Submitted:
28 October 2025
Posted:
29 October 2025
You are already at the latest version
Abstract
ATP hydrolysis drives essential processes across biology, from nucleic acid translocation and conformational switching to signal transduction. The GHKL ATPase family—DNA Gyrase B, Heat Shock Protein 90 (Hsp90), Histidine Kinases, and MutL homologs—shares a Bergerat-fold that couples nucleotide binding and hydrolysis to conformational changes, dimerization, and signaling. Despite their diverse roles, GHKL proteins rely on common ATP-dependent principles. Within this family, MutLalpha (MLH1-PMS2 in humans, Mlh1-Pms1 in yeast) is central to eukaryotic mismatch repair, where it provides the endonuclease activity needed for strand incision and coordinates interactions with other repair partners. MutLalpha exemplifies how the Bergerat-fold has been adapted to regulate DNA interactions, partner communication, and protein turnover on DNA. By examining MutLalpha through the lens of other GHKL proteins, we can clarify how ATP binding and hydrolysis drive its conformational dynamics, nuclease activation, and regulation within its pathway, highlighting how conserved mechanistic strategies are repurposed across biological systems.
Keywords:
DNA mismatch repair
; DNA repair
; ATPases
; MutL
; GHKL
1. Enigmatic Features of MutL Homolog ATPase Activity
Mismatch repair (MMR) is a conserved pathway that preserves genome stability by correcting replication errors, a process that depends on MutL proteins and their ATPase activity [1,2,3,4,5,6,7,8,9]. Mutation studies have long established that loss of this activity compromises MMR, yet how MutL proteins functionally use ATP binding and hydrolysis in repair remains unresolved. Unlike helicases or polymerases, which use ATP as fuel for processive motion, MutL’s ATPase activity appears to act primarily as a molecular signal, but how this signal licenses MutL’s activities is not well understood. MutL belongs to a broader ATPase family whose shared mechanisms may provide insight into how it uses ATP in MMR.
The MMR pathway prevents the accumulation of mutations that could compromise genome integrity, by removing and resynthesizing mispaired nucleotides that escape polymerase proofreading. The pathway begins when a MutS homolog scans DNA for mispaired bases and, upon recognition, recruits a MutL homolog to direct downstream processing [10]. In Escherichia coli, MutL acts as a molecular matchmaker, linking MutS to the endonuclease MutH, which incises hemimethylated GATC sites on the newly synthesized strand. In contrast, many bacteria, including Bacillus subtilis, and all eukaryotes lack MutH and instead rely on MutL homologs with intrinsic endonuclease activity to introduce nicks into the nascent strand, which may already contain discontinuities to potentially serve as strand discrimination marks [11,12,13,14,15,16]. In all cases, the nick generated by MutH or MutL provides an entry point for mismatch excision and subsequent resynthesis by a DNA polymerase [10,11,17,18].
Mismatch excision in E. coli is carried out by exonucleases (ExoI, VII, X, or RecJ) and the UvrD helicase [19,20,21]. In eukaryotes, mismatch removal has been proposed to occur through at least three pathways, including: (i) excision by EXO1 followed by gap filling by Pold or Pole; (ii) strand displacement synthesis by Pold coupled with flap cleavage by a flap endonuclease (FEN1 in humans, Rad27 in yeast); or (iii) iterative nicking by MutLα (a heterodimer of the MutL homologs MLH1 with PMS2 in humans, or Mlh1 with Pms1 in yeast) followed by oligonucleotide removal through strand displacement synthesis or excision by EXO1 [10,22,23] (Figure 1).
A central unresolved question in MMR is how MutL and MutLα’s essential, but not well understood, ATPase activity contributes to repair events. Biochemical studies have shown that nucleolytic B. subtilis MutL and eukaryotic MutLα can nick DNA nonspecifically in vitro even without ATPase activity [24,25,26], whereas in vivo, MutLα ATPase activity is indispensable [5,6,7,8,9]. This discrepancy suggests that ATP hydrolysis may not be strictly required to catalyze the incision itself but could play multiple roles in the repair context. For example, ATP could play a role in stabilizing MutL/MutLα’s association with DNA or partner proteins, licensing or positioning its endonuclease activity at the correct strand or site, and/or enabling recycling and reset to permit multiple rounds of nicking for mismatch removal.
Insight may come from its membership in the GHKL ATPase family, named for DNA Gyrase B, Heat Shock Protein 90 (Hsp90), Histidine Kinases, and MutL, whose members share a conserved ATP-binding domain structure [27,28]. Across these otherwise diverse proteins, ATP binding and hydrolysis are consistently used to orchestrate conformational switching, dimerization, and pathway signaling. Viewing MutLα through this broader GHKL paradigm provides a framework for identifying the general principles by which ATP hydrolysis drives its roles in eukaryotic mismatch repair.
1.1. MutL Homolog Architecture and ATPase-Driven Conformational Changes
The MMR pathway and MutL proteins are largely conserved across biology. Members of the MutL family function primarily as dimers, forming homodimers in bacteria and, in eukaryotes, heterodimers in which MLH1 pairs with PMS2 (in mammals) or Pms1 (in yeast) as defined above to form MutLα, with PMS1 (in mammals) or Mlh2 (in yeast) to form MutLβ, or with MLH3 to form MutLγ. The nomenclature can be confusing: human PMS2 is orthologous to yeast Pms1, while human PMS1 is orthologous to yeast Mlh2. Among these complexes, MutLα is the major contributor to eukaryotic MMR [2,10,30,31,32,33,34,35]. Across organisms, each MutL homolog subunit contains a globular amino (N)-terminal domain and a globular carboxy (C)-terminal domain connected by an intrinsically disordered linker (Figure 2A). The C-terminal domains are believed to be constitutively dimerized in the absence of nucleotide, forming a stable dimer [2,24,36,37,38,39,40,41,42]. In contrast, the N-terminal domains are separated in the apo state but dimerize upon ATP binding, a conformational switch thought to be essential for MMR initiation [4,5,39]. These N-terminal regions contain a conserved ATP-binding pocket amongst the GHKL family called the Bergerat-fold, which features a dynamic “lid” region (Figure 2A-C). Mutations to this fold disrupt conformational changes and elevate mutation rates, underscoring the critical role of ATPase activity and this signature fold [4,5,6,43,44].
Because MutL and MutLα’s functions require ATPase activity in vivo, multiple experimental approaches have probed how ATP binding stabilizes and reorganizes the protein. The role of ATP in MutL stabilization was first shown by crystallography in E. coli, where fragments of MutL could only be crystallized in the presence of nonhydrolyzable ADP-PnP but not in the apo form, indicating nucleotide binding may be required to order the ATPase domains [40,46]. Proteolysis assays reinforced this hypothesis, where ATP binding stabilized MutL proteins, in Saccharomyces cerevisiae, Thermus thermophilus, and humans against cleavage, which is consistent with ATP-induced compaction [5,45,47,48].
Global rearrangements in MutL proteins were also observed in the presence of ATP. Full-length E. coli MutL eluted later from size-exclusion columns when nucleotide-bound than when in the apo form, which may be consistent with compaction into a clamp-like state [39,49]. Evidence of ATP-dependent compaction or clamp closure is also supported by biochemical, structural, and single molecule data [50,51]. Additionally, using single molecule atomic force microscopy experiments, which directly visualized MutL homologs were directly visualized in open, one “arm” closed states (where one subunit was compacted), and semi- and fully condensed states, with ATP binding shifting the equilibrium toward closed, condensed conformations [45,52] (Figure 2D).
Genetic and mechanistic evidence links these structural transitions to repair fidelity. In E. coli, ATPase-deficient mutants failed to close the clamp, abolished hydrolysis, and exhibited elevated mutagenesis [2]. In yeast, deletion of linker segments thought to mediate clamp opening and closing likewise reduced ATPase activity, impaired nuclease cycling, and resulted in increased mutation rates [25,53]. The functional significance of these transitions was tested more directly in engineered yeast MutLα complexes with chemically inducible dimerization domains. When artificially “locked” in a closed state, the complexes displayed elevated ATPase and nuclease activity, whereas variants unable to close showed nuclease defects. Despite opposite effects on biochemical activity, both perturbations led to increased mutagenesis in vivo, highlighting that regulated cycles of clamp opening and closure are critical for MMR [54].
A recurring feature of MutL is its asymmetric use of ATP. Even though bacterial MutL proteins are homodimers, the two sites appear nonequivalent: one protomer tends to stabilize the closed clamp, while the other hydrolyzes ATP more rapidly [2,5,39,40]. This asymmetry may act as a timing mechanism, ensuring that ATP binding signals clamp closure while hydrolysis in only one subunit is sufficient to destabilize the complex. In eukaryotes, the asymmetry is even more pronounced. Using yeast MutLα, it has been demonstrated that Mlh1 binds ATP with higher affinity and primarily drives clamp formation, whereas Pms1 has faster turnover (Figure 2D). In vivo, this division of labor has clear consequences because mutations impairing ATP binding in Mlh1 sharply increase mutation rates, while equivalent mutations in yeast Pms1 have more modest effects, demonstrating that the two subunits are not interchangeable [5].
Together, these findings suggest that ATP binding and hydrolysis regulate MutL and MutLα’s clamp-like transitions and thereby influence repair efficiency. However, the precise relationship between these conformational changes and specific repair steps remains unclear. It is not clear whether ATP primarily licenses a productive repair state, resets the clamp after engagement, or balances both functions.
1.2. Higher-Order MutL Assemblies and Partner Interactions
Beyond the dimeric clamp, MutL homologs can form higher-order assemblies. In E. coli, size-exclusion chromatography revealed ~300 kDa species in the absence of ATP, consistent with multimers, while ATP analogs shifted the protein to ~139 kDa dimers [40,55,56]. Crosslinking experiments similarly confirm apo multimers, while ADP-PnP stabilizes dimers [39]. DNA binding assays reveal cooperative behavior. Both E. coli MutL and S. cerevisiae MutLα bind longer DNA with higher apparent affinity and sigmoidal isotherms [57,58]. Sucrose density gradient sedimentation analysis also suggests that there is a surplus of human MutLα relative to the mismatch recognition protein MutSα on mismatched DNA [59]. Functionally, MutLα and MutLγ show increased endonuclease activity on longer substrates over shorter DNA [60,61]. Direct visualization by atomic force and electron microscopy experiments corroborate this model, with multiple MutLα/MutLγ particles binding to a single DNA molecule in yeast and human systems, consistent with cooperative assembly [58,61,62].
Although ATP clearly stabilizes dimerization, its influence on DNA binding appears more nuanced. For yeast MutLα, ATP marginally reduces affinity for DNA, suggesting that its primary role may be to regulate conformational state and partner interactions rather than DNA engagement itself [25,26]. DNA binding assays with E. coli MutL show somewhat increased affinity in the presence of nonhydrolyzable ATP analogs, consistent with nucleotide-driven stabilization of its DNA-bound state, with hydrolysis potentially releasing the protein from DNA [40]. For both bacterial MutL and eukaryotic MutLα, DNA binding maps to the N-terminal domains, which also harbor the ATPase sites, highlighting a possible close physical and functional coupling between nucleotide state and DNA interactions [40,41,63].
Consistent with this, MutL proteins participate in higher-order complexes with other MMR factors. Interactions with MutS proteins appear to be nucleotide-dependent but do not strictly require MutL’s intrinsic ATPase activity; even ATPase-deficient variants retain capacity to associate with MutS complexes, though these interactions may be less efficient in some cases [8,29,34,43,55,56,64,65,66,67,68,69,70]. These data do not resolve whether MutL proteins undergo an ATP-dependent step immediately after recruitment to DNA by MutS proteins. A possibility given that DNA itself stimulates E. coli MutL and human and yeast MutLα’s ATPase activity [25,40,71]. In contrast, interactions with MutH and processivity clamps are more directly tied to MutL’s nucleotide state. In E. coli, MutL’s ATPase activity stimulates MutH, while in yeast and human systems, MutLα’s endonuclease activity is largely inactive until stimulated by PCNA, which also enhances MutLα’s ATPase activity [13,14,15,25,26,65,71]. In B. subtilis, MutL functions are similarly regulated by the b clamp [2,72,73].
