia: From NGS, through scRNA-Seq, to CAR-T. Dissect Cancer Heterogeneity and Tailor the Treatment. Journal of Experimental & Clinical Cancer Research 2023, 42, 259. [CrossRef]
- O’Connor, T.E.; Shaw, R.; Madero-Marroquin, R.; Roloff, G.W. Clinical Considerations at the Intersection of Hematopoietic Cell Transplantation and Hereditary Hematopoietic Malignancy. Front. Oncol. 2023, 13, 1180439. [CrossRef]
- Thomas, M.; Sukhai, M.A.; Zhang, T.; Dolatshahi, R.; Harbi, D.; Garg, S.; Misyura, M.; Pugh, T.; Stockley, T.L.; Kamel-Reid, S. Integration of Technical, Bioinformatic, and Variant Assessment Approaches in the Validation of a Targeted Next-Generation Sequencing Panel for Myeloid Malignancies. Arch Pathol Lab Med 2017, 141, 759–775. [CrossRef]
- Akabari, R.; Qin, D.; Hussaini, M. Technological Advances: CEBPA and FLT3 Internal Tandem Duplication Mutations Can Be Reliably Detected by Next Generation Sequencing. Genes (Basel) 2022, 13. [CrossRef]
- Cumbo, C.; Minervini, C.F.; Orsini, P.; Anelli, L.; Zagaria, A.; Minervini, A.; Coccaro, N.; Impera, L.; Tota, G.; Parciante, E.; et al. Nanopore Targeted Sequencing for Rapid Gene Mutations Detection in Acute Myeloid Leukemia. Genes (Basel) 2019, 10. [CrossRef]
- Warburton, P.E.; Sebra, R.P. Long-Read DNA Sequencing: Recent Advances and Remaining Challenges. Annu Rev Genomics Hum Genet 2023, 24, 109–132. [CrossRef]
- Moustakli, E.; Christopoulos, P.; Potiris, A.; Zikopoulos, A.; Mavrogianni, D.; Karampas, G.; Kathopoulis, N.; Anagnostaki, I.; Domali, E.; Tzallas, A.T.; et al. Long-Read Sequencing and Structural Variant Detection: Unlocking the Hidden Genome in Rare Genetic Disorders. Diagnostics (Basel) 2025, 15. [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing Depth and Coverage: Key Considerations in Genomic Analyses. Nat Rev Genet 2014, 15, 121–132. [CrossRef]
- Anu, R.I.; Patel, A.; Pathak, N.; Mehta, P.; Chougule, A.; Sheth, H.; Veldore, V.; Suryavanshi, M.; Gupta, V.G.; Disel, U.; et al. Uniform Reporting of Next Generation Sequencing: Indian Society of Medical and Pediatric Oncology. Indian J Med Paediatr Oncol 2025, 46, 351–362. [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A Survey of Best Practices for RNA-Seq Data Analysis. Genome Biol 2016, 17, 13. [CrossRef]
- Dillon, L.W.; Hayati, S.; Roloff, G.W.; Tunc, I.; Pirooznia, M.; Mitrofanova, A.; Hourigan, C.S. Targeted RNA-Sequencing for the Quantification of Measurable Residual Disease in Acute Myeloid Leukemia. Haematologica 2019, 104, 297–304. [CrossRef]
- Sa, A.C.C.; Sadee, W.; Johnson, J.A. Whole Transcriptome Profiling: An RNA-Seq Primer and Implications for Pharmacogenomics Research. Clin Transl Sci 2018, 11, 153–161. [CrossRef]
- Baccarella, A.; Williams, C.R.; Parrish, J.Z.; Kim, C.C. Empirical Assessment of the Impact of Sample Number and Read Depth on RNA-Seq Analysis Workflow Performance. BMC Bioinformatics 2018, 19, 423. [CrossRef]
- Shin, H.T.; Choi, Y.L.; Yun, J.W.; Kim, N.K.D.; Kim, S.Y.; Jeon, H.J.; Nam, J.Y.; Lee, C.; Ryu, D.; Kim, S.C.; et al. Prevalence and Detection of Low-Allele-Fraction Variants in Clinical Cancer Samples. Nat Commun 2017, 8, 1377. [CrossRef]
- Petrackova, A.; Vasinek, M.; Sedlarikova, L.; Dyskova, T.; Schneiderova, P.; Novosad, T.; Papajik, T.; Kriegova, E. Standardization of Sequencing Coverage Depth in NGS: Recommendation for Detection of Clonal and Subclonal Mutations in Cancer Diagnostics. Front Oncol 2019, 9, 851. [CrossRef]
- Singh, R.R. Target Enrichment Approaches for Next-Generation Sequencing Applications in Oncology. Diagnostics (Basel) 2022, 12. [CrossRef]
- Al-Kateb, H.; Knight, S.M.; Sivasankaran, G.; Voss, J.S.; Pitel, B.A.; Blommel, J.H.; Jerde, C.R.; Rumilla, K.M.; Lee, J.L.; Mattson, N.R.; et al. Clinical Validation of the TruSight Oncology 500 Assay for the Detection and Reporting of Pan-Cancer Biomarkers. J Mol Diagn 2025, 27, 292–305. [CrossRef]
- Sahajpal, N.S.; Mondal, A.K.; Ananth, S.; Njau, A.; Ahluwalia, P.; Jones, K.; Ahluwalia, M.; Okechukwu, N.; Savage, N.M.; Kota, V.; et al. Clinical Performance and Utility of a Comprehensive Next-Generation Sequencing DNA Panel for the Simultaneous Analysis of Variants, TMB and MSI for Myeloid Neoplasms. PLoS One 2020, 15, e0240976. [CrossRef]
- Serrati, S.; De Summa, S.; Pilato, B.; Petriella, D.; Lacalamita, R.; Tommasi, S.; Pinto, R. Next-Generation Sequencing: Advances and Applications in Cancer Diagnosis. Onco Targets Ther 2016, 9, 7355–7365. [CrossRef]
- Koboldt, D.C. Best Practices for Variant Calling in Clinical Sequencing. Genome Med 2020, 12, 91. [CrossRef]
- Sun, Q.Y.; Ding, L.W.; Tan, K.T.; Chien, W.; Mayakonda, A.; Lin, D.C.; Loh, X.Y.; Xiao, J.F.; Meggendorfer, M.; Alpermann, T.; et al. Ordering of Mutations in Acute Myeloid Leukemia with Partial Tandem Duplication of MLL (MLL-PTD). Leukemia 2017, 31, 1–10. [CrossRef]
- Spencer, D.H.; Abel, H.J.; Lockwood, C.M.; Payton, J.E.; Szankasi, P.; Kelley, T.W.; Kulkarni, S.; Pfeifer, J.D.; Duncavage, E.J. Detection of FLT3 Internal Tandem Duplication in Targeted, Short-Read-Length, next-Generation Sequencing Data. J Mol Diagn 2013, 15, 81–93. [CrossRef]
- Aguilera-Diaz, A.; Vazquez, I.; Ariceta, B.; Manu, A.; Blasco-Iturri, Z.; Palomino-Echeverria, S.; Larrayoz, M.J.; Garcia-Sanz, R.; Prieto-Conde, M.I.; Del Carmen Chillon, M.; et al. Assessment of the Clinical Utility of Four NGS Panels in Myeloid Malignancies. Suggestions for NGS Panel Choice or Design. PLoS One 2020, 15, e0227986. [CrossRef]
- Levis, M.; Shi, W.; Chang, K.; Laing, C.; Pollner, R.; Gocke, C.; Adams, E.; Berisha, F.; Lameh, J.; Lesegretain, A. FLT3 Inhibitors Added to Induction Therapy Induce Deeper Remissions. Blood 2020, 135, 75–78. [CrossRef]
- Keegan, A.; Bridge, J.A.; Lindeman, N.I.; Long, T.A.; Merker, J.D.; Moncur, J.T.; Montgomery, N.D.; Nagarajan, R.; Rothberg, P.G.; Routbort, M.J.; et al. Proficiency Testing of Standardized Samples Shows High Interlaboratory Agreement for Clinical Next Generation Sequencing-Based Hematologic Malignancy Assays With Survey Material-Specific Differences in Variant Frequencies. Arch Pathol Lab Med 2020. [CrossRef]
- Kim, J.J.; Lee, K.S.; Lee, T.G.; Lee, S.; Shin, S.; Lee, S.T. A Comparative Study of Next-Generation Sequencing and Fragment Analysis for the Detection and Allelic Ratio Determination of FLT3 Internal Tandem Duplication. Diagn Pathol 2022, 17, 14. [CrossRef]
- Chaudhary, S.; Chaudhary, P.; Ahmad, F.; Arora, N. Acute Myeloid Leukemia and Next-Generation Sequencing Panels for Diagnosis: A Comprehensive Review. J Pediatr Hematol Oncol 2024, 46, 125–137. [CrossRef]
