Submitted:
27 October 2025
Posted:
28 October 2025
You are already at the latest version
Abstract
In this study, an alternative methodology for the aqueous synthesis of chalcones is presented. This methodology is ecologically sound, since it occurs at room temperature and utilizes only KOH (a green base). In addition, it showed a broad applicability, as evidenced by the preparation of 37 derivatives in good to excellent yields (up to 98%). Moreover, most compounds were recovered pure enough through straightforward fil-tration and water washing. The development of the reaction is accompanied by a study that demonstrates that the yield of the products and the reaction mechanism are directly affected by the solubility and physical state of the starting materials.
Keywords:
aqueous synthesis of chalcones
; green synthesis of organic compounds
; sustainable chemistry
; flavonoids
; Claisen-Schmidt condensation
1. Introduction
Chalcones are classified as flavonoids, a diverse group of secondary metabolites derived from plants, that exhibit significant biological activities [1,2]. They function as intermediary metabolic products in the synthesis of various flavonoids, including flavanones, flavones and flavanols, among others [3]. Additionally, these compounds are present in different plant issues, where they fulfill critical functions, for example: pigmentation, attractor or repellent of insects, antifeedants, allelochemicals and UV light protectors, etc [4,5].
Chalcones have an interesting pharmacological profile which gives them a high probability of becoming therapeutic agents [6,7]. The α,β-unsaturated carbonyl system present in chalcones imparts them electrophilic properties, so like in nature, chalcones are important intermediaries for the synthesis of more complex molecules [8,9,10,11,12,13,14]. Numerous reports illustrate the pharmacological properties of chalcones in the literature, including antioxidants [15,16], anti-inflammatory [17], antiallergic [18,19], anticancer [14,20,21], antimicrobic [22,23] and anti-diabetic,[24] to mention a few. This versatility confers upon the chalcone framework, the privileged structure denomination and underscores the necessity for the continuous development of new synthetic methodologies for their obtention [25,26,27]. Some examples of chalcones with important biological properties are shown in Figure 1. Licochalcone A and Marachalcone A are Natural Products obtained from plants, while compounds 1Ae and 1Ge are products of laboratory that were synthesized with the methodology here presented. Compound 1Ae possess important antiproliferative properties; for example is active against human A549 cells (lung cancer) [28], against human HCT116 cells (colon cancer), and against CAL51 cells (breast cancer) [29]. Compound 1Ge possess anti-inflammatory activity [30].
Conventional methodologies employed for the synthesis of organic substances and active pharmaceutical ingredients (APIs) generate a substantial volume of waste and necessitate substantial energy consumption. A significant proportion of these methodologies are corrosive and hazardous [33] This is not unexpected, as they were developed during a period that did not prioritize environmental considerations. Contemporary methods in organic synthesis are striving to adhere to the principles of sustainability and green chemistry, recognizing the imperative to safeguard our environment and natural resources. Consequently, traditional methodologies are being superseded by more sustainable alternatives. In this context, a water-promoted methodology for the synthesis of chalcones is presented. This method is ecologically sound and demonstrates the potential and limitations of water to promoting the synthesis of organic molecules; thus, contributing to novel ecological trends.
2. Results and Discussion
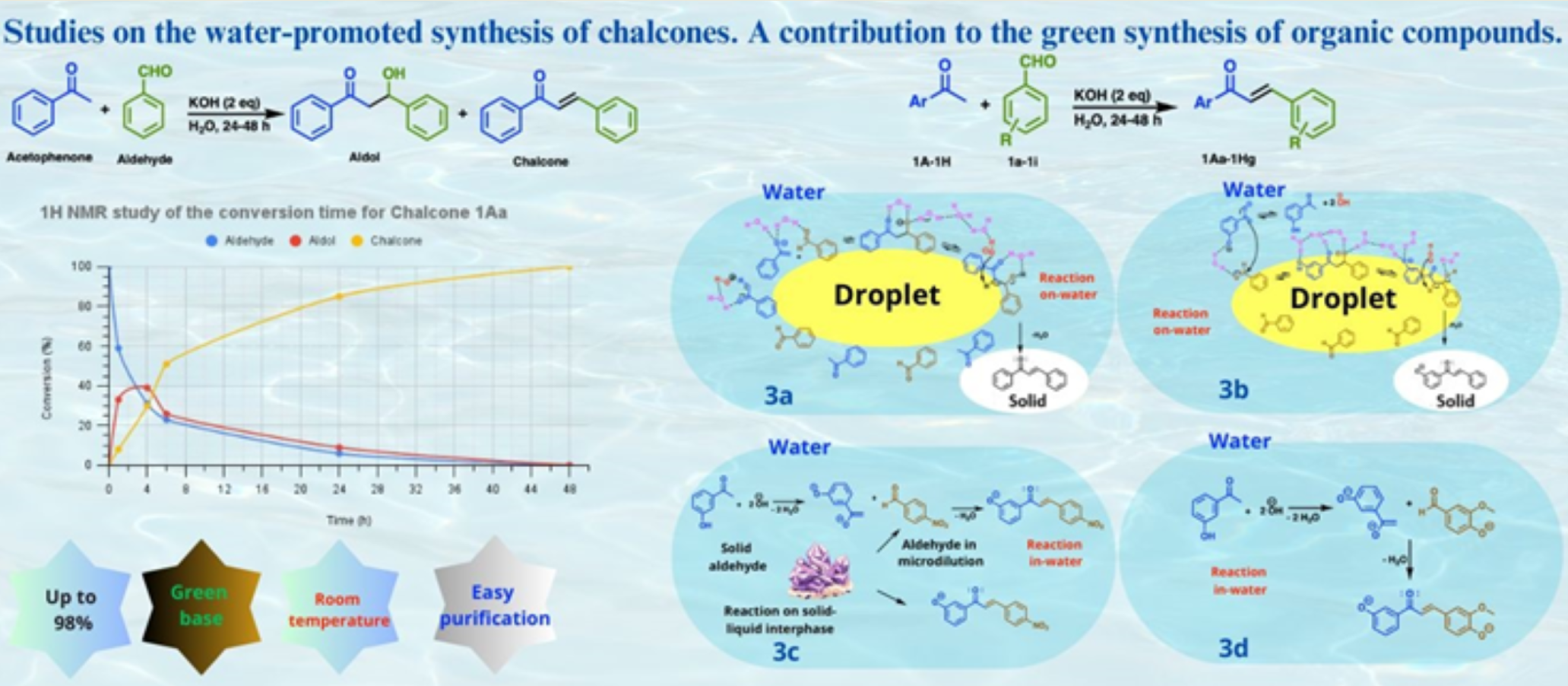
It was previously theorized, that a chemical reaction could not occur unless reagents were liquid, or in solution [34,35] This theory led to the extensive use of organic solvents and the limited or non-use of water in organic product manufacturing. Water is incapable of dissolving most organic compounds. However, it should be noted that many organic solvents are volatile and toxic, and they frequently constitute most of the waste produced during a reaction. Consequently, the employment of water or environmentally friendly solvents has emerged as the prevailing approach in the development of novel methodologies. Water’s utilization in the synthesis of chalcones has been minimal, with alcohols being the most prevalent solvents. A comprehensive review of the extant literature revealed five distinct water-promoted methodologies for synthesizing chalcones, all of which operate under the Claysen-Schmidt condensation conditions, employing aromatic ketones and aromatic aldehydes as starting materials (Scheme 1).
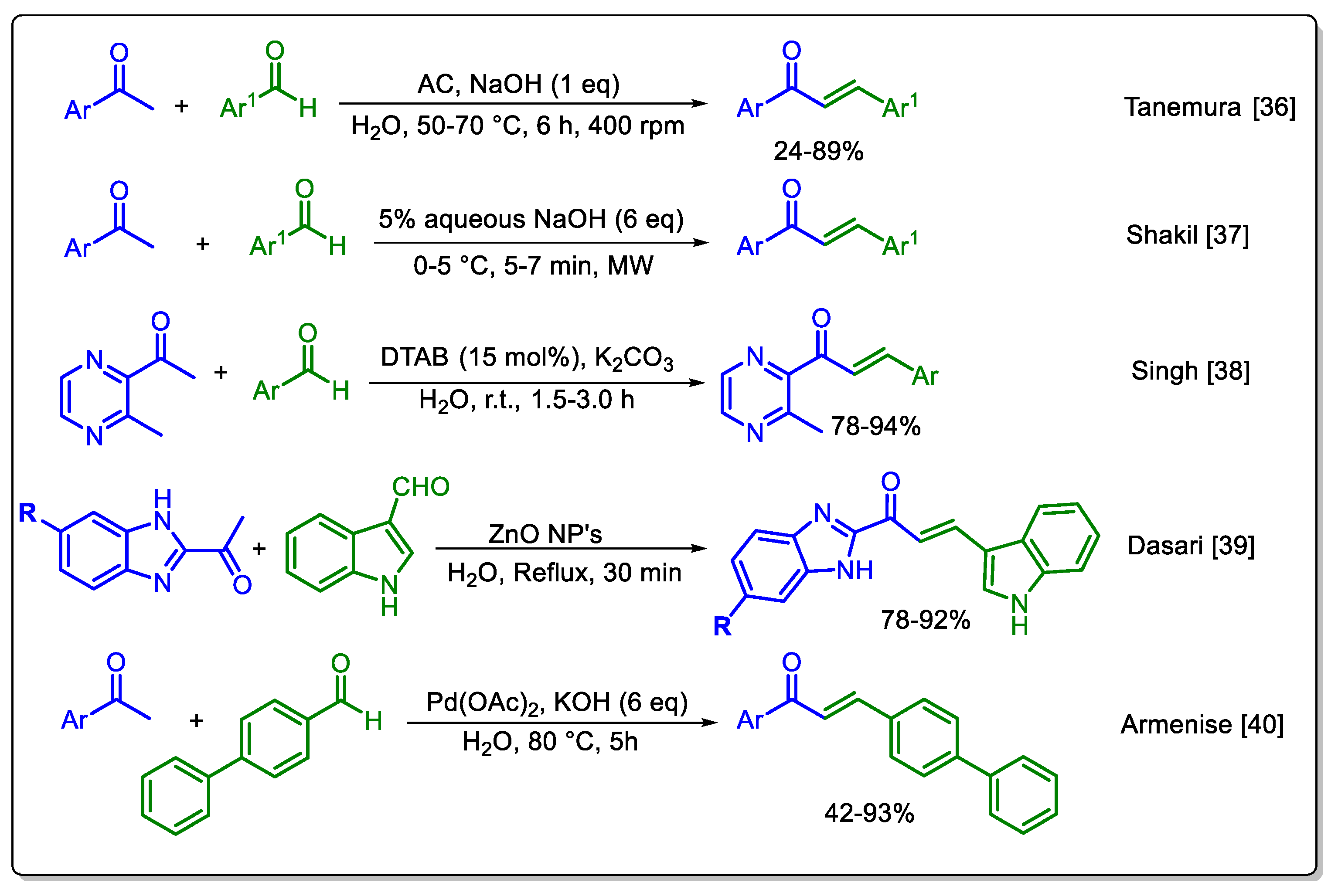
In these methodologies, a variety of catalysts are employed, including, but not limited to activated charcoal (AC), metallic salts such as ZnO and Pd(OAc)2 and dodecyltrimethylammonium bromide (DTAB), a surfactant agent and phase transference catalyst. The temperature range of the reactions extends from 0 °C to reflux, and the use of strong bases as NaOH, KOH and K2CO3 is employed. Under these conditions, reaction time is reduced, though the most optimal result was obtained when MW energy was used, since time is reduced to minutes. Conversely, the methodology herein introduced operates at ambient temperature and is more straightforward, as it does not necessitate specialized equipment, specific catalysts, or additives, solely requiring potassium hydroxide, a green base. Furthermore, the method is highly versatile. As the reactions occur in an aqueous environment and the majority of compounds are obtained by straightforward filtration, the generation of waste is significantly reduced.