Together, these observations suggest that ATP not only governs MutL’s clamp dynamics but also modulates its interactions with key partners such as MutH, and PCNA. What remains unresolved is how these roles are integrated on DNA: does ATP binding primarily regulate MutL’s interactions with partner proteins, or do partner proteins reshape MutL’s ATPase cycle to control when and where the protein acts along a DNA contour? Framing MutL’s ATPase activity in the broader context of the GHKL family, where ATP consistently acts as a molecular switch to regulate complex processes, may help clarify how these multiple functions coordinate in MMR.
2. ATP Hydrolysis and the GHKL ATPase Paradigm
The questions surrounding how ATP regulates MutL’s clamp cycling and partner interactions can be better framed in the context of the broader GHKL family. In this family, ATP binding and hydrolysis serve as molecular switches that trigger conformational transitions, licensing active states, and resetting them for subsequent rounds. GHKL proteins are unified by a Bergerat-fold ATP-binding motif, whose hallmark “lid” region opens and closes to regulate nucleotide access and product release, linking nucleotide state to downstream activity (reviewed in [27,28]) (Figure 2C).
Despite carrying out diverse cellular functions, including DNA topology control, protein folding, signal transduction, and MMR, GHKL proteins share the strategy of ATP-dependent clamp cycling. Structural variations in the ATP-binding pocket and lid control how each member translates nucleotide binding and hydrolysis into distinct outputs, with ATP binding generally driving large conformational rearrangements that enable dimerization, higher-order assembly, and activity [39,40,45,57,65,67,68]. These ATP-dependent transitions allow GHKL proteins to propagate conformational states across protein complexes and coordinate pathway-specific events, which may provide a useful framework for understanding how MutLα uses ATP in MMR.
3. Mechanistic Insights from Other GHKL Family Members
3.1. DNA Gyrase B
Among GHKL proteins, DNA gyrase B (GyrB) is the most extensively studied and provides a well-defined example for how ATP hydrolysis can coordinate large conformational changes with catalytic turnover. GyrB is a subunit of bacterial DNA gyrase, a type II topoisomerase composed of two DNA gyrase A (GyrA) and two GyrB subunits [74,75]. During replication and transcription, torsional strain ahead of the replication fork generates positive supercoiling that can inhibit processivity [76,77]. DNA gyrase relieves this strain through a strand-passage mechanism involving transient cleavage of one DNA duplex (the G-segment), passage of a second duplex (the T-segment) through the break, and resealing. This introduces negative supercoils and maintains topological homeostasis [78,79] (Figure 3A).
ATP binding and hydrolysis by GyrB drive a conformational cycle that closely parallels MutL and MutLα’s clamp transitions. DNA is first bound by the GyrA subunit; upon binding two ATP molecules, the N-terminal domains of GyrB dimerize, locking the enzyme into a closed state competent for strand passage [80,81]. A double-strand break is introduced into the G-segment, the T-segment is passed through, and the linking number of the DNA is altered [82,83,84,85]. After passage, the T-segment is released, and ATP hydrolysis at the GyrB N-terminal domains resets the enzyme for another round of catalysis. Biochemical studies support this model. FRET and proteolysis assays show that apo GyrB is an open clamp, ATP binding induces compaction into a closed state essential for DNA capture, and structural analyses confirm these transitions [80,85,86,87,88,89]. Pull-down assays with ATPase mutants further demonstrate that dimerization is strictly ATP-dependent. Once ATP is bound, the B-gate closes to capture the T-segment, and subsequent hydrolysis reopens the gate for recycling [90,91,92].
Substrate length also influences the cycle, highlighting another parallel with MutL proteins. Longer DNA fragments stimulate more efficient ATP hydrolysis [98]. MutL homologs likewise bind more stably to extended substrates and show enhanced endonuclease activity on long DNA [58,60,61]. The underlying reasons for this similarity may differ between proteins, though. For DNA gyrase, increased flexibility of longer DNA could enhance strand passage, whereas for MutL homologs, enhanced activity may involve multiple complexes bound on the same substrate or direct manipulation of DNA, for which there is recent evidence [60,62,99]. But, in both cases, ATP-driven conformational changes are regulated by DNA length, ensuring that enzymatic activity is coordinated with substrate context.
Together, these data suggest that ATP acts as a signal in GyrB where binding ATP licenses DNA capture by the gyrase, while hydrolysis resets the enzyme for successive rounds. This “capture-and-reset” cycle offers a compelling parallel to MutL and MutLα’s clamp-like dynamics, raising the possibility that ATP hydrolysis plays a similar role in resetting DNA-bound MutL and MutLα complexes during MMR.
3.2. Heat Shock Protein 90
Heat shock protein 90 (Hsp90) illustrates how the GHKL ATPase fold can be adapted to protein quality control rather than DNA transactions, yet it still deploys ATP in a way that may mirror MutL and MutLα’s activities. Hsp90 is a highly conserved molecular chaperone present in bacteria and eukaryotes, where it promotes folding of a wide range of client proteins under both basal and stress conditions [100,101,102,103]. Proper Hsp90 activity is essential for cell viability. Polypeptide clients that fail to engage productively are often degraded, leading to loss of critical cellular functions, and interactome studies underscore its role as a central hub that links ATP-driven conformational dynamics to proteome stability [104,105]. Hsp90 functions as a homodimer, with each monomer containing three domains: an N-terminal ATPase domain, a middle domain that binds clients, and a C-terminal dimerization domain [106,107,108].
ATP binding and hydrolysis drive large conformational transitions between open and closed states (Figure 3B). In the apo form, Hsp90 remains open, where it can bind to a client protein [95,109]. After client binding, Hsp90 will bind ATP inducing a closed conformation, with dimerized N-terminal domains that license client folding [100,109,110,111,112,113,114]. ATP hydrolysis acts as a molecular switch that primes Hsp90 for client protein release [95,115,116]. Following ATP hydrolysis, the N-terminal domains remain bound to ADP. The conformation of this ADP-bound state remains debated: some studies suggest ADP stabilizes a compact form, while others propose it represents an intermediate between the apo and fully closed states [111,117,118]. Nonetheless, the consensus is that the ADP-bound state promotes client release and the chaperone resets by reopening the dimer upon ADP release. Thus, ATP establishes the active state, while hydrolysis defines the reset step, following the same “capture-and-reset” logic observed in GyrB.
This ATP-driven cycle is strongly modulated by co-chaperones. Aha1, one of the best studied, stimulates ATPase activity by inducing conformational changes that accelerate dimer closure and thereby enhance client processing [119,120,121,122]. Functional studies using denatured firefly luciferase underscored the importance of this regulation. Here, inhibitors disrupting the Aha1-Hsp90 interaction reduced ATPase activity and impaired refolding capacity, directly linking nucleotide turnover to enzymatic function [120]. Enzymatic cycling between ATP and ADP in the binding site thus serves as a molecular signal that couples client engagement with release [117,118].
While MutL/MutLα and GyrB use ATP to regulate clamp cycles on DNA, Hsp90 similarly clamps polypeptide substrates with ATP binding allowing the protein to engage with the client, with subsequent hydrolysis resetting the chaperone. These parallels may underscore a unifying principle of the GHKL family in that ATP binding could establish an active state, while hydrolysis triggers release and reset.
3.3. Histidine Kinases
Histidine kinases represent the signaling branch of the GHKL family. They are primarily transmembrane proteins and form the sensory module of two-component systems, which are widespread in bacteria and also present in eukaryotes, where their mechanisms are less understood [123,124,125]. Through their N-terminal transmembrane domains, histidine kinases detect environmental changes such as nutrient availability, pH, oxygen, or light [126,127,128,129,130]. Upon stimulus detection, the cytoplasmic C-terminal domains, which harbor the Bergerat-fold, use ATP to autophosphorylate a conserved histidine [131,132]. The phosphoryl group is then transferred to a response regulator, usually a transcription factor, to modulate gene expression [133,134,135] (Figure 3C).
Histidine kinases typically function as asymmetric homodimers. Their N-terminal transmembrane helices form helical hairpins, while the cytoplasmic domains bind and hydrolyze ATP to drive phosphorylation [125,131]. Like other GHKL proteins, they undergo nucleotide-dependent conformational transitions. Notably, ion mobility-mass spectrometry of the histidine kinase ExsG, which assembles into a higher-order homohexamer rather than the canonical dimer, revealed distinct open and closed subunit states, with ATP binding favoring the open, active conformation [136]. NMR studies of light-responsive histidine kinases similarly showed that ATP stabilizes secondary structure. In that study, the authors found that when bound to ATP in an active state, the histidine kinase is structurally stabilized compared to an ADP bound state [137]. These findings highlight the conservation of ATP-driven conformational switching across GHKLs, but also the divergence in how conformations are linked to activity. In contrast to MutL or GyrB, where the closed state is generally the productive one, histidine kinases couple their open state to signaling.
Viewed in the broader GHKL context, histidine kinases illustrate several shared principles. ATP binding promotes structural rearrangements at the dimer interface, much like how MutL/MutLα may clamp DNA or Hsp90 clamps clients. ATP cycles also license and reset activity where binding enables autophosphorylation, and hydrolysis eventually returns the kinase to a sensing-competent state. What distinguishes histidine kinases is how these transitions are wired directly into signaling. Here, ATP binding not only drives conformational change but also enables transfer of the phosphoryl group as the signal itself, whereas in MutL proteins, GyrB, and Hsp90, ATP hydrolysis transmits conformational signals within protein-DNA or protein-client assemblies. In all cases, the Bergerat-fold found in GHKL proteins converts ATP binding and hydrolysis into a communication mechanism, adapted to the demands of each biological system.
4. MutLα in MMR: Old Puzzles, New Views
Despite decades of study, MutL/MutLα’s ATPase cycle and how it contributes to efficient MMR remains incompletely understood. Mutational studies leave no doubt that ATP is required, but the central mechanistic questions persist: does ATP binding primarily act as a licensing step that enables clamp closure and partner engagement, does hydrolysis function as a reset mechanism that disengages MutL/MutLα from DNA, or do both steps cooperate in a coordinated cycle? As outlined in Section 1, structural and functional studies accentuate these questions rather than resolve them, highlighting subunit asymmetry, higher-order assemblies, and context-dependent roles for ATP. Viewed through the broader GHKL paradigm, these features suggest that MutL proteins may use ATP at multiple points, both to establish active states and to reset them, making ATP hydrolysis a potential recurring signal rather than a single trigger in MMR.
Like other GHKL proteins, MutL homologs may use ATP as a licensing signal that activates their functions only in the proper context. In many family members, ATP binding is induced by specific contexts, such as DNA, partner proteins, or environmental signals, ensuring that activity is engaged only when appropriate. MutL may follow a similar logic, where its latent activities remain suppressed until interactions with DNA or downstream repair factors stimulate ATP binding, with subsequent hydrolysis promoting recycling on DNA. Viewed through this GHKL framework, past biochemical, structural, and biophysical data suggest a model in which eukaryotic MutLα employs ATP both to license activity and to drive turnover during MMR.
Structural and biochemical studies indicate that following mismatch recognition by MutSα or MutSβ, MutLα is recruited through contacts mediated by the MLH1 subunit [138,139,140]. There are conflicting models for whether this recruitment occurs directly at the mismatch site or away from it through sliding-clamp activity [3,50,62,70,141,142,143,144,145]. This recruitment, together with the asymmetric ATPase properties of the heterodimer (discussed in Section 1) and evidence that DNA binding stimulates ATP turnover, suggests a model in which MLH1 may serve as an anchor for the initiation complex (Figure 4). In such a model, ATP binding by MLH1 may stabilize MutLα on DNA, while PMS2/Pms1 remains poised to engage activating partners. Such asymmetric clamp closure parallels other GHKL proteins, including GyrB and Hsp90, where ATP binding locks the complex into an active conformation primed for function.
Upon interactions with PCNA, MutLα‘s endonuclease activity is stimulated through interactions between the PMS2/Pms1 subunit and PCNA [25,26,65,70,71]. In minimal biochemical systems containing MutLα, PCNA, and DNA, ATP is not required for nonspecific nicking [25,26]. However, it has not been explicitly tested whether ATP plays a more stimulatory or regulatory role in fully reconstituted systems. Following endonuclease activation, PMS2/Pms1 may itself bind ATP, consistent with evidence that PCNA stimulates MutLα ATPase activity. This ATP binding could act as a signal for enzymatic reset, paralleling other GHKL proteins in which ATP binding licenses catalytic activity and hydrolysis or nucleotide release resets the complex for another cycle.