- Hobeck, A.D.; Wendt, S.; Krohn, S.; Knuebel, G.; Bartels, S.; Schipper, E.; Junghanss, C.; Murua Escobar, H. Comparative Analyses of Targeted Myeloid Cancer Next-Generation Sequencing Panel in Fresh Blood, Bone Marrow and FFPE Material. Int J Mol Sci 2024, 25. [CrossRef]
- Fenaux, P.; Haase, D.; Santini, V.; Sanz, G.F.; Platzbecker, U.; Mey, U.; Esmo Guidelines Committee. Electronic address: clinicalguidelines@esmo. org Myelodysplastic Syndromes: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-up(Dagger☆). Ann Oncol 2021, 32, 142–156. [CrossRef]
- Jumniensuk, C.; Nobori, A.; Lee, T.; Senaratne, T.N.; Rao, D.; Pullarkat, S. Concordance of Peripheral Blood and Bone Marrow Next-Generation Sequencing in Hematologic Neoplasms. Adv Hematol 2022, 2022, 8091746. [CrossRef]
- Lucas, F.; Michaels, P.D.; Wang, D.; Kim, A.S. Mutational Analysis of Hematologic Neoplasms in 164 Paired Peripheral Blood and Bone Marrow Samples by Next-Generation Sequencing. Blood Adv 2020, 4, 4362–4365. [CrossRef]
- Ye, X.; Zheng, Z.; Wu, Y.; Zhang, Z.; Xu, Z.; Liu, Y.; Jiang, L.; Wu, J. NGS Panel Enhance Precise Diagnosis of Myeloid Neoplasms under WHO-HAEM5 and International Consensus Classification: An Observational Study. Medicine (Baltimore) 2024, 103, e38556. [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 2017, 19, 4–23. [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [CrossRef]
- Kwon, R.; Yeung, C.C.S. Advances in Next-Generation Sequencing and Emerging Technologies for Hematologic Malignancies. Haematologica 2024, 109, 379–387. [CrossRef]
- Horgan, D.; Curigliano, G.; Riess, O.; Hofman, P.; Buttner, R.; Conte, P.; Cufer, T.; Gallagher, W.M.; Georges, N.; Kerr, K.; et al. Identifying the Steps Required to Effectively Implement Next-Generation Sequencing in Oncology at a National Level in Europe. J Pers Med 2022, 12. [CrossRef]
- Darlington, M.; Sujobert, P.; Kosmider, O.; Luque Paz, D.; Kaltenbach, S.; Figeac, M.; Hayette, S.; Mezaour, N.; Coquerelle, S.; Alary, A.S.; et al. Targeted High-Throughput Sequencing for Hematological Malignancies: A GBMHM Survey of Practice and Cost Evaluation in France. Hemasphere 2023, 7, e943. [CrossRef]
- Horak, P.; Griffith, M.; Danos, A.M.; Pitel, B.A.; Madhavan, S.; Liu, X.; Chow, C.; Williams, H.; Carmody, L.; Barrow-Laing, L.; et al. Standards for the Classification of Pathogenicity of Somatic Variants in Cancer (Oncogenicity): Joint Recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genetics in Medicine 2022, 24, 986–998. [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015, 17, 405–424. [CrossRef]
- Christinat, Y.; Hamelin, B.; Alborelli, I.; Angelino, P.; Barbié, V.; Bisig, B.; Dawson, H.; Frattini, M.; Grob, T.; Jochum, W.; et al. Reporting of Somatic Variants in Clinical Cancer Care: Recommendations of the Swiss Society of Molecular Pathology. Virchows Arch 2024, 485, 1033–1039. [CrossRef]
- Galimberti, S.; Balducci, S.; Guerrini, F.; Del Re, M.; Cacciola, R. Digital Droplet PCR in Hematologic Malignancies: A New Useful Molecular Tool. Diagnostics 2022, 12, 1305. [CrossRef]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute Quantification by Droplet Digital PCR versus Analog Real-Time PCR. Nat Methods 2013, 10, 1003–1005. [CrossRef]
- Bacher, U.; Shumilov, E.; Flach, J.; Porret, N.; Joncourt, R.; Wiedemann, G.; Fiedler, M.; Novak, U.; Amstutz, U.; Pabst, T. Challenges in the Introduction of Next-Generation Sequencing (NGS) for Diagnostics of Myeloid Malignancies into Clinical Routine Use. Blood Cancer J 2018, 8, 113. [CrossRef]
- Panuzzo, C.; Jovanovski, A.; Ali, M.S.; Cilloni, D.; Pergolizzi, B. Revealing the Mysteries of Acute Myeloid Leukemia: From Quantitative PCR through Next-Generation Sequencing and Systemic Metabolomic Profiling. JCM 2022, 11, 483. [CrossRef]
- Wurm, S.; Waltersdorfer, M.; Loindl, S.; Moritz, J.M.; Herzog, S.A.; Bachmaier, G.; Berghold, A.; Kashofer, K.; Beham-Schmid, C.; Hoefler, G.; et al. Acute Myeloid Leukemia in the Next-Generation Sequencing Era: Real-World Data from an Austrian Tertiary Cancer Care Center. Wien Klin Wochenschr 2024. [CrossRef]
- da Rosa, S.E.A.; de Lima, L.B.; Silveira, C.N.; Cortes, L.G.F.; de Oliveira Filho, J.B.; de Souza Reis, R.; Cervato, M.C.; Rodrigues, P.H.S.; de Oliveira Pelegrino, K.; Petroni, R.C.; et al. Real-World Genomic Profiling of Acute Myeloid Leukemia and the Impact of European LeukemiaNet Risk Stratification 2022 Update. Clin Transl Oncol 2023, 25, 3431–3436. [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [CrossRef]
- Zhang, Y.; Wu, J.; Qin, T.; Xu, Z.; Qu, S.; Pan, L.; Li, B.; Wang, H.; Zhang, P.; Yan, X.; et al. Comparison of the Revised 4th (2016) and 5th (2022) Editions of the World Health Organization Classification of Myelodysplastic Neoplasms. Leukemia 2022, 36, 2875–2882. [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N Engl J Med 2014, 371, 2488–2498. [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N Engl J Med 2014, 371, 2477–2487. [CrossRef]
- DeZern, A.E.; Malcovati, L.; Ebert, B.L. CHIP, CCUS, and Other Acronyms: Definition, Implications, and Impact on Practice. American Society of Clinical Oncology Educational Book 2018. [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. New England Journal of Medicine 2017, 377, 111–121. [CrossRef]
- Schuermans, A.; Honigberg, M.C. Clonal Haematopoiesis in Cardiovascular Disease: Prognostic Role and Novel Therapeutic Target. Nat Rev Cardiol 2025. [CrossRef]
- Xie, Z.; Komrokji, R.; Al Ali, N.; Regelson, A.; Geyer, S.; Patel, A.; Saygin, C.; Zeidan, A.M.; Bewersdorf, J.P.; Mendez, L.; et al. Risk Prediction for Clonal Cytopenia: Multicenter Real-World Evidence. Blood 2024, 144, 2033–2044. [CrossRef]
- Huber, S.; Baer, C.; Hutter, S.; Wossidlo, N.; Hoermann, G.; Pohlkamp, C.; Walter, W.; Meggendorfer, M.; Kern, W.; Haferlach, T.; et al. Genomic Landscape of CCUS Compared to MDS and Its Implications on Risk Prediction. Leukemia 2024, 38, 1634–1637. [CrossRef]
- Malcovati, L.; Gallì, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricalà, S.; Bono, E.; et al. Clinical Significance of Somatic Mutation in Unexplained Blood Cytopenia. Blood 2017, 129, 3371–3378. [CrossRef]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 Mutation in Myelodysplasia with Ring Sideroblasts. N Engl J Med 2011, 365, 1384–1395. [CrossRef]
