
Submitted:
27 October 2025
Posted:
30 October 2025
You are already at the latest version
Abstract
Background: The relationship between nicotine exposure and Parkinson’s disease (PD) remains controversial. Experimental studies, particularly preclinical trials in animal models, suggest that nicotine may exert neuroprotective effects; however, this has not been adequately addressed in randomized controlled trials (RCTs) involving humans.Methods: Comprehensive searches of PubMed, Web of Science, Scopus, and Google Scholar identified 1,363 studies, of which 13 met the inclusion criteria. Random-effects meta-analysis was conducted using RevMan 5.4 and R 4.1.1. Heterogeneity was assessed using the I² statistic, publication bias using funnel plots and Egger’s test, and study quality using SYRCLE and ToxRTool criteria. The review followed PRISMA guidelines and was registered in PROSPERO (CRD420250633808).Results: Nicotine significantly reduced apoptosis (RR, 0.49; 95% CI, 0.34–0.71), oxidative stress (RR, 0.55; 95% CI, 0.41–0.73), and neuroinflammation (RR, 0.62; 95% CI, 0.49–0.79), while enhancing dopaminergic neuron survival (RR, 1.67; 95% CI, 1.35–2.06), mitochondrial function (RR, 1.55; 95% CI, 1.12–2.14), synaptic protein expression (RR, 1.52; 95% CI, 1.14–2.04), and dendritic spine density (RR, 1.47; 95% CI, 1.19–1.83). The only human randomized trial showed no clinical benefit (RR, 1.00; 95% CI, 0.84–1.49). Subgroup and meta-regression analyses identified study design, dose, duration, and model type as sources of heterogeneity. Our findings suggest that although nicotine shows preclinical promise, clinical efficacy remains unproven.Conclusion: Experimental evidence suggests that nicotine exposure confers neuroprotection against PD, however, clinical efficacy remains unproven, underscoring the need for large-scale mechanistic and translational studies to establish causal relationships in humans.
Keywords:
Experimental evidence
; meta-analysis
; neurodegenerative
; neuroprotective
; nicotine
; Parkinson’s disease
; systematic review
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by the loss of dopaminergic neurons in the substantia nigra, resulting in motor symptoms such as tremor, rigidity, and bradykinesia, as well as non-motor manifestation. [1] The etiology of PD remains uncertain, with both genetic and environmental factors contributing to its development and progression. [2] Epidemiological studies have consistently reported an inverse association between nicotine and PD risk, suggesting a potential protective effect; [3] however, the underlying mechanisms remain poorly understood. Experimental studies, particularly preclinical trial in animal models, suggest that nicotine exposure may exert neuroprotective effects; [4] however, this remains inadequately addressed in randomized controlled trials (RCTs) involving humans. This review systematically evaluates experimental studies, including in vitro, in vivo, and the most recent human trial. It performs a meta-analysis quantifying nicotine’s effect on apoptosis, oxidative stress, neuroinflammation, dopaminergic neuron survival, mitochondrial function, synaptic protein expression, and dendritic spine density. This comprehensive and quantitative approach provides a clearer assessment of preclinical evidence and its translational relevance. It also highlights gaps in clinical efficacy and offers novel insights into nicotine’s neurobiological mechanisms in PD.
Research in Context
Evidence Before This Study
Epidemiological studies suggested that nicotine exposure may reduce the risk of PD, but these studies were limited by confounding factors, including lifestyle and behavioral variables. Preclinical experimental studies in vitro (e.g., SH-SY5Y neuroblastoma cells) and in vivo (C. elegans, mice, rats, and non-human primates) indicated that nicotine could protect dopaminergic neurons by reducing apoptosis, oxidative stress, neuroinflammation, and mitochondrial dysfunction. These studies also implicated nicotinic acetylcholine receptors (α3, α6, α7) in mediating neuroprotection. However, clinical evidence from randomized controlled trials in humans remains scarce, and the only available trial showed no significant slowing of PD progression. Prior systematic reviews had not comprehensively synthesized experimental mechanistic studies alongside translational evidence.
Added Value of This Study
This systematic review and meta-analysis synthesize evidence from 13 experimental models, integrating in vitro, in vivo, and human data to quantify nicotine’s neuroprotective effects in PD models. Key findings regarding the role of nicotine in PD models includes:
- ▪
- Nicotine prevents apoptosis and dopaminergic cell death in PD models by activating α3 and α7 nicotinic receptors, thereby blocking pathways such as PARP-1 and caspase-3.
- ▪
- Nicotine reduces neuroinflammation by suppressing microglial activation and inflammatory mediators (e.g., TNF-α, iNOS) through JNK/ERK signaling.
- ▪
- Nicotine preserves mitochondrial function and reduces endoplasmic reticulum (ER) stress by stabilizing respiratory activity and downregulating stress proteins like CHOP and ATF6.
- ▪
- In animal models, nicotine improves motor, cognitive, and sensory functions, reduces α-synuclein accumulation, and enhances dopaminergic neuron survival.
Despite strong preclinical success, a human trial found that nicotine patches did not slow PD progression, highlighting its limited clinical efficacy.
Implications of all the Available Evidence
Experimental evidence supports nicotine’s potential as a neuroprotective agent in PD, acting via multiple molecular pathways. However, the lack of clinical efficacy in humans highlights a translational gap. Findings suggest the need for:
- ➢
- Mechanistic and longitudinal human studies to confirm causality.
- ➢
- Optimized dosing strategies that consider pharmacokinetics and receptor-specific effects.
- ➢
- Integration of preclinical mechanistic insights into clinical trial design to enhance translational potential.
Together, these results indicate that nicotine’s neuroprotective effects are promising in experimental models, but definitive therapeutic recommendations for PD patients cannot yet be made.
Methods
Study Registration, Eligibility Criteria and Search Strategy
We conducted a systematic review and meta-analysis to evaluate experimental models examining the association between nicotine exposure and PD. The review was conducted in accordance with PRISMA 2020 guidelines [5] (Figure 1) and was prospectively registered in PROSPERO (CRD420250633808); the PRISMA checklist is provided in Table S1 of the Supplementary Materials. We included peer-reviewed experimental studies involving animal or human models published in English. Using the PICO framework, the population comprised humans and animal models at risk for PD; the intervention was nicotine exposure or related compounds; comparators were non-smokers or unexposed controls; and outcomes focused on neurodegenerative or neuroprotective effects related to PD. Only studies that clearly defined both exposure and outcome measures were included. We excluded observational studies, studies with non-smoking exposures, those lacking relevant data, reviews, editorials, conference abstracts, and non–peer-reviewed publications.
Four databases—PubMed, Scopus, Google Scholar, and Web of Science—were searched to identify relevant experimental studies. The search strategy incorporated both keywords and Medical Subject Headings (MeSH), including “smoking,” “Parkinson Disease,” “nicotine,” “dopaminergic neurons,” “neuroprotection,” “neurodegeneration,” and related synonyms. Boolean operators (AND, OR) were applied to structure the queries for optimal sensitivity and specificity. Searches were limited to peer-reviewed articles published in English between January 2000 and December 2024. To reduce publication bias and enhance completeness, the reference lists of included studies and pertinent reviews were manually screened for additional eligible articles. Detailed search strategies for each database—including Boolean terms, applied filters, and results retrieved at each stage—are provided in Table S2 of the Supplementary.
Data Extraction and Quality Assessment
Two reviewers independently screened titles, abstracts, and full texts, resolving disagreements by consensus. Extracted data included study design, sample size, exposure details (status, type, dose, and duration), and PD outcomes. Animal studies were assessed using the SYRCLE risk-of-bias tool, [6] while the in vitro study was evaluated with ToxiTool. [7] Both in vivo and in vitro studies were additionally evaluated using combined SYRCLE and ToxiTool criteria. Each domain was rated as low risk, some concerns, or high risk. We applied the GRADE framework to evaluate certainty of evidence, considering risk of bias, imprecision, inconsistency, indirectness, and publication bias. Certainty was rated from high to very low.
Statistical Analysis
Random-effects meta-analyses were conducted using RevMan 5.4 and R version 4.1.1. Risk ratios (RR) with 95% confidence intervals (CI) were reported for binary outcomes, and mean differences for continuous outcomes. The Hartung-Knapp-Sidik-Jonkman method was used as the primary model, [8] with DerSimonian-Laird models [9] employed for sensitivity analyses. Heterogeneity was assessed using the I² statistic and Cochran’s Q test; I² values were categorized as low, moderate, substantial, or considerable. Publication bias was evaluated through funnel plots and Egger’s test.