Acetophenone 1A and salicylaldehyde 1a were the starting materials utilized during the screening of the reaction. Initially, potassium hydroxide (1 eq) and water (drinking or distilled, 2 mL) were tested to promote the reaction (Table 1, entry 1), which was performed at room temperature. As this reaction occurs independently of a surfactant agent, vigorous stirring was required to promote the formation of organic droplets; consequently, the stirring rate was set at 850 rpm. Fortunately, after 4 days, a solid precipitate was obtained from the reaction. Subsequent analysis of the solid by 1H-NMR allowed its identification as the chalcone 1Aa. The experiment was repeated (entry 2), but now using 1 mL of water, and chalcone 1Aa was obtained in excellent yield (88%). In comparison, when using 1 mL of water, stirring and filtering was more difficult and a slurry was formed in the vial; therefore, 2 mL of water was established for the reaction.
Subsequent to demonstrating the capacity to obtain a chalcone under these conditions, the impact of varying bases was examined. However, after a seven-day reaction period in the presence of weaker bases, such as Na2CO3 or K2CO3, no product formation was observed (entries 3 and 4). In an effort to reduce the reaction time, the effects of varying quantities of strong base were examined. Notably, the use of 2 eq of KOH resulted in a significant reduction of the reaction time to 2 days (entry 5). A similar outcome was observed when NaOH was employed in equal proportion (entry 7). In addition, the effect of 3 eq of KOH (entry 6), and KOH in the presence of piperidine (0.1 mol, entry 8) were tested; however, the reaction time remained unaltered (2 d). To ascertain whether the reaction rate could be enhanced, an additional experiment was conducted at 60 °C (entry 9). Unfortunately, the presence of multiple products was detected by TLC, which precluded a conclusive determination, and the optimized conditions for the reaction were identified in entry 5.
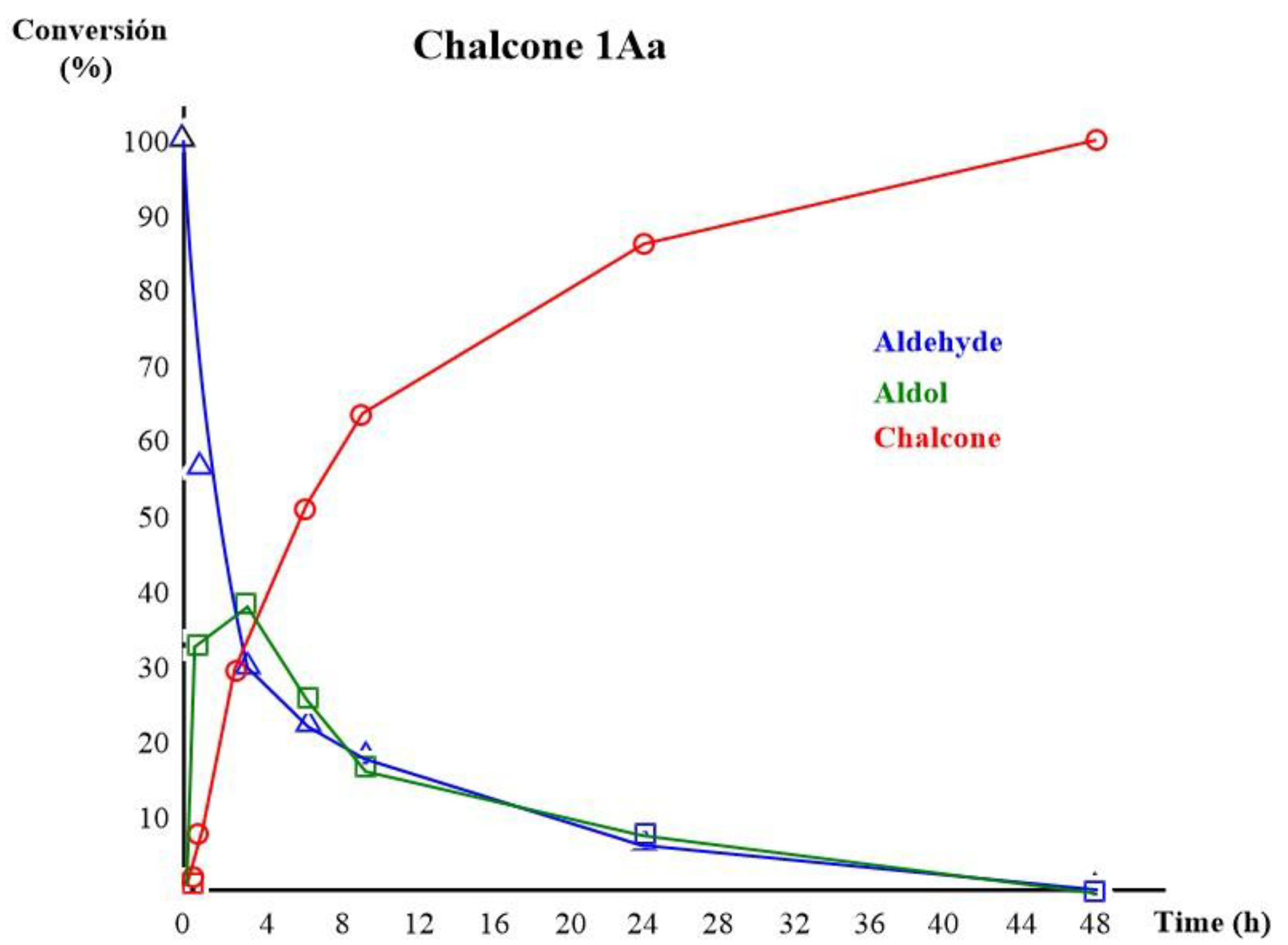
A 1H NMR study was conducted to verify the conversion time of starting materials to product, under the best conditions (Figure 2). Initially, the aldol:chalcone ratio was determined to be 8:33 (1 h). At the 4-hour mark, the aldol concentration peaked at 39% while the chalcone concentration continued to rise, reaching 30%. At 6 hours, the aldol concentration (26%) was half the chalcone concentration (51%), indicating the rapid conversion of aldol to chalcone and its enhanced stability. At 24 hours, the reaction was still in progress, with a huge concentration of chalcone (85%) and the continued presence of aldol (9%). However, at 48 hours, the aldol was undetectable, and the chalcone had reached a concentration of 100%. This experimental data enabled the determination of the reaction time to be 48 hours.
Figure 2.
1H NMR study of the conversion time for Chalcone 1Aa.
Following the establishment of the reaction conditions (see Table 1, entry 5), the scope was investigated, and a total of nine distinct series of chalcones (1Aa-1Ja) were prepared (see Table 2). During the experimental phase, it was observed that the yield and the compound’s acquisition were less influenced by the electronic effects of substituents on aromatic rings compared to their physical state and hydrophilicity/lipophilicity. Fortunately, it was observed that the majority of products were obtained in good to excellent yields (66-98%), with the exception of compound 1Dd (entry 19, 40%). Notably, reaction was successfully scaled up to 50 mmol with chalcone 1Aa, resulting in 87% yield (entry 39). An important advantage of this methodology is that most products were obtained in a pure state through simple filtration and water washing; in addition, it can be extended to the preparation of heterochalcones (1Ia, 1Ja). It is noteworthy that certain 2-OH-chalcones in series 1F, 1G, and 1H exhibit a propensity to cyclize, resulting in the formation of flavanones. However, the present study focuses exclusively on the introduction of stable chalcones [12].
In this study, starting materials (SM) in both solid and liquid states were utilized (see Table 2 and Table 3). While liquid starting materials were all water-insoluble (Lipophilic, L), among the solid materials, hydroxylated ketones (1C, 1E, 1H) were water-soluble, (Hydrophilic Solid HS), while nitrated ketone (1D) and nitrated aldehyde (1d) were water-insoluble (Lipophilic Solid, LS). As previously mentioned, the physical state of reactants and their water-solubility are significant factors because they affect the composition of the reaction system, the mechanism, the yield and the reaction possibility itself.
In concordance with these considerations, six distinct water/starting material-starting material (W/SM-SM) reaction systems can be delineated (Table 3). These include the following: water/lipophilic liquid-lipophilic liquid (W/L-L), water/lipophilic liquid-lipophilic solid (W/L-LS), water/lipophilic liquid-hydrophilic solid (W/L-HS), water/hydrophilic solid-lipophilic solid (W/HS-LS), water/lipophilic solid-lipophilic solid (W/LS-LS) and water/hydrophilic solid-hydrophilic solid (W/HS-HS). As previously stated, the majority of products were obtained in good yields, indicating that water can facilitate reactions, irrespective of the hydrophilicity/lipophilicity, or physical state of SM. However, in instances where both SM are solid and lipophilic, a water/solid system (W/LS-LS) is formed, resulting in a suspension. Among the six systems that have been described, this last one presents the most significant challenges to the progression of the reaction, given that both SM are solid and lipophilic. In a theory, this reaction should not occur; however, product 1Dd was obtained in low yield (40%, entry 19).
According to the system formed, different mechanisms can be proposed, (Table 3, Figure 3). In the W/L-L system, both SM (aldehyde and ketone) are liquid, lipophilic, and follow the classical on-water mechanism (Figure 3a). The proximity of the SM molecules to each other enables their susceptibility to water pressure, thereby facilitating the reaction process. In the interphase, hanging hydroxyls assist in enolate and chalcone formation by extracting the α-protons from the ketone and the aldol respectively [34,41] The resulting chalcone product, due to its solid and water-insoluble nature, precipitates upon formation. During the experimental phase, it was observed that when an LS was mixed with a lipophilic liquid L, both reactants were incorporated into a lipophilic droplet, and a W/L-LS system was formed. This system exhibited a mechanism analogous to that of W/L-L (Figure 3a). In the case of the W/L-HS system, since the ketone is hydrophilic, the hydroxyl deprotonation and enolate formation elapses into the water (Figure 3b). Subsequently, the reaction progresses to the interphase, where the process culminates in the formation of the chalcone. Hanging hydroxyls facilitate the hydrogen removal from the aldol in the interphase, where both SM are present. Therefore, this could be regarded as a variation of the on-water mechanism described for the W/L-L system.
Aqueous reactions were observed to proceed even when both reactants are solid. In the W/LS-HS system, where one reactant is hydrophilic and the other is lipophilic, no problems were observed, even though the lipophilic solid was dispersed and forming a heterogeneous solid-liquid suspension (Figure 3c). The mechanism in this case could elapse in-water, by micro-solubilization of the lipophilic reactant [42], or in the solid-liquid interphase. For the W/LS-LS system, the mechanism could elapse in the same way. Finally, the W/HS-HS system reaction proceeds in water, showing the possibility of carrying the Claisen-Schmidt condensation out in this solvent, despite the excess of water that in a theory, should avoid the reaction advance. (Figure 3d) [42].
3. Materials and Methods
The melting points were determined using a Fisher-Jones apparatus and are not corrected. 1H-NMR, 13C and DEPTQ spectra were recorded at 25°C on a Bruker Ascend™ 600 MHz (1H NMR and 151 MHz DEPTQ, 13C). Chemical shifts are expressed as δ (ppm) values relative to TMS as internal standard and coupling constants (J) values are given in Hertz. The majority of spectra were recorded in CDCl3. Multiplicities are described as s (singlet), d (doublet), dd (doublet double), dt (triplet double), t (triplet), q (quartet), or m (multiplet). All reagents were obtained from Sigma-Aldrich. Mass Spectra were recorded on a JEOL JMS-700 MStation at a voltage of 70 eV. The chromatography solvents were of technical grade, and they were purified and distilled prior to use.
Synthetic Procedure. Into a glass vial of 5 mL, water (either drinking water or distilled water, 1 mL), the corresponding acetophenone (0.5 mmol), the corresponding benzaldehyde (0.5 mmol) and sodium hydroxide (1N solution, 1 mmol, 1 mL) were introduced. The vial cap was placed, and the mixture was stirred at a rate of 850 rpm for a period of 48 h. The progression of the reaction was monitored by TLC. The solid obtained was filtered, washed with water (either drinking water or distilled water, 2x1 mL) and allowed to dry overnight at room temperature. Finally, the solid was introduced into a desiccator for a period of two days.