The fate of MutLα after ATP hydrolysis remains unresolved. One model proposes that hydrolysis opens the clamp and dissociates the N-terminal domains, leading to complete release of the protein from DNA. In this case, MutLα leaves behind a nick as an entry point for excision or strand-displacement synthesis, or potentially recycles to the same mismatch site to introduce additional nicks that amplify the repair signal [22,146,147,148]. An alternative model may involve iterative nicking when multiple MutLα complexes are assembled on DNA. In this model, hydrolysis-induced resetting could release the protein that performed the incision while other MutLα proteins remain bound and become primed to nick again or all members of the complex recycle but remain bound to the DNA. This is at least partially supported by DNA binding assays with yeast proteins where ternary complexes putatively consisting of MutSα, MutLα, and PCNA persist on substrates with streptavidin-bumpered ends in the presence of ATP and magnesium [70]. A related possibility is that MutLα, alone or in complex with MutSα/β exploits sliding-clamp properties to reset without full dissociation, thereby facilitating repeated rounds of incision. Such a mechanism echoes broader themes across the GHKL family, where ATP functions not only as a license for activity but also as a reset signal that drives iterative, context-dependent cycles of function. At present, however, these models remain speculative, and more explicit experimental tests will be required to distinguish among them. It will also be important to determine whether different eukaryotic MutLα proteins vary in how they utilize ATP, and how these modes compare with bacterial MutL homologs and their subunits.
5. Broader Implications and Future Directions
Beyond the role of MutLα in canonical MMR, other MutL complexes also display ATP-dependent activities whose mechanistic basis remains unresolved. For example, genetic studies highlight the importance of ATP for MutLα during meiosis, yet biochemical work shows that MutLα can be activated on DNA junctions even in the absence of nucleotide [149,150]. This raises the question of whether ATP plays an analogous role for MutLγ as it does for MutLα. MutLβ is even less well understood, with its biological role in MMR and meiosis only beginning to be elucidated [32,35,151,152,153,154], but its conservation across eukaryotes suggests that ATPase activity may contribute to functions not yet fully defined. Considering these gaps through the broader GHKL framework provides a way to generate new hypotheses about how distinct MutL complexes adapt ATP usage to their specialized contexts.
More broadly, lessons from MMR can inform our understanding of other GHKL proteins beyond MutL homologs. Several GHKL ATPases act on chromatin: microrchidia (MORC) proteins use their ATPase domains in chromatin remodeling and gene regulation [155,156,157,158], and SMCHD1, which has also been proposed to adopt the shared GHKL ATPase fold, may employ clamp-like dynamics to reshape chromatin architecture [159,160,161]. Similarly, topoisomerase VI subunits such as the meiotic factor TOP6BPL retain the conserved GHKL lid and may couple ATP hydrolysis to conformational transitions and DNA processing in ways reminiscent of MutL [162]. Recent structural work has further revealed that the neurodegenerative disease-linked protein sacsin contains a Bergerat-fold very similar to Hsp90 proteins, which may suggest chaperone-like activities [163]. By explicitly comparing across this family, we can uncover both shared mechanistic strategies and unique adaptations that clarify a wide spectrum of biological pathways.
Advances in structural and computational tools now make it possible to probe these questions with unprecedented resolution. Cryo-EM and single-molecule approaches can directly visualize conformational states and DNA transactions, while deep-learning-based structure prediction platforms such as AlphaFold offer new opportunities to model elusive intermediates and higher-order assemblies. Integrating these approaches with classical genetics and biochemistry will be key not only to defining how ATP hydrolysis is wired into the diverse activities of known GHKL proteins, but also to uncovering conserved activities that may guide the identification of new family members and unanticipated biological roles.
Author Contributions
Writing—original draft preparation, J.M.P., C.M.M.; writing—review and editing, J.M.P., C.M.M; funding acquisition, C.M.M. All authors have read and agreed to the published version of the manuscript.
Funding
J.M.P. and C.M.M. supported by the National Institute of General Medical Sciences of the NIH (Grant R35GM142651).
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We thank all members of the Manhart lab for helpful discussions about the content of this review. We also thank the Department of Chemistry and the College of Science and Technology at Temple University for their support.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Lahue, R.S.; Modrich, P.; Au, K.G. DNA Mismatch Correction in a Defined System. Science 1989, 245, 160. [Google Scholar] [CrossRef]
- Guarné, A.; Ramon-Maiques, S.; Wolff, E.M.; Ghirlando, R.; Hu, X.; Miller, J.H.; Yang, W. Structure of the MutL C-Terminal Domain: A Model of Intact MutL and Its Roles in Mismatch Repair. EMBO J. 2004, 23, 4134–4145. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Foster, P.L.; Brooks, P.; Fishel, R. The Coordinated Functions of the E. Coli MutS and MutL Proteins in Mismatch Repair. Mol. Cell 2003, 12, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Tomer, G.; Buermeyer, A.B.; Nguyen, M.M.; Liskay, R.M. Contribution of Human Mlh1 and Pms2 ATPase Activities to DNA Mismatch Repair*. J. Biol. Chem. 2002, 277, 21801–21809. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.C.; Shcherbakova, P.V.; Kunkel, T.A. Differential ATP Binding and Intrinsic ATP Hydrolysis by Amino-Terminal Domains of the Yeast Mlh1 and Pms1 Proteins. J. Biol. Chem. 2002, 277, 3673–3679. [Google Scholar] [CrossRef]
- Tran, P.T.; Liskay, R.M. Functional Studies on the Candidate ATPase Domains of Saccharomyces Cerevisiae MutLα. Mol. Cell. Biol. 2000, 20, 6390–6398. [Google Scholar] [CrossRef]
- Junop, M.S.; Yang, W.; Funchain, P.; Clendenin, W.; Miller, J.H. In Vitro and in Vivo Studies of MutS, MutL and MutH Mutants: Correlation of Mismatch Repair and DNA Recombination. DNA Repair 2003, 2, 387–405. [Google Scholar] [CrossRef]
- Aronshtam, A.; Marinus, M.G. Dominant Negative Mutator Mutations in the mutL Gene of Escherichia Coli. Nucleic Acids Res. 1996, 24, 2498–2504. [Google Scholar] [CrossRef]
- Johnson, J.R.; Erdeniz, N.; Nguyen, M.; Dudley, S.; Liskay, R.M. Conservation of Functional Asymmetry in the Mammalian MutLα ATPase. DNA Repair 2010, 9, 1209–1213. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu. Rev. Genet. 2015, 49, 291–313. [Google Scholar] [CrossRef]
- Putnam, C.D. Strand Discrimination in DNA Mismatch Repair. DNA Repair 2021, 105, 103161. [Google Scholar] [CrossRef] [PubMed]
- Sriramachandran, A.M.; Petrosino, G.; Méndez-Lago, M.; Schäfer, A.J.; Batista-Nascimento, L.S.; Zilio, N.; Ulrich, H.D. Genome-Wide Nucleotide-Resolution Mapping of DNA Replication Patterns, Single-Strand Breaks, and Lesions by GLOE-Seq. Mol. Cell 2020, 78, 975–985.e7. [Google Scholar] [CrossRef] [PubMed]
- Kadyrov, F.A.; Dzantiev, L.; Constantin, N.; Modrich, P. Endonucleolytic Function of MutLα in Human Mismatch Repair. Cell 2006, 126, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Kadyrov, F.A.; Holmes, S.F.; Arana, M.E.; Lukianova, O.A.; O’Donnell, M.; Kunkel, T.A.; Modrich, P. Saccharomyces Cerevisiae MutLalpha Is a Mismatch Repair Endonuclease. J. Biol. Chem. 2007, 282, 37181–37190. [Google Scholar] [CrossRef]
- Pluciennik, A.; Dzantiev, L.; Iyer, R.R.; Constantin, N.; Kadyrov, F.A.; Modrich, P. PCNA Function in the Activation and Strand Direction of MutLα Endonuclease in Mismatch Repair. Proc. Natl. Acad. Sci. 2010, 107, 16066–16071. [Google Scholar] [CrossRef]
- Burgers, P.M.J.; Kunkel, T.A. Eukaryotic DNA Replication Fork. Annu. Rev. Biochem. 2017, 86, 417–438. [Google Scholar] [CrossRef]
- Williams, J.S.; Kunkel, T.A. Ribonucleotides in DNA: Origins, Repair and Consequences. DNA Repair 2014, 19, 27–37. [Google Scholar] [CrossRef]
- Hsieh, P.; Yamane, K. DNA Mismatch Repair: Molecular Mechanism, Cancer, and Ageing. Mech. Ageing Dev. 2008, 129, 391–407. [Google Scholar] [CrossRef]
- Modrich, P. Mechanisms in E. Coli and Human Mismatch Repair (Nobel Lecture). Angew. Chem. Int. Ed. 2016, 55, 8490–8501. [Google Scholar] [CrossRef]
- Modrich, P. Mechanisms and Biological Effects of Mismatch Repair. Annu. Rev. Genet. 1991, 25, 229–253. [Google Scholar] [CrossRef]
- Putnam, C.D. Evolution of the Methyl Directed Mismatch Repair System in Escherichia Coli. DNA Repair 2016, 38, 32–41. [Google Scholar] [CrossRef]
- Goellner, E.M.; Putnam, C.D.; Kolodner, R.D. Exonuclease 1-Dependent and Independent Mismatch Repair. DNA Repair 2015, 32, 24–32. [Google Scholar] [CrossRef]
- Bowen, N.; Kolodner, R.D. Reconstitution of Saccharomyces Cerevisiae DNA Polymerase ε-Dependent Mismatch Repair with Purified Proteins. Proc. Natl. Acad. Sci. 2017, 114, 3607–3612. [Google Scholar] [CrossRef]
- Pillon, M.C.; Lorenowicz, J.J.; Uckelmann, M.; Klocko, A.D.; Mitchell, R.R.; Chung, Y.S.; Modrich, P.; Walker, G.C.; Simmons, L.A.; Friedhoff, P.; et al. Structure of the Endonuclease Domain of MutL: Unlicensed to Cut. Mol. Cell 2010, 39, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Furman, C.M.; Manhart, C.M.; Alani, E.; Finkelstein, I.J. Intrinsically Disordered Regions Regulate Both Catalytic and Non-Catalytic Activities of the MutLα Mismatch Repair Complex. Nucleic Acids Res. 2019, 47, 1823–1835. [Google Scholar] [CrossRef] [PubMed]
- Piscitelli, J.M.; Witte, S.J.; Sakinejad, Y.S.; Manhart, C.M. Mlh1-Pms1 ATPase Activity Is Regulated Distinctly by Self-Generated Nicks and Strand Discrimination Signals in Mismatch Repair. Nucleic Acids Res. 2025, 53, gkae1253. [Google Scholar] [CrossRef]
- Dutta, R.; Inouye, M. GHKL, an Emergent ATPase/Kinase Superfamily. Trends Biochem. Sci. 2000, 25, 24–28. [Google Scholar] [CrossRef]