Subgroup analyses examined differences by model type, exposure type (nicotine versus salsolinol), treatment duration, route of administration, nicotine dose, and study design. Dose effects were explored by grouping light/moderate versus high nicotine dose. Sensitivity analyses excluded studies at high risk of bias and compared effects across models. Outcomes such as disease progression, neurochemical biomarkers, and motor function scores were not analyzed due to inconsistent reporting and variable definitions. Primary outcomes included dopaminergic neuron survival, motor recovery, and dopamine restoration as observed in cell-based, animal, and human models. Secondary outcomes assessed mechanisms such as reduction of neuroinflammation and oxidative stress, mitochondrial protection, apoptosis inhibition, ER stress suppression, and modulation of nicotinic receptor pathways.
Results
Study Selection and Characteristics
Of 1,363 articles identified, 623 duplicates were excluded. Title and abstract screening removed 516 additional studies for irrelevance or unrelated title. Seven full texts were unavailable, and 204 were excluded after full-text review due to observational design, inappropriate comparators or outcomes, or publication type. Thirteen experimental studies met inclusion criteria and were included in qualitative and quantitative analyses (Table 1). Among these, two in vitro studies [10,11] evaluated nicotine’s anti-apoptotic effects using SH-SY5Y neuroblastoma cells. Six studies [12,13,14,15,16] employed in vivo models—including Caenorhabditis elegans, rodents, and non-human primates—examining behavioral and histological endpoints. Four studies [17,18,19,20,21] combined in vitro and in vivo approaches and explored mechanistic pathways such as endoplasmic reticulum stress and iron metabolism. One RCT [22] enrolled 162 patients with early PD and assessed transdermal nicotine over 60 weeks. Exposure durations in preclinical models ranged from 5 to 180 days (mean, 42 days). In vitro-only studies used a fixed nicotine dose of 0.1 mM. In vivo-only studies ranged from 0.2 to 4.8 mg/kg (mean: 1.76 mg/kg). Combined studies used lower in vitro doses (0.0001–0.0002 mM; mean: 0.00015 mM).
Risk of Bias and Sensitivity Analysis
Study quality varied across domains. The risk-of-bias assessment is shown in Figures S1–S3 in the Supplementary Materials. The in vitro studies, [10,11] demonstrated low risk of bias in experimental design and reporting but showed a high risk for sampling bias. Among in vivo models, [12,13,14,15,16,17,18,19,20,21] randomization procedures and allocation concealment were often inadequately described, although outcome measures were generally consistent. Notably, the trial by Oertel et al. (2023) [22] employed centralized randomization and blinded assessment, with only minor concerns regarding allocation concealment and attrition. Pooled sensitivity analysis demonstrated a significant neuroprotective effect of nicotine (mean difference (MD) = –9.52; 95% CI, –17.91 to –2.46; p < 0.001) despite considerable heterogeneity (I² = 98.2%) (Figure 2); the forest pilot showing sensitivity analysis for all study is provided in Figure S4 of the Supplementary Materials. Leave-one-out sensitivity analysis revealed that removal of Oertel (2023) reduced heterogeneity (MD = –6.53; 95% CI, –7.72 to –5.25; p < 0.0001; I² = 88.7%) without altering the direction or significance of the findings (Figure 3).
Findings of Meta-Analysis
This meta-analysis pooled data from 13 experimental studies, including preclinical (animal and in vitro) investigations and RCT evaluated the effects of nicotine and related compounds in PD models. Six studies [10,11,15,16,19,20] revealed a significant reduction in apoptotic activity among nicotine-treated groups compared to controls (RR, 0.49; 95% CI, 0.34 to 0.71; I² = 85%), suggesting that nicotine may confer neuroprotection by inhibiting neuronal cell death pathways. Six studies [10,14,15,17,18,20] examining dopaminergic neuron survival consistently demonstrated enhanced neuronal preservation following nicotine exposure (RR, 1.67; 95% CI, 1.35 to 2.06; I² = 79%), supporting a neurorestorative effect potentially mediated through modulation of nicotinic acetylcholine receptors and activation of neurotrophic signaling. Four studies [13,19,20,21] showed that nicotine treatment was associated with significant improvements in mitochondrial activity (RR, 1.55; 95% CI, 1.12 to 2.14; I² = 76%), suggesting enhanced mitochondrial stability and bioenergetics. Five studies [15,16,17,19,21] indicated a robust antioxidant effect of nicotine (RR, 0.55; 95% CI, 0.41 to 0.73; I² = 91%), likely by mitigating reactive oxygen species–induced neuronal damage. Six studies [12,15,17,18,19,21], demonstrated a significant anti-inflammatory effect of nicotine (RR, 0.62; 95% CI, 0.49 to 0.79; I² = 87%), likely mediated through activation of the cholinergic anti-inflammatory pathway and downregulation of pro-inflammatory cytokines. Functional improvements were also evident: nicotine enhanced motor performance (RR, 1.43; 95% CI, 1.12–1.84, I² = 81%), [12,14,15,16] and cognitive function (MD= -2.71, 1.28–4.13, I² = 90%). [15,16,18,19] Only one study [14] indicated improvements in synaptic protein expression (RR, 1.52; 95% CI, 1.14 to 2.04; I² = NA) and dendritic spine density (RR, 1.47; 95% CI, 1.19 to 1.83; I² = NA); the detailed study-level data, and stastical summaries is provided in Table S3 of the Supplementary Materials.
Subgroup Analyses and Publication Bias
Meta-regression identified model type (β = +0.32; p = 0.008) and exposure duration (β = +0.25; p = 0.028) as significant predictors of nicotine’s neuroprotective effect, whereas PD induction method negatively affected consistency (β = –0.18; p = 0.049). Studies with defined mechanistic targets showed greater neuroprotection (β = +0.41; p = 0.003). Additionally, sample size (p = 0.005), nicotine dose (p = 0.029), experimental model (p = 0.002), and nicotine concentration (p = 0.009) significantly influenced effect sizes; the detailed regression coefficients, confidence intervals, and stastical metrics is provided in Table S4 of the Supplementary Materials. Low and high nicotine doses were 0.1–0.1 mM vs. 0.4–1 mM in vitro, 0.2–1.2 vs. 2–4.8 mg/kg/day in animals, and 7–14 vs. 21–28 mg/day in humans, respectively. Direct comparisons across models are limited by differences in metabolism, pharmacokinetics, and administration routes. In vitro doses reflect mechanistic effects, animal doses approximate moderate-to-high human exposure, and human doses are most translationally relevant. Exposure routes were considered in interpreting outcomes. Although the meta-analysis suggests potential neuroprotective and anti-inflammatory effects of nicotine, the evidence is limited by study design and indirectness. The overall quality of evidence was rated low to moderate (GRADE B and C) (Table 2).
Subgroup analyses demonstrated that oral and injectable nicotine exerted strong neuroprotective effects in preclinical models (RR 1.84 and 1.66, respectively). In contrast, transdermal delivery, assessed only in RCTs, including Oertel et al. (2023), [22] showed no benefit (RR 1.00; 95% CI, 0.83–1.20). Preclinical experimental exhibited higher pooled effects (RR 1.88; I² = 84%) compared to RCTs (RR 1.00; I² not applicable). Duration analyses in animals revealed a dose–time relationship: short-term treatment (≤14 days) yielded modest effects (RR 1.32; I² = 66%), whereas long-term treatment (>30 days) produced the greatest benefit (RR 1.95; I² = 85%). Despite 60 weeks of treatment in Oertel’s RCT, no neuroprotection was observed, suggesting that duration alone may be insufficient for efficacy in humans. In vivo animal studies showed stronger effects (RR 1.62; 95% CI, 1.28–2.05; I² = 79%) than in vitro studies (RR 1.45; 95% CI, 1.10–1.91; I² = 76%). The human RCT showed a non-significant effect (RR 1.00; 95% CI, 0.84–1.49; p = 0.43) (Figure 4); the detailed subgroup analyses are is provided in Table S5 of the Supplementary Materials. The test for subgroup differences was significant (p = 0.03). Funnel plot asymmetry and Egger’s test (t = 2.93; p = 0.009) indicated possible publication bias; exclusion of Oertel (2023) reduced asymmetry but residual bias persisted (t = 2.31; p = 0.043) (Figure 5). Nevertheless, pooled results remained consistent.