(E)-1-phenyl-3-(phenyl)prop-2-en-1-one (1Aa). This compound was obtained pure enough after filtration to afford white crystals (91.6 mg, 88 %), m.p. 56-58 °C (lit, 56-57 °C) [21]. 1H NMR (600 MHz, CDCl3), δ (ppm) 8.02 (d, 2H, J = 7.3 Hz), 7.81 (d, 1H, J = 15.7 Hz), 7.67-7.63 (m, 2H), 7.59 (t, 1H, J = 7.4 Hz), 7.53 (d, 1H, J = 15.7 Hz), 7.51 (t, 2H, J = 7.6 Hz), 7.44-7.40 (m, 3H). 13C NMR (150 MHz, CDCl3), δ (ppm) 190.8, 145.0, 138.5, 135.1, 132.9, 130.7, 129.1, 128.8, 128.7, 128.6, 122.4.
(E)-3-(3-fluorophenyl)-1-phenylprop-2-en-1-one (1Ab) This compound was obtained pure enough after filtration to afford yellow crystals (91.5 mg, 81 %), m.p. 47-49 °C (lit, 48 °C) [43]. 1H NMR (600 MHz, CDCl3), δ (ppm) 8.03 (d, 2H, J = 8.5 Hz), 7.91 (d, 2H, J = 15.9 Hz), 7.66 (d, 1H, J = 15.9 Hz), 7.66 (s, 1H), 7.60 (t, 1H, J = 7.3 Hz), 7.52 (t, 2H, J = 7.6 Hz), 7.42-7.37 (m, 1H), 7.21 (t, 1H, J = 7.6 Hz), 7.17-7.11 (m, 1H). DEPTQ (150 MHz, CDCl3), δ (ppm) 190.4, 164.1, 143.4, 138.2, 137.4, 133.1, 130.7, 129.1, 128.9, 128.8, 124.7, 123.5, 117.6, 114.7
(E)-3-(4-fluorophenyl)-1-phenylprop-2-en-1-one (1Ac). This compound was obtained pure enough after filtration to afford white crystals (99.4 mg, 88 %), m.p. 81-83 °C (lit, 79 °C) [43]. 1H NMR (600 MHz, CDCl3), δ (ppm) 8.01 (d, 2H, J = 8.5 Hz), 7.77 (d, 1H, J = 15.7 Hz), 7.64 (dd, 2H, J = 8.6, 5.4 Hz), 7.59 (t, 1H, J = 7.4 Hz), 7.51 (t, 2H, J = 7.6 Hz), 7.45 (d, 1H, J = 15.7 Hz), 7.11 (t, 2H, J = 8.6 Hz). DEPTQ (150 MHz, CDCl3), δ (ppm) 190.5, 165.1, 143.7, 138.3, 133.0, 131.4, 130.5, 129.1, 128.8, 122.1, 116.4.
(E)-3-(4-nitrophenyl)-1-phenylprop-2-en-1-one (1Ad). This compound was obtained pure enough after filtration to afford yellow crystals (103.8 mg, 82 %), m.p. 156-158 °C (lit, 157-159 °C) [44]. 1H NMR (600 MHz, CDCl3), δ (ppm) 8.28 (d, 2H, J = 8.8 Hz), 8.03 (d, 2H, J = 8.5 Hz), 7.82 (d, 1H, J = 15.8 Hz), 7.79 (d, 2H, J = 8.6 Hz), 7.64 (d, 1H, J = 15.7 Hz), 7.63 (t, 1H, J = 8.5 Hz), 7.54 (t, 2H, J = 7.7 Hz). 13C NMR (150 MHz, CDCl3), δ (ppm) 189.6, 148.5, 141.5, 140.0, 133.3, 130.4, 128.9, 128.8, 125.7, 124.3, 124.2.
(E)-3-(4-methoxyphenyl)-1-phenylprop-2-en-1-one (1Ae). This compound was obtained pure enough after filtration to afford white crystals (93 mg, 78 %), m.p. 73-75 °C (lit, 77-78 °C) [21]. 1H NMR (600 MHz, CDCl3), δ (ppm) 8.01 (d, 2H, J = 8.5 Hz), 7.79 (d, 1H, J = 15.6 Hz), 7.61 (d, 2H, J = 8.6 Hz), 7.58 (t, 1H, J = 7.4 Hz), 7.51 (t, 2H, J = 7.6 Hz), 7.42 (d, 1H, J = 15.6 Hz), 6.95 (d, 2H, J = 8.8 Hz), 3.87 (s, 3H). DEPTQ (150 MHz, CDCl3), δ (ppm) 190.8, 161.9, 144.9, 138.7, 132.7, 130.4, 128.7, 128.6, 127.9, 120.1, 114.6, 55.6.
(E)-1-(4-chlorophenyl)-3-phenylprop-2-en-1-one (1Ba). This compound was obtained pure enough after filtration to afford a yellow solid (104.4 mg, 86 %), m.p. 99-101 °C (lit, 94-95 °C) [45]. 1H NMR (600 MHz, CDCl3), δ (ppm) 7.96 (d, 2H, J = 8.5 Hz), 7.81 (d, 1H, J = 15.7 Hz), 7.64 (dd, 2H, J = 6.4, 3.5 Hz), 7.48 (d, 2H, J = 8.6 Hz), 7.47 (d, 1H, J = 15.5 Hz), 7.45-7.40 (m, 3H). DEPTQ (150 MHz, CDCl3), δ (ppm) 189.4, 145.5, 139.4, 136.7, 134.9, 130.9, 130.1, 129.2, 129.1, 128.7, 121.8.
(E)-1-(4-chlorophenyl)-3-(3-fluorophenyl)prop-2-en-1-one (1Bb). This compound was obtained pure enough after filtration to afford a white solid (105.6 mg, 81 %), m.p. 99-101 °C (lit, m.p. not reported) [45]. 1H NMR (600 MHz, CDCl3), δ (ppm) 7.96 (d, 2H, J = 8.6 Hz), 7.76 (d, 1H, J = 15.7 Hz), 7.48 (d, 2H, J = 8.7 Hz), 7.46 (d, 1H, J = 15.8 Hz), 7.42-7.37 (m, 2H), 7.34 (dd, 1H, J = 8.6, 2.4 Hz), 7.15-7.09 (m, 1H). DEPTQ (150 MHz, CDCl3), δ (ppm) 188.96, 164.07, 143.90, 139.64, 137.14, 136.43, 130.74, 130.07, 129.18, 124.73, 122.88, 117.76, 114.74.
(E)-1-(4-chlorophenyl)-3-(4-fluorophenyl)prop-2-en-1-one (1Bc). This compound was obtained pure enough after filtration to afford a white solid (104.3 mg, 80 %), m.p. 120-122 °C (lit, 119-123 °C) [45]. 1H NMR (600 MHz, CDCl3), δ (ppm) 7.96 (d, 2H, J = 8.5 Hz), 7.78 (d, 1H, J = 15.7 Hz), 7.64 (dd, 2H, J = 8.6, 5.4 Hz), 7.48 (d, 2H, J = 8.5 Hz), 7.40 (d, 1H, J = 15.7 Hz), 7.12 (t, 2H, J = 8.5 Hz). DEPTQ (150 MHz, CDCl3), δ (ppm) 189.1, 165.2, 144.1, 139.5, 136.6, 131.2, 130.6, 130.0, 129.1, 121.4, 116.4.
(E)-1-(4-chlorophenyl)-3-(4-nitrophenyl)prop-2-en-1-one (1Bd). This compound was obtained pure enough after filtration to afford yellow crystals (95 mg, 66%), m.p. 161-163 °C (lit, 164-166 °C) [44]. 1H NMR (600 MHz, CDCl3), δ (ppm) 8.28 (d, 2H, J = 8.7 Hz), 7.98 (d, 2H, J = 8.5 Hz), 7.83 (d, 1H, J = 15.6 Hz), 7.79 (d, 2H, J = 8.8 Hz), 7.59 (d, 1H, J = 15.7 Hz), 7.51 (d, 2H, J = 8.5 Hz). DEPTQ (150 MHz, CDCl3), δ (ppm) 188.5, 148.9, 142.1, 141.0, 140.1, 136.0, 130.1, 129.3, 129.1, 125.4, 124.4.
(E)-1-(4-chlorophenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (1Be). This compound was obtained pure enough after filtration to afford a white solid (107.7 mg, 79 %), m.p. 120-122 °C (lit, 119-123 °C) [46]. 1H NMR (600 MHz, CDCl3), δ (ppm) 7.95 (d, 2H, J = 8.6 Hz), 7.78 (d, 1H, J = 15.6 Hz), 7.60 (d, 2H, J = 8.8 Hz), 7.47 (d, 2H, J = 8.6 Hz), 7.35 (d, 1H, J =15.5 Hz), 6.94 (d, 2H, J = 8.8 Hz), 3.86 (s, 3H). DEPTQ (150 MHz, CDCl3), δ (ppm) 189.3, 162.0, 145.3, 139.1, 137.0, 130.5, 130.0, 129.0, 127.6, 119.4, 114.6, 55.6.
(E)-1-(3-hydroxyphenyl)-3-phenylprop-2-en-1-one (1Ca). This compound was purified by column chromatography (Hexane 10 : AcOEt 1) to afford a white solid (85.2 mg, 76 %), m.p. 134-136 °C (lit, 130-132 °C) [47]. 1H NMR (600 MHz, CDCl3), δ (ppm) 7.80 (d, 1H, J = 15.7 Hz), 7.66-7.62 (m, 2H), 7.58 (d, 1H, J = 8.2 Hz), 7.53 (s, 1H), 7.50 (d, 1H, J = 15.7 Hz), 7.43-7.40 (m, 3H), 7.38 (t, 1H, J = 7.9 Hz), 7.11 (ddd, 1H, J = 8.1, 2.6, 0.87 Hz). DEPTQ (150 MHz, CDCl3), δ (ppm) 190.6, 156.4, 145.4, 139.9, 135.0, 130.8, 130.1, 129.1, 128.7, 122.2, 121.2, 120.4, 115.3.
(E)-3-(3-fluorophenyl)-1-(3-hydroxyphenyl)prop-2-en-1-one (1Cb). This compound was obtained pure enough after filtration to afford a white solid (94.5 mg, 78 %), m.p. 120-122 °C (lit, 121-123 °C) [47]. 1H NMR (600 MHz, CDCl3), δ (ppm) 7.76 (d, 1H, J = 15.7 Hz), 7.58 (s, 1H), 7.57 (d, 1H, J = 7.7 Hz), 7.48 (d, 1H, J = 15.7 Hz), 7.41-7.35 (m, 3H), 7.33 (d, 1H, J = 7.3 Hz), 7.14-7.08 (m, 2H). DEPTQ (150 MHz, CDCl3), δ (ppm) 190.4, 164.1, 156.6, 143.9, 139.5, 137.3, 130.7, 130.1, 124.7, 123.3, 121.2, 120.7, 117.7, 115.3, 114.8.
(E)-3-(4-fluorophenyl)-1-(3-hydroxyphenyl)prop-2-en-1-one (1Cc). This compound was purified by column chromatography (Hexane 10 : AcOEt 1) to afford a white solid (100.5 mg, 83 %), m.p. 123-125 °C (lit, 124-126 °C) [47]. 1H NMR (600 MHz, CDCl3), δ (ppm) 7.78 (d, 1H, J = 15.7 Hz), 7.63 (dd, 2H, J = 8.8, 5.4 Hz), 7.57 (d, 1H, J = 7.6 Hz), 7.54 (s, 1H), 7.42 (d, 1H, J = 15.7 Hz), 7.38 (t, 1H, J = 7.8 Hz), 7.14-7.08 (m, 3H). DEPTQ (150 MHz, CDCl3), δ (ppm) 190.4, 165.2, 156.4, 144.1, 139.8, 131.3, 130.6 (2C), 130.1, 121.9, 121.2, 120.5, 116.4 2(C), 115.3.
(E)-1-(3-hydroxyphenyl)-3-(4-nitrophenyl)prop-2-en-1-one (1Cd). This compound was purified by column chromatography (Hexane 8 : AcOEt 1) to afford a yellow solid (103.7 mg, 77%), m.p. 176-178 °C (lit, 177-180 °C) [48]. 1H NMR (600 MHz, CDCl3), δ (ppm) 8.28 (d, 2H, J = 8.8 Hz), 7.81 (d, 1H, J = 15.8 Hz), 7.78 (d, 2H, J = 8.7 Hz), 7.59 (d, 1H, J = 15.7 Hz), 7.63-7.57 (m, 1H), 7.50 (s, 1H), 7.41 (t, 1H, J = 7.9 Hz), 7.11 (dd, 1H, J = 8.0, 2.5 Hz). DEPTQ (150 MHz, CDCl3), δ (ppm) 189.4, 158.47, 148.68, 141.80, 140.71, 139.08, 130.27 (CH, C-6′), 129.10, 125.88, 124.40, 121.36, 120.75, 115.23.