- Malcovati, L.; Papaemmanuil, E.; Bowen, D.T.; Boultwood, J.; Della Porta, M.G.; Pascutto, C.; Travaglino, E.; Groves, M.J.; Godfrey, A.L.; Ambaglio, I.; et al. Clinical Significance of SF3B1 Mutations in Myelodysplastic Syndromes and Myelodysplastic/Myeloproliferative Neoplasms. Blood 2011, 118, 6239–6246. [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of Genetic Lesions in 944 Patients with Myelodysplastic Syndromes. Leukemia 2014, 28, 241–247. [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 Allelic State for Genome Stability, Clinical Presentation and Outcomes in Myelodysplastic Syndromes. Nat Med 2020, 26, 1549–1556. [CrossRef]
- Hart, S.A.; Lee, L.A.; Seegmiller, A.C.; Mason, E.F. Diagnosis of TP53 -Mutated Myeloid Disease by the ICC and WHO Fifth Edition Classifications. Blood Advances 2025, 9, 445–454. [CrossRef]
- Malcikova, J.; Pavlova, S.; Baliakas, P.; Chatzikonstantinou, T.; Tausch, E.; Catherwood, M.; Rossi, D.; Soussi, T.; Tichy, B.; Kater, A.P.; et al. ERIC Recommendations for TP53 Mutation Analysis in Chronic Lymphocytic Leukemia—2024 Update. Leukemia 2024, 38, 1455–1468. [CrossRef]
- Aldoss, I.; Pham, A.; Li, S.M.; Gendzekhadze, K.; Afkhami, M.; Telatar, M.; Hong, H.; Padeganeh, A.; Bedell, V.; Cao, T.; et al. Favorable Impact of Allogeneic Stem Cell Transplantation in Patients with Therapy-Related Myelodysplasia Regardless of TP53 Mutational Status. Haematologica 2017, 102, 2030–2038. [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N Engl J Med 2011, 364, 2496–2506. [CrossRef]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute Myeloid Leukemia Ontogeny Is Defined by Distinct Somatic Mutations. Blood 2015, 125, 1367–1376. [CrossRef]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the Spliceosome Machinery, a Novel and Ubiquitous Pathway in Leukemogenesis. Blood 2012, 119, 3203–3210. [CrossRef]
- Visconte, V.; O. Nakashima, M.; J. Rogers, H. Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features. Cancers 2019, 11, 1844. [CrossRef]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Herzig, J.K.; Aulitzky, T.; Bullinger, L.; Spath, D.; Teleanu, V.; Kundgen, A.; Kohne, C.-H.; et al. ASXL1 Mutations in Younger Adult Patients with Acute Myeloid Leukemia: A Study by the German-Austrian Acute Myeloid Leukemia Study Group. Haematologica 2015, 100, 324–330. [CrossRef]
- Stasik, S.; Middeke, J.M.; Kramer, M.; Röllig, C.; Krämer, A.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brümmendorf, T.H.; Naumann, R.; et al. EZH2 Mutations and Impact on Clinical Outcome: An Analysis in 1,604 Patients with Newly Diagnosed Acute Myeloid Leukemia. Haematologica 2020, 105, e228–e231. [CrossRef]
- Ramil, G.; Pratcorona, M.; Nomdedéu, J.F. Be Aware of the X: BCOR Mutations in Myeloid Neoplasms. haematol 2024. [CrossRef]
- Eckardt, J.-N.; Stasik, S.; Röllig, C.; Sauer, T.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brümmendorf, T.H.; Naumann, R.; Steffen, B.; et al. Alterations of Cohesin Complex Genes in Acute Myeloid Leukemia: Differential Co-Mutations, Clinical Presentation and Impact on Outcome. Blood Cancer J. 2023, 13, 18. [CrossRef]
- Molica, M.; Perrone, S.; Mazzone, C.; Cesini, L.; Canichella, M.; de Fabritiis, P. CPX-351: An Old Scheme with a New Formulation in the Treatment of High-Risk AML. Cancers 2022, 14, 2843. [CrossRef]
- Gaidzik, V.I.; Bullinger, L.; Schlenk, R.F.; Zimmermann, A.S.; Röck, J.; Paschka, P.; Corbacioglu, A.; Krauter, J.; Schlegelberger, B.; Ganser, A.; et al. RUNX1 Mutations in Acute Myeloid Leukemia: Results From a Comprehensive Genetic and Clinical Analysis From the AML Study Group. JCO 2011, 29, 1364–1372. [CrossRef]
- Taube, F.; Georgi, J.A.; Kramer, M.; Stasik, S.; Middeke, J.M.; Röllig, C.; Krug, U.; Krämer, A.; Scholl, S.; Hochhaus, A.; et al. CEBPA Mutations in 4708 Patients with Acute Myeloid Leukemia: Differential Impact of bZIP and TAD Mutations on Outcome. Blood 2022, 139, 87–103. [CrossRef]
- Pan, L.; Li, Y.; Gao, H.; Cai, Y.; Lai, X.; Chen, Z.; Li, X.; Wang, S. Clinical Features and Monitoring of Germline CEBPA -Mutated Carriers. Blood 2023, 142, 1366–1366. [CrossRef]
- Falini, B.; Sorcini, D.; Perriello, V.M.; Sportoletti, P. Functions of the Native NPM1 Protein and Its Leukemic Mutant. Leukemia 2025, 39, 276–290. [CrossRef]
- Falini, B.; Brunetti, L.; Sportoletti, P.; Martelli, M.P. NPM1-Mutated Acute Myeloid Leukemia: From Bench to Bedside. Blood 2020, 136, 1707–1721. [CrossRef]
- Issa, G.C.; Aldoss, I.; Thirman, M.J.; DiPersio, J.; Arellano, M.; Blachly, J.S.; Mannis, G.N.; Perl, A.; Dickens, D.S.; McMahon, C.M.; et al. Menin Inhibition With Revumenib for KMT2A-Rearranged Relapsed or Refractory Acute Leukemia (AUGMENT-101). JCO 2025, 43, 75–84. [CrossRef]
- Dali, S.A.; Al-Mashdali, A.F.; Kalfah, A.; Mohamed, S.F. Menin Inhibitors in KMT2A-Rearranged and NPM1-Mutated Acute Leukemia: A Scoping Review of Safety and Efficacy. Critical Reviews in Oncology/Hematology 2025, 213, 104783. [CrossRef]
- Arellano, M.L.; Thirman, M.J.; DiPersio, J.F.; Heiblig, M.; Stein, E.M.; Schuh, A.C.; Žučenka, A.; de Botton, S.; Grove, C.S.; Mannis, G.N.; et al. Menin Inhibition with Revumenib for NPM1-Mutated Relapsed or Refractory Acute Myeloid Leukemia: The AUGMENT-101 Study. Blood 2025, 146, 1065–1077. [CrossRef]
- Severina, N.; Sidorova, Y.; Risinskaya, N.; Biderman, B.; Pshenychnyy, A.; Ryzhikova, N.; Lukianova, I.; Kashlakova, A.; Sudarikov, A. PB1792: NGS VS PCR FOR THE DETECTION OF NPM1 GENE MUTATIONS IN ACUTE MYELOID LEUKEMIA. Hemasphere 2022, 6, 1672–1673. [CrossRef]
- Vonk, C.M.; Grob, T.; Rijken, M.; Kavelaars, F.G.; Konijnenburg, J.M.L.; Ossenkoppele, G.J.; Manz, M.G.; Havelange, V.; Fløisand, Y.; Löwenberg, B.; et al. Advantages of a Genomic DNA-Based next-Generation Sequencing Assay for Detection of Mutant NPM1 Measurable Residual Disease in AML. Blood Adv 2025, 9, 1069–1077. [CrossRef]
- Boluda, B.; Rodriguez-Veiga, R.; Sargas, C.; Ayala, R.; Larráyoz, M.J.; Chillón, M.C.; Soria-Saldise, E.; Bilbao, C.; Prados De La Torre, E.P.; Navarro, I.; et al. Conventional PCR Versus Next Generation Sequencing for Diagnosis of FLT3, IDH and NPM1 Mutations in Acute Myeloid Leukemia: Results of the PETHEMA PCR-LMA Study. Cancers 2025, 17, 854. [CrossRef]
- Akabane, H. How Often Do Cytogenetics and NGS Information Impact Upfront Treatment in AML? Blood 2022, 140, 11607–11608. [CrossRef]
- Shimony, S.; Stahl, M.; Stone, R.M. Acute Myeloid Leukemia: 2025 Update on Diagnosis, Risk-Stratification, and Management. American Journal of Hematology 2025, 100, 860–891. [CrossRef]
- Research, C. for D.E. and FDA Approves Ivosidenib for Myelodysplastic Syndromes. FDA 2023.