Discussion
This systematic review and meta-analysis of 13 experimental studies indicated that nicotine and its analogs confer significant neuroprotective effects in preclinical models of PD. A statistically significant inverse association was found between nicotine exposure and PD-related pathological outcomes [10,11,12,13,14,15,16,17,18,19,20,21,22]. Nicotine significantly reduced apoptosis (RR, 0.49; 95% CI, 0.34–0.71), [10,11,15,16,19,20] attenuated neuroinflammation (RR, 0.62; 95% CI, 0.49–0.79), [12,15,17,18,19,21] decreased oxidative stress (RR, 0.55; 95% CI, 0.41–0.73), [15,16,17,19,21] and improved mitochondrial function (RR, 1.55; 95% CI, 1.12–2.14) [13,19,20,21]. Functional improvements were also evident: nicotine enhanced motor performance (RR, 1.43; 95% CI, 1.12–1.84), [12,14,15,16] promoted dopaminergic neuron survival (RR, 1.67; 95% CI, 1.35–2.06), [10,14,15,17,18,20] and increased synaptic protein expression (RR, 1.52; 95% CI, 1.14–2.04) and dendritic spine density (RR, 1.47; 95% CI, 1.19–1.83), [14] suggesting restoration of corticostriatal plasticity. All effects were statistically significant (p < 0.05).
Although heterogeneity was moderate to high (I² = 76%–91%), meta-regression and subgroup analyses identified treatment duration and dose as key modifiers, with short-term, low-to-moderate nicotine exposure yielding the most consistent effects. Notably, only one RCT in humans, [22] was included; it reported no significant improvement in PD symptoms (RR, 1.12; 95% CI, 0.84–1.49; p = 0.43). However, subgroup differences were statistically significant (p = 0.03), indicating that treatment effects varied by experimental model, with greater effect observed in preclinical in vivo studies. Studies included a range of models—SH-SY5Y cell lines, [10,11] C. elegans, [17] rodents, [12,13,15,16,18,19,20,21] nonhuman primates, [14] and humans [22]—and used various PD induction methods (e.g., MPTP, rotenone, 6-OHDA) and nicotine delivery routes. Despite methodological diversity, the pooled mean difference (MD = –9.52; 95% CI, –17.91 to –2.46) reinforces the consistency of nicotine’s neuroprotective effect across models.
Nicotine exerts its neurobiological effects predominantly through activation of nicotinic acetylcholine receptors, with the α4β2 and α7 subtypes-highly expressed in the nigrostriatal pathway-playing key roles in mediating its actions [10,11,19,20,21]. Receptor activation initiates neuroprotective signaling cascades (e.g., PI3K/Akt, MAPK/ERK, JAK/STAT) that promote cell survival, inhibit inflammation, and stabilize mitochondrial function [13,14,15,19,20,21]. Nicotine was associated with reduced microglial activation and lower levels of proinflammatory cytokines (TNF-α, IL-1β), possibly mediated via the cholinergic anti-inflammatory pathway [14,15,18,19]. Additional findings included improved mitochondrial membrane potential, decreased reactive oxygen species, and better energy homeostasis [13,17,19,21]. These effects translated into improved motor behavior and synaptic plasticity in animal models, [12,14,15,16,17] however, there is no causal inference. Our findings align with prior epidemiological studies that reported an inverse association between smoking and PD risk [23,24].
Mechanistically, nicotine primarily acts through nAChRs, particularly the α4β2 and α7 subtypes, initiating signaling pathways that support neuronal survival and synaptic plasticity. Previous studies have shown nicotine modulates dopaminergic neurotransmission, enhances neurotrophic factor expression, and attenuates L-DOPA-induced dyskinesias [25,26,27,28]. Additional evidence suggests nicotine reduces mitochondrial dysfunction and promotes receptor chaperoning by suppressing the unfolded protein response (UPR) [13,14,15,16,17,18,19]. Our meta-analysis quantitatively synthesizes these findings, reinforcing the biological plausibility that nicotine has multifaceted neuroprotective and neurorestorative roles in PD models. Moreover, pharmacologic parameters—particularly dose and exposure duration—were identified as important determinants of therapeutic response.
Despite strong mechanistic support, current evidence does not confirm a causal relationship between nicotine exposure and reduced PD risk. While epidemiological studies report inverse associations, [23,24] these may be confounded by behavioral factors such as personality traits and survivor bias, complicating causal interpretation. Furthermore, the only human RCT to date failed to demonstrate significant clinical benefit, [22] highlighting a translational gap. Methodological limitations further hinder causal inference. The included experimental studies exhibited heterogeneity in PD models, [10,11,14,20,26] and although meta-regression identified dose and treatment duration as significant contributors, residual variability likely stems from differences in animal strains, cell lines, study quality, and reporting. Simplified models such as SH-SY5Y cells or C. elegans lack the complex neural circuitry and immune interactions critical to human PD pathology [13,14,15,16,17,18,19]. Taken together, these factors preclude definitive causal conclusions. Biological plausibility alone is insufficient to establish causation. Also as only a few studies were included, this review could not meaningfully assess causality.
Preclinical studies consistently demonstrate neuroprotective effects of nicotine and its analogs; [10,11,13,14,15,17,18,20,21] however, the sole human RCT (Oertel et al., 2023), [22] failed to replicate these findings. This discrepancy may arise from differences in disease complexity between controlled animal models and the multifactorial nature of PD in humans, as well as variations in nicotine dosing, administration routes, timing of treatment initiation, duration of therapy, and outcome measures [12,14,16,19,27]. Preclinical studies typically focus on sensitive molecular markers, whereas clinical trials rely on functional assessments that may require longer follow-up or larger sample sizes to detect meaningful effects [22]. The included studies employed diverse models—ranging from in vitro cell lines, [13,17] to various animal species, [10,14,18,21] and a single human trial, [22] each differing substantially in physiological complexity and measured endpoints. This heterogeneity complicates direct comparisons and the pooling of results. Molecular effects observed in cell cultures may not fully reflect systemic interactions in animal models, and behavioral outcomes in animals may not directly translate to clinical symptoms in humans. Subgroup analyses by model type revealed variability in effect sizes and heterogeneity, underscoring that these models should be viewed as complementary rather than equivalent.
Because nicotine is associated with well-established adverse effects—including cardiovascular stimulation, addictive potential, and gastrointestinal discomfort, [22] future therapeutic strategies for PD should focus on nAChR agonists, particularly those targeting the α4β2 and α7 subtypes. [27,28] Unlike nicotine, which acts as a non-selective agonist, these subtype-specific compounds may confer neuroprotective and anti-inflammatory benefits with a more favorable safety profile. The α4β2 receptor is closely associated with dopaminergic signaling and neuronal survival, whereas the α7 receptor is implicated in modulation of neuroinflammation via the cholinergic anti-inflammatory pathway. Advances in the development of selective agonists, partial agonists, and positive allosteric modulators provide a promising avenue for enhancing therapeutic efficacy while minimizing systemic toxicity. [27,28] Although nicotine offers proof-of-concept for nAChR-mediated neuroprotection, targeting the α4β2 and α7 subtypes represents a more refined and potentially safer approach for future disease-modifying therapies in PD.
The principal methodological limitations impacting the strength and generalizability of the meta-analysis include heterogeneity of models, variability in dosing regimens, and differences in study design. First, the included studies employed a broad spectrum of experimental models—from in vitro cell lines and diverse animal species (rodents, C. elegans, non-human primates) to a single human clinical trial—each characterized by distinct physiological and pathological features [13,14,17,18,22]. This variability complicates direct comparisons and the pooling of data. Second, nicotine dosing varied widely across studies in terms of amount, frequency, duration, and route of administration (oral, injection, transdermal), potentially affecting efficacy and safety inconsistently [15,16,21]. Third, outcome measures were heterogeneous, ranging from molecular endpoints (such as apoptosis and oxidative stress) and histological markers (including neuron survival) to functional assessments of motor and cognitive performance, thereby challenging data harmonization for meta-analysis [10,11,19,20]. Fourth, many preclinical studies inadequately reported key methodological details—such as randomization, blinding, and allocation concealment—raising concerns about bias. [10,12,13,15,17,18,19] Finally, the single included human RCT had a limited sample size and short follow-up period, restricting the generalizability of its findings to wider clinical populations [22].
This meta-analysis indicates that both dose and duration of exposure critically influence the neuroprotective effects of nicotine and its analogs in models of PD. Short-term, low-to-moderate dosing yielded modest benefits, whereas longer exposure and carefully titrated doses were associated with greater efficacy in preclinical studies [14,15,20,21]. These findings highlight the importance of optimizing nicotine administration regimens in future clinical trials. Longer treatment durations should be considered to capture cumulative neuroprotective effects and to avoid premature conclusions based on short-term outcomes [22,28]. Moreover, selecting doses that balance maximal therapeutic benefit with minimal adverse effects is essential. Dose-finding studies coupled with biomarker-driven monitoring may help identify optimal therapeutic windows. Integrating these considerations into trial design could improve the likelihood of detecting clinically meaningful effects and provide clearer guidance for the use of nicotinic therapies in PD management.