(E)-1-(3-hydroxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (1Ce). This compound was purified by column chromatography (Hexane 10 : AcOEt 1) to afford a yellow solid (101.7 mg, 80%), m.p. 141-143 °C (lit, 142-144 °C) [48]. 1H NMR (600 MHz, CDCl3), δ (ppm) 8.28 (d, 2H, J = 8.8 Hz), 7.81 (d, 1H, J = 15.8 Hz), 7.78 (d, 2H, J = 8.7 Hz), 7.59 (d, 1H, J = 15.7 Hz), 7.63-7.57 (m, 1H), 7.50 (s, 1H), 7.41 (t, 1H, J = 7.9 Hz), 7.11 (dd, 1H, J = 8.0, 2.5 Hz). DEPTQ (150 MHz, CDCl3), δ (ppm) 190.7, 162.0, 156.5, 145.3, 140.1, 130.5, 130.0, 127.7, 121.0, 120.2, 120.0, 115.3, 114.6, 55.6.
(E)-3-(4-hydroxy-3-methoxyphenyl)-1-(3-hydroxyphenyl)prop-2-en-1-one (1Cf). This compound was purified by column chromatography (Hexane 10 : AcOEt 1) to afford a yellow solid (92 mg, 68%), m.p. 158-160 (lit, m.p. not reported) [49]. 1H NMR (600 MHz, CDCl3), δ (ppm) 7.69 (d, 1H, J = 15.4 Hz), 7.54-7.52 (m, 1H), 7.49 (d, 1H, J = 15.5 Hz), 7.40 (t, 1H, J = 2.1 Hz), 7.34-7.28 (m, 2H), 7.19 (dd, 1H, J = 8.2, 2.0 Hz), 7.01 (dd, 1H, J = 8.0, 2.6 Hz), 6.82 (d, 1H, J = 8.0 Hz), 3.9 (s. 3H). 13C NMR (150 MHz, CDCl3), δ (ppm) 192.7, 159.1, 151.1, 149.4, 147.1, 141.1, 130.8, 128.2, 124.9, 121.1, 120.9, 120.1, 116.6, 115.8, 112.1, 56.5.
(E)-1-(4-nitrophenyl)-3-phenylprop-2-en-1-one (1Da). This compound was obtained pure enough after filtration to afford a yellow solid (117.8 mg, 93 %), m.p. 148-150 °C (lit, 143-144 °C) [45]. 1H NMR (600 MHz, CDCl3), δ (ppm) 8.36 (d, 2H, J = 8.9 Hz), 8.16 (d, 2H, J = 8.9 Hz), 7.86 (d, 1H, J = 15.7 Hz), 7.70-7.64 (m, 2H), 7.49 (d, 1H, J = 15.7 Hz), 7.49-7.43 (m, 3H). DEPTQ (150 MHz, CDCl3), δ (ppm) 189.2, 150.3, 147.0, 143.3, 134.5, 131.4, 129.6, 129.3, 128.9, 124.0, 121.6.
(E)-3-(4-fluorophenyl)-1-(4-nitrophenyl)prop-2-en-1-one (1Dc). This compound was obtained pure enough after filtration to afford a yellow solid (133 mg, 98 %), m.p. 208-210 °C (lit, 210-212 °C) [50]. 1H NMR (600 MHz, CDCl3), δ (ppm) 8.35 (d, 2H, J = 8.9 Hz), 8.13 (d, 2H, J = 8.9 Hz), 7.83 (d, 1H, J = 15.6 Hz), 7.63 (d, 2H, J = 8.7 Hz), 7.36 (d, 1H, J = 15.6 Hz), 6.96 (d, 2H, J = 8.8 Hz), 3.88 (s, 3H). DEPTQ (151 MHz, CDCl3), δ (ppm) 189.0, 165.5, 163.8, 150.3, 145.6, 143.2, 130.9, 129.5, 124.0, 121.2, 116.5.
(E)-3-(4-methoxyphenyl)-1-(4-nitrophenyl)prop-2-en-1-one (1De). This compound was obtained pure enough after filtration to afford a mustard yellow solid (110.5 mg, 78 %), m.p. 182-183 °C (lit, 178-180 °C) [50]. 1H NMR (600 MHz, CDCl3), δ (ppm) 8.35 (d, 2H, J = 8.9 Hz), 8.13 (d, 2H, J = 8.9 Hz), 7.83 (d, 1H, J = 15.6 Hz), 7.63 (d, 2H, J = 8.7 Hz), 7.36 (d, 1H, J = 15.6 Hz), 6.96 (d, 2H, J = 8.8 Hz), 3.88 (s, 3H). DEPTQ (150 MHz, CDCl3), δ (ppm) 189.2, 165.2, 150.2, 146.9, 143.7, 130.8, 129.4, 127.3, 124.0, 119.2, 114.8, 55.64.
(E)-1-(5-chloro-2-hydroxyphenyl)-3-(4-fluorophenyl)-prop-2-en-1-one (1Ec). This compound was obtained pure after filtration to afford a yellow solid (132.8 mg; 96 %), m.p. 158-160 °C, (lit, 158-160 °C) [51]. 1H NMR (600 MHz, CDCl3) δ 12.65 (s, 1H), 7.90 (d, J = 15.4 Hz, 1H), 7.85 (d, J = 2.6 Hz, 1H), 7.71 – 7.64 (m, 3H), 7.48 (d, J = 15.4 Hz, 1H), 7.44 (dd, J = 8.9, 2.5 Hz, 1H), 7.14 (t, J = 8.6 Hz, 2H), 6.99 (d, J = 8.9 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 193.1, 165.0 (d, J = 253.4 Hz), 162.6, 145.7, 136.7, 131.3 (d, J = 8.9 Hz), 131.2 (d, J = 3.6 Hz), 129.3, 124.1, 121.1, 120.8, 119.7 (d, J = 3.1 Hz), 116.9 (d, J = 22.1 Hz).
(E)-1-(5-chloro-2-hydroxyphenyl)-3-(2-methoxyphenyl)-prop-2-en-1-one (1Eg). This compound was purified by column chromatography (hexane: ethyl acetate, 95:5) to afford a yellow solid (119.8 mg, 83%), m.p. 132-134 °C, (lit, m.p. not reported) [52]. 1H NMR (600 MHz, CDCl3) δ 12.78 (s, 1H), 8.18 (d, J = 15.6 Hz, 1H), 7.79 (d, J = 2.6 Hz, 1H), 7.61 (d, J = 15.5 Hz, 2H), 7.39 – 7.32 (m, 2H), 6.95 (t, J = 7.5 Hz, 1H), 6.90 (dd, J = 8.6, 6.4 Hz, 2H), 3.88 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 193.38, 162.05, 159.18, 142.19, 135.94, 132.61, 129.78, 128.93, 123.41, 123.37, 120.88, 120.81, 120.16, 120.04, 111.37, 55.67.
(E)-1-(5-chloro-2-hydroxyphenyl)-3-(3-methoxyphenyl)-prop-2-en-1-one (1Eh). This compound was purified by column chromatography (hexane: ethyl acetate, 95:5) to afford a yellow solid (94.7 mg, 67%), m.p. 96-98 °C (lit, m.p. not reported) [52]. 1H NMR (600 MHz, CDCl3) δ 12.70 (s, 1H), 7.91 (d, J = 15.4 Hz, 1H), 7.87 (d, J = 2.6 Hz, 1H), 7.55 (d, J = 15.4 Hz, 1H), 7.45 (dd, J = 8.9, 2.5 Hz, 1H), 7.37 (t, J = 7.9 Hz, 1H), 7.29 (d, J = 7.6 Hz, 1H), 7.18 (t, J = 2.1 Hz, 1H), 7.02 (dd, J = 5.6, 2.1 Hz, 1H), 7.00 (d, J = 8.9 Hz, 1H), 3.89 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 192.8, 162.1, 160.0, 146.5, 136.2, 135.7, 130.1, 128.8, 123.6, 121.5, 120.6, 120.3, 119.7, 117.0, 113.8, 55.5.
(E)-1-(5-chloro-2-hydroxy-4-methylphenyl)-3-(3-fluorophenyl)-prop-2-en-1-one (1Fb). This compound was purified by column chromatography (hexane: ethyl acetate, 95:5) to afford a yellow solid (120.6 mg, 83%), m.p. 138-140 °C.1H NMR (600 MHz, CDCl3) δ 12.5 (d, J = 1.1 Hz, 1H), 7.8 (d, J = 15.5 Hz, 1H), 7.8 (s, 1H), 7.5 (d, J = 15.4 Hz, 1H), 7.4 (t, J = 1.5 Hz, 1H), 7.4 – 7.3 (m, 1H), 7.3 (dt, J = 9.5, 2.0 Hz, 1H), 7.1 (tt, J = 7.1, 1.6 Hz, 1H), 6.9 (s, 1H), 2.3 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 192.1, 163.1 (d, J = 247.3 Hz), 162.1, 145.9, 144.4 (d, J = 2.8 Hz), 136.7 (d, J = 7.7 Hz), 130.7 (d, J = 8.1 Hz), 129.2, 124.8 (d, J = 3.1 Hz), 124.3, 120.9, 120.7, 118.9, 117.9 (d, J = 21.5 Hz), 114.8 (d, J = 22.0 Hz), 20.9. HRMS (FAB+): M/Z [M+H]+ observed for [C16H12ClFO2]+: 290.0431, estimated: 289.0446.
(E)-1-(5-chloro-2-hydroxy-4-methylphenyl)-3-(4-fluorophenyl)-prop-2-en-1-one (1Fc). This compound was obtained pure after filtration to afford a yellow solid (136.6 mg, 94%), m.p. 194-198 °C, (lit, m.p. not reported) [53,54].1H NMR (600 MHz, ) δ 12.6 (s, 1H), 7.9 (d, J = 15.4 Hz, 1H), 7.9 (s, 1H), 7.7 (dd, J = 8.5, 5.5 Hz, 3H), 7.5 (d, J = 15.4 Hz, 1H), 7.2 (t, J = 8.5 Hz, 2H), 6.9 (s, 1H), 2.4 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 192.7, 164.9 (d, J = 253.2 Hz), 162.5, 146.2, 145.1, 131.2 (d, J = 8.9 Hz), 129.6, 124.7, 121.1, 119.9 (d, J = 2.1 Hz), 119.5, 116.8 (d, J = 22.1 Hz), 21.3.
(E)-1-(5-chloro-2-hydroxy-4-methylphenyl)-3-(4-methoxyphenyl)-prop-2-en-1-one (1Fe). This compound was obtained pure after filtration to afford an orange solid (145.3 mg, 96%), m.p. 110-112 °C, (lit, m.p. not reported) [55]. 1H NMR (600 MHz, CDCl3) δ 12.8 (s, 1H), 7.9 (d, J = 15.3 Hz, 1H), 7.8 (s, 1H), 7.6 (d, J = 8.6 Hz, 1H), 7.4 (d, J = 15.3 Hz, 1H), 7.0 (d, J = 8.8 Hz, 1H), 6.9 (s, 1H), 3.9 (s, 1H), 2.4 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 192.8, 162.7, 162.5, 146.4, 145.7, 131.2, 129.6, 127.8, 124.6, 121.1, 119.6, 117.7, 115.1, 56.0, 21.3.
(E)-1-(5-chloro-2-hydroxy-4-methylphenyl)-3-(2-methoxyphenyl)-prop-2-en-1-one (1Fg). This compound was purified by column chromatography (hexane: ethyl acetate, 95:5) to afford a yellow solid (113.5 mg, 75%), m.p. 178-180 °C, 1H NMR (600 MHz, CDCl3) δ 12.8 (s, 1H), 8.2 (d, J = 15.6 Hz, 1H), 7.9 (s, 1H), 7.7 (ddd, J = 15.5, 7.9, 1.6 Hz, 2H), 7.4 (ddd, J = 8.7, 7.4, 1.7 Hz, 1H), 7.0 (t, J = 8.2 Hz, 1H), 7.0 (d, J = 8.3 Hz, 1H), 6.9 (s, 1H), 3.9 (s, 3H), 2.4 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 193.0, 162.0, 159.1, 145.3, 141.6, 132.4, 129.6, 129.3, 124.1, 123.5, 120.9, 120.5, 120.2, 119.2, 111.3, 55.7, 20.8.