- Bergerat, A.; de Massy, B.; Gadelle, D.; Varoutas, P.-C.; Nicolas, A.; Forterre, P. An Atypical Topoisomerase II from Archaea with Implications for Meiotic Recombination. Nature 1997, 386, 414–417. [Google Scholar] [CrossRef]
- Srivatsan, A.; Bowen, N.; Kolodner, R.D. Mispair-Specific Recruitment of the Mlh1-Pms1 Complex Identifies Repair Substrates of the Saccharomyces Cerevisiae Msh2-Msh3 Complex. J. Biol. Chem. 2014, 289, 9352–9364. [Google Scholar] [CrossRef] [PubMed]
- Räschle, M.; Marra, G.; Nyström-Lahti, M.; Schär, P.; Jiricny, J. Identification of hMutLβ, a Heterodimer of hMLH1 and hPMS1. J. Biol. Chem. 1999, 274, 32368–32375. [Google Scholar] [CrossRef]
- Li, G.M.; Modrich, P. Restoration of Mismatch Repair to Nuclear Extracts of H6 Colorectal Tumor Cells by a Heterodimer of Human MutL Homologs. Proc. Natl. Acad. Sci. 1995, 92, 1950–1954. [Google Scholar] [CrossRef]
- Pannafino, G.; Alani, E. Coordinated and Independent Roles for MLH Subunits in DNA Repair. Cells 2021, 10, 948. [Google Scholar] [CrossRef]
- Wang, T.-F.; Kleckner, N.; Hunter, N. Functional Specificity of MutL Homologs in Yeast: Evidence for Three Mlh1-Based Heterocomplexes with Distinct Roles during Meiosis in Recombination and Mismatch Correction. Proc. Natl. Acad. Sci. 1999, 96, 13914–13919. [Google Scholar] [CrossRef]
- Plotz, G.; Raedle, J.; Brieger, A.; Trojan, J.; Zeuzem, S. hMutSα Forms an ATP-Dependent Complex with hMutLα and hMutLβ on DNA. Nucleic Acids Res. 2002, 30, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Furman, C.; Elbashir, R.; Alani, E. Expanded Roles for the MutL Family of DNA Mismatch Repair Proteins. Yeast Chichester Engl. 2021, 38, 39–53. [Google Scholar] [CrossRef]
- Kosinski, J.; Steindorf, I.; Bujnicki, J.M.; Giron-Monzon, L.; Friedhoff, P. Analysis of the Quaternary Structure of the MutL C-Terminal Domain. J. Mol. Biol. 2005, 351, 895–909. [Google Scholar] [CrossRef]
- Kosinski, J.; Hinrichsen, I.; Bujnicki, J.M.; Friedhoff, P.; Plotz, G. Identification of Lynch Syndrome Mutations in the MLH1–PMS2 Interface That Disturb Dimerization and Mismatch Repair. Hum. Mutat. 2010, 31, 975–982. [Google Scholar] [CrossRef]
- Gueneau, E.; Dherin, C.; Legrand, P.; Tellier-Lebegue, C.; Gilquin, B.; Bonnesoeur, P.; Londino, F.; Quemener, C.; Le Du, M.-H.; Márquez, J.A.; et al. Structure of the MutLα C-Terminal Domain Reveals How Mlh1 Contributes to Pms1 Endonuclease Site. Nat. Struct. Mol. Biol. 2013, 20, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Ban, C.; Yang, W. Crystal Structure and ATPase Activity of MutL: Implications for DNA Repair and Mutagenesis. Cell 1998, 95, 541–552. [Google Scholar] [CrossRef]
- Ban, C.; Junop, M.; Yang, W. Transformation of MutL by ATP Binding and Hydrolysis: A Switch in DNA Mismatch Repair. Cell 1999, 97, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Arana, M.E.; Holmes, S.F.; Fortune, J.M.; Moon, A.F.; Pedersen, L.C.; Kunkel, T.A. Functional Residues on the Surface of the N-Terminal Domain of Yeast Pms1. DNA Repair 2010, 9, 448–457. [Google Scholar] [CrossRef]
- Wu, H.; Zeng, H.; Lam, R.; Tempel, W.; Kerr, I.D.; Min, J. Structure of the Human MLH1 N-Terminus: Implications for Predisposition to Lynch Syndrome. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 981–985. [Google Scholar] [CrossRef]
- Spampinato, C.; Modrich, P. The MutL ATPase Is Required for Mismatch Repair. J. Biol. Chem. 2000, 275, 9863–9869. [Google Scholar] [CrossRef]
- Hu, X.; Machius, M.; Yang, W. Monovalent Cation Dependence and Preference of GHKL ATPases and Kinases. FEBS Lett. 2003, 544, 268–273. [Google Scholar] [CrossRef]
- Sacho, E.J.; Kadyrov, F.A.; Modrich, P.; Kunkel, T.A.; Erie, D.A. Direct Visualization of Asymmetric Adenine Nucleotide-Induced Conformational Changes in MutLα. Mol. Cell 2008, 29, 112–121. [Google Scholar] [CrossRef]
- DePristo, M.A.; de Bakker, P.I.W.; Blundell, T.L. Heterogeneity and Inaccuracy in Protein Structures Solved by X-Ray Crystallography. Structure 2004, 12, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K.; Nishida, M.; Nakagawa, N.; Masui, R.; Kuramitsu, S. Bound Nucleotide Controls the Endonuclease Activity of Mismatch Repair Enzyme MutL. J. Biol. Chem. 2008, 283, 12136–12145. [Google Scholar] [CrossRef]
- Räschle, M.; Dufner, P.; Marra, G.; Jiricny, J. Mutations within the hMLH1 and hPMS2 Subunits of the Human MutLα Mismatch Repair Factor Affect Its ATPase Activity, but Not Its Ability to Interact with hMutSα. J. Biol. Chem. 2002, 277, 21810–21820. [Google Scholar] [CrossRef] [PubMed]
- Niedziela-Majka, A.; Maluf, N.K.; Antony, E.; Lohman, T.M. Self-Assembly of Escherichia Coli MutL and Its Complexes with DNA. Biochemistry 2011, 50, 7868–7880. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-W.; Han, X.-P.; Han, C.; London, J.; Fishel, R.; Liu, J. MutS Functions as a Clamp Loader by Positioning MutL on the DNA during Mismatch Repair. Nat. Commun. 2022, 13, 5808. [Google Scholar] [CrossRef]
- Niedziela-Majka, A.; Maluf, N.K.; Antony, E.; Lohman, T.M. Self-Assembly of Escherichia Coli MutL and Its Complexes with DNA. Biochemistry 2011, 50, 7868–7880. [Google Scholar] [CrossRef] [PubMed]
- Claeys Bouuaert, C.; Keeney, S. Distinct DNA-Binding Surfaces in the ATPase and Linker Domains of MutLγ Determine Its Substrate Specificities and Exert Separable Functions in Meiotic Recombination and Mismatch Repair. PLoS Genet. 2017, 13, e1006722. [Google Scholar] [CrossRef]
- Plys, A.J.; Rogacheva, M.V.; Greene, E.C.; Alani, E. The Unstructured Linker Arms of Mlh1–Pms1 Are Important for Interactions with DNA during Mismatch Repair. J. Mol. Biol. 2012, 422, 192–203. [Google Scholar] [CrossRef]
- Furman, C.M.; Wang, T.-Y.; Zhao, Q.; Yugandhar, K.; Yu, H.; Alani, E. Handcuffing Intrinsically Disordered Regions in Mlh1–Pms1 Disrupts Mismatch Repair. Nucleic Acids Res. 2021, 49, 9327–9341. [Google Scholar] [CrossRef]
- Grilley, M.; Welsh, K.M.; Su, S.S.; Modrich, P. Isolation and Characterization of the Escherichia Coli mutL Gene Product. J. Biol. Chem. 1989, 264, 1000–1004. [Google Scholar] [CrossRef]
- Drotschmann, K.; Aronshtam, A.; Fritz, H.J.; Marinus, M.G. The Escherichia Coli MutL Protein Stimulates Binding of Vsr and MutS to Heteroduplex DNA. Nucleic Acids Res. 1998, 26, 948–953. [Google Scholar] [CrossRef]
- Bende, S.M.; Grafström, R.H. The DNA Binding Properties of the MutL Protein Isolated from Escherichia Coli. Nucleic Acids Res. 1991, 19, 1549. [Google Scholar] [CrossRef]
- Hall, M.C.; Wang, H.; Erie, D.A.; Kunkel, T.A. High Affinity Cooperative DNA Binding by the Yeast Mlh1-Pms1 Heterodimer1. J. Mol. Biol. 2001, 312, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.; Lee, G.S.; Gu, L.; Yang, W.; Li, G.-M. Mispair-Bound Human MutS–MutL Complex Triggers DNA Incisions and Activates Mismatch Repair. Cell Res. 2021, 31, 542–553. [Google Scholar] [CrossRef]
- Witte, S.J.; Rosa, I.M.; Collingwood, B.W.; Piscitelli, J.M.; Manhart, C.M. The Mismatch Repair Endonuclease MutLα Tethers Duplex Regions of DNA Together and Relieves DNA Torsional Tension. Nucleic Acids Res. 2023, 51, 2725–2739. [Google Scholar] [CrossRef] [PubMed]
- Manhart, C.M.; Ni, X.; White, M.A.; Ortega, J.; Surtees, J.A.; Alani, E. The Mismatch Repair and Meiotic Recombination Endonuclease Mlh1-Mlh3 Is Activated by Polymer Formation and Can Cleave DNA Substrates in Trans. PLOS Biol. 2017, 15, e2001164. [Google Scholar] [CrossRef] [PubMed]
- Bradford, K.C.; Wilkins, H.; Hao, P.; Li, Z.M.; Wang, B.; Burke, D.; Wu, D.; Smith, A.E.; Spaller, L.; Du, C.; et al. Dynamic Human MutSα–MutLα Complexes Compact Mismatched DNA. Proc. Natl. Acad. Sci. 2020, 117, 16302–16312. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.C.; Shcherbakova, P.V.; Fortune, J.M.; Borchers, C.H.; Dial, J.M.; Tomer, K.B.; Kunkel, T.A. DNA Binding by Yeast Mlh1 and Pms1: Implications for DNA Mismatch Repair. Nucleic Acids Res. 2003, 31, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Schofield, M.J.; Nayak, S.; Scott, T.H.; Du, C.; Hsieh, P. Interaction of Escherichia Coli MutS and MutL at a DNA Mismatch. J. Biol. Chem. 2001, 276, 28291–28299. [Google Scholar] [CrossRef]
- Lee, S.D.; Alani, E. Analysis of Interactions Between Mismatch Repair Initiation Factors and the Replication Processivity Factor PCNA. J. Mol. Biol. 2006, 355, 175–184. [Google Scholar] [CrossRef]
- Gu, L.; Hong, Y.; McCulloch, S.; Watanabe, H.; Li, G.-M. ATP-Dependent Interaction of Human Mismatch Repair Proteins and Dual Role of PCNA in Mismatch Repair. Nucleic Acids Res. 1998, 26, 1173–1178. [Google Scholar] [CrossRef]
- Prolla, T.A.; Pang, Q.; Alani, E.; Kolodner, R.D.; Liskay, R.M. MLH1, PMS1, and MSH2 Interactions During the Initiation of DNA Mismatch Repair in Yeast. Science 1994, 265, 1091–1093. [Google Scholar] [CrossRef]
- Plotz, G.; Welsch, C.; Giron-Monzon, L.; Friedhoff, P.; Albrecht, M.; Piiper, A.; Biondi, R.M.; Lengauer, T.; Zeuzem, S.; Raedle, J. Mutations in the MutSalpha Interaction Interface of MLH1 Can Abolish DNA Mismatch Repair. Nucleic Acids Res. 2006, 34, 6574–6586. [Google Scholar] [CrossRef]
- Selmane, T.; Schofield, M.J.; Nayak, S.; Du, C.; Hsieh, P. Formation of a DNA Mismatch Repair Complex Mediated by ATP. J. Mol. Biol. 2003, 334, 949–965. [Google Scholar] [CrossRef]
- Bowers, J.; Tran, P.T.; Joshi, A.; Liskay, R.M.; Alani, E. MSH-MLH Complexes Formed at a DNA Mismatch Are Disrupted by the PCNA Sliding Clamp1. J. Mol. Biol. 2001, 306, 957–968. [Google Scholar] [CrossRef]
- Genschel, J.; Kadyrova, L.Y.; Iyer, R.R.; Dahal, B.K.; Kadyrov, F.A.; Modrich, P. Interaction of Proliferating Cell Nuclear Antigen with PMS2 Is Required for MutLα Activation and Function in Mismatch Repair. Proc. Natl. Acad. Sci. 2017, 114, 4930–4935. [Google Scholar] [CrossRef]
- Pillon, M.C.; Miller, J.H.; Guarné, A. The Endonuclease Domain of MutL Interacts with the β Sliding Clamp. DNA Repair 2011, 10, 87–93. [Google Scholar] [CrossRef]
- Almawi, A.W.; Scotland, M.K.; Randall, J.R.; Liu, L.; Martin, H.K.; Sacre, L.; Shen, Y.; Pillon, M.C.; Simmons, L.A.; Sutton, M.D.; et al. Binding of the Regulatory Domain of MutL to the Sliding β-Clamp Is Species Specific. Nucleic Acids Res. 2019, 47, 4831–4842. [Google Scholar] [CrossRef]
- Rajakumari, K.; Aravind, K.; Balamugundhan, M.; Jagadeesan, M.; Somasundaram, A.; Brindha Devi, P.; Ramasamy, P. Comprehensive Review of DNA Gyrase as Enzymatic Target for Drug Discovery and Development. Eur. J. Med. Chem. Rep. 2024, 12, 100233. [Google Scholar] [CrossRef]