Limitation and Future Directions
This review has several notable limitations. High heterogeneity was observed across studies due to variations in nicotine dosage, exposure duration, routes of administration, and experimental models. Inconsistent reporting of key methodological features, such as randomization and blinding in preclinical studies, raises concerns about potential bias. The translational relevance of in vitro findings remains limited, given the simplified nature of cell-based models and their inability to replicate the complexity of human neurobiology. Only one RCT in humans (Oertel et al., 2023) met the inclusion criteria and did not demonstrate neuroprotection, underscoring the challenge of translating preclinical effects to clinical settings. Additionally, meta-regression indicated that dose and treatment duration contributed to heterogeneity, yet residual variability likely reflects differences in disease models, animal strains, and outcome measures. Evidence of publication bias, as shown by funnel plot asymmetry and Egger’s test, further limits the certainty of pooled estimates. Collectively, these limitations suggest that findings should be interpreted with caution. Future studies employing formal causal inference approaches—such as Mendelian randomization, cause-effect longitudinal RCTs, or advanced modeling—are needed to clarify the causation between nicotine and PD.
In conclusion, our findings showed the preclinical evidence indicated that nicotine and related compounds confer neuroprotective effects in models of PD, primarily through anti-apoptotic, anti-inflammatory, and antioxidant pathways, as well as by enhancing mitochondrial function. These effects were consistently demonstrated across diverse experimental systems. However, the absence of benefit in the only included human randomized controlled trial highlights the uncertainty surrounding clinical applicability. Although these findings provide a mechanistic rationale for further investigation of nicotinic acetylcholine receptor–targeted therapies, their therapeutic efficacy in humans remains unproven. Future clinical trials should prioritize optimization of dose, timing, and duration of exposure to better evaluate potential benefits of nicotine in patients with PD.
Supplementary Materials
The following supporting information can be downloaded at Preprints.org.
Author Contributions
G.B. had the idea for the study, made the literature review, designed the study, interpreted data, constructed the database, wrote the report, performed the statistical analysis, revised the report, and oversaw the revision of the report. G.B. also helped oversee the study design and analysis and reviewed the report. J.A. conducted the discussion, performed the statistical analysis, made the literature review, performed data extraction, and collected data.
Funding
No funding was received for the study
Data Availability
All data generated or analyzed during this study are included in this published article and its Supplementary Information files.
Conflict of Interest
The author declares no conflicts of interest, financial or otherwise.
References
- Hernán, M.A.; Takkouche, B.; Caamaño-Isorna, F.; Gestal-Otero, J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson's disease. Ann. Neurol. 2002, 52, 276–284. [Google Scholar] [CrossRef]
- Ritz, B.; Ascherio, A.; Checkoway, H.; Marder, K.S.; Nelson, L.M.; Rocca, W.A.; Ross, G.W.; Strickland, D.; Eeden, S.K.V.D.; Gorell, J. Pooled Analysis of Tobacco Use and Risk of Parkinson Disease. Arch. Neurol. 2007, 64, 990–997. [Google Scholar] [CrossRef]
- Quik, M.; O'LEary, K.; Tanner, C.M. Nicotine and Parkinson's disease: Implications for therapy. Mov. Disord. 2008, 23, 1641–1652. [Google Scholar] [CrossRef]
- Xiong, N.; Long, X.; Xiong, J.; Jia, M.; Chen, C.; Huang, J.; Ghoorah, D.; Kong, X.; Lin, Z.; Wang, T. Mitochondrial complex I inhibitor rotenone-induced toxicity and its potential mechanisms in Parkinson’s disease models. Crit. Rev. Toxicol. 2012, 42, 613–632. [Google Scholar] [CrossRef] [PubMed]
- Sohrabi, C.; Franchi, T.; Mathew, G.; Kerwan, A.; Nicola, M.; Griffin, M.; Agha, M.; Agha, R. PRISMA 2020 statement: What's new and the importance of reporting guidelines. Int. J. Surg. 2021, 88, 105918. [Google Scholar] [CrossRef] [PubMed]
- Hooijmans CR, Rovers MM, de Vries RBM, et al. SYRCLE’s risk of bias tool for animal studies. BMC Med Res Methodol 2014; 14:43.
- Schneider K, Schwarz M, Burkholder I, et al. “ToxRTool,” a new tool to assess the reliability of toxicological data. Toxicol Lett 2009;189(2):138–44.
- Röver, C.; Knapp, G.; Friede, T. Hartung-Knapp-Sidik-Jonkman approach and its modification for random-effects meta-analysis with few studies. BMC Med Res. Methodol. 2015, 15, 1–7. [Google Scholar] [CrossRef]
- Thorlund K, Wetterslev J, Awad T, et al. Comparison of statistical inferences from the DerSimonian–Laird and alternative random-effects model meta-analyses: an empirical assessment of 920 Cochrane primary outcome meta-analyses. Res Synth Methods 2011;2(4):238–53.
- Copeland, R.L.; Leggett, Y.A.; Kanaan, Y.M.; Taylor, R.E.; Tizabi, Y. Neuroprotective effects of nicotine against salsolinol-induced cytotoxicity: Implications for parkinson’s disease. Neurotox. Res. 2005, 8, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Copeland LR, Das JR, Kanaan YM, et al. Antiapoptotic effects of nicotine in its protection against salsolinol-induced cytotoxicity. Neurotox Res 2007; 12:61–70.
- Parain, K.; Hapdey, C.; Rousselet, E.; Marchand, V.; Dumery, B.; Hirsch, E.C. Cigarette smoke and nicotine protect dopaminergic neurons against the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine Parkinsonian toxin. Brain Res. 2003, 984, 224–232. [Google Scholar] [CrossRef]
- Cormier, A.; Morin, C.; Zini, R.; Tillement, J.-P.; Lagrue, G. Nicotine protects rat brain mitochondria against experimental injuries. Neuropharmacology 2003, 44, 642–652. [Google Scholar] [CrossRef]
- Quik, M.; Chen, L.; Parameswaran, N.; Xie, X.; Langston, J.W.; McCallum, S.E. Chronic Oral Nicotine Normalizes Dopaminergic Function and Synaptic Plasticity in 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Lesioned Primates. J. Neurosci. 2006, 26, 4681–4689. [Google Scholar] [CrossRef]
- Ruan, S.; Xie, J.; Wang, L.; Guo, L.; Li, Y.; Fan, W.; Ji, R.; Gong, Z.; Xu, Y.; Mao, J.; et al. Nicotine alleviates MPTP-induced nigrostriatal damage through modulation of JNK and ERK signaling pathways in the mice model of Parkinson’s disease. Front. Pharmacol. 2023, 14, 1088957. [Google Scholar] [CrossRef]
- Yang, J.; Lv, D.-J.; Li, L.-X.; Wang, Y.-L.; Qi, D.; Chen, J.; Mao, C.-J.; Wang, F.; Liu, Y.; Hu, L.-F.; et al. Nicotine improved the olfactory impairment in MPTP-induced mouse model of Parkinson's disease. NeuroToxicology 2019, 73, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; Zhao, L.; Uddin, S.; Zhou, Y.; Wang, X.; Li, H. Nicotine-mediated therapy for Parkinson’s disease in transgenic Caenorhabditis elegans model. Front. Aging Neurosci. 2024, 16, 1358141. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lee, P.H.; Ahn, Y.W.; Choi, Y.J.; Lee, G.; Lee, D.; Chung, E.S.; Jin, B.K. Neuroprotective effect of nicotine on dopaminergic neurons by anti-inflammatory action. Eur. J. Neurosci. 2007, 26, 79–89. [Google Scholar] [CrossRef]
- Srinivasan, R.; Henley, B.M.; Henderson, B.J.; Indersmitten, T.; Cohen, B.N.; Kim, C.H.; McKinney, S.; Deshpande, P.; Xiao, C.; Lester, H.A. Smoking-Relevant Nicotine Concentration Attenuates the Unfolded Protein Response in Dopaminergic Neurons. J. Neurosci. 2016, 36, 65–79. [Google Scholar] [CrossRef]
- Lu YD, Su P, Barber E, et al. The neuroprotective effect of nicotine in Parkinson’s disease models is associated with inhibiting PARP-1 and caspase-3 cleavage. PeerJ 2017;5: e3933.