(E)-1-(5-chloro-2-hydroxy-4-methylphenyl)-3-(2-fluorophenyl)-prop-2-en-1-one (1Fi). This compound was purified by column chromatography (hexane: ethyl acetate, 95:5) to afford a yellow solid (81.4 mg, 56%), m.p. 138-140 °C.1H NMR (600 MHz, CDCl3) δ 12.6 (s, 1H), 8.0 (d, J = 15.6 Hz, 1H), 7.8 (s, 1H), 7.7 (ddt, J = 15.6, 7.6, 1.4 Hz, 2H), 7.4 (ddd, J = 8.0, 5.0, 1.8 Hz, 1H), 7.2 (td, J = 7.5, 1.1 Hz, 1H), 7.2 (ddd, J = 10.9, 8.3, 1.1 Hz, 1H), 6.9 (s, 1H), 2.4 (d, J = 0.7 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ 192.4, 162.8 (d, J = 255.6 Hz), 162.0, 145.8, 138.7 (d, J = 2.2 Hz), 132.5 (d, J = 8.9 Hz), 130.1 (d, J = 2.8 Hz), 129.3, 124.7 (d, J = 3.5 Hz), 124.3, 122.7 (d, J = 11.2 Hz), 122.3 (d, J = 7.7 Hz), 120.6, 118.9, 116.4 (d, J = 22.1 Hz), 20.9. HRMS (FAB+): M/Z [M+H]+ observed for [C16H12ClFO2]+: 290.0431, estimated: 289.0446.
(E)-1-(4-fluoro-2-hydroxyphenyl)-3-phenylprop-2-en-1-one (1Ga). This compound was obtained pure after filtration to afford a yellow solid (78.7 mg, 65%), m.p. 110-112 °C, (lit, m.p. not reported) [56]. 1H NMR (600 MHz, CDCl3) δ 13.2 (d, J = 1.5 Hz, 1H), 7.9 (dtd, J = 15.1, 5.9, 3.0 Hz, 1H), 7.7 (dd, J = 6.6, 2.9 Hz, 2H), 7.6 (d, J = 15.5 Hz, 1H), 7.4 (dd, J = 4.8, 1.9 Hz, 3H), 6.7 (dd, J = 10.4, 2.5 Hz, 1H), 6.7 (td, J = 8.5, 2.6 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 192.6, 167.5 (d, J = 257.0 Hz), 166.2 (d, J = 14.4 Hz), 145.8, 134.5, 132.0 (d, J = 11.9 Hz), 131.1, 129.1, 128.7, 119.9, 117.1 (d, J = 2.1 Hz), 107.2 (d, J = 2.1 Hz), 105.2 (d, J = 23.3 Hz).
(E)-1-(4-fluoro-2-hydroxyphenyl)-3-(3-fluorophenyl)-prop-2-en-1-one (1Gb). This compound was obtained pure after filtration to afford a yellow solid (93.7 mg,72%), m.p. 142-144 °C, (lit, m.p. not reported) [57]. 1H NMR (600 MHz, CDCl3) δ 13.1 (d, J = 1.5 Hz, 1H), 7.9 (dd, J = 9.0, 6.3 Hz, 1H), 7.9 (d, J = 15.4 Hz, 1H), 7.6 (d, J = 15.4 Hz, 1H), 7.4 – 7.4 (m, 2H), 7.4 (dq, J = 9.7, 1.4 Hz, 1H), 7.2 – 7.1 (m, 1H), 6.7 (dd, J = 10.3, 2.5 Hz, 1H), 6.7 (ddd, J = 9.0, 8.0, 2.5 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 192.3, 167.6 (d, J = 257.6 Hz), 166.3 (d, J = 14.3 Hz), 163.1 (d, J = 247.2 Hz), 144.2 (d, J = 2.7 Hz), 136.7 (d, J = 7.6 Hz), 132.0 (d, J = 11.8 Hz), 130.7 (d, J = 8.1 Hz), 124.8 (d, J = 2.9 Hz), 121.2, 117.9 (d, J = 21.4 Hz), 117.0 (d, J = 1.9 Hz), 114.7 (d, J = 22.0 Hz), 107.3 (d, J = 23.1 Hz), 105.2 (d, J = 23.6 Hz).
(E)-1-(4-fluoro-2-hydroxyphenyl)-3-(4-methoxyphenyl)-prop-2-en-1-one (1Ge). This compound was obtained pure after filtration to afford a yellow solid (98 mg, 72%), m.p. 148-150 °C, (lit, m.p. not reported) [57]. 1H NMR (600 MHz, CDCl3) δ 13.3 (s, 1H), 7.9 (td, J = 8.5, 2.6 Hz, 1H), 7.9 (d, J = 15.3 Hz, 1H), 7.6 (d, J = 8.7 Hz, 2H), 7.4 (d, J = 15.3 Hz, 1H), 7.0 (d, J = 8.7 Hz, 2H), 6.7 (dd, J = 7.8, 2.6 Hz, 1H), 6.7 (td, J = 8.5, 2.6 Hz, 1H), 3.9 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 192.5, 167.3 (d, J = 256.6 Hz), 166.1(d, J = 14.2 Hz), 162.1, 145.6, 131.8 (d, J = 11.7 Hz), 130.6, 127.2, 117.3, 117.2 (d, J = 2.2 Hz), 114.6, 107.0 (d, J = 22.8 Hz), 105.1 (d, J = 23.4 Hz), 55.5.
(E)-1-(4-fluoro-2-hydroxyphenyl)-3-(2-methoxyphenyl)-prop-2-en-1-one (1Gg). This compound was obtained pure after filtration to afford a yellow solid (117 mg, 86%), m.p. 138-140 °C. 1H NMR (600 MHz, CDCl3) δ 13.3 (d, J = 1.4 Hz, 1H), 8.2 (d, J = 15.6 Hz, 1H), 7.9 (dd, J = 8.9, 6.4 Hz, 1H), 7.7 (d, J = 15.6 Hz, 1H), 7.6 (dd, J = 7.7, 1.7 Hz, 1H), 7.4 (ddd, J = 8.3, 7.4, 1.7 Hz, 1H), 7.0 (td, J = 7.5, 1.0 Hz, 1H), 7.0 (dd, J = 8.3, 1.0 Hz, 1H), 6.7 (dd, J = 10.4, 2.5 Hz, 1H), 6.7 (ddd, J = 8.8, 8.0, 2.6 Hz, 1H), 3.9 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 193.2, 167.3 (d, J =256.5 Hz), 166.2 (d, J = 14.3 Hz), 159.1, 141.5, 132.3, 132.0 (d, J = 12.0 Hz), 129.8, 123.5, 120.8, 120.6, 117.3 (d, J = 2.4 Hz), 111.4, 107.0 (d, J = 22.7 Hz), 106.9, 105.1 (d, J = 23.6 Hz), 55.6. HRMS (FAB+): M/Z [M+H]+ observed for [C16H13FO3]+: 272.2827, estimated: 273.0914.
(E)-1-(4-fluoro-2-hydroxyphenyl)-3-(3-methoxyphenyl)-prop-2-en-1-one (1Gh). This compound was obtained pure after filtration to afford a yellow solid (115.7 mg, 85%), m.p. 128-130 °C, (lit, m.p. not reported) [57]. 1H NMR (600 MHz, CDCl3) δ 13.2 (d, J = 1.3 Hz, 1H), 7.9 (dd, J = 9.0, 6.4 Hz, 1H), 7.9 (d, J = 15.4 Hz, 1H), 7.5 (d, J = 15.4 Hz, 1H), 7.4 (t, J = 7.9 Hz, 1H), 7.3 (d, J = 8.2 Hz, 2H), 7.2 (t, J = 2.4 Hz, 1H), 7.0 (dd, J = 8.3, 2.6 Hz, 1H), 6.7 (dd, J = 10.4, 2.6 Hz, 1H), 6.7 (ddd, J = 9.0, 8.0, 2.5 Hz, 1H), 3.9 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 192.6, 167.5 (d, J = 257.0 Hz), 166.2 (d, J = 14.3 Hz), 160.0, 145.7, 135.8, 132.0 (d, J = 11.7 Hz), 130.1, 121.3, 120.2, 117.1 (d, J = 2.2 Hz), 116.7, 113.8, 107.2 (d, J = 22.7 Hz), 105.2 (d, J = 23.3 Hz), 55.4.
(E)-1-(4-fluoro-2-hydroxyphenyl)-3-(2-fluorophenyl)-prop-2-en-1-one (1Gi). This compound was obtained pure after filtration to afford a solid (97.6 mg, 75%), m.p. 110-112 °C. 1H NMR (600 MHz, CDCl3) δ 13.1 (d, J = 1.4 Hz, 1H), 8.0 (d, J = 15.7 Hz, 1H), 7.9 (dd, J = 8.9, 6.4 Hz, 1H), 7.7 (d, J = 15.7 Hz, 1H), 7.6 (td, J = 7.6, 1.7 Hz, 1H), 7.4 (dddd, J = 8.7, 7.2, 5.1, 1.7 Hz, 1H), 7.2 (td, J = 7.6, 1.1 Hz, 1H), 7.2 (ddd, J = 11.0, 8.3, 1.2 Hz, 1H), 6.7 (dd, J = 10.4, 2.5 Hz, 1H), 6.7 (ddd, J = 8.8, 8.0, 2.6 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 192.7, 167.6 (d, J = 257.1 Hz), 166.3 (d, J = 14.4 Hz), 161.9 (d, J = 255.3 Hz), 138.6 (d, J = 1.7 Hz), 132.4 (d, J = 8.9 Hz), 132.1 (d, J = 11.8 Hz), 130.3 (d, J = 2.8 Hz), 124.7 (d, J = 3.6 Hz), 122.7 (d, J = 8.2 Hz), 122.6 (d, J = 11.3 Hz), 117.1 (d, J = 2.1 Hz), 116.5 (d, J = 22.1 Hz), 107.2 (d, J = 22.7 Hz), 105.2 (d, J = 23.6 Hz). HRMS (FAB+): M/Z [M+H]+ observed for [C15H10F2O2]+: 260.2427, estimated: 261.0730.
(E)-1-(5-fluoro-2-hydroxyphenyl)-3-(4-fluorophenyl)-prop-2-en-1-one (1Hc). This compound was obtained pure after filtration to afford a yellow solid (123.6 mg, 95%), m.p. 184-186 °C, (lit, m.p not reported) [58]. 1H NMR (600 MHz, CDCl3) δ 12.5 (s, 1H), 7.9 (d, J = 15.4 Hz, 1H), 7.7 (dd, J = 8.7, 5.4 Hz, 2H), 7.6 (dd, J = 9.0, 3.0 Hz, 1H), 7.5 (d, J = 15.4 Hz, 1H), 7.3 (ddd, J = 8.2, 3.2, 1.4 Hz, 2H), 7.1 (t, J = 8.6 Hz, 2H), 7.0 (dd, J = 9.1, 4.6 Hz, 1H); (151 MHz, CDCl3) δ 192.7 (d, J = 2.7 Hz), 164.5 (d, J = 253.2 Hz), 159.8, 154.9 (d, J = 238.4 Hz), 145.0, 130.8 (d, J = 8.5 Hz), 130.6 (d, J = 3.3 Hz), 124.0 (d, J = 23.4 Hz), 119.9 (d, J = 7.3 Hz), 119.4 (d, J = 6.0 Hz), 119.3 (d, J = 2.3 Hz), 116.4 (d, J = 22.0 Hz), 114.5 (d, J = 23.3 Hz).