- Vanden Broeck, A.; Lotz, C.; Ortiz, J.; Lamour, V. Cryo-EM Structure of the Complete E. Coli DNA Gyrase Nucleoprotein Complex. Nat. Commun. 2019, 10, 4935. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.; Chen, C.; Ge, H.; Xie, X.S. Mechanism of Transcriptional Bursting in Bacteria. Cell 2014, 158, 314–326. [Google Scholar] [CrossRef]
- Wu, H.Y.; Shyy, S.H.; Wang, J.C.; Liu, L.F. Transcription Generates Positively and Negatively Supercoiled Domains in the Template. Cell 1988, 53, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.O.; Cozzarelli, N.R. A Sign Inversion Mechanism for Enzymatic Supercoiling of DNA. Science 1979, 206, 1081–1083. [Google Scholar] [CrossRef]
- Stelljes, J.T.; Weidlich, D.; Gubaev, A.; Klostermeier, D. Gyrase Containing a Single C-Terminal Domain Catalyzes Negative Supercoiling of DNA by Decreasing the Linking Number in Steps of Two. Nucleic Acids Res. 2018, 46, 6773–6784. [Google Scholar] [CrossRef]
- Hartmann, S.; Gubaev, A.; Klostermeier, D. Binding and Hydrolysis of a Single ATP Is Sufficient for N-Gate Closure and DNA Supercoiling by Gyrase. J. Mol. Biol. 2017, 429, 3717–3729. [Google Scholar] [CrossRef]
- Brino, L.; Urzhumtsev, A.; Mousli, M.; Bronner, C.; Mitschler, A.; Oudet, P.; Moras, D. Dimerization of Escherichia Coli DNA-Gyrase B Provides a Structural Mechanism for Activating the ATPase Catalytic Center. J. Biol. Chem. 2000, 275, 9468–9475. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, Z.; Mitchenall, L.A.; Maxwell, A.; Deng, J.; Zhang, H.; Zhou, Y.; Chen, Y.; Wang, D.-C.; Zhang, X.-E.; et al. The Dimer State of GyrB Is an Active Form: Implications for the Initial Complex Assembly and Processive Strand Passage. Nucleic Acids Res. 2011, 39, 8488–8502. [Google Scholar] [CrossRef]
- Witz, G.; Stasiak, A. DNA Supercoiling and Its Role in DNA Decatenation and Unknotting. Nucleic Acids Res. 2010, 38, 2119–2133. [Google Scholar] [CrossRef]
- Kampranis, S.C.; Maxwell, A. Conversion of DNA Gyrase into a Conventional Type II Topoisomerase. Proc. Natl. Acad. Sci. 1996, 93, 14416–14421. [Google Scholar] [CrossRef] [PubMed]
- Gubaev, A.; Klostermeier, D. DNA-Induced Narrowing of the Gyrase N-Gate Coordinates T-Segment Capture and Strand Passage. Proc. Natl. Acad. Sci. 2011, 108, 14085–14090. [Google Scholar] [CrossRef]
- Germe, T.R.; Bush, N.G.; Baskerville, V.M.; Saman, D.; Benesch, J.L.; Maxwell, A. Rapid, DNA-Induced Interface Swapping by DNA Gyrase. eLife 12. [CrossRef]
- Gross, C.H.; Parsons, J.D.; Grossman, T.H.; Charifson, P.S.; Bellon, S.; Jernee, J.; Dwyer, M.; Chambers, S.P.; Markland, W.; Botfield, M.; et al. Active-Site Residues of Escherichia Coli DNA Gyrase Required in Coupling ATP Hydrolysis to DNA Supercoiling and Amino Acid Substitutions Leading to Novobiocin Resistance. Antimicrob. Agents Chemother. 2003, 47, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Kampranis, S.C.; Maxwell, A. Conformational Changes in DNA Gyrase Revealed by Limited Proteolysis. J. Biol. Chem. 1998, 273, 22606–22614. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Parente, A.C.; Bryant, Z. Structural Dynamics and Mechanochemical Coupling in DNA Gyrase. J. Mol. Biol. 2016, 428, 1833–1845. [Google Scholar] [CrossRef]
- Roca, J. Topoisomerase II: A Fitted Mechanism for the Chromatin Landscape. Nucleic Acids Res. 2009, 37, 721–730. [Google Scholar] [CrossRef]
- Berger, J.M.; Gamblin, S.J.; Harrison, S.C.; Wang, J.C. Structure and Mechanism of DNA Topoisomerase II. Nature 1996, 379, 225–232. [Google Scholar] [CrossRef]
- Roca, J.; Wang, J.C. The Capture of a DNA Double Helix by an ATP-Dependent Protein Clamp: A Key Step in DNA Transport by Type II DNA Topoisomerases. Cell 1992, 71, 833–840. [Google Scholar] [CrossRef]
- Ruan, S.; Tu, C.-H.; Bourne, C.R. Friend or Foe: Protein Inhibitors of DNA Gyrase. Biology 2024, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Albakova, Z. HSP90 Multi-Functionality in Cancer. Front. Immunol. 2024, 15. [Google Scholar] [CrossRef]
- Genest, O.; Wickner, S.; Doyle, S.M. Hsp90 and Hsp70 Chaperones: Collaborators in Protein Remodeling. J. Biol. Chem. 2019, 294, 2109–2120. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, R.; Kifayat, S.; Pooniya, K.K.; Kularia, S.; Adimalla, B.S.; Sanapalli, B.K.R.; Sanapalli, V.; Sigalapalli, D.K. Bacterial Histidine Kinase and the Development of Its Inhibitors in the 21st Century. Antibiotics 2024, 13, 576. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yu, C.; Wu, H.; Li, G.; Li, C.; Hong, W.; Yang, X.; Wang, H.; You, X. Recent Advances in Histidine Kinase-Targeted Antimicrobial Agents. Front. Chem. 2022, 10. [Google Scholar] [CrossRef]
- Maxwell, A.; Gellert, M. The DNA Dependence of the ATPase Activity of DNA Gyrase. J. Biol. Chem. 1984, 259, 14472–14480. [Google Scholar] [CrossRef]
- Collingwood, B.W.; Bhalkar, A.N.; Manhart, C.M. The Mismatch Repair Factor Mlh1-Pms1 Uses ATP to Compact and Remodel DNA. bioRxiv, 6333; .81. [Google Scholar] [CrossRef]
- Young, J.C.; Moarefi, I.; Hartl, F.U. Hsp90. J. Cell Biol. 2001, 154, 267–274. [Google Scholar] [CrossRef]
- Wei, H.; Zhang, Y.; Jia, Y.; Chen, X.; Niu, T.; Chatterjee, A.; He, P.; Hou, G. Heat Shock Protein 90: Biological Functions, Diseases, and Therapeutic Targets. MedComm 2024, 5, e470. [Google Scholar] [CrossRef]
- Ritossa, F. A New Puffing Pattern Induced by Temperature Shock and DNP in Drosophila. Experientia 1962, 18, 571–573. [Google Scholar] [CrossRef]
- Wiech, H.; Buchner, J.; Zimmermann, R.; Jakob, U. Hsp90 Chaperones Protein Folding in Vitro. Nature 1992, 358, 169–170. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, J.; Mikami, I.; Tominaga, Y.; Kuchenbecker, K.M.; Lin, Y.-C.; Bravo, D.T.; Clement, G.; Yagui-Beltran, A.; Ray, M.R.; Koizumi, K.; et al. Inhibition of Hsp90 Leads to Cell Cycle Arrest and Apoptosis in Human Malignant Pleural Mesothelioma. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2008, 3, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Kolhe, J.A.; Babu, N.L.; Freeman, B.C. The Hsp90 Molecular Chaperone Governs Client Proteins by Targeting Intrinsically Disordered Regions. Mol. Cell 2023, 83, 2035–2044.e7. [Google Scholar] [CrossRef]
- Lopez, A.; Dahiya, V.; Delhommel, F.; Freiburger, L.; Stehle, R.; Asami, S.; Rutz, D.; Blair, L.; Buchner, J.; Sattler, M. Client Binding Shifts the Populations of Dynamic Hsp90 Conformations through an Allosteric Network. Sci. Adv. 7. [CrossRef]
- Chiosis, G.; Digwal, C.S.; Trepel, J.B.; Neckers, L. Structural and Functional Complexity of HSP90 in Cellular Homeostasis and Disease. Nat. Rev. Mol. Cell Biol. 2023, 24, 797–815. [Google Scholar] [CrossRef]
- Li, J.; Buchner, J. Structure, Function and Regulation of the Hsp90 Machinery. Biomed. J. 2013, 36, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Southworth, D.R.; Agard, D.A. Client-Loading Conformation of the Hsp90 Molecular Chaperone Revealed in the Cryo-EM Structure of the Human Hsp90:Hop Complex. Mol. Cell 2011, 42, 771–781. [Google Scholar] [CrossRef]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal Structure of an Hsp90–Nucleotide–P23/Sba1 Closed Chaperone Complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef]
- Shiau, A.K.; Harris, S.F.; Southworth, D.R.; Agard, D.A. Structural Analysis of E. Coli Hsp90 Reveals Dramatic Nucleotide-Dependent Conformational Rearrangements. Cell 2006, 127, 329–340. [Google Scholar] [CrossRef]
- Chiosis, G.; Digwal, C.S.; Trepel, J.B.; Neckers, L. Structural and Functional Complexity of HSP90 in Cellular Homeostasis and Disease. Nat. Rev. Mol. Cell Biol. 2023, 24, 797–815. [Google Scholar] [CrossRef]
- Prodromou, C.; Panaretou, B.; Chohan, S.; Siligardi, G.; O’Brien, R.; Ladbury, J.E.; Roe, S.M.; Piper, P.W.; Pearl, L.H. The ATPase Cycle of Hsp90 Drives a Molecular ‘Clamp’ via Transient Dimerization of the N-Terminal Domains. EMBO J. 2000, 19, 4383–4392. [Google Scholar] [CrossRef]
- Prodromou, C. The ‘Active Life’ of Hsp90 Complexes. Biochim. Biophys. Acta BBA - Mol. Cell Res. 2012, 1823, 614–623. [Google Scholar] [CrossRef]
- Reidy, M.; Masison, D.C. ATP Plays a Structural Role in Hsp90 Function. Nat. Commun. 2025, 16, 6710. [Google Scholar] [CrossRef]
- Zuehlke, A.; Johnson, J.L. Hsp90 and Co-Chaperones Twist the Functions of Diverse Client Proteins. Biopolymers 2010, 93, 211–217. [Google Scholar] [CrossRef]
- Giannoulis, A.; Feintuch, A.; Barak, Y.; Mazal, H.; Albeck, S.; Unger, T.; Yang, F.; Su, X.-C.; Goldfarb, D. Two Closed ATP- and ADP-Dependent Conformations in Yeast Hsp90 Chaperone Detected by Mn(II) EPR Spectroscopic Techniques. Proc. Natl. Acad. Sci. 2020, 117, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Reidy, M.; Garzillo, K.; Masison, D.C. Nucleotide Exchange Is Sufficient for Hsp90 Functions in Vivo. Nat. Commun. 2023, 14, 2489. [Google Scholar] [CrossRef] [PubMed]
- Siligardi, G.; Hu, B.; Panaretou, B.; Piper, P.W.; Pearl, L.H.; Prodromou, C. Co-Chaperone Regulation of Conformational Switching in the Hsp90 ATPase Cycle. J. Biol. Chem. 2004, 279, 51989–51998. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.K.; Hutt, D.M.; Tait, B.; Guy, N.C.; Sivils, J.C.; Ortiz, N.R.; Payan, A.N.; Komaragiri, S.K.; Owens, J.J.; Culbertson, D.; et al. Management of Hsp90-Dependent Protein Folding by Small Molecules Targeting the Aha1 Co-Chaperone. Cell Chem. Biol. 2020, 27, 292–305.e6. [Google Scholar] [CrossRef]
- Wolmarans, A.; Lee, B.; Spyracopoulos, L.; LaPointe, P. The Mechanism of Hsp90 ATPase Stimulation by Aha1. Sci. Rep. 2016, 6, 33179. [Google Scholar] [CrossRef]
- Oroz, J.; Blair, L.J.; Zweckstetter, M. Dynamic Aha1 Co--chaperone Binding to Human Hsp90. Protein Sci. Publ. Protein Soc. 2019, 28, 1545–1551. [Google Scholar] [CrossRef]
- Ulrich, L.E.; Zhulin, I.B. The MiST2 Database: A Comprehensive Genomics Resource on Microbial Signal Transduction. Nucleic Acids Res. 2010, 38, D401–D407. [Google Scholar] [CrossRef]
- Marina, A.; Waldburger, C.D.; Hendrickson, W.A. Structure of the Entire Cytoplasmic Portion of a Sensor Histidine-Kinase Protein. EMBO J. 2005, 24, 4247–4259. [Google Scholar] [CrossRef]
- Bhate, M.P.; Molnar, K.S.; Goulian, M.; DeGrado, W.F. Signal Transduction in Histidine Kinases: Insights from New Structures. Structure 2015, 23, 981–994. [Google Scholar] [CrossRef]