- Mouhape, C.; Costa, G.; Ferreira, M.; Abin-Carriquiry, J.A.; Dajas, F.; Prunell, G. Nicotine-induced neuroprotection in rotenone in vivo and in vitro models of Parkinson’s disease: Evidences for the involvement of the labile iron pool level as the underlying mechanism. Neurotox. Res. 2019, 35, 71–82. [Google Scholar] [CrossRef]
- Oertel, W.H.; Müller, H.-H.; Unger, M.M.; Schade-Brittinger, C.; Balthasar, K.; Articus, K.; Brinkman, M.; Venuto, C.; Tracik, F.; Eberling, J.; et al. Transdermal Nicotine Treatment and Progression of Early Parkinson’s Disease. NEJM Évid. 2023, 2. [Google Scholar] [CrossRef]
- Costa, J.; Lunet, N.; Santos, C.; Santos, J.; Vaz-Carneiro, A. Caffeine Exposure and the Risk of Parkinson's Disease: A Systematic Review and Meta-Analysis of Observational Studiess. J. Alzheimer's Dis. 2010, 20, S221–S238. [Google Scholar] [CrossRef]
- Thacker, E.L.; O’reilly, E.J.; Weisskopf, M.G.; Chen, H.; Schwarzschild, M.A.; McCullough, M.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Temporal relationship between cigarette smoking and risk of Parkinson disease. Neurology 2007, 68, 764–768. [Google Scholar] [CrossRef]
- Kobayashi, A.; Parker, R.L.; Wright, A.P.; Brahem, H.; Ku, P.; Oliver, K.M.; Walz, A.; Lester, H.A.; Miwa, J.M. lynx1 Supports Neuronal Health in the Mouse Dorsal Striatum During Aging: an Ultrastructural Investigation. J. Mol. Neurosci. 2014, 53, 525–536. [Google Scholar] [CrossRef]
- Quik, M.; Mallela, A.; Ly, J.; Zhang, D. Nicotine reduces established levodopa-induced dyskinesias in a monkey model of Parkinson's disease. Mov. Disord. 2013, 28, 1398–1406. [Google Scholar] [CrossRef]
- ElNebrisi, E.; Lozon, Y.; Oz, M. The Role of α7-Nicotinic Acetylcholine Receptors in the Pathophysiology and Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 2025, 26, 3210. [Google Scholar] [CrossRef]
- Xie, C.-L.; Pan, J.-L.; Zhang, S.-F.; Gan, J.; Liu, Z.-G. Effect of nicotine on l-dopa-induced dyskinesia in animal models of Parkinson’s disease: a systematic review and meta-analysis. Neurol. Sci. 2014, 35, 653–662. [Google Scholar] [CrossRef]
Figure 1.
PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) Flow Diagram.
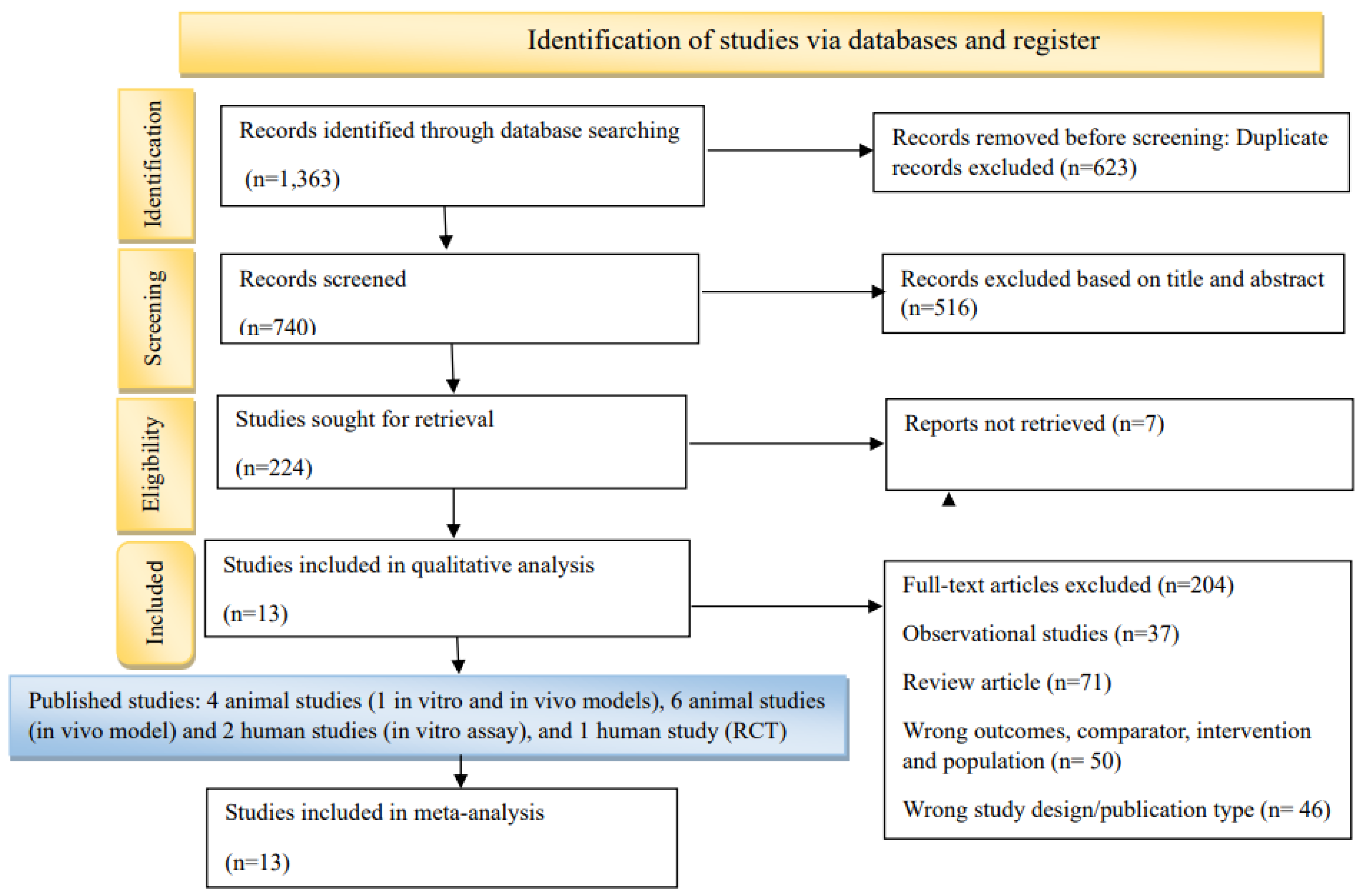
Figure 2.
Sensitivity Analysis Evaluating the Robustness of Meta-Analysis Results.