(E)-1-(5-fluoro-2-hydroxyphenyl)-3-(4-methoxyphenyl)-prop-2-en-1-one (1He). This compound was purified by column chromatography (hexane: ethyl acetate, 95:5) to afford a yellow solid 115.7 mg, 85%), m.p. 126-128 °C, (lit, m.p. not reported) [57]. 1H NMR (600 MHz, CDCl3) δ 12.7 (s, 1H), 7.9 (d, J = 15.3 Hz, 1H), 7.6 (d, J = 8.8 Hz, 2H), 7.6 (dd, J = 9.1, 3.1 Hz, 1H), 7.4 (d, J = 15.4 Hz, 1H), 7.2 (ddd, J = 9.1, 7.7, 3.1 Hz, 1H), 7.0 (ddd, J = 9.2, 7.5, 4.6 Hz, 1H), 7.0 (d, J = 8.7 Hz, 2H), 3.9 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 192.8, 162.3, 159.7 (d, J = 1.4 Hz), 154.9 (d, J = 238.2 Hz), 146.3, 130.8, 127.1, 123.6 (d, J = 23.7 Hz), 119.8 (d, J = 7.2 Hz), 119.7 (d, J = 6.1 Hz), 117.0, 114.6, 114.5 (d, J = 23.5 Hz), 55.5.
(E)-1-(furan-2-yl)-3-phenylprop-2-en-1-one (1Ia). This compound was obtained pure enough after filtration to afford a white solid (80.3 mg, 81%), m.p. 80 °C (lit, m.p. not reported) [59]. 1H NMR (600 MHz, CDCl3), δ (ppm) 8.28 (d, 2H, J = 8.8 Hz), 8.03 (d, 2H, J = 8.5 Hz), 7.82 (d, 1H, J = 15.8 Hz), 7.79 (d, 2H, J = 8.6 Hz), 7.64 (d, 1H, J = 15.7 Hz), 7.63 (t, 1H, J = 8.5 Hz), 7.54 (t, 2H, J = 7.7 Hz). 13C NMR (150 MHz, CDCl3), δ (ppm) 178.2, 153.9, 146.6, 144.2, 135.0, 130.8, 129.1, 128.7, 121.4, 117.6, 112.7.
(E)-3-phenyl-1-(pyridin-2-yl)prop-2-en-1-one (1Ja). This compound was obtained pure enough after filtration to afford a white solid (89.4 mg, 85%), m.p. 70-72 °C (lit, 72-74 °C) [60]. 1H NMR (600 MHz, CDCl3), δ (ppm) 8.74 (d, 1H, J = 4.7 Hz), 8.30 (d, 1H, J = 16.0 Hz), 8.18 (d, 1H, J = 7.8 Hz), 7.94 (d, 1H, J = 16.0 Hz), 7.86 (td, 1H, J = 7.7, 1.6 Hz), 7.76-7.70 (m, 2H), 7.50-7.44 (m, 1H), 7.43-7.37(m, 3H). 13C NMR (150 MHz, CDCl3), δ (ppm) 189.5, 154.2, 148.9, 144.8, 137.0, 135.2, 130.6, 128.9 (2C), 126.9, 123.0, 120.9.
4. Conclusions
The Claisen-Schmidt condensation can be performed in aqueous media, under basic conditions. Under the conditions outlined in this study, a substantial number of compounds can be prepared with yields ranging from 40% to 98%, independent of the electronic nature of the substituents present in the starting materials. The yield was found to be predominantly influenced by the hydrophilicity/lipophilicity of the starting materials and their physical state. Consequently, this methodology is not applicable when both starting materials are lipophilic and water insoluble. The results obtained herein demonstrate that water can and should replace volatile and contaminant organic solvents for the preparation of a large quantity of chalcones. Developed methodology fulfills at least six of the twelve principles of green chemistry: a) prevention, b) less hazardous chemical synthesis, c) designing safer chemicals, d) safer solvents and auxiliaries and e) design for energy efficiency and catalysis. These principles are consistent with Goal n.12 of the 2030 Agenda for Sustainable Development, which aims to ensure sustainable consumption and production patterns by reducing chemicals and waste, and by promoting sustainable consumption and production.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Author Contributions
Quirino Torres Sauret: data curation, investigation, methodology, writing-original-draft. Daisy Gabriela Murillo Yanes: data curation, investigation, methodology. Luis Fernando Roa de la Fuente: data curation, formal analysis, supervision, validation. Roberto Martínez: conceptualization, data curation, formal analysis, investigation, methodology, supervision, validation. Jorge R. Juárez Posadas: data curation, formal analysis, supervision, validation. Manuel Velasco Ximello: conceptualization, data curation, formal analysis, investigation, methodology, supervision, validation. Erika M. Ramos Rivera: data curation, formal analysis, investigation, validation. Oswaldo I. Hernández Abreu: conceptualization, data curation, formal analysis, investigation, methodology, supervision, validation, resources. Rosalía Torralba: data curation, formal analysis, investigation. Roxana Martínez-Pascual: data curation, formal analysis, investigation. Omar Santiago Sosa: data curation, formal analysis, investigation. Cuauhtémoc Alvarado Sánchez: conceptualization, data curation, funding acquisition, project administration, resources, supervision, validation, writing-reviewing & editing, investigation, writing original draft.
Funding
This work was supported by the Secretaría de Ciencias, Humanidades, Tecnología e Inovación (SECIHTI, México), under grant number CF-2023-I-1700. E. M. thanks SECIHTI (México), for scholarship number 447166.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article and Supplementary Materials. Further inquiries can be directed to the corresponding author.
Acknowledgments
The authors thank SECIHTI for financial support (CF-2023-I-1700). E. M. thanks SECIHTI (México), for scholarship (447166). The authors acknowledge Chantal Seoane for editing the English language text.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Miranda, C. L.; Maier, C. S.; Stevens, J. F. Flavonoids. In eLS; John Wiley & Sons, Ltd, 2012. [CrossRef]
- Rosa, G. P.; Seca, A. M. L.; Barreto, M. D. C.; Silva, A. M. S.; Pinto, D. C. G. A. Chalcones and Flavanones Bearing Hydroxyl and/or Methoxyl Groups: Synthesis and Biological Assessments. Appl. Sci. 2019, 9, 2846. [Google Scholar] [CrossRef]
- Ninomiya, M.; Koketsu, M. Minor Flavonoids (Chalcones, Flavanones, Dihydrochalcones, and Aurones). In Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes; Ramawat, K. G., Mérillon, J.-M., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2013. [Google Scholar] [CrossRef]
- Elkanzi, N. A. A.; Hrichi, H.; Alolayan, R. A.; Derafa, W.; Zahou, F. M.; Bakr, R. B. Synthesis of Chalcones Derivatives and Their Biological Activities: A Review. ACS Omega 2022, 7, 27769–27786. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, H.; Santos, C.; Cavaleiro, J.; Silva, A. Chalcones as Versatile Synthons for the Synthesis of 5- and 6-Membered Nitrogen Heterocycles. Curr. Org. Chem. 2014, 18, 2750–2775. [Google Scholar] [CrossRef]
- Salehi, B.; Quispe, C.; Chamkhi, I.; El Omari, N.; Balahbib, A.; Sharifi-Rad, J.; Bouyahya, A.; Akram, M.; Iqbal, M.; Docea, A. O.; Caruntu, C.; Leyva-Gómez, G.; Dey, A.; Martorell, M.; Calina, D.; López, V.; Les, F. Pharmacological Properties of Chalcones: A Review of Preclinical Including Molecular Mechanisms and Clinical Evidence. Front. Pharmacol. 2021, 11, 592654. [Google Scholar] [CrossRef]
- Mezgebe, K.; Melaku, Y.; Mulugeta, E. Synthesis and Pharmacological Activities of Chalcone and Its Derivatives Bearing N -Heterocyclic Scaffolds: A Review. ACS Omega 2023, 8, 19194–19211. [Google Scholar] [CrossRef]
- Aoki, N.; Muko, M.; Ohta, E.; Ohta, S. C-Geranylated Chalcones from the Stems of Angelica Keiskei with Superoxide-Scavenging Activity. J. Nat. Prod. 2008, 71, 1308–1310. [Google Scholar] [CrossRef] [PubMed]
- Nasir Abbas Bukhari, S.; Jasamai, M.; Jantan, I.; Ahmad, W. Review of Methods and Various Catalysts Used for Chalcone Synthesis. Mini-Rev. Org. Chem. 2013, 10, 73–83. [Google Scholar] [CrossRef]
- Donaire-Arias, A.; Poulsen, M. L.; Ramón-Costa, J.; Montagut, A. M.; Estrada-Tejedor, R.; Borrell, J. I. An Improved High-Yield and Substituent-Independent Protocol for an Old Structure. Molecules 2023, 28, 7576. [Google Scholar] [CrossRef]
- Radwan, M. A. A.; Alshubramy, M. A.; Abdel-Motaal, M.; Hemdan, B. A.; El-Kady, D. S. Synthesis, Molecular Docking and Antimicrobial Activity of New Fused Pyrimidine and Pyridine Derivatives. Bioorganic Chem. 2020, 96, 103516. [Google Scholar] [CrossRef]
- Torres-Sauret, Q.; Vilchis-Reyes, M. A.; Martínez, R.; Romero-Ceronio, N.; Alarcon-Matus, E.; Hernández-Abreu, O.; Vázquez Cancino, R.; Alvarado Sánchez. , C. Crossing Borders: On-Water Synthesis of Flavanones. ChemistrySelect 2022, 7, e202202567. [Google Scholar] [CrossRef]
- El-Hashash, M. A. E-A.; Gohma, S. M.; El-Arab, E. Z. Utility of Pyrazolylchalcone Synthon to Synthesize Azolopyrimidines under Grindstone Technology. Chem Pharm Bull (Tokyo). 2017, 65, 90–96. [Google Scholar] [CrossRef]
- Gomha, S. M.; Abdallah, M. A.; Abbas, I. M.; Kazem, M. S. H. Synthesis, Cytotoxicity Evaluation, Molecular Docking and Utility of Novel Chalcones as Precursors for Heterocycles Incorporating Pyrazole Moiety. Med. Chem. Shariqah United Arab Emir. 2018, 14, 344–355. [Google Scholar] [CrossRef]
- Janković, T.; Turković, N.; Kotur-Stevuljević, J.; Vujić, Z.; Ivković, B. Differences in Antioxidant Potential of Chalcones in Human Serum: In Vitro Study. Chem. Biol. Interact. 2020, 324, 109084. [Google Scholar] [CrossRef]
- Bale, A. T.; Salar, U.; Khan, K. M.; Chigurupati, S.; Fasina, T.; Ali, F.; Ali, M.; Nanda, S. S.; Taha, M.; Perveen, S. Chalcones and Bis-Chalcones Analogs as DPPH and ABTS Radical Scavengers. Lett. Drug Des. Discov. 2021, 18, 249–257. [Google Scholar] [CrossRef]
- Mahapatra, D. K.; Bharti, S. K.; Asati, V. Chalcone Scaffolds as Anti-Infective Agents: Structural and Molecular Target Perspectives. Eur. J. Med. Chem. 2015, 101, 496–524. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yoshimura, M.; Yamaguchi, F.; Kouchi, T.; Tsuji, R.; Saito, M.; Obata, A.; Kikuchi, M. Anti-Allergic Activity of Naringenin Chalcone from a Tomato Skin Extract. Biosci. Biotechnol. Biochem. 2004, 68, 1706–1711. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, C.; Shinoda, K.; Yoshimura, M.; Watanabe, Y.; Obata, A.; Nakayama, T. Naringenin Chalcone Suppresses Allergic Asthma by Inhibiting the Type-2 Function of CD4 T Cells. Allergol. Int. 2010, 59, 67–73. [Google Scholar] [CrossRef]
- Ouyang, Y.; Li, J.; Chen, X.; Fu, X.; Sun, S.; Wu, Q. Chalcone Derivatives: Role in Anticancer Therapy. Biomolecules 2021, 11, 894. [Google Scholar] [CrossRef]
- Syam, S.; Abdelwahab, S. I.; Al-Mamary, M. A.; Mohan, S. Synthesis of Chalcones with Anticancer Activities. Molecules 2012, 17, 6179–6195. [Google Scholar] [CrossRef] [PubMed]
- Okolo, E. N.; Ugwu, D. I.; Ezema, B. E.; Ndefo, J. C.; Eze, F. U.; Ezema, C. G.; Ezugwu, J. A.; Ujam, O. T. New Chalcone Derivatives as Potential Antimicrobial and Antioxidant Agent. Sci. Rep. 2021, 11, 21781. [Google Scholar] [CrossRef]
- Henry, E. J.; Bird, S. J.; Gowland, P.; Collins, M.; Cassella, J. P. Ferrocenyl Chalcone Derivatives as Possible Antimicrobial Agents. J. Antibiot. (Tokyo) 2020, 73, 299–308. [Google Scholar] [CrossRef]
- M. Gomha, S.; M. Riyadh, S.; M. Abdalla, M. Solvent-Drop Grinding Method: Efficient Synthesis, DPPH Radical Scavenging and Anti-Diabetic Activities of Chalcones, Bis-Chalcones, Azolines, and Bis-Azolines. Curr. Org. Synth. 2015, 12, 220–228. [Google Scholar] [CrossRef]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Farooq, S.; Ngaini, Z. Recent Synthetic Methodologies for Chalcone Synthesis (2013-2018). Curr. Organocatalysis 2019, 6, 184–192. [Google Scholar] [CrossRef]
- Rammohan, A.; Reddy, J. S.; Sravya, G.; Rao, C. N.; Zyryanov, G. V. Chalcone Synthesis, Properties and Medicinal Applications: A Review. Environ. Chem. Lett. 2020, 18, 433–458. [Google Scholar] [CrossRef]
- Stantliff, T. M.; Hill, A.; Kuo, M. E.; Neal, H. E.; Harrod, T. C.; Goens, K.; Mashuta, M.; Christianson, A. M.; Krzysiak, A. J. Flexibility in the Bridge of Chalcone Derivatives Is Important for the Inhibition of Cellular Growth. Bioorg. Med. Chem. Lett. 2023, 95, 129467. [Google Scholar] [CrossRef] [PubMed]
- Iftikhar, S.; Khan, S.; Bilal, A.; Manzoor, S.; Abdullah, M.; Emwas, A.-H.; Sioud, S.; Gao, X.; Chotana, G. A.; Faisal, A.; Saleem, R. S. Z. Synthesis and Evaluation of Modified Chalcone Based P53 Stabilizing Agents. Bioorg. Med. Chem. Lett. 2017, 27, 4101–4106. [Google Scholar] [CrossRef]
- Neha, K.; Wakode, S. Contemporary Advances of Cyclic Molecules Proposed for Inflammation. Eur. J. Med. Chem. 2021, 221, 113493. [Google Scholar] [CrossRef]
- Eucerin: Nuestras investigaciones| Base de Datos de Ingredientes. https://www.eucerin.es/nuestras-investigaciones/base-de-datos-de-ingredientes/licochalcone-a (accessed 2024-08-20).