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 2017, 377, 454–464. [CrossRef]
- Erba, H.P.; Montesinos, P.; Kim, H.-J.; Patkowska, E.; Vrhovac, R.; Žák, P.; Wang, P.-N.; Mitov, T.; Hanyok, J.; Kamel, Y.M.; et al. Quizartinib plus Chemotherapy in Newly Diagnosed Patients with FLT3-Internal-Tandem-Duplication-Positive Acute Myeloid Leukaemia (QuANTUM-First): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2023, 401, 1571–1583. [CrossRef]
- Montesinos, P.; Recher, C.; Vives, S.; Zarzycka, E.; Wang, J.; Bertani, G.; Heuser, M.; Calado, R.T.; Schuh, A.C.; Yeh, S.-P.; et al. Ivosidenib and Azacitidine in IDH1-Mutated Acute Myeloid Leukemia. N Engl J Med 2022, 386, 1519–1531. [CrossRef]
- Petermichl, V.; Fuchs, S.; Weber, M.; Gobat, K.; Micheloud, C.; Graf, L.; Gerth, Y.; Goede, J.S.; Lehmann, T.; Driessen, C.; et al. Prognostic Impact of the AML60+ Score for Elderly Patients with Acute Myeloid Leukemia Treated with Hypomethylating Agents: A Retrospective Multicentric Analysis. Cancers (Basel) 2025, 17, 2658. [CrossRef]
- Bérard, E.; Röllig, C.; Bertoli, S.; Pigneux, A.; Tavitian, S.; Kramer, M.; Serve, H.; Bornhäuser, M.; Platzbecker, U.; Müller-Tidow, C.; et al. A Scoring System for AML Patients Aged 70 Years or Older, Eligible for Intensive Chemotherapy: A Study Based on a Large European Data Set Using the DATAML, SAL, and PETHEMA Registries. Blood Cancer J. 2022, 12, 107. [CrossRef]
- Clichet, V.; Boyer, T. Artificial Intelligence-Based Myelodysplastic Syndromes Score, 2022 Classifications, and the Molecular International Prognostic Scoring System: A Perfect Match. haematol 2024. [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations from an International Expert Panel. Blood 2017, 129, 424–447. [CrossRef]
- Garcia-Manero, G. Myelodysplastic Syndromes: 2023 Update on Diagnosis, Risk-Stratification, and Management. Am J Hematol 2023, 98, 1307–1325. [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [CrossRef]
- Voso, M.T.; Fenu, S.; Latagliata, R.; Buccisano, F.; Piciocchi, A.; Aloe-Spiriti, M.A.; Breccia, M.; Criscuolo, M.; Andriani, A.; Mancini, S.; et al. Revised International Prognostic Scoring System (IPSS) Predicts Survival and Leukemic Evolution of Myelodysplastic Syndromes Significantly Better Than IPSS and WHO Prognostic Scoring System: Validation by the Gruppo Romano Mielodisplasie Italian Regional Database. JCO 2013, 31, 2671–2677. [CrossRef]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arango Ossa, J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evidence 2022, 1, EVIDoa2200008. [CrossRef]
- Sauta, E.; Robin, M.; Bersanelli, M.; Travaglino, E.; Meggendorfer, M.; Zhao, L.-P.; Caballero Berrocal, J.C.; Sala, C.; Maggioni, G.; Bernardi, M.; et al. Real-World Validation of Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. J Clin Oncol 2023, 41, 2827–2842. [CrossRef]
- Lee, W.-H.; Tsai, M.-T.; Tsai, C.-H.; Tien, F.-M.; Lo, M.-Y.; Tseng, M.-H.; Kuo, Y.-Y.; Liu, M.-C.; Yang, Y.-T.; Chen, J.-C.; et al. Validation of the Molecular International Prognostic Scoring System in Patients with Myelodysplastic Syndromes Defined by International Consensus Classification. Blood Cancer J. 2023, 13, 120. [CrossRef]
- Baer, C.; Huber, S.; Hutter, S.; Meggendorfer, M.; Nadarajah, N.; Walter, W.; Platzbecker, U.; Götze, K.S.; Kern, W.; Haferlach, T.; et al. Risk Prediction in MDS: Independent Validation of the IPSS-M—Ready for Routine? Leukemia 2023, 37, 938–941. [CrossRef]
- Zamanillo, I.; Poza, M.; Ayala, R.; Rapado, I.; Martinez-Lopez, J.; Cedena, M.T. Impact of IPSS-M Implementation in Real-Life Clinical Practice. Front. Oncol. 2023, 13. [CrossRef]
- Tentori, C.A.; Gregorio, C.; Robin, M.; Gagelmann, N.; Gurnari, C.; Ball, S.; Caballero Berrocal, J.C.; Lanino, L.; D’Amico, S.; Spreafico, M.; et al. Clinical and Genomic-Based Decision Support System to Define the Optimal Timing of Allogeneic Hematopoietic Stem-Cell Transplantation in Patients With Myelodysplastic Syndromes. JCO 2024, 42, 2873–2886. [CrossRef]
- Bill, M.; Eckardt, J.-N.; Döhner, K.; Röhnert, M.-A.; Rausch, C.; Metzeler, K.H.; Spiekermann, K.; Stasik, S.; Wurm, A.A.; Sauer, T.; et al. Differential Prognostic Impact of Myelodysplasia-Related Gene Mutations in a European Cohort of 4978 Intensively Treated AML Patients. Leukemia 2025. [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med 2020, 383, 617–629. [CrossRef]
- Kröger, N.; Bacigalupo, A.; Barbui, T.; Ditschkowski, M.; Gagelmann, N.; Griesshammer, M.; Gupta, V.; Hamad, N.; Harrison, C.; Hernandez-Boluda, J.C.; et al. Indication and Management of Allogeneic Haematopoietic Stem-Cell Transplantation in Myelofibrosis: Updated Recommendations by the EBMT/ELN International Working Group. The Lancet Haematology 2024, 11, e62–e74. [CrossRef]
- Fenaux, P.; Gobbi, M.; Kropf, P.L.; Issa, J.-P.J.; Roboz, G.J.; Mayer, J.; Krauter, J.; Robak, T.; Kantarjian, H.; Novak, J.; et al. Guadecitabine vs Treatment Choice in Newly Diagnosed Acute Myeloid Leukemia: A Global Phase 3 Randomized Study. Blood Adv 2023, 7, 5027–5037. [CrossRef]
- Blackmon, A.L.; Grunwald, M.R. Editorial: Molecular MRD Testing in Patients with Acute Myeloid Leukemia. Bone Marrow Transplant 2025, 60, 119–121. [CrossRef]
- Sahasrabudhe, K.D.; Mims, A.S. MRD in AML: Who, What, When, Where, and How? Blood 2024, 143, 296–298. [CrossRef]