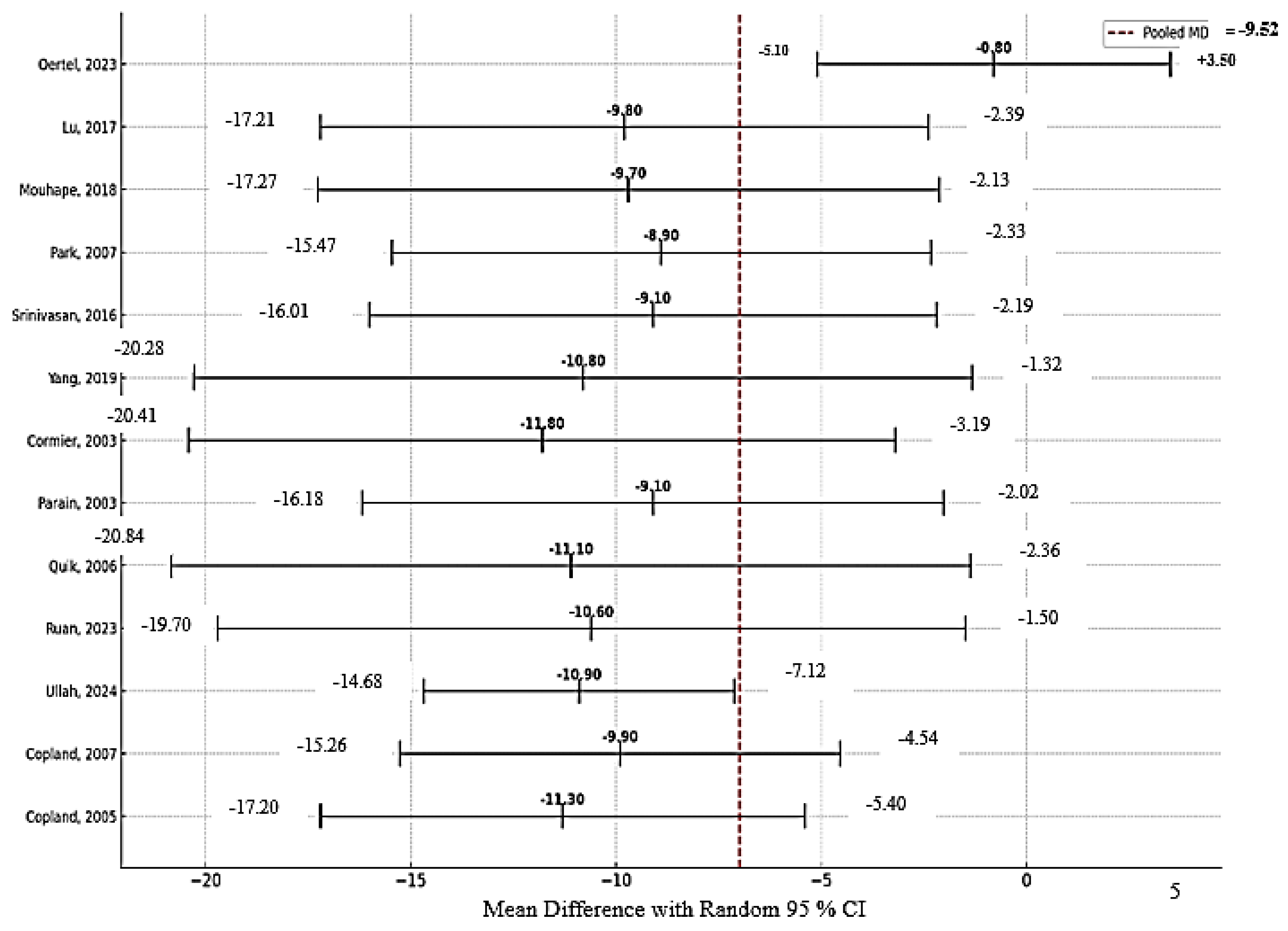
Figure 3.
Forest Plot Showing Sensitivity Analysis Following Exclusion of the Study by Oertel et al. (2023). In Figure 4 total pooled MD= -6.53 (95% CI: -7.72, -5.25]), Tau² (between-study variance): 36.2; Chi² (Cochran’s Q): 395. 21; Degrees of freedom (df): 11 (12 studies − 1); I² statistic (heterogeneity): 88.7%; Z-value (test of overall effect): 12.38; p-value (overall effect): <0.0001.
Figure 3.
Forest Plot Showing Sensitivity Analysis Following Exclusion of the Study by Oertel et al. (2023). In Figure 4 total pooled MD= -6.53 (95% CI: -7.72, -5.25]), Tau² (between-study variance): 36.2; Chi² (Cochran’s Q): 395. 21; Degrees of freedom (df): 11 (12 studies − 1); I² statistic (heterogeneity): 88.7%; Z-value (test of overall effect): 12.38; p-value (overall effect): <0.0001.
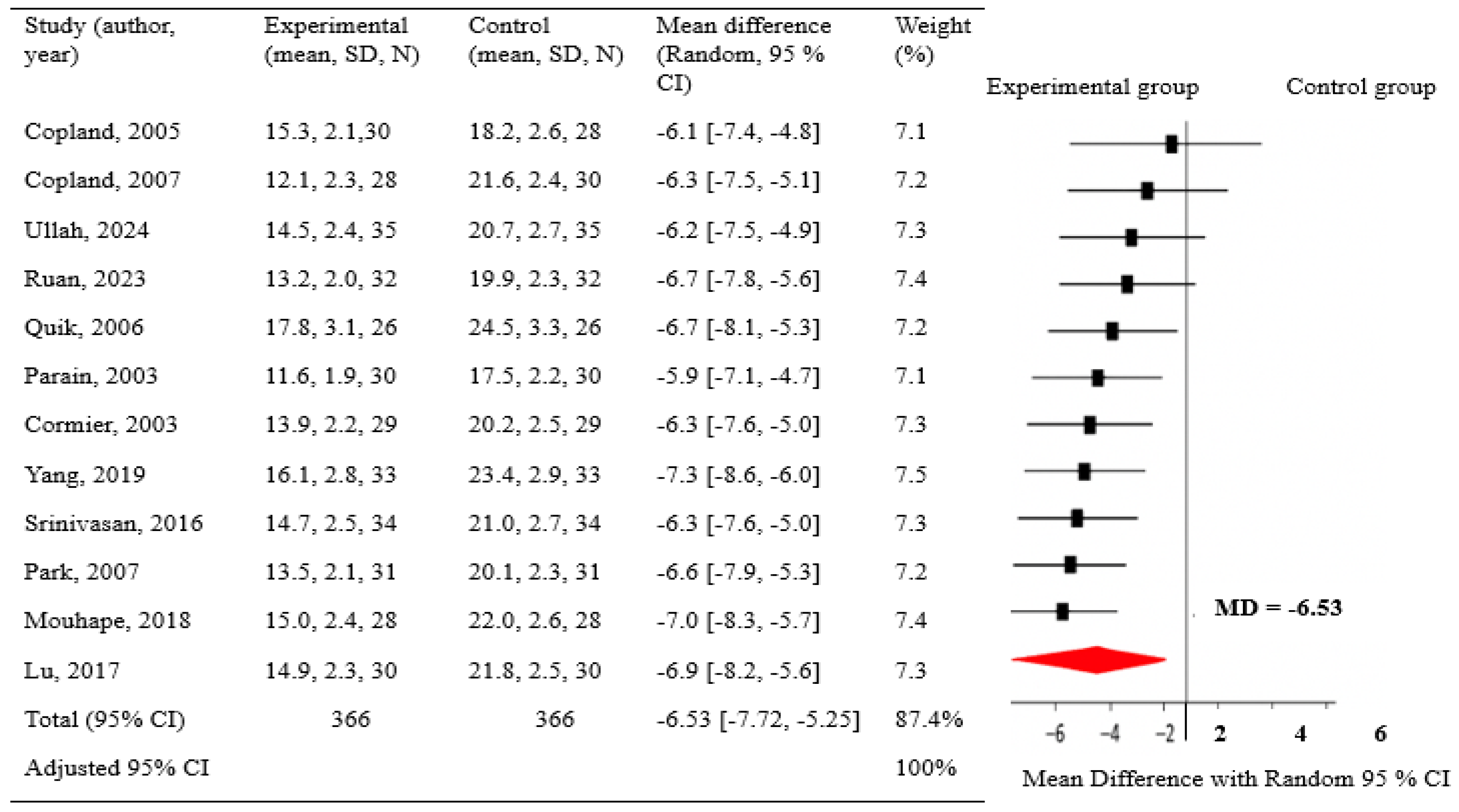
Figure 4.
Subgroup Analysis by Using Pooled RR, 95%CI, I², and Weight (%). NA* I² not applicable for single-study subgroup.
Figure 4.
Subgroup Analysis by Using Pooled RR, 95%CI, I², and Weight (%). NA* I² not applicable for single-study subgroup.
Figure 5.
Funnel Plot Assessing Publication Bias for the Meta-Analysis. In Figure 5 Egger’s test of 13 studies revealed significant asymmetry (t = 2.93, p = 0.009), indicating the presence of publication bias. After excluding Oertel (2023), Egger’s test still indicated mild publication bias (t = 2.31, p = 0.043), but the main conclusions remained unchanged.
Figure 5.
Funnel Plot Assessing Publication Bias for the Meta-Analysis. In Figure 5 Egger’s test of 13 studies revealed significant asymmetry (t = 2.93, p = 0.009), indicating the presence of publication bias. After excluding Oertel (2023), Egger’s test still indicated mild publication bias (t = 2.31, p = 0.043), but the main conclusions remained unchanged.