- Mei-Ing Chung; Mei-Hsun Lai; Ming-Hong Yen; Ru-Rong Wu; Chun-Nan Lin. Phenolics from Hypericum Geminiflorum. Phytochemistry 1997, 44, 943–947. [Google Scholar] [CrossRef]
- Marotta, L.; Rossi, S.; Ibba, R.; Brogi, S.; Calderone, V.; Butini, S.; Campiani, G.; Gemma, S. The Green Chemistry of Chalcones: Valuable Sources of Privileged Core Structures for Drug Discovery. Front. Chem. 2022, 10. [Google Scholar] [CrossRef]
- Kitanosono, T.; Kobayashi, S. Reactions in Water Involving the “On-Water” Mechanism. Chem. – Eur. J. 2020, 26, 9408–9429. [Google Scholar] [CrossRef]
- Shaik, A. B.; Bhandare, R. R.; Nissankararao, S.; Edis, Z.; Tangirala, N. R.; Shahanaaz, S.; Rahman, M. M. Design, Facile Synthesis and Characterization of Dichloro Substituted Chalcones and Dihydropyrazole Derivatives for Their Antifungal, Antitubercular and Antiproliferative Activities. Molecules 2020, 25, 3188. [Google Scholar] [CrossRef] [PubMed]
- Tanemura, K.; Rohand, T. Activated Charcoal-Mediated Synthesis of Chalcones Catalyzed by NaOH in Water. Tetrahedron Lett. 2021, 71, 152918. [Google Scholar] [CrossRef]
- Shakil, N. A.; Singh, M. K.; Kumar, J.; Sathiyendiran, M.; Kumar, G.; Singh, M. K.; Pandey, R. P.; Pandey, A.; Parmar, V. S. Microwave Synthesis and Antifungal Evaluations of Some Chalcones and Their Derived Diaryl-Cyclohexenones. J. Environ. Sci. Health Part B 2010, 45, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Kitawat, B. S.; Singh, M.; Kale, R. K. Robust Cationic Quaternary Ammonium Surfactant-Catalyzed Condensation Reaction for ( E )-3-Aryl-1-(3-Alkyl-2-Pyrazinyl)-2-Propenone Synthesis in Water at Room Temperature. ACS Sustain. Chem. Eng. 2013, 1, 1040–1044. [Google Scholar] [CrossRef]
- Dasari, G. K.; Sunkara, S.; Gadupudi, P. C. R. Green and Ecofriendly Synthesis of Indole-condensed Benzimidazole Chalcones in Water and Their Antimicrobial Evaluations. J. Heterocycl. Chem. 2020, 57, 1201–1210. [Google Scholar] [CrossRef]
- Armenise, N.; Malferrari, D.; Ricciardulli, S.; Galletti, P.; Tagliavini, E. Multicomponent Cascade Synthesis of Biaryl-Based Chalcones in Pure Water and in an Aqueous Micellar Environment. Eur. J. Org. Chem. 2016, 2016, 3177–3185. [Google Scholar] [CrossRef]
- Shen, Y. R.; Ostroverkhov, V. Sum-Frequency Vibrational Spectroscopy on Water Interfaces: Polar Orientation of Water Molecules at Interfaces. Chem. Rev. 2006, 106, 1140–1154. [Google Scholar] [CrossRef]
- Cortes-Clerget, M.; Yu, J.; Kincaid, J. R. A.; Walde, P.; Gallou, F.; Lipshutz, B. H. Water as the Reaction Medium in Organic Chemistry: From Our Worst Enemy to Our Best Friend. Chem. Sci. 2021, 12, 4237–4266. [Google Scholar] [CrossRef]
- Antonio-Arias, J. E.; Díaz-Oliva, V. del C.; Romero-Ceronio, N.; Gómez-Rivera, A.; Aguilar-Mariscal, H.; Fuente, L. F. R. la; Lobato-García, C. E. Monomodal vs Multimodal Microwave Irradiation Applied in the Synthesis of Fluorochalcones. American Journal of Organic Chemistry 2018, 8, 8–12. [Google Scholar]
- Masmoudi, N.; Chtourou, M. Green and Ultrasound-Assisted Synthesis of 1,3-Diaryl-2-Propenones Catalyzed by Amberlyte IRA-410 and Amberlyte IRA-400 Basic Resins. Lett. Org. Chem. 2023, 20, 362–369. [Google Scholar] [CrossRef]
- Tang, L.; Gao, Y.; Chen, J.; Yang, L.; Xiao, B.; Shen, G.; Ouyang, Y.; Han, W. Ligand-Free Palladium-Catalyzed Substoichiometric Base Mediated Carbonylation of Aryl Iodides with Alkenylboronic Acids under Ambient Conditions. Synlett 2023, 34, 1280–1284. [Google Scholar] [CrossRef]
- Karaman, İ.; Gezegen, H.; Gürdere, M. B.; Dingil, A.; Ceylan, M. Screening of Biological Activities of a Series of Chalcone Derivatives against Human Pathogenic Microorganisms. Chem. Biodivers. 2010, 7, 400–408. [Google Scholar] [CrossRef]
- Zhao, P.-L.; Liu, C.-L.; Huang, W.; Wang, Y.-Z.; Yang, G.-F. Synthesis and Fungicidal Evaluation of Novel Chalcone-Based Strobilurin Analogues. J. Agric. Food Chem. 2007, 55, 5697–5700. [Google Scholar] [CrossRef]
- Chintakrindi, A. S.; Gohil, D. J.; Kothari, S. T.; Chowdhary, A. S.; Kanyalkar, M. A. Design, Synthesis and Evaluation of Chalcones as H1N1 Neuraminidase Inhibitors. Med. Chem. Res. 2018, 27, 1013–1025. [Google Scholar] [CrossRef]
- Jacob, K. C.; Jadhav, G. V.; Vakharia, M. N. Synthesis of 2-Hydroxy-4-Methyl-5-Chlorochalcones and Their Derivatives. Pesticides 1972, 6, 94–96. [Google Scholar]
- Qiu, X. Y.; Li, S. Z.; Shi, A. R.; Yue, Q. L. Synthesis, Characterized and Biological Activities of Chalcone Derivatives. Adv. Mater. Res. 2012, 535–537, 2540–2543. [Google Scholar] [CrossRef]
- Balaji, S.; Manikandan, V.; Senbagam, R.; Vijayakumar, R.; Rajarajan, M.; Vanangamudi, G.; Thirunarayanan, G. Synthesis, Evaluation of Antimicrobial Activities of some (e)-1-(5-chloro- 2-hydroxyphenyl)-3-phenylprop-2-en-1-one compounds. Med. Sci. 2016, 5. [Google Scholar]
- Ammaji, S.; Masthanamma, S.; Bhandare, R. R.; Annadurai, S.; Shaik, A. B. Antitubercular and Antioxidant Activities of Hydroxy and Chloro Substituted Chalcone Analogues: Synthesis, Biological and Computational Studies. Arab. J. Chem. 2022, 15. [Google Scholar] [CrossRef]
- Dofe, V. S.; Sarkate, A. P.; Kathwate, S. H.; Gill, C. H. Synthesis, Antimicrobial Activity and Anti-Biofilm Activity of Novel Tetrazole Derivatives. Heterocycl. Commun. 2017, 23, 325–330. [Google Scholar] [CrossRef]
- Dofe, V. S.; Sarkate, A. P.; Lokwani, D. K.; Shinde, D. B.; Kathwate, S. H.; Gill, C. H. Novel O-Alkylated Chromones as Antimicrobial Agents: Ultrasound Mediated Synthesis, Molecular Docking and ADME Prediction. J. Heterocycl. Chem. 2017, 54, 2678–2685. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Vijeender, K.; Reddy, K. V. New Synthesis of Flavanones Catalyzed by L-Proline. Tetrahedron Lett. 2005, 46, 6991–6993. [Google Scholar] [CrossRef]
- Devia, A. C.; Ferretti, F. H.; Ponce, C. A.; Tomás, F. Conformational Equilibrium and Intramolecular Hydrogen Bond of 4′X and 4X Substituted 2′(OH)Chalcones. J. Mol. Struct. THEOCHEM 1999, 493, 187–197. [Google Scholar] [CrossRef]
- Muller, B. M.; Mai, J.; Yocum, R. A.; Adler, M. J. Impact of Mono- and Disubstitution on the Colorimetric Dynamic Covalent Switching Chalcone/Flavanone Scaffold. Org. Biomol. Chem. 2014, 12, 5108–5114. [Google Scholar] [CrossRef] [PubMed]
- Jesus, A. R.; Marques, A. P.; Rauter, A. P. An Easy Approach to Dihydrochalcones via Chalcone in Situ Hydrogenation. Pure Appl. Chem. 2016, 88, 349–361. [Google Scholar] [CrossRef]
- De, R.; Savarimuthu, A.; Ballav, T.; Singh, P.; Nanda, J.; Hasija, A.; Chopra, D.; Bera, M. K. DBU-Catalyzed Rearrangement of Secondary Propargylic Alcohols: An Efficient and Cost-Effective Route to Chalcone Derivatives. Synlett 2020, 31, 1587–1592. [Google Scholar] [CrossRef]
- Ciupa, A.; Mahon, M. F.; Bank, P. A. D.; Caggiano, L. Simple Pyrazoline and Pyrazole “Turn on” Fluorescent Sensors Selective for Cd2+ and Zn2+ in MeCN. Org. Biomol. Chem. 2012, 10, 8753–8757. [Google Scholar] [CrossRef]
Figure 1.
Chalcones with biological activity. Anti-inflammatory [7,30,31], cytotoxic [32], and anticancer [28,29].