- Purcell, E.B.; Siegal-Gaskins, D.; Rawling, D.C.; Fiebig, A.; Crosson, S. A Photosensory Two-Component System Regulates Bacterial Cell Attachment. Proc. Natl. Acad. Sci. 2007, 104, 18241–18246. [Google Scholar] [CrossRef]
- Bortolotti, A.; Vazquez, D.B.; Almada, J.C.; Inda, M.E.; Drusin, S.I.; Villalba, J.M.; Moreno, D.M.; Ruysschaert, J.M.; Cybulski, L.E. A Transmembrane Histidine Kinase Functions as a pH Sensor. Biomolecules 2020, 10, 1183. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Hao, B.; Mansy, S.S.; Gonzalez, G.; Gilles-Gonzalez, M.A.; Chan, M.K. Structure of a Biological Oxygen Sensor: A New Mechanism for Heme-Driven Signal Transduction. Proc. Natl. Acad. Sci. 1998, 95, 15177–15182. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Chavez, R.; Alvarez, A.F.; Romeo, T.; Georgellis, D. The Physiological Stimulus for the BarA Sensor Kinase. J. Bacteriol. 2010, 192, 2009–2012. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.F.; Georgellis, D. The Role of Sensory Kinase Proteins in Two-Component Signal Transduction. Biochem. Soc. Trans. 2022, 50, 1859–1873. [Google Scholar] [CrossRef]
- Kansari, M.; Idiris, F.; Szurmant, H.; Kubař, T.; Schug, A. Mechanism of Activation and Autophosphorylation of a Histidine Kinase. Commun. Chem. 2024, 7, 1–13. [Google Scholar] [CrossRef]
- Casino, P.; Miguel-Romero, L.; Marina, A. Visualizing Autophosphorylation in Histidine Kinases. Nat. Commun. 2014, 5, 3258. [Google Scholar] [CrossRef]
- Szurmant, H.; White, R.A.; Hoch, J.A. Sensor Complexes Regulating Two-Component Signal Transduction. Curr. Opin. Struct. Biol. 2007, 17, 706–715. [Google Scholar] [CrossRef]
- Zapf, J.; Sen, U.; Madhusudan; Hoch, J. A.; Varughese, K.I. A Transient Interaction between Two Phosphorelay Proteins Trapped in a Crystal Lattice Reveals the Mechanism of Molecular Recognition and Phosphotransfer in Signal Transduction. Structure 2000, 8, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Casino, P.; Rubio, V.; Marina, A. Structural Insight into Partner Specificity and Phosphoryl Transfer in Two-Component Signal Transduction. Cell 2009, 139, 325–336. [Google Scholar] [CrossRef]
- Wojnowska, M.; Yan, J.; Sivalingam, G.N.; Cryar, A.; Gor, J.; Thalassinos, K.; Djordjevic, S. Autophosphorylation Activity of a Soluble Hexameric Histidine Kinase Correlates with the Shift in Protein Conformational Equilibrium. Chem. Biol. 2013, 20, 1411–1420. [Google Scholar] [CrossRef]
- Dikiy, I.; Edupuganti, U.R.; Abzalimov, R.R.; Borbat, P.P.; Srivastava, M.; Freed, J.H.; Gardner, K.H. Insights into Histidine Kinase Activation Mechanisms from the Monomeric Blue Light Sensor EL346. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 4963–4972. [Google Scholar] [CrossRef]
- Groothuizen, F.S.; Winkler, I.; Cristóvão, M.; Fish, A.; Winterwerp, H.H.; Reumer, A.; Marx, A.D.; Hermans, N.; Nicholls, R.A.; Murshudov, G.N.; et al. MutS/MutL Crystal Structure Reveals That the MutS Sliding Clamp Loads MutL onto DNA. eLife 2015, 4, e06744. [Google Scholar] [CrossRef]
- Mendillo, M.L.; Hargreaves, V.V.; Jamison, J.W.; Mo, A.O.; Li, S.; Putnam, C.D.; Woods, V.L.; Kolodner, R.D. A Conserved MutS Homolog Connector Domain Interface Interacts with MutL Homologs. Proc. Natl. Acad. Sci. 2009, 106, 22223–22228. [Google Scholar] [CrossRef] [PubMed]
- Plotz, G.; Raedle, J.; Brieger, A.; Trojan, J.; Zeuzem, S. N-Terminus of hMLH1 Confers Interaction of hMutLalpha and hMutLbeta with hMutSalpha. Nucleic Acids Res. 2003, 31, 3217–3226. [Google Scholar] [CrossRef]
- Gorman, J.; Plys, A.J.; Visnapuu, M.-L.; Alani, E.; Greene, E.C. Visualizing One-Dimensional Diffusion of Eukaryotic DNA Repair Factors along a Chromatin Lattice. Nat. Struct. Mol. Biol. 2010, 17, 932–938. [Google Scholar] [CrossRef]
- Gorman, J.; Wang, F.; Redding, S.; Plys, A.J.; Fazio, T.; Wind, S.; Alani, E.E.; Greene, E.C. Single-Molecule Imaging Reveals Target-Search Mechanisms during DNA Mismatch Repair. Proc. Natl. Acad. Sci. 2012, 109, E3074–E3083. [Google Scholar] [CrossRef]
- Hao, P.; LeBlanc, S.J.; Case, B.C.; Elston, T.C.; Hingorani, M.M.; Erie, D.A.; Weninger, K.R. Recurrent Mismatch Binding by MutS Mobile Clamps on DNA Localizes Repair Complexes Nearby. Proc. Natl. Acad. Sci. 2020, 117, 17775–17784. [Google Scholar] [CrossRef] [PubMed]
- Qiu, R.; Sakato, M.; Sacho, E.J.; Wilkins, H.; Zhang, X.; Modrich, P.; Hingorani, M.M.; Erie, D.A.; Weninger, K.R. MutL Traps MutS at a DNA Mismatch. Proc. Natl. Acad. Sci. 2015, 112, 10914–10919. [Google Scholar] [CrossRef]
- Liu, J.; Hanne, J.; Britton, B.M.; Bennett, J.; Kim, D.; Lee, J.-B.; Fishel, R. Cascading MutS and MutL Sliding Clamps Control DNA Diffusion to Activate Mismatch Repair. Nature 2016, 539, 583–587. [Google Scholar] [CrossRef]
- Guan, J.; Lu, C.; Jin, Q.; Lu, H.; Chen, X.; Tian, L.; Zhang, Y.; Ortega, J.; Zhang, J.; Siteni, S.; et al. MLH1 Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the cGAS-STING Pathway. Cancer Cell 2021, 39, 109–121.e5. [Google Scholar] [CrossRef]
- Borsellini, A.; Lebbink, J.H.G.; Lamers, M.H. MutL Binds to 3’ Resected DNA Ends and Blocks DNA Polymerase Access. Nucleic Acids Res. 2022, 50, 6224–6234. [Google Scholar] [CrossRef]
- Kadyrov, F.A.; Genschel, J.; Fang, Y.; Penland, E.; Edelmann, W.; Modrich, P. A Possible Mechanism for Exonuclease 1-Independent Eukaryotic Mismatch Repair. Proc. Natl. Acad. Sci. 2009, 106, 8495–8500. [Google Scholar] [CrossRef] [PubMed]
- Kadyrova, L.Y.; Gujar, V.; Burdett, V.; Modrich, P.L.; Kadyrov, F.A. Human MutLγ, the MLH1–MLH3 Heterodimer, Is an Endonuclease That Promotes DNA Expansion. Proc. Natl. Acad. Sci. 2020, 117, 3535–3542. [Google Scholar] [CrossRef] [PubMed]
- Rogacheva, M.V.; Manhart, C.M.; Chen, C.; Guarne, A.; Surtees, J.; Alani, E. Mlh1-Mlh3, a Meiotic Crossover and DNA Mismatch Repair Factor, Is a Msh2-Msh3-Stimulated Endonuclease. J. Biol. Chem. 2014, 289, 5664–5673. [Google Scholar] [CrossRef] [PubMed]
- Flores-Rozas, H.; Kolodner, R.D. The Saccharomyces Cerevisiae MLH3 Gene Functions in MSH3-Dependent Suppression of Frameshift Mutations. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 12404–12409. [Google Scholar] [CrossRef]
- Duroc, Y.; Kumar, R.; Ranjha, L.; Adam, C.; Guérois, R.; Md Muntaz, K.; Marsolier-Kergoat, M.-C.; Dingli, F.; Laureau, R.; Loew, D.; et al. Concerted Action of the MutLβ Heterodimer and Mer3 Helicase Regulates the Global Extent of Meiotic Gene Conversion. eLife 2017, 6, e21900. [Google Scholar] [CrossRef]
- Campbell, C.S.; Hombauer, H.; Srivatsan, A.; Bowen, N.; Gries, K.; Desai, A.; Putnam, C.D.; Kolodner, R.D. Mlh2 Is an Accessory Factor for DNA Mismatch Repair in Saccharomyces Cerevisiae. PLOS Genet. 2014, 10, e1004327. [Google Scholar] [CrossRef]
- Harfe, B.D.; Minesinger, B.K.; Jinks-Robertson, S. Discrete in Vivo Roles for the MutL Homologs Mlh2p and Mlh3p in the Removal of Frameshift Intermediates in Budding Yeast. Curr. Biol. CB 2000, 10, 145–148. [Google Scholar] [CrossRef]
- Ling, E.M.; Baslé, A.; Cowell, I.G.; van den Berg, B.; Blower, T.R.; Austin, C.A. A Comprehensive Structural Analysis of the ATPase Domain of Human DNA Topoisomerase II Beta Bound to AMPPNP, ADP, and the Bisdioxopiperazine, ICRF193. Struct. England1993 2022, 30, 1129–1145.e3. [Google Scholar] [CrossRef]
- Li, S.; Yen, L.; Pastor, W.A.; Johnston, J.B.; Du, J.; Shew, C.J.; Liu, W.; Ho, J.; Stender, B.; Clark, A.T.; et al. Mouse MORC3 Is a GHKL ATPase That Localizes to H3K4me3 Marked Chromatin. Proc. Natl. Acad. Sci. 2016, 113, E5108–E5116. [Google Scholar] [CrossRef]
- Douse, C.H.; Bloor, S.; Liu, Y.; Shamin, M.; Tchasovnikarova, I.A.; Timms, R.T.; Lehner, P.J.; Modis, Y. Neuropathic MORC2 Mutations Perturb GHKL ATPase Dimerization Dynamics and Epigenetic Silencing by Multiple Structural Mechanisms. Nat. Commun. 2018, 9, 651. [Google Scholar] [CrossRef]
- Robert, T.; Nore, A.; Brun, C.; Maffre, C.; Crimi, B.; Guichard, V.; Bourbon, H.-M.; de Massy, B. The TopoVIB-Like Protein Family Is Required for Meiotic DNA Double-Strand Break Formation. Science 2016, 351, 943–949. [Google Scholar] [CrossRef]
- Gurzau, A.D.; Blewitt, M.E.; Czabotar, P.E.; Murphy, J.M.; Birkinshaw, R.W. Relating SMCHD1 Structure to Its Function in Epigenetic Silencing. Biochem. Soc. Trans. 2020, 48, 1751–1763. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.C.; Inoue, K.; Kim, S.; Perera, L.; Shaw, N.D. A Ubiquitin-like Domain Is Required for Stabilizing the N-Terminal ATPase Module of Human SMCHD1. Commun. Biol. 2019, 2, 1–10. [Google Scholar] [CrossRef]
- Chen, K.; Dobson, R.C.J.; Lucet, I.S.; Young, S.N.; Pearce, F.G.; Blewitt, M.E.; Murphy, J.M. The Epigenetic Regulator Smchd1 Contains a Functional GHKL-Type ATPase Domain. Biochem. J. 2016, 473, 1733–1744. [Google Scholar] [CrossRef]
- Corbett, K.D.; Berger, J.M. Structure of the Topoisomerase VI--B Subunit: Implications for Type II Topoisomerase Mechanism and Evolution. EMBO J. 2003, 22, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Ménade, M.; Kozlov, G.; Trempe, J.-F.; Pande, H.; Shenker, S.; Wickremasinghe, S.; Li, X.; Hojjat, H.; Dicaire, M.-J.; Brais, B.; et al. Structures of Ubiquitin-like (Ubl) and Hsp90-like Domains of Sacsin Provide Insight into Pathological Mutations. J. Biol. Chem. 2018, 293, 12832–12842. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Eukaryotic mismatch repair and potential roles for MutLα’s ATPase activity. Mismatch recognition is carried out by MutSα (primarily base-base mismatches and small insertion/deletion loops) and MutSβ (larger insertion/deletion mismatches, with some overlapping specificity) [29]. Both recruit MutLα (blue and yellow complex) to the mismatch site. MutSα/β (pink and grey complex) are known to bind ATP for recruitment. Whether ATP binding by MutLα is required immediately after its recruitment or instead modulates subsequent MutLα-MutS interactions (double-headed arrow), remains unclear. MutLα’s ATPase activity is stimulated by DNA and PCNA (purple), suggesting potential functions in licensing or positioning endonuclease activity (indicated by lightning bolt), coordinating interactions with repair factors, or enabling protein turnover and multiple rounds of nicking. While this schematic highlights the eukaryotic pathway, analogous ATP-regulated MutL mechanisms may also operate in bacteria that lack MutH and methyl-directed strand discrimination.