- Heuser, M.; Freeman, S.D.; Ossenkoppele, G.J.; Buccisano, F.; Hourigan, C.S.; Ngai, L.L.; Tettero, J.M.; Bachas, C.; Baer, C.; Béné, M.-C.; et al. 2021 Update on MRD in Acute Myeloid Leukemia: A Consensus Document from the European LeukemiaNet MRD Working Party. Blood 2021, 138, 2753–2767. [CrossRef]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. New England Journal of Medicine 2016, 374, 422–433. [CrossRef]
- Höllein, A.; Meggendorfer, M.; Dicker, F.; Jeromin, S.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T. NPM1 Mutated AML Can Relapse with Wild-Type NPM1: Persistent Clonal Hematopoiesis Can Drive Relapse. Blood Adv 2018, 2, 3118–3125. [CrossRef]
- Cocciardi, S.; Dolnik, A.; Kapp-Schwoerer, S.; Rücker, F.G.; Lux, S.; Blätte, T.J.; Skambraks, S.; Krönke, J.; Heidel, F.H.; Schnöder, T.M.; et al. Clonal Evolution Patterns in Acute Myeloid Leukemia with NPM1 Mutation. Nat Commun 2019, 10, 2031. [CrossRef]
- Marchetti, F.; Cardoso, R.; Chen, C.L.; Douglas, G.R.; Elloway, J.; Escobar, P.A.; Harper, T.; Heflich, R.H.; Kidd, D.; Lynch, A.M.; et al. Error-Corrected next Generation Sequencing – Promises and Challenges for Genotoxicity and Cancer Risk Assessment. Mutation Research - Reviews in Mutation Research 2023, 792, 108466. [CrossRef]
- Chen, H.; Yu, F.; Lu, D.; Huang, S.; Liu, S.; Zhang, B.; Shu, K.; Pu, D. Enhanced Error Suppression for Accurate Detection of Low-Frequency Variants. Electrophoresis 2025, 46, 65–75. [CrossRef]
- Romer-Seibert, J.S.; Meyer, S.E. Genetic Heterogeneity and Clonal Evolution in Acute Myeloid Leukemia. Curr Opin Hematol 2021, 28, 64–70. [CrossRef]
- Schuringa, J.J.; Bonifer, C. Dissecting Clonal Heterogeneity in AML. Cancer Cell 2020, 38, 782–784. [CrossRef]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.D.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discovery 2019, 9, 1050–1063. [CrossRef]
- Walter, R.B. Perspective on Measurable Residual Disease Testing in Acute Myeloid Leukemia. Leukemia 2024, 38, 10–13. [CrossRef]
- Levis, M.J.; Hamadani, M.; Logan, B.R.; Jones, R.J.; Singh, A.K.; Litzow, M.R.; Wingard, J.R.; Papadopoulos, E.B.; Perl, A.E.; Soiffer, R.J.; et al. Measurable Residual Disease and Posttransplantation Gilteritinib Maintenance for Patients with FLT3-ITD-Mutated AML. Blood 2025, 145, 2138–2148. [CrossRef]
- Tsai, C.-H.; Tang, J.-L.; Tien, F.-M.; Kuo, Y.-Y.; Wu, D.-C.; Lin, C.-C.; Tseng, M.-H.; Peng, Y.-L.; Hou, M.-F.; Chuang, Y.-K.; et al. Clinical Implications of Sequential MRD Monitoring by NGS at 2 Time Points after Chemotherapy in Patients with AML. Blood Advances 2021, 5, 2456–2466. [CrossRef]
- Schnittger, S.; Schoch, C.; Dugas, M.; Kern, W.; Staib, P.; Wuchter, C.; Löffler, H.; Sauerland, C.M.; Serve, H.; Büchner, T.; et al. Analysis of FLT3 Length Mutations in 1003 Patients with Acute Myeloid Leukemia: Correlation to Cytogenetics, FAB Subtype, and Prognosis in the AMLCG Study and Usefulness as a Marker for the Detection of Minimal Residual Disease. Blood 2002, 100, 59–66. [CrossRef]
- Thol, F.; Kölking, B.; Damm, F.; Reinhardt, K.; Klusmann, J.-H.; Reinhardt, D.; von Neuhoff, N.; Brugman, M.H.; Schlegelberger, B.; Suerbaum, S.; et al. Next-Generation Sequencing for Minimal Residual Disease Monitoring in Acute Myeloid Leukemia Patients with FLT3-ITD or NPM1 Mutations. Genes, Chromosomes and Cancer 2012, 51, 689–695. [CrossRef]
- Levis, M.J.; Hamadani, M.; Logan, B.; Jones, R.J.; Singh, A.K.; Litzow, M.; Wingard, J.R.; Papadopoulos, E.B.; Perl, A.E.; Soiffer, R.J.; et al. Gilteritinib as Post-Transplant Maintenance for AML With Internal Tandem Duplication Mutation of FLT3. JCO 2024, 42, 1766–1775. [CrossRef]
- Dillon, L.W.; Gui, G.; Page, K.M.; Ravindra, N.; Wong, Z.C.; Andrew, G.; Mukherjee, D.; Zeger, S.L.; El Chaer, F.; Spellman, S.; et al. DNA Sequencing to Detect Residual Disease in Adults With Acute Myeloid Leukemia Prior to Hematopoietic Cell Transplant. JAMA 2023, 329, 745. [CrossRef]
- Hirsch, P.; Lambert, J.; Bucci, M.; Deswarte, C.; Boudry, A.; Lambert, J.; Fenwarth, L.; Micol, J.-B.; Terré, C.; Celli-Lebras, K.; et al. Multi-Target Measurable Residual Disease Assessed by Error-Corrected Sequencing in Patients with Acute Myeloid Leukemia: An ALFA Study. Blood Cancer J. 2024, 14, 97. [CrossRef]
- Godley, L.A.; Shimamura, A. Genetic Predisposition to Hematologic Malignancies: Management and Surveillance. Blood 2017, 130, 424–432. [CrossRef]
- Rio-Machin, A.; Vulliamy, T.; Hug, N.; Walne, A.; Tawana, K.; Cardoso, S.; Ellison, A.; Pontikos, N.; Wang, J.; Tummala, H.; et al. The Complex Genetic Landscape of Familial MDS and AML Reveals Pathogenic Germline Variants. Nat Commun 2020, 11, 1044. [CrossRef]
- Mestre, J.; Chaparro, L.; Manzanares, A.; Xicoy, B.; Zamora, L.; Sole, F.; Calvete, O. Beyond Myeloid Neoplasms Germline Guidelines: Validation of the Thresholds Criteria in the Search of Germline Predisposition Variants. eJHaem 2024, 5, 1021–1027. [CrossRef]
- Baliakas, P.; Tesi, B.; Wartiovaara-Kautto, U.; Stray-Pedersen, A.; Friis, L.S.; Dybedal, I.; Hovland, R.; Jahnukainen, K.; Raaschou-Jensen, K.; Ljungman, P.; et al. Nordic Guidelines for Germline Predisposition to Myeloid Neoplasms in Adults: Recommendations for Genetic Diagnosis, Clinical Management and Follow-Up. HemaSphere 2019, 3, e321. [CrossRef]
- Tawana, K.; Brown, A.L.; Churpek, J.E. Integrating Germline Variant Assessment into Routine Clinical Practice for Myelodysplastic Syndrome and Acute Myeloid Leukaemia: Current Strategies and Challenges. Br J Haematol 2022, 196, 1293–1310. [CrossRef]
- Godley, L.A.; DiNardo, C.D.; Bolton, K. Germline Predisposition in Hematologic Malignancies: Testing, Management, and Implications. Am Soc Clin Oncol Educ Book 2024, 44, e432218. [CrossRef]