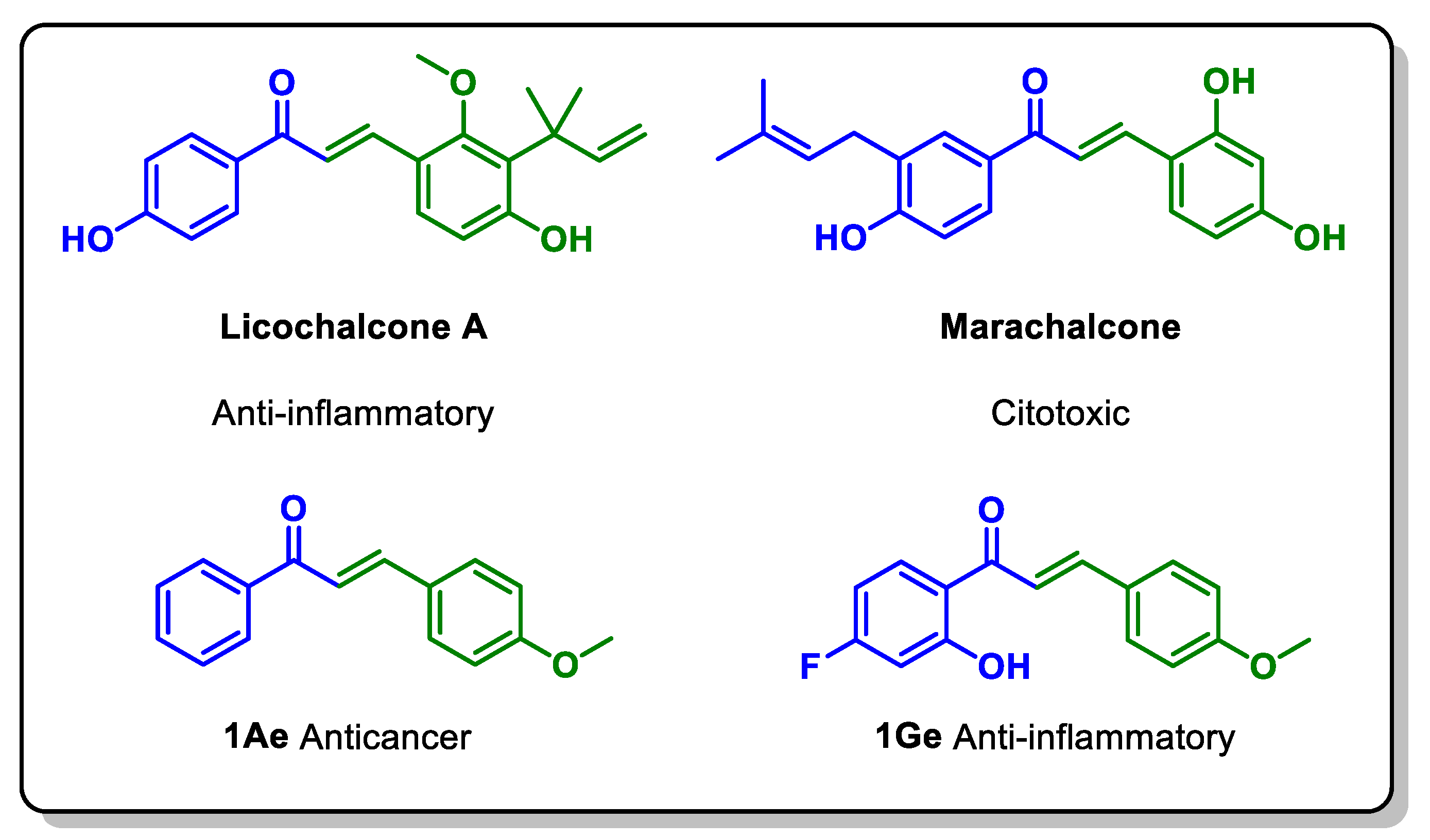
Figure 3.
Mechanism proposal.
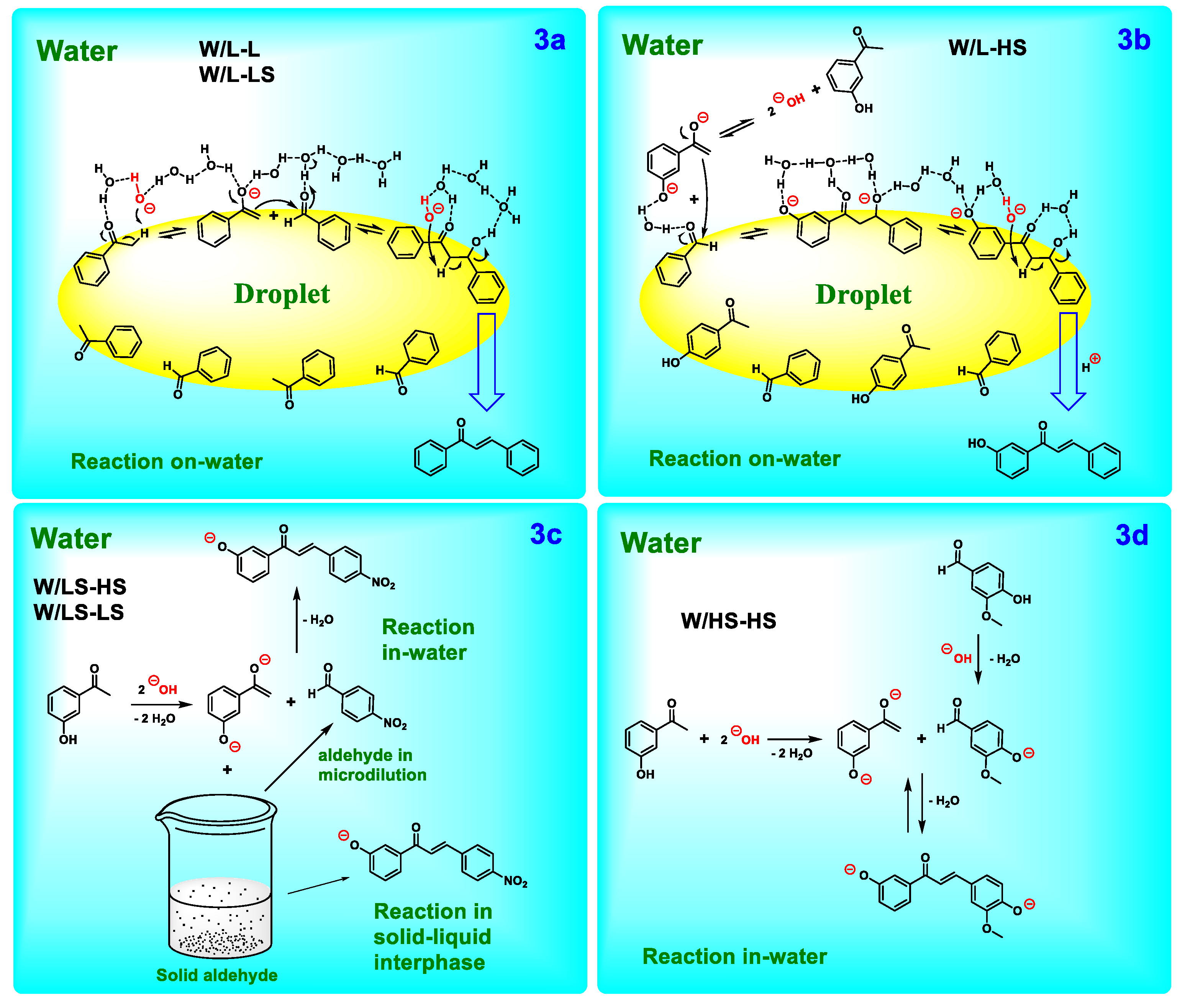

Table 1.
Screening for the aqueous synthesis of chalcones.
 | ||||
| Entry | Base | Base (eq) | Time (h) | Yield (%) |
| 1 | KOH | 1 | 4 | ND |
| 2 1 | KOH | 1 | 4 | 88 |
| 3 | Na2CO3 | 2 | 7 | 0 |
| 4 | K2CO3 | 2 | 7 | 0 |
| 5 | KOH | 2 | 2 | 88 |
| 6 | KOH | 3 | 2 | 88 |
| 7 | NaOH | 2 | 2 | 88 |
| 8 | KOH/piperidine | 2/0.1 M | 2 | 85 |
| 9 2 | KOH | 2 | 1 | - |
Reaction was performed at 0.5 mmol scale with 2 mL of water. Stirring was adjusted at 850 rpm, temperature 25-40 °C. 1 1 mL of water, 2 reactions performed at 60 °C. Not Determined (ND).
Table 2.
Scope for the aqueous synthesis of chalcones.
 | |||
| Entry | Ar | R |
Chalcone Yield (%) |
| 1 | 1A Ph | 1a H | 1Aa (88) |
| 2 | 1b 3ʹ-F | 1Ab (81) | |
| 3 | 1c 4ʹ-F | 1Ac (88) | |
| 4 | 1d 4ʹ-NO2 | 1Ad (82) | |
| 5 | 1e 4ʹ-OMe | 1Ae (78) | |
| 6 |
1B 4-Cl-Ph |
1a H | 1Ba (86) |
| 7 | 1b 3ʹ-F | 1Bb (81) | |
| 8 | 1c 4ʹ-F | 1Bc (80) | |
| 9 | 1d 4ʹ-NO2 | 1Bd (66) | |
| 10 | 1e 4ʹ-OMe | 1Be (79) | |
| 11 |
1C 3-OH-Ph |
1a H | 1Ca (76) |
| 12 | 1b 3ʹ-F | 1Cb (78) | |
| 13 | 1c 4ʹ-F | 1Cc (83) | |
| 14 | 1d 4ʹ-NO2 | 1Cd (77) | |
| 15 | 1e 4ʹ-OMe | 1Ce (80) | |
| 16 | 1f 3ʹ-OMe,4ʹ-OH | 1Cf (68) | |
| 17 |
1D 4-NO2-Ph |
1a H | 1Da (93) |
| 18 | 1c 4ʹ-F | 1Dc (98) | |
| 19 | 1d 4ʹ-NO2 | 1Dd (40) | |
| 20 | 1e 4ʹ-OMe | 1De (78) | |
| 21 |
1E 2-OH-5-Cl-Ph |
1c 4ʹ-F | 1Ec (96) |
| 22 | 1g 2ʹ-OMe | 1Eg (83) | |
| 23 | 1h 3ʹ-OMe | 1Eh (67) | |
| 24 |
1F 2-OH-4-Me-5-Cl-Ph |
1b 3ʹ-F | 1Fb (83) |
| 25 | 1c 4ʹ-F | 1Fc (94) | |
| 26 | 1e 4ʹ-OMe | 1Fe (96) | |
| 27 | 1g 2ʹ-OMe | 1Fg (75) | |
| 28 | 1i 2ʹ-F | 1Fi (56) | |
| 29 |
1G 2-OH-4-F-Ph |
1a H | 1Ga (65) |
| 30 | 1b 3ʹ-F | 1Gb (72) | |
| 31 | 1e 4ʹ-OMe | 1Ge (72) | |
| 32 | 1g 2ʹ-OMe | 1Gg (86) | |
| 33 | 1h 3ʹ-OMe | 1Gh (85) | |
| 34 | 1i 2ʹ-F | 1Gi (75) | |
| 35 | 1H 2-OH-5-F-Ph | 1c 4ʹ-F | 1Hc (95) |
| 36 | 1e 4ʹ-OMe | 1He (85) | |
| 37 | 1I Furane | 1a H | 1Ia (81) |
| 38 | 1J Pyridine | 1a H | 1Ja (85) |
| 39 1 | 1A | 1a H | 1Aa (87) |
Reaction was performed at 0.5 mmol scale with 2 mL of water. Stirring was adjusted at 850 rpm, temperature 25-40 °C, time of 48 h. 1 50 mmol scale reaction.
Table 3.
W/SM-SM reaction systems.
| Ketone | Aldehyde | System | ||
| Liquid (L) |
1A,1B,1G,1I | L | 1a-1c, 1e, 1g,1h | W/L-L |
| LS | 1d | W/L-LS | ||
| Lipophilic Solid (LS) |
1D |
L |
1a-1c, 1e, 1g,1h | W/L-LS |
| LS | 1d | W/LS-LS | ||
| Hydrophilic Solid (HS) |
1C, 1E-1H |
L |
1a-1c, 1e, 1g,1h | W/L-HS |
| LS | 1d | W/HS-LS | ||
| HS | 1f | W/HS-HS | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