Figure 1.
Eukaryotic mismatch repair and potential roles for MutLα’s ATPase activity. Mismatch recognition is carried out by MutSα (primarily base-base mismatches and small insertion/deletion loops) and MutSβ (larger insertion/deletion mismatches, with some overlapping specificity) [29]. Both recruit MutLα (blue and yellow complex) to the mismatch site. MutSα/β (pink and grey complex) are known to bind ATP for recruitment. Whether ATP binding by MutLα is required immediately after its recruitment or instead modulates subsequent MutLα-MutS interactions (double-headed arrow), remains unclear. MutLα’s ATPase activity is stimulated by DNA and PCNA (purple), suggesting potential functions in licensing or positioning endonuclease activity (indicated by lightning bolt), coordinating interactions with repair factors, or enabling protein turnover and multiple rounds of nicking. While this schematic highlights the eukaryotic pathway, analogous ATP-regulated MutL mechanisms may also operate in bacteria that lack MutH and methyl-directed strand discrimination.
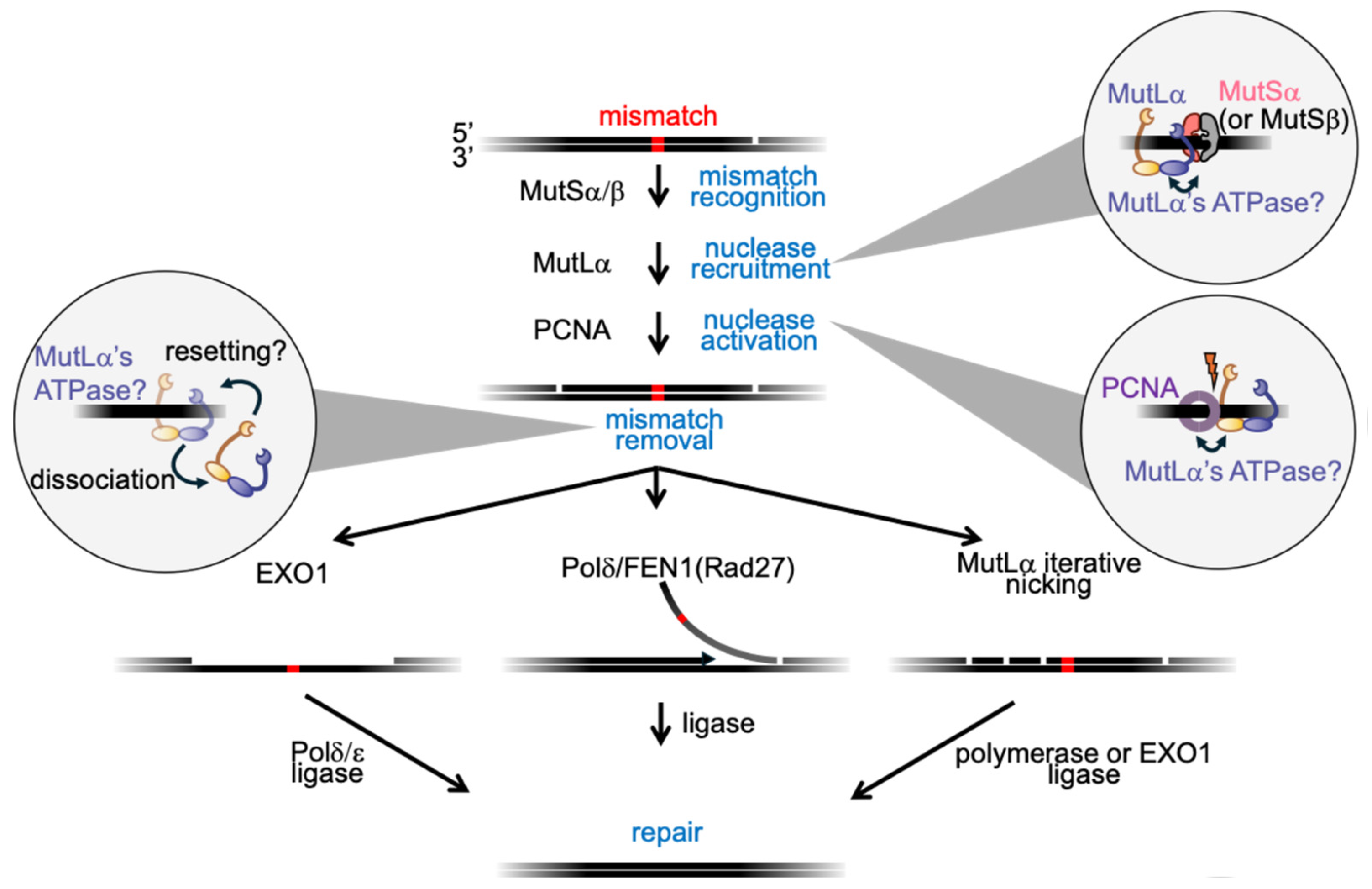
Figure 2.
MutLα domain architecture and conserved ATP-binding fold. (A) Composite full-length model of the MutLα heterodimer assembled from available crystal structures. The human MLH1 N-terminal domain (blue; PDB: 4P7A) is connected by an intrinsically disordered region to the dimeric C-terminal domains of yeast Mlh1 (blue)-Pms1 (yellow) (PDB: 4E4W). The yeast Pms1 N-terminal domain (yellow; PDB: 3H4L) is similarly positioned and connected to its C-terminal domain by an intrinsically disordered region. This composite highlights the conserved organization of N-terminal ATPase domains flexibly linked to constitutively dimerized C-terminal domains. The Bergerat-fold ATPase site is shown in green and the endonuclease motif in purple in the C-terminal domain of yeast Pms1. (B) Magnified view of the structure of the yeast Pms1 N-terminal domain showing the conserved Bergerat-fold (green) bound to an ATP analog (phosphoaminophosphonic acid–adenylate ester). (C) Schematic of the Bergerat-fold (adapted from [27] as found in E. coli MutL with conserved glycine residues indicated (red), other conserved residues are indicated including a catalytic residue (blue). The ATP-“lid” is highlighted in yellow. (D) Model for the ATPase cycle of MutLα, illustrating asymmetrical usage of ATP (black circles) by the two subunits, domain dimerization, and compaction mediated by intrinsically disordered regions upon ATP binding. Adapted from models in [40,45].
Figure 2.
MutLα domain architecture and conserved ATP-binding fold. (A) Composite full-length model of the MutLα heterodimer assembled from available crystal structures. The human MLH1 N-terminal domain (blue; PDB: 4P7A) is connected by an intrinsically disordered region to the dimeric C-terminal domains of yeast Mlh1 (blue)-Pms1 (yellow) (PDB: 4E4W). The yeast Pms1 N-terminal domain (yellow; PDB: 3H4L) is similarly positioned and connected to its C-terminal domain by an intrinsically disordered region. This composite highlights the conserved organization of N-terminal ATPase domains flexibly linked to constitutively dimerized C-terminal domains. The Bergerat-fold ATPase site is shown in green and the endonuclease motif in purple in the C-terminal domain of yeast Pms1. (B) Magnified view of the structure of the yeast Pms1 N-terminal domain showing the conserved Bergerat-fold (green) bound to an ATP analog (phosphoaminophosphonic acid–adenylate ester). (C) Schematic of the Bergerat-fold (adapted from [27] as found in E. coli MutL with conserved glycine residues indicated (red), other conserved residues are indicated including a catalytic residue (blue). The ATP-“lid” is highlighted in yellow. (D) Model for the ATPase cycle of MutLα, illustrating asymmetrical usage of ATP (black circles) by the two subunits, domain dimerization, and compaction mediated by intrinsically disordered regions upon ATP binding. Adapted from models in [40,45].
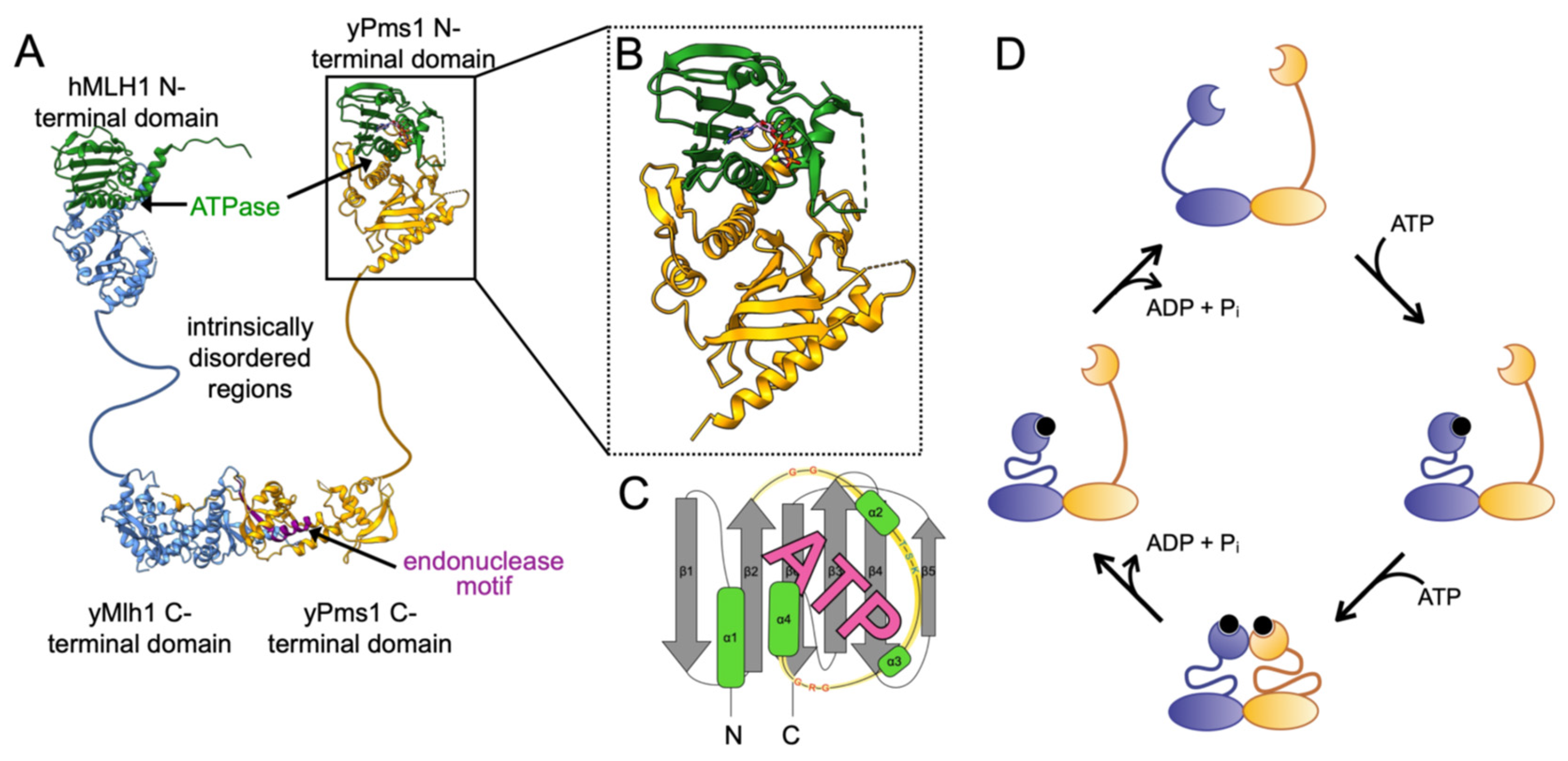
Figure 3.