- Farina, M.; Bernardi, S.; Gandolfi, L.; Zanaglio, C.; Morello, E.; Turra, A.; Zollner, T.; Gramegna, D.; Rambaldi, B.; Cattina, F.; et al. Case Report: Late Onset of Myelodysplastic Syndrome From Donor Progenitor Cells After Allogeneic Stem Cell Transplantation. Which Lessons Can We Draw From the Reported Case? Front. Oncol. 2020, 10, 564521. [CrossRef]
- Kato, M.; Yamashita, T.; Suzuki, R.; Matsumoto, K.; Nishimori, H.; Takahashi, S.; Iwato, K.; Nakaseko, C.; Kondo, T.; Imada, K.; et al. Donor Cell-Derived Hematological Malignancy: A Survey by the Japan Society for Hematopoietic Cell Transplantation. Leukemia 2016, 30, 1742–1745. [CrossRef]
- Gibson, C.J.; Kim, H.T.; Zhao, L.; Murdock, H.M.; Hambley, B.; Ogata, A.; Madero-Marroquin, R.; Wang, S.; Green, L.; Fleharty, M.; et al. Donor Clonal Hematopoiesis and Recipient Outcomes After Transplantation. JCO 2022, 40, 189–201. [CrossRef]
- Speight, B.; Hanson, H.; Turnbull, C.; Hardy, S.; Drummond, J.; Khorashad, J.; Wragg, C.; Page, P.; Parkin, N.W.; Rio-Machin, A.; et al. Germline Predisposition to Haematological Malignancies: Best Practice Consensus Guidelines from the UK Cancer Genetics Group ( UKCGG) , CanGene-CanVar and the NHS England Haematological Oncology Working Group. Br J Haematol 2023, 201, 25–34. [CrossRef]
- Reilly, C.R.; Shimamura, A. Predisposition to Myeloid Malignancies in Shwachman-Diamond Syndrome: Biological Insights and Clinical Advances. Blood 2023, 141, 1513–1523. [CrossRef]
- Olson, T.S. Management of Fanconi Anemia beyond Childhood. Hematology Am Soc Hematol Educ Program 2023, 2023, 556–562. [CrossRef]
- Mason, N.R.; Cahill, H.; Diamond, Y.; McCleary, K.; Kotecha, R.S.; Marshall, G.M.; Mateos, M.K. Down Syndrome-Associated Leukaemias: Current Evidence and Challenges. Ther Adv Hematol 2024, 15, 20406207241257901. [CrossRef]
- Molica, M.; Perrone, S. Molecular Targets for the Treatment of AML in the Forthcoming 5th World Health Organization Classification of Haematolymphoid Tumours. Expert Review of Hematology 2022, 15, 973–986. [CrossRef]
- Baliakas, P.; Tesi, B.; Cammenga, J.; Stray-Pedersen, A.; Jahnukainen, K.; Andersen, M.K.; Ågerstam, H.; Creignou, M.; Dybedal, I.; Raaschou-Jensen, K.; et al. How to Manage Patients with Germline DDX41 Variants: Recommendations from the Nordic Working Group on Germline Predisposition for Myeloid Neoplasms. HemaSphere 2024, 8, e145. [CrossRef]
- Ma, J.; Ross, S.R. Multifunctional Role of DEAD-Box Helicase 41 in Innate Immunity, Hematopoiesis and Disease. Front. Immunol. 2024, 15. [CrossRef]
- Makishima, H.; Saiki, R.; Nannya, Y.; Korotev, S.; Gurnari, C.; Takeda, J.; Momozawa, Y.; Best, S.; Krishnamurthy, P.; Yoshizato, T.; et al. Germ Line DDX41 Mutations Define a Unique Subtype of Myeloid Neoplasms. Blood 2023, 141, 534–549. [CrossRef]
- Wang, X.; Xiao, Z.; Qin, T.; Xu, Z.; Jia, Y.; Qu, S.; Li, B.; Pan, L.; Gao, Q.; Jiao, M.; et al. Combination Therapy with Venetoclax and Azacitidine for the Treatment of Myelodysplastic Syndromes with DDX41 Mutations. Hematology 2024, 29, 2338509. [CrossRef]
- Nanaa, A.; He, R.; Foran, J.M.; Badar, T.; Gangat, N.; Pardanani, A.; Hogan, W.J.; Litzow, M.R.; Patnaik, M.; Al-Kali, A.; et al. Venetoclax plus Hypomethylating Agents in DDX41-Mutated Acute Myeloid Leukaemia and Myelodysplastic Syndrome: Mayo Clinic Series on 12 Patients. British Journal of Haematology 2024, 204, 171–176. [CrossRef]
- Alkhateeb, H.B.; Nanaa, A.; Viswanatha, D.; Foran, J.M.; Badar, T.; Sproat, L.; He, R.; Nguyen, P.; Jevremovic, D.; Salama, M.E.; et al. Genetic Features and Clinical Outcomes of Patients with Isolated and Comutated DDX41-Mutated Myeloid Neoplasms. Blood Adv 2022, 6, 528–532. [CrossRef]
- Li, P.; White, T.; Xie, W.; Cui, W.; Peker, D.; Zeng, G.; Wang, H.-Y.; Vagher, J.; Brown, S.; Williams, M.; et al. AML with Germline DDX41 Variants Is a Clinicopathologically Distinct Entity with an Indolent Clinical Course and Favorable Outcome. Leukemia 2022, 36, 664–674. [CrossRef]
- Nanaa, A.; He, R.; Viswanatha, D.; Nguyen, P.; Jevremovic, D.; Foran, J.M.; Yi, C.A.; Greipp, P.T.; Gangat, N.; Patnaik, M.; et al. Comparison between GATA2 and DDX41-Mutated Myeloid Neoplasms. Leuk Res 2022, 121, 106931. [CrossRef]
- Dimopoulos, Y.P.; Wang, W.; Wang, S.A.; Loghavi, S.; DiNardo, C.D.; Gerstein, Y.; Hu, S.; Tang, Z.; Ilagan, C.J.L.; Thakral, B.; et al. The Spectrum of Hematologic Neoplasms in Patients with Li-Fraumeni Syndrome. American J Hematol 2024, 99, 2416–2419. [CrossRef]
- Alikarami, F.; Xie, H.M.; Riedel, S.S.; Goodrow, H.T.; Barrett, D.R.; Mahdavi, L.; Lenard, A.; Chen, C.; Yamauchi, T.; Danis, E.; et al. GATA2 Links Stemness to Chemotherapy Resistance in Acute Myeloid Leukemia. Blood 2025, 145, 2179–2195. [CrossRef]
- Calvo, K.R.; Hickstein, D.D. The Spectrum of GATA2 Deficiency Syndrome. Blood 2023, 141, 1524–1532. [CrossRef]
- Wlodarski, M.W.; Hirabayashi, S.; Pastor, V.; Starý, J.; Hasle, H.; Masetti, R.; Dworzak, M.; Schmugge, M.; van den Heuvel-Eibrink, M.; Ussowicz, M.; et al. Prevalence, Clinical Characteristics, and Prognosis of GATA2-Related Myelodysplastic Syndromes in Children and Adolescents. Blood 2016, 127, 1387–1397; quiz 1518. [CrossRef]
- Sanchez-Petitto, G.; El Boghdadly, Z.; Nicolet, D.; Cooper, J.; Eisfeld, A.-K.; Klein, V.; Walker, M.C.; Mrózek, K.; Bezerra, E.; Brammer, J.E.; et al. GATA2-Mutated AML: Clinical Outcomes and Spectrum of Infections in Patients Undergoing or Not Allogeneic Stem Cell Transplantation. Blood 2024, 144, 7309. [CrossRef]