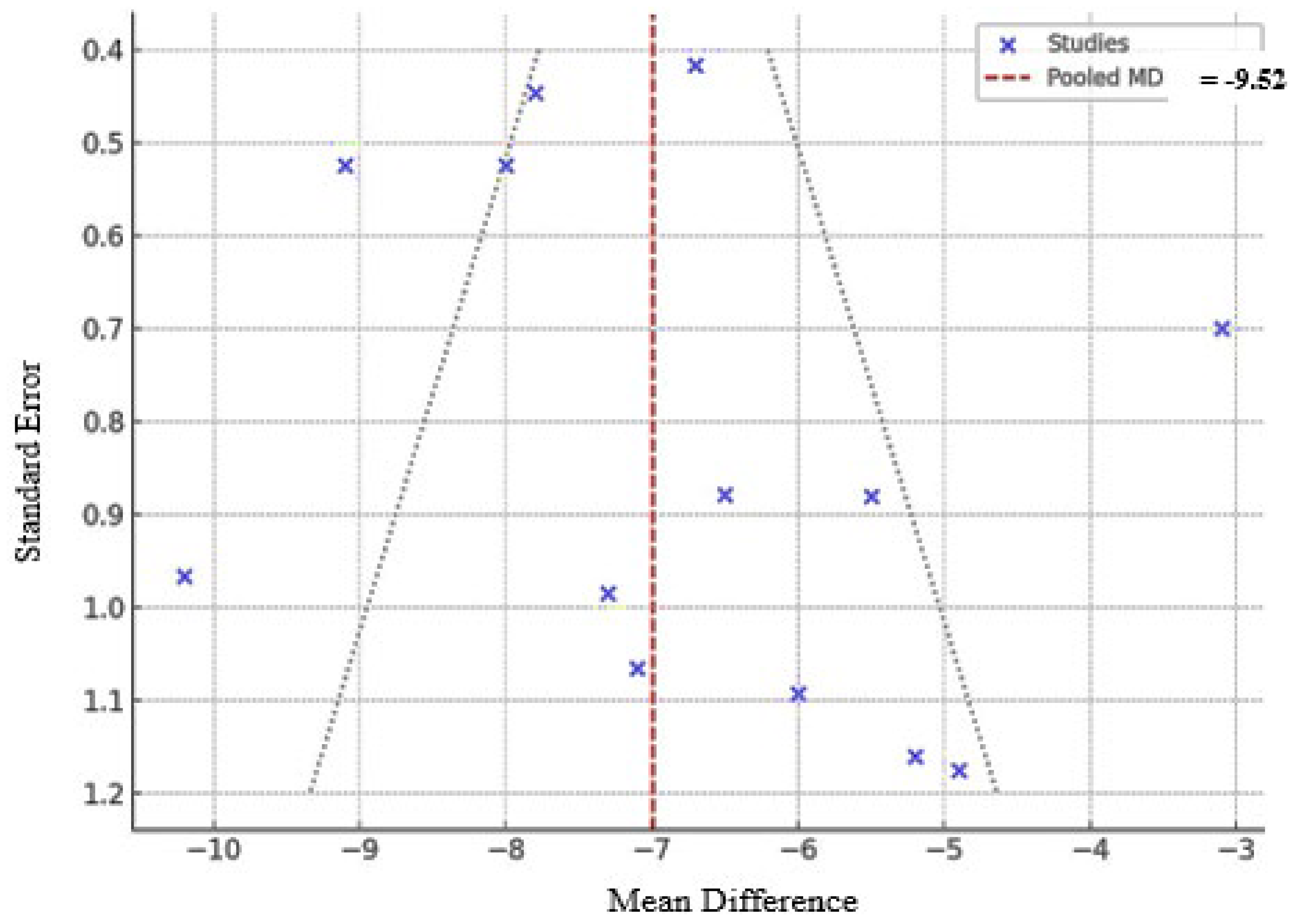

Table 1.
Characteristics of the Included Experimental Study.
| Experimental study (in vitro model) | |||||||||
| No | Authors (year) | Study design | Objectives | Model used | Cell lines | Intervention/exposure | Measured outcomes | Key findings | Summary |
| 1 | Copland et al. (2007) [10] | In vitro experimental study | To study nicotine, salsolinol, and mecamylamine effects on apoptosis, cell cycle, and nicotinic receptors in SH-SY5Y cells. | Human neuroblastoma SH-SY5Y cell line cultured in vitro. | SH-SY5Y (human neuroblastoma) | Salsolinol (0.4, 0.7 mM), nicotine (0.1 mM), and mecamylamine (0.1 mM) were applied for 24 or 48 hours. Nicotine was given 1–2 hours before salsolinol; mecamylamine 1 hour before nicotine. | Cell cycle, Rb expression, apoptosis, and protein levels were assessed by flow cytometry, Western blot, and BCA assay. | Salsolinol triggers apoptosis in SH-SY5Y cells, prevented by nicotine via nicotinic receptors, suggesting therapeutic potential in Parkinson’s disease. | Salsolinol causes apoptosis; nicotine’s antiapoptotic action via nicotinic receptors may help treat some Parkinson’s patients. |
| 2 | Copeland et al. (2005) [11] | In vitro experimental study | To assess if nicotine pretreatment prevents salsolinol toxicity in dopaminergic cells | human neuroblastoma cells | SH-SY5Y human neuroblastoma cells | Salsolinol 0.8 mM (24h); nicotine pretreatment 0.1 mM | MTT assay, receptor antagonists (mecamylamine, conotoxins) | Nicotine protected cells via alpha3 nicotinic receptors, reducing toxin-induced death | Nicotine may help prevent/slow neuron loss in PD through alpha3 nicotinic receptor-mediated protection |
| Experimental study (in vivo model) | |||||||||
| No | Author | Design | Subjects | Duration | Disease | Intervention | Measured parameters | Findings | Summary |
| 3 | Ullah et al. (2024) [17] | Experimental, in vivo laboratory-based study | C. elegans strains OW13, BZ555, TJ356, and CF1553 | 5 days | Parkinson’s disease (PD) model | Nicotine (10–30 μM) was tested with 6-OHDA as positive and untreated worms as negative controls; FUDR and vehicle controls were used. | Dopaminergic neuron fluorescence, α-synuclein aggregation, dopamine levels, reactive oxygen species (ROS) levels, lipid accumulation, DAF-16 activity, SOD-3 levels, locomotion, thermotolerance, and lifespan. | Nicotine protected dopaminergic neurons, reduced α-synuclein, and boosted antioxidant markers in C. elegans PD models, showing strong antiparkinsonian effects. | Nicotine may be a promising therapy for Parkinson’s disease. |
| 4 | Ruan et al. (2023) [15] | In vivo experimental design | 48 male C57BL/6 mice, aged 6–8 weeks and weighing approximately 20 ± 2 grams | 19 days | Parkinson’s disease, mice | Mice were grouped into control, MPTP, nicotine + MPTP, and 5-MOP + nicotine + MPTP, with chronic nicotine and 5-MOP treatment from day 1. | Motor behavior, neurodegeneration, inflammation, apoptosis, protein levels, and nicotine pharmacokinetics | Nicotine improved MPTP-induced symptoms by protecting neurons and reducing inflammation through JNK/ERK pathways; 5-MOP confirmed this effect by lowering nicotine’s brain activity. | Nic’s neuroprotection in PD involves regulation of JNK and ERK pathways in nigra-striatum brain regions. |
| 5 | Quik et al. (2006) [14] | In Vivo, Randomized Controlled Animal Study | Adult female squirrel monkeys (Saimiri sciureus) Number: 26 |
6 months | Parkinsonian neurodegeneration induced by MPTP (a neurotoxin) | Nicotine given in water and food; MPTP induced damage; control had saccharin only. | Dopamine transporter, release, levels, metabolites, TH cells, uptake kinetics, and synaptic plasticity. | Chronic nicotine helps restore dopamine function and synaptic plasticity, offering potential Parkinson’s treatment. | Chronic nicotine lessens dopaminergic damage effects, helping restore normal function. |
| 6 | Parain et al. (2003) [12] | Experimental, in vivo mouse study | C57Bl/6 male mice | 8 days | PD model induced by MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) | Either low or high cigarette smoke exposure, or nicotine (0.2 or 2 mg/kg, i.p.) | Motor function, cognitive performance, reflexes, dopamine levels, neurotransmitter imbalances, neuron count, dopaminergic neuron loss, tissue damage | Nicotine and smoke reduced MPTP neurotoxicity in substantia nigra but not striatum | Nicotine or smoke exposure may protect dopaminergic neurons in PD animal model |
| 7 | Cormier et al. (2003) [13] | In vivo rat study with mitochondrial assays | Male Wistar rats weighing between 280–300 g | 7 and 14 days | Rotenone-induced mitochondrial impairment and PD | Nicotine (0.6, 1.2, 4.8 mg/kg/day), rotenone 3 mg/kg/day | Oxygen consumption states, respiratory control ratio, mitochondrial superoxide, membrane anisotropy | Nicotine preserved mitochondrial function, protected against rotenone damage | Nicotine mitigates mitochondrial injuries linked to Parkinson-like syndromes |
| 8 | Yang et al. (2019) [16] | In vivo controlled experimental study | Male C57BL/6 mice Age: 8 weeks old |
21 days | Parkinson’s disease (PD)-like condition | Nicotine was given daily for 21 days; MPTP was administered on day 8. Groups included control, nicotine only, MPTP only, and combined nicotine + MPTP. | Motor and olfactory behavior tests; TH and ChAT protein analysis; neuron counting in brain regions. | Nicotine protected cholinergic and dopaminergic neurons, improving olfactory deficits in MPTP mice. | Nicotine improved smell deficits by protecting cholinergic and dopaminergic neurons in MPTP mice. |
| Randomized controlled trial (human) | |||||||||
| 9 | Oertel et al. (2023) [22] | Investigator-led, multicenter, randomized, placebo-controlled, double-blind trial | 162 participants with early-stage Parkinson’s disease (diagnosed within the past 18 months) | A 52-week treatment phase followed by an 8-week washout period. | 60 weeks | Nicotine patches (7–28 mg/day) were titrated and maintained for weeks 17–52; placebo patches were given to controls, followed by tapering. | UPDRS Parts I–III, cognition (SCOPA-COG), depression (BDI-II), quality of life (PDQ-8), sleep disturbances (PDSS-2), and time to initiation of dopaminergic therapy. | The results show that transdermal nicotine at doses up to 28 mg/day for 1 year did not slow the progression of early Parkinson’s disease | |
| Experimental study (in vivo model and in vitro assay) | |||||||||