ATPase cycles of DNA gyrase, Hsp90, and histidine kinase. (A) DNA gyrase. DNA gyrase A subunits (GyrA) bind supercoiled DNA, with the G-segment (grey) positioned for cleavage and the T-segment (black) destined for strand passage. DNA gyrase B (GyrB) binds two molecules of ATP (black circles), driving dimerization of its N-terminal domains and concerted cleavage of the G-segment by GyrA [74,93]. The T-segment is then passed through the double-strand break, released, and the enzyme resets as ATP hydrolysis opens the GyrB N-terminal domains for another catalytic cycle. Adapted from model in [85]. (B) Hsp90. The N-terminal domains of the homodimer bind ATP following engagement of a client polypeptide (brown), leading to dimerization and formation of a closed state. Co-chaperones such as Aha1 (green) stimulate ATP hydrolysis, releasing inorganic phosphate while ADP (grey circles) remains bound. The folded client and ADP are subsequently released, resetting the chaperone. Adapted from models in [94,95] (C) Histidine kinase. Membrane phospholipids are depicted in green. ATP binding promotes interactions between the C-terminal ATPase domains and the phosphotransfer domains, leading to autophosphorylation of a conserved histidine. Hydrolysis and phosphotransfer then relay the phosphoryl group to a response regulator, typically a transcription factor (teal). Phosphoryl transfer resets the kinase for another round of sensing and signaling. Adapted from models in [96,97].
Figure 3.
ATPase cycles of DNA gyrase, Hsp90, and histidine kinase. (A) DNA gyrase. DNA gyrase A subunits (GyrA) bind supercoiled DNA, with the G-segment (grey) positioned for cleavage and the T-segment (black) destined for strand passage. DNA gyrase B (GyrB) binds two molecules of ATP (black circles), driving dimerization of its N-terminal domains and concerted cleavage of the G-segment by GyrA [74,93]. The T-segment is then passed through the double-strand break, released, and the enzyme resets as ATP hydrolysis opens the GyrB N-terminal domains for another catalytic cycle. Adapted from model in [85]. (B) Hsp90. The N-terminal domains of the homodimer bind ATP following engagement of a client polypeptide (brown), leading to dimerization and formation of a closed state. Co-chaperones such as Aha1 (green) stimulate ATP hydrolysis, releasing inorganic phosphate while ADP (grey circles) remains bound. The folded client and ADP are subsequently released, resetting the chaperone. Adapted from models in [94,95] (C) Histidine kinase. Membrane phospholipids are depicted in green. ATP binding promotes interactions between the C-terminal ATPase domains and the phosphotransfer domains, leading to autophosphorylation of a conserved histidine. Hydrolysis and phosphotransfer then relay the phosphoryl group to a response regulator, typically a transcription factor (teal). Phosphoryl transfer resets the kinase for another round of sensing and signaling. Adapted from models in [96,97].
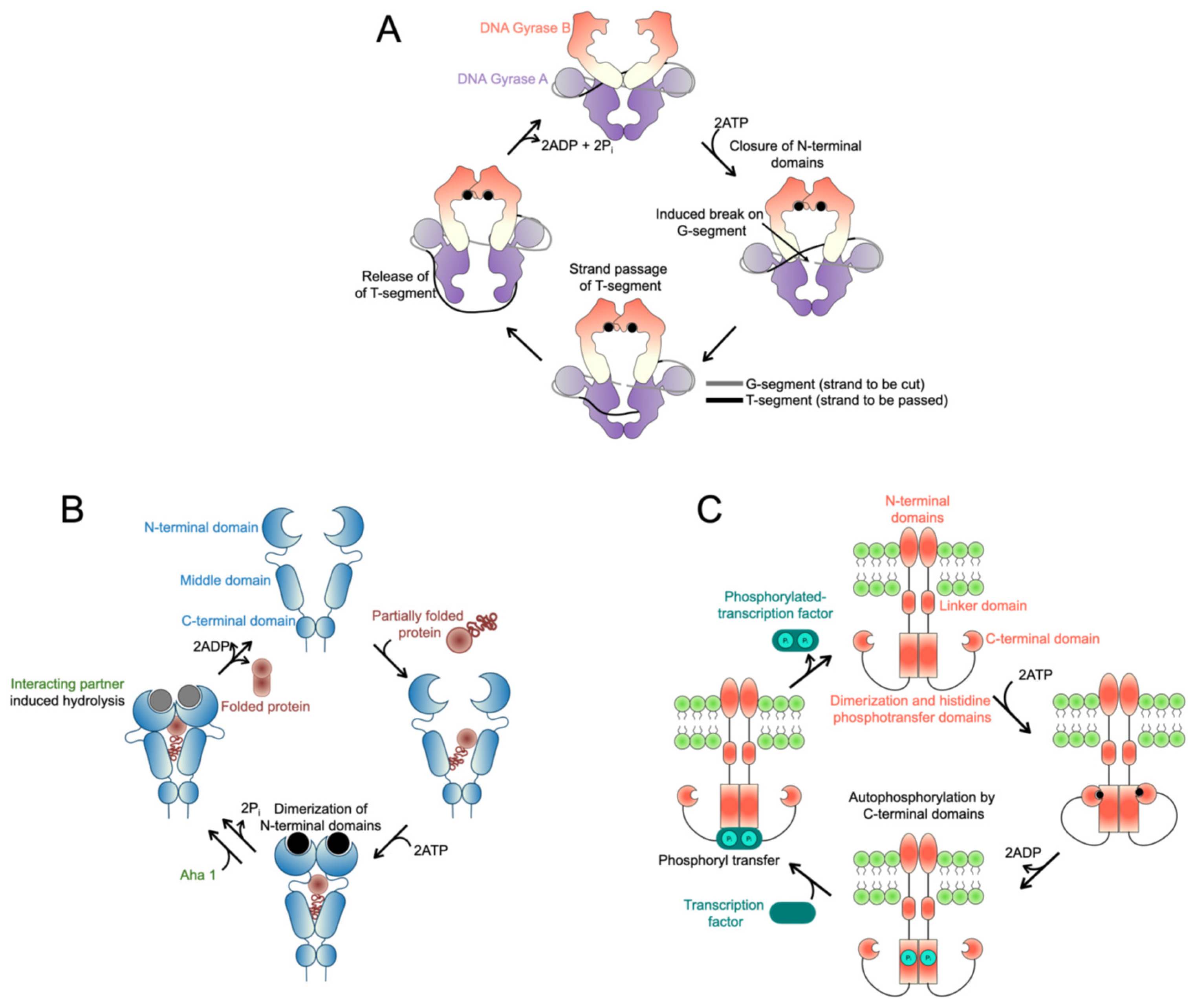
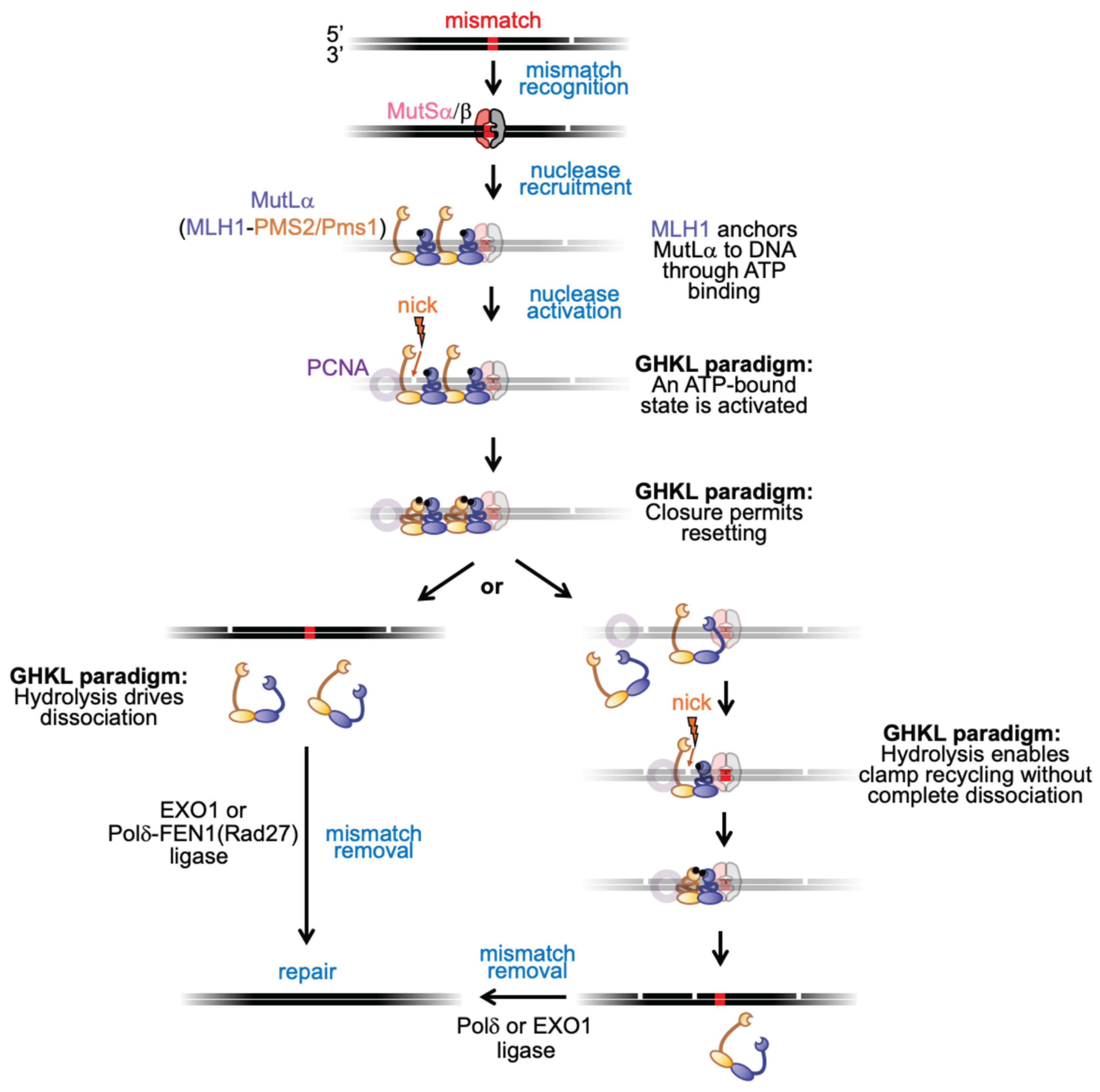
Figure 4.
Model for MutLα’s ATPase roles in eukaryotic MMR through the GHKL paradigm. Mismatch recognition by MutSα/β triggers MutLα recruitment (MutS activities not shown in detail). To highlight MutLα ATPase clamp dynamics, MutS and DNA are greyed. MutLα ATPase is stimulated by DNA, with MLH1 (blue) positioned as the MutS-interacting anchor and PMS2/Pms1 (yellow) interacting with PCNA (purple, greyed). PCNA stimulates both endonuclease (lightning bolt) and ATPase activities. In the GHKL paradigm, ATP hydrolysis resets the clamp, leading either to iterative nicking without dissociation or to complete dissociation and recycling which could result in iterative nicking if the pathway restarts. Mismatches can be removed by Pold or EXO1.
Figure 4.
Model for MutLα’s ATPase roles in eukaryotic MMR through the GHKL paradigm. Mismatch recognition by MutSα/β triggers MutLα recruitment (MutS activities not shown in detail). To highlight MutLα ATPase clamp dynamics, MutS and DNA are greyed. MutLα ATPase is stimulated by DNA, with MLH1 (blue) positioned as the MutS-interacting anchor and PMS2/Pms1 (yellow) interacting with PCNA (purple, greyed). PCNA stimulates both endonuclease (lightning bolt) and ATPase activities. In the GHKL paradigm, ATP hydrolysis resets the clamp, leading either to iterative nicking without dissociation or to complete dissociation and recycling which could result in iterative nicking if the pathway restarts. Mismatches can be removed by Pold or EXO1.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