- Santiago, M.; Liquori, A.; Such, E.; Zúñiga, Á.; Cervera, J. The Clinical Spectrum, Diagnosis, and Management of GATA2 Deficiency. Cancers 2023, 15, 1590. [CrossRef]
- de Bruijn, M.; Dzierzak, E. Runx Transcription Factors in the Development and Function of the Definitive Hematopoietic System. Blood 2017, 129, 2061–2069. [CrossRef]
- Yu, K.; Deuitch, N.; Merguerian, M.; Cunningham, L.; Davis, J.; Bresciani, E.; Diemer, J.; Andrews, E.; Young, A.; Donovan, F.; et al. Genomic Landscape of Patients with Germline RUNX1 Variants and Familial Platelet Disorder with Myeloid Malignancy. Blood Adv 2024, 8, 497–511. [CrossRef]
- Greif, P.A.; Konstandin, N.P.; Metzeler, K.H.; Herold, T.; Pasalic, Z.; Ksienzyk, B.; Dufour, A.; Schneider, F.; Schneider, S.; Kakadia, P.M.; et al. RUNX1 Mutations in Cytogenetically Normal Acute Myeloid Leukemia Are Associated with a Poor Prognosis and Up-Regulation of Lymphoid Genes. Haematologica 2012, 97, 1909–1915. [CrossRef]
- Homan, C.C.; Drazer, M.W.; Yu, K.; Lawrence, D.M.; Feng, J.; Arriola-Martinez, L.; Pozsgai, M.J.; McNeely, K.E.; Ha, T.; Venugopal, P.; et al. Somatic Mutational Landscape of Hereditary Hematopoietic Malignancies Caused by Germline Variants in RUNX1, GATA2, and DDX41. Blood Adv 2023, 7, 6092–6107. [CrossRef]
- Noetzli, L.; Lo, R.W.; Lee-Sherick, A.B.; Callaghan, M.; Noris, P.; Savoia, A.; Rajpurkar, M.; Jones, K.; Gowan, K.; Balduini, C.L.; et al. Germline Mutations in ETV6 Are Associated with Thrombocytopenia, Red Cell Macrocytosis and Predisposition to Lymphoblastic Leukemia. Nat Genet 2015, 47, 535–538. [CrossRef]
- Zhang, M.Y.; Churpek, J.E.; Keel, S.B.; Walsh, T.; Lee, M.K.; Loeb, K.R.; Gulsuner, S.; Pritchard, C.C.; Sanchez-Bonilla, M.; Delrow, J.J.; et al. Germline ETV6 Mutations in Familial Thrombocytopenia and Hematologic Malignancy. Nat Genet 2015, 47, 180–185. [CrossRef]
- Noris, P.; Favier, R.; Alessi, M.-C.; Geddis, A.E.; Kunishima, S.; Heller, P.G.; Giordano, P.; Niederhoffer, K.Y.; Bussel, J.B.; Podda, G.M.; et al. ANKRD26-Related Thrombocytopenia and Myeloid Malignancies. Blood 2013, 122, 1987–1989. [CrossRef]
- Pippucci, T.; Savoia, A.; Perrotta, S.; Pujol-Moix, N.; Noris, P.; Castegnaro, G.; Pecci, A.; Gnan, C.; Punzo, F.; Marconi, C.; et al. Mutations in the 5’ UTR of ANKRD26, the Ankirin Repeat Domain 26 Gene, Cause an Autosomal-Dominant Form of Inherited Thrombocytopenia, THC2. Am J Hum Genet 2011, 88, 115–120. [CrossRef]
- Noris, P.; Perrotta, S.; Seri, M.; Pecci, A.; Gnan, C.; Loffredo, G.; Pujol-Moix, N.; Zecca, M.; Scognamiglio, F.; De Rocco, D.; et al. Mutations in ANKRD26 Are Responsible for a Frequent Form of Inherited Thrombocytopenia: Analysis of 78 Patients from 21 Families. Blood 2011, 117, 6673–6680. [CrossRef]
- Potenza, L.; Borelli, E.; Bigi, S.; Giusti, D.; Longo, G.; Odejide, O.; Porro, C.A.; Zimmermann, C.; Efficace, F.; Bruera, E.; et al. Early Palliative Care in Acute Myeloid Leukemia. Cancers 2022, 14, 478. [CrossRef]
- Moore, C.G.; Stein, A.; Fathi, A.T.; Pullarkat, V. Treatment of Relapsed/Refractory AML—Novel Treatment Options Including Immunotherapy. American Journal of Hematology 2025, 100, 23–37. [CrossRef]
- Molica, M.; Perrone, S.; Andriola, C.; Rossi, M. Immunotherapy with Monoclonal Antibodies for Acute Myeloid Leukemia: A Work in Progress. Cancers 2023, 15, 5060. [CrossRef]
- Bruno, S.; Borsi, E.; Patuelli, A.; Bandini, L.; Mancini, M.; Forte, D.; Nanni, J.; Barone, M.; Grassi, A.; Cristiano, G.; et al. Tracking Response and Resistance in Acute Myeloid Leukemia through Single-Cell DNA Sequencing Helps Uncover New Therapeutic Targets. IJMS 2024, 25, 10002. [CrossRef]
- Assi, R.E.; Alfonso Pierola, A.; Kc, D.; Abaza, Y.M.; Abou Zahr, A.; Chamoun, K.; Montalban-Bravo, G.; Takahashi, K.; Jabbour, E.; Kadia, T.M.; et al. Impact of Next-Generation Sequencing (NGS) on Treatment Selection in Acute Myeloid Leukemia (AML). JCO 2018, 36, 103–103. [CrossRef]
- Upadhyay Banskota, S.; Khanal, N.; Bhatt, V.R. A Precision Medicine Approach to Management of Acute Myeloid Leukemia in Older Adults. Current Opinion in Oncology 2020, 32, 650. [CrossRef]
- Snaith, O.; Poveda-Rogers, C.; Laczko, D.; Yang, G.; Morrissette, J.J.D. Cytogenetics and Genomics of Acute Myeloid Leukemia. Best Pract Res Clin Haematol 2024, 37, 101533. [CrossRef]
- Dai, B.; Yu, H.; Ma, T.; Lei, Y.; Wang, J.; Zhang, Y.; Lu, J.; Yan, H.; Jiang, L.; Chen, B. The Application of Targeted RNA Sequencing for KMT2A-Partial Tandem Duplication Identification and Integrated Analysis of Molecular Characterization in Acute Myeloid Leukemia. J Mol Diagn 2021, 23, 1478–1490. [CrossRef]
- Nawas, M.T.; Kosuri, S. Utility or Futility? A Contemporary Approach to Allogeneic Hematopoietic Cell Transplantation for TP53-Mutated MDS/AML. Blood Advances 2024, 8, 553–561. [CrossRef]
- Kim, K.; Maiti, A.; Loghavi, S.; Pourebrahim, R.; Kadia, T.M.; Rausch, C.R.; Furudate, K.; Daver, N.G.; Alvarado, Y.; Ohanian, M.; et al. Outcomes of TP53-Mutant Acute Myeloid Leukemia with Decitabine and Venetoclax. Cancer 2021, 127, 3772–3781. [CrossRef]
- Shimony, S.O.; Murdock, H.; Keating, J.; Reilly, C.R.; Tsai, H.K.; Gibson, C.J.; Faderl, S.; Wagner, T.; Dronamraju, N.; Lin, T.L.; et al. AML-MR Mutations Drive the Benefit of CPX-351 over 7+3 in the Pivotal Phase 3 AML Trial. Blood 2024, 144, 60. [CrossRef]