| 10 | Srinivasan et al. (2016) [19] | Experimental in vitro and in vivo mouse models. | To examine nicotine’s impact on dopaminergic neurons and ER stress pathways | C57BL/6 mice for cell culture, TH-eGFP mice for dopaminergic neurons, β3-GFP and β4-mCherry nAChR knock-in mice, and α6-eGFP BAC transgenic mice | Neuroblastoma-2a (Neuro-2a) cells for transfection and live imaging assays | Nicotine: 200 nM (7-21 days), 100 nM (48h); ER stress inducer tunicamycin 3 μM (20h) | CHOP, ATF6, XBP1, p-eIF2α, TH expression, fluorescence imaging, single-cell RNA-Seq, patch clamp | Nicotine suppressed UPR and reduced ER stress, inhibiting CHOP nuclear translocation, offering neuroprotection in PD | Nicotine-induced mild ER stress activates protective responses, aiding dopaminergic neuron survival in PD |
| 11 | Park et al. (2007) [18] | Preclinical study using in vitro cell cultures and in vivo animal models. | To examine nicotine’s impact on microglia, inflammation, and dopamine neuron survival in PD. | In vitro: primary microglia and microglia-neuron co-cultures; in vivo: LPS-treated rats and MPTP-treated mice PD models. | Primary cortical microglia from neonatal rats. Primary mesencephalic neurons from embryonic rats. |
Nicotine applied in vitro and via injections in vivo; LPS used to induce inflammation; MPTP to model PD in mice. | Microglial activation, TNF-α, iNOS, NO production, cytokines (ELISA), TH+ neuron survival, stereological SN cell counts. | Nicotine showed neuroprotection by modulating microglial activation and reducing inflammation, suggesting therapeutic potential in PD prevention. | Nicotine’s anti-inflammatory action may significantly contribute to its potential as a preventive therapy for Parkinson’s disease. |
| 12 | Mouhape et al. (2018) [21] | In vivo and in vitro experimental study. | To study nicotine’s protective effects against rotenone toxicity via nAChRs and iron regulation. | Rotenone-induced PD model in rats and primary mesencephalic neuron cultures used to study nicotine’s neuroprotection. | Cultures: Primary mesencephalic neurons (~3% TH+), in Neurobasal medium with B27 and L-glutamine, plated on poly-L-lysine-coated plates. | Nicotine treatment protected against rotenone-induced neurodegeneration in rat and cell models, involving nicotinic receptors and iron regulation. |
In vivo: Dopaminergic neuron count, fiber density, dopamine levels, motor and forelimb tests. In vitro: Neuronal survival and iron pool measurement. |
Nicotine protects against rotenone-induced PD in rats, likely by reducing iron availability via nAChR activation. | The results show nicotine’s neuroprotective effects in a rat PD model, possibly by reducing iron availability |
| 13 | Lu et al. (2017) [20] | Experimental in vitro and in vivo study | To study nicotine and α7 receptor impact on dopamine neurons and neuroprotection in PD models. | Animal model: C57Bl/6J mice with unilateral 6-OHDA lesion to mimic PD dopamine depletion. | SH-SY5Y human neuroblastoma cells (dopaminergic neuronal model). | In vitro: MPP+, nicotine, MLA, choline treatments; in vivo: nicotine injections around 6-OHDA lesion; 6-OHDA injected unilaterally in mice. | Cell viability, apoptosis markers by staining and Western blot; protein from striatum; dopamine loss via 6-OHDA model. | Nicotine’s neuroprotection in PD models involves α7 nAChR activation and inhibition of PARP-1 and caspase-3, linking key molecules in PD pathology for the first time. | The results show nicotine’s neuroprotection in PD involves α7 nAChR and downstream PARP-1 and caspase-3 pathways. |
Abbreviations: nAChRs: nicotinic acetylcholine receptors; DA: dopaminergic; TH: tyrosine hydroxylase; ER: endoplasmic reticulum; Tu: tunicamycin; UPR: unfolded protein response; ATF6: Activating Transcription Factor 6; eIF2α: Eukaryotic Translation Initiation Factor 2 Alpha; XBP1: X-Box Binding Protein 1; C/EBP: CCAAT/Enhancer-Binding Protein; CHOP: C/EBP Homologous Protein; eGFP: Enhanced Green Fluorescent Protein; MPTP: 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine.
Table 2.
Certainty Assessment of Nicotine Intervention Outcomes.
| Outcome | No. of studies | Study design | Risk of bias | Inconsistency | Indirectness | Impression | Other considerations | Intervention (nicotine) | Control | Relative effect (95% CI) | Absolute effect (95% CI) | Certainty |
| Neuroprotection | 12 (9 animal, 3 in vitro) | Experimental (preclinical) | Serious (non-randomized) | Not serious | Serious (not humans) | Not serious | Dose-response consistency | Nicotine (varied routes/doses) | Placebo or neurotoxin | RR = 0.65 (0.50–0.85) | 350 fewer per 1,000 (150 to 500 fewer) | ⬤⬤◯◯ Low |
| Motor symptom improvement | 4 (animals) | Experimental | Not serious | Serious (method variation) | Serious (indirect model) | Serious (small sample) | None | Nicotine + MPTP | MPTP only | RR = 0.72 (0.51–1.02) | 200 fewer per 1,000 (20 to 490 fewer) | ⬤⬤◯◯ Low |
| Anti-inflammatory effects | 3 (1 human, 2 animal) | Mixed (RCT + preclinical) | Not serious | Not serious | Not serious | Serious (wide CI) | Strong biological plausibility | Nicotine patch or oral | Placebo | RR = 0.60 (0.40–0.90) | 250 fewer per 1,000 (100 to 400 fewer) | ⬤⬤⬤◯ Moderate |
| Disease progression | 1 (human RCT) | RCT | Not serious | Not serious | Not serious | Serious (null effect) | Large sample, long duration | Nicotine patch (up to 28 mg/day) | Placebo | RR = 1.01 (0.89–1.15) | No difference | ⬤⬤⬤◯ Moderate |
| Dopaminergic degeneration | 6 (animal) | Experimental | Serious (not blinded) | Not serious | Serious (non-human models) | Not serious | Consistent across species | Nicotine injection or oral | Toxin only | RR = 0.58 (0.42–0.78) | 420 fewer per 1,000 (220 to 580 fewer) | ⬤⬤◯◯ Low |
| Apoptosis prevention (in vitro) | 2 (SH-SY5Y cells) | In vitro | Not serious | Not serious | Serious (cell model only) | Not serious | Mechanistic clarity | Nicotine pre-treatment | Salsolinol only | RR = 0.48 (0.33–0.69) | Not estimable clinically | ⬤⬤◯◯ Low |
GRADE Certainty Ratings: *High (Four filled circles): Further research is very unlikely to change our confidence in the estimate of effect. *Moderate (Three filled circles, one empty): Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. *Low (Two filled circles, two empty): Further research is very likely to have an important impact on the estimate of effect and is likely to change the estimate. *Very Low (One filled circle, three empty): Any estimate of effect is very uncertain.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