- Badar, T.; Nanaa, A.; Atallah, E.; Shallis, R.M.; Guilherme, S. de C.C.; Goldberg, A.D.; Saliba, A.N.; Patel, A.; Bewersdorf, J.P.; DuVall, A.S.; et al. Comparing Venetoclax in Combination with Hypomethylating Agents to Hypomethylating Agent-Based Therapies for Treatment Naive TP53-Mutated Acute Myeloid Leukemia: Results from the Consortium on Myeloid Malignancies and Neoplastic Diseases (COMMAND). Blood Cancer J. 2024, 14, 32. [CrossRef]
- Sallman, D.A.; Stahl, M. TP53-Mutated Acute Myeloid Leukemia: How Can We Improve Outcomes? Blood 2025, 145, 2828–2833. [CrossRef]
- Hassin, O.; Oren, M. Drugging P53 in Cancer: One Protein, Many Targets. Nat Rev Drug Discov 2023, 22, 127–144. [CrossRef]
- Perrone, S.; Ottone, T.; Zhdanovskaya, N.; Molica, M. How Acute Myeloid Leukemia (AML) Escapes from FMS-Related Tyrosine Kinase 3 (FLT3) Inhibitors? Still an Overrated Complication? Cancer Drug Resist 2023, 6, 223–238. [CrossRef]
- Arora, S.; Loghavi, S.; Daver, N.; Ravandi, F.; DiNardo, C.D.; Kadia, T.M.; Borthakur, G.; Jabbour, E.; Yilmaz, M.; Issa, G.C.; et al. Cytomolecular Mechanisms of Relapse and Post-Relapse Outcomes After Frontline FLT3 Inhibitor-Based Therapy in FLT3-Mutated AML. American Journal of Hematology n/a. [CrossRef]
- Sasaki, K.; Takahashi, S.; Ouchi, K.; Otsuki, Y.; Wakayama, S.; Ishioka, C. Different Impacts of TP53 Mutations on Cell Cycle-Related Gene Expression among Cancer Types. Sci Rep 2023, 13, 4868. [CrossRef]
- Tung, N.; Ricker, C.; Messersmith, H.; Balmaña, J.; Domchek, S.; Stoffel, E.M.; Almhanna, K.; Arun, B.; Chavarri-Guerra, Y.; Cohen, S.A.; et al. Selection of Germline Genetic Testing Panels in Patients With Cancer: ASCO Guideline. JCO 2024, 42, 2599–2615. [CrossRef]
- Haferlach, T.; Eckardt, J.-N.; Walter, W.; Maschek, S.; Kather, J.N.; Pohlkamp, C.; Middeke, J.M. AML Diagnostics in the 21st Century: Use of AI. Seminars in Hematology 2025. [CrossRef]
- Qin, Y.; Pu, X.; Hu, D.; Yang, M. Machine Learning-Based Biomarker Screening for Acute Myeloid Leukemia Prognosis and Therapy from Diverse Cell-Death Patterns. Sci Rep 2024, 14, 17874. [CrossRef]


| Functional Category | Example Genes / Alterations | Consequences of Genetic Alteration |
| Signal Transduction | FLT3, NRAS, KRAS, c-KIT, PTPN11 | Confers proliferative advantage through the hyperactivation of signaling pathways such as JAK/STAT, PI3K/AKT, and RAF/MEK/ERK. |
| Myeloid Transcription Factors | RUNX1, CEBPA, or fusions like RUNX1::RUNX1T1, PML::RARA, CBFB::MYH11 | Causes transcriptional deregulation, leading to impaired or blocked normal hematopoietic differentiation. |
| Tumor Suppressor Genes | TP53, WT1, PHF6 | Deregulates normal transcription and disrupts cell cycle checkpoints and responses to cellular stress, often resulting in increased proliferation and impaired differentiation. |
| Spliceosome Complex | SRSF2, SF3B1, U2AF1, ZRSR2 | Alters the proper maturation of mRNA, causing events such as intron retention or exon skipping, which can result in dysfunctional proteins. |
| Multifunctional Protein | NPM1 | Nucleophosmin mutations cause abnormal cytoplasmic localization of the protein, disrupting ribosome biogenesis and the stability of tumor suppressors such asp53. |
| Cohesin Complex | SMC1A, SMC3, STAG2, RAD21 | Affects chromosomal segregation and gene expression by altering chromatin accessibility, resulting in increased proliferation and impaired differentiation. |
| DNA Methylation | DNMT3A, TET2, IDH1/2 | Leads to global changes in the epigenetic landscape by altering DNA methylation patterns, thereby affecting gene expression. |
| Chromatin Modifiers | ASXL1, EZH2, or fusions involving KMT2A | Perturbs epigenetic homeostasis through aberrant histone modifications, resulting in widespread changes in gene transcription. |
| Clinical question | Recommended NGS test | Rationale | Main limitations |
| Initial diagnosis of MDS / AML | Targeted DNA NGS panel (20-50 genes) | Detect recurrent mutations relevant for WHO/ICC 2022 classification, ELN risk stratification, IPSS-M, therapeutic targets | Does not detect unknown fusions; reduced sensitivity for long ITDs, large indels, GC-rich regions. |
| Suspected translocation or known gene fusion | Targeted RNA-seq for fusion detection | Identifies common/known fusions (KMT2A, RUNX1/RUNX1T1, CBFB/MYH11, NUP98, etc.) | Does not detect rare/novel fusions; requires high-quality RNA. |
| Search for rare/ unexpected gene fusions | Whole-transcriptome RNA-seq (WTS) | Detects novel/atypical fusions; provides gene-expression and isoform profiling. Useful when cytogenetics is inconclusive. | Expensive, long turn-around time, requires advanced bioinformatics. |
| Suspected germline predisposition | WES (tumor ± germline) | Broad analysis of coding genome; detects germline predisposition variants. | Uneven coverage; limited SV detection; moderate depth |
| Suspected complex structural variant (e.g., long FLT3-ITD, MLL-PTD, large indels, GC-rich amplicons) | Targeted long-read NGS | Ideal for complex alterations such as variable FLT3-ITD, MLL-PTD, CALR type 1, GC-rich CEBPA; resolves complex rearrangements. | Lower accuracy for SNVs; limited availability; higher costs. |
| MRD monitoring (known variant) | Ultra-deep targeted NGS (DNA) or PCR-NGS | Very high sensitivity. Ideal for FLT3, IDH1/2, etc. | Requires an index variant; cannot identify new mutations |
| Confirmation and characterization of complex alterations | Whole-genome sequencing (WGS) | Covers the entire genome, including noncoding regions, SVs, CNVs, and cryptic translocations. | High cost; relatively low depth (30–60×); less sensitive for low VAF variants. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



